
















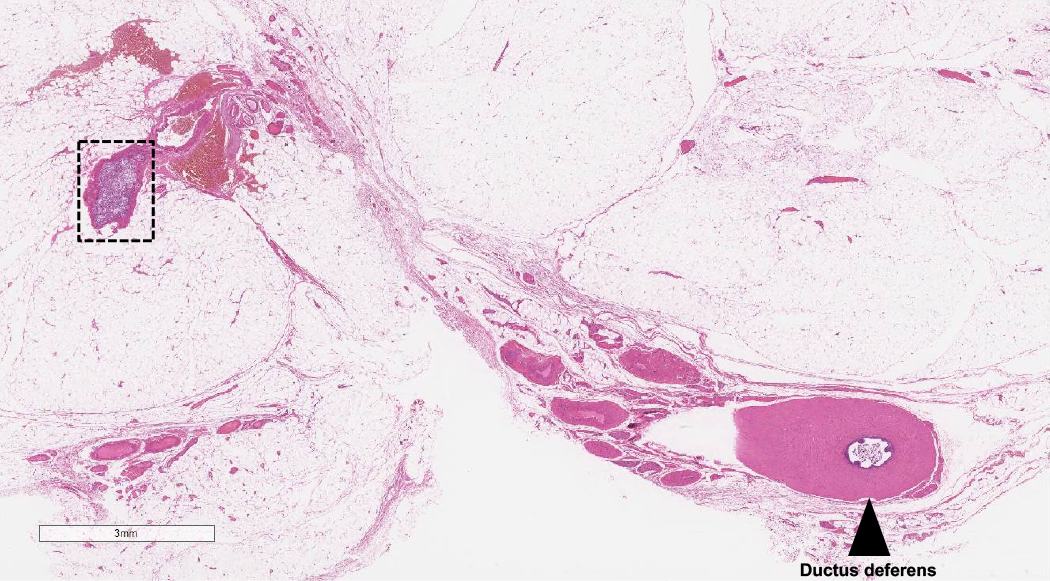
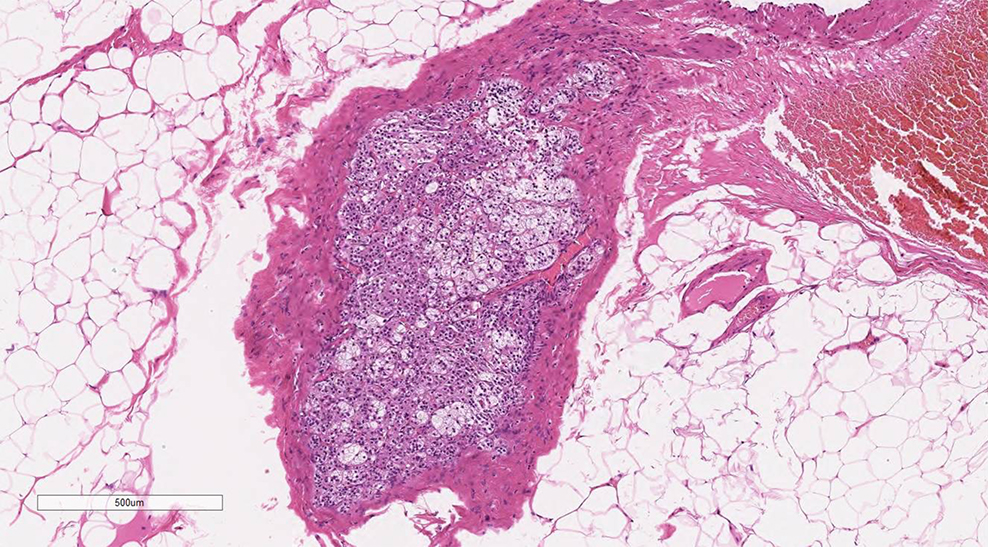
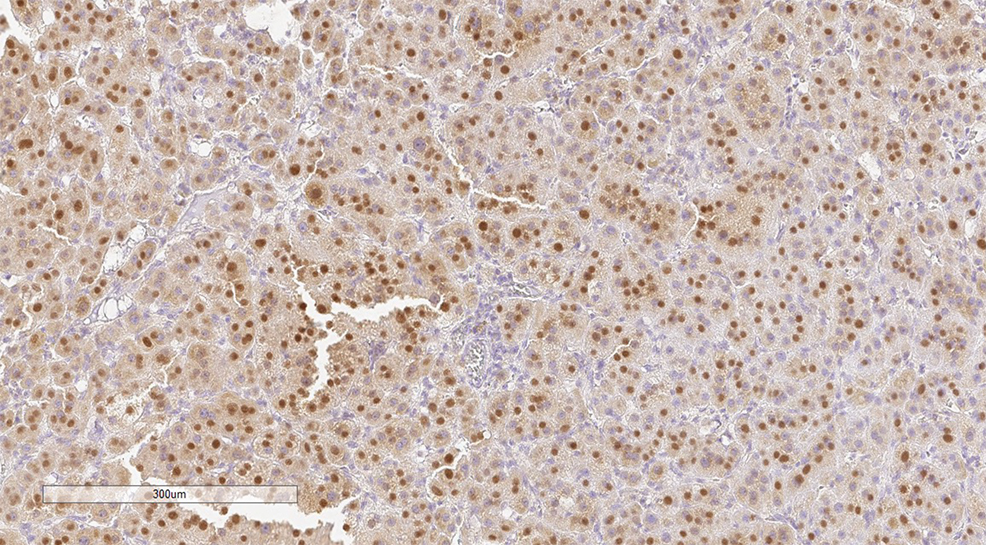
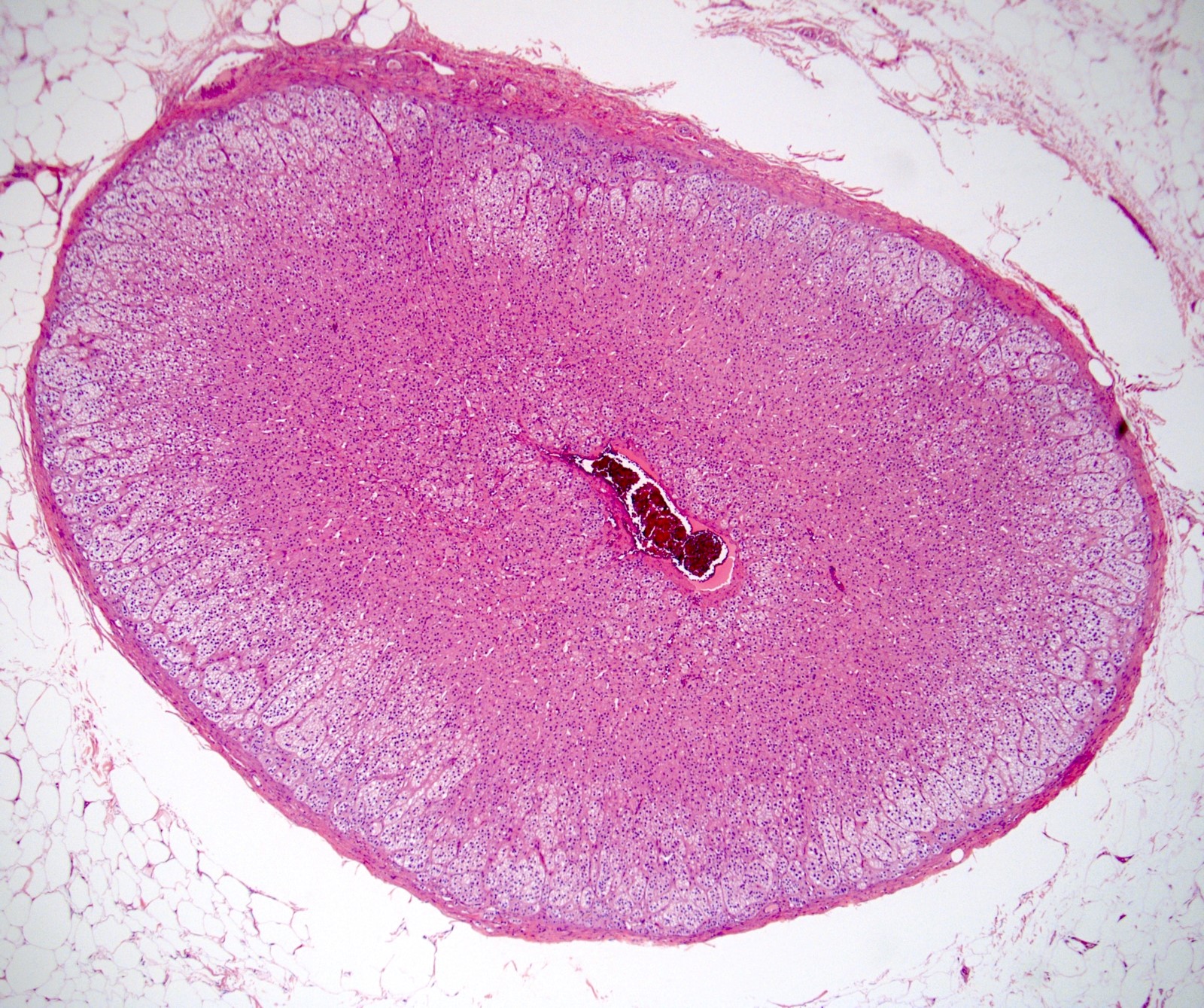











































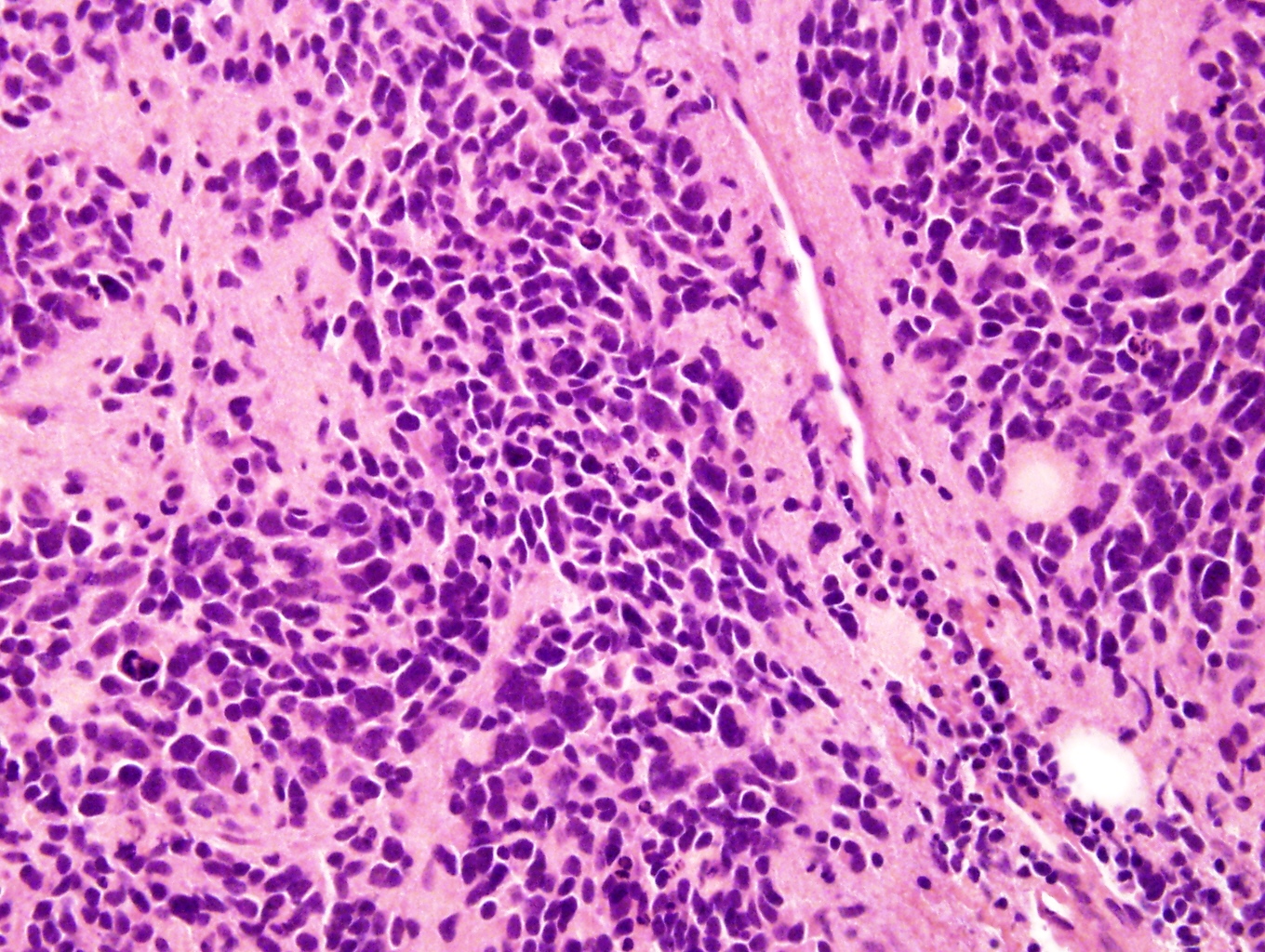





























Primary adrenal insufficiency
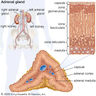
Definition / general
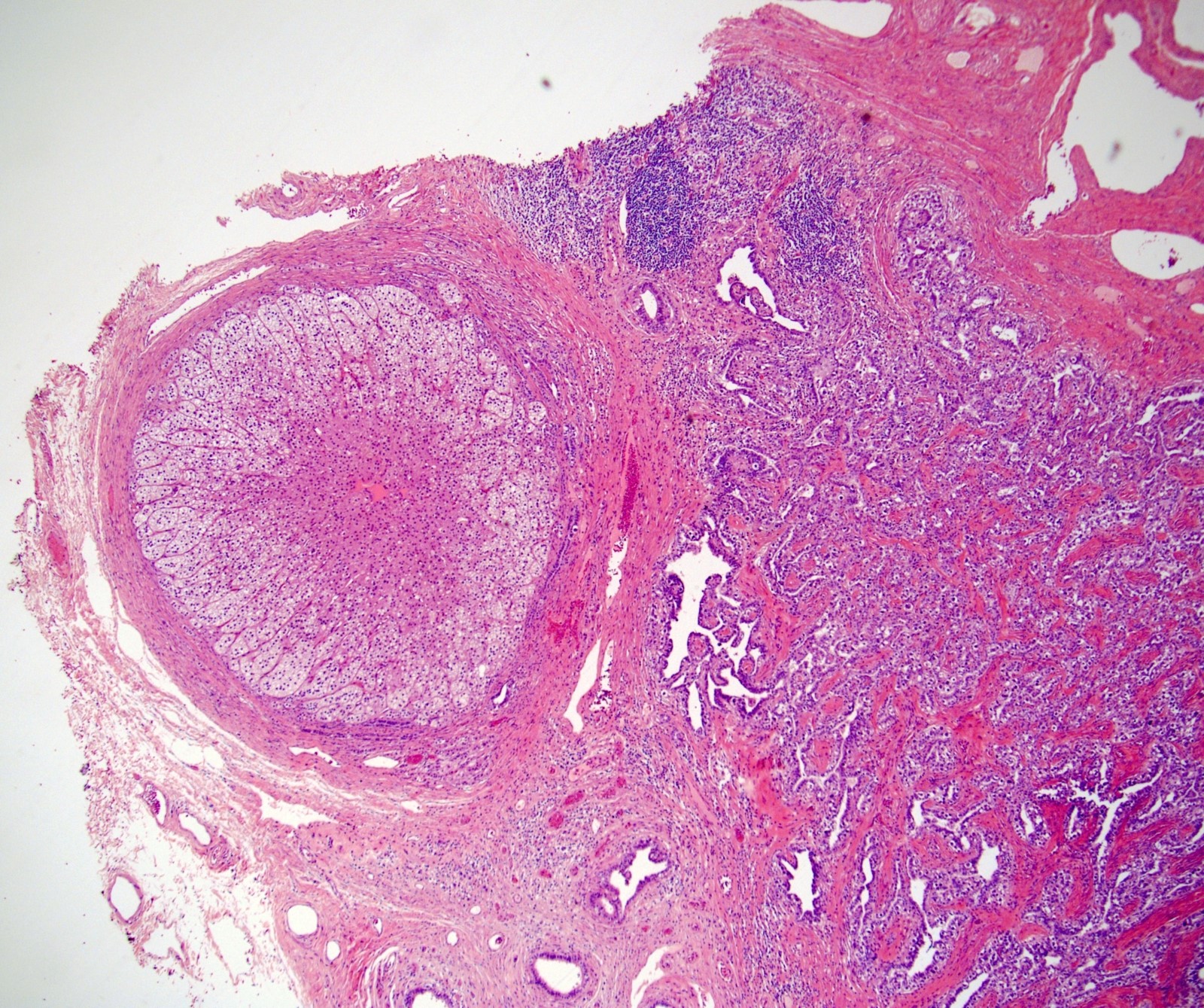
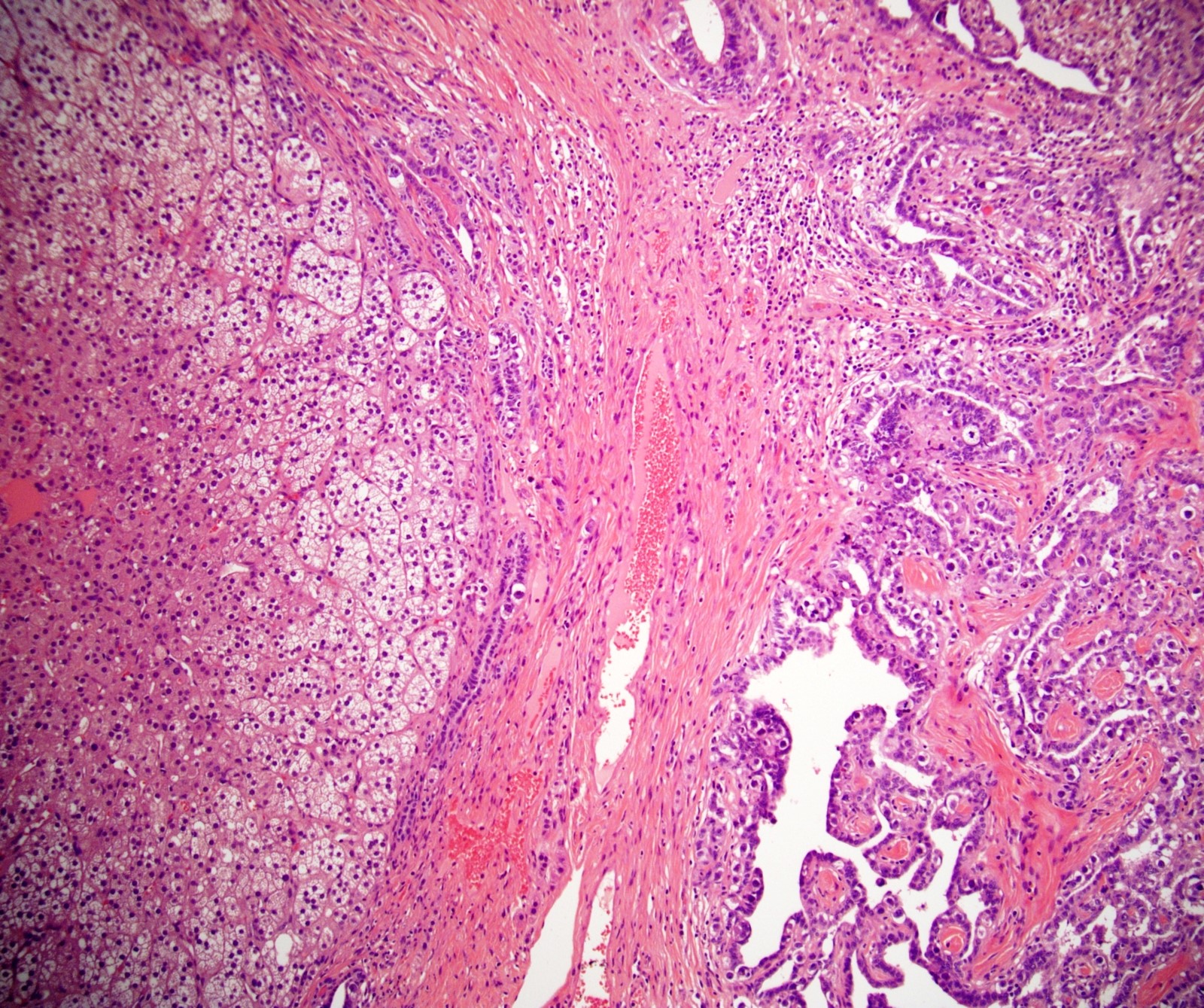
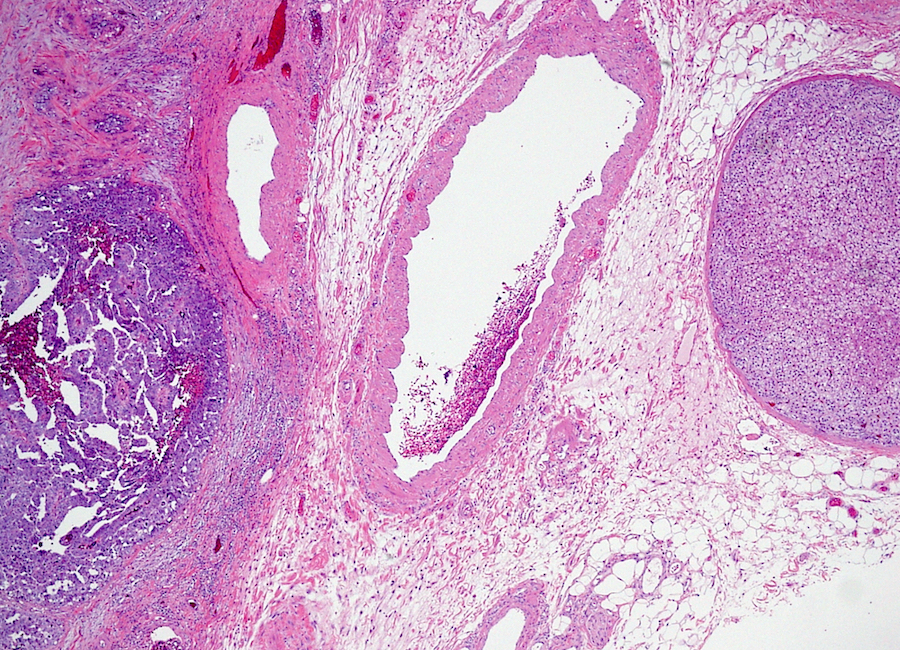
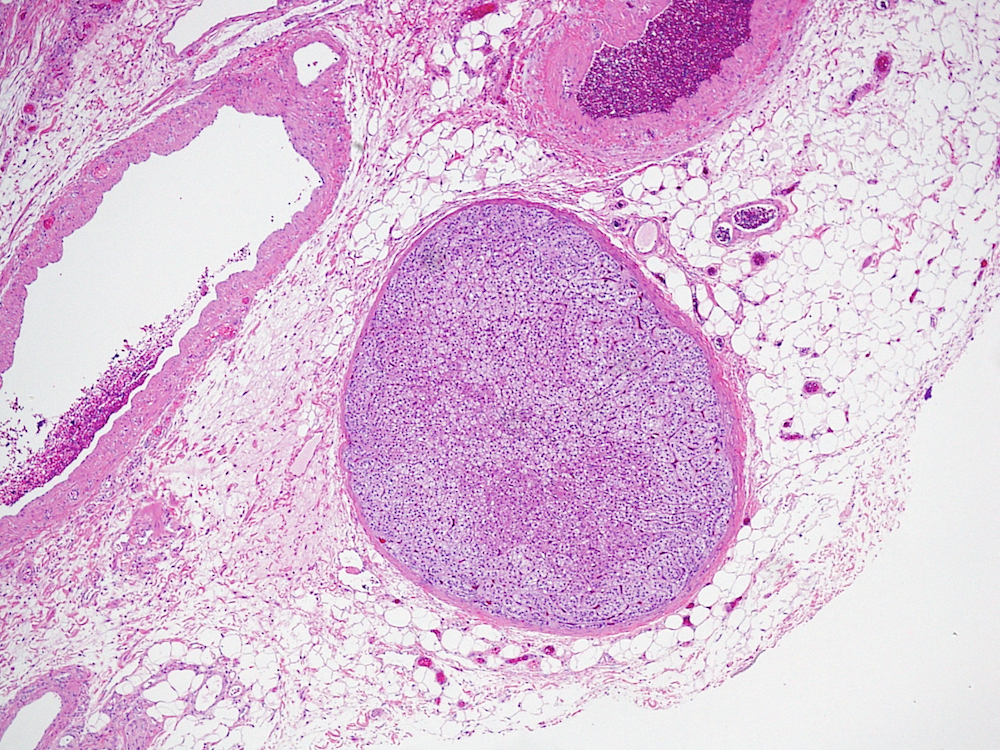
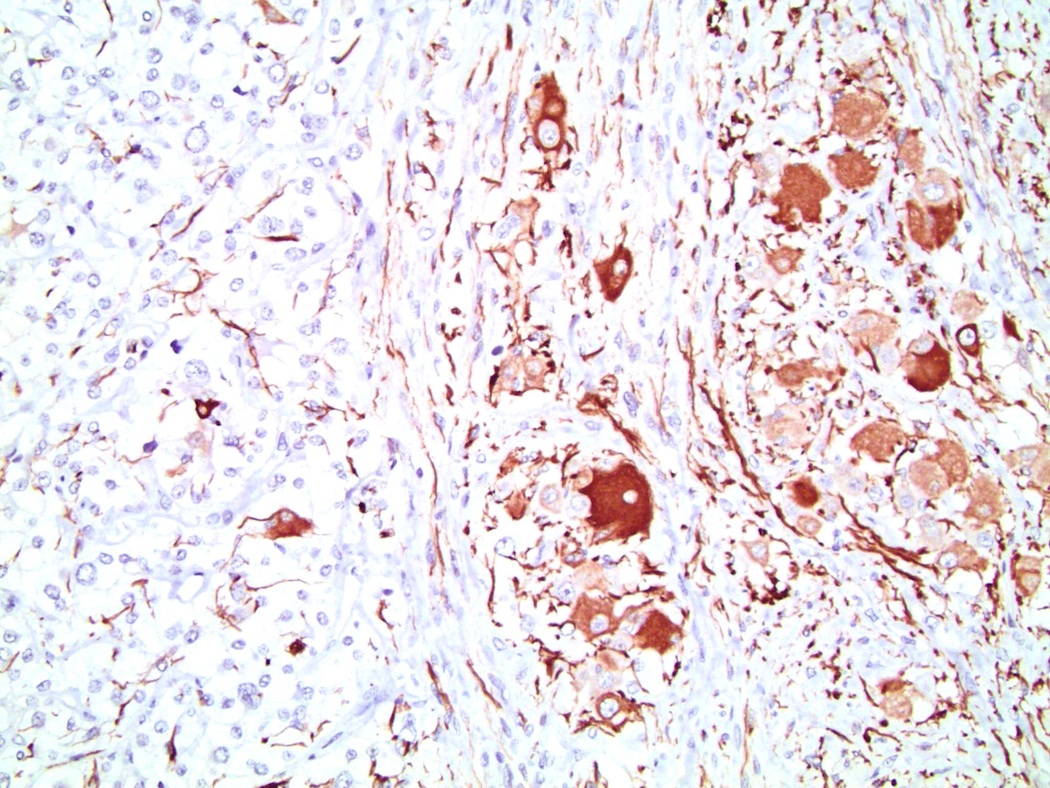
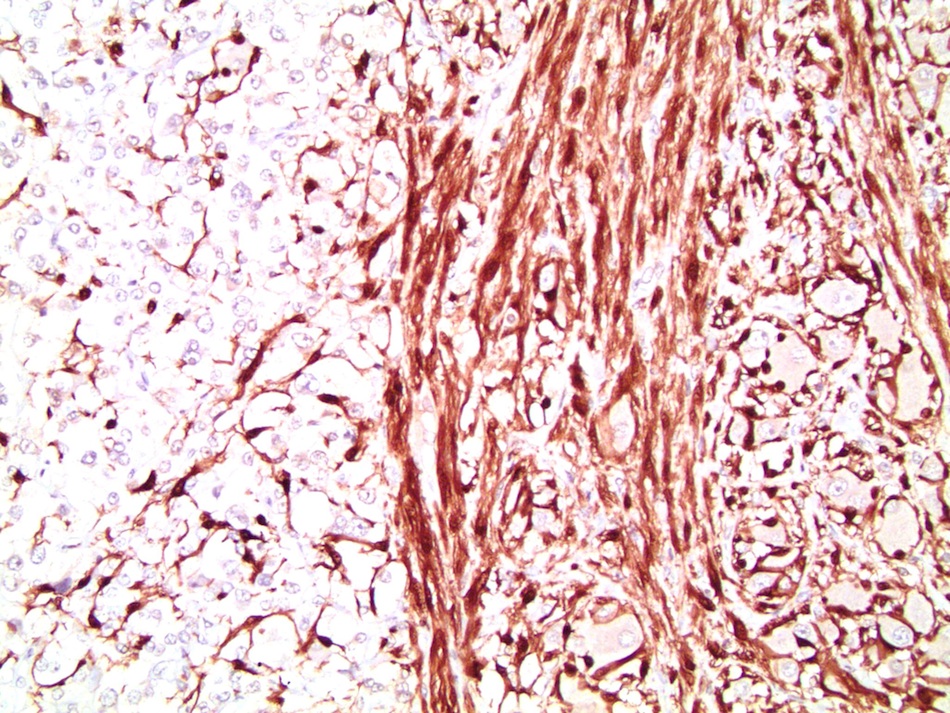
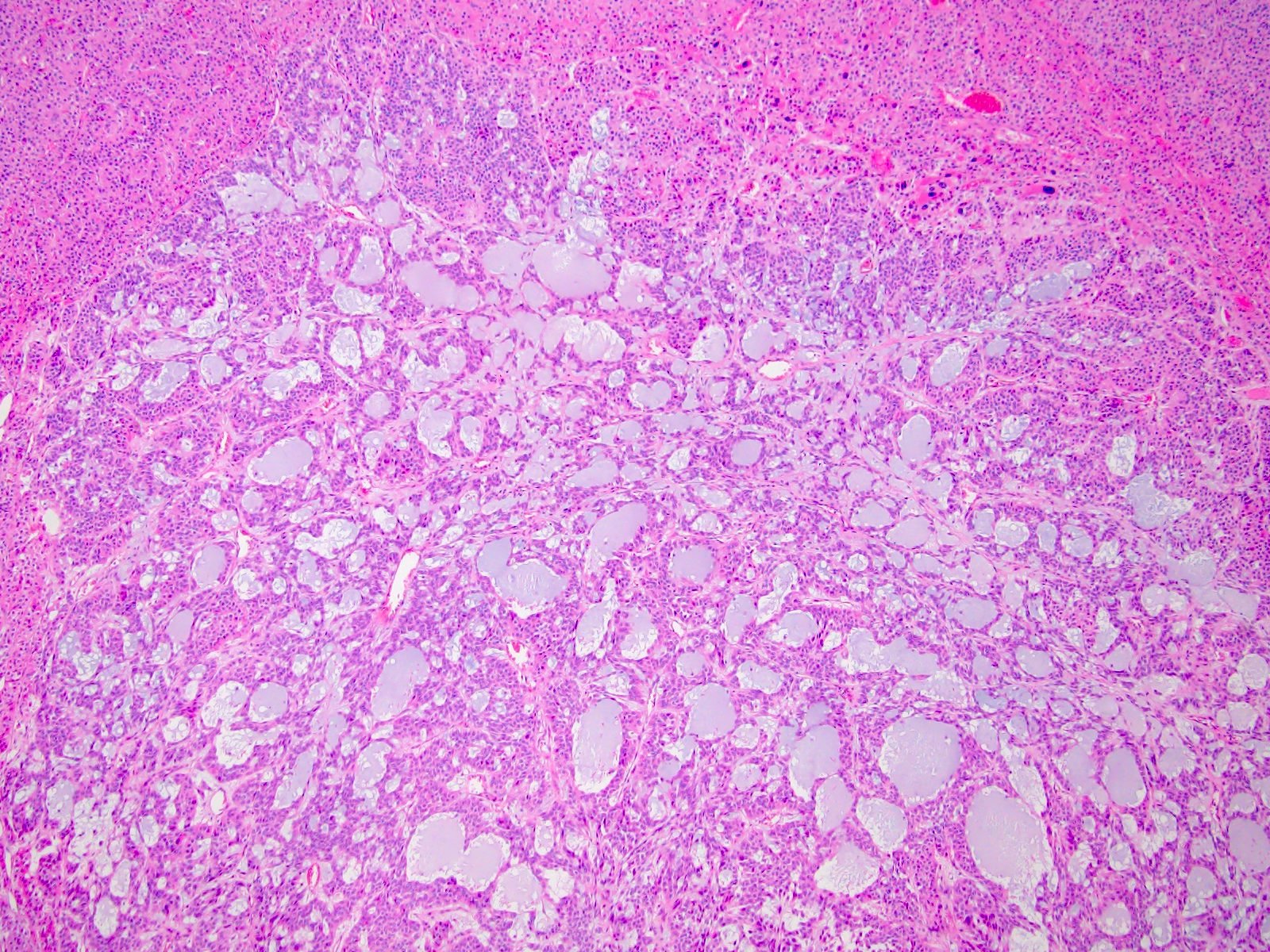
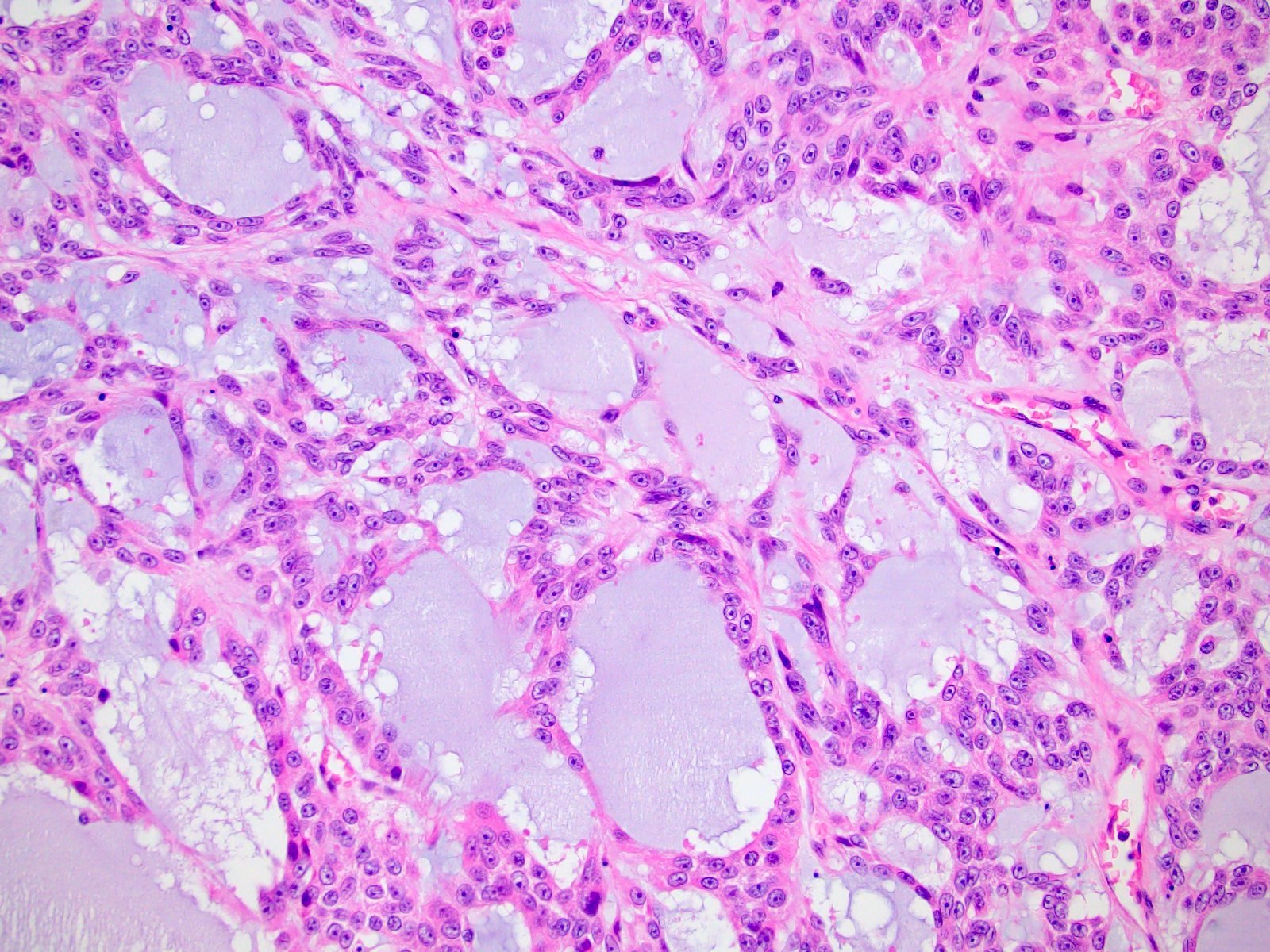
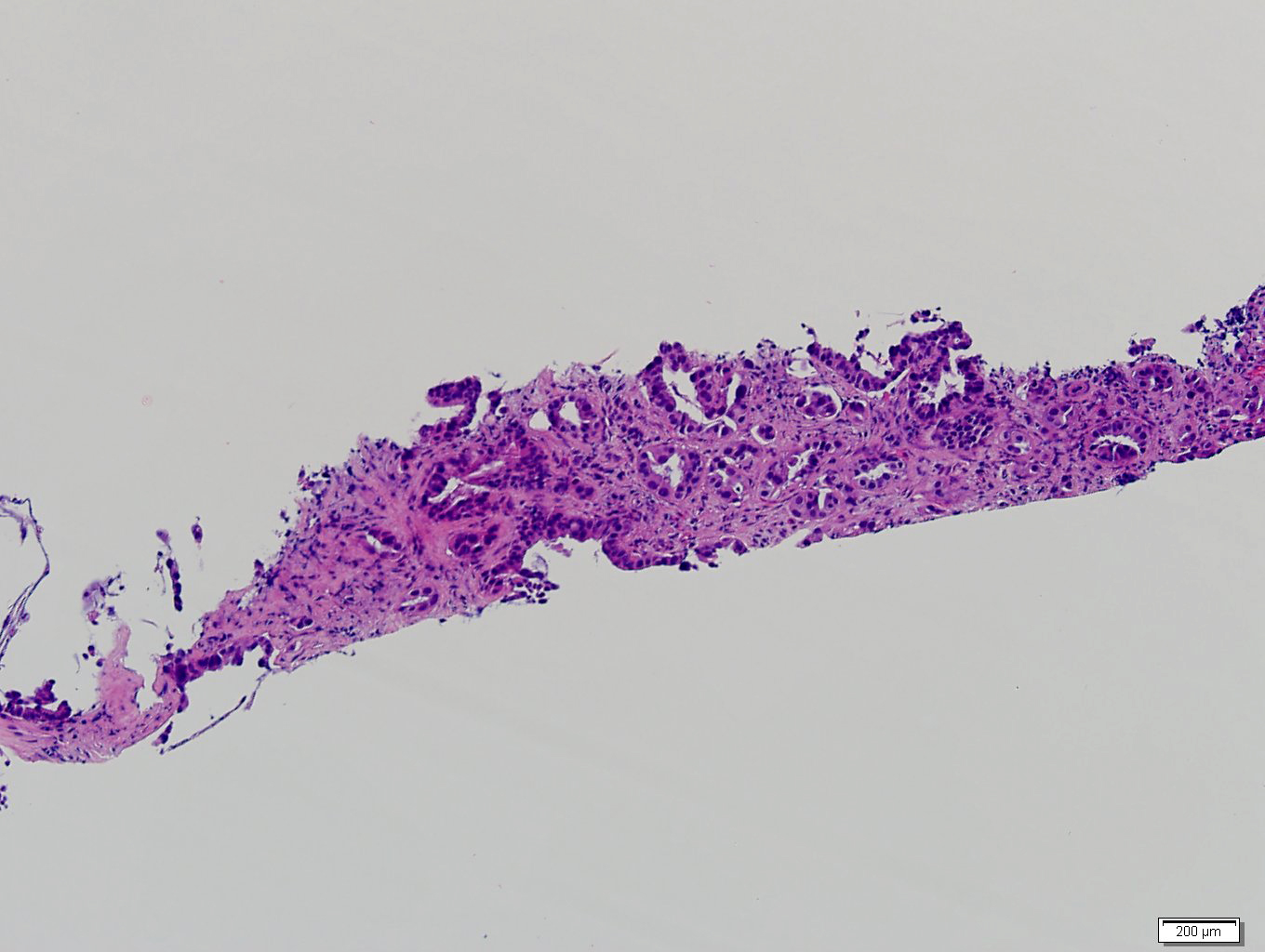
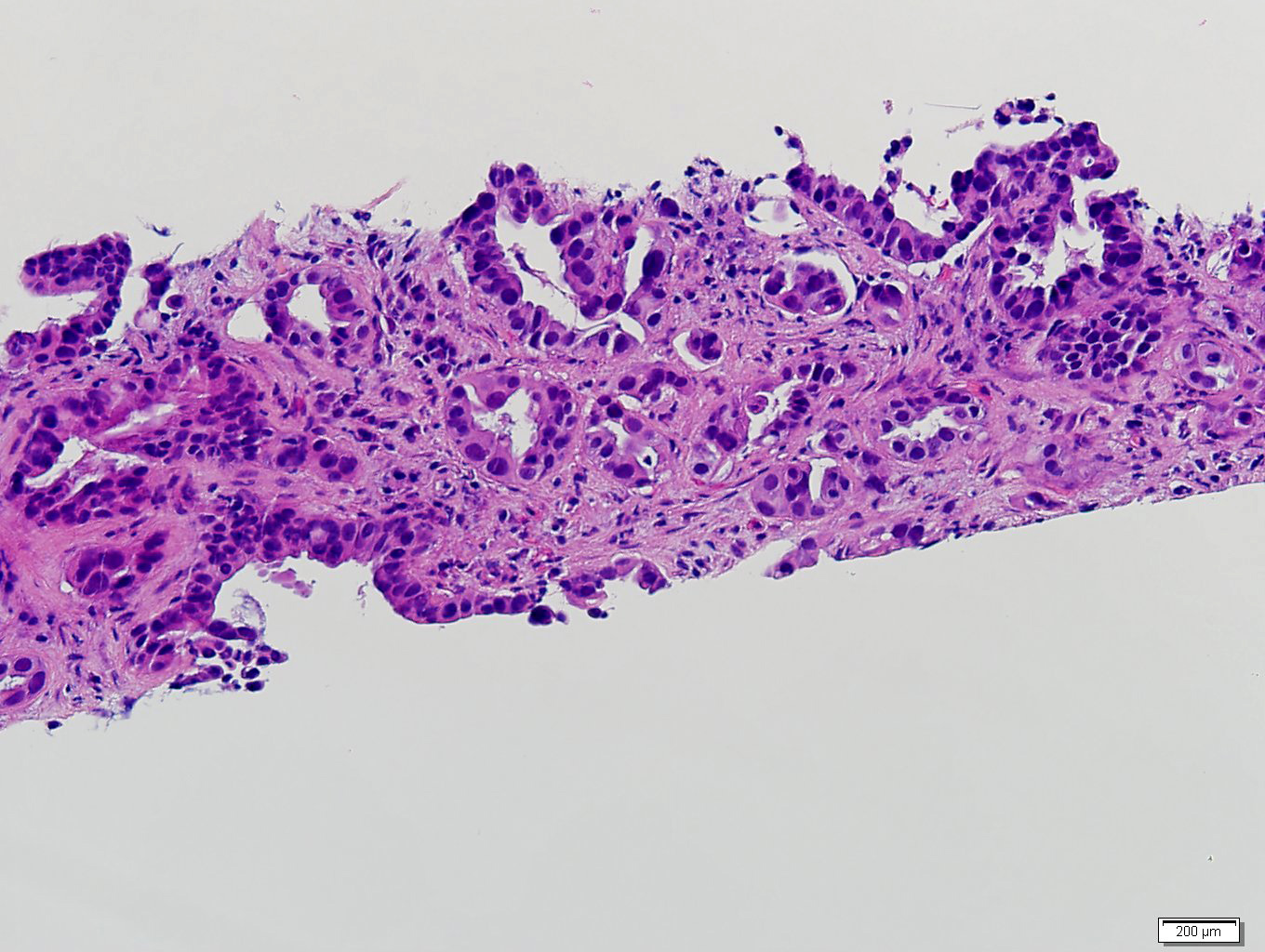
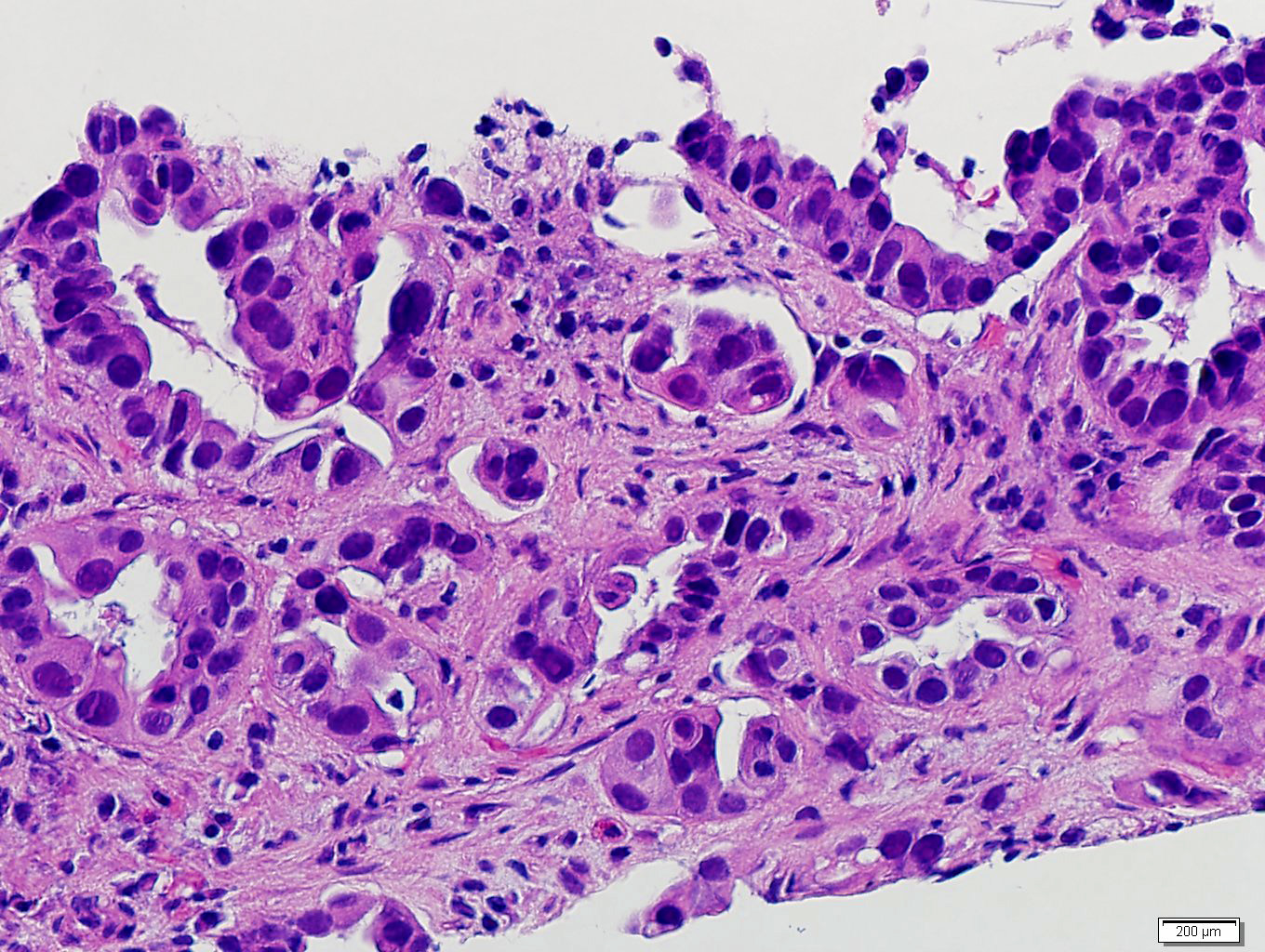
Etiology
Treatment
- Often insidious in onset, patients may present in shock due to increased stress
- Patients usually live normal lives after diagnosis (depending on cause); may be at higher risk for heart failure, hypertension or osteopenia
- Patients with chronic adrenal insufficiency (primary or secondary) and acute stress require immediate increase in steroids
Etiology
- Rapid withdrawal of exogenous steroids (i.e. no taper) or failure to increase steroids with acute stress
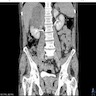
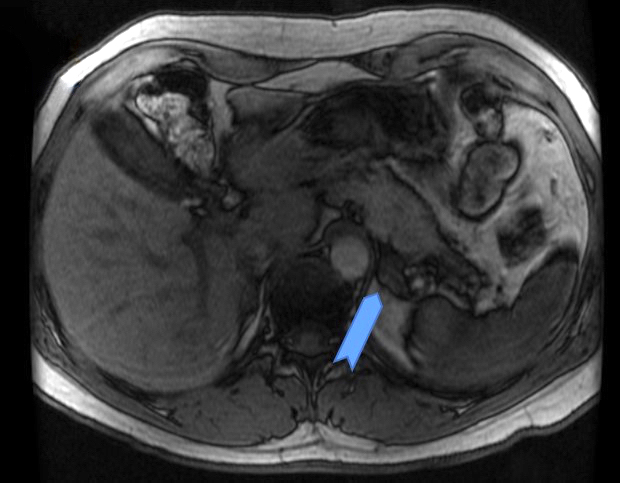
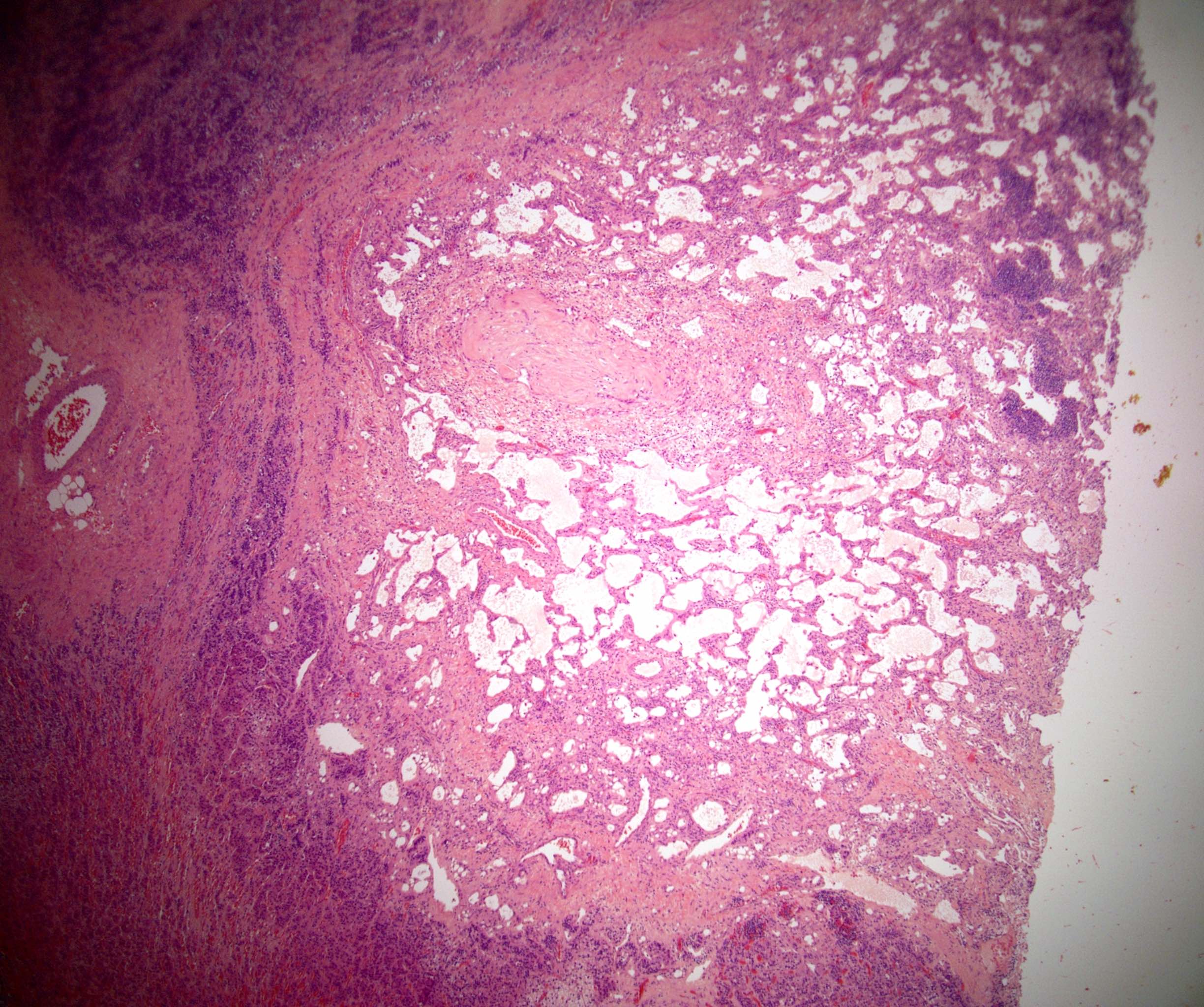
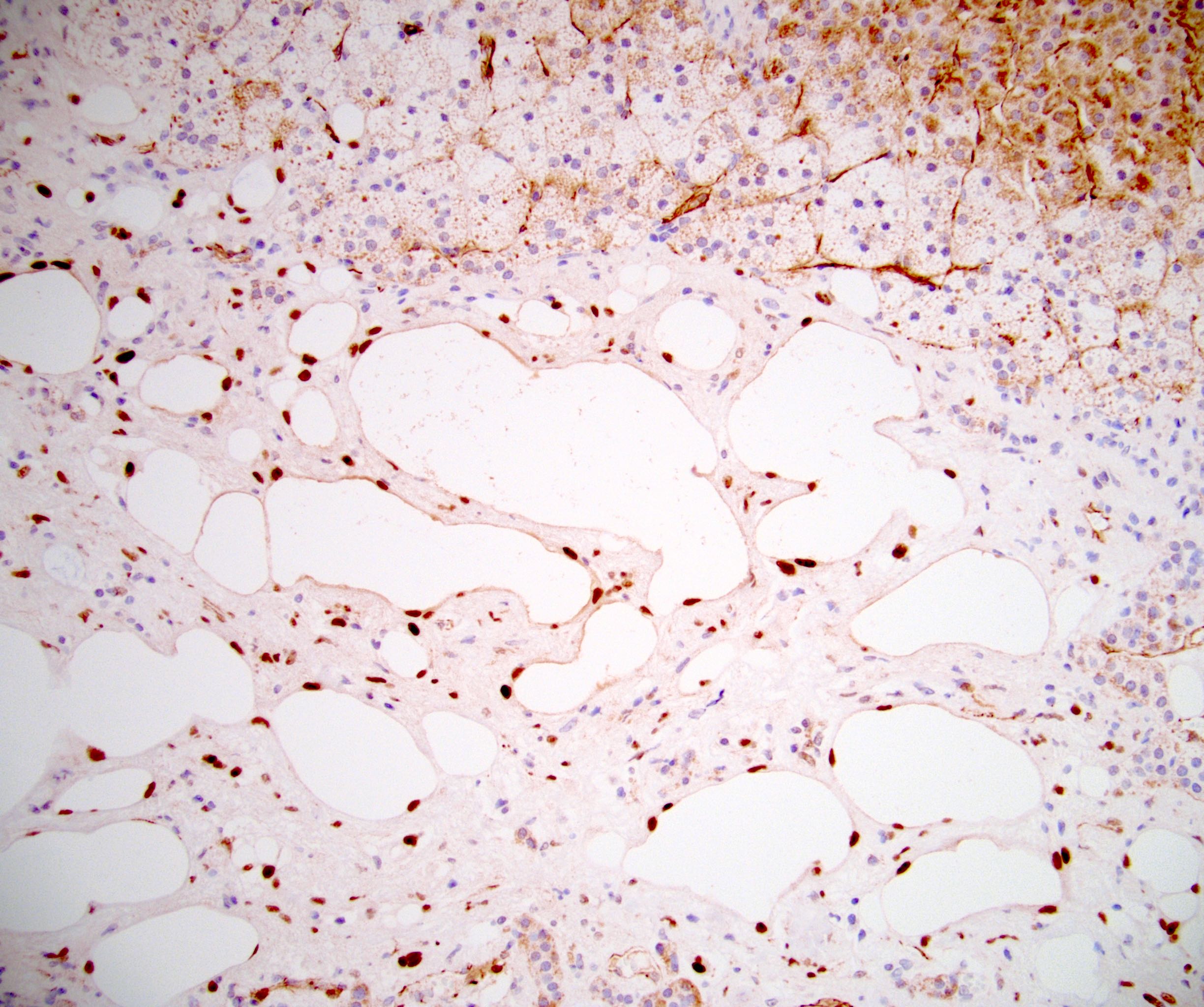
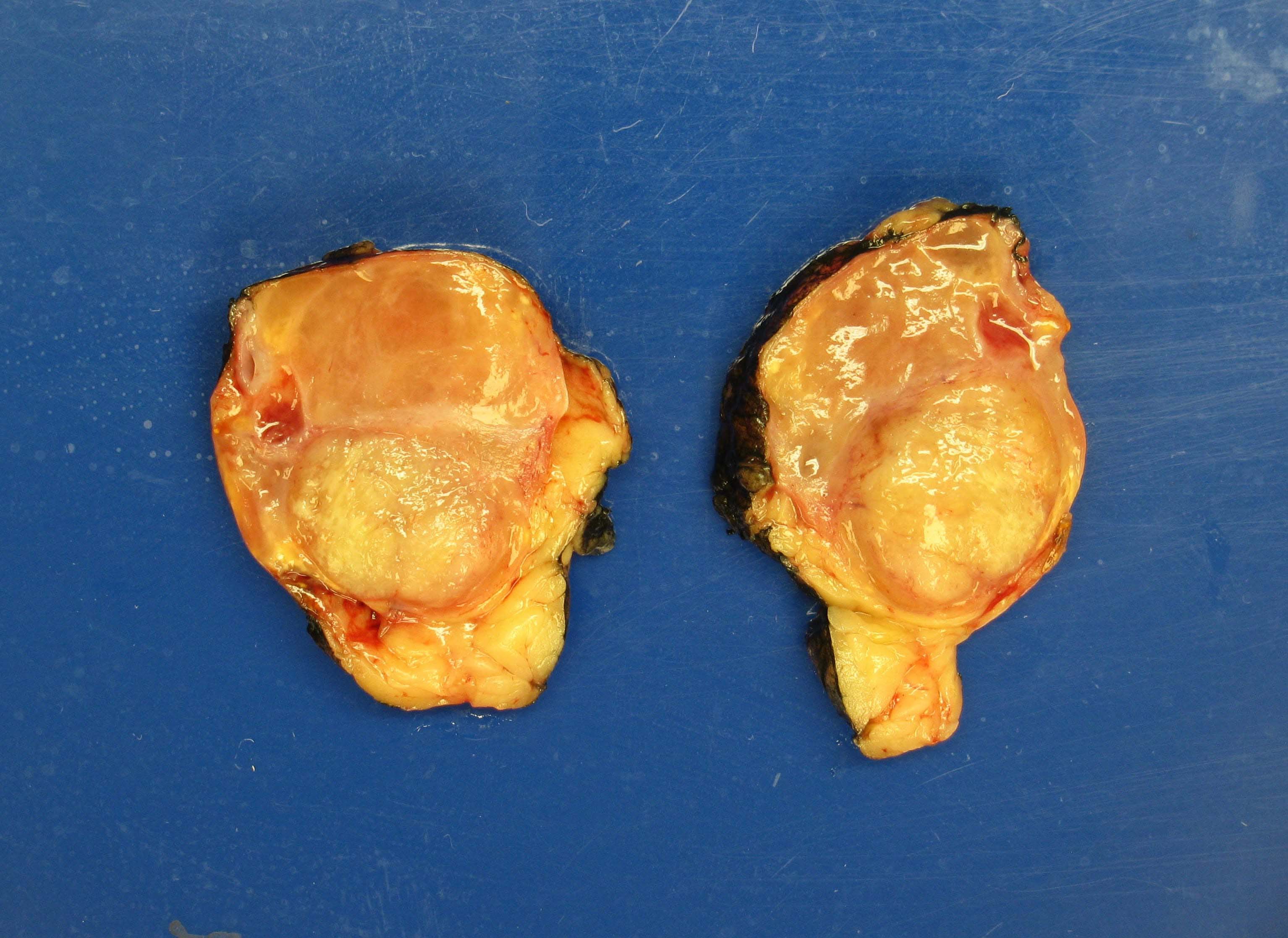
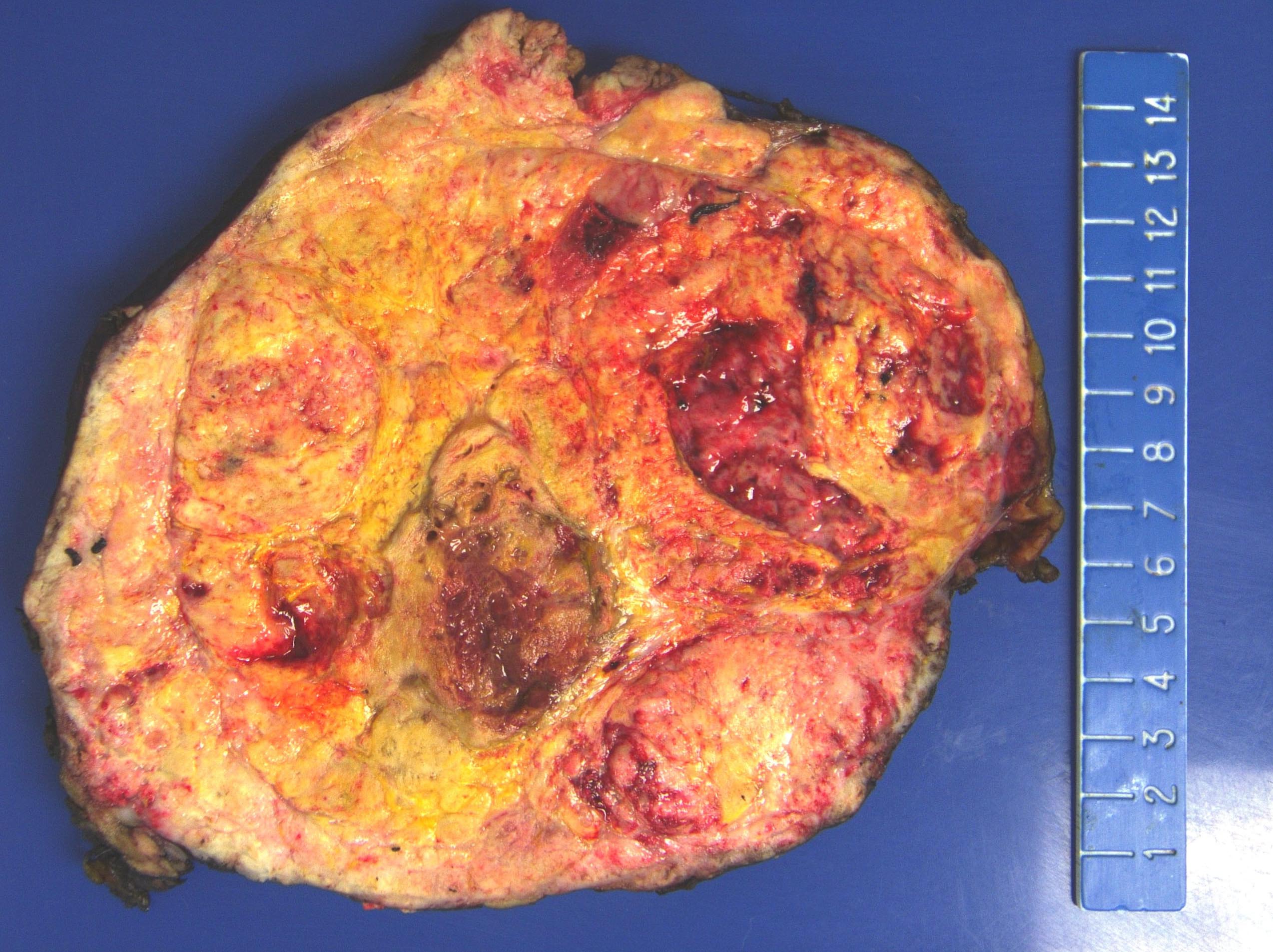
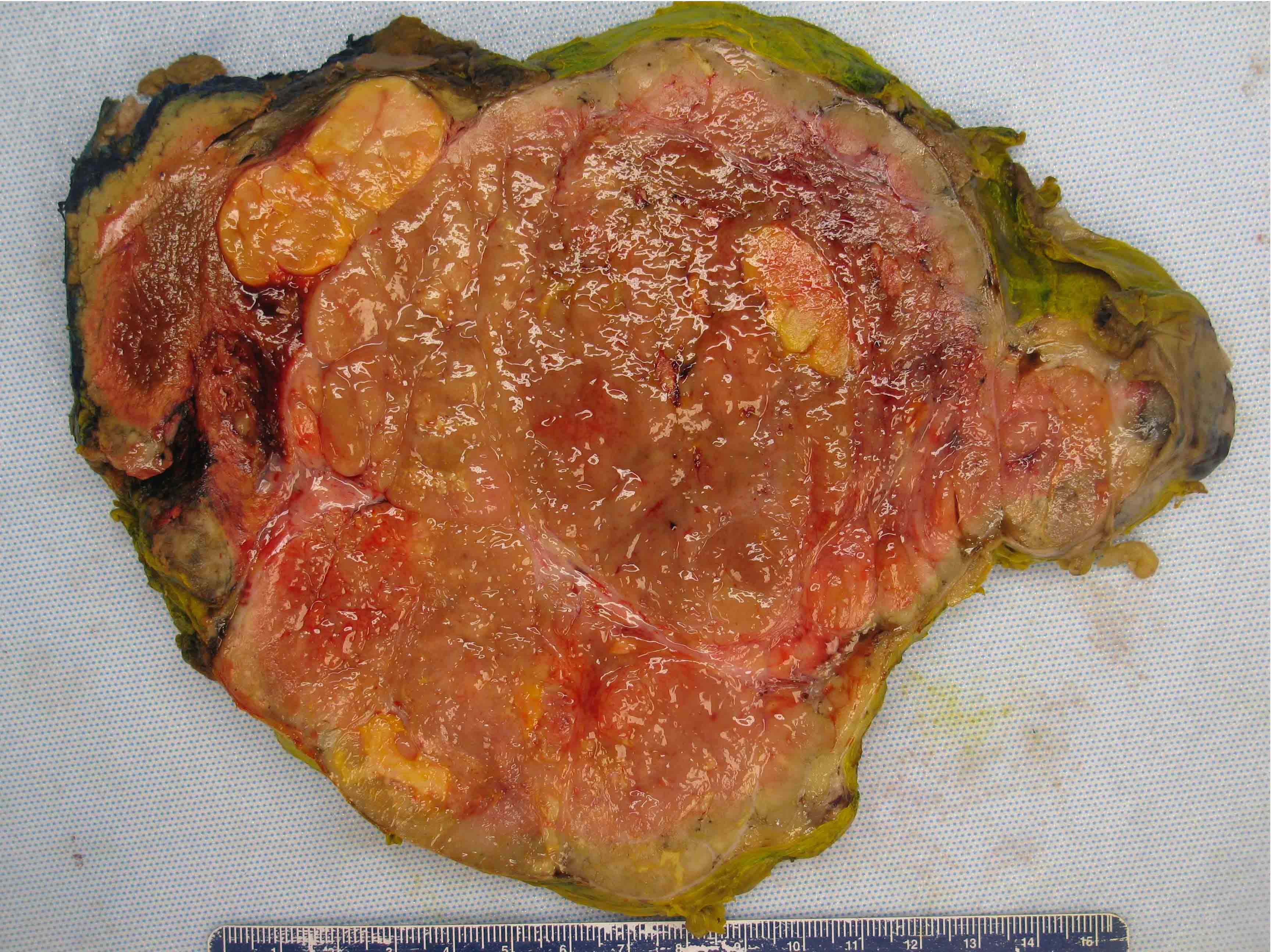
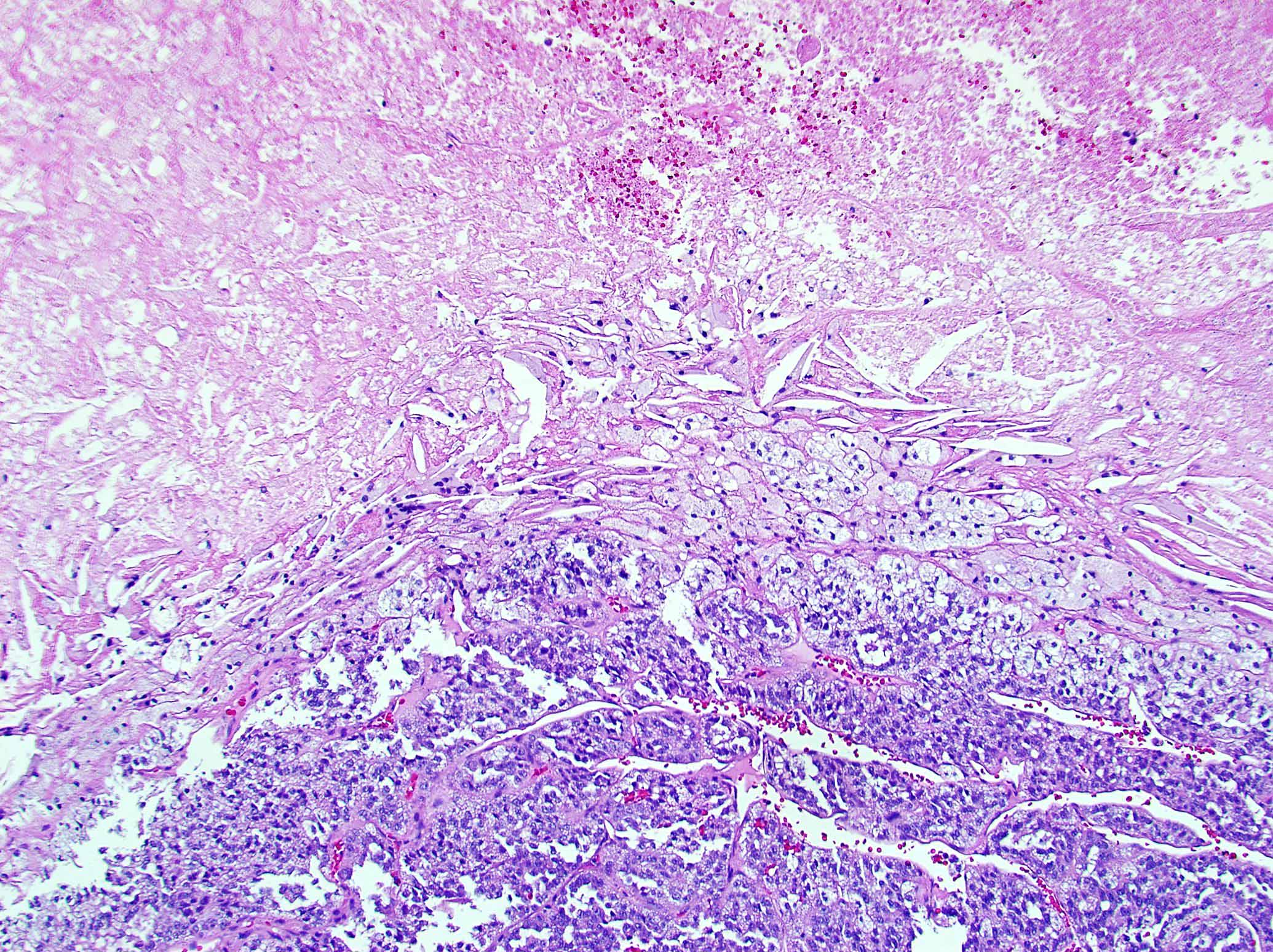
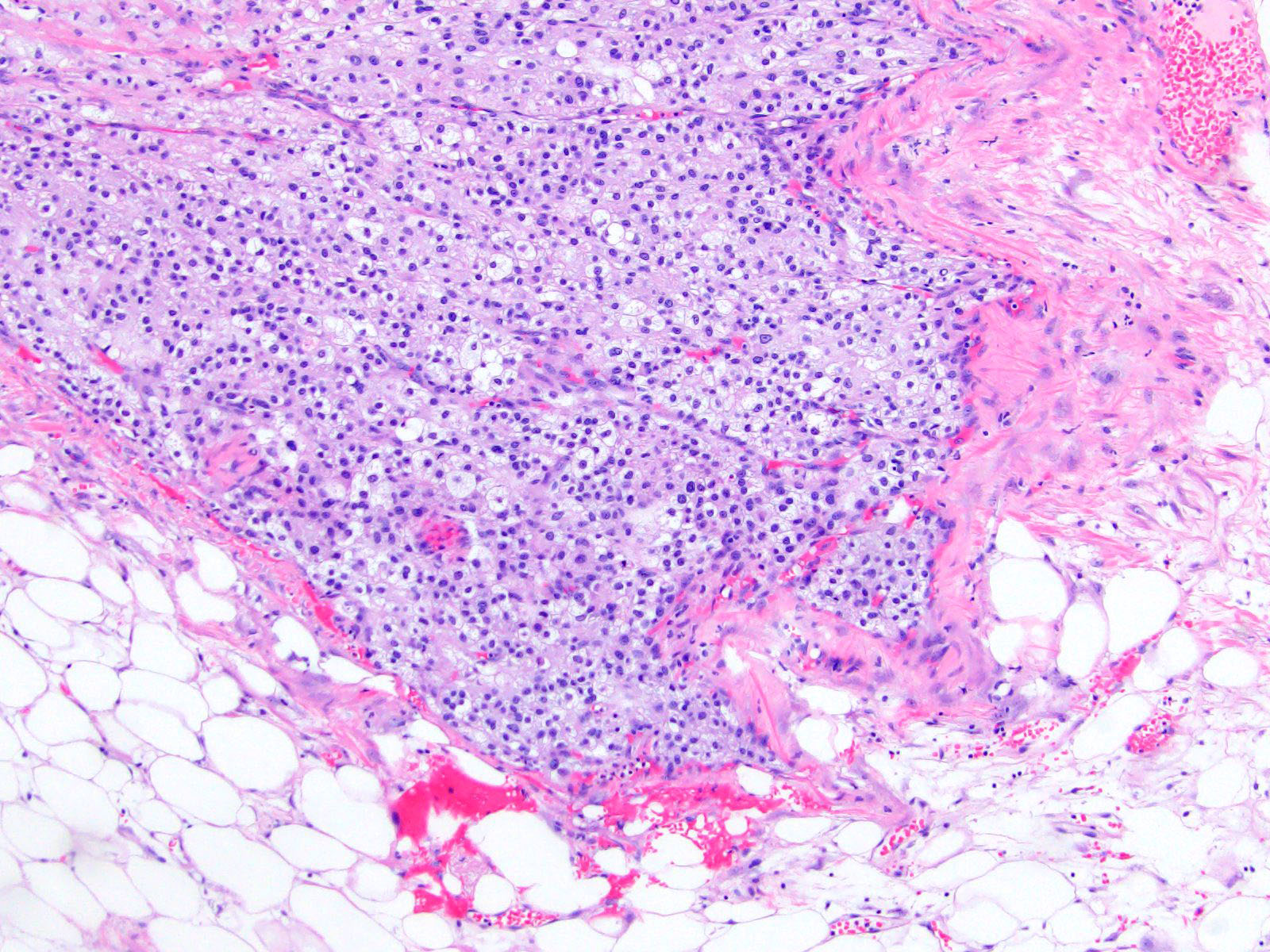
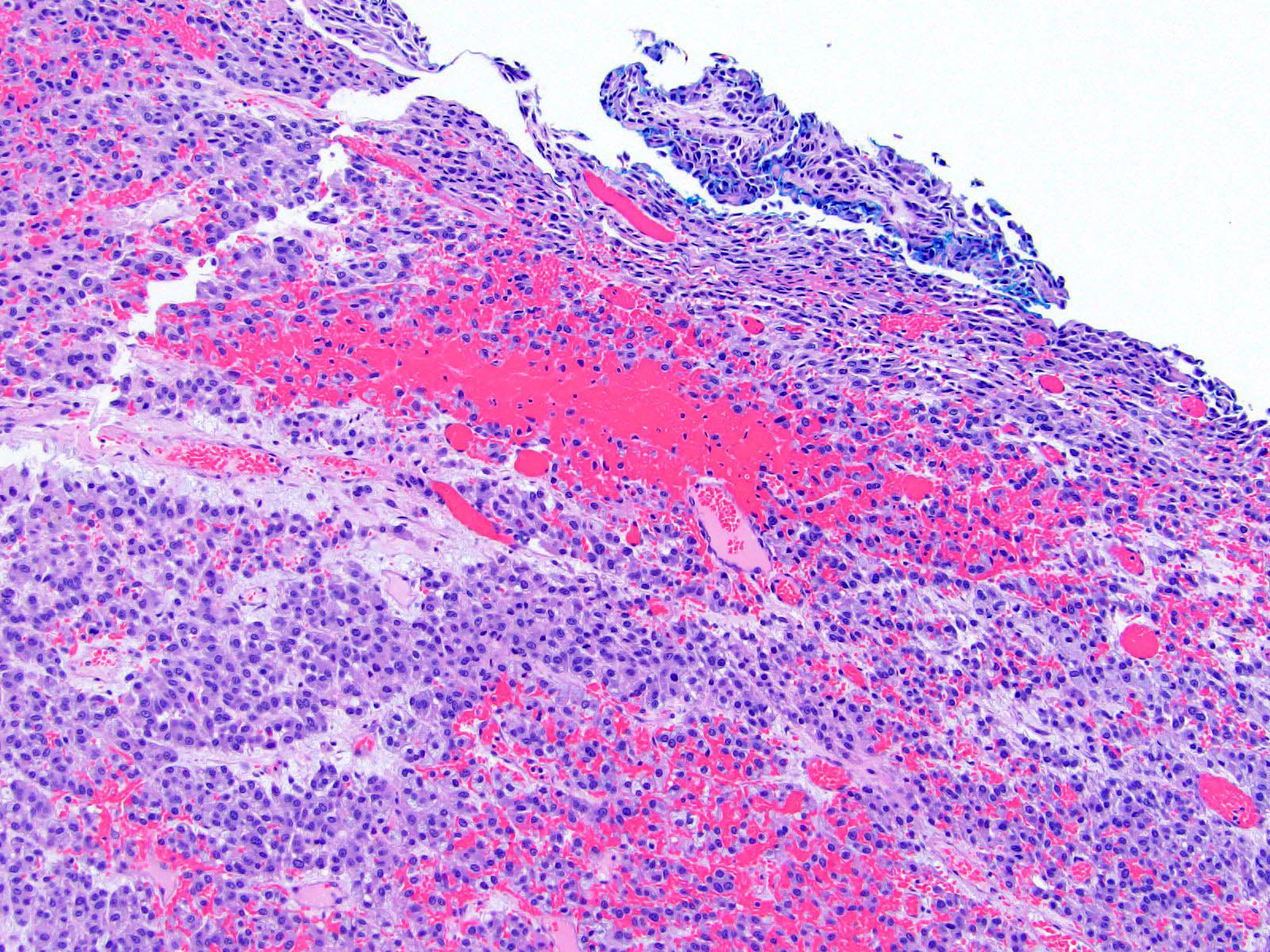
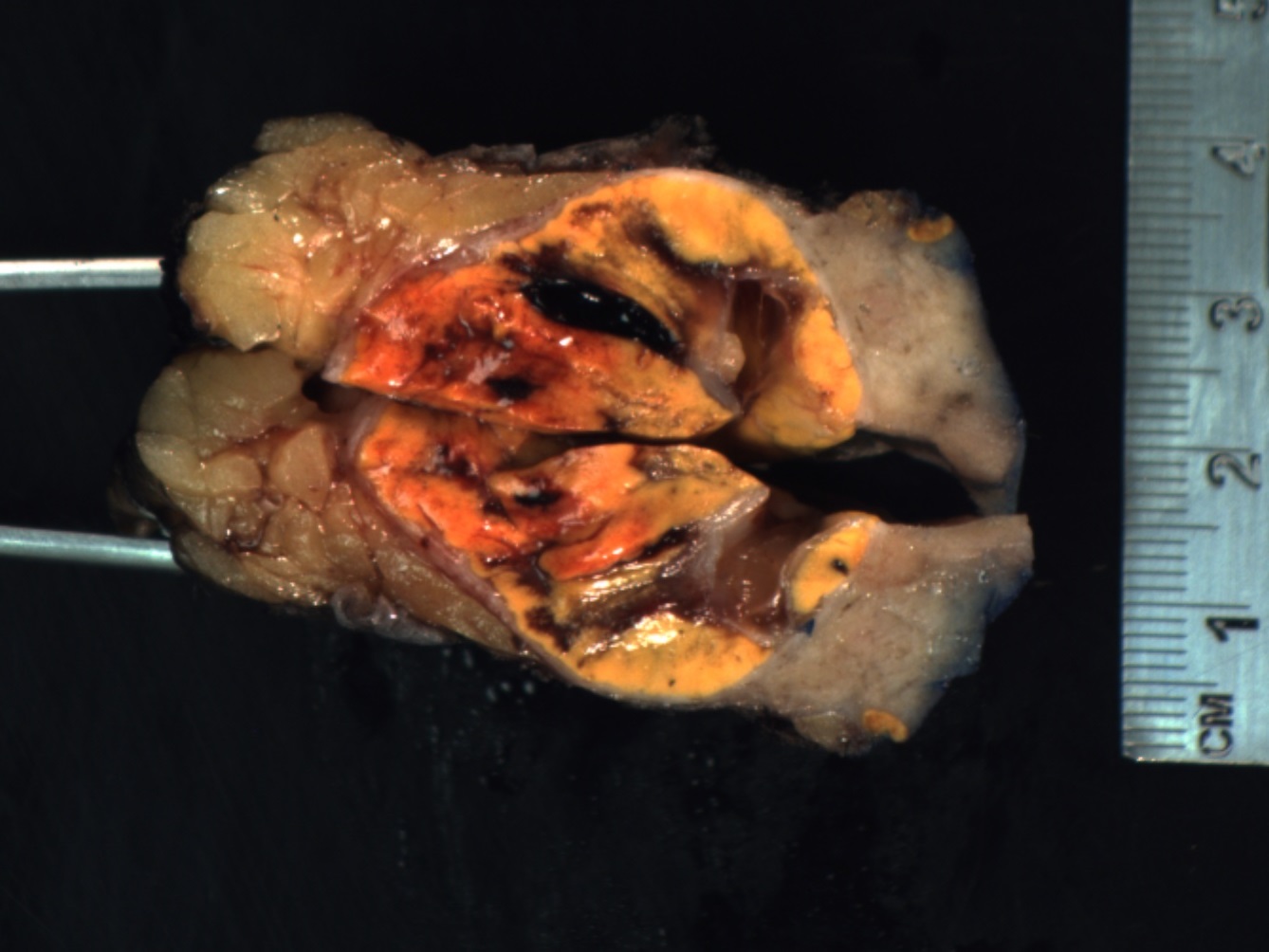
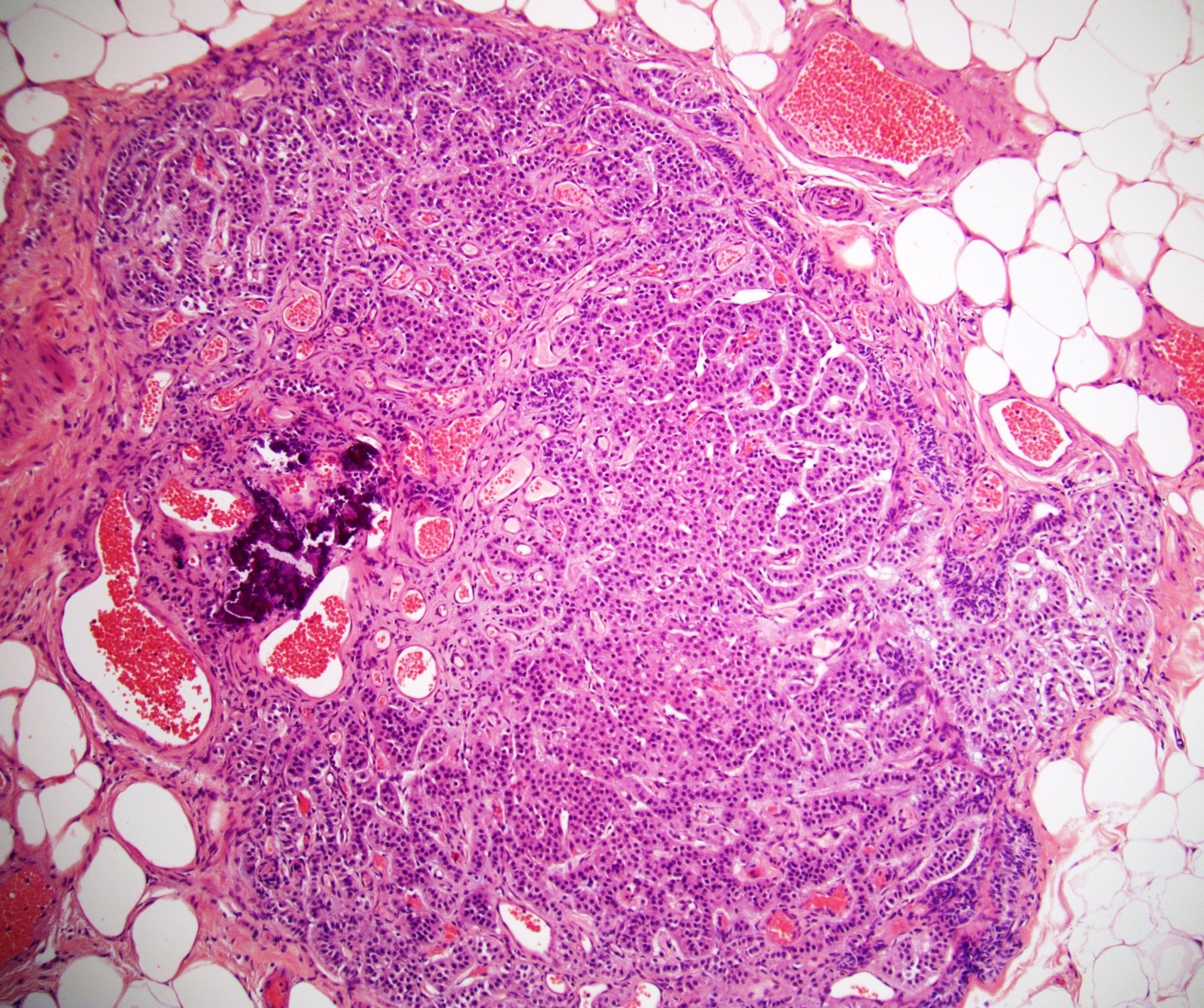
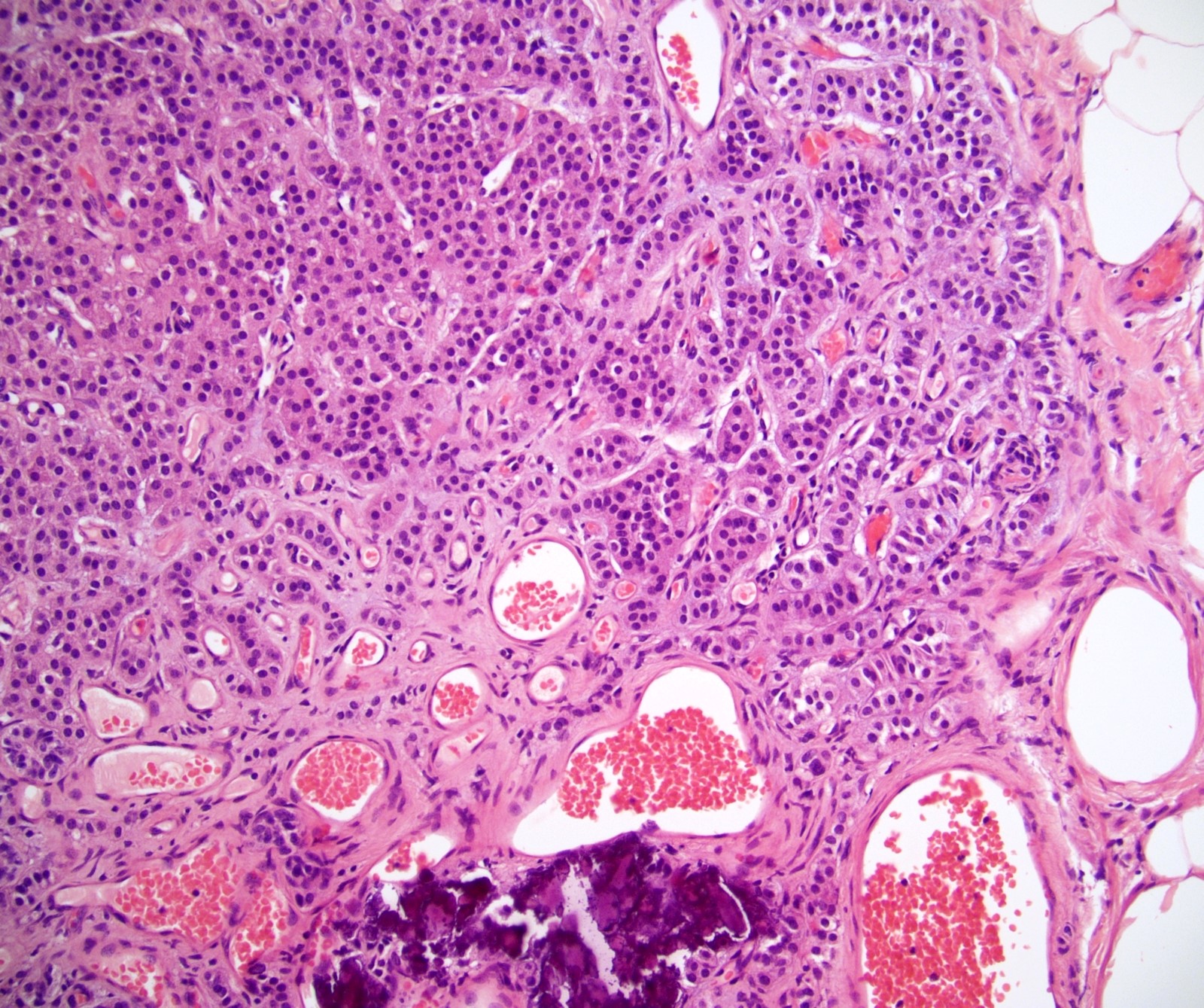
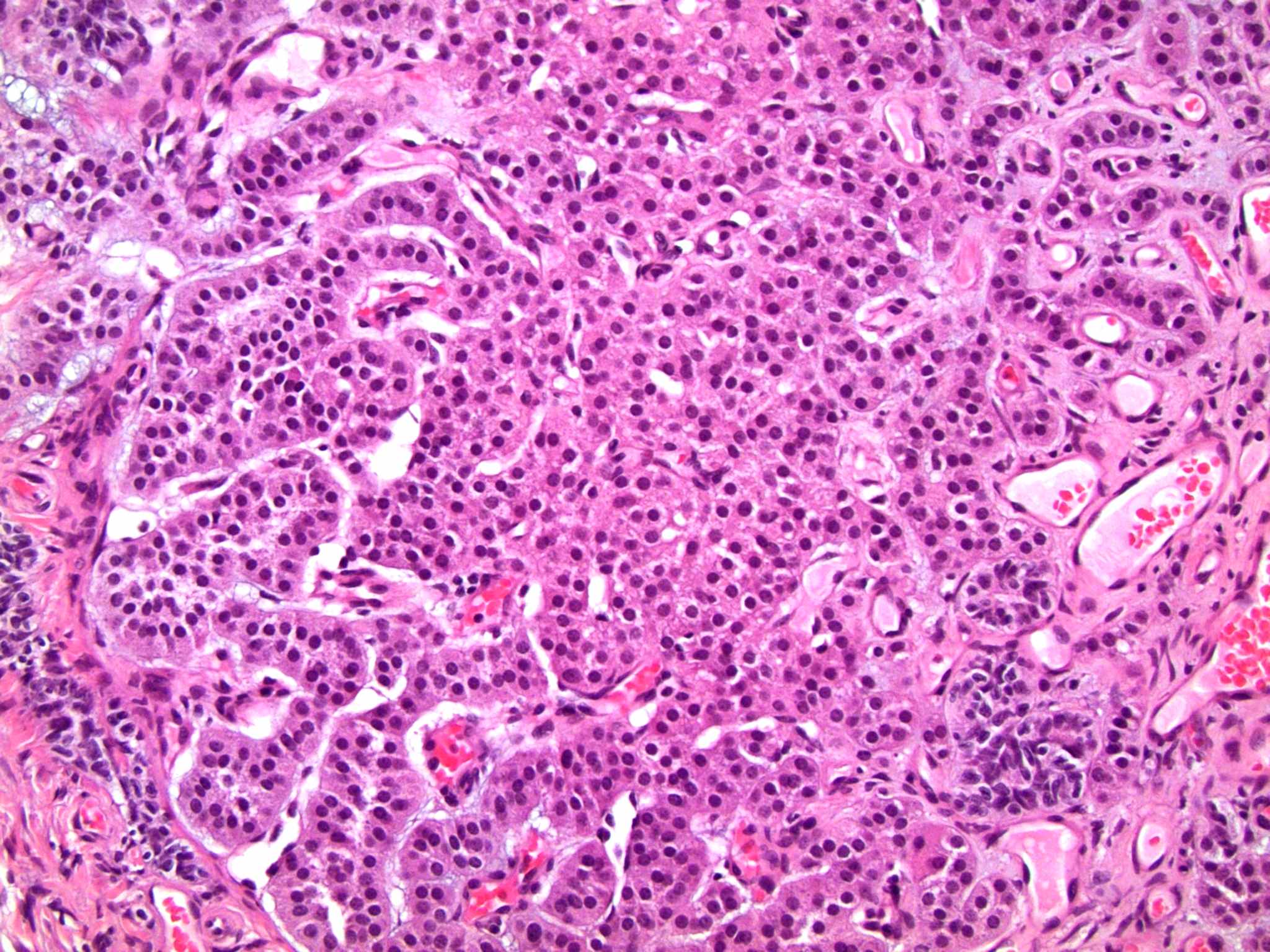
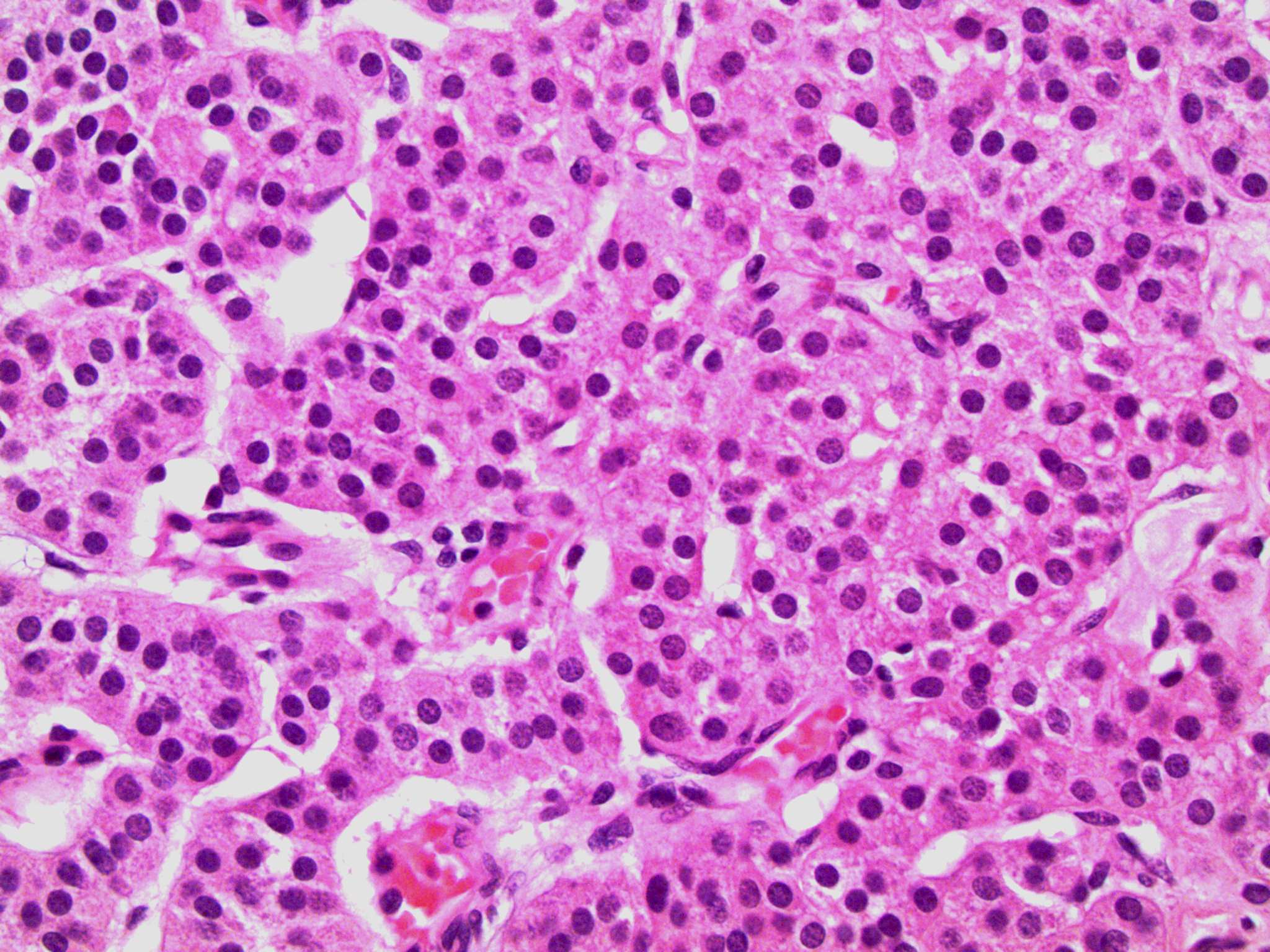
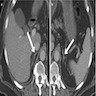
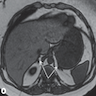
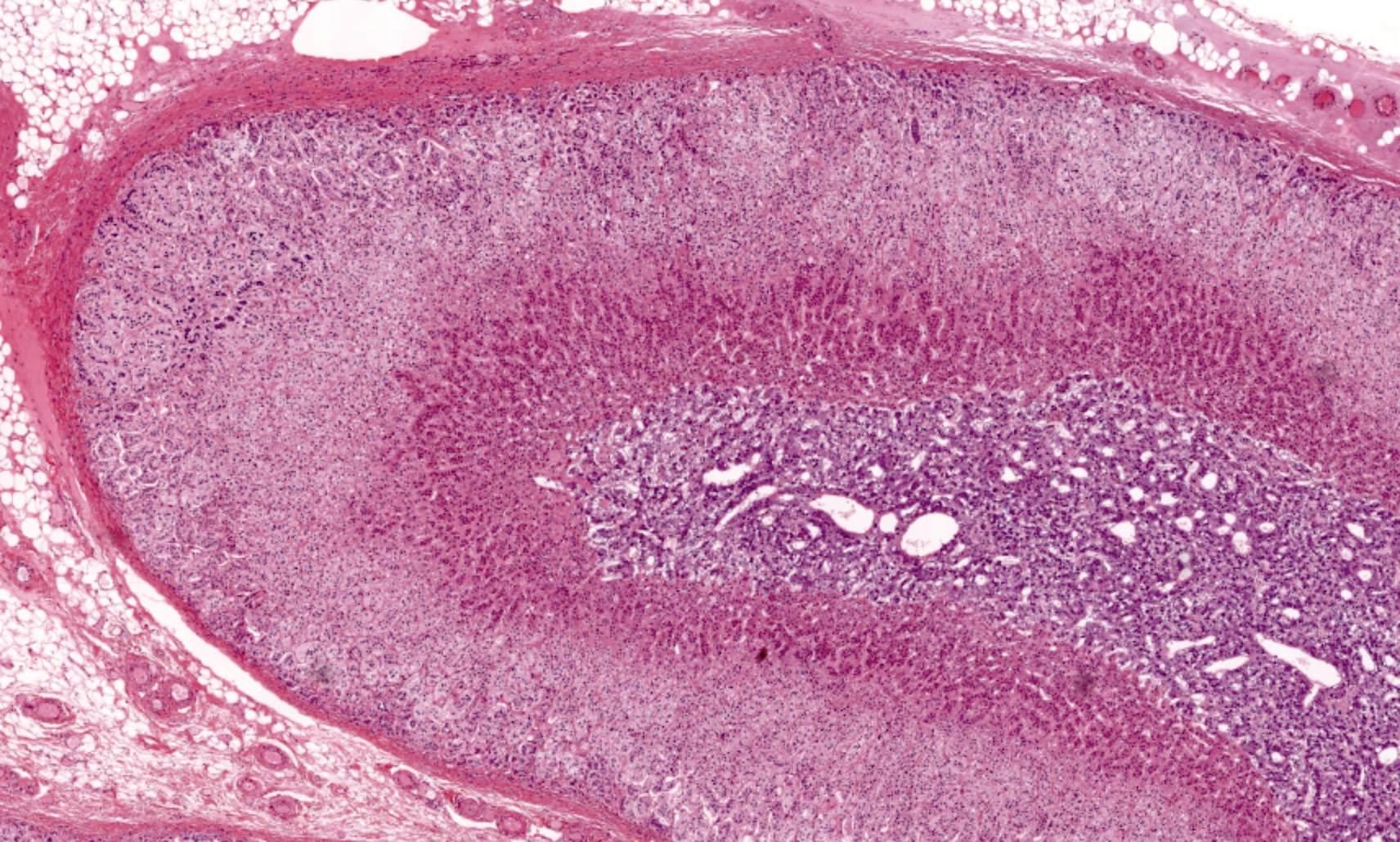
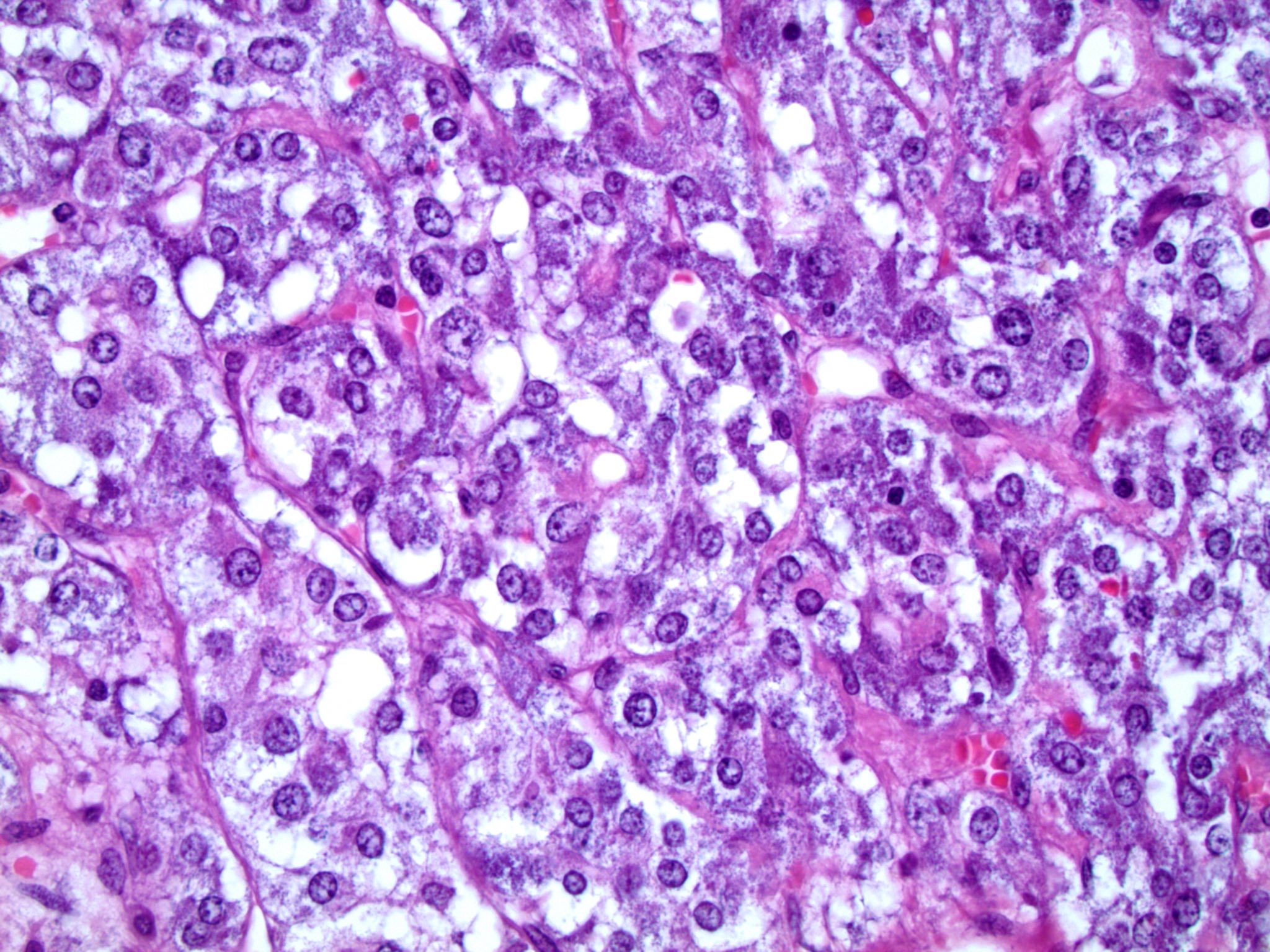
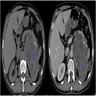
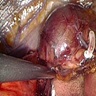
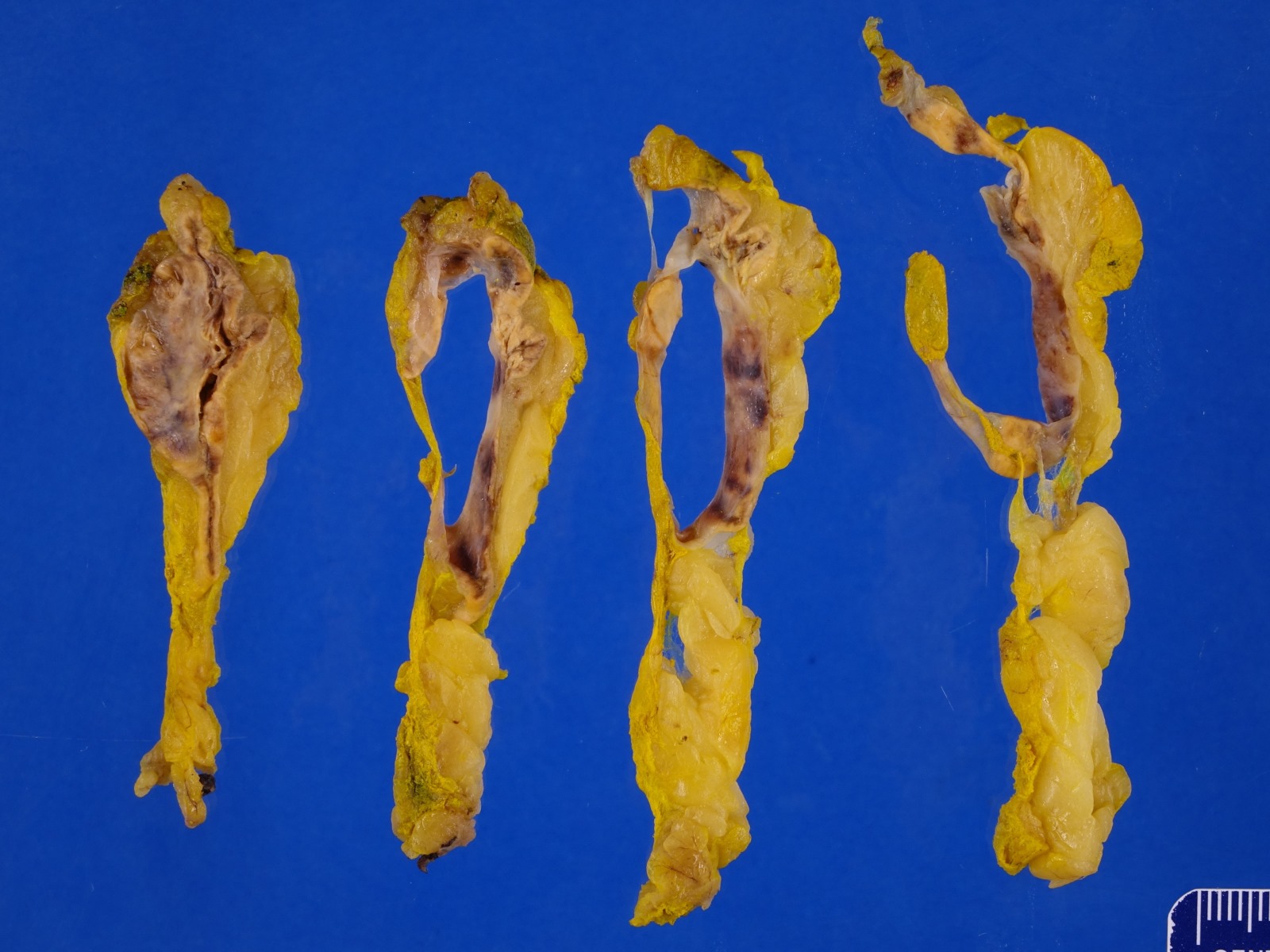
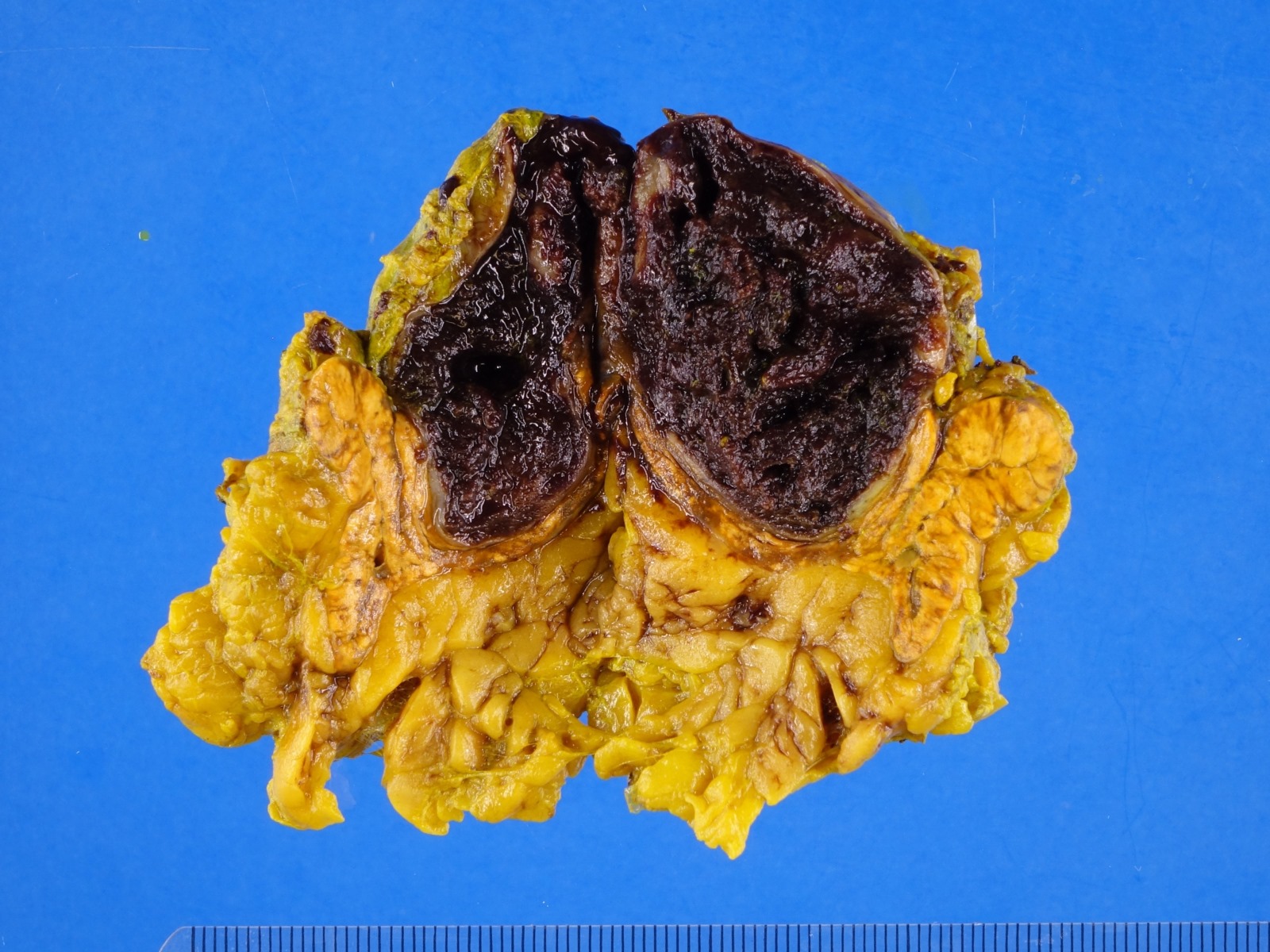
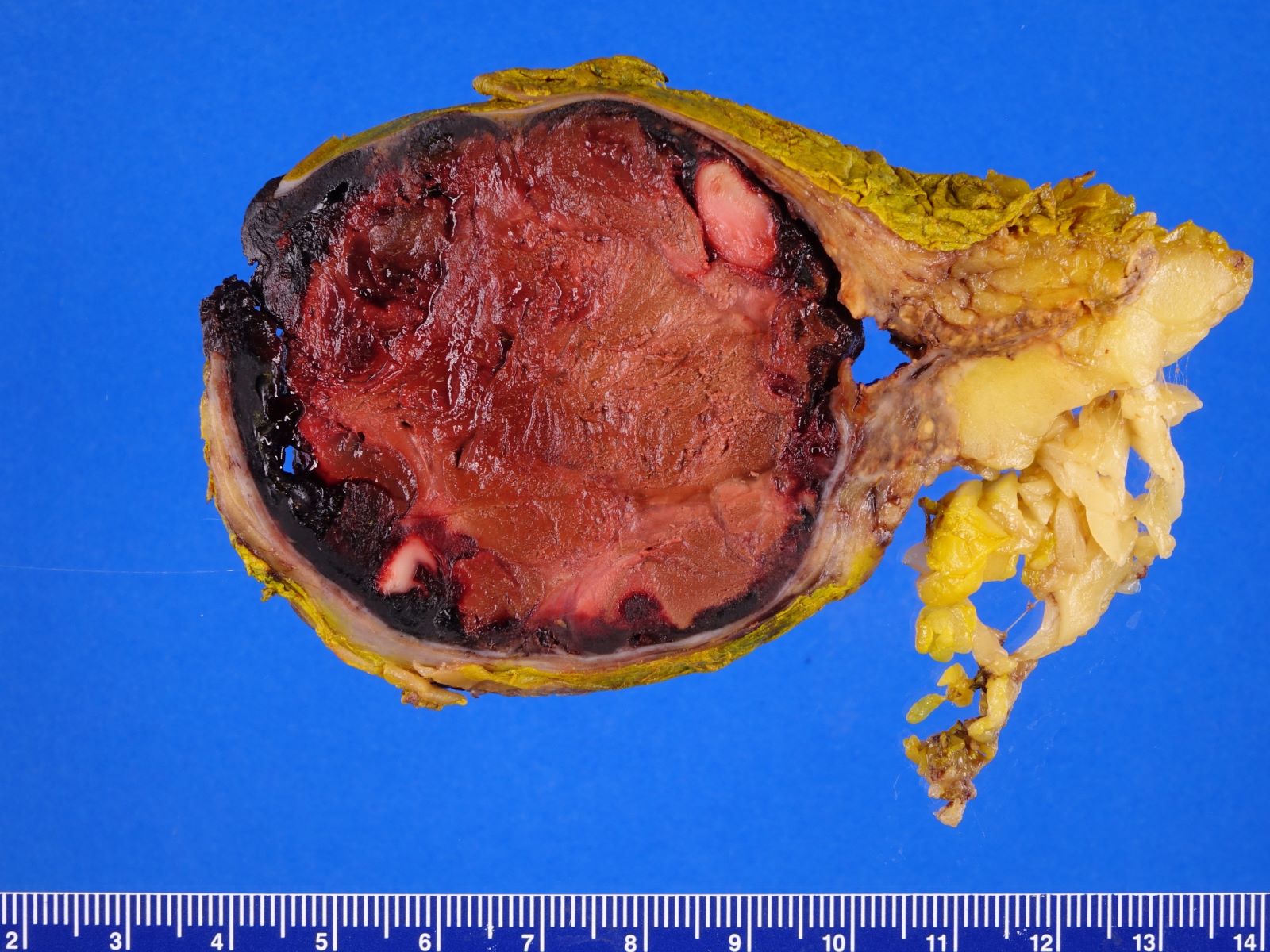
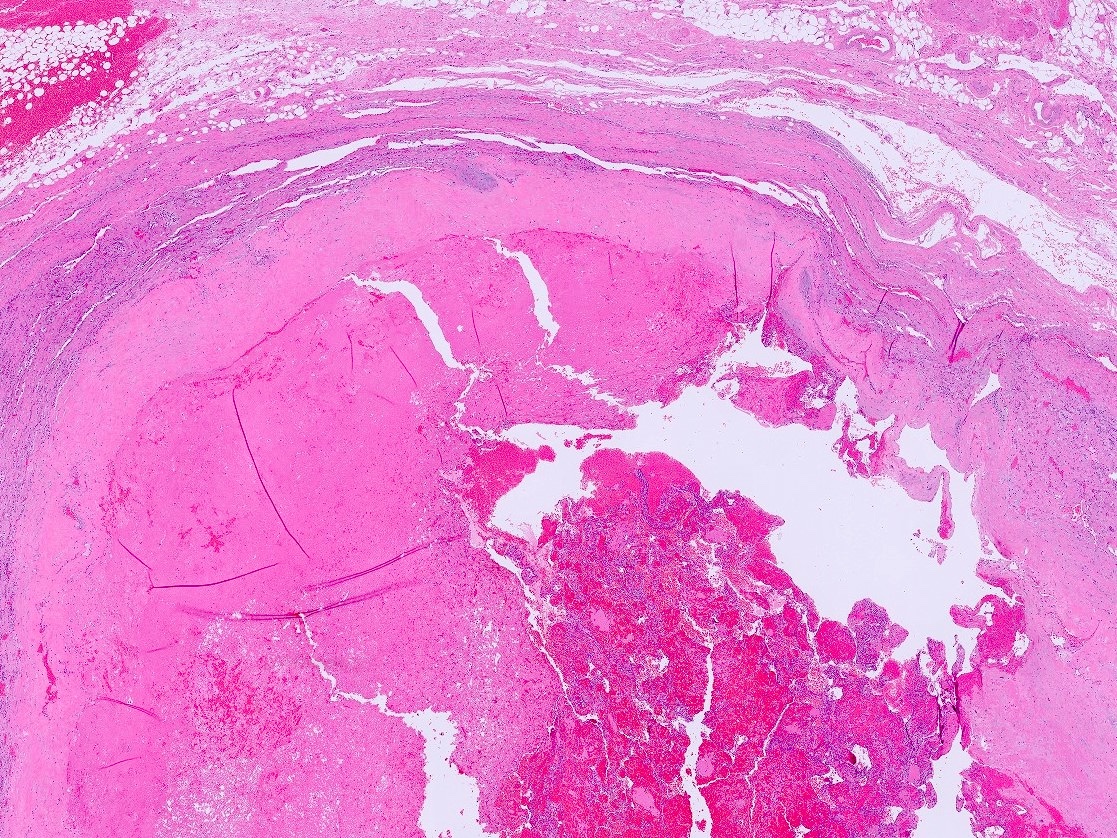
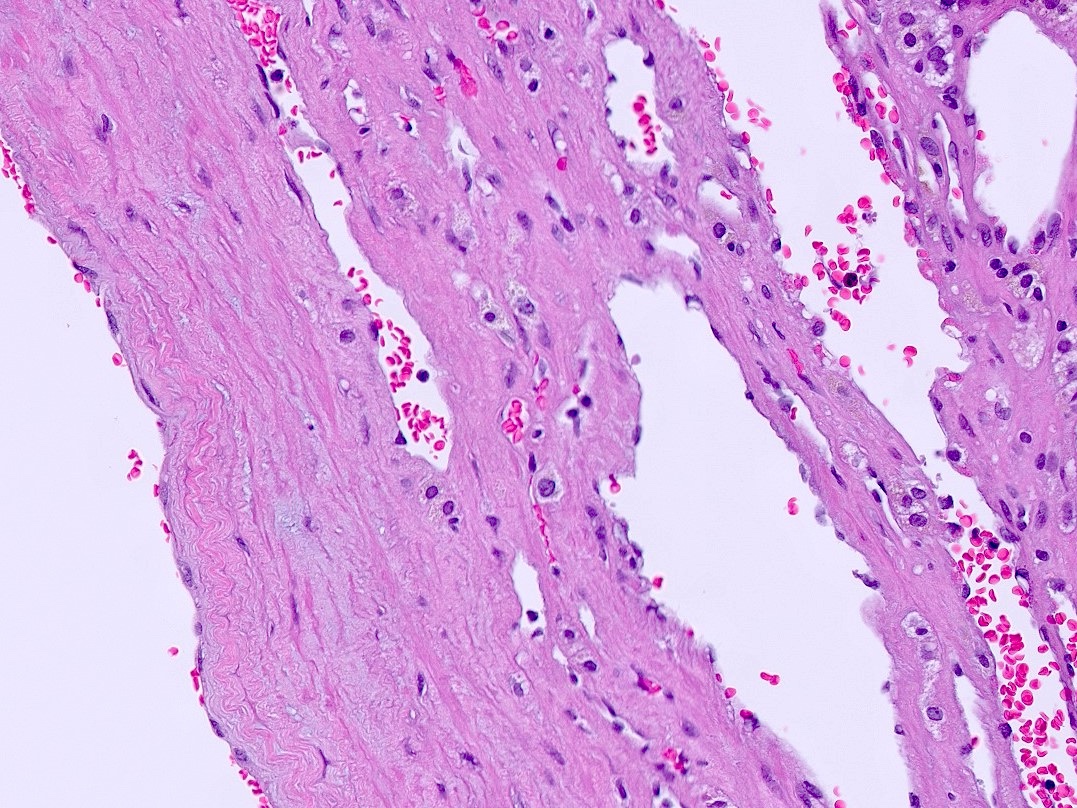
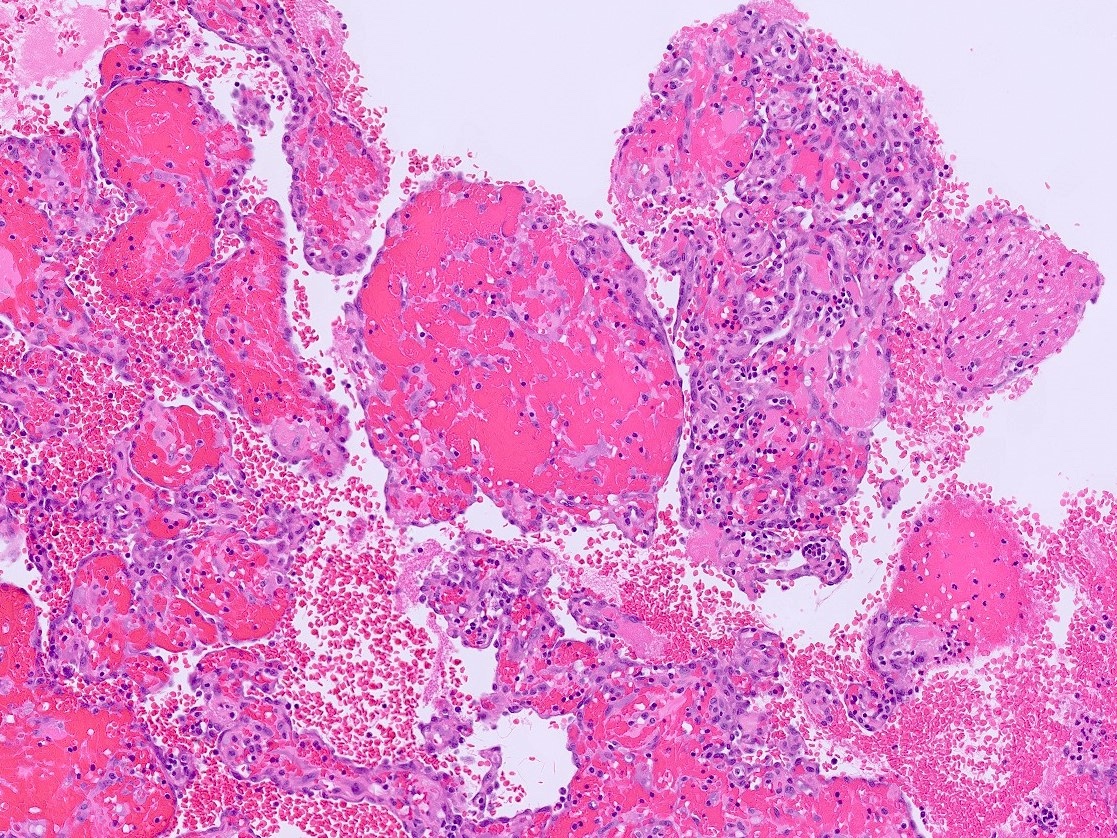
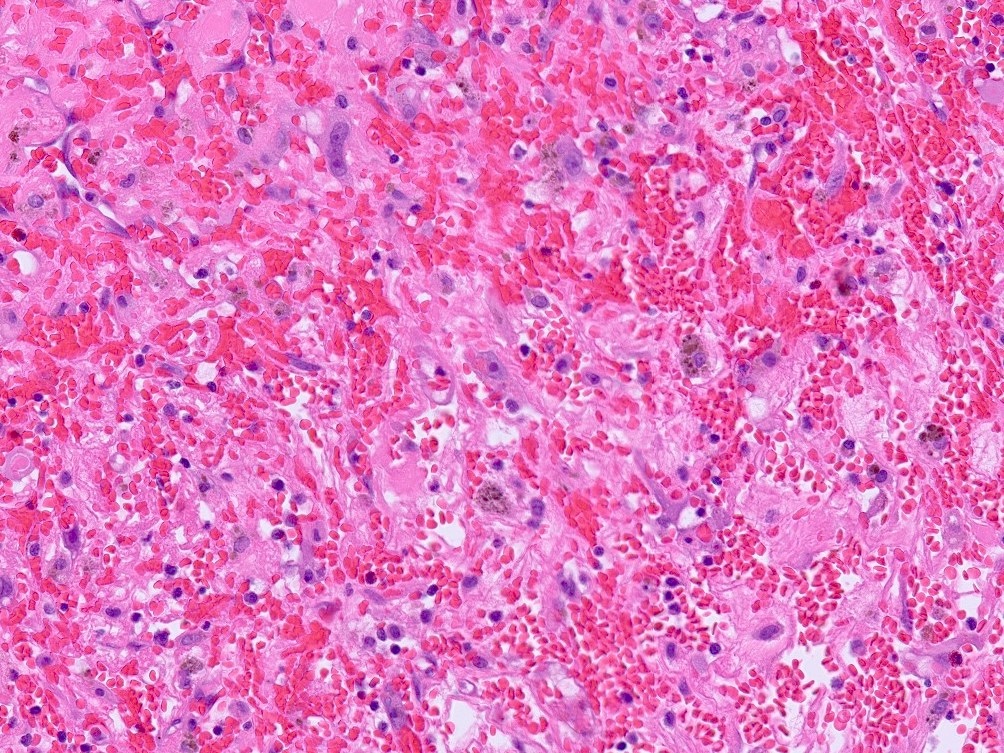
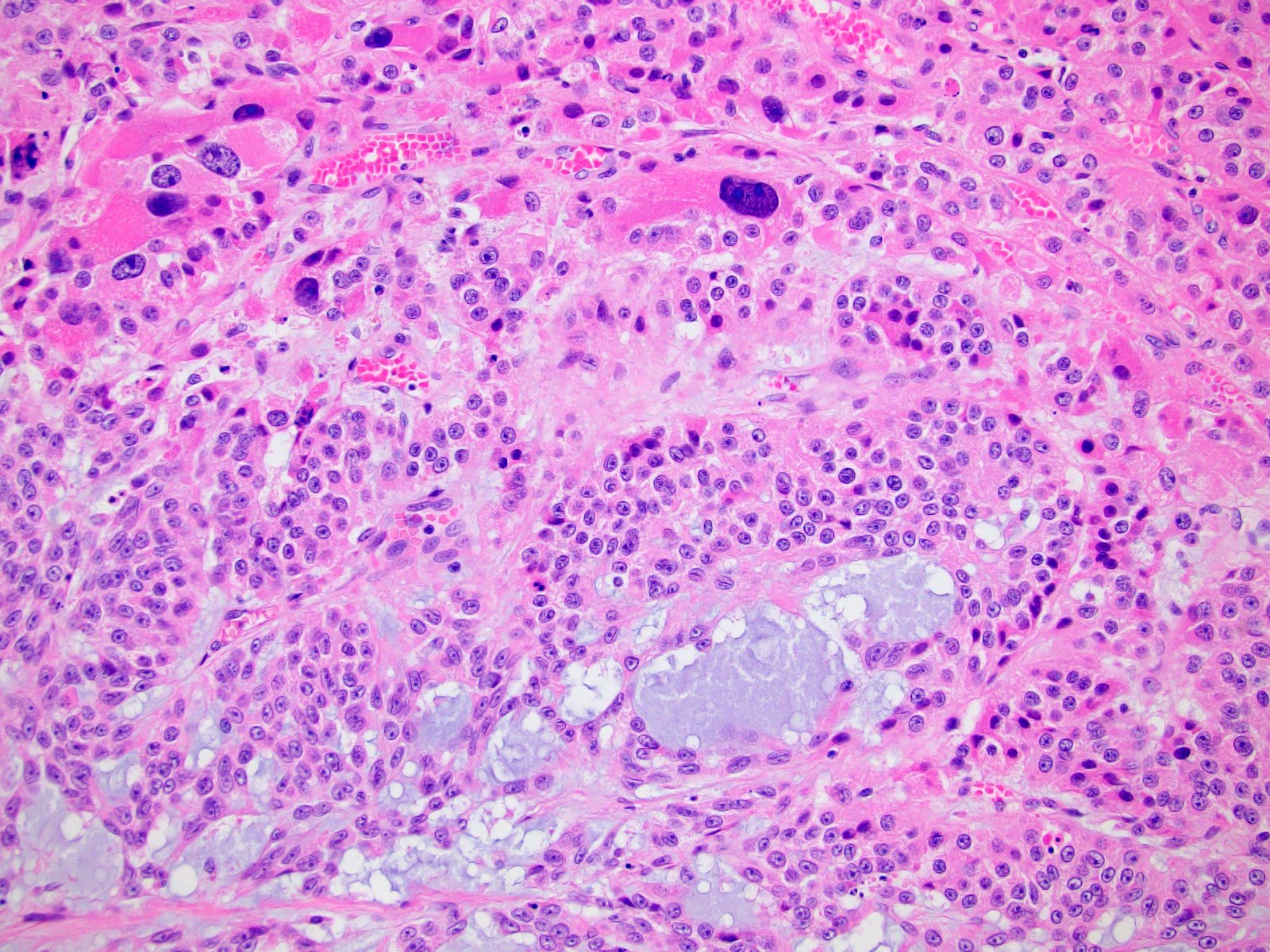
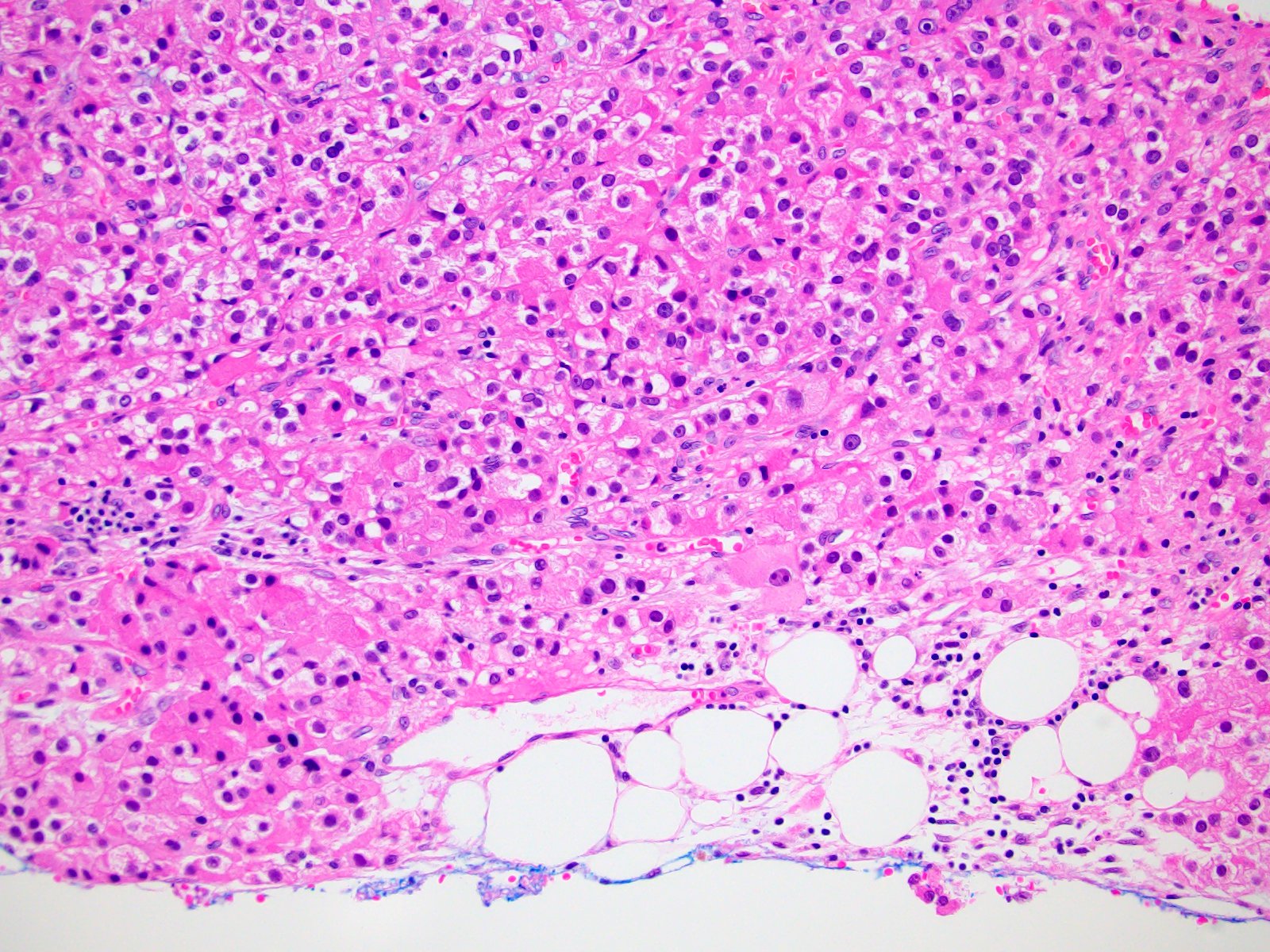
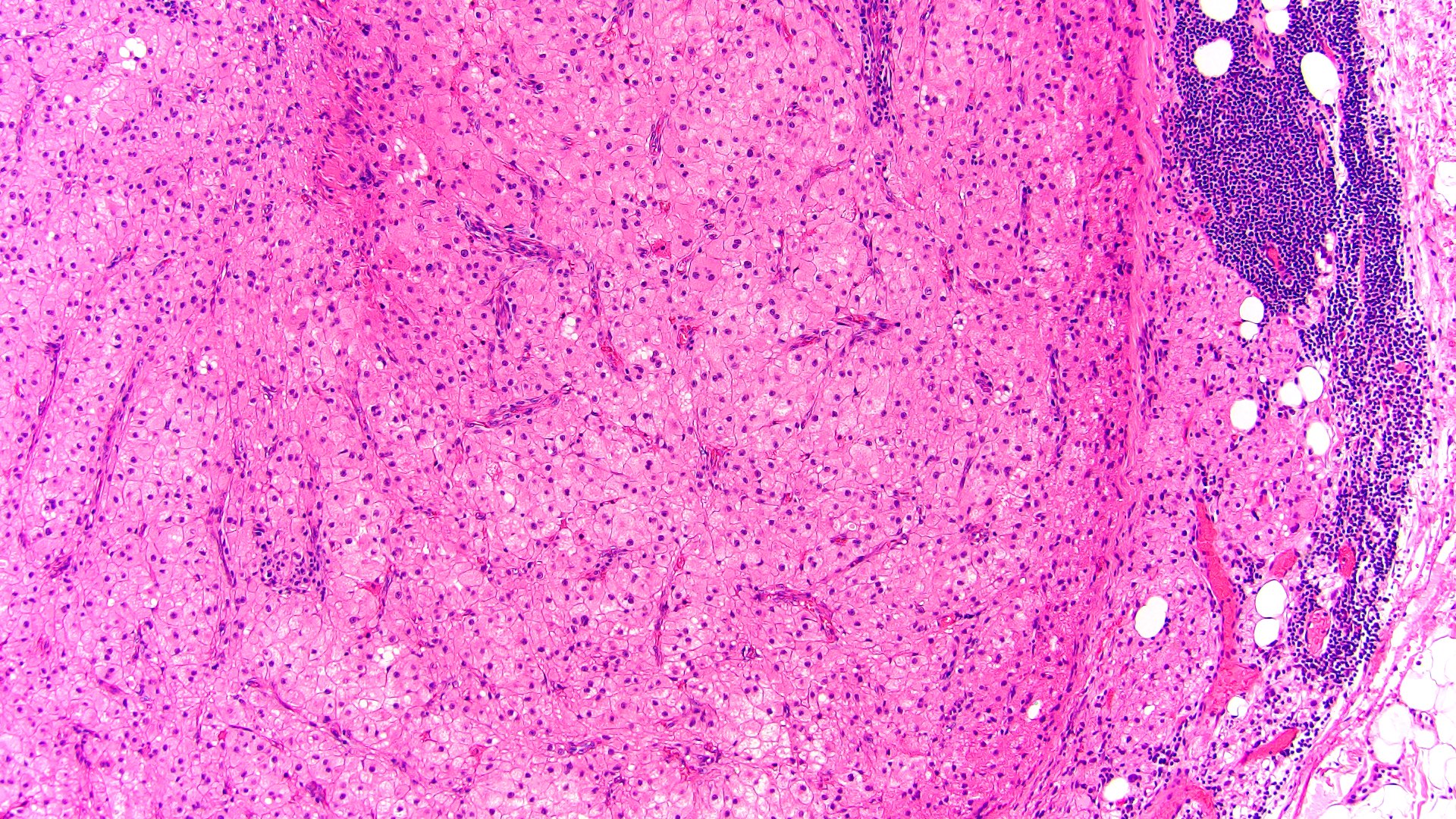
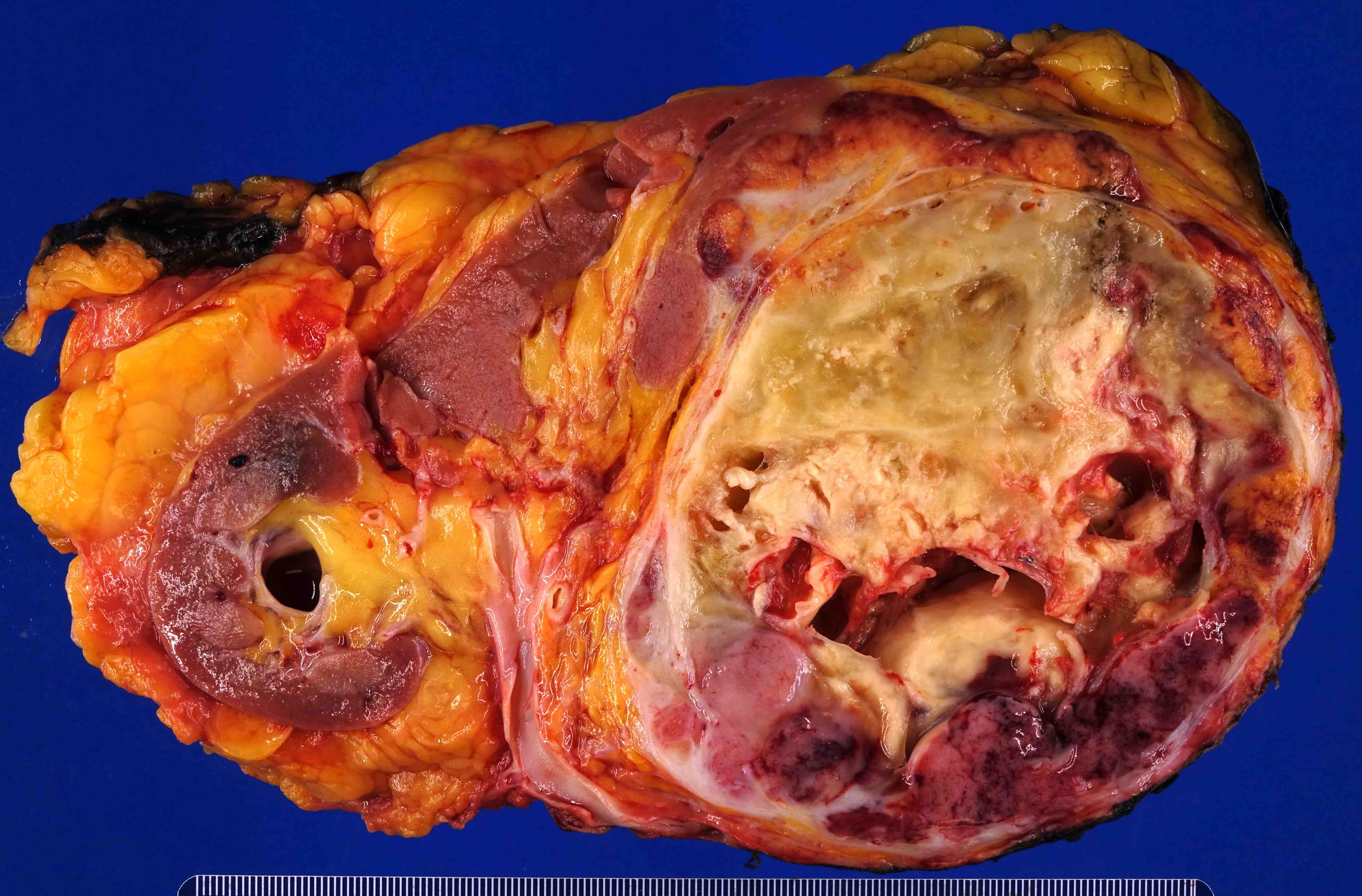
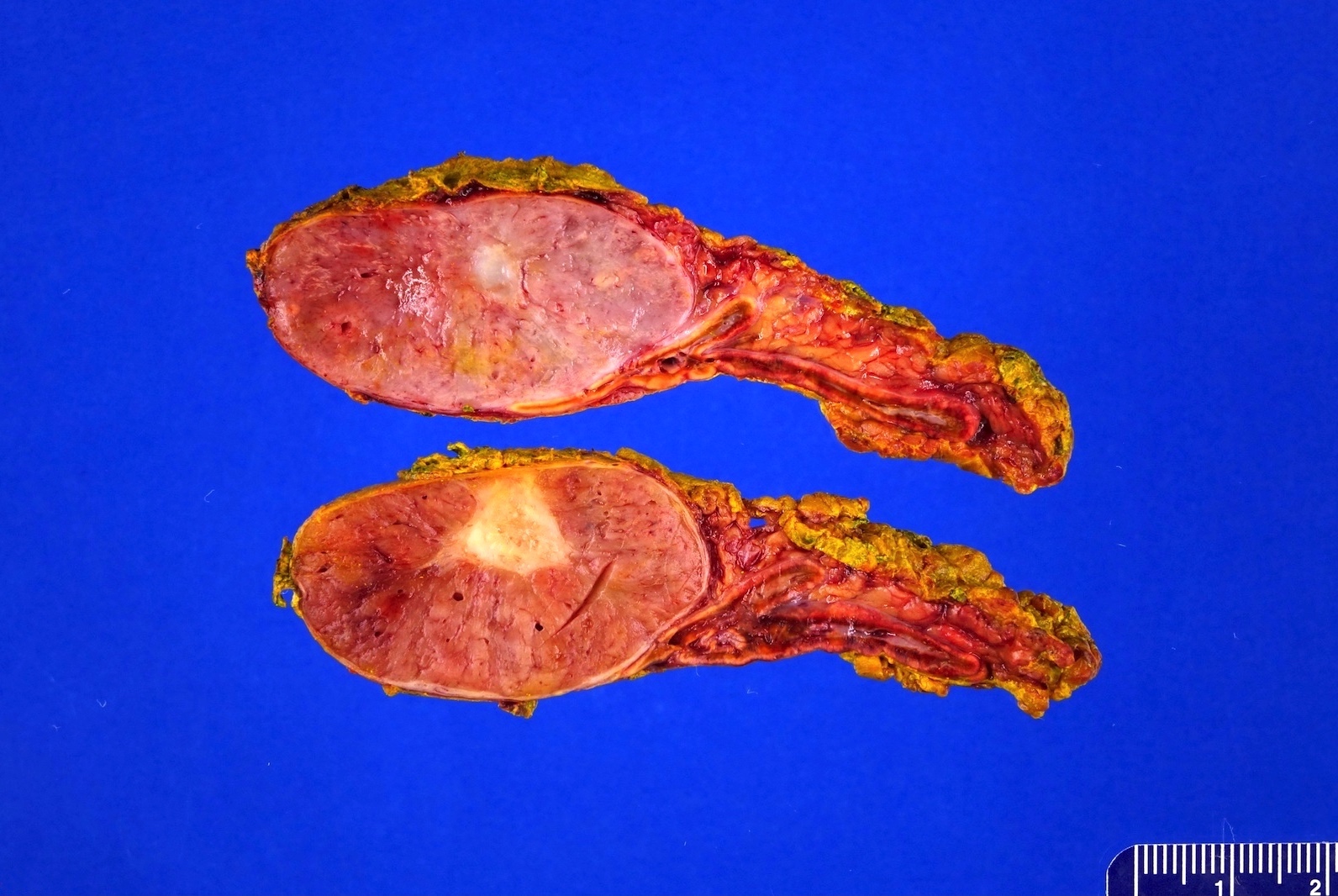
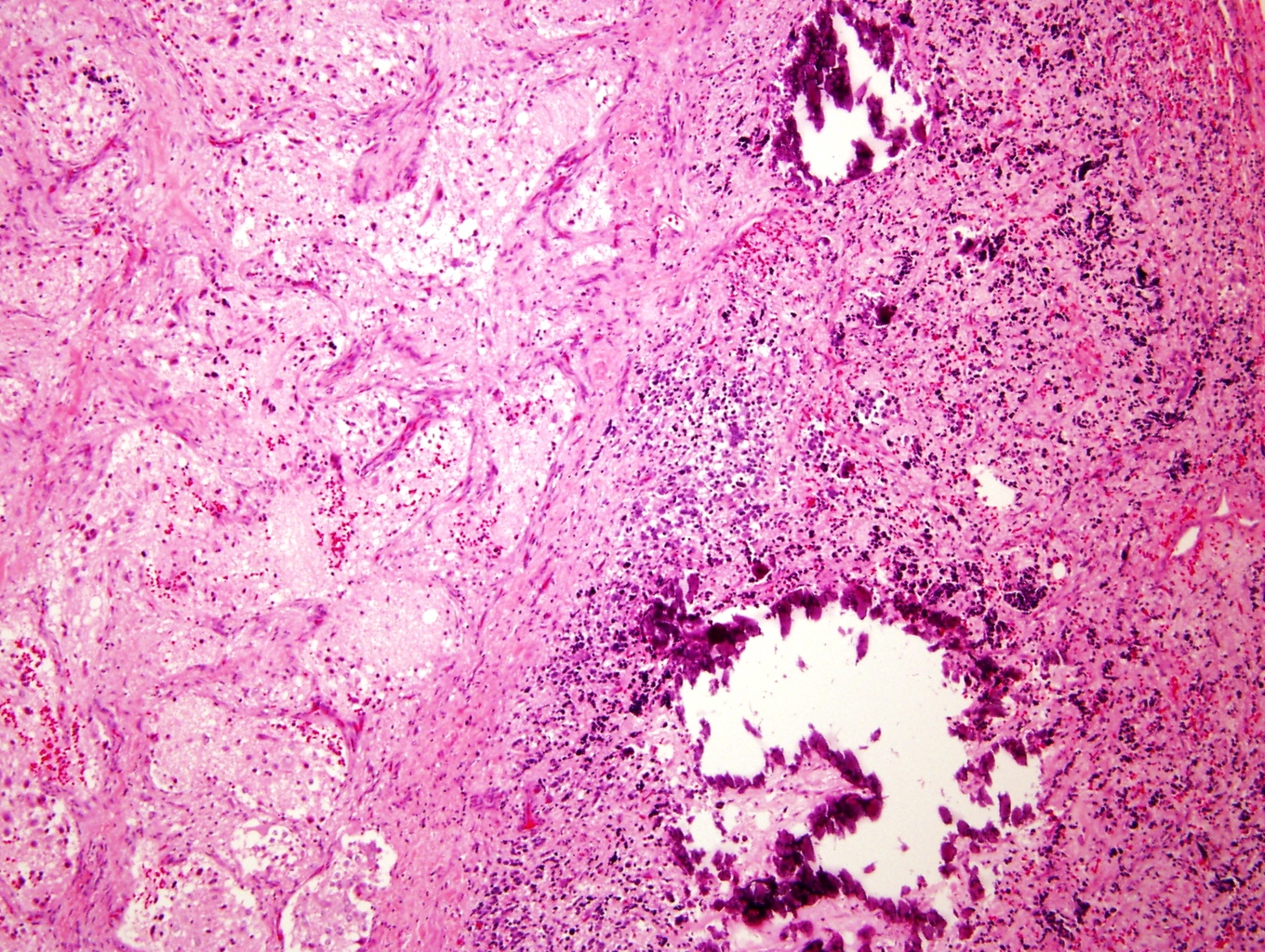
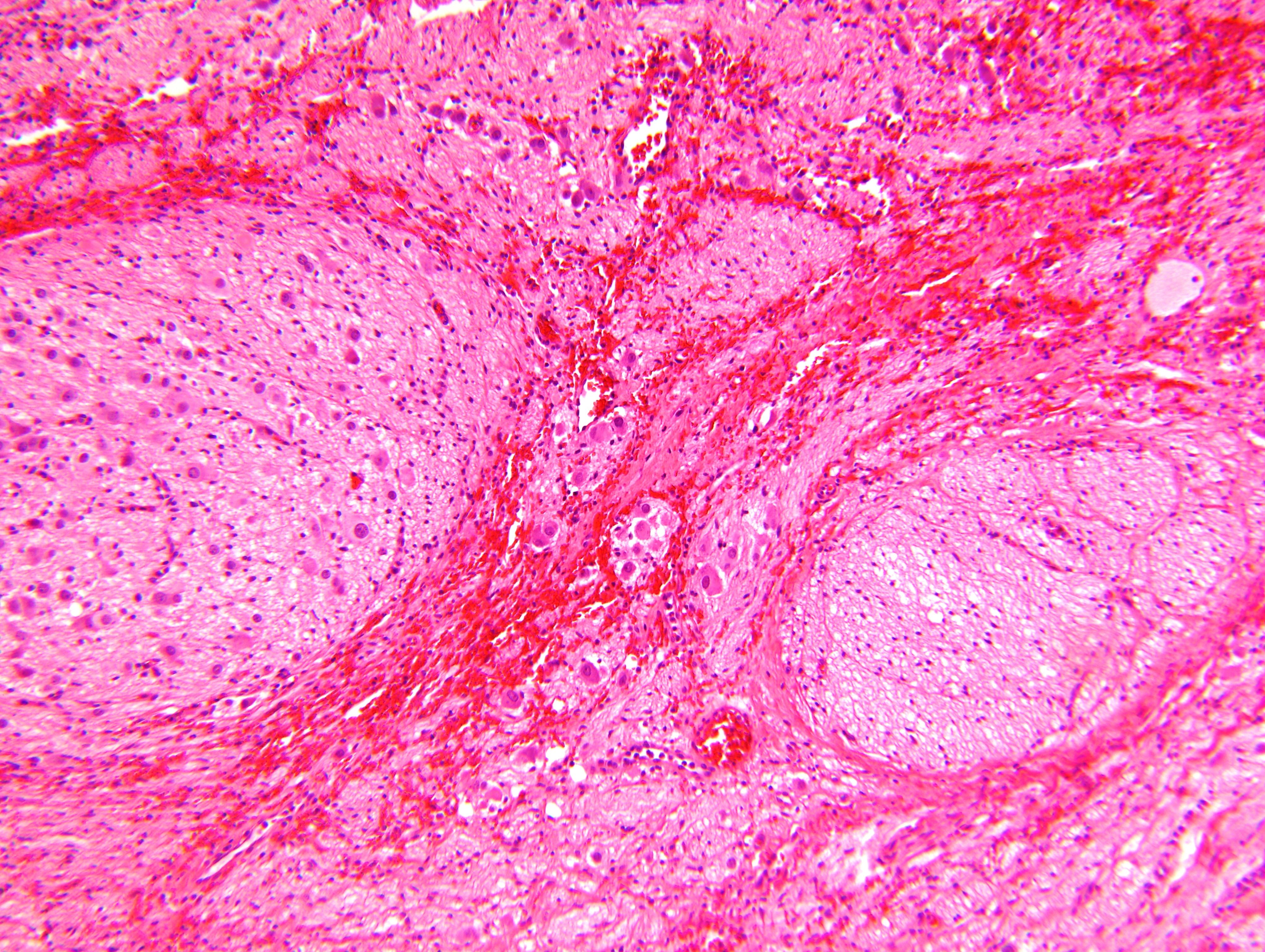
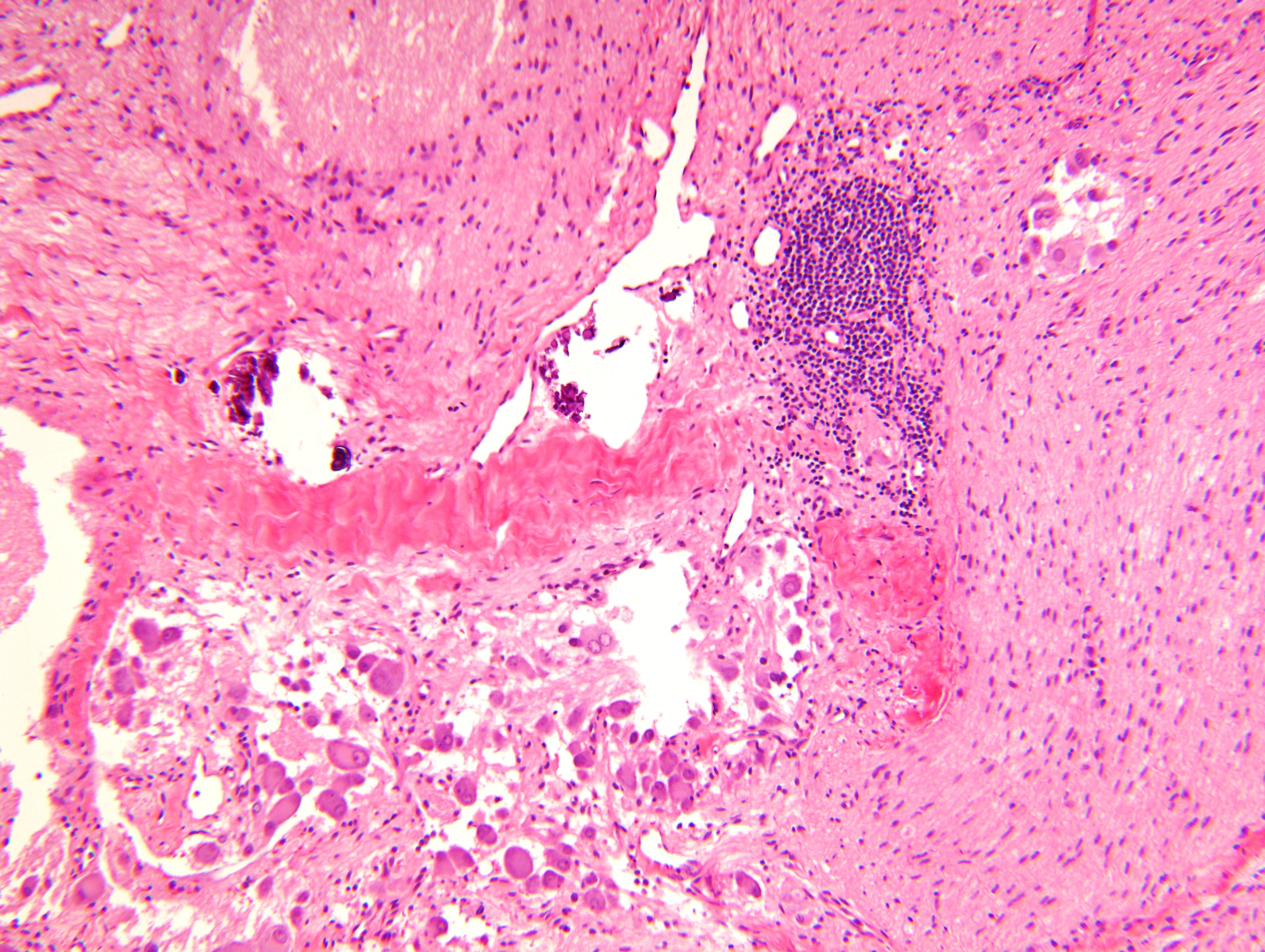
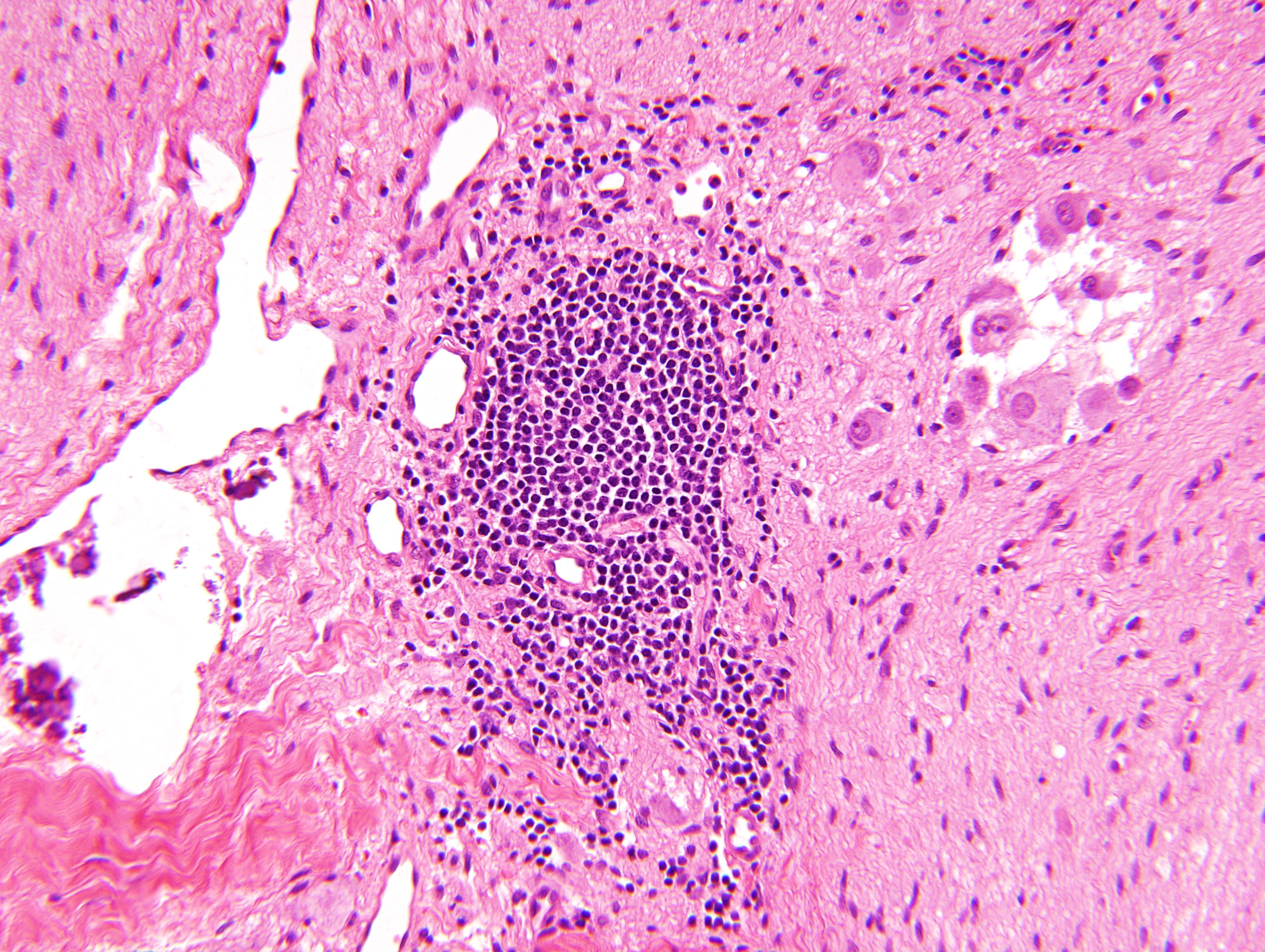
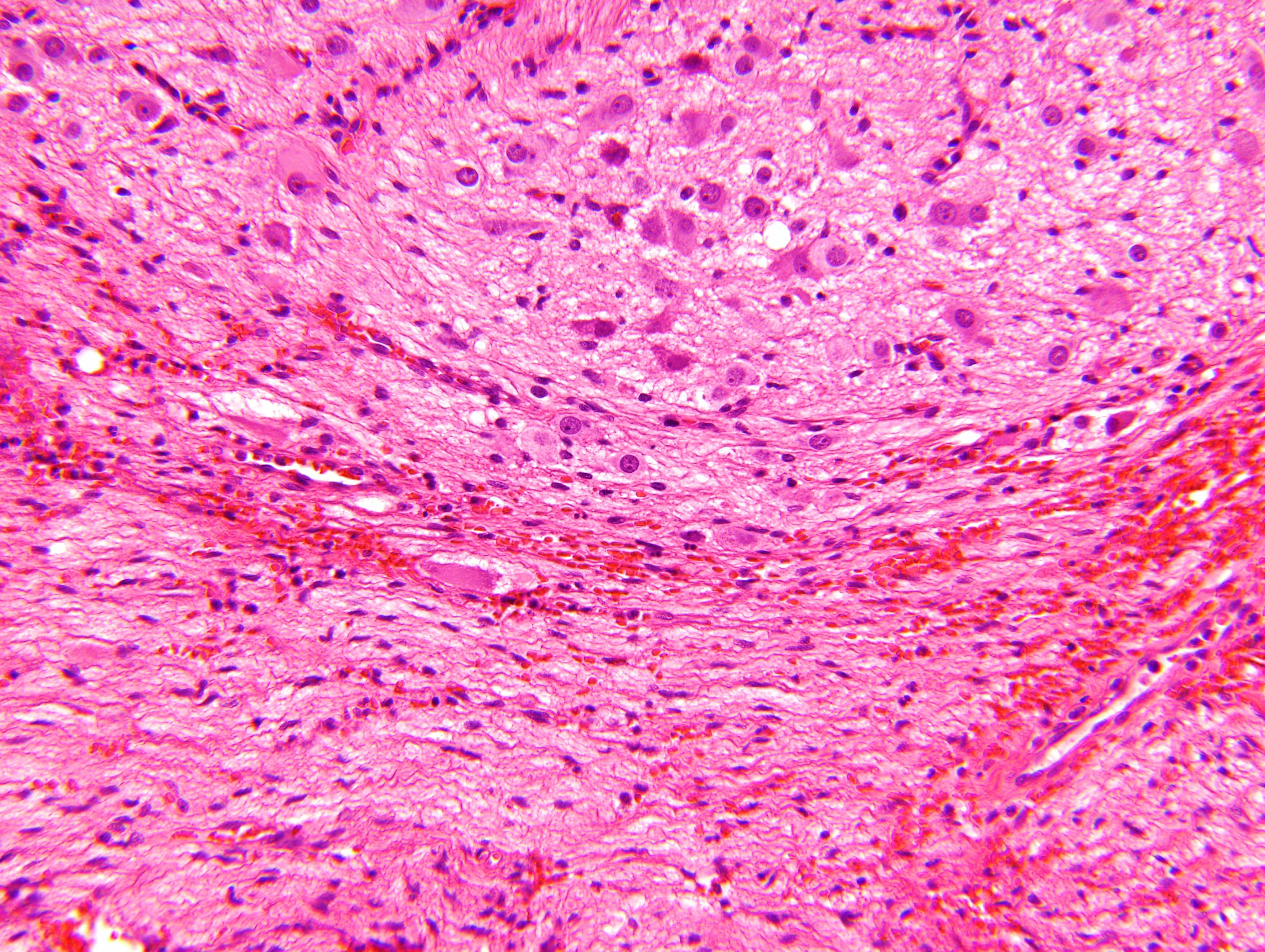
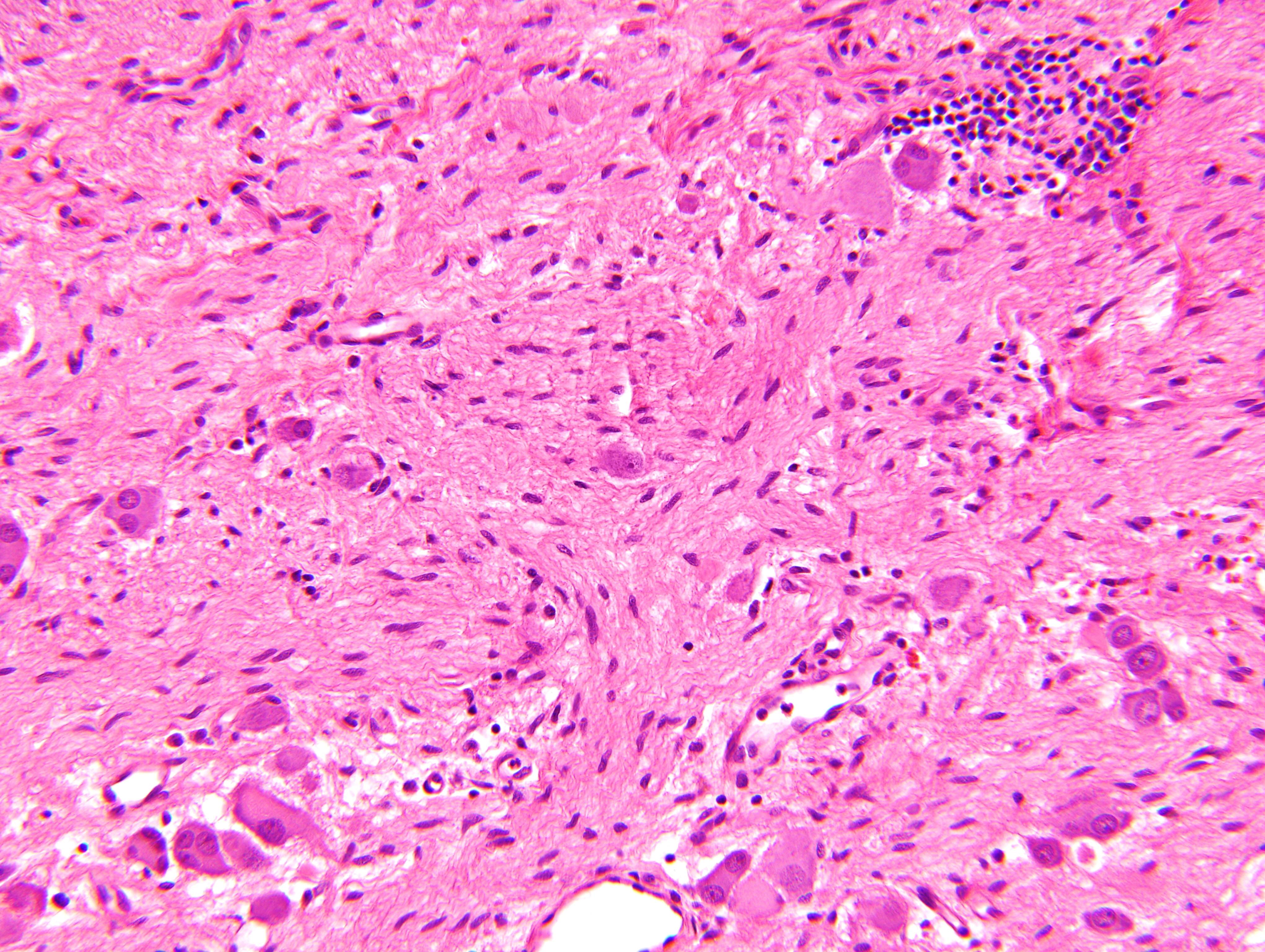
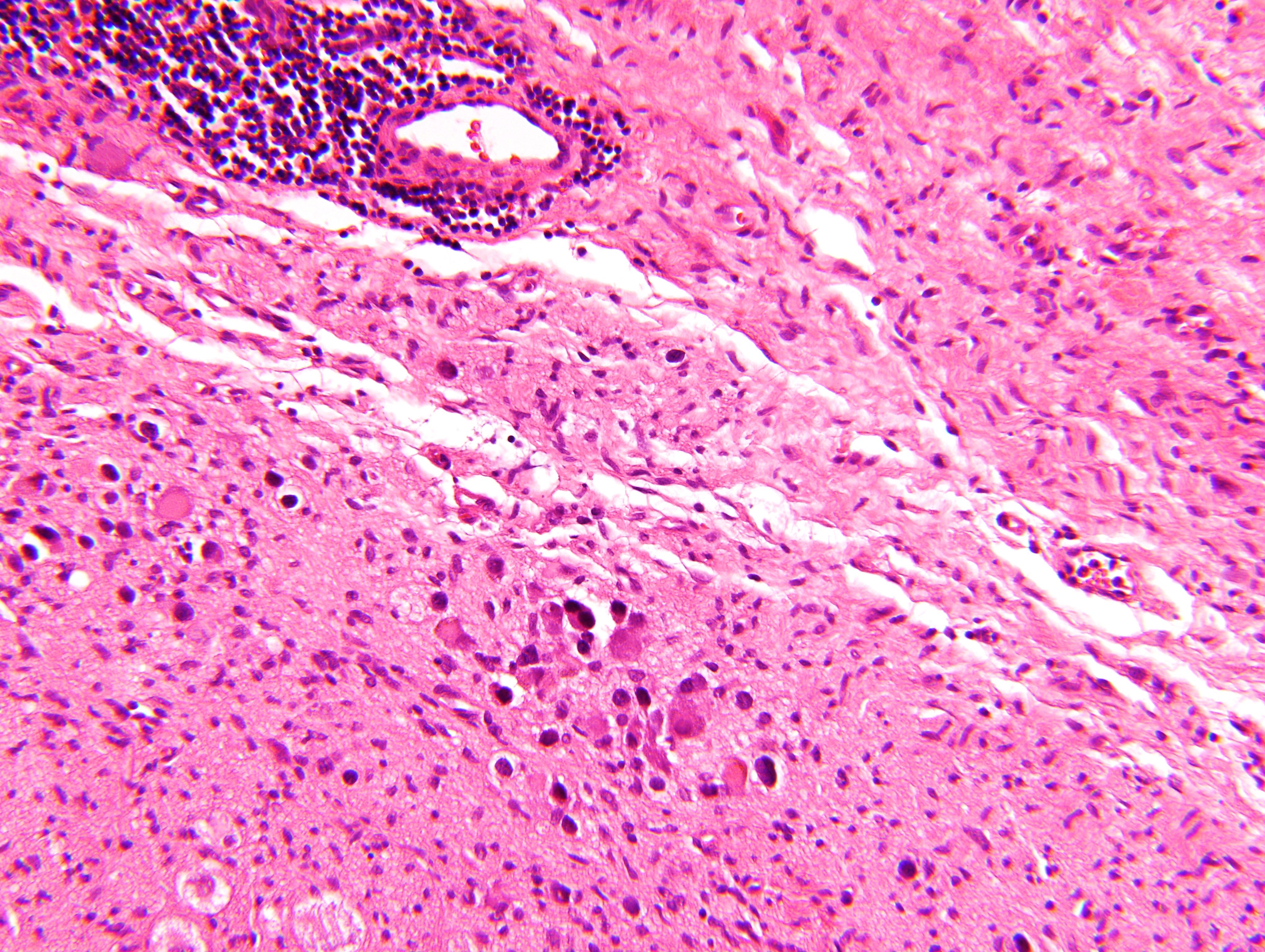
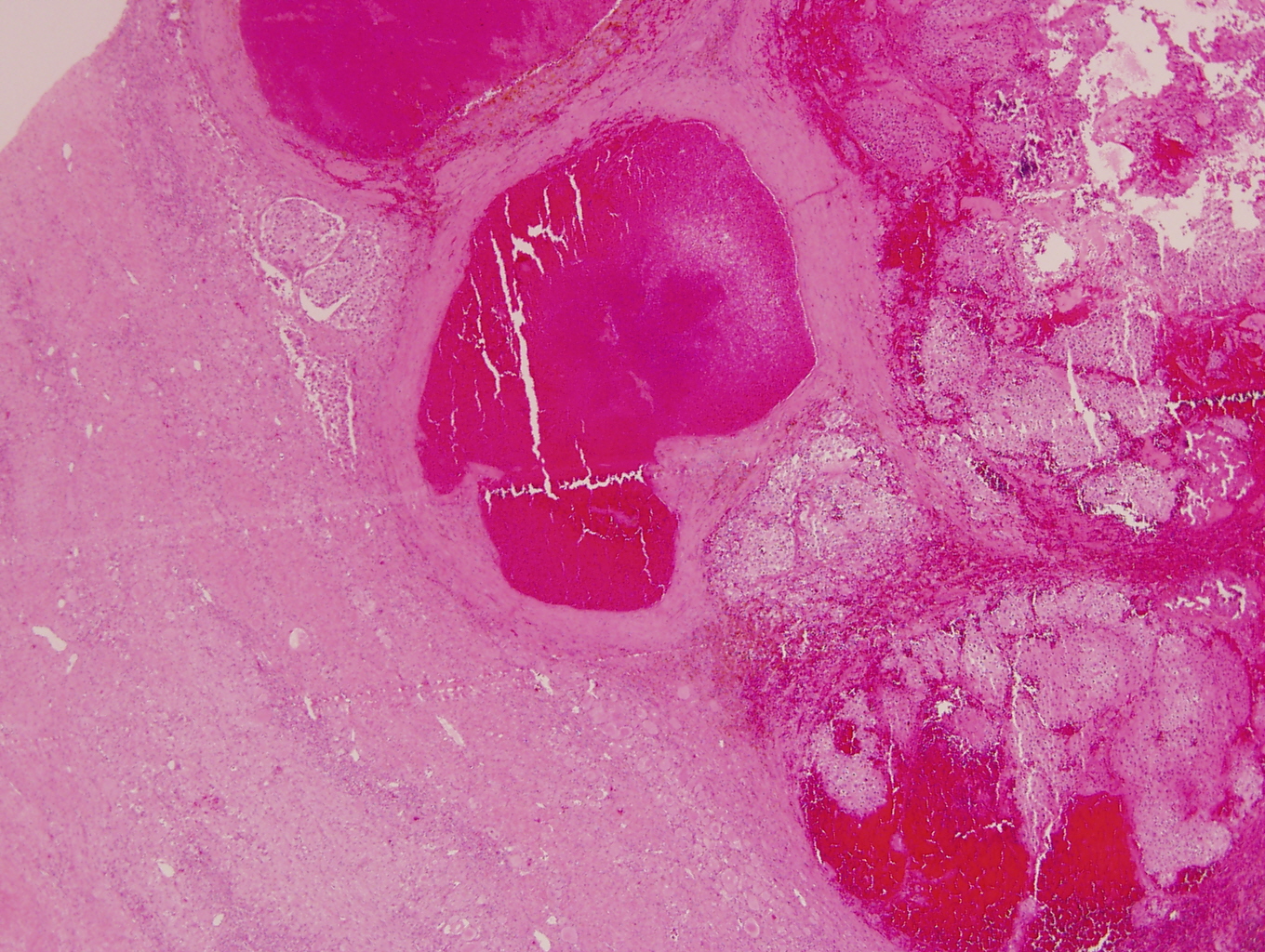
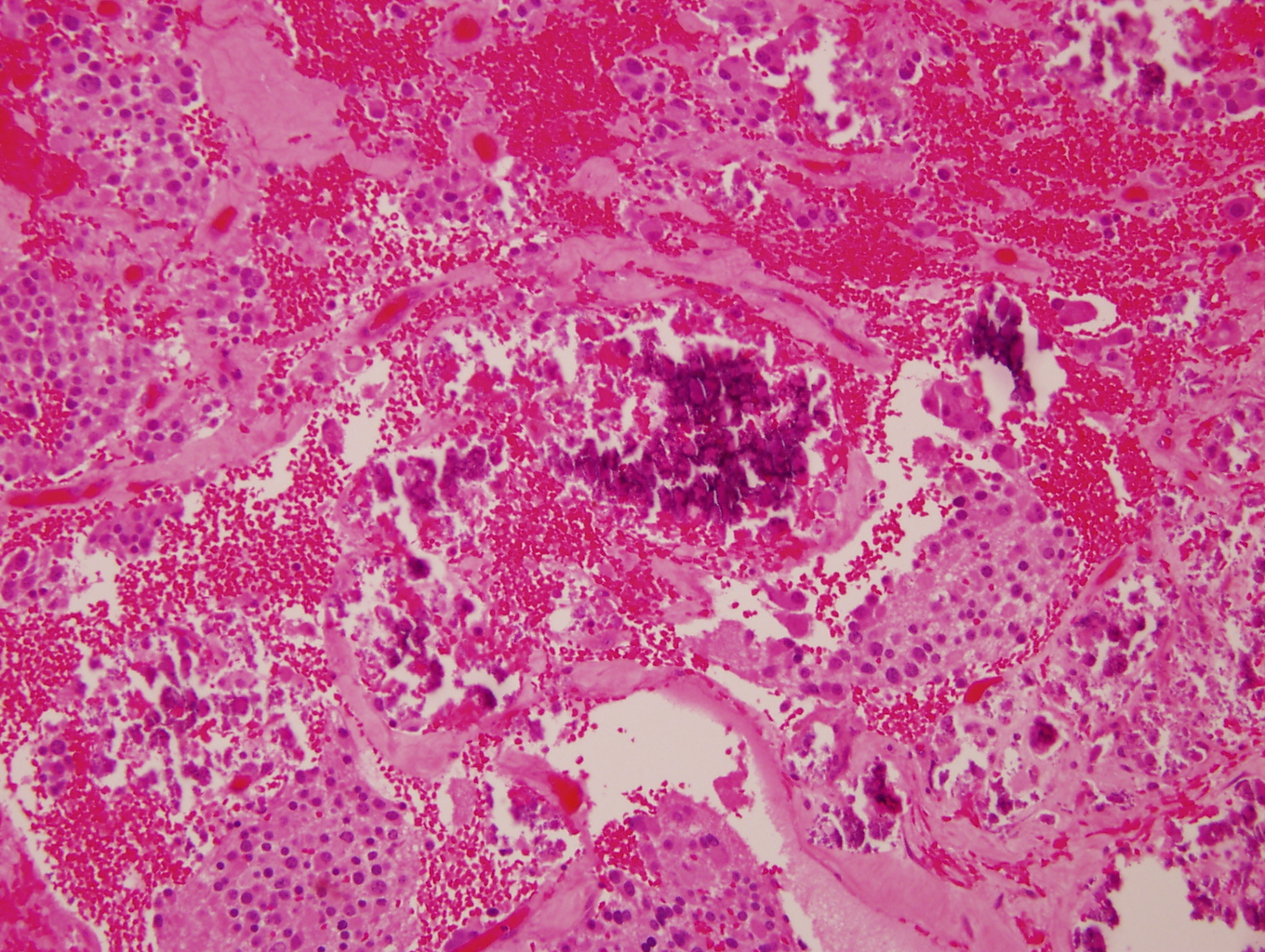
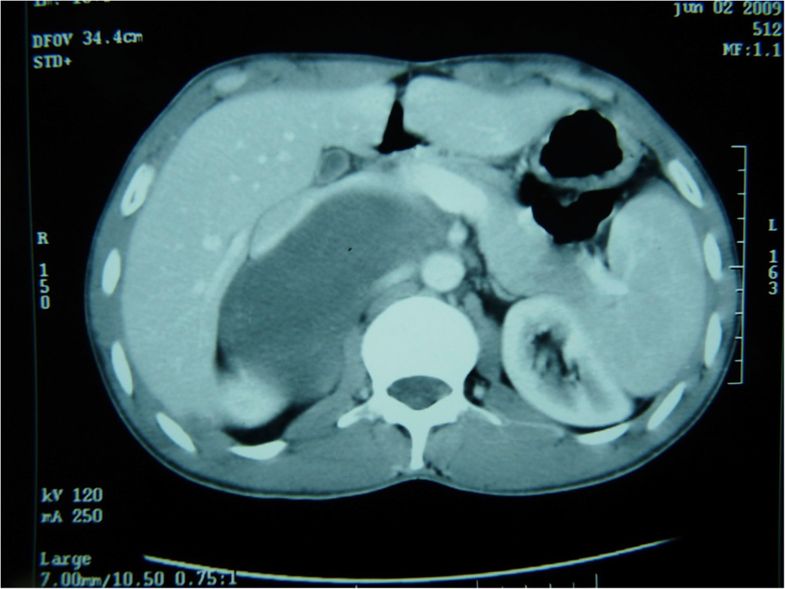
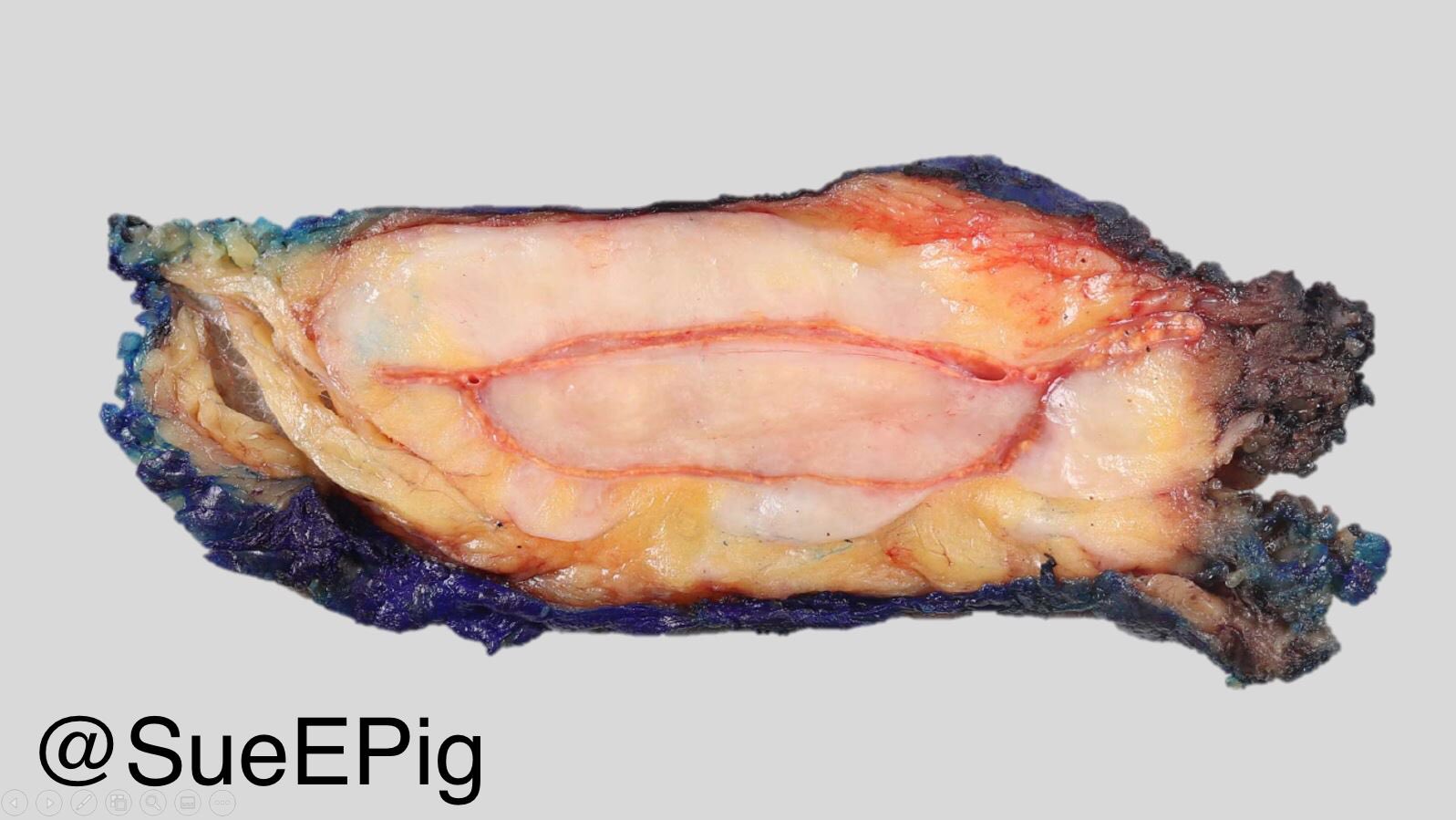
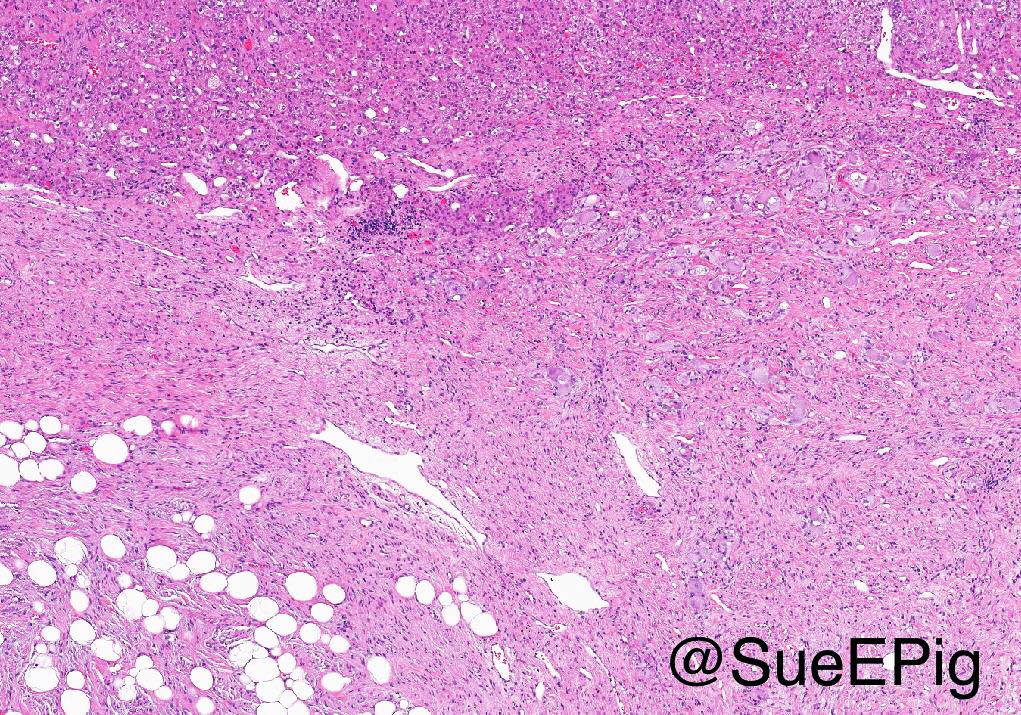
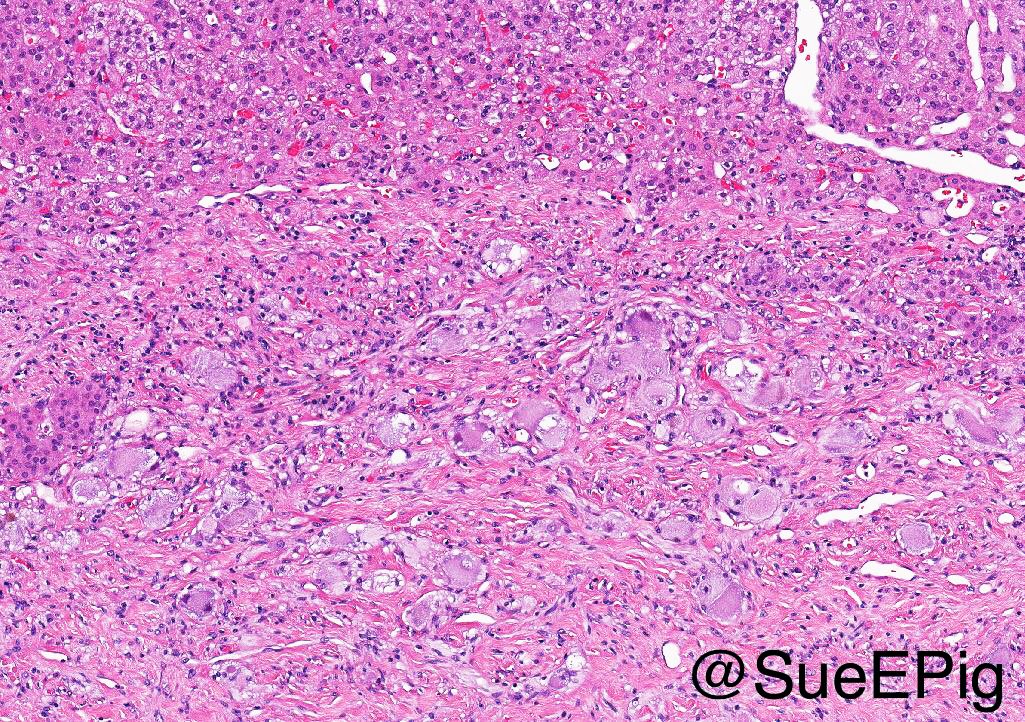
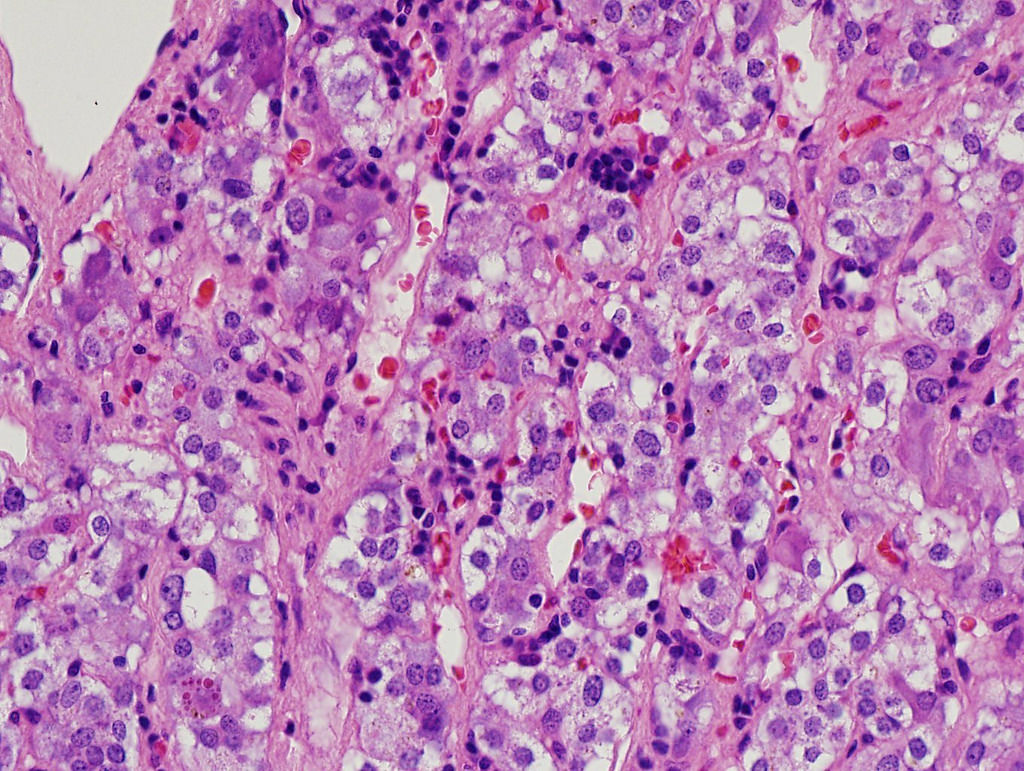
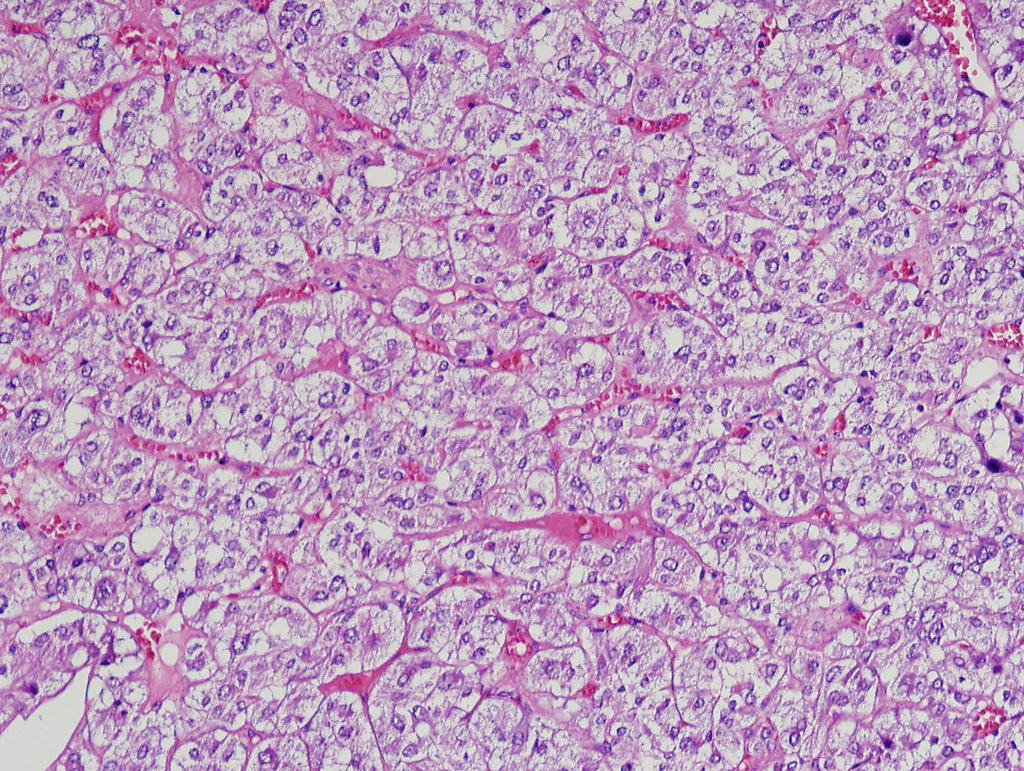
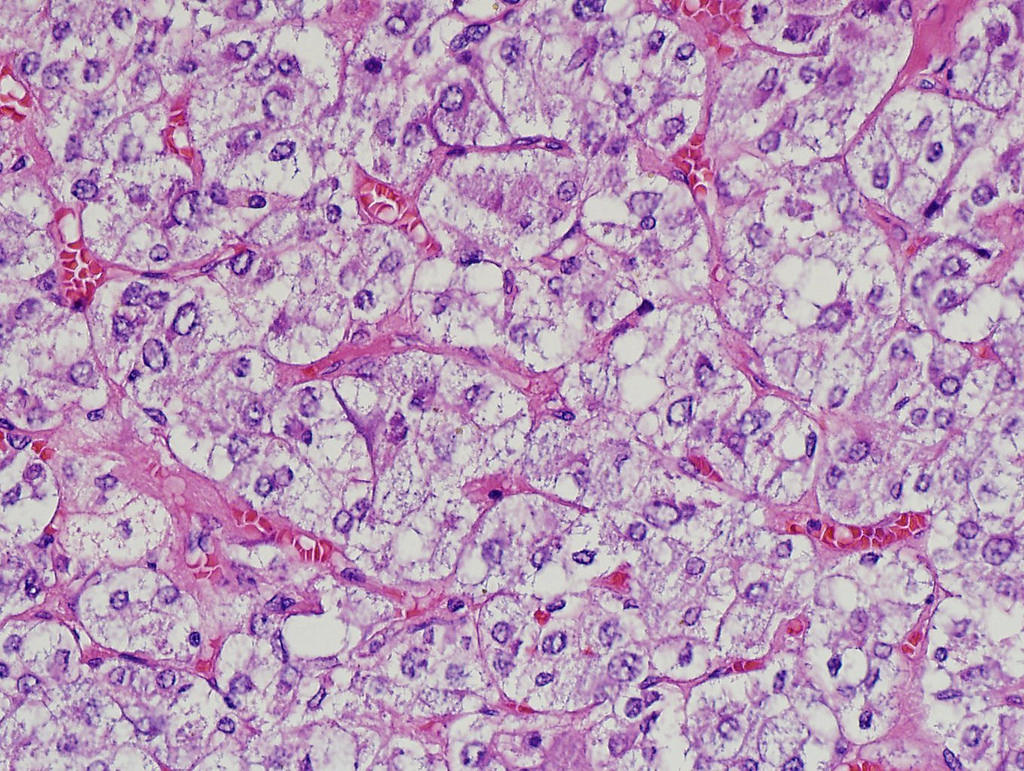
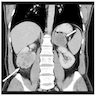
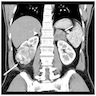
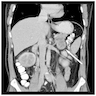
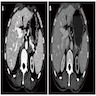
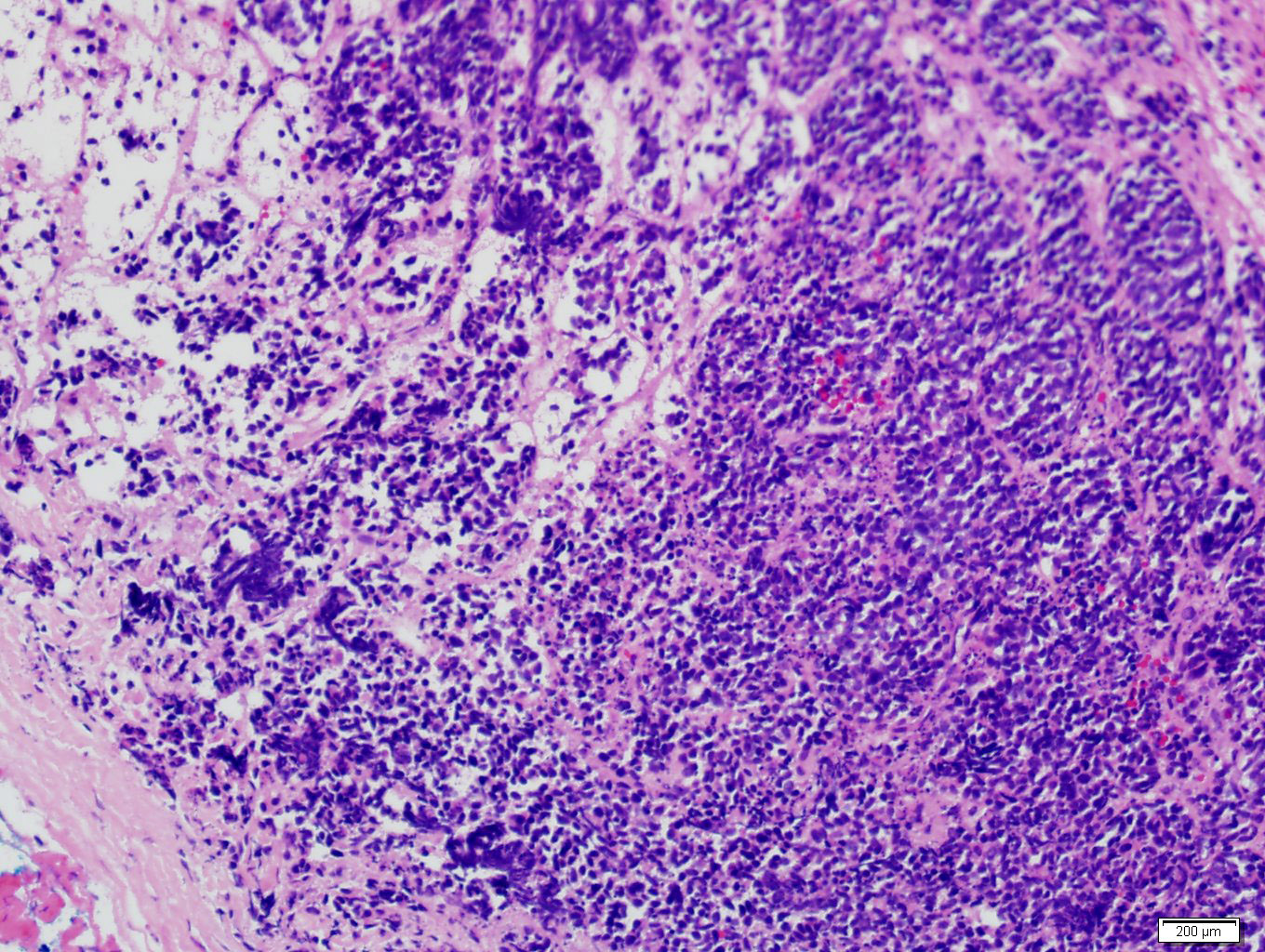
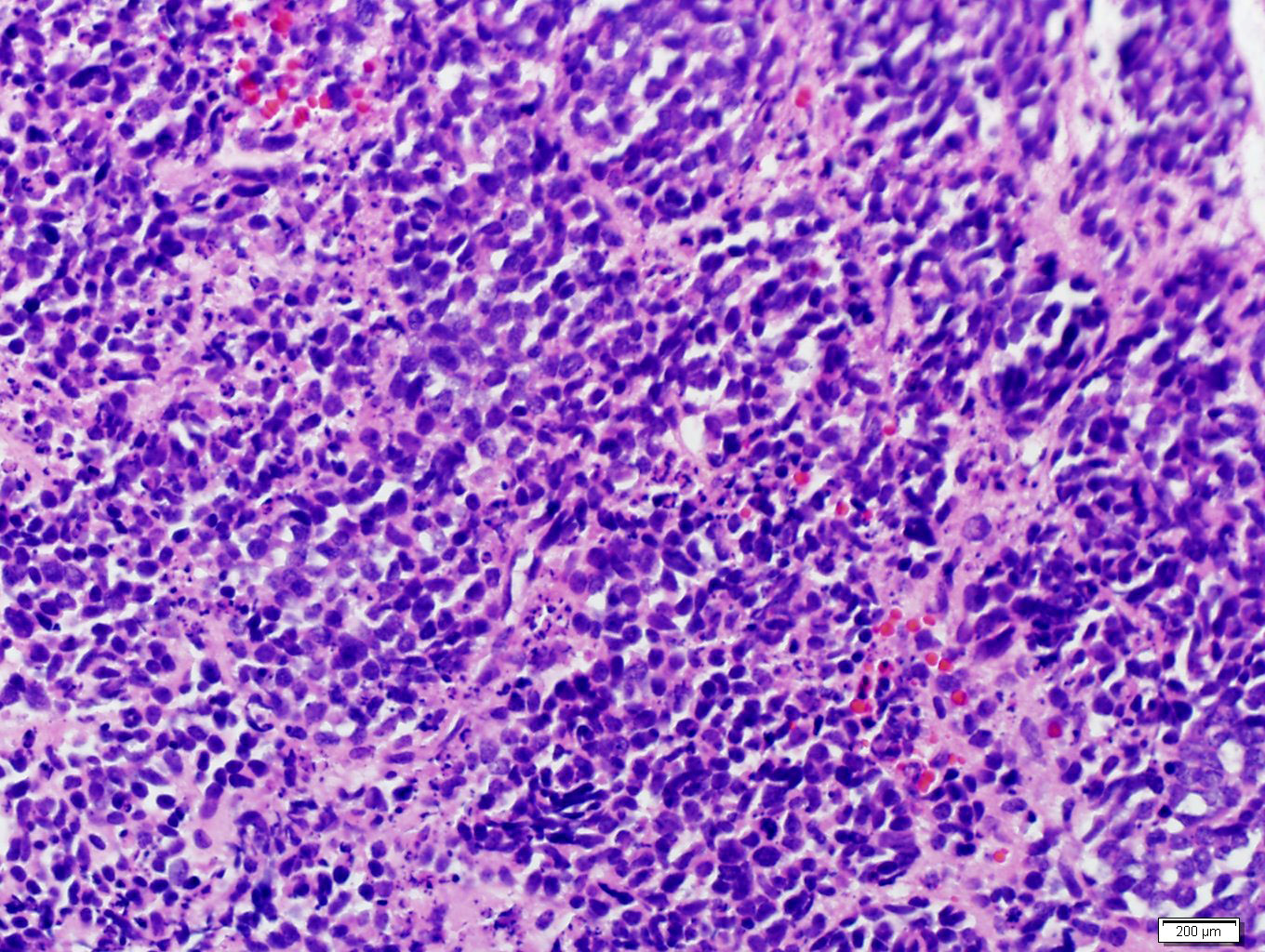
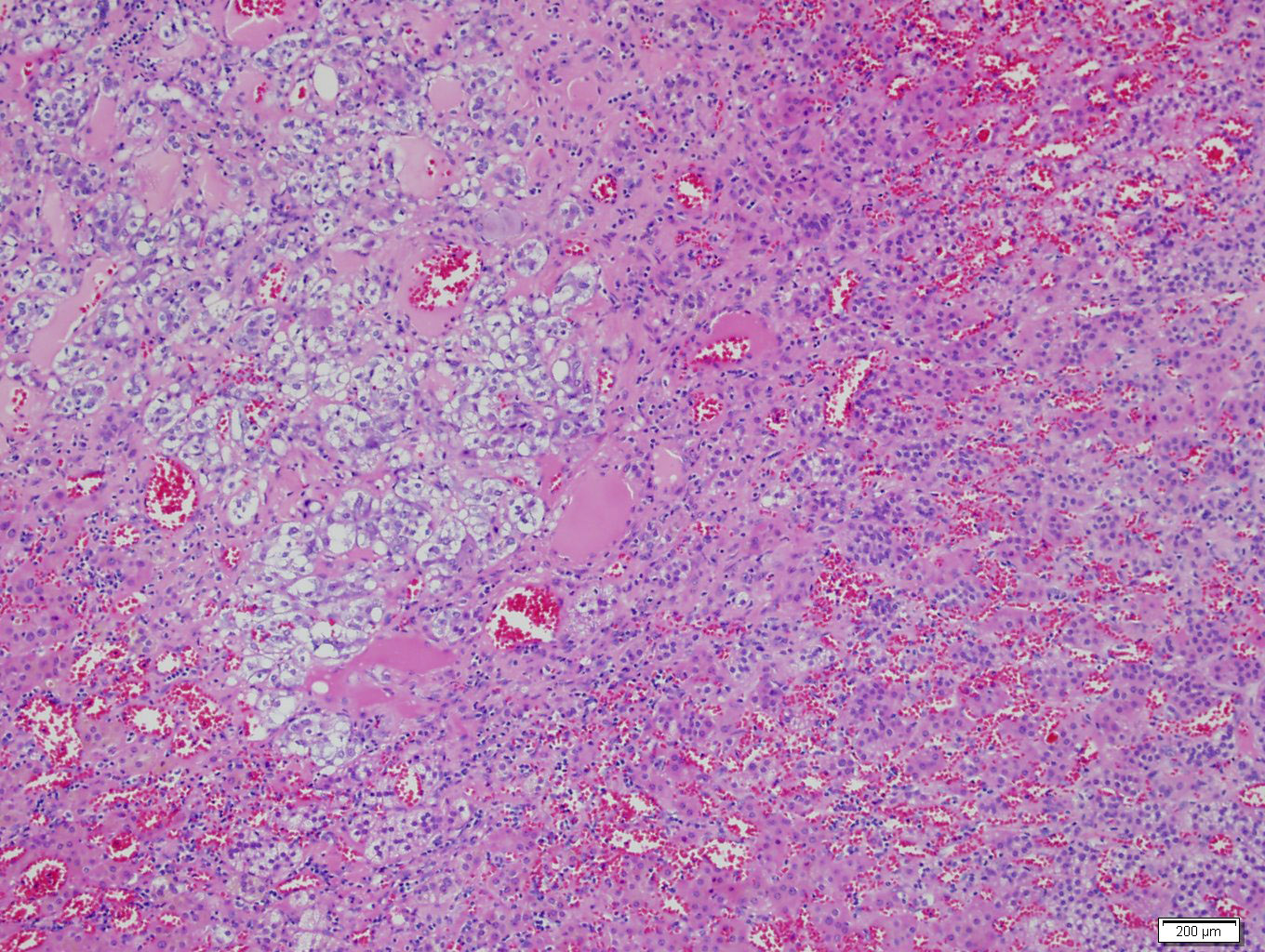
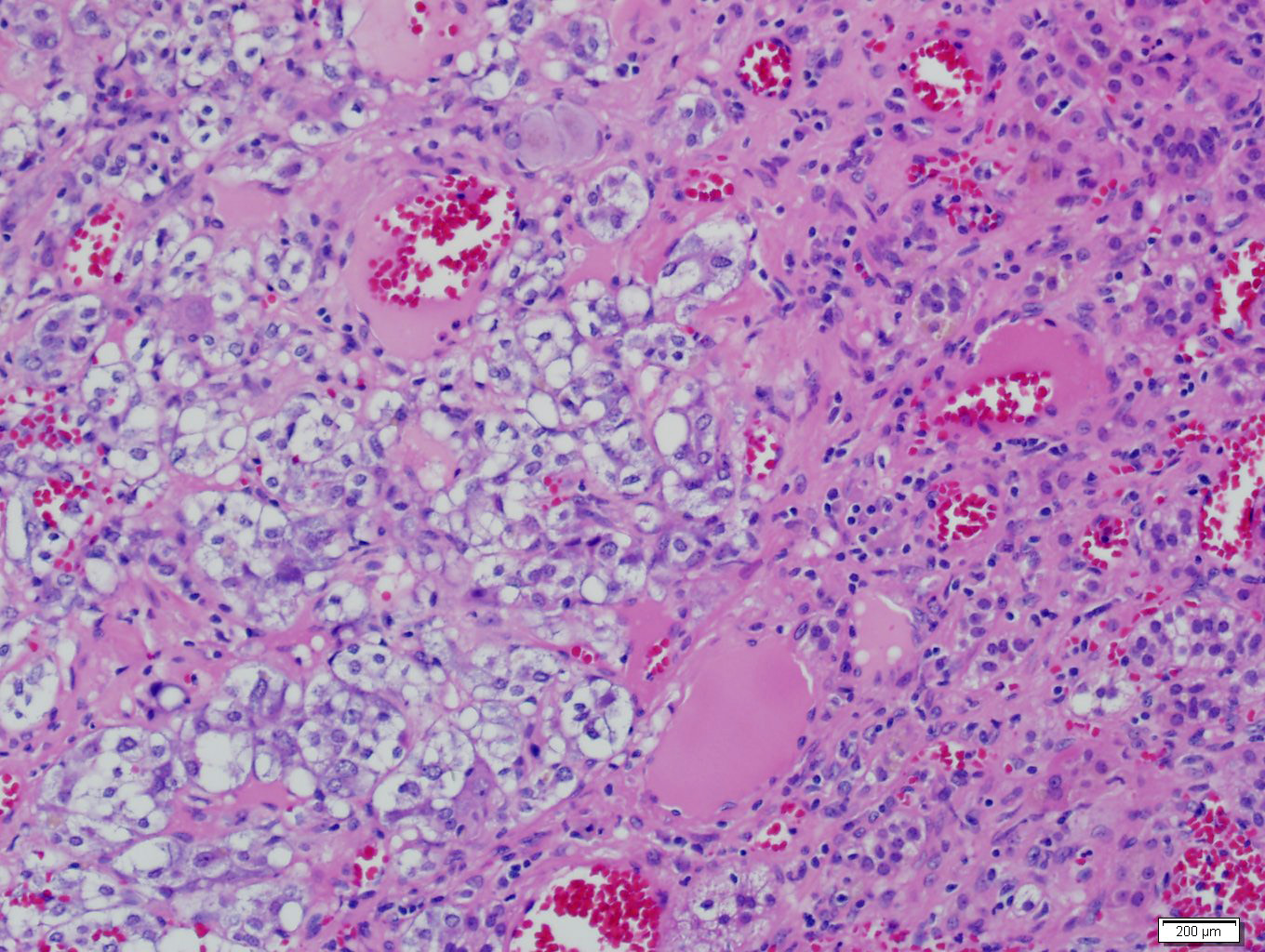
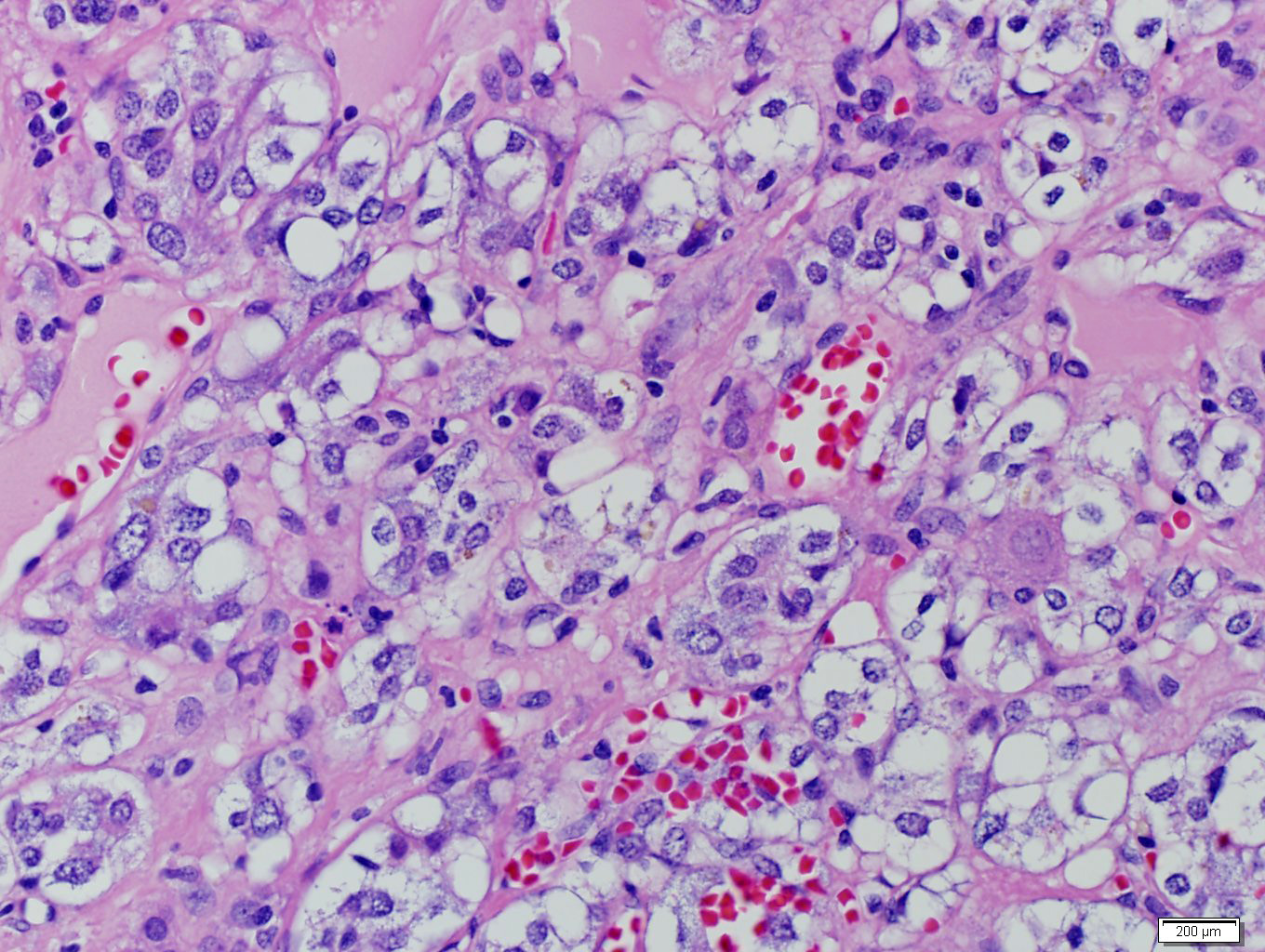
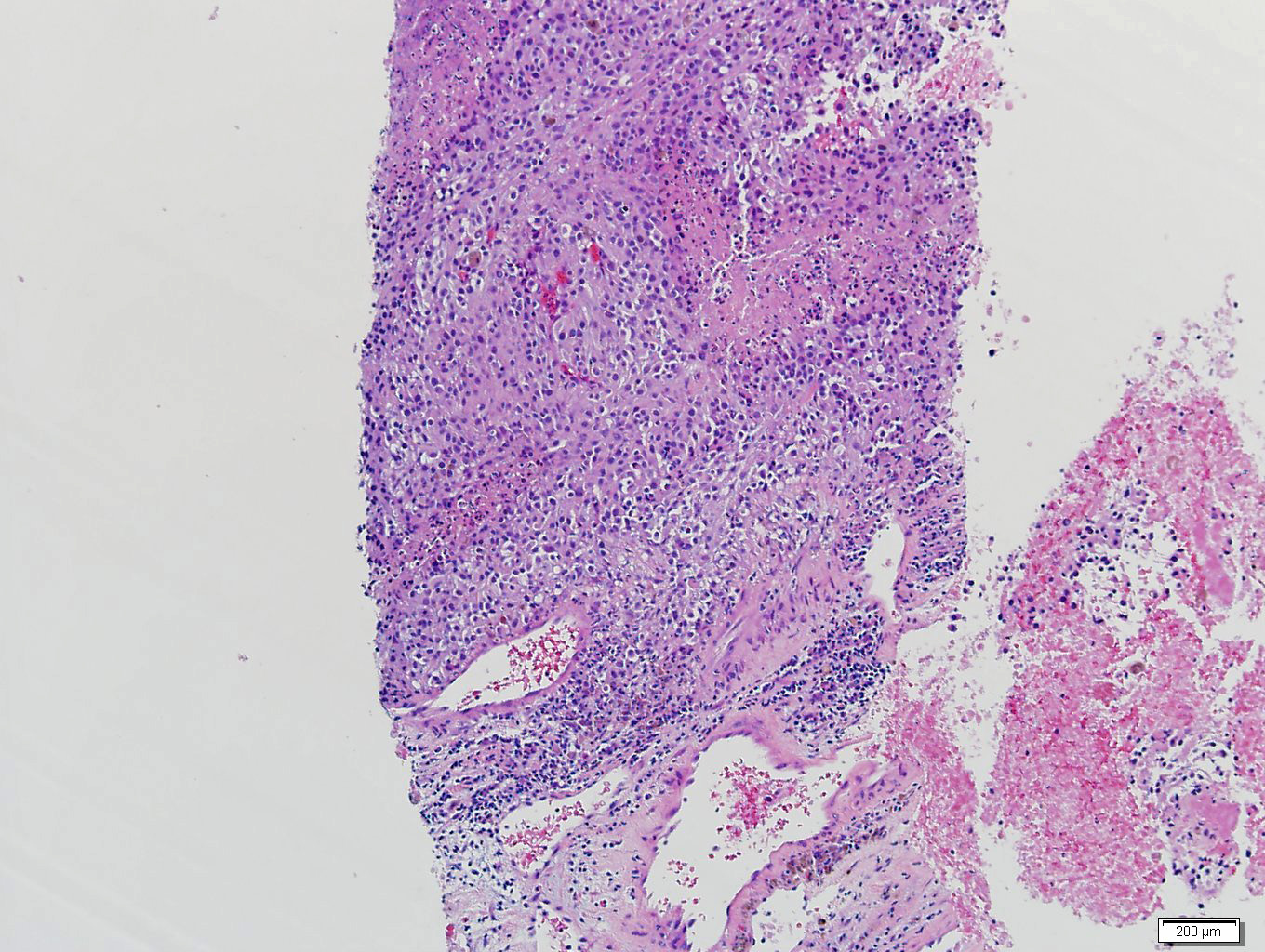
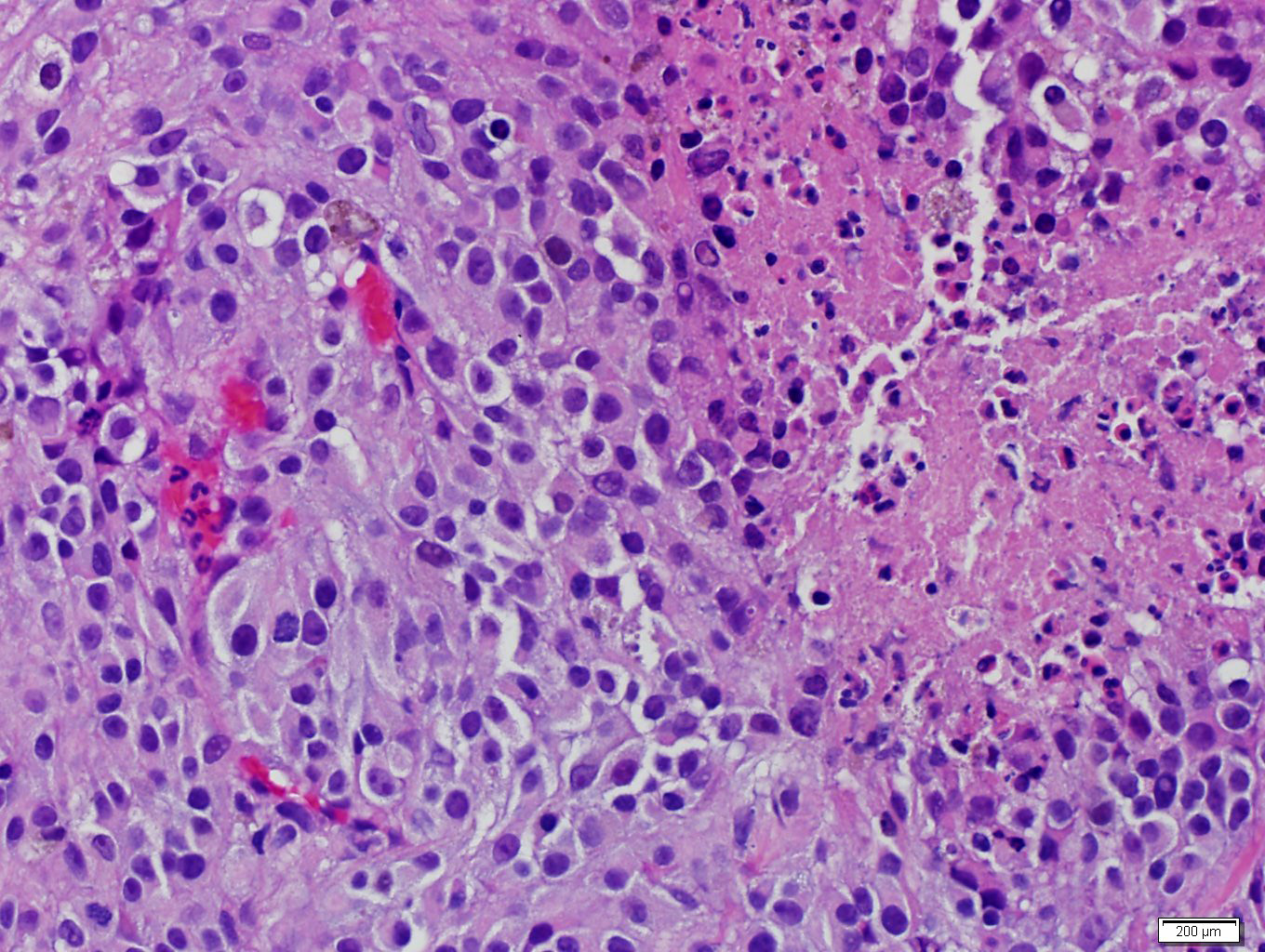
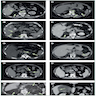
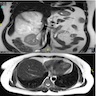
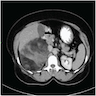
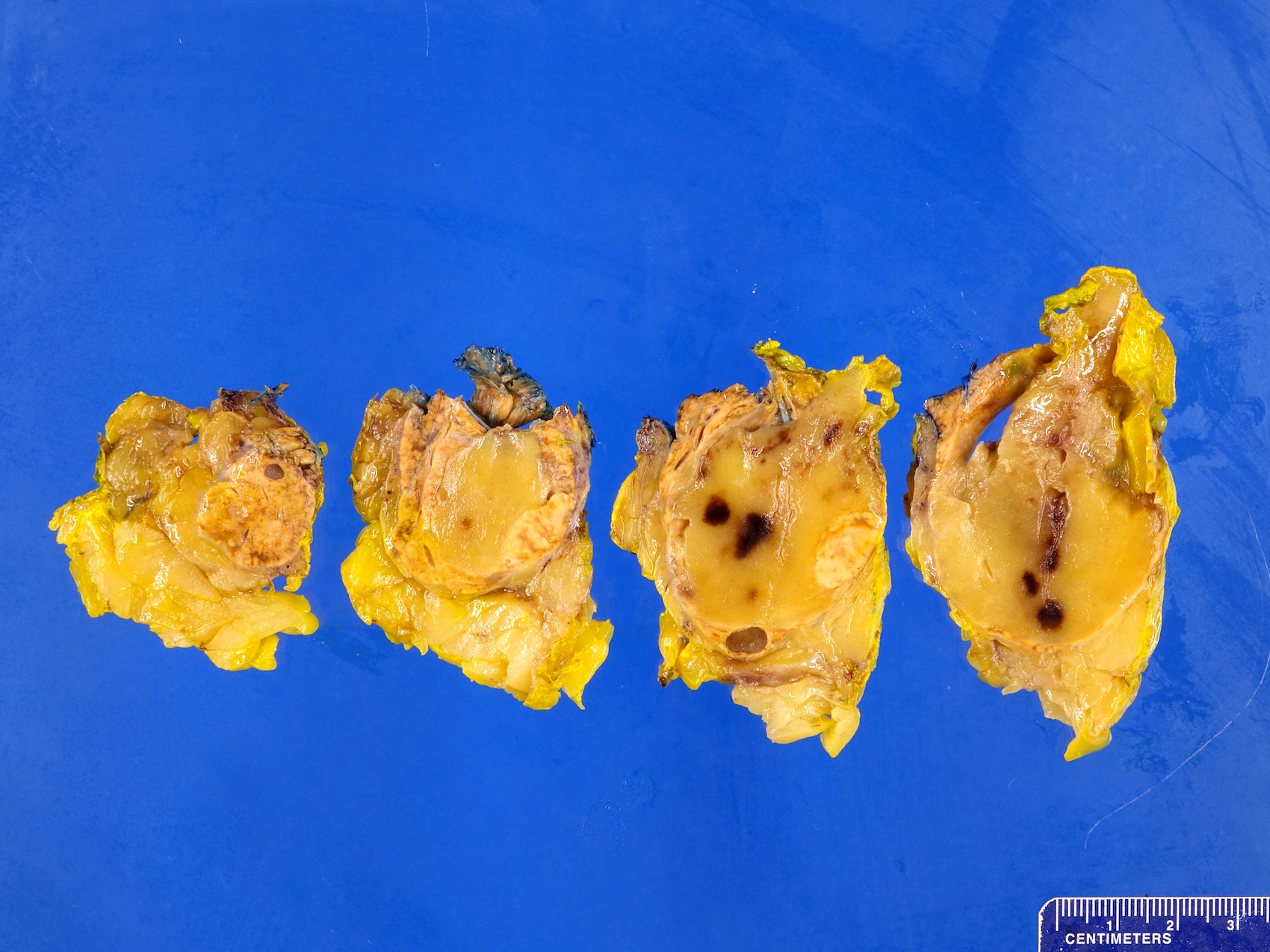
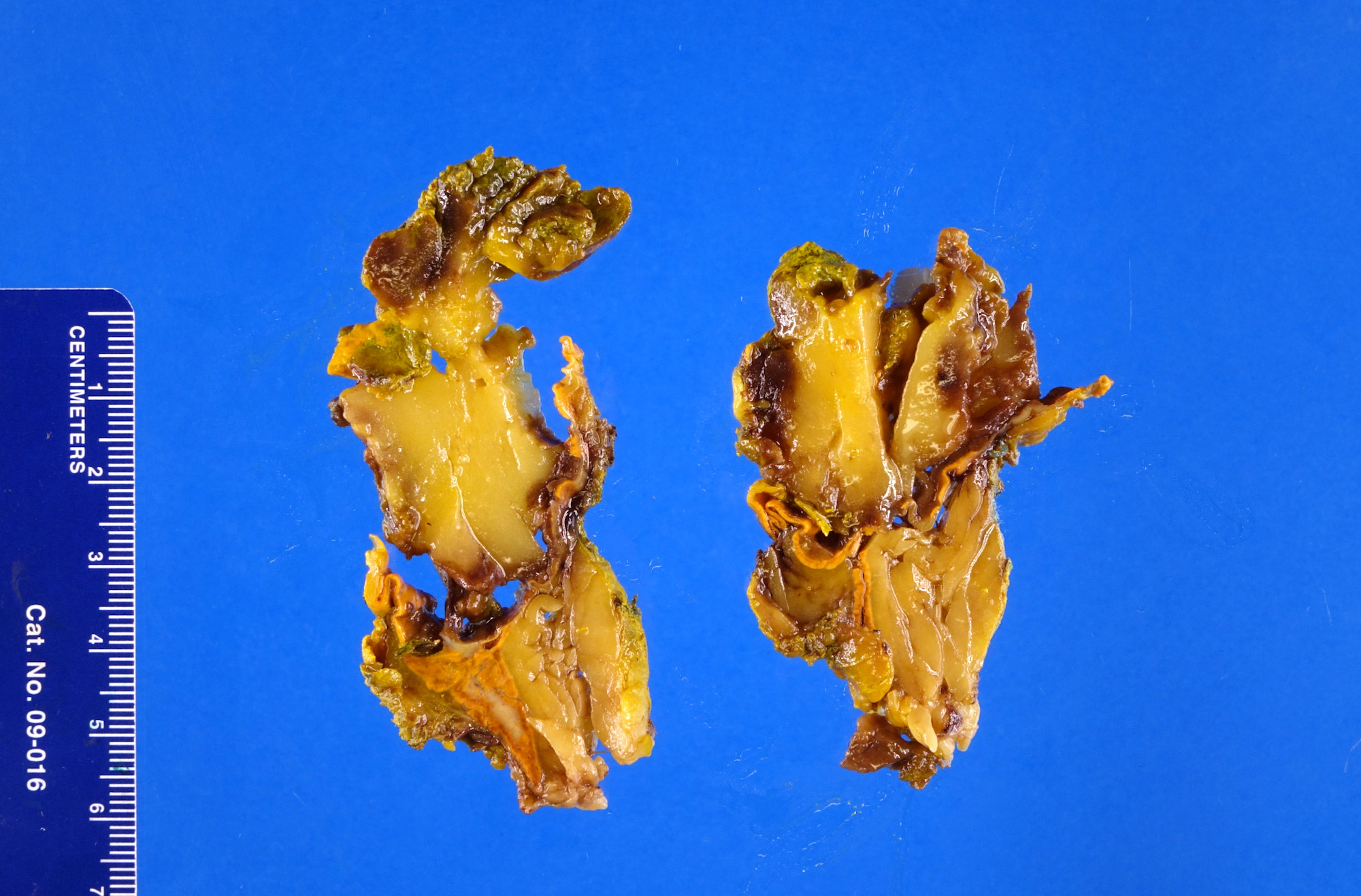
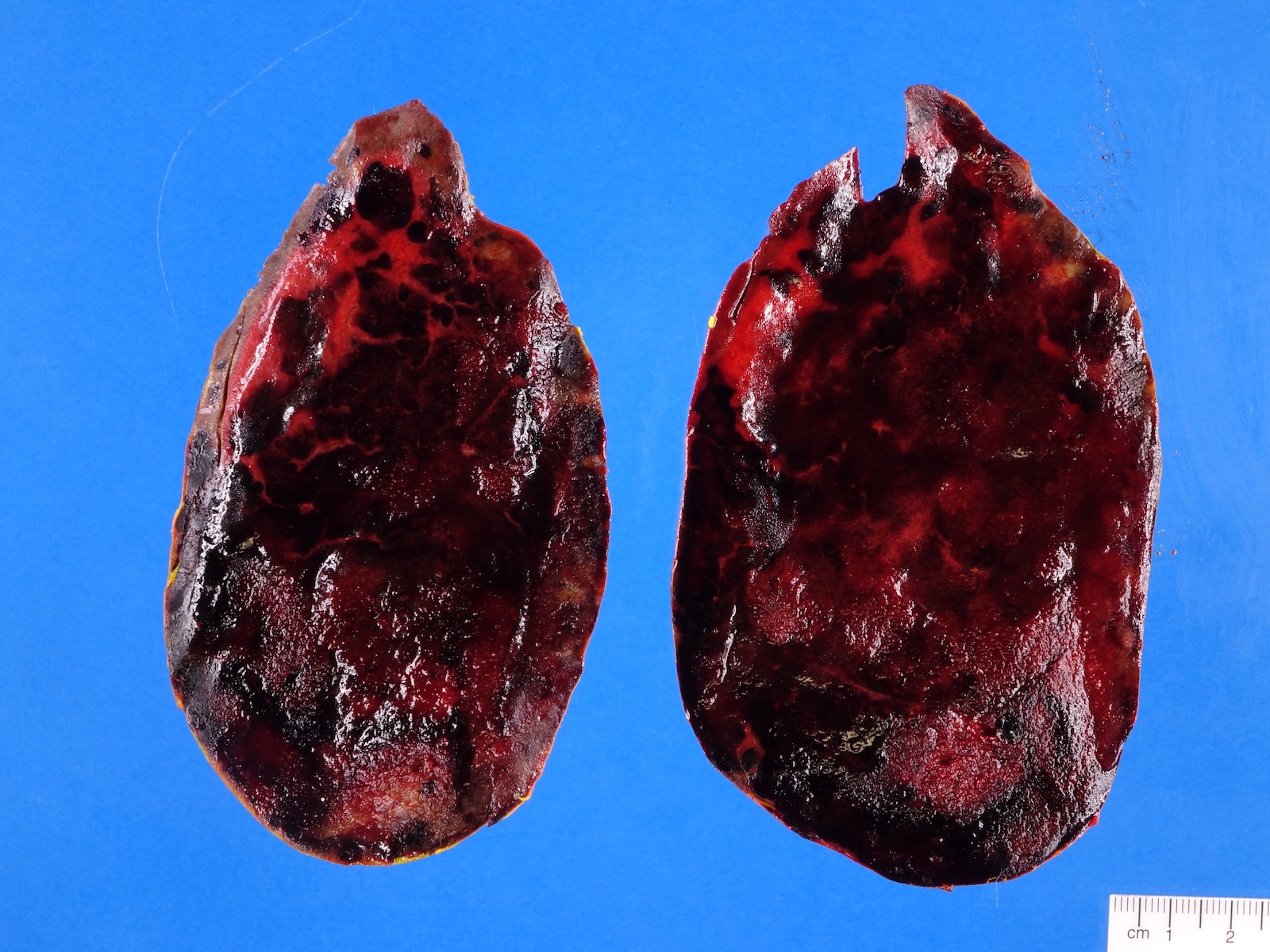
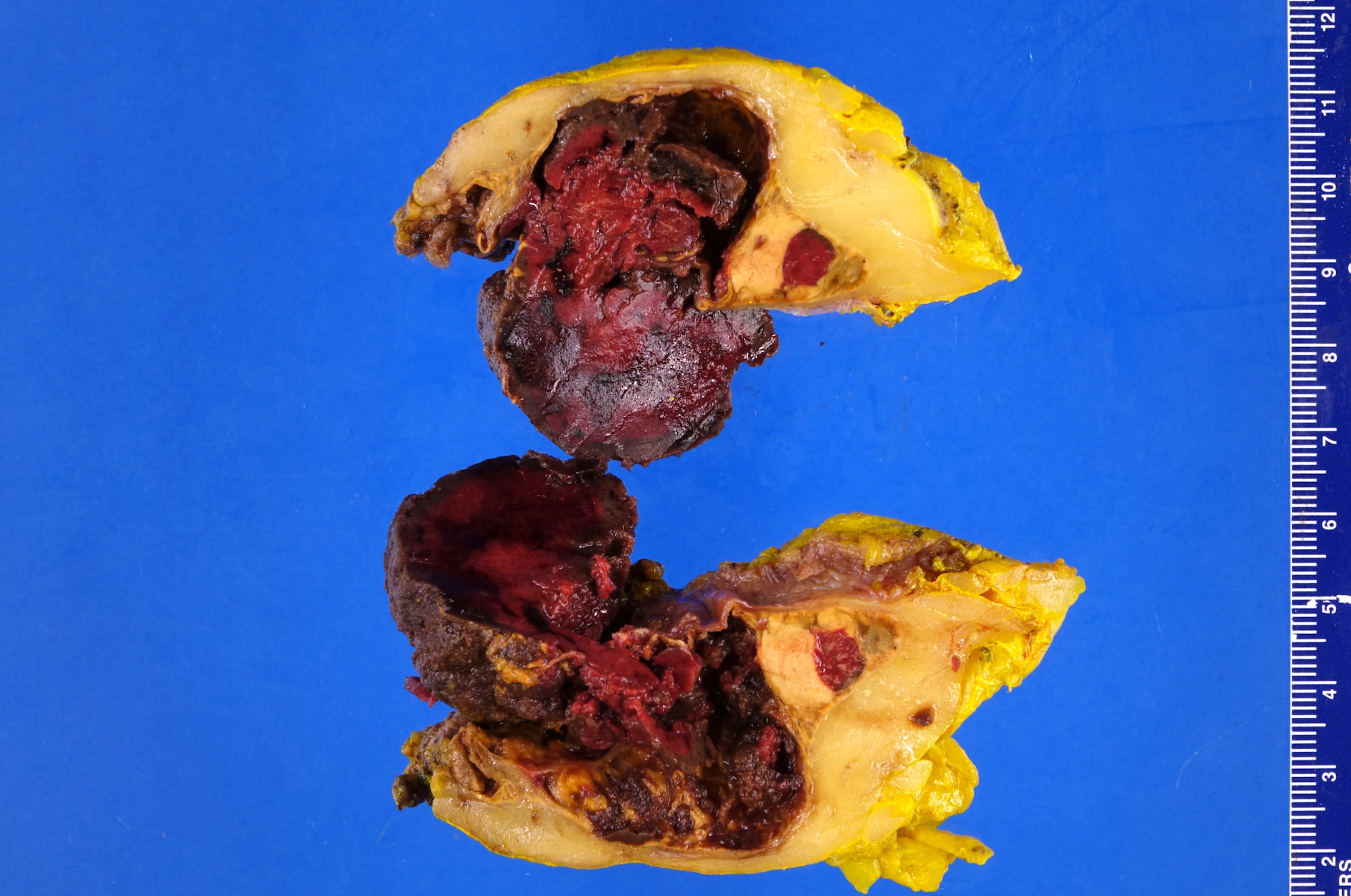
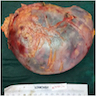
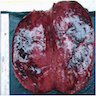
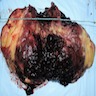
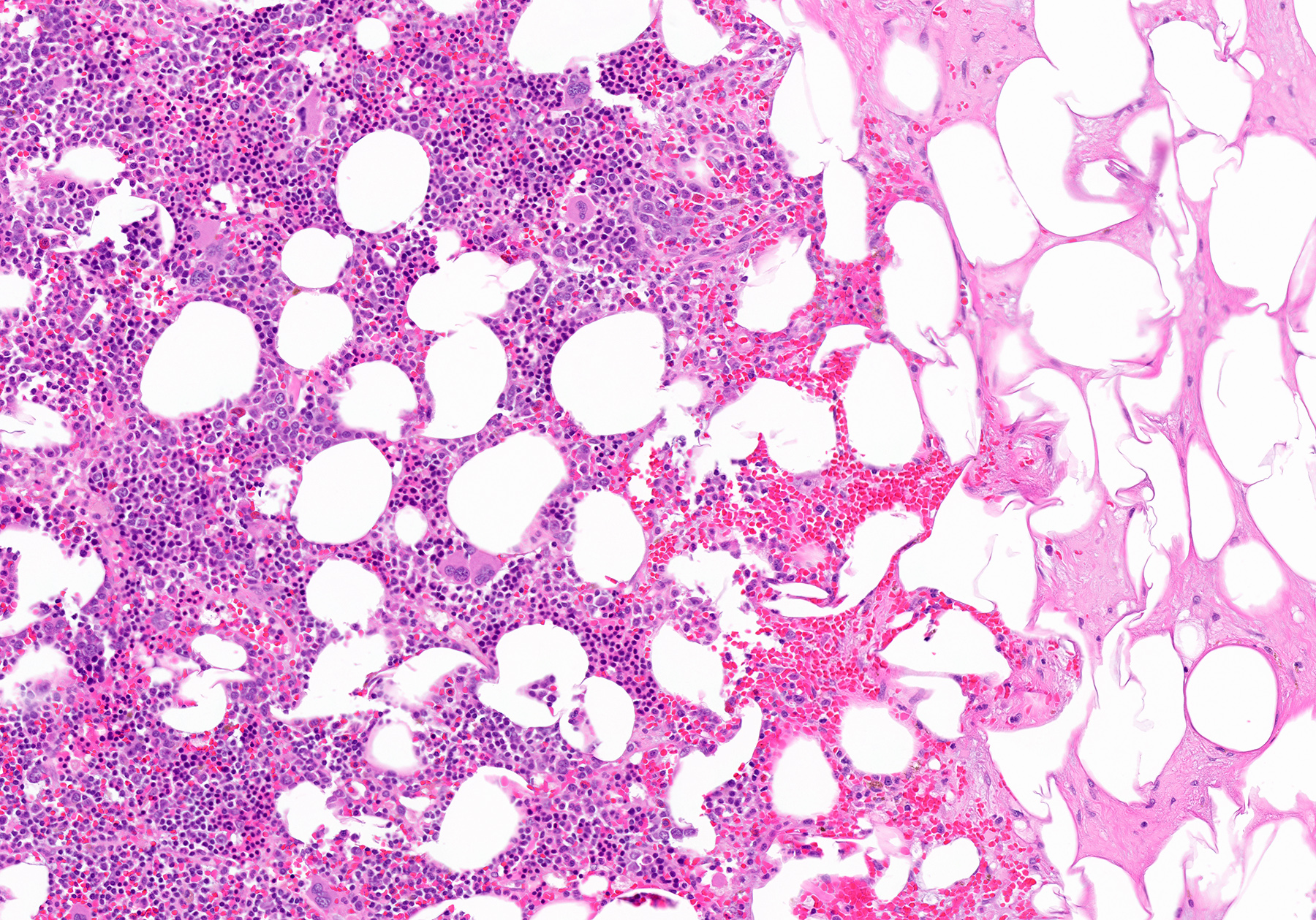
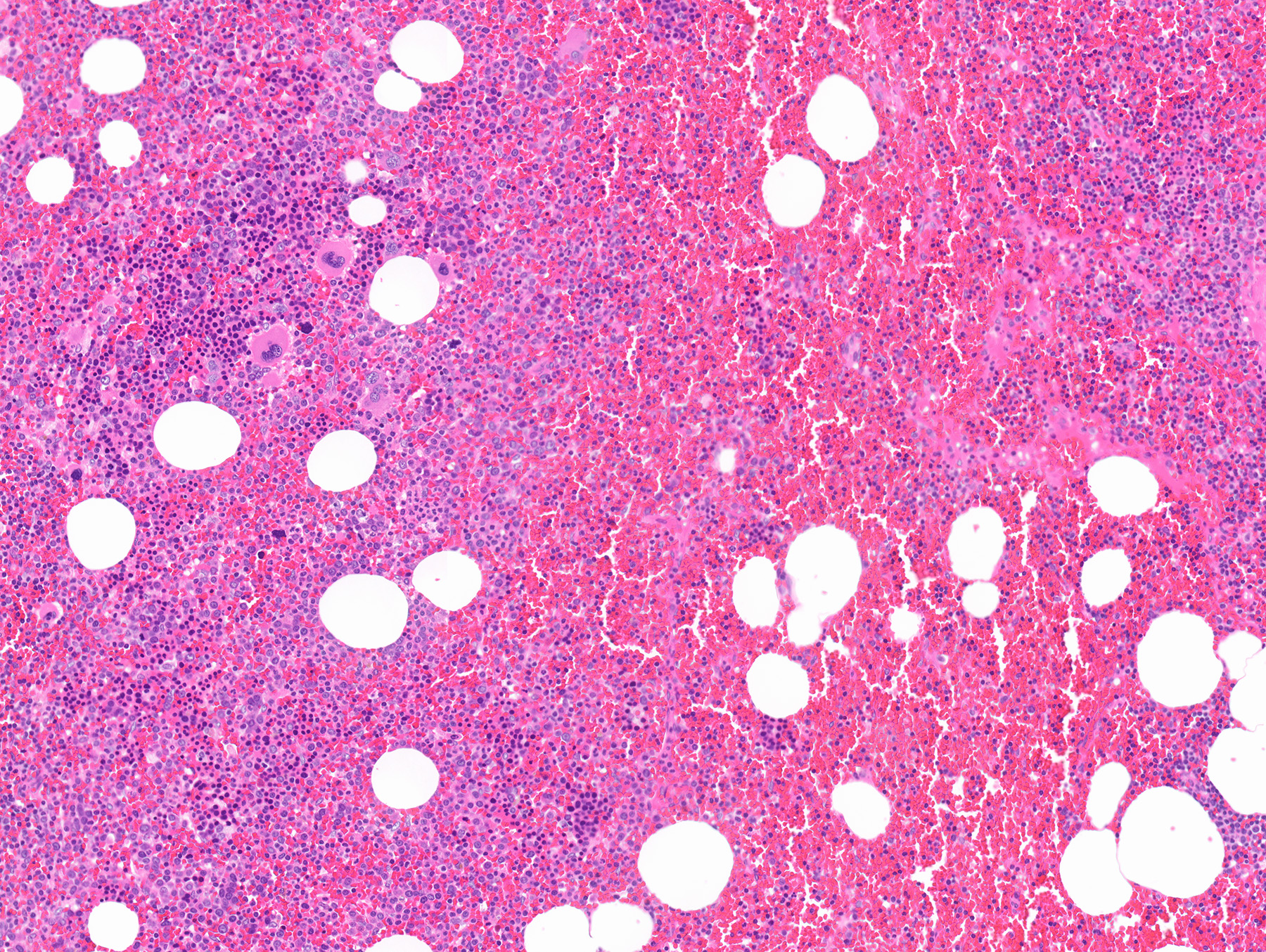
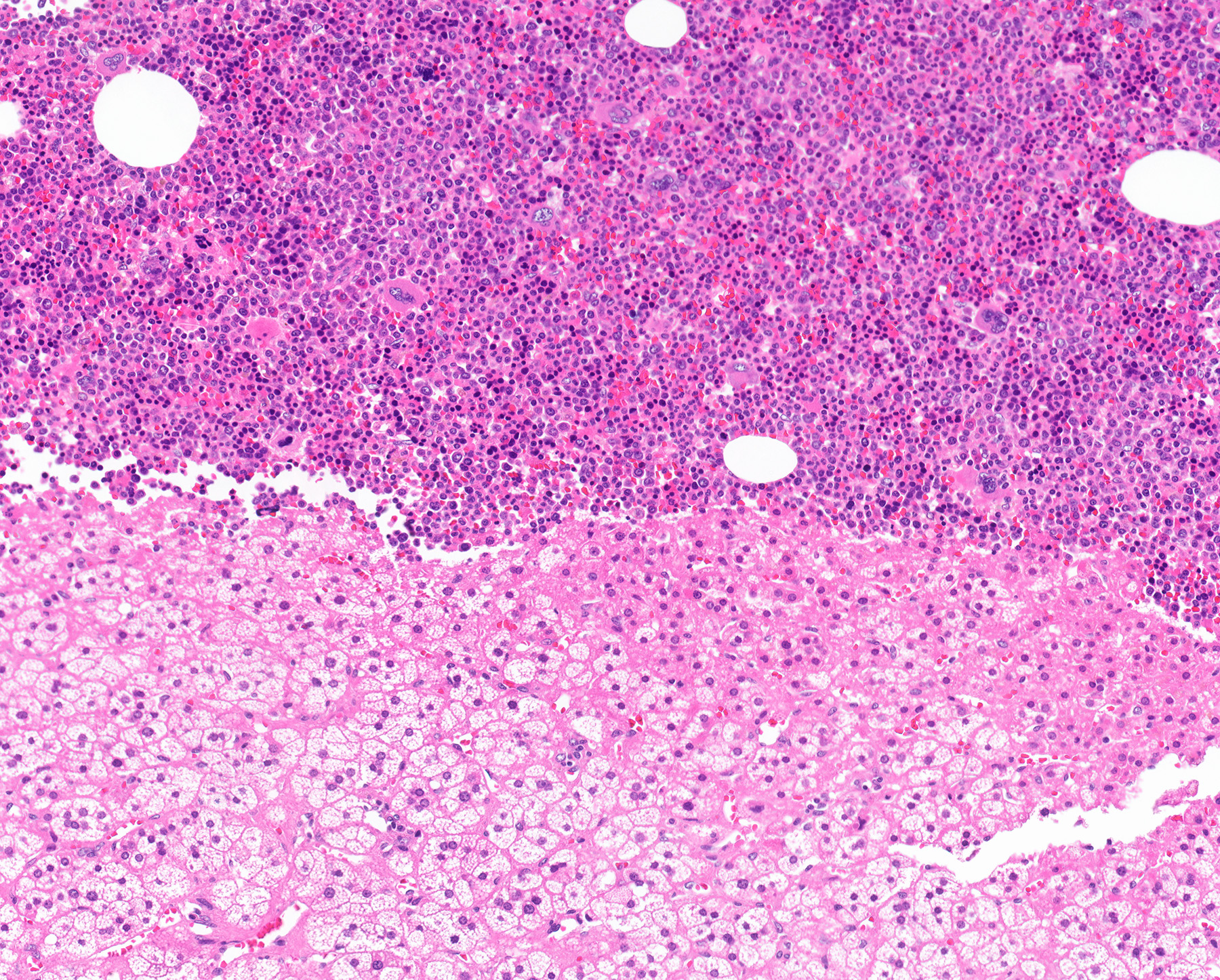
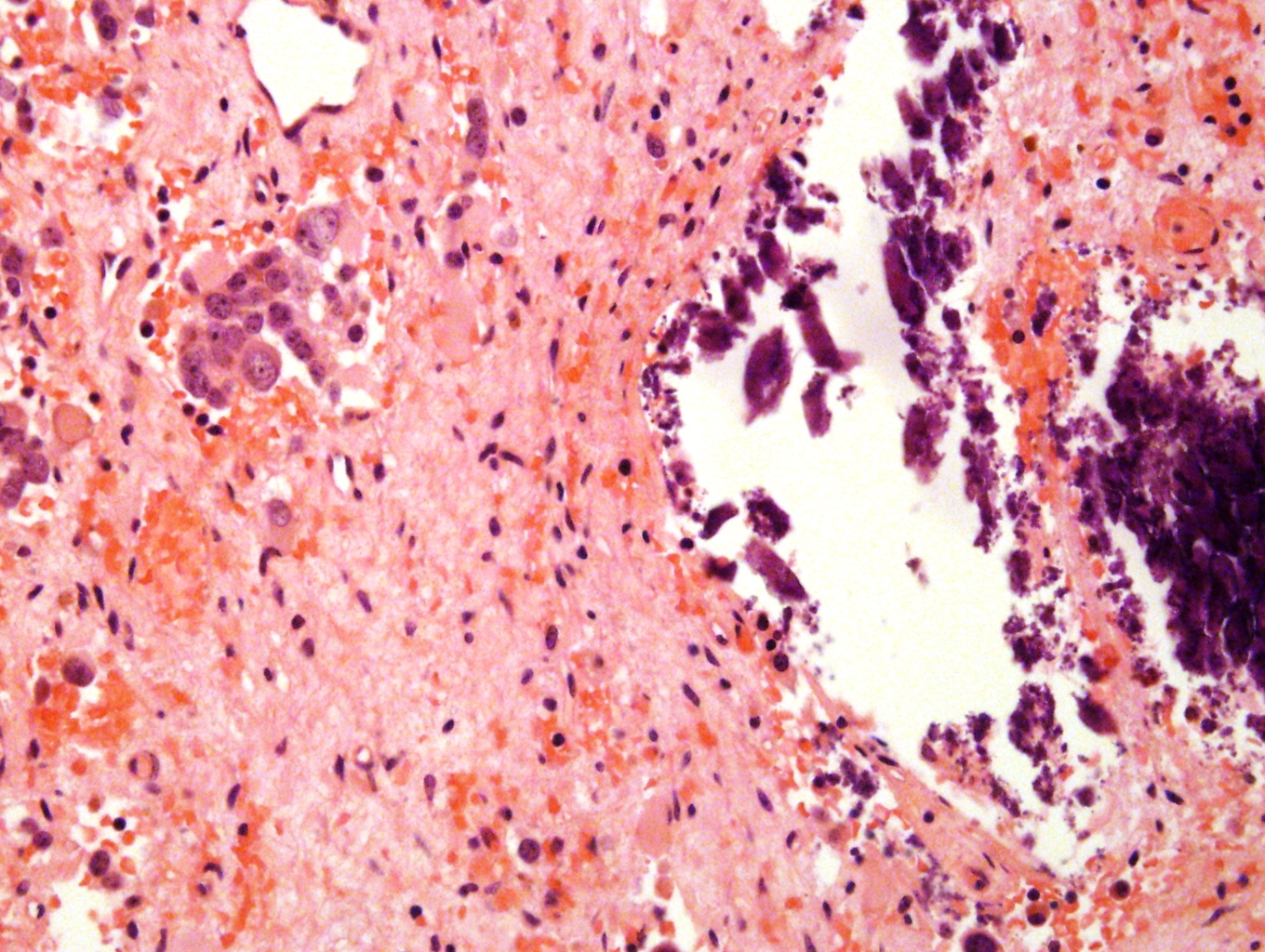
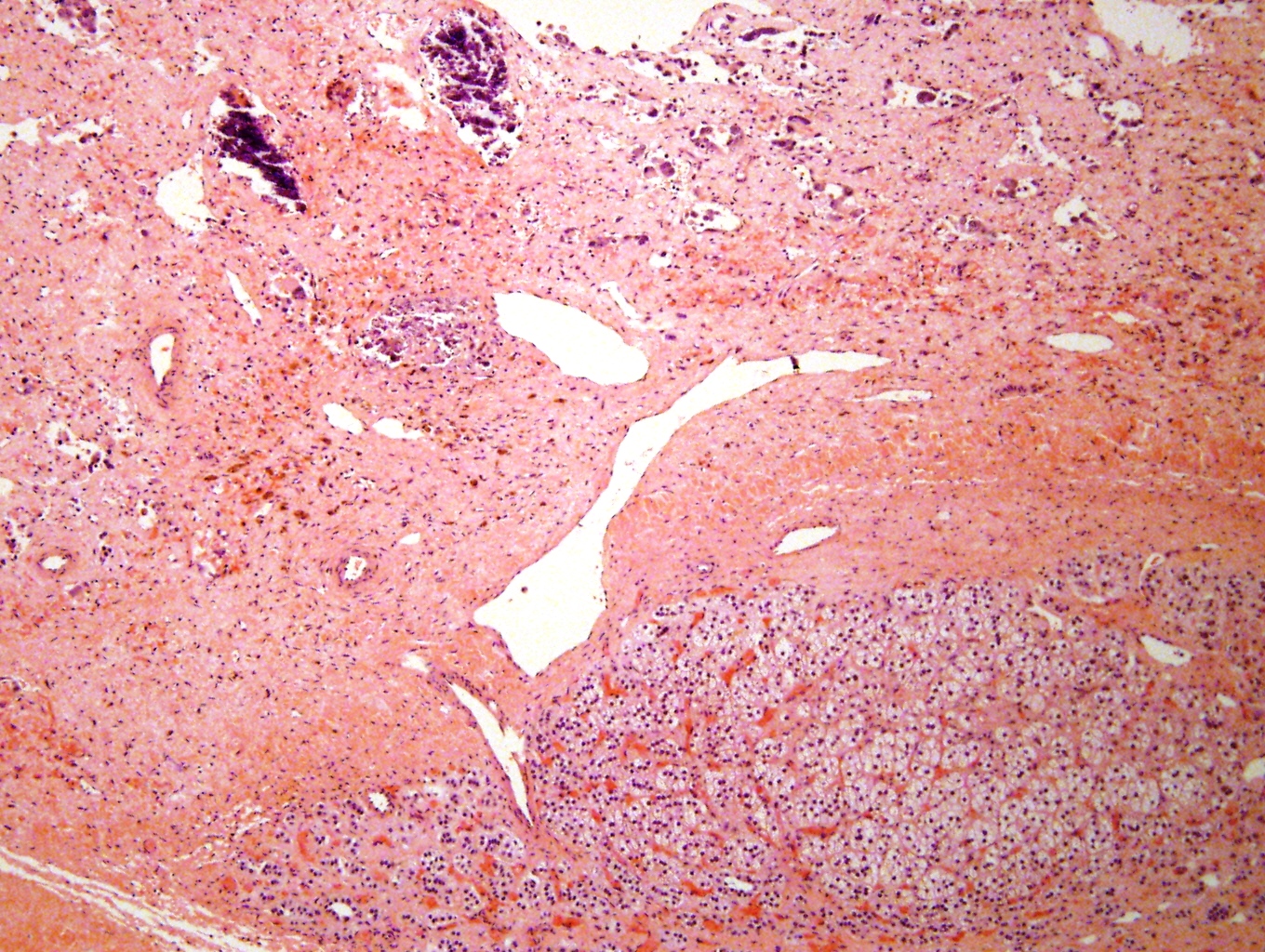
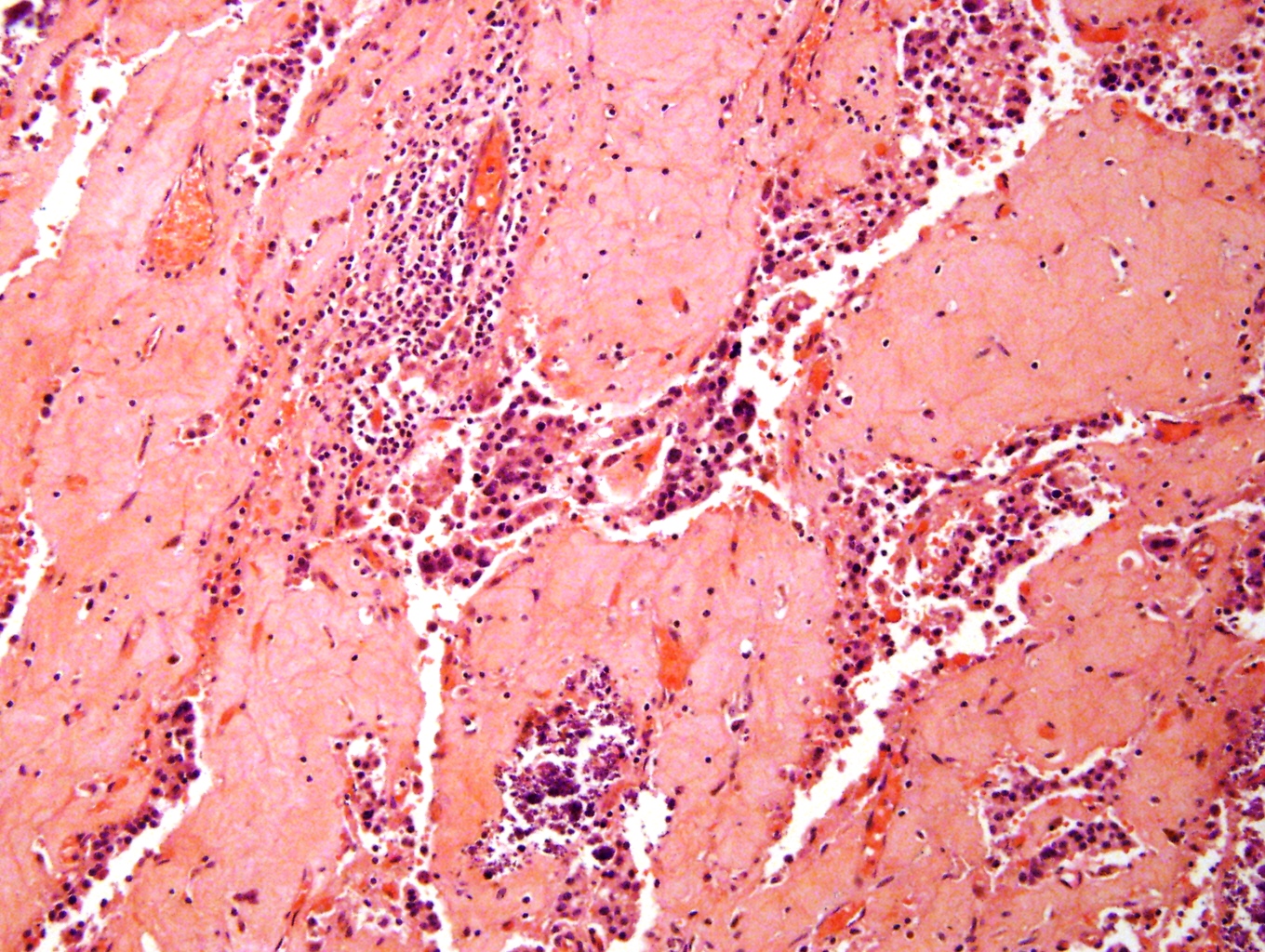
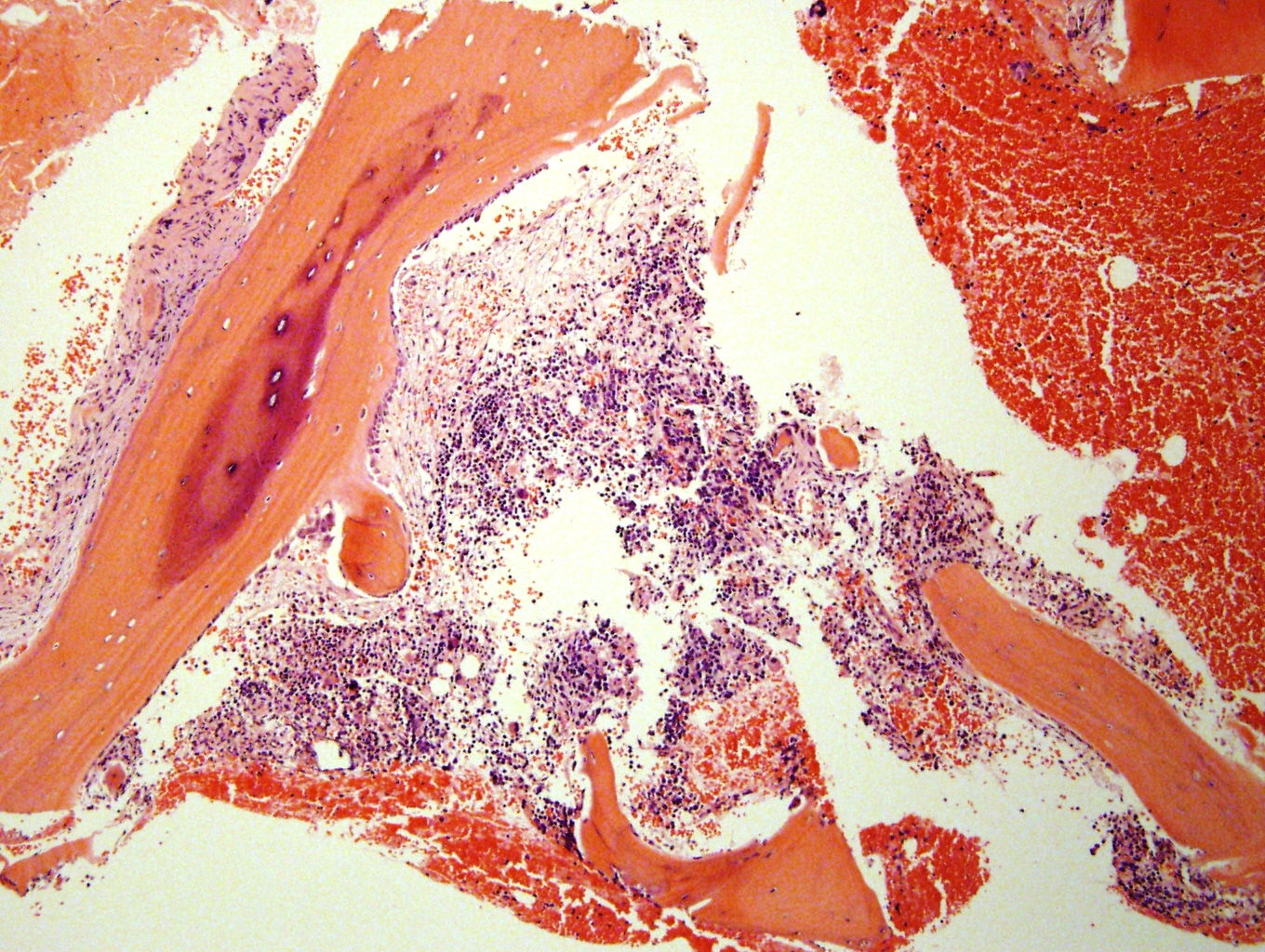
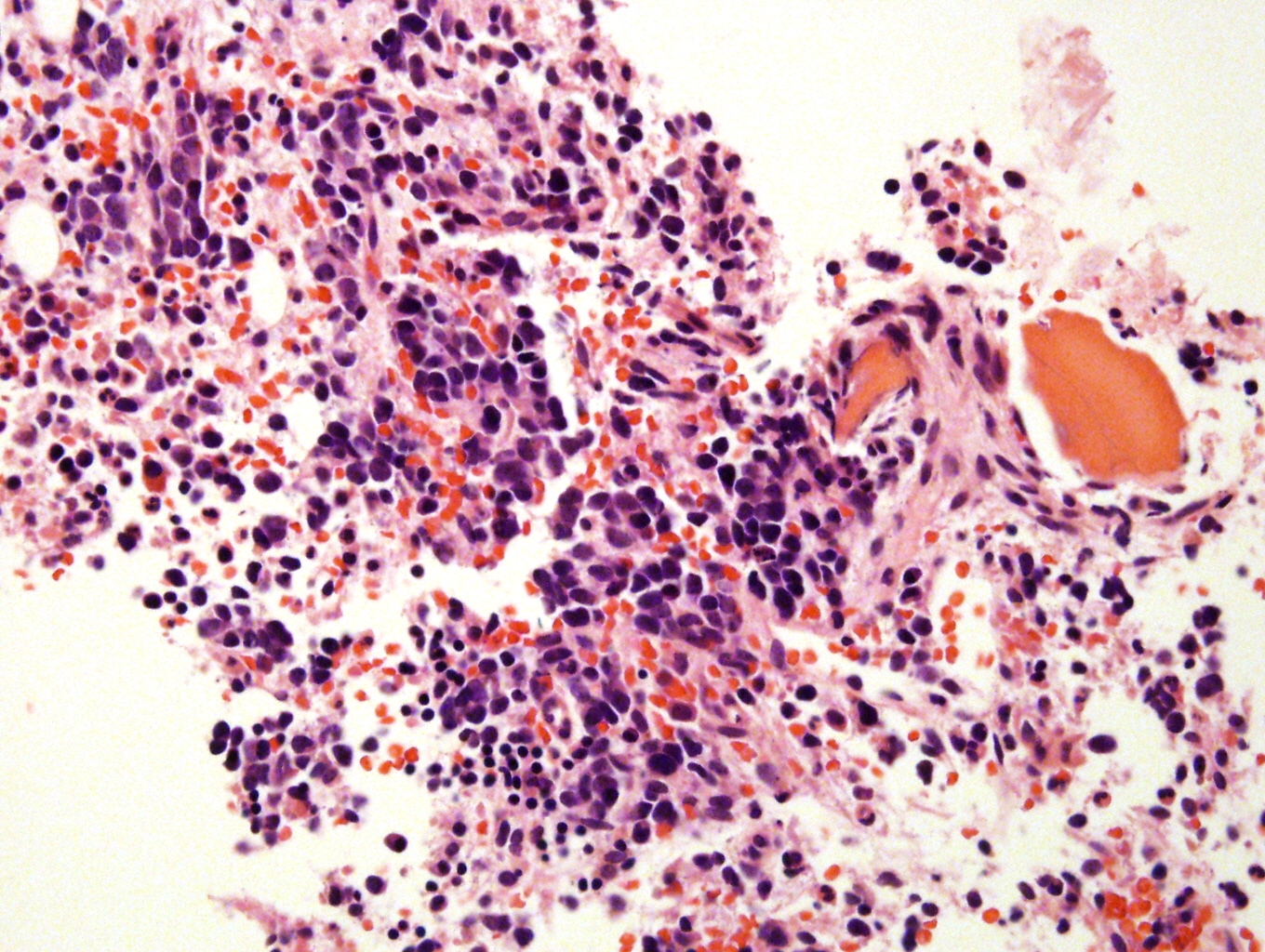
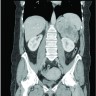
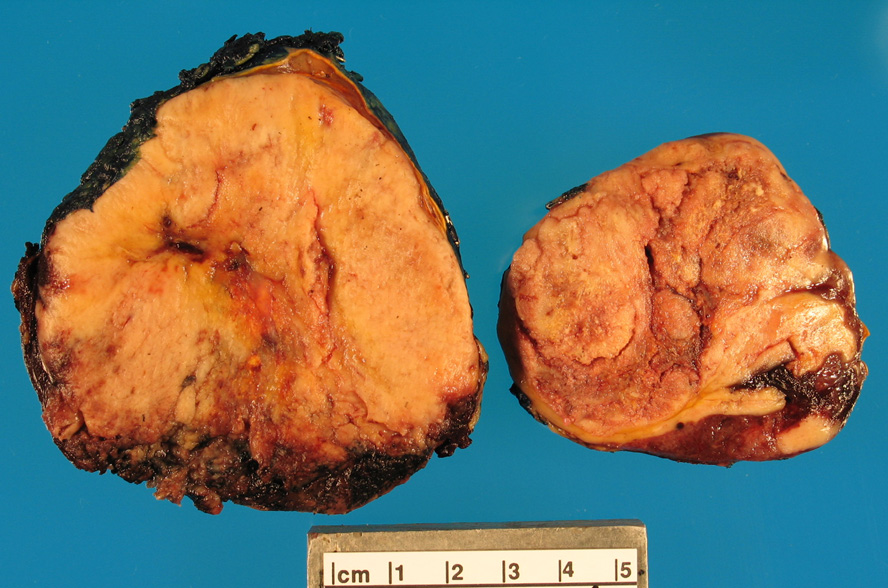
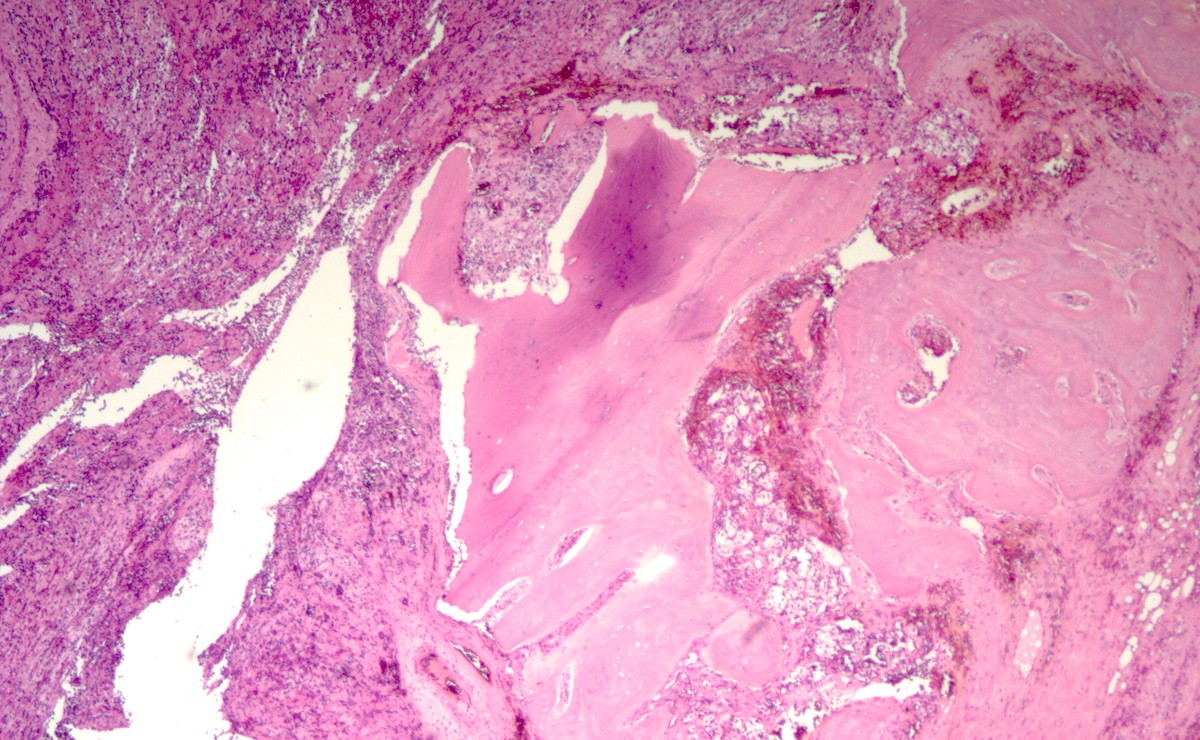
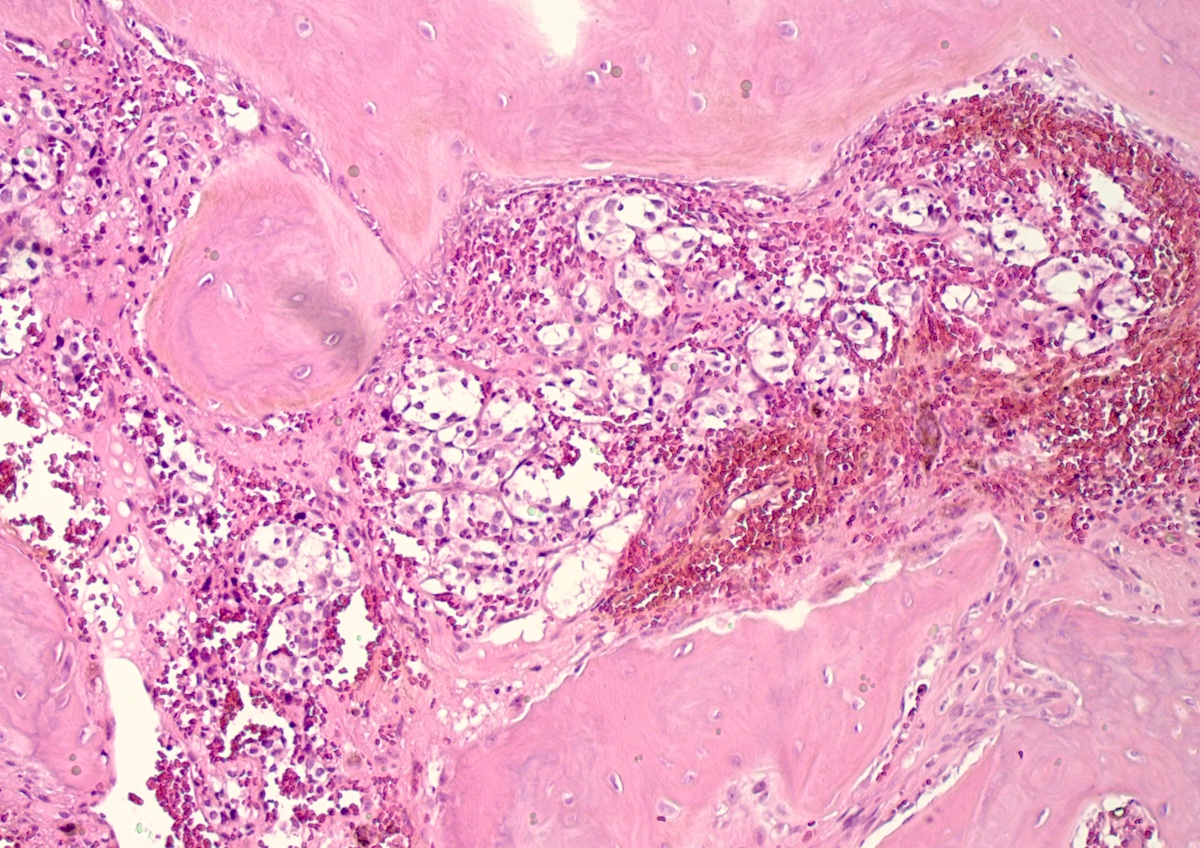
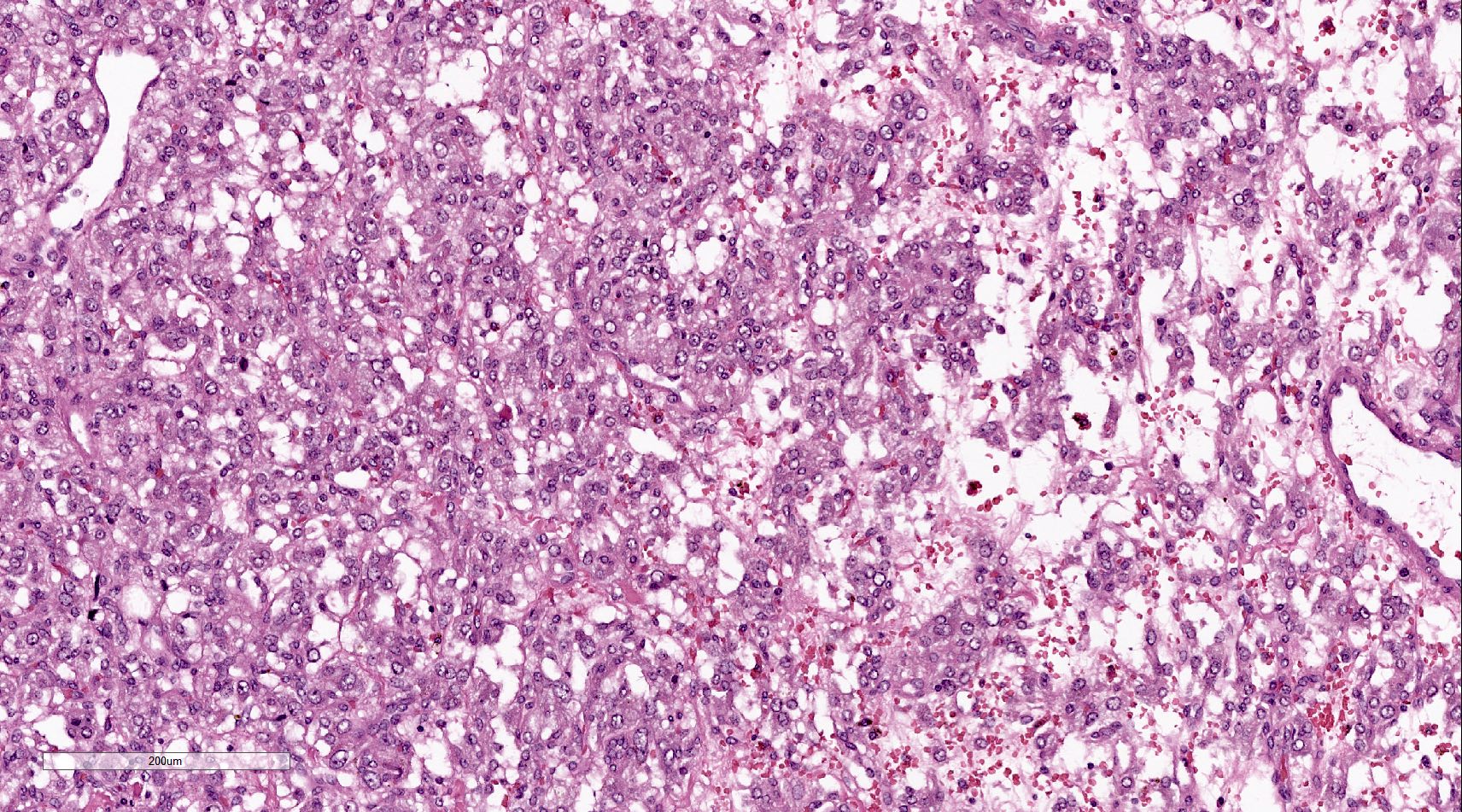
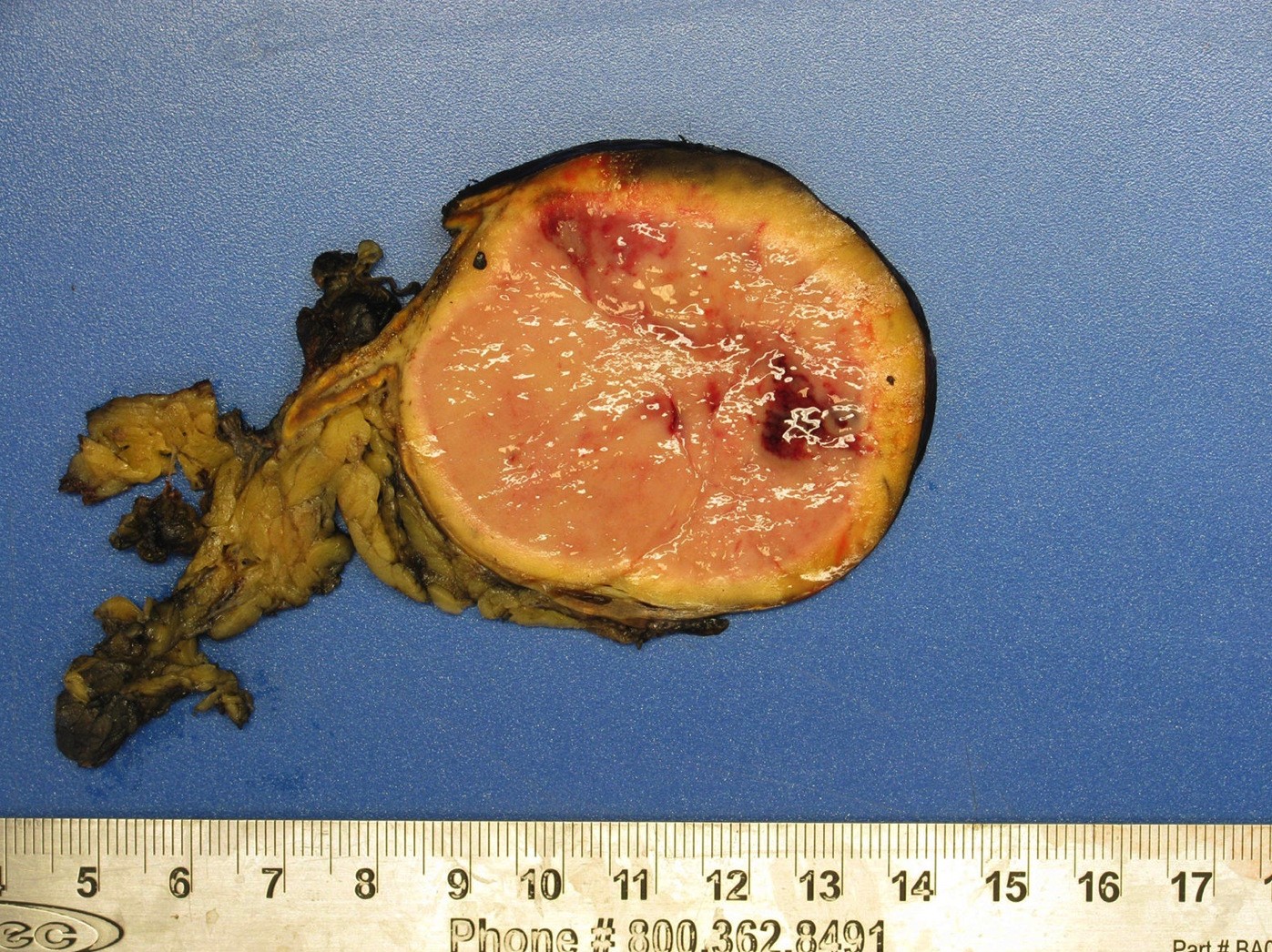
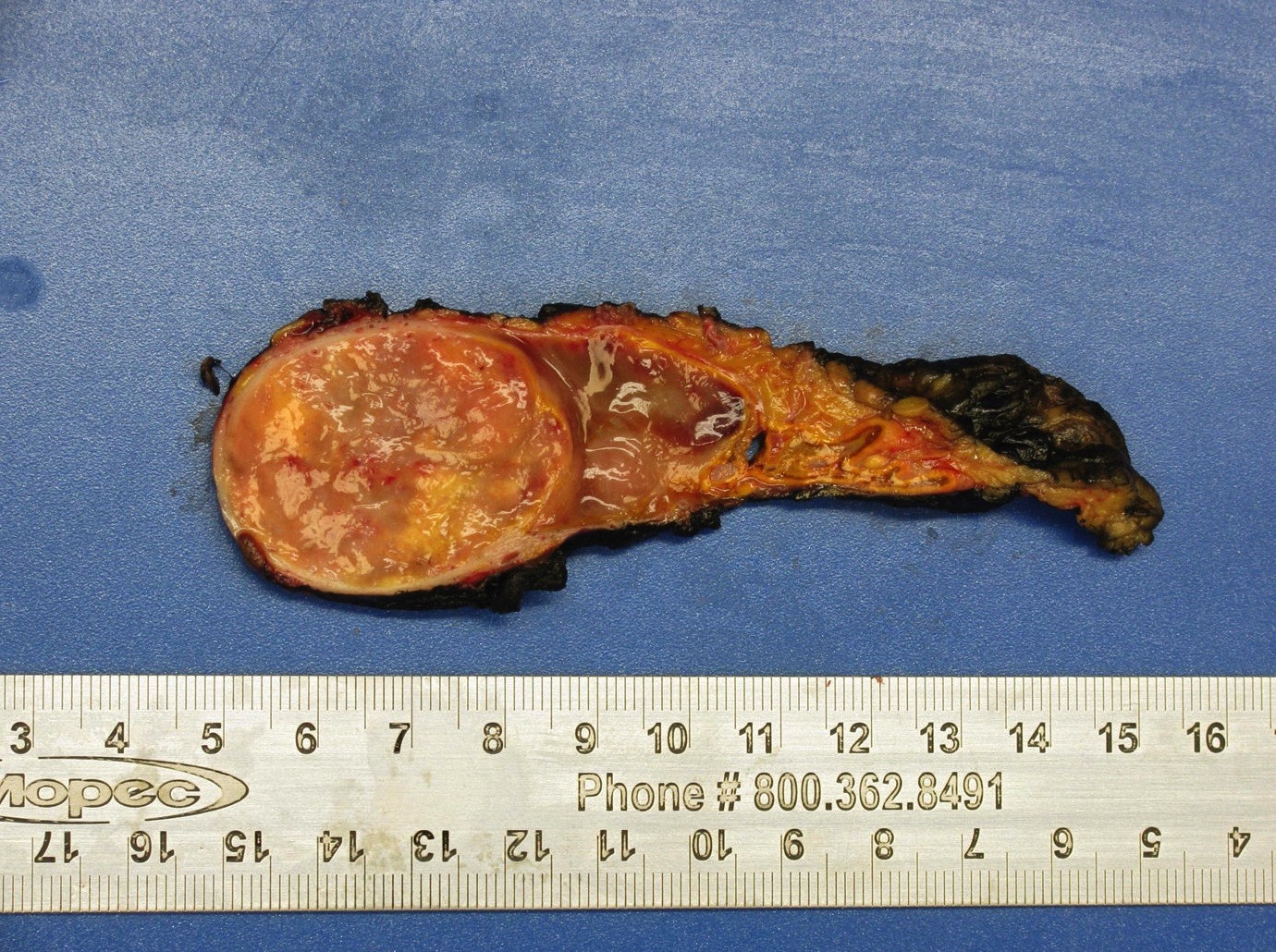
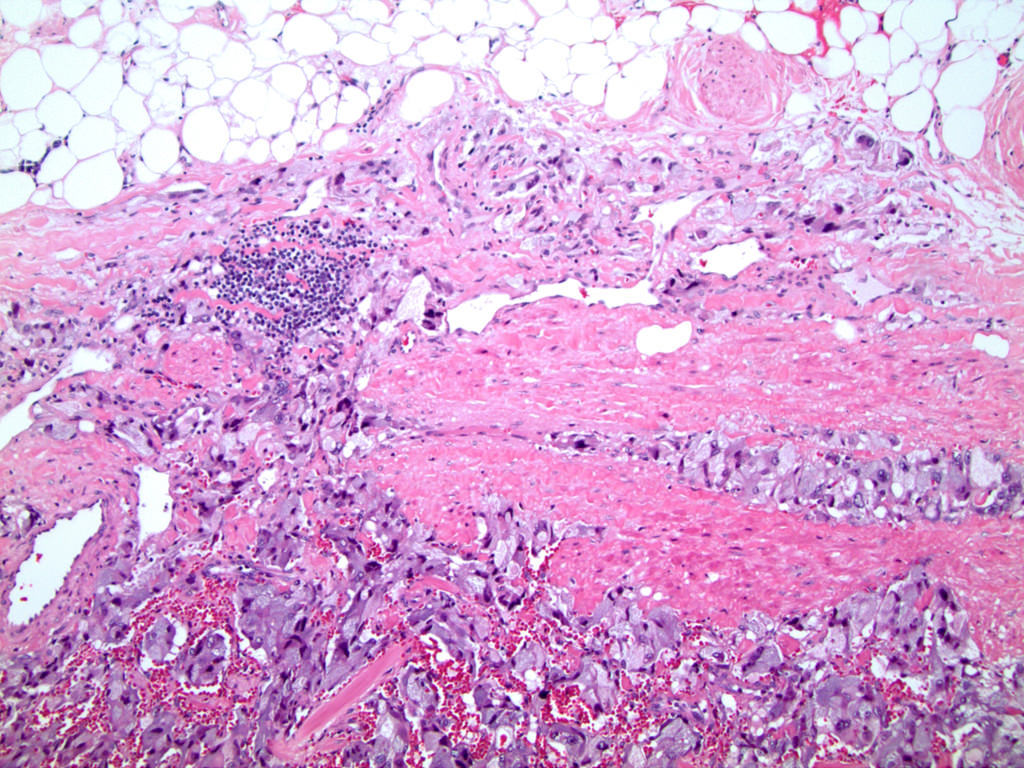
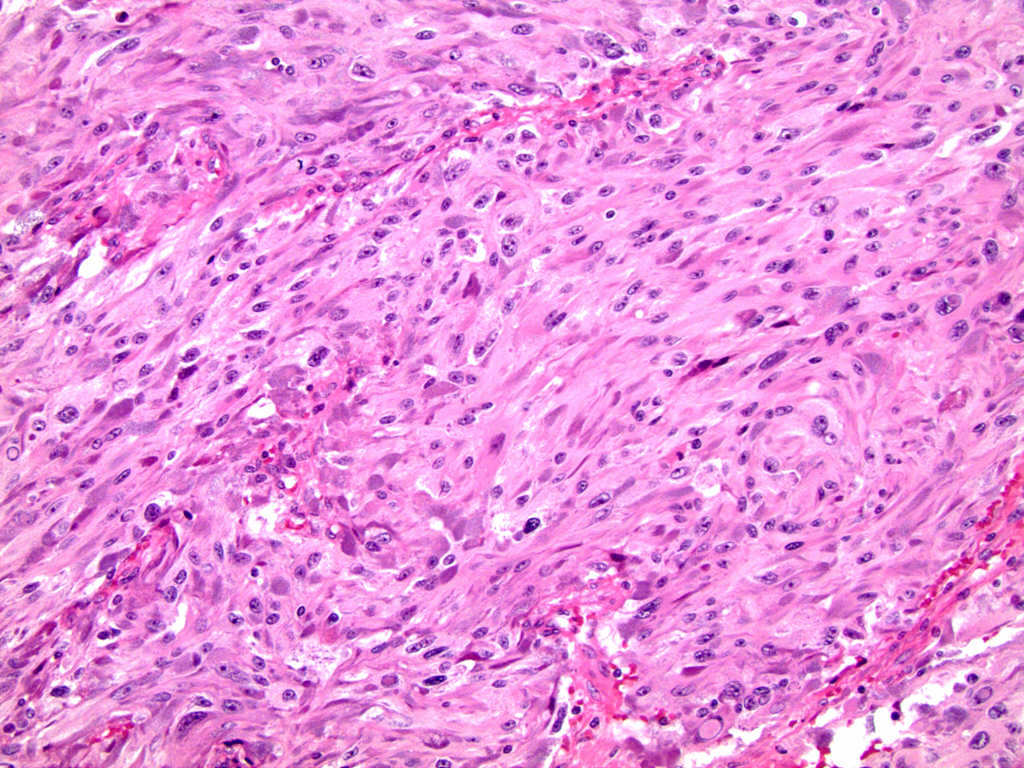
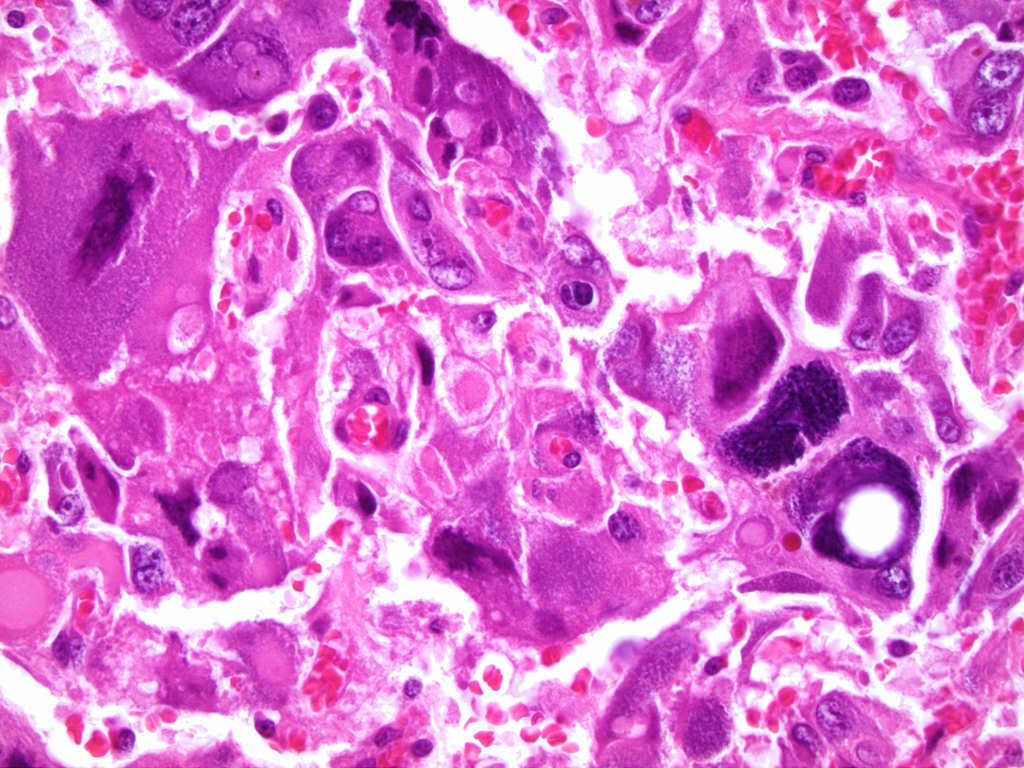
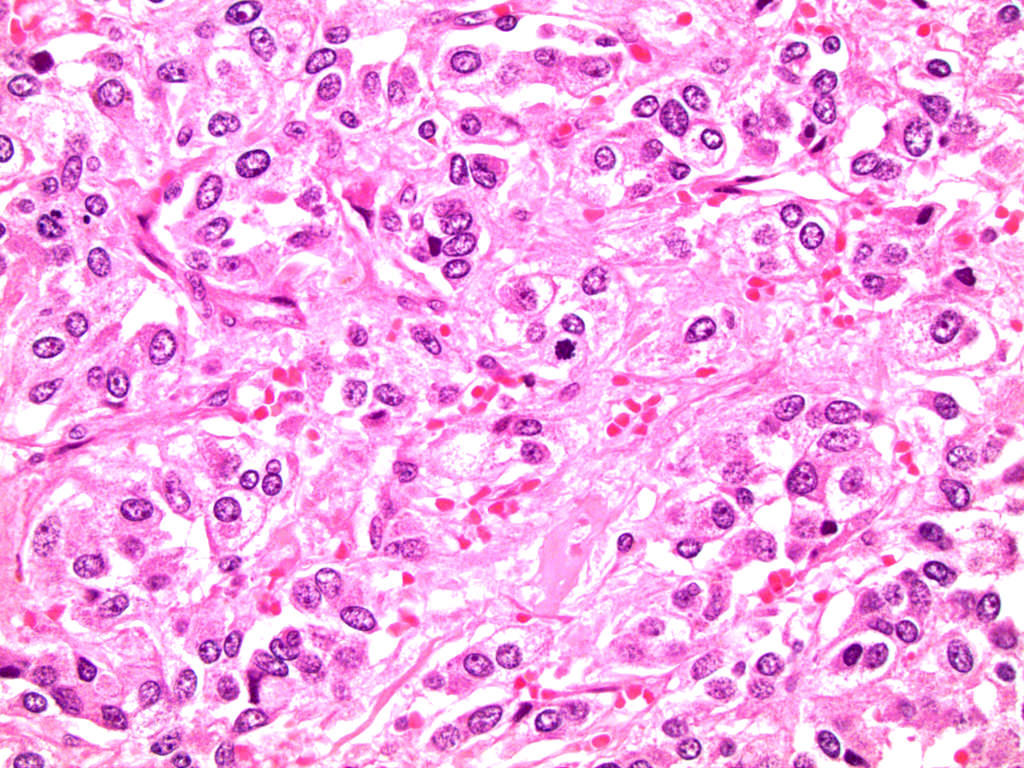
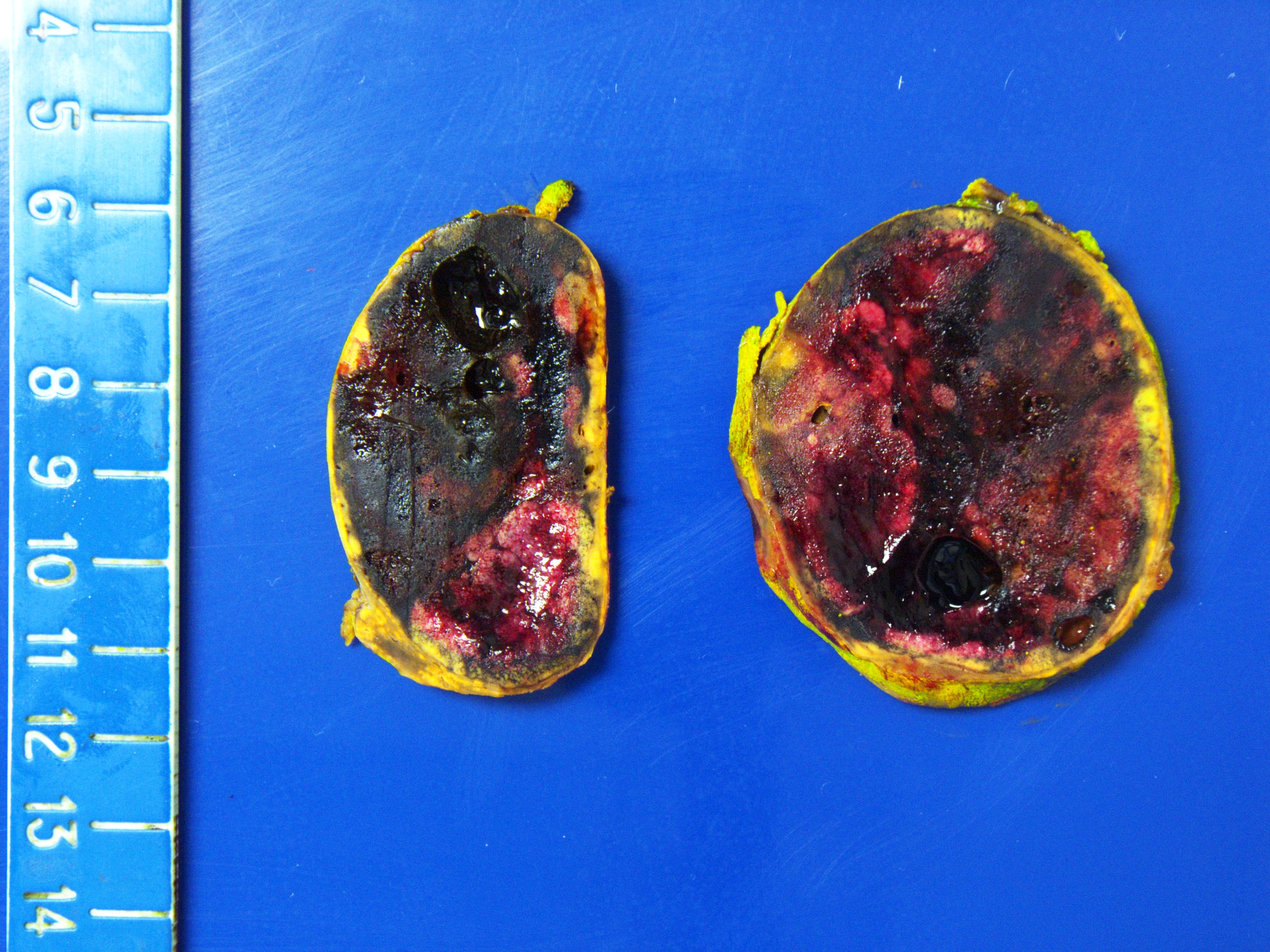
- Massive adrenal hemorrhage destroying adrenal cortex due to anticoagulation, coagulopathy and newborns with physiologic deficiencies in prothrombin time
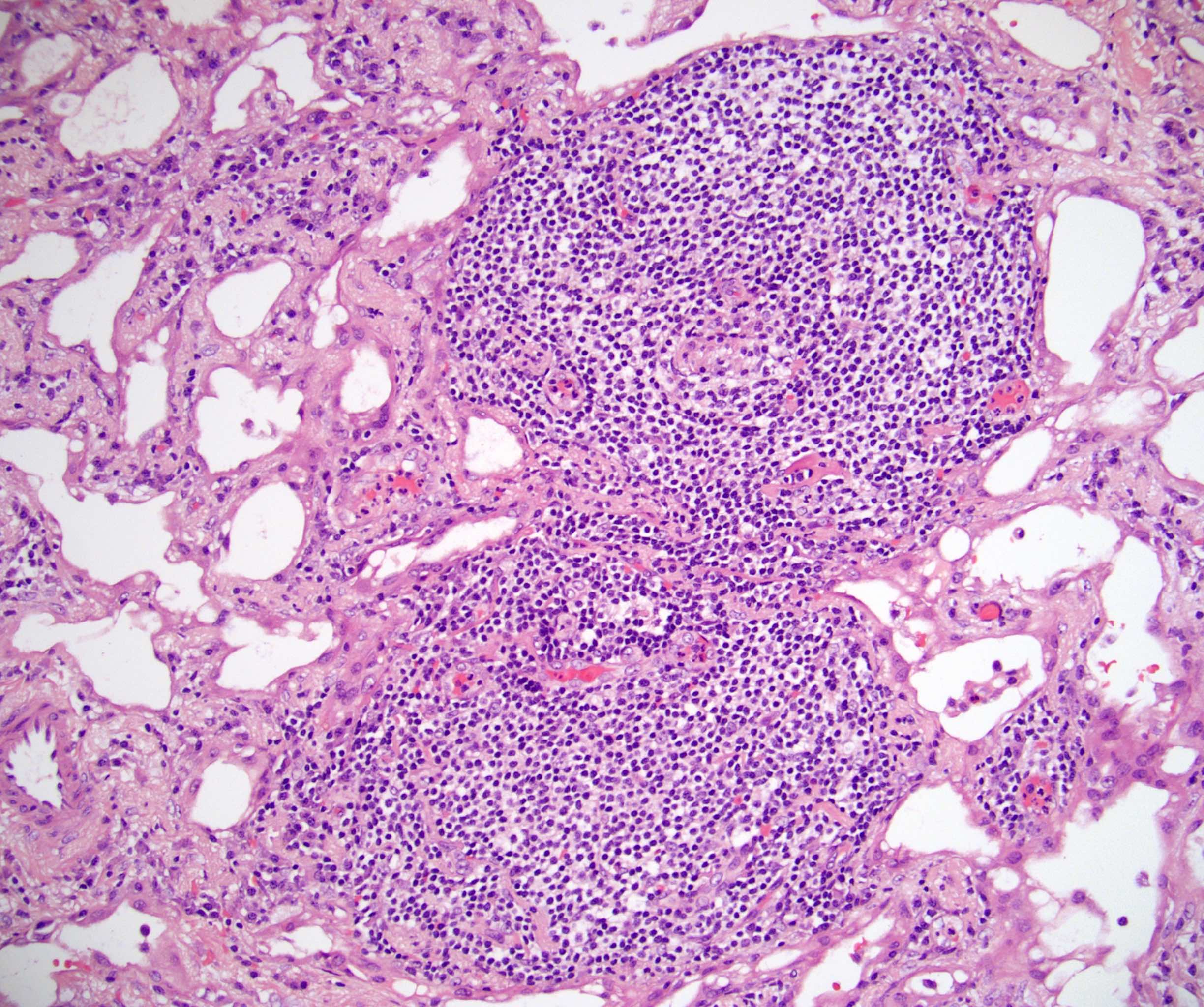
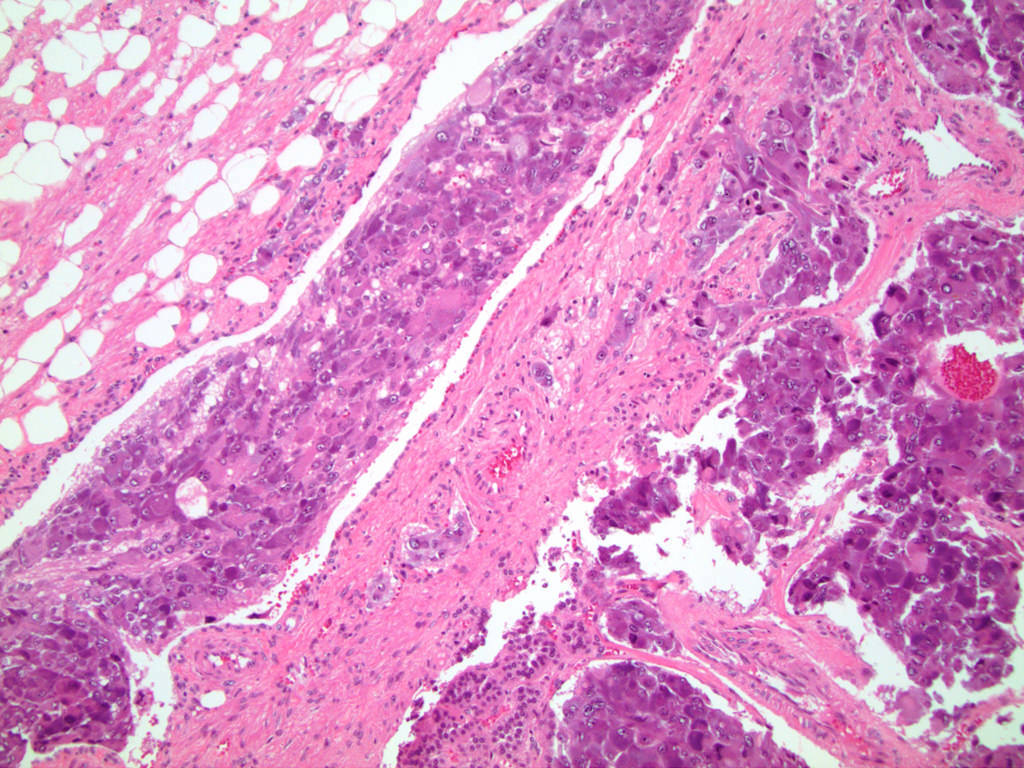
- Hypotension / shock that causes mild or massive corticomedullary necrosis, including Waterhouse-Friderichsen syndrome
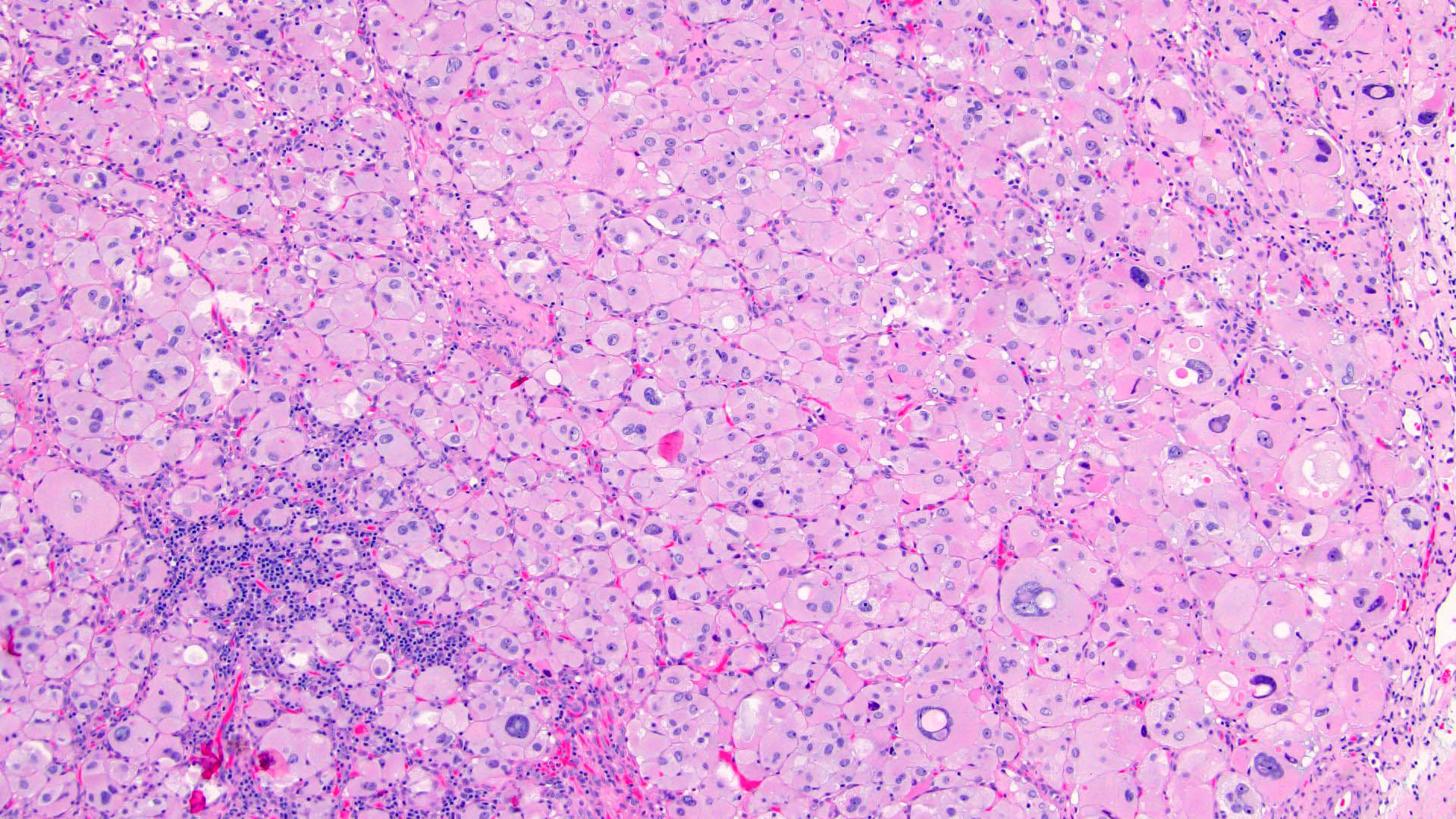
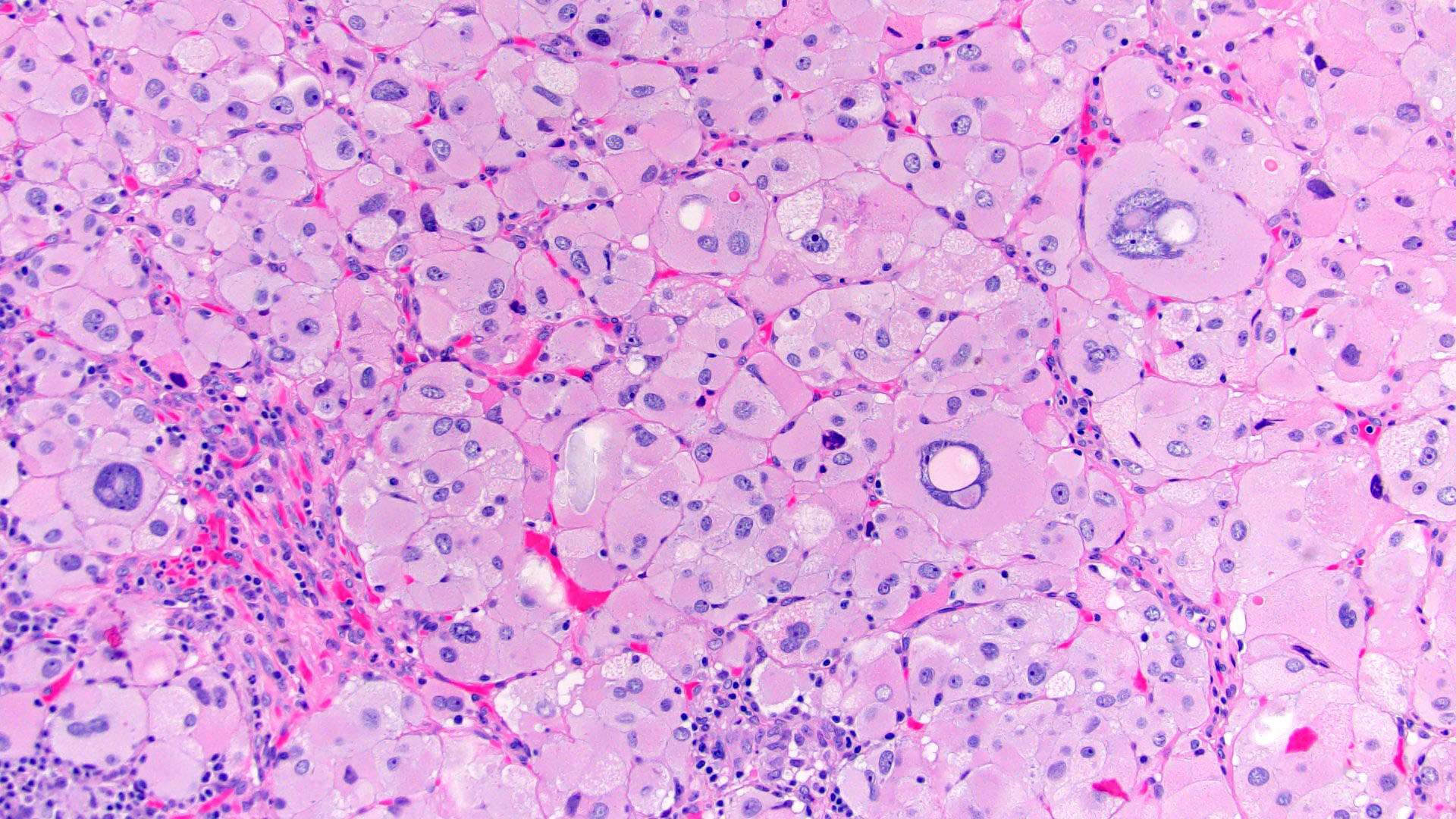
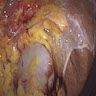
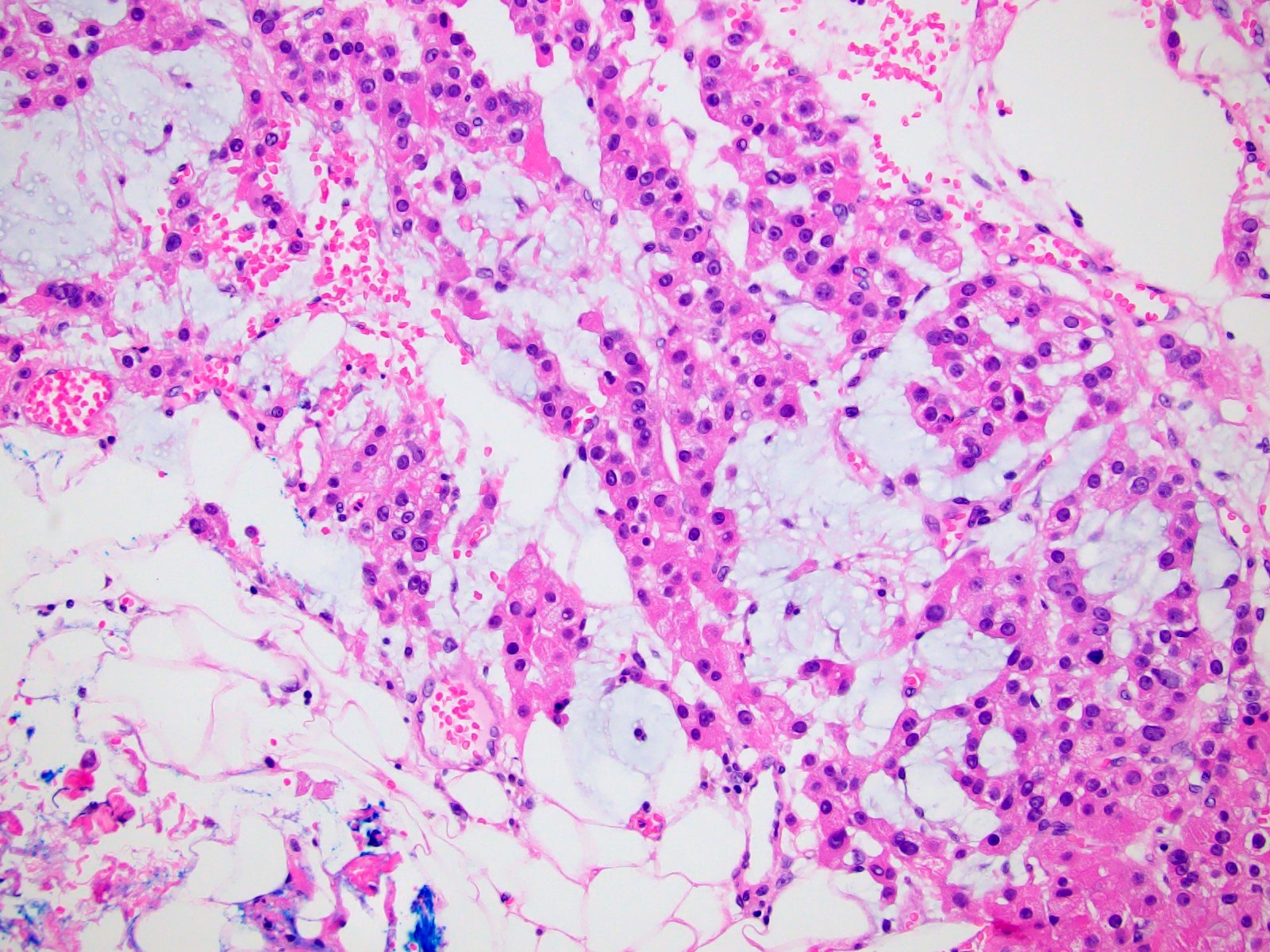
- Infections that destroy substantial adrenal cortical tissue
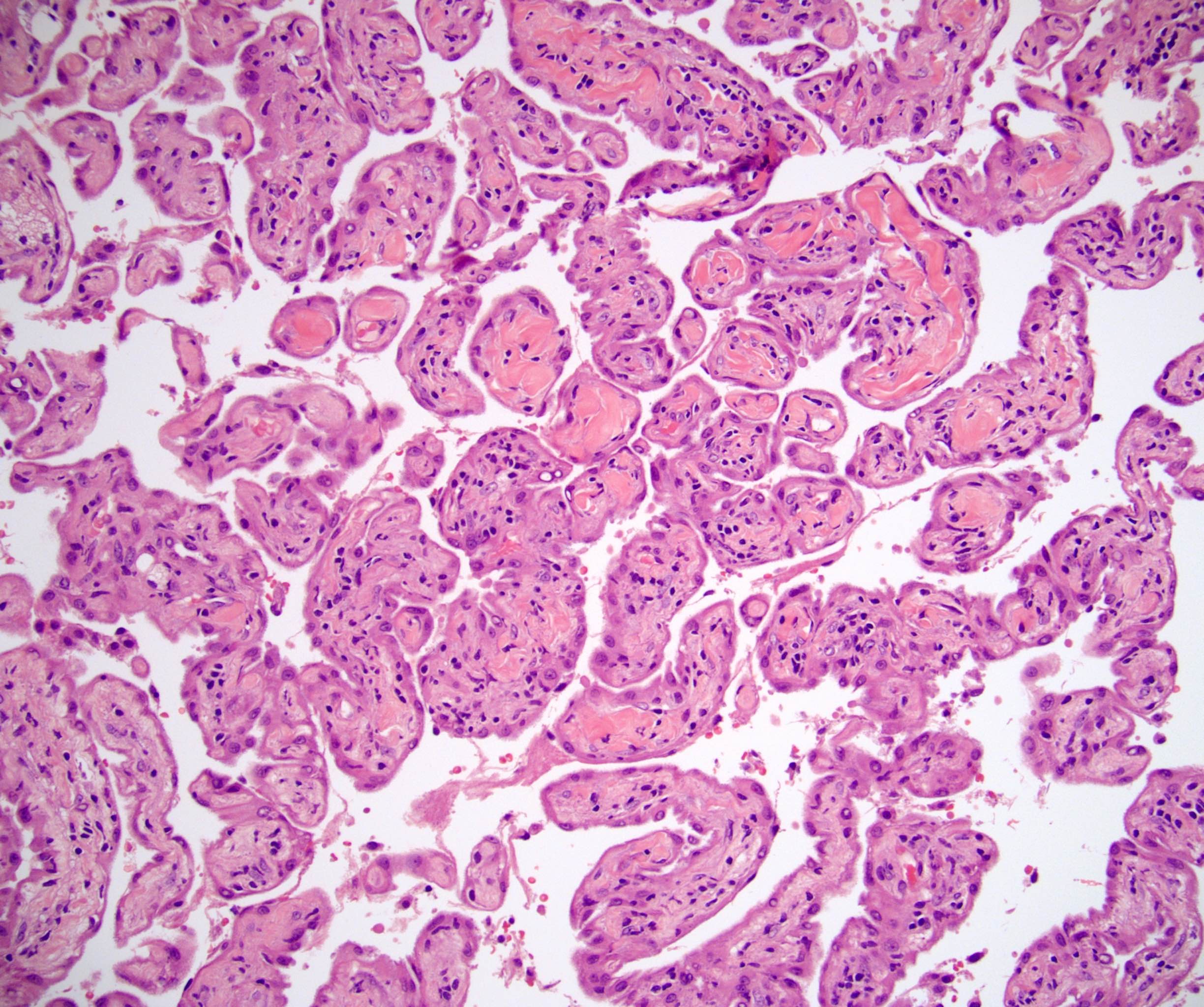
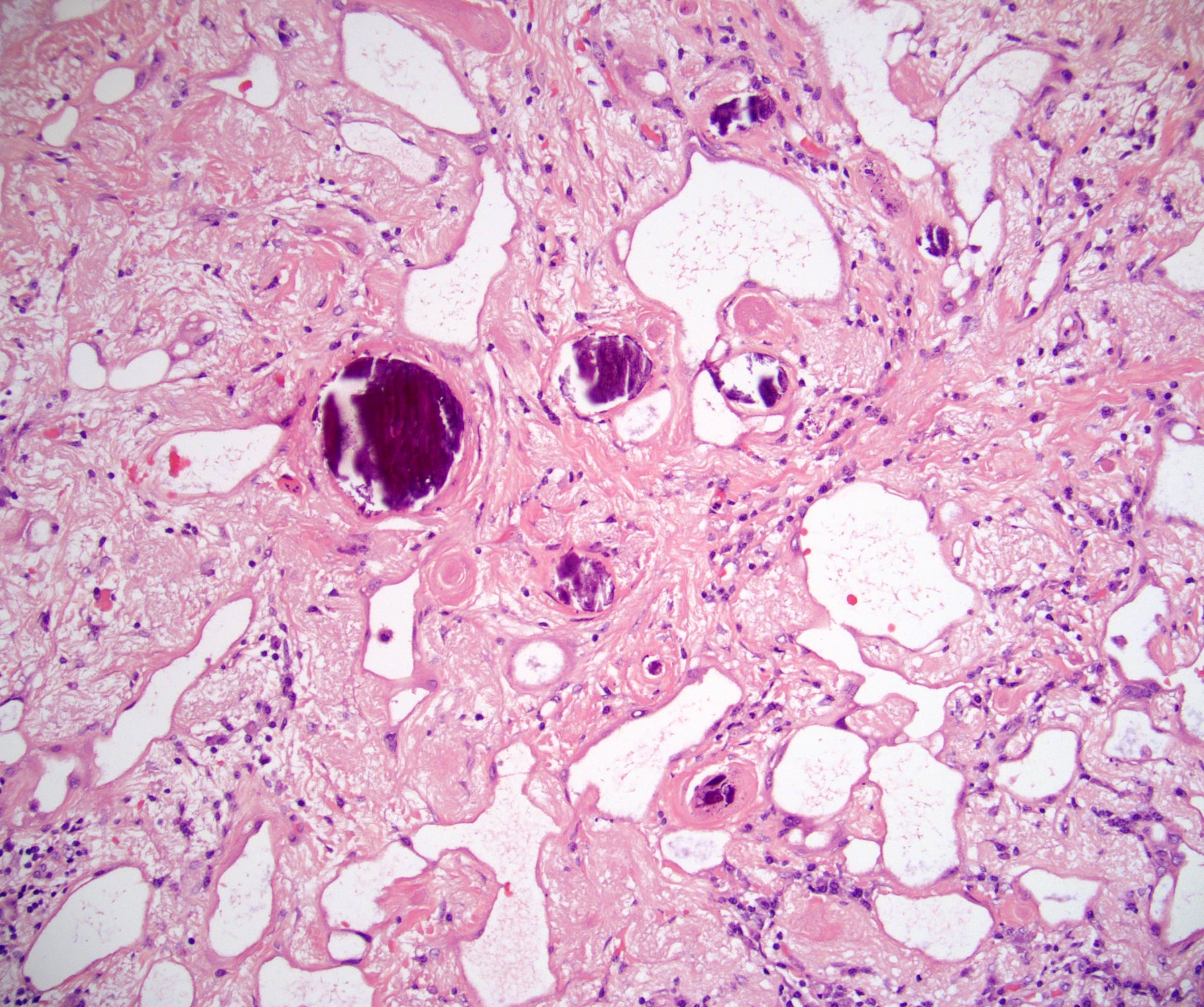
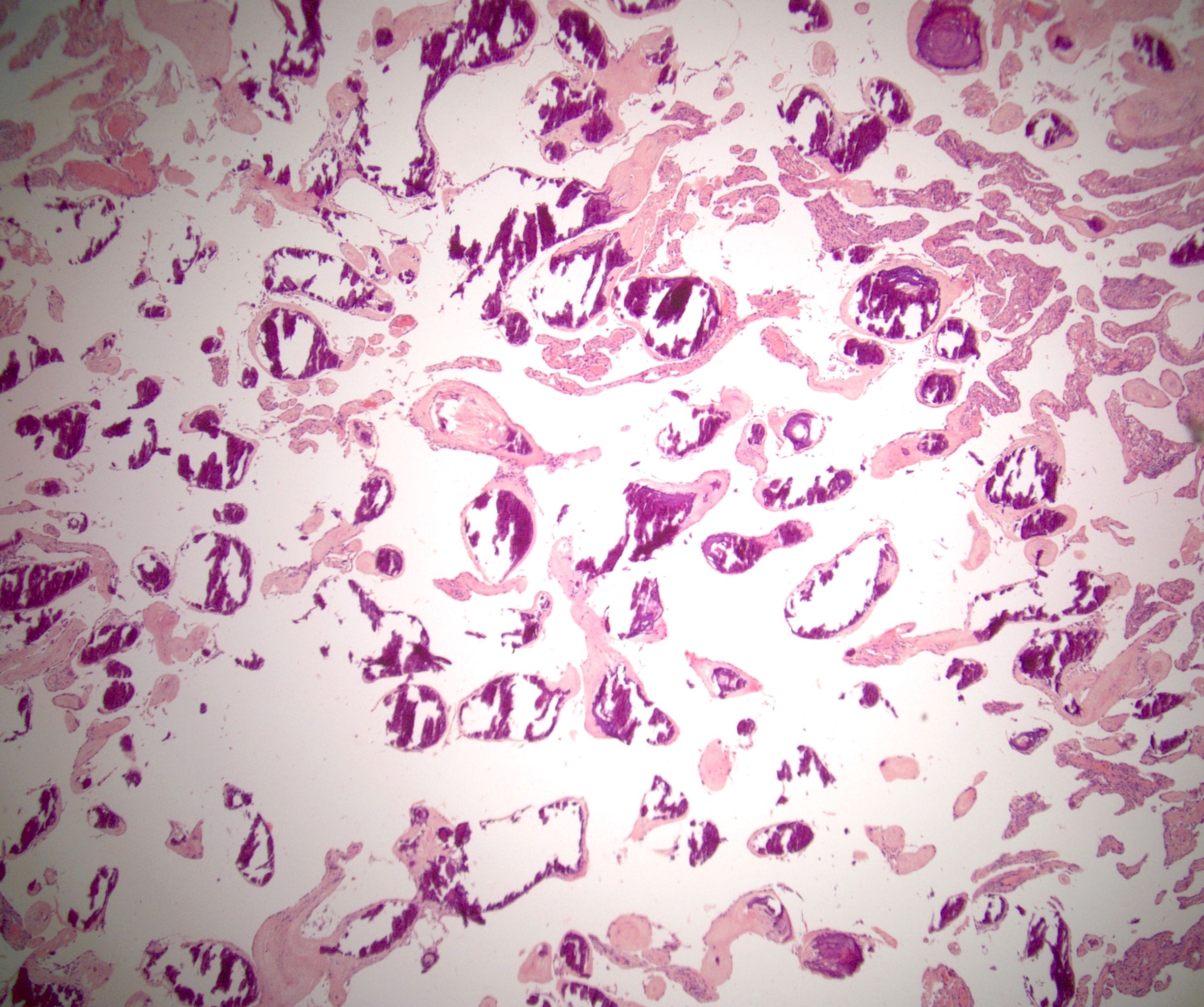
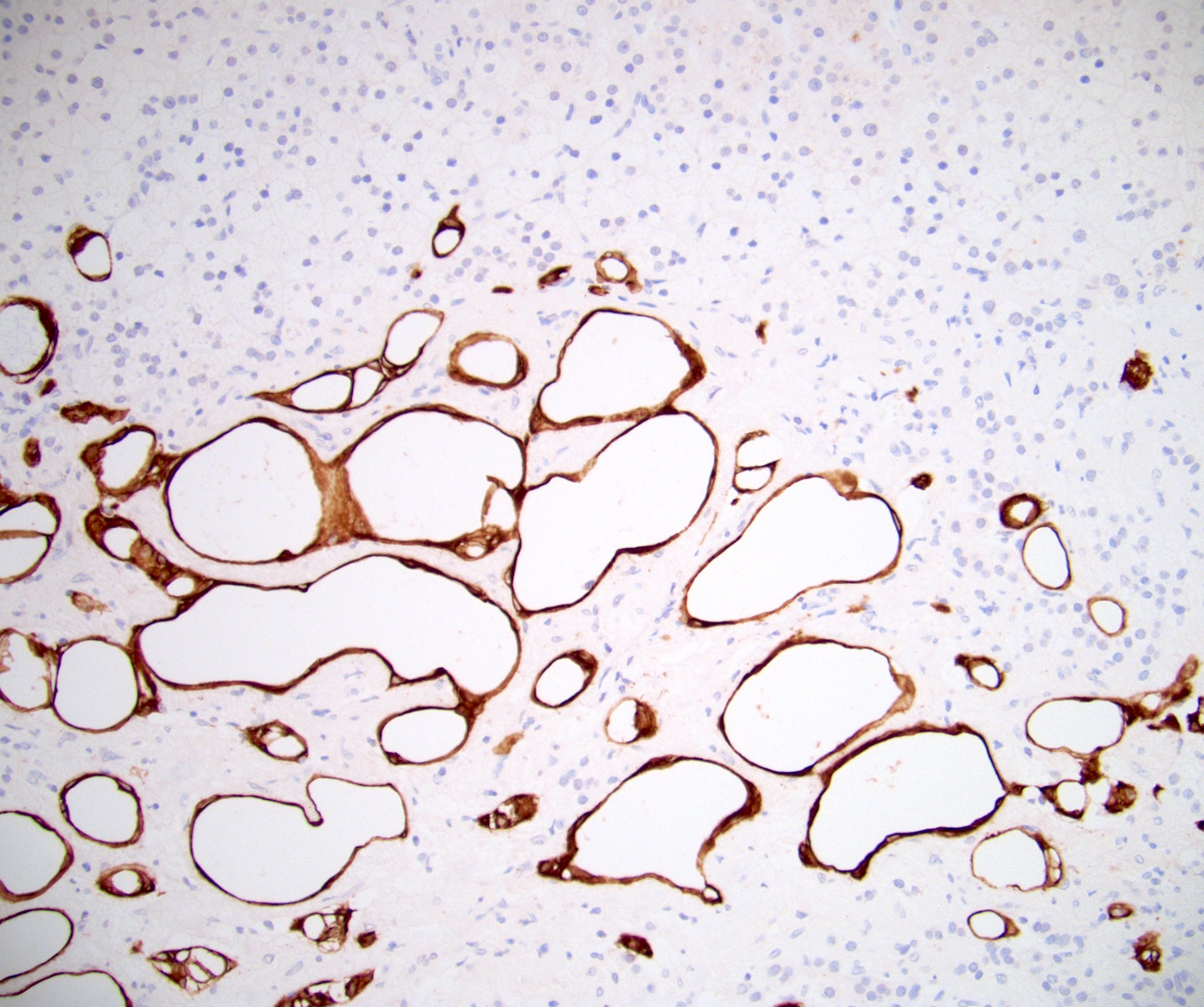
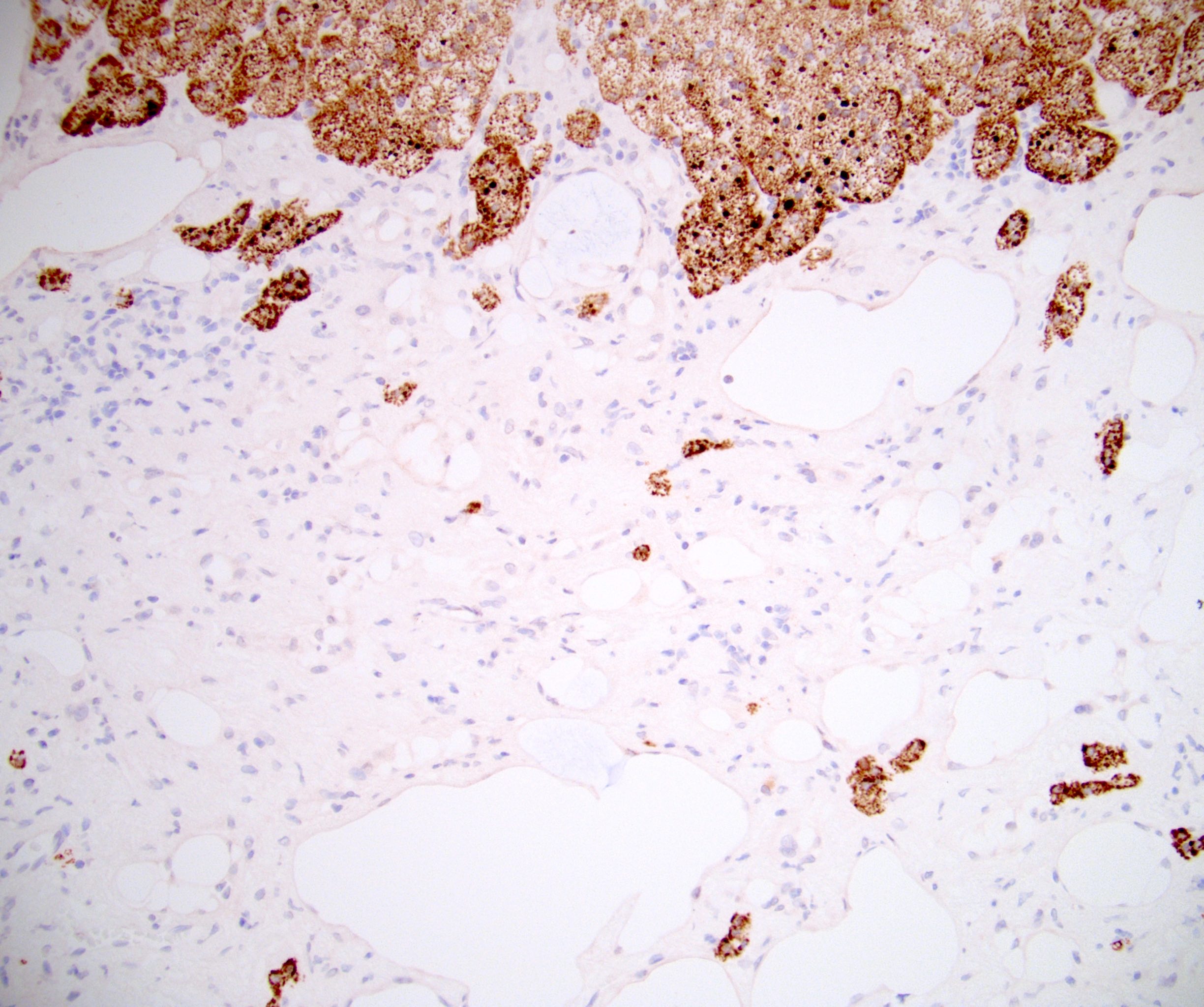
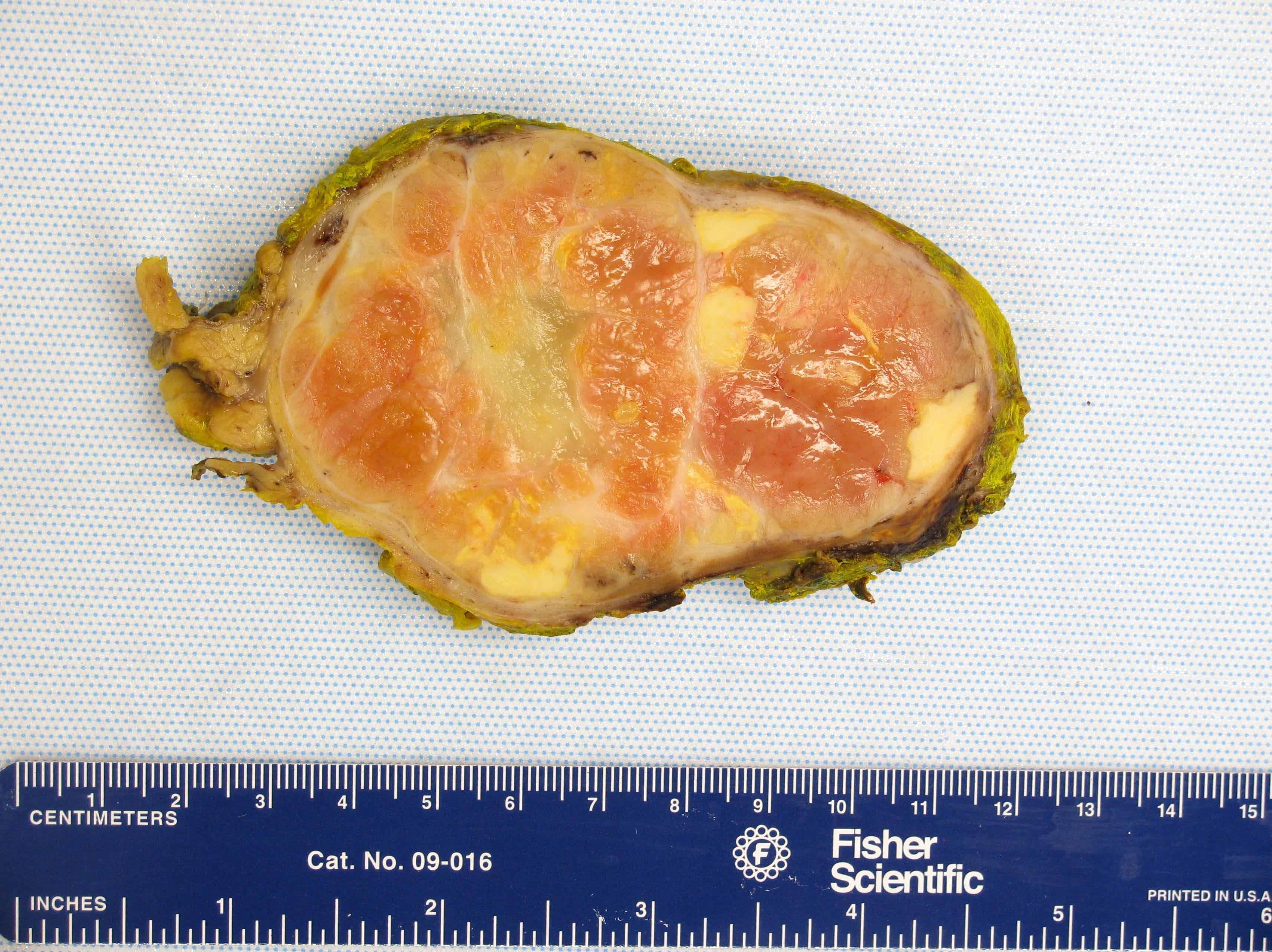
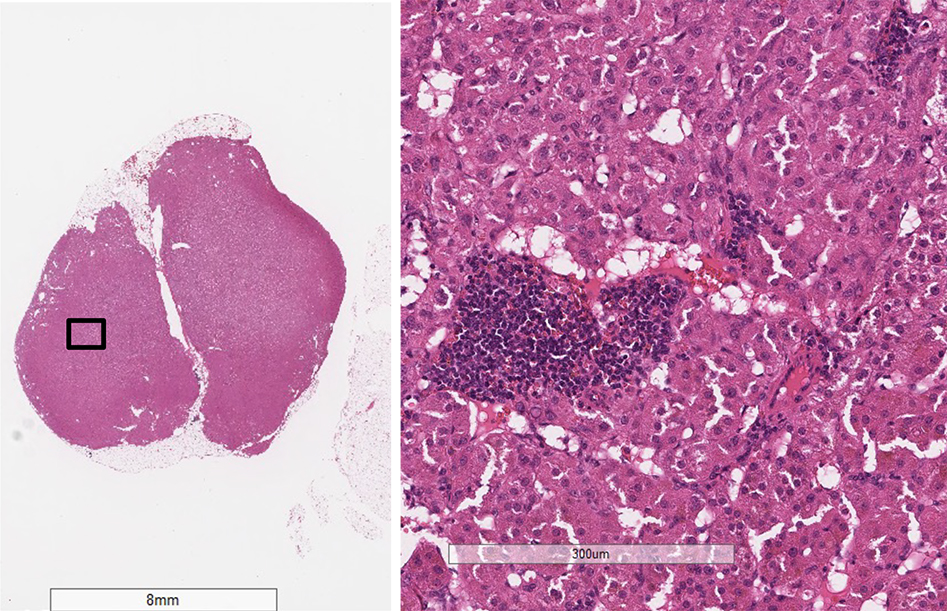
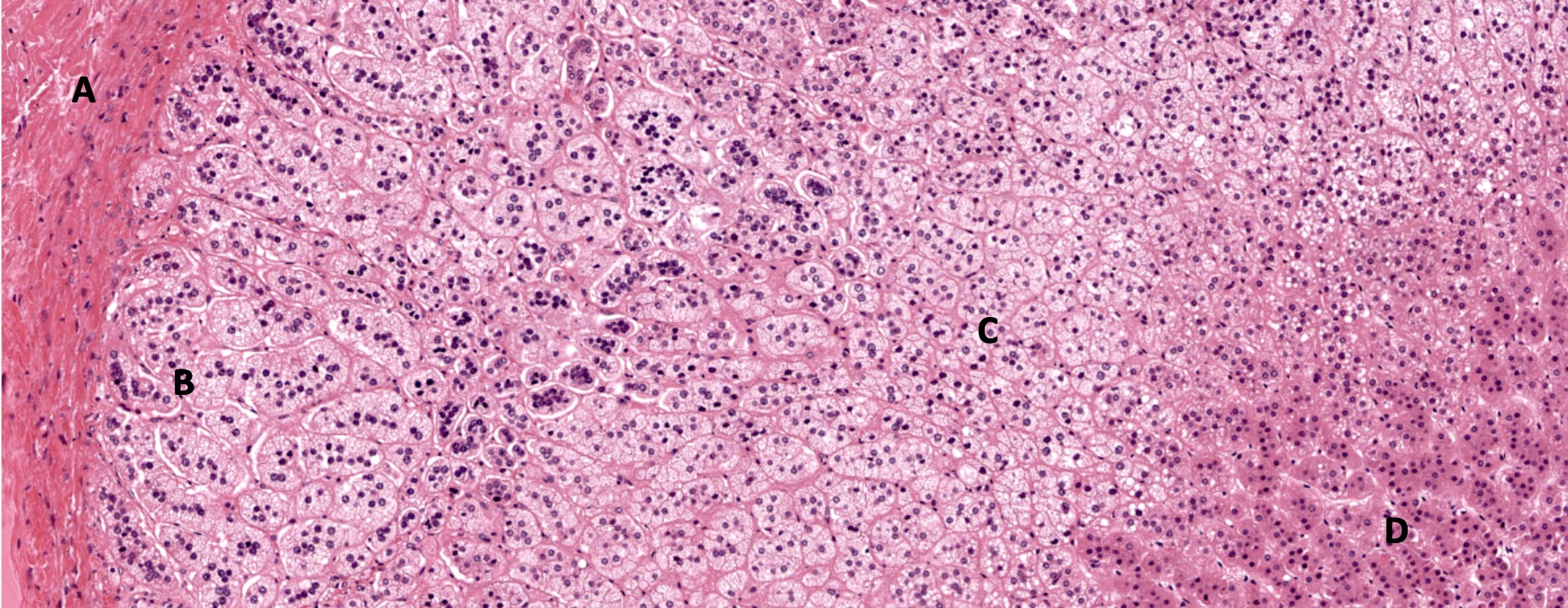
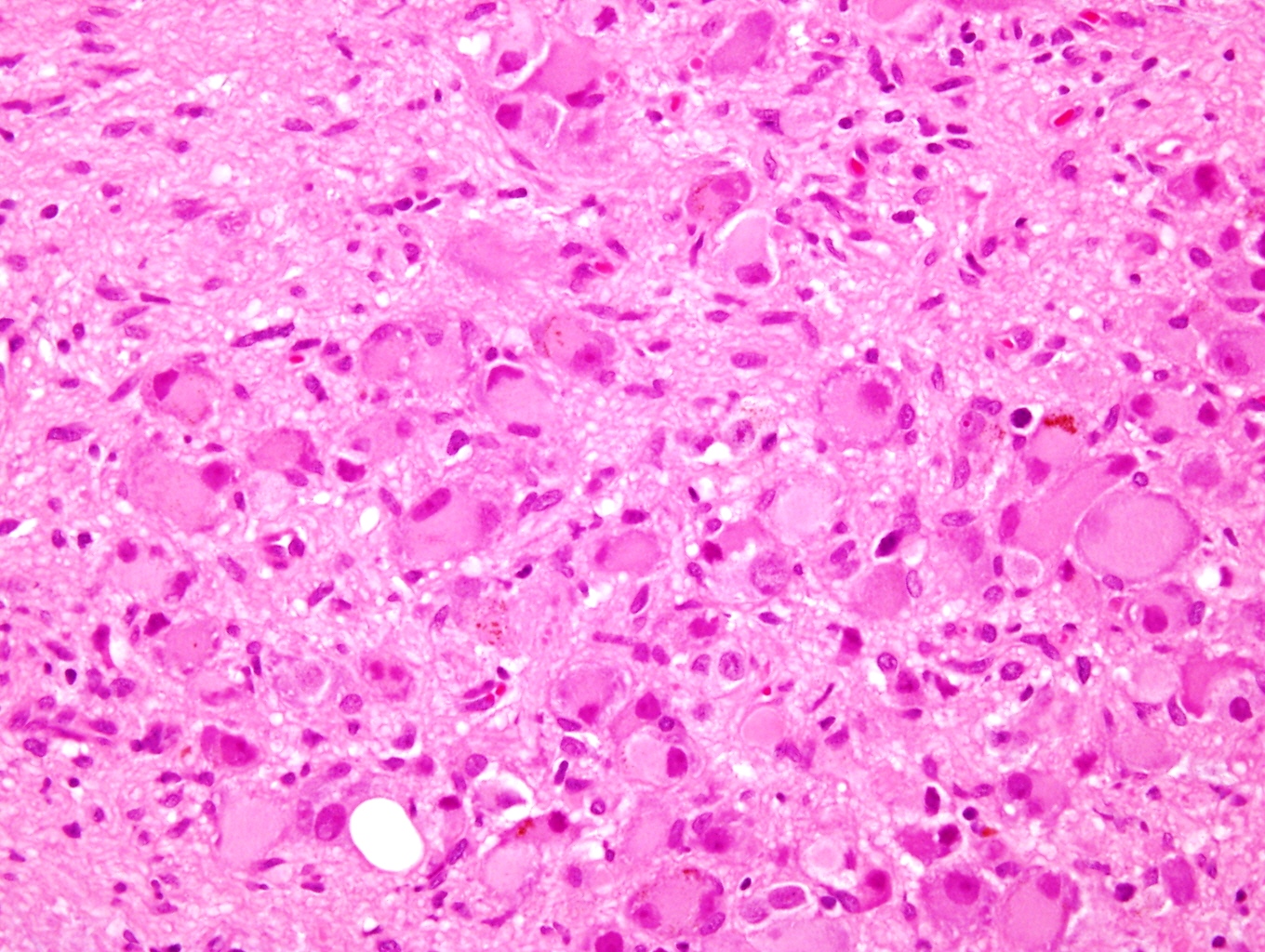
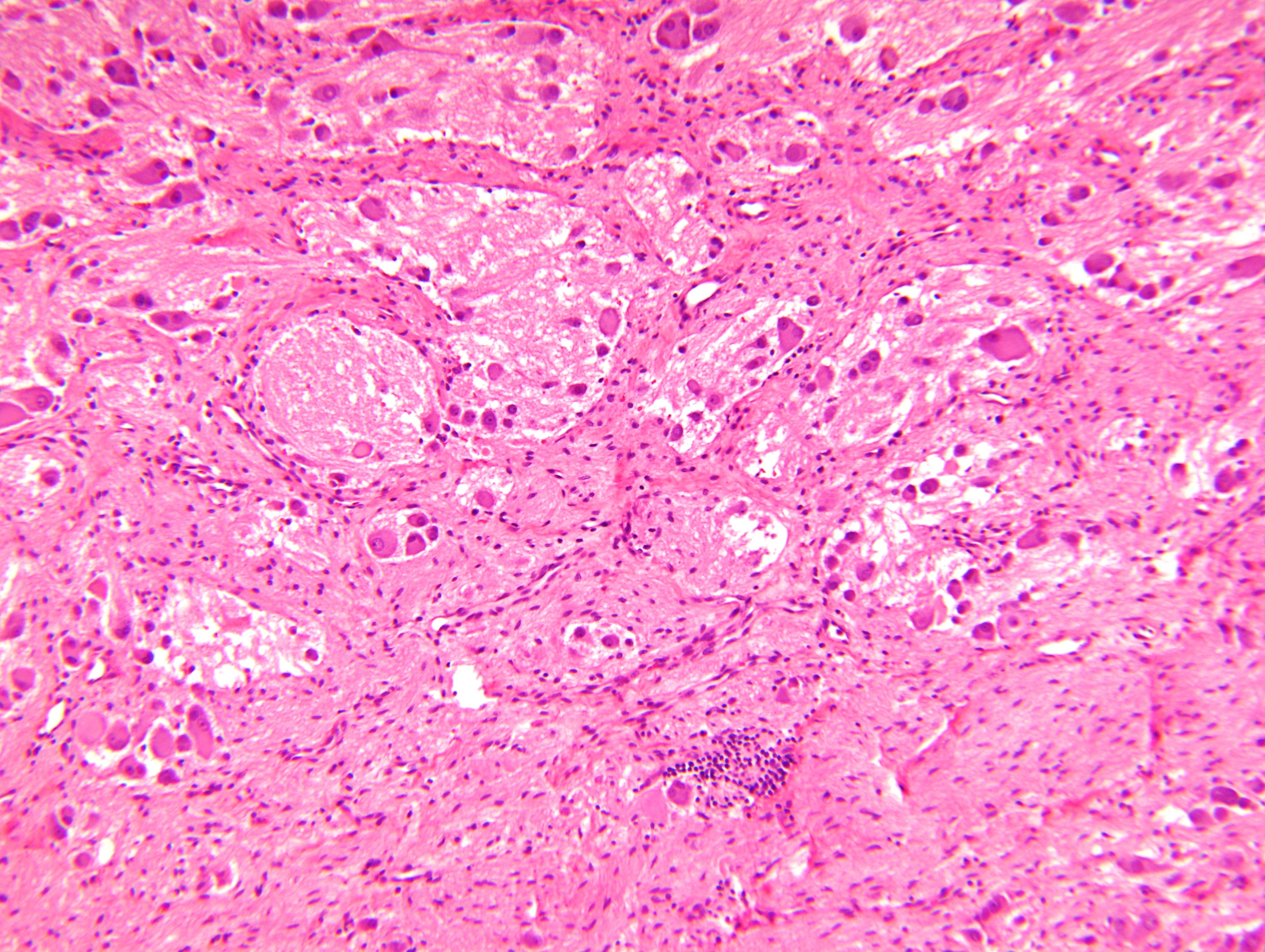
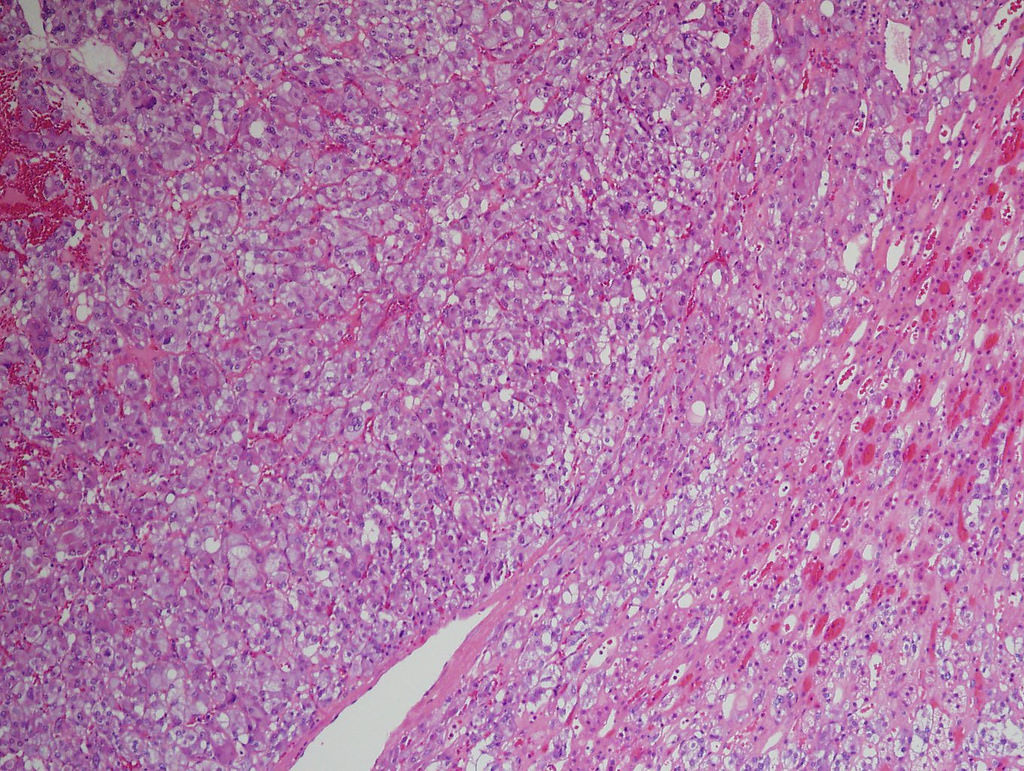
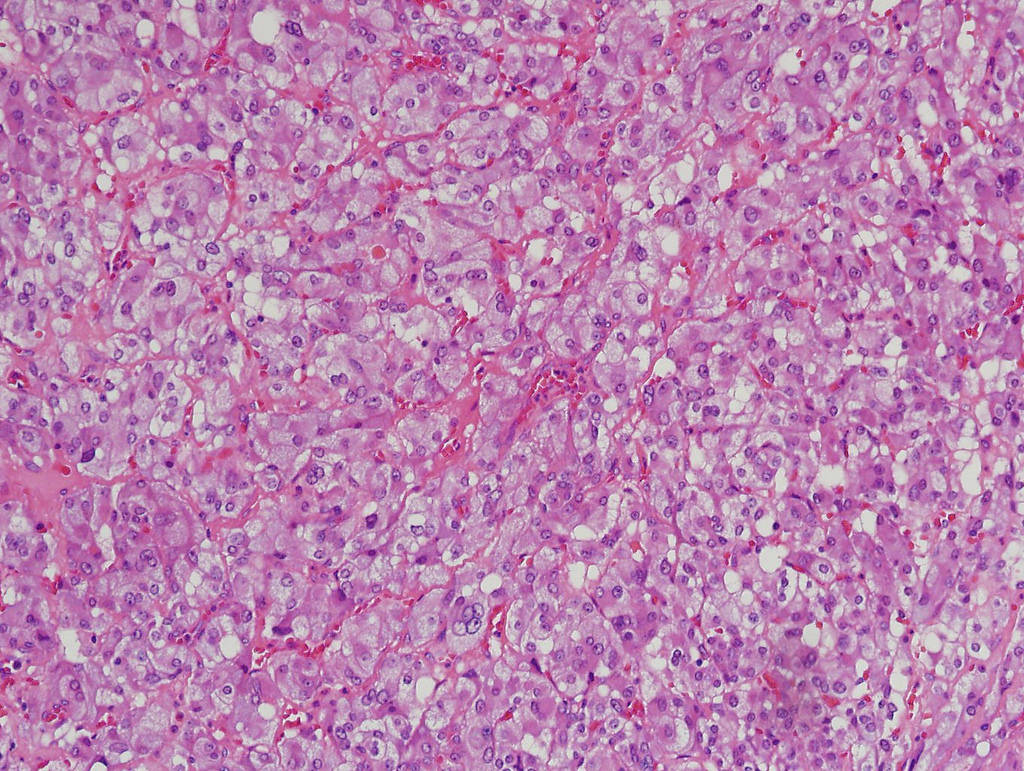
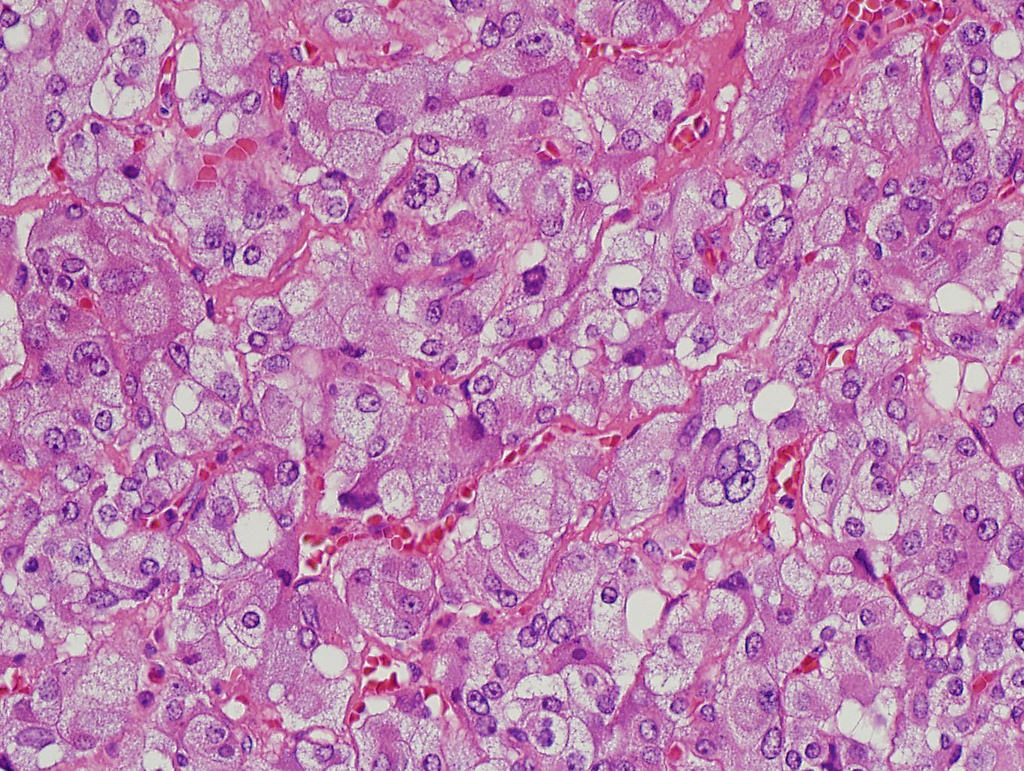
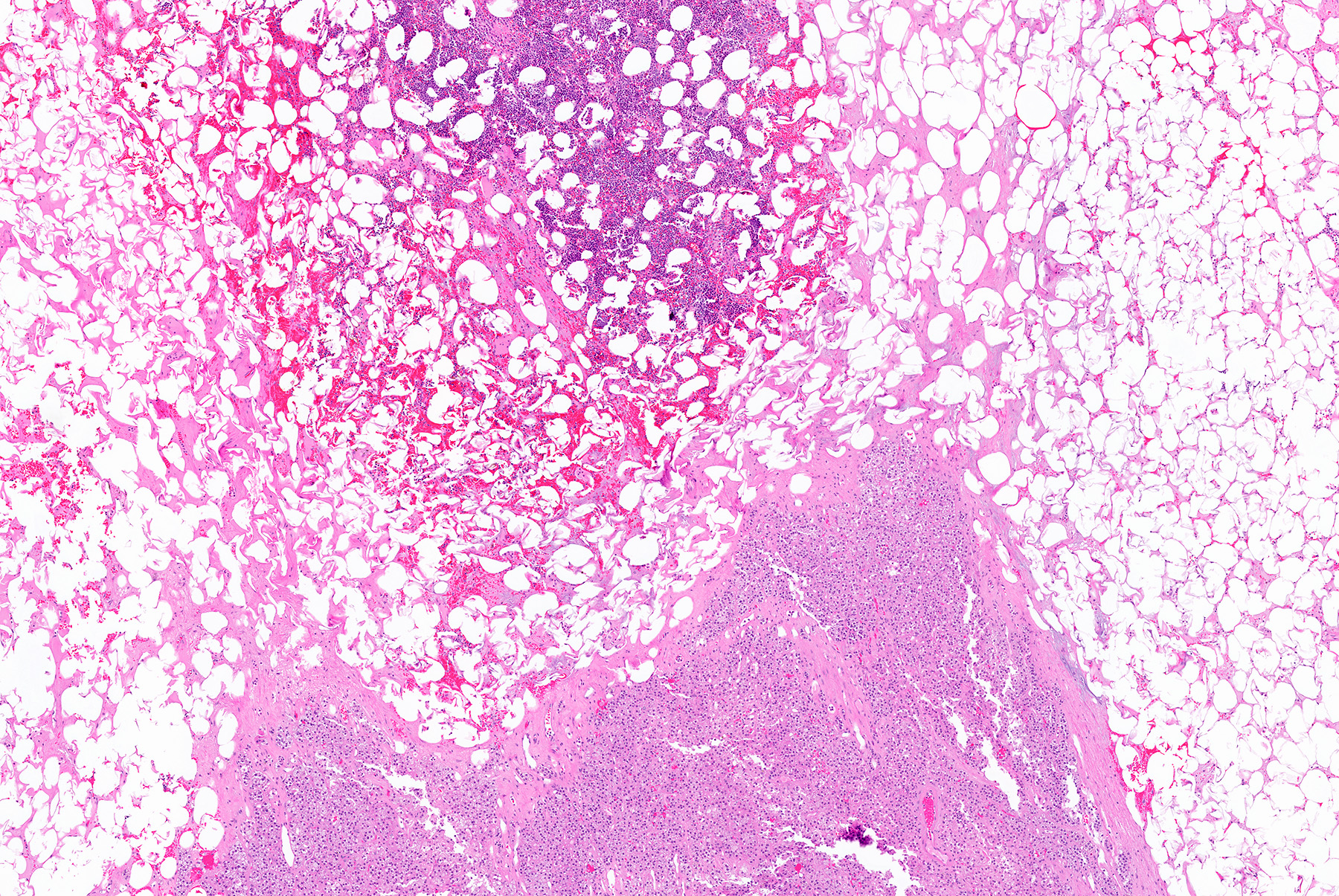
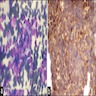
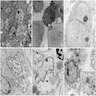
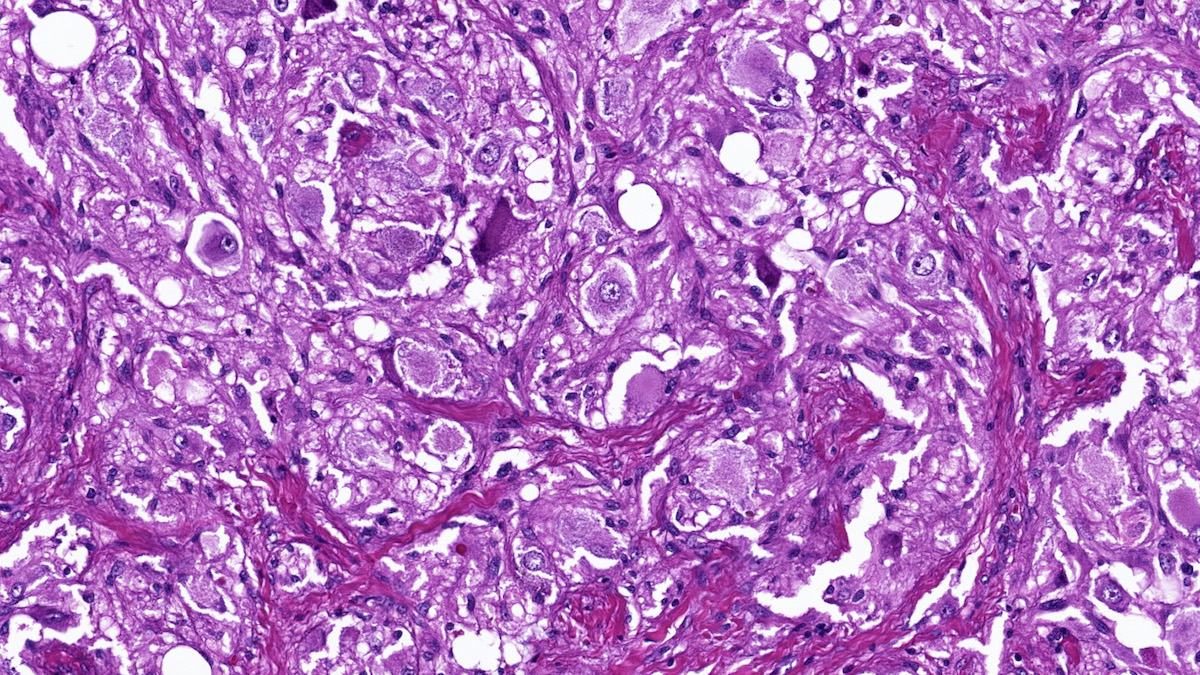
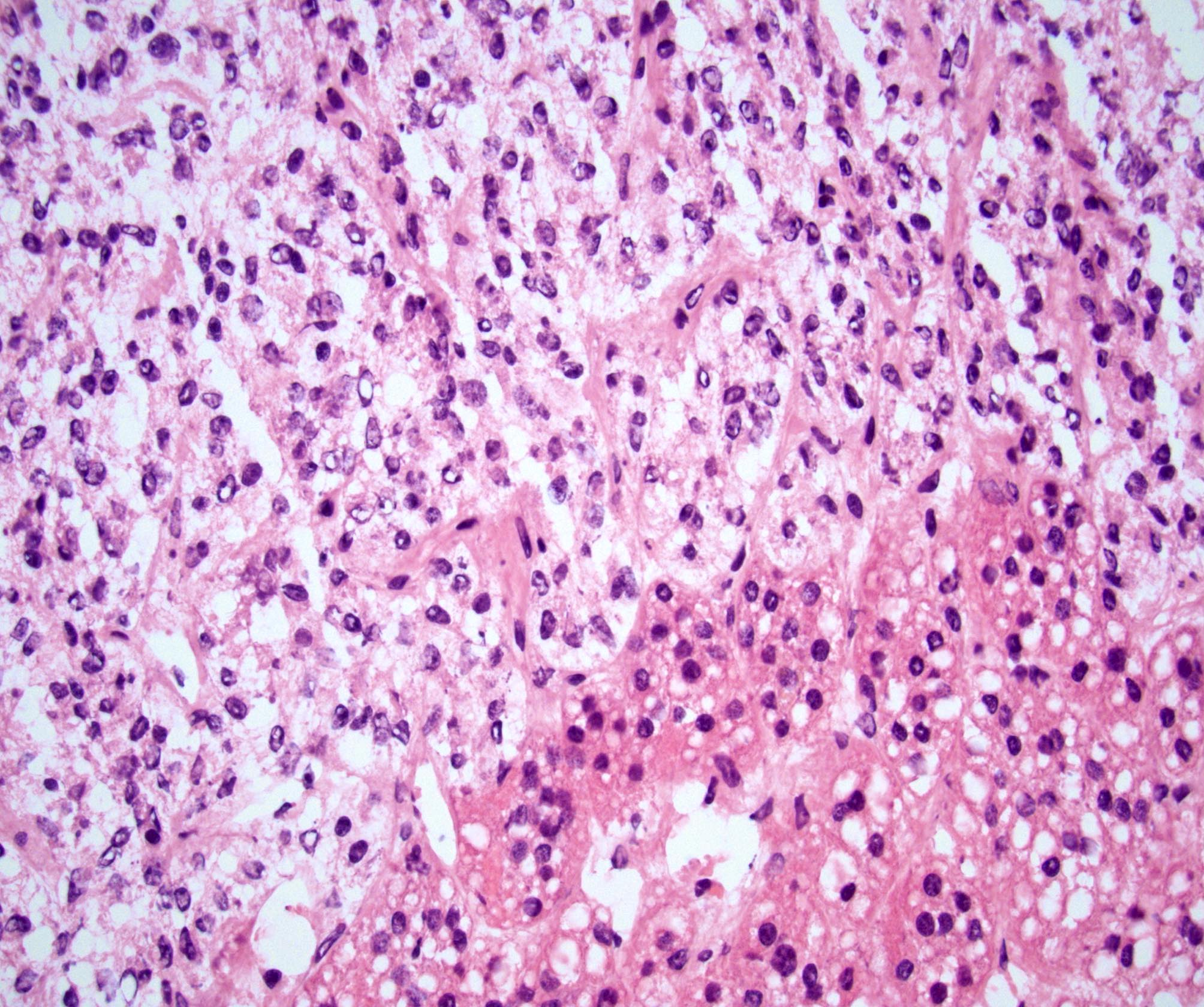
- Amyloidosis
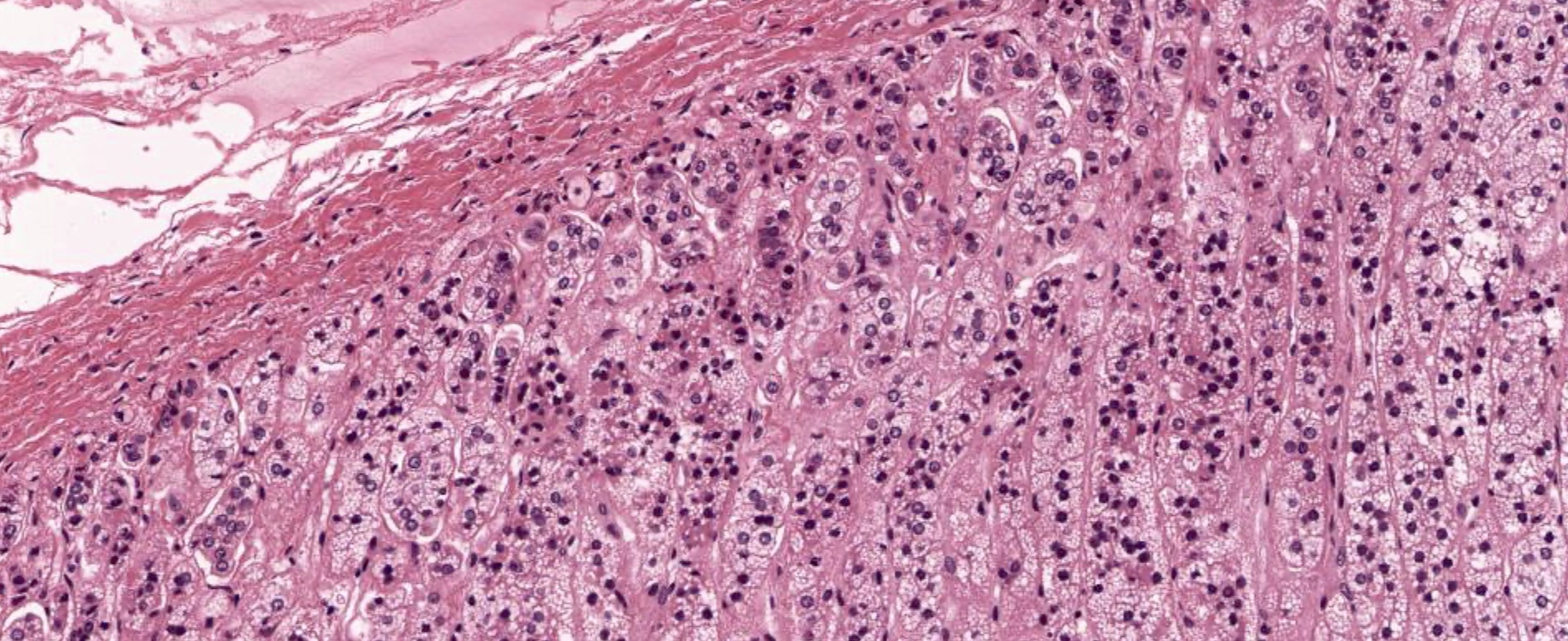
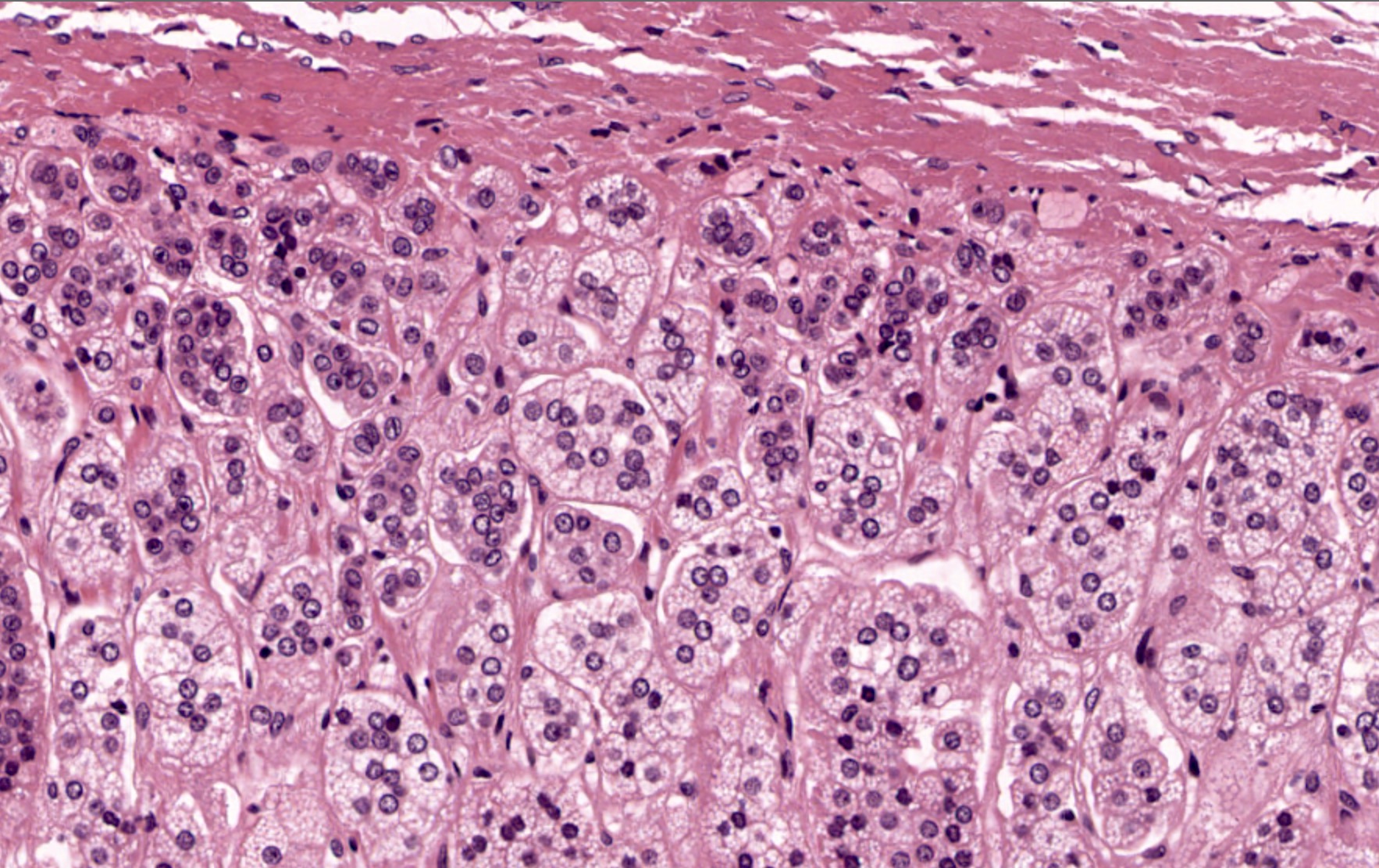
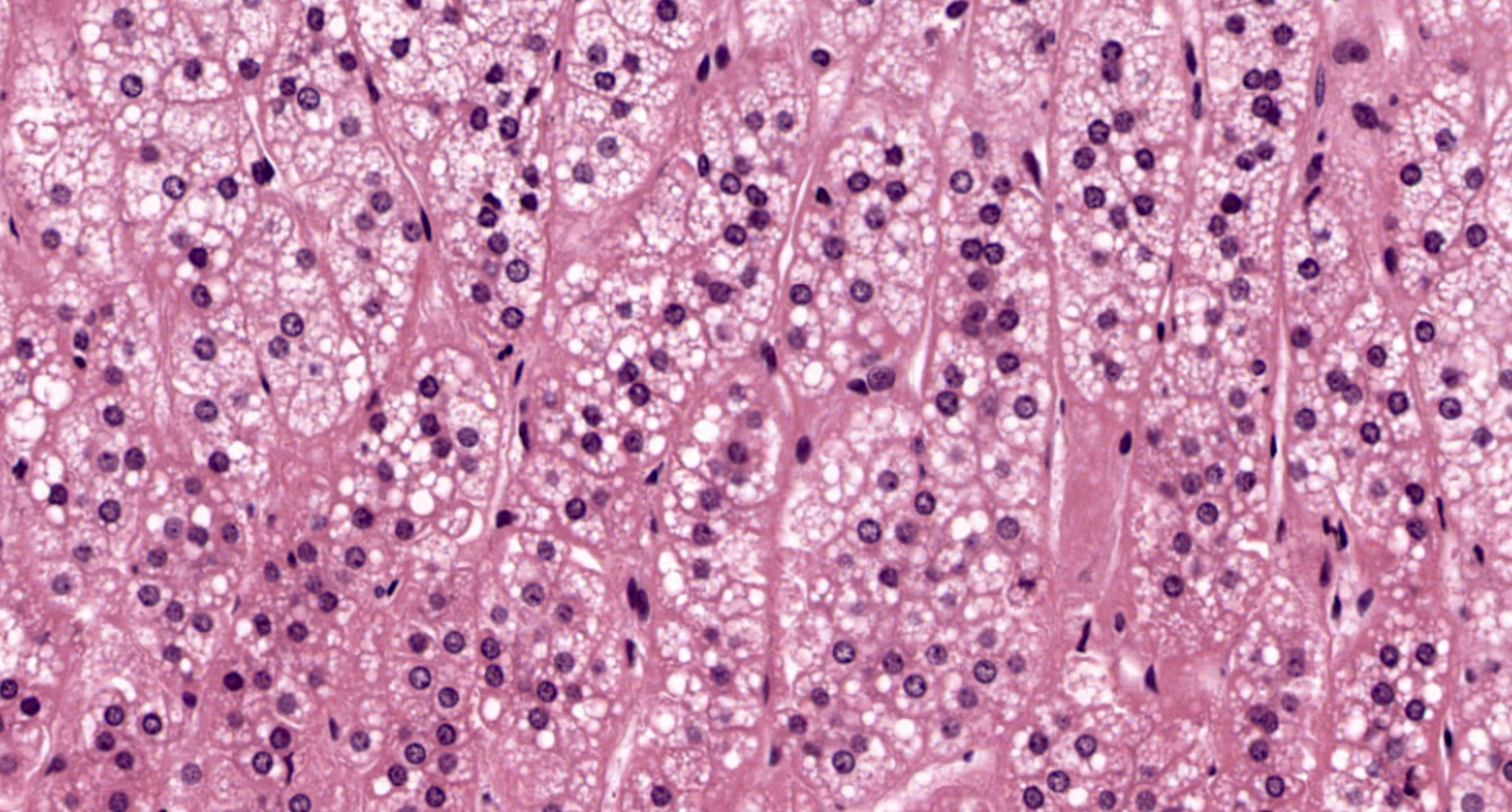
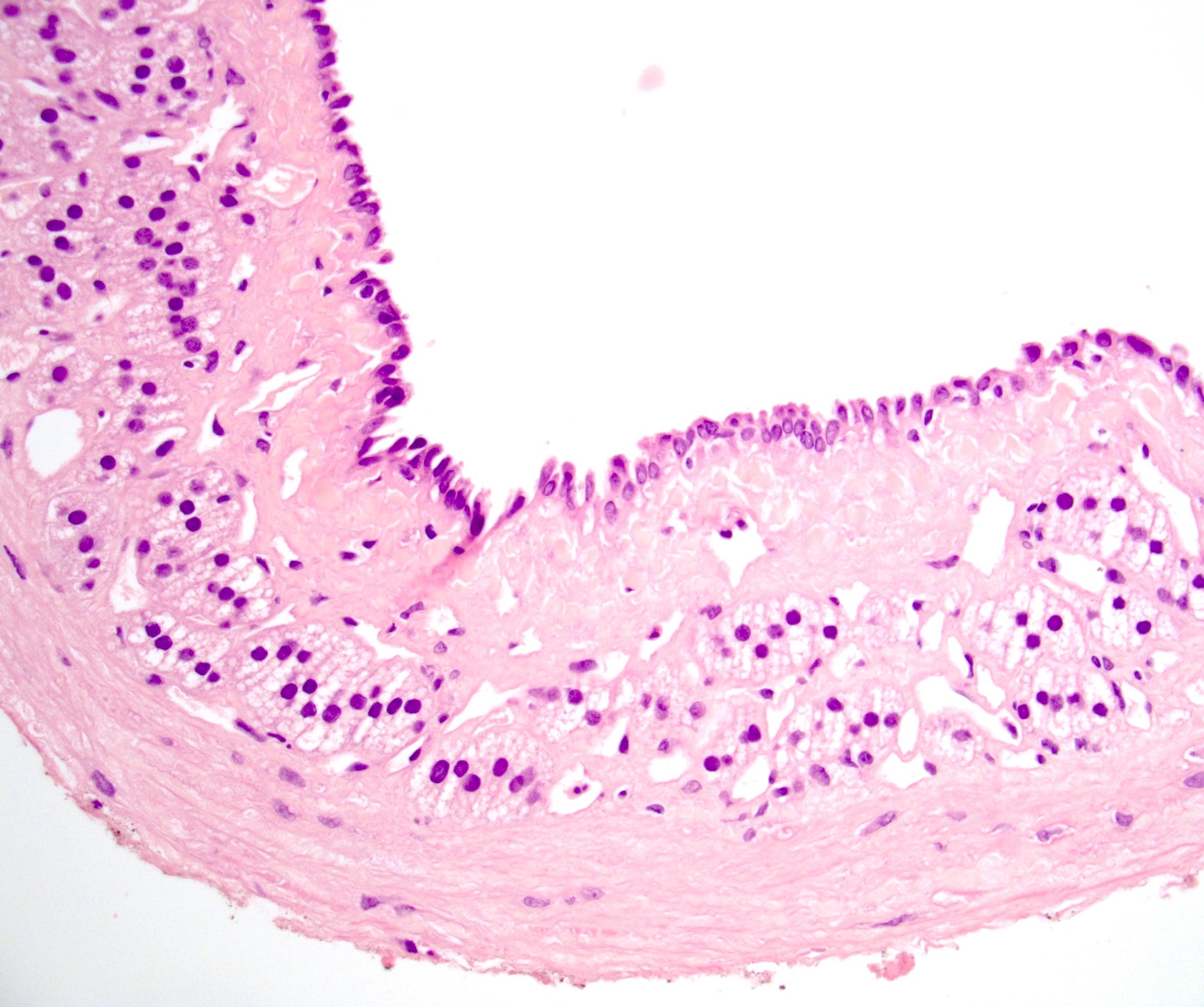
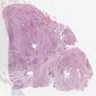
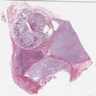
- Rarely causes cortical hypofunction, only if extensive bilateral involvement
- Usually associated with systemic amyloidosis-AA type
- 68% of consecutive autopsies had adrenal amyloid deposits, often multinodular and probably due to aging
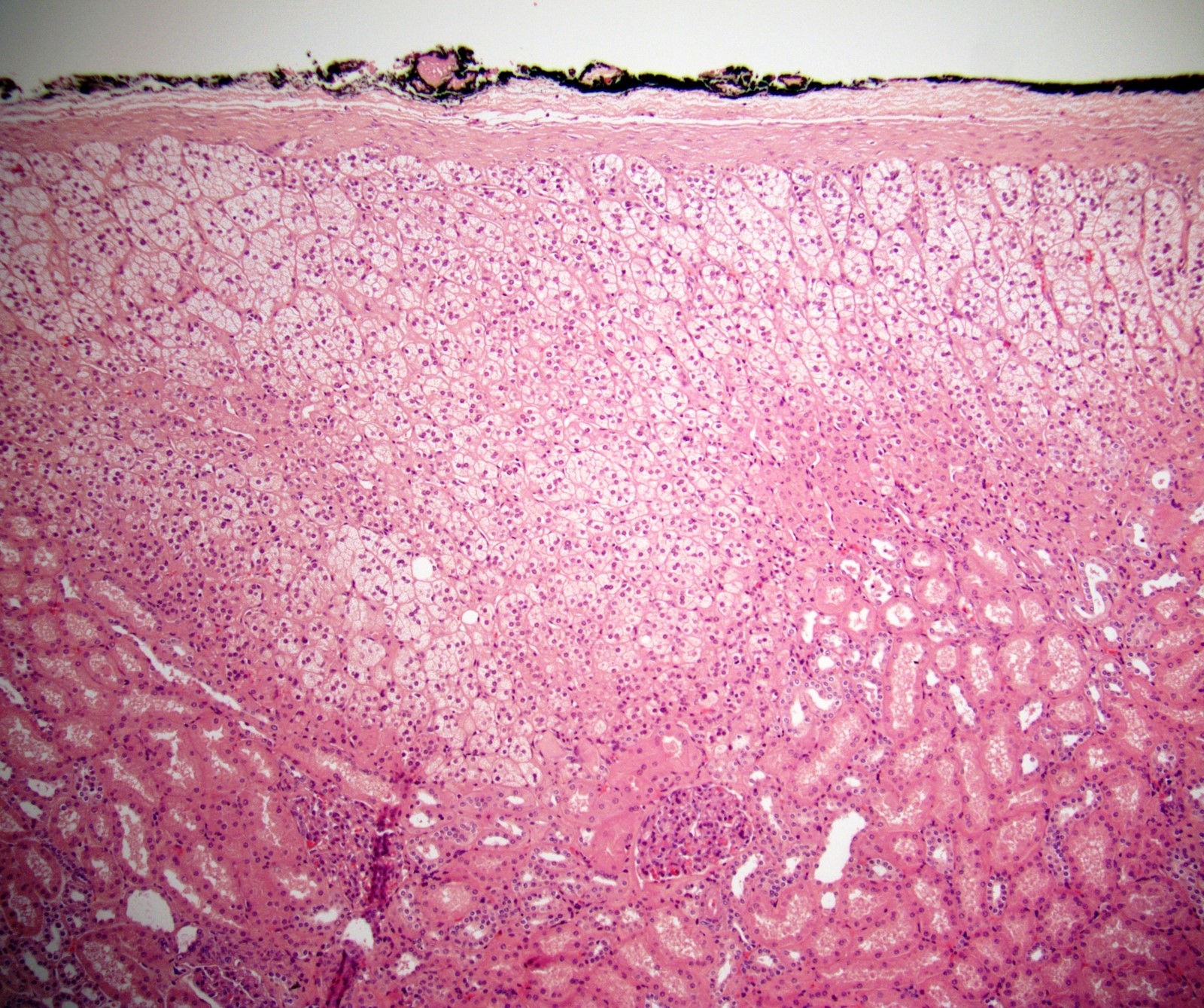
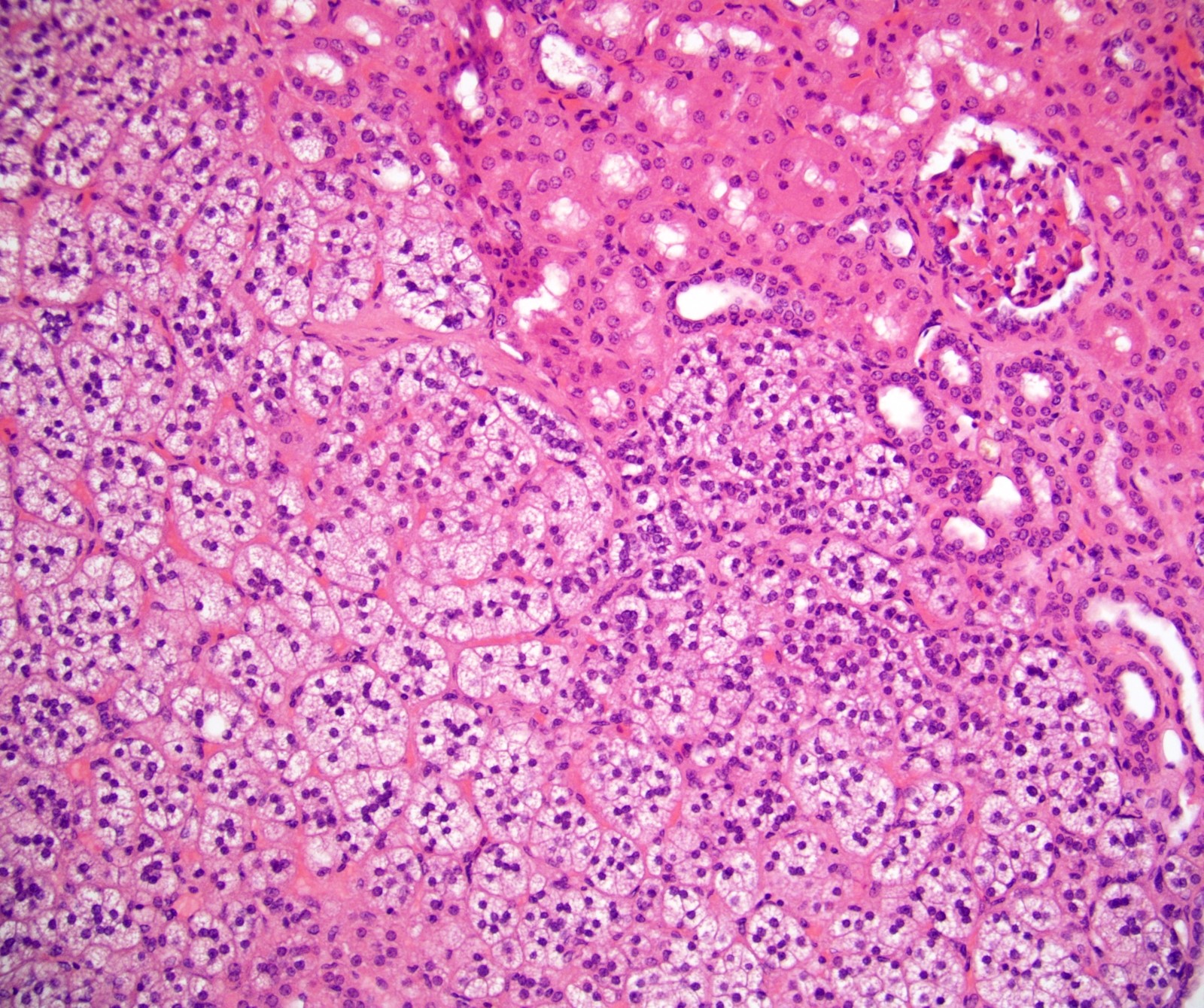
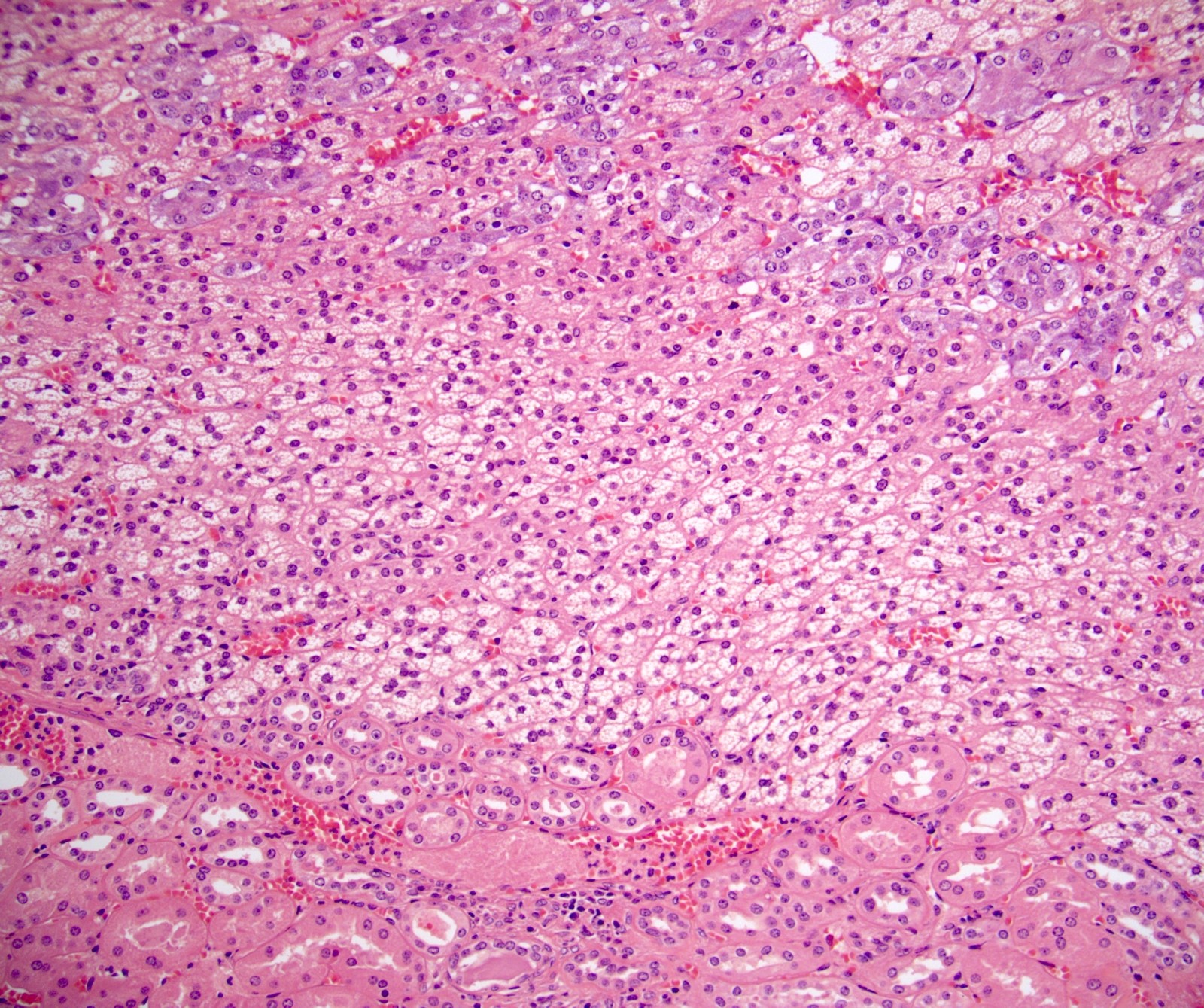
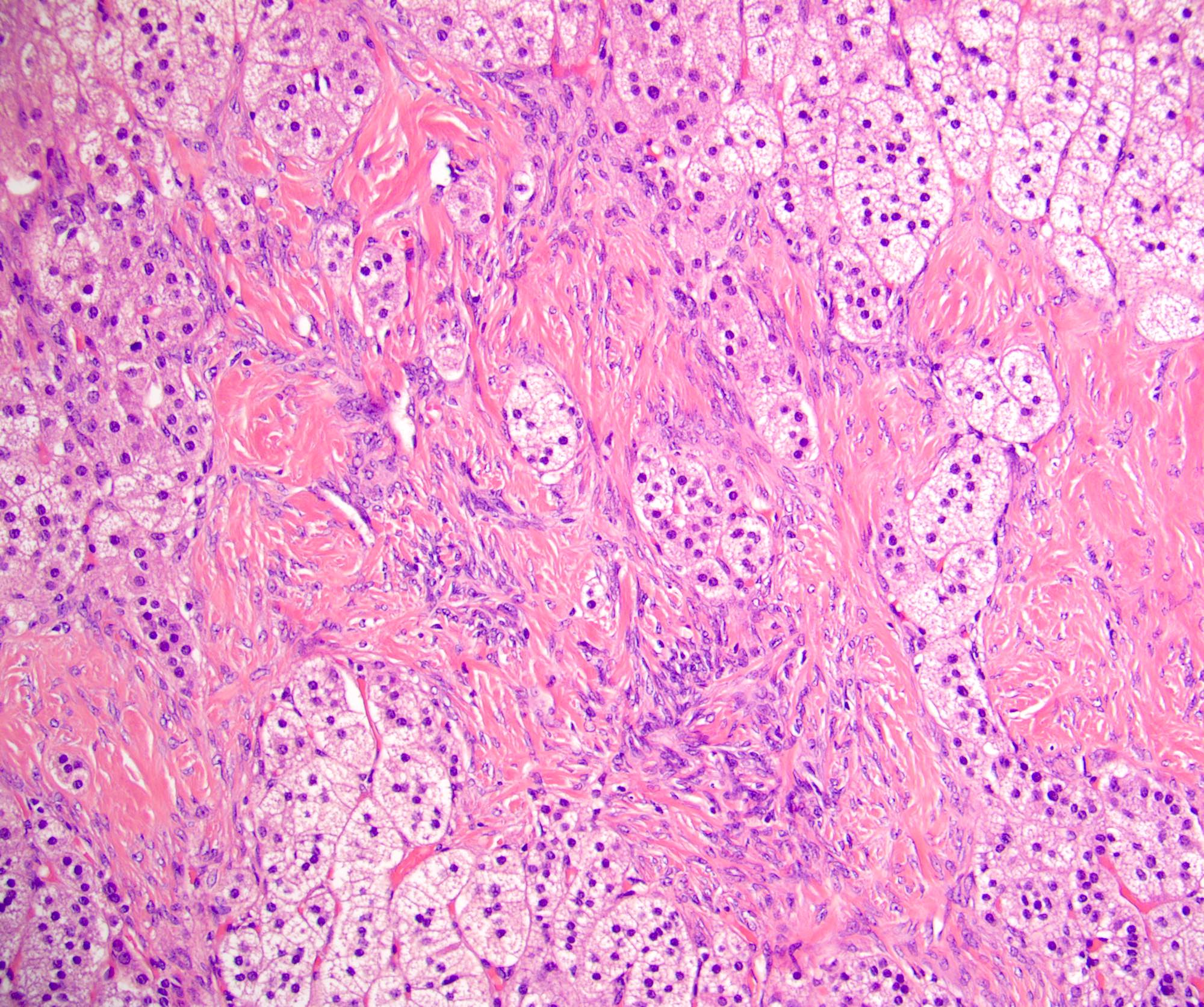
- Typically affects zona fasciculata and reticularis
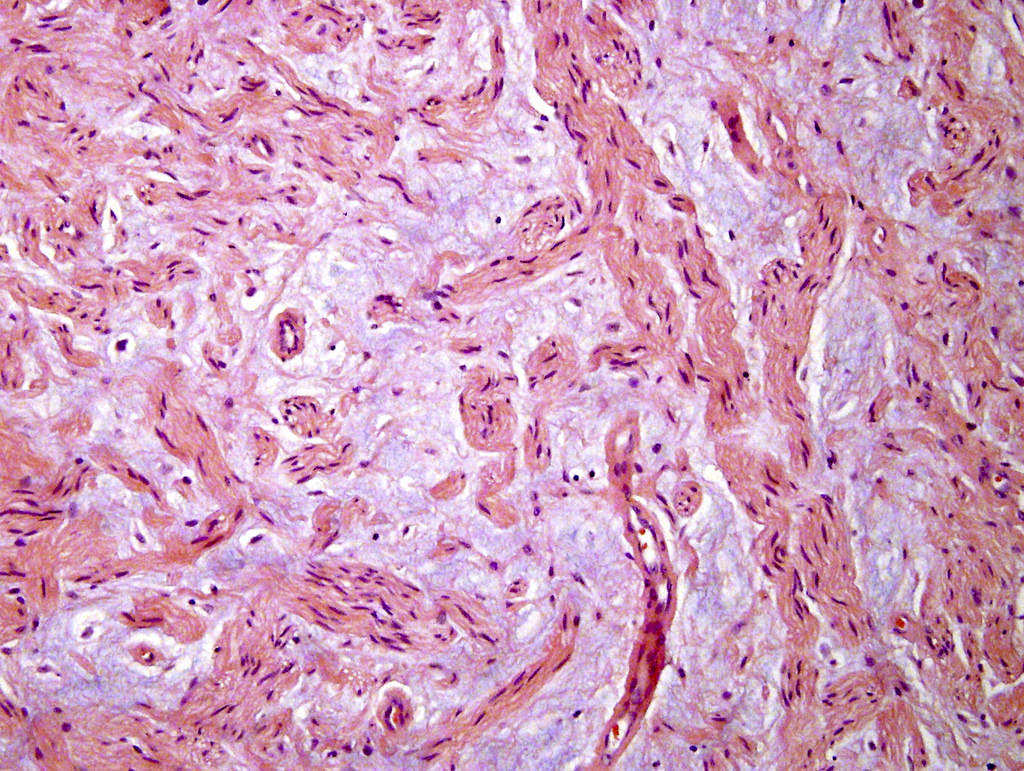
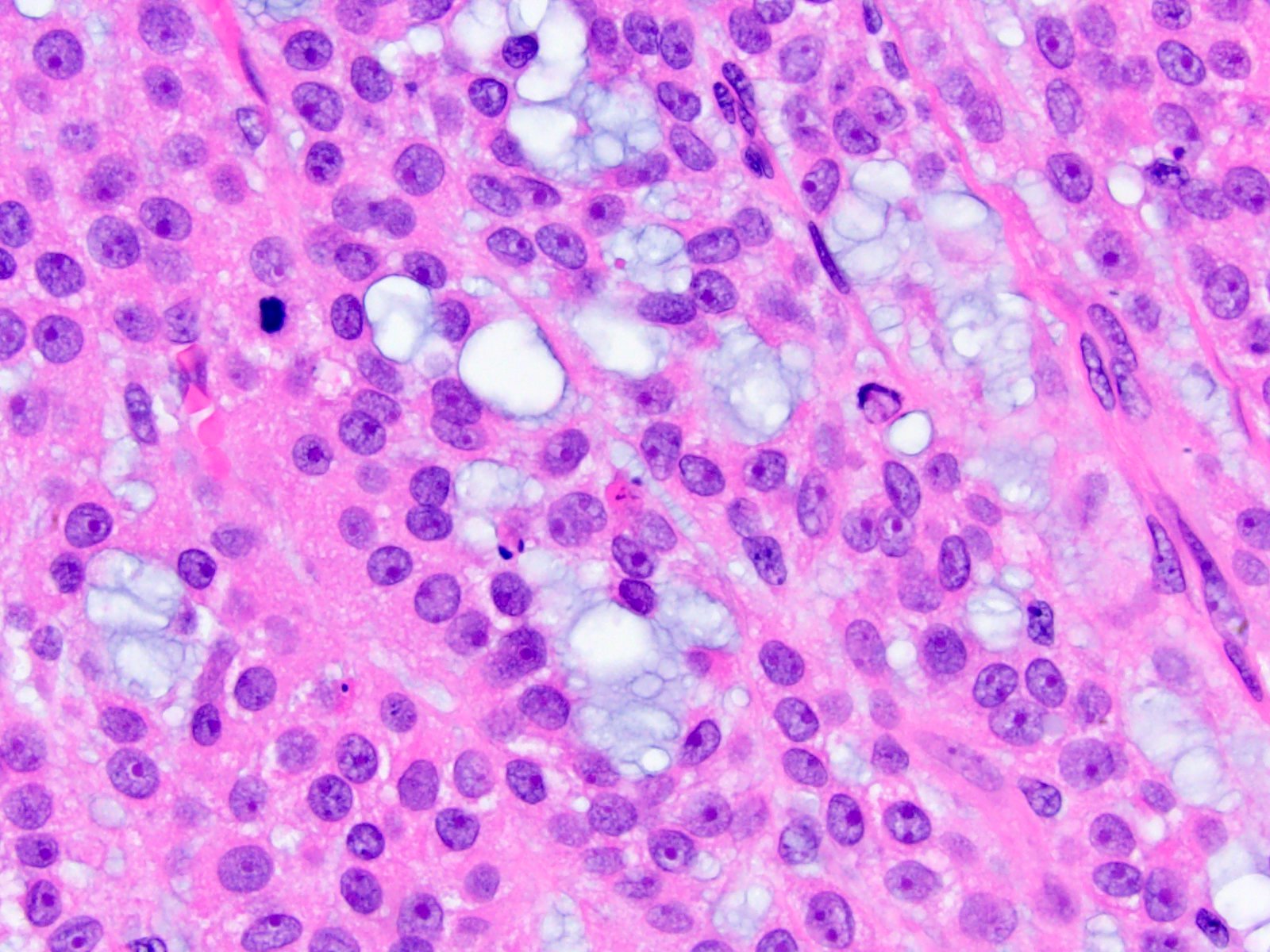
- Acellular salmon-pink amorphous material is present between cortical cells, which ultimately become atrophic
- Amyloidosis-AL type is typically deposited intravascularly
- Drugs
- Aminoglutethimide: inhibits enzyme converting cholesterol to pregnenolone, causes decrease in cortisol and aldosterone
- Metapyrone: inhibits 11 beta hydroxylase, which inhibits cortisol and aldosterone synthesis
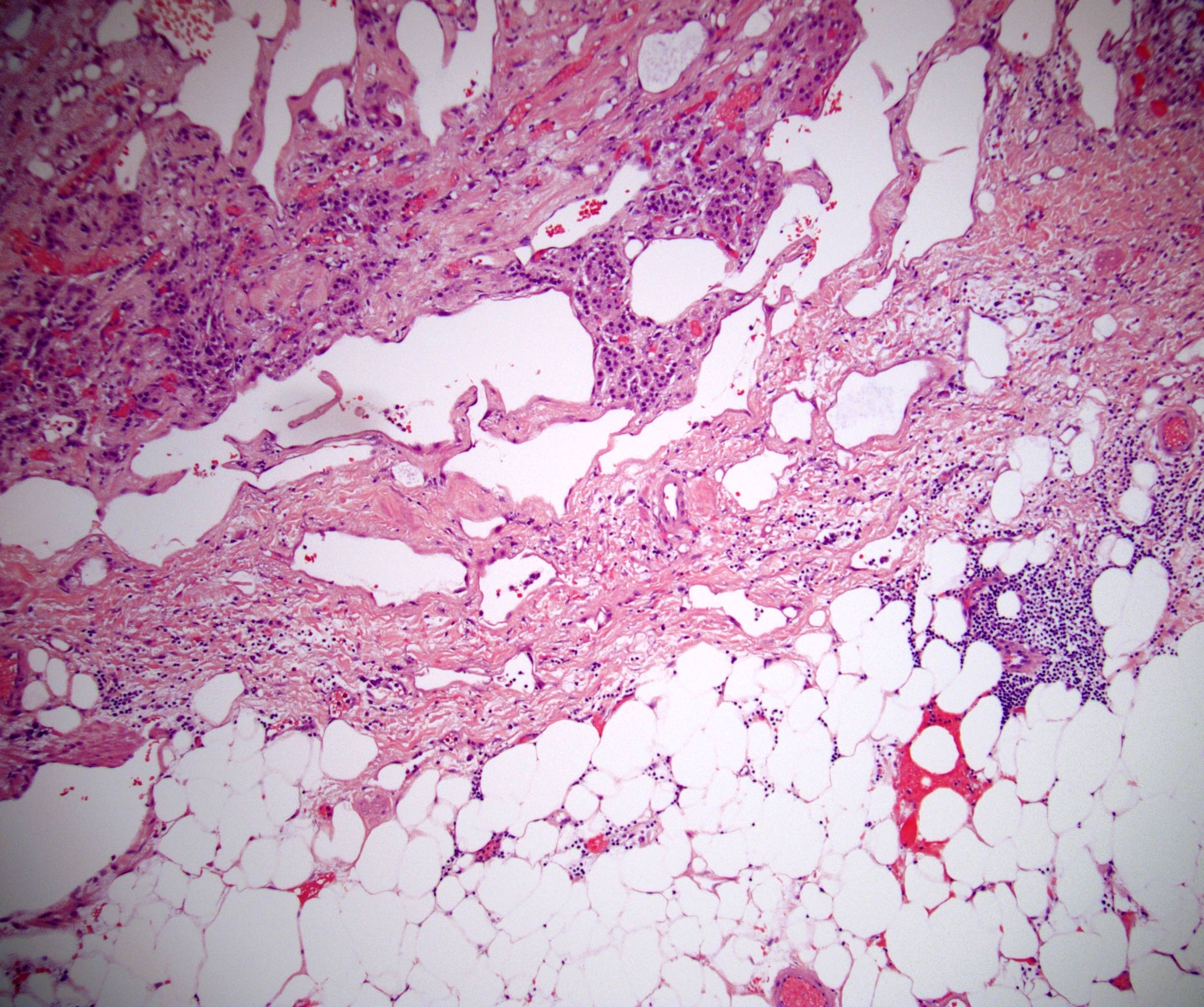
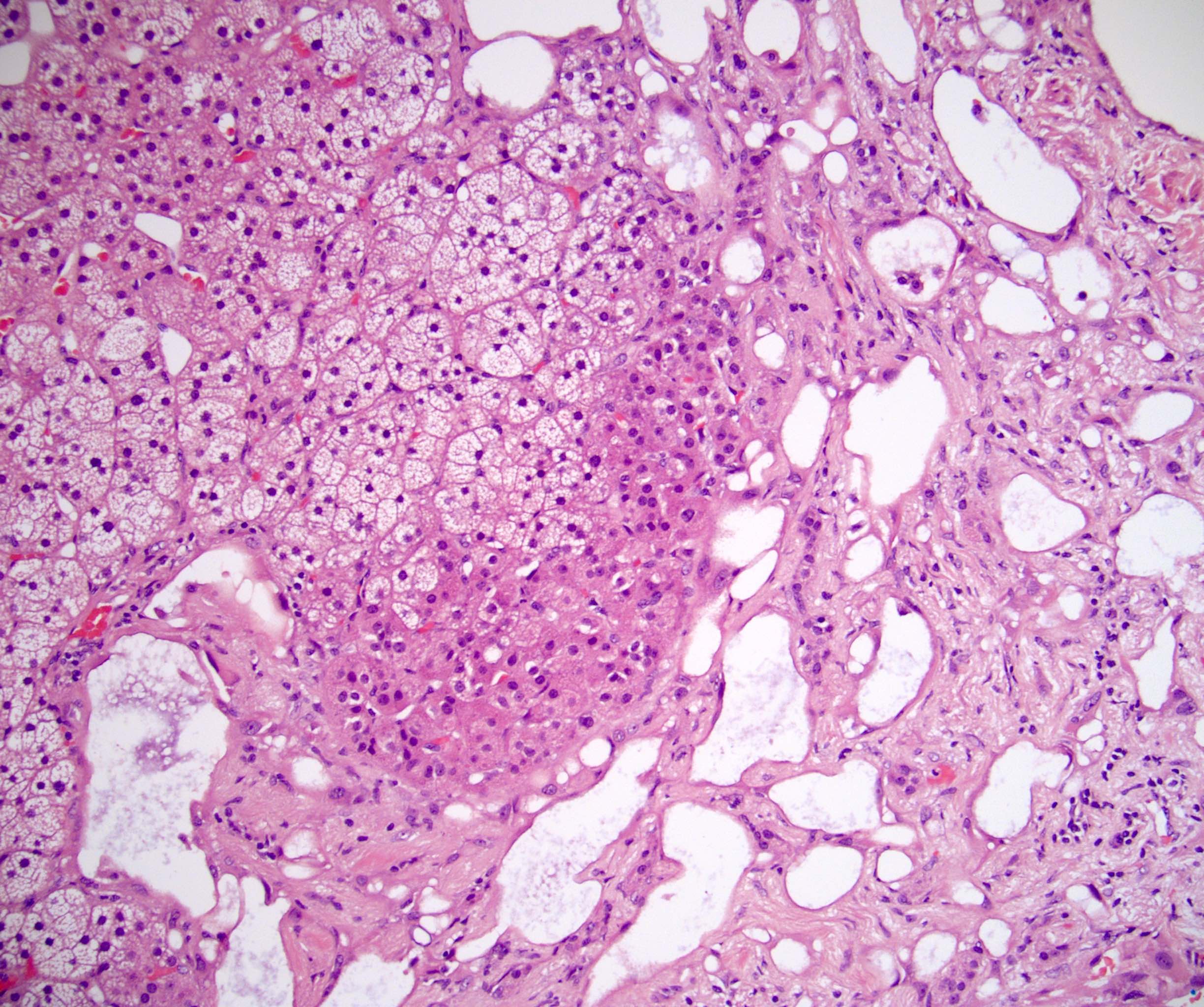
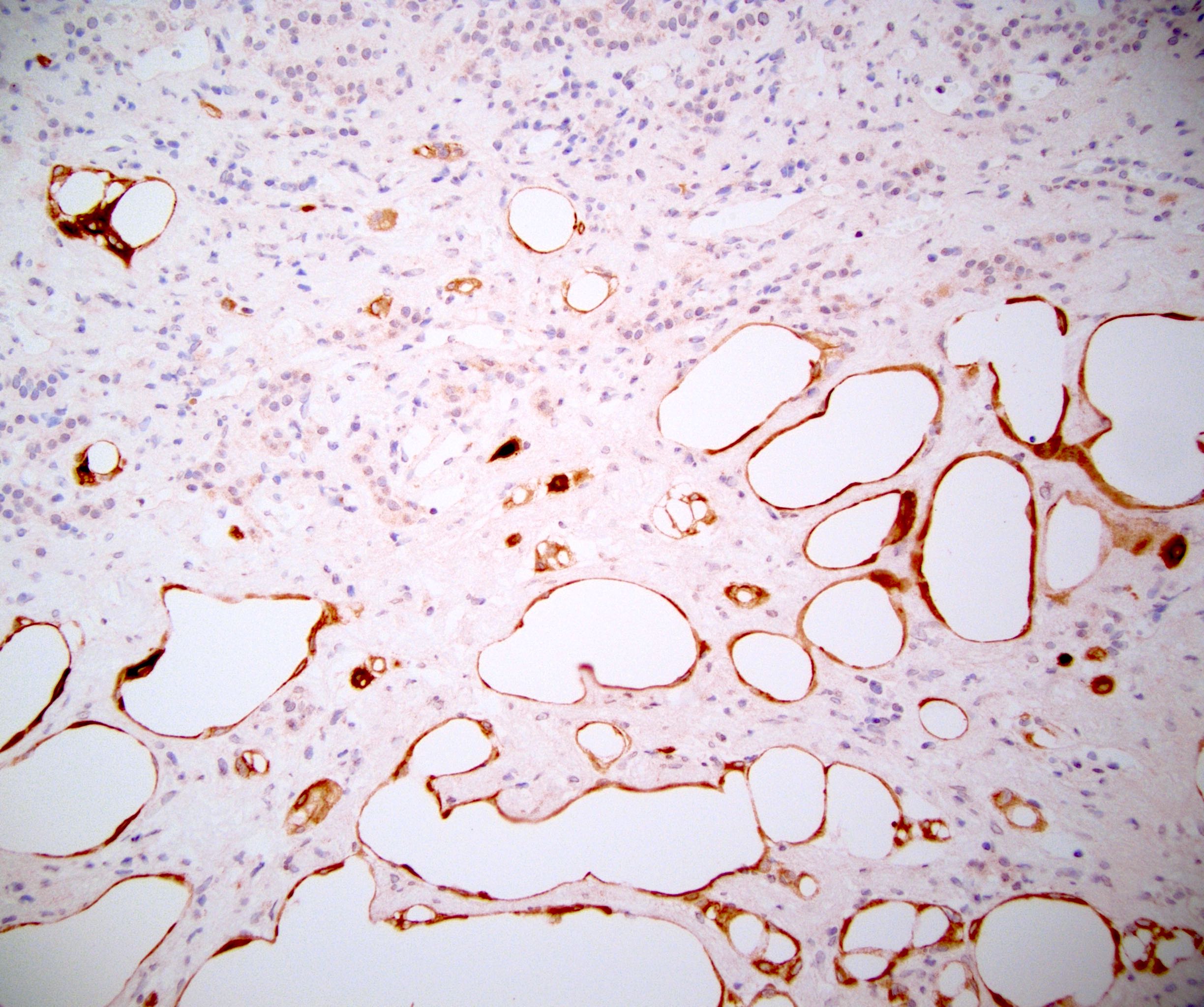
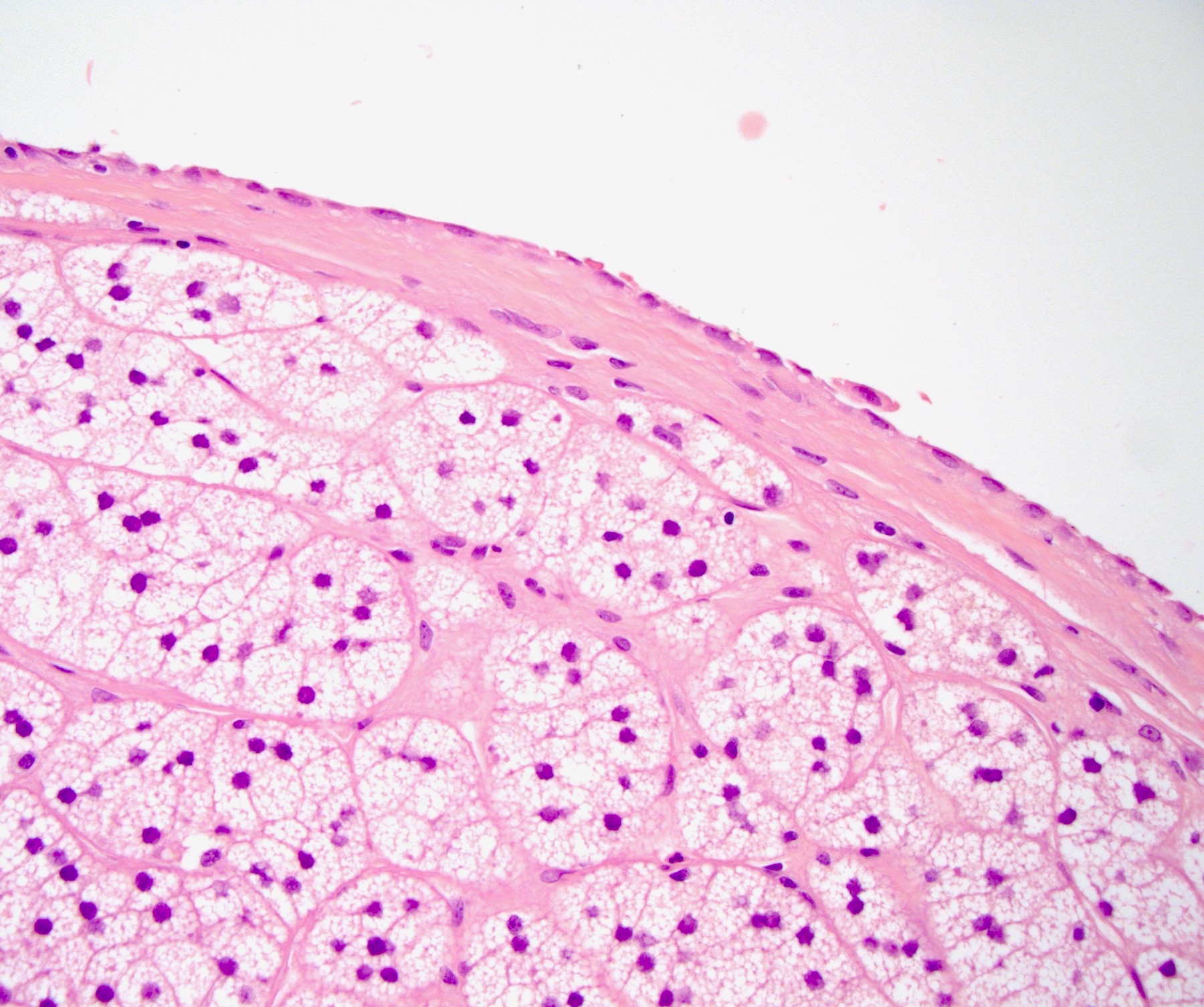
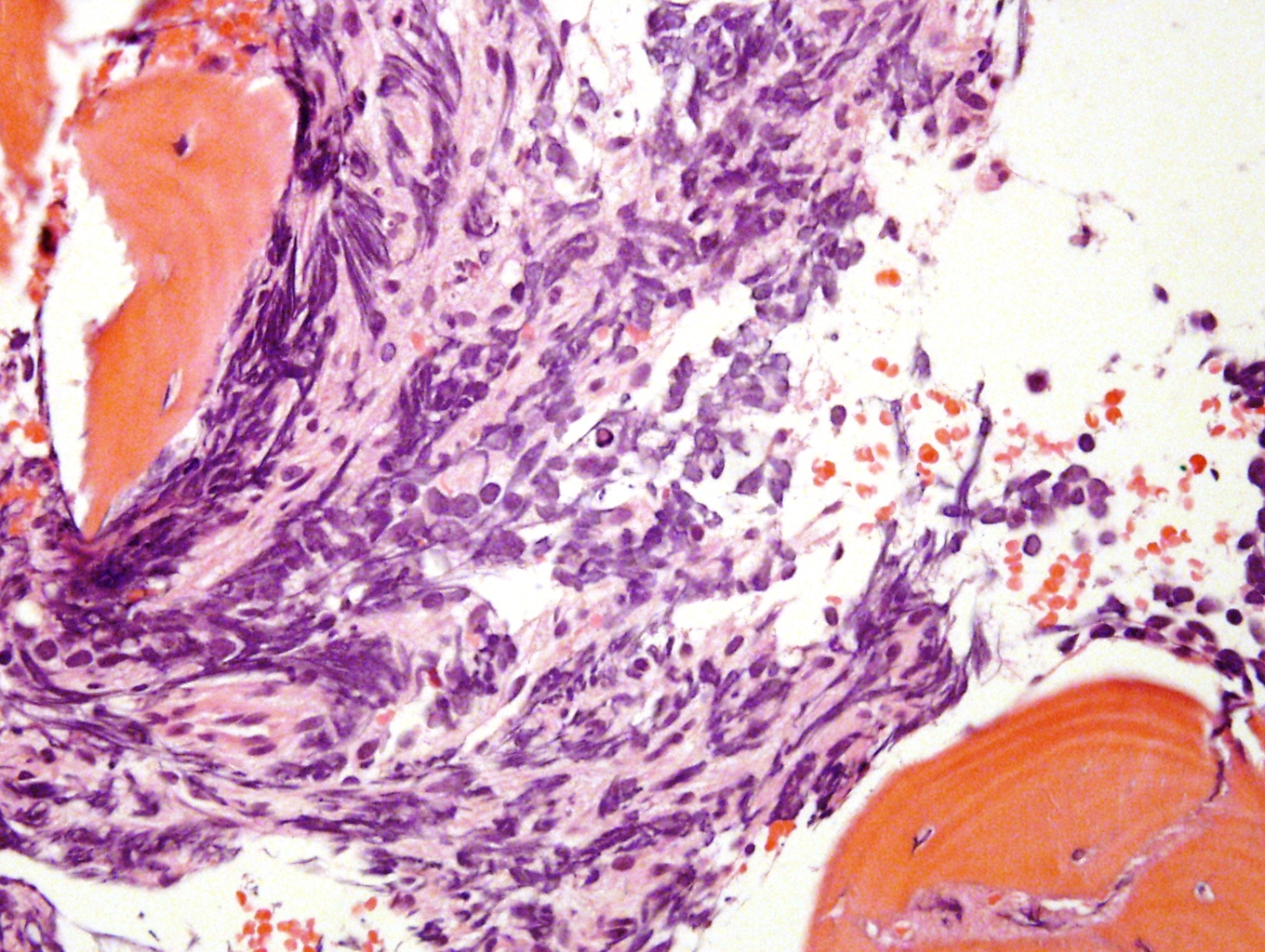
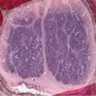
- Mitotane: cytotoxic to zona fasciculata and reticularis, produces medical adrenalectomy; produces atrophic adrenal glands with fibrosis and residual islands of cortical cells
- Radiation
- May cause fibrosis, although cortex is relatively radioresistant
- High doses (> 5000 roentgens) to abdomen, pelvis or lumbar region may cause hyaline fibrosis of reticularis and reduction of fasciculata, although does not necessarily affect cortical function
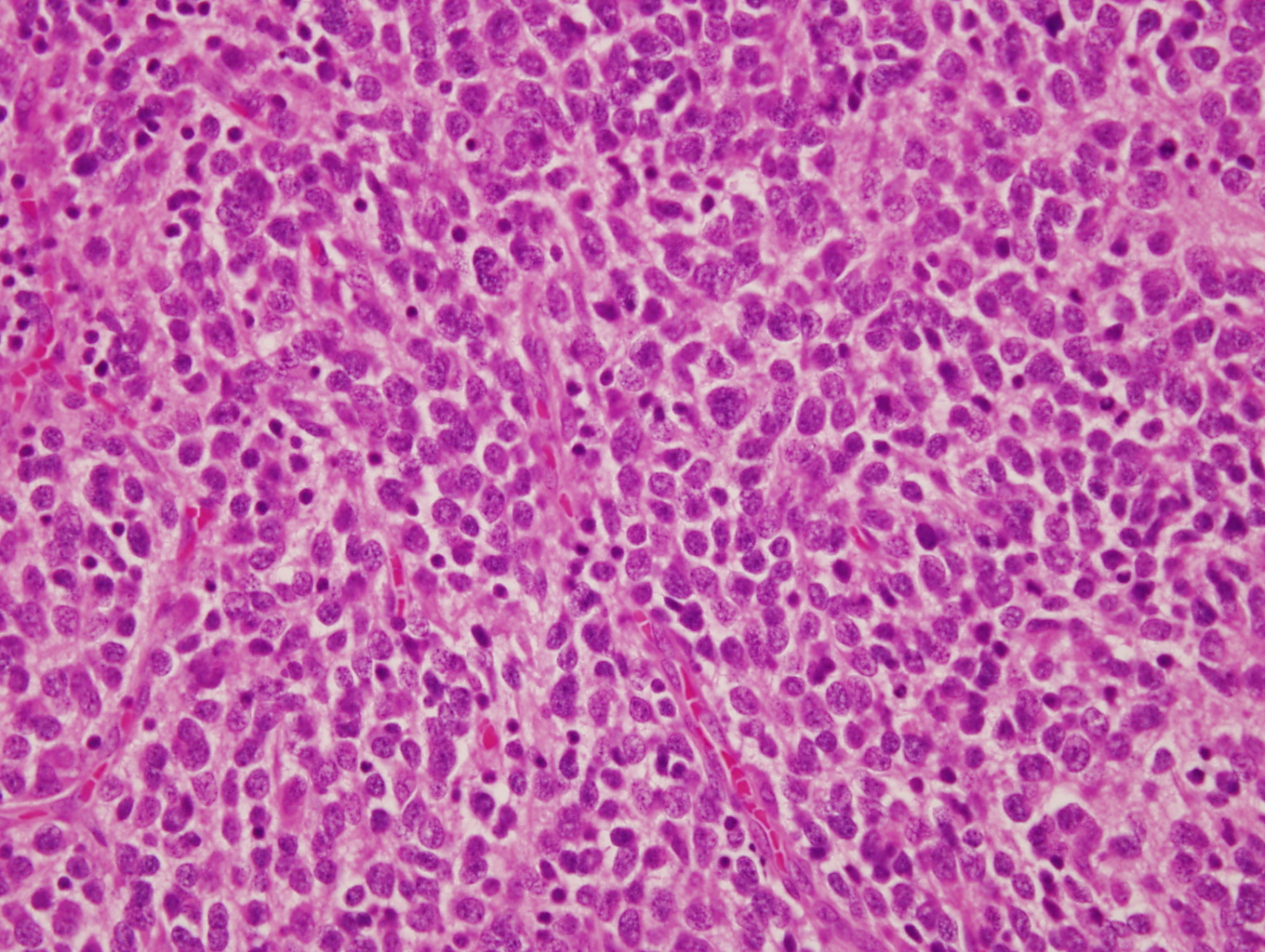
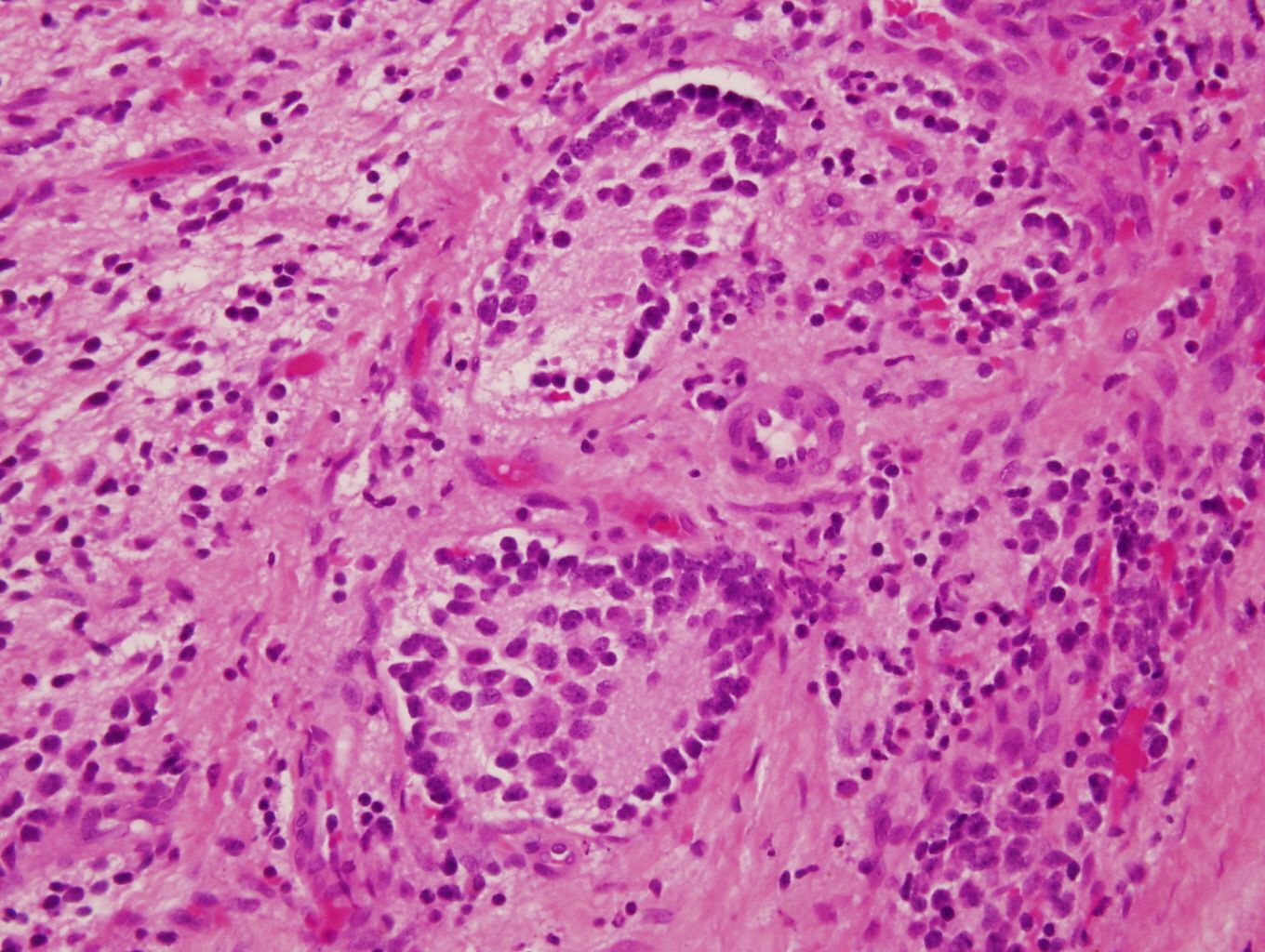
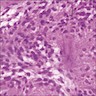
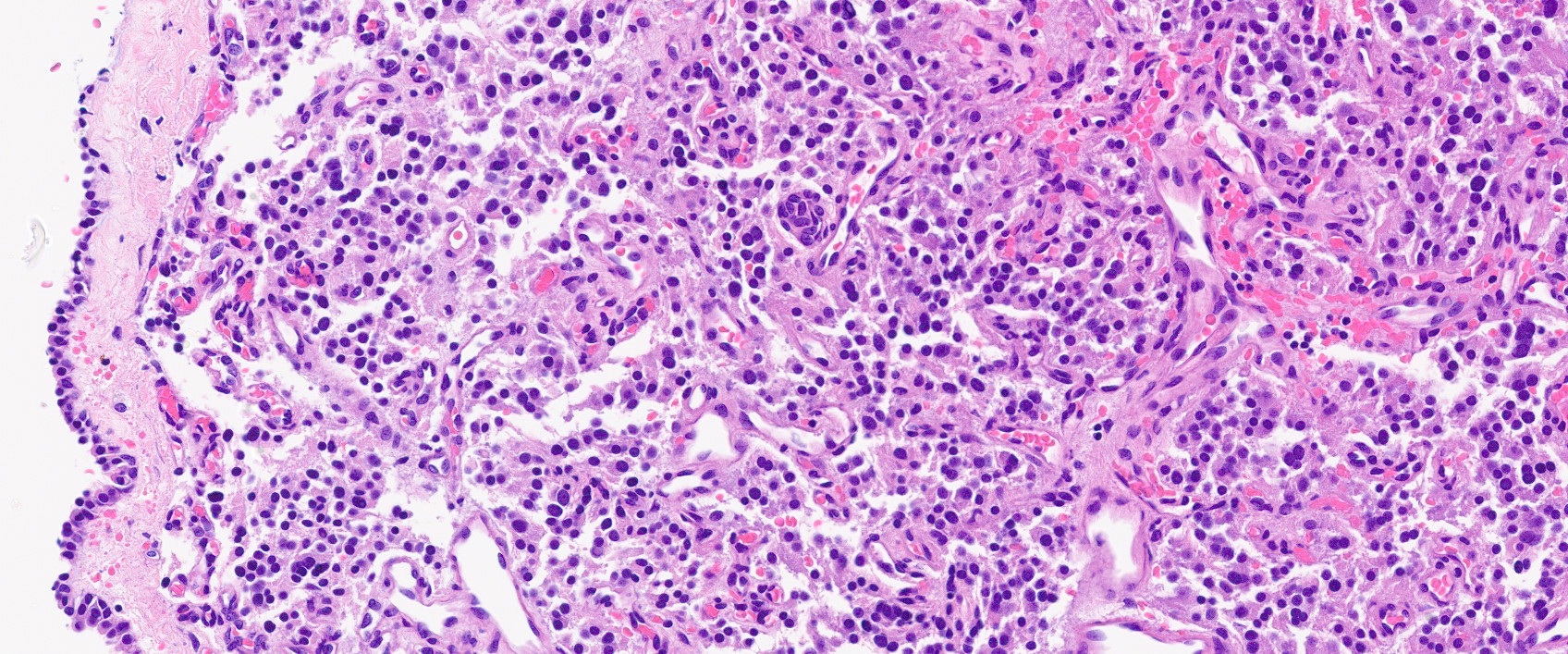
- Autoimmune disorders (autoimmune adrenalitis or polyglandular autoimmune syndromes)
Treatment
- Glucocorticoids, mineralocorticoids and IV fluids
- In chronic patients, must give steroid boost during infections, prior to surgery or during pregnancy