
12 August 2015 - Case of the Week #361
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. A. Eftekhar Javadi, Tehran University of Medical Sciences (Iran), for contributing this case and Jennifer R. Kaley, M.D., University of Arkansas for Medical Sciences for contributing the discussion.
Advertisement
Case of the Week #361
Clinical history:
A 17 year old girl presented with a seizure. She had myoclonus in her extremities which was progressive and now has generalized myoclonus epilepsy. She had declined in intellectual function and is now intellectually disabled. No genetic testing was done.
A skin biopsy from the axilla was taken.
Microscopic (histologic) images :

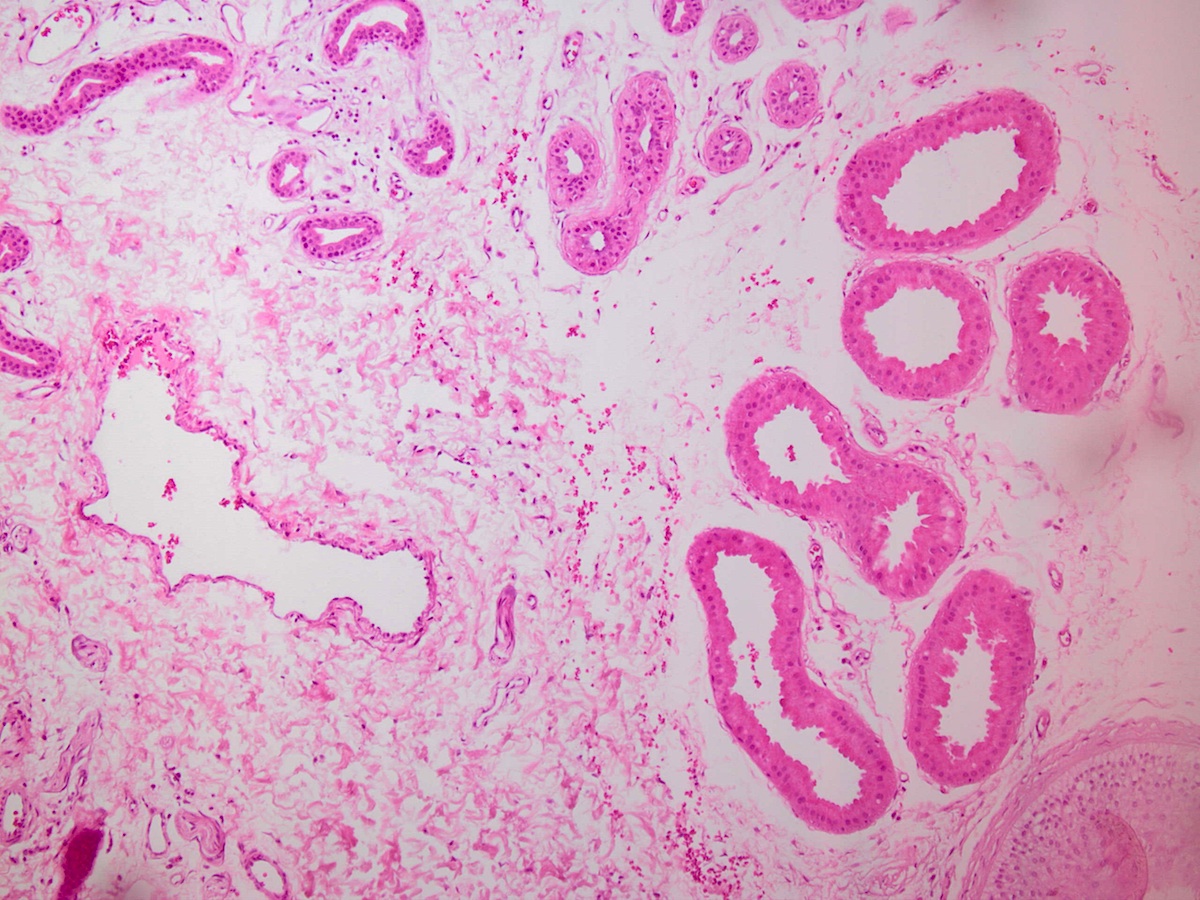

What is your diagnosis?
Diagnosis: Lafora body disease
Special stains:
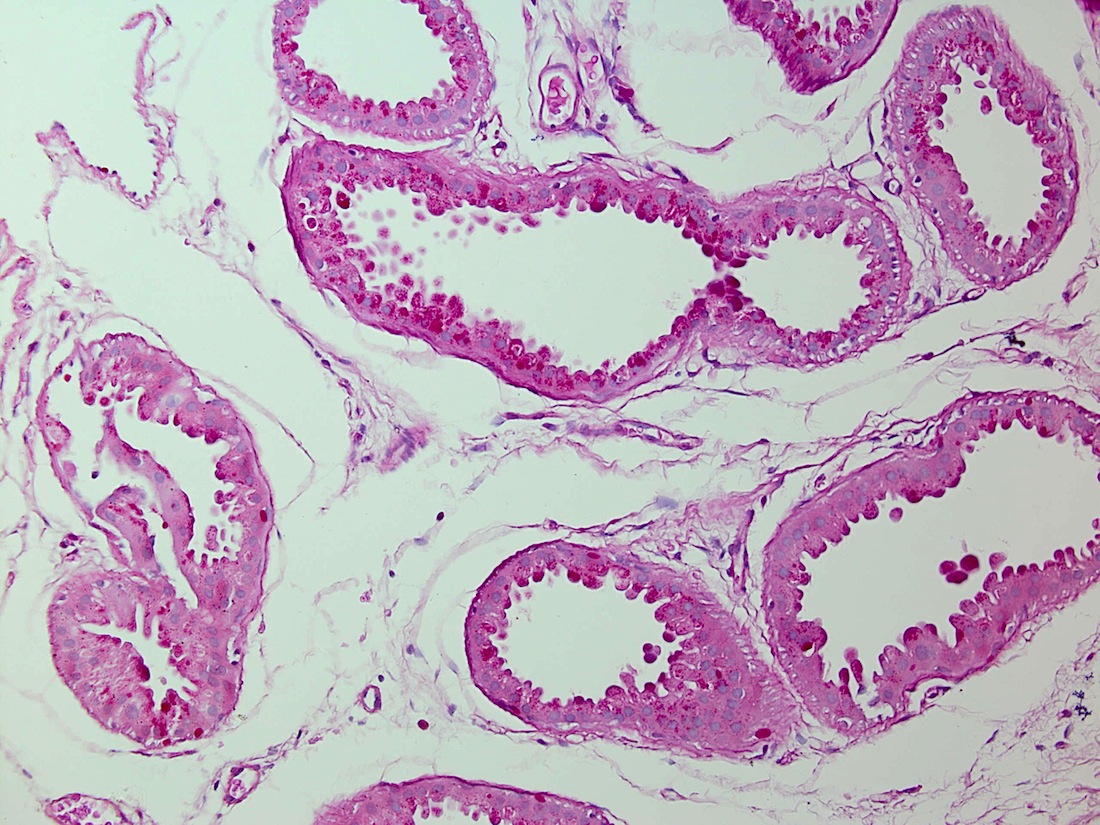
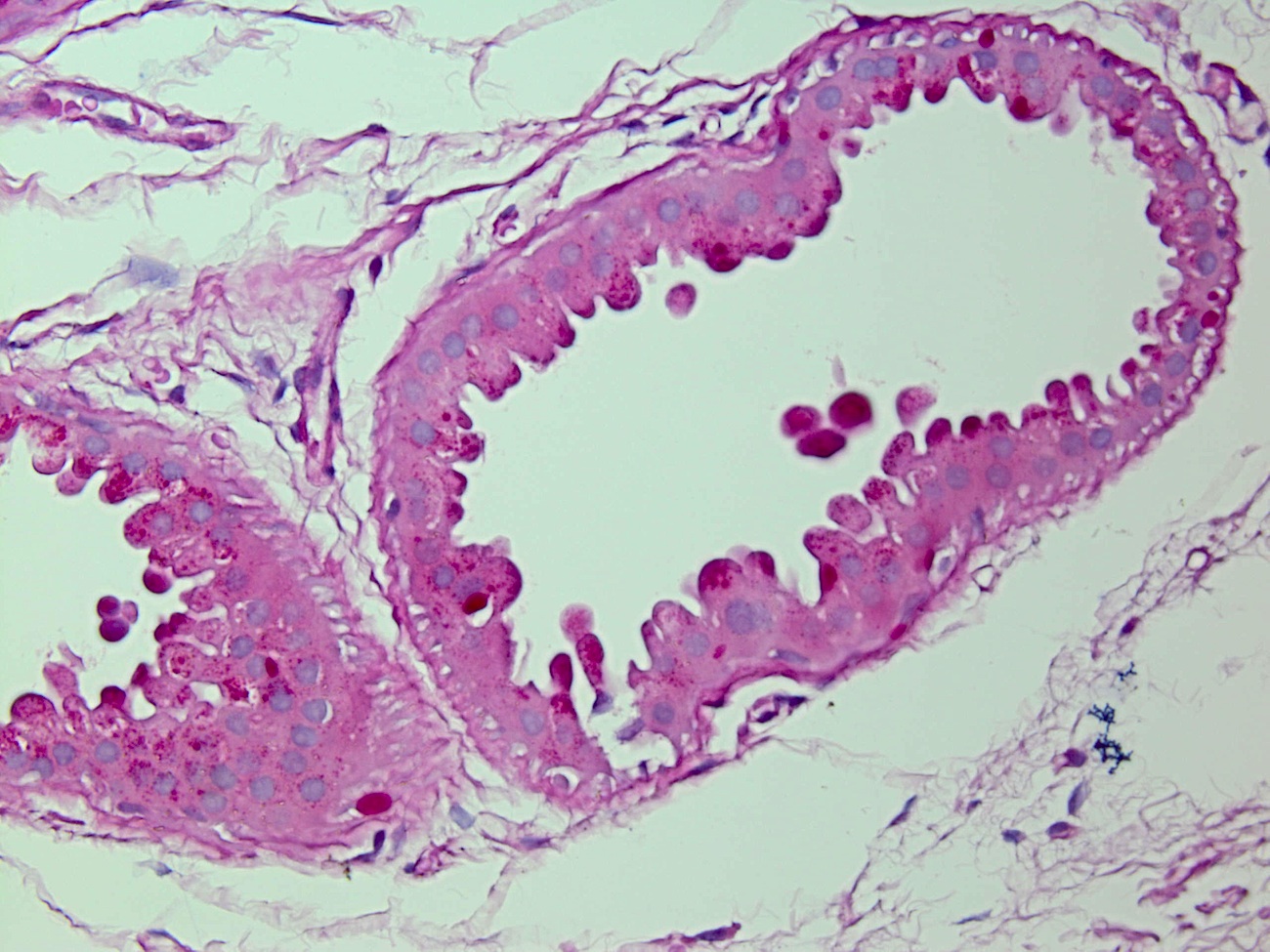


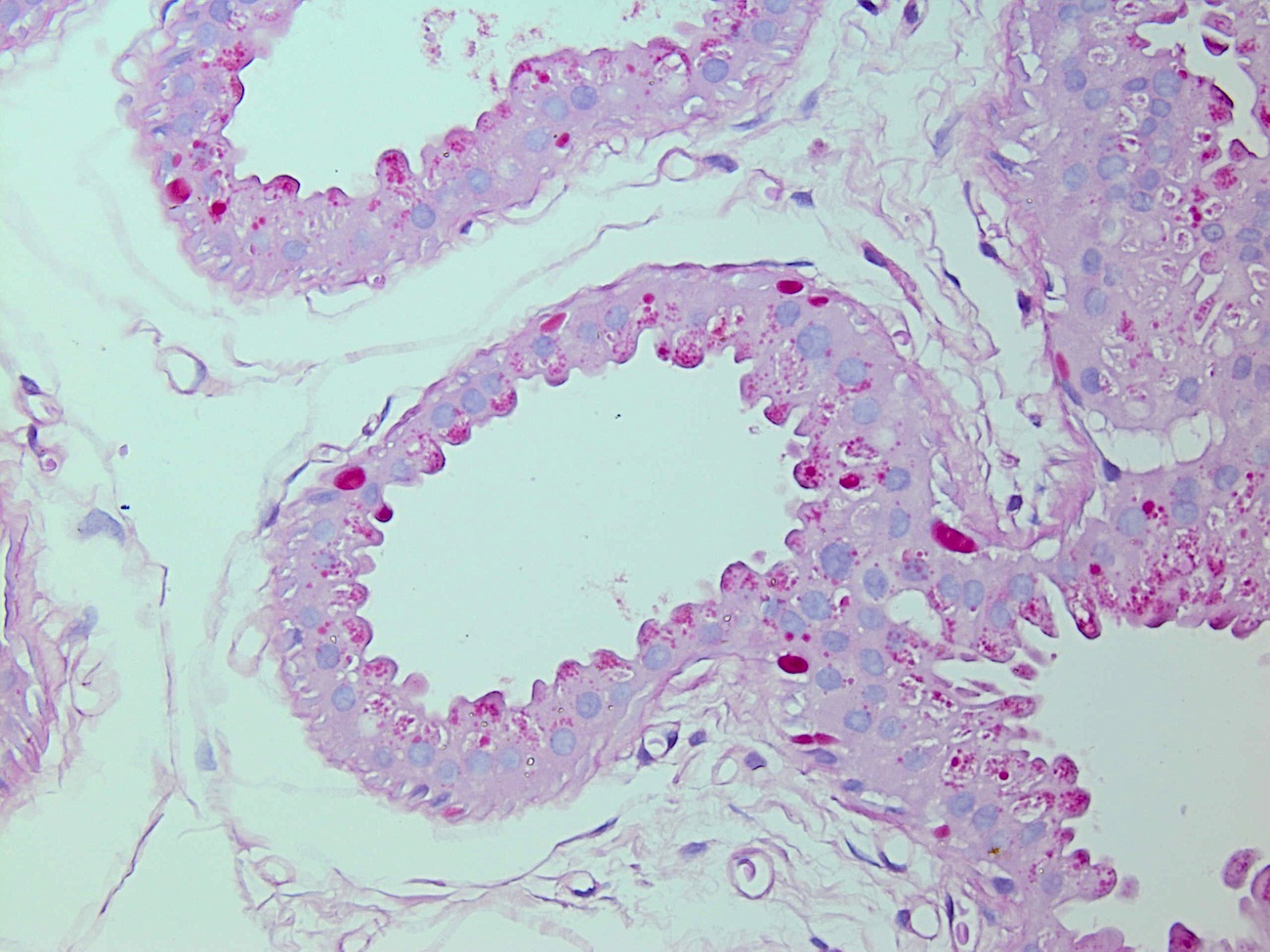
Discussion:
Biopsies showed apocrine glands with PAS positive intracytoplasmic inclusions.
Lafora body disease (LD) is a rare and severe form of progressive myoclonic epilepsy. This autosomal recessive disorder, with onset occurring between the ages of 10 and 18, is characterized by a course of generalized seizures followed by progressively worsening myoclonus, cognitive decline leading to dementia and eventual death within 10 years (Brain 2012;135:2684).
LD is due to EPM2A, EPM2B or NHLRC1 mutations (J Neurol Sci 2013;325:170, Brain 2012;135:2684) occurring on chromosome 6q23-27 (Ann Pathol 2013;33:84) and resulting in loss of function of proteins laforin or malin (J Biol Chem 2013;288:9482). A particularly aggressive course was reported in a patient demonstrating a homozygous deletion encompassing the entire NHLRC1 gene, thus demonstrating the mutational heterogeneity of LD (J Neurol Sci 2013;325:170). A recently described, early onset variant of this disease, presenting at age 5 and demonstrating a more protracted clinical course with patients living into the fourth decade, has been attributed to mutations of the PRDM8 gene (Brain 2012;135:2684). Although the pathogenesis is currently not completely understood, neuronatin, which regulates glycogen synthesis, was recently identified as a substrate of malin. Insoluble aggregates of neuronatin have been demonstrated in the cortical area of LD brain biopsy samples (J Biol Chem 2013;288:9482).
While the clinical course is distinctive, the diagnosis must usually be confirmed by demonstrating pathognomonic Lafora bodies, which are aggregates of polyglucosan, poorly constructed glycogen molecules with long strands that make them insoluble (Brain 2012;135:2684). Skin biopsy is the preferred method for diagnosis and shows PAS positive inclusions in peripheral cells of eccrine sweat ducts, axillary apocrine sweat glands or peripheral nerves (Neurology 1981;31:1564, Arch Dermatol 1987;123:1667, Am J Dermatopathol 1991:410, Arch Neurol 1986;43:296). While not necessary for diagnosis, electron microscopy reveals fine pale staining filaments, fine dark staining granules and dark rimmed vacuoles within these non membrane bound inclusions (Neurology 1981;31:1564).
Of interest, one report of autopsy findings in a patient with LD described Lafora bodies in various endocrine organs including the thyroid, anterior pituitary, hypothalamus and pancreas, thus raising the possibility of endocrinologic abnormalities in LD patients (Pediatr Neurol 2014;51:713).
Most affected individuals die within ten years of onset, usually from status epilepticus or from complications related to nervous system degeneration. Treatment is symptomatic. Antiepileptic drugs (AEDs) are effective against generalized seizures and gastrostomy feedings can decrease the risk of aspiration pneumonia when disease is advanced (GeneReviews: Progressive Myoclonus Epilepsy, Lafora Type [Accessed 16 December 2022]).
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. A. Eftekhar Javadi, Tehran University of Medical Sciences (Iran), for contributing this case and Jennifer R. Kaley, M.D., University of Arkansas for Medical Sciences for contributing the discussion.


- Scheuer's Liver Biopsy Interpretation (9th ed) by Jay Lefkowitch. 2015, 440 pages, $165 list.
- Biopsy Interpretation of the Kidney and Adrenal Gland by Satish Tickoo, Ying-bei Chen and Debra Zynger. 2015, 368 pages, 400+ illus, $162 list.
For more information, visit our Books page.
Case of the Week #361
Clinical history:
A 17 year old girl presented with a seizure. She had myoclonus in her extremities which was progressive and now has generalized myoclonus epilepsy. She had declined in intellectual function and is now intellectually disabled. No genetic testing was done.
A skin biopsy from the axilla was taken.
Microscopic (histologic) images :
What is your diagnosis?
Click here for diagnosis and discussion:
Diagnosis: Lafora body disease
Special stains:
PAS+ stain
Discussion:
Biopsies showed apocrine glands with PAS positive intracytoplasmic inclusions.
Lafora body disease (LD) is a rare and severe form of progressive myoclonic epilepsy. This autosomal recessive disorder, with onset occurring between the ages of 10 and 18, is characterized by a course of generalized seizures followed by progressively worsening myoclonus, cognitive decline leading to dementia and eventual death within 10 years (Brain 2012;135:2684).
LD is due to EPM2A, EPM2B or NHLRC1 mutations (J Neurol Sci 2013;325:170, Brain 2012;135:2684) occurring on chromosome 6q23-27 (Ann Pathol 2013;33:84) and resulting in loss of function of proteins laforin or malin (J Biol Chem 2013;288:9482). A particularly aggressive course was reported in a patient demonstrating a homozygous deletion encompassing the entire NHLRC1 gene, thus demonstrating the mutational heterogeneity of LD (J Neurol Sci 2013;325:170). A recently described, early onset variant of this disease, presenting at age 5 and demonstrating a more protracted clinical course with patients living into the fourth decade, has been attributed to mutations of the PRDM8 gene (Brain 2012;135:2684). Although the pathogenesis is currently not completely understood, neuronatin, which regulates glycogen synthesis, was recently identified as a substrate of malin. Insoluble aggregates of neuronatin have been demonstrated in the cortical area of LD brain biopsy samples (J Biol Chem 2013;288:9482).
While the clinical course is distinctive, the diagnosis must usually be confirmed by demonstrating pathognomonic Lafora bodies, which are aggregates of polyglucosan, poorly constructed glycogen molecules with long strands that make them insoluble (Brain 2012;135:2684). Skin biopsy is the preferred method for diagnosis and shows PAS positive inclusions in peripheral cells of eccrine sweat ducts, axillary apocrine sweat glands or peripheral nerves (Neurology 1981;31:1564, Arch Dermatol 1987;123:1667, Am J Dermatopathol 1991:410, Arch Neurol 1986;43:296). While not necessary for diagnosis, electron microscopy reveals fine pale staining filaments, fine dark staining granules and dark rimmed vacuoles within these non membrane bound inclusions (Neurology 1981;31:1564).
Of interest, one report of autopsy findings in a patient with LD described Lafora bodies in various endocrine organs including the thyroid, anterior pituitary, hypothalamus and pancreas, thus raising the possibility of endocrinologic abnormalities in LD patients (Pediatr Neurol 2014;51:713).
Most affected individuals die within ten years of onset, usually from status epilepticus or from complications related to nervous system degeneration. Treatment is symptomatic. Antiepileptic drugs (AEDs) are effective against generalized seizures and gastrostomy feedings can decrease the risk of aspiration pneumonia when disease is advanced (GeneReviews: Progressive Myoclonus Epilepsy, Lafora Type [Accessed 16 December 2022]).
