
- Acute megakaryoblastic leukemia (AMKL, M7)
- Up to 10% of AML in children, 5% or less of adult AML (Orphanet (May 2004): Acute megakaryoblastic leukemia [Accessed 6 April 2018])
- See also Myeloid leukemia associated with Down syndrome
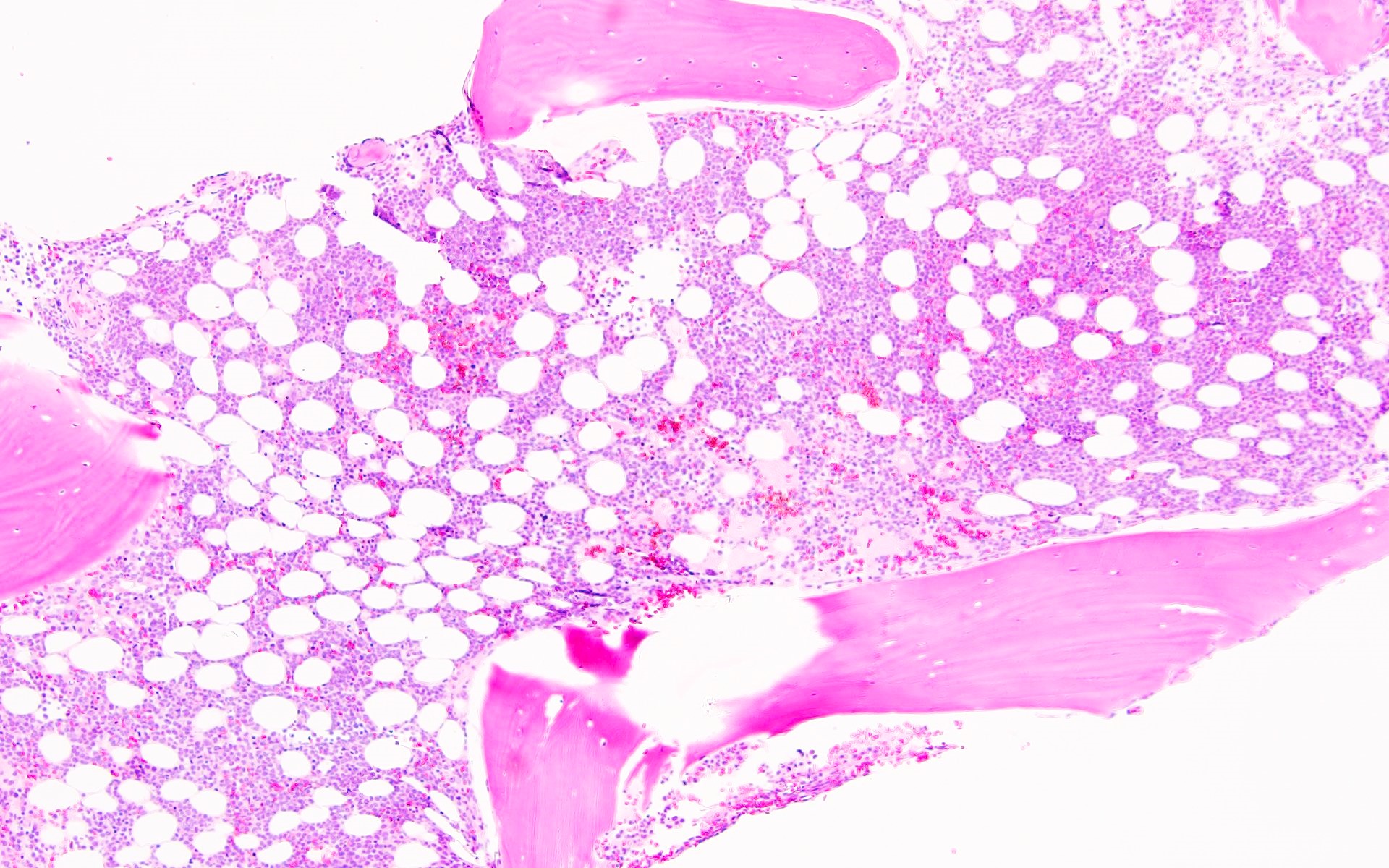
- Associated with marrow fibrosis due to megakaryoblast secretion of fibrogenic cytokines, which makes marrow aspiration difficult
- In adults, median age 57 years, 59% have prior hematologic disorder or myelodysplastic syndrome (Blood 2006;107:880)
- 19% had prior chemotherapy, classify now as AML-MRC (myelodysplasia related changes) or t-AML
- Survival: poor, median overall survival is 6 months
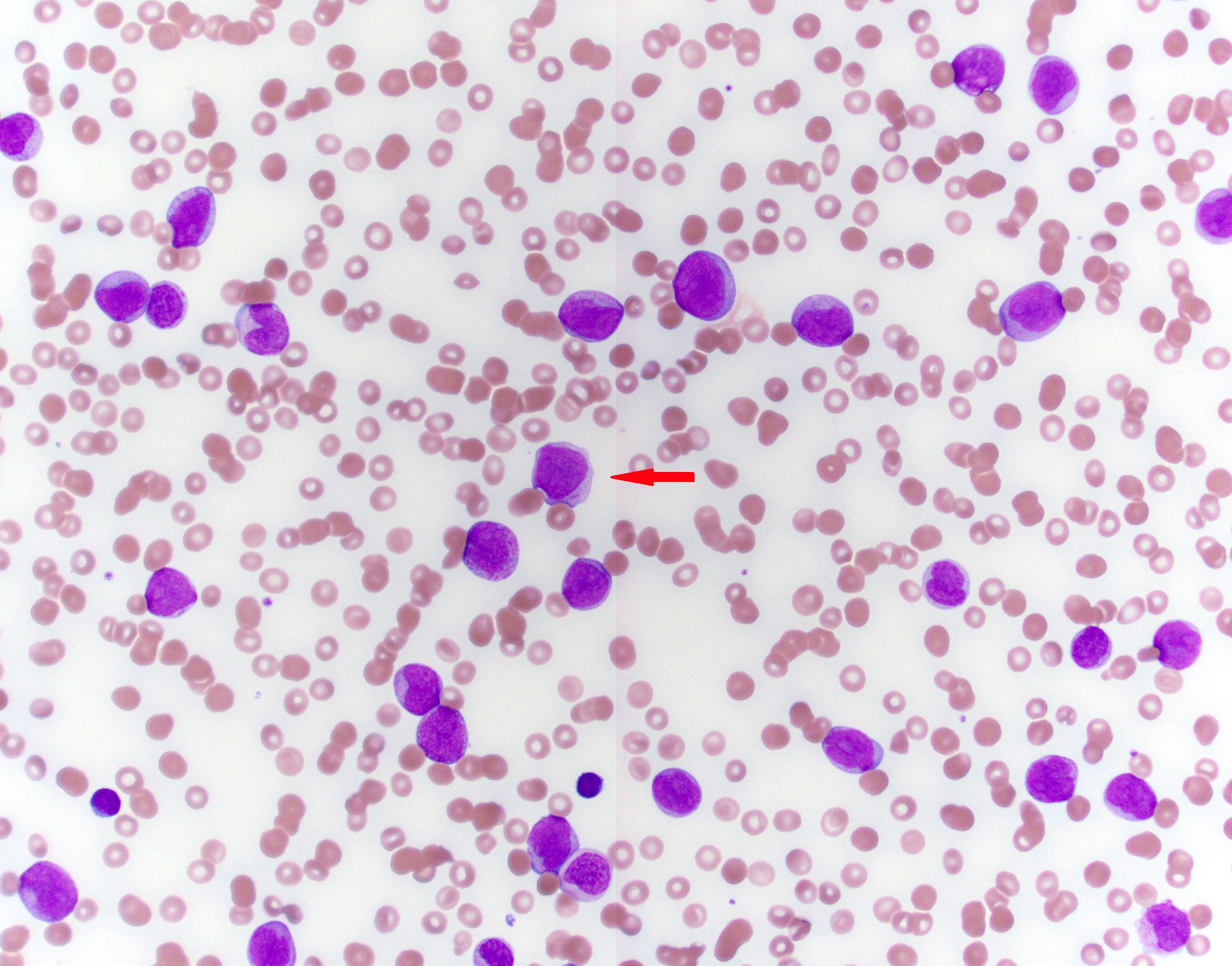
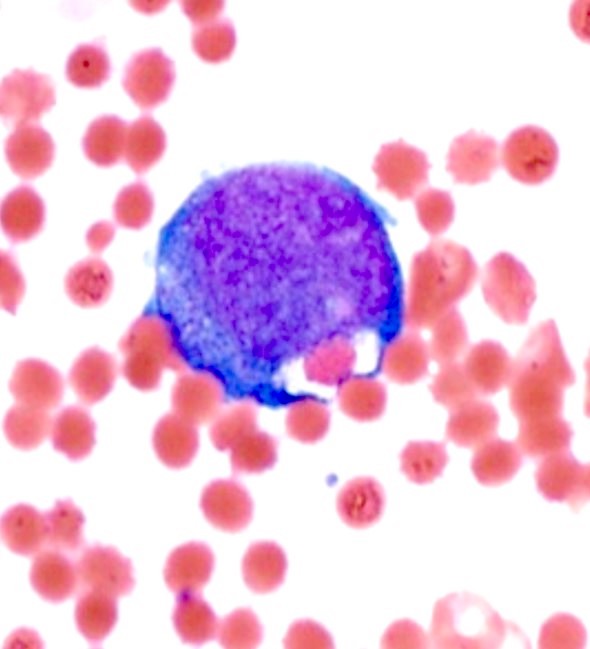
- Peripheral blood: often contains micromegakaryocytes and atypical platelets
- Down Syndrome (DS): 150x increased risk of AML compared to non-Down children age 0 - 4 years; 70% are AML M7 compared to 3 - 6% in non-Down children
- DS children ages 0 - 3 years: ALL vs AML risk is 1:1.2 compared to 4:1 for non-DS children
- Thrombocytopenia, may have thrombocytosis, dysplastic features in neutrophils, erythroids, megakaryocytes and platelets
- Infrequent hepatosplenomegaly
- Associated with germ cell tumors in young boys
- WHO 2008: 20%+ blasts
- 50%+ blasts of megakaryocytic lineage are present in bone marrow
- Must exclude AML-MRC (myelodysplasia related changes), AML with t(1;22)(p13;q13); inv(3)(q21;q26.2); t(3;3)(q21;q26.2) and Down syndrome related
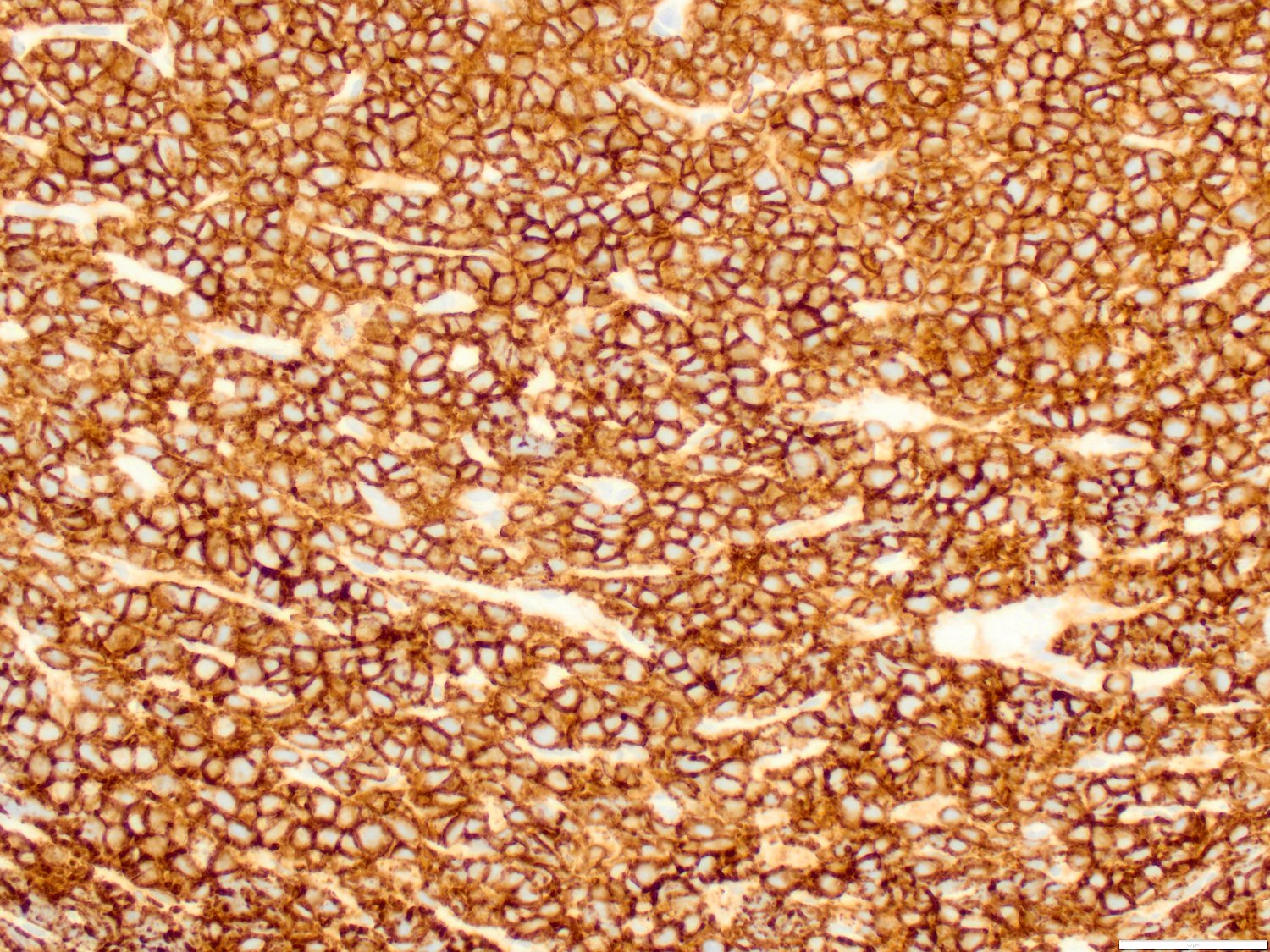
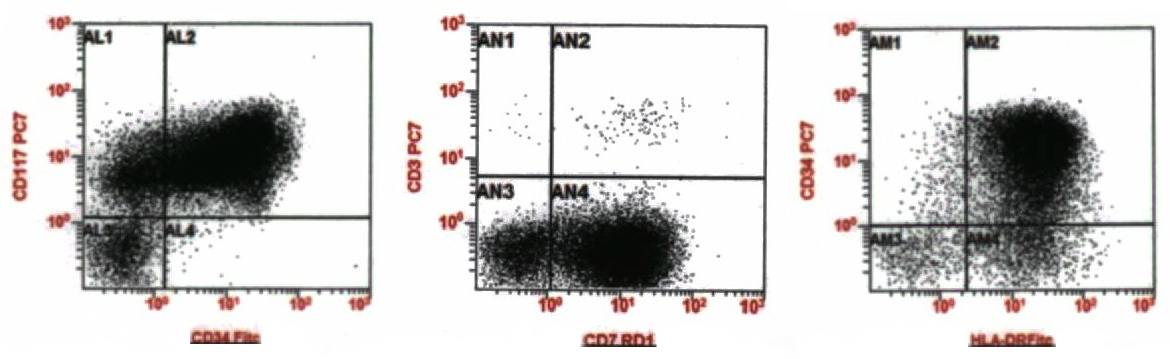
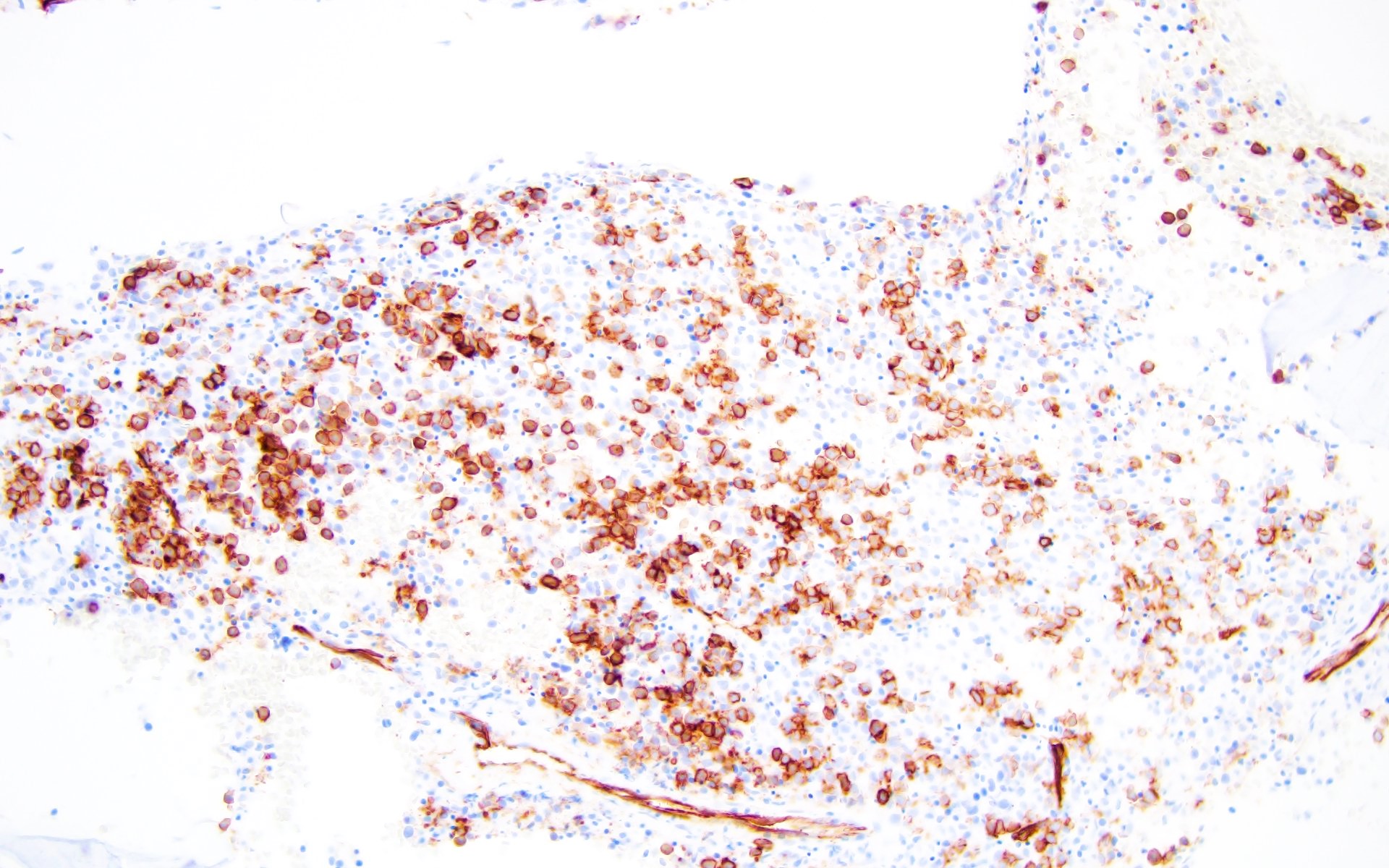
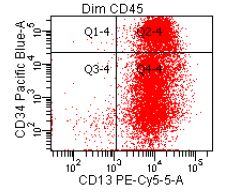
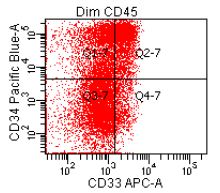
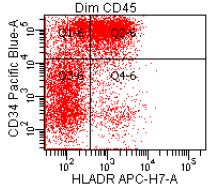
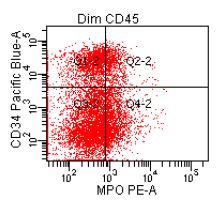
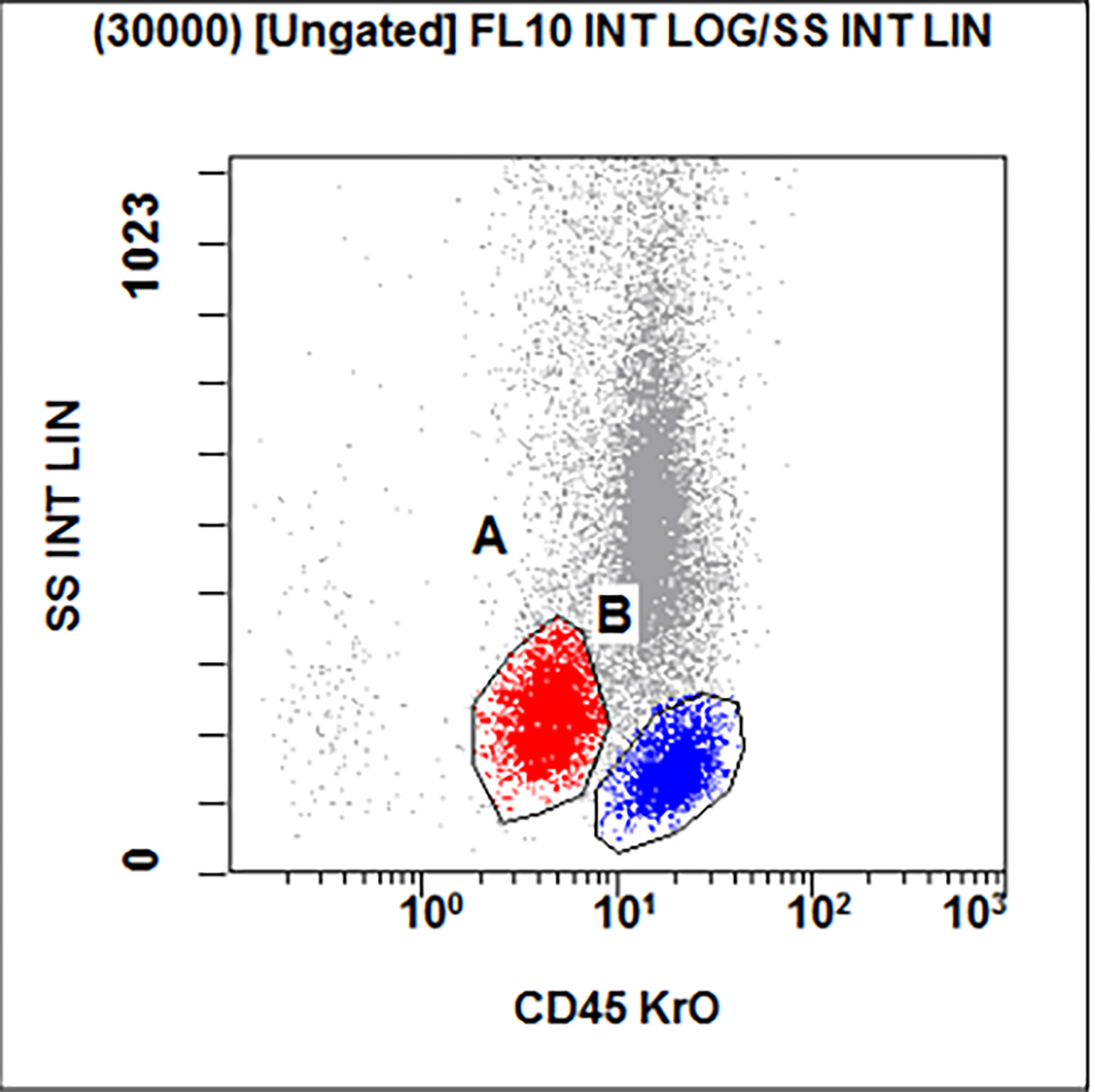
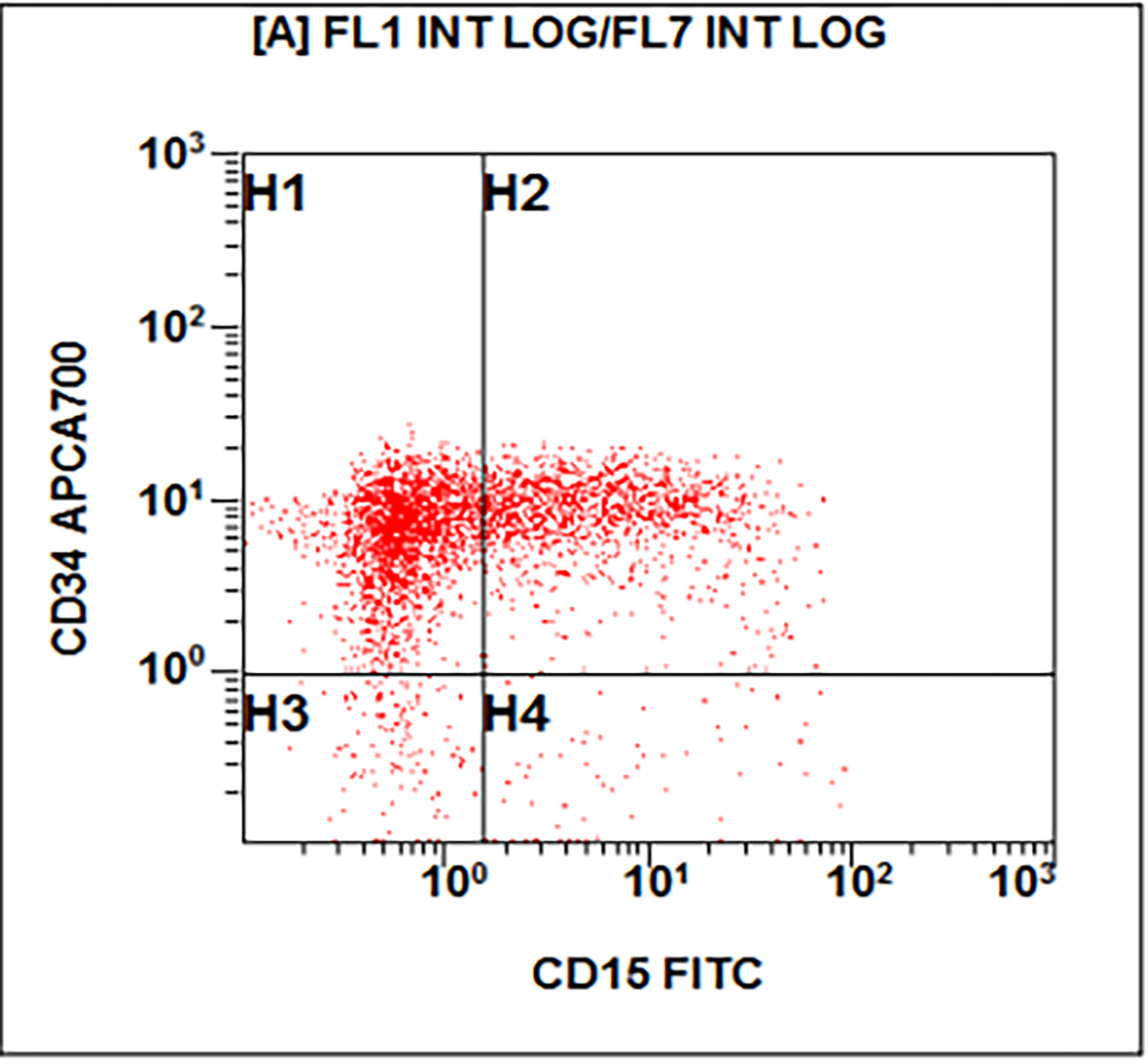
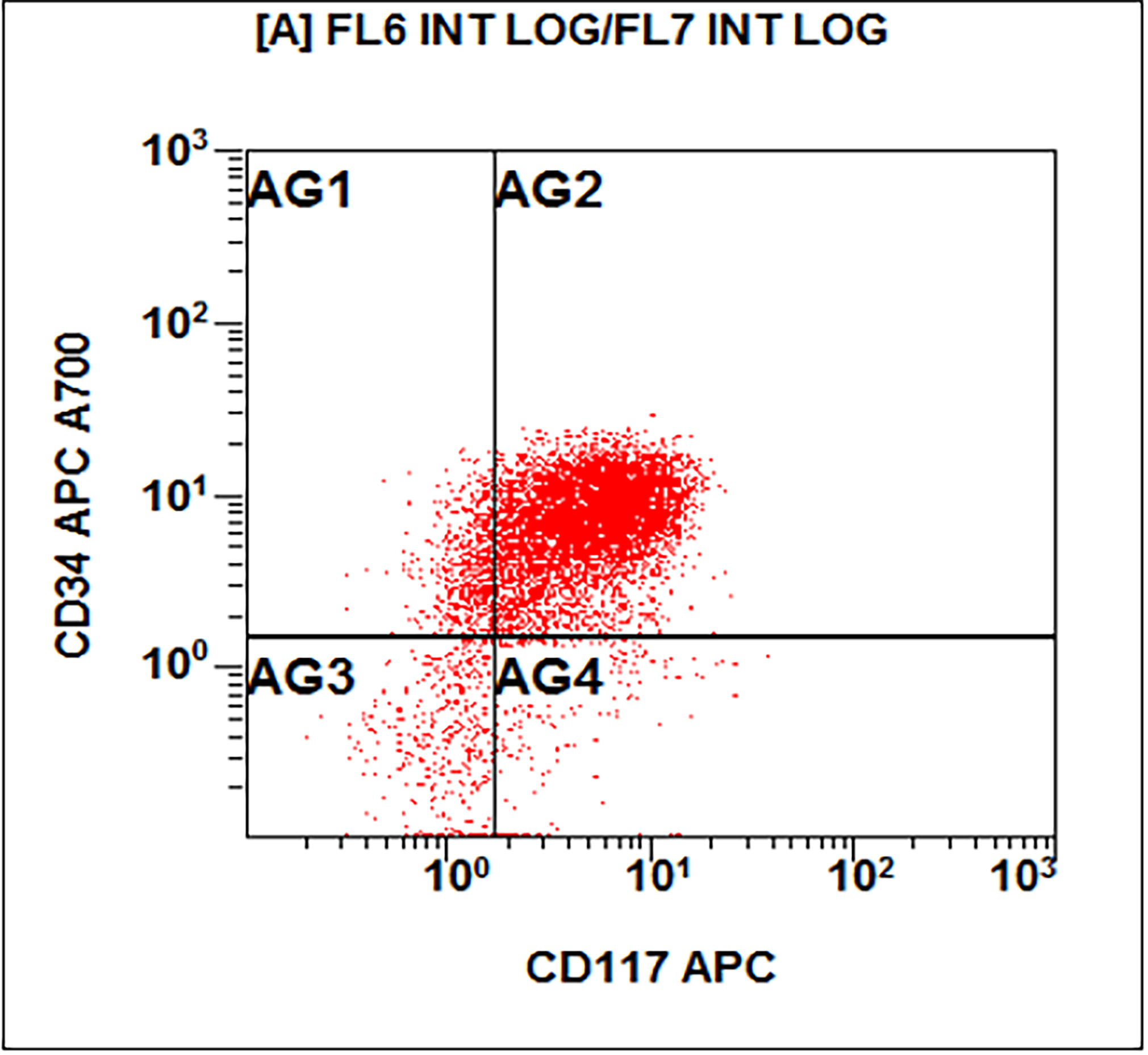
- Megakaryocytic lineage is based on CD41+, CD61+ or positive platelet peroxidase reaction on EM
- 17 month old girl with bi-allelic deletions within 13q14 and transient trisomy 21 with absence of GATA1 (Pediatr Blood Cancer 2011;57:516)
- Young boy with coexisting mediastinal germ cell tumor (Clin Transl Oncol 2007;9:329)
- 25 year old man with findings on FNA and CSF (Cytojournal 2011;8:17)
- 25 year old man with i(12p) related disease after primary mediastinal germ cell tumor (J Korean Med Sci 2011;26:1099)
- 58 year old woman with coexisting myeloid sarcoma of femur (Srp Arh Celok Lek 2011;139:805)
- Diagnosis 15 years after kidney transplantation (Ann Hematol 2011;90:843)
- "Cannibalistic" phagocytosis in acute megakaryoblastic leukemia (AML M7) with t(10;17)(p15;q22) (Leuk Lymphoma 2010;51:1944)
- Coexistence of meningeal infiltration and multiple lymphadenopathy as the initial presentation of de novo adult acute megakaryoblastic leukemia (Leuk Res 2011;35:e50)
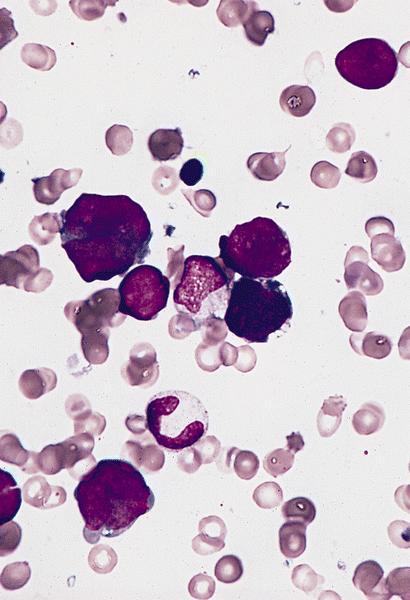
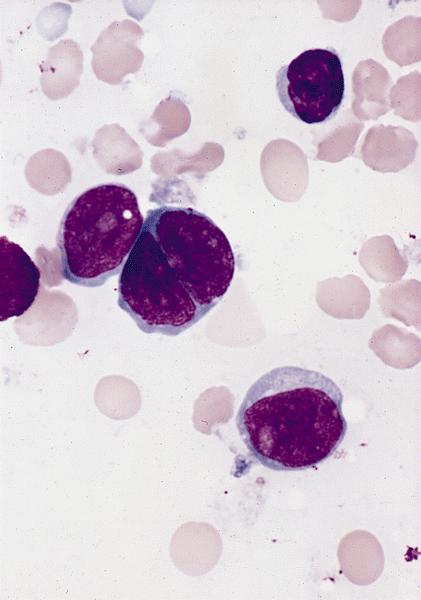
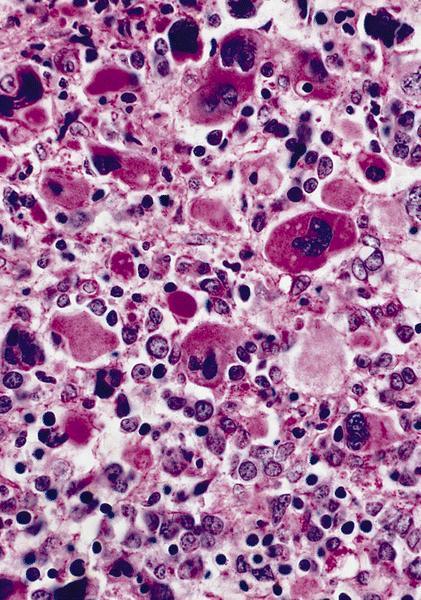
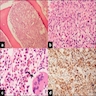
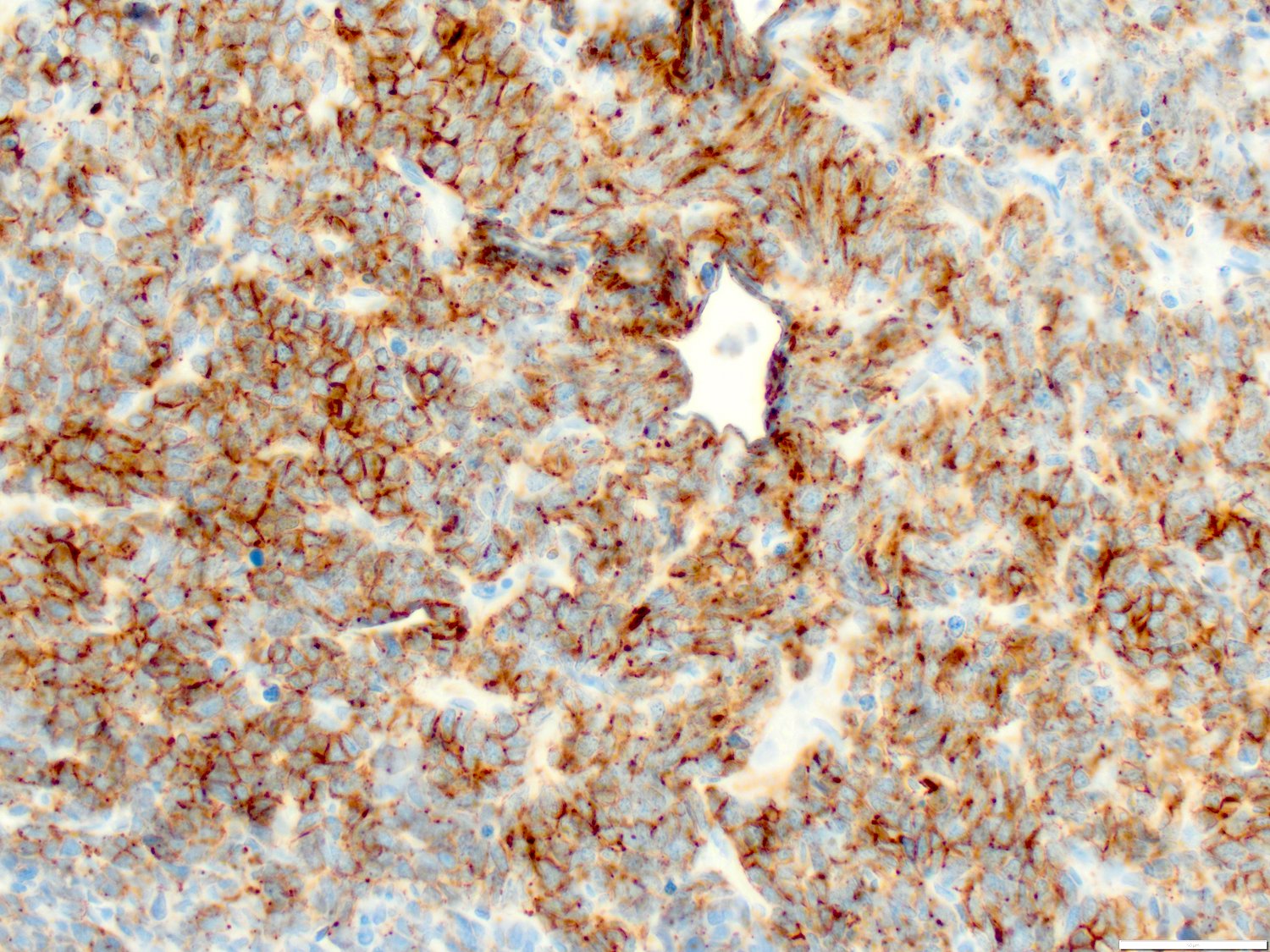
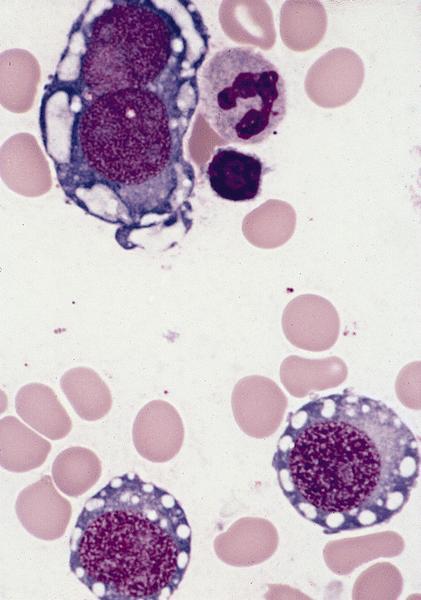
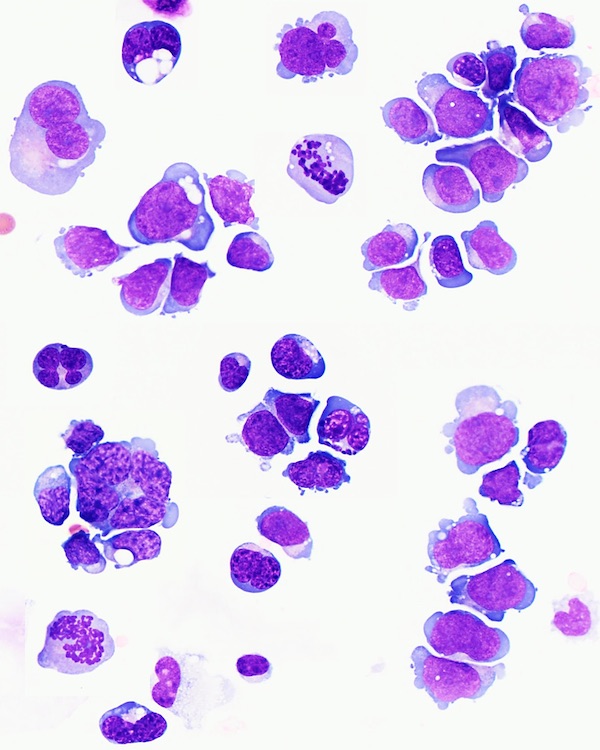
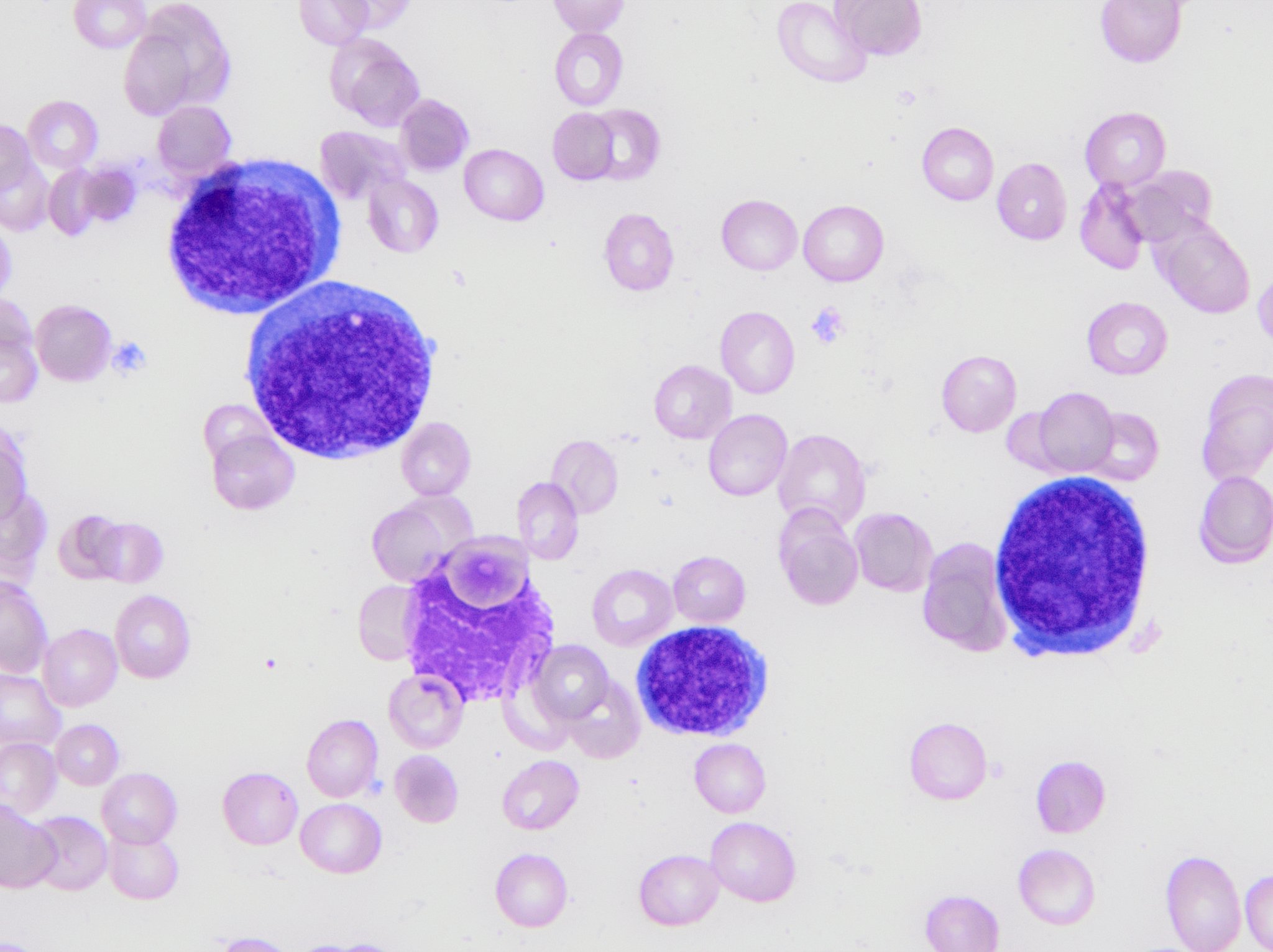
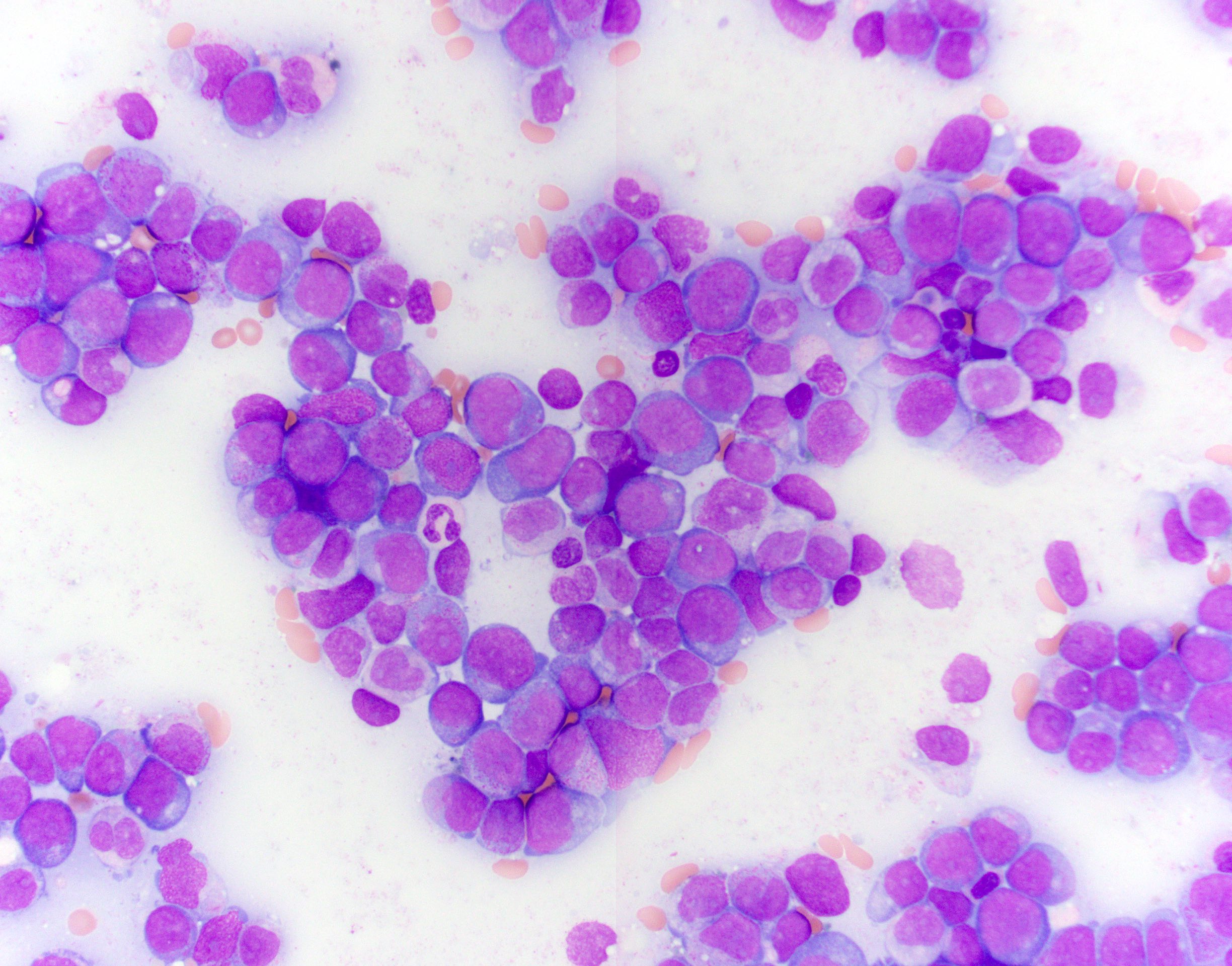
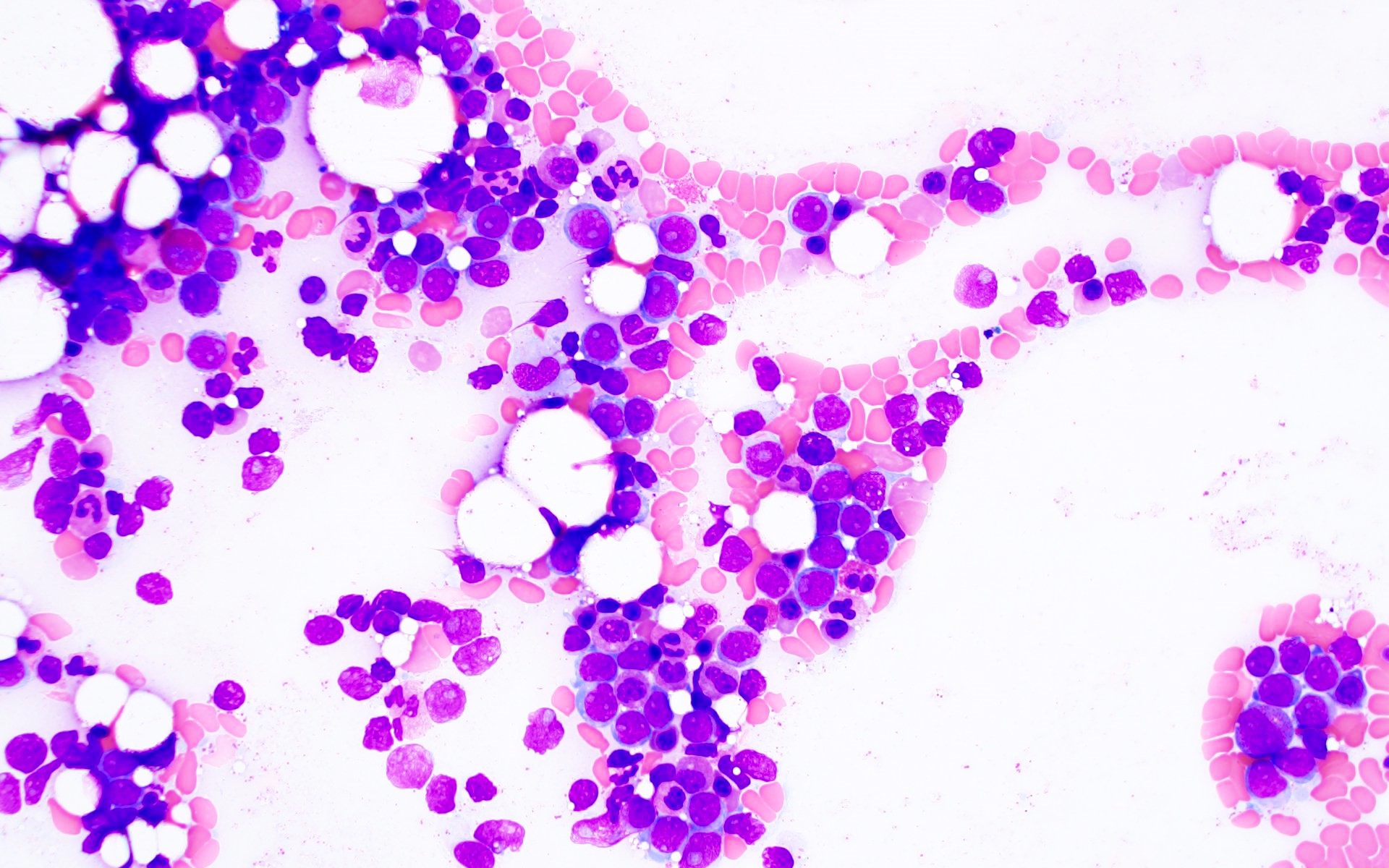
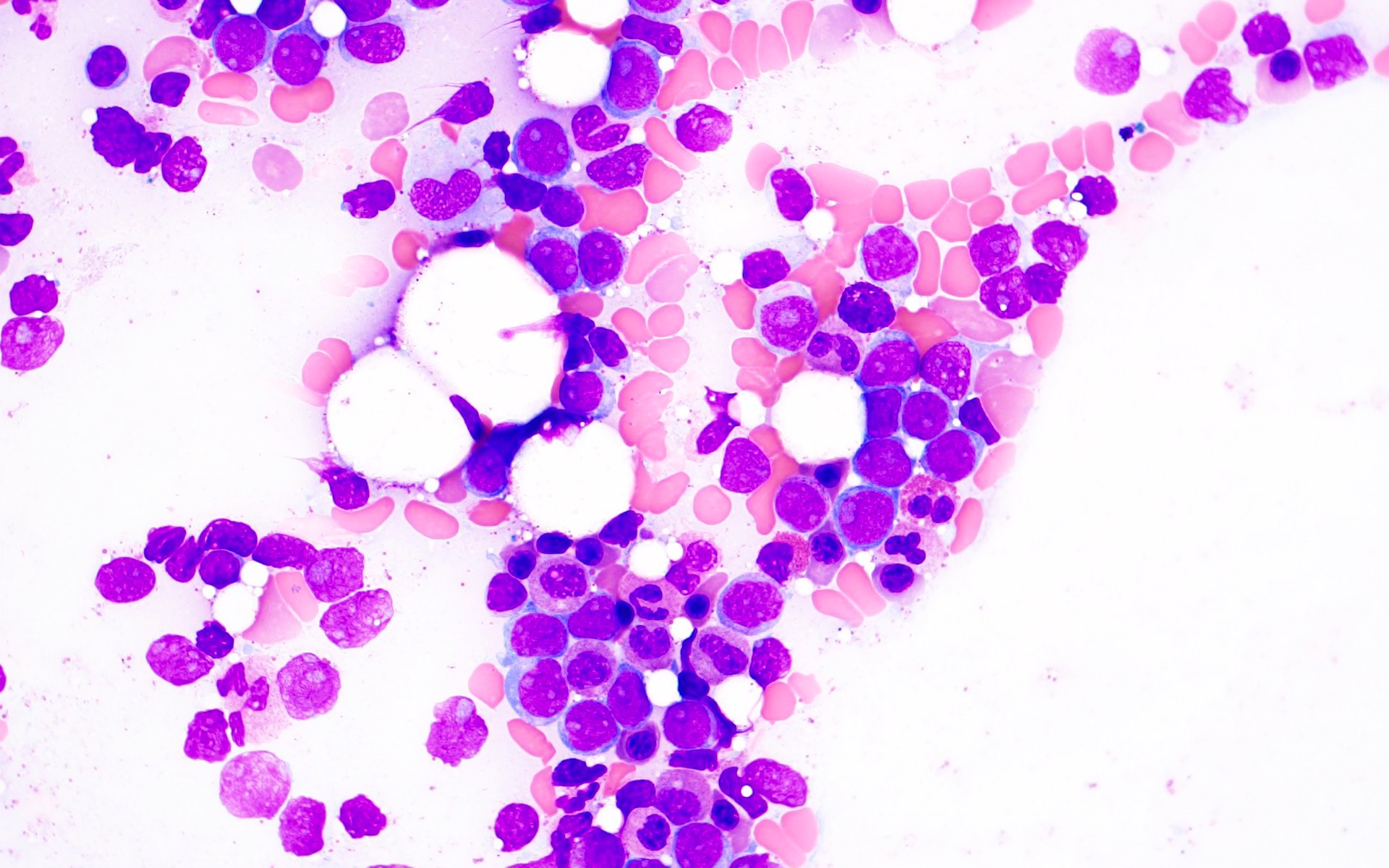
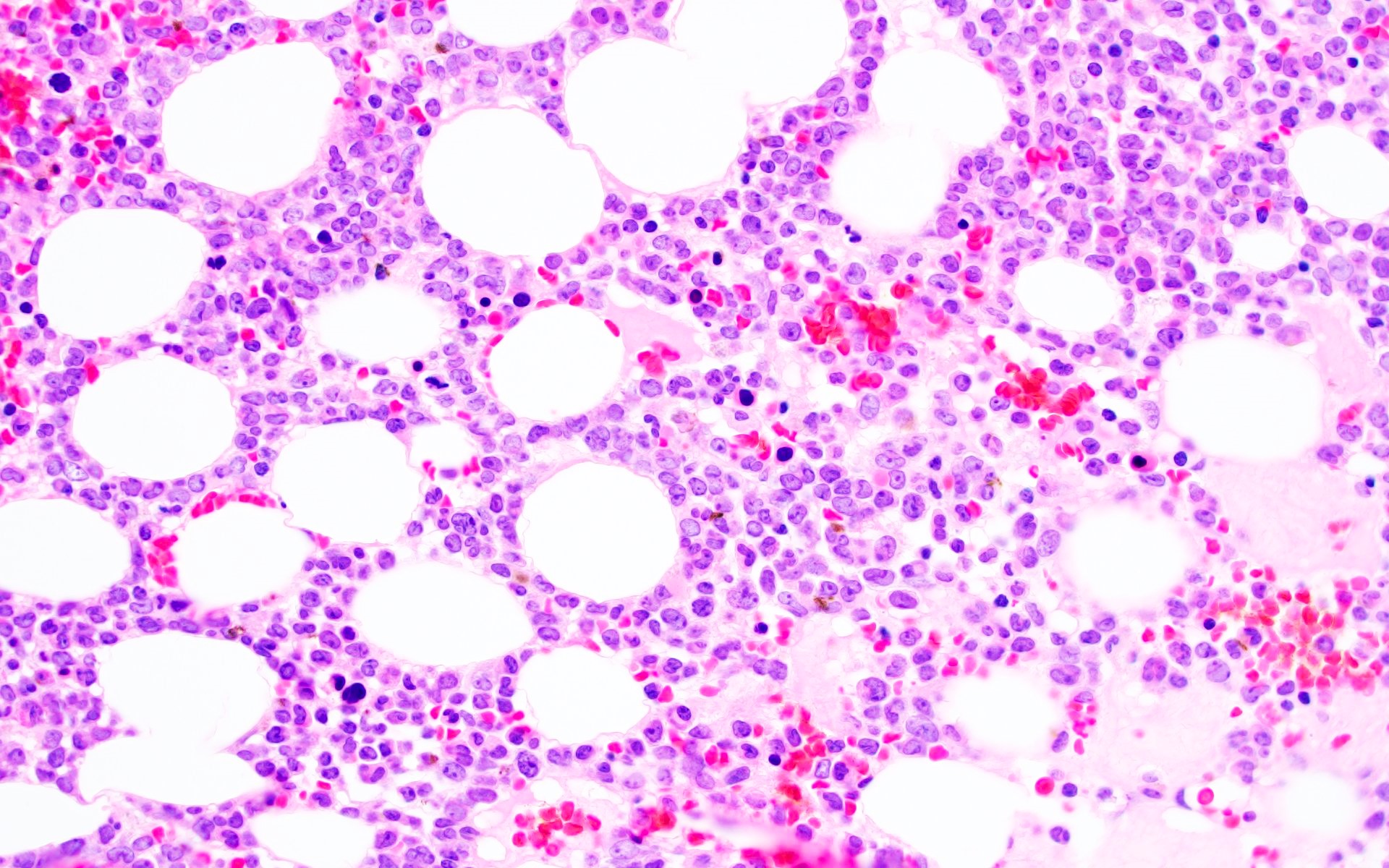
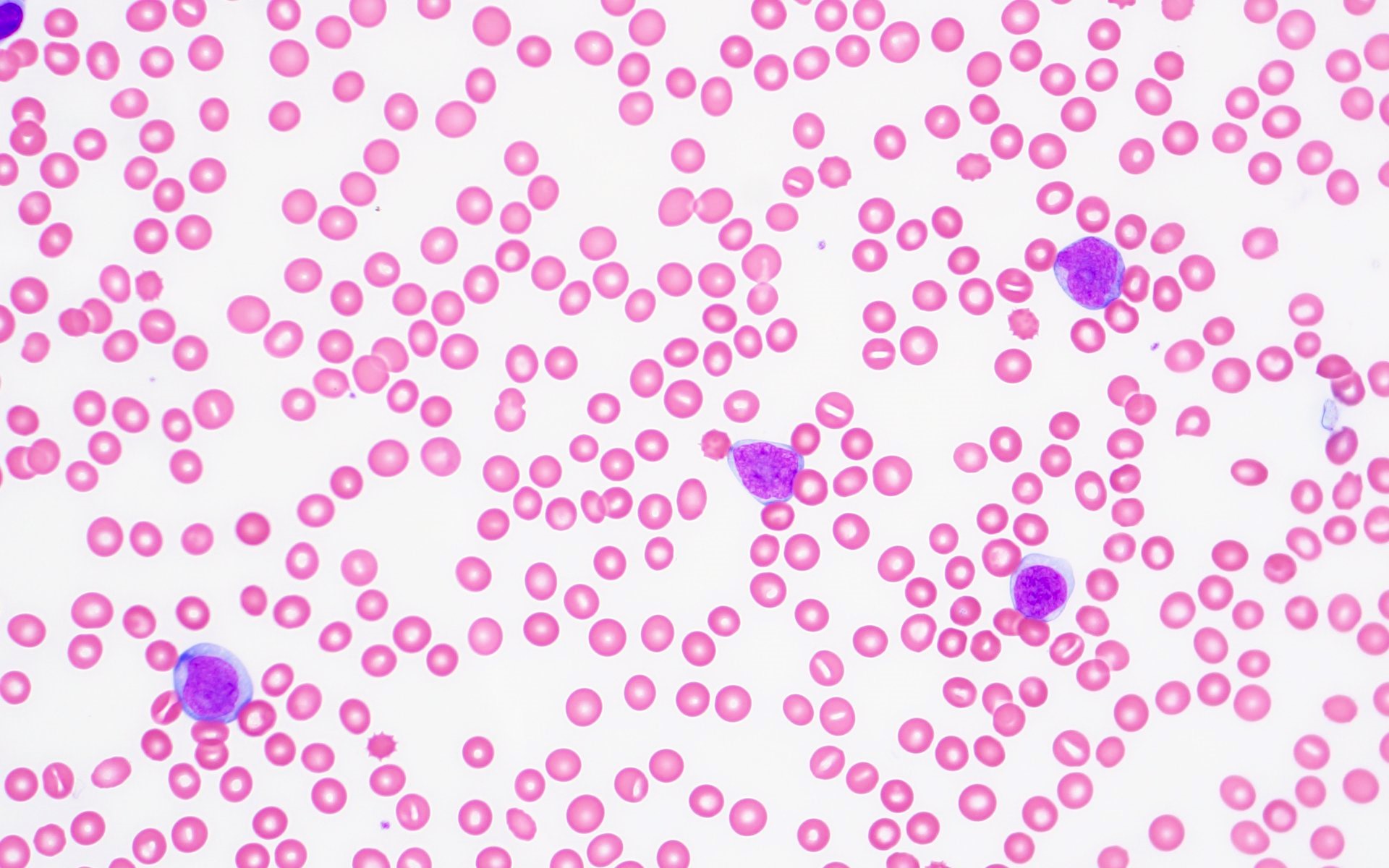
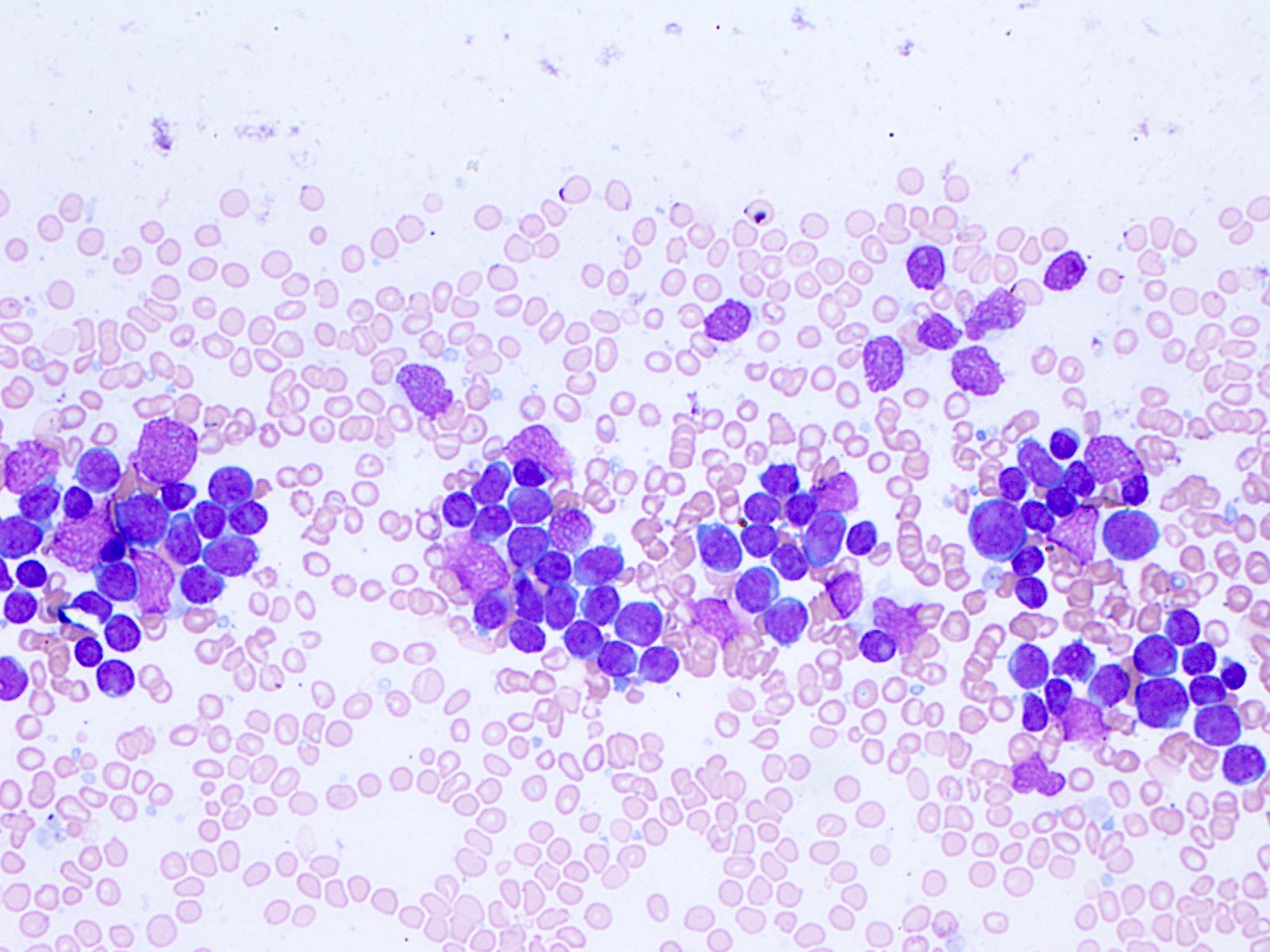
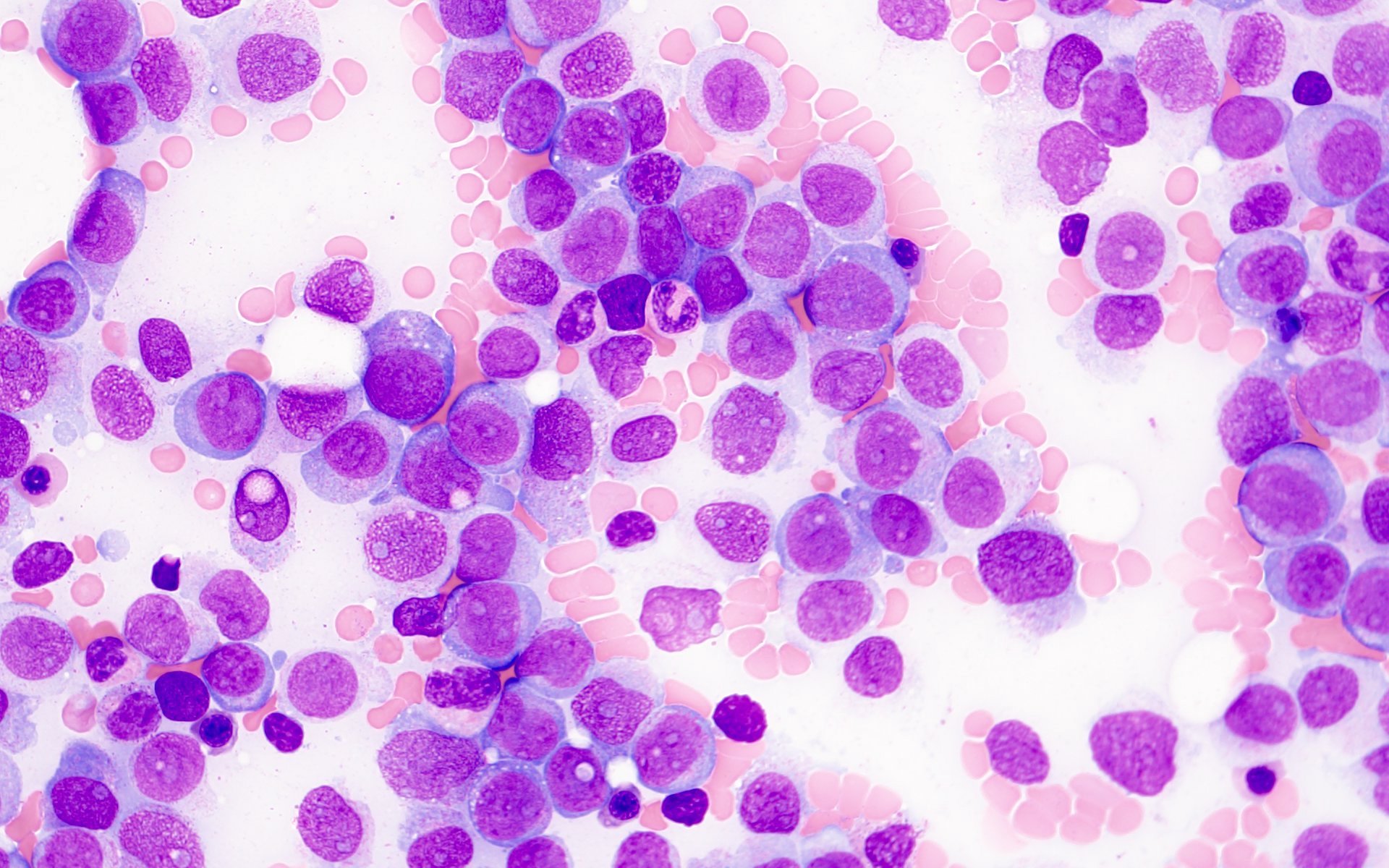
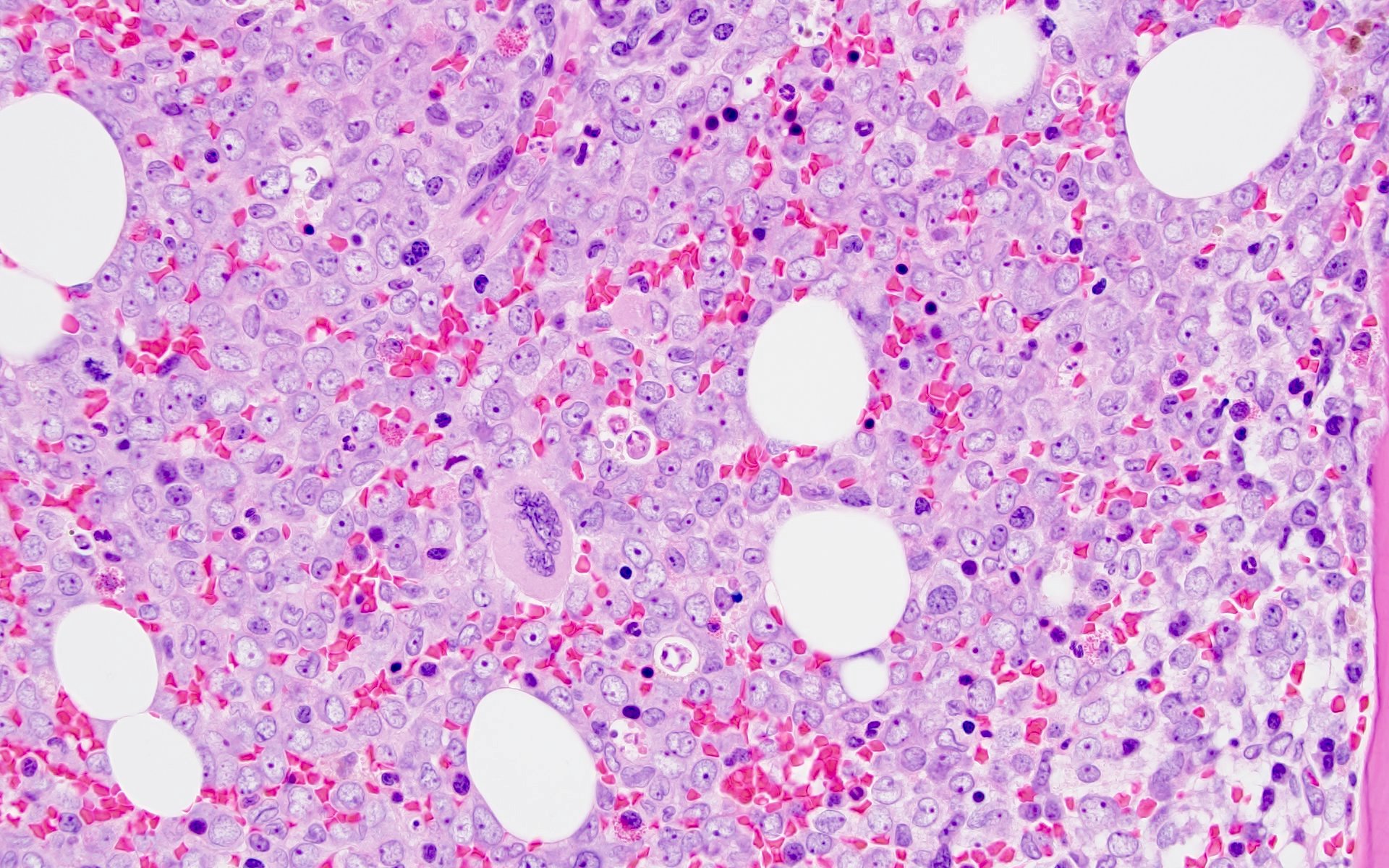
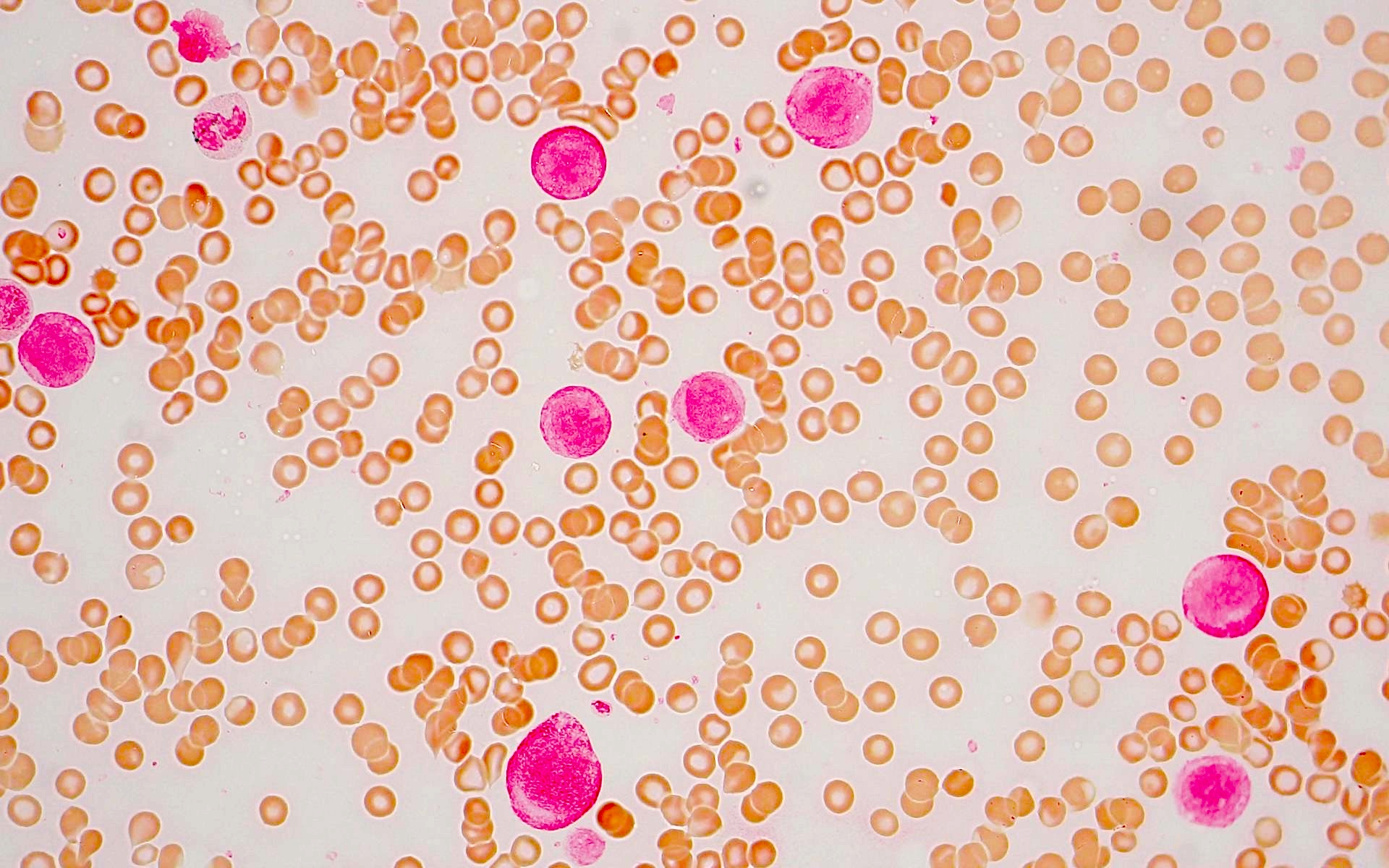
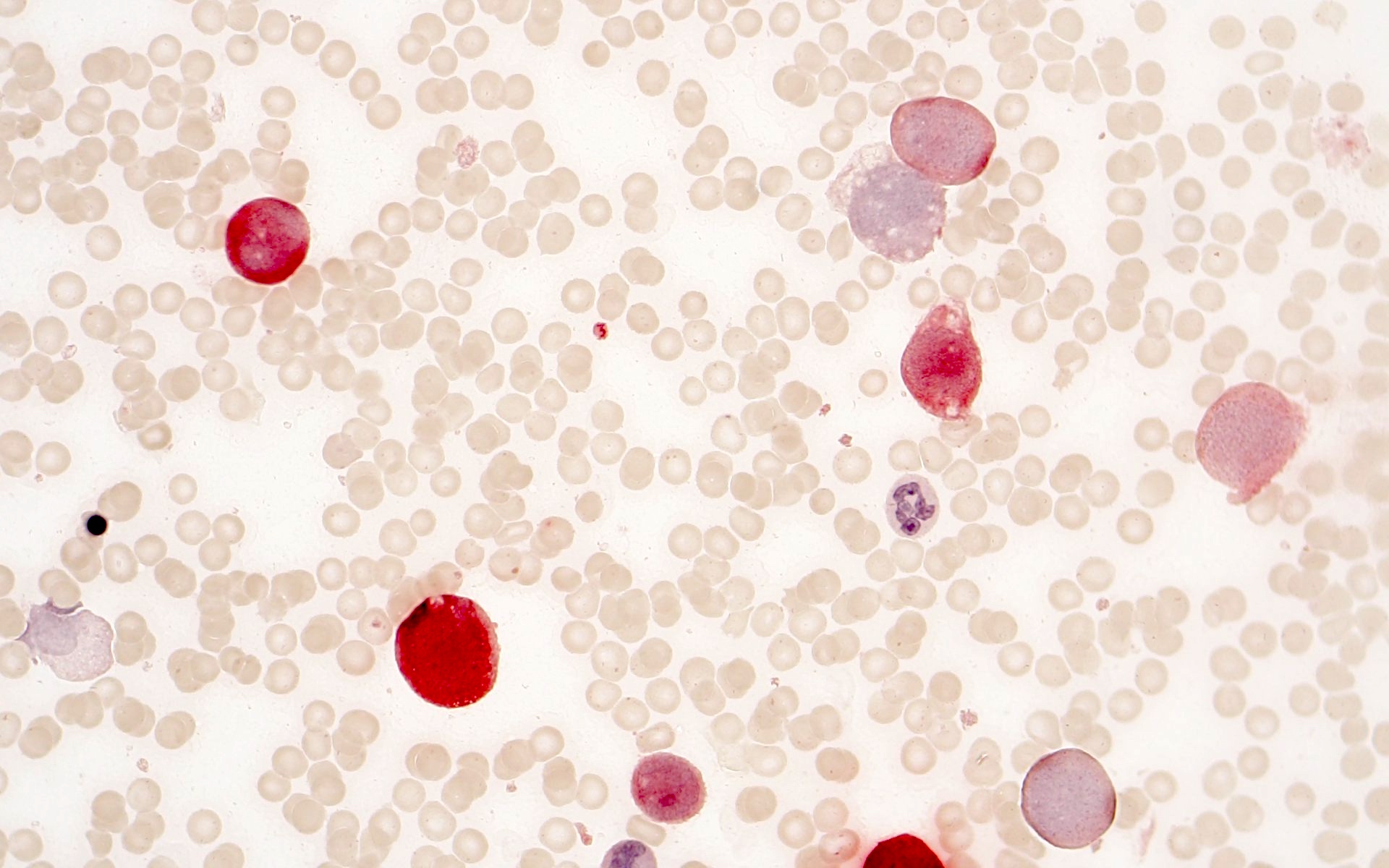
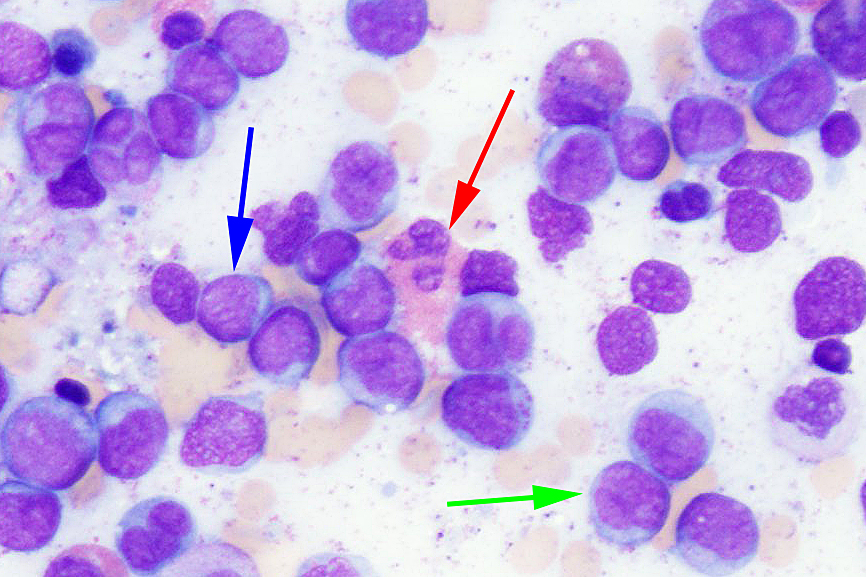
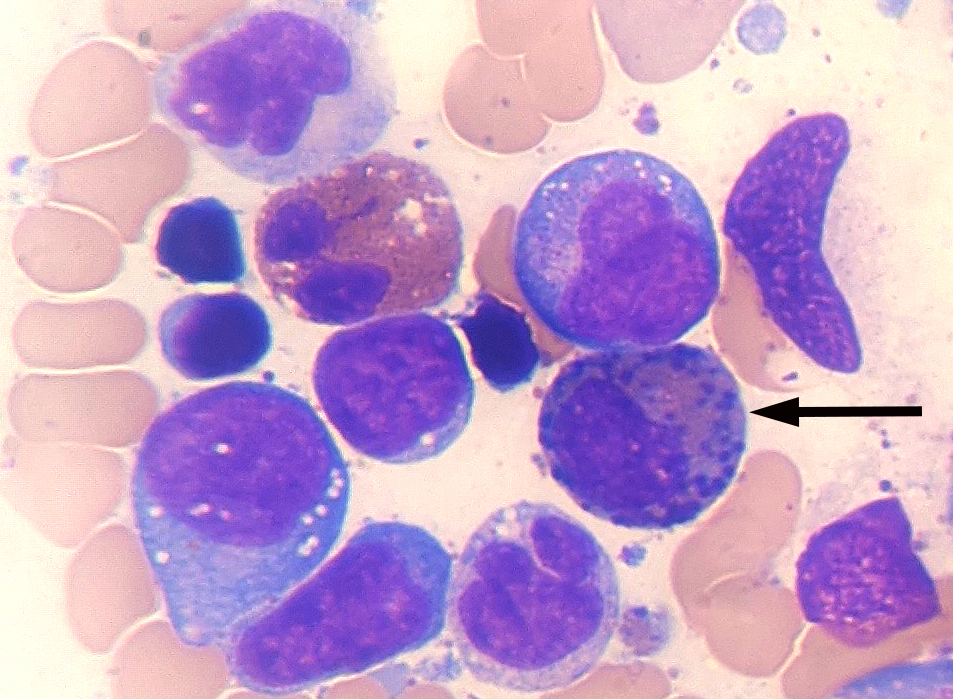
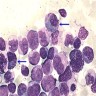
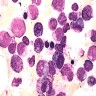
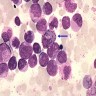
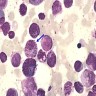
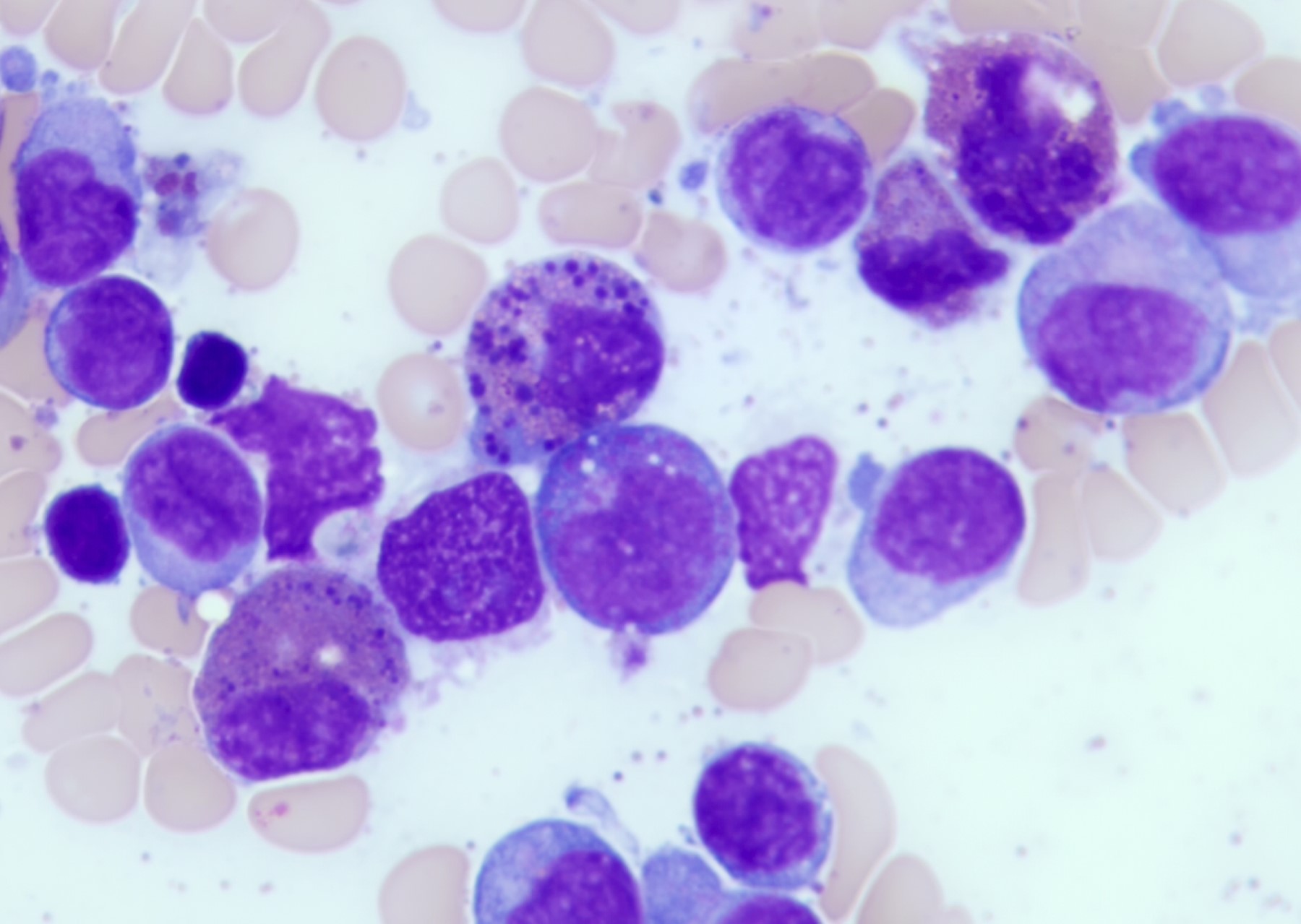
- Megakaryoblasts (often better morphology on biopsy than smear) are medium / large cells with blue vacuolated, agranular, eosinophilic cytoplasm containing fine granules, cytoplasmic projections (blebs and pseudopods) resembling platelets, irregular cytoplasmic borders and cytoplasmic zoning; may occur in clusters
- Nuclei are round or slightly indented with finely reticular, dense chromatin and 1 - 3 nucleoli
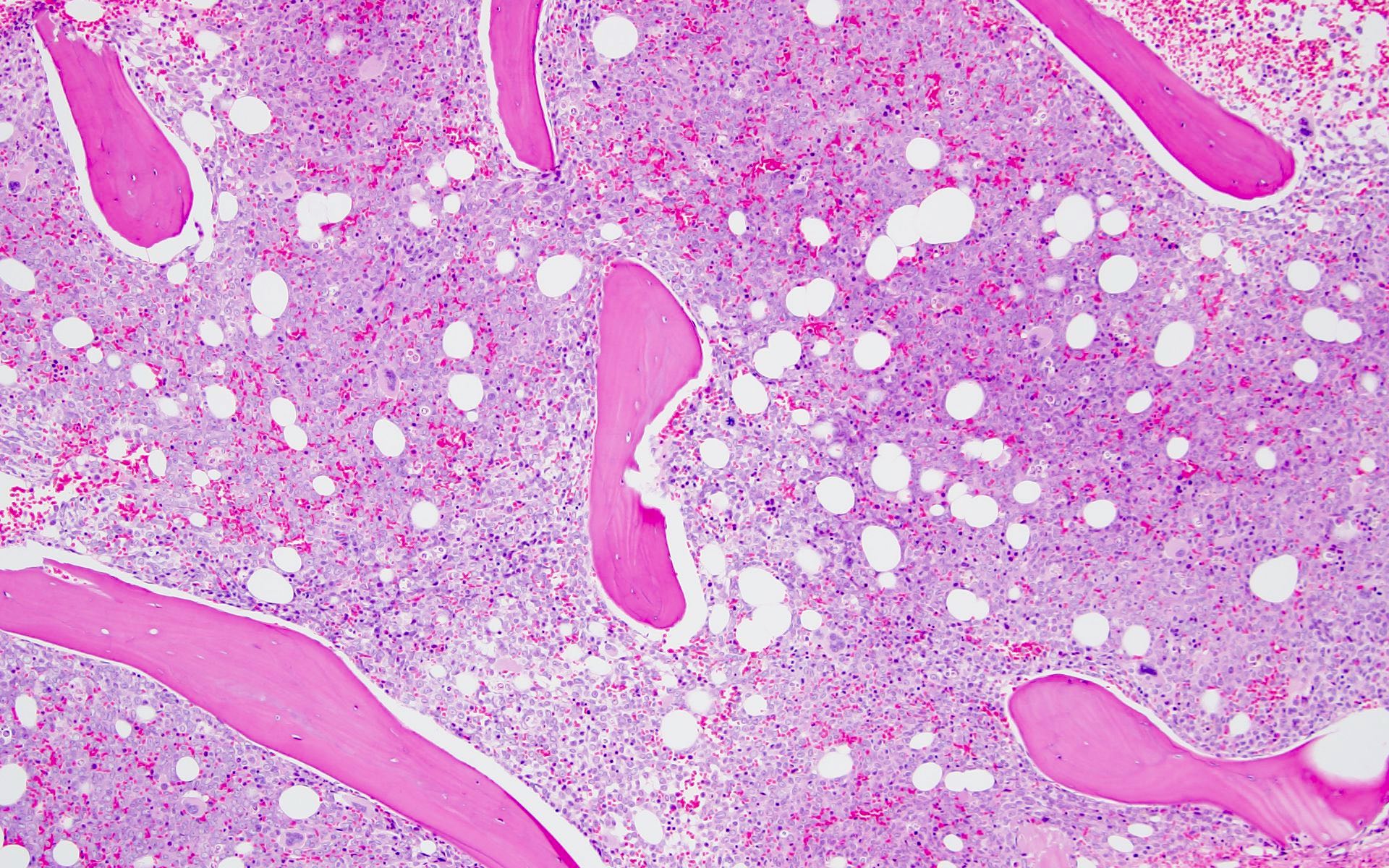
- Myelofibrosis or increased marrow reticulin is common; may also have small lymphoid-like blasts
AFIP images
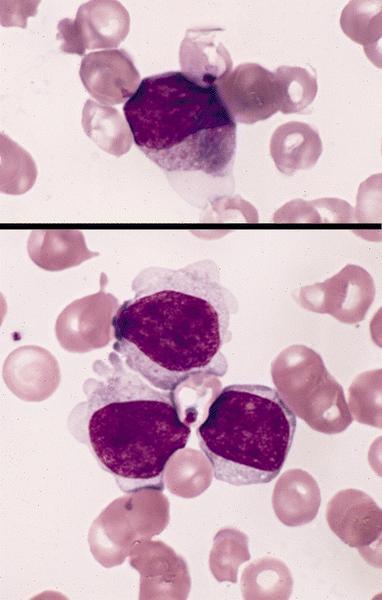
Abundant cytoplasm
Promegakaryocytes and large blasts
Touch prep shows 3 blasts
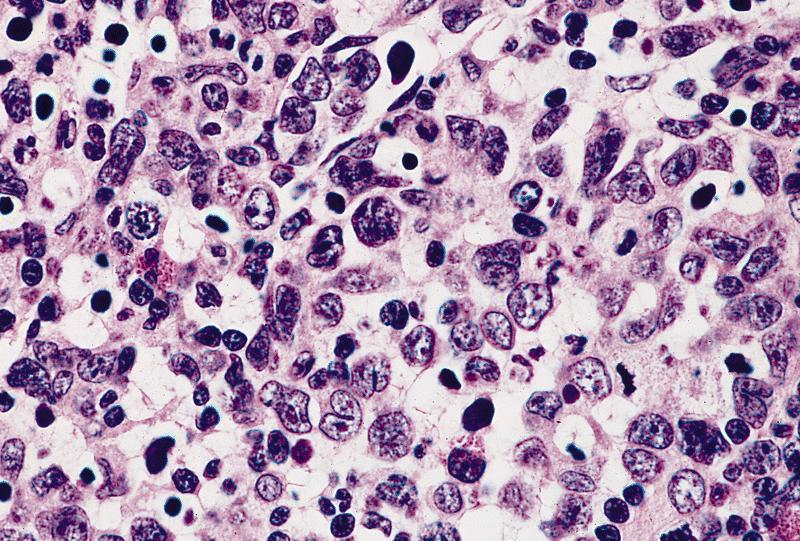
Extensive infiltration by blasts
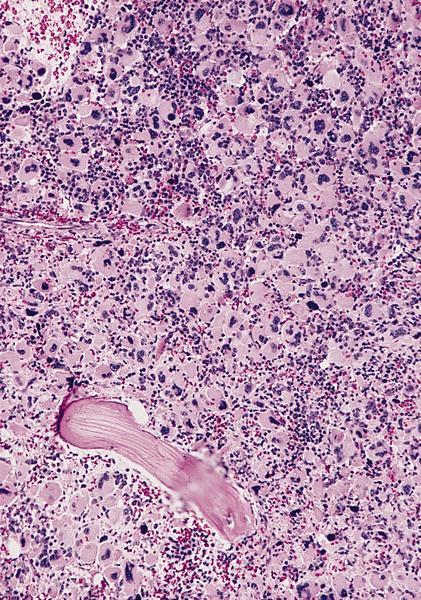
Marked proliferation of megakaryocytes
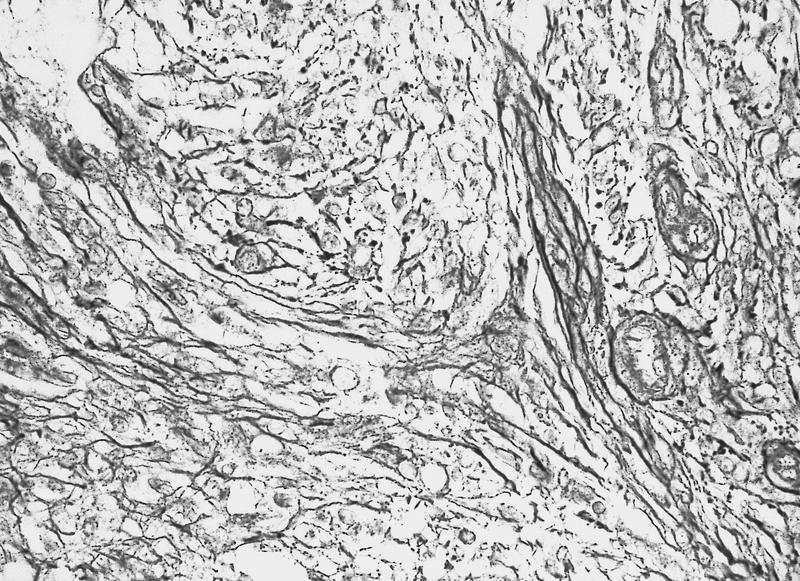
Reticulin stain shows marked increase in reticulin fibers
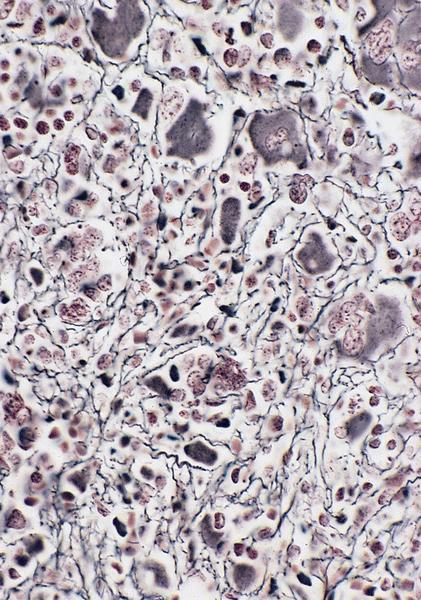
PAS+
Images hosted on other servers:
Bone marrow
biopsy: extensive
infiltration
by blasts
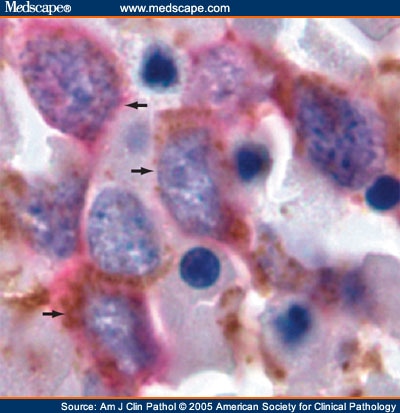
CD34: red, podocalyxin: brown
Images hosted on other servers:
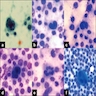

FNA and CSF
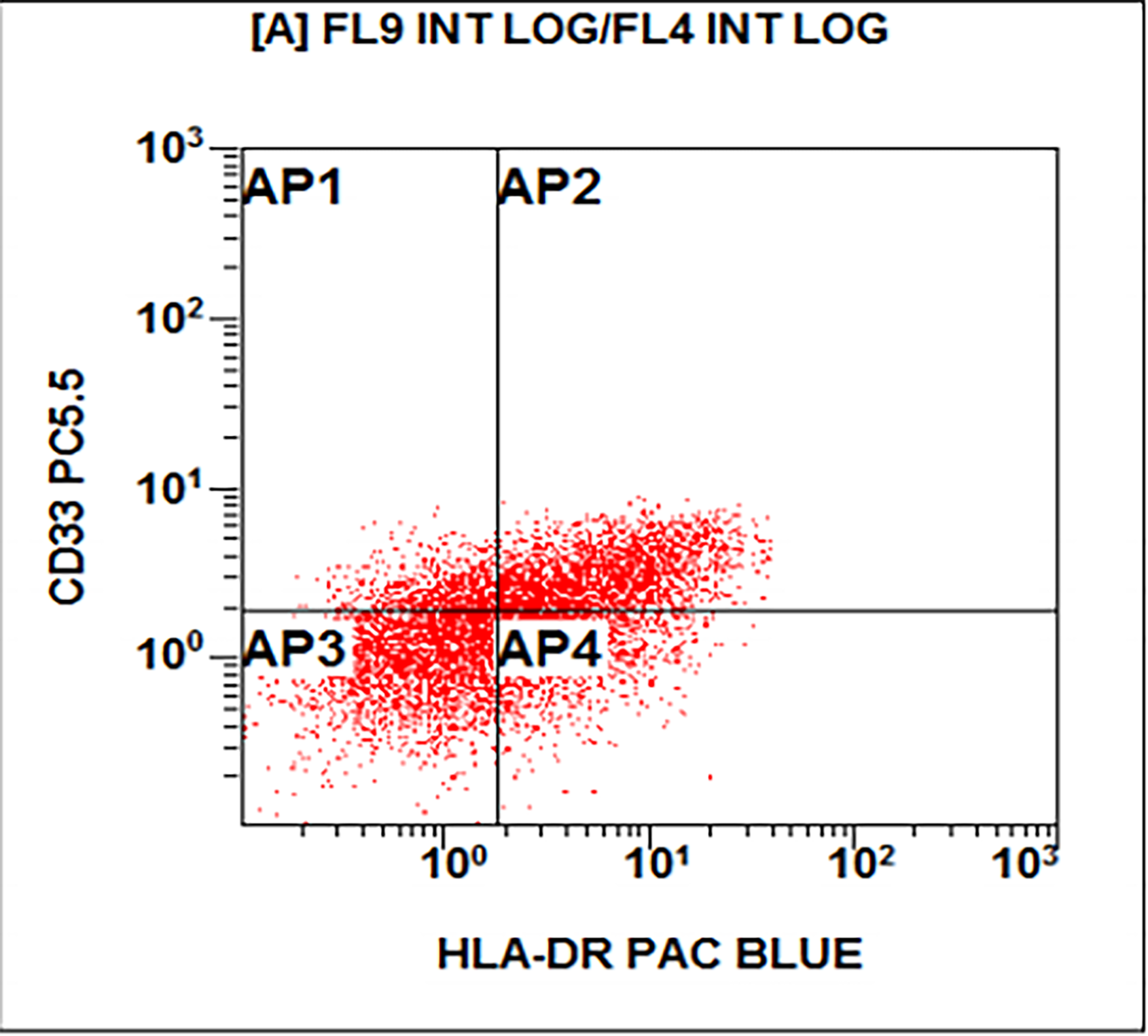
- CD41 and CD61 (megakaryocyte specific), CD42b (Mod Pathol 2005;18:603), CD34, CD36, factor VIII and von Willebrand factor
- Variable CD13, CD33, CD71, alpha naphthyl acetate esterase, PAS and HLA-DR
- Rarely positive for alpha-1-antitrypsin, alpha-1-antichymotrypsin or lysozyme (Am J Surg Pathol 1987;11:883)
- Megakaryoblasts have demarcation membranes and “bulls-eye” alpha granules with peroxidase activity in nuclear envelope and endoplasmic reticulum, but not in granules and Golgi complex
- Rare type of acute myeloid leukemia (megakaryoblastic) with a megakaryocytic phenotype (AMKL)
- Described as an entity under the category of acute myeloid leukemia with recurrent genetic abnormalities in the revised 2016 WHO Classification (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Acute megakaryoblastic leukemia with t(1;22)(p13.3;q13.1) resulting in fusion of RBM15 and MKL1 genes
- Rare subtype of AML with recurrent genetic abnormalities and differentiation along the megakaryocytic lineage
- Defined by t(1:22)(p13.3;q13.1); RBM15-MKL1
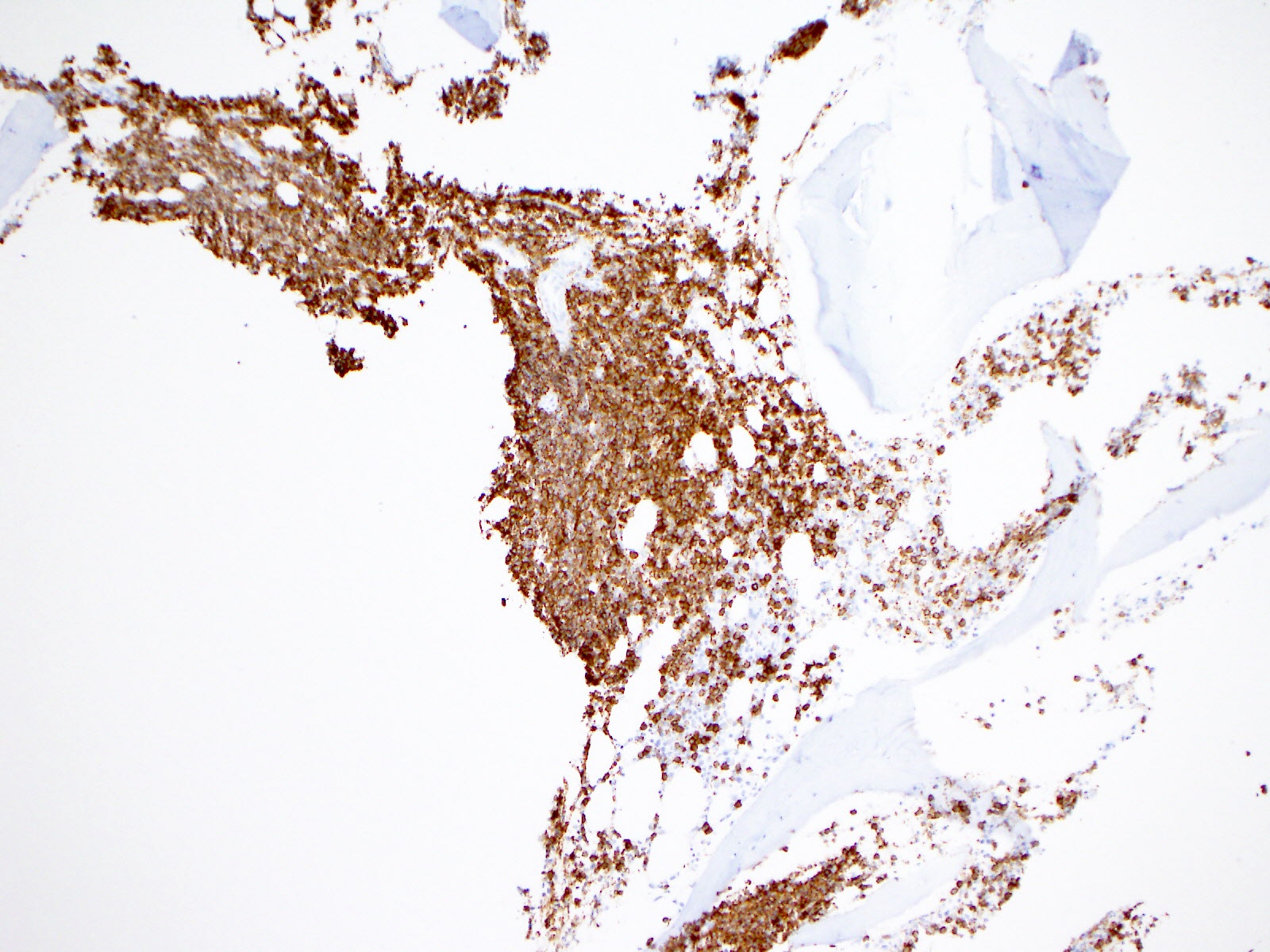
- Blasts are positive for platelet specific markers CD41a+, CD42b+, CD61+
- Occurs most commonly de novo in infants and young children (aged ≤ 3 years) without Down syndrome
- Severe disease often with poor prognosis
- Acute myeloid leukemia (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1
- AMKL with t(1:22)(p13.3;q13.1); RBM15-MKL1
- RBM15 and MKL1 are also referred to as OTT and MAL, respectively
- Abbreviations RBM15, MKL1, OTT and MAL stand for RNA binding motif protein 15, megakaryoblastic leukemia 1, OneTwoTwo and megakaryoblastic acute leukemia
- ICD-O: 9911/3 - Acute myeloid leukemia (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1
- Represents < 1% of all cases of AML
- Occurs most commonly in infants and young children (aged ≤ 3 years) without Down syndrome
- Most cases occur in the first 6 months of life (median patient age: 4 months)
- Female predominance
- Congenital cases have been described (Am J Hematol 2015;90:963, Cancer Genet Cytogenet 1988;34:277, J Pediatr Hematol Oncol 1999;21:428)
- Bone marrow, spleen and liver
- Lymph node involvement can also occur
- Postulated normal counterpart: myeloid progenitor cell with predominant megakaryocytic differentiation
- RBM15-MKL1 fusion gene arising from the t(1;22)(p13.3;q13.1) is thought to be the initiating event (Blood 2015;126:943, Nat Genet 2001;28:220)
- RBM15 encodes a protein with RNA recognizing motifs and a split end paralogue and orthologue C terminal (SPOC) domain that interact with the SMRT and NCoR corepressor complexes and the transcription factor downstream of Notch signaling named RBPJ (EMBO J 2002;21:5417, Genes Dev 2003;17:1909)
- Product of MKL1 acts as a transcriptional coactivator of serum response factor (SRP), a transcription factor that regulates genes involved in cell growth, proliferation, differentiation and genes controlling the actin cytoskeleton (Blood 2010;116:1942)
- Fusion of RBM15 and MKL1 may influence chromatin organization, differentiation induced by the HOX pathway and extracellular signaling pathways (Proc Natl Acad Sci USA 2001;98:5776)
- Majority of cases present with marked hepatosplenomegaly
- Patients have anemia and usually thrombocytopenia and moderately elevated white cell count
- Can present as a liver mass mimicking hepatoblastoma or severe hepatic failure (Pediatr Blood Cancer 2020;67:e28111, Glob Pediatr Health 2017;4:2333794X16689011)
- Lymphadenopathy and ascites have also been described upon presentation (Blood 1991;78:748)
- Bone marrow biopsy
- Cytogenetics
- Molecular genetics
- Immunophenotyping
- Karyotype analysis showing evidence of t(1;22)(p13.3:q13.1) or molecular genetic evidence demonstrating RBM15-MKL1 fusion
- Some studies have suggested that patients treated with intensive AML chemotherapy respond well, with long disease free survival (Leukemia 2000;14:216, Leuk Lymphoma 2003;44:49)
- However, the overall evidence suggests that it has a worse prognosis compared with other pediatric acute megakaryocytic leukemia, including those associated with Down syndrome (Leukemia 2000;14:216, Blood 1991;78:748, Blood 2015;126:1575, Ann Hematol 2015;94:1327)
- 10 day old girl presented with an epistaxis (Am J Hematol 2015;90:963)
- 1 month old girl with RBM15-MKL1 presenting as severe hepatic failure (Glob Pediatr Health 2017;4:2333794X16689011)
- 2 month old twin girls presented with fever and poor feeding and subsequently developed hepatosplenomegaly (J Pediatr Hematol Oncol 1999;21:428)
- 4 month old girl with AMKL where detection of RBM15-MKL1 fusion was useful for diagnosis and monitoring of minimal residual disease (Acta Med Okayama 2014;68:119)
- 11 month old Caucasian boy with AML mimicking hepatoblastoma (Pediatr Blood Cancer 2020;67:e28111)
- Chemotherapy (combination of an anthracycline and cytarabine)
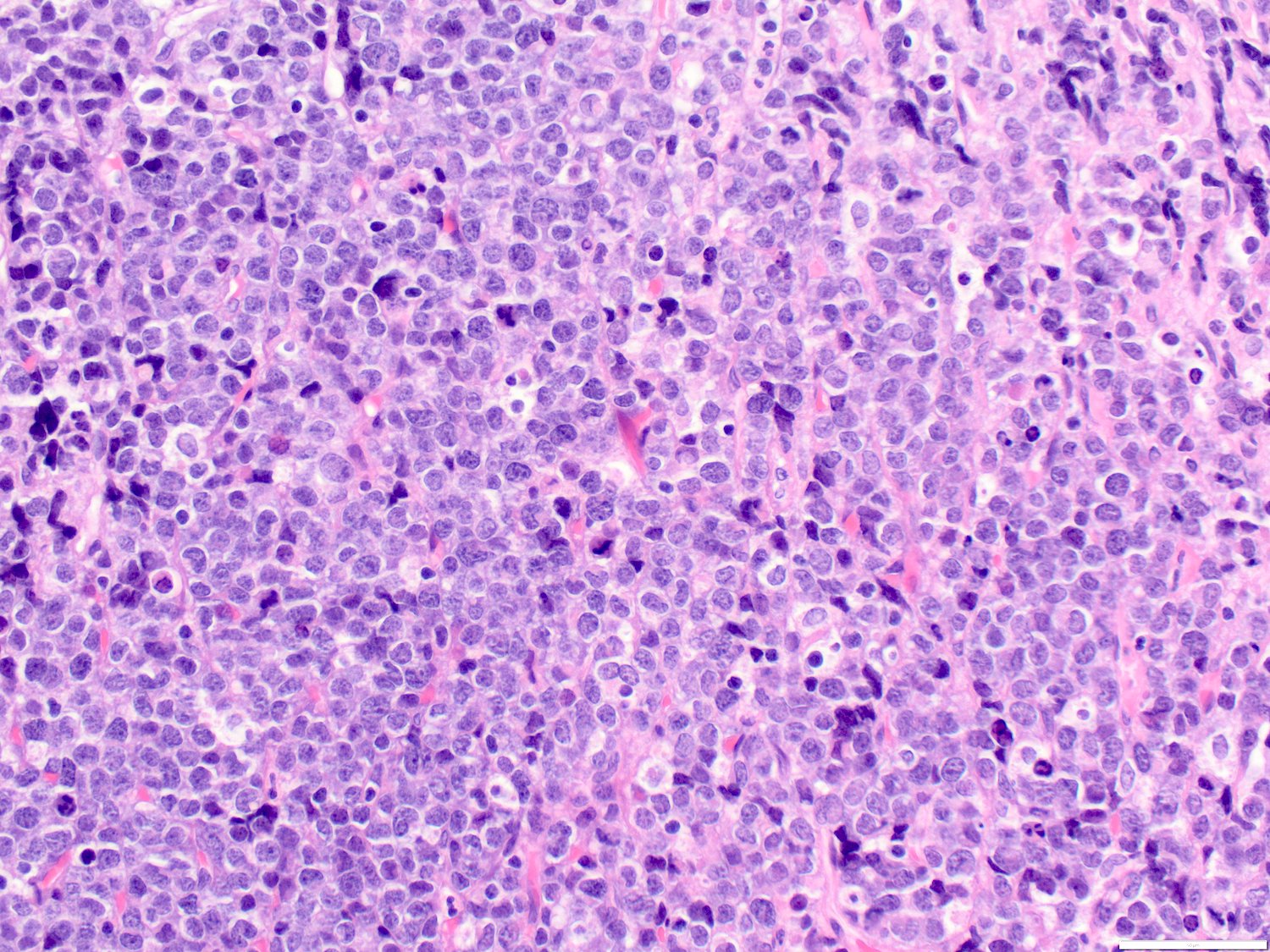
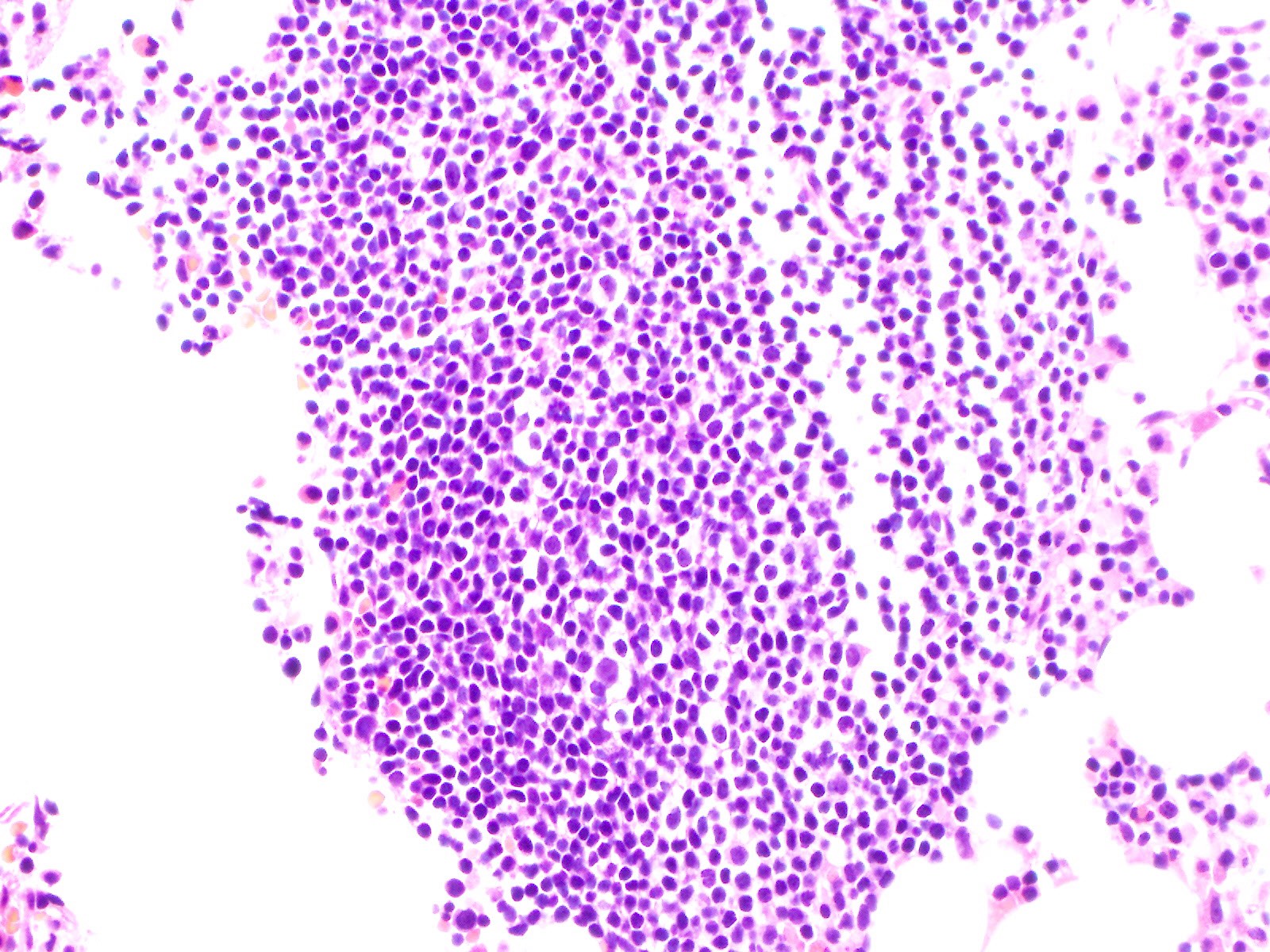
- Mixture of small and large megakaryoblasts may be found in the bone marrow and peripheral blood together with more undifferentiated blast cells with high N:C ratio
- Megakaryoblasts are most often medium sized to large (12 - 18 μm)
- Nucleus is round, slightly irregular / indented with fine reticular chromatin and 1 - 3 nucleoli
- Cytoplasm is basophilic, often agranular, with distinct blebs or pseudopod formation
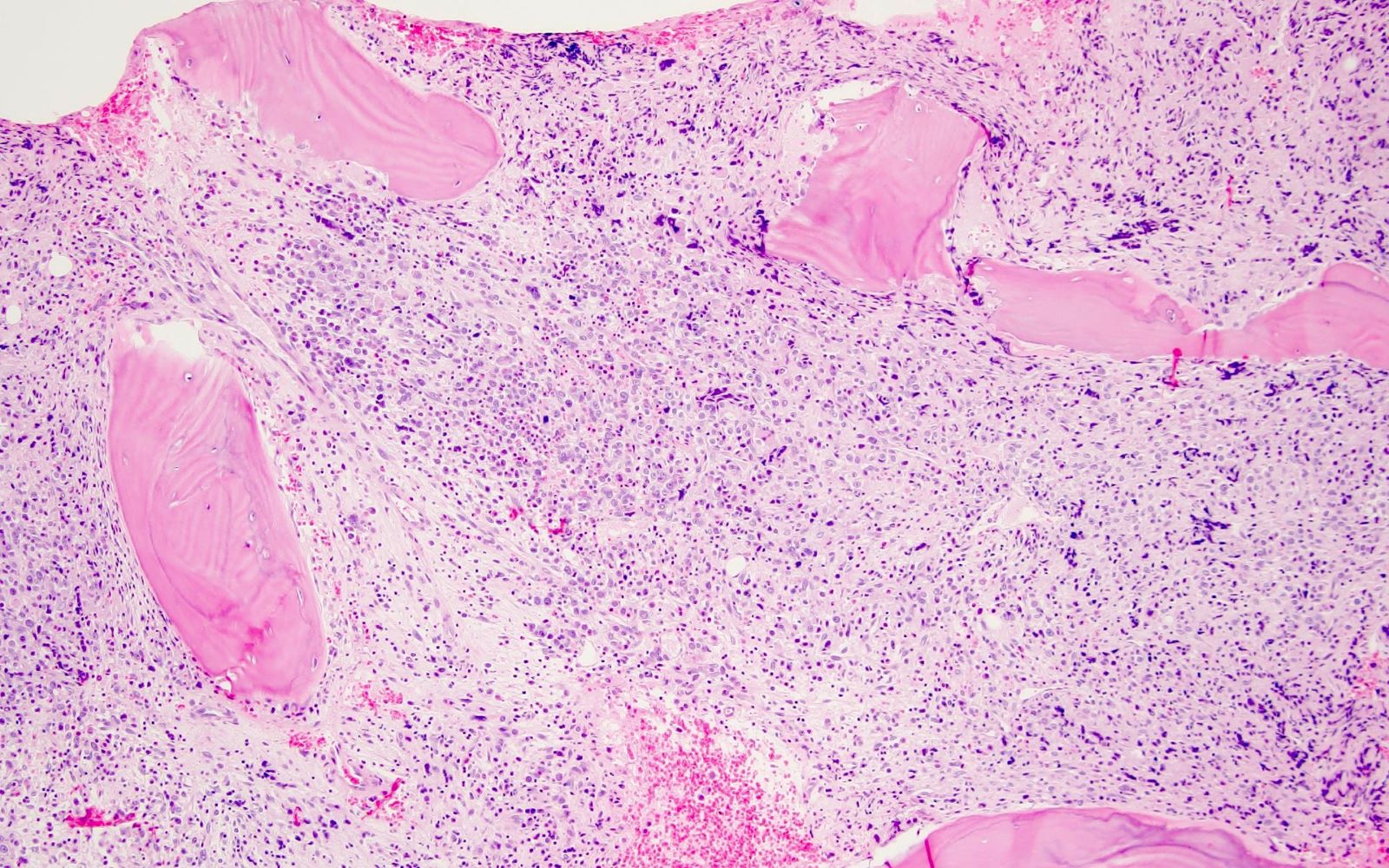
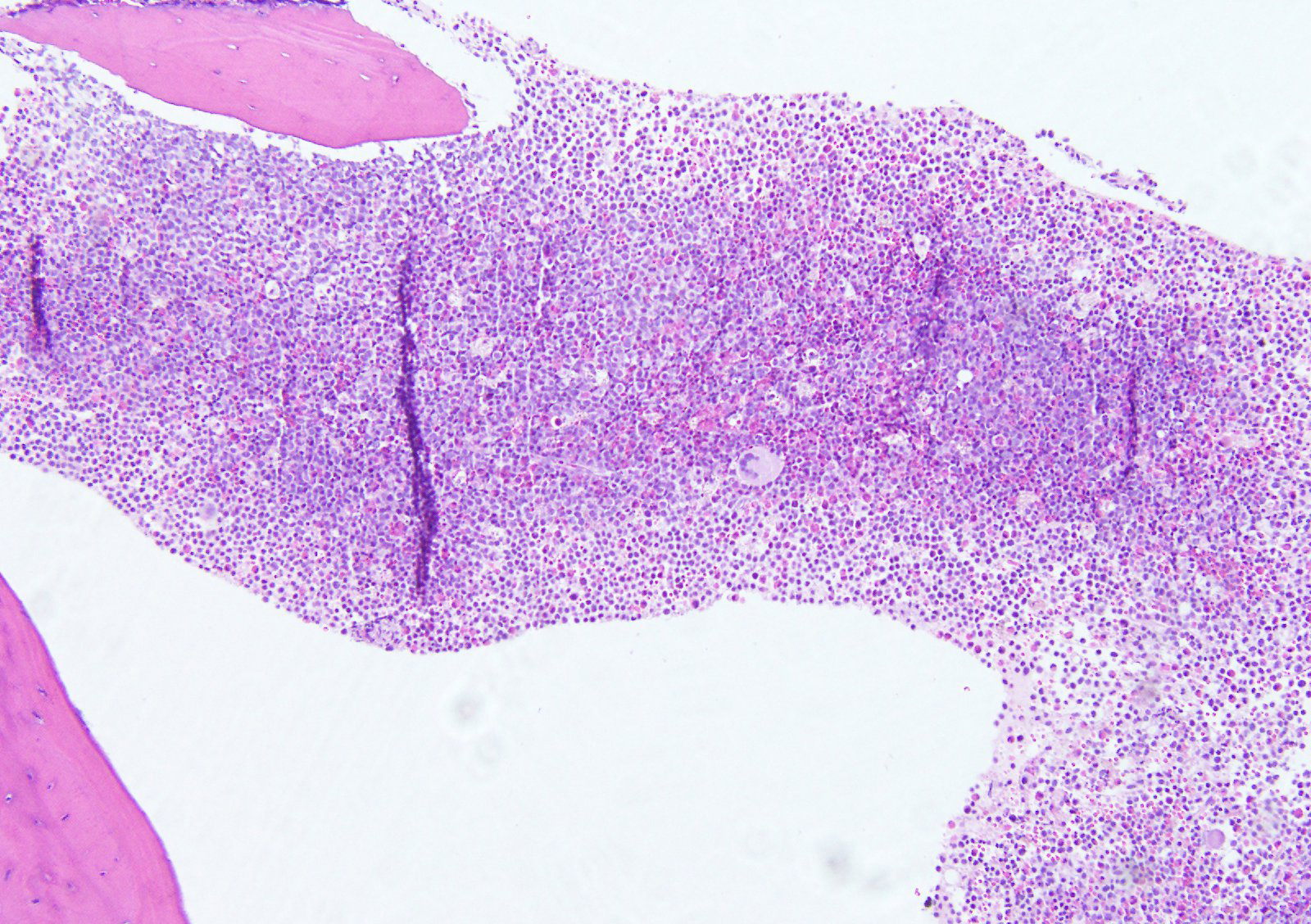
- Bone marrow is normocellular to hypercellular, with varying degrees of reticulin and fibrosis
- Dense fibrosis may result in a pattern of infiltration mimicking metastatic tumor (Leukemia 2000;14:216, Blood 1991;78:748)
- Establishing the presence of ≥ 20% blasts on bone marrow aspirate may prove difficult due to extensive fibrosis; making correlation with bone marrow biopsy findings crucial
- Micromegakaryocytes are commonly present
- Dysplastic erythroid and granulocytic cells are usually not seen
- TdT
- Myeloperoxidase antibodies
- CD34
- CD45
- HLA-DR
- Cytochemical staining for Sudan Black B and myeloperoxidase is consistently negative
- Blasts are CD41a+, CD42b+, CD61+
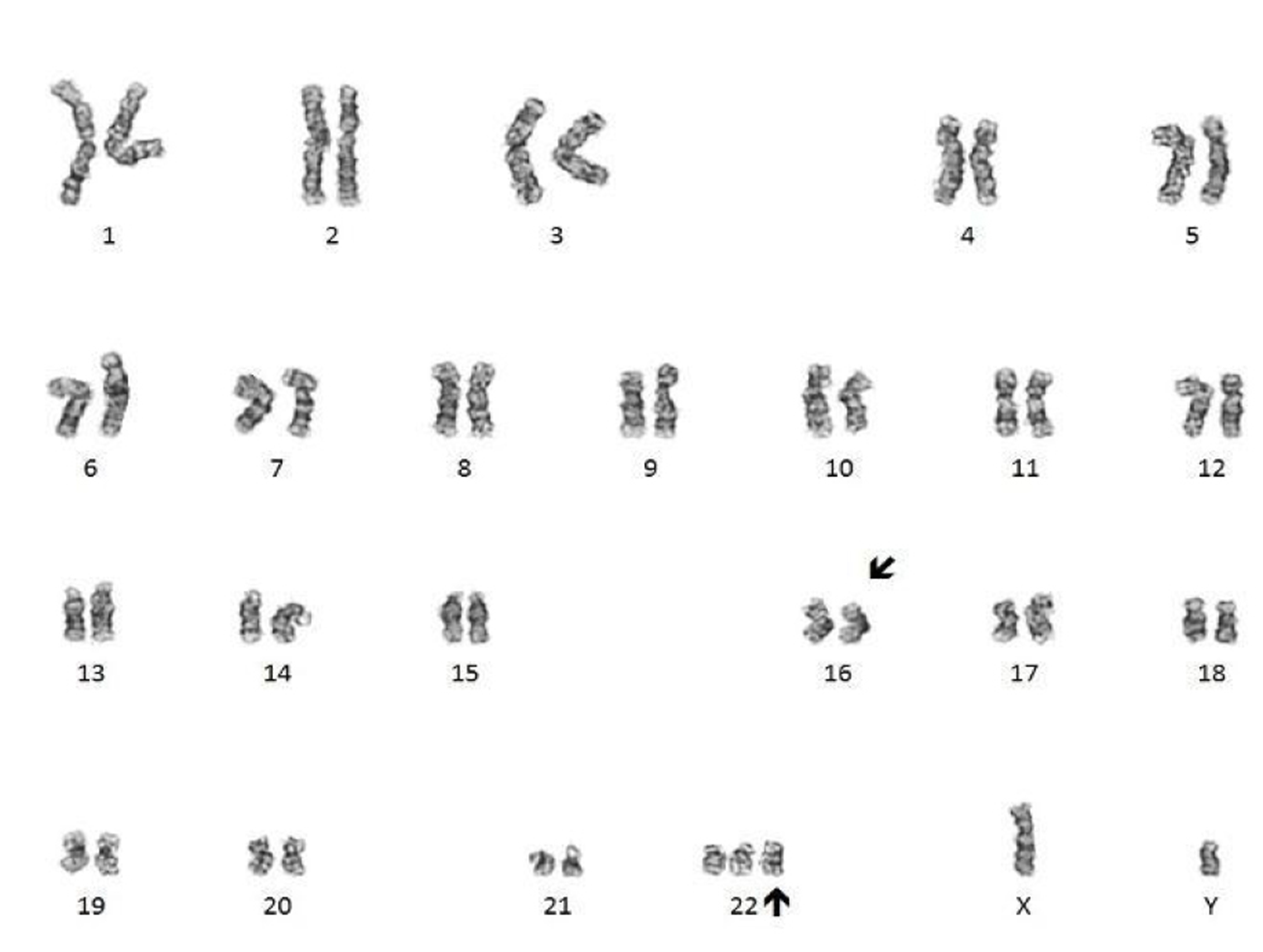
- Translocation t(1;22)(p13.3;q13.1) is visible on karyotype (Nat Genet 2001;28:220)
- Detection of RBM15-MRTFA (MKL1) fusion transcript via RT-PCR
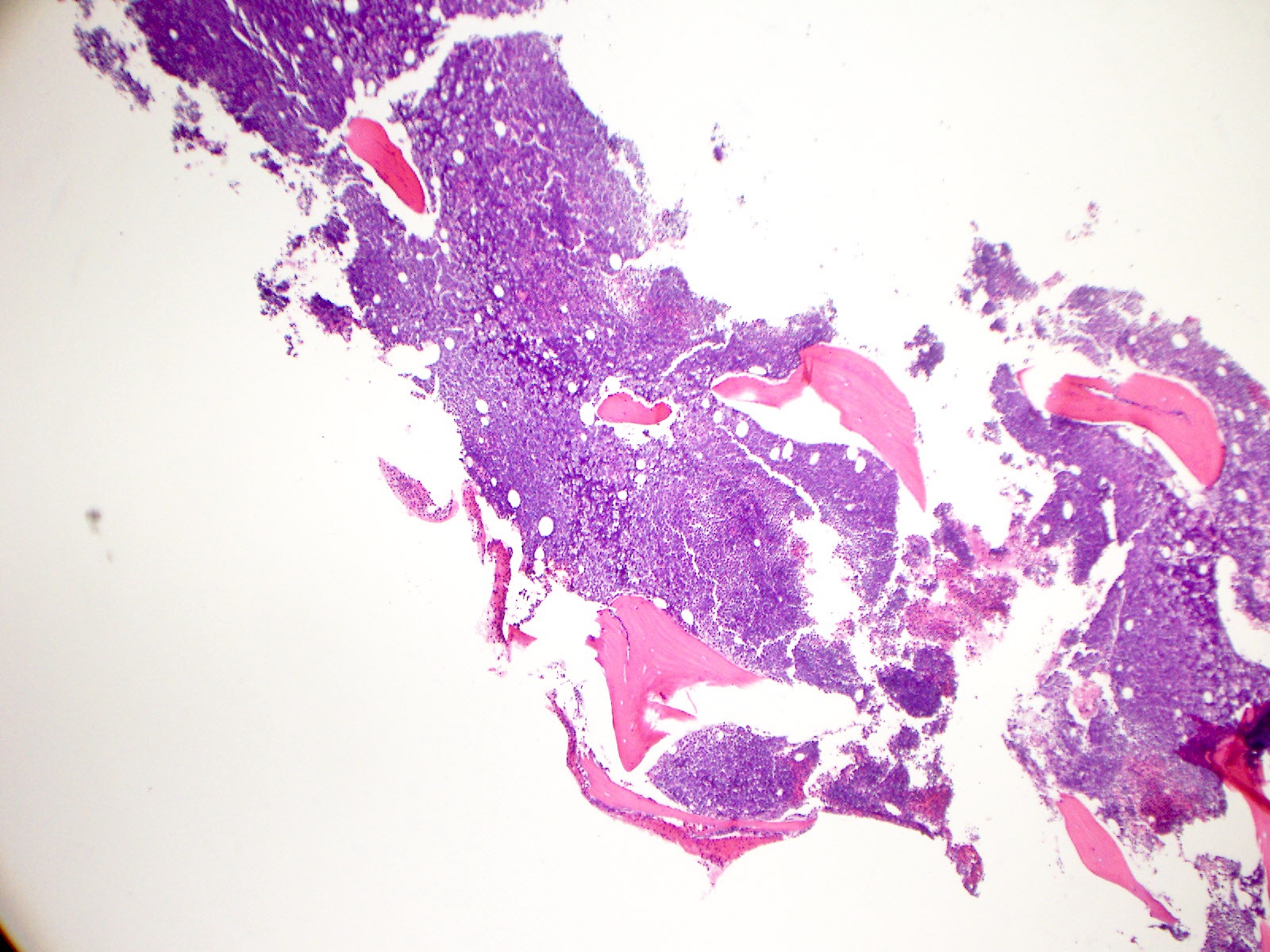
- Right posterior iliac crest, core biopsy, aspirate smear, touch imprint and clot particle:
- Acute myeloid leukemia (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1 (see comment)
- Comment: The bone core biopsy demonstrates extensive replacement of marrow by blast cells in areas arranged into clusters and aggregates surrounded by fibrotic stroma. The marrow aspirate smears reveal a heteromorphic population of blasts. Blast phenoptyping with immunohistochemical stains and flow cytometry analysis reveal positivity for markers of megakaryocytic maturation. Cytogenetics evaluation reveals t(1;22)(p13.3;q13.1), confirming above diagnosis.
- Non-Down syndrome associated acute megakaryoblastic leukemia:
- AMKL with CBFA2T3-GLIS2 fusion
- AMKL with NUP98-KDM5A fusion
- Acute myeloid leukemia associated with Down syndrome:
- GATA1 mutation is present
- t(1;22) translocation is not present
- Hepatoblastoma
- DEK-NUP214
- KMT2A-MLLT3
- PML-RARA
- RBM15-MKL1
- RUNX1-RUNX1T1
- Children aged 5 - 10
- Elderly females
- Elderly males
- Infants with Down syndrome
- Infants without Down syndrome
Comment Here
Reference: AML (megakaryoblastic) with t(1;22)(p13.3;q13.1)
- CD13
- CD36
- CD41
- CD61
- Myeloperoxidase
- Acute monoblastic and acute monocytic leukemia (AML M5)
- 10% of AML cases
- High incidence of bleeding disorders (including DIC), organomegaly, lymphadenopathy, gingival hyperplasia, CNS and other tissue involvement (monocytes infiltrate)
- May present with acute respiratory failure (Am J Respir Crit Care Med 2003;167:1329)
- M5a versus M5b: analysis of features is controversial; (a) similar features (Leuk Res 2008;32:269); (b) different expression of CD68 and CD11c (Zhongguo Shi Yan Xue Ye Xue Za Zhi 2006;14:1079); (c) different features (Zhongguo Shi Yan Xue Ye Xue Za Zhi 2006;14:654)
- Similar prognosis as other subtypes (J Clin Oncol 2004;22:1276)
- 80% or more nonerythroid bone marrow cells are monocyte lineage (monoblasts, promonocytes and monocytes)
- A minor neutrophil component < 20%
- Monoblast granules and monocytes are strongly positive for nonspecific esterase and lysozyme, but negative for myeloperoxidase
- More mature monocyte lineage cells may be weakly myeloperoxidase positive
- Note: if NSE negative, confirm monocyte lineage with immunostains
- At least two monocytic markers: CD4, CD14, CD11b (80 - 90%, Am J Clin Pathol 1997;107:283), CD11c (50%), CD36, CD64 (Am J Clin Pathol 1998;110:797), CD68 and HLA-DR
- Myeloid markers: CD13 (often very bright), CD33 (often very bright), CD15 and CD65
- CD34 (30%), CD117 and MPO (more often M5b and less often M5a)
- Abberant expression: CD7, CD56 (25 - 40%)
- Lysozyme, CD68 and CD163
- 11q23 translocations in 19%, trisomy 8 in 17%
- Acute erythroid leukemia (AML-M6)
- A neoplastic proliferation of immature cells (undifferentiated or proerythroblastic in appearance) committed exclusively to the erythroid lineage (> 80% of the bone marrow cells are erythroid, with ≥ 30% proerythroblasts), with no evidence of a significant myeloblastic component (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Note: per WHO 2016, the category of acute erythroleukemia M6a no longer exists; cases previously classified as M6a are either myelodysplastic syndrome (MDS) or AML depending on the myeloblast percentage in bone marrow or peripheral blood
- A rare aggressive acute myeloid leukemia (Am J Hematol 2017;92:292)
- Erythroid precursors > 80% and proerythroblasts ≥ 30% of the bone marrow cells
- Myeloblasts are not increased
- Therapy related cases should be diagnosed as therapy related myeloid neoplasms
- Acute myeloid leukemia, M6 type
- Acute erythroid leukemia
- Erythroleukemia
- Pure erythroid leukemia M6b
- Di Guglielmo disease
- Acute erythraemic myelosis
- Acute erythraemia
- ICD-10: C94.00 - acute erythroid leukemia, not having achieved remission
- Extremely rare, < 1% of all AML (Am J Hematol 2017;92:292)
- Any age, can be congenital (Int J Hematol 2017;106:711)
- Both de novo and secondary (Mod Pathol 2018;31:705, Mod Pathol 2011;24:375)
- Bone marrow or peripheral blood (Am J Hematol 2017;92:292)
- Mass lesion (erythroblastic sarcoma) in tissue such as central nervous system, orbit, liver, ovary, etc. (Haematologica 2020 Jan 16 [Epub ahead of print])
- Anemia (Am J Hematol 2017;92:292)
- Circulating erythroblasts
- Mass (Haematologica 2020 Jan 16 [Epub ahead of print])
- Erythroblasts > 80% in bone marrow or peripheral blood with proerythroblasts ≥ 30%
- Myeloblasts are not increased (< 5%)
- For extramedullary mass lesion, sheets of erythroid precursor cells with proerythroblasts ≥ 30%
- Usually poor with median survival of 1.4 to 3 months (Mod Pathol 2018;31:705, Mod Pathol 2011;24:375)
- 3 month old girl with low grade fever and cough, pure erythroid leukemia and bilateral ovarian erythroid sarcoma (Hum Pathol 2011;42:749)
- 15 month old boy with a history of failure to thrive, vomiting and high fever, prolonged diarrhea and pure erythroid leukemia (Leukemia 2011;25:1510)
- 2 year old girl presenting with headache, vomiting and a de nova erythroid sarcoma in third ventricle (Haematologica 2020 Jan 16 [Epub ahead of print])
- 44 year old woman with progressive fatigue, pancytopenia and pure erythroid leukemia (Clin Case Rep 2019;7:1829)
- 48 year old man with shortness of breath, weight loss and pure erythroid leukemia (Hematol Rep 2015;7:5674)
- AML treatment protocol (Am J Hematol 2017;92:292)
- Radiation therapy can be considered for localized mass lesion in orbit or CNS (Haematologica 2020 Jan 16 [Epub ahead of print])
- Bone marrow transplant
- Bone marrow biopsy: sheets or clusters of immature cells, tumor cells may show intrasinusoidal growth pattern (Am J Hematol 2017;92:292)
- Bone marrow aspirate: erythroid precursors > 80% and proerythroblasts ≥ 30% of bone marrow cells
- Proerythroblasts have round nuclei, fine chromatin and one or more prominent nucleoli, deeply basophilic cytoplasm with vacuoles, often agranular
- Ring sideroblasts are common
- Myeloblasts are not increased
- Dysmegakaryopoiesis is common and dysgranulopoiesis is infrequent
- Mass in tissue: poorly differentiated tumor cells (Haematologica 2020 Jan 16 [Epub ahead of print])
Contributed by Huifei Liu, M.D., Ph.D.
Erythroblastic sarcoma
E-cadherin
CD43
AFIP images

Bone marrow smear (Wright-Giemsa)
Contributed by Huifei Liu, M.D., Ph.D.
CSF erythroblasts
- Anemia, thrombocytopenia or pancytopenia (Am J Hematol 2017;92:292)
- Circulating erythroblasts
Contributed by Huifei Liu, M.D., Ph.D.
Circulating erythroblasts
- Erythroid specific markers: E-cadherin (positive in majority cases), hemoglobin A and glycophorin A (usually positive but can be negative in poorly differentiated erythroblasts) (Am J Hematol 2017;92:292)
- Not lineage-specific markers: CD36, CD43, CD71, CD117, GATA1, Ferritin H
- These markers are unusually positive; however, they can be positive in other type of hematopoietic neoplasms
- CD36 is commonly positive on monocytic leukemia and acute megakaryocytic leukemia
- CD43 is positive in most AML, T-ALL as well as some B-ALL; it is a very useful marker for extramedullary poorly differentiated tumor to confirm hematopoietic lineage
- CD71 can be positive in other leukemic blasts although its expression levels in PEL should be very bright
- CD117 is positive in most AML and some T-ALL
- GATA1 is a transcription factor for erythroid development and it is positive in both PEL and acute megakaryocytic leukemia (Am J Clin Pathol 2017;147:420)
- Ferritin H is expressed in erythroid precursors as well as macrophages
- Special stains: PAS (usually in block-like pattern), alpha naphthyl acetate esterase, acid phosphatase
- Myeloid markers (CD13, CD33, myeloperoxidase) (Am J Hematol 2017;92:292)
- Megakaryocytic makers (CD41 and CD61), usually negative but may be partially expressed in some cases
- CD34, CD45, HLA-DR
- Note: these markers are generally negative but can be weakly or partially positive in some cases
- Often complex karyotype, frequently -5/del(5q) and -7/del(7q) (Am J Hematol 2017;92:292)
- t(1;16)(p31;q24) has only been described in a total of 5 cases of pure erythroid leukemia, with fusion of NFIA/CBFA2T3 confirmed in 2 cases of pure erythroid leukemia, likely a pure erythroid leukemia specific rearrangement (Haematologica 2020;105:e194, Leukemia 2013;27:980)
- t(11;20)(p11;q11)/ZMYND8-RELA was reported in 1 case of pure erythroid leukemia (Am J Hematol 2017;92:292)
- p53 mutation was detected in a significant portion of adult cases (Nat Genet 2019;51:694)
- Bone marrow, right, biopsy, clot section, aspirate smears, touch imprint and peripheral blood smear:
- Pure erythroid leukemia, blasts 95% of total nucleated cells (see comment)
- Comment:
- Peripheral blood:
- Red blood cells: Normocytic normochromic anemia with slight anisopoikilocytosis.
- White blood cells: Leukopenia with few large proerythroblasts. The blasts have scant basophilic cytoplasm, round nuclei, open chromatin and few have prominent nucleoli. Cytoplasmic vacuoles are noted in some blasts.
- Platelets: Decreased with rare giant forms.
- Bone marrow biopsy:
- Decalcification procedures performed to allow for histological assessment of submitted tissue.
- Gross measurement: 0.5 cm
- Quality: Adequate
- Cellularity: 100%
- Megakaryocytes: Rare
- Infiltrate: The marrow is nearly completely replaced by sheets of immature cells with high nuclear to cytoplasmic ratio, open chromatin and some with prominent nucleoli.
- Bone marrow clot:
- Quality: Inadequate, blood only, no particles with many immature cells
- Bone marrow smears:
- Quality / cellularity: Adequate
- Granulocytes: Markedly decreased
- Erythrocytes: Markedly increased erythroid precursors with increased proerythroblasts.
- Megakaryocytes: Not seen
- Lymphocytes: Decreased, few small and mature cells.
- bone marrow differential count: 500 total cells counted
- Blasts percentage (0-5): 95
- Promyelocytes percentage (2-8): 0
- Myelocytes percentage (5-20): 0
- Metamyelocytes percentage (13-32): 0
- Granulocytes percentage (7-30): 2
- Monocytes percentage (0-5): 0
- Lymphocytes percentage (3-17): 3
- Plasma cells percentage (0-2): 0
- Eosinophils percentage (0-4): 0
- Basophils percentage (0-1): 0
- Pronormoblasts percentage (1-8): 80
- Normoblasts percentage (7-32): 15
- M:E ratio (2-4): N/A
- Bone marrow touch imprint:
- Quality: Adequate, similar to smear with increased erythroid blasts.
- Flow cytometric analysis:
- Aberrant erythroid blasts, 95.2% of total nucleated cells (F19-160).
- Pending tests:
- Cytogenetic karyotype and FISH.
- Peripheral blood:
- Other type of acute leukemia, especially acute megakaryocytic leukemia
- Other poorly differentiated malignancy or small blue round cell tumor
- Reactive erythroid hyperplasia
- Pure erythroid leukemia should not be diagnosed as AML with myelodysplasia related change (AML with MRC) even if there is prior history of myeloid neoplasm, significant dysplasia of 2 lineages and a defining cytogenetic abnormality
- AML with MRC requires minimal 20% myeloblasts while pure erythroid leukemia should not have a significant component of myeloblasts
- CD117
- CD34
- Myeloperoxidase
- E-cadherin
- CD71
A 7 year old boy presented with leukocytosis (55K), anemia (Hb 7.5 g/dl) and thrombocytopenia (52k). Peripheral blood smear (see photo) shows many mononucleated cells of variable sizes with high nuclear:cytoplasmic ratio, round nuclei, open chromatin and deep blue cytoplasm. Some large cells have conspicuous nucleolus. By flow cytometric immunophenotyping, these cells are positive for CD36, CD117 (heterogeneous) and negative for CD34, CD45, HLA-DR and all other lymphoid, myeloid and monocytic markers. Which of the following sentences is correct regarding this disease?
- These cells are positive for CD61
- These cells show reactivity with alpha-naphthyl acetate esterase, acid phosphatase and PAS
- These cells most likely have a normal karyotype (46, XY)
- A percentage of ≥ 20% of these cells in blood or bone marrow is required for diagnosis as pure erythroid leukemia
- The prognosis for this patient is good
Comment Here
Reference: Acute erythroid leukemia (AML-M6)
- Defined as < 20% bone marrow cellularity, 20% or more are blasts (WHO); other definitions range up to 40% cellularity
- Up to 10% of AML cases
- Median age 67 years (Leuk Res 1996;20:563)
- Rare blasts in peripheral blood
- Often has smoldering course although intensive chemotherapy may cause complete remission
- 78 year old man and woman with minimally differentiated hypoplastic leukemia (Nihon Ronen Igakkai Zasshi 1997;34:70)
AFIP images
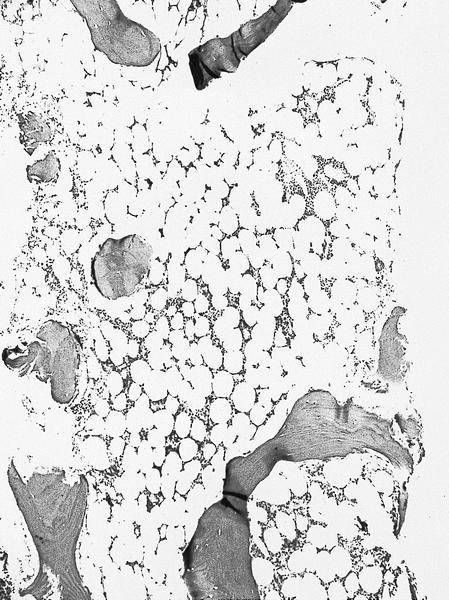
Markedly hypocellular marrow
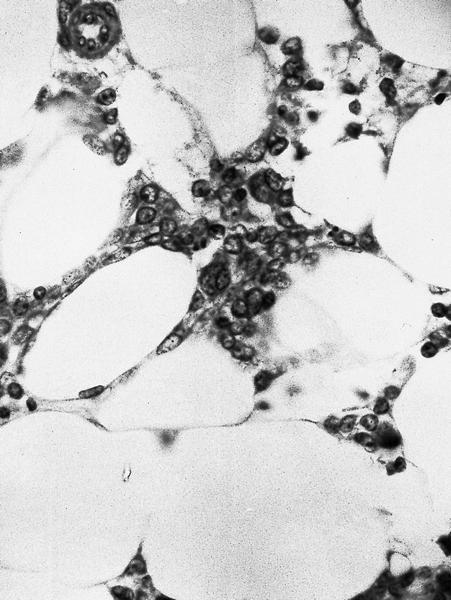
Cells in interstitium are predominantly blasts
- Aplastic anemia: no excess blasts, interstitial bone marrow cells are plasma cells, lymphocytes and mast cells, not blasts
- Refractory anemia with excess blasts
- Substance abuse: see Arch Pathol Lab Med 2005;129:e35
- Subtype of acute myeloid leukemia (AML) with recurrent genetic abnormality
- Required for diagnosis, the CEBPA mutations must be biallelic
- Associated with more favorable prognosis
- CEBPA mutations must be biallelic, not just a single mutation, for diagnosis
- May represent a germline predisposition syndrome and germline testing may be considered in patients with persistent CEBPA mutations following morphologic remission or in patients with family history of leukemia
- ICD-10: C92.0 - acute myeloblastic leukemia
- Accounts for 4 - 9% of AML diagnoses in children and young adults
- Less common in older patients
- Reference: Blood 2009;113:6558
- Peripheral blood and bone marrow
- Hematopoietic progenitor cells require biallelic mutations of CEBPA
- Possible continued clonal evolution
- Reference: Sci Adv 2019;5:eaaw4304
- A subset of patients carry an underlying germline mutation, leading to predisposition to develop AML
- Associated with lower frequency of lymphadenopathy and myeloid sarcoma
- Routine CBC with differential, bone marrow biopsy with flow cytometry, chromosome analysis and either targeted CEBPA molecular testing or next generation sequencing (massively parallel sequencing) for myeloid mutations
- Typically present with relatively higher hemoglobin levels (still anemic), lower platelet counts and lower lactate dehydrogenase than CEBPA wildtype AML
- PET scan identified hypermetabolic bone marrow
- Favorable prognosis similar to AML with inv(16)(p13.1q22) or t(8;21)(q22;q22.1)
- FLT3-ITD and GATA2 mutation status is of uncertain prognostic significance
- Reference: Eur J Haematol 2015;94:439
- 18 year old woman and 30 year old man (siblings) diagnosed with AML within 2 weeks of each other; father has a history of AML (N Engl J Med 2004;351:2403)
- 19 year old man presenting with leukocytosis, bruising and petechiae is diagnosed with AML with biallelic CEBPA (SH/EAHP Workshop 2017: Familial Acute Myeloid Leukemia with Germline CEBPA Mutation [Accessed 15 September 2021])
- 33 year old man with relapsed AML following stem cell transplant (ASH Image Bank: AML with Biallelic CEBPA Mutation [Accessed 15 September 2021])
- 36 year old woman at 35 weeks gestation presents with pancytopenia and is diagnosed with AML with biallelic CEBPA mutations (Blood Adv 2017;1:500)
- 52 year old woman with 18% blasts on peripheral blood smear with cytogenetic findings of biallelic CEBPA mutations (Annals of Case Reports ReDelve 2018;2018:1)
- Treated with similar induction and consolidation methods as other AMLs: 7+3 (cytarabine and anthracycline) and consolidation with cytarabine or azacitidine
- May benefit from stem cell transplant (cannot use family member with CEBPA mutation if germline)
- Relapsed patients have favorable prognosis as well (Blood 2013;122:1576)
- No distinctive morphologic features
- Typically, AML with or without maturation morphology
- Multilineage dysplasia is present in 26% of cases of de novo AML with mutated CEBPA without an associated adverse prognosis; does not change classification
- Reference: Haematologica 2017;102:529
Contributed by Kristin Karner, M.D.
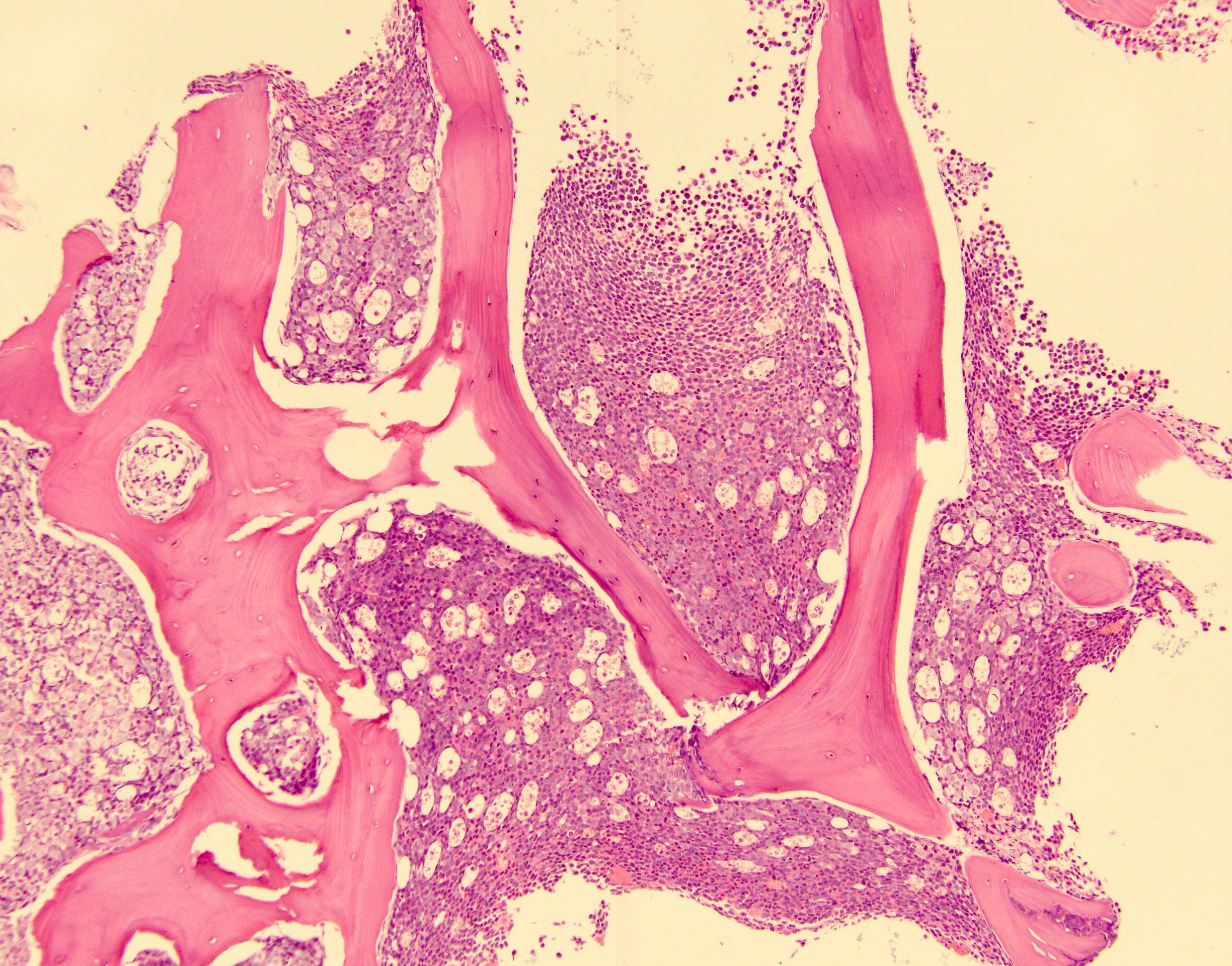
Blasts infiltrating the marrow
Blasts on aspirate smear
- Varying amount of peripherally circulating blasts with or without maturation
Contributed by Kristin Karner, M.D.
Blasts in peripheral blood
- Depending on the blast immunophenotype, immunohistochemistry for CD34 is typically positive
Contributed by Jessica Corean, M.D.
Typical immunophenotype
- Biallelic mutations of CEBPA required for diagnosis, disrupting the N and C terminus of CEBPA (J Clin Oncol 2010;28:570, Blood 2009;113:3088)
- In familial cases, typically 1 mutation is germline, the other is somatic
- CCAAT / enhancer binding protein alpha (CEBPA)
- Mutation types include mutations in the encoding gene and promoter hypermethylation (Haematologica 2011;96:384)
- Normal karyotype in 70% of cases
- FLT3-ITD seen in 5 - 9% of cases
- GATA2 seen in 39% of cases
- Subset of cases have abnormal karyotype, del(9q) is common but does not make a diagnosis of AML with myelodysplasia related changes
- Other reports of del(11q), which should be diagnosed as AML with myelodysplasia related changes
- Patients should be evaluated for familial / germline syndromes (Blood 2015;126:1214)
- Bone marrow aspirate, particle clot section and core biopsy:
- Acute myeloid leukemia with biallelic mutations of CEBPA (84.3% blasts by morphology)
- Microscopic description:
- Peripheral blood smear:
- Differential (100 cells)
- Neutrophils: 8%
- Lymphocytes: 7%
- Monocytes: 1%
- Eosinophils: 0%
- Basophils: 0%
- Metamyelocytes: 1%
- Blasts: 83%
- 1 nRBC/100 leukocytes
- Erythrocyte number: decreased
- Erythrocyte morphology: macrocytic, marked anisopoikilocytosis with ovalocytes, moderate polychromasia
- Leukocyte number: increased
- Leukocyte morphology: normal segmentation and granulation of neutrophils; blasts are small to intermediate in size with fine nuclear chromatin, nucleoli and scant basophilic cytoplasm
- Platelet number: decreased
- Platelet morphology: normal
- Differential (100 cells)
- Aspirate:
- Aspiration differential count (300 cells)
- Blasts: 84%
- Promyelocytes: 2%
- Myeloids: 6%
- Erythroids: 4%
- Lymphocytes: 4%
- Specimen quality: adequate
- Spicules: present
- Trilineage hematopoiesis: scant residual
- Erythroid maturation: decreased, full spectrum maturation
- Myeloid maturation: blast morphology is similar to that described in the peripheral blood
- Megakaryocyte morphology: 1 hyperlobated megakaryocyte identified in touch preparation
- Aspiration differential count (300 cells)
- Core biopsy:
- Bone trabeculae: normal
- Cellularity: 80%
- Trilineage hematopoiesis: scant residual
- Erythroid maturation and localization: markedly decreased
- Myeloid maturation and localization: sheets of blasts
- Megakaryocyte number: rare
- Megakaryocyte histotopography: rare hyperlobated megakaryocyte
- Peripheral blood smear:
- Flow cytometry studies:
- Interpretation: increased atypical CD34 positive myeloblasts (partial CD2, CD7 positive, CD11b positive, CD33 negative), representing approximately 89% of the leukocytes, consistent with acute myeloid leukemia
- Chromosome results: 46,XY[20]
- Myeloid mutation panel by NGS result:
- CEBPA c.332_339del, p.Ala111fs (NM_004364.4) variant frequency: 48.3%
- CEBPA c.759dup, p.Lys254fs (NM_004364.4) variant frequency: 47.3%
- TET2 c.2746C>T, p.Gln916* (NM_001127208.2) variant frequency: 48.9%
- TET2 c.4201G>T, p.Glu1401* (NM_001127208.2) variant frequency: 43.6%
- WT1 c.1410_1411ins34, p.Arg471fs (NM_024426.4) variant frequency: 1.0%
- Acute myeloid leukemia with myelodysplasia related changes:
- Present: history of myelodysplastic syndrome or myelodysplastic / myeloproliferative neoplasm; myelodysplastic syndrome related cytogenetic abnormality or multilineage dysplasia
- Absence of prior cytotoxic or radiation therapy for an unrelated disease and recurrent cytogenetic abnormality found in AML
- Acute myeloid leukemia with (other) recurrent genetic abnormalities:
- Through FISH, chromosome analysis and mutation analysis, AML defining recurrent genetic abnormalities can be identified
- This includes t(8;21), inv(16) or t(16;16), PML-RARA, t(9;11), t(6;9), inv(3) or t(3;3), t(1;22), BCR-ABL1, mutated NPM1, mutated RUNX1
- Therapy related myeloid neoplasms:
- Onset of 2 - 10 years post cytotoxic chemotherapy or radiation administered for a previous neoplastic or nonneoplastic disease
- Acute myeloid leukemia, NOS:
- With the absence of pertinent history and recurrent cytogenetic abnormalities, AML is classified as NOS
- The karyotype must not show any aberrations
- This has a poor prognosis
- This is typically a therapy related myeloid neoplasm
- This may be germline associated
Comment Here
Reference: AML with biallelic mutation of CEBPA
Which of the following is the most frequently encountered flow cytometry immunophenotype for AML with biallelic CEBPA mutation?
- Positive for CD33 and CD117, negative for CD34 and HLA-DR
- Positive for CD41, CD61 and CD42b
- Positive for CD33, CD13 (low expression), CD117, CD123, CD4, CD36, CD64, negative for HLA-DR
- Positive for CD13, CD7, CD34 and HLA-DR
Comment Here
Reference: AML with biallelic mutation of CEBPA
- Acute myeloid leukemia (AML) with BCR::ABL1 fusion is a de novo AML in which BCR::ABL1 is detected at initial diagnosis without evidence of chronic myeloid leukemia (CML)
- BCR::ABL1 fusion is detected at the time of diagnosis in a de novo AML with BCR::ABL1
- No CML morphologic features prior to or at diagnosis or posttherapy
- Morphologic spectrum, includes AML with minimal differentiation, without maturation or with monocytic differentiation
- Additional chromosomal aberrations are infrequently seen
- Cryptic deletions within immunoglobulin and T cell receptor accompanied by losses of the IKZF1 or CDNK2A / B genes
- Acute myeloid leukemia with t(9;22)(q34.1;q11.2)
- ICD-O: 9912/3 - acute myeloid leukemia with BCR::ABL1
- ICD-11: 2A60.0 & XH6FZ7 - acute myeloid leukemia with recurrent genetic abnormalities & acute myeloid leukemia with BCR::ABL1
- AML with BCR::ABL1 is a rare entity and accounts for 0.5 - 3% of all AML cases
- It is seen more commonly in males (Haematologica 1996;81:423, Leuk Res 2004;28:579, Am J Clin Pathol 2007;127:642)
- Peripheral blood and bone marrow are mostly involved
- Extramedullary site involvement is rare (Leuk Res 2008;32:1476)
- Philadelphia (Ph) chromosome and the chimeric BCR::ABL1 fusion gene result from t(9;22)(q34;q11) or its variants, which encodes a constitutively active tyrosine kinase with oncogenic properties (Br J Haematol 2013;161:541)
- 3 different breakpoint cluster regions in the BCR gene (M-bcr, m-bcr and µ-bcr) are reported: 8.5 kb hybrid mRNA (b2a2 or b3a2) encodes the 210 kDa protein (p210), 7.5 kb hybrid mRNA (e1a2) encodes the 190 kDa protein (p190) and 9 kb hybrid mRNA (c3a2) encodes the 230 kDa protein (p230) (Genomics 1995;27:67, Oncogene 2002;21:8652)
- ~70 - 80% cases of AML with BCR::ABL1 harbor p210 transcripts (Am J Clin Pathol 2007;127:642)
- Unknown
- Present with leukocytosis and anemia or thrombocytopenia
- Patients with AML with BCR::ABL1 in comparison to CML myeloid blast phase (CML MBP) have lower peripheral blood basophil percentage, absolute basophil count and are, less commonly, found to have splenomegaly (Am J Clin Pathol 2007;127:642)
- Essential diagnostic criteria
- Myeloid neoplasm with > 20% blasts expressing myeloid immunophenotype (based on immunohistochemistry or flow cytometry) in the peripheral blood or bone marrow
- Detection of BCR::ABL1 at the time of initial diagnosis
- Lack of CML features prior to or at diagnosis or after therapy
- Desirable diagnostic criteria
- Presence of t(9;22)(q34.1;q11.2) on conventional karyotyping
- Determination of the BCR::ABL1 transcript subtype and establishment of a baseline level of BCR::ABL1 transcript for monitoring treatment response
- Leukocytosis
- Anemia or thrombocytopenia
- FDG avid extramedullary lesions may be seen (Leuk Res 2008;32:1476)
- AML with BCR::ABL1 has an unfavorable prognosis with a median survival of < 9 months (Haematologica 1996;81:423, Am J Clin Pathol 2007;127:642)
- 28 year old woman presented with leukocytosis and was diagnosed with AML with e1a2 BCR::ABL1 fusion (Rev Bras Hematol Hemoter 2017;39:379)
- 68 year old man with a history of dementia presented with leukocytosis and was diagnosed with AML with BCR::ABL1 (Hemasphere 2020;4:e484)
- 77 year old woman who presented with pancytopenia underwent a bone marrow biopsy and was diagnosed with AML with BCR::ABL1 (Leuk Res Rep 2020;15:100233)
- 78 year old man with no prior history of CML presented with leukocytosis and was diagnosed with AML with BCR::ABL1 (BMC Cancer 2019;19:50)
- Standard AML induction chemotherapy (anthracycline / cytosine arabinoside containing regimen) response is poor (Leukemia 1998;12:1881)
- In addition, single agent tyrosine kinase inhibitors (TKI) alone are also not effective; however, a single case report indicated achieving complete molecular remission in a patient treated with dasatinib and chemotherapy (Ann Hematol 2016;95:1211, Medicine (Baltimore) 2018;97:e12949)
- Venetoclax and TKI combination regimens have shown good response in some studies (Acta Haematol 2020;143:567)
- Outcome of patients < 50 years of age with TKI pretreated BCR::ABL1 positive AML receiving allogeneic stem cell transplant is relatively favorable (Am J Hematol 2018;93:31)
- Survival status postallogeneic transplantation appears similar to intermediate risk AML, with one report demonstrating 3 year overall survival of 73% (Bone Marrow Transplant 2021;56:232)
- AML with BCR::ABL1 has a broad morphologic spectrum, which includes AML with minimal differentiation (FAB M0), without maturation (FAB M1) or with monocytic differentiation (FAB M5) (Haematologica 1996;81:423, Leuk Lymphoma 2013;54:138)
- AML with BCR::ABL1 demonstrates a marrow cellularity of 80%, which is lower than the 98% median marrow of CML MBP (Am J Clin Pathol 2007;127:642)
- In comparison to CML MBP, the bone marrow in AML with BCR::ABL1 shows lower myeloid/erythroid ratio at diagnosis (median: 2.0 versus 4.8) and a lower bone marrow aspirate basophil percentage (median: 0% versus 4.5%) (Am J Clin Pathol 2007;127:642)
- Dwarf megakaryocytes are rarely seen (Leuk Lymphoma 2013;54:138)
Contributed by Barina Aqil, M.D.
Increased blasts
Atypical mononuclear cells
Blast morphology
CD34 positive blasts
- Leukocytosis
- Variable anemia, with normocytic or macrocytic red blood cells
- Variable thrombocytopenia
- Left shifted granulocytes may be seen with circulating blasts, which may be ≥ 20% by definition
- Blasts may be myeloblasts or monoblasts / promonocytes
- CML MBP cases have a higher peripheral blood basophil percentage (median: 2.5%) than AML with BCR::ABL1 (median: 0%) and a higher peripheral blood absolute basophil count (Am J Clin Pathol 2007;127:642)
Contributed by Barina Aqil, M.D.
Circulating blasts
- CD34, HLA-DR and myeloid antigens (CD117, CD13 and CD33)
- Expression of CD7, CD19 and TdT may be seen (Leukemia 1998;12:1881, Am J Clin Pathol 2007;127:642)
- Myeloperoxidase may be positive or negative depending on the type of blasts
Contributed by Barina Aqil, M.D.
Dim CD45+ myeloid blasts
Partial CD34+ and CD117+
Partial CD13+
Dim CD33+
Partial HLA-DR+
Predominantly MPO-
- Fluorescence in situ hybridization (FISH) and chromosome analysis identified t(9;22)(q34.1;q11.2) that defines this subtype of AML
- Additional chromosomal aberrations, such as trisomy 8, an additional Ph chromosome, trisomy 19 and isochromosome 17q seen in CML MBP are infrequently seen in AML with BCR::ABL1 (Am J Clin Pathol 2007;127:642)
- Del(7q) / -7 abnormalities can be seen (Am J Clin Pathol 2007;127:642, Leuk Lymphoma 2013;54:138, Leukemia 1998;12:1881)
- Cryptic deletions within immunoglobulin and T cell receptor genes are seen and are frequently accompanied by losses of the IKZF1 or CDNK2A / B genes (BMC Genomics 2010;11:41, Br J Haematol 2013;161:541)
- RUNX1 mutation is common in AML with BCR::ABL1 and occurs in ~40% of cases (Genes Chromosomes Cancer 2021;60:426)
- Mutations of NPM1, FLT3 or DNMT3A are not commonly detected (Genes Chromosomes Cancer 2021;60:426)
- Other mutated genes include ASXL1, BCOR, IDH1 / IDH2 and SRSF2; each of these occur in 10 - 15% of cases (Leuk Lymphoma 2013;54:138, Leukemia 2017;31:2211, Genes Chromosomes Cancer 2021;60:426)
Contributed by Barina Aqil, M.D.

BCR::ABL1 fusion by FISH
- Peripheral blood, bone marrow aspirate and core biopsy:
- Acute myeloid leukemia with BCR::ABL1 (see comment)
- Comment: The cytogenetic and FISH studies showed t(3;9;22) resulting in rearrangement of BCR::ABL1. The overall findings are consistent with acute myeloid leukemia with BCR::ABL1. The peripheral blood neutrophils show no significant shift to immaturity and the marrow myeloid to erythroid ratio is not elevated. Given these findings, a de novo acute myeloid leukemia with BCR::ABL1 is favored over blast phase of chronic myeloid leukemia with BCR::ABL1.
- Peripheral blood: The peripheral blood smear shows pancytopenia. There is macrocytic anemia with anisopoikilocytosis including ovalocytes, teardrop forms and rare circulating nucleated red blood cells. Occasional circulating blasts are noted. The blasts are intermediate to large in size with high nuclear to cytoplasmic ratio and dispersed chromatin. Neutrophils show toxic changes but without significant shift to immaturity. Eosinophils are present but not increased. Platelets are decreased in number with unremarkable morphology.
- Bone marrow aspirate: The bone marrow aspirate smears show increased blasts showing similar morphology to those seen in the peripheral blood smear. The erythroid precursors are decreased but show progressive mature with unremarkable morphology. Megakaryocytes are present with unremarkable morphology.
- Bone marrow core biopsy: The bone marrow core biopsy shows hypercellular marrow (~80% cellular). Myeloid precursors are increased with a shift to immaturity including frequent blasts. The blasts are intermediate to large in size with dispersed chromatin, present scattered and in aggregates throughout the biopsy. Erythroid precursors are relatively decreased but show progressive maturation. Megakaryocytes are decreased with unremarkable morphology.
- Cytogenetic analysis reported an abnormal male karyotype: 46,XY,t(3;9;22)(p21; q34.1; q11.2)[3]/48, idem,+8,+12[17]. The t(3;9;22) is a 3 way variant translocation resulting in BCR::ABL1 fusion. Gains of chromosomes 8 and 12 in related abnormal clone 2 represent clonal evolution.
- FISH analysis was positive for the BCR::ABL1 fusion with a signal pattern consistent with a 3 way translocation (1F2R2G, 66%).
- Acute myeloid leukemia with other recurrent cytogenetic abnormality:
- Presence of another cytogenetic abnormality, which is defining for de novo AML subtype in addition to t(9;22)(q34.1;q11.2), takes precedence over the diagnosis of AML with BCR::ABL1 (Br J Haematol 2011;152:713, Leuk Lymphoma 2013;54:138)
- Therapy related AML or AML with myelodysplasia related changes (AML MRC):
- BCR::ABL1 may be acquired secondarily in AML transforming from previous myelodysplastic neoplasm or in relapsed / refractory AML after therapy that previously lacked BCR::ABL1 (Mod Pathol 2018;31:1141)
- Chronic myeloid leukemia (CML), myeloid blast phase:
- CML MBP cases have a higher peripheral blood basophil percentage (median: 2.5%) than AML with BCR::ABL1 (median: 0%), a higher peripheral blood absolute basophil count and increased frequency of splenomegaly
- Bone marrow in AML with BCR::ABL1 shows lower myeloid/erythroid ratio at diagnosis (median: 2.0 versus 4.8) (Am J Clin Pathol 2007;127:642)
- Additional chromosomal alterations that are typical of myeloid blast phase of CML are less commonly seen in AML with BCR::ABL1
- Although genes reported to be altered in the transformed stages (BP), including TP53, RB1, MYC, CDKN2A, NRAS, KRAS, RUNX1 (also known as AML1), TET2, CBL, ASXL1, IDH1 and IDH2, are similar to AML with BCR::ABL1, no IKZF1 deletions together with cryptic deletions within the immunoglobulin and T cell receptor genes are usually seen (Leuk Lymphoma 2013;54:138, Genes Chromosomes Cancer 2021;60:426)
- Mixed phenotype acute leukemia (MPAL) with BCR::ABL1:
- AML with BCR::ABL1 may aberrantly express lymphoid antigens (CD7 and CD19)
- Additional cytogenetic abnormalities are observed less in MPAL with BCR::ABL1 (13 - 30%) as compared to AML with BCR::ABL1 (50 - 60%) and CML MBP (70 - 80%) (Int J Hematol 2011;94:552, Eur J Haematol 2014;93:297, Am J Clin Pathol 2007;127:642, Leuk Lymphoma 2013;54:138, Br J Haematol 2013;161:541, Genes Chromosomes Cancer 2021;60:426)
- Cryptic deletions within immunoglobulin and T cell receptor genes with losses of the IKZF1 or CDNK2A / B genes are also detected in MPAL with BCR::ABL1 as in AML with BCR::ABL1
- Mutations in TP53, NRAS, IDH2, FLT3, PHF6, ASXL1, ETV6 and DNMT3A occur in higher frequency in MPAL unlike AML with BCR::ABL1; in contrast, NPM1 mutations are not found in MPAL, similar to AML with BCR::ABL1 (Exp Hematol 2016;44:740, Nat Commun 2018;9:2670, Blood Adv 2018;2:3526)
Which of the following is true of acute myeloid leukemia (AML) with BCR::ABL1?
- Detection of BCR::ABL1 can occur posttherapy
- It has no co-occurrence with t(8;21) / RUNX1::RUNX1T1
- It is a myeloid neoplasm with < 20% myeloid blasts
- There can be a prior history of myelodysplastic neoplasm or myeloproliferative / myelodysplastic neoplasm
Comment Here
Reference: AML with BCR::ABL1
- Cryptic deletions within the immunoglobulin and T cell receptor genes
- High frequency of splenomegaly
- Increased peripheral basophil percentage
- Presence of additional chromosomal aberrations (trisomy 8, trisomy 19 and isochromosome 17q)
Comment Here
Reference: AML with BCR::ABL1
- Not a specific WHO designated entity but has specific prognostic and treatment ramifications
- FLT3 (FMS-like tyrosine kinase 3) gene encodes a membrane bound receptor tyrosine kinase and is located on chromosome 13q12
- 2 mutation types in leukemia: internal tandem duplication (ITD), usually within the juxtamembrane domain, and a missense point mutation on the tyrosine kinase domain (TKD)
- Can be found in ~30% of cytogenetically normal acute myeloid leukemia (AML) (Br J Haematol 2017;179:530)
- Up to 40% of acute promyelocytic leukemia (APL) patients have FLT3 mutations
- ~20% of AML cases with t(9;11)(p21.3;q23.3) have point mutations in FLT3
- High percentage (~70%) of AML with t(6;9)(p23;q34.1) have FLT3 mutations (Leukemia 2006;20:1295)
- Higher prevalence in younger patients with AML (< 60 years old) (Ann Hematol 2017;96:1993)
- There are several new FLT3 targeted drugs that show promising results, which necessitate rapid analysis of the FLT3 status in patients with AML
- FMS-like tyrosine kinase 3
- More prevalent in patients < 60 years old
- Found in ~31% of patients with acute promyelocytic leukemia (22% FLT3-ITD mutation and 9% FLT3 D835) (Haematologica 2011;96:1470)
- Can be found in about 30% (28 - 34% FLT3-ITD and 11 - 14% FLT3-TKD) of cytogenetically normal AML (Br J Haematol 2017;179:530)
- White blood cell count is increased compared with AML without FLT3 mutation (Dis Markers 2013;35:581)
- FLT3 mutations are generally detected in the clinical laboratory by PCR and electrophoresis based product sizing as well as using next generation sequencing (NGS) platforms
- FLT3-ITD is an independent adverse prognostic indicator in AML
- Favorable outcome if mutated nucleophosmin (NPM1) without FLT3-ITD or with FLT3-ITDlow (allelic ratio < 0.5)
- Intermediate outcome if mutated NPM1 and FLT3-ITDhigh (allelic ratio ≥ 0.5) or wild type NPM1 without FLT3-ITD or with FLT3-ITDlow (without adverse risk genetic lesions)
- Adverse outcome if wild type NPM1 and FLT3-ITDhigh
- Prognostic impact of FLT3-TKD mutations is uncertain (Leukemia 2019;33:299)
- Acute promyelocytic leukemia with FLT3 mutations appear to represent a subset of APL patients who have a higher risk of relapse (Am J Hematol 2010;85:956)
- 4 and 9 month old boys, previously healthy, born at full term (Case Rep Oncol 2020;13:266)
- 26 year old man developed GVHD after treatment (Case Rep Oncol Med 2020;2020:4936846)
- 28 year old woman in the 26th week of gestation (BMC Pregnancy Childbirth 2019;19:394)
- 60 year old man diagnosed with FLT3-ITD positive AML in second remission (Clin Case Rep 2019;7:2579)
- 73 year old woman with FLT3 and NPM1 mutations (Case Rep Hematol 2016;2016:1259759)
- FLT3 targeted therapies including lestaurtinib, sorafenib, midostaurin, quizartinib, crenolanib and gilteritinib (Ther Adv Hematol 2019;10:2040620719827310)
- No specific microscopic findings associated with FLT3 mutation
- No correlation with a single, specific French-American-British (FAB) subtype was found (Blood 2002;100:59)
- When FLT3 occurs with other mutations or cytogenetic abnormalities, the characteristic microscopic features seen with those abnormalities are usually present
Contributed by Etan Marks, D.O.



Bone marrow core biopsy
CD34 bone marrow
Images hosted on other servers:

Peripheral smear of AML
- Similar to other acute myeloid leukemias with ≥ 20% myeloblasts or ≥ 20% promyelocytes
Contributed by Etan Marks, D.O.
Large irregular myeloblasts

Back to back myeloblasts
- Aberrant expression of CD2 has been associated with FLT3-ITD mutations in acute promyelocytic leukemia (Haematologica 2011;96:1470)
- CD135 is the receptor for the cytokine Flt3 ligand (Cytometry B Clin Cytom 2013;84:390)
- CD34 is often positive but can be negative
- CD117
- HLA-DR is often positive, unless it is found in a case of acute promyelocytic leukemia
- FLT3-ITD mutations consist of a duplicated coding sequence usually derived from the juxtamembrane domain inserted in tandem (ITD) (Haematologica 2011;96:1470, Cytometry B Clin Cytom 2013;84:390)
- These in frame insertion mutations range from 3 to > 200 bp in length and result in disruption of the autoinhibitory function
- FLT3-TKD point mutations occur in the activation loop of the kinase domain, most commonly at residue aspartate 835 (D835)
- FLT3 is usually reported as an addendum following the initial diagnosis of AML

FLT3 report
- Same as other AMLs
- Growth factor related increased myeloblasts
- Myelodysplastic syndrome with increased blasts
- Acute lymphoblastic leukemia
- 5q deleted AML
- Mutated FLT3 and wild type NPM1
- Wild type FLT3 and mutated NPM1
- Wild type NPM1 and biallelic mutated CEBPA
Which AML with cytogenetic abnormalities has the highest frequency of FLT3 mutations?
- Acute promyelocytic leukemia
- AML with 5q deletion
- AML with t(6;9)(p23;q34.1)
- AML with t(9;11)(p21.3;q23.3)
- Acute myeloid leukemia (AML) with t(8;16)(p11.2;p13.3) / KAT6A::CREBBP is a rare AML with recurrent cytogenetic abnormality
- Detection of KAT6A::CREBBP or t(8;16)(p11.2;p13.3) is required by cytogenetic or molecular studies for diagnosis
- Should not fulfill the diagnostic criteria for AML with defining genetic abnormalities, AML myelodysplasia related (AML MR), AML postcytotoxic therapy (AML pCT) or mixed phenotype acute leukemia (MPAL)
- Morphologically, blasts often show myelomonocytic or monocytic differentiation with some demonstrating erythrophagocytosis
- Blasts have characteristic immunophenotype as detected by flow cytometry
- Acute myeloid leukemia with t(8;16)(p11;p13)
- Incidence is ~0.2% of AMLs
- AML with t(8;16)(p11;p13) is a rare entity that may arise as a congenital, de novo or therapy related neoplasm, status postchemotherapy or radiation therapy (RT) (Leukemia 2008;22:1567, Leukemia 2009;23:934)
- Presents in all the age groups ranging from neonatal period in children to adults, with a median age of 60 years
- Peripheral blood, bone marrow and occasionally extramedullary disease (leukemia cutis) (Leukemia 2009;23:934, Ann Hematol 2019;98:1149, Leuk Res 2013;37:32)
- t(8;16) results in a fusion of the KAT6A gene located on chromosome 8p11 with the CREBBP gene located on chromosome 16p13 (Nat Genet 1996;14:33)
- Adult cases are usually therapy related whereas pediatric cases are often de novo (Leukemia 2009;23:934, Leuk Res 2013;37:32, Blood 2013;122:2704, Am J Clin Pathol 2022;157:701)
- Presents with bleeding tendency and disseminated intravascular coagulopathy (Leukemia 2009;23:934, Blood 2013;122:2704, Leuk Res 2013;37:32, Leukemia 2008;22:1567, Am J Clin Pathol 2022;157:701)
- Presence of ≥ 20% myeloid blasts in the bone marrow or peripheral blood per WHO 2022 but the blast requirement for the diagnosis is > 10% according to the 2022 International Consensus Classification (Virchows Arch 2023;482:27)
- Detection of KAT6A::CREBBP / t(8;16)(p11.2;p13.3)
- Not meeting the diagnostic criteria for AML with other defining genetic abnormalities, AML MR, AML pCT or MPAL
- Elevated white blood cell count with increased blast percentages (≥ 10% per ICC; ≥ 20% per WHO 5th edition) (Virchows Arch 2023;482:27, Am J Clin Pathol 2022;157:701)
- Disseminated intravascular coagulation
- Overall poor prognosis in adults
- Spontaneous remission has been reported in children diagnosed in the early neonatal period; some cases with spontaneous remission though subsequently relapse (Blood 2013;122:2704)
- Allogeneic stem cell transplantation has shown a significantly better outcome in adults with KAT6A::CREBBP without prior cytotoxic therapy or myeloid neoplasm (Br J Haematol 2021;192:832)
- Full term female infant was noted at birth to have numerous blue maculopapular skin lesions over the body and found to have occasional circulating blasts (Pediatr Blood Cancer 2017;64:e26450)
- 23 year old man presented with pain and night sweats; circulating blasts were noted along with thrombocytopenia (Blood 2016;128:314)
- Woman in mid 40s with a history of celiac disease presented with shortness of breath and was found to have 16% circulating blasts (BMJ Case Rep 2023;16:e253812)
- Initial studies had reported median overall survival (OS) of 4.7 - 8.5 months in AML with t(8;16) on chemotherapy (Leuk Res 2013;37:32)
- With the introduction of stem cell transplant (SCT), OS relatively improved to 18.2 months and was described even better in patients with de novo AML or noncomplex karyotype (Ann Hematol 2019;98:1149, Br J Haematol 2021;192:832)
- Cases with de novo AML and t(8;16) who received allo-SCT in first clinical remission (CR) achieved improved rates, suggesting that an early transplant in first CR is likely beneficial in these patients (Ann Hematol 2019;98:1149, Br J Haematol 2021;192:832)
- Venetoclax in combination with hypomethyling agents has shown a good response (Blood 2019;133:7)
- Morphologically, blasts often show myelomonocytic or monocytic differentiation (Clin Case Rep 2014;2:333)
- Blasts are positive for myeloperoxidase (MPO) and alpha naphthyl butyrate esterase (ANB)
- Erythrophagocytosis by the leukemic blasts is seen in some cases (Leukemia 2009;23:934, Clin Case Rep 2014;2:333)
Contributed by Barina Aqil, M.D.

Increased blasts
Increased blasts
Atypical mononuclear cells
Blast morphology
- Blasts are medium to large in size with irregular / folded nuclei, fine chromatin, prominent nucleoli and moderate to abundant cytoplasm
- Some blasts show cytoplasmic granules or vacuolization
- Blasts often show myelomonocytic or monocytic differentiation with prominent cytoplasmic granulation with or without Auer rods
Contributed by Barina Aqil, M.D.

Circulating blasts
Circulating blasts, MPO negative
Circulating blasts, ANB positive
- Distinct phenotypic profile characterized by
- Distinguishing from maturing myeloid elements is possible by the brighter expression of CD33 on the leukemic blasts (Am J Clin Pathol 2022;157:701)
Contributed by Barina Aqil, M.D.

Blast phenotype
- t(8;16) leads to a fusion product involving KAT6A and CREBBP (Br J Haematol 2020;190:133)
- Recurrent mutations are identified in ASXL1, FLT3, TP53, RUNX1, EZH2, SRSF2, CBL and TET2 (Br J Haematol 2021;192:832, Am J Clin Pathol 2022;157:701)
- Concomitant cytogenetic abnormalities and even complex karyotypes can be seen (Ann Hematol 2019;98:1149)
- Peripheral blood, bone marrow aspirate and left posterior iliac crest bone marrow core biopsy:
- Acute myeloid leukemia with KAT6A::CREBBP / t(8;16)(p11;p13) extensively involving a hypercellular bone marrow (see comment)
- Comment: The blasts are positive for MPO and CD68 indicating a myelomonocytic lineage. Some of the blasts show erythrophagocytosis.
- Peripheral blood smear:
- Peripheral blood smear shows normochromic normocytic anemia with mild anisopoikilocytosis. Neutrophils are decreased with unremarkable morphology. There are occasional medium to large blasts with irregular / folded nuclei, fine chromatin, prominent nucleoli and moderate to abundant granular cytoplasm. Rare Auer rods are seen. The platelets are markedly decreased.
- Bone marrow aspirate smear / touch preparation:
- Bone marrow aspirate smears and touch preparations are cellular and adequate for interpretation. The myeloid series show left shifted maturation with increased blasts with similar morphology as described in the peripheral blood smear. The erythroid precursors show progressive maturation. Megakaryocytes are decreased.
- Bone marrow core biopsy (decalcified) / particle clot:
- Unilateral bone marrow core biopsy is hypercellular for age (80 - 90% cellular) with extensive replacement by blasts that are medium to large in size with fine chromatin and pinpoint nucleoli. The erythroid precursors show progressive maturation. Megakaryocytes are decreased but show unremarkable morphology.
- AML with monocytic differentiation:
- Acute promyelocytic leukemia:
- Presentation with coagulopathy (DIC) and blasts with prominent cytoplasmic granulation can be similar to AML with KAT6A::CREBBP but phenotype and molecular studies are helpful in resolving this differential
- AML with KMT2A rearrangement:
- Extramedullary involvement is seen in 33% of adult patients
- Morphology is similar to AML with KAT6A::CREBPP with myelomonocytic or monocytic blasts
- Erythrophagocytosis is not seen
- Blasts show monocytic differentiation with expression of CD33, CD65, CD4, CD15, HLA-DR, lysozyme and variable CD34 and CD117
- Presence of KMT2A rearrangement with any one of the described > 80 fusion partners
- AML with NPM1 mutation:
- Morphology may be similar to AML with KAT6A:CREBPP with myelomonocytic or monocytic blasts
- Blast with cup shaped nuclear morphology is considered to be highly specific for this entity
- Multilineage dysplasia is commonly seen
- Immunophenotype can be quite variable; however, the majority of blasts predominantly are negative for CD34 but with expression of CD117 and CD33
- NPM1 mutation detection is an essential diagnostic criterion
- Concomitant FLT3 internal tandem duplication (ITD) mutation evaluation is important for risk assessment
- Patients with AML with NPM1 mutation along with FLT3 ITD mutation have poorer prognosis than AML with NPM1 mutation alone
What is the characteristic blast phenotype in acute leukemia with KAT6A::CREBBP, which is shown in the image above?
- CD13+, CD33+, CD11b+, CD14-, CD64-, CD34+
- CD34-, CD117-, CD13+, CD33+, CD11b+, CD64+
- CD34+, CD117-, CD13+, CD33+, CD11b-, CD64+
- CD34+, CD117+, CD13+, CD33+, CD64+
Comment Here
Reference: AML with KAT6A::CREBBP
A 42 year old man with no significant past medical history presented with fatigue and weight loss for 2 months. Laboratory workup showed leukocytosis (WBC 23.0 K/uL), anemia (8.0 g/dL) and occasional circulating blasts in peripheral blood. The bone marrow biopsy showed > 20% myeloid blasts with coexpression of CD64 and partial CD11b. This finding (see image) is characteristically seen in ~75% cases of which disease?
- AML with CEBPA
- AML with DEK::NUP214
- AML with KAT6A::CREBBP
- AML with NPM1
Comment Here
Reference: AML with KAT6A::CREBBP
- Acute myeloid leukemia (AML) with CBFA2T3(RUNX1T3)::GLIS2 is a rare subtype of pediatric AML
- Pediatric acute myeloid leukemia with dismal prognosis
- CBFA2T3::GLIS2 is the most common alteration seen in non-Down syndrome acute megakaryoblastic leukemia (non-DS AMKL)
- Often expressing the unique RAM phenotype by flow cytometry with bright CD56 and lack of HLA-DR and CD38 expression
- Exclusively seen in pediatric population, especially < 5 years of age with predominance in infants (Cancer Cell 2012;22:683, Blood 2013;121:3469, J Hematol Oncol 2017;10:26)
- Comprises almost 30% of pediatric non-DS AMKL (Nat Genet 2017;49:451, Cancer Cell 2012;22:683)
- Peripheral blood, bone marrow, lymph node, 1 reported case with a paraspinal mass (Cancer Cell 2012;22:683, EJHaem 2023;4:765)
- Cryptic inversion on chromosome 16 [inv(16)(p13.3q24.3)] results in the joining of CBFA2T3, a member of the ETO family of nuclear corepressors, to GLIS2, a member of the GLI family of transcription factors (Cancer Cell 2012;22:683)
- CBFA2T3 and GLIS2 genes are both localized, in inverted orientation, on each arm of chromosome 16 close to the telomeres, in 16q24.3 and 16p13.3, respectively (Cancer Cell 2012;22:683)
- CBFA2T3::GLIS2 fusion transcript leads to upregulation of BMP2 (bone morphogenetic proteins) and GATA3, a downstream target of Hedgehog signaling as well as JAK-STAT pathway (J Exp Med 2012;209:2017, Development 2004;131:1165, Cancer Cell 2012;22:683)
- Not known
- Median age of diagnosis is 1.26 - 1.5 years of age (Leukemia 2016;30:2077, Blood 2016;127:3424)
- Case reports have described leukocytosis and extensive lymphadenopathy (Leuk Res Rep 2021;17:100287, EJHaem 2023;4:765)
- Extramedullary involvement is more frequent in patients expressing the CBFA2T3::GLIS2 chimeric gene (25%) compared to the frequency reported for pediatric AML in general (Blood 2013;121:3469, Blood 2013;122:170)
- It is seen in non-Down syndrome patients
- Diagnostic criteria per 2022, World Health Organization (WHO) Classification of Tumors requires ≥ 20% myeloid blasts in the peripheral blood or bone marrow and detection of CBFA2T3::GLIS2 (Leukemia 2022;36:1703)
- International Consensus Classification (ICC), 2022 requires ≥ 10% myeloid blasts in the bone marrow or peripheral blood (Blood 2022;140:1200)
- Must exclude the diagnostic criteria for AML with other defining genetic abnormalities, AML MR (AML myelodysplasia related), AML pCT (postcytotoxic therapy) or MPAL (mixed phenotype acute leukemia)
- This entity is recognize by the WHO 5th edition under AML with other defined genetic alterations but is not a specific entity within the ICC
- Leukocytosis with increased blasts (≥ 10% per ICC; ≥ 20% per WHO 5th edition) (Leukemia 2022;36:1703, Blood 2022;140:1200)
- No differences are reported with reference to white blood cell count (WBC), bone marrow blast percentage, platelet count or hemoglobin levels as compared to other AML subtypes (Leukemia 2016;30:2077)
- CBFA2T3::GLIS2 positive AML patients tend to have a higher percentage of bone marrow blasts at diagnosis compared to fusion negative patients (Blood 2016;127:3424, Genes Chromosomes Cancer 2017;56:394)
- CBFA2T3::GLIS2 AML has dismal outcome (Nat Genet 2017;49:451)
- 5 year overall survival is ~30% (Cancer Cell 2012;22:683)
- 5 month old girl with leukocytosis, eosinophilia and paraspinal mass (EJHaem 2023;4:765)
- 12 month old girl presented with bruising and found to have marked leukocytosis and skin nodules (Cold Spring Harb Mol Case Stud 2022;8:a006220)
- 1 year old girl presented with fever, cytopenias and hepatosplenomegaly (Leuk Lymphoma 2023;64:2042)
- 2 year old monozygotic twins presented simultaneously with RAM AML (Pediatr Transplant 2018;22:e13291)
- 5 year old boy with extensive mesenteric and retroperitoneal lymphadenopathy (Leuk Res Rep 2021;17:100287)
- Patients may benefit from intensive chemotherapy followed by allogeneic stem cell transplant as postremission intensification strategy (Pediatr Transplant 2018;22:e13291)
- Overexpression of the folate receptor 1 (FOLR1) gene has shown great result with targeted therapy (J Clin Invest 2022;132:e157101)
- Navitoclax or DT2216 treatment in combination with low dose cytarabine has been found effective in murine models (Blood Adv 2024;8:112)
- No specific morphological features have been found associated with this fusion gene (Blood 2013;121:3469)
- In AMKL CBFA2T3::GLIS2 positive patients, megakaryoblasts are reported as the predominant component of the blast population in bone marrow or peripheral blood (Br J Haematol 2019;184:337)
- Megakaryoblasts are large cells with abundant cytoplasm and prominent nucleoli; they often display cytoplasmic blebs or pseudopods
- In one of the pediatric studies, 50% (N = 10) of the fusion positive patients had a non-FAB M7 subtype, others were distributed as follows: M5 (15%), M0 (15%), M1 (10%), M2 (5%), M4 (5%) (Blood 2013;121:3469)
Contributed by Barina Aqil, M.D.
Atypical mononuclear cells

Blast morphology

Increased blasts
Blasts
- Circulating blasts as previously described
Contributed by Barina Aqil, M.D.

Circulating blasts
- RAM phenotype was named after the patient's initials in original study (Leukemia 2016;30:2077)
- Bright CD56 expression (at minimum 2 log10 units greater than normal myeloid progenitors)
- Dim to negative expression of CD45, CD38 and lack of HLA-DR
- Reported cases have shown aberrant expression of T and B cell markers, such as cytoplasmic CD3 and CD19 (EJHaem 2023;4:765, Leuk Lymphoma 2023;64:462)
- Concomitant cytogenetic abnormalities are rare in patients with CBFA2T3::GLIS2 (Br J Haematol 2019;184:337)
- In cases of abnormal karyotype, complex aberrations, trisomy 21 and hyperdiploidy have been noted (Cancer Cell 2012;22:683, Nat Genet 2017;49:451, Genes Chromosomes Cancer 2017;56:394)
- Somatic mutations are significantly lower than other subgroups of AMKL (Cancer Cell 2012;22:683, Nat Genet 2017;49:451)
- GATA1, KIT and FLT3 mutations are less frequently noted in CBFA2T3::GLIS2 positive than in negative cases (Br J Haematol 2019;184:337)
- RAS and JAK-STAT pathway mutations are reported more often (Cancer Cell 2012;22:683, Nat Genet 2017;49:451)
- Peripheral blood, bone marrow aspirate and left posterior iliac crest bone marrow core biopsy:
- Acute myeloid leukemia with CBFA2T3::GLIS2 extensively involving a hypercellular (virtually 100%) bone marrow (see comment)
- Flow cytometric immunophenotyping of the bone marrow aspirate reveals a blast population (~90% of cellular events) with a dim to negative CD45 expression that is positive for CD34, bright positive for CD56 and negative for CD38 and HLA-DR.
- Comment: The overall morphologic, immunophenotypic and genetic findings are consistent with a diagnosis of acute myeloid leukemia with CBFA2T3::GLIS2.
- Peripheral blood smear: The peripheral blood smear shows predominantly circulating blasts (90%) that are large in size with abundant cytoplasm and frequent prominent nucleoli with occasional cytoplasmic blebs. Normocytic normochromic anemia is present with rare nucleated red blood cells. There is absolute neutropenia and lymphopenia. Thrombocytopenia with rare large platelets.
- Bone marrow aspirate smear: The majority of the cells present are blasts with similar morphology as seen in the peripheral blood. Background hematopoiesis is markedly decreased. Rare megakaryocytes are seen.
- Bone marrow core biopsy (decalcified) / particle clot: The bone marrow is markedly hypercellular for age (~100%) and is composed almost entirely of blasts. Very rare myeloid and erythroid precursors are present. The particle clot shows multiple bone marrow particles composed predominantly of blasts.
- AML with minimal differentiation (M0):
- AML with RUNX1::RUNX1T1:
- Mixed phenotype acute leukemia, T / myeloid:
- Early T precursor lymphoblastic leukemia / lymphoma:
- Acute megakaryoblastic leukemia (M7):
- Myeloid proliferations associated with Down syndrome:
- Transient abnormal myelopoiesis (TAM) and myeloid leukemia associated with Down syndrome (ML-DS) are two clonal conditions seen in patients with trisomy 21
- TAM presents usually in the first 7 days of life while ML-DS onset is 1.6 years (Br J Haematol 2018;182:200, J Clin Oncol 2014;32:3021, Blood 2017;129:3304)
- Blast morphology ranges from undifferentiated myeloid blasts to megakaryoblasts
- Blasts express CD117 with variable expression of CD34, CD36, CD41/42b or CD235a (glycophorin A)
- Exon 2/3 somatic GATA1 mutations are seen (Blood 2011;118:2222, Nat Genet 2013;45:1293, Cancer Cell 2019;36:123)
- In ML-DS additional mutations are also identified, such as cohesin complex, EZH2, KANSL1 or JAK3 (Nat Genet 2013;45:1293, Cancer Cell 2019;36:123)
What characteristic phenotype panel is helpful in diagnosing acute myeloid leukemia with CBFA2T3::GLIS2?
- CD34, CD56, CD38, HLA-DR
- CD34, CD117, CD13, CD33
- CD34, CD117, CD41, CD61
- CD34, TdT, CD19, CD79a
Comment Here
Reference: AML with RUNX1T3(CBFA2T3)::GLIS2
- Acute myeloid leukemia (AML) with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22); CBFB-MYH11 is a subtype of AML with recurrent genetic abnormalities characterized by myelomonocytic differentiation and presence of abnormal eosinophils
- Inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) results in the formation of an abnormal core binding factor beta subunit / myosin heavy chain 11 (CBFB-MYH11) fusion gene
- Most cases show myelomonocytic differentiation with presence of abnormal eosinophils
- Diagnosis of AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) can be made even when the blast percentage is less than 20%
- Prognosis is good compared with other AMLs
- Acute myelomonocytic leukemia with abnormal eosinophils
- French American British (FAB) M4Eo
- Acute myeloid leukemia with CBFB-MYH11
- 7% of AMLs in the pediatric age group (J Clin Oncol 2010;28:2674)
- < 5% of AMLs in older adults (Blood 2006;108:3280)
- Most cases present with involvement of the blood and bone marrow
- Occasional cases present as myeloid sarcoma (Am J Clin Pathol 2020;153:333)
- Both inv(16)(p13.1;q22) and t(16;16)(p13.1;q22) result in a core binding factor beta subunit / myosin heavy chain 11 (CBFB-MYH11) fusion gene encoding an abnormal CBFB-MYH11 fusion protein (Semin Hematol 2015;52:215)
- CBFB-MYH11 fusion protein causes aberrant self renewal and suppresses differentiation
- Additional mutations promote leukemogenesis
- Fatigue resulting from anemia
- Easy bruising from thrombocytopenia
- Mass effects of myeloid sarcoma, such as intestinal obstruction
- Higher white blood cell count at diagnosis of AML with inv(16)(p.13.1q22) and t(16;16)(p.13.1;q22) compared with other AMLs (J Clin Oncol 2005;23:5705)
- Diagnosis is based on a combination of morphology, flow cytometry, conventional cytogenetics, real time PCR and FISH analysis
- Anemia and thrombocytopenia
- Overall prognosis is good compared to other AMLs (Blood 2003;102:462)
- KIT and FLT3 mutations are associated with worse prognosis
- Trisomy 22 is associated with a more favorable outcome
- High levels of minimal residual disease after induction are associated with worse outcomes (Am Soc Clin Oncol Educ Book 2018;38:555)
- 30 year old man presented to emergency department with 2 weeks history of fever, abdominal pain and fatigue (Clin Med Insights Blood Disord 2017;10:1179545X17700858)
- 30 year old woman presented with a 2 week history of fever, cough with dyspnea and chest pain, generalized weakness and erythematous papules over the dorsum of her right hand and bleeding per vaginum (Indian J Pathol Microbiol 2016;59:104)
- 32 year old woman was first seen for gingivitis, hypertrophic gingiva, metrorrhagia and purpura (Hematol Oncol 2017;35:385)
- 47 year old man presented with intermittent low grade fever and progressive fatigue (Mol Cytogenet 2020;13:4)
- Induction chemotherapy with 3 days of anthracycline and 7 days of cytarabine (7+3) (Am Soc Clin Oncol Educ Book 2018;38:555)
- Cytarabine or HDAC inhibitors are used for consolidation therapy
- Gemtuzumab ozogamicin (toxin conjugated monoclonal antibody to CD33) may improve outcomes when combined with standard chemotherapy
- Hypercellular bone marrow core biopsy and aspirate clot sections with presence of immature cells; increased eosinophilic cells can be seen
Contributed by Ameet R. Kini, M.D., Ph.D. and Maryam F. Raouf, M.D.
Increased blasts and promonocytes
Abnormal eosinophilic cell
Hypercellular bone marrow
Images hosted on other servers:
AML with abnormal eosinophils (arrow)
Abnormal eosinophils with prominent purple granules
Abnormal eosinophils cluster in patient with AMML
Abnormal immature eosinophil found in bone marrow
Abnormal eosinophil with basophilic granules
Images hosted on other servers:
AML with inv(16)
- Increased blasts showing nuclei with fine chromatin and high nuclear to cytoplasmic ratio
- Some blasts display Auer rods
- Blasts are usually > 20% but the diagnosis is made even when blasts are < 20%, provided inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) is seen
- Increased eosinophils at all stages of maturation, with the earlier stages displaying dual granulation (Haematologica 2002;87:886)
- Increased immature monocytic cells (promonocytes)
- Presence of blasts and immature monocytic cells
- Eosinophils are not usually increased (Blood 1986;68:1242)
- Abnormal eosinophils are faintly positive for naphthol AS-D chloroacetate esterase
- Myeloblasts are positive for myeloperoxidase
- Monocytic cells are positive for alpha naphthyl butyrate esterase (nonspecific esterase)
- Immunohistochemical stains show positivity for CD34 and CD117 in the blasts
- Negative for nonhematopoietic markers and most lymphoid markers
Contributed by Ameet R. Kini, M.D., Ph.D. and Maryam F. Raouf, M.D.
Blasts with dim CD45
Blasts with CD34, CD15
Increased blasts with HLA-DR, CD33
Blasts with CD13, CD117
- Conventional cytogenetics shows presence of inv(16)(p13.1;q22) or t(16;16)(p13.1;q22)
- FISH analysis may be required to detect cryptic cases and shows the CBFB-MYH11 fusion gene (Genes Chromosomes Cancer 1998;22:87)
- In rare cases, real time PCR can detect the CBFB-MYH11 transcript when cytogenetics and FISH analyses are negative
- Real time PCR is used to assess minimal / measurable residual disease (MRD)
Contributed by Ameet R. Kini, M.D., Ph.D. and Maryam F. Raouf, M.D.
Inversion of chromosome 16

CBFB FISH
AML diagnosis
Targeted therapy for inv(16)
AML diagnosis and treatment
MRD in AML
- Bone marrow, right posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint hypercellular bone marrow (95%) with involvement by acute myeloid leukemia with inv(16)(p13.1;q22) (see comment)
- Comment: Bone marrow core biopsy is hypercellular for age (95%) Immature cells are increased. Erythroid precursors are decreased. Mature myeloid cells are decreased. Megakaryocytes are adequate and appear normal in morphology.
- Aspirate clot section is adequate and shows features similar to the core biopsy.
- Bone marrow aspirate smear is adequate. Blasts are increased (30%) and show a high nuclear to cytoplasmic ratio with fine chromatin. Immature monocytic cells (promonocytes) are also increased. Scattered eosinophils and eosinophil precursors are seen throughout the aspirate smear. Some of the eosinophilic cells show dual granulation. Megakaryocytes are adequate in number and show normal morphology. Cytochemical stains show that many of the blasts are positive for myeloperoxidase. Alpha naphthyl butyrate esterase (nonspecific esterase) is positive in the immature monocytic cells.
- Touch preparation findings are similar to the core biopsy.
- Flow cytometry shows a large abnormal myeloid blast population with expression of CD34, CD15 (subset), CD33, CD13, HLA-DR and CD117. There is also an immature monocytic population showing characteristic expression of CD64, CD11b and CD11c, and possible absence of CD14. Gating on the lymphocytes shows no evidence of a monoclonal B cell population or T cell abnormality based on the markers assayed.
- Cytogenetic analysis shows presence of a pericentric inversion of chromosome 16. FISH analysis using a dual color break apart probe shows rearrangement of CBFB, consistent with inv(16)(p13.1;q22).
- Chronic eosinophilic leukemia:
- Blasts are < 20%
- inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) is absent
- Myeloid / lymphoid neoplasms with eosinophilia and gene rearrangement:
- Acute myeloid leukemia with t(8:21)(q22;q22.1); RUNX1-RUNX1T1:
- Eosinophils do not usually display morphologic or cytochemical abnormalities
- inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) is absent
An otherwise healthy 15 year old child presents to clinic with fatigue. The patient’s mother states her child has decreased energy and seems to bruise easily. CBC shows anemia and thrombocytopenia. The bone marrow biopsy is hypercellular. The bone marrow aspirate smear (figure) shows the presence of blasts and increased abnormal eosinophils. Cytochemical stains show a dual population of blasts with a subset expressing myeloperoxidase and another subset nonspecific esterase. What is the most likely diagnosis?
- Acute lymphoblastic leukemia
- Acute megakaryoblastic leukemia
- Acute myeloid leukemia with inv(16)
- Acute promyelocytic leukemia with t(15;17)
- Hypereosinophilic syndrome
Comment Here
Reference: AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22)
- BCR-ABL1
- CBFB-MYH11
- KMT2A-MLLT3
- PML-RARA
- RUNX1-RUNX1T1
- A distinct acute myeloid leukemia (AML) subcategory recognized by presence of the pathognomonic cytogenetic abnormality inv(3)(q21q26.2) or t(3;3)(q21;q26.2) [inv(3) / t(3;3)] (Blood 2016;127:2391)
- Recognized as a separate entity in the acute myeloid leukemia (AML) category of the 2008 WHO classification and updated in the 2016 revision (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue, 4th Edition, 2008, Blood 2016;127:2391)
- May present as de novo disease or evolve from preexisting myelodysplastic syndrome (Cancer Genet 2019;230:28)
- Diagnosis requires ≥ 20% blasts (in peripheral blood or bone marrow) and is often clinically characterized by anemia, normal to elevated platelet counts and bone marrow hyperplasia with multilineage dysplasia of nonblast cells, particularly dysmegakaryocytopoiesis (Cancer Genet 2019;230:28)
- Aggressive form of AML with very poor prognosis and poor response to conventional induction chemotherapy (Cancer Genet 2019;230:28)
- Aggressive form of AML with very poor prognosis and poor response to conventional induction chemotherapy (Cancer Genet 2019;230:28)
- Diagnosed by presence of the pathognomonic cytogenetic abnormality inv(3)(q21q26.2) or t(3;3)(q21;q26.2) [inv(3) / t(3;3)] (Blood 2016;127:2391)
- End result is juxtaposition of the distal GATA2 enhancer with the MECOM (EVI1) proto-oncogene (Blood 2016;127:2391)
- May present as de novo disease or evolve from preexisting myelodysplastic syndrome (Cancer Genet 2019;230:28)
- AML with inv(3)
- Acute myeloid leukemia with GATA2, MECOM
- AML with RPN1 / EVI1
- 3q21q26 syndrome (Hematology 2015;20:435)
- AML with RPN1-MECOM
- ICD-0: 9869/3 - acute myeloid leukemia with inv(3)(q21;q26.2) or t(13.3)(q21;q26.2); RPN1-EVI1 (NIH: Acute Myeloid Leukemia with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM [Accessed 16 July 2020])
- ICD-10: C92.0 - acute myeloblastic leukemia
- More frequent in people younger than 60 years (median age of 50 years) (Hematology 2015;20:435)
- Slight male predominance (54%) (Hematology 2015;20:435)
- 1 - 2% of all forms of AML (Cancer Genet 2019;230:28)
- Bone marrow
- Peripheral blood
- See Etiology
- Recurrent cytogenetic abnormality, inv(3)(q21q26.2) / t(3;3)(q21;q26.2), causes a juxtaposition of the distal GATA2 enhancer with MECOM (EVI1) (Blood 2016;127:2391)
- These rearrangements (not to be thought of as a fusion) result in the proto-oncogene MECOM (EVI1) at 3q26 (Blood 2016;127:2391, Cancer Cell 2014;25:415)
- Simultaneous GATA2 haploinsufficiency also results (Blood 2016;127:2391)
- Core mRNA splicing factor SF3B1 and RAS / RTK signaling pathways have been found to be important in the pathogenesis of this AML subtype (Blood 2019;134:917, Blood 2015;125:133)
- Secondary karyotypic abnormalities, including monosomy 7 (most common), complex karyotype and 5q deletion, are commonly present (Cancer Genet 2019;230:28, Hematology 2015;20:435, J Clin Oncol 2010;28:3890)
- Presence of at least 20% blasts in peripheral blood or bone marrow
- Often presents with anemia
- Elevated white blood cell count (sometimes low)
- Normal or elevated platelet count
- Association with central diabetes insipidus (Hematology 2015;20:435)
- Elevated LDH
- Requires enumeration of 20% blasts on peripheral blood smear or marrow aspirate differential count(s)
- Flow cytometry or immunohistochemical stains may assist in blast phenotyping
- Juxtaposition of GATA2 and MECOM, inv(3)(q21q26.2) or t(3;3)(q21;q26.2) [inv(3) / t(3;3)] can be detected by classic cytogenetics, FISH and other molecular / genetic studies including chromosome microarray, next generation sequencing, RT-PCR, etc.
- CBC with anemia
- Leukocytosis (sometimes leukopenia) with 20% blasts
- Normal or elevated platelet count
- Elevated LDH
- Flow cytometry analysis reveals increased dim CD45 events (blasts)
- inv(3)(q21q26.2) or t(3;3)(q21;q26.2) [inv(3) / t(3;3)] finding in AML itself represents a very poor prognosis and predicts poor response to conventional induction chemotherapy (Hematology 2015;20:435, J Clin Oncol 2010;28:3890)
- Age greater than 60 years at diagnosis indicates poor prognosis (Hematology 2015;20:435)
- High WBC count at diagnosis confers poor prognosis (Hematology 2015;20:435)
- Presence of monosomy 7 has been reported to be an indicator of poor prognosis (J Clin Oncol 2010;28:3890)
- However, this has not proven to be a consistent finding in other studies (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Young woman with AML and concurrent central diabetes insipidus in the presence of inversion(3)(q21q26) (Blood Cells Mol Dis 2019;76:45)
- 25 year old woman with germline GATA2 mutation and acquired NRAS Q61K mutation who presents with rapid progression to AML (Leuk Res Rep 2019;12:100176)
- 77 year old man with a variant of AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) (MECOM) rearrangement (Mol Cytogenet 2015;8:68)
- Case series of AML with double inv(3)(q21q26.2) (J Clin Oncol 2010;28:3890)
- This subtype of AML is well known to show poor response to standard induction chemotherapy (such as the 7 + 3 regimen consisting of cytarabine and an anthracycline), with remission rates of only 10 - 30% and overall survival of < 10% at 8 - 10 months (Hematology 2015;20:435, J Clin Oncol 2010;28:3890, Clin Lymphoma Myeloma Leuk 2020;20:24)
- Combination of lenalidomide and a hypomethylating agent (azacitidine of decitabine), has shown promise with improved overall response rates, particularly when used as first line therapy (J Clin Oncol 2010;28:3890)
- Stem cell transplantation does not appear to conclusively improve overall survival in this subtype of AML (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- 20% blasts in the peripheral blood with or without dysplastic features
- Hypercellular (often) bone marrow with > 20% blasts (Int J Lab Hematol 2019;41:380)
- Multilineage dysplasia with variable bone marrow fibrosis (usually little or no reticulin fibrosis) (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Prominence of mono or bilobed megakaryocytes in the bone marrow (Hematology 2015;20:435)
- Micromegakaryocytes have been described (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Binuclear granulocytes (pseudo-Pelger-Hüet neutrophils) (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Morphological features often resemble AML without maturation and acute myelomonocytic leukemia (Blood 2016;127:2391)
- However, a study of Asian patients found acute monoblastic and monocytic leukemia in nearly half of the patients (Int J Lab Hematol 2019;41:380)
Contributed by Alexa J. Siddon, M.D.
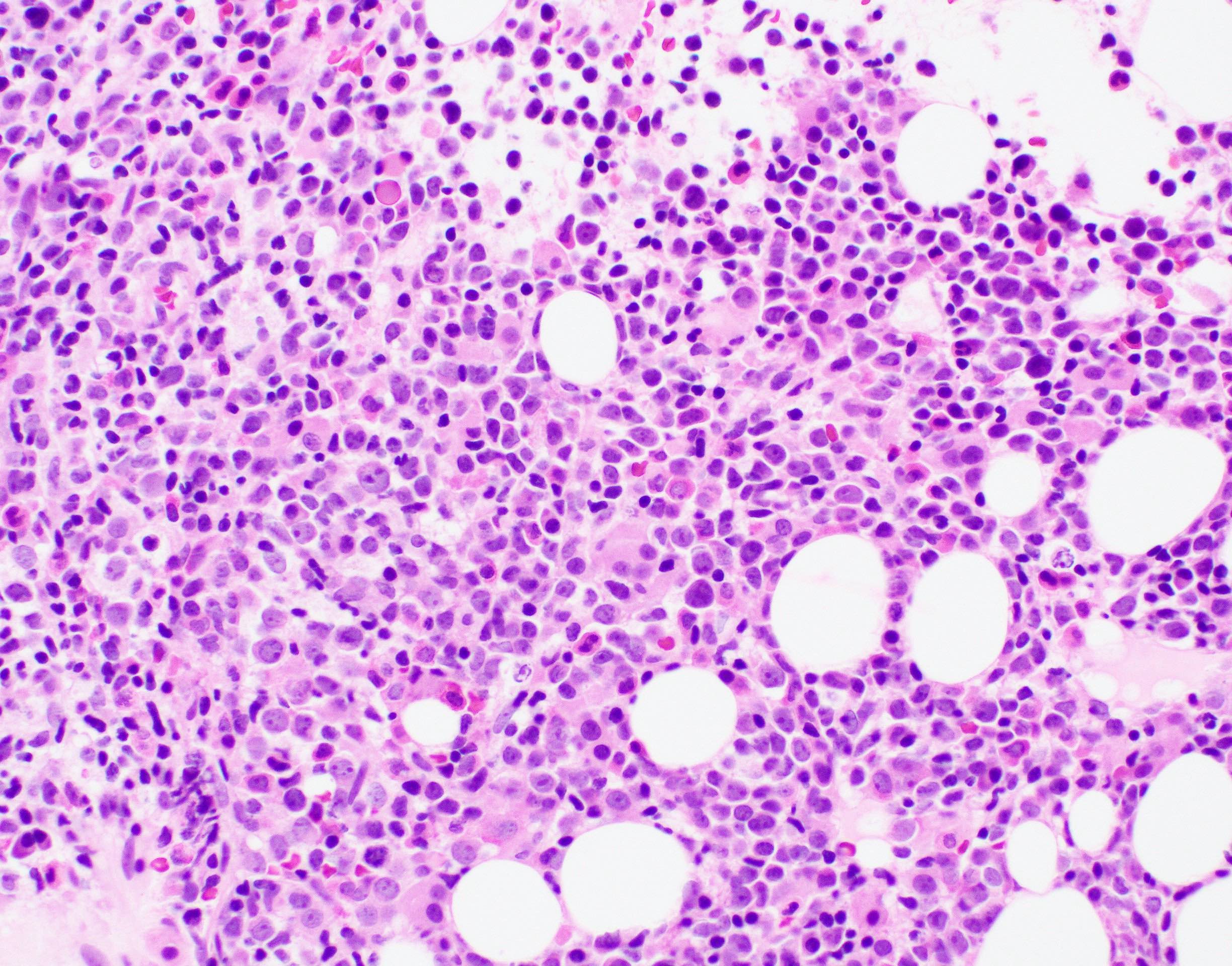
Hypercellular marrow with increased blasts
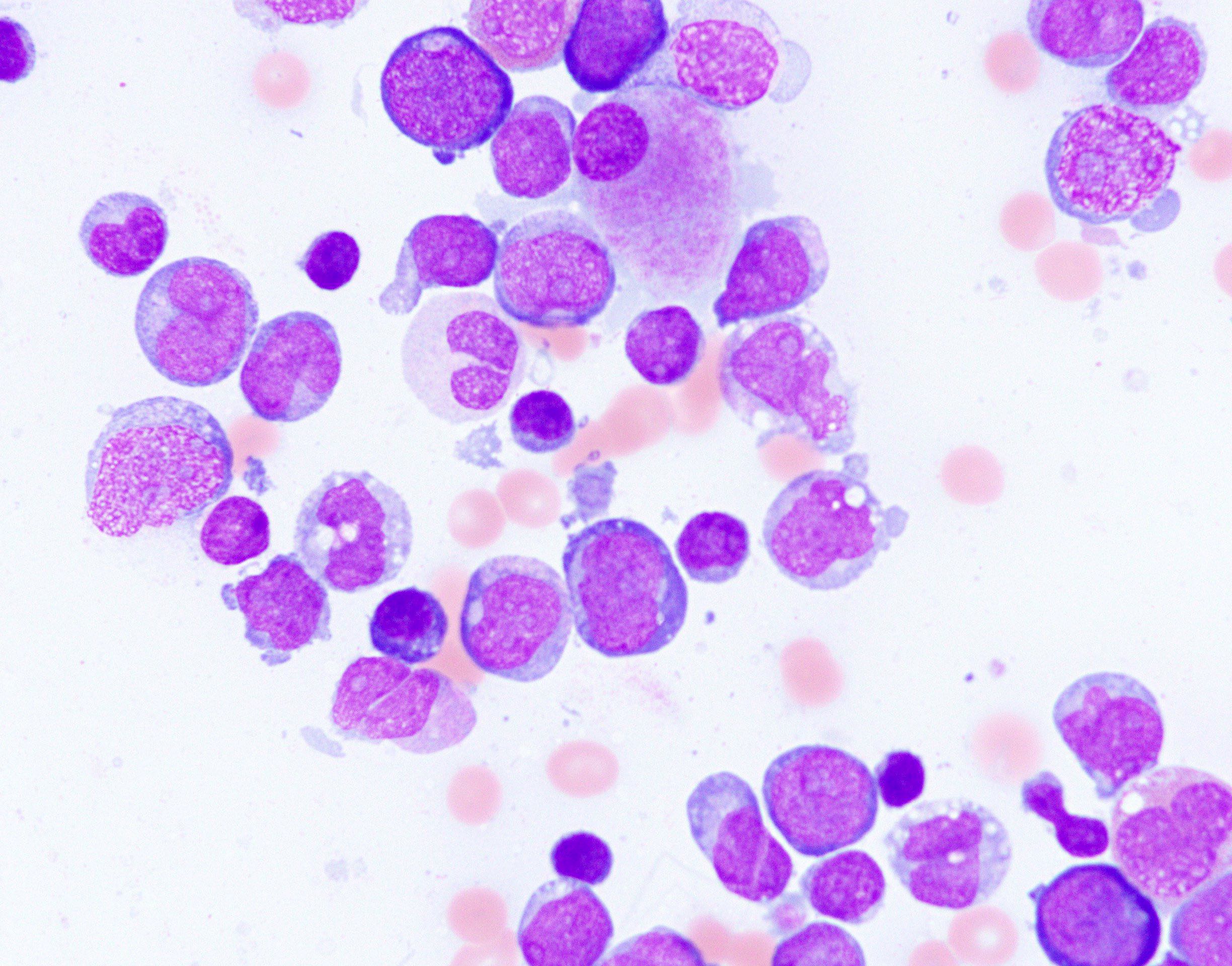
Aspirate with hypolobated megakaryocyte
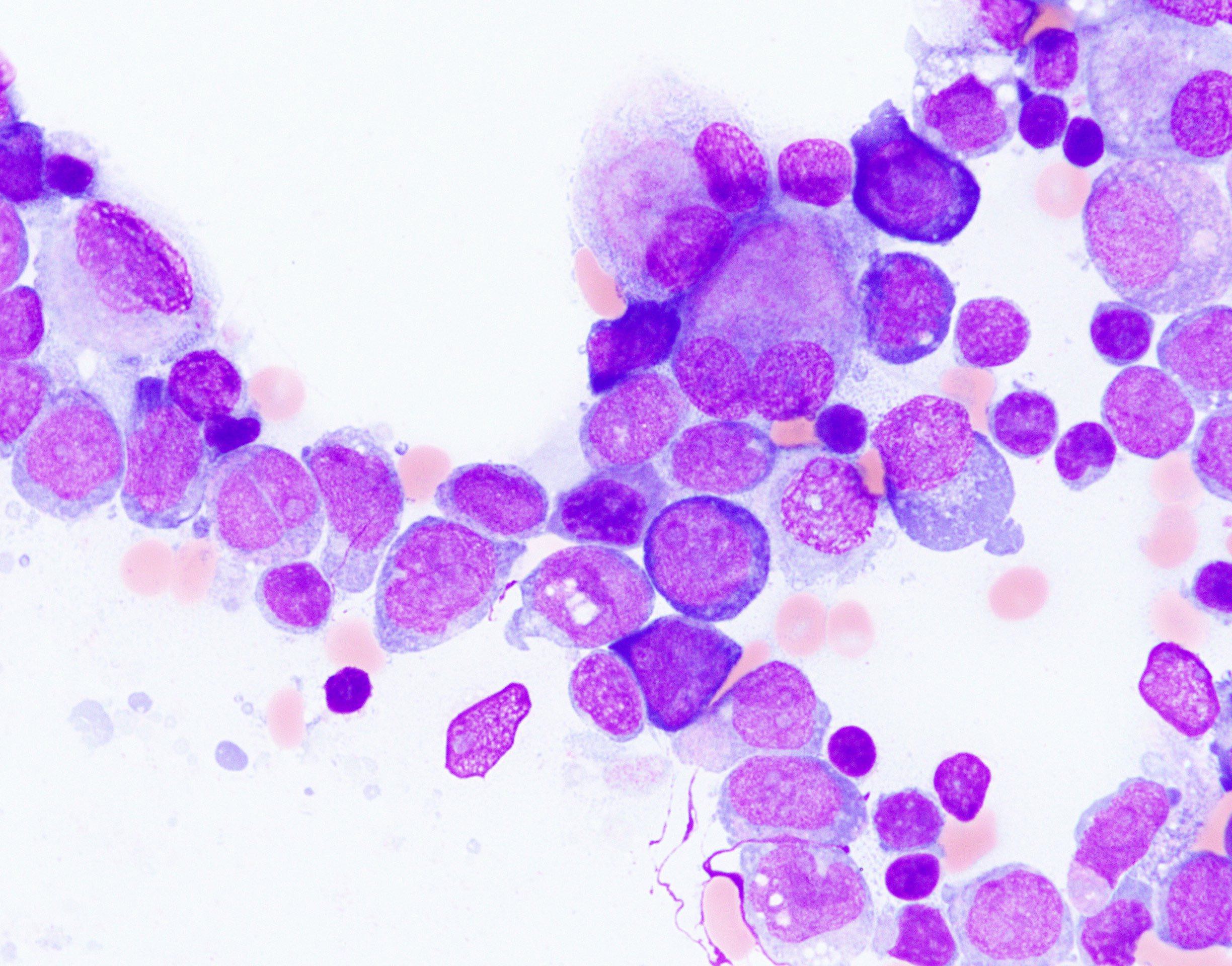
Aspirate with blasts
- Anemia with erythroid dysplasia
- Leukocytosis or leukopenia
- Binuclear granulocytes (pseudo-Pelger-Hüet neutrophils) (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Thrombocytosis or thrombocytopenia
- No characteristic immunophenotypic profile
- Myeloperoxidase (33 - 42.3% of cases) (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Expression of panmyeloid and immature markers: CD34, CD13, CD33, CD117, CD38 and HLA-DR (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- Aberrant expression of CD7, a lymphoid marker (42.3 - 60%) (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- CD56 (20%) (Hematology 2015;20:435, Int J Lab Hematol 2019;41:380)
- CD41, a megakaryocytic marker in 15.8% of patients (Hematology 2015;20:435)
- Less frequent expression in patients of Asian origin (Int J Lab Hematol 2019;41:380)
- inv(3)(q21q26.2) or t(3;3)(q21;q26.2) [inv(3) / t(3;3)]
- Increased expression of EVI1 (MECOM)
- Relative haploinsufficiency of GATA2
- Associated monosomy 7, complex karyotype and 5q deletion
- Gene mutations identified include those of NRAS, GATA2, SF3B1, FLT3-ITD, KIT D816, PTPN11, CBL, KRAS and BCR-ABL1 and CEBPA (Blood 2015;125:133, Int J Lab Hematol 2019;41:380)
Images hosted on other servers:

Double inv(3)(q21q26) and monosomy 7

FISH with EVI1 breakapart probe
- Right posterior iliac crest, core biopsy, aspirate smear, touch imprint and clot particle:
- Acute myeloid leukemia with inv(3)(q21.3;q26.2); GATA2, MECOM (see comment)
- Comment: The bone core biopsy demonstrates an increase in blasts and prominent dysmegakaryocytopoiesis. The presence of inv(3)(q21.3;q26.2) confirms above diagnosis.
- Other AML with recurrent cytogenetic abnormalities:
- Presence of other subtype defining cytogenetic abnormalities different from inv(3) / t(3;3)
- MDS with inv(3):
- Blast count < 20%
- CD41 positivity on immunophenotypic studies is a key part of the diagnostic criteria
- Deletion of chromosome 5q is the most frequently associated cytogenetic abnormality
- Expression of CD7 is not consistent with the entity
- Expression of MECOM (EVI1) is commonly upregulated
- GATA2 fusion with MECOM (EVI1) is the characteristic molecular feature
Comment Here
Reference: AML with inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2)
- Anemia and thrombocytopenia are hallmark findings on the peripheral smear
- Bone marrow is often hypocellular with extensive fibrosis
- Erythroid dysplasia is uncommon unlike in other subtypes of AML
- Finding of pseudo-Pelger-Hüet neutrophils suggest an alternative diagnosis
- Increased megakaryocytic dysplasia is commonly observed on bone marrow aspiration / biopsy
Comment Here
Reference: AML with inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2)
- 10% of AML cases; 5% of childhood leukemias
- Any age, 20% are < 25 years and 40% are 60 years+
- Anemia, thrombocytopenia, neutropenia; variable number of blasts in peripheral blood
- Variable prognosis
- Criteria for diagnosis: 20%+ nonerythroid cells in peripheral blood or bone marrow are myeloblasts; monocytic precursors are < 20% in bone marrow and granulocytes are 10%+ of cells
- Enzyme cytochemistry: most blasts are positive for myeloperoxidase or Sudan Black B, and chloroacetate esterase
- 7 year old boy with myeloid sarcoma with acute myeloblastic leukemia (J Coll Physicians Surg Pak 2011;21:369)
- 30 year old man with t(5;11) (Cancer Genet Cytogenet 2007;172:154)
- 35 year old pregnant woman (Rinsho Ketsueki 2011;52:18)
- Three patients with variant t(8;21) (Cancer Genet Cytogenet 2010;199:31)
- Usually hypercellular marrow
- Full range of myeloid maturation through maturing neutrophils, often with abnormal segmentation and 10%+ bone marrow cells with variable degree of dysplasia
- Auer rods in 70% of blasts; myeloblasts with or without azurophilic granules
- Erythroid and megakaryocyte precursors may have dysplastic changes
- Often increased eosinophilic precursors without cytological and cytochemical abnormalities of inv(16)(p13.1q22)
- Basophils may be increased, rarely mast cell hyperplasia (Indian J Pathol Microbiol 2007;50:655)
AFIP images
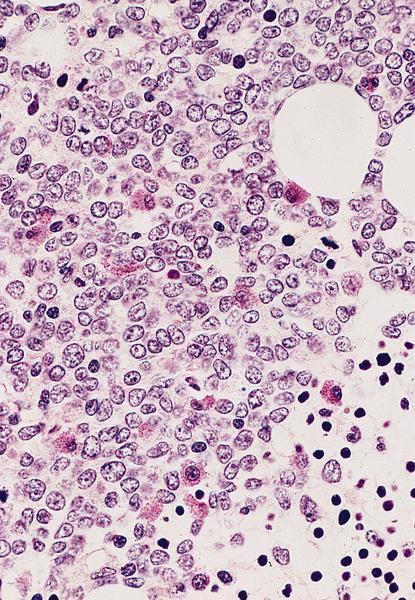
Bone marrow
biopsy: markedly
hypercellular
marrow
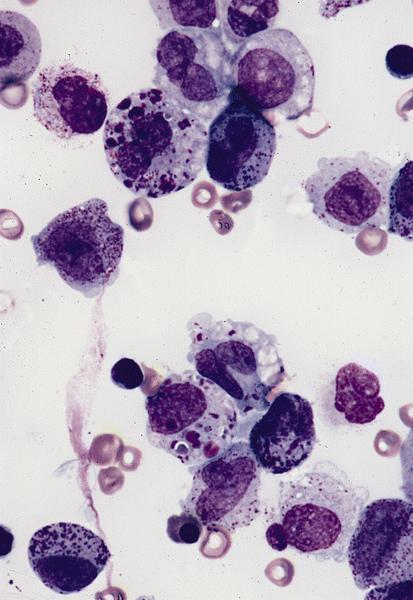
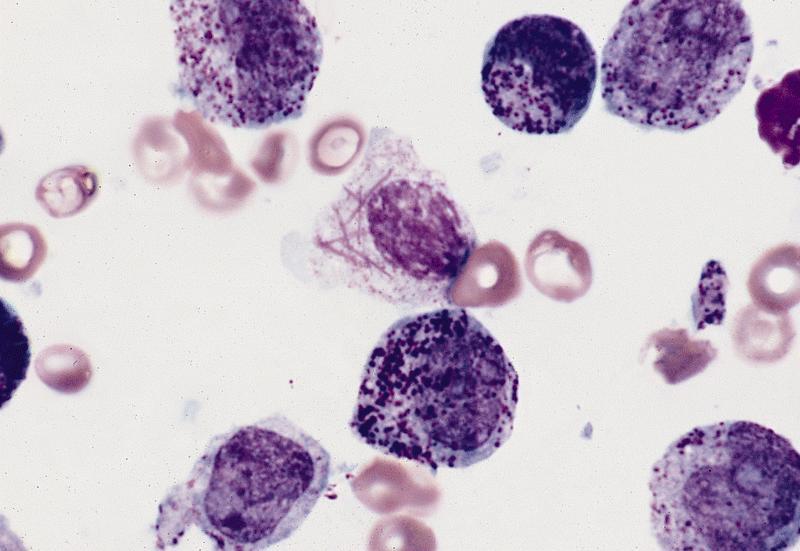
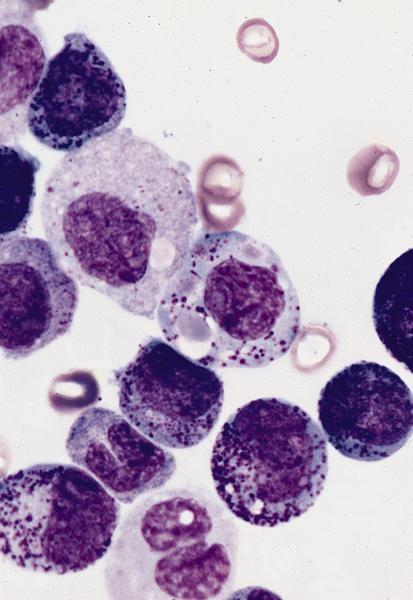
Diagnosed as AML with maturation because no t(15;17) and no DIC but FISH not done, so may actually be acute promyelocytic leukemia
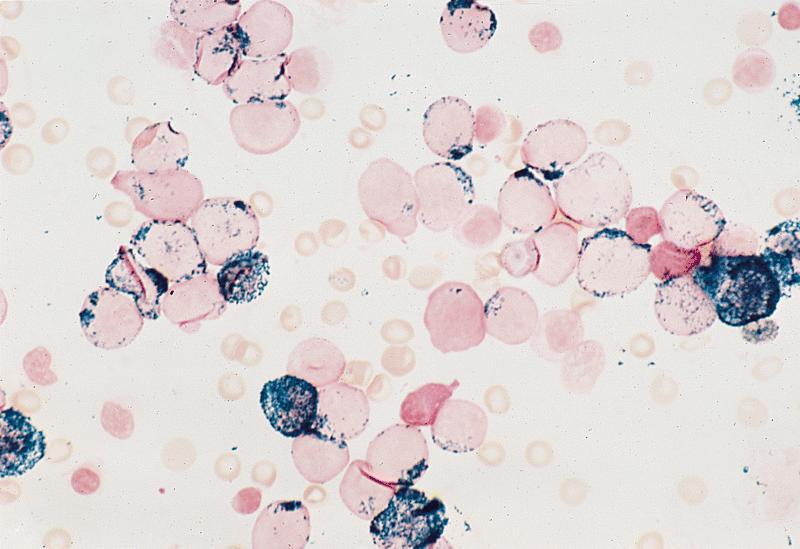
Myeloperoxidase positive blasts
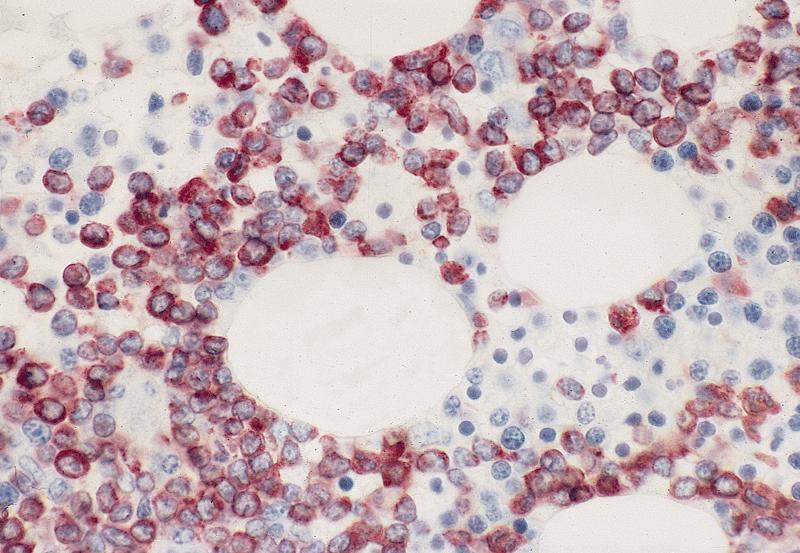
Erythroid cells are negative
Bone marrow smears (Wright-Giemsa):
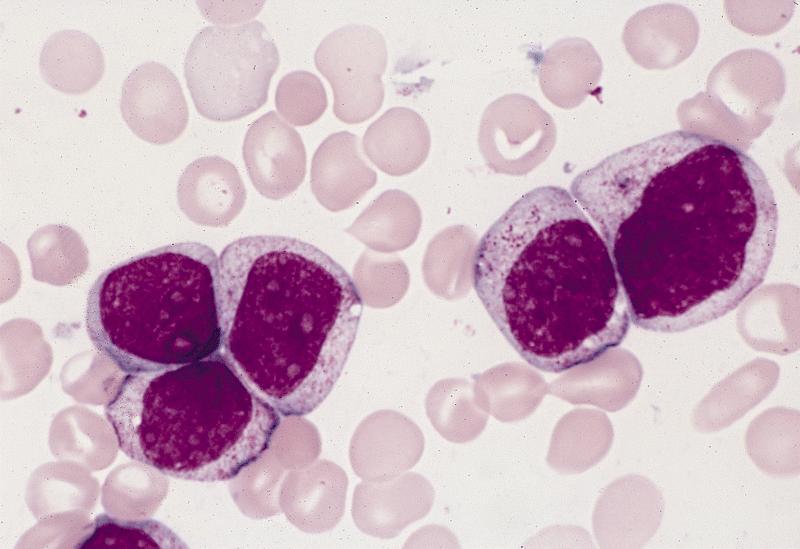
Type III myeloblasts
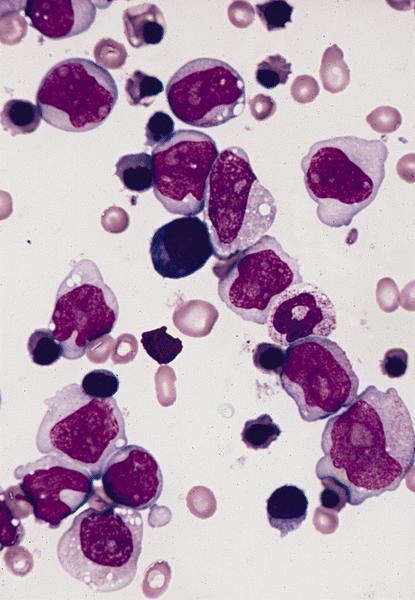
Myeloblasts,
promyelocytes,
myelocytes and
neutrophils
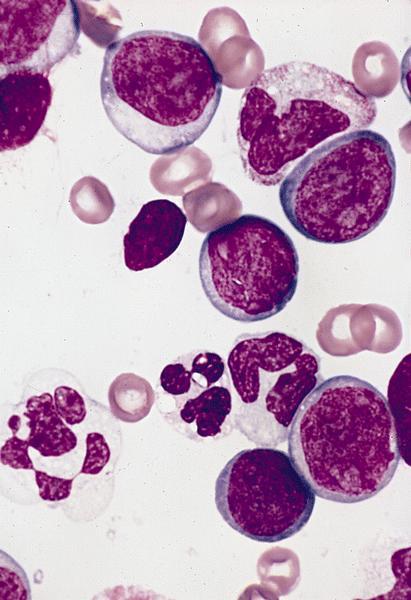
Several myeloblasts and maturing forms
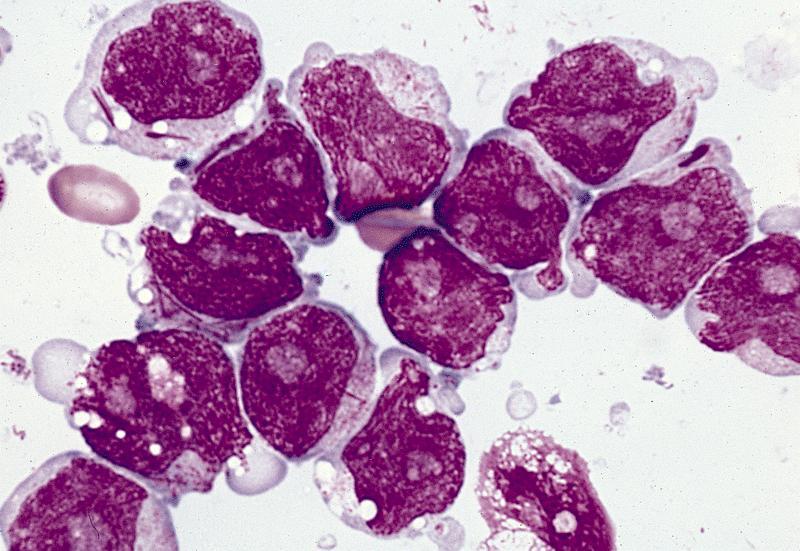
Pseudopods, cytoplasm and prominent Auer rods
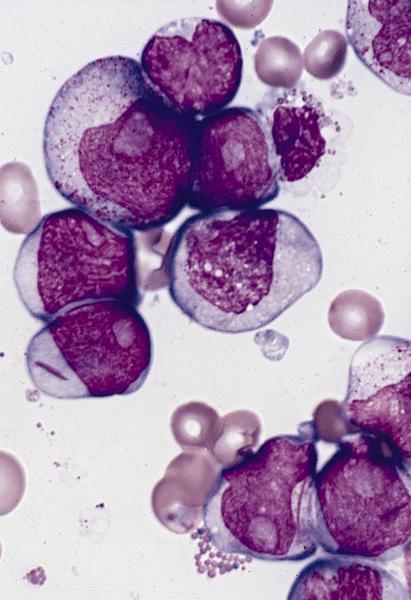
Several blasts have prominent nucleoli and Auer rods
AFIP images
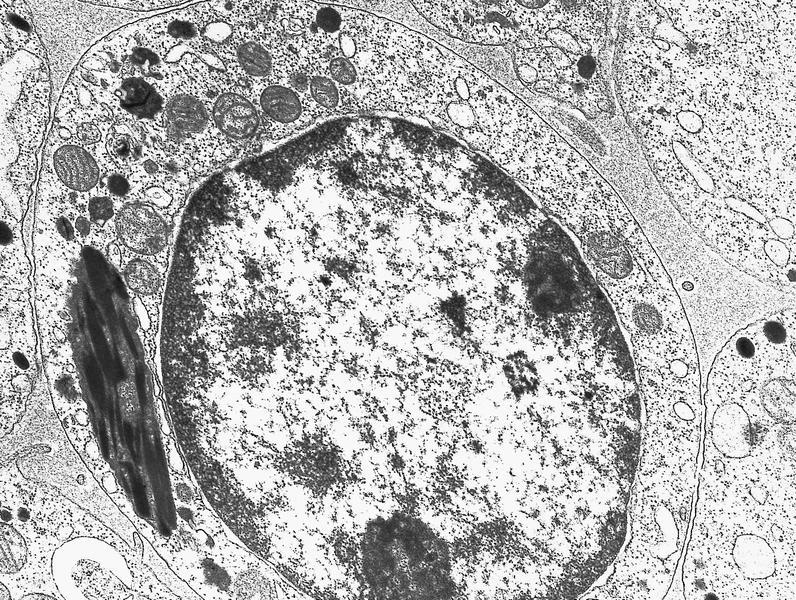
Numerous primary granules and fusion of Auer rods
- t(8;21) cases classified as AML with t(8;21); RUNX1-RUNX1T1; may be only genetic abnormality or part of more complex abnormalities (Cytometry B Clin Cytom 2008;74:25)
- FLT3 mutations associated with HLA-DR negative patients (Leuk Res 2007;31:921)
- No definitive evidence of myeloid differentiation by morphology and light microscopy cytochemistry; need immunohistochemistry, flow cytometry, or EM cytochemistry to characterize as myeloid
- Criteria for diagnosis: nongranular blasts; less than 3% of blasts are positive for myeloperoxidase (MPO), Sudan Black B (SBB) or naphthol-ASD-chloroacetate esterase (CAE) by enzyme cytochemistry, although blasts may express myeloperoxidase by EM or immunohistochemistry; blasts do not express classic lymphocyte antigens, but may aberrantly express some lymphocyte antigens
- 5% or less of AML cases; any age, mostly infants or older adults
- Typically presents with anemia, thrombocytopenia, neutropenia, marrow failure, but may have leukocytosis with markedly increased blasts
- Children (Blood 2007;109:2314) and adults (Br J Haematol 2001;113:737) may have poorer outcome than other AML subtypes
- Enzyme cytochemistry: negative for nonspecific esterase, although may have nonspecific weak or focal reaction distinct from monocytic cells
- 58 year old man with minimally differentiated acute myelogenous leukemia (AML-M0) granulocytic sarcoma in oral cavity (Oral Oncol 2002;38:516)
- Subtelomeric t(5;9)(q35.3;q34.3) and deletion of RB1 gene (Cancer Genet Cytogenet 2008;181:36)
- Deletion of (15)(q11.2q22) (Cancer Genet Cytogenet 2008;184:57)
- Medium sized blasts, round or slightly indented nuclei with dispersed chromatin
- One or two nucleoli, agranular cytoplasm with varying degree of basophilia and no Auer rods
- Rarely small blasts with condensed chromatin and scant cytoplasm that may resemble lymphoblasts
- Occasionally, residual normal population of maturing neutrophils may be present
- Bone marrow: markedly hypercellular with poorly differentiated blasts
AFIP images
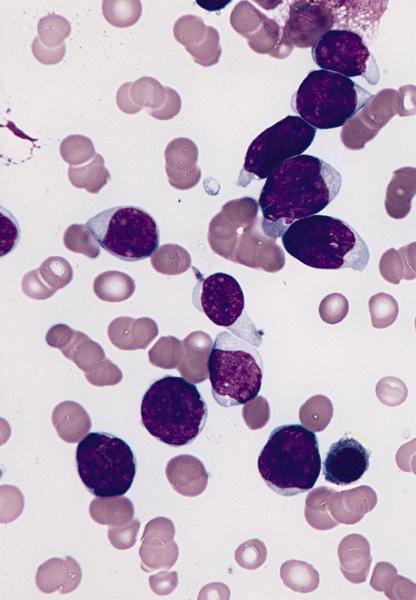
Bone marrow smear
(Wright-Giemsa):
no differentiated
features
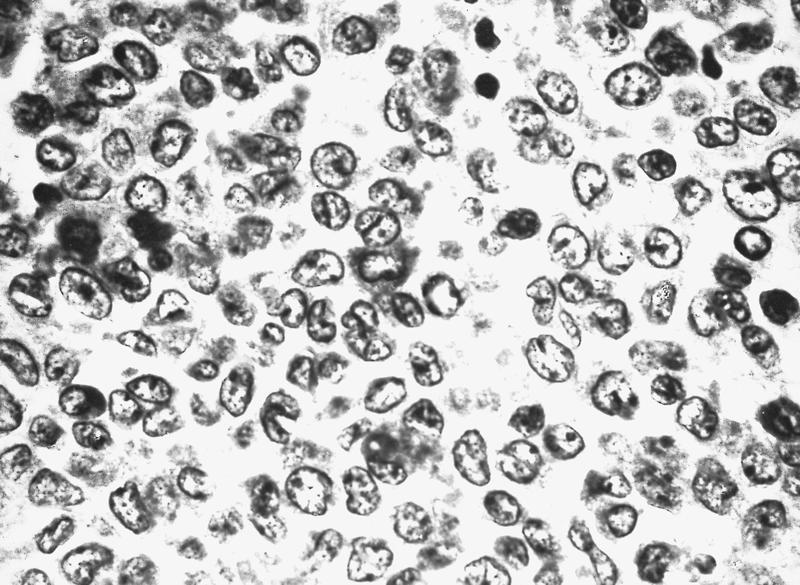
Bone marrow biopsy:
complete replacement
of marrow by blasts
without differentiation
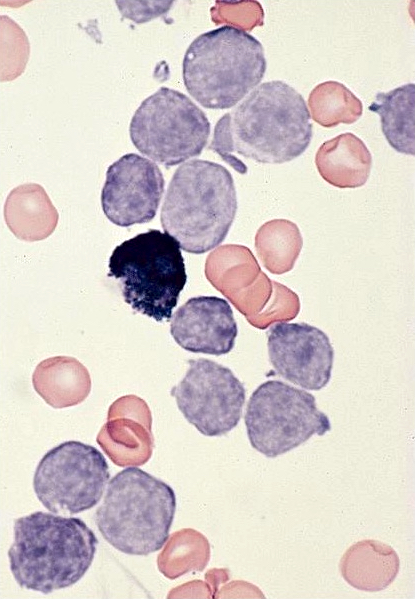
Myeloblasts are negative for myeloperoxidase
Images hosted on other servers:
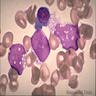
Bone marrow smear
(Wright-Giemsa):
no differentiated
features
Blasts are positive for myeloperoxidase by IHC
- Pediatric AML-M0 is usually CD33 bright, TdT-, CD34- and CD13-/weak (Am J Clin Pathol 2000;113:193, Clinical Flow Wiki: Acute Minimally Differentiated Leukemia (M0) [Accessed 3 April 2018])
Images hosted on other servers:

Various gating plots
- CD13, CD33 (Am J Clin Pathol 2001;115:876), CD34, CD117 (Am J Clin Pathol 2002;117:380), HLA-DR and CD38
- Variable expression of myeloperoxidase, Sudan Black B, TdT (50%), CD2, CD4, CD7 (40%) and CD16 / CD56
- CD11b (usually), CD14 (usually), CD15 (usually), CD36 (usually), CD41, CD61, CD64 (usually, Arch Pathol Lab Med 2007;131:748) and most lymphocyte antigens(CD5, cCD3, cCD79a, cCD22)
- Glycophorin A
- Often complex chromosomal abnormalities; tend to have more -5/del(5q), -7/del(7q), +8 and del(11q), but these should be reclassified as AML-MRC
- AML1 / CBFA / RUNX1 (27%) and strong association with trisomy 13 and FLT3 mutation (16 - 22%, Haematologica 2007;92:1123)
- More often trisomy 21 and hypodiploidy than other AML, although outcome is similar (Blood 2007;109:2314)
- Resembles myeloblasts; may show focal myeloperoxidase+ granules
AFIP images
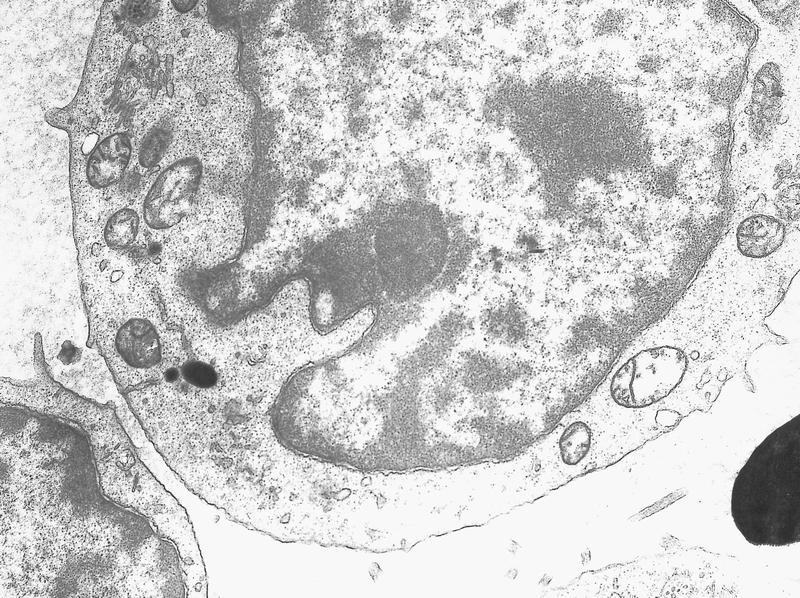
Granules are myeloperoxidase positive
Images hosted on other servers:
Granules are myeloperoxidase positive
- Acute leukemia of ambiguous lineage
- ALL
- AML-MRC
- Mixed phenotypic leukemia
- AML-M7 (megakaryoblastic)
- Leukemic phase of large cell lymphoma
- Acute myeloid leukemia (AML) with mutations in the NPM1 gene, a common recurrent genetic abnormality found in a subset of patients with AML
- One of the most common recurrent genetic abnormalities in AML
- Associated with better prognosis
- Associated with monocytic and myelomonocytic morphology
- NPM1 immunohistochemistry showing cytoplasmic localization may be used as a surrogate for PCR
- Acute myeloid leukemia with cytoplasmic nucleophosmin
- ICD-11: 2A60.0 - acute myeloid leukemia with recurrent genetic abnormalities
- Typically presents de novo in adults
- F > M
- Bone marrow
- Extramedullary involvement may occur
- Most common sites are gingiva, lymph nodes and skin
- Exon 12 mutation leading to aberrant localization of nucleophosmin and nucleophosmin associated proteins in the cytoplasm; while not completely understood, this may cause several hypothetical leukemogenic events (Leukemia 2017;31:798)
- NPM1 mutation causes HOX gene upregulation which supports the leukemic state in NPM1 mutant AML (Cancer Cell 2018;34:499)
- Unknown at this time
- NPM1 mutations are one of the most common recurrent genetic lesions in AML
- Occurs in approximately 35% of AML cases overall and 50 - 60% of cases with normal karyotype (Blood Rev 2018;32:167
- Patients often present with anemia and thrombocytopenia, although platelet counts are usually higher than other AML subtypes (Blood 2005;106:3740
- Requisite blast percentage for a diagnosis of AML is ≥ 20% myeloblasts, monoblasts / promonocytes or megakaryoblasts in the peripheral blood or bone marrow
- NPM1 mutation
- Detection via molecular techniques
- Aberrant cytoplasmic expression detected via immunohistochemistry
- AML with mutated NPM1, normal karyotype and absence of FLT3 internal tandem duplication mutation has characteristically favorable prognosis
- In younger patients, the prognosis is similar to AML with t(8;21) or AML with inv(16)
- NPM1 and FLT3 tyrosine kinase domain paired mutations offer higher overall and event free survival (Blood 2008;111:2527)
- Myeloid immunophenotype is associated with poorer relapse free survival and overall survival and is associated with more CD34 expression (Blood Adv 2019;3:3322)
- 12 and 15 year old girls presenting with NPM1 AML and cup-like blasts (J Pediatr Hematol Oncol 2018;40:e237)
- 24 year old woman, 33 year old man and 74 year old woman presenting with NPM1 mutated AML M5 with spontaneous remission (Clin Case Rep 2015;3:955)
- 46 year old man and 51 year old woman presenting with leukemia cutis (J Cutan Pathol 2020 Mar 20 [Epub ahead of print])
- 68 year old woman presenting with transformation of NPM1 mutated chronic eosinophilic leukemia to acute myeloid leukemia (Leuk Res Rep 2018;9:45)
- 74 year old man presenting with AML with NPM1 mutation and BCR-ABL fusion (Case Rep Hematol 2019;2019:6707506)
- Most patients receive conventional induction and consolidation chemotherapy with cytarabine and an anthracycline
- Patients do not receive allogeneic stem cell transplant
- Possible role of all trans retinoic acid (ATRA) as an adjuvant to induction therapy
- ATRA and arsenic work synergistically to degrade cytoplasmic NPM1, inducing apoptosis, growth arrest and differentiation
- Also shown to sensitize NPM1 mutant leukemic cells to daunorubicin
- ATRA and arsenic work synergistically to degrade cytoplasmic NPM1, inducing apoptosis, growth arrest and differentiation
- Addition of gemtuzumab ozogamicin (GO), a CD33 antigen targeting immunoconjugate, has shown an increase in the likelihood of NPM1 mutation negative minimal residual disease (Leukemia 2017;31:798)
- Wide range of morphologies are seen in AML with NPM1 mutation (N Engl J Med 2005;352:254)
- There is a strong association between both acute myelomonocytic and acute monocytic leukemia and NPM1 mutation (Blood 2007;109:874)
- NPM1 mutations are also detected in AML with and without maturation and in pure erythroid leukemia
- Cup-like nuclei are highly associated with the presence of NPM1 and FLT3 internal tandem duplication mutations (Cancer 2009;115:5481)
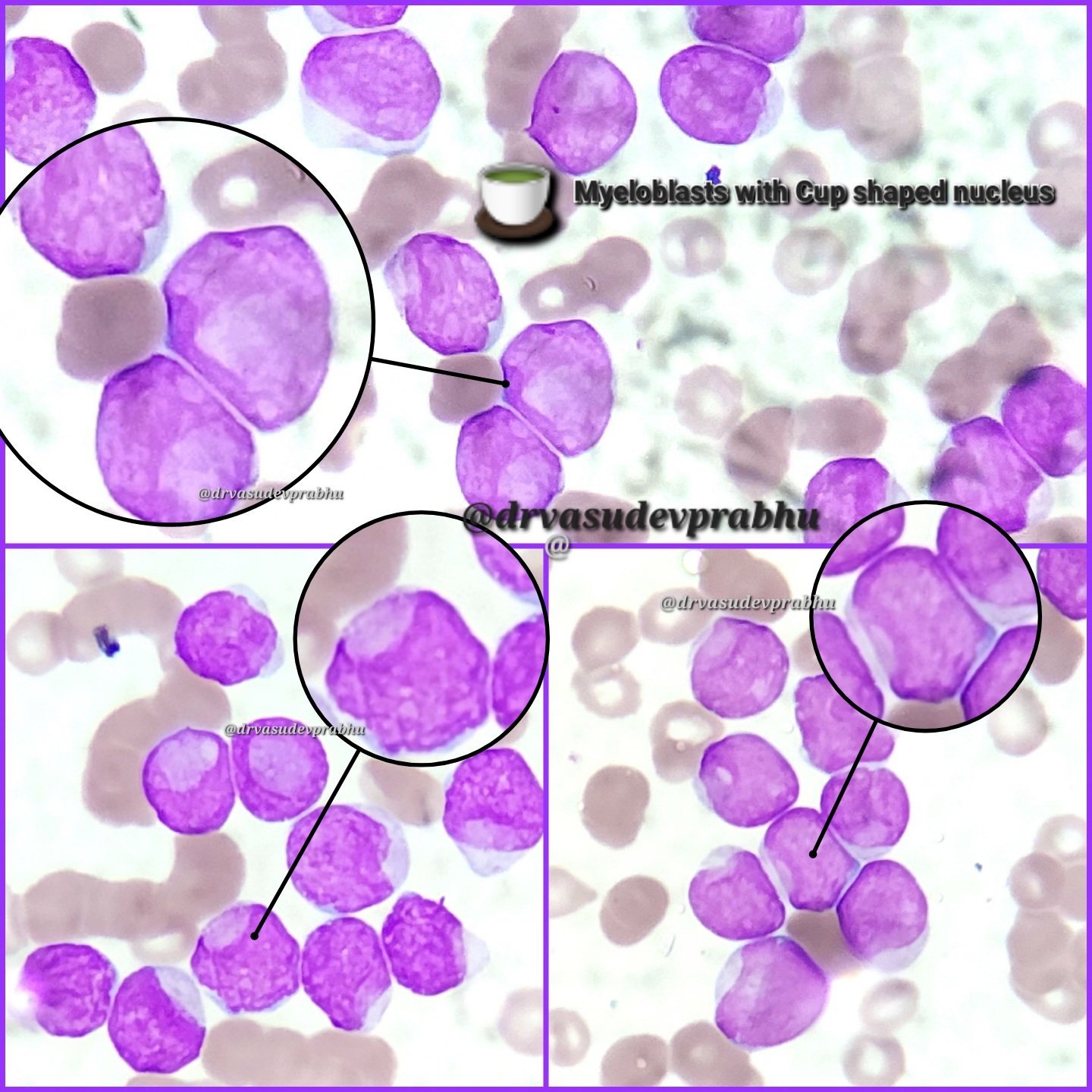
Contributed by Kamran M. Mirza, M.D., Ph.D.
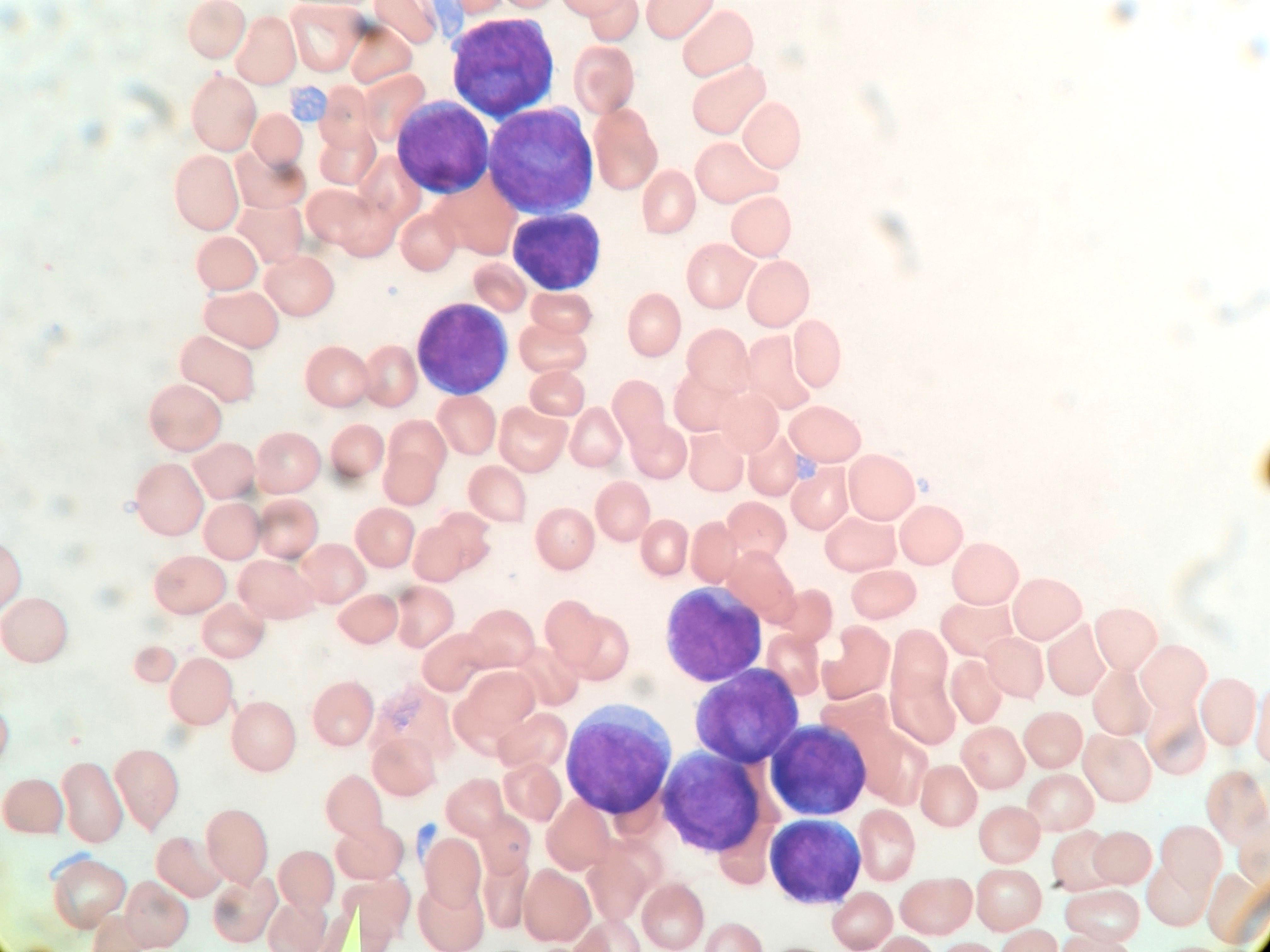
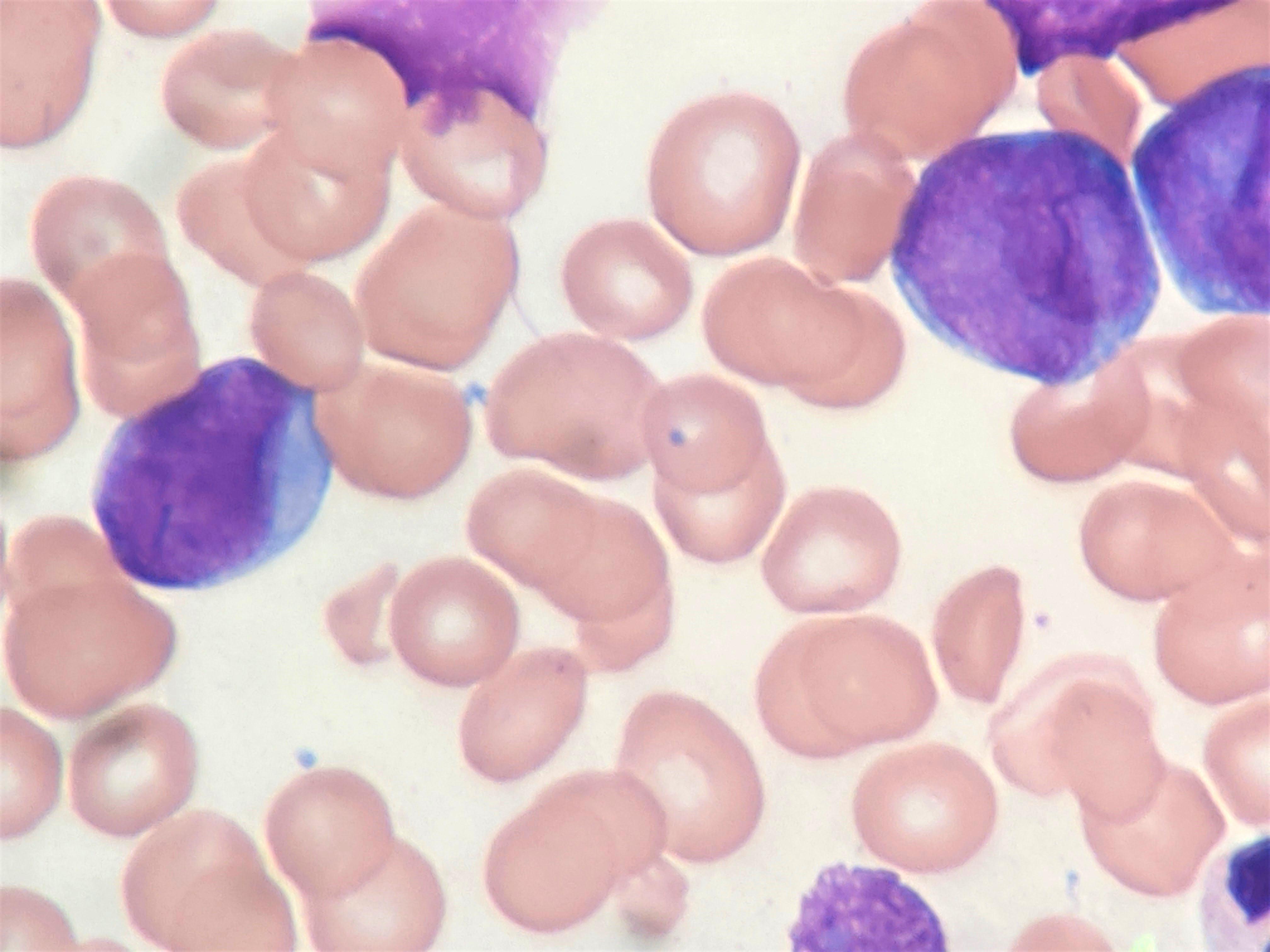
Blasts with cup-like indentations
- Cytoplasmic NPM1
- CD34 is negative in most cases
- Three phenotypic groups (Blood Adv 2019;3:3322):
- 4 base pair insertion in exon 12 of NPM1; detected via reverse transcription PCR
- Bone marrow, left posterior iliac crest, bone marrow core biopsy, aspirate, clot and peripheral blood:
- Acute myeloid leukemia with mutated NPM1
- Pancytopenia with rare circulating blasts (see comment)
- Comment: Peripheral smear shows pancytopenia with macrocytic anemia and mild anisopoikilocytosis with microcytes, macrocytes and occasional elliptocytes. Rare circulating blasts are present.
- The bone marrow core biopsy is of sufficient length for evaluation. There are multiple patchy hypercellular areas showing shows increased numbers of blasts in clusters with markedly decreased maturing / mature myeloid forms. Few scattered megakaryocytes with normal morphology are apparent. Erythropoiesis is decreased, although maturing precursors are still present in clusters.
- The aspirate smear shows that many of the blasts have a folded nuclei with often bilobed nuclei with prominent nucleoli in a few blasts reminiscent of cup-like nuclear invaginations in a proportion of blasts (8 - 9%) and moderate amount of gray-blue cytoplasm. Auer rods are not present. A differential count yields 83% blasts. Few residual erythroid precursors and occasional megakaryocytes are present without significant dysplasia in either of these 2 cell lines. Very few residual maturing myeloid forms are present. Iron stain shows decreased sideroblastic iron and adequate storage iron. Cytochemistry stains (alpha naphthyl butyrate esterase and myeloperoxidase) are performed and show the following: the blasts are myeloperoxidase positive (5 - 10% of blasts) and alpha naphthyl butyrate (monocyte) esterase is negative in all the blasts.
- Flow cytometry analysis shows an abnormal myeloid blast population expressing dim CD45, dim CD56 (subset), dim CD38, CD34, dim CD33, CD117 and dim CD13. The blasts are negative for HLA-DR.
- NPM1 mutation analysis confirms the presence of NPM1 mutation
- AML NOS, acute myelomonocytic leukemia:
- Lacks NPM1 mutation
- Other AML with recurrent genetic abnormalities:
- Lacks NPM1 mutation
- Blasts are frequently CD34 positive
- It has poor prognosis
- It is most frequently associated with normal karyotypes
- It is rare
Comment Here
Reference: AML with mutated NPM1
- Coin on coin nuclear morphology
- Dwarf megakaryocytes
- Immature eosinophils with large, violet granules
- Monocytic and myelomonocytic morphology
Comment Here
Reference: AML with mutated NPM1
- Due to the new classifications, this topic is now covered here
- Prior to WHO 2022 and ICC 2022:
- Provisional entity in the 2016 WHO classification
- De novo acute leukemia diagnosed with ≥ 20% blasts in blood or bone marrow
- All morphological subtypes of AML, NOS may harbor RUNX1 mutation, with the highest frequency being AML with minimal differentiation
- Diagnosis of AML with mutated RUNX1 excludes cases that fulfill criteria for other specific AML with recurrent genetic abnormalities, therapy related myeloid neoplasms or AML with myelodysplasia related changes (Blood 2016;128:462, Leukemia 2016;30:2282)
- Unfavorable prognostic implication, with mutated RUNX1 being associated with inferior survival than wildtype (Leukemia 2016;30:2282, J Clin Oncol 2012;30:3109)
- Provisional WHO entity describing a de novo acute myeloid leukemia
- Cases possess monoallelic RUNX1 mutations (typically frameshift or missense)
- Blast morphology most compatible with M0
- Cases do not meet diagnostic criteria for other AML with genetic abnormality
- Highest incidence in men and patients over 60 years of age
- Associated gene mutations include those of SRSF2 (39%), ASXL1 (36%), DNMT3A (19%), IDH2 (17%) and SF3B1 (17%) (Leukemia 2018;32:295)
- Other coexisting gene mutations include those of FLT3-ITD, TET2, RAS, PTPN11, p53, JAK2 and EZH2 genes (Leukemia 2018;32:295)
- ICD-O: 9861/3 - Acute myeloid leukemia, NOS (NIH: Acute Myeloid Leukemia, NOS [Accessed 16 July 2020])
- Reportedly accounts for 4 - 16% of cases of AML (Blood 2016;128:462)
- Higher incidence in men (60%) (Blood 2016;128:462, Leukemia 2016;30:2282, Int J Mol Sci 2017;18:1618, J Clin Oncol 2017;35:934)
- Higher incidence in AML in patients older than 60 years (10 - 20%) (Int J Mol Sci 2017;18:1618, J Clin Oncol 2017;35:934)
- RUNX1 mutations may be seen at increased frequency in patients with AML and a history of radiation exposure or inherited disorders such as Fanconi anemia and congenital neutropenia (Leukemia 2016;30:2282)
- However, these AML may be better classified under alternative categories of therapy related myeloid neoplasm or myeloid neoplasm with germline predisposition
- Peripheral blood
- Bone marrow
- Runt related transcription factor 1 (RUNX1) is known to play a key role in hematopoiesis as a key regulator of genes involved in the differentiation of myeloid cells (Int J Mol Sci 2017;18:1618)
- RUNX1 mutation leads to upregulation of those genes normally expressed in primitive hematopoietic cells and B cell progenitors and downregulation of promoters of myelopoiesis (J Clin Oncol 2012;30:3109)
- Mutation of RUNX1 therefore predisposes to the development of AML with more immature / undifferentiated morphology (M0 in the French-American-British [FAB] classification) (Leukemia 2016;30:2282, Int J Mol Sci 2017;18:1618)
- Associated gene mutations include those of SRSF2 (39%), ASXL1 (36%), DNMT3A (19%), IDH2 (17%) and SF3B1 (17%) (Leukemia 2018;32:295)
- Other coexisting gene mutations include those of FLT3-ITD, TET2, RAS, PTPN11, p53, JAK2 and EZH2 genes (Leukemia 2018;32:295)
- Anemia
- Leukopenia
- Thrombocytopenia
- Low LDH
- Bone marrow aspiration / biopsy to enumerate ≥ 20% blasts
- Flow cytometric evaluation of peripheral blood and bone marrow aspirate to confirm blast phenotype
- Next generation sequencing (NGS) or mutational analysis to evaluate for any RUNX1 alterations
- Presence of mutated RUNX1 gene is by itself a poor prognostic factor in this provisional subtype of AML (Blood 2016;128:462, Leukemia 2016;30:2282, N Engl J Med 2016;374:2209)
- Presence of > 1 RUNX1 mutation correlates with even worse prognosis (Leukemia 2018;32:295)
- AML with mutated RUNX1 is characterized by lower rates of response to induction chemotherapy, higher relapse rates and a poor long term clinical outlook (J Clin Oncol 2017;35:934, N Engl J Med 2016;374:2209, Blood 2017;129:424)
- It has a worse prognosis than AML without mutation of RUNX1
- In contrast, AML with RUNX1 / RUNX1T1 fusion has a more favorable outcome (Sci Rep 2018;8:11293)
- Age 60 years and above indicates poor prognosis (Blood 2011;117:2348)
- Higher WBC count is a marker of poor prognosis (Blood 2011;117:2348)
- Associated comutations including those involving FLT3-ITD, ASXL1, SRSF2, DNMT3A and PHF6 confer a worse prognosis (Leukemia 2016;30:2282, J Clin Oncol 2017;35:934, Blood 2011;117:2348, Leuk Lymphoma 2020;61:1395)
- Associated ASXL1 and ≥ 2 additional mutations correlated with shorter overall survival (Leukemia 2018;32:295)
- Comutation of IDH2 confers a favorable prognosis (Leukemia 2016;30:2282)
- 23 year old woman with isolated bone marrow non-Langerhans cell histiocytosis preceding AML with mutated RUNX1 (Am J Clin Pathol 2019;151:638)
- AML with mutated RUNX1 is currently treated with standard induction chemotherapy, including regimens consisting of cytarabine and an anthracycline
- However, overall response rate, progression free survival and overall survival are known to be poor in the AML subtype (Leukemia 2016;30:2282, Blood 2011;117:2348)
- Use of hypomethylating agent may negate the effect of RUNX1 in elderly patients (Int J Mol Sci 2017;18:1618)
- Stem cell transplantation is a therapeutic option for suitable patients
- However, it may not significantly improve survival (Blood 2011;117:2348)
- Use of BET protein inhibitors in combination with agents such as venetoclax, decitabine or cytarabine, has shown promise in vitro (Leukemia 2018;32:295)
- Majority of cases (~60%) show the M0 (FAB classification) morphology, with very immature / undifferentiated cells (Int J Mol Sci 2017;18:1618, Blood 2011;117:2348)
- Blasts have variable size, high nucleus to cytoplasmic ratio and variably prominent nucleoli with no evidence of differentiation
- M2 and M1 morphology are the next most common (Blood 2011;117:2348)
- RUNX1 mutated AMLs typically demonstrate high bone marrow blast counts (Blood 2011;117:2348)
Contributed by Genevieve M. Crane, M.D., Ph.D.
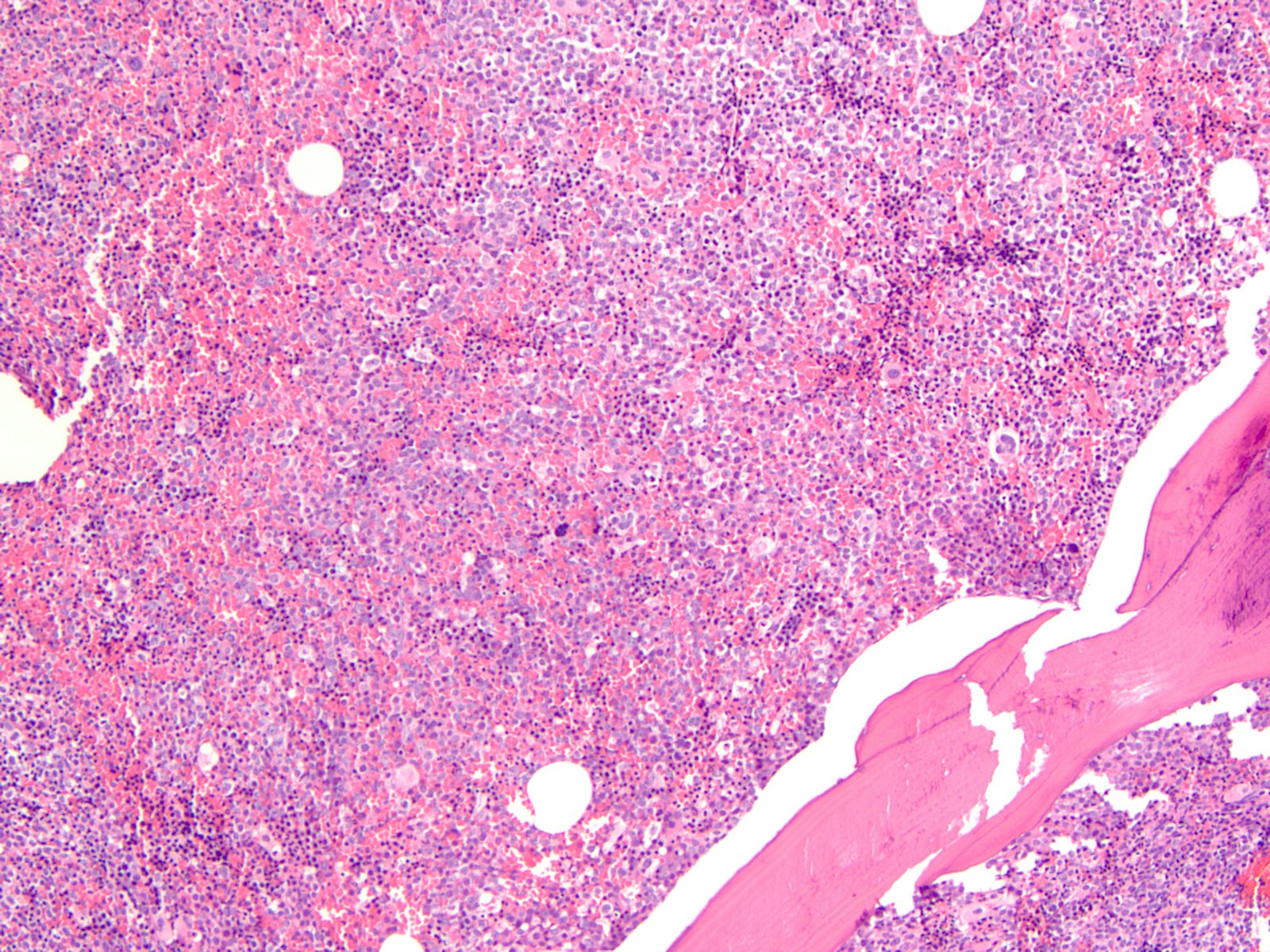
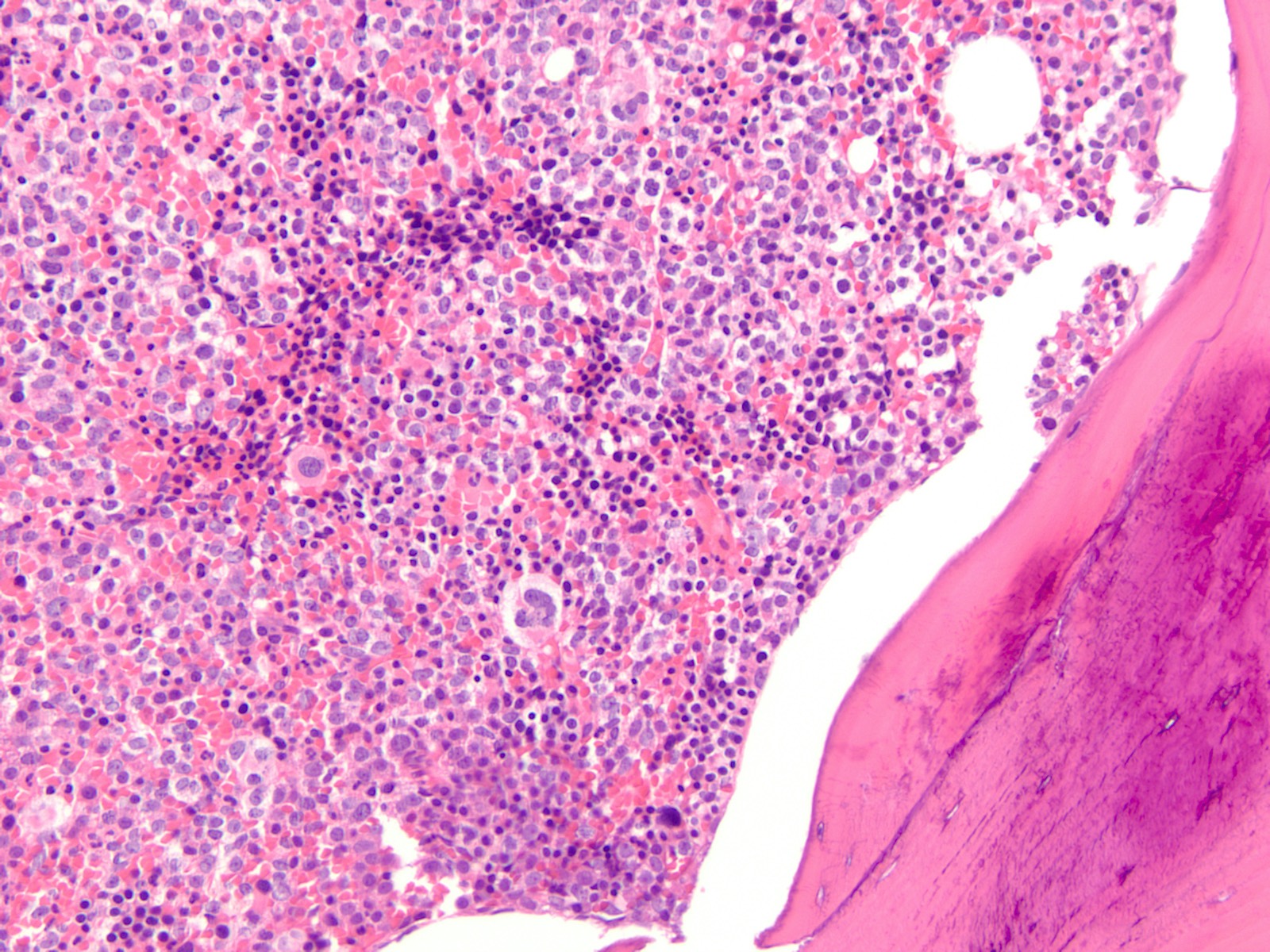
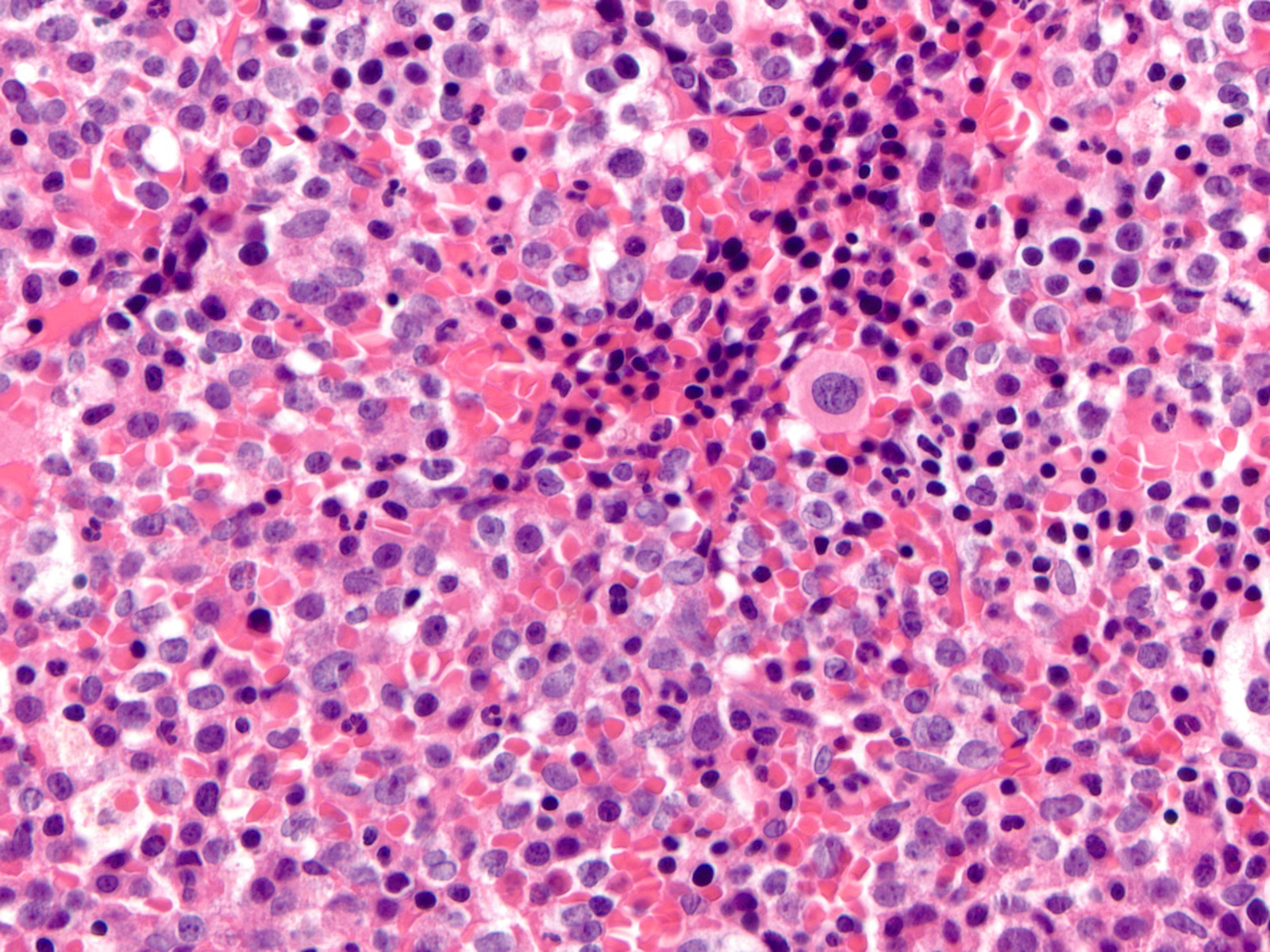
Bone marrow core biopsy
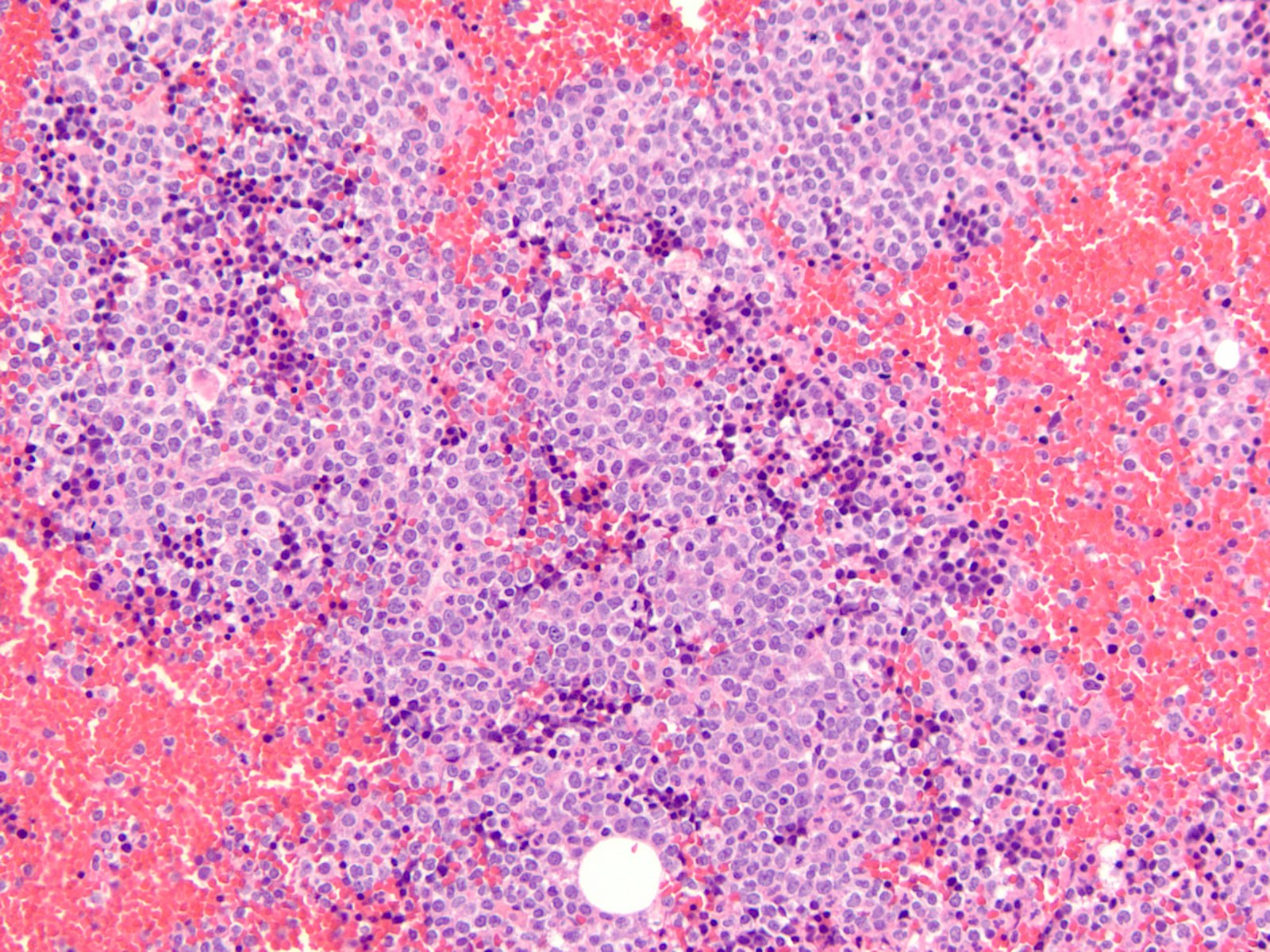
Bone marrow clot
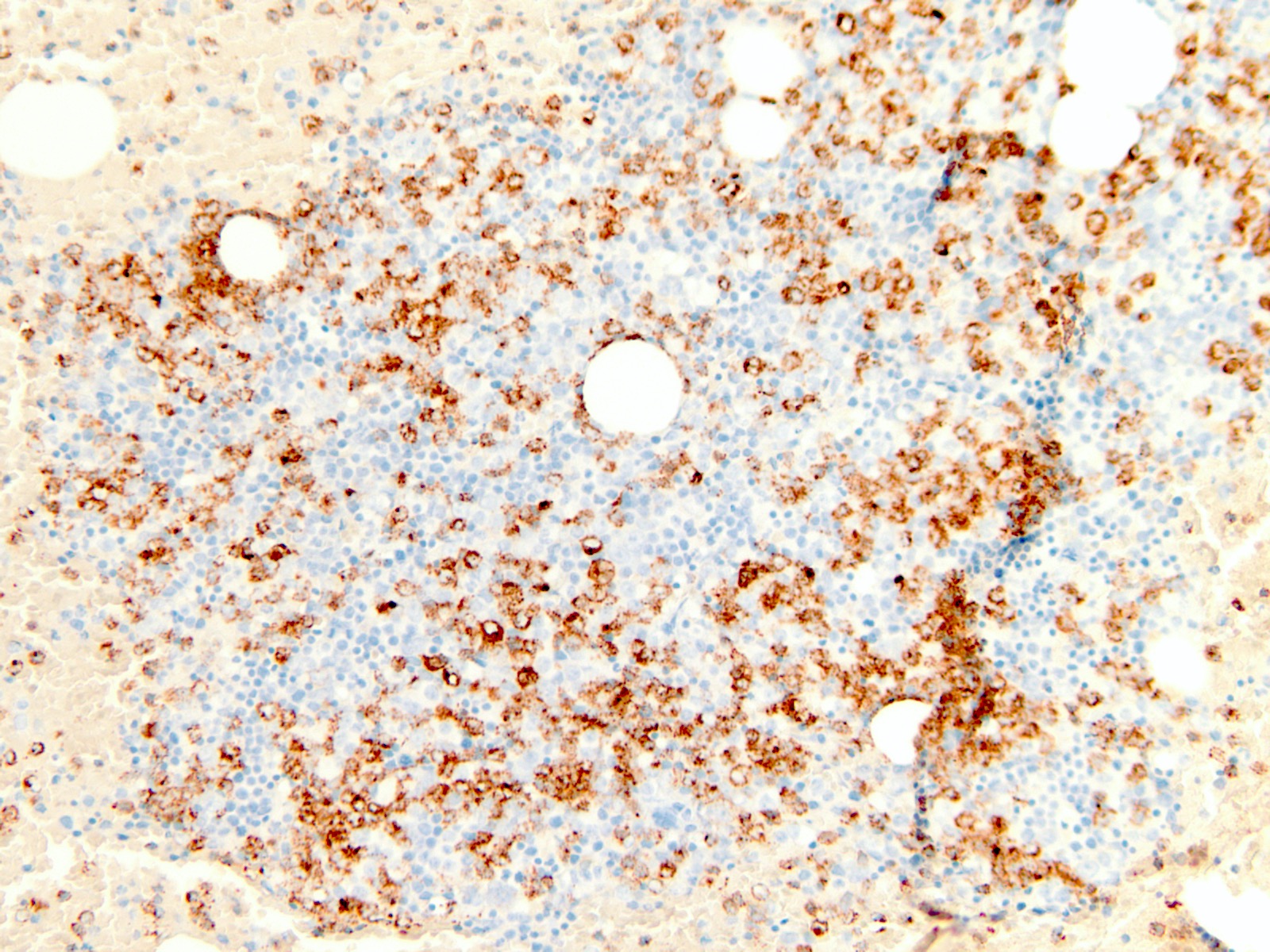
Bone marrow clot, CD34
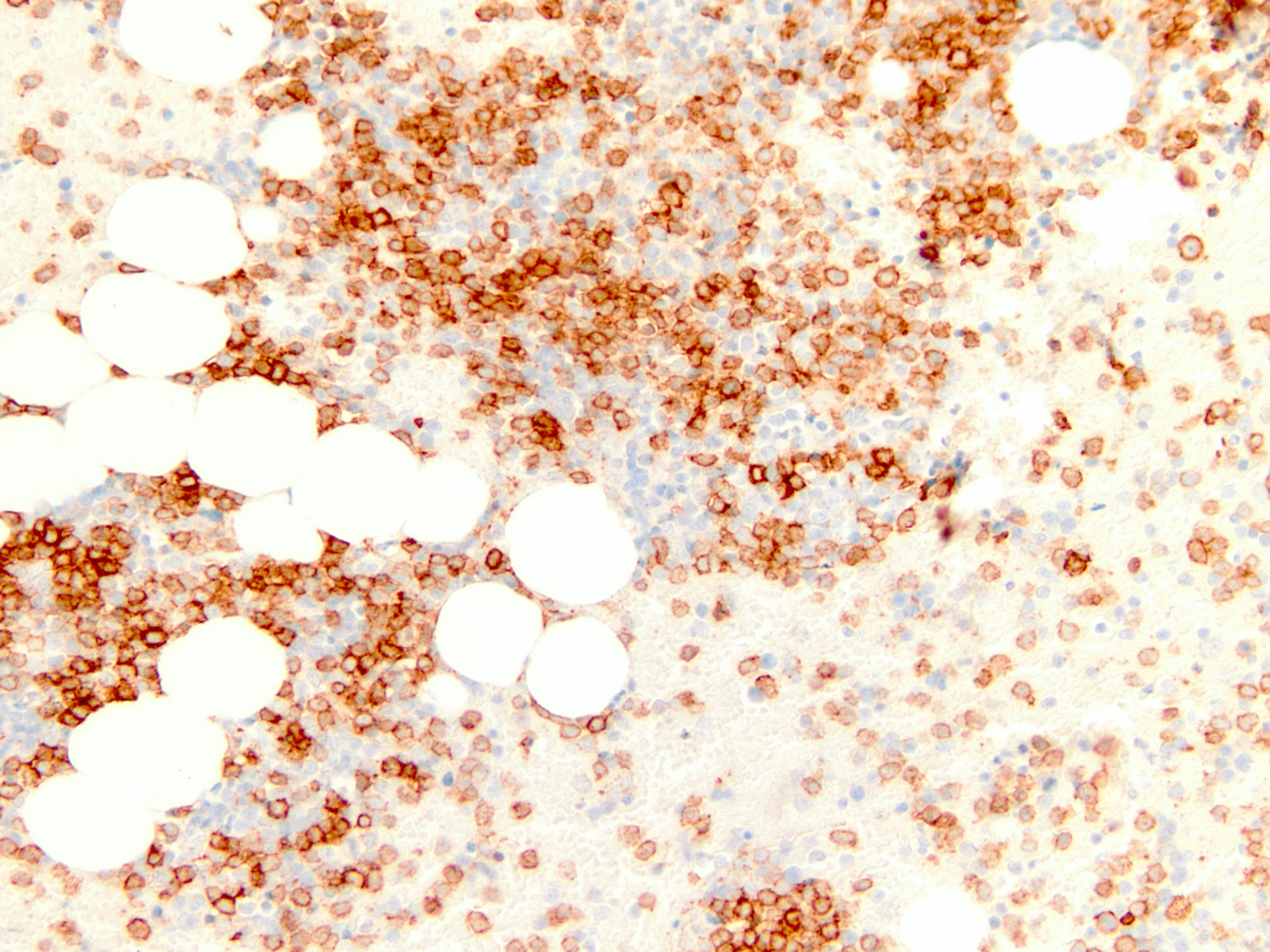
Bone marrow clot, CD117
Contributed by Genevieve M. Crane, M.D., Ph.D.
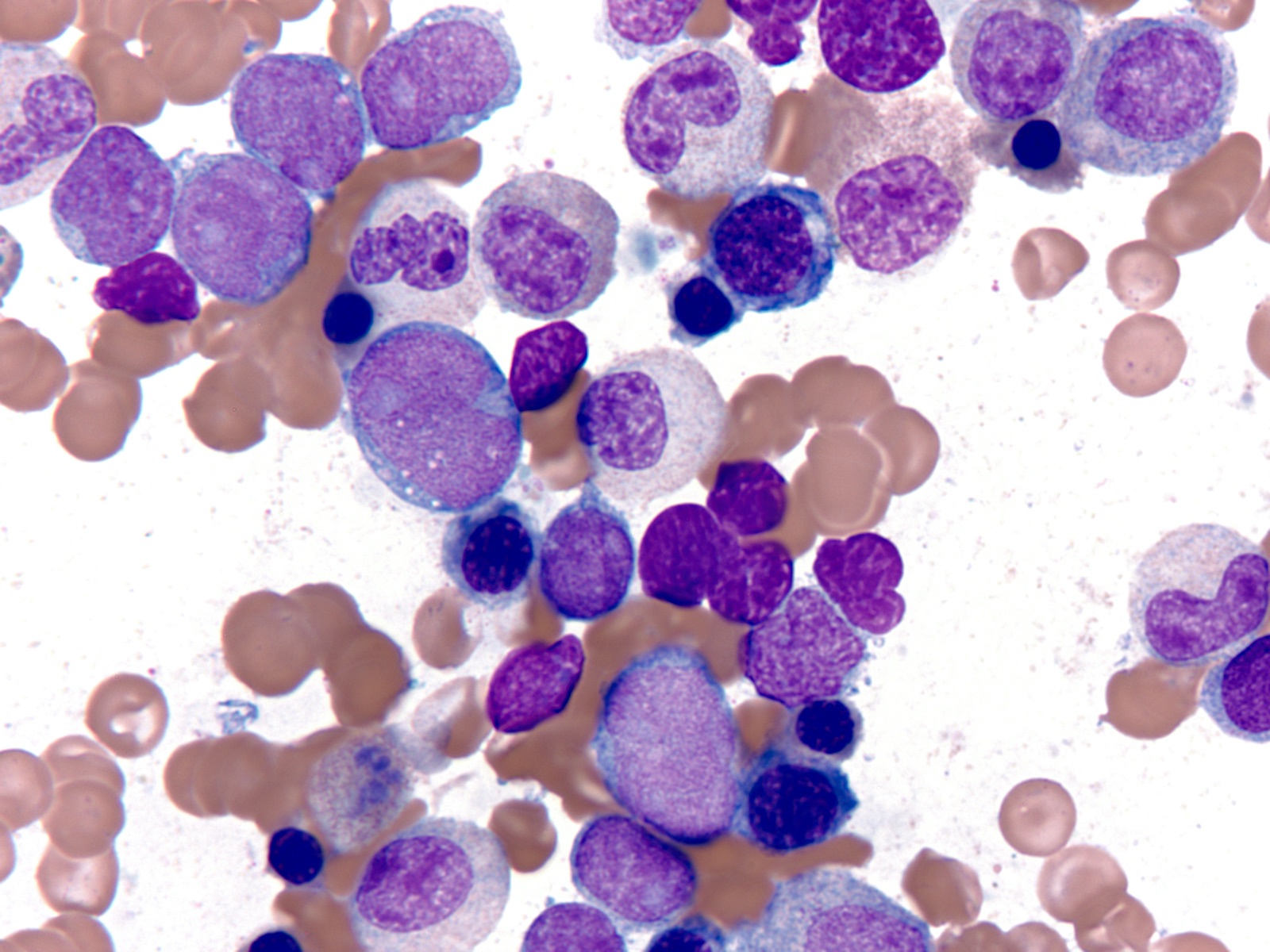
Bone marrow aspirate with increased blasts
- Lower hemoglobin, white blood cell and blast counts compared with wild type RUNX1
- Thrombocytopenia
Contributed by Genevieve M. Crane, M.D., Ph.D.
Peripheral blood smear with increased blasts
- Blasts typically express CD13, CD34 and HLA-DR (Blood 2016;128:462)
- There is variable expression of CD33, monocytic markers and myeloperoxidase (Blood 2016;128:462)
- CD34 higher in RUNX1 mutated blasts compared with RUNX1 wild type (Blood 2011;117:2348)
- Stains for B and T cell differentiation are negative
- Mutation of RUNX1 (located on chromosome 21q22.12) is disease defining (Blood 2016;128:462)
- Most RUNX1 mutations are monoallelic, within the runt homology domain (RHO) and the transactivation domain (TAD)
- RUNX1 mutations are typically frameshift or missense
- Known associated gene mutations include those of SRSF2, ASXL1, DNMT3A, IDH2, SF3B1, FLT3-ITD, TET2, RAS, PTPN11, p53, JAK2 and EZH2 genes (Leukemia 2018;32:295)
- Right posterior iliac crest, bone core biopsy and aspirate smear:
- Acute myeloid leukemia with mutated RUNX1 (see comment)
- Comment: The peripheral blood smear reveals pancytopenia with circulating blasts. The blasts are intermediate in size with high N:C ratio and variably prominent nucleoli. The marrow core biopsy and aspirate smear are packed with sheets of blasts which demonstrate minimal to no myeloid differentiation. Flow cytometry analysis of the marrow aspirate reveals the blast phenotype to be dim CD45, with expression of CD34 and CD13 and absence of B and T lineage markers. Next generation sequencing demonstrates the presence of a missense pathogenic RUNX1 mutation with a variant allele frequency of 65%. FISH panel and cytogenetic analysis are normal.
- AML, NOS, M0:
- Absence of RUNX1 as the defining molecular abnormality
- AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1:
- Characterized by the RUNX1-RUNX1T1 translocation / fusion product rather than the RUNX1 mutation seen in AML with mutated RUNX1
- Common loss of a sex chromosome or deletion 9q
- Good prognosis compared to AML with mutated RUNX1
- Therapy related AML
- AML with myelodysplasia related changes:
- In the presence of a RUNX1 mutation, this diagnosis takes precedence if the following 3 criteria are met (Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 2017, 4th Edition):
- ≥ 20% bone marrow blasts
- History of MDS or MDS / MPN or MDS related cytogenetic abnormality or multilineage dysplasia (> 50% of cells in at least 2 lineages but not meeting criteria for AML with mutated NPM1 or biallelic mutation of CEBPA)
- Absence of prior cytotoxic therapy or radiation, absence of cytogenetic abnormality defining of another AML category
- In the presence of a RUNX1 mutation, this diagnosis takes precedence if the following 3 criteria are met (Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 2017, 4th Edition):
- Myeloid neoplasm with germline predisposition:
- Patients with a germline mutation in RUNX1 show an autosomal dominant familial platelet disorder and have a predisposition to developing MDS / AML; typically younger than sporadic AML patients (Ther Adv Hematol 2013;4:254, Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 2017, 4th Edition)
- M0 (AML with minimal maturation)
- M1 (AML without maturation)
- M3 (AML with PML-RARA)
- M4 (acute myelomonocytic leukemia)
- M7 (acute megakaryocytic leukemia)
- ASXL1 and SRSF2 are the most commonly comutated genes
- Comutation with FLT3-ITD negates the poor prognostic impact of RUNX1
- Incidence is higher in people younger than 50 years
- It is associated with a better prognosis than AML with RUNX1-RUNX1T1 fusion
- It has lower frequency of CD34 positivity in blast cells than other subtypes of AML
Comment Here
Reference: AML with mutated RUNX1
- ≥ 20% myeloid blasts in the blood or bone marrow with 1 or more of the following: features of myelodysplasia, prior history of myelodysplastic syndrome (MDS) or MDS / myeloproliferative neoplasm (MPN) or MDS related cytogenetic abnormalities
- Requires absence of prior cytotoxic / radiation therapy and absence of a recurrent cytogenetic abnormality
- In de novo acute myeloid leukemia (AML) with mutated NPM1 or biallelic mutation of CEBPA, multilineage dysplasia alone or del(9q) alone are insufficient
- Includes refractory anemia with excess blasts in transformation defined by French-American-British (FAB) classification
- AML with morphological features of myelodysplasia in > 50% of the cells of 2 or more lineages or a prior history of MDS or MDS / MPN or MDS related cytogenetic abnormalities
- Generally poorer prognosis with lower remission rates and shorter overall survival than other AML subtypes (Am J Clin Pathol 2020;154:731)
- Commonly presents with severe pancytopenia
- Acute myeloid leukemia with multilineage dysplasia
- Acute myeloid leukemia with prior myelodysplastic syndrome
- Acute myeloid leukemia with myelodysplasia related changes (AML-MRC)
- ICD-O: 9895/3 - acute myeloid leukemia with myelodysplasia related changes
- Mainly elderly patients; median age 70 years (Am J Hematol 2020;95:612)
- Rare in children
- Up to 48% of AML (Surg Pathol Clin 2010;3:1153)
- Blood and bone marrow
- Frequently presents with severe pancytopenia
- Cases with 20 - 29% blasts may have a slowly progressive course with less marked cytopenias (Am J Hematol 2014;89:E193)
- Made based on the following (Surg Pathol Clin 2010;3:1153):
- Microscopic features, including bone marrow blast percentage and the presence and degree of dysplasia
- Immunophenotyping to confirm myeloid lineage
- Results of cytogenetic studies
- Clinical correlation to evaluate for prior history of MDS or MDS / MPN and exclude prior cytotoxic or radiation therapy for an unrelated disease
- Generally poorer prognosis with lower remission rates and shorter overall survival than other AML subtypes (Am J Clin Pathol 2020;154:731)
- Diagnosis of AML-MRC solely based on multilineage dysplasia or previously untreated MDS or MDS / MPN have significantly better outcomes than those with MDS defining cytogenetic abnormalities or disease secondary to a previously treated MDS or MDS / MPN (Am J Hematol 2020;95:612)
- In de novo AML cases lacking myelodysplasia related cytogenetic abnormalities, select dysplastic features may carry more prognostic significance than others with micromegakaryocytes and hypogranulated myeloid cells identified as significantly associated with shorter event free survival (Mod Pathol 2015;28:965)
- 47 year old man with a history of Crohn's disease and new onset acute myeloid leukemia with myelodysplasia related changes (Case Rep Oncol 2018;11:573)
- 54 year old man with a prior history of acute myeloid leukemia with myelodysplasia related changes presenting with new onset pleural effusion with leukemic blasts (Diagn Cytopathol 2011;39:451)
- 77 year old man with acute myeloid leukemia with myelodysplasia related changes (Intern Med 2011;50:3037)
- 77 year old man with acute myeloid leukemia with myelodysplasia related changes demonstrating mixed lineage phenotype (Blood 2016;128:1663)
- 84 year old woman with acute myeloid leukemia with myelodysplasia related changes showing basophilic differentiation (Am J Hematol 2014;89:1082)
- Intensive induction chemotherapy followed by allogeneic stem cell transplant (Expert Opin Emerg Drugs 2021;26:245)
- Chemotherapy with hypomethylating agents and with or without venetoclax (Blood 2019;133:7)
- Liposomal formulation of daunorubicin and cytarabine (J Clin Oncol 2018;36:2684)
- Blasts 20% or more
- Dysplasia in > 50% of the cells in 2 or more lineages
- Dyserythropoiesis: nuclear budding, megaloblastosis, multinucleation, nuclear budding, irregular nuclear contours, cytoplasmic vacuoles, karyorrhexis, ring sideroblasts
- Dysgranulopoiesis: hypogranular cytoplasm, hyposegmented nucleus, abnormal nuclear segmentation
- Dysmegakaryopoiesis: micromegakaryocytes, nuclear hyposegmentation, nuclear hypersegmentation, separated nuclear lobes
- References: Surg Pathol Clin 2010;3:1153, Am J Clin Pathol 2020;154:731
Contributed by Srishti Gupta, M.B.B.S., M.D. and Elizabeth Courville, M.D.
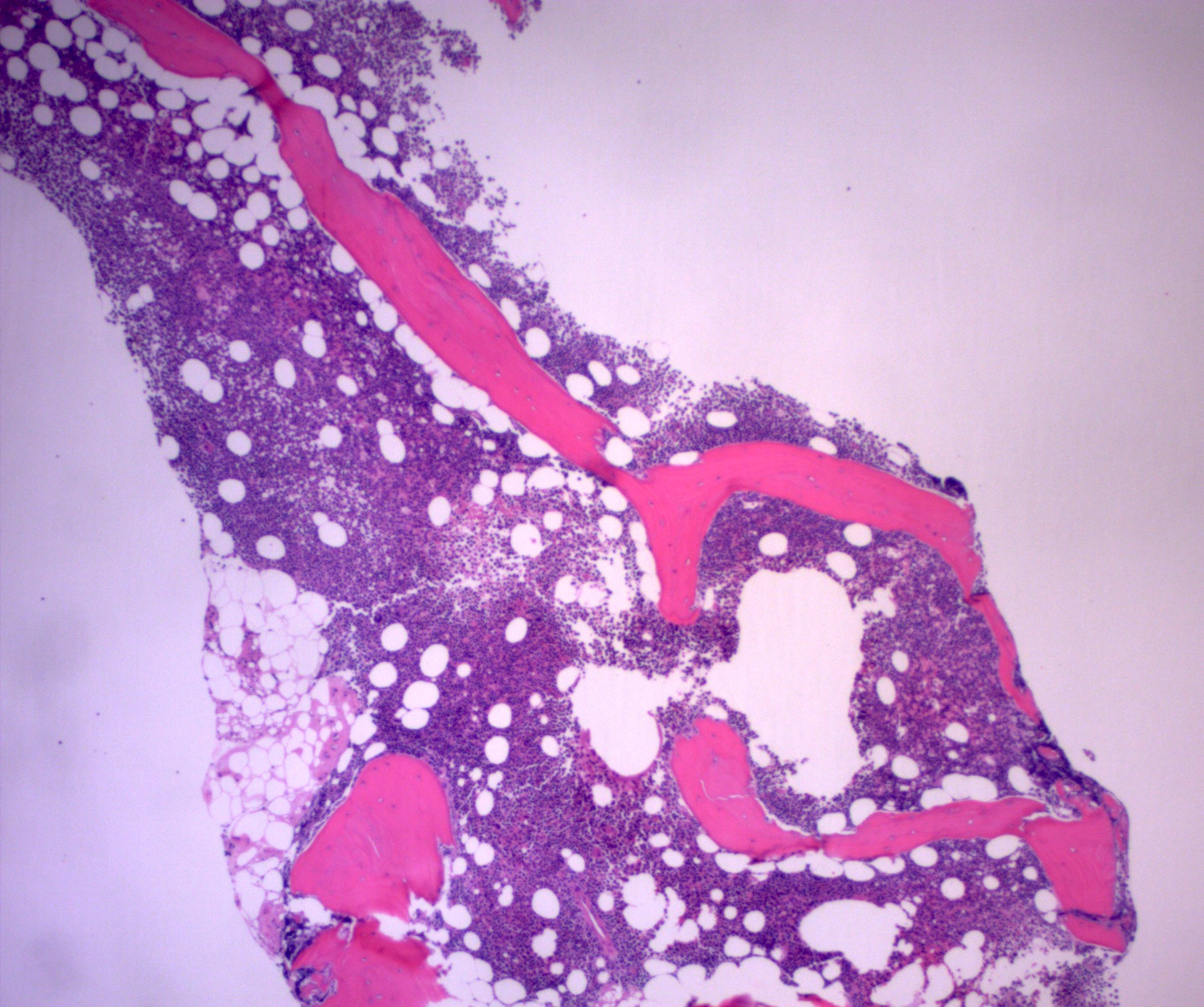
Hypercellular bone marrow
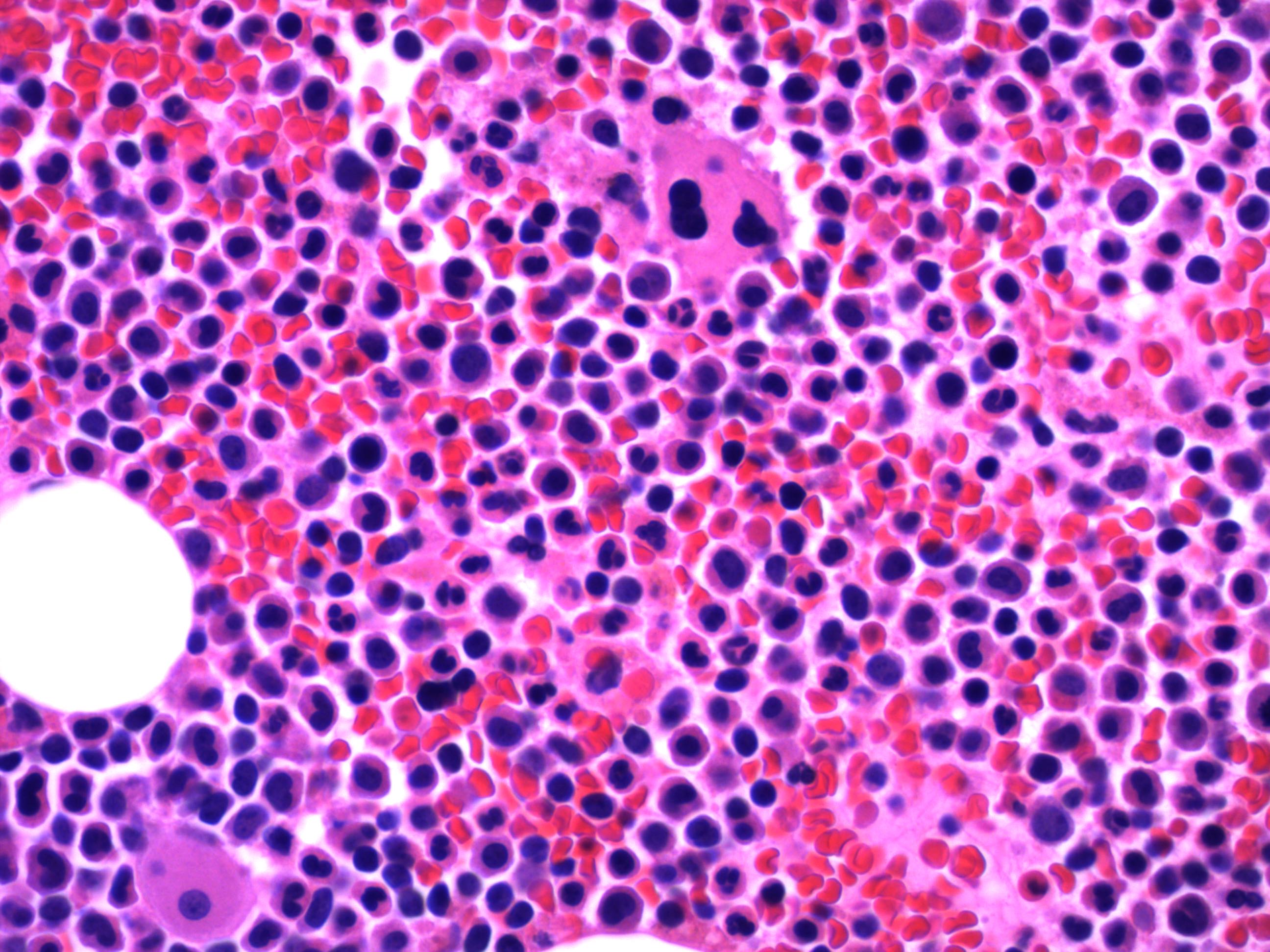
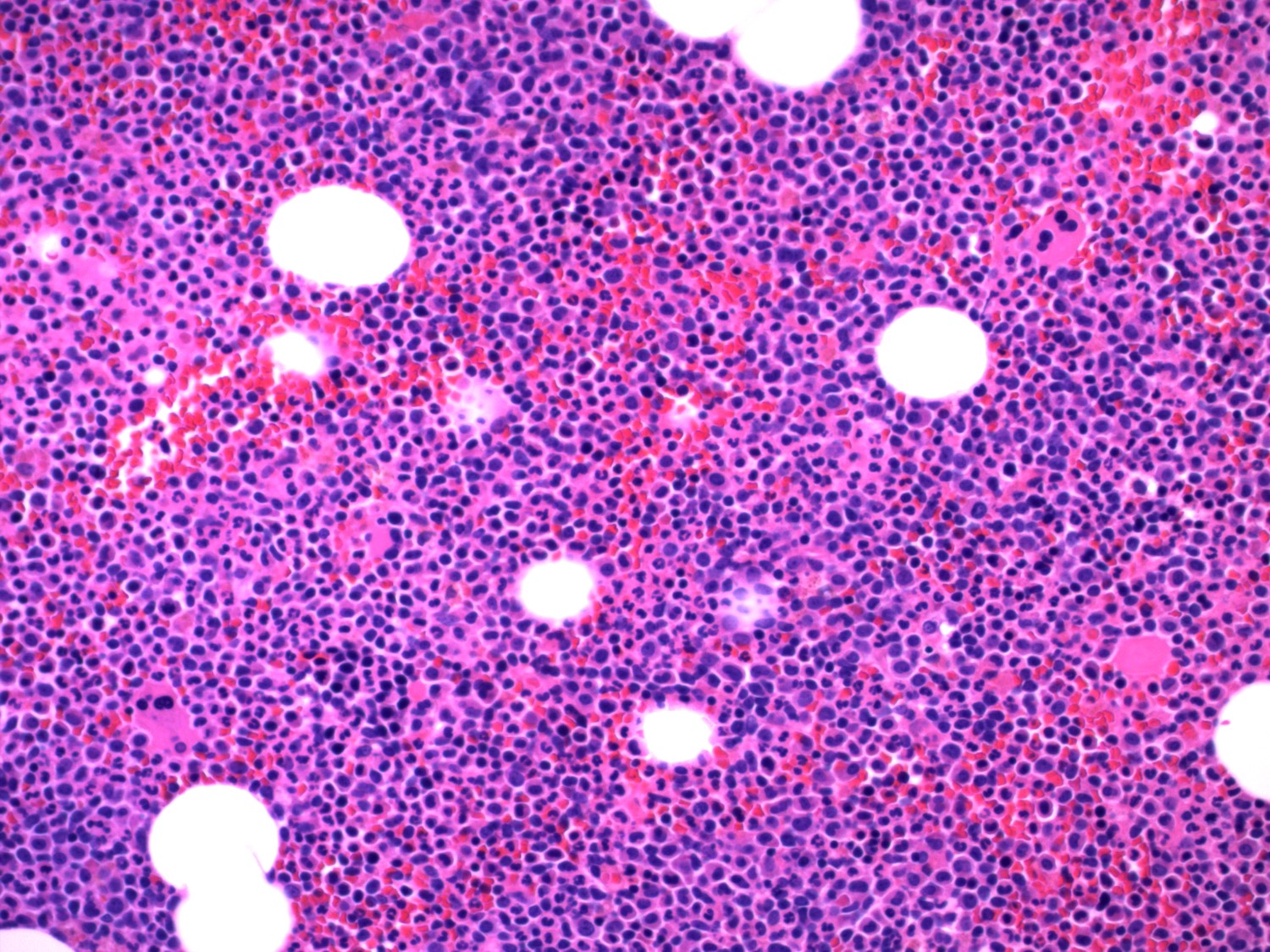
Dysmegakaryopoiesis

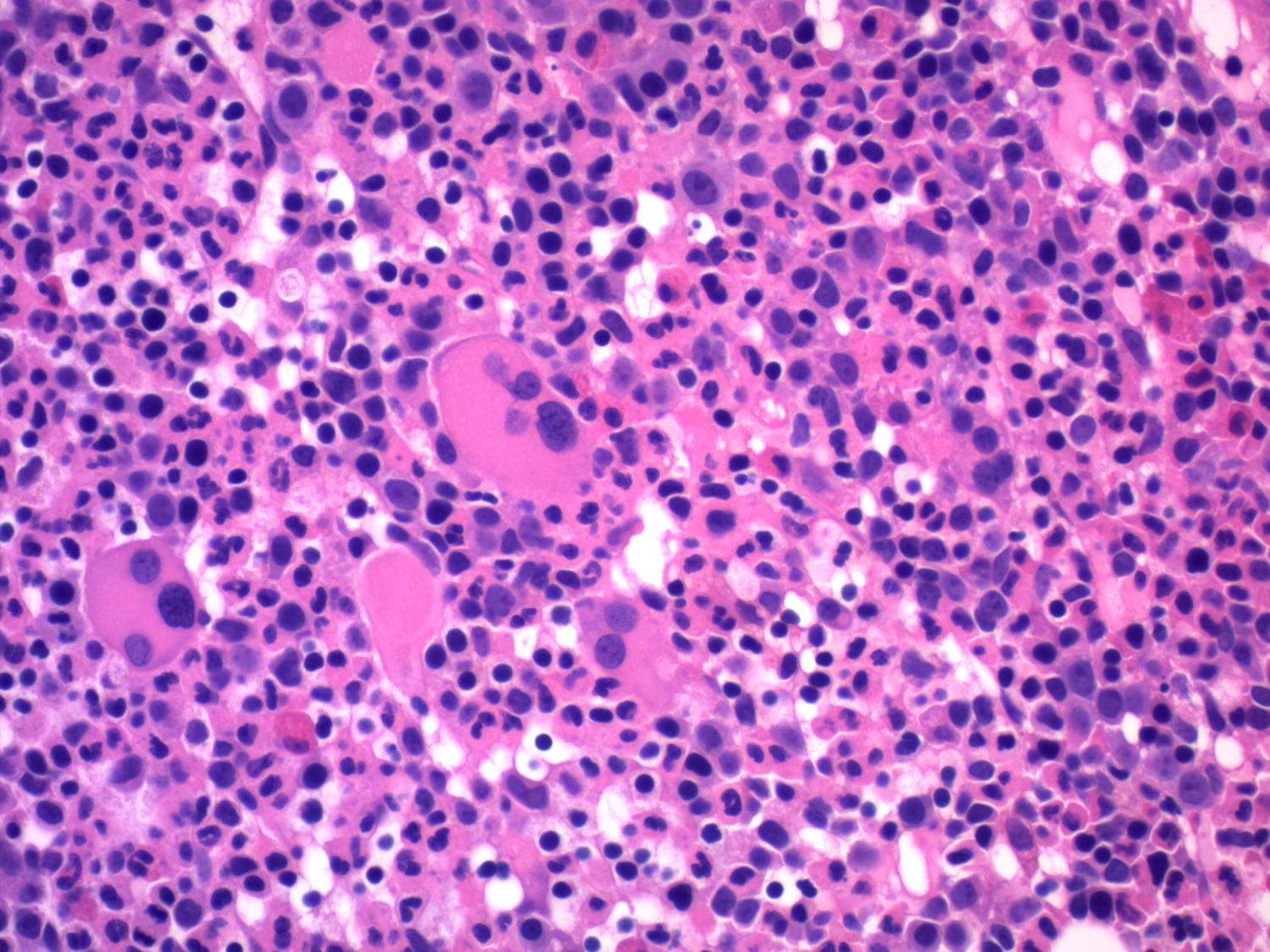
Dysmegakaryopoiesis

CD34 positive blasts

Hypercellular bone marrow
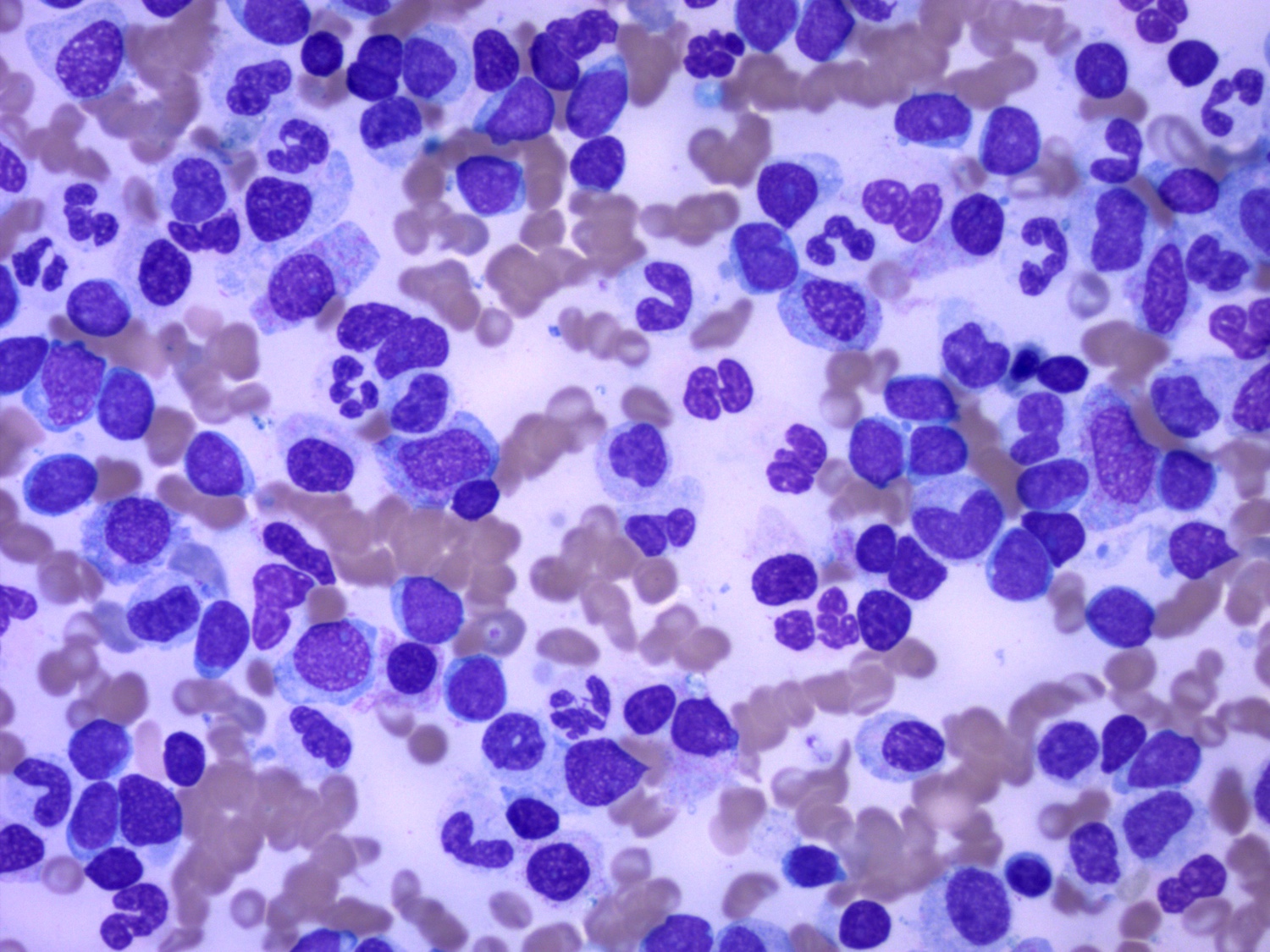
Dysmyelopoiesis and blasts

Myeloid dysplasia with blasts
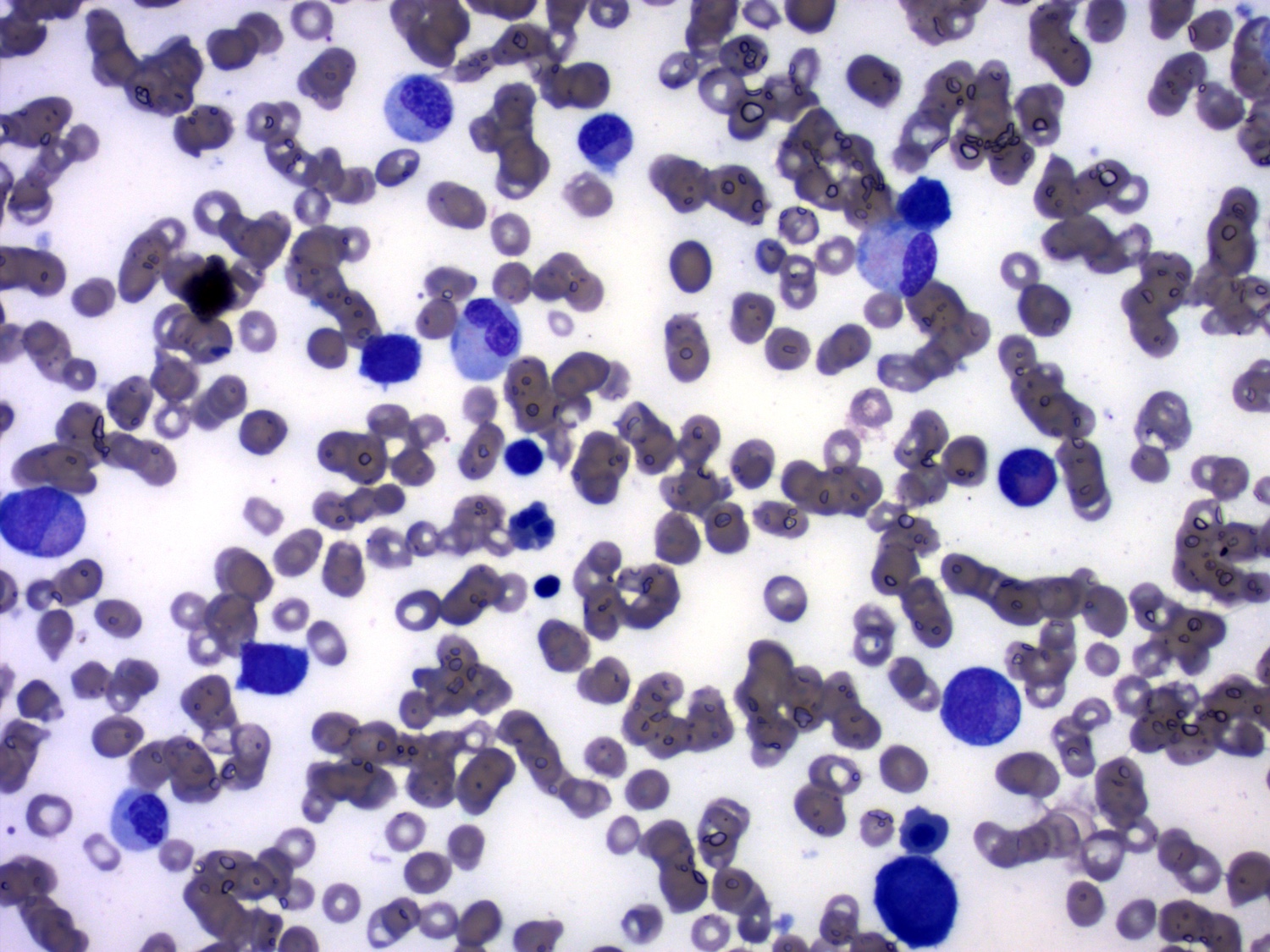
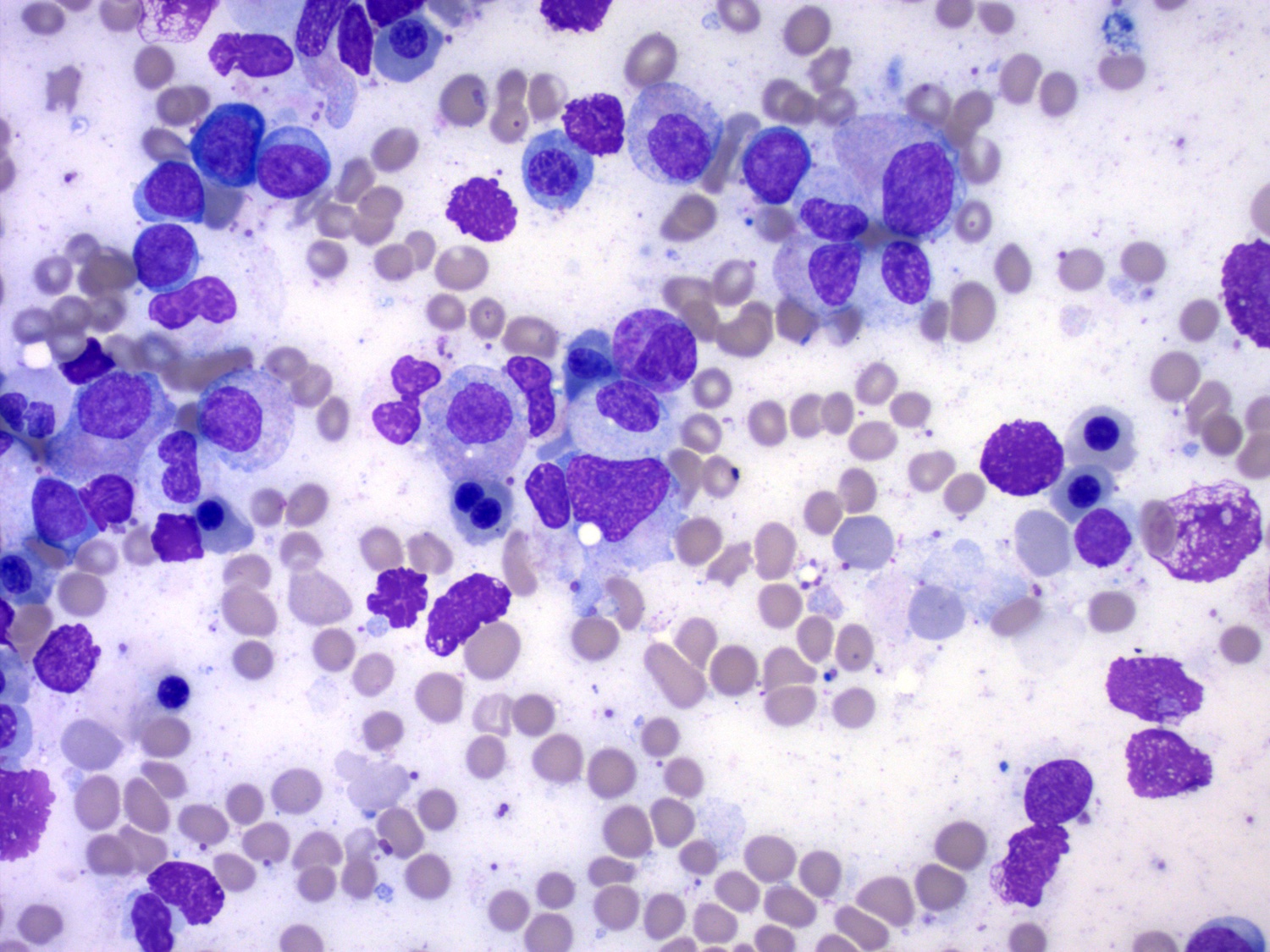
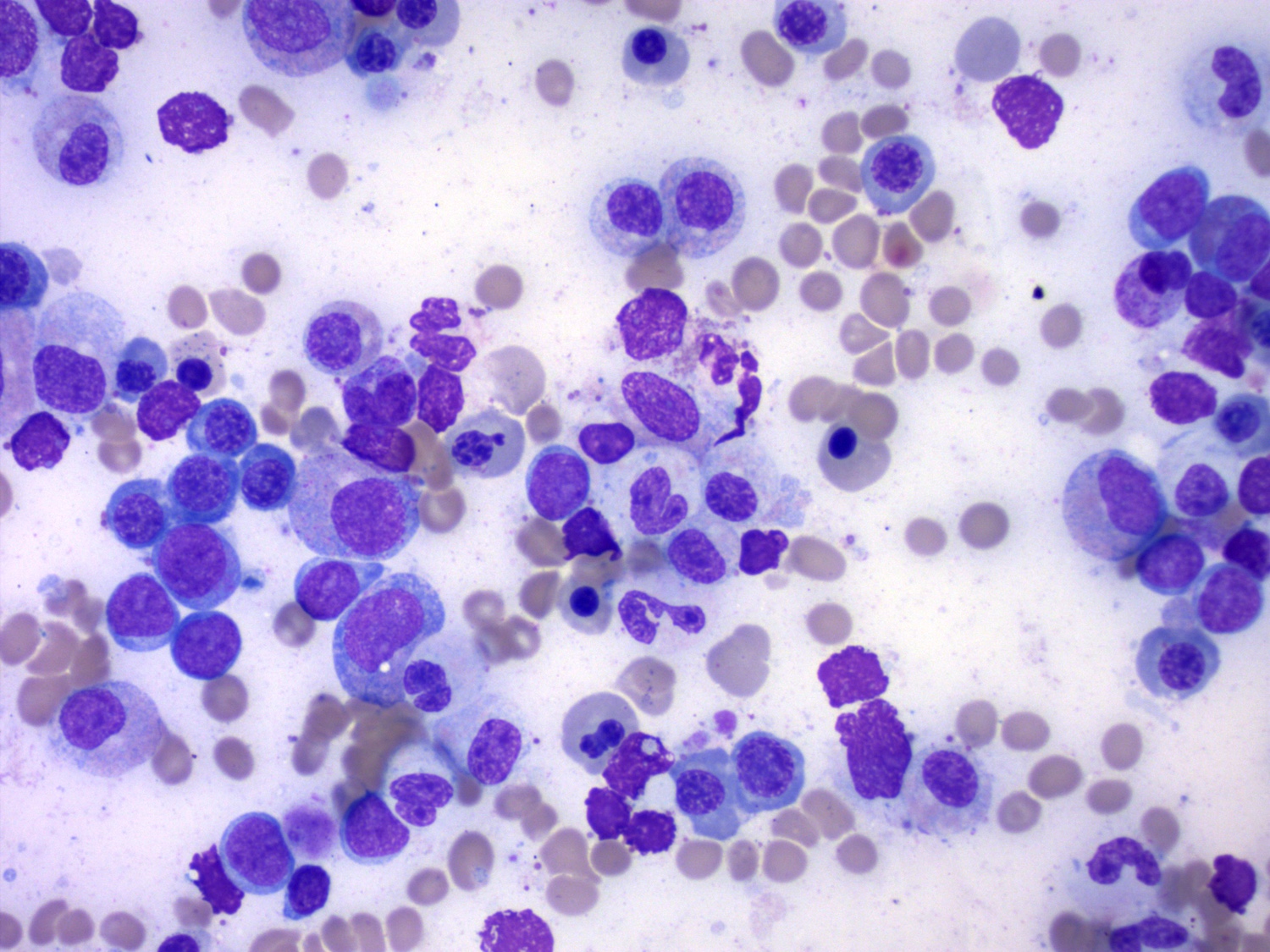
Bilineage dysplasia
- Nucleated red cells with dysplastic features in the form of nuclear budding and nuclear irregularities
- Increased red blood cell anisopoikilocytosis
- Large, hypogranular platelets
- Circulating blasts may be present
- Hypogranular or hypolobated myeloid elements
- Cytopenias
- Reference: Jaffe: Hematopathology, 2nd Edition, 2016
Contributed by Srishti Gupta, M.B.B.S., M.D. and Elizabeth Courville, M.D.
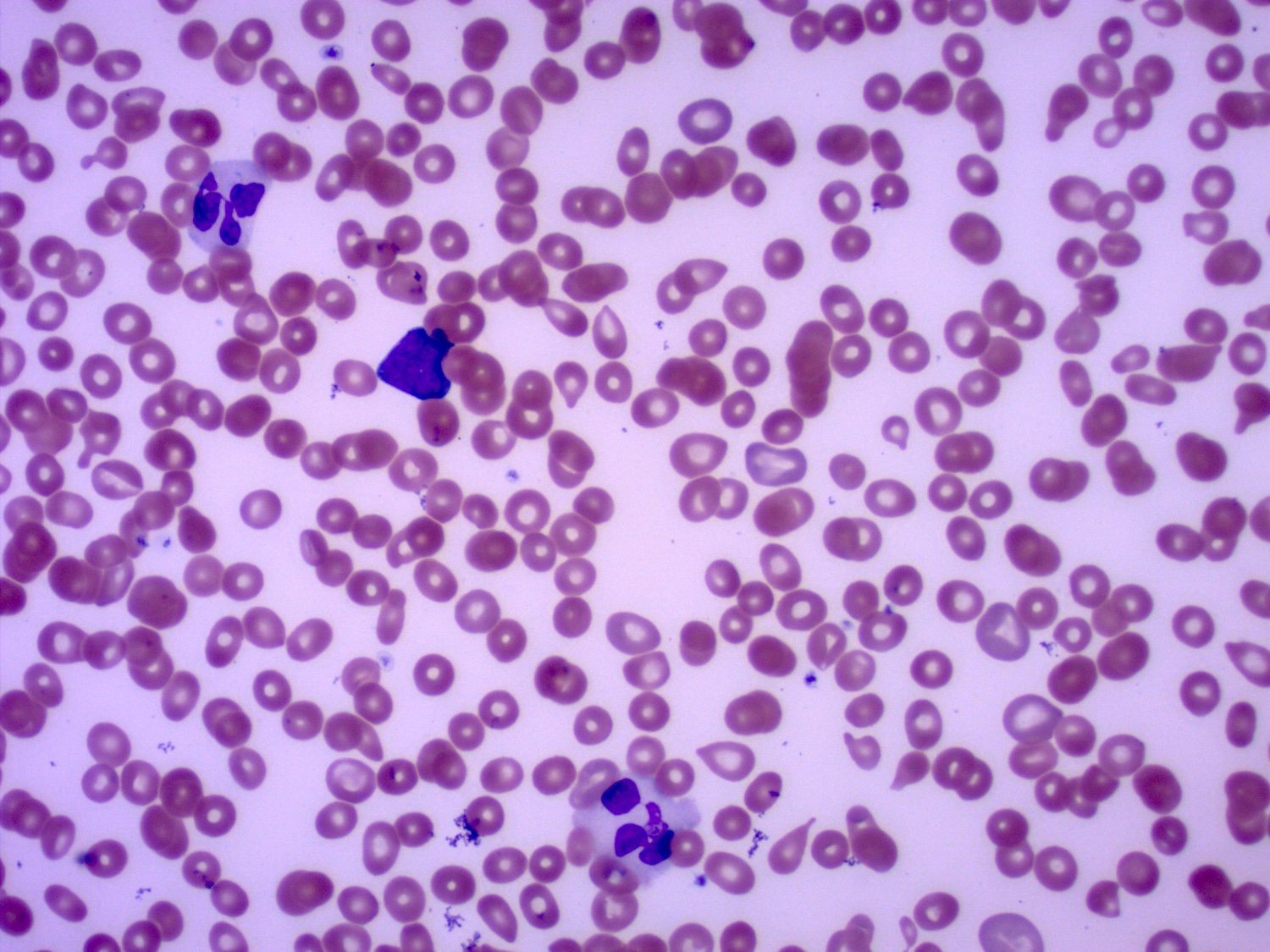
Hypogranular neutrophils, blast
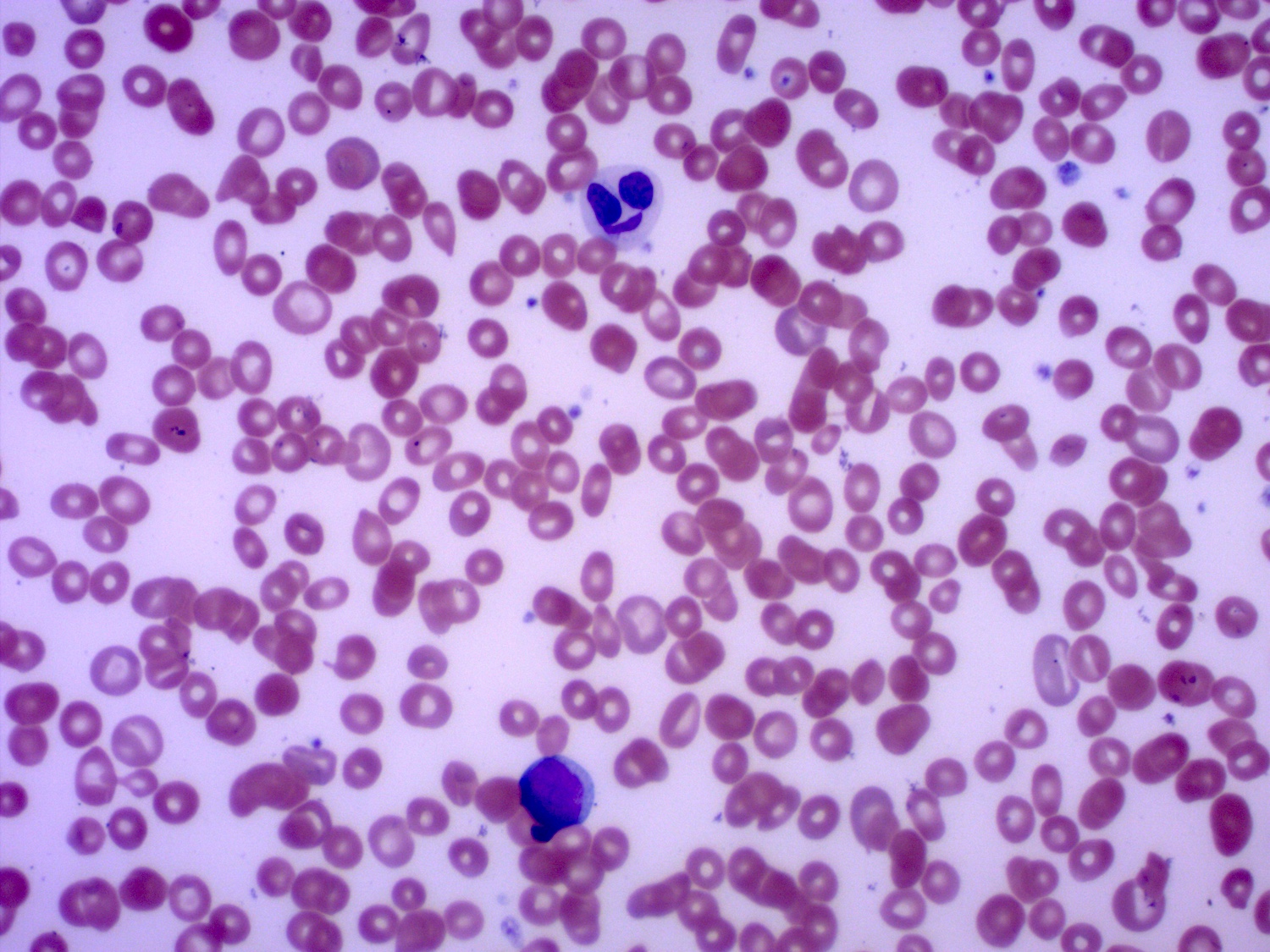
Hypogranular, hypolobate neutrophil, blast
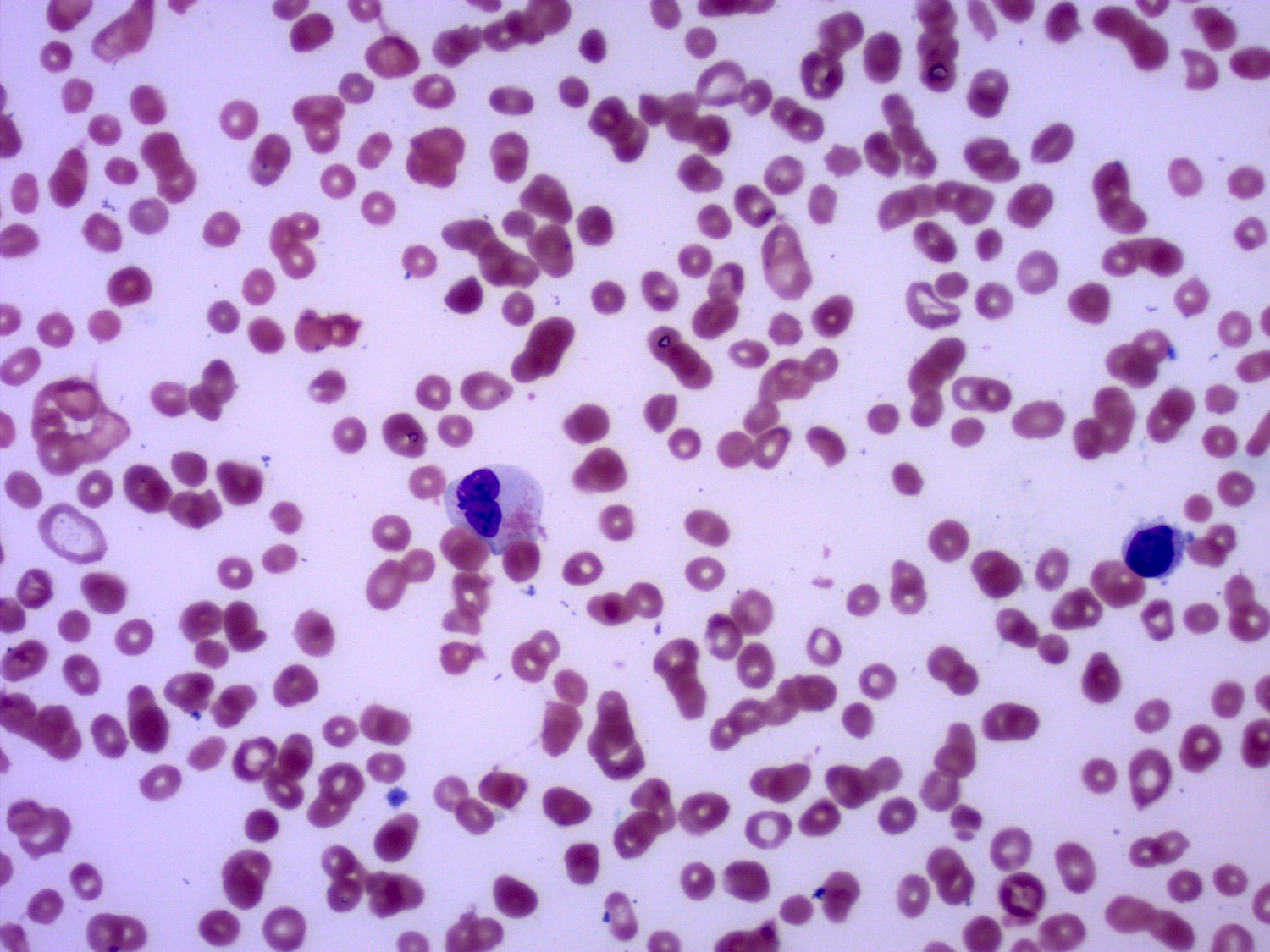
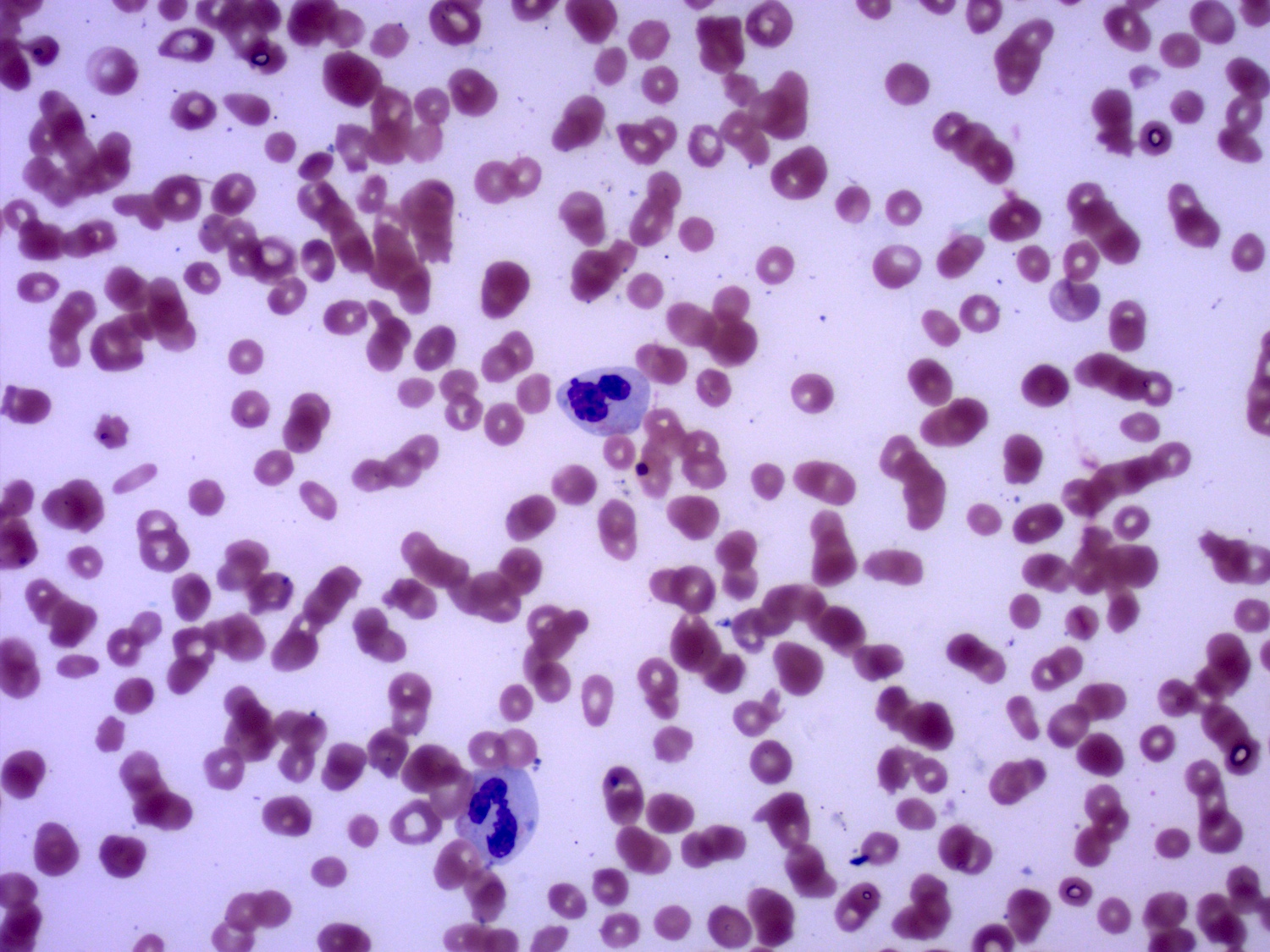
Hypogranular, hypolobate neutrophil
- CD61 may highlight micromegakaryocytes
- CD34 and CD117 may help with blast percentage estimation on the core biopsy
- Reference: Jaffe: Hematopathology, 2nd Edition, 2016
- Variable immunophenotypic features
- Immunophenotypic features are those reported in myelodysplastic syndromes, including reduced side scatter reflecting hypogranularity of neutrophils, aberrant differentiation patterns, loss of hematogones and aberrant expression of CD56 or CD7 (Acta Haematol 2019;141:232)
- Complex karyotypes, loss of chromosome 7 / del(7q), del(5q) and unbalanced translocations involving 5q are the most common cytogenetic abnormalities
- Balanced translocations are less common but frequently involve 5q32-33
- Presence of a specific genetic abnormality characteristic of AML with recurrent genetic abnormalities as defined in the WHO excludes a diagnosis of AML-MRC
- NPM1 mutation and biallelic mutation of CEBPA are fairly uncommon and are rarely associated with MDS related cytogenetic abnormalities
- MDS related mutations (including in U2AF1, ASXL1, TP53) are more frequent in this entity than in AML, NOS (Mod Pathol 2015;28:706)
Table 1: MDS related cytogenetic abnormalities (Am J Clin Pathol 2020;154:731)
| Balanced translocation |
|
| Unbalanced translocation |
|
| Complex cytogenetic abnormality | 3 or more unrelated cytogenetic abnormalities, excluding the recurrent cytogenetic abnormalities seen in AML |
Contributed by Eli Williams, Ph.D.

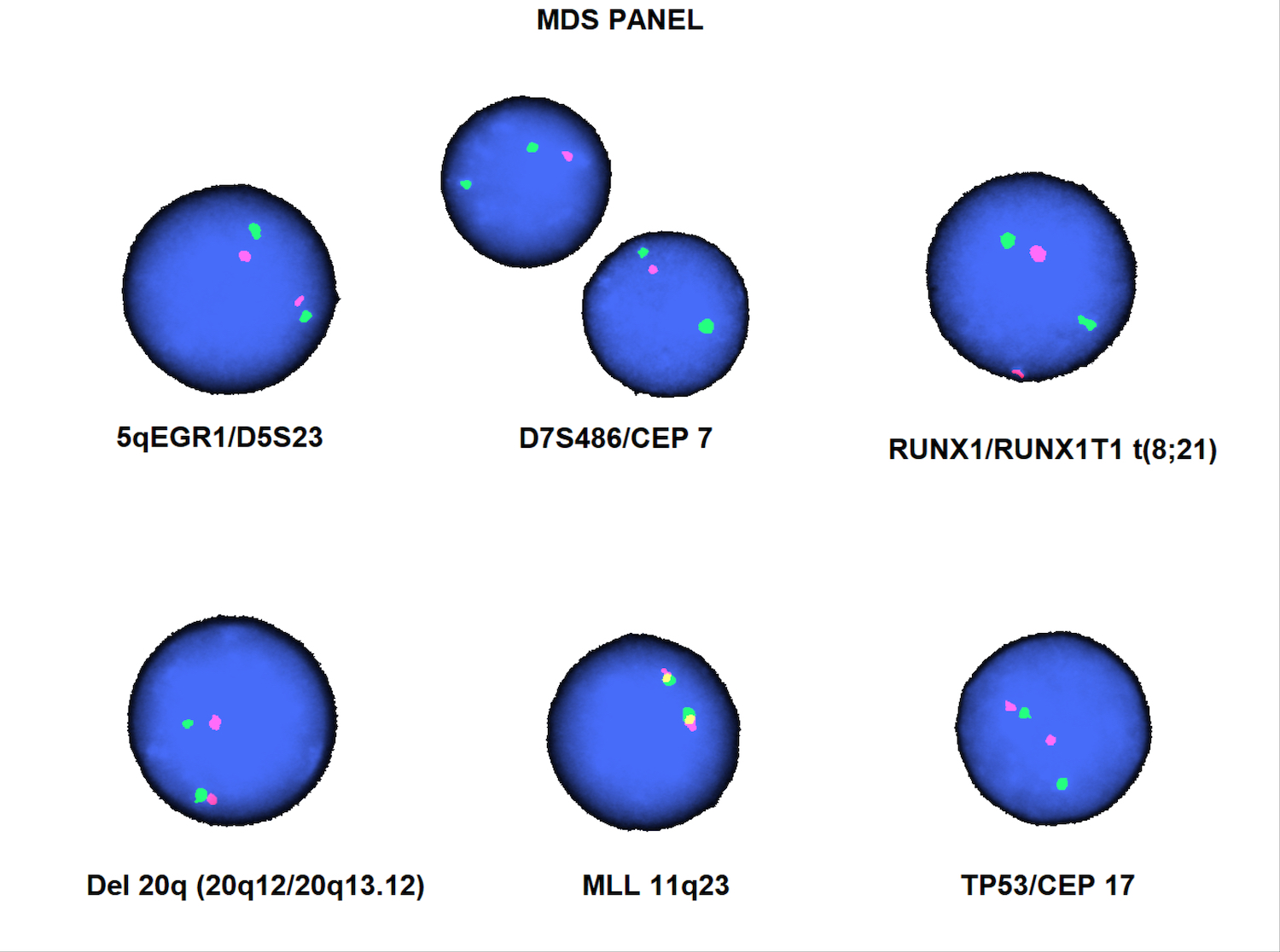
Deletion of 7(q)
- Posterior iliac crest, left / right, bone marrow aspirate, clot section and biopsy:
- Acute myeloid leukemia with myelodysplasia related changes (see comment)
- Comment: The patient's specimen demonstrates a hypercellular marrow with myeloid predominant trilineage hematopoiesis and increased blasts (> 20% by aspirate count and immunohistochemical stains). The peripheral blood shows leukopenia, thrombocytopenia and normocytic normochromic anemia. Flow cytometry analysis identified a large population of abnormal myeloid blasts. Overall, the findings in the current specimen are consistent with acute myeloid leukemia. Given the patient's history of myelodysplastic syndrome, acute myeloid leukemia with myelodysplastic related changes is a compatible diagnosis if a therapy related AML is clinically excluded and a defined recurrent cytogenetic abnormality is not identified (see WHO). Correlation with cytogenetic studies is recommended.
- Therapy related acute myeloid leukemia:
- Characterized by history of cytotoxic chemotherapy or relevant radiation therapy for an unrelated disease
- AML with recurrent cytogenetic abnormalities:
- AML with t(8;21), inv(3) or t(6;9) may show evidence of multilineage dysplasia
- These entities are defined by the presence of defined recurrent cytogenetic abnormalities
- Acute panmyelosis with myelofibrosis:
- Marrow is hypercellular and shows panmyelosis within a diffusely fibrotic stroma
- No prior history of MDS or MDS / MPN
- No AML-MRC defining cytogenetic abnormalities
- Distinction from AML-MRC with myelofibrosis may be difficult
- Acute megakaryoblastic leukemia:
- No prior history of MDS or MDS / MPN
- No AML-MRC defining cytogenetic abnormalities
- No accompanying granulocytic or erythroid dysplasia
- Pure erythroid leukemia:
- Characterized by erythroid precursors representing > 80% of the bone marrow cells, with ≥ 30% proerythroblasts
- Some of the cases previously classified as erythroleukemia (erythroid / myeloid type) may be categorized as AML-MRC according to the current classification system (Blood 2016;127:2391)
- Pure erythroid leukemia should not be diagnosed as AML with myelodysplasia related changes, even if there is a history of a prior myeloid neoplasm, as AML-MRC by definition requires myeloblasts whereas the neoplastic cells in pure erythroid leukemia are erythroid blasts
A patient is diagnosed with de novo myeloid neoplasm. There is multilineage dysplasia (> 50% in 2 of the lineages) and > 20% myeloid blasts, as you can see in the above image from the bone marrow aspirate. The karyotype is normal and an NPM1 mutation is identified. What is the most appropriate diagnosis?
- Acute erythroid leukemia
- Acute myeloid leukemia with myelodysplasia related changes
- Acute myeloid leukemia, NOS
- Therapy related myeloid neoplasm (acute myeloid leukemia)
- Biallelic mutation of CEBPA
- History of below the knee amputation for diabetic complications
- History of cytotoxic chemotherapy for breast carcinoma
- History of myelodysplastic syndrome (MDS)
Comment Here
Reference: AML with myelodysplasia related changes
- Acute myeloid leukemia (AML) with other KMT2A rearrangements: subclassification of AML defined by the International Consensus Classification (ICC) as AML with rearrangement of the KMT2A gene other than t(9;11) / MLLT3::KMT2A
- Recurrent translocations recognized in this category include AMLs with t(4;11) AFF1::KMT2A; t(6;11) / AFDN::KMT2A; t(10;11) / MLLT10::KMT2A; t(10;11) / TET1::KMT2A; t(11;19) / KMT2A::ELL; t(11;19) / KMT2A::MLLT1
- In the 5th edition of WHO, the category of AML with KMT2A rearrangement includes any KMT2A gene rearrangement, regardless of the partner gene
- KMT2A rearrangement is a recurrent genetic finding identified in both acute lymphoblastic leukemias / lymphomas (ALL) and AML
- KMT2A rearrangement has an inverse association with patient age, with a large subset of infantile cases of AML presenting with KMT2A rearrangement
- AML with KMT2A rearrangement most commonly presents with a monocytic phenotype, with myelomonocytic and myeloid phenotypes less frequent
- KMT2A has numerous partner genes; by International Consensus Classification (ICC) 2022 definition, AML with other KMT2A rearrangements include t(4;11) AFF1::KMT2A; t(6;11) / AFDN::KMT2A; t(10;11) / MLLT10::KMT2A; t(10;11) / TET1::KMT2A; t(11;19) / KMT2A::ELL; t(11;19) / KMT2A::MLLT1
- Overall prognosis is poor but prognosis varies somewhat with fusion gene
- KMT2A rearrangement can be identified by various testing methods including conventional karyotype, FISH, PCR and NGS
- KMT2A gene was previously named mixed lineage leukemia (MLL)
- WHO 5th edition classification: acute myeloid leukemia with KMT2A rearrangement
- ICC 2022 classification: acute myeloid leukemia with other KMT2A rearrangements
- ICD-O: 9897/3 - acute myeloid leukemia with KMT2A rearrangement
- ICD-10
- ICD-11: 2A60.0 & XH1E41 - acute myeloid leukemia with recurrent genetic abnormalities & acute myeloid leukemia, 11q23 abnormalities
- KMT2A rearrangement
- Incidence has an inverse relationship with age (Blood 2003;102:2395)
- Most frequently encountered genetic abnormality in pediatric population, accounting for 20% of pediatric AML cases
- Less common among adult population, accounting for 2 - 3% of adult AML cases
- Has increased prevalence among patients with AML post-cytotoxic therapy (AML pCT) (Cytometry B Clin Cytom 2022;102:123)
- By definition, AML with other KMT2A rearrangement involves bone marrow and peripheral blood
- Prevalence of extramedullary disease is higher than that of non-KMT2A rearranged leukemia in both adults and pediatric populations with a high frequency of fusion partners (Proc Natl Acad Sci U S A 2020;117:26340)
- Extramedullary disease most frequently manifests in the liver, spleen or CNS; less commonly as myeloid sarcoma in other locations
- Data is limited for low frequency fusion partners
- Myeloid precursor cell undergoes genetic event resulting in rearrangement of KMT2A gene on chromosome 11
- KMT2A rearrangement in AML usually manifests as a translocation
- Rearrangement of KMT2A disrupts its usual role in regulating gene expression during early development and hematopoiesis; this disruption will lead to clonal expansion of the myeloid cell lineage harboring the KMT2A (Cancer Lett 2019;458:56)
- Clonal expansion will result in increased myeloid lineage blasts, further developing into bone marrow replacement and subsequent pancytopenias
- Definitive etiology is not well understood
- Risk factors in adult patients include cytotoxic chemotherapy and other nonspecific DNA damaging events
Images hosted on other servers:

Frequent KMT2A partner genes
- WHO criteria for AML with KMT2A rearrangement
- Essential
- Myeloid neoplasm with increased peripheral blood or bone marrow blasts (may be < 20%)
- Blasts express a myeloid immunophenotype, not fulfilling immunophenotypic criteria for mixed phenotype AML
- Presence of KMT2A rearrangement
- Not fulfilling diagnostic criteria for myeloid neoplasm post-cytotoxic therapy
- Desirable
- Identification of the KMT2A fusion partner
- ICC 2022 criteria for AML with other KMT2A rearrangement (Blood 2022;140:1200)
- Overall criteria similar to the WHO, except importantly t(9;11) / MLLT3::KMT2A is categorized as a separate entity
- Recognized recurrent fusion partners are described in other sections
- Identification of KMT2A rearrangement is critical for this diagnosis (see Molecular / cytogenetics description for further description)
- At least 10% myeloblasts in the peripheral blood or bone marrow sampling are required for diagnosis by ICC classification
- Essential
- Anemia and thrombocytopenia likely present at diagnosis
- WBC will vary case to case; ranges from leukopenia to leukocytosis and correlates with number of circulating blasts
- In general, prognosis of KMT2A rearranged AML is poor with high risk of relapse; however, prognosis varies upon fusion partner to KMT2A gene (Blood Cancer J 2021;11:162)
- Few partner genes may give intermediate to good prognosis
- Overall median survival in patients with KMT2A rearrangement is 0.9 years
- Additional cytogenetic abnormalities and mutations, especially in myelodysplasia associated genes, worsens prognosis
- 31 weeks gestational age female neonate with AFF1::KMT2A fusion AML (Oncol Lett 2022;24:283)
- 6 year old girl with KMT2A::MLLT10 fusion AML (Cancer Genet 2023;276:36)
- 7 year old boy with KMT2A::MLLT1 fusion AML (Cytogenet Genome Res 2019;157:213)
- 43 year old woman with KMT2A::SEPT5 fusion AML (SAGE Open Med Case Rep 2018;6:2050313X17750334)
- 63 year old woman with hepatic gastrointestinal stromal tumor (GIST) and concomitant KMT2A::ELL fusion AML (World J Clin Cases 2020;8:1251)
- Intensive chemotherapy as induction followed by additional consolidation therapy
- Trials utilizing novel agents such as MEK inhibitors, demethylating agents and other targeted therapies are ongoing (Front Pharmacol 2022;13:749472)
- Hematopoietic stem cell transplant
- Increased myeloid lineage blasts (percentage will vary case to case)
- WHO 5th edition does not have a blast requirement and the ICC requires at least 10% blasts in peripheral blood or bone marrow
- On the aspirate smears, crush prep or touch prep, blast morphology will vary by phenotype
- Myeloblasts will have open chromatin, high N:C ratio, prominent nucleoli and possible Auer rods in the cytoplasm
- Monoblasts will have open chromatin, lower N:C ratio, possible cytoplasmic vacuoles and larger cell size
- AML with monocytic differentiation tends to have a spectrum of monocyte differentiation, with mature monocytes, promonocytes and monoblasts (Haematologica 2009;94:994)
- Unless there are coinciding myelodysplasia related gene mutations, significant dysplasia is not expected
- On core biopsies, varying degrees of bone marrow involvement by blasts will be seen
- Blasts will appear as immature cells with intermediate to large cell size, open chromatin and usually with prominent nucleoli
- Pattern of involvement may be interstitial, aggregate or sheets
- Degree of bone marrow effacement by leukemia will impact background trilineage hematopoiesis
- Trilineage hematopoiesis will be affected by treatment in follow up bone marrow biopsies
Contributed by William Morrow, M.D.

Aspirate with increased blasts

Aspirate with increased monoblasts

Increased monoblasts

Clot section blast aggregate

Core biopsy blast sheets
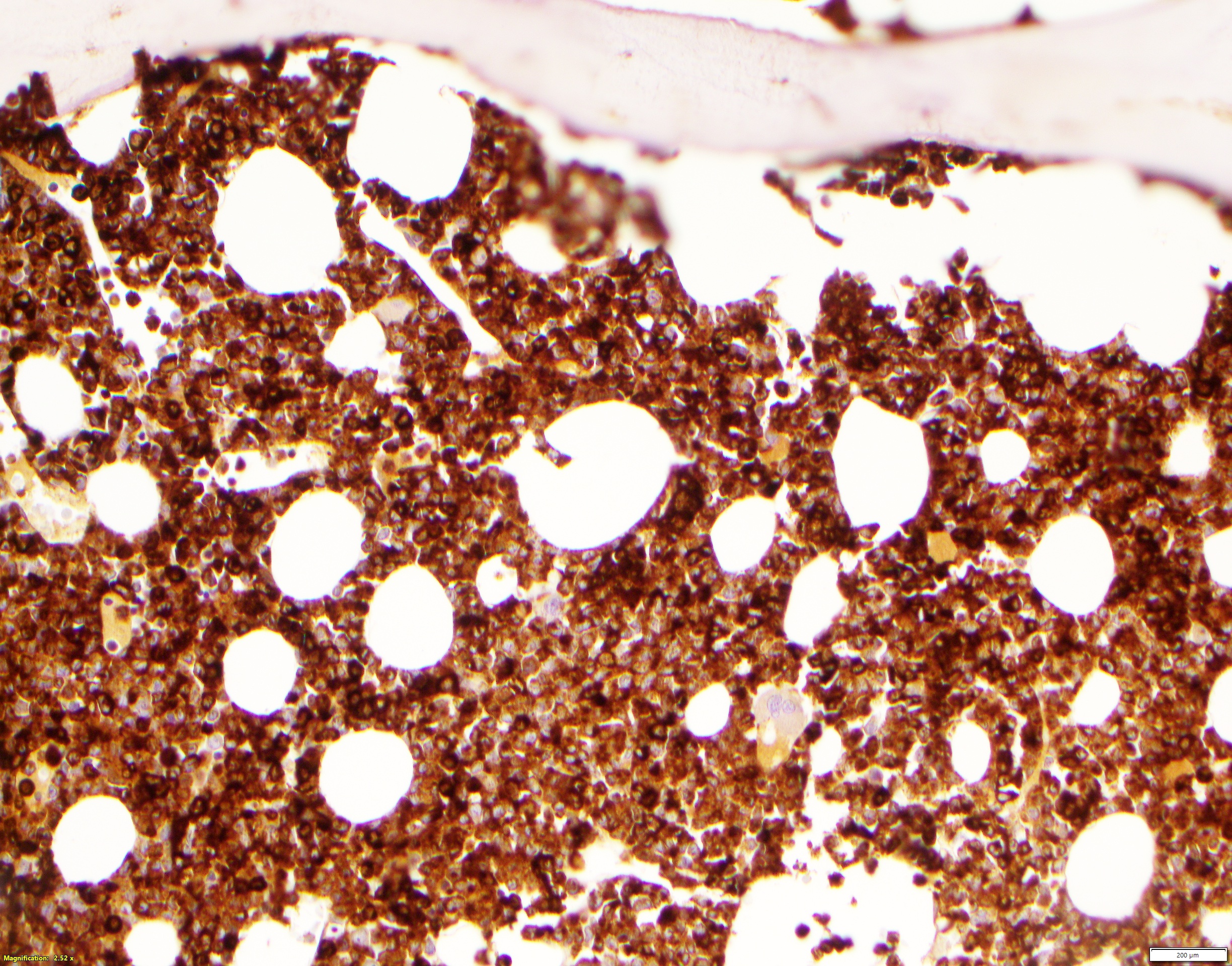
Muramidase (lysozyme) IHC

CD4 IHC
Images hosted on other servers:

Acute myeloid leukemia
- Increased circulating blasts, frequently with monocytic features, such as increased cytoplasm, vacuoles, cleaved or grooved nuclei
- Variable degree of cytopenias depending on level of bone marrow involvement
- Typically a normocytic, normochromic anemia with otherwise unremarkable red blood cell morphology
- Typically a thrombocytopenia with unremarkable platelet morphology
- No features of dysplasia in granulocytes unless there is a coinciding myelodysplasia related gene mutation(s)
- Reference: Haematologica 2009;94:994
Contributed by William Morrow, M.D.

Circulating blasts
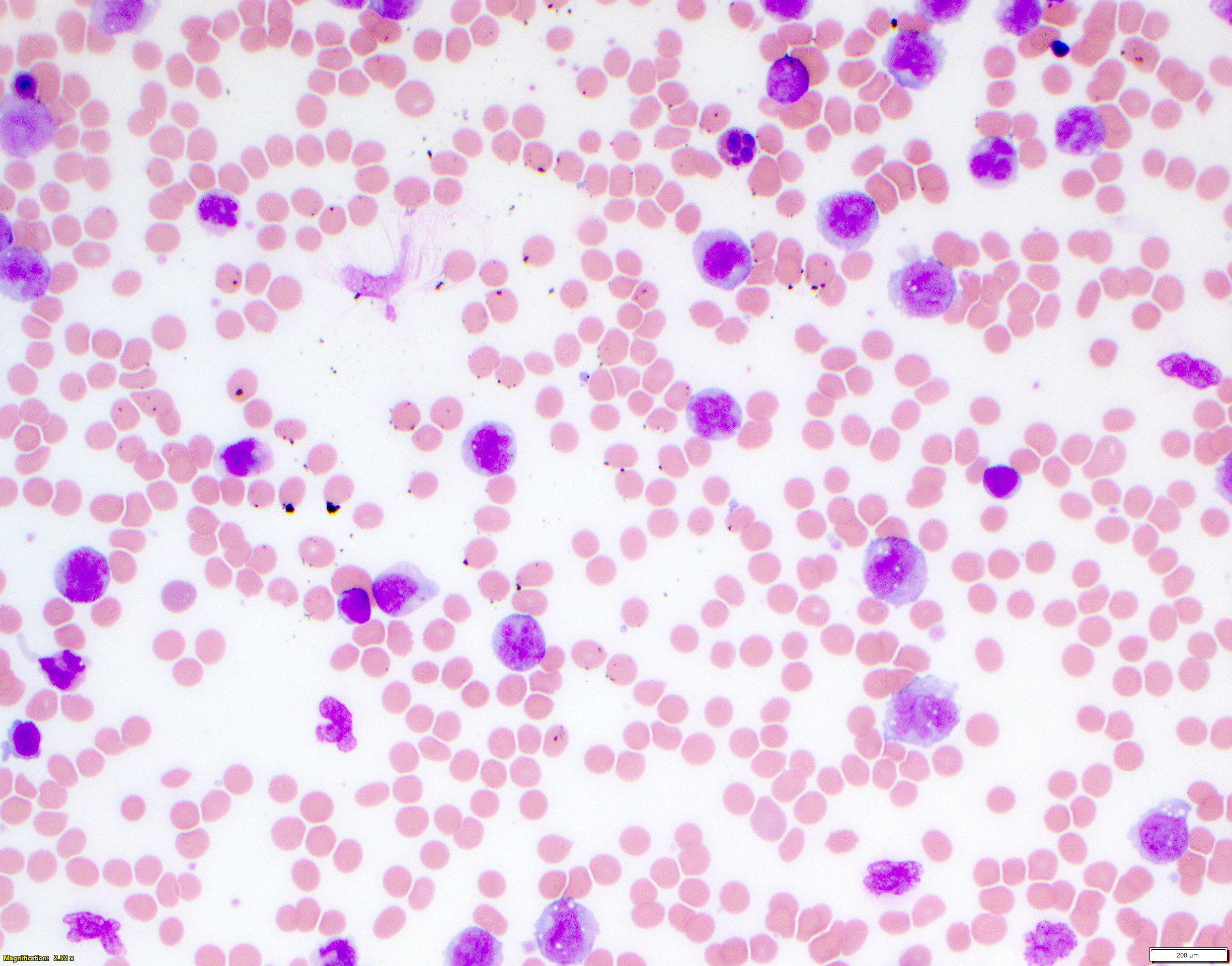
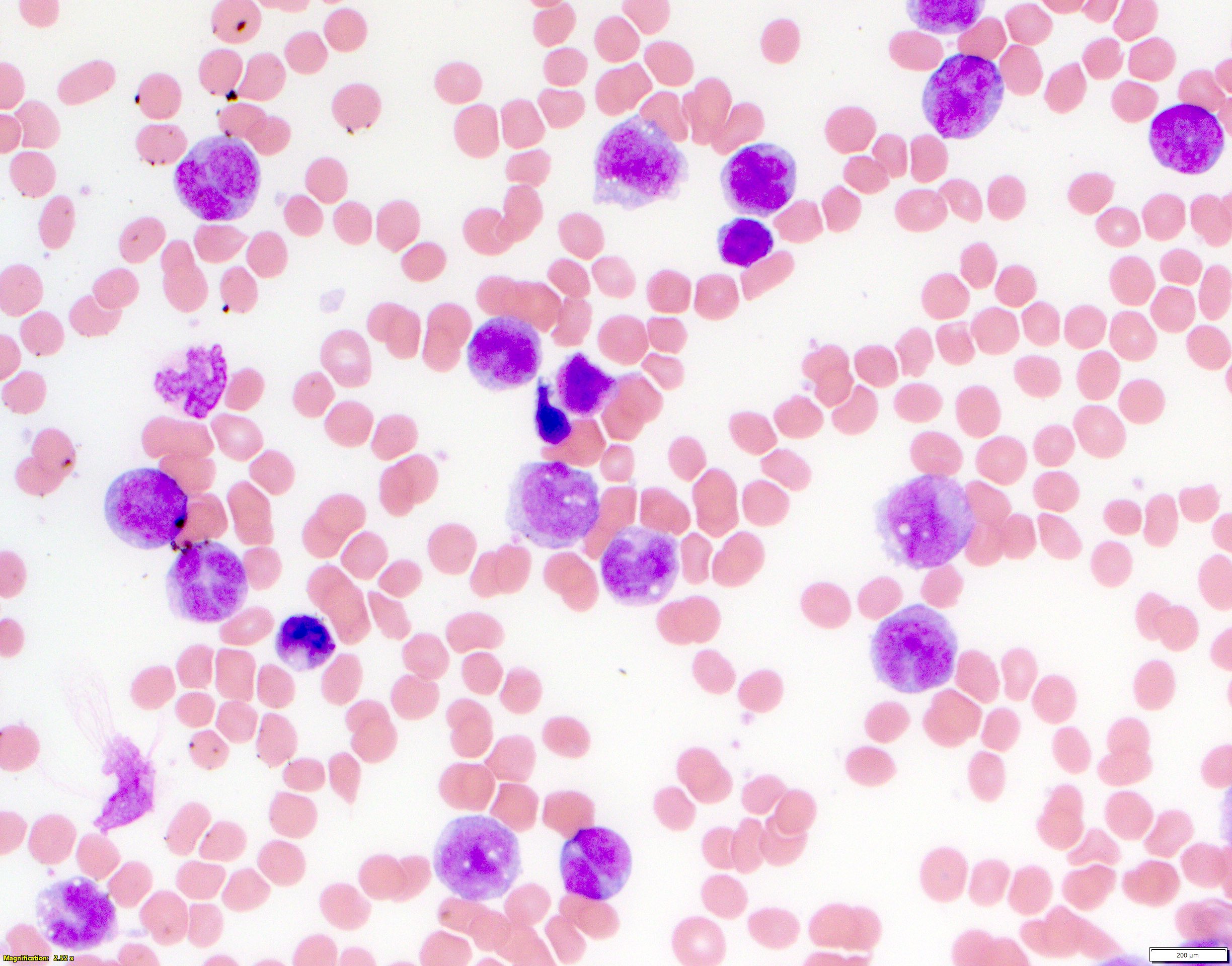
Circulating monoblasts
- Immunophenotype by flow cytometry varies but most commonly represents monocytic differentiation, with a smaller subset of cases having myelomonocytic or myeloid immunophenotype
- Cases with monocytic differentiation will be positive for CD33, CD13, CD64, CD4, HLA-DR, CD14 (variable) and negative for CD34, CD117 and MPO
- Cases with myeloid differentiation will be positive for CD34, MPO, CD33, HLA-DR and negative for CD64, CD4 and CD14
- CD123 is positive in 90% of cases with KMT2A rearrangement (Cytometry B Clin Cytom 2022;102:123)
- Markers of lymphoblastic differentiation, such as TdT, CD3, CD19, CD10 are almost universally negative
Contributed by William Morrow, M.D.

Flow CD33 versus CD64

Flow CD11b versus CD64

Flow CD33 versus CD34
- KMT2A gene (lysine methyltransferase 2A) is located on chromosome 11q23.3
- More than 130 different partners have been recognized in association with KMT2A; AFF1, MLLT1, MLLT3, MLLT10, MLLT4 and ELL are the most frequently recognized partners in order of decreasing frequency (BMC Med Genomics 2020;13:106)
- These genes represent over 90% of KMT2A rearrangement cases and are specifically recognized by the ICC 2022 classification for KMT2A with other rearrangement
- Due to the various different breakpoints in the KMT2A gene, comprehensive testing for KMT2A rearrangement can be challenging
- Different types of structural rearrangements can be the underlying cause of KMT2A fusions, including insertions, deletions, inversions and translocations
- Several different molecular and cytogenetic testing modalities can identify KMT2A rearrangement
- FISH is routinely performed using the aspirate material collected during the bone marrow biopsy procedure; KMT2A break apart probes (with a probe labeling 3’KMT2A and a probe labeling 5’KMT2A) are routinely ordered as part of a FISH panel for a diagnostic leukemia marrow
- Some rearrangements may be cryptic by FISH, such as KMT2A::USP2
- Routine karyotype can detect many translocations involved in KMT2A rearrangement (such as t(9;11)); however, some translocations may be cryptic (such as t(10;11) MLLT10::KMT2A)
- Additional cytogenetic alterations can coincide with KMT2A rearrangement
- PCR can be used to detect specific fusion transcripts
- Some available panels include up to 20 fusion genes, including all the recurrent fusions identified
- Myeloid next generation sequencing (NGS) panels may have KMT2A gene incorporated into a fusion panel and can detect KMT2A
- FISH is routinely performed using the aspirate material collected during the bone marrow biopsy procedure; KMT2A break apart probes (with a probe labeling 3’KMT2A and a probe labeling 5’KMT2A) are routinely ordered as part of a FISH panel for a diagnostic leukemia marrow
- Of note, partial tandem duplication (PTD) of KMT2A may be detected by NGS; PTD of the KMT2A gene does not qualify as KMT2A rearrangement for either classification scheme
Contributed by William Morrow, M.D.
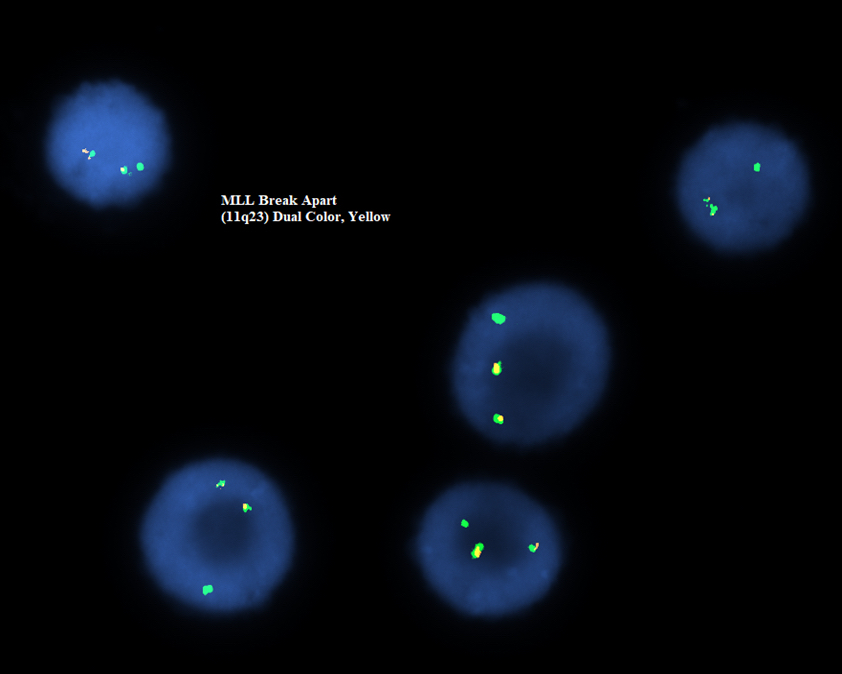
KMT2A break apart FISH
Images hosted on other servers:

Cryptic KMT2A::MLLT4 fusion

FISH of KMT2A::SEPT5 fusion
- Bone marrow, biopsy:
- Acute myeloid leukemia with other KMT2A rearrangement (see comment)
- Comment: Increased blasts (50%) in a hypercellular marrow (90%). The blasts are morphologically and immunophenotypically consistent with myeloblasts. Concurrent FISH studies identified KMT2A rearrangement (see FISH report for full details). Concurrent karyotype studies identified t(10;11) (see karyotype report for full details). A myeloid NGS panel recognized a KMT2A::MLLT10. Overall, these findings are consistent with acute myeloid leukemia with KMT2A rearrangement (WHO 5th edition) or acute myeloid leukemia with other KMT2A rearrangement (ICC 2022).
- Acute myeloid leukemia with t(9;11) / MLLT3::KMT2A:
- Differentiated by defining KMT2A rearrangement
- Classified separately by the ICC
- Shares classification by the WHO 5th edition
- Acute myeloid leukemia with other defining genetic abnormalities:
- Lacks KMT2A rearrangement
- Presence of other defining genetic feature
- Acute lymphoblastic leukemia with KMT2A rearrangement:
- Differentiated by immunophenotype and cell lineage
- Acute monocytic leukemia:
- Myeloid leukemia with monocytic features lacking KMT2A rearrangement
- Chronic myelomonocytic leukemia with KMT2A rearrangement:
- No increase in blasts
- Dysplastic features
- Rare and difficult to diagnose
- Increased blasts and KMT2A rearrangement better classified as AML
A patient with newly diagnosed acute myeloid leukemia had cytogenetics, FISH and myeloid NGS testing performed. FISH identified findings seen in the above image and myeloid NGS identified a KMT2A::AFDN fusion. Per the ICC 2022 guidelines, which of the following is the best classification of this patient's disease?
- Acute myeloid leukemia, NOS
- Acute myeloid leukemia with myelodysplasia related cytogenetics
- Acute myeloid leukemia with other KMT2A translocation
- Acute myeloid leukemia with t(9;11) / MLLT3::KMT2A
- B lymphoblastic leukemia with t(v;11q23.3) / KMT2A rearrangement
Comment Here
Reference: AML with other KMT2A rearrangements
- Eosinophilic
- Lymphoblastic
- Monocytic
- Myeloid
- Myelomonocytic
Comment Here
Reference: AML with other KMT2A rearrangements
- Not part of WHO classification
- Frequent in acute promyelocytic leukemia; up to 25% of AML-M2 (Acta Paediatr Jpn 1990;32:651)
- Giant granules may be due to fusion of primary granules or small dense vesicles
- See also Bone marrow nonneoplastic chapter
- 16 year old girl with AML-M5a and t(10;11) (Clin Lab Haematol 2000;22:303)
- Giant cytoplasmic granules
AFIP images
Specific granules
resemble
Chediak-Higashi
anomaly
- May be associated with double minutes (Leukemia 2002;16:152)
- Peroxidase positive granules with a dense matrix but no obvious crystalline structure, may contain membranous lamellae or tubular structures (Cancer Res 1980;40:4473, Sangre (Barc) 1994;39:135)
- Rare type of acute myeloid leukemia (AML)
- AML with myelodysplastic syndrome (MDS) related changes (WHO, 2017)
- Balanced translocation meeting criteria for AML with MDS related changes
- Blasts > 20% and no prior therapy
- Described as an entity under the category of AML with other rare recurring translocations in International Consensus Classification (ICC) (Blood 2022;140:1200)
- AML with fusion of PRDM16 and RPN1
- Rare subtype of AML with rare genetic abnormalities and differentiation
- Defined by t(1;3)(p36;q21) (PRDM16::RPN1)
- Associated with dysplasia, especially dysmegakaryopoiesis (Cancer Genet Cytogenet 2010;203:187, Genes Chromosomes Cancer 2003;36:313)
- Associated with peripheral thrombocytosis
- Associated with poor prognosis
- Acute myeloid leukemia with t(1;3)(p36;q21) (PRDM16::RPN1)
- AML with t(1;3)(p36;q21) (PRDM16::RPN1)
- ICD-O: 9895/3 - acute myeloid leukemia with myelodysplasia related changes
- Rare, occurs mainly in elderly patients (WHO, 2017) (Cancer Genet Cytogenet 2010;203:187)
- Wide age range (30 - 80 years); M = F (Cancer Genet Cytogenet 2010;203:187)
- t(1;3)(p36;q21) feature constitutes ≤ 1% of overall MDS cases (WHO, 2017)
- Bone marrow, spleen and liver
- Lymph node involvement can also occur
- Genes at break point of translocation include RPN1 at 3q21.3 and group of genes at PRDM16 at 1p36.3 (Br J Haematol 2012;156:76)
- Related to chromosome 3 abnormalities, such as inv(3)(q21q26), t(3;3)(q21;q26), ins(3;3)(q26;q21q26) and t(1;3)
- t(1;3) show dysmegakaryopoietic features similar to the 3q21q26 syndrome
- PRDM16 gene (MEL1, MDS1::EVI1-like gene) is activated as a result of translocation of the gene next to RPN1 gene at 3q21 (Genes Chromosomes Cancer 2003;36:313)
- PRDM16 gene at 1p36.3 (the breakpoints are located within a 90 kb region in the 5' region of PRDM16)
- RPN1 gene at 3q21 (the breakpoints are located within a 60 kb region centromeric to the breakpoint cluster region of the 3q21q26 / inv(3) syndrome)
- Aberrant expression of PRDM16 as a result of RPN1 translocation is thought to be associated with pathogenesis of this AML subtype
- Aberrant expression of PRDM16 as a result of RPN1 translocation
- Majority of cases present
- Anemia (common)
- Thrombocytosis and leukocytosis
- Pancytopenia
- Some cases present as MDS and then progress to AML (WHO, 2017)
- Translocation (1;3) can be the sole chromosomal abnormality at initial diagnosis or can be associated with other chromosomal abnormalities
- Peripheral thrombocytosis is a distinct feature (Cancer Genet Cytogenet 2010;203:187)
- t(1;3) was first reported in 1984 (Blood 1984;64:553)
- Aberrant expression of PRDM16 as a result of RPN1 translocation is associated with various other hematological conditions (Cancer Genet Cytogenet 2010;203:187)
- MDS
- Myelodysplastic / myeloproliferative neoplasms (MDS / MPN)
- Myeloproliferative neoplasms (MPN)
- AML
- Acute lymphoblastic leukemia (ALL)
- Prognosis is poor; median survival is ~6 months
- WHO diagnostic criteria (WHO, 2017)
- > 20% peripheral blood or bone marrow blasts
- Any of the following:
- History of MDS or MDS / MPN
- MDS related cytogenetic abnormality
- Multilineage dysplasia
- Absence of both of the following:
- Prior cytotoxic or radiation therapy
- Recurrent cytogenetic abnormalities of AML
| WHO revised 4th edition | WHO 5th edition | ICC classification |
|
|
|
- Peripheral blood
- Bone marrow biopsy
- Cytogenetics
- Karyotype analysis showing (1;3)(p36;q21) (PRDM16::RPN1)
- Molecular genetics
- Immunophenotyping
- Peripheral blood
- Anemia, thrombocytosis and leukocytosis
- Bone marrow
- Usually hypercellular marrow
- Blasts
- Molecular studies
- Translocation can be detected by karyotyping or AML FISH PRDM16::RPN1 t(1;3)(p36.3;q21.3) (Mayo Clinic Laboratories: AMLMF [Accessed 15 February 2023])
- Independent of t(1;3)(p36.3;q21.2) abnormality, prognosis has been shown to be worse in AML (Mod Pathol 2015;28:965, Leuk Res 1995;19:121)
- Very few known complete remission outcomes in AML with t(1;3)(p36.3;q21.2) (Cancer Genet Cytogenet 2010;203:187)
- Survival period was worse following detection of t(1;3)(p36.3;q21.2) abnormality in AML (21.3 months) than in acute lymphoblastic leukemia with t(1;3)(p36.3;q21.2) (78.6 months) (Cancer Genet Cytogenet 2010;203:187)
- High expression of PRDM16 correlated with poor prognosis and decreased 5 year survival (Genes Chromosomes Cancer 2017;56:800)
- 6 year old girl with t(1;3)(p36;q21) following treatment for AML (Cancer Genet Cytogenet 2009;191:59)
- 59 year old woman and 66 and 82 year old men presenting with (1;3)(p36;q21) (PRDM16::RPN1) (Genes Chromosomes Cancer 2003;36:313)
- AML with t(1;3) and extreme thrombocytosis (Cancer Genet Cytogenet 2010;203:187)
- No specific treatments for this specific translocation have been reported
- Chemotherapy
- Conventional chemotherapy has had little success in achieving complete remission (Cancer Genet Cytogenet 2010;203:187)
- Stem cell transplantation
- Presentation as AML (M1 - M5) has been reported; however, presentation as AML M4 (French American British [FAB] classification) is common (Cancer Genet Cytogenet 2010;203:187)
- Prominent monocytic component has been reported in some cases
- Multilineage dysplasia especially dysmegakaryopoiesis (Cancer Genet Cytogenet 2010;203:187, Genes Chromosomes Cancer 2003;36:313)
- At least 50% dysplasia across 2 different lineages (WHO, 2017)
- Megakaryocytes
- Megakaryocytic hyperplasia and dysplasia
- Peripheral thrombocytosis is a distinct feature
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
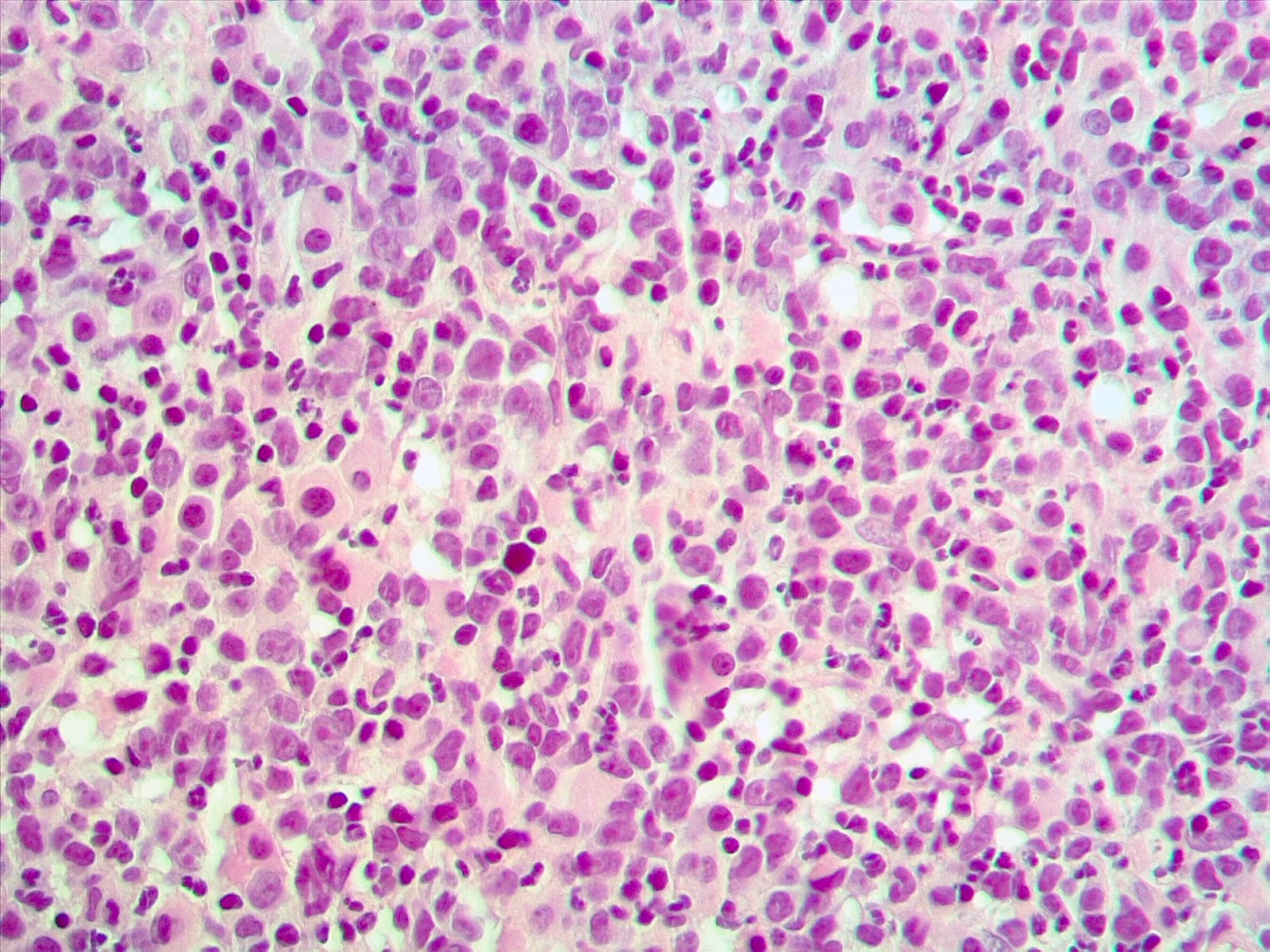
Numerous blasts
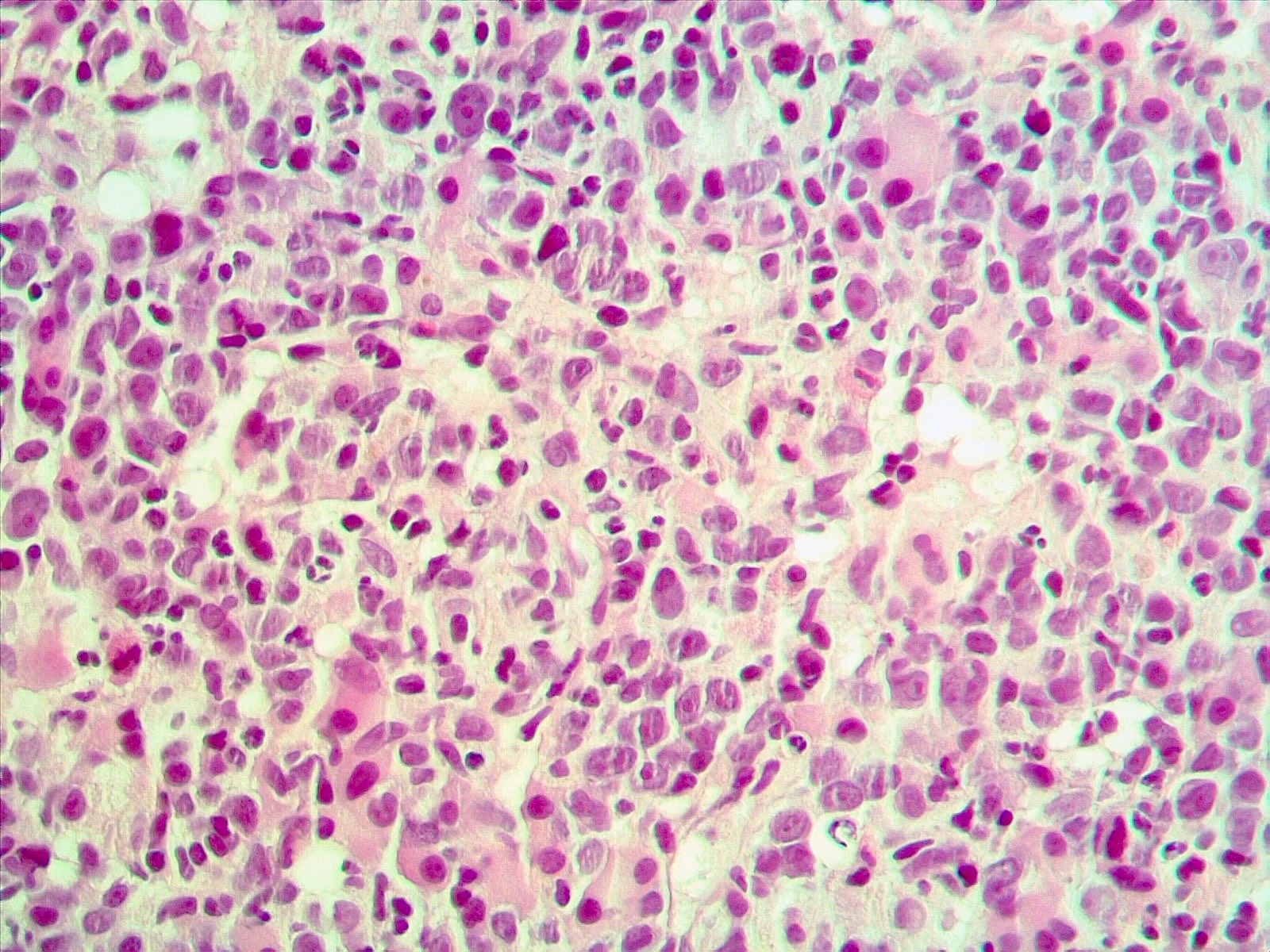
Numerous blasts and myelodysplasia related changes
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
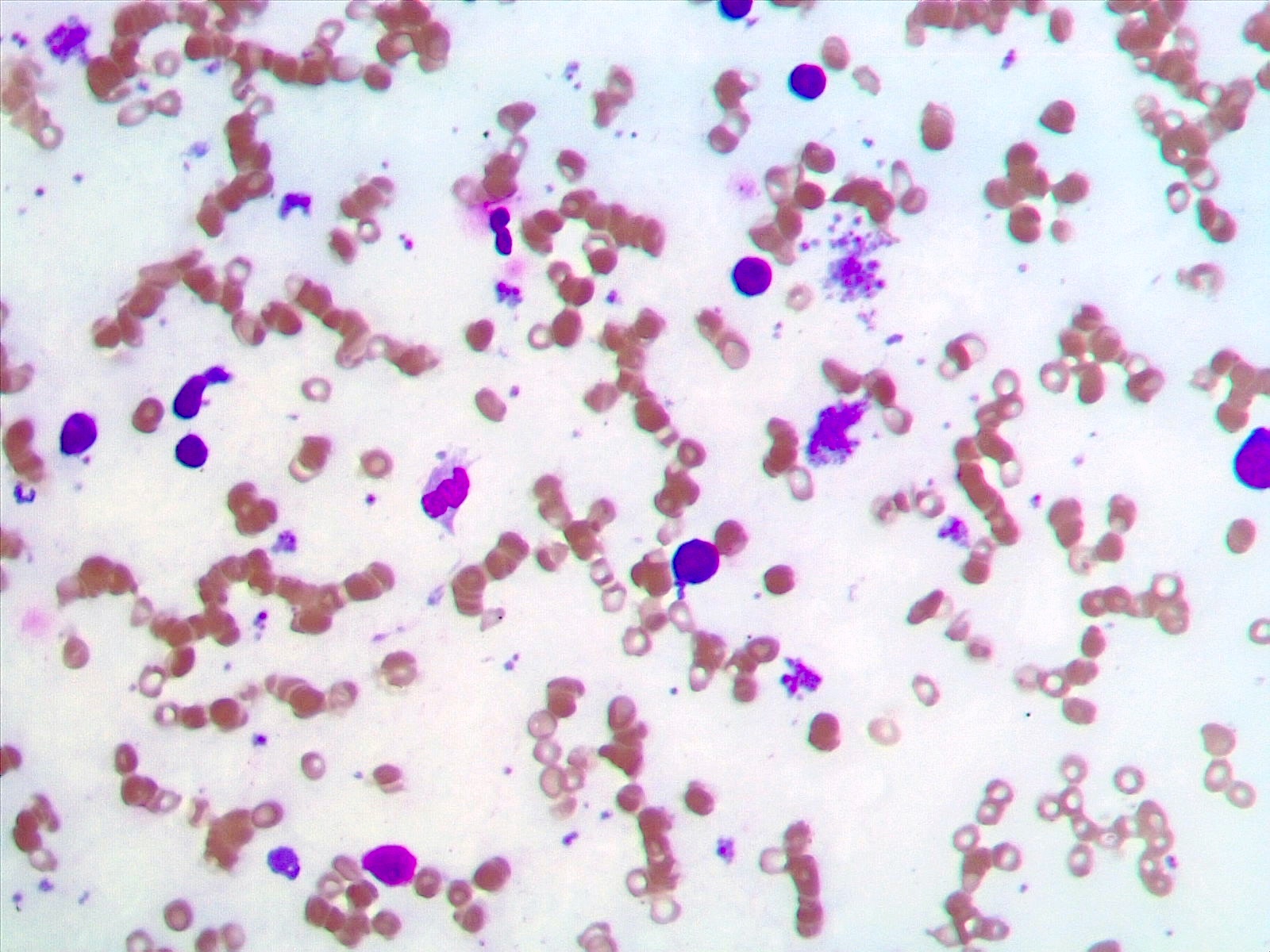
Scattered blasts
- Variable and possibly related to the patient's distinct AML FAB type
- Usually positive
- Variable positivity
- Possible abnormal expression of CD56, CD72
- Other myeloid markers may be positive
- Reference: Cancer Genet Cytogenet 2010;203:187
- Variable and possibly related to the patient's distinct AML FAB type
- Usually negative
- Reference: Cancer Genet Cytogenet 2010;203:187
- Translocation can be detected by karyotyping or AML FISH PRDM16::RPN1 t(1;3)(p36.3;q21.3) (Mayo Clinic Laboratories: AMLMF [Accessed 15 February 2023])
- Right posterior iliac crest, core biopsy, aspirate smear, touch imprint and clot particle:
- Acute myeloid leukemia with t(1;3)(p36;q21) (see comment)
- Comment: The bone core biopsy demonstrates extensive replacement of marrow by blast cells in areas arranged into clusters and aggregates surrounded by fibrotic stroma. The marrow aspirate smears reveal an increased population of blasts. Blast phenotyping with immunohistochemical stains and flow cytometry analysis reveal positivity for myeloid markers. Cytogenetics evaluation reveals t(1;3)(p36;q21). Overall, the results support the diagnosis of AML with t(1;3)(p36;q21).
- MDS with excess blasts:
- Blasts < 20%
- May show other MDS related cytogenetic or molecular abnormalities
- May show t(1;3)(p36;q21); these cases may progress to AML
- Myeloproliferative neoplasms:
- Blasts < 20%
- May show t(1;3)(p36;q21); these cases may progress to AML
A 60 year old woman presented with fatigue and weight loss. The patient's initial laboratory workup showed leukocytosis, anemia and thrombocytosis. A bone marrow biopsy was performed, which showed increased blasts and multilineage dysplasia. A karyotype showed with t(1;3)(p36;q21). Which of the following is true about this entity?
- Blasts show a distinct phenotype characteristic of this entity
- Case shows overexpression of the PRDM16 gene
- Frequently associated with NPM1 mutation
- Has an intermediate prognosis
Comment Here
Reference: AML with t(1;3)(p36;q21)
- Included in WHO 2008 classification under AML with recurrent genetic abnormalities
- First identified by Rowley and Potter in 1976
- 0.7 - 1.8% of AML
- Occurs in both children and adults (2 - 66 years old), both sexes equally; median age children 13 years, adults 35 years
- Associated with multilineage dysplasia and basophilia (> 2% in marrow and peripheral blood in 44 - 62% of cases)
- In adults, median white blood cell count is 12x109/L, lower than other AML
- In children, lower hemoglobin values and higher WBC counts than adults
- Associated with dysplasia and a high frequency of FLT3 gene mutations (Am J Clin Pathol 2004;122:348)
- Anemia, thrombocytopenia, blasts in peripheral blood
- Often pancytopenia, fatigue, bleeding, DIC and increased incidence of infections
- Poor prognosis
- < 50% remission after chemotherapy; often dead within 1 year after diagnosis (Leukemia 2006;20:1295)
- Allogeneic hematopoietic stem cell transplantation
- Minimal residual disease: monitor by real time RT-PCR (Leukemia 2005;19:1338)
- Current clinical trials include anti-CD33 and FLT3 inhibitors
- 20%+ blasts; intermediate features between AML with maturation (AML-M2), acute promyelocytic leukemia (APL) and acute myelomonocytic leukemia (AML-M4)
- Often ringed sideroblasts (Am J Clin Pathol 1997;107:430)
- Unilineage or multilineage dysplasia (67%) (Arch Pathol Lab Med 2008;132:1835)
- t(6;9)(p23;q34) is often sole clonal abnormality; some have complex karyotype
- t(6;9) produces chimeric fusion gene between DEK (6p23) and NUP214 (9q34; formerly known as CAN)
- DEK-NUP214 fusion gene encodes a messenger RNA involved in leukemogenesis
- Also FLT3-ITD (69% pediatric and 78% adult cases), often additional chromosomal abnormalities; FLT3-TDK uncommon
- Patients with FLT3-ITD have higher WBC, higher bone marrow blasts and lower rates of complete remission
- AML with t(8;21)(q22;q22.1) is defined by a genetic rearrangement that results in the fusion of RUNX1 and RUNX1T1, has characteristic morphologic and immunophenotypic features and is associated with a generally favorable prognosis
- AML with t(8;21)(q22;q22.1) is classified in the category of AML with recurrent genetic abnormalities and is diagnostic of AML regardless of blast count
- t(8;21)(q22;q22.1) is a balanced translocation that results in the fusion of RUNX1 and RUNX1T1
- Blasts have distinct morphologic and immunophenotypic features, including abundant basophilic cytoplasm containing azurophilic granules with occasional Auer rods and frequent aberrant expression of B cell markers, such as CD19
- Diagnosis is established by detection of t(8;21) by real time PCR or cytogenetics
- Prognosis is generally favorable, while coexisting KIT mutations adversely affect outcomes
- AML with RUNX1-RUNX1T1
- AML with AML1-ETO
- Usually classified as AML M2 subtype in the previous French American British (FAB) classification system
- ICD-10: C92.00 - Acute myeloblastic leukemia, not having achieved remission
- Typically occurs in younger patients and comprises approximately 1 - 5% of all AML cases
- Peripheral blood
- Bone marrow
- Myeloid sarcoma soft tissue / organs
- t(8;21)(q22;q22.1) results in fusion of the RUNX1 gene on chromosome 21q22.1 with RUNX1T1 gene on chromosome 8q22
- RUNX1 is a member of the core binding factor family of transcription factors, which is critical for the establishment of normal hematopoiesis (Blood 2017;129:2070)
- t(8;21), RUNX1-RUNX1T1 fusion gene expresses a chimeric protein that disrupts the normal function of the core binding factor and predisposes to acute myeloid leukemia (Cell Rep 2013;4:1131)
- Most cases show additional chromosomal abnormalities and cooperating driver mutations that promote transformation to acute myeloid leukemia (Blood 2012;119:e67)
- Established by detection of t(8;21) by real time PCR or cytogenetics
- AML with t(8;21)(q22;q22.1) has a relatively favorable outcome and is associated with high rate of complete remission
- Presence of KIT mutations adversely affects prognosis (J Clin Oncol 2006;24:3904)
- 28 year old man with headache was found to have intracranial myeloid sarcoma (Onco Targets Ther 2020;13:237)
- 65 year old woman with epistaxis (Case Rep Hematol 2019;2019:1312630)
- 2 patients with AML also found to have associated systemic mastocytosis (Exp Mol Pathol 2019;108:131)
- Typically chemotherapy with induction, followed by intensive cytarabine consolidation
- Blast cells characteristically have slightly basophilic cytoplasm containing abundant azurophilic granules and perinuclear hofs
- Occasionally, large pseudo Chediak-Higashi granules and thin Auer rods may be seen; Auer rods may be seen in neutrophils
- Mature eosinophils that are morphologically normal are often increased in number in the bone marrow
- Mature granulocytes may show dysplastic changes with pseudo Pelger-Huet anomaly
- Generally no dysplasia in the erythroid and megakarocytic lineages
Contributed by Sean Gu, M.D., Ph.D., Alexa J. Siddon, M.D. and AFIP images
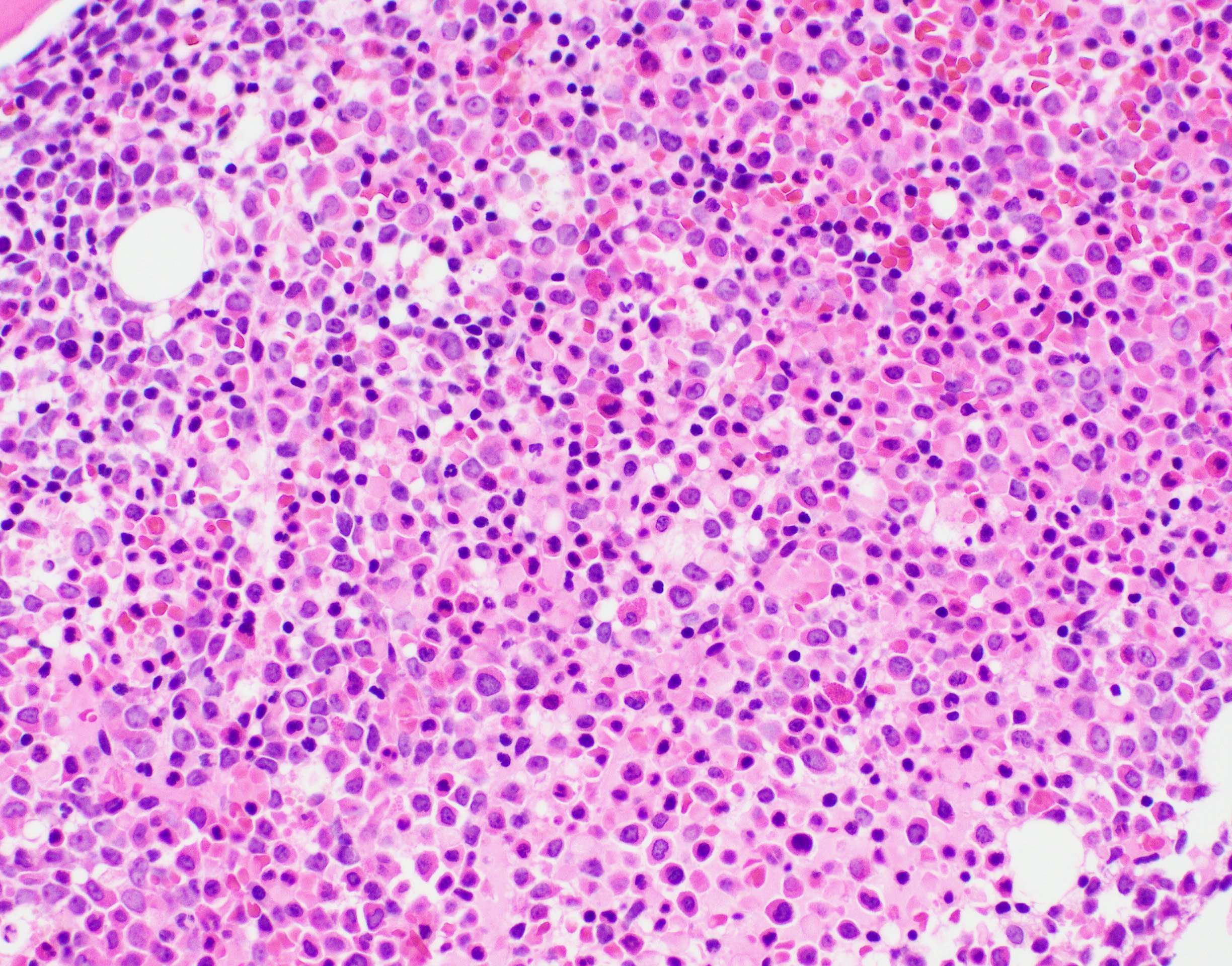
Left shift
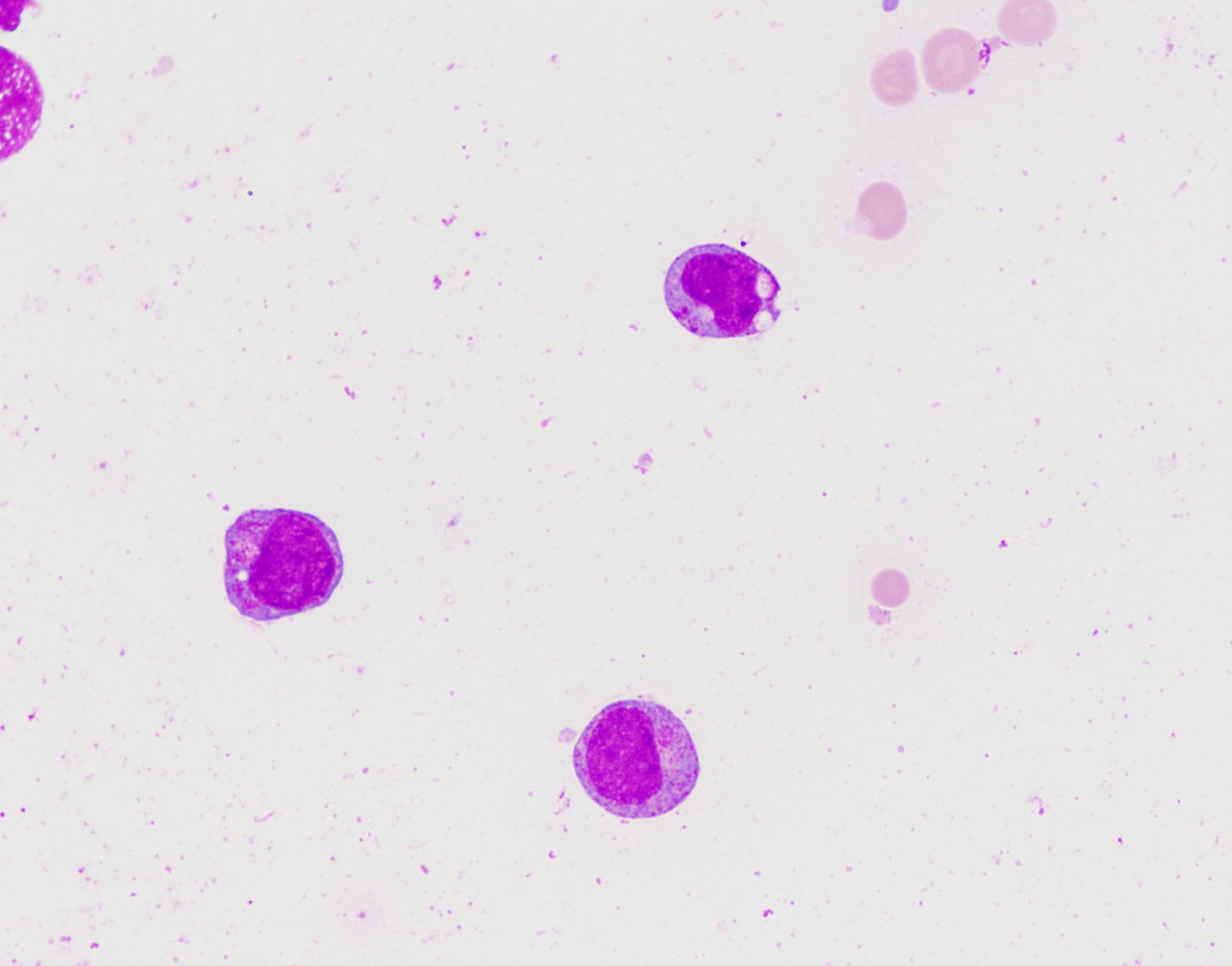
Blasts
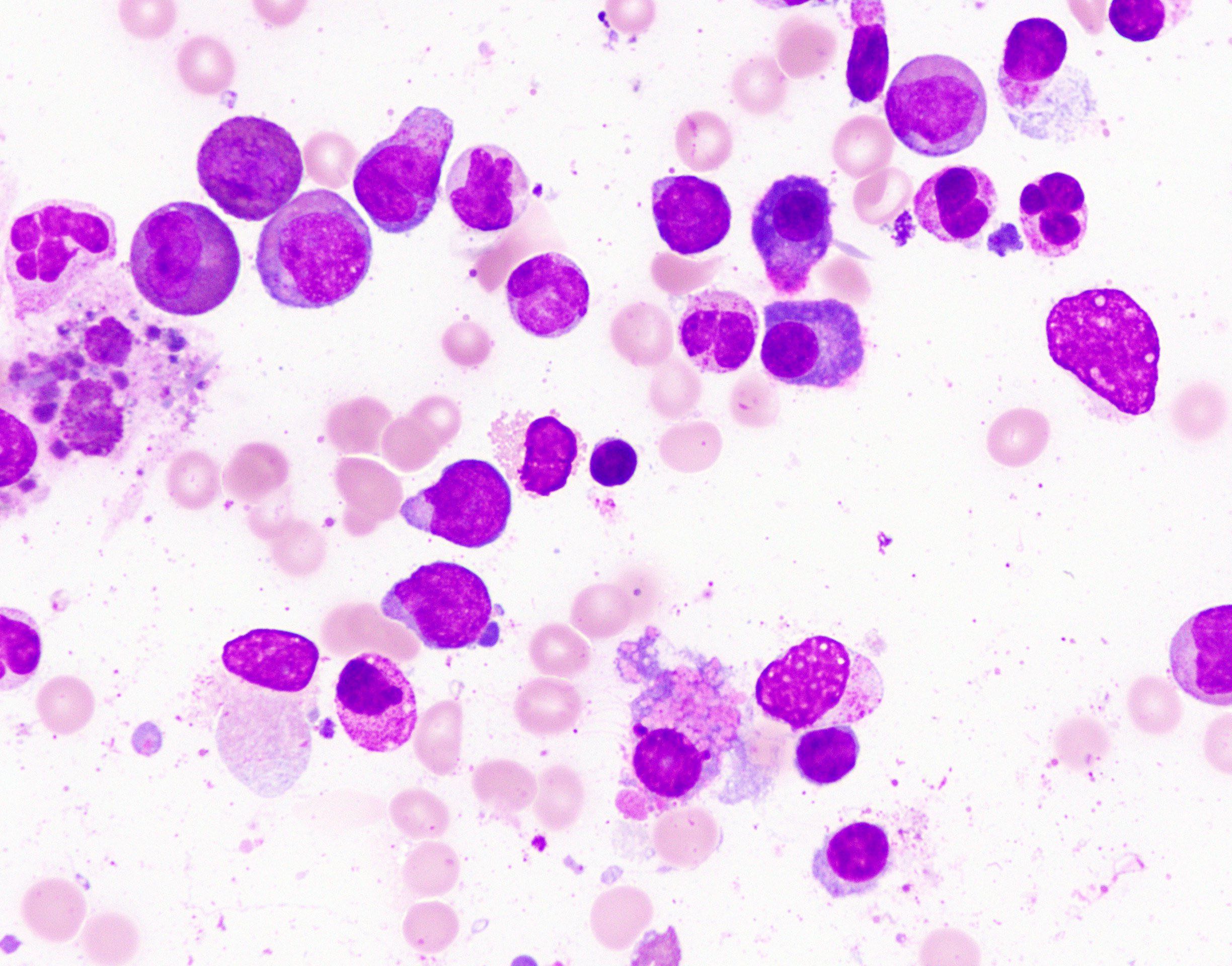
Aspirate morphology
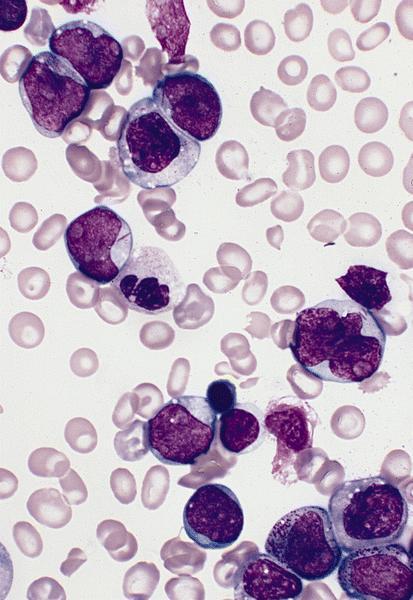
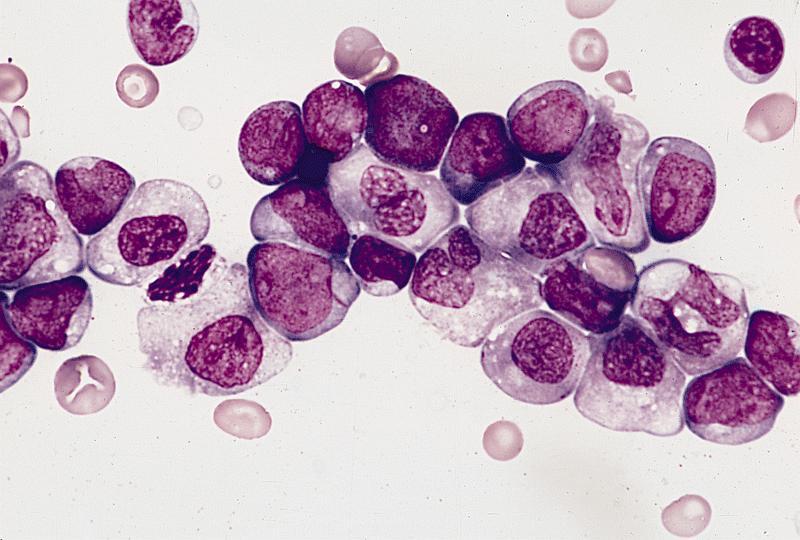
Bone marrow smear (Wright-Giemsa)
Images hosted on other servers:
Myeloblast in AML with t(8;21)
- High levels of CD34, CD13, HLA-DR and myeloperoxidase and weak CD33 expression
- Frequent aberrant expression of B cell markers CD19 and PAX5 (Blood 1992;80:470, Am J Clin Pathol 2006;126:235)
- Expression of CD56 is associated with concomitant KIT mutations and worse prognosis (Blood 1997;90:1643)
Contributed by Genevieve M. Crane, M.D., Ph.D.
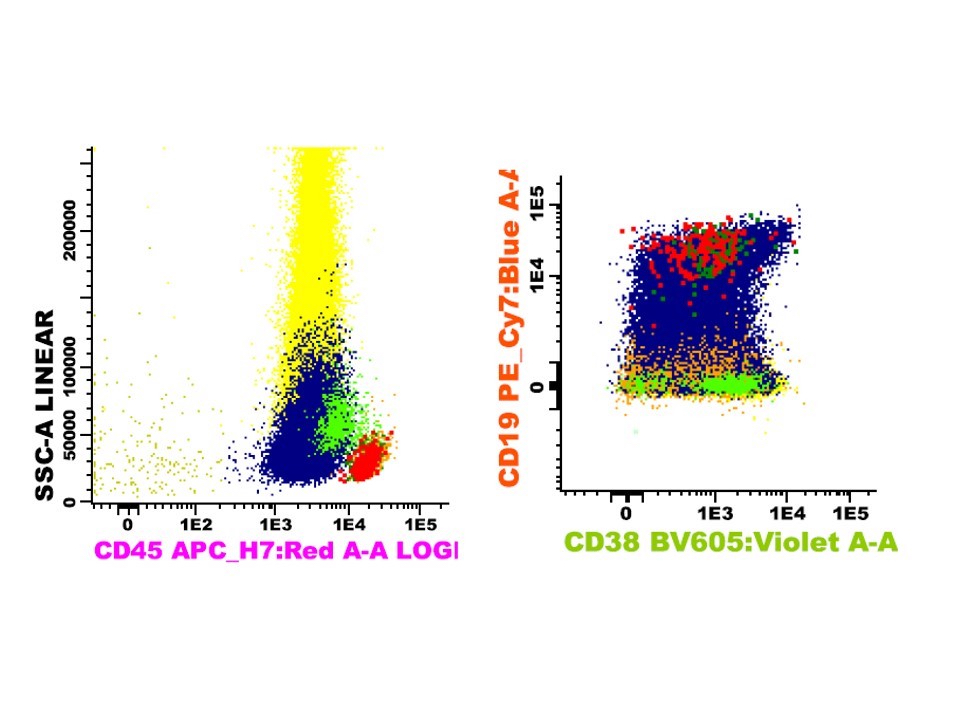
Partial CD19 expression in AML with t(8;21)
- Demonstration of t(8;21) by real time PCR or cytogenetics
AFIP images
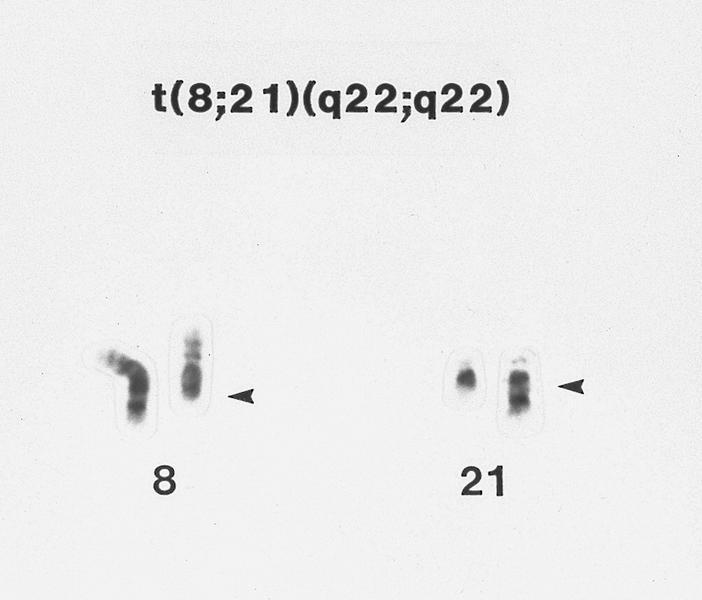
t(8;21) with abnormal chromosomes
Images hosted on other servers:

FISH probes showing
2 abnormal yellow
fusions made of red
and green probes
- Left posterior iliac crest, core biopsy and aspirate smear:
- Acute myeloid leukemia with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 (see comment)
- Comment: The bone core biopsy demonstrates an increase in blasts, which are noted to have prominent eosinophilic granules on the aspirate. The presence of t(8;21) by PCR confirms the diagnosis.
- Other AML with recurrent cytogenetic abnormalities:
- AML with t(15;17) may morphologically resemble AML with t(8;21) but would have different flow and molecular findings
- AML with myelodysplasia related changes:
- Lacks t(8;21), often has complex karyotype
- AML with maturation:
- Lacks t(8;21)
- Myelodysplastic syndromes:
- If there are < 20% blasts and no t(8;21), the findings including dysplasia and Auer rods would be compatible with a myelodystplastic syndrome
- AML with t(8;21) generally has a poor prognosis
- AML with t(8;21) frequently aberrantly expresses the lymphoid markers CD19 and PAX5
- Auer rods are rare in AML with t(8;21)
- Presence of a concomitant KIT mutation is a good prognostic finding
- Translocation is best identified by microarray
Comment Here
Reference: AML with t(8;21)(q22;q22)
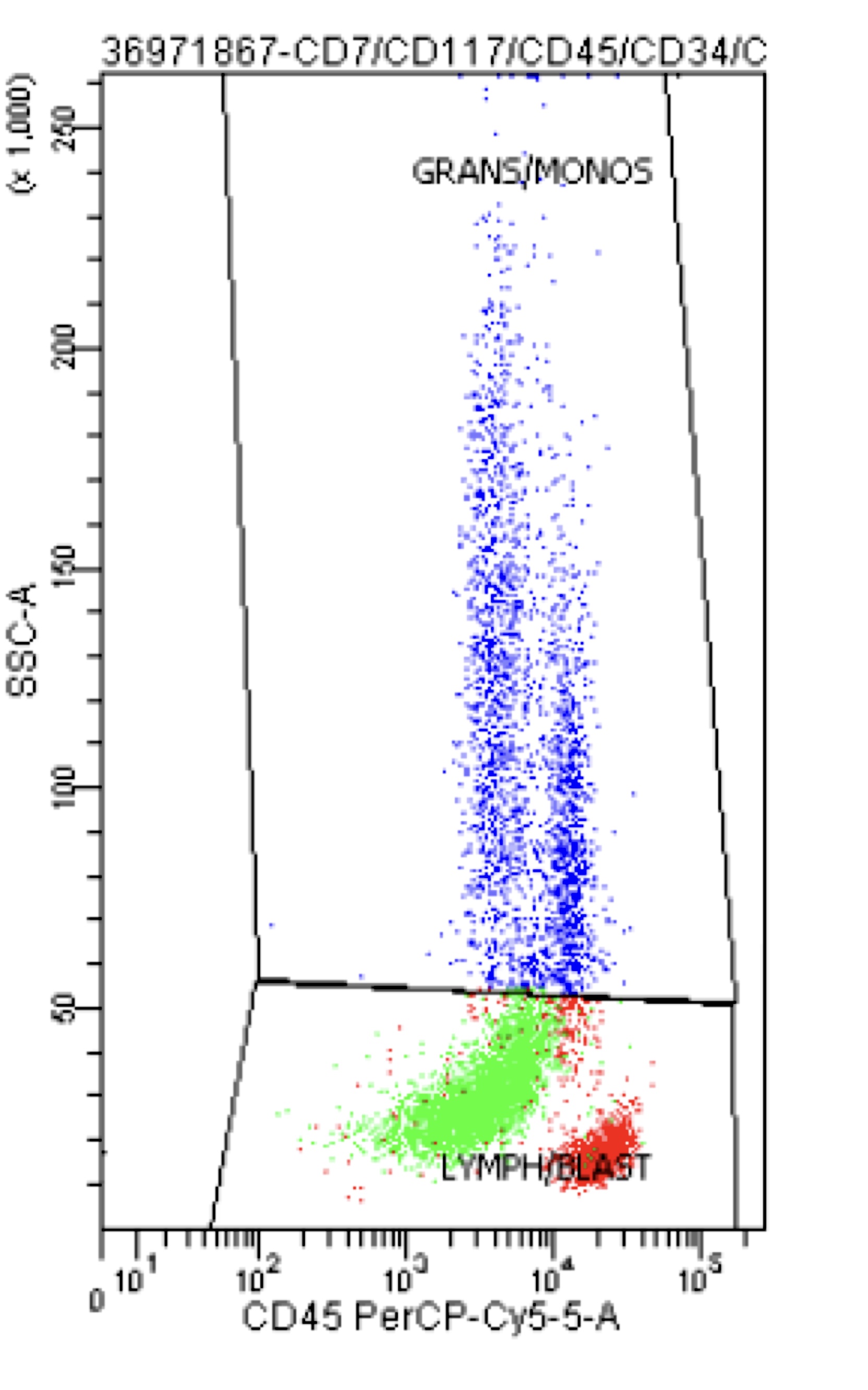
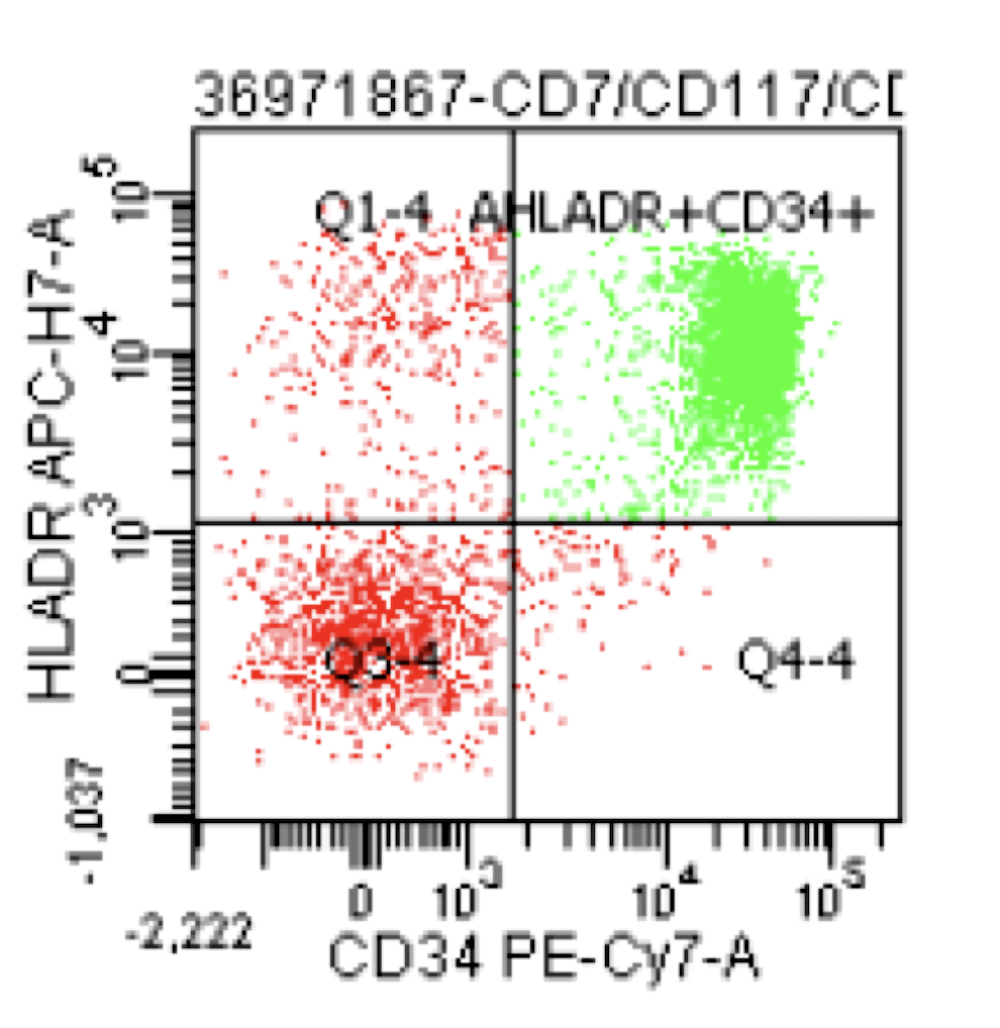
A 26 year old woman is referred to the emergency department when she is noted to have 32% blasts in peripheral blood. Her smear shows large basophilic blasts with prominent eosinophilic granules and rare Auer rods. What will the flow cytometry immunophenotype likely show?
- CD34-, CD117+, HLADR-, MPO++, CD33,
- CD34+, CD33dim+, MPO+, CD71-, CD19dim+
- CD34-, CD13+, CD11b+, CD14+, CD64+, HLA-DR+
- CD34+, CD117+, CD33+, CD19-, CD3-, MPO-
- CD34+, CD19+, CD20-, CD10+, TdT+
- MLL gene at 11q23 (HRX) encodes a DNA binding protein that positively regulates expression of target genes, including multiple HOX genes, by methylation of histone H3 lys4 / chromatin remodeling (OMIM: Lysine Specific Methyltransferase 2A; KMT2A [Accessed 9 February 2022], Atlas of Genetics and Cytogenetics [Accessed 9 February 2022])
- 9p22 is MLLT3 / AF9 gene
- Included in WHO 2008 as AML with recurrent cytogenetics
- 3 - 5% of AML (Anticancer Res 2005;25(3B):1931)
- 9 - 12% of childhood AML; 2% of adult AML
- Overlaps other categories, including high percentage of topo II inhibitor, therapy related AML; also ALL and biphenotypic leukemia (Blood 2003;102:2395)
- Symptoms: DIC, extramedullary myeloid sarcoma and tissue infiltration (gingiva, skin)
- Translocations occur in AML (intermediate prognosis) and ALL (poor prognosis) (Blood Cells Mol Dis 2008;40:192)
- Prognosis may be superior to other AML with 11q23 translocations
- MLL partial tandem duplication AML: prognosis varies from poor to similar to other AML (Br J Haematol 2006;135:438, Blood 2007;109:5164)
- Also present in 93% of normal cord blood samples at low levels (Leuk Res 2006;30:1091)
- Associated with normal karyotype or trisomy 11
- 60 year old woman with therapy related ALL with MLL gene rearrangement following treatment of breast cancer (Korean J Lab Med 2010;30:255)
- MLL insertion with MLL-MLLT3 gene fusion in acute leukemia (Cancer Genet Cytogenet 2008;183:53)
- 20%+ blasts / blast equivalents (monoblasts / monocytes) in peripheral blood or bone marrow, usually myelomonocytic or monocytic (AML M4, M5) and occasionally AML with (M2) or without (M1) maturation
- Monoblasts are large cells with abundant, moderate to intensively basophilic cytoplasm, pseudopods, azurophilic granules, vacuoles; round nuclei, lacy chromatin and one or more prominent nucleoli
- Promonocytes have basophilic cytoplasm with granules and occasional large azurophilic granules, vacuoles; irregular and delicately convoluted nuclei
- Myeloperoxidase
- Children: CD34 and CD117 negative
- Adults: variable CD14, often negative / dim
- Involves MLL mixed lineage or myeloid / lymphoid leukemia gene, present in both AML and ALL; also called HTRX1, HRX and ALL1
- FISH is more sensitive than conventional cytogenetics in detecting MLL; may also detect 11q22-25 rearrangements that are MLL negative (Am J Clin Pathol 2004;122:298)
Variant MLL translocations in AML: - > 80 translocations described, most commonly MLLT2(AF4) causing ALL; and MLLT3(AF9) causing AML
- Mixed phenotype acute leukemia has t(v;11q23)
- AML-MRC if t(2;11)(p21;q23) or t(11;16)(q23;p13.3)
- Diagnose as t-AML if history of cytotoxic therapy
- Other common translocations: 6q27 (MLLT4), 10p12 (MLLT10), 19p13.1 (ELL) and 19p13.3 (MLLT1)
- 10 - 20% of AML cases, 44% in one Brazil hospital (Sao Paulo Med J 2006;124:45)
- Usually adults (median age 46 years), present with anemia, thrombocytopenia and neutropenia; may have leukocytosis with markedly increased blasts
- 4% of childhood AML
- Criteria for diagnosis: 90%+ of nonerythroid cells in marrow are myeloblasts; < 10% granulocytic elements; 3%+ of blasts must be positive for myeloperoxidase or Sudan Black B and / or Auer rods by enzyme cytochemistry
- Enzyme cytochemistry: 3%+ of blasts are positive for myeloperoxidase or Sudan Black B (confirm by immunohistochemistry if only 3 - 10% positive for MPO by enzyme cytochemistry); chloroacetate esterase positive
- Poor prognosis
- 4 year old child with t(6;9) and basophilia (Ann Biol Clin (Paris) 2003;61:352)
- 44 year old man with large and small blasts (Arch Pathol Lab Med 2004;128:448)
- Presenting with arterial thromboembolism (Leuk Res 2007;31:869)
- Typically markedly hypercellular marrow, but normocellular and hypocellular cases occur
- Very immature cells, usually round with few azurophilic cytoplasmic granules or Auer rods
- Nuclei are round or indented; little maturation beyond myeloblast stage
- Cells may may resemble lymphoblasts and not appear myeloid
AFIP images
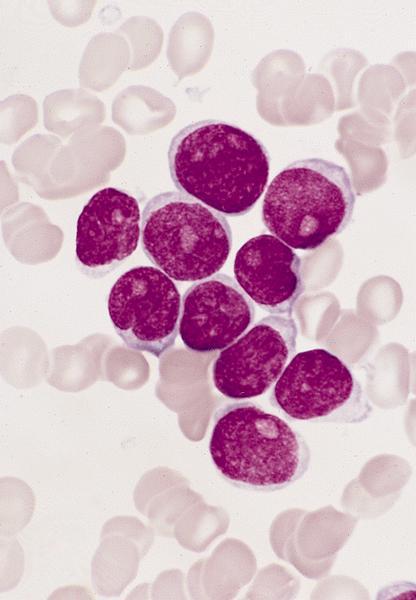
Blasts show mild size variation
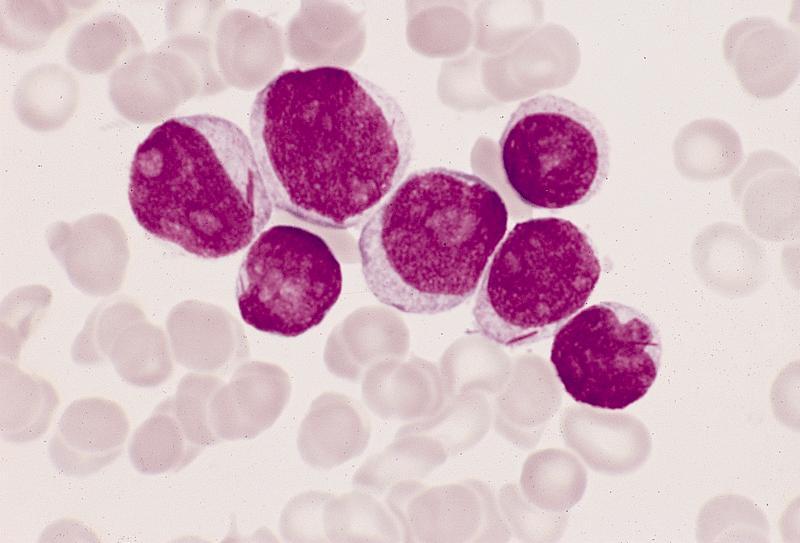
Blasts show more variation in size and number of nucleoli
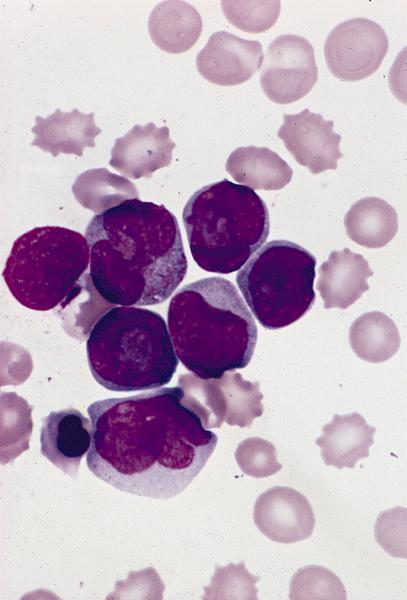
Myeloblasts have irregular nuclei
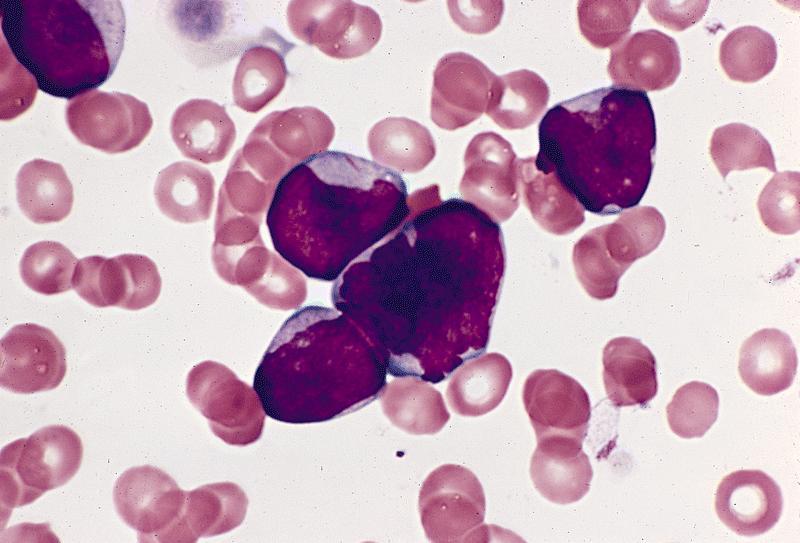
Myeloblasts have marked size variation
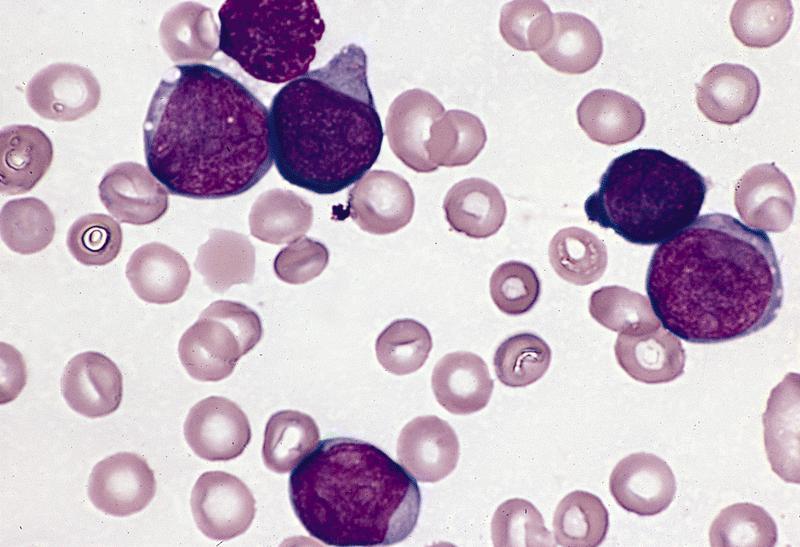
Some variation in size
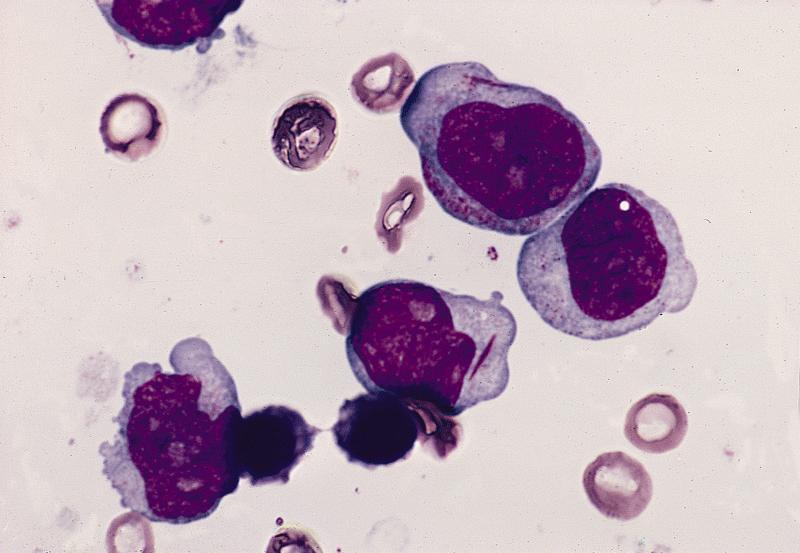
Large myeloblasts
with abundant
eosinophilic
cytoplasm
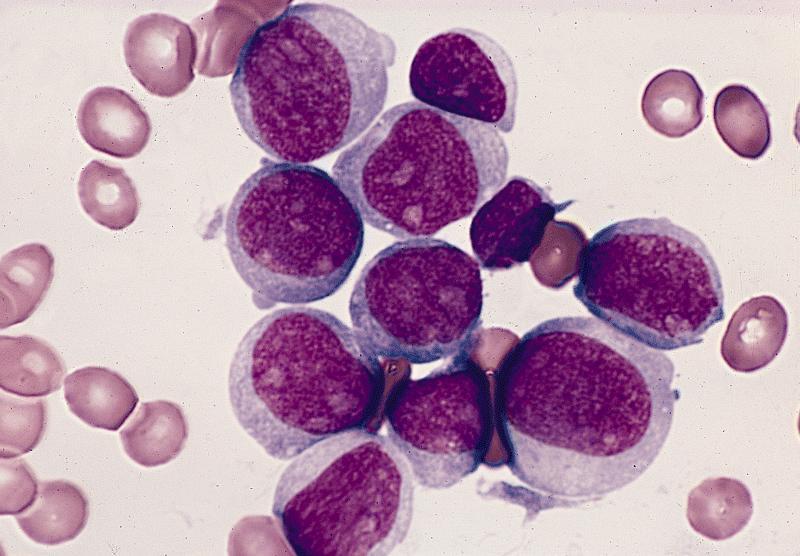
Agranular myeloblasts
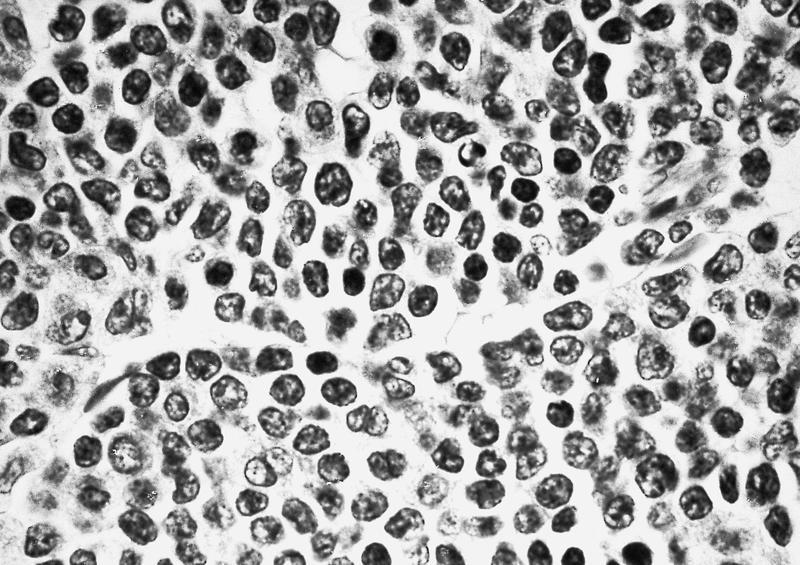
Bone marrow biopsy
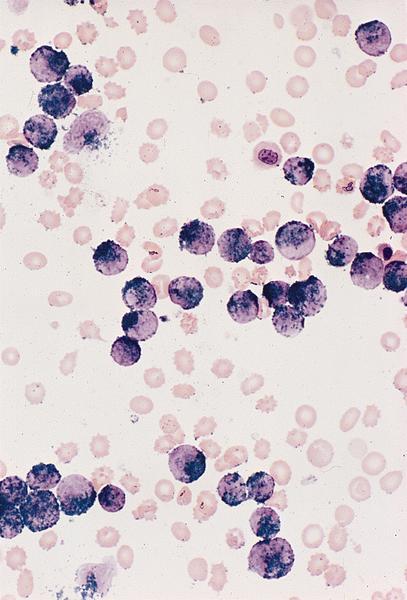
Myeloblasts are myeloperoxidase+
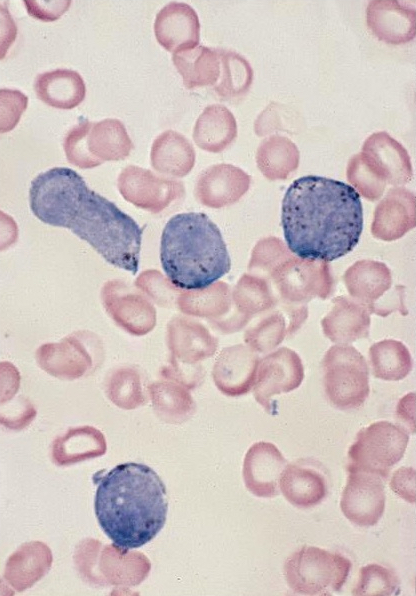
Numerous granules are Sudan black B+
AFIP images
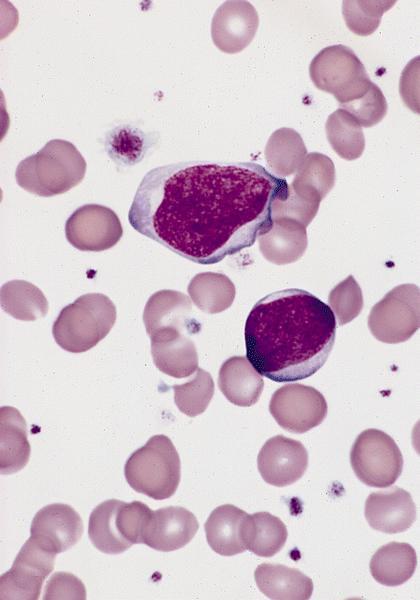
Peripheral blood smear
- May have heterogeneous features (Ultrastruct Pathol 1995;19:9)
AFIP images
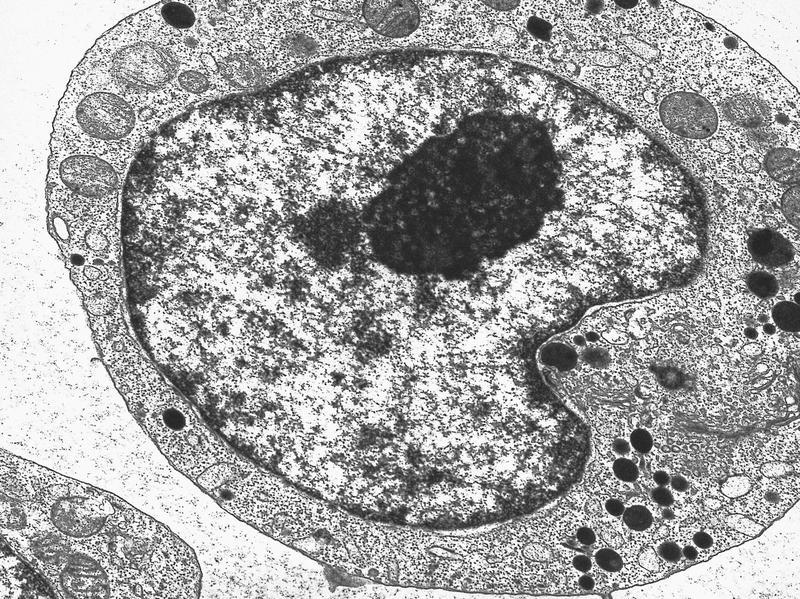
Numerous electron dense granules in Golgi region
- Associated with t(8;21)
- FLT3 ITD in 22% (Ai Zheng 2007;26:58)
- FLT3 mutations associated with HLA-DR negative patients (Leuk Res 2007;31:921)
- Acute myeloid leukemia (AML) with predominance of abnormal promyelocytes and fusion of the promyelocytic leukemia (PML) gene with the retinoic acid receptor alpha (RARA) gene
- Acute myeloid leukemia with predominance of abnormal promyelocytes, characterized by t(15;17)(q24.1;q21.2) and leading to PML::RARA fusion
- Young to middle aged adults are most commonly affected
- 2 morphologic variants are hypergranular and microgranular
- Early diagnosis and treatment are imperative due to the high risk of coagulopathy and disseminated intravascular coagulation (DIC)
- Standard treatment includes all trans retinoic acid (ATRA) plus arsenic trioxide (ATO), with or without chemotherapy; excellent prognosis and high cure rates
- Reference: Arch Pathol Lab Med 2015;139:1308
- Acute promyelocytic leukemia with PML::RARA fusion (World Health Organization Classification, 5th Edition)
- Acute promyelocytic leukemia (APL) with t(15;17)(q24.1;q21.2) / PML::RARA (2022 International Consensus Classification)
- Formerly called AML M3 by the French American British classification
- ICD-O: 9866/3 - acute promyelocytic leukemia with PML::RARA
- ICD-11: 2A60.0 & XH1A50 - acute myeloid leukemia with recurrent genetic abnormalities & acute promyelocytic leukemia, t(15;17)(q22;q11-12)
- Relatively uncommon acute myeloid leukemia subtype
- Young and middle aged adults are most commonly affected (20 - 59 years)
- Accounts for 5 - 8% of acute myeloid leukemia cases in younger patients
- Higher incidence in Latin American population (Best Pract Res Clin Haematol 2003;16:357, Cancer 2012;118:5811)
- M ≈ F
- Peripheral blood and bone marrow
- Extramedullary involvement is rarely seen
- Characterized by balanced translocation t(15;17)(q24.1;q21.2) leading to PML::RARA fusion and a PML::RARA oncoprotein
- PML::RARA oncoprotein suppresses gene expression by recruitment of a number of transcriptional repressors, which leads to block in differentiation and malignant transformation of myeloid cells (PLoS One 2014;9:e104906)
- Secondary chromosomal and genetic abnormalities contribute to the phenotype
- Unknown in majority of cases
- Prior exposure to chemotherapy or radiotherapy in some cases (Cancer 2015;121:2393, Clin Lymphoma Myeloma Leuk 2022;22:e7)
- Leukopenia is a common presenting sign
- Leukocytosis in the microgranular variant
- Symptoms of pancytopenia are fatigue, weakness, infection, bleeding
- High risk of coagulopathy and disseminated intravascular coagulation, especially in microgranular acute promyelocytic leukemia (Arch Pathol Lab Med 2015;139:1308, Blood Cancer J 2021;11:123)
- Early diagnosis and treatment are imperative
- Established by detection of t(15;17) / PML::RARA fusion by RT-PCR, conventional cytogenetics or FISH
- RT-PCR is the most sensitive method of detection, as the translocation can be cryptic by karyotype and FISH
- Blast count can be < 20%
- Reference: Arch Pathol Lab Med 2015;139:1308
- Complete blood count showing pancytopenia
- Leukocytosis in microgranular acute promyelocytic leukemia
- Prolonged prothrombin time (PT) or activated partial thromboplastin clotting time (aPTT)
- Elevated D dimer and fibrin degradation products
- Decreased fibrinogen (Blood Cancer J 2021;11:123)
- White blood cell (WBC) and platelet count at presentation are useful prognostic factors for segregating patients into low, intermediate and high risk categories
- Low risk: WBC < 10,000/mL and platelets > 40,000/mL
- Intermediate risk: WBC < 10,000/mL and platelets < 40,000/mL
- High risk: WBC > 10,000/mL (Blood Cancer J 2021;11:123)
- With current treatment regimens, cure rates are > 90% (N Engl J Med 2013;369:111, J Clin Oncol 2017;35:605, Blood 2017;129:1275)
- 37 and 60 year old women with acute promyelocytic leukemia (Leuk Res Rep 2022;18:100353)
- 42 year old woman with acute promyelocytic leukemia with myelofibrosis (Medicine (Baltimore) 2021;100:e24567)
- 62 year old man with ZBTB16::RARA positive atypical promyelocytic leukemia (Medicina (Kaunas) 2022;58:520)
- 67 year old man with extramedullary acute promyelocytic leukemia (Front Oncol 2022;12:886436)
- 71 year old man with acute promyelocytic leukemia and chronic lymphocytic leukemia (Hematol Oncol Stem Cell Ther 2019;12:161)
- All trans retinoic acid plus arsenic trioxide is currently the standard of care for low and intermediate risk acute promyelocytic leukemia patients
- In high risk patients, chemotherapy is added to all trans retinoic acid and arsenic trioxide
- If arsenic trioxide is not available, combination of all trans retinoic acid and chemotherapy is used for all risk categories (Blood Cancer J 2021;11:123)
- Bone marrow hypercellular
- 2 cytomorphologic variants of acute promyelocytic leukemia are hypergranular (classic) and microgranular (hypogranular)
- Hypergranular acute promyelocytic leukemia
- Abnormal promyelocytes vary in size and shape, frequently with kidney shaped or bilobed nuclei
- Cytoplasm packed with large granules, staining bright pink, red or purple
- Auer rods, frequently in bundles
- Myeloblasts with Auer rods can be seen
- Microgranular acute promyelocytic leukemia
- Predominantly bilobed nuclei
- Hypogranular appearing cytoplasm due to submicroscopic size of azurophilic granules
- Most cases have some abnormal promyelocytes with prominent granules or Auer rods (Arch Pathol Lab Med 2015;139:1308)
- ZBTB16::RARA t(11;17) acute promyelocytic leukemia has some characteristic morphologic features (Blood 2000;96:1287)
- More regular nucleus, not bilobed; occasionally more condensed nuclear chromatin compared with classic acute promyelocytic leukemia
- Coarse cytoplasmic granules or less often, fine or no granules
- No bundles of Auer rods
- Increased number of hypogranular pelgeroid neutrophils
Contributed by Anamarija M. Perry, M.D., @sanamloghavi on Twitter and AFIP
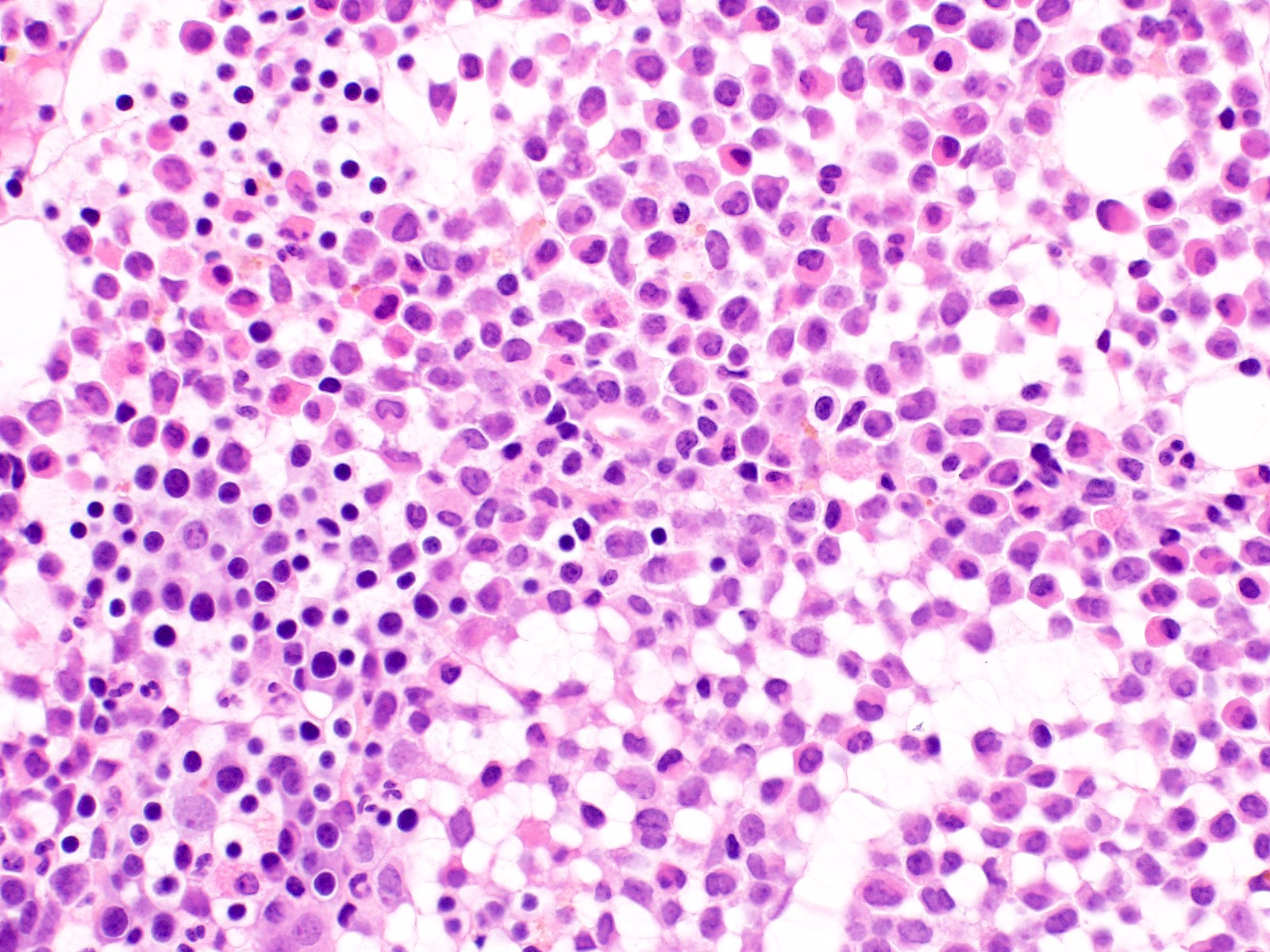
Hypercellular bone marrow
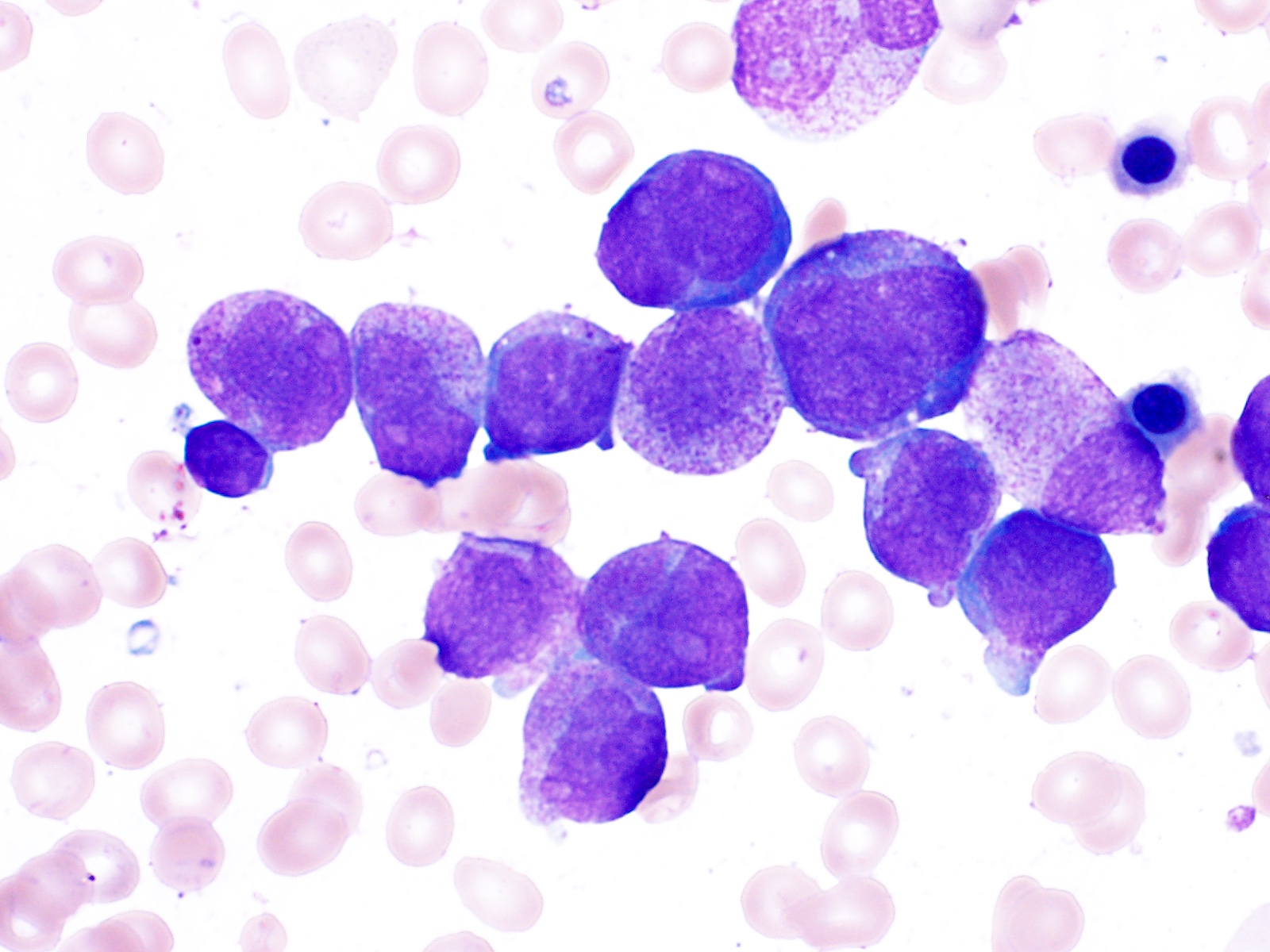
Abnormal promyelocytes
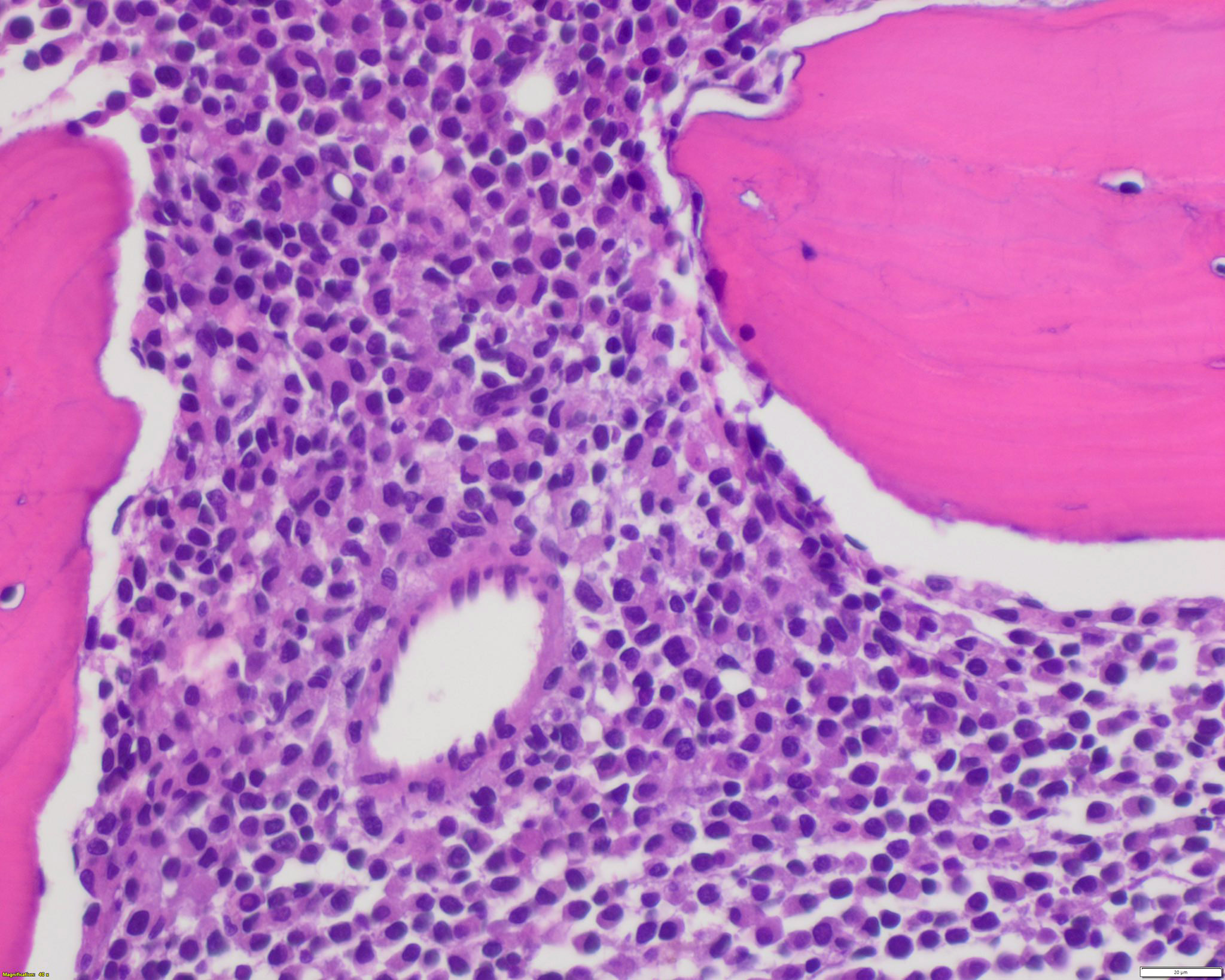 Contributed by @sanamloghavi on Twitter (see original post here)">
Contributed by @sanamloghavi on Twitter (see original post here)">
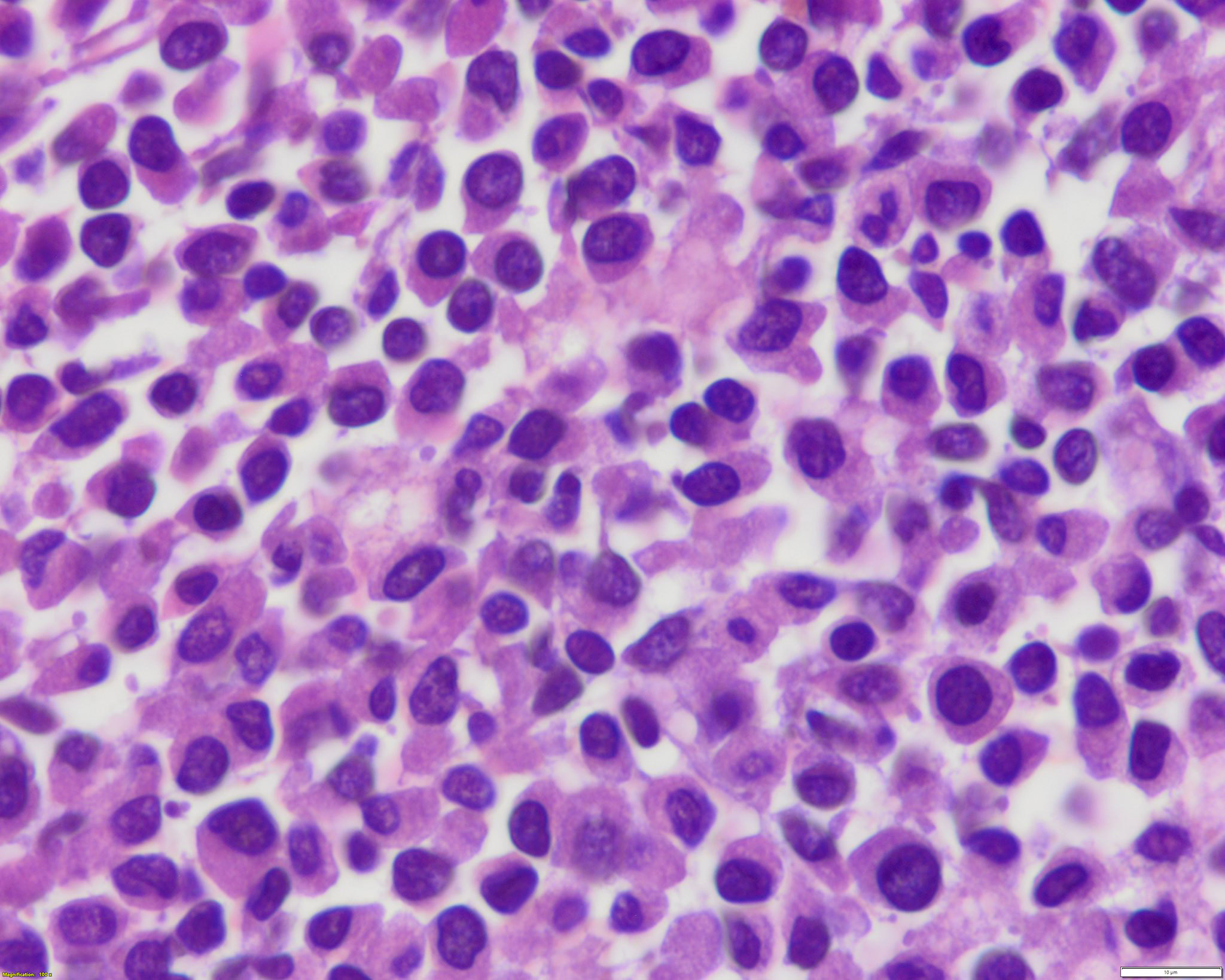
APL with PML::RARA
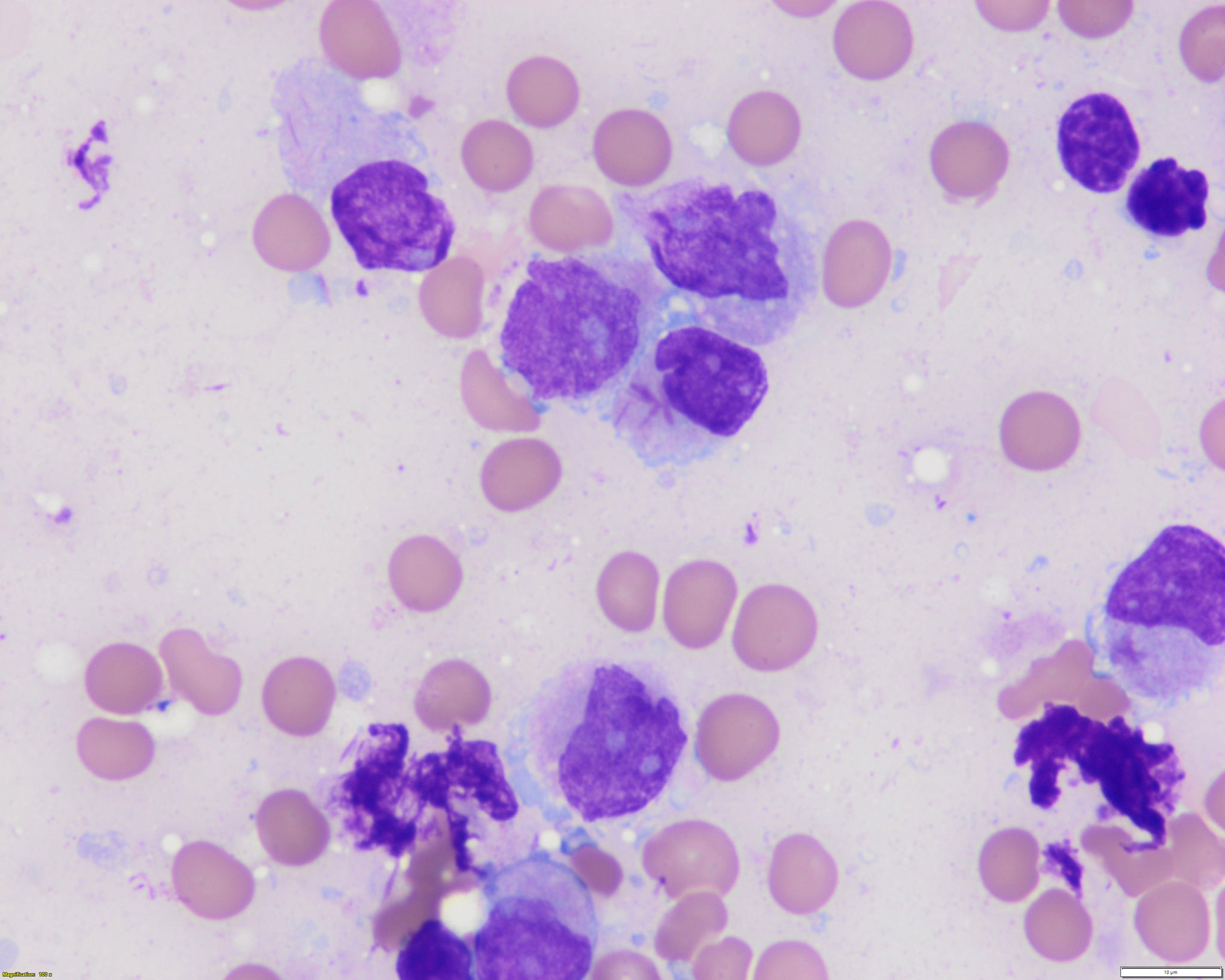 Contributed by @sanamloghavi on Twitter (see original post here)">
Contributed by @sanamloghavi on Twitter (see original post here)">

APL with PML::RARA
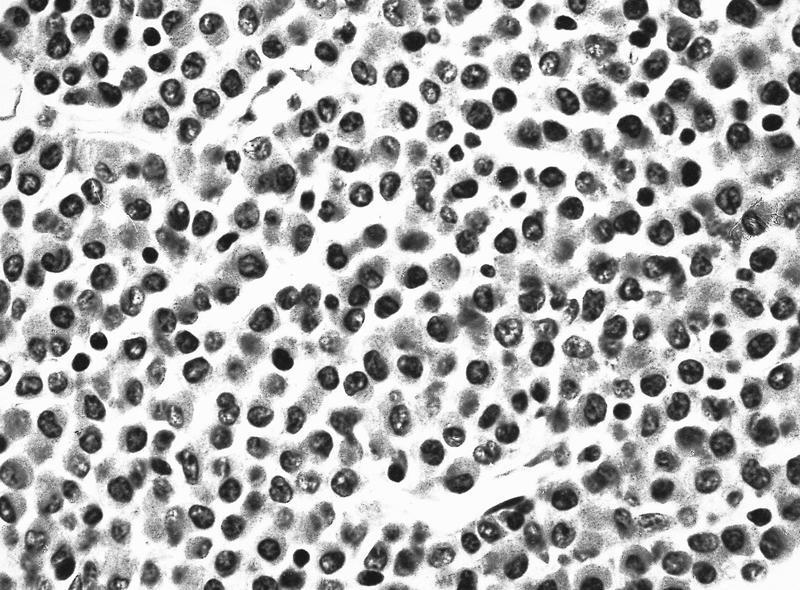
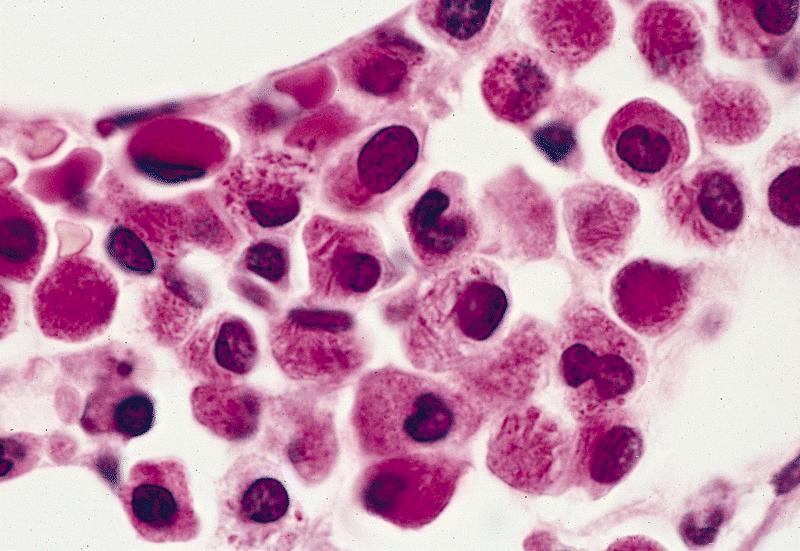
Bone marrow biopsy
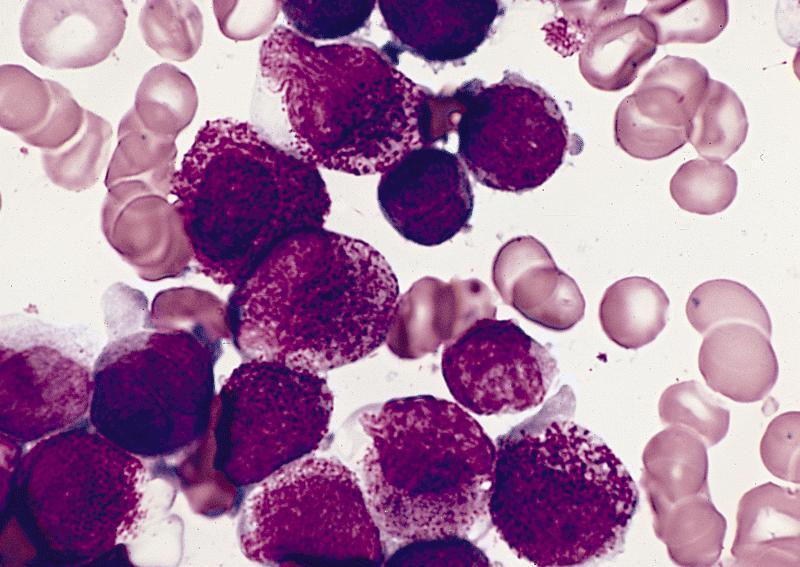
Bone marrow smear (Wright-Giemsa)
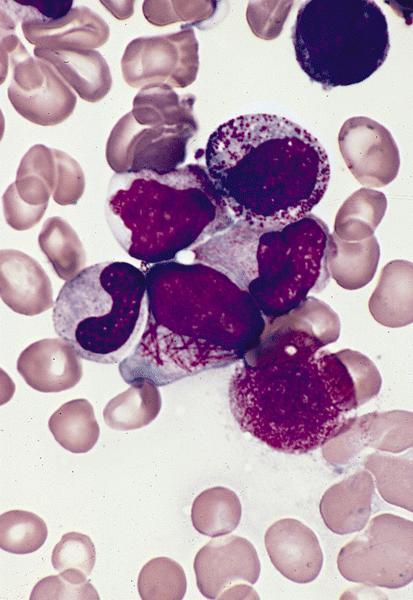
Treatment related
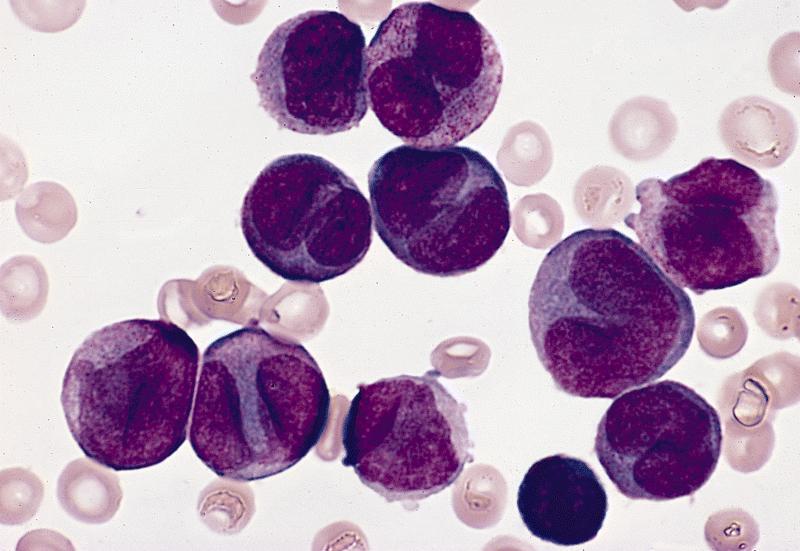
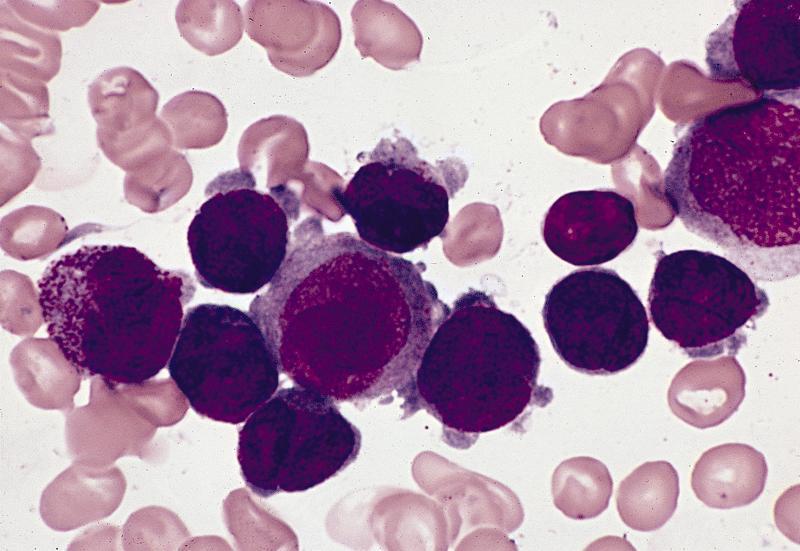
Microgranular variant
Images hosted on other servers:

APL, classic variant

APL, microgranular variant
- In classic variant, pancytopenia is frequently seen with occasional circulating abnormal promyelocytes
- In microgranular acute promyelocytic leukemia, leukocytosis with numerous abnormal promyelocytes
Contributed by Anamarija M. Perry, M.D.
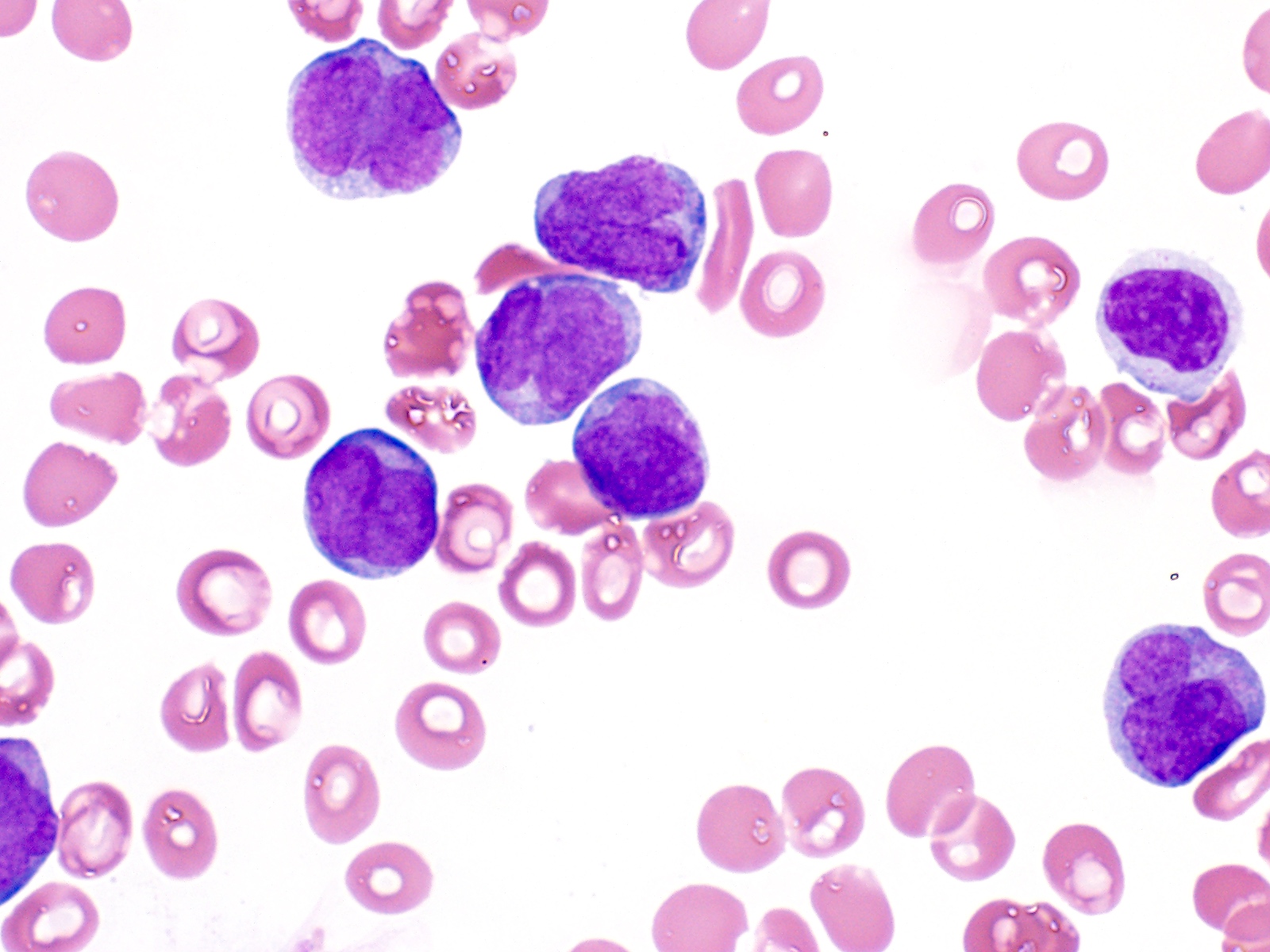
Microgranular variant
- Hypergranular acute promyelocytic leukemia
- Abnormal promyelocytes with high forward and side scatter
- Low or absent expression of CD34 and HLA-DR
- Positive for CD13, CD33, CD64, CD117 and cytoplasmic myeloperoxidase
- Negative for CD11a, CD11b, CD15, CD18, CD65, CD66b and CD66c
- CD56 seen in 10% of cases; CD2 rarely expressed, frequently with CD34 and CD56
- Lymphoid markers (e.g., CD5, CD7, CD19, CD22, cytoplasmic CD3) and TdT rarely expressed (Best Pract Res Clin Haematol 2003;16:369, Haematologica 1999;84:405, J Clin Oncol 2000;18:1295, Blood 2006;108:3484, Cytometry B Clin Cytom 2022;102:283)
- Microgranular acute promyelocytic leukemia
- Abnormal promyelocytes have lower side scatter and are located in the blast gate on the CD45 versus side scatter plot
- Similar phenotype to hypergranular acute promyelocytic leukemia with more common expression of CD2 and CD34
- Expression of CD2 in acute promyelocytic leukemia is associated with FLT3 internal tandem duplication (ITD) mutation (Am J Hematol 2001;67:34, Pol J Pathol 2012;63:8, Cytometry B Clin Cytom 2022;102:283)
Contributed by Anamarija M. Perry, M.D.
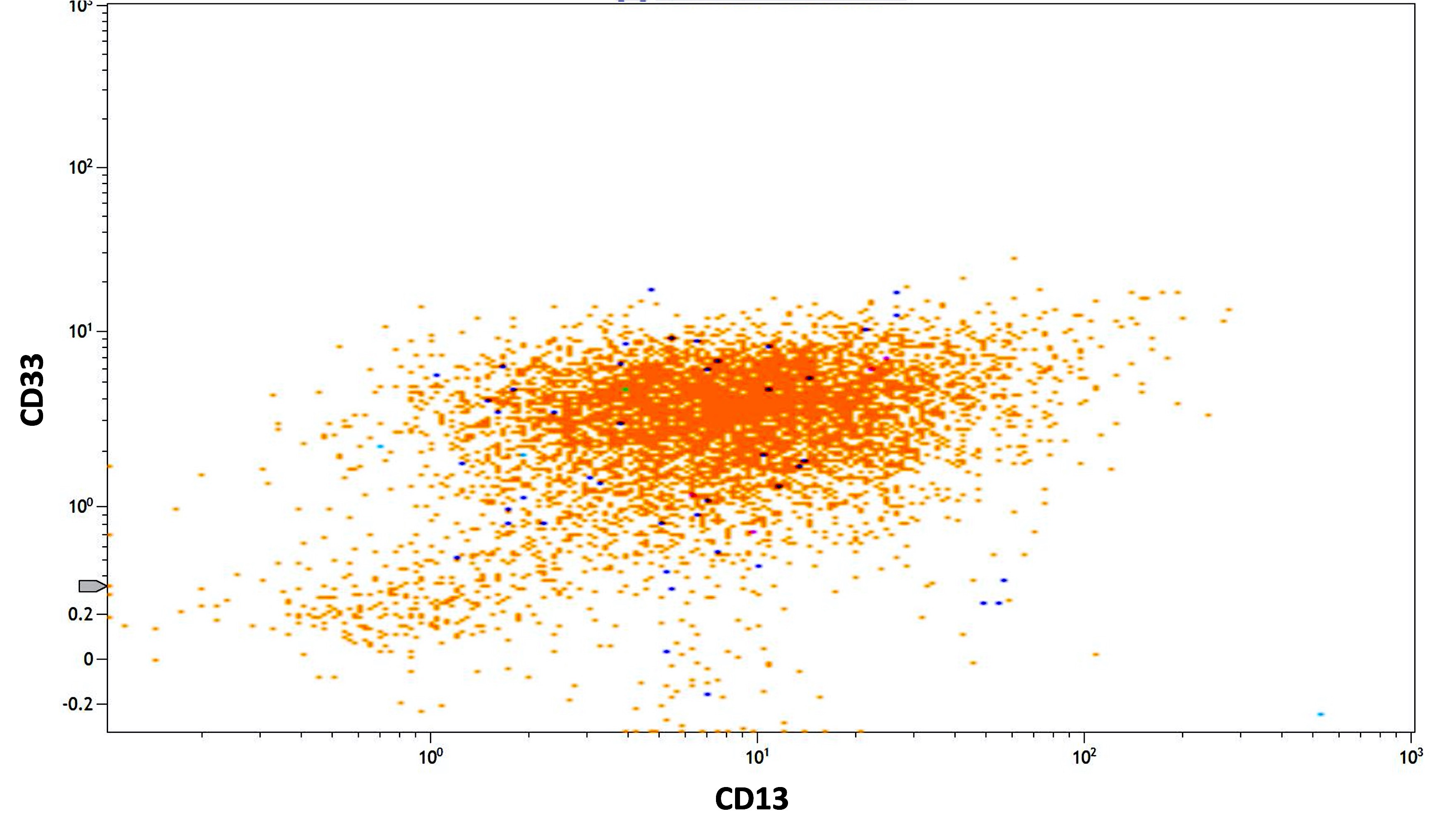
Classic variant, CD13+ and CD33+
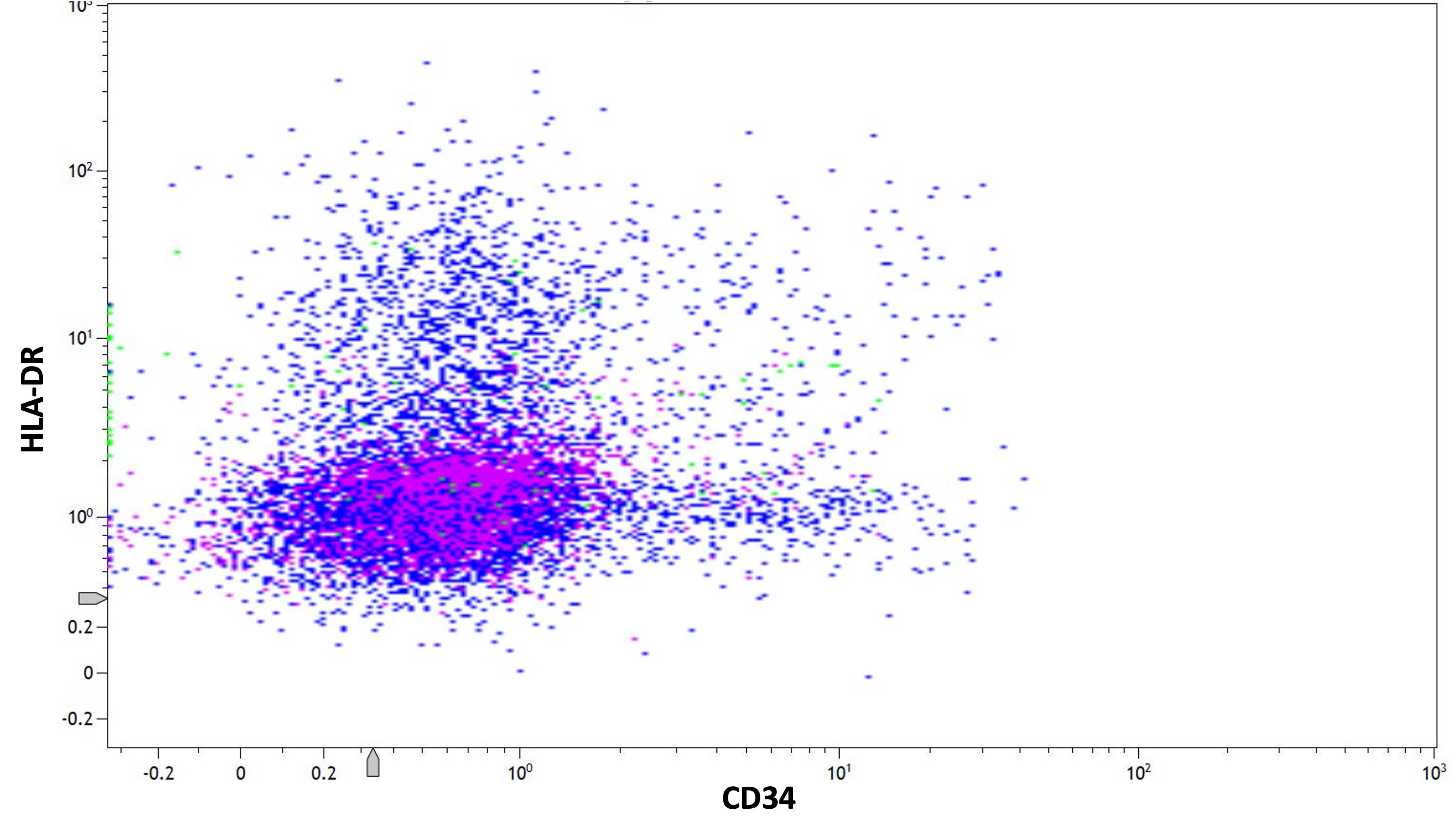
Classic variant, HLA-DR- and CD34-
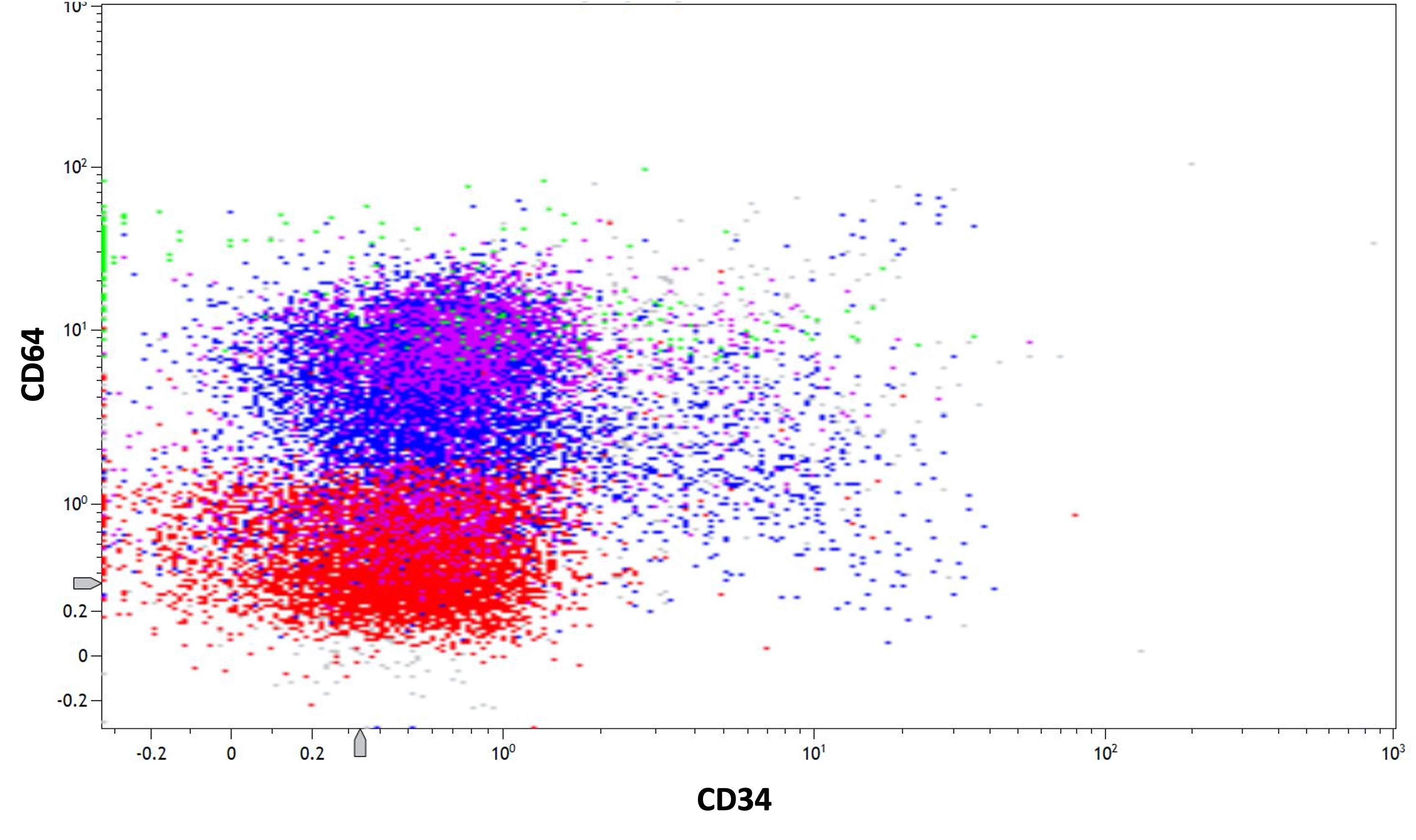
Classic variant, CD34- and CD64+
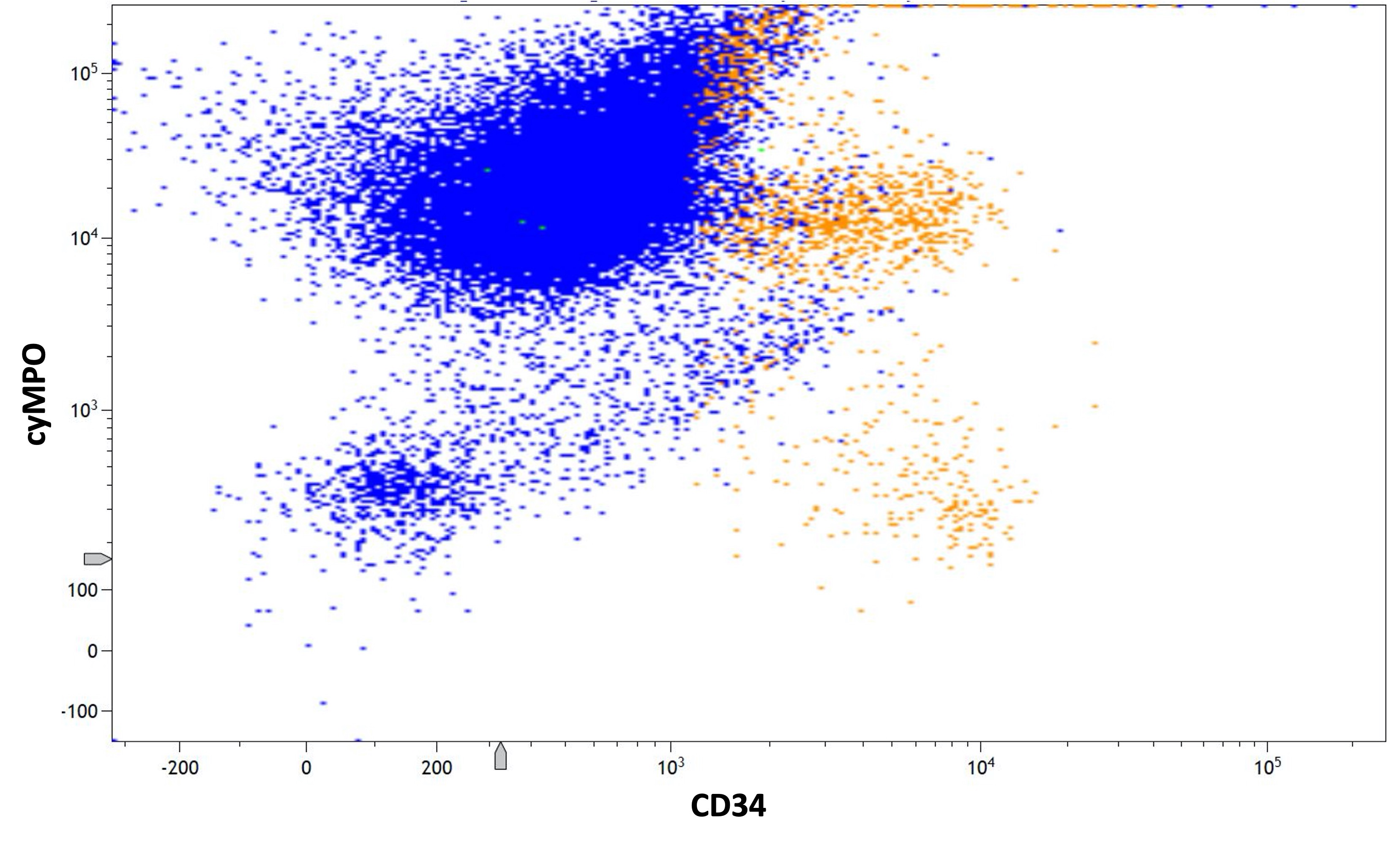
Classic variant, CD34- and cyMPO+
Microgranular variant, moderate CD45
Microgranular variant, CD2+ and CD34+
AFIP images
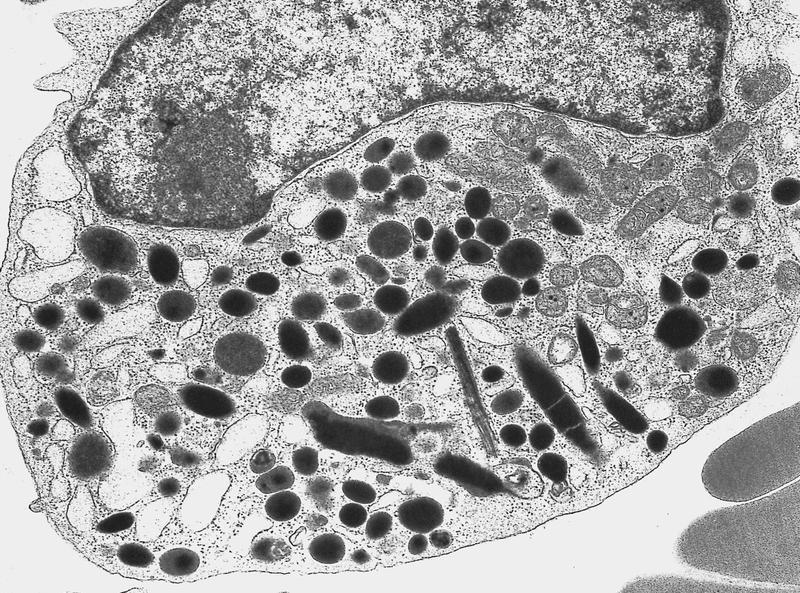
Cytoplasm and nucleus
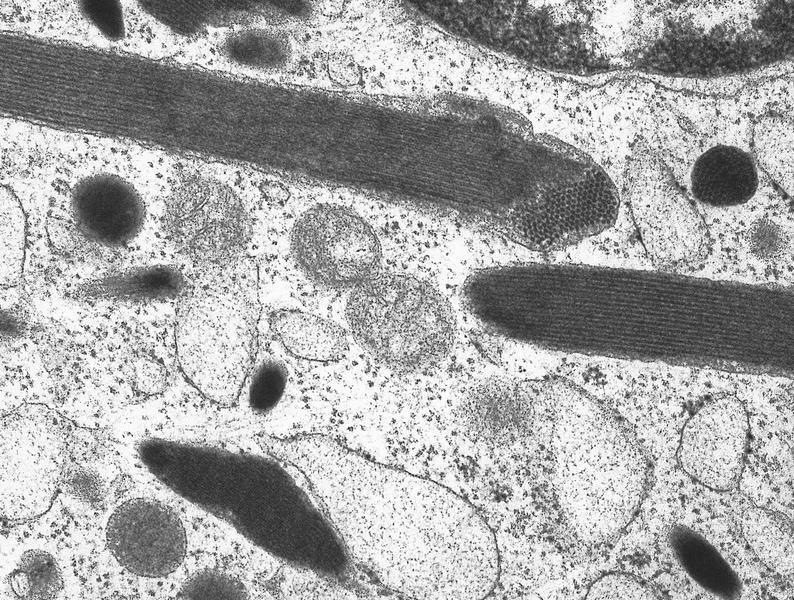
Cross section of Auer rod
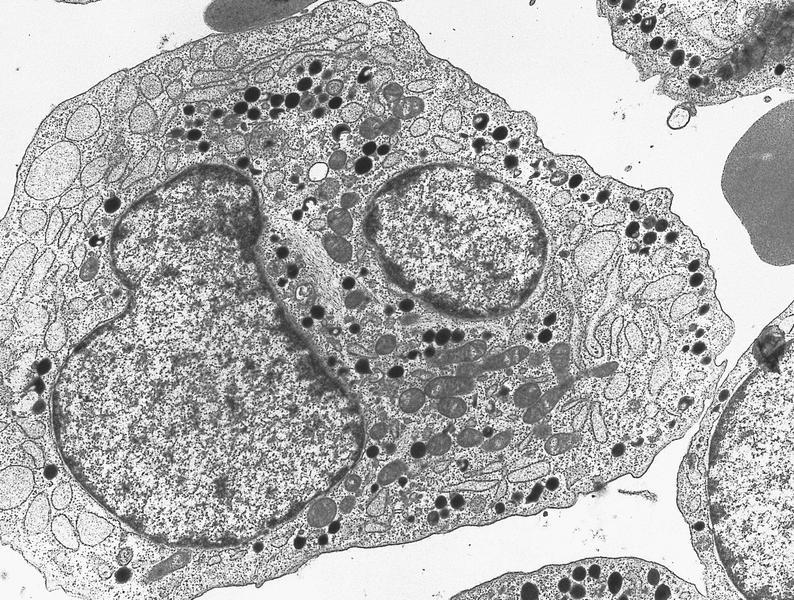
Microgranular variant
Small granules and stellate array
- t(15;17)(q24.1;q21.2) leading to PML::RARA fusion
- PML::RARA can be detected in 90 - 95% of cases (Nature 1990;347:558)
- 3 PML::RARA transcript isoforms (depending on the PML breakpoint location on chromosome 15): long (in intron 6, Bcr1), variant (in exon 6, Bcr2) and short (in intron 3, Bcr3) (Cancers (Basel) 2020;12:624)
- Occasional cases have cryptic t(15;17)(q24.1;q21.2) or complex rearrangements involving an additional chromosome (Leukemia 2016;30:1672)
- Deletion 7q and trisomy 8 are the most common frequent additional cytogenetic alterations (Cancers (Basel) 2020;12:624)
- Variant RARA translocations (Hematol Oncol Stem Cell Ther 2020;13:189, Cancers (Basel) 2020;12:967, Arch Pathol Lab Med 2015;139:1308, Front Oncol 2022;12:871590)
- Detected in ~5% of cases, involve RARA gene at 17q21.2 and different partners
- Should be diagnosed as acute promyelocytic leukemia with variant RARA translocations
- Partner genes
- ZBTB16 at 11q23 has a poor response to all trans retinoic acid and arsenic trioxide, characteristic morphology (see Microscopic (histologic) description)
- NUMA1 at 11q13
- NPM1 at 5q35
- STAT5B at 17q21 has a poor response to all trans retinoic acid and arsenic trioxide
- PRKAR1A at 17q24
- FIP1L1 at 4q12
- BCOR at Xp11
- OBFC2A at 2q32
- TBLR1 at 3q26
- GTF2I at 7q11
- IRF2BP2 at 1q42
- FNDC3B at 3q26
- STAT3 at 17q21
- HNRNPC at 14q11
- NUP98
- TNRC18
- Most common somatic mutations include FLT3 (including FLT3 ITD and FLT3 p.D835), WT1, NRAS, KRAS, USP9X, ARID1A and EED (Leukemia 2016;30:1672, Leukemia 2019;33:1387)
Contributed by Anamarija M. Perry, M.D. and Lina Shao, Ph.D.
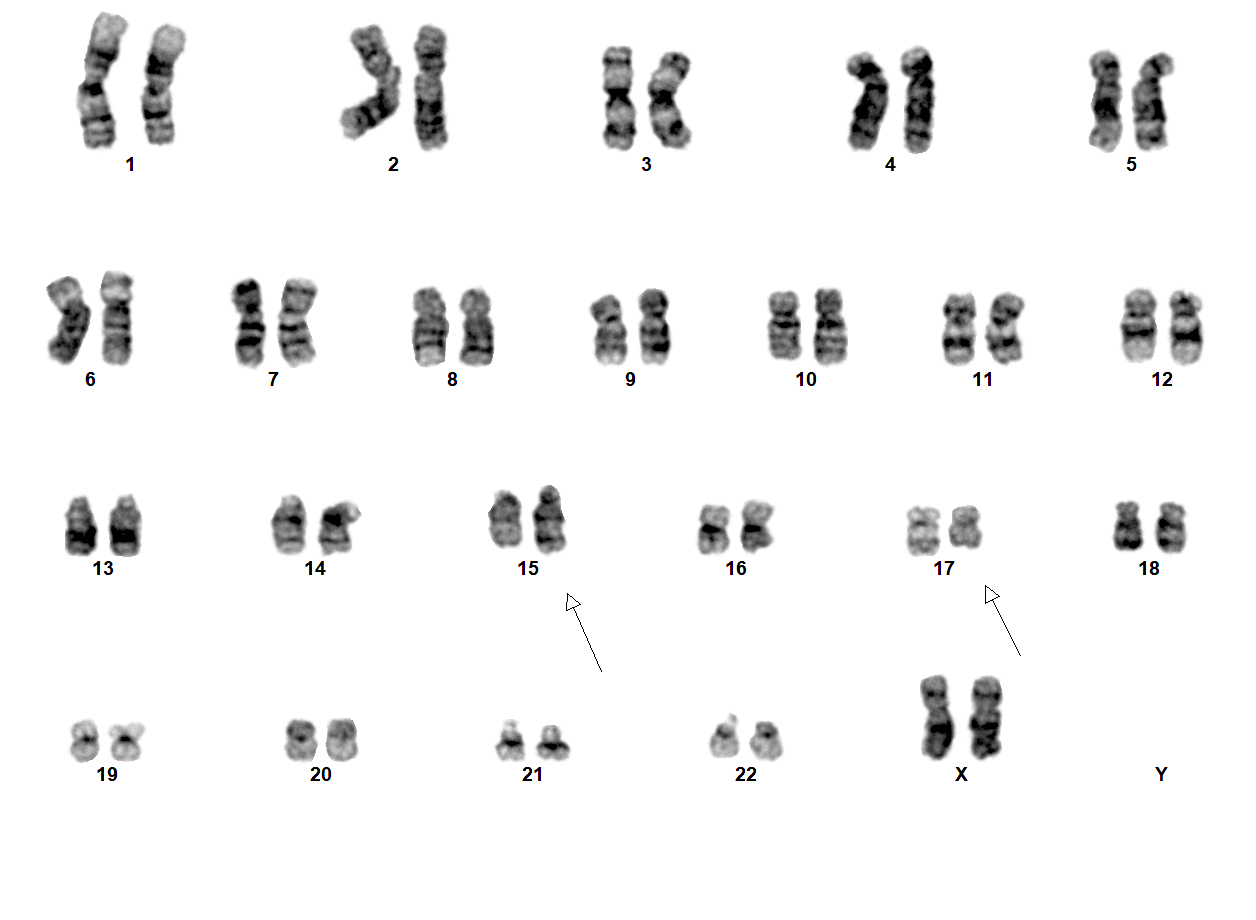
Karyotype

FISH study
- Bone marrow, core biopsy, touch imprints, aspirate clot, aspirate smears and peripheral blood smears:
- Acute promyelocytic leukemia with PML::RARA
- Adequacy: The core biopsy represents ~7 mm of evaluable bone marrow. The aspirate materials are adequate.
- Cellularity: Hypercellular, ~90% on average.
- Erythroid elements: Decreased quantity. Full spectrum of maturation. No dysplasia.
- Myeloid elements: Predominance of atypical promyelocytes in bone marrow. The neoplastic promyelocytes are large cells and most of them are heavily granulated. Many feature a lobulated nucleus and some have overlapping nuclear lobes. Rare cells with numerous (10 - 15) Auer rods are found in the bone marrow and blood smears. There is little granulocytic maturation beyond the promyelocyte stage.
- Megakaryocytes: Adequate number. No definite dysplasia.
- Ancillary studies: Fluorescence in situ hybridization analysis of the bone marrow aspirate is positive for PML::RARA gene fusion.
- Complete blood count:
- White blood cell (K/uL): 0.9
- Red blood cell (M/uL): 3.06
- Hemoglobin (g/dL): 10.1
- Hematocrit (%): 27.1
- Mean corpuscular volume (fl): 88.6
- Red cell distribution width (%): 14.5
- Platelet (K/uL): 106
- Peripheral blood: Absolute leukopenia with occasional leukemic promyelocytes noted. Slight red cell anisocytosis and moderate poikilocytosis with some ovalocytes but no schistocytes. A few nucleated red cells noted on scan. Slight to moderate thrombocytopenia with some variation in platelet size and occasional hypogranular forms.
- Acute myeloid leukemia with monocytic differentiation:
- Can resemble the microgranular variant of acute promyelocytic leukemia with bilobed nuclei and apparent lack of granules
- In microgranular acute promyelocytic leukemia, usually some classic cells with coarse cytoplasmic granules or bundles of Auer rods are seen
- Other than CD64, acute promyelocytic leukemia is usually negative for monocytic markers (e.g., CD14 and CD36)
- Acute myeloid leukemia with mutated NPM1 (Hematol Oncol 2012;30:109, Cytometry B Clin Cytom 2022;102:283):
- Subset of these cases can have overlapping immunophenotypic features with microgranular acute promyelocytic leukemia
- CD2 and CD34 are commonly expressed in acute promyelocytic leukemia and usually negative in AML with mutated NPM1
- When expressed simultaneously, CD13 and CD64 are highly sensitive and specific for acute promyelocytic leukemia
A 27 year old woman presents with fatigue of ~2 weeks' duration and pancytopenia with marked leukopenia. Bone marrow biopsy and aspiration were performed and representative morphology is shown in the image above. Which of the following studies should be done to confirm the diagnosis suspected on morphology?
- FISH for MYC rearrangement
- FISH for PML::RARA fusion
- Mutation analysis for FLT3
- Mutation analysis for NPM1
- RT-PCR for BCR::ABL1
Comment Here
Reference: APL with PML::RARA
- CD34+, CD13+, CD33+, MPO+, CD2-
- CD34+, CD13+, CD33+, MPO-, CD2-
- CD34+, HLA-DR-, CD13+, CD33+, CD64-
- CD34-, HLA-DR-, CD13+, CD33+, CD64+
- CD34-, HLA-DR+, CD13+, CD33+, CD64-
Comment Here
Reference: APL with PML::RARA
- First described (basophilic leukemia) in 1906 by Joachim (Dtsch Arch Klin Med 1906;87:437)
- Acute myeloid leukemia (AML) with primary differentiation to basophils
- To qualify as a type of AML, not otherwise specificed (NOS), other specifically described groups (i.e. AML with myelodysplasia related changes, therapy related AML or AML with recurrent genetic abnormalities) must be excluded
- Wide age range, 1 day - 82 years (Leuk Lymphoma 1999;32:269)
- Rapid clinical progression
- First step in diagnosis is the recognition of the presence of coarse basophilic granules
- Immunophenotyping and electron microscopy features are essential to identify basophilic lineage
- Crucial to differentiate basophilic cells from mast cells
- Diagnosis of acute basophilic leukemia (J Clin Oncol 2011;29:e623):
- Circulating peripheral blood and bone marrow immature basophils / blasts
- Positive with metachromatic staining (toluidine blue), acid phosphatase (diffuse pattern), some cases with periodic acid Schiff (PAS) positivity in large blocks
- Negative for myeloperoxidase (MPO), Sudan black B (SBB) or naphthol ASD chloroacetate esterase (CAE)
- Circulating peripheral blood and bone marrow immature basophils / blasts
- Basophilic leukemia (no longer used)
- Rare disease
- ~ 4 - 5% of all cases of acute nonlymphocytic leukemia and < 2% of all hematopoietic malignancies (Leuk Lymphoma 1999;32:269)
- Peripheral blood, bone marrow, skin, organomegaly
| Immunophenotypic profile of basophils, plasmacytoid dendritic cells (pDC) and mast cells | ||||
| CD203c | Positive | Positive | Negative | Positive |
| CD123 | Positive | Positive / Negative | Positive | Negative |
| HLA-DR | Negative | Positive / Negative | Positive (high) | Positive (low) |
| CD34 | Negative | Negative | Negative | Negative |
| CD117 | Negative | Negative | Negative | Positive |
| CD38 | Positive | Positive / Negative | Negative | Positive (low) |
| CD33 | Positive | Positive / Negative | Negative | Positive (moderate) |
| CD64 | Negative | Positive / Negative | Negative | Negative |
- Wide age range: 1 day - 82 years (Leuk Lymphoma 1999;32:269)
- Typically presents with bone marrow failure
- Variable circulating blasts
- Cutaneous involvement (due to high histamine levels): pruritus, edema, urticarial rashes, focal hyperpigmentation
- GI symptoms: nausea, vomiting, diarrhea, dyspepsia, abdominal swelling or ulcers
- Organomegaly and lytic lesions
- Prerequisite for the diagnosis of acute basophilic leukemia (ABL) and chronic basophilic leukemia (CBL) (Leukemia 2017;31:788)
- Presence of hyperbasophilia (HB) (absolute basophil count exceeding 1,000 per microliter of peripheral blood (PB), present for over at least 8 weeks)
- Percentage of basophils must be ≥ 40% of total leukocytes
- Basophils must belong to the malignant clone as evidenced by
- (Immature) morphology of basophils
- Type of underlying neoplasm (myeloid), if present
- Presence of cytogenetic or molecular alterations is helpful for establishing clonality
- It is important to distinguish ABL from CBL:
- ABL: HB criteria + basophils ≥ 40% of nucleated bone marrow or PB cells + myeloblasts +
metachromatic blasts ≥ 20%
- Primary ABL: no preceding / underlying bone marrow neoplasm
- Secondary ABL: known preceding / underlying bone marrow neoplasm
- CBL: HB criteria + basophils ≥ 40% of nucleated bone marrow or PB cells + myeloblasts +
metachromatic blasts < 20%
- Primary CBL: no preceding / underlying bone marrow neoplasm
- Secondary CBL: known preceding / underlying bone marrow neoplasm
- ABL: HB criteria + basophils ≥ 40% of nucleated bone marrow or PB cells + myeloblasts +
metachromatic blasts ≥ 20%
- CT scan for spleen size and lymph node status
- Complete blood count - white blood cell, hemoglobin, platelets, differential leukocyte count (basophil)
- Serum chemistry, including serum tryptase level
- Allergy diagnostics, including total IgE
- Inflammation markers (C reactive protein (CRP), erythrocyte sedimentation rate (ESR))
- Bone marrow histology (biopsy, clot and smears) and immunohistochemistry
- Chromosome analysis and extended FISH panel for myelodysplastic syndromes (MDS) or myeloproliferative neoplasms (MPN)
- Molecular studies / next generation sequencing panel:
- BCR-ABL1 to be excluded by real time PCR or FISH
- JAK2 V617F by real time PCR
- JAK2 exon 12
- MPL mutation
- CALR mutation
- FIP1L1-PDGFRA by real time PCR or FISH
- Reference: Leukemia 2017;31:788
- Poor prognosis
- Survival time ranges from 2 to > 36 months in chronic basophilic leukemia (CBL); 2 to 16 months in acute basophilic leukemia (ABL) (Leukemia 2017;31:788)
- 44 year old woman with trisomy 19 and t(9;22) and acute basophilic leukemia (Case Rep Pathol 2011;2011:269491)
- 63 year old man presenting with infectious pneumopathy (Am J Hematol 2004;76:134)
- 63 year old woman with history of MDS with isolated del(5q) (Br J Haematol 2019;185:817)
- 63 year old woman with FLT3 ITD+ acute basophilic leukemia with rare complex karyotype presenting with acute respiratory failure (Rev Romana Med Lab 2018;26:87)
- 65 year old man with leukocytosis; acute basophilic leukemia with t(16;21)(p11;q22) generating the FUS‐ERG fusion gene (Clin Case Rep 2017;5:1938)
- 74 year old woman with a history of acute myeloid leukemia (Blood 2014;123:3071)
- 82 year old woman with acute basophilic leukemia associated with loss of gene ETV6 and protean complications (J Clin Oncol 2011;29:e623)
- Acute basophilic leukemia with add(3)(q12) accompanied by histamine excess symptoms (Ann Hematol 2017;96:1197)
- Acute basophilic leukemia with U2AF1 mutation (Blood Cells Mol Dis 2019;76:63)
- Stem cell transplant (SCT)
- If SCT is not possible, polychemotherapy, targeted drugs, experimental drugs or palliative cytoreductive treatment
- Histamine related symptoms (if HB is present): prophylactic histamine receptor (HR) antagonists (HR1 and HR2 blocker), proton pump inhibitors and steroids
- Combination of daunorubicin / idarubicin and cytarabine (Oncol Lett 2014;8:2513)
- Bone marrow biopsy / aspirate
- Hypercellular bone marrow with medium sized blasts with high nuclear to cytoplasmic ratio, an oval, round or bilobed nucleus, dispersed chromatin with 1 - 3 prominent nucleoli
- Moderate amounts of basophilic cytoplasm that contains a variable number of coarse basophilic granules
- Cytoplasmic vacuolation may be present
- Mature basophils are usually sparse
- Dysplastic features in the erythroid precursors may be present
Contributed by Pallavi Khattar, M.D.
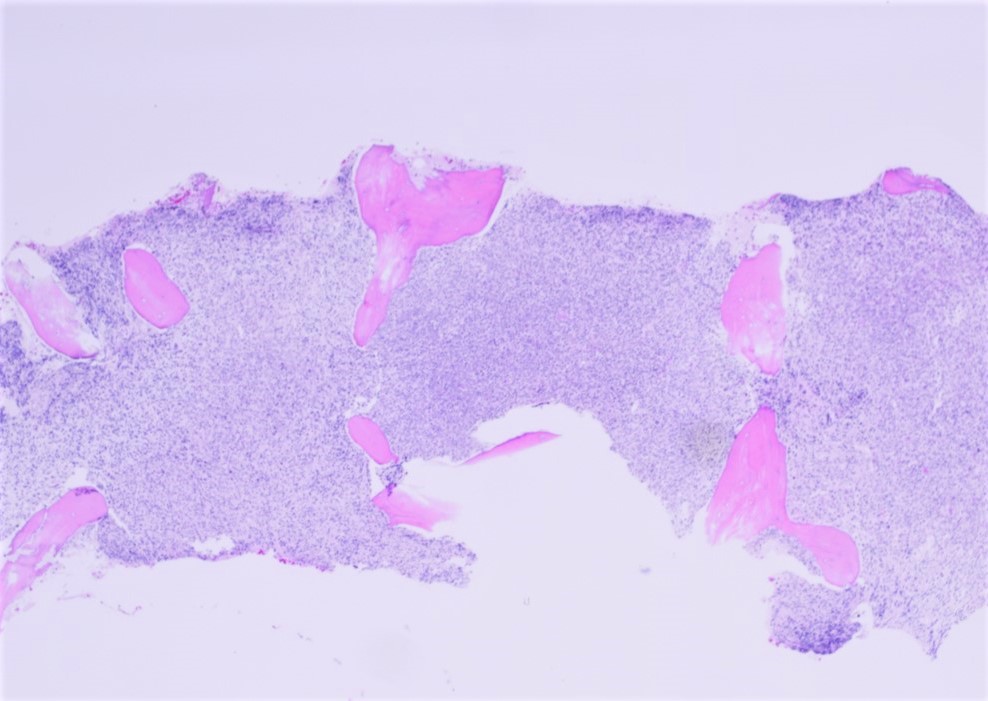
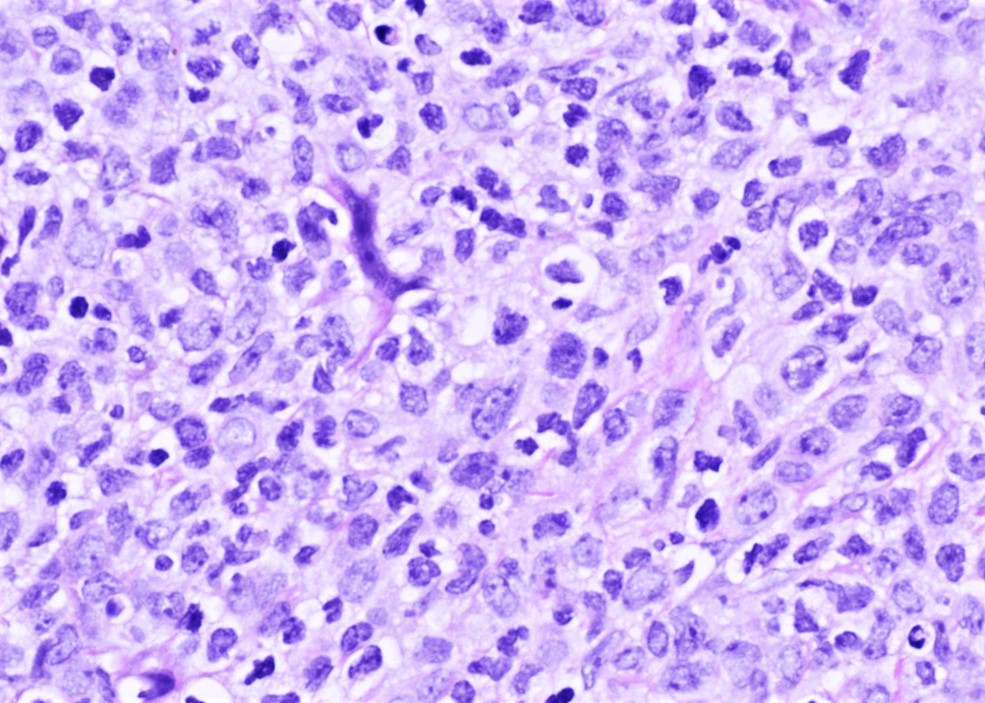
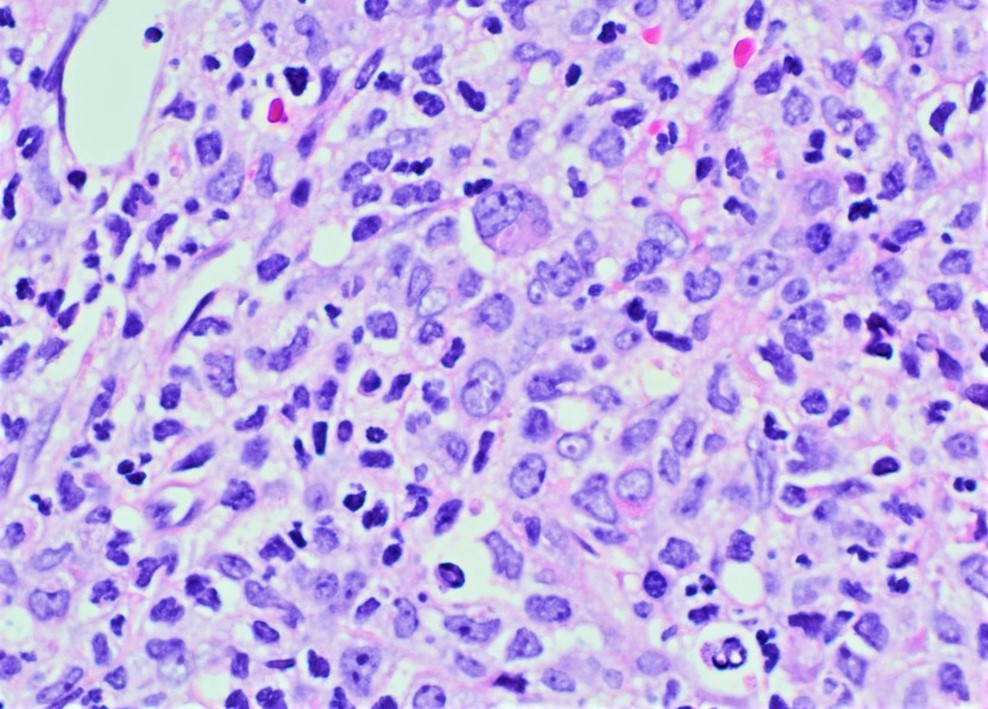
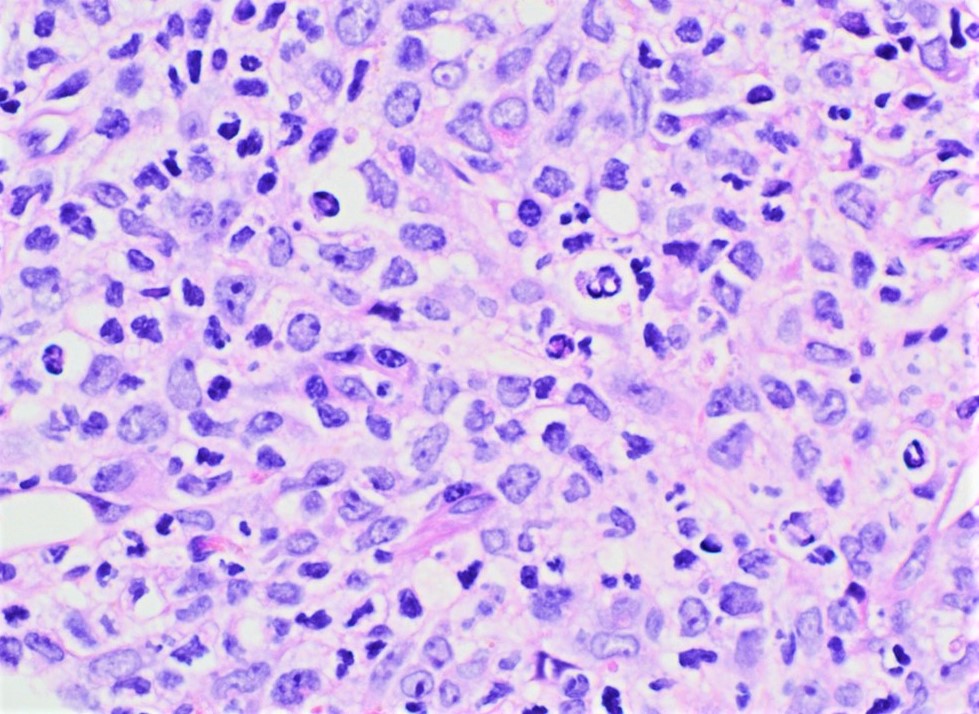
Bone marrow biopsy
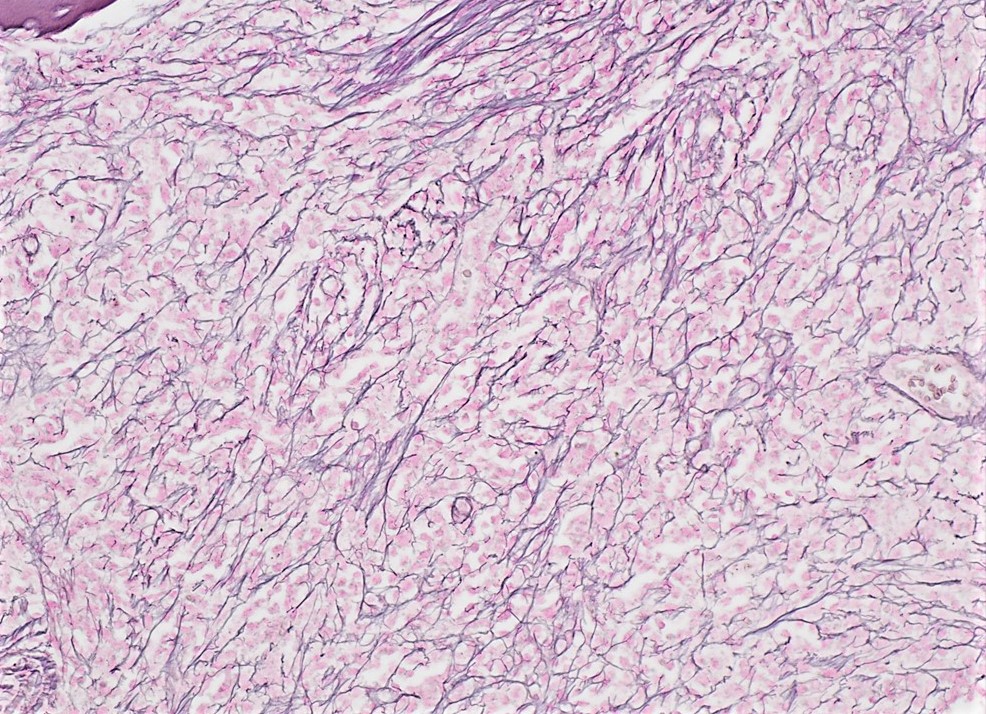
Bone marrow biopsy - reticulin stain
- CD13, CD33, CD123, CD203c, CD11b, CD34, CD9, CD25, CD22 (subset), TdT (subset), HLA-DR, Basogranulin (BB1), 2D7a (Leukemia 2017;31:788)
- Cytochemical stains: toluidine blue (metachromatic staining), acid phosphatase (diffuse pattern), periodic acid-Schiff (PAS) (large block-like pattern)
- CD117, tryptase, most monocytic markers and most lymphoid markers
- Cytochemical stains: myeloperoxidase (MPO), Sudan black B (SBB), naphthol AS-D chloroacetate esterase (CAE)
- Immunophenotypic profile of basophils (See Diagrams / tables)
- Positive: CD45, CD9, CD13, CD33 , CD11b, CD25, CD123 (IL3 R alpha chain), Fcε receptor type I (IgE-R), Bsp-1, CD203c (ectoenzyme ENPP3)
- Negative: CD117, CD25, monocytic markers (except CD11b), lymphoid markers (except CD22)
- Reference: Leukemia 2017;31:788
- Granules contain an electron dense particulate substance and are internally bisected, e.g. have a theta character (θ) or contain crystalline material arranged in a pattern of scrolls or lamellae
- Coexistence of basophil and mast cell granules may be identified in the same immature cells
- No consistent chromosomal abnormality identified; exclude cases with t(9;22) BCR-ABL
- Complex karyotype may be present (Cancer Genet Cytogenet 2008;182:46, Cancer Genet Cytogenet 1991;51:215)
- Recurrent t(X;6)(p11.2;q23.3) resulting in MYB-GATA1 appears to occur in male infants (Blood 2011;117:5719)
- t(3;6)(q21;p21) and abnormalities involving 12p (Bibl Haematol 1978;45:142)
- Bone marrow, right posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Consistent with acute myeloid leukemia, NOS favor acute basophilic leukemia (see comment)
- Comment: Hypercellular marrow (100%) is essentially replaced by sheets of immature myeloid cells (approximately 70% of total cellularity) with increased basophils. Bone marrow aspirate smears show blasts, 25%; atypical immature basophils, 38%; mature basophils, 7%. The blasts often had basophil-like dark, coarse granules. Special stains demonstrate that the blasts were positive for metachromatic toluidine blue but negative for myeloperoxidase (MPO). Flow cytometric analysis demonstrates immature myeloid cells (comprises 30% of total) expressing CD13, CD33, CD34, CD38 and HLA-DR and heterogeneous population of basophils (approximately 35% of total) express CD11b, CD13, CD33, CD22, CD123 and CD203. No monotypic B cells or immunophenotypically abnormal T cells are detected. Additional cytogenetic and molecular studies are in progress and will be reported separately.
- Peripheral blood: Anemia, thrombocytopenia and leukocytosis. Hemoglobin was 8.9 g/dl, platelet 61/ 109/L and total leukocyte count 15 /109/L. The smear revealed a large population of basophils with a variable degree of maturation ranging from blasts with coarse basophilic granules to mature basophil granulocytes, some of which had signs of degranulation.
- Bone marrow biopsy: Quality adequate. Hypercellular marrow (100%), large population of basophils with a variable degree of maturation ranging from blasts with coarse basophilic granules to mature basophil granulocytes, some of which had signs of degranulation. Bone marrow residual myeloid as well as erythroid lineages were markedly reduced to absent. Rare unremarkable megakaryocytes were seen. There are no granulomas. The bony trabeculae show evidence of remodeling. Stainable iron is present (iron stain). There is a diffuse increase in reticulin fibers with focal collagen fibrosis; (reticulin stain, trichrome stain).
- Bone marrow clot section: Quality adequate. Cellularity - 100% morphologic features are similar to those observed in the core biopsy.
- Bone marrow aspirate: Quality adequate. The smears show blast cells, 25%; atypical immature basophils, 38%; mature basophils, 7%; normal mature neutrophils, 11%; lymphocytes, 7%; monocytes, 2% and erythroid cells, 10%. The blasts often had basophil-like dark, coarse granules and basophils had characteristic confluent vacuoles merged with the cell membrane suggestive of degranulation.
- Special Stains: Blasts were toluidine blue metachromatic but negative for myeloperoxidase.
- Flow Cytometry Analysis: Gating - mononuclear cells by forward and side light scatter.
- Interpretation: Immature myeloid blasts (comprises 30% of total) were positive for CD13, CD33, CD34, CD38 and HLA-DR. In addition, heterogeneous population of basophils (approximately 35% of total) express CD11b, CD22, CD13, CD33, CD123 and CD203. The immature myeloid blasts were negative for TdT, CD10, CD14, CD61, CD64, CD2, sCD3, cCD3, CD4, CD5, CD8, CD16, CD56, CD19, CD20, CD22, CD23, cCD79a, CD103, CD117, FMC7, kappa and lambda.
- Reactive conditions with a massive expansion of polyclonal basophils
- Acute promyelocytic leukemia with basophilic differentiation (Br J Haematol 1982;50:201, Leukemia 1994;8:1441)
- AML with t(6;9)(p23;q34.1) DEK-NUP214:
- Seen in older children; rare in infants
- Often associated with basophilia, multilineage dysplasia and poor prognosis (Br J Haematol 2014;166:254)
- Myeloproliferative and myelodysplastic neoplasm with basophilia (MPN basophils; MDS basophils):
- Accelerated phase of chronic myeloid leukemia (CML)
- Blast phase of myeloproliferative neoplasm
- JAK2 mutated MPN, MDS / MPN overlap diseases (Am J Clin Pathol 2015;144:188)
- AML with t(8;21) RUNX1-RUNX1T1 and basophilia:
- Seen in younger patients
- Blasts are large with abundant basophilic cytoplasm containing numerous azurophilic granules and perinuclear hofs
- Often with large granules (pseudo-Chédiak-Higashi granules)
- Usually associated with high rate of complete remission and longterm disease survival (Rinsho Ketsueki 2017;58:991)
- Lymphoblastic leukemia with prominent coarse granules (rare)
- Mast cell leukemia:
- Expresses CD117, tryptase and CD25 (Diagn Pathol 2018;13:14)
- CD25
- CD117
- CD123
- CD203c
Comment Here
Reference: Acute basophilic leukemia
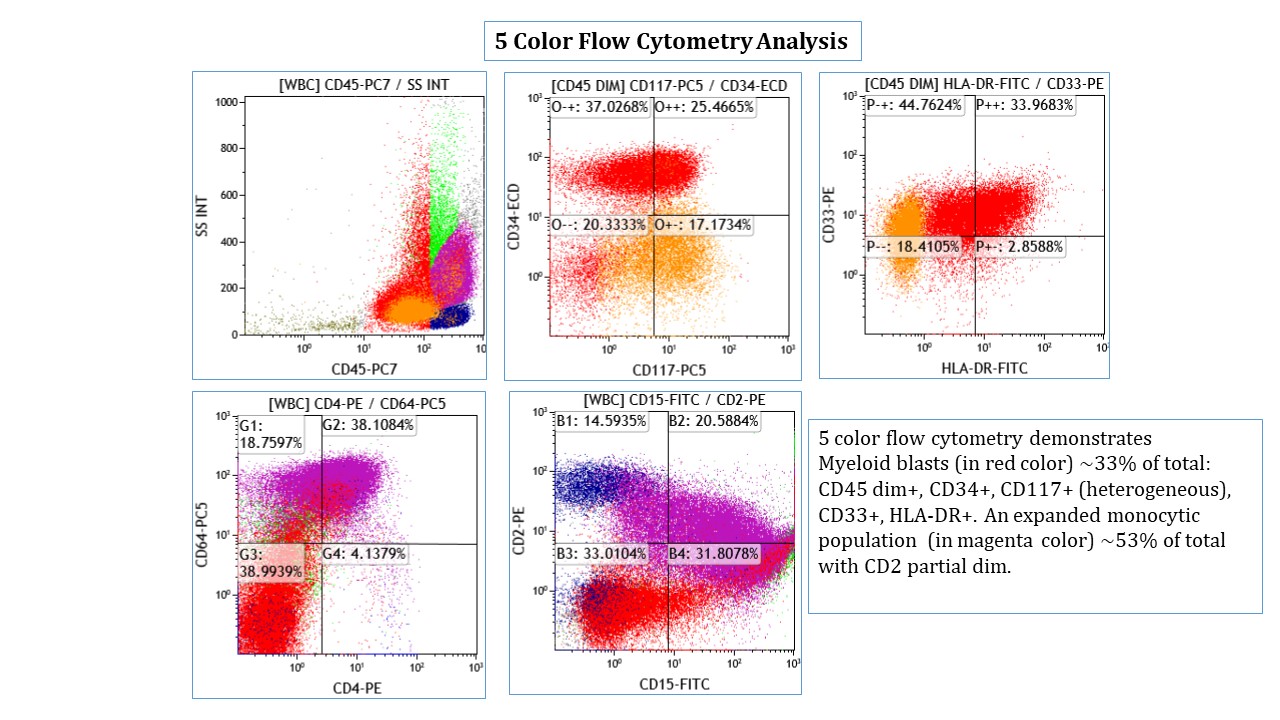
Which of the following is positive for CD34 and CD117 immunostain?
- Basophil
- Eosinophil
- Mast cell
- Myeloblast
Comment Here
Reference: Acute basophilic leukemia
- Highly aggressive neoplasm of precursor lymphoid (blast) cells
- 2 main subtypes based on lymphoid lineage
- B lymphoblastic leukemia / lymphoma (B-ALL / LBL)
- T lymphoblastic leukemia / lymphoma (T-ALL / LBL)
- Characterized by neoplastic proliferation of clonal precursor B cells or T cells that typically have blastic cytomorphology
- Lymphoblastic lymphoma generally refers to tissue mass lesion, while leukemia refers to bone marrow or blood involvement
- B-ALL / LBL has a better prognosis in children than in adults
- Childhood B lineage ALL / LBL has a better prognosis than childhood T lineage ALL / LBL
- Immature cell markers, such as CD34 and TdT, can help to differentiate lymphoblasts from Burkitt lymphoma which, is considered a mature high grade B cell lymphoma that mimics lymphoblastic lymphoma / leukemia
- Lymphoblastic leukemia / lymphoma
- Acute lymphoblastic leukemia (ALL)
- Precursor B cell lymphoblastic leukemia / lymphoma or precursor T lymphoblastic leukemia / lymphoma
- Separation of lymphoblastic leukemias and lymphomas based on a measure of disease burden / stage
- Cases with tissue involvement + replacement of < 25% marrow involvement by lymphoid blasts = lymphoblastic lymphoma (LBL)
- Cases with ≥ 25% marrow involvement = lymphoblastic leukemia
- Estimated annual incidence in the United States approximately 1.6 cases per 100,000 population (Blood Cancer J 2017;7:e577)
- Approximately 75 - 80% of ALL / LBL occurs in children, while the remainder occurs as aggressive disease in adults (J Natl Compr Canc Netw 2015;13:1240)
- Among LBL, T cell > B cell phenotype (~90% versus 10%) (Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017)
- Among ALL, B cell > T cell phenotype
- Children: 85% versus 15%
- Adults: 75% versus 25%
- T-ALL / LBL:
- More common in adolescents than children
- M:F = 2.2:1
- B-ALL / LBL:
- Bimodal age distribution with highest peak in children < 6 years old
- Second lower incidence peak in adults > 65 years old (Eur J Cancer Care (Engl) 2005;14:53)
- Slight male predominance, M:F = 1.2 - 1.8:1
- Bone marrow, peripheral blood or nodal involvement
- Anterior mediastinum
- Particularly T-LBL
- Compromised cardiac function and superior vena cava syndrome may result
- Less frequent sites involved
- Skin
- Soft tissues
- Central nervous system
- Clonal proliferation of precursor B cells in the bone marrow
- Clonal proliferation of precursor T cells in the thymus (thymocytes)
- Specific sequential pathologic mechanisms that follow appear to be variable, based on the varying genetic aberrations identified
- Idiopathic and likely multifactorial
- Chromosomal aberrations with gene mutations and rearrangements commonly cited
- Specific risk factors seen in a minority of patients:
- Prenatal radiation exposure (Xradiation)
- Postnatal exposure to high doses of radiation (e.g. therapeutic radiation was previously used for tinea capitis and thymus enlargement)
- Genetic conditions, such as Down syndrome, neurofibromatosis and ataxia telangiectasia
- Inherited genetic polymorphisms
- Reference: Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017
- B-ALL / LBL
- Common presentation of fever, fatigue, bone or joint pain, bleeding or anorexia (signs of bone marrow infiltration)
- May present with widespread lymphadenopathy, hepatosplenomegaly, involvement of skin, soft tissue and testes, with predilection for the central nervous system
- Mediastinal mass uncommon
- T-ALL / LBL
- Most commonly presents with anterior mediastinal mass with or without superior vena cava syndrome
- Lymph node involvement on Xray / CT / MRI showing thymic enlargement or pleural effusion, resulting in symptoms of respiratory distress
- Generalized lymphadenopathy and hepatosplenomegaly also frequent
- Reference: Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017
- Multimodal pathologic evaluation with some combination of morphology, flow cytometry, immunohistochemistry, cytogenetics, FISH, PCR, NGS and basic clinical laboratory tests
- Nodal excisional biopsy may be the initial diagnostic procedure, although marrow involvement is frequently present simultaneously
- If marrow involvement suspected, initial approach may consist of complete blood count and peripheral blood smear, followed by bone marrow aspiration and biopsy
- Complete blood count
- Almost universally shows anemia and thrombocytopenia
- ~25% have severely decreased platelet count (< 20,000/uL)
- Total white blood cell (WBC) count variable but most commonly presents with neutropenia
- 50% show WBC count < 10,000/uL
- 30% show WBC count 5,000 - 50,000/uL
- 20% show WBC count > 50,000/uL
- 20% show preleukemic state with cytopenia < 1 year before overt leukemia
- Rare reports of asymptomatic hypereosinophilia (Cancer 1984;54:1058), most commonly seen in the setting of t(5;14) (q31;q32) or relatively less common recurrent abnormalities, such as translocation t(7;12)(q22;p13); also reported in the setting of hyperdiploidy (Blood 2020;135:974, Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017)
- Almost universally shows anemia and thrombocytopenia
- Lymphadenopathy
- T-LBL may show an anterior mediastinal mass due to thymic enlargement or presence of pleural effusion on Xray
Images hosted on other servers:

T-LBL mediastinal involvement
- Prognosis generally determined by lineage (B cell or T cell phenotype), plus cytogenetic or molecular abnormalities detected
- B-ALL / LBL
- Unfavorable prognostic factors:
- Infancy and adult age of diagnosis
- High white blood cell count
- Slow response to initial therapy
- Central nervous system involvement at the time of diagnosis
- Minimal residual disease after therapy
- Unfavorable prognostic factors:
- T-ALL / LBL
- Worse prognosis than B-ALL / LBL among pediatric population
- Better prognosis than B-ALL / LBL among adult population
- Reference: Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017
- 2 year old boy with no significant medical history presented with lower extremity pain and fever (Ochsner J 2019;19:260)
- 31 year old man with unprecedented medical history who presented with constant back pain not responsive to pain meds, night sweats and fever (Stem Cell Investig 2019;6:17)
- 42 year old woman who presented with a 1 month history of paleness and fatigue (BMC Cancer 2018;18:1143)
- 44 year old woman with a secondary thymoma after chemotherapy treatment for T acute lymphoblastic leukaemia / lymphoma (Open Access Maced J Med Sci 2018;6:2373)
- 51 year old man presented with an increase in superficial lymph nodes (Medicine (Baltimore) 2018;97:e12743)
- Multiagent chemotherapeutic regimen stratified based on epidemiology, presentation and other patient specific associated characteristics of the disease, as well as presence of specific cytogenetic abnormalities and therapeutic response to initial induction (Pediatr Int 2018;60:4)
- Intrathecal prophylactic therapy and radiation for those presenting with central nervous system involvement (Biol Blood Marrow Transplant 2016;22:575)
- Allogeneic stem cell / bone marrow transplantation in those demonstrating high risk findings (Am J Hematol 2017;92:18)
- Targeted tyrosine kinase inhibitors or JAK inhibitors may be used in patients with Ph-like ALL (Best Pract Res Clin Haematol 2018;31:351)
- CAR-T cell immunotherapy in relapsed patients (Curr Hematol Malig Rep 2018;13:396)
- Supportive therapy for cytopenias
- In tissue masses or marrow, effacement of architecture by round blue cells raises the differential diagnoses of ALL / LBL versus neuroblastoma, Ewing sarcoma and other mimickers
- Proliferation of typically small to medium sized primitive cells but some tumor cells may appear as larger blasts containing cytoplasmic vacuoles
- B and T cell precursors are morphologically indistinguishable
- In nodal involvement, blasts usually display a diffuse growth pattern
- Partial nodal involvement of T-LBL may have a predominantly interfollicular distribution
- Lymphoblasts may be oval in shape and display an indented nucleus with finely dispersed chromatin and inconspicuous nucleoli
- Revised criteria to the original French American British (FAB) classification of ALL cytologic subtypes
- L1 and L2 subtype segregation not shown to correlate with clinical or biologic behavior; thus, most commonly used for descriptive purpose at this time
- L1 subtype: most common, with intermediate size, uniform features, scant basophilic cytoplasm, slightly condensed chromatin with inconspicuous / absent nucleoli
- L2 subtype: more heterogeneous appearance, with slightly more cytoplasm and more prominent nucleoli, resembling myeloblasts
- L3 subtype: now corresponds to leukemic phase of Burkitt lymphoma, a high grade mature B cell lymphoma (Blood Cancer J 2017;7:e577)
- L1 and L2 subtype segregation not shown to correlate with clinical or biologic behavior; thus, most commonly used for descriptive purpose at this time
Contributed by Patricia Tsang, M.D.
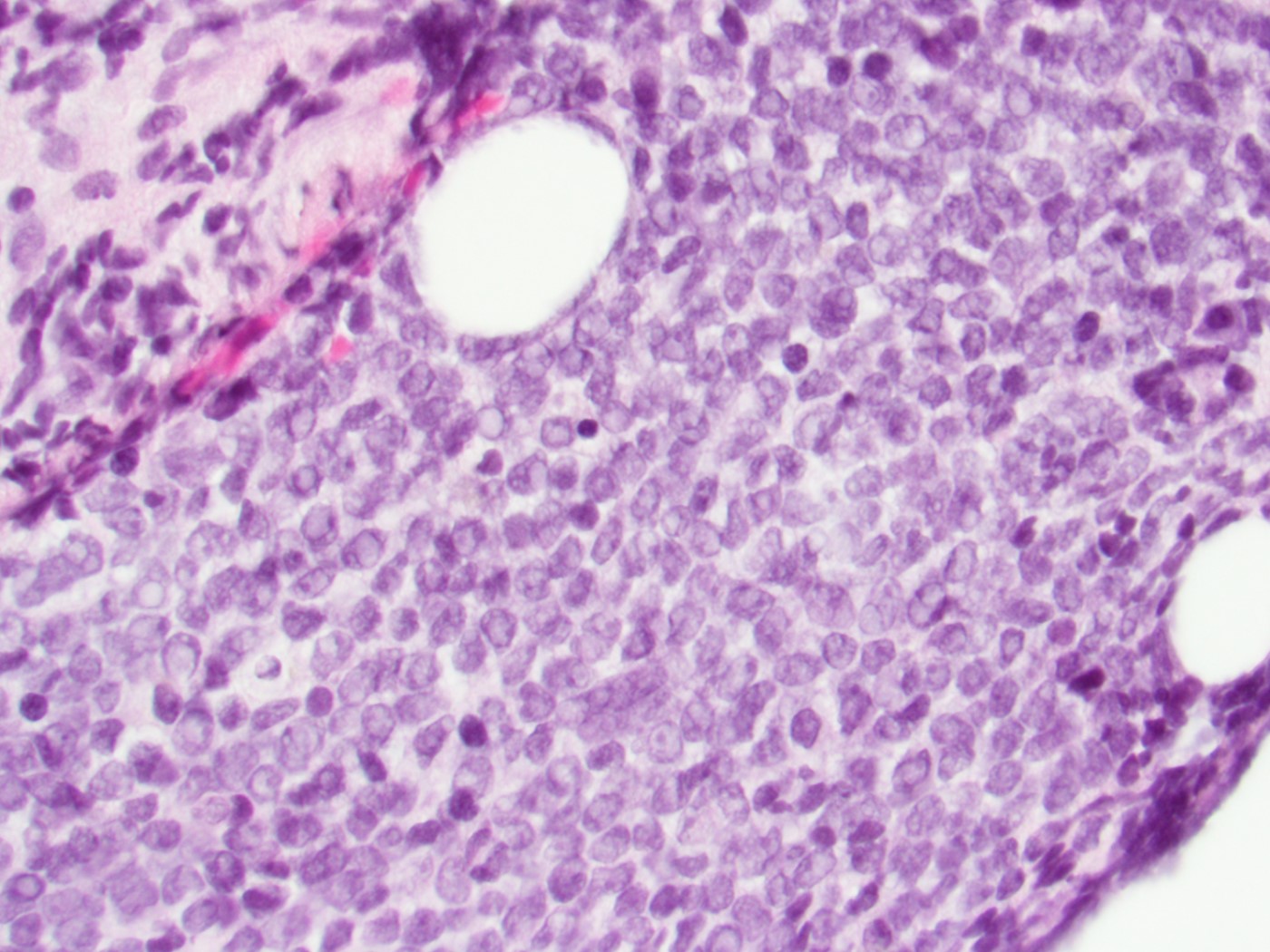
B-LBL retroperitoneal fibroadipose
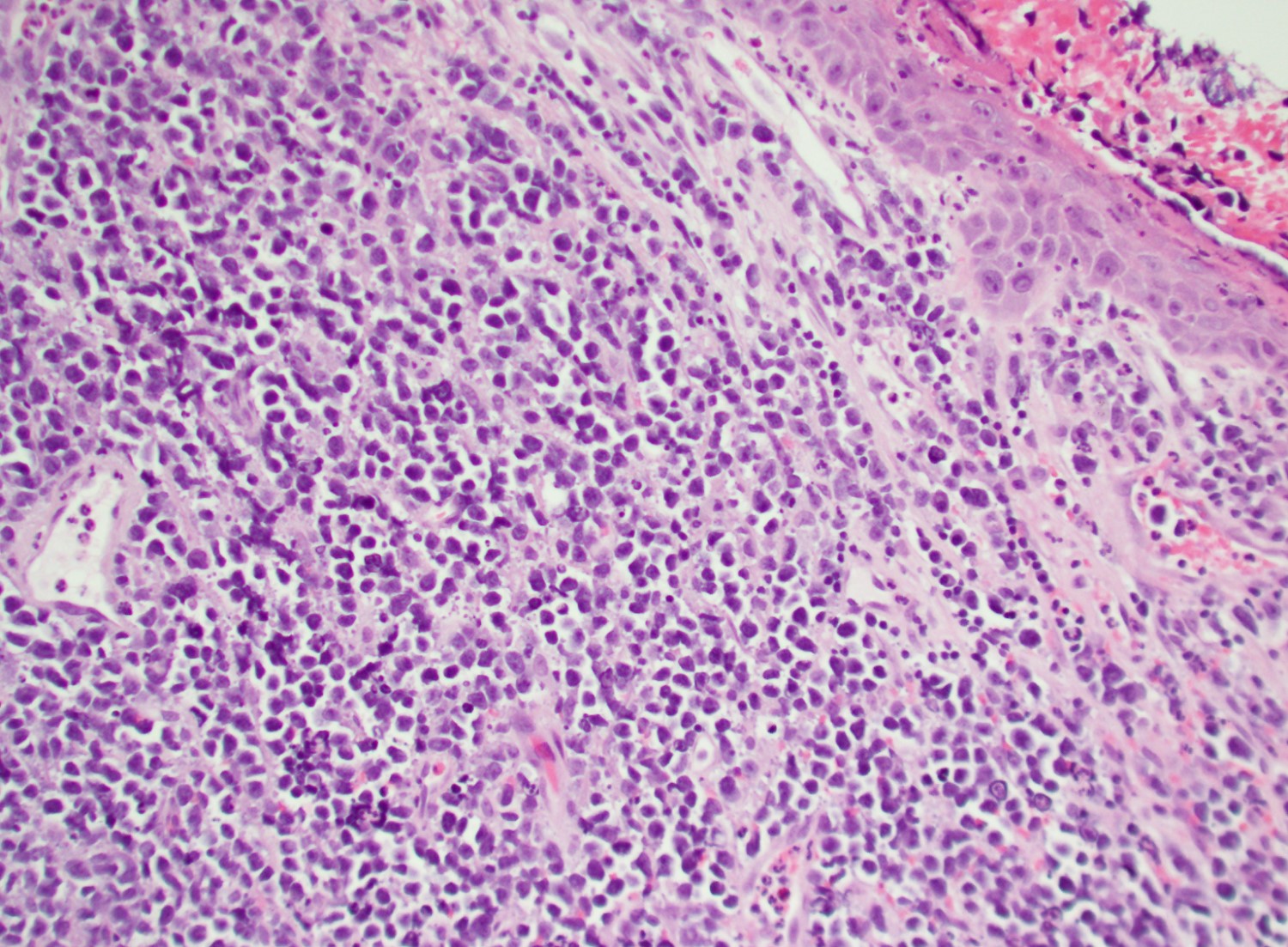
B-LBL tongue
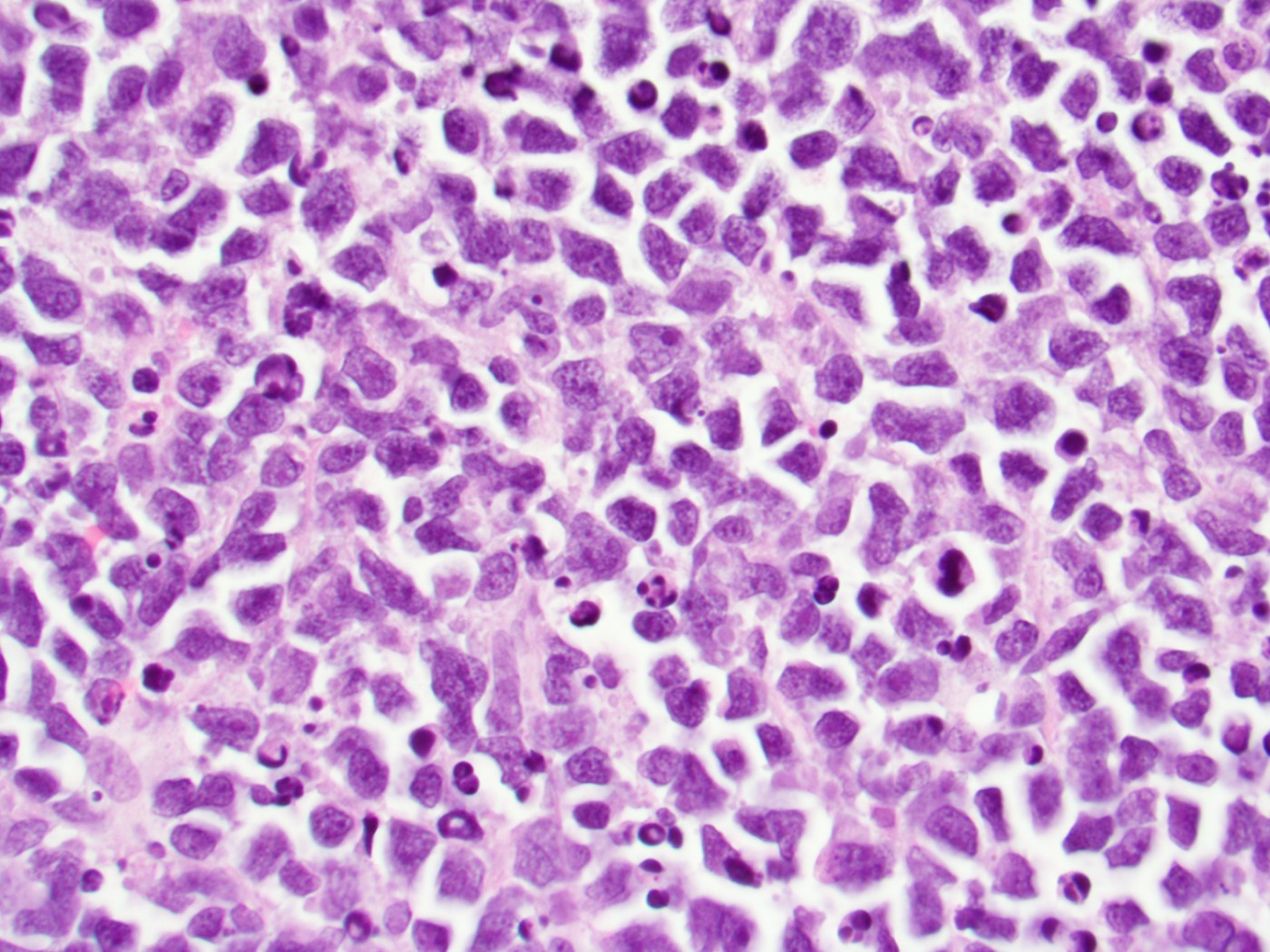
B-LBL tongue
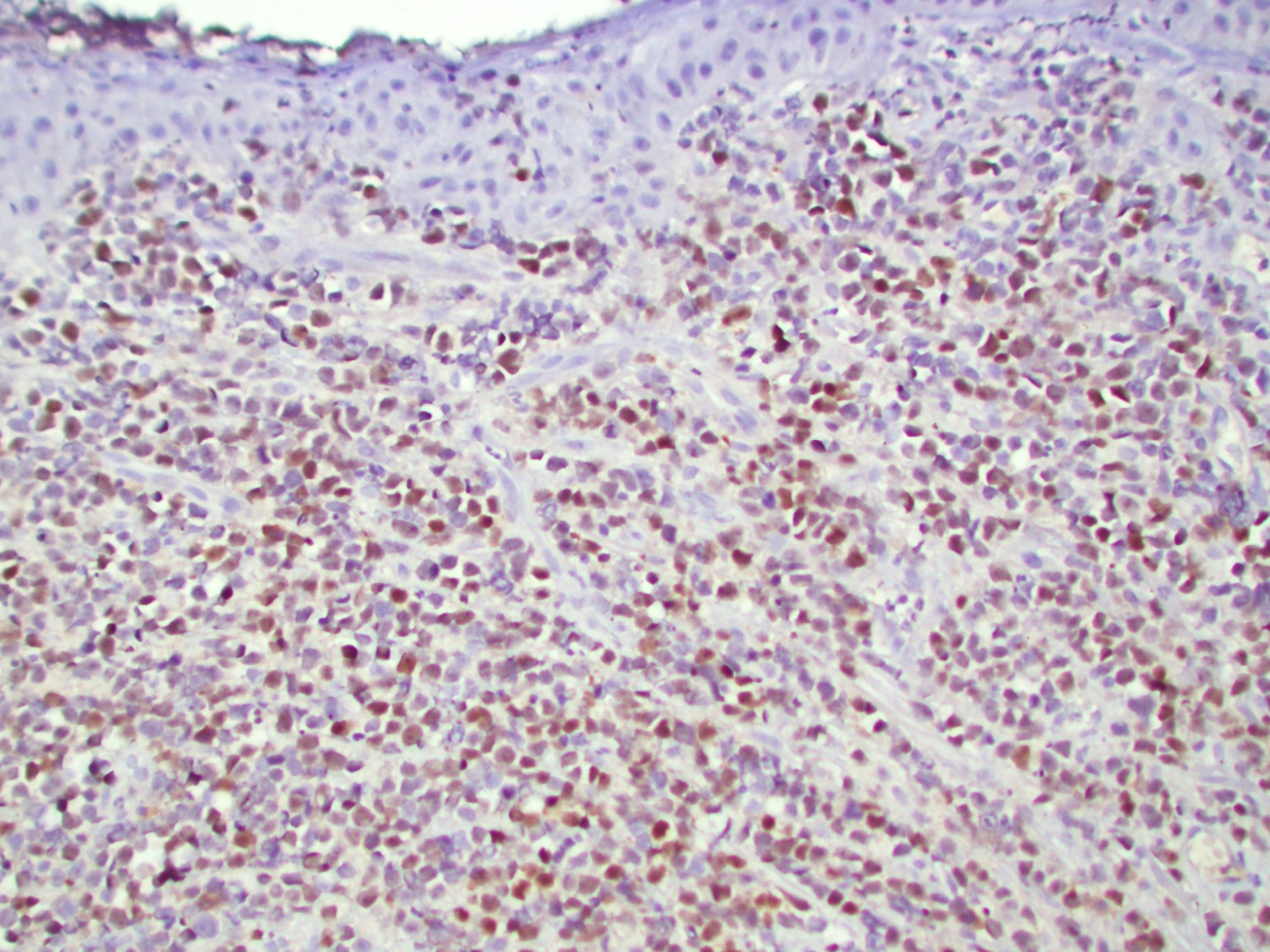
B-LBL tongue TdT
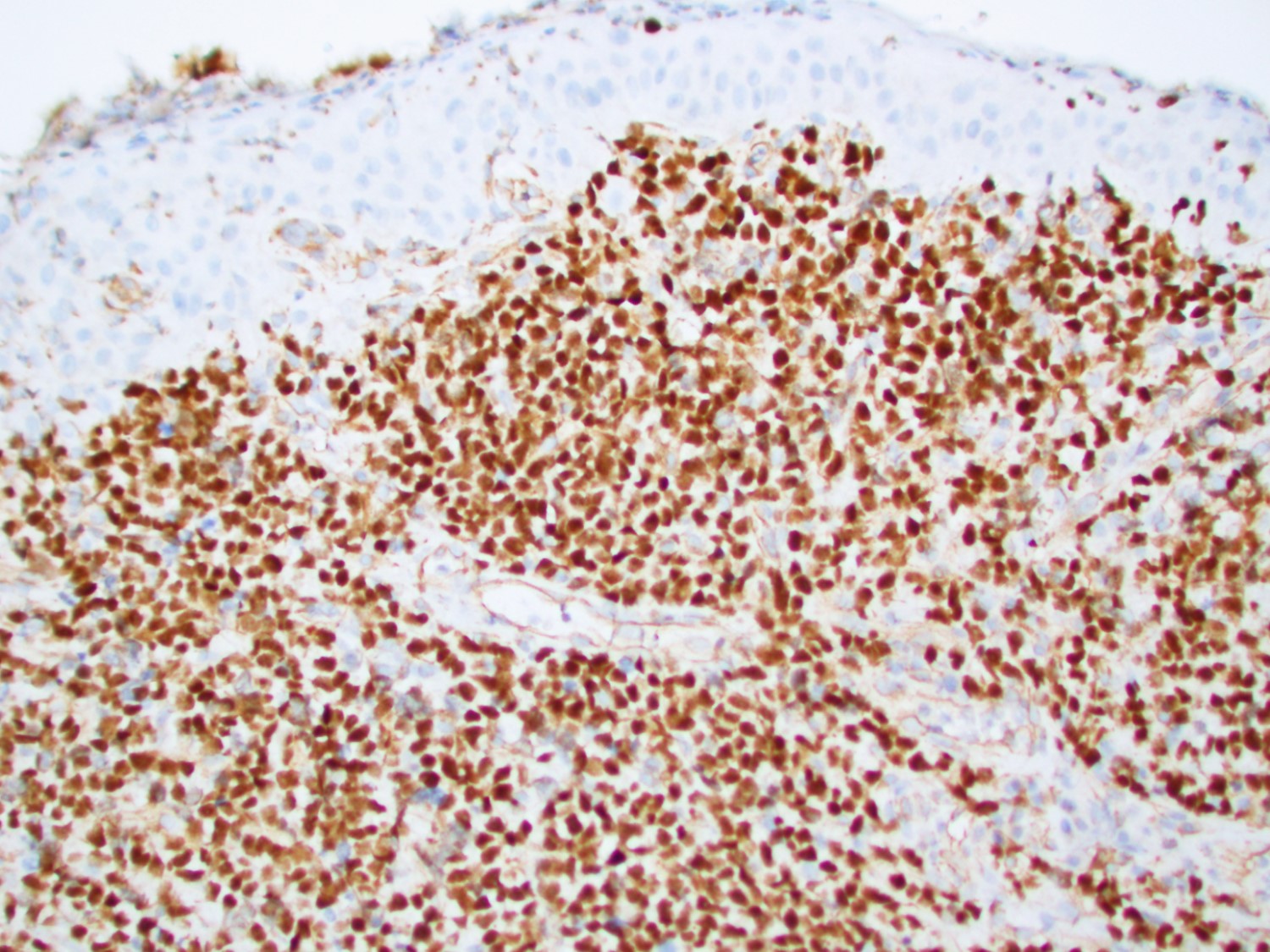
B-LBL tongue PAX5
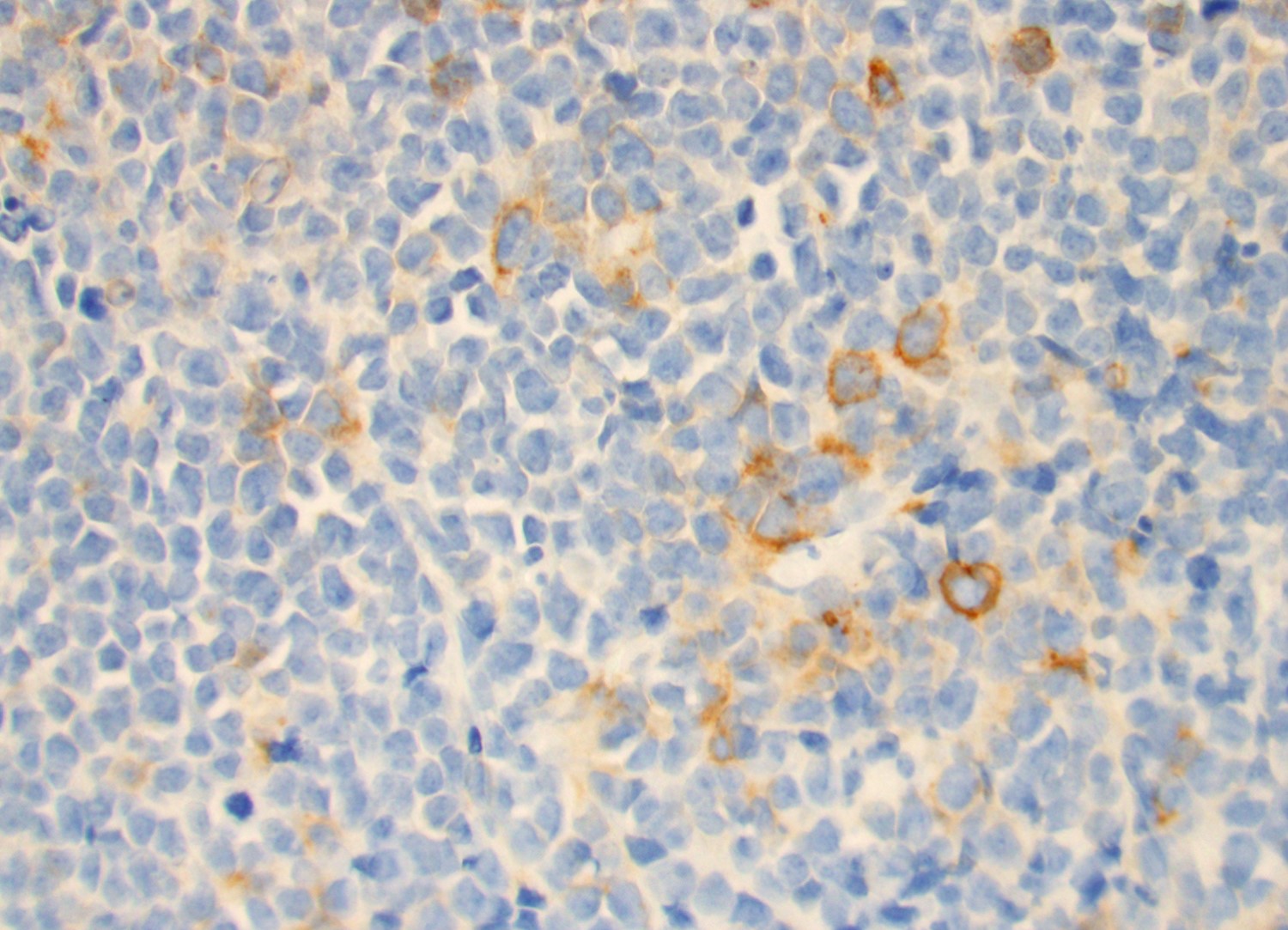
B-LBL tongue CD20
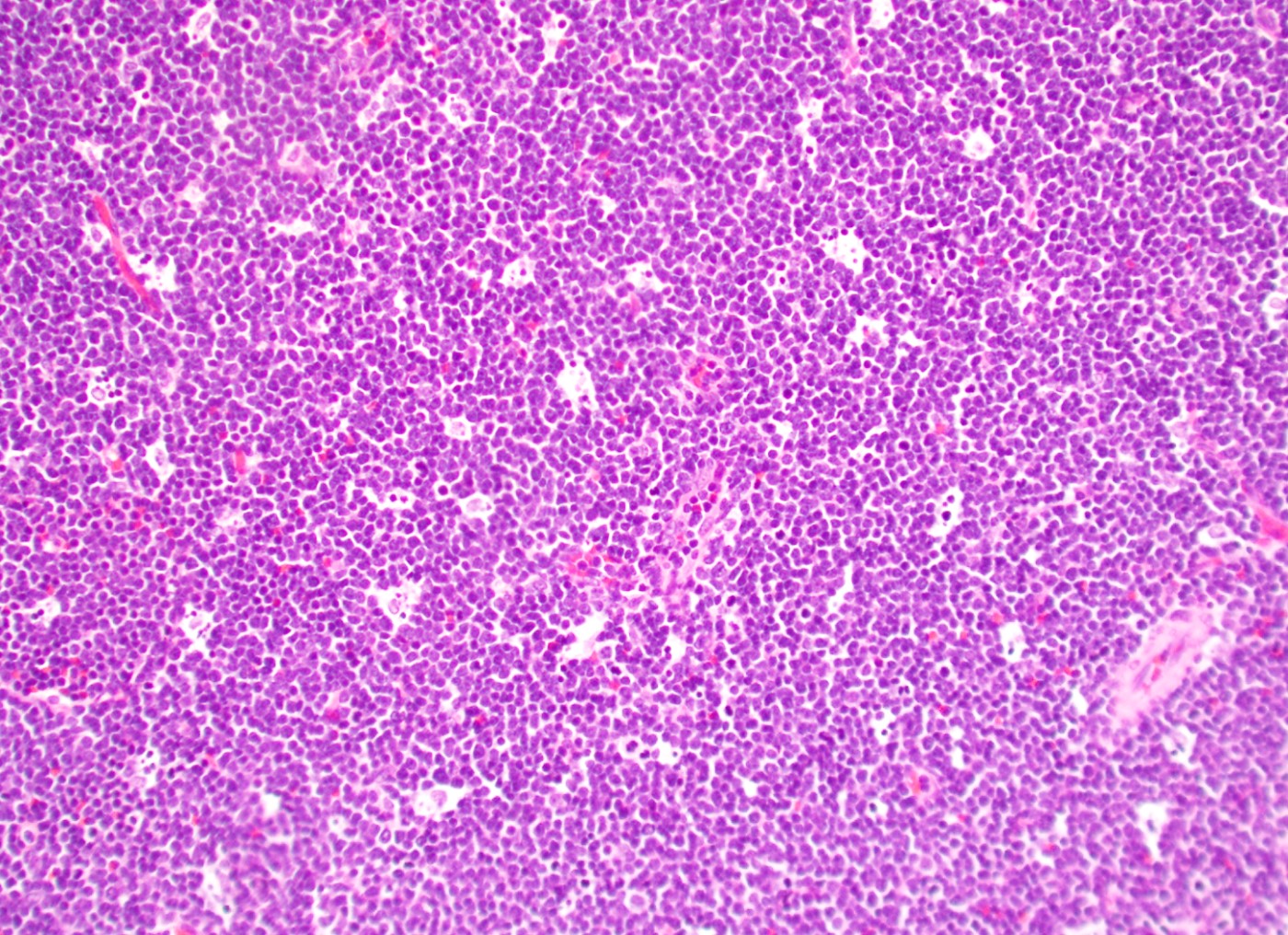
T-LBL starry sky
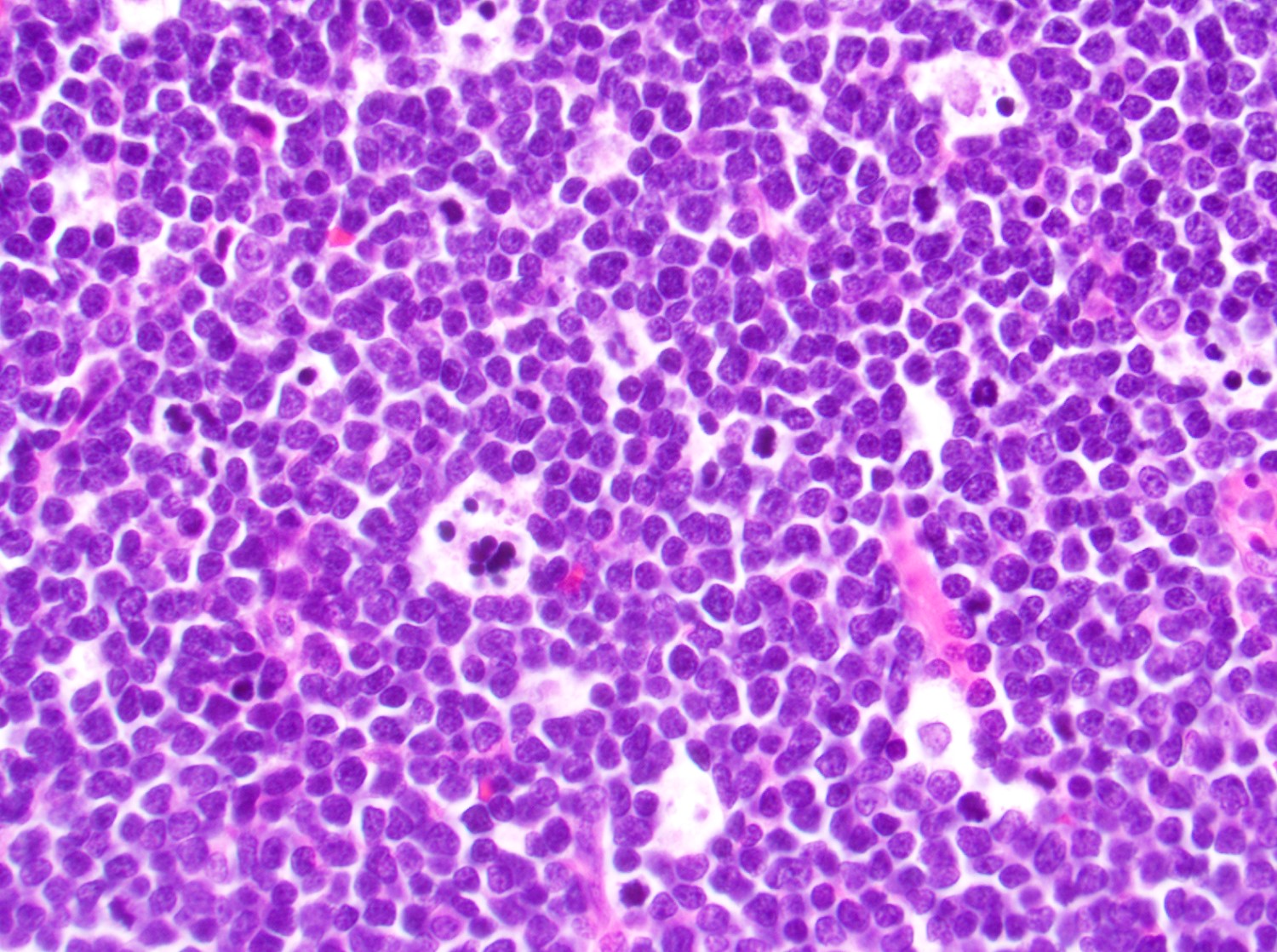
T-LBL lymph node
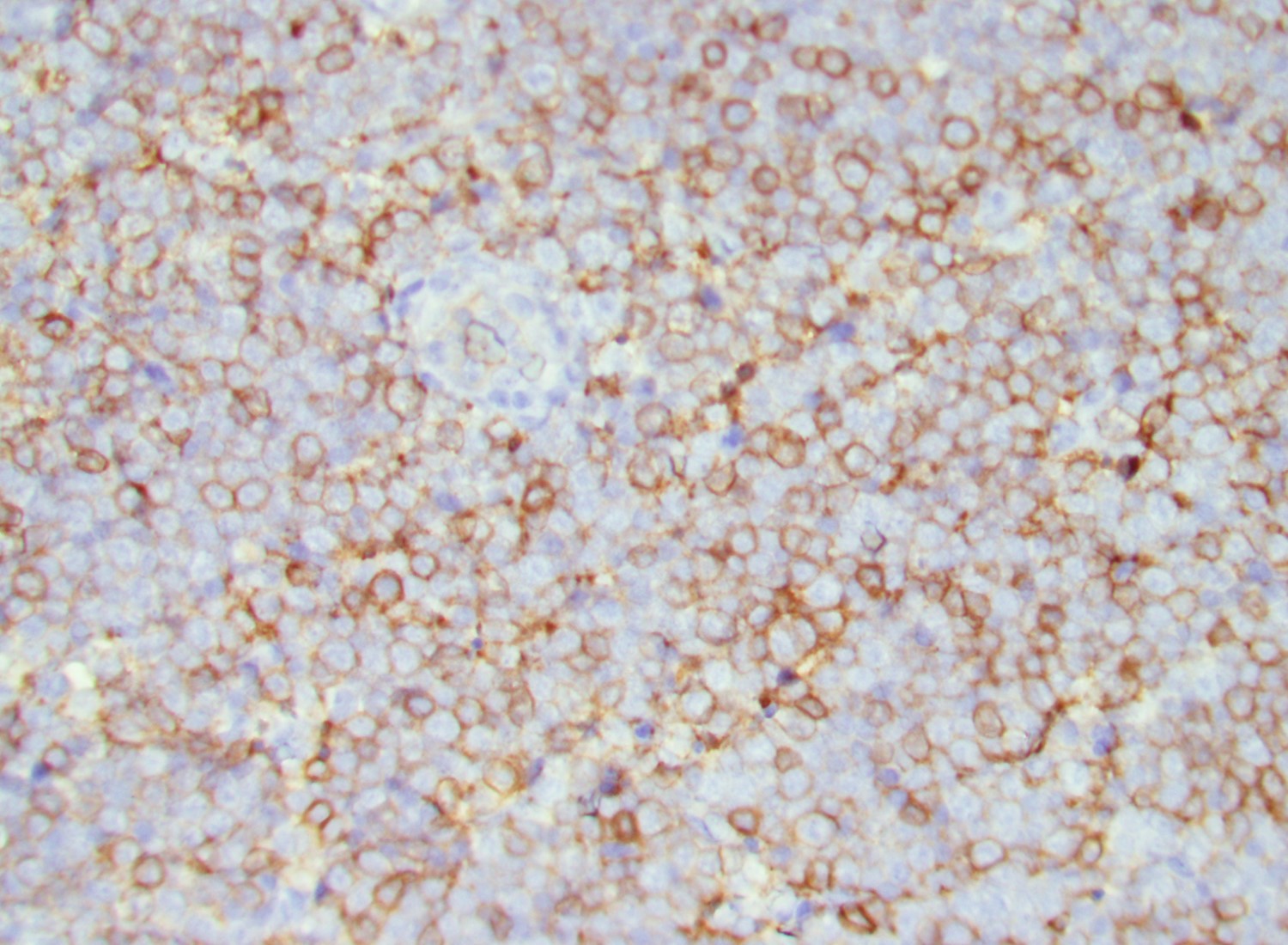
T-LBL CD1a
- Lymphoblasts may have variable cell sizes and typically show a high N/C ratio
- May range from small blasts with a condensed chromatin pattern and inconspicuous nucleoli to larger blasts with an open chromatin pattern, prominent nucleoli and bluish cytoplasm
- Reference: Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017
- Predominance of lymphoblasts
Contributed by Patricia Tsang, M.D.
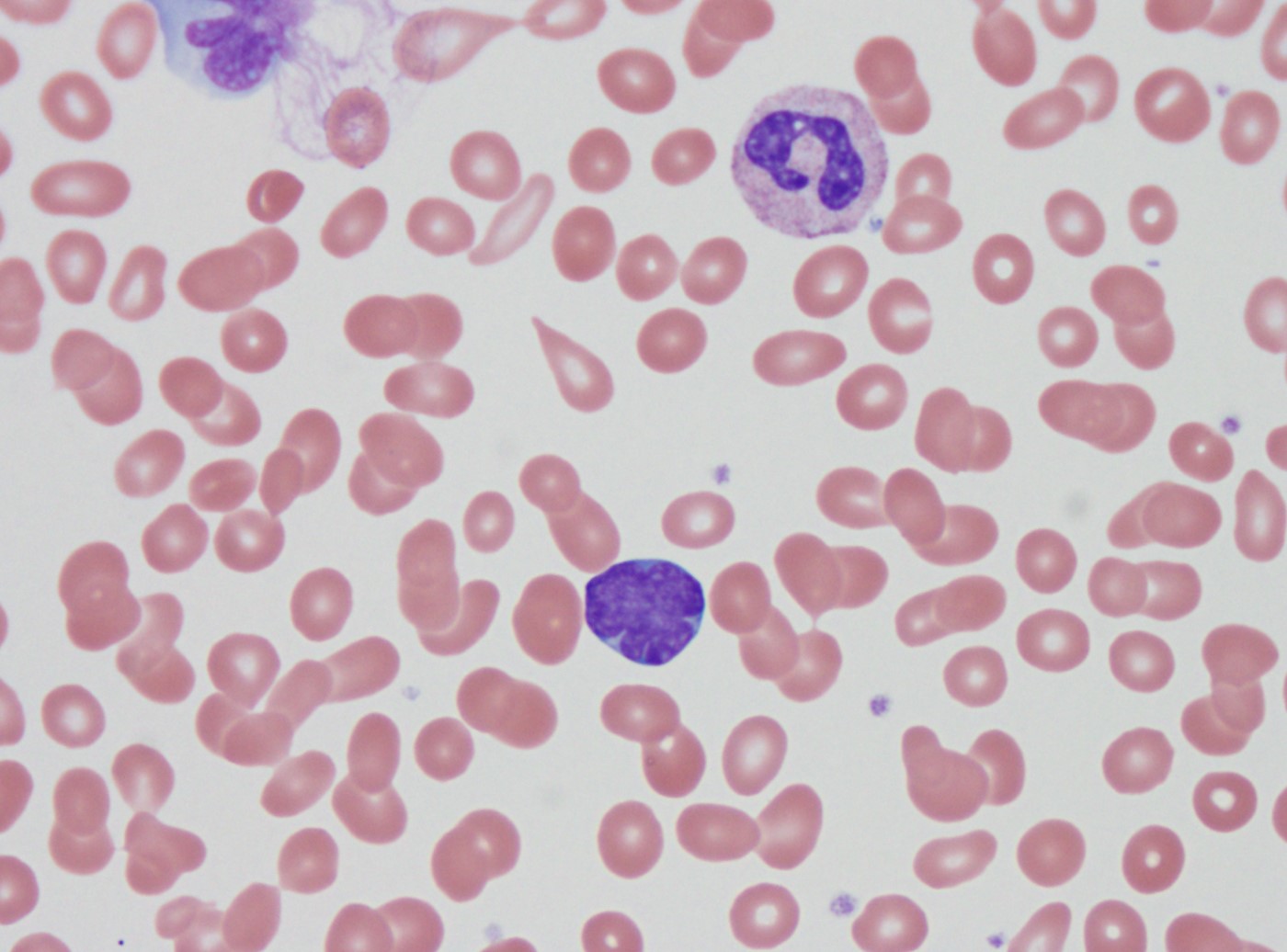
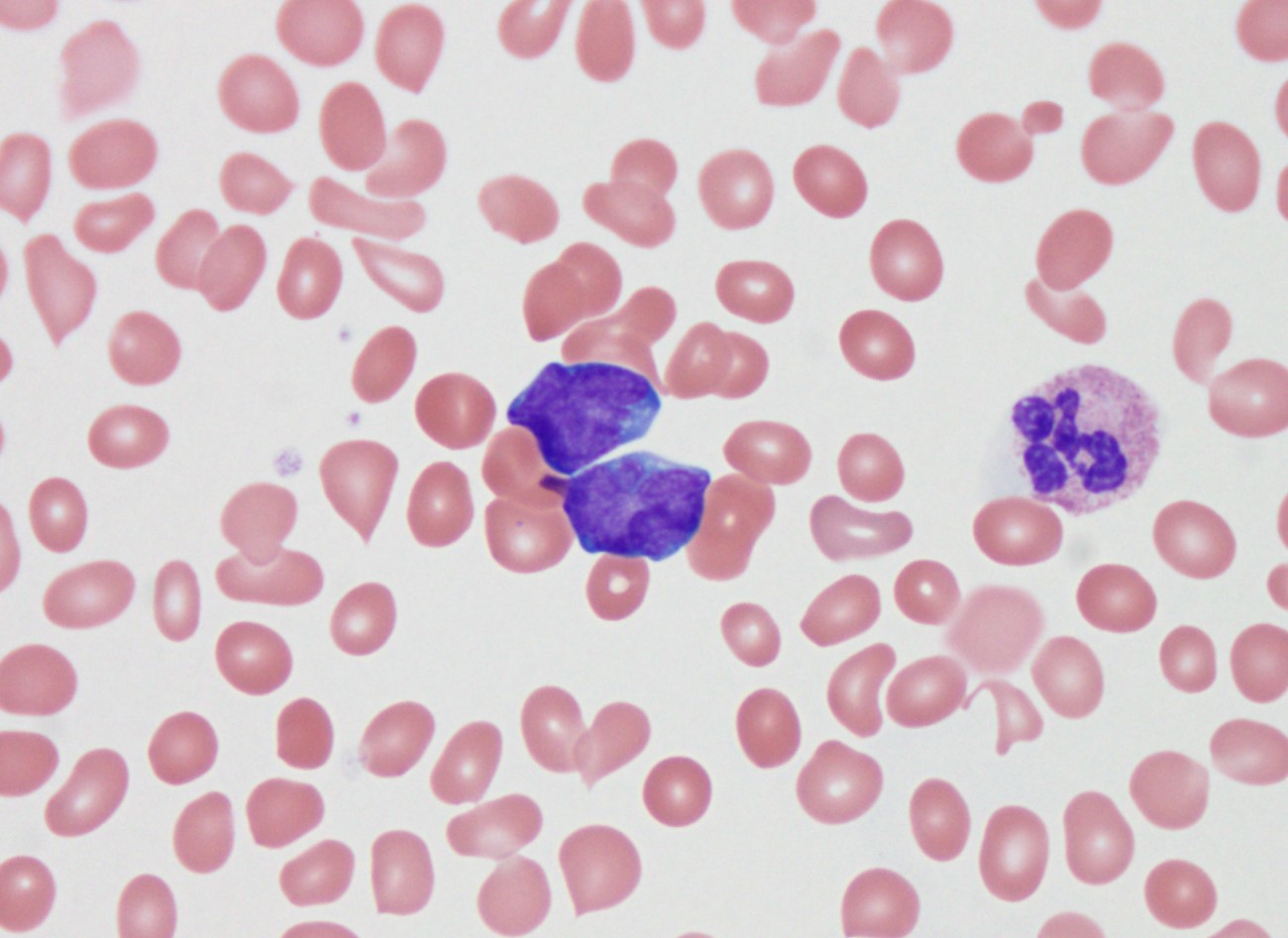
T lymphoblast on blood smear
- B-ALL / LBL
- T-ALL / LBL
- Reference: Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017
- Myeloperoxidase and lysozyme should not be expressed
- B-ALL / LBL: surface immunoglobulin is usually absent
Contributed by Patricia Tsang, M.D.
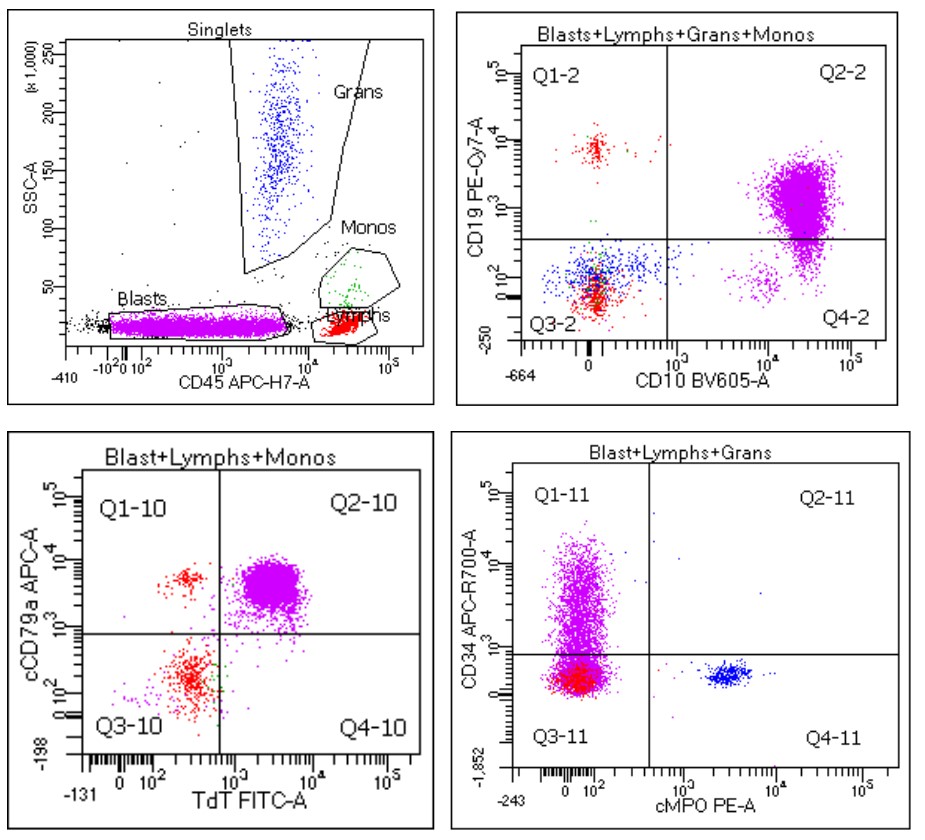
B lymphoblastic lymphoma
- Most B-ALL show clonal IgH (immunoglobulin heavy locus) gene rearrangement; simultaneous T cell receptor (TCR) gene rearrangement present in up to 70%
- Recurrent genetic abnormalities in B-ALL often have prognostic significance:
- t(12;21) TEL-AML: very favorable prognosis; most common rearrangement in childhood B-ALL / LBL (25%), with cures of > 90%
- Hyperdiploidy: favorable prognosis; trisomies 4, 10 and 17 have the best prognosis
- t(9;22) BCR-ABL1 (Philadelphia chromosome): very poor prognosis; incidence increases with age, 3% of childhood cases and 25% of adult
- t(v;11q23) MLL gene rearrangement: poor prognosis
- Hypodiploidy: poor prognosis
- BCR-ABL1-like IGH-CRLF2 translocation (and less commonly EPOR): CRLF2 translocation frequent in Down syndrome; standard risk in children and high risk in adolescents and adults; more frequent in Hispanics and Native Americans
- t(5;14) IL3-IGH: uncertain prognostic significance; reactive eosinophilia is characteristic
- Most T-ALL / LBL harbor T cell receptor gene rearrangement; simultaneous IGH gene rearrangement present in about 20%
- In T-ALL / LBL, recurrent cytogenetic abnormalities can also be seen:
- TCR loci at 14q11.2, 7p35 and 7p14-15, with various partner genes
- Del (9p) with loss of tumor suppressor gene CDKN2A occurs in at least 30%
- NOTCH1 gene mutations can lead to shorter survival in adult T-ALL / LBL
- Reference: Swerdlow: WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues (Medicine), 4th Edition, 2017
- Base of tongue, biopsy:
- B lymphoblastic lymphoma, not otherwise specified (see microscopic description and FISH)
- Microscopic description: Biopsy of the tongue shows benign squamous mucosa with an extensive and diffuse subepithelial infiltrate of medium sized to large neoplastic cells, some containing a prominent nucleolus. Mitoses and apoptotic bodies are readily observed. Immunohistochemistry demonstrates that the neoplastic cells are of precursor B cell origin (CD45+, PAX5+, CD79a+, TdT+, CD10+, CD43+, CD34 subset +, CD20 few scattered cells +). Not expressed are CD117, CD3, CD30, myeloperoxidase, lysozyme, EBV (EBER) and MYC. Proliferative fraction is > 95% based on Ki67.
- FISH: Interphase FISH analysis performed on formalin fixed paraffin embedded tissue shows the following results
- Positive for tetraploidy, consistent with a favorable prognostic implication in B-ALL / LBL.
- Negative for MYC-IGH t(8;14), BCR-ABL t(9;22), BCL2 rearrangement and BCL6 rearrangement.
- No specific genetic abnormalities were identified in the FISH ALL panel to fulfill the diagnostic criteria of B-ALL/LBL with recurrent genetic abnormalities.
- Burkitt lymphoma, a high grade B cell lymphoma:
- Hematogones, non-clonal B cell precursors in bone marrow, can be confused with B-ALL, especially in minimal residual disease:
- Typically smaller than lymphoblasts
- Have characteristic immunophenotypic expression pattern by flow cytometry
- Thymoma with reactive thymic T cells exhibiting CD4+ CD8+ immature T cell phenotype, resembling T-LBL in anterior mediastinal mass:
- Immunohistochemistry highlights the neoplastic epithelial cell component
- Lobulated architecture
- May be associated with myasthenia gravis in adults
- Incidental indolent T lymphoproliferative proliferations (Cytometry B Clin Cytom 2020;98:282)
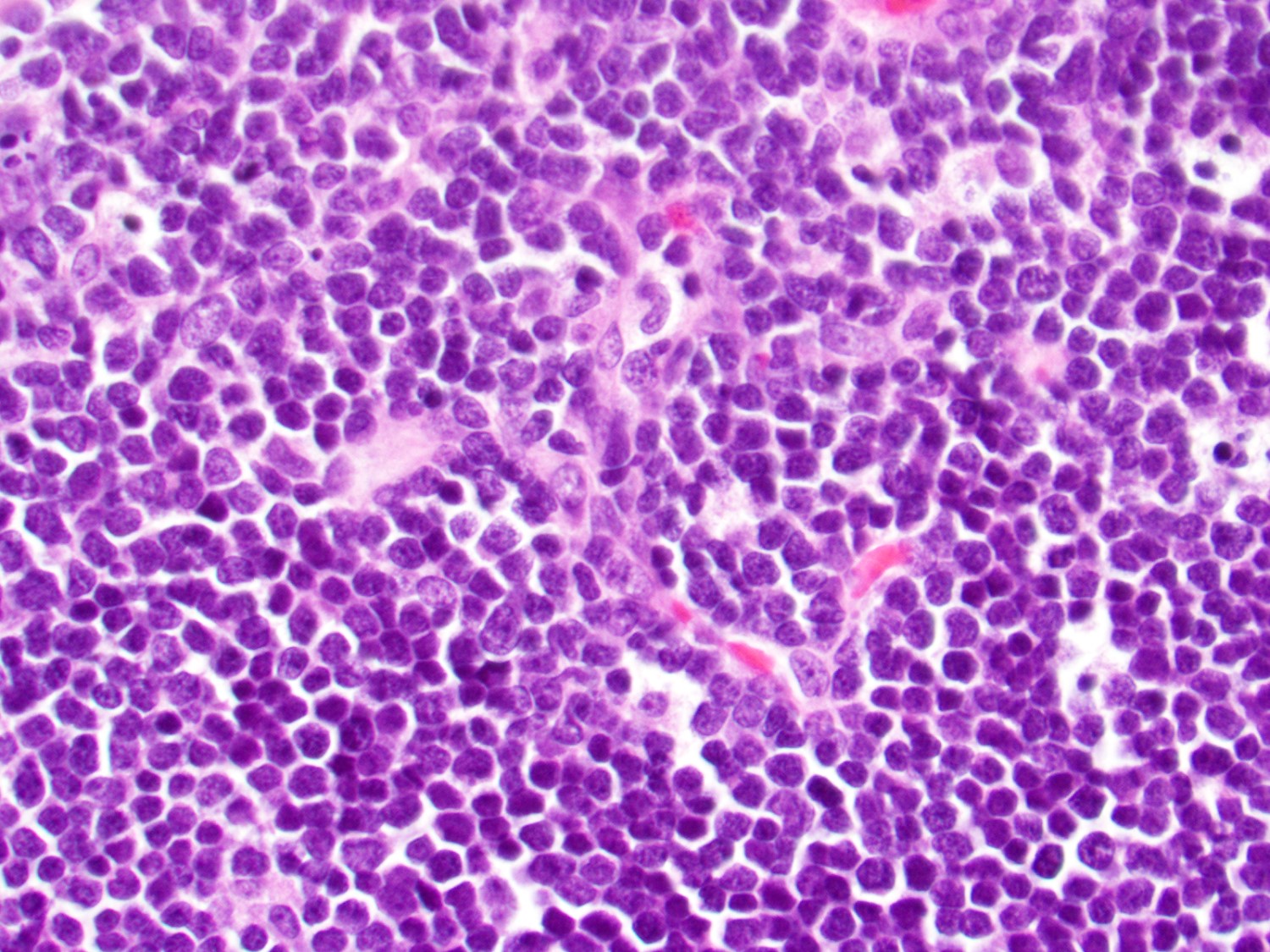
A 4 year old boy is brought to the physician by his mother due to a 5 week history of lethargy, a progressively enlarging left sided neck mass and a 5 day onset of unexplainable bilateral diffuse lower leg petechial hemorrhage. On exam, the left posterior cervical lymph node is enlarged and nontender to palpation. A lymph node biopsy shows a predominance of interfollicular infiltrate containing numerous blast cells and focal necrosis. Immunohistochemistry is positive for PAX5, CD10 and TdT. Upon further workup, which of the following translocations would be associated with a good prognosis?
- t(12;21)
- t(14;18)
- t(8;14)
- t(9;22)
- BCR-ABL1
- CRLF2
- ETV6-RUNX1
- FLT3
- As per the current 5th edition of WHO classification (2022), acute myelomonocytic leukemia (AMML) is a distinct entity within acute myeloid leukemia (AML), defined by differentiation category (Leukemia 2022;36:1703)
- Acute myeloid leukemia, not otherwise specified (AML, NOS) according to the International Consensus Classification (ICC)
- Acute myelomonocytic leukemia shows evidence of both granulocytic and monocytic differentiation
- Does not meet the criteria for inclusion in any of the other AML groups
- Blasts / blast equivalents include myeloblasts, monoblasts and promonocytes and must comprise at least 20% of blood or bone marrow cells
- Granulocytic differentiation is present but cells of monocytic lineage must be at least 20% of cells
- AML M4 (FAB)
- AMMoL
- AMML
- AMML represents 5 - 10% of all cases of AML (Blood 2013;121:2424)
- AMML is seen in all age groups but is more common in older individuals (> 60 years of age)
- Median age is ~50 years
- 3% of childhood leukemia (Orphanet: Acute Myelomonocytic Leukemia [Accessed 21 November 2022])
- M:F = 1.4:1
- Bone marrow, peripheral blood
- Extramedullary infiltration in the spleen, lymph nodes, tonsils, skin or sites like the brain, testis and ovaries has been rarely noted (Am J Clin Pathol 2006;125:783, Case Rep Oncol Med 2019;2019:4189275)
- Proliferation of a malignant hematopoietic stem cell which retains the capacity to differentiate into both granulocytic and monocytic cells
- Genetic drivers of AML include both chromosomal alterations and gene mutations
- Exact causes are unknown
- Signs and symptoms relate to anemia, thrombocytopenia and increased number of infections
- Early symptoms include fever, fatigue, weakness, weight loss, shortness of breath
- Also can present as organomegaly, lymphadenopathy and other tissue infiltration (monocytic infiltrate)
- Reference: Curr Oncol 2022;29:6245
- Diagnosis is based on integration of morphology, immunohistochemistry, cytochemistry, flow cytometry studies, cytogenetics and molecular findings
- Immunophenotyping is useful to confirm that both myeloid and monocytic populations are present
- Genetic testing excludes AML with recurrent genetic abnormalities which have myelomonocytic morphology
- Criteria for diagnosis:
- Blasts / blast equivalents comprise ≥ 20% of bone marrow cells
- Blasts / blast equivalents include myeloblasts, monoblasts and promonocytes
- Cells of monocytic lineage (monoblasts, promonocytes, monocytes) overall comprise at least 20% of cells
- AMML is similar to but distinct from acute monoblastic / monocytic leukemia, which requires ≥ 80% of blasts / blast equivalents to be of monocytic lineage
- Additional criteria: ≥ 3% of blasts should be positive for MPO by cytochemical stain (indicates myeloid lineage)
- Monocytic lineage may be ascertained by nonspecific esterase stain (not always present) or monocytic immunophenotype (e.g., expression of CD14, CD11c, CD64, CD163, lysozyme)
- No recurrent genetic abnormalities identified (Leukemia 2022;36:1703)
- AMML of AML, defined by differentiation, morphologically resembles AML with CBFB::MYH11 fusion (formerly known as FAB M4Eo), AML with KMT2A rearrangement and some cases of AML with NPM1 mutation
- Blasts / blast equivalents comprise ≥ 20% of bone marrow cells
- Leukocytosis is common
- Anemia, thrombocytopenia
- Poor prognostic factors:
- Adverse mutation profile, including wild type NPM1 and FLT3 ITDhigh, mutated RUNX1 and mutated TP53 (Am J Hematol 2018;93:1267)
- Complex karyotype
- 54 year old woman with pure erythroid leukemia subsequent to acute myelomonocytic leukemia (Medicine (Baltimore) 2021;100:e25528)
- 65 year old woman presented with de novo acute myeloid leukemia with complex karyotype (AML M4) (Mol Cytogenet 2013;6:18)
- 82 year old woman with acute myelomonocytic leukemia with uncommon morphological features (Ann Hematol 2020;99:351)
- Standard induction chemotherapy with cytarabine given twice daily for 10 days combined with an anthracycline drug for 3 days
- Consolidation therapy often with higher doses of same drugs used for induction or an allogenic stem cell transplant
- Targeted therapy if respective mutations are present (e.g., FLT3 or IDH1 / IDH2 inhibitors)
- Reference: Curr Oncol 2022;29:6245
- By definition, blasts and blast equivalents comprise at least 20% of cells in marrow or blood, while cells of monocytic lineage (monoblasts, promonocytes and monocytes) are also at least 20% of cells
- Myeloblasts have high N:C ratios, round nuclei and variably granular cytoplasm; Auer rods occasionally seen
- Monoblasts are large cells with abundant, moderately to intensely basophilic cytoplasm
- May have pseudopod formation, scattered fine azurophilic granules and vacuoles
- Often with round nuclei with lacy chromatin and one or more large nucleoli
- Promonocytes have more irregular nuclei, typically with delicate folds and abundant, less basophilic cytoplasm which is more obviously granulated with occasional large azurophilic granules and vacuoles
- Reference: Haematologica 2009;94:994
Contributed by Ramya Gadde, M.D., Rose Beck, M.D., Ph.D. and AFIP
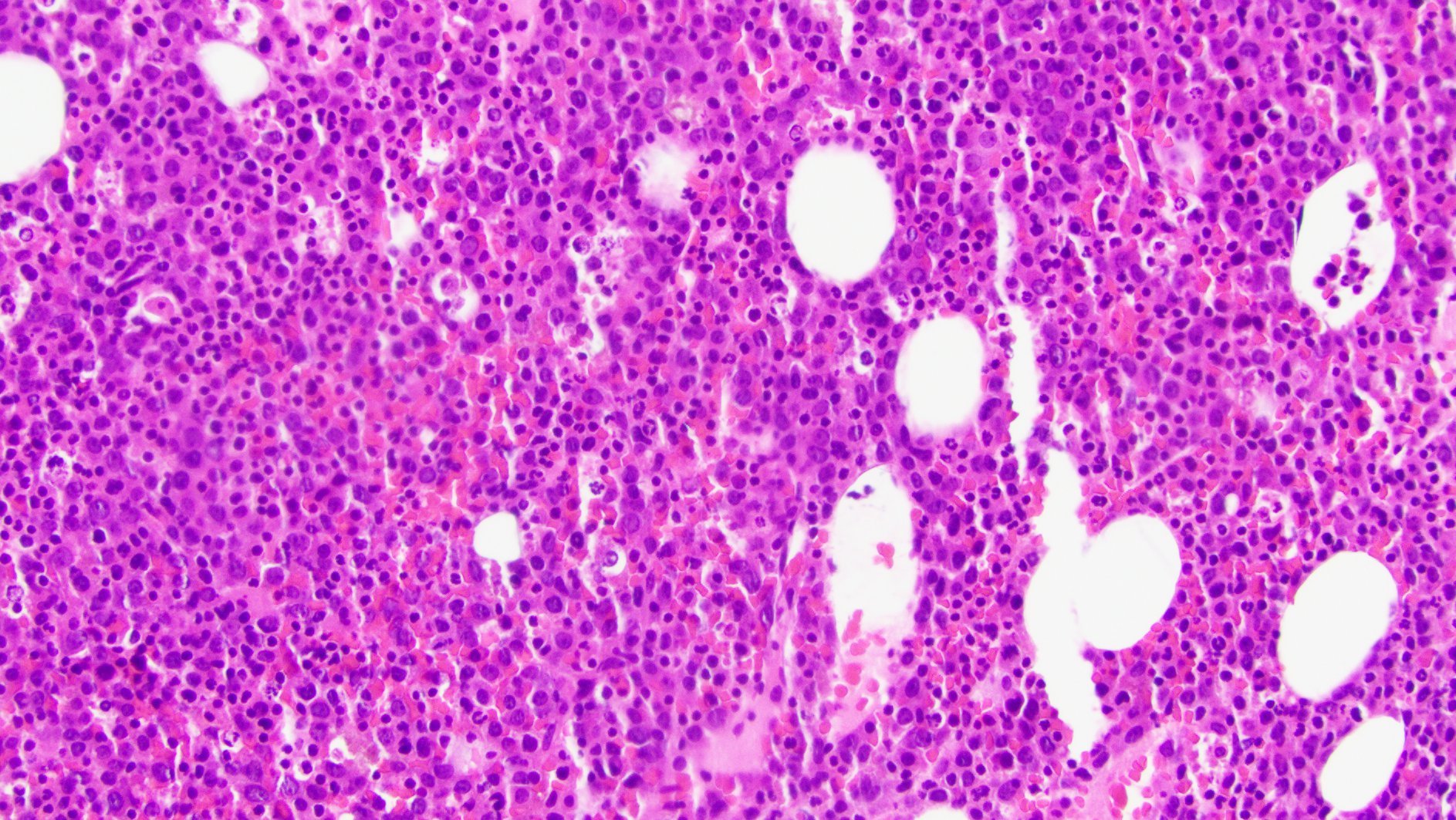
Increased myelomonocytic population
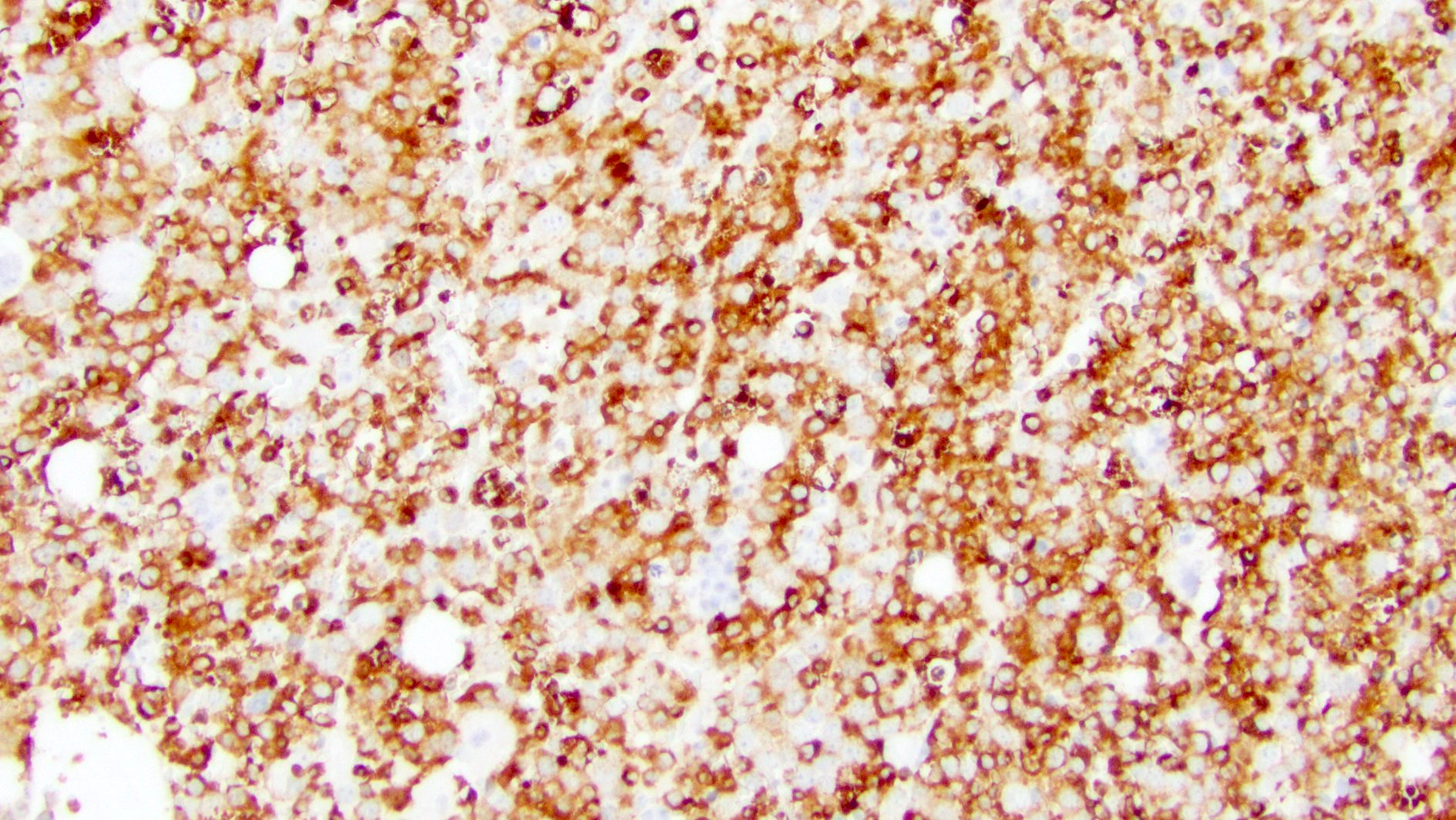
Lysozyme immunostain
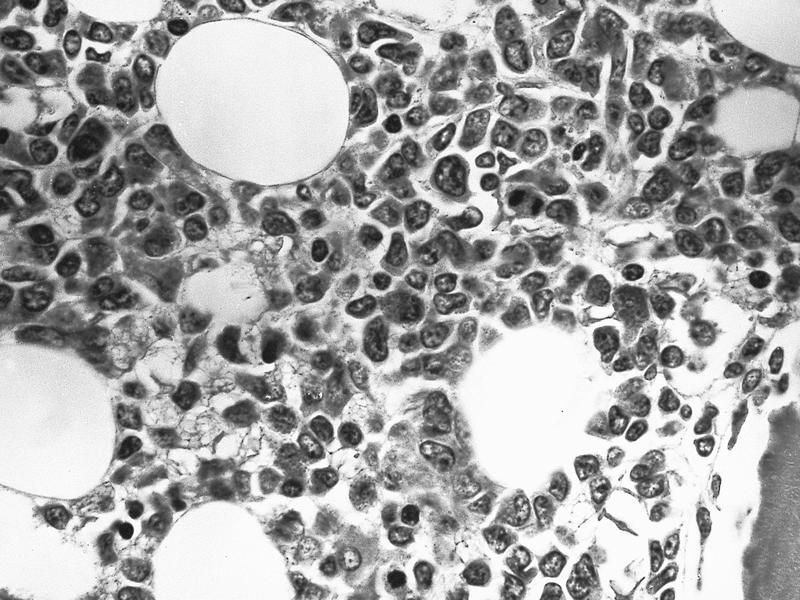
Bone marrow biopsy

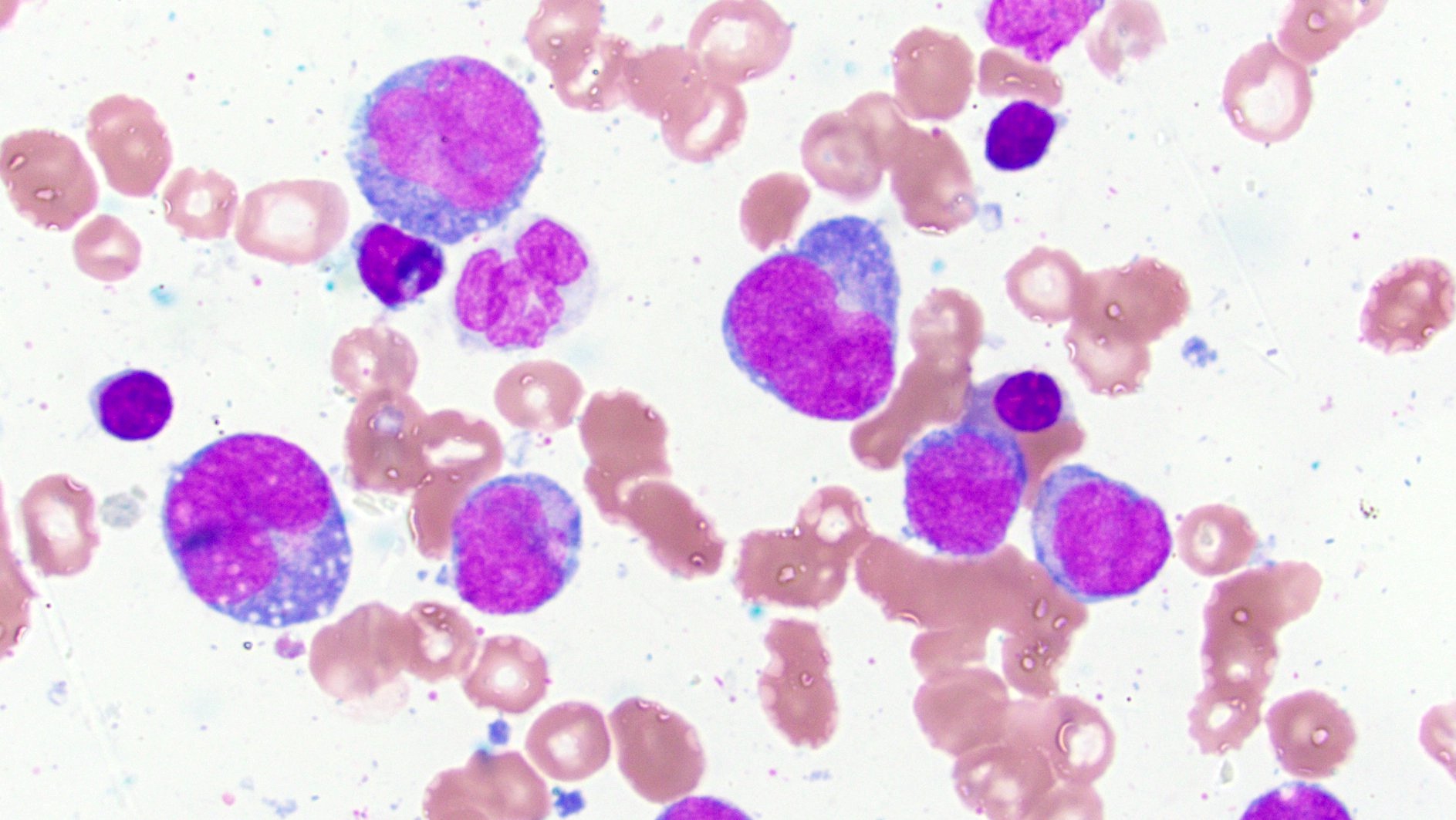
Increased blasts and blast equivalents
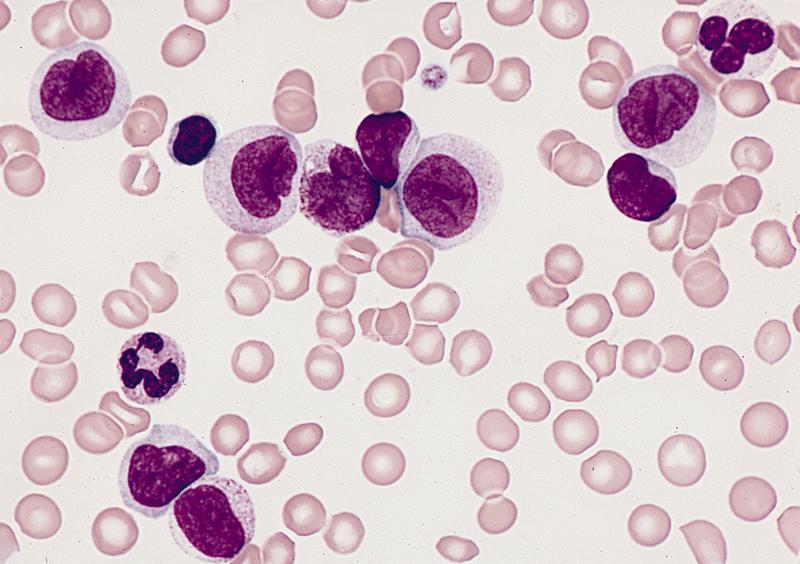
Mixture of monocytes and neutrophils
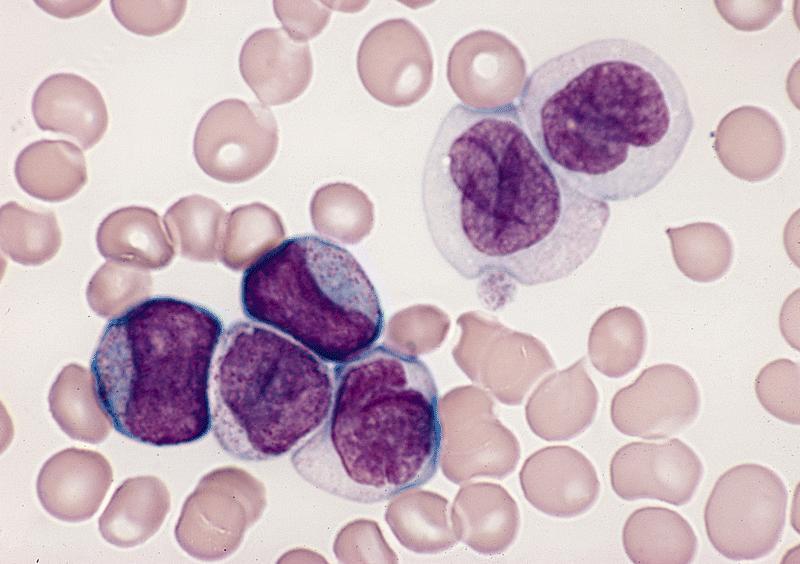
Promonocytes
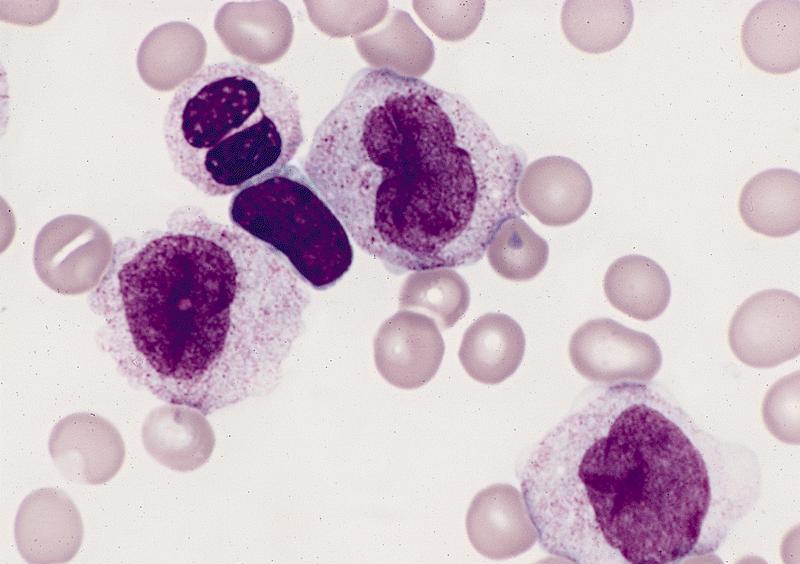
Promonocytes and dysplastic neutrophil
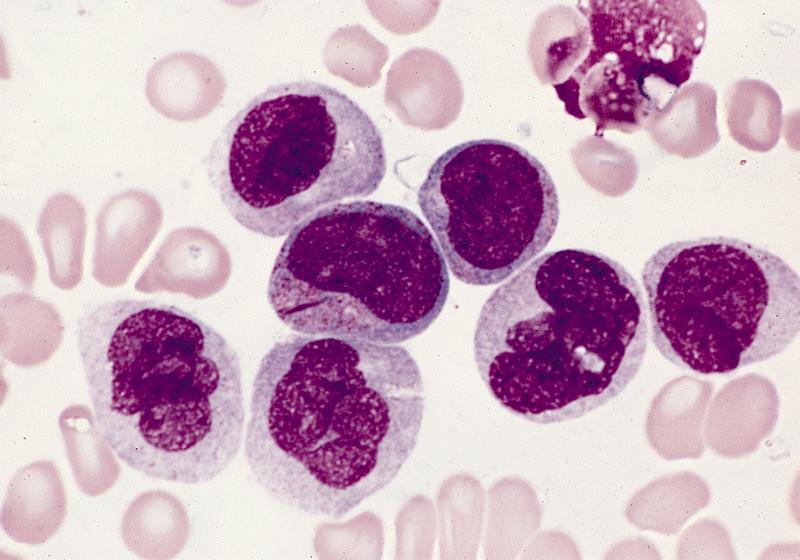
Promonocytes and myelocytes
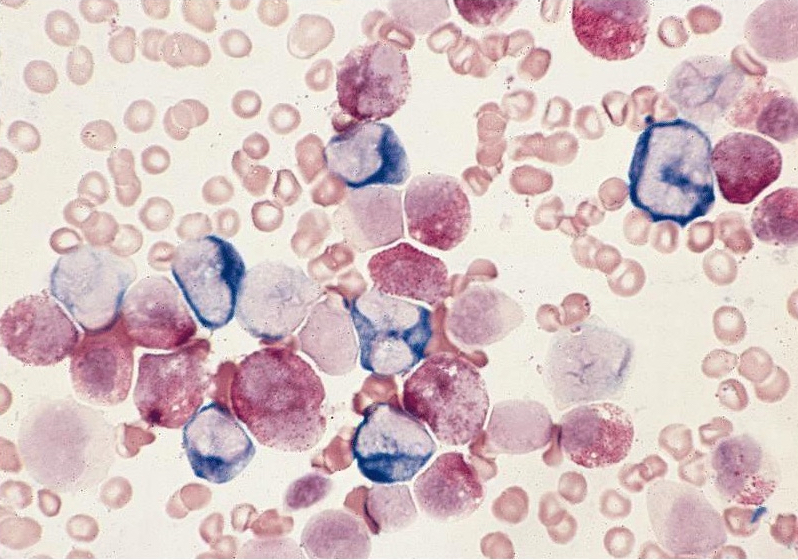
Chloroacetate and nonspecific esterase stains
- Peripheral blood often has leukocytosis with blasts, may show increased monocytes and is often more mature than the monocytic cells observed in bone marrow
AFIP images
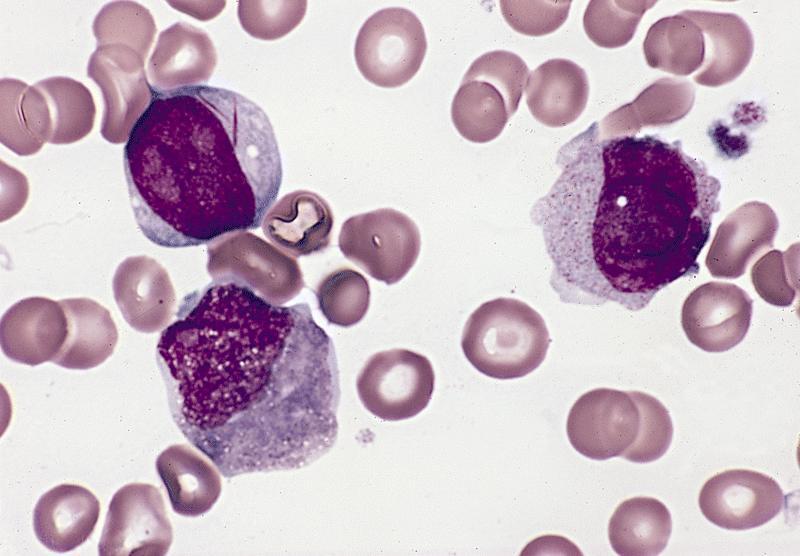
Myeloblast, neutrophilic myelocyte and promonocyte
- Expression of markers may vary between monocytic and myeloid populations
- Variably express myeloid antigens CD13, CD33, CD65, CD15
- CD34 and CD117 typically present on the myeloblast population, which may be small
- Monocytic cells typically express CD14, CD64, CD11b, CD11c, CD4, CD36, CD68 (PGM1), CD163, lysozyme
- Most cases express HLA-DR
- Coexpression of CD15, CD36 and CD64 is characteristic of monocytic differentiation
- Reference: Am J Clin Pathol 2004;122:865
- CD41, CD61, glycophorin A
- Lymphoid markers (minority of cases may have aberrant expression of CD7)
- Myeloid and monocytic cells share a common progenitor
- In the progression of myelomonocytic pathway, increase in the expression of CD15, CD33 and CD64 markers are noted
- In general, myeloblasts show dim to moderate CD45, low side scatter and express CD13, CD33, CD34, CD117 and HLA-DR
- Monoblasts and promonocytes are CD45 bright with side scatter slightly higher than myeloblasts
- References: Clin Lab Med 2017;37:753, Methods Mol Biol 2019;2032:281, Methods Cell Biol 2004;75:665, Cytometry B Clin Cytom 2017;92:218
Contributed by Ramya Gadde, M.D. and Rose Beck, M.D., Ph.D.
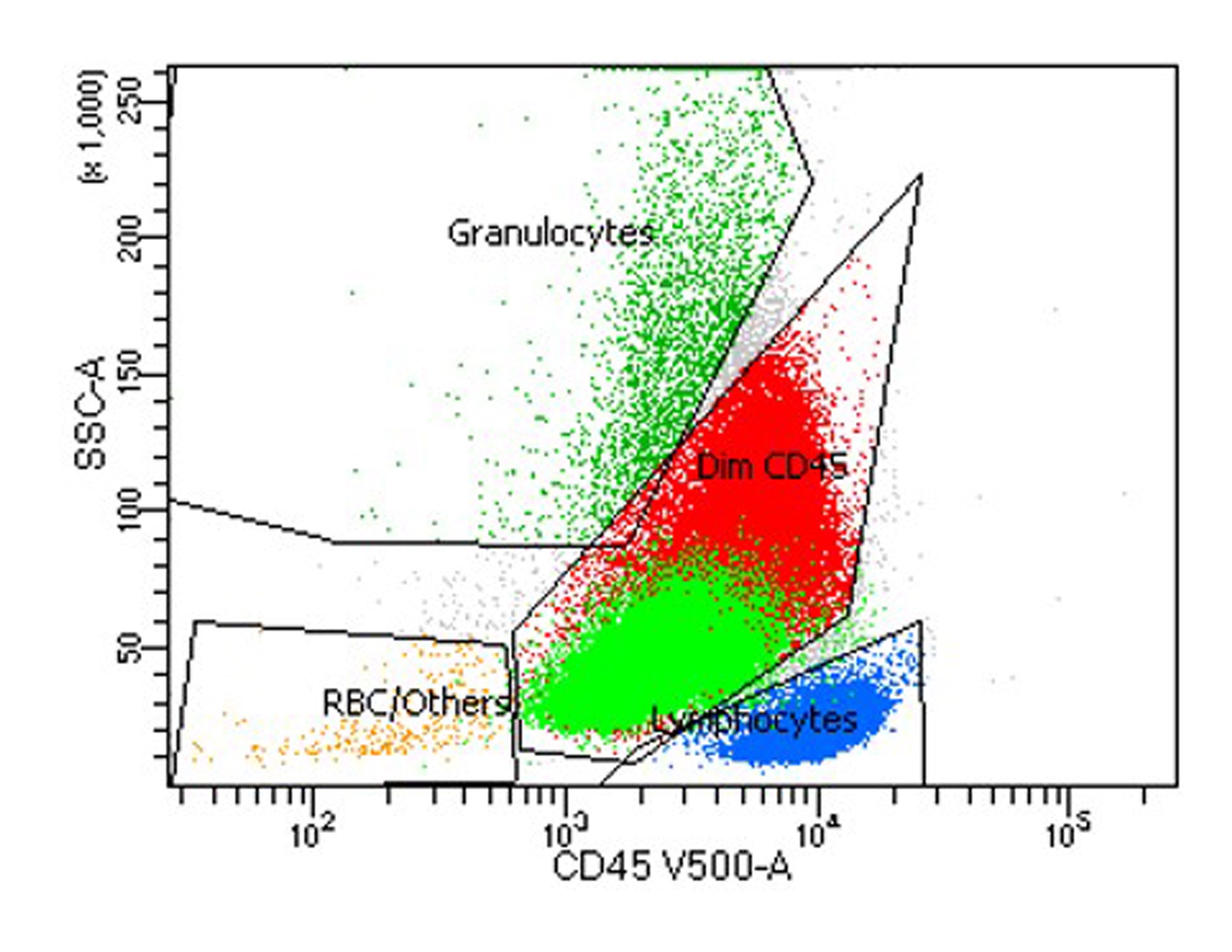

Blast and monocytic population
- Nonspecific cytogenetic abnormalities in most cases, such as +8, monosomy 7 or del (7q), monosomy 5 or del(5q)
- Bone marrow, core biopsy with touch imprint, clot section, aspirate smears, left iliac crest:
- Acute myelomonocytic leukemia (see comment)
- Comment: Hypercellular bone marrow for age (80 - 90%) with involvement by acute myeloid leukemia, defined by differentiation. Immature myelomonocytic cells are increased. Erythroid precursors are decreased. Mature myeloid cells are decreased. Megakaryocytes are adequate and appear normal in morphology.
- Bone marrow aspirate smear is adequate. Blasts / blast equivalents including myeloblasts, monoblasts and promonocytes are increased (50%). Megakaryocytes are adequate in number and show normal morphology.
- Flow cytometry studies demonstrate an abnormal myeloid blast population with expression of dim CD45, CD34, CD15 (subset), CD33, CD13, HLA-DR and CD117. There is also an immature monocytic population with CD64, CD11b, CD11c, CD13, CD14, CD33 and CD64 expression.
- AML with maturation:
- ≥ 3% blasts positive for MPO (by immunophenotyping or cytochemistry) or Sudan Black B by cytochemistry
- Maturing cells of the granulocytic lineage constitute ≥ 10% of the nucleated bone marrow cells
- Monocyte lineage cells constitute < 20% of bone marrow cells
- Expression of 2 or more myeloid associated antigens, such as MPO, CD13, CD33 and CD117 (Leukemia 2022;36:1703)
- AML with NPM1 mutation:
- Blast count < 20% is acceptable; presence of blasts with cup shaped nuclei and mutations in NPM1 gene is characteristic (Leukemia 2022;36:1703, Acta Haematol 2019;141:232)
- AML with KMT2A rearrangement:
- Blast count < 20% is acceptable; presence of KMT2A::MLL gene rearrangements is characteristic (Acta Haematol 2019;141:232)
- AML with CBFB::MYH11 fusion:
- Blast count < 20% is acceptable; presence of abnormal eosinophils, eosinophil precursors and CBFB::MYH11 fusion [inv(16)(p13.1;q22) or t(16;16)(p13.1;q22)] is characteristic (Acta Haematol 2019;141:232)
- Chronic myelomonocytic leukemia:
- Absolute and relative monocytosis; blasts are < 20% of the cells in blood and bone marrow
- Features of both a myeloproliferative neoplasm and a myelodysplastic syndrome are present
- G-CSF related transient atypical monocytosis (Clin Lab Haematol 2004;26:359):
- Recent administration of growth factor; lacks clonal cytogenetic and molecular alterations (Blood 2011;118:4283)
- Leukemoid reaction:
- Leukocytosis with neutrophilia and left shift; high leukocyte alkaline phosphatase (LAP) score
- Lacks clonal cytogenetic and molecular alterations
- Acute monocytic leukemia:
- ≥ 80% monocytes or their precursors (monoblasts or promonocytes)
- < 20% maturing granulocytic cells (Leukemia 2022;36:1703)
- Acute promyelocytic leukemia, microgranular variant:
- Presence of micro / hypogranular promyelocytes; PML::RARA translocation or a variant RARA translocation is characteristic
- Therapy related AML:
- Known history of cytotoxic therapy or radiation therapy administered for neoplastic and nonneoplastic conditions
What is the most likely overall morphology seen in this bone marrow?
- Acute myelomonocytic leukemia
- Acute promyelocytic leukemia
- AML with maturation
- AML without maturation
- CD34+, CD117+, CD33+, HLA-DR+
- CD34-, CD117+, CD33+, HLA-DR-
- CD34-, CD117-, CD14+, CD33+, HLA-DR+
- CD34+, CD117+, CD33+, HLA-DR+ (myeloblast population) and CD34-, CD117-, CD14+, CD33+, HLA-DR+ (monocytic population)
Comment Here
Reference: AMML
- Rare subtype of acute myeloid leukemia (AML), NOS (Blood 2016;127:2391)
- Accounts for 2% of all acute leukemias (Acta Med Port 2013;26:613)
- Does not fulfill criteria for AML with recurrent genetic abnormalities, myelodysplastic related changes or other AML entity
- Median survival is 2 - 9 months
- Acute panmyeloid proliferation with increased blasts (≥ 20% of cells in the bone marrow or peripheral blood) and fibrosis of the bone marrow
- Marrow is diffusely fibrotic, showing panmyelosis, with immature granulocytic, megakaryocytic and erythroid cells
- Weakness, fatigue, fever, bone pain, pancytopenia; usually no marked splenomegaly
- Also called acute (malignant) myelofibrosis, acute (malignant) myelosclerosis, acute myelodysplasia with myelofibrosis
- ICD-10: C94.4 - acute panmyelosis with myelofibrosis
- Mostly adults; very rare in pediatric population
- Patients acutely sick at presentation with fever and bone pain
- Rare, rapid onset and aggressive course
- References: Acta Med Port 2013;26:613, Blood 2016;127:2391
- Bone marrow
- Involves granulocytic, megakaryocytic and erythroid cells, resulting in panmyelosis; marrow becomes fibrotic but with a high blast count of > 20%
- Acute onset of disease, pancytopenia and bone marrow fibrosis in the absence of splenomegaly
- Peripheral blood shows pancytopenia, which is usually marked
- Rare erythroblast can be seen, without presence of teardrop shaped cells
- Dysplastic changes in myeloid cells are frequent
- Abnormal platelets may be seen
- Bone marrow biopsy is often suboptimal or unsuccessful due to degree of fibrosis; on histology the bone marrow biopsy shows diffuse stromal fibrosis, with increased erythrocyte precursors, megakaryocyte precursors and granulocyte precursors (panmyelosis), which show dysplastic features
- Degree of presence of blasts in acute panmyelosis with myelofibrosis (APMF) is uncertain but recent studies have reported 20 - 25%
- Degree of myelofibrosis is also variable but most patients have 3/3 reticulin fibrosis (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised Edition, 2017)
- Complete blood count with differential (CBC w/ diff)
- Leukocytosis, lymphocytosis and atypical lymphocytes (if leukemic involvement)
- Comprehensive blood chemistry (CMP)
- Including liver and kidney analysis
- Lactate dehydrogenase (LDH)
- Elevated (indicator of tumor lysis)
- Serum beta 2 microglobulin
- Variable (indicator of tumor mass)
- Reference: Clin Adv Hematol Oncol 2018;16:619
- Overall rapidly progressive clinical course, with a median survival of < 1 year
- 16 year old boy admitted with severe pancytopenia and rapidly progressive bone marrow fibrosis (Acta Med Port 2013;26:613)
- 45 year old man with abrupt onset of rapidly progressing low backache, weakness, pancytopenia without organomegaly (Mediterr J Hematol Infect Dis 2013;5:e2013042)
- 66 year old man with suspicion of acute myeloid leukemia was admitted and treated for acute myeloid leukemia, NOS and later found to have APMF with EVI1 amplification (Cancer Genet 2012;205:255)
- Treated as acute myeloid leukemia: 3+7 induction regimen with idarubicin and cytarabine, followed by high dose intermittent ARA C (Mediterr J Hematol Infect Dis 2013;5:e2013042)
- Hematopoietic cell transplantation (Biol Blood Marrow Transplant 2019;25:2113)
- Hypercellular marrow with erythroblasts, immature granulocytes, megakaryocytes (Clin Adv Hematol Oncol 2018;16:619)
- Prominent megakaryocytic abnormalities with variation in size and dysplastic changes, immature granulocytes with dysplasia and immature erythrocytes (Leuk Lymphoma 2004;45:681)
- Megakaryocytes with nonlobulated or hypolobulated nuclei are often present (Leuk Lymphoma 2004;45:681)
- Abnormal platelets may be seen
- Usually marked fibrosis (reticulin > collagen) (Clin Adv Hematol Oncol 2018;16:619)
- Aspirate smear is often hypocellular due to marked fibrosis (Clin Adv Hematol Oncol 2018;16:619)
Contributed by Julie Feldstein, M.D. and AFIP
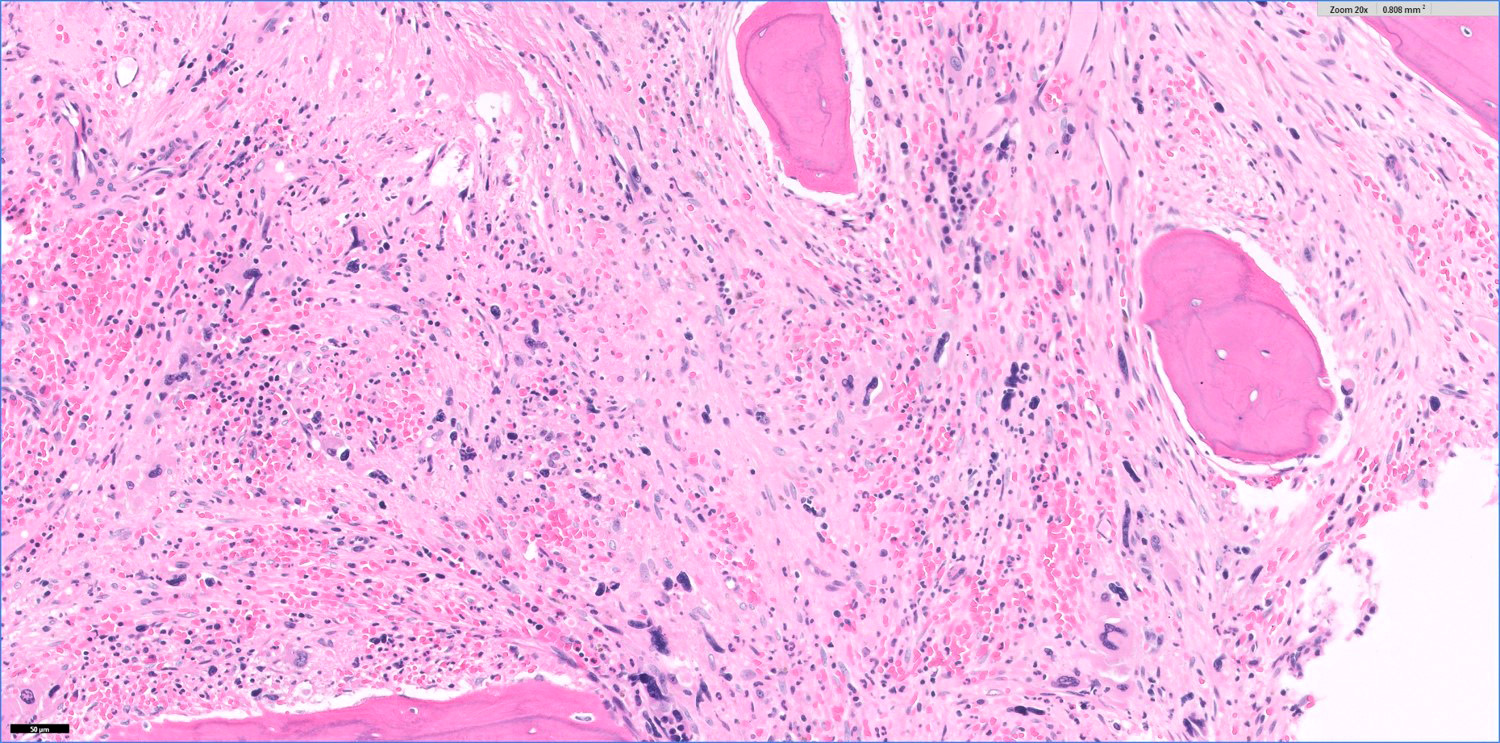
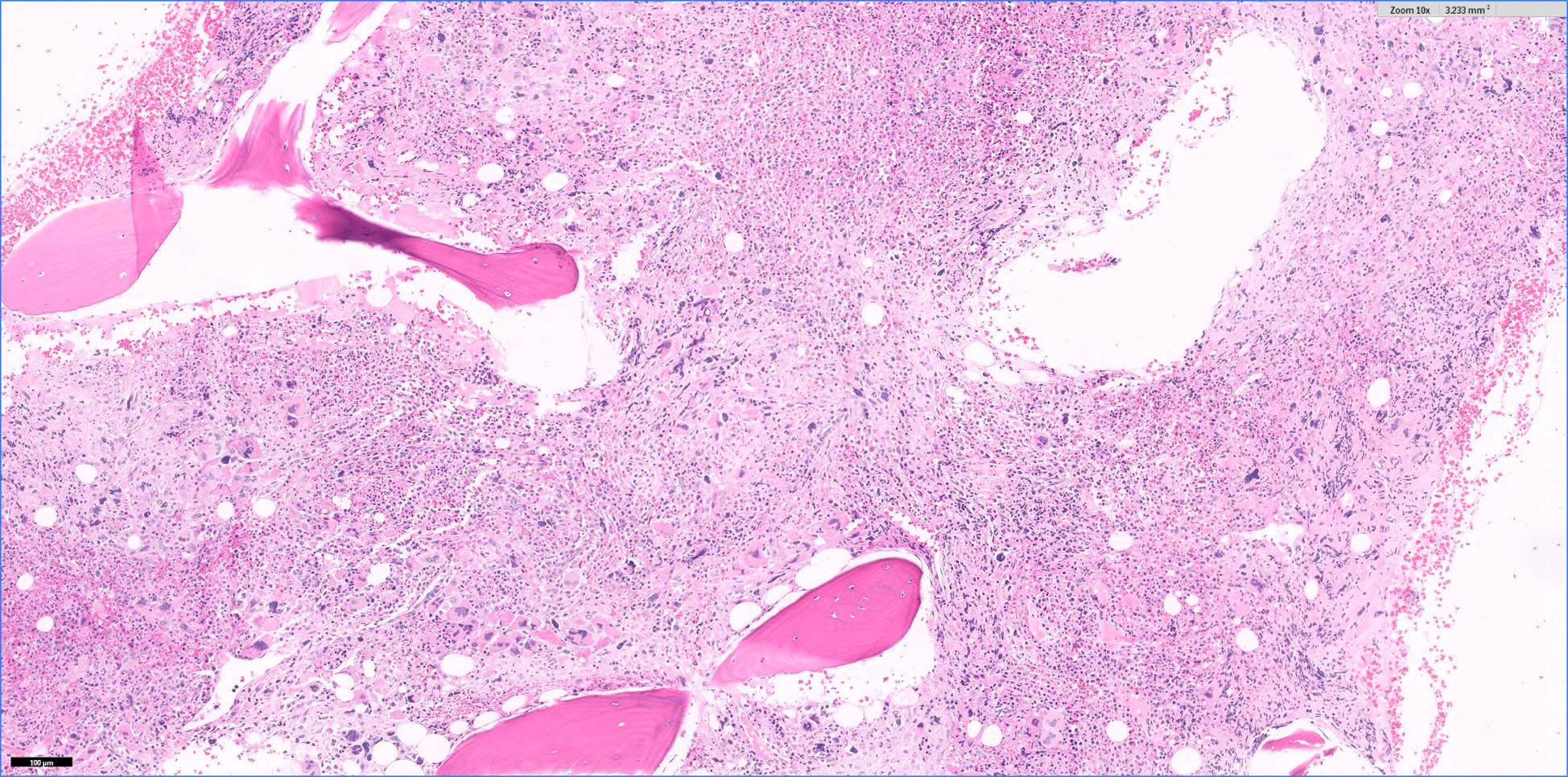
Bone marrow trephine biopsy
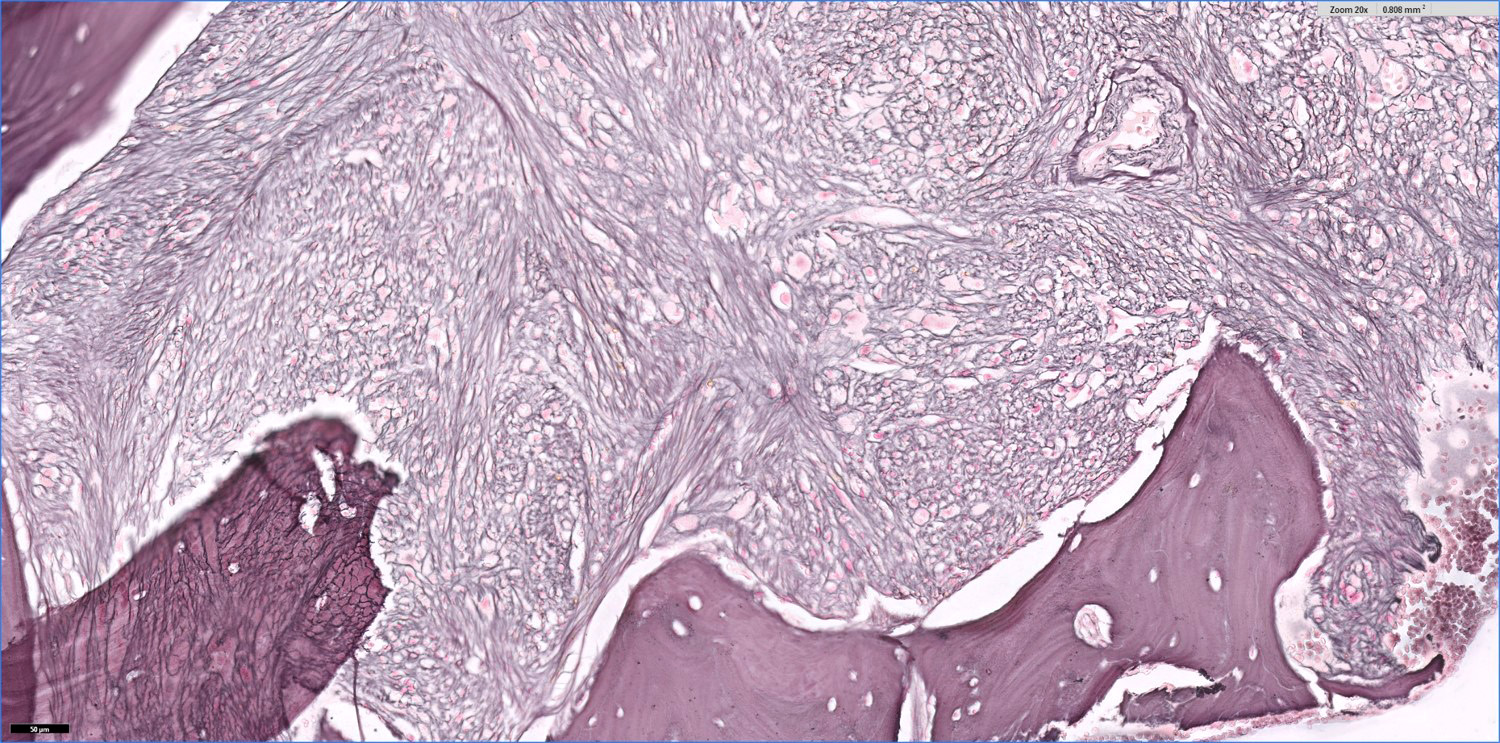
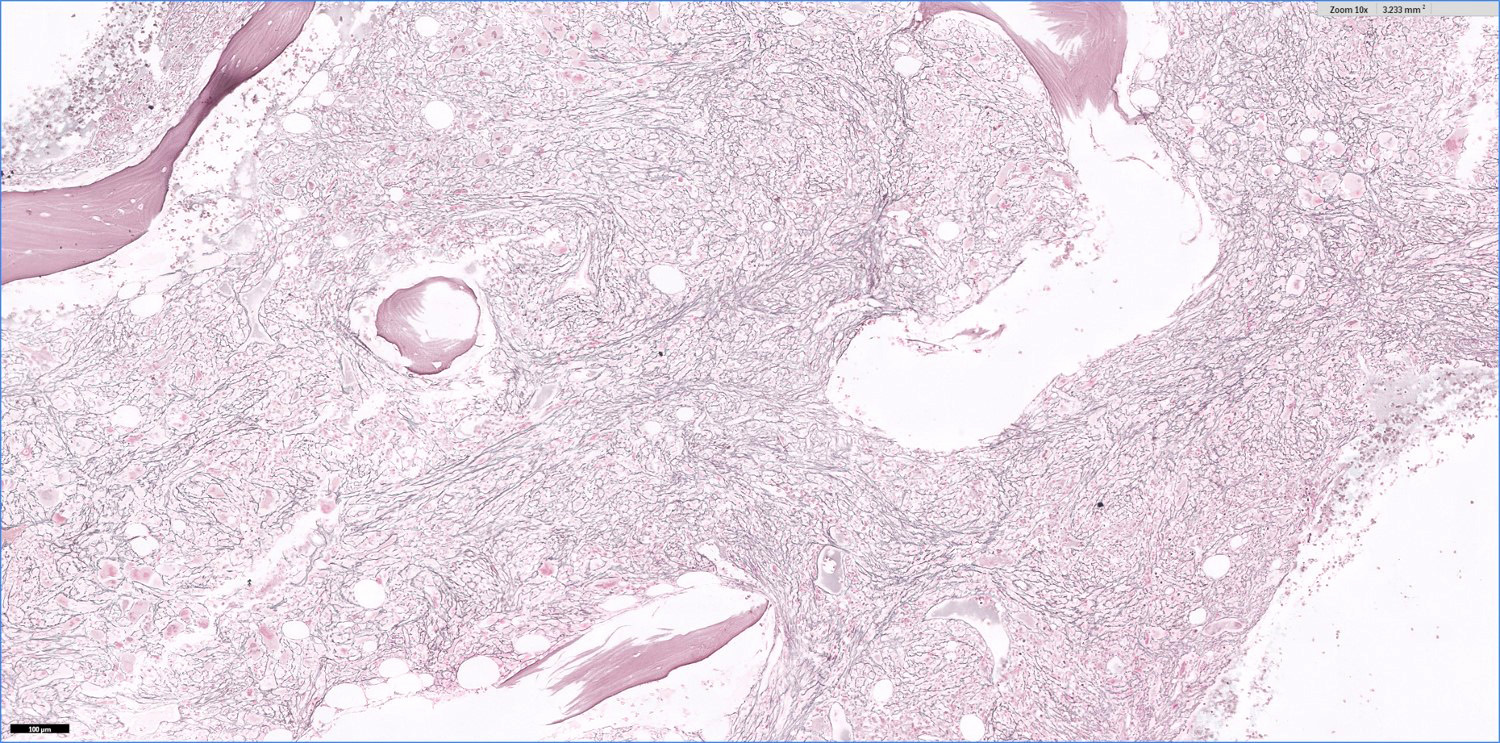
Reticulin
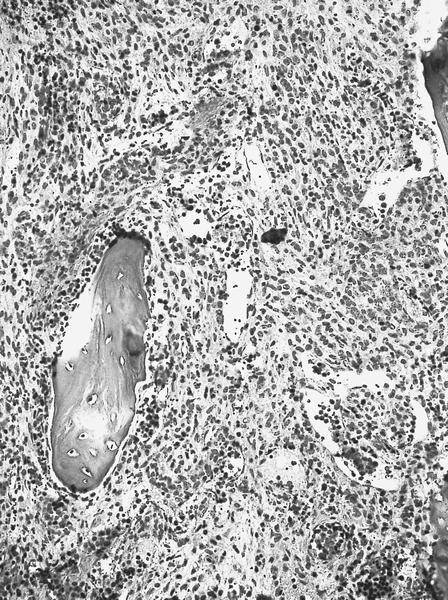

Bone marrow biopsy
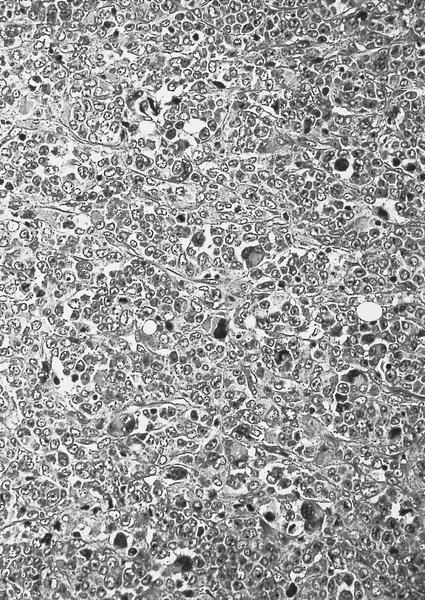
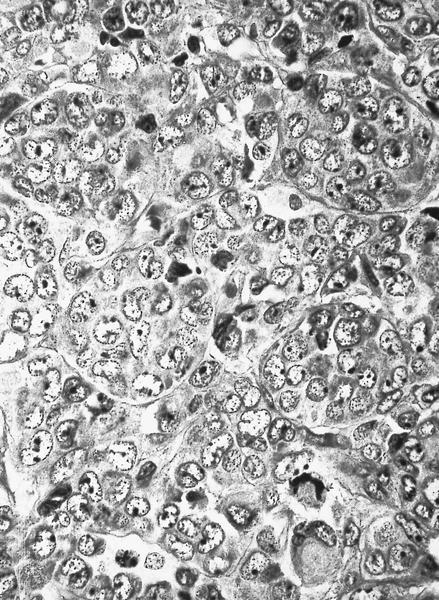
Lymph node biopsy
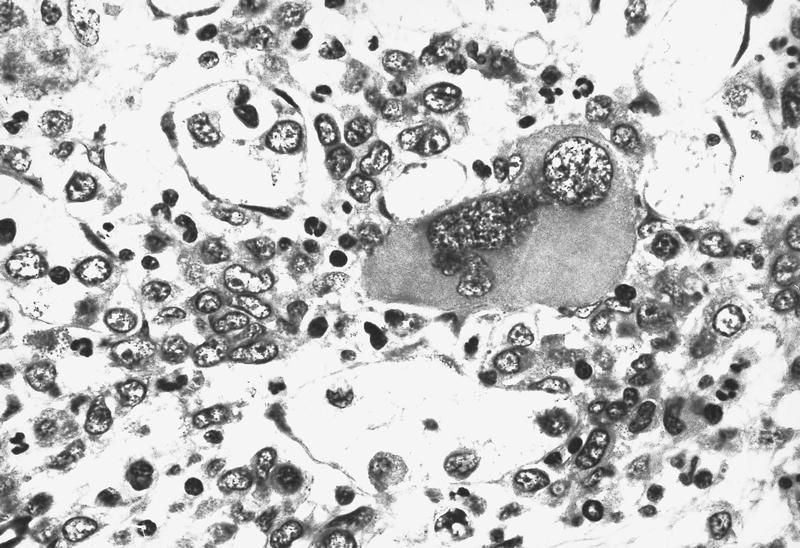
Bone marrow smear (Wright-Giemsa)
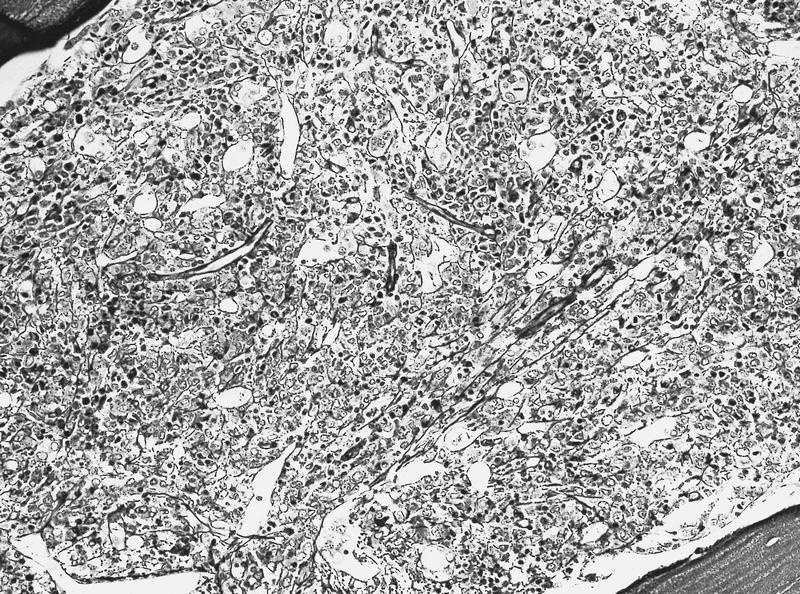
Stains
AFIP images
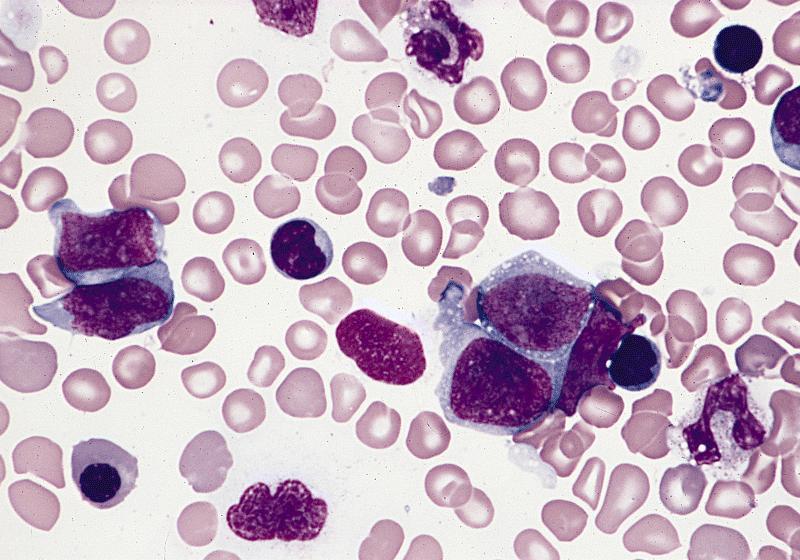
Peripheral smear (Wright-Giemsa)
- Blasts: CD34, HLA-DR (Leuk Lymphoma 2004;45:1873)
- Erythroblasts: glycophorin A, hemoglobin A, CD71, E-cadherin
- Granulocytes: myeloperoxidase, CD13, CD33 and CD117
- Monocytes: lysozyme, CD68
- Megakaryocytes: CD31, CD41, CD61, PAS and factor VIII
- CD42b and MPO are usually negative in blasts (Mod Pathol 2005;18:603)
- Von Willebrand factor
- Percentage of blasts varies between 3 and 30% (Mod Pathol 2005;18:603)
- Distinct population of myeloid blasts positive for CD34, CD33 and CD117
- Some cases have shown an aberrant coexpression of CD7 and CD13 antigens
- Myeloid blasts in acute panmyelosis with myelofibrosis typically lack megakaryocytic associated antigens, such as CD41 and CD61 (Mod Pathol 2005;18:603)
- Del(5q) and t(5q) (Ann Hematol 2004;83:513)
- Loss of chromosome 7 or del(7q) (Ann Hematol 2004;83:513)
- Nonspecific cytogenetic abnormalities reported in most cases with sufficient available material
- JAK2 mutations are typically absent (Ann Hematol 2004;83:513)
- A. Posterior iliac crest, bone marrow biopsy and aspirate:
- Acute panmyelosis (see comment)
- Reticulin fibrosis
- Comment: This is a hypercellular bone marrow for age featuring left shifted myeloid maturation with increased blasts. Numerous megakaryocytes with severe dysplastic features and focal clustering are seen. Flow cytometry shows left shifted myeloid maturation with increased blasts (~18%). The findings raise the differential diagnosis of acute panmyelosis with fibrosis versus MDS with excess blasts - 2. Clinical correlation regarding extramedullary hematopoiesis as well as molecular and cytogenetic results is recommended.
- Bone marrow biopsy:
- Iron: present (biopsy and smear)
- Reticulin: increased (2 - 3 of 3)
- Microscopic description:
- The biopsy is adequate and consists of trabecular bone and bone marrow elements. The cellularity ranges from 70 to 90% and consists of left shifted myeloids admixed with erythroid precursors. Numerous megakaryocytes are present and show marked dysplastic features, such as hyperchromatic and monolobated forms.
- CD34 immunostain is performed on formalin fixed paraffin embedded (FFPE) block A1, which highlights some of numerous dysplastic megakaryocytes as well as blasts, estimated around 15 - 20%.
- Bone marrow aspirate:
- Differential count:
- M:E = 4.6:1
- Blasts: 15%, promyelocytes: 6%, myelocytes / metamyelocytes: 15%, bands / segs: 34%, erythroids: 18%, lymphocytes: 14%, eosinophils: 1%, plasma cells: 1%, monocytes: 3%
- The aspirate smear is hypoparticulate and hypocellular. There is a moderate increase in blasts (13%). The blasts are small to intermediate in size, with scant cytoplasm and high N/C ratio. Mild dysplastic changes are noted in the myeloid precursors in the form of abnormal segmentation and few pseudo Pelger-Huët cells as well as occasional binucleated erythroids.
- Differential count:
- Acute megakaryoblastic leukemia:
- No prominent changes in granulocytes or erythroid cells (Ann Hematol 2004;83:513)
- Chronic idiopathic myelofibrosis:
- Marked splenomegaly and prominent dysplasia
- Metastatic carcinoma with desmoplasia (Ann Hematol 2004;83:513)
- Myelodysplastic syndrome with myelofibrosis:
- Lacks high percentage of blasts
- Post polycythemia vera and post essential thrombocythemia myelofibrosis
- Acute myeloid leukemia with fibrosis:
- Proliferative process is mostly of 1 cell type (such as myeloblasts) (Ann Hematol 2004;83:513)
- Primary myelofibrosis:
- Contains more numerous blasts and the megakaryocytes in primary myelofibrosis show distinctive cytologic atypia
- Primary myelofibrosis contains more numerous blasts and the megakaryocytes in primary myelofibrosis show distinctive cytologic atypia
- Secondary myelofibrosis after essential thrombocythemia (ET), polycythemia vera (PV):
- Usually with high hemoglobin and hematocrit (hemoglobin levels > 18.5 g/dL [hematocrit, 55.5%] in men or > 16.5 g/dL [hematocrit, 49.5%] in women)
- Associated with JAK2 mutations
- Autoimmune myelofibrosis:
- Associated with reactive deposition of fibrous connective tissue in the bone marrow and extramedullary hematopoiesis
- Hepatosplenomegaly often present (Clin Adv Hematol Oncol 2018;16:619)
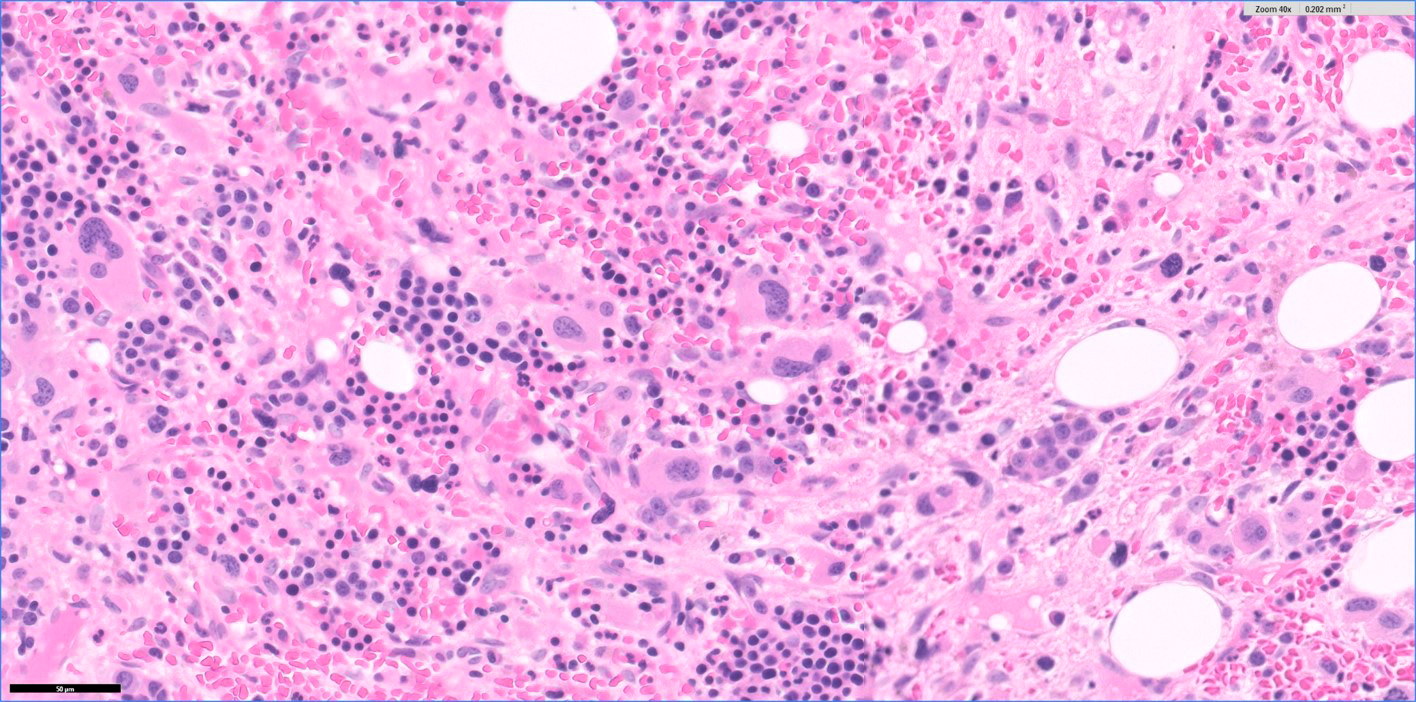

A 67 year old man previously in good state of health presented with fatigue, fever, bone pain and shortness of breath without splenomegaly. A complete blood count demonstrated anemia and thrombocytosis. A bone marrow biopsy shows a hypercellular marrow with panmyelosis, increased immature cells, dysplastic megakaryocytes and 2+ reticulin fibrosis. No other genetic abnormalities are found. What is the most likely diagnosis?
- Acute myeloid leukemia
- Acute panmyelosis with myelofibrosis
- Chronic myelogenous leukemia
- Myelodysplastic syndrome
Comment Here
Reference: Acute panmyelosis with myelofibrosis
- ABL
- CALR
- Del5;t5
- JAK2
Comment Here
Reference: Acute panmyelosis with myelofibrosis
- Acute leukemias in which the morphologic, cytochemical and immuno-phenotypic features of the blasts:
- Lack sufficient evidence to classify as myeloid or lymphoid origin
- Or, have morphologic and / or immunophenotypic characteristics of both myeloid and lymphoid cells
- Or, have both B and T lineages (acute bilineal leukemia and acute biphenotypic leukemia)
Acute leukemias of ambiguous lineage include the following:- Acute undifferentiated acute leukemia
- Mixed phenotype acute leukemia with t(9;22)(q34;q112.); BCR-ABL1
- Mixed phenotype acute leukemia with t(v;11q23); MLL rearranged
- Mixed phenotype acute leukemia, B/myeloid, NOS
- Mixed phenotype acute leukemia, T/myeloid, NOS
- Mixed phenotype acute leukemia, NOS rare types
- Other acute leukemias of ambiguous lineage
- < 4% of all acute leukemias, more frequent in adults
- Unknown
- Related to bone marrow failure: fatigue, infections, bleeding
- Acute undifferentiated leukemia
- Leukemic cells lack any differentiating features
- Acute biphenotypic and acute bilineal leukemias
- May present as one subtype of AML with features of ALL (B, T or B and T)
- Leukemias lack specific lineage markers
- cCD79a, cCD22, CD3 and MPO
- Generally don’t express more than one lineage associated marker
- Often express HLA-DR, CD34, CD38, may express TdT and CD7
Bilineal acute leukemia
- Dual population of blasts, each with distinct lineage: positive for myeloid, lymphoid or B and T-cell markers (Leukemia 2007;21:2264)
- May evolve into biphenotypic acute leukemia (Atlas of Genetics and Cytogenetics: Biphenotypic Acute Leukaemia (BAL) [Accessed 13 April 2018])
Biphenotypic acute leukemia
- Blasts co-express myeloid and T or B lineage markers
- Or, concurrent B and T lineage markers
- Rarely co-express markers for myeloid, T and B lineages
- Co-expression of one or two cross-lineage (nonspecific) markers is not sufficient for biphenotypic leukemia; e.g. myeloid antigen positive ALL or Lymphoid antigen positive AML
- Lineage switch after therapeutic intervention
- Possible expansion of pre-existing minor population of blasts of different lineage following therapeutic suppression of the major population
- Possible lineage instability
- High degree of cytogenetic abnormalities
- 1/3 of cases have Ph chromosome
- t(4;11)(q21;q23) or 11q23, typically have CD10(–) precursor B lymphoid component
- T/myeloid biphenotypic or bilineal leukemia can have complex karyotype
- Multipotent progenitor stem cell
- Unfavorable, particularly in adults
- t(4;11) or Ph particularly unfavorable
- Usually aggressive chemotherapy or bone marrow transplant
- 5 year old boy with t(9;17)(p11;q11) (Leuk Lymphoma 1997;25:179)
- 20 year old woman with myeloid, B cell and NK phenotype (Arch Pathol Lab Med 2003;127:E93)
- 80 year old man with blasts coexpressing CD79a and myeloid markers (Arch Pathol Lab Med 2003;127:356)
- Due to transformation of essential thrombocythemia (Am J Hematol 2006;81:624)
AFIP images
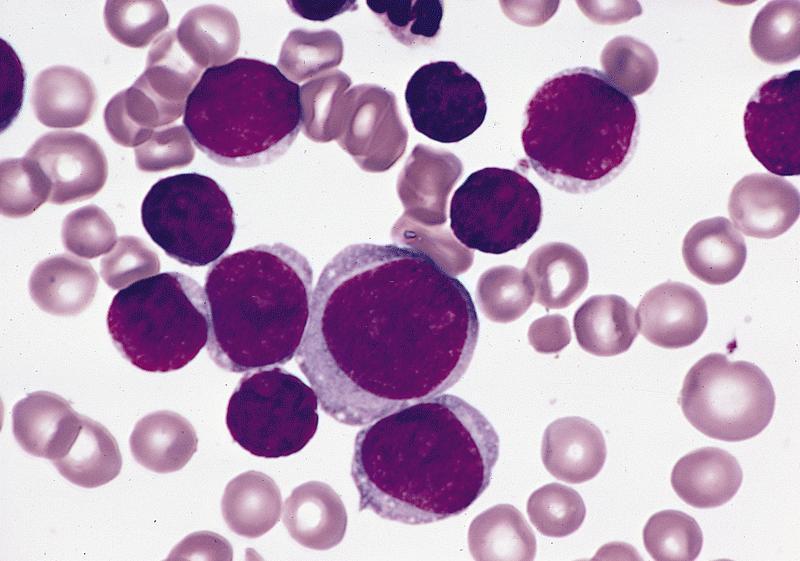
Child with t(4;11)(q21;q23)
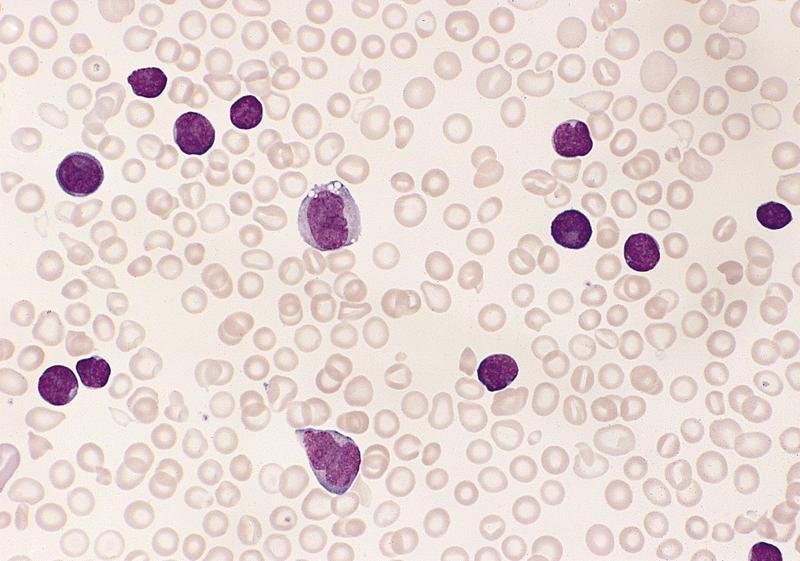
Lymphoblasts and monoblasts
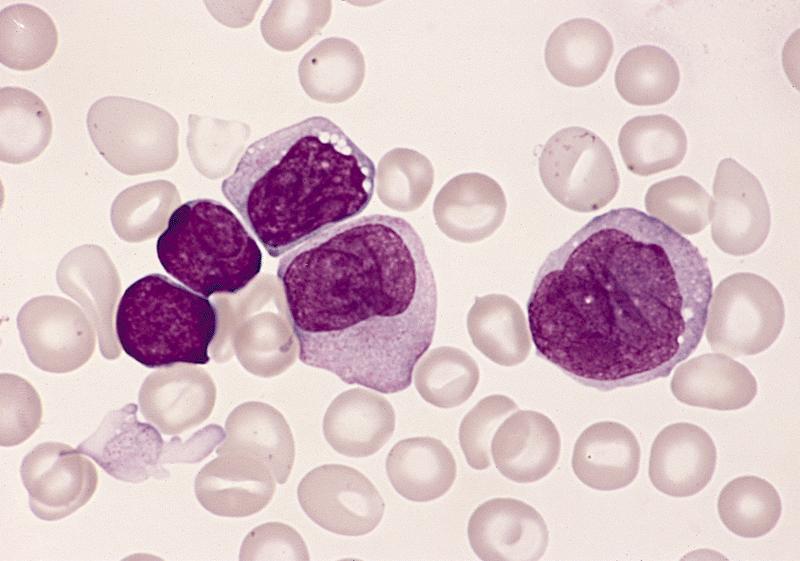
Monoblasts, promonocytes and lymphoblasts
AFIP images
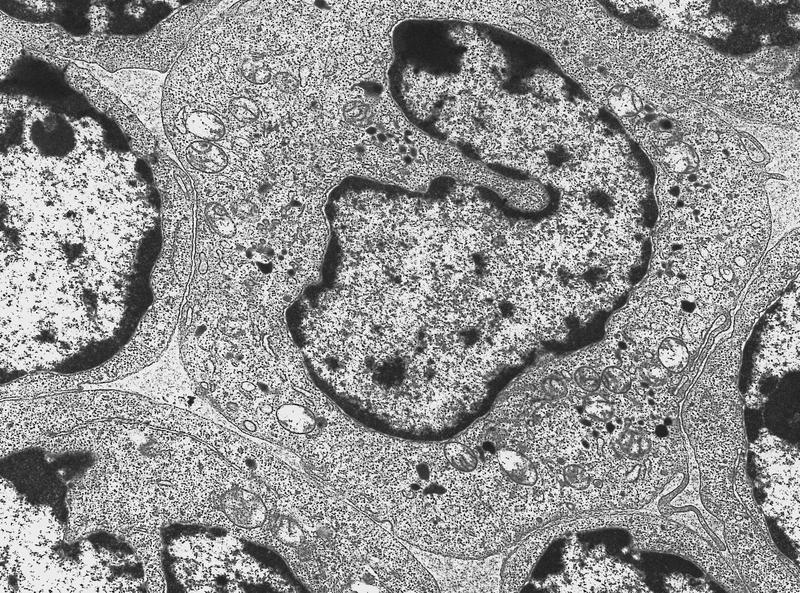
Monocytoid blast
- Many cases have IgH and TCR rearrangements or deletions
- For biphenotypic acute leukemia
- Myeloid antigen positive ALL
- Lymphoid antigen positive AML
- For undifferentiated acute leukemia
- Minimally differentiated AML
- Unusual precursor-B-cell or T-cell ALL
- Leukemic disorder with myelodysplastic / myeloproliferative features present at initial diagnosis
- Leukocytosis characterized by increased morphologically dysplastic neutrophils and their precursors
- Clonal hematopoietic stem cell disorder that manifests with leukocytosis (always > 13 × 109/L, frequently > 25 × 109/L)
- Features are distinct from other myelodysplastic / myeloproliferative neoplasms (chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML), myelodysplastic / myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS / MPN-RS-T))
- Does not meet WHO diagnostic criteria for BCR-ABL1 positive chronic myeloid leukemia, primary myelofibrosis, polycythemia vera or essential thrombocythemia
- No Philadelphia chromosome / BCR-ABL1 fusion gene
- No PDGFRA, PDGFRB or FGFR1 rearrangements
- No PCM1-JAK2 translocation
- Philadelphia chromosome negative (Ph1-) atypical CML
- ICD-O: 9876/3 - Atypical chronic myeloid leukemia, BCR-ABL1 negative
- Rare subtype of MDS / MPN
- Incidence: ~1 - 2 cases per every 100 BCR-ABL1 positive CML
- Median age at presentation: ~70 years (Am J Hematol 2017;92:542)
- M:F = ~1:1, with slight male predominance (Ther Adv Hematol 2015;6:308)
- Peripheral blood and bone marrow always involved
- Splenic and hepatic involvement common
- No specific pattern of cytogenetic or molecular abnormalities
- Karyotypic abnormalities common (80% of cases)
- Molecular abnormalities common:
- SETBP1, ETNK1, ASXL1, TET2, RAS and RUNX1 mutations commonly seen (Biomark Res 2017;5:33, Leukemia 2013;27:1852, Haematologica 2014;99:e244, Blood 2015;125:499, Blood 2015;125:422, Int J Hematol 2015;101:229, Am J Hematol 2017;92:542)
- Presence of CALR, MPL and JAK2 mutations tend to exclude the diagnosis of aCML
- Unknown currently
- Moderate anemia
- Thrombocytopenia
- Splenomegaly
- Leukocytosis
- References: Haematologica 2006;91:1566, Ann Oncol 2000;11:441, J Clin Oncol 2001;19:2915, Blood 1991;78:205, Blood 2014;123:2645
- WHO diagnostic criteria:
- Peripheral blood leukocytosis ~13 × 109/L, due to increased numbers of neutrophils and their precursors (i.e. promyelocytes, myelocytes and metamyelocytes), with neutrophil precursors constituting ≥ 10% of the leukocytes
- Dysgranulopoiesis, which may include abnormal chromatin clumping or projection, abnormal segmentation or hypogranularity
- No or minimal absolute basophilia; basophils constitute < 2% of the peripheral blood leukocytes
- No or minimal absolute monocytosis; monocytes constitute < 10% of the peripheral blood leukocytes
- Hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages
- < 20% blasts in the blood and bone marrow
- No evidence of PDGFRA, PDGFRB or FGFR1 rearrangement or of PCM1-JAK2 translocation
- WHO criteria for BCR-ABL positive chronic myeloid leukemia, primary myelofibrosis, polycythemia vera or essential thrombocythemia are not met
- History of MPN, the presence of MPN features in the bone marrow or MPN associated mutations (e.g. JAK2, CALR or MPL) should be excluded
- References: PLoS One 2014;9:e98258, Haematologica 2006;91:1566, Ann Oncol 2000;11:441, Blood 1991;78:205, Blood 2014;123:2645
- Increased LDH
- Splenomegaly
- Poor prognosis
- Median survival = 15 months, 5 year overall survival = 25% (Ther Adv Hematol 2015;6:308)
- Poorer outcomes associated with (Haematologica 2006;91:1566, Ann Oncol 2000;11:441):
- Age > 65 years
- White blood cells > 50 × 109/L
- Hemoglobin < 10 g/dL
- Thrombocytopenia
- ~30 - 40% of aCML transforms to acute myeloid leukemia (Blood 2014;123:2645)
- 19 year old man with hepatosplenomegaly and leukocytosis (Ann Hematol 2020;99:1145)
- 30 year old son and 59 year old father with SETBP1 positive aCML (Braz J Med Biol Res 2015;48:583)
- 68 year old woman with fever and generalized bone pain (Clin Case Rep 2018;6:915)
- No standard treatment
- Allogeneic stem cell transplant (SCT) is only treatment that achieves long term remission (Blood 2017;129:838, Blood 2013;122:1707)
- Hypomethylating agents for non-SCT candidates (Blood 2017;129:838)
- Other therapies, such as mutation targeted agents, are becoming more readily available (Blood 2017;129:838, Blood 2013;122:1707)
- Bone marrow (Jaffe: Hematopathology, 2nd Edition, 2016, Blood 2014;123:2645):
- Hypercellular marrow with increased granulocytic proliferation
- Increased myeloid:erythroid ratio > 10:1
- May have increased blasts but < 20%
- Granulocytic dysplasia often marked
- Dyserythropoiesis can be present in up to half of cases
- Dysmegakaryopoiesis common, including small or micromegakaryocytes and hypolobated forms
- Reticulin fibrosis increased in some patients
- Syndrome of abnormal chromatin clumping (Haematologica 1990;75:532, Nouv Rev Fr Hematol 1995;37:245):
- Considered a variant of aCML
- Characterized by high percentage of granulocytes and precursors with exaggerated chromatin clumping identified in peripheral blood or bone marrow
Contributed by Ling Zhang, M.D.
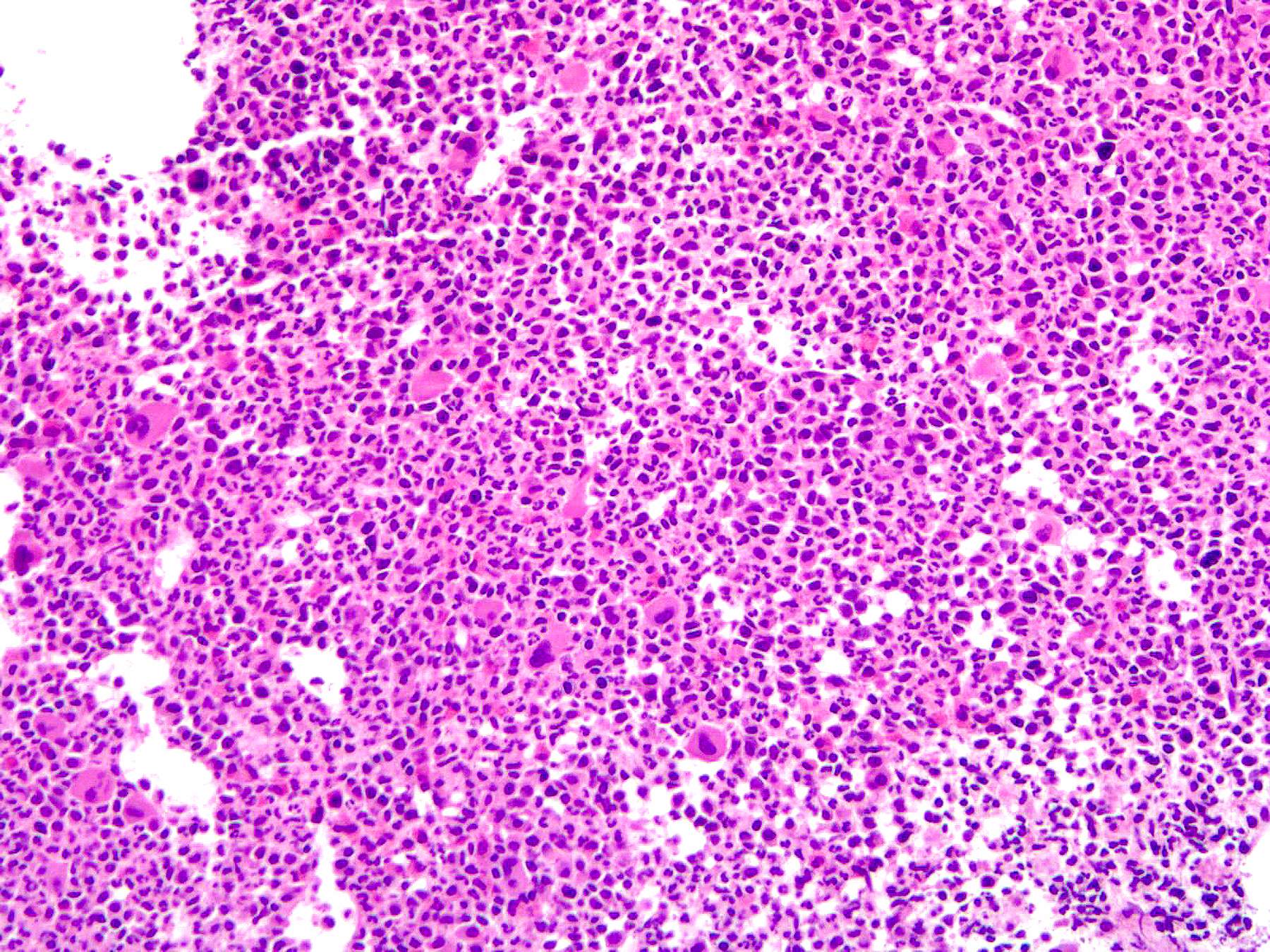
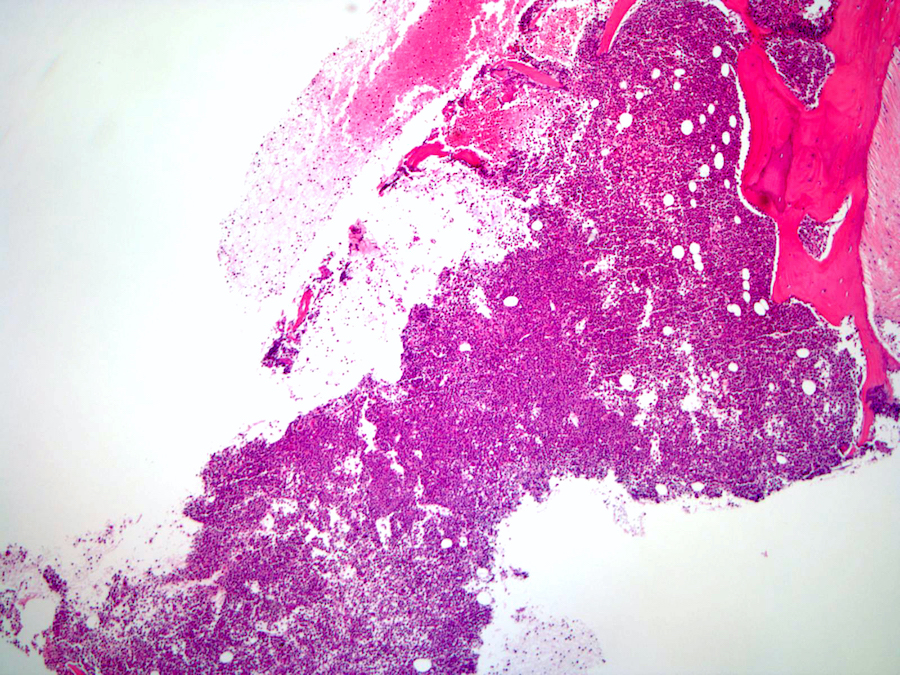
Bone marrow biopsy
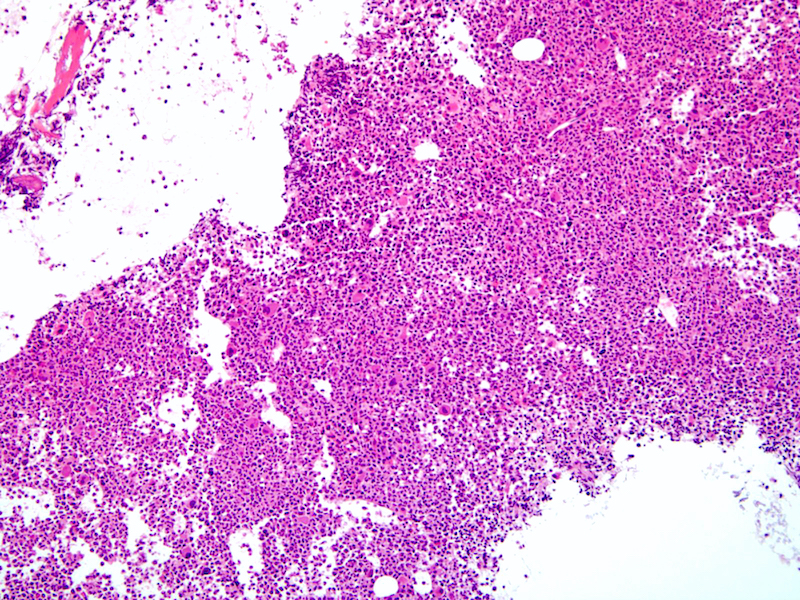
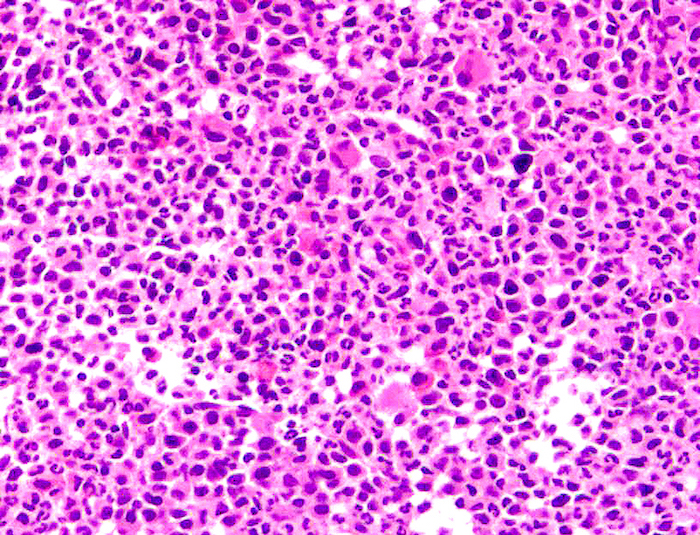
Bone marrow biopsy
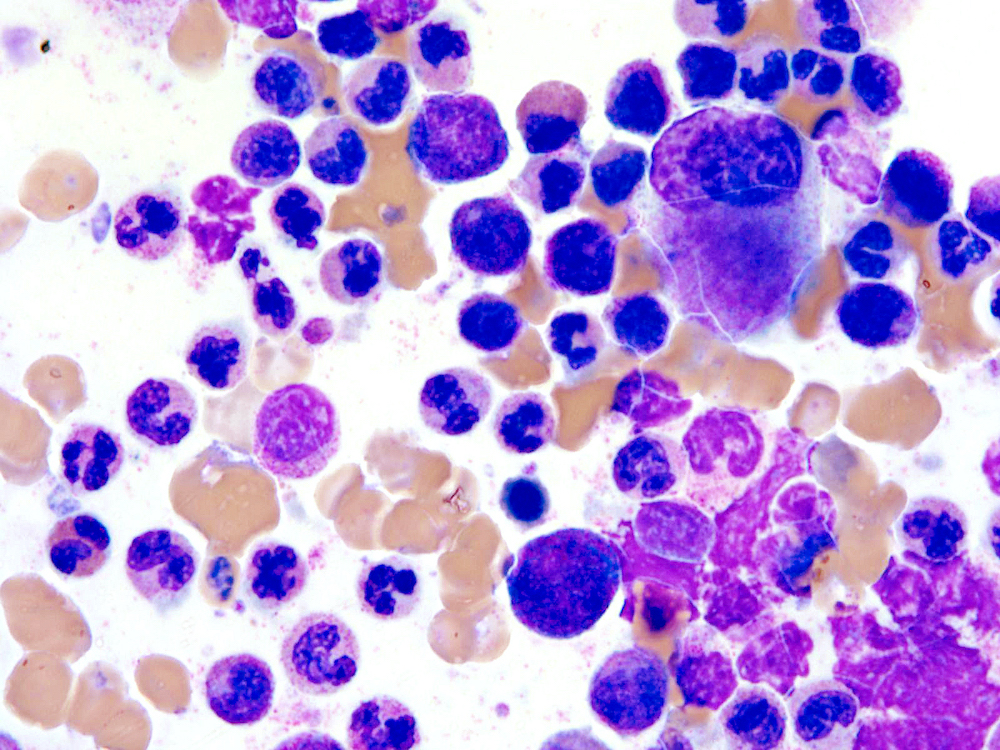
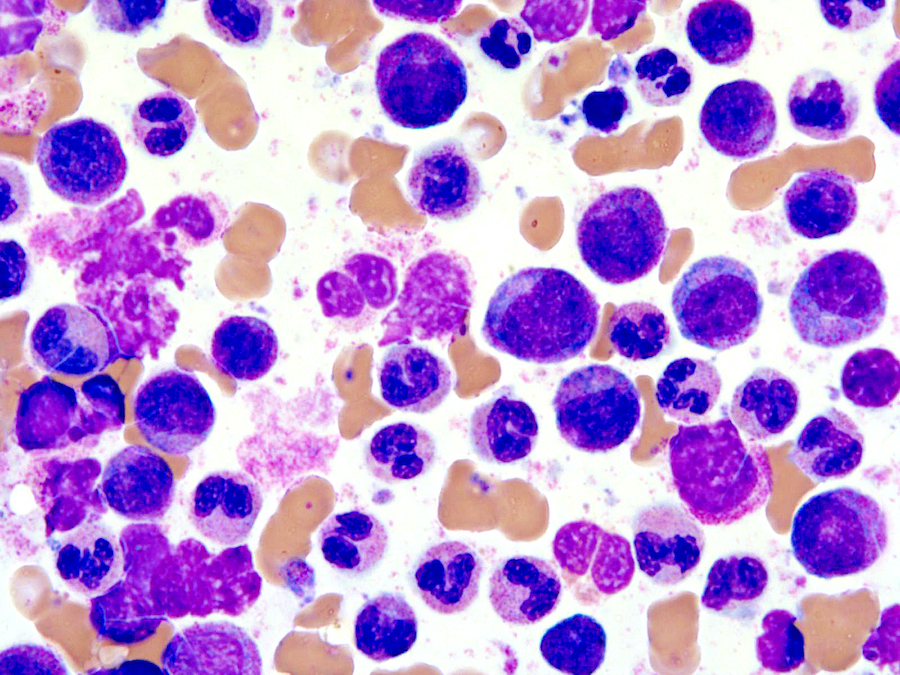
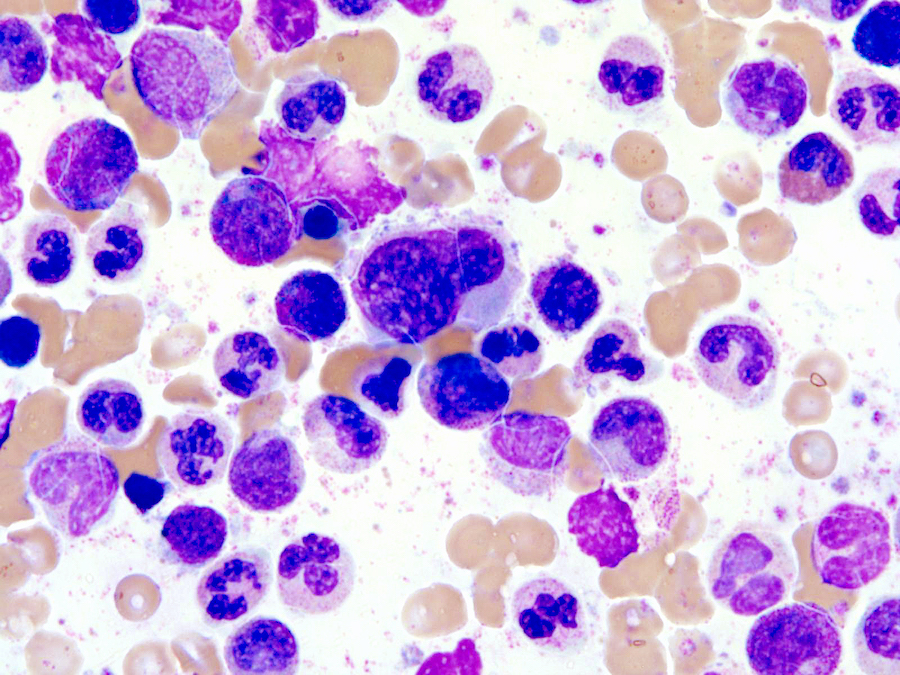
Bone marrow aspirate smear
- Leukocytosis with white blood cells (WBC) ≥ 13 × 109/L with median values of 24 - 96 × 109/L
- < 20% blasts in peripheral blood and bone marrow; usually < 5%
- Neutrophil precursors (promyelocytes, myelocytes and metamyelocytes) comprising > 10% of WBC
- Prominent dysgranulopoiesis
- Acquired Pelger-Huët abnormality
- Clumped chromatin
- Hypersegmentation of nuclei
- Abnormal nuclear projections
- Cytoplasmic hypogranularity
- Absent or minimal monocytosis; < 10% of leukocytes
- Absent or minimal basophilia; < 2% of leukocytes
- Anemia and thrombocytopenia are common
- References: PLoS One 2014;9:e98258, Haematologica 2006;91:1566, Ann Oncol 2000;11:441, Blood 1991;78:205, Blood 2014;123:2645
Contributed by Ling Zhang, M.D.
Peripheral blood smear
- No specific immunophenotype
- Granulocytes positive for MPO, CD33, CD13, CD15
- Monocytes positive for CD14 and CD68; monocytosis on biopsy should call into question a diagnosis of aCML
- References: J Clin Oncol 2001;19:2915, Blood 1991;78:205
- No specific flow cytometry findings
- Cytogenetics
- Karyotypic abnormalities present in up to 80% of cases
- Most common abnormalities:
- Gain of chromosome 8
- del(q20)
- Rarely i17q
- Abnormalities of 12, 13, 14, 17 and 19 also reported
- Molecular study
- NGS mutation findings:
- SETBP1, ETNK1, ASXL1, TET2, RAS, RUNX1 and PTPN1 detected
- Most cases have more than 1 mutation
- Mutations of SETBP1 on chromosome 18q21.1 seen in ~10 - 25% cases
- ETNK1 mutations slightly less common than SETBP1 (seen in ~8% of cases)
- CSF3R mutation present in < 10% of aCML
- Presence of MPN associated mutations (JAK2, CALR or MPL) tend to exclude diagnosis of aCML
- References: Biomark Res 2017;5:33, Leukemia 2013;27:1852, Haematologica 2014;99:e244, Blood 2015;125:499, Blood 2015;125:422, Int J Hematol 2015;101:229, Am J Hematol 2017;92:542, N Engl J Med 2013;368:1781
- NGS mutation findings:
- Peripheral blood:
- Mild normocytic anemia
- Marked leukocytosis with neutrophilia (70%) and circulating immature granulocytic precursors (22%)
- Dysplastic neutrophils
- No circulating blasts or mature monocytosis identified
- Mild thrombocytopenia
- Bone marrow, right posterior iliac crest, core biopsy, clot section and aspirate smear:
- Hypercellular bone marrow with marked granulocytic hyperplasia and dysgranulopoiesis consistent with atypical chronic myeloid leukemia (see comment)
- Comment: Of note, there is a marked leukocytosis with overt dysmyelopoiesis. FISH study is reportedly negative for t(9;22) / BCR-ABL1. Gene mutation profile showed a mutation involving the SETBP1 gene (VAF of 66%). Karyotyping was reportedly normal female karyotype, 46,XX[20]. The overall findings are consistent with atypical chronic myeloid leukemia.
- Peripheral smear: Manual review of the peripheral blood shows normochromic, normocytic anemia with mild thrombocytopenia. RBCs: mild normochromic, normocytic with minimal anisopoikilocytosis. WBCs: marked leukocytosis with markedly increased dysplastic neutrophils and left shifted myeloid precursors. Platelets: thrombocytopenia with rare large forms.
- Bone marrow biopsy: Quality: adequate. Cellularity: 90%. The bone marrow shows hypercellularity (90%) with marked granulocytic hyperplasia and diminished erythroid precursors. Megakaryocytes are increased and many of them are smaller than normal in size. The M:E ratio is increased. Reticulin stain highlights mild reituculin fibrosis (MF1). Iron stores are decreased to absent.
- Bone marrow clot section: Quality: adequate. Cellularity: 90% morphologic features are similar to those observed in the core biopsy.
- Bone marrow aspirate: Quality: adequate. Granulocytes: markedly increased; left shifted maturation without overt increase in blasts; marked dysplasia (abnormal chromatin pattern, hyposegmented nuclei or hypogranular cytoplasm). Erythrocytes: markedly decreased with progressive maturation, mild dysplasia (slight nuclear irregularities, basophilic stippling of cytoplasm or N:C maturation asynchrony). Megakaryocytes: adequate in number; dysplastic (variable in size, increased hypolobated forms, frequently with nuclear hyperchromasia). Blasts: overall ~2% of nucleated cells. No ring sideroblasts are present.
- Myeloproliferative neoplasm (MPN):
- No dysplasia (except for abnormal megakaryocytes)
- Cytosis (see CML and CNL below for differential findings)
- BCR-ABL fusion
- JAK2, CALR, MPL, CSF3R mutations
- Chronic myeloid leukemia (CML):
- Presence of Philadelphia chromosome / BCR-ABL fusion gene
- Dysplasia is minimal in CML
- Basophilia > 2% in CML
- Chronic neutrophilic leukemia (CNL):
- Leukocytosis comprised mostly of neutrophils
- No significant left shift in proliferating neutrophils
- CSF3R mutation more common in CNL and favor this entity
- Myelodysplastic syndrome (MDS):
- Dysplasia in ≥ 1 lineage
- No leukocytosis
- Cytopenias predominate
- Myeloid / lymphoid neoplasms with eosinophilia and gene rearrangement:
- Chronic myelomonocytic leukemia (CMML):
- Monocytosis, > 10% of WBCs
- More severe dysplasia in aCML
- Higher survival
- Myelodysplastic / myeloproliferative neoplasm, unclassifiable (MDS / MPN, U):
- Leukocytosis and dysplastic changes in MDS / MPN, U raises differential diagnosis of aCML
- Presence of SETBP1 mutations favor the diagnosis of aCML
- Often lower WBC count when compared to aCML
- Distinction cannot be made in a subset of cases
- Clinically they are indistinguishable
- Significant blood and bone marrow morphological overlapping features
- aCML carries a worse prognosis (overall survival of 12.4 months versus 21.8 months for MDS / MPN, U)
- Reference: Jaffe: Hematopathology, 2nd Edition, 2016
A 71 year old man presented to his primary care physician with complaints of fatigue and nausea. Upon further workup, he was found to have normocytic anemia, thrombocytopenia and marked leukocytosis (70 × 109/L) with no monocytosis. Imaging studies demonstrated a mildly enlarged spleen with no discernible lesions. A bone marrow biopsy was performed showing no increase in blasts with representative images shown above. FISH studies for BCR-ABL were negative. Gene mutation profile demonstrated a mutation involving the ETNK1 gene. Which of the following is the most likely diagnosis?
- Chronic myeloid leukemia (CML)
- Chronic neutrophilic leukemia (CNL)
- Acute myeloid leukemia (AML)
- Chronic myelomonocytic leukemia (CMML)
- Atypical chronic myeloid leukemia (aCML)
Comment Here
Reference: Atypical chronic myeloid leukemia
- CSF3R
- JAK2
- PCM-JAK2
- PDGFR
- SETBP1
Comment Here
Reference: Atypical chronic myeloid leukemia
- Introduced as a provisional entity in the WHO 2017 (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Does not have BCR-ABL1 translocation but shows a pattern of gene expression similar to that seen in ALL with BCR-ABL1
- Exhibits National Cancer Institute (NCI) high risk features: (a) WBC ≥ 50,000/microL or age ≥ 10 years; up to 13 years if treated on a COG protocol; (b) persistent, postinduction, minimal residual disease; and (c) genetic alterations in kinase signaling including cytokine receptors and tyrosine kinases (Pediatr Blood Cancer 2017;64:10.1002)
- Important to recognize because:
- Associated with a poor prognosis
- Some are sensitive to tyrosine kinase inhibitors similar to BCR-ABL1 (Eur J Cancer 2017;82:203, Cancer Cell 2016;29:186)
- Use of tyrosine kinase inhibitors with treatment protocols may improve outcome (Eur J Cancer 2017;82:203)
- Identified in 15 - 20% of patients with B ALL (Lancet Oncol 2009;10:125, N Engl J Med 2009;360:470)
- ICD-10: C91.00 - acute lymphoblastic leukemia not having achieved remission
- Relatively common subtype, representing 7 - 25% of B ALL patients (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Reported in both pediatric and adult patients
- More common in Down syndrome patients; uniquely shows CRLF2 translocation
- Harbors a large number of kinase activating gene rearrangements primarily involving the ABL class, JAK / STAT or RAS pathway associated signaling pathways
- Common genes involved include ABL1, ABL2, CRLF2, CSF1R, EPOR, NTRK3, PDGFRB, JAK1, JAK2, JAK3, FLT3, IL7R, SH2B3 and IKZF1
- These genes encode proteins involved in B cell development, proliferation and differentiation, cell cycle regulation and cell signaling
- Overexpression of cytokine receptors such as cytokine receptor-like factor 2 (CRLF2) occurs in both ALL with BCR-ABL1-like / Philadelphia-like and other B ALL categories (Eur J Cancer 2017;82:203)
- B ALL in general usually presents with symptoms related to bone marrow suppression by lymphoblasts
- Patients can present with anemia, leucopenia or thrombocytopenia or a combination of these
- Symptoms include bruising or bleeding due to thrombocytopenia, pallor or fatigue due to anemia and recurrent infections caused by neutropenia / leucopenia or bone pains
- May also present with lymphadenopathy (> 10 mm in single dimension of the lymph node), hepatomegaly or splenomegaly
- B ALL with BCR-ABL1-like and other B ALL with recurrent genetic abnormalities show no unique clinical presentation or microscopic findings
- Further, similar to some other B ALL with recurrent genetic abnormalities, B ALL with BCR-ABL1-like shows no unique immunophenotypic profile (Pediatr Blood Cancer 2017;64:10.1002)
- Essentially a diagnosis of exclusion
- Identifying patients with B ALL with BCR-ABL1-like can influence the management of the patient
- Those with PDGFRB translocation can benefit from tyrosine kinase inhibitors, while patients with JAK translocations may benefit from JAK inhibitors (Pediatr Blood Cancer 2017;64:10.1002)
- Adults tend to have poor outcome even with high intensity chemotherapy regimens
- B ALL with BCR-ABL1-like patients do not show a BCR-ABL1 fusion protein expressed from t(9;22)(q34;q11.2); however, they have a gene expression profile similar to ALL with BCR-ABL1
- Almost none of the genetic alterations are detected by standard genetic diagnostic methods such as conventional karyotyping and FISH because these genetic alterations are diverse and often cryptic
- To date, there are over 60 different identified rearrangements and approximately 16 different targetable genes
- Patients can only be diagnosed by gene expression panels (genome wide Affymetrix gene expression arrays):
- Currently 2 gene expression signatures have been verified for diagnosis of B ALL with BCR-ABL1-like / Ph-like
- BCR-ABL1-like gene expression array consists of 110 expression probe sets and was originally developed to classify B ALL in a population based Dutch / German cohort
- Gene expression of a group of cases were similar to ALL with BCR-ABL1 and therefore named the category ALL with BCR-ABL1-like
- These had an overall unfavorable outcome (Lancet Oncol 2009;10:125)
- Philadelphia-like signature was first demonstrated in cases of B ALL with IKZF1 deletions in a high risk United States cohort (N Engl J Med 2009;360:470)
- Differences between these 2 gene expression signatures can be explained by their different origins and discovery cohorts
- Few reference laboratories are currently offering low density microarray classifiers (8 or 15 gene) that may help screen patients
- One of these reference laboratories is Tricore reference laboratories (BCR-ABL1 like ALL (Ph-like ALL): TriCore Reference Laboratories [Accessed 8 May 2019])
- Some institutions and reference laboratories have developed multiplex FISH testing that may help screen cases
- Some institutions such as St. Jude utilize transcriptome and whole exome sequencing
- Flowcytometry:
- There is evidence that B-ALL with BCR-ABL1-like present as common-ALL on IPT, however this maturation stage is not distinct of the entity and is common in other B-ALL subt
- ypes B-ALL with BCR-ABL1-like patients with CRLF translocations show high levels of surface expression of the protein on flow cytometry, which can be used as a screening tool
- Prognostic factors include age, white blood cell count, immunophenotype, genetics and detection of measurable residual disease
- Numerous stratification schemes to assess risk in B ALL
- NCI risk group:
- NCI standard risk group: WBC < 50,000/microL and age 1 to < 10 years
- NCI high risk group: WBC ≥ 50,000/microL or age ≥ 10 years (up to 13 years if treated on a COG protocol)
- One scheme stratifies patients into standard risk and high risk based on age (< 10 years = standard, > 10 years = high) and WBC count (< 50,000/mm3 = standard, > 50,000/mm3 = high)
- Recurrent genetic abnormalities are identified in the majority of B ALL cases
- Prognosis of B ALL with BCR-ABL1-like is generally unfavorable
- 10 year old boy treated with tyrosine kinase inhibitor for EBF1-PDGFRB fusion (J Clin Oncol 2013;31:e413)
- 16 year old boy treated with tyrosine kinase inhibitor for EBF1-PDGFRB fusion (Haematologica 2013;98:e146)
- 16 year old girl and 29 year old man with B ALL and novel ABL1 fusion genes (Br J Haematol 2011;153:43)
- 17 year old girl with novel JAK2 F694L mutation treated with JAK2 inhibitor and stem cell transplant (Pediatr Blood Cancer 2017;64:10.1002)
- 20 year old patient with a relapse of Philadelphia chromosome negative ALL who responded to tyrosine kinase inhibitor therapy (Oncol Res Treat 2018;41:550)
- B ALL with BCR-ABL1-like is managed by standard combination chemotherapy
- Effective treatment modality for B ALL since the 1950s
- Usually administered in 3 distinct phases (induction, consolidation and maintenance) and should include intrathecal treatment, which is directed to the central nervous system
- Addition of tyrosine kinase inhibitors in B ALL with BCR-ABL1-like depends on the specific genetic alteration and has been shown to improve patient's outcome
- Both imatinib and dasatinib specifically inhibit ABL1, ABL2, PDGFRB and CSF1R kinases; ruxolitinib inhibits JAK / CRLF2 / EPOR alterations (Eur J Cancer 2017;82:203)
- Sensitivity to ruxolitinib in patients with JAK class aberrations is promising and has been successful in 2 JAK class fusion positive patients and 1 JAK2 mutated ALL (Eur J Cancer 2017;82:203)
- Adults tend to have poor outcome even with high intensity chemotherapy regimens; therefore, bone marrow transplantation becomes an important treatment option
- Blasts have scant agranular cytoplasm, coarse to fine chromatin, often with indistinct nucleoli
- No Auer rods and no dysplastic myeloid cells
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
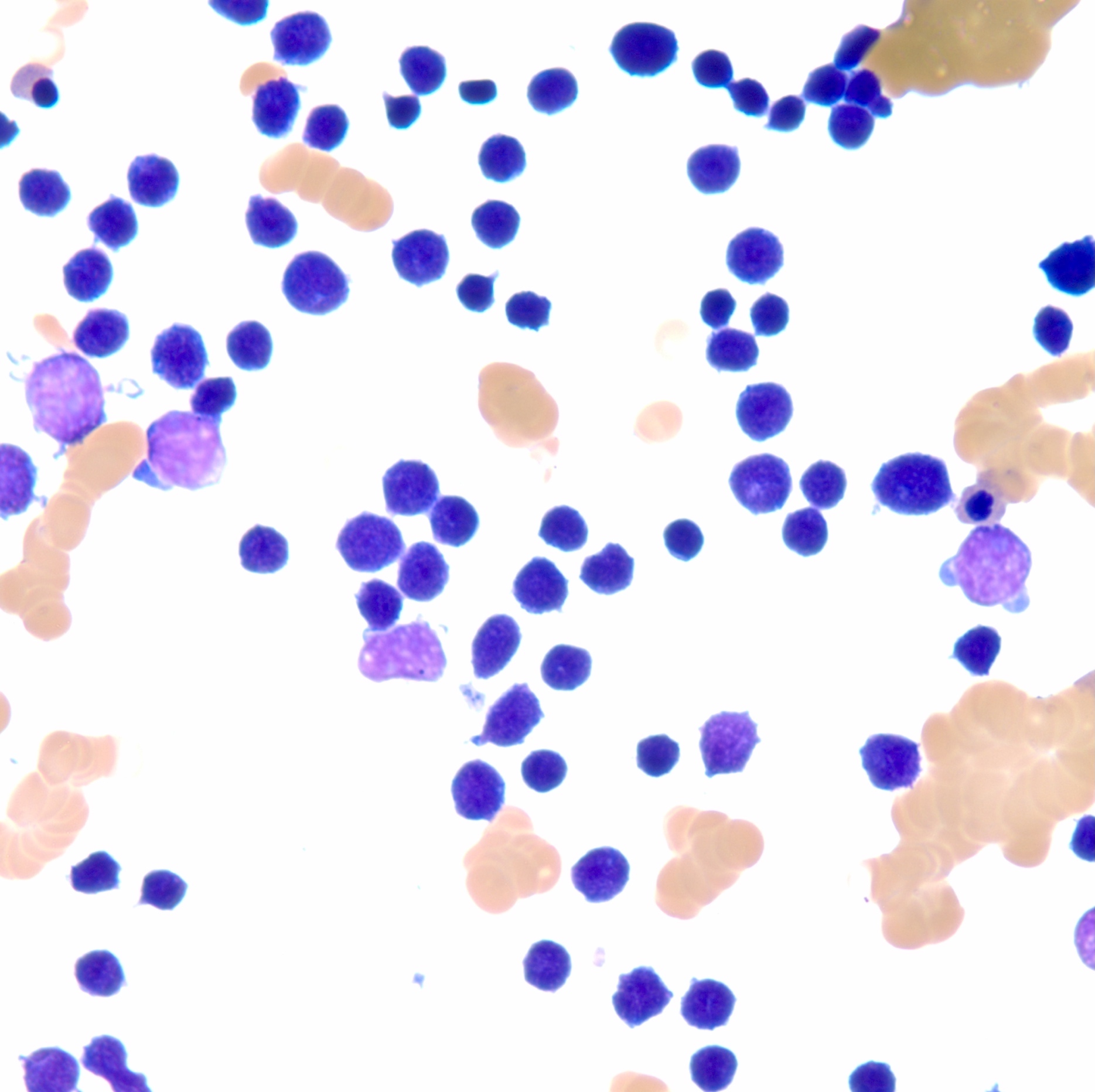
Lymphoblasts
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
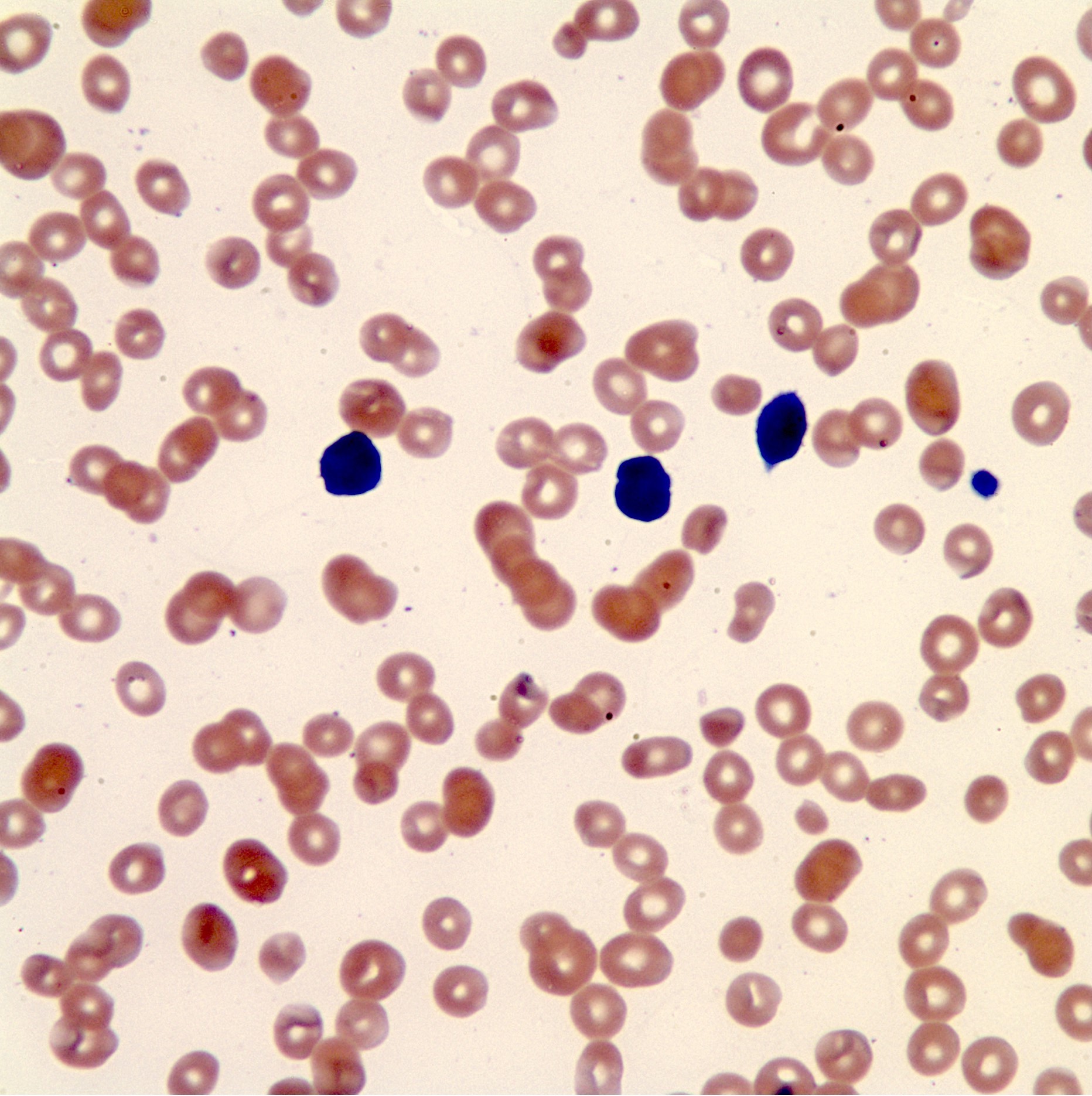
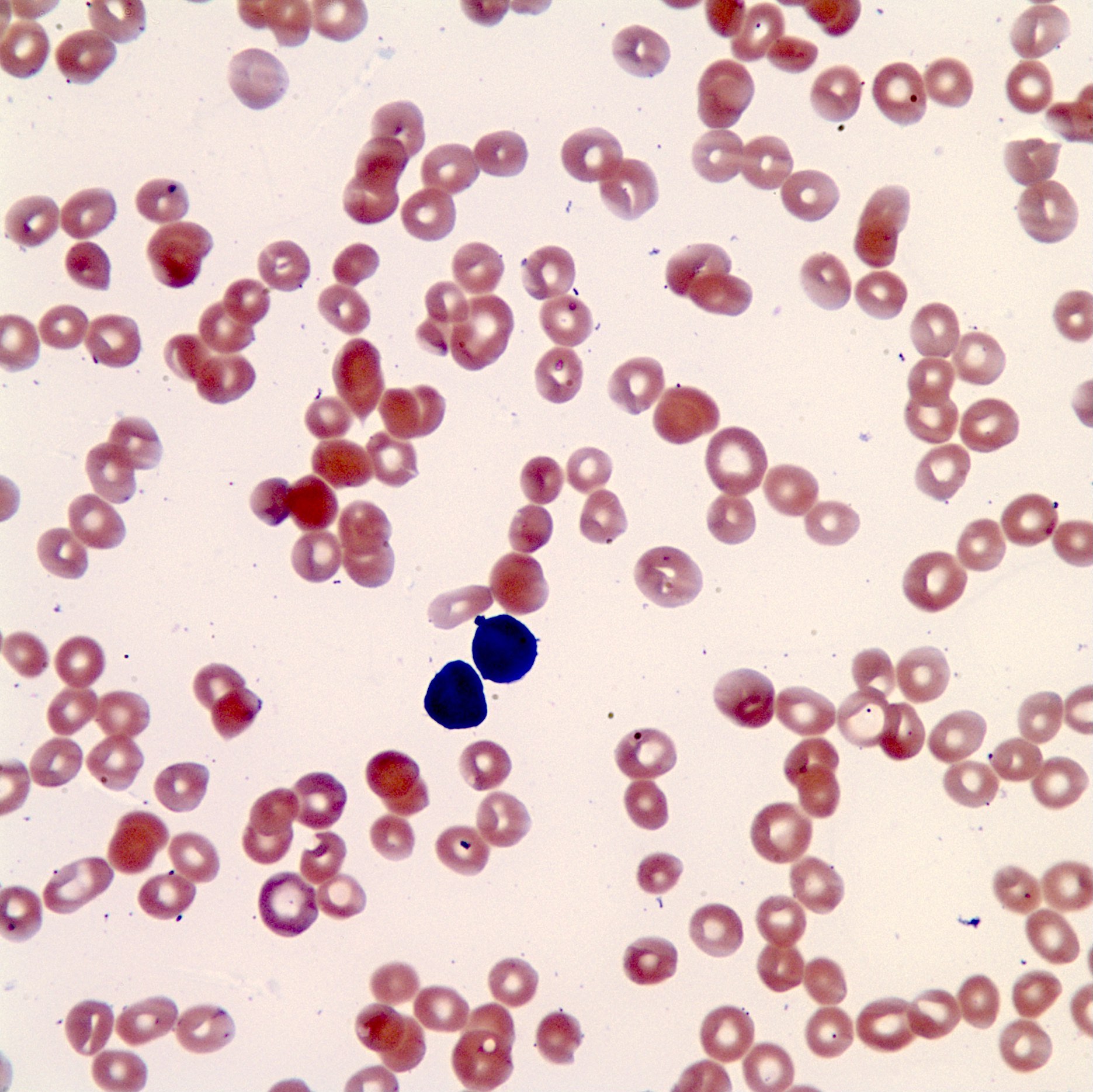
Lymphoblasts
- Gene expression profiling is the gold standard for diagnosis of B ALL with BCR-ABL1-like / Ph-like (Lancet Oncol 2009;10:125, N Engl J Med 2009;360:470)
- Genetic alterations in B ALL with BCR-ABL1-like are harder to detect by standard genetic diagnostic approaches (Eur J Cancer 2017;82:203)
- Common gene rearrangements include CRLF2, EPOR and IGH
- Down syndrome patients uniquely show CRLF2 translocation
- ABL1 oncogene translocation with ABL2, PDGFRB, NTRK3, TYK2, CSF1R and JAK2 have all been reported
- Many cases of B ALL with BCR-ABL1-like may additionally show other deletions or mutations that have a clear role in leukemogenesis such as IKZFA and CDKN2A / B
- FISH and karyotyping are helpful in ruling out other B ALL entities, especially ALL with recurrent genetic abnormalities
- Multiplex FISH testing can be useful in ruling out B ALL with recurrent genetic abnormalities
- Identifying patients with B ALL with BCR-ABL1-like can influence patients' prognosis and management of the patient
- PDGFRB translocation: may benefit from tyrosine kinase inhibitors
- JAK translocations: may benefit from JAK inhibitors
- B lymphoblastic leukemia / lymphoma, NOS: diagnosis should only be rendered after the exclusion of all other entities
- Burkitt lymphoma / leukemia
- B lymphoblastic leukemia / lymphoma with recurrent genetic abnormalities: must perform genetic testing including karyotyping, FISH or gene expression array; the entities in this group include:
- B ALL / LBL with t(9;22)(q34;q11.2); BCR-ABL1
- B ALL / LBL with t(v;11q23.3); KMT2A rearrangement
- B lymphoblastic leukemia / lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1
- B lymphoblastic leukemia / lymphoma with hyperdiploidy
- B lymphoblastic leukemia / lymphoma with hypodiploidy
- B lymphoblastic leukemia / lymphoma with t(5;14)(q31.1;q32.3); IL3-IGH
- B lymphoblastic leukemia / lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1
- Flow cytometry
- Gene expression profiling
- Karyotyping
- Multiplex FISH
Comment Here
Reference: BCR-ABL1-like
- NCI high risk features
- NCI intermediate risk features
- NCI low risk features
- NCI risk features is not applicable
Comment Here
Reference: BCR-ABL1-like
- Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a highly aggressive hematologic malignancy derived from plasmacytoid dendritic cells (pDC) (Curr Hematol Malig Rep 2018;13:477)
- Nonneoplastic counterpart: plasmacytoid dendritic cells (Adv Anat Pathol 2009;16:392)
- Over 80% of patients present with skin lesions; other common sites include the bone marrow and lymph nodes
- Expression of CD123 and TCF4 is required for diagnosis; in addition, most cases are positive for CD4, CD56 and TCL1
- Distinction from acute myeloid leukemia (AML) in particular is critical for appropriate therapy selection
- Blastic plasmacytoid dendritic cell neoplasm
- Agranular CD4+ / CD56+ hematodermic neoplasm / tumor
- Agranular CD4+ NK cell leukemia
- CD56+ TdT+ blastic NK cell lymphoma
- ICD-O: 9727/3 - blastic plasmacytoid dendritic cell neoplasm
- Rare, representing < 1% of hematologic malignancies (Leuk Res 2018;73:21)
- Incidence is 0.04 cases per 100,000 in U.S.
- M:F = 3:1
- Most patients are in fifth or sixth decades of life but the disease can affect any age group, including pediatric
- Skin, bone marrow, lymph nodes and spleen are most common but any anatomic site can be involved
- CSF involvement at presentation in up to 10% of patients, more at relapse (Oncotarget 2016;7:10174)
- Postulated cell of origin is pDC, which plays an important role in bridging the innate and adaptive immune systems (Curr Hematol Malig Rep 2018;13:477)
- Monoallelic and biallelic 12p / ETV6 deletions seem to represent an early pathogenic event (Leuk Res 2018;73:86)
- BPDCN specific transcriptional network regulated by the E-box transcription factor TCF4 (also known as E2-2) that plays a master regulatory role in BPDCN cells and appears to be in turn regulated by the bromodomain and extraterminal domain (BET) protein BRD4 (Cancer Cell 2016;30:764)
- Epigenetic dysregulation is very common and results from mutations in genes involved in DNA methylation, histone methylation and chromatin remodeling (Haematologica 2019;104:729)
- Frequent NR3C1 haploinsufficiency linked to aberrant overexpression of a novel long noncoding RNA (lncRNA) gene, lincRNA-3q, whose product is involved in G1/S cell cycle transition through E2F (Blood 2016;127:3040)
- BPDCN cells are highly dependent on BCL2 for survival (Cancer Discov 2017;7:156)
- Neoplastic cells in BPDCN have features of nonactivation state and seem to arise in a background of immunodeficiency (Blood Cancer J 2019;9:99)
- No known etiology
- Frequent association with other myeloid malignancies, including chronic myelomonocytic leukemia and myelodysplastic syndrome
- No bacterial or viral pathogen (Blood Adv 2020;4:1006)
Contributed by Joseph Khoury, M.D.
Markers expressed by pDCs
- Skin lesions, with heterogeneous manifestations of bruise-like, violaceous maculopapular lesions, nodules, plaques or exanthema
- ~10% of patients have an overt leukemic presentation (Am J Hematol 2013;88:1055)
- B symptoms
- Lymphadenopathy
- Splenomegaly
- Malignant infiltrate of blastoid cells; need to distinguish from mature pDC proliferations (MPDCP)
- Confirmation of pDC immunophenotype by flow cytometry or immunohistochemistry
- Cytogenetic and molecular studies usually not required for diagnosis
- Cytopenias are common but some patients have normal CBC findings
- No known pathologic or molecular prognostic factors
- 62 year old woman with BPDCN exhibiting unusual lymphoid features and macrovacuoles (Ann Hematol 2019;98:2221)
- 63 year old man with disseminated blastic plasmacytoid dendritic cell neoplasm (Blood 2015;126:558)
- Tagraxofusp, a CD123 targeted immunotoxin, is the first FDA approved treatment for BPDCN (N Engl J Med 2019;380:1628)
- ALL type induction chemotherapy regimens are associated with better outcomes compared with AML type regimens
- Allogeneic stem cell transplant is recommended for patients who achieve first complete remission and are fit (Hematol Oncol Clin North Am 2020;34:621)
- Diffuse infiltrate seen on bone marrow biopsy (trephine) and clot preparation with replacement of hematopoietic elements
- On Wright-Giemsa preparations, the neoplastic cells are elongated with a tapered lightly basophilic agranular cytoplasm that ends with a tail shaped structure; the nucleus has blastoid features with open chromatin and a prominent nucleolus
- In some cases, neoplastic cells have cytoplasmic microvacuoles with a peculiar arrangement along the cytosolic aspect of the cell membrane resembling a pearl necklace
- Skin involvement is characterized by infiltrates of immature neoplastic cells with blastoid morphology involving the superficial and deep dermis, often with extension into the subcutis; the epidermis is typically spared; mitotic figures are usually identified readily
Contributed by Joseph Khoury, M.D.
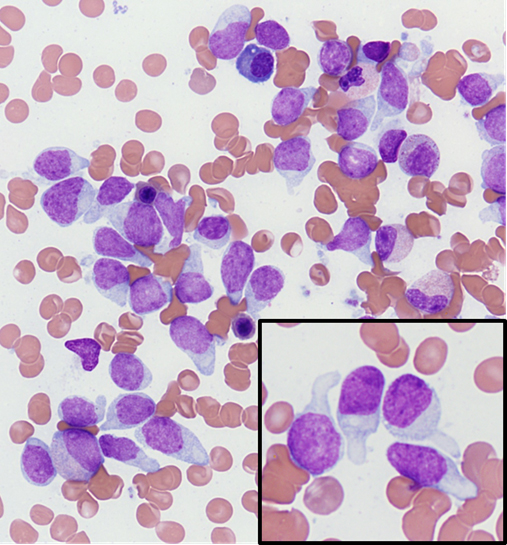
Cytomorphology of BPDCN cells

Bone marrow involvement by BPDCN
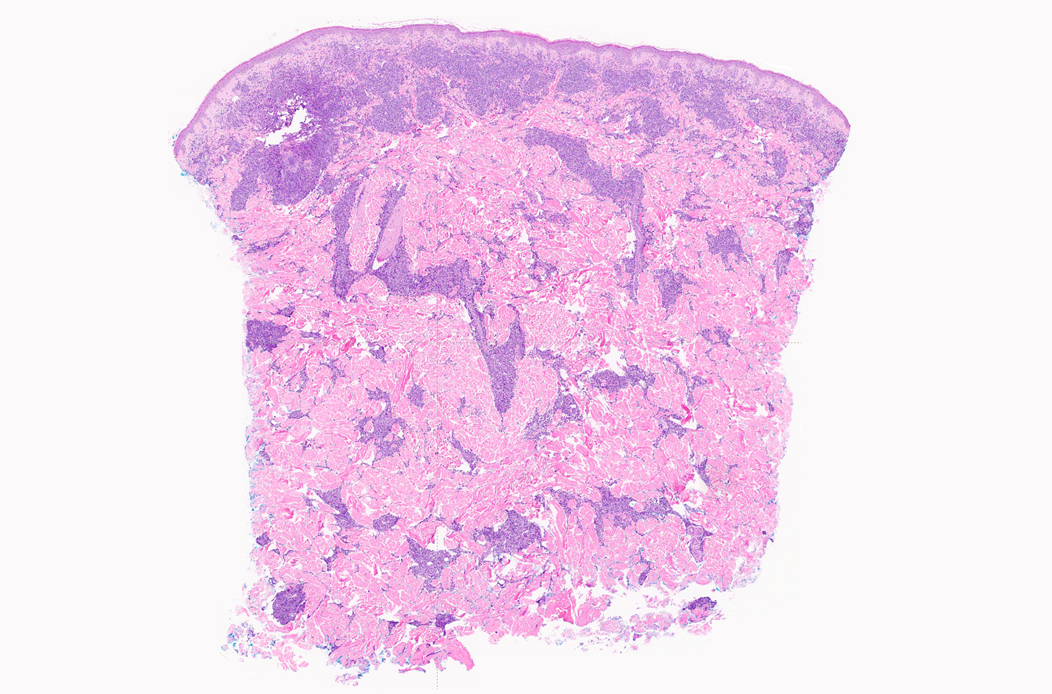
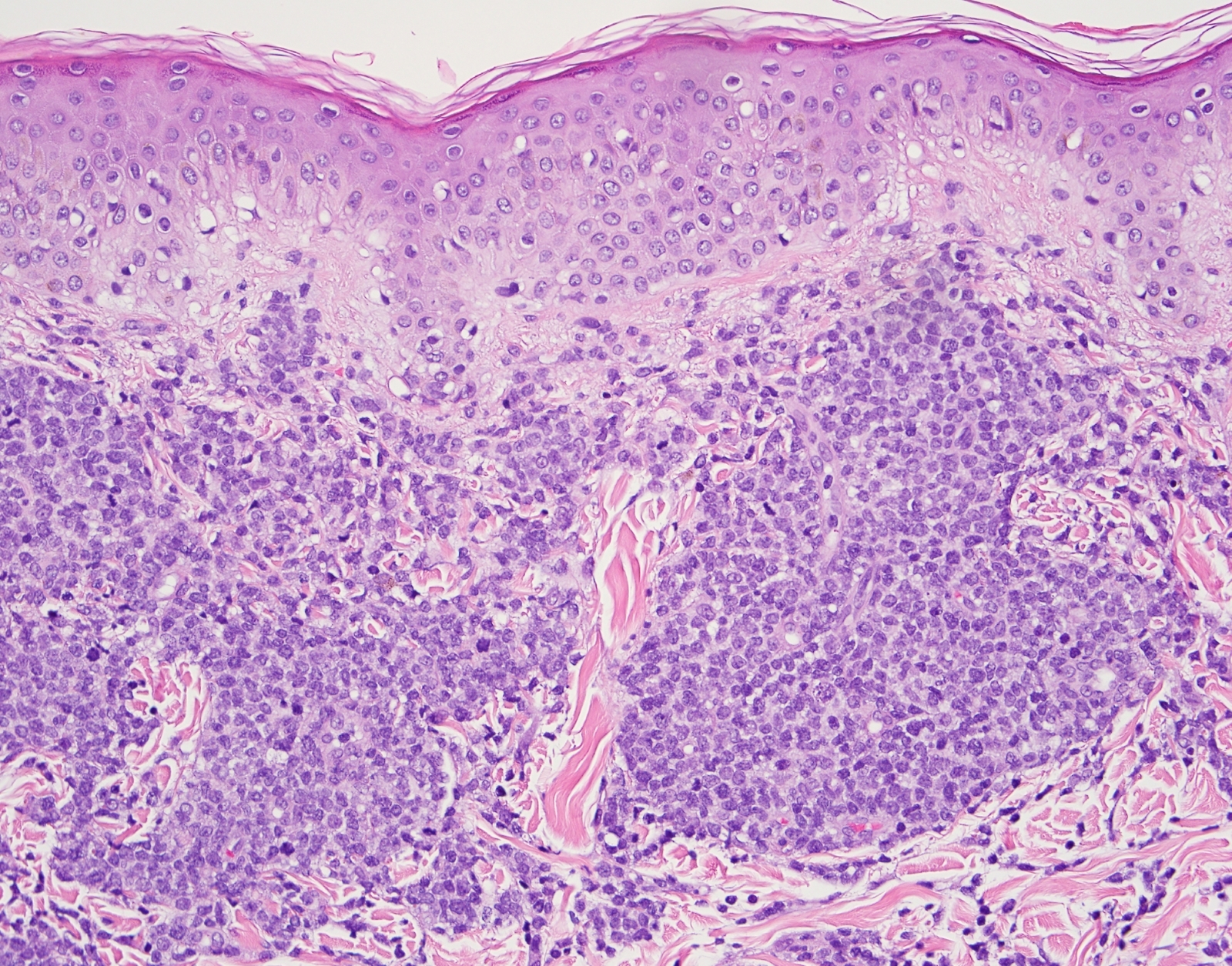
Skin involvement by BPDCN
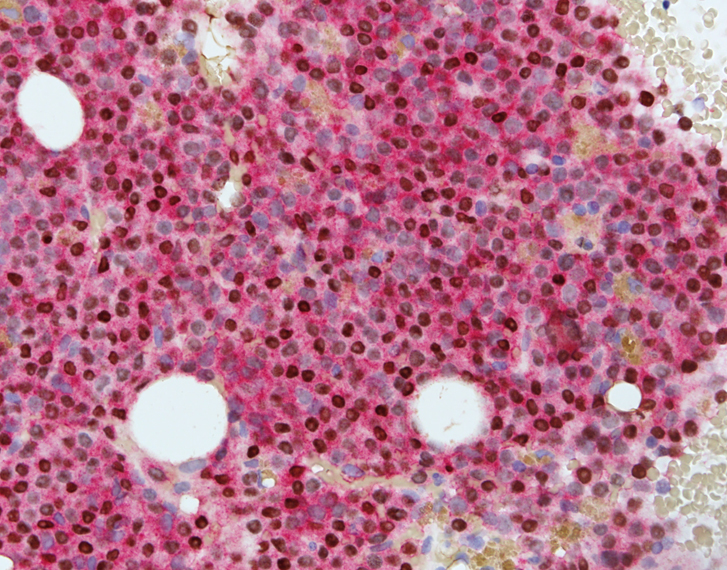
TCF4 / CD123 dual color stain in bone marrow
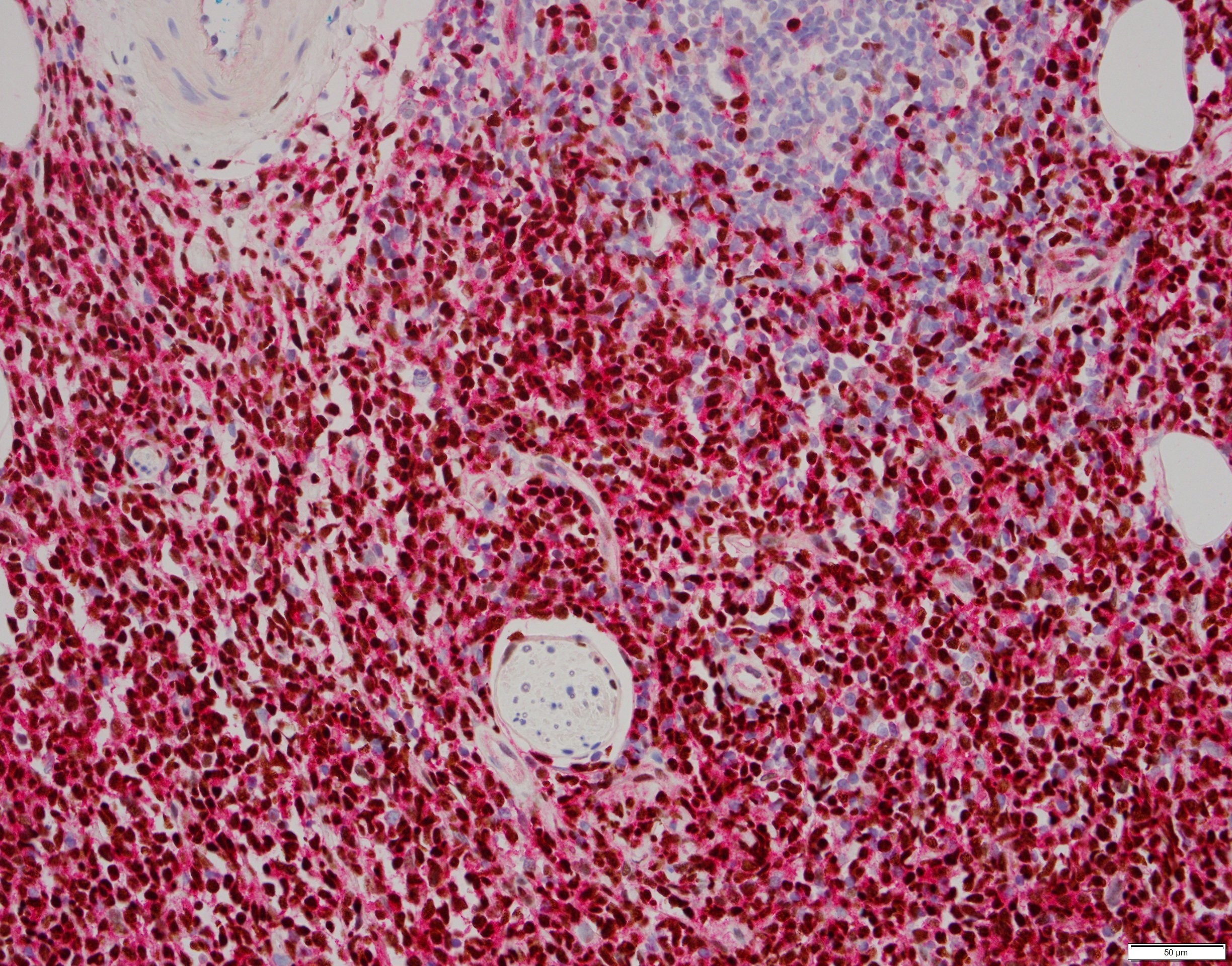
TCF4/CD123 dual color stain in skin
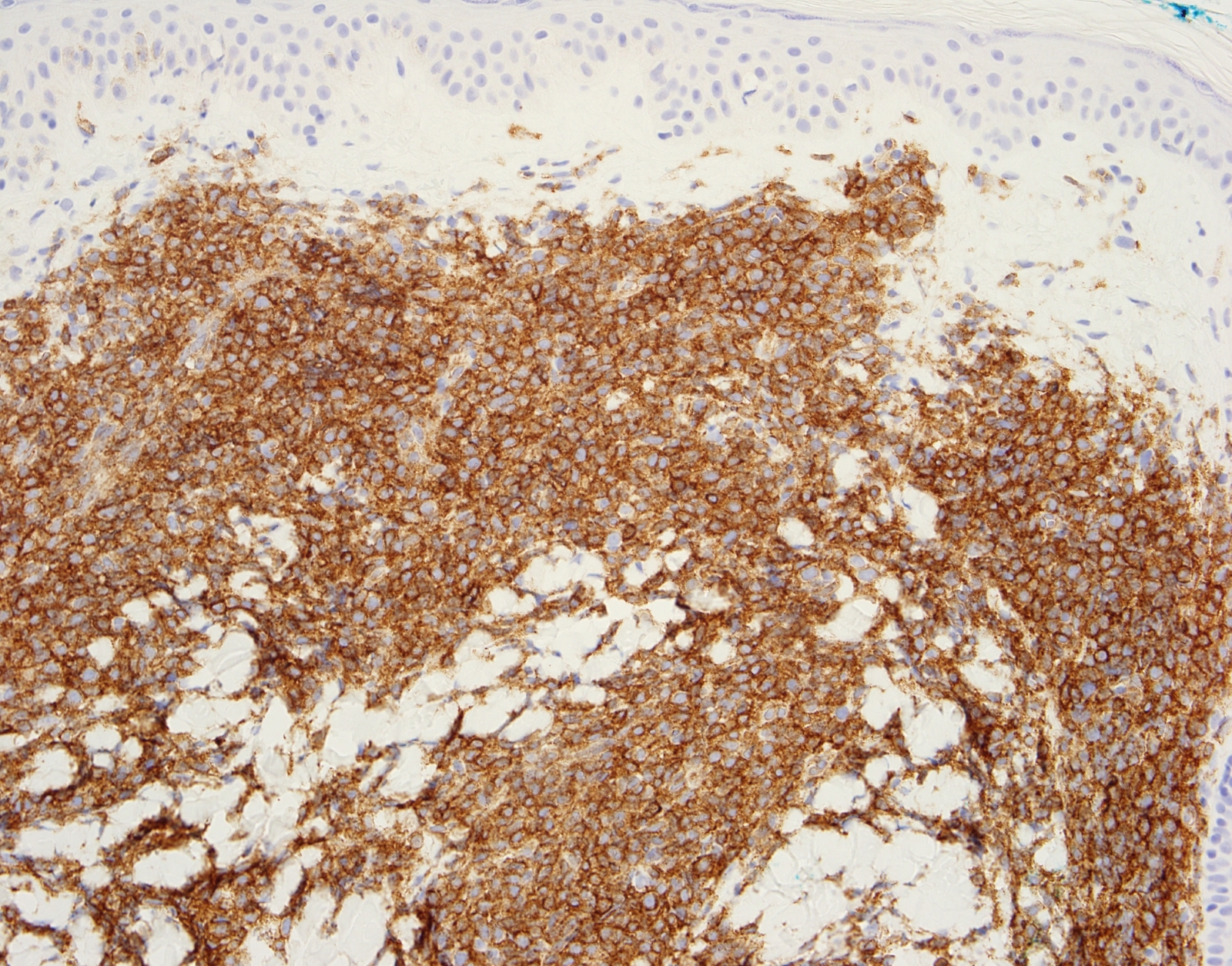
CD123
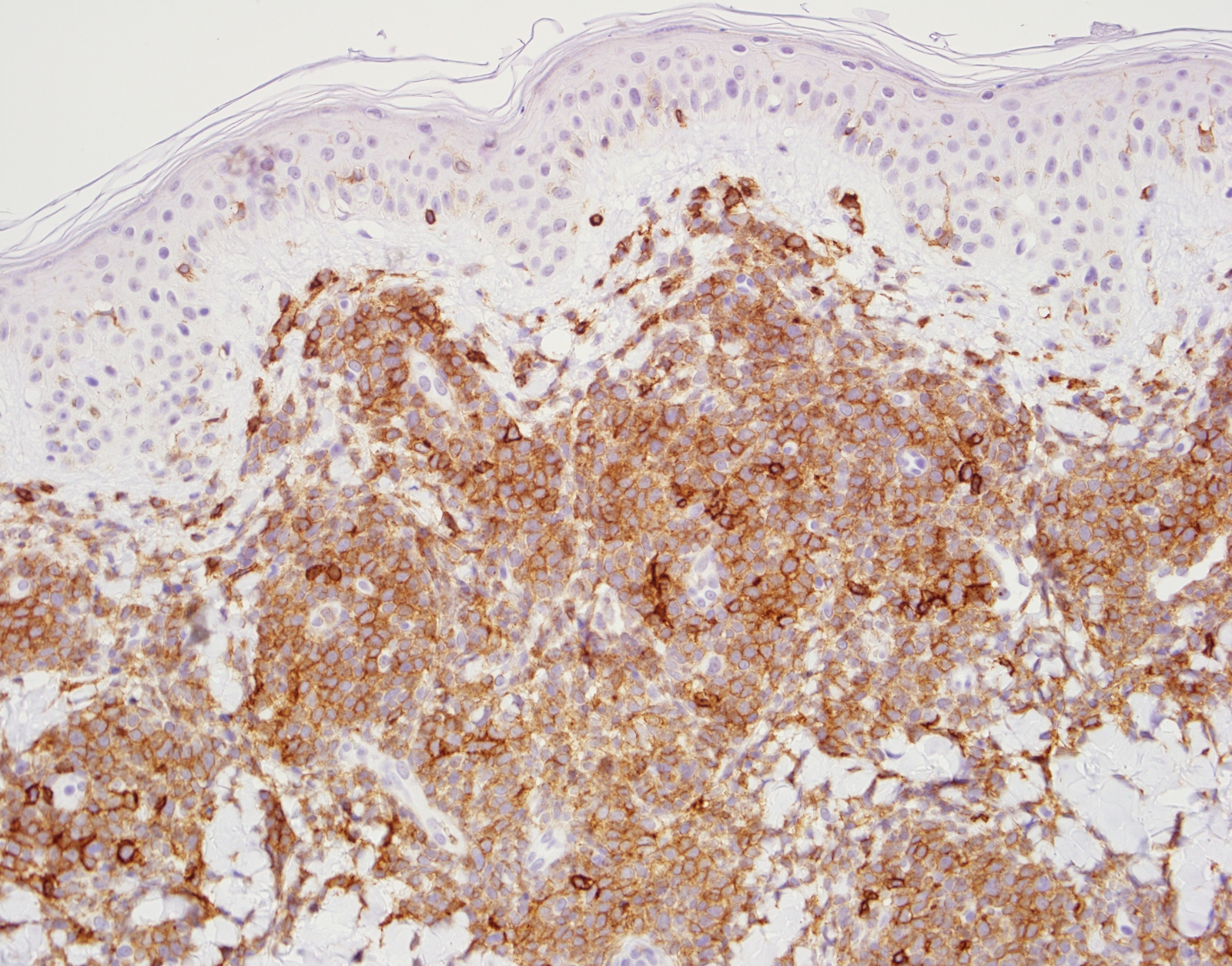
CD4

CD56
- Lineage specific markers, including CD3, CD19, CD20, PAX5, lysozyme, myeloperoxidase
- CD34
- MNDA (Am J Surg Pathol 2016;40:502)
- 3 primary diagnostic aims (Haematologica 2020 Apr 2 [Epub ahead of print]):
- Distinguish between BPDCN and other types of acute leukemia, mainly AML
- Detect mature pDC proliferations in the context of other myeloid neoplasms, primarily chronic myelomonocytic leukemia
- Detection of measurable / minimal residual disease (MRD)
- Neoplastic cells fall into the blast gate on CD45 / SSC
- Detection of MRD requires careful distinction between BPDCN cells and nonneoplastic pDCs
Contributed by Joseph Khoury, M.D.
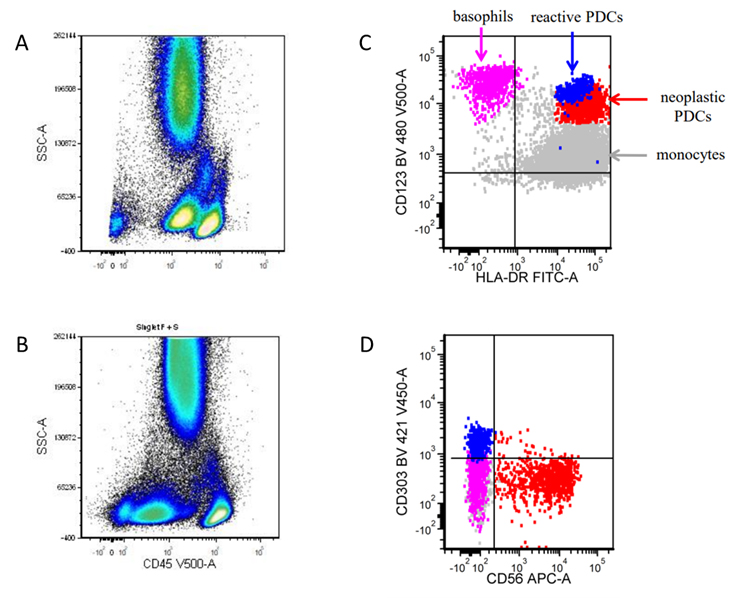
Flow cytometry findings in BPDCN
- Complex karyotype is common
- Many adult patients have MDS associated chromosomal abnormalities and gene mutational profile
- Gene rearrangements detected commonly: ETV6, MYC
- Structural chromosomal losses, commonly involving 13q14 (RB1), 9p21.3 (CDKN2A/CDKN2B), 12p13.2-p13.1 (CDKN1B, ETV6), 7p12.2 (IKZF1)
- Mutations detected commonly: TET2, ASXL1, IDH1, IDH2, DNMT3A, EZH2, SF3B1, SRSF2, ZRSR2, FLT3, KIT, KRAS, NRAS
- Bone marrow, aspiration, clot and core biopsy:
- Diagnosis: blastic plasmacytoid dendritic cell neoplasm (see comment)
- Comment:
- Immunohistochemistry: positive for TCF4 / CD123 coexpression
- Flow cytometry: large population of neoplastic cells, positive for CD123, CD4, CD56, CD303 (decreased), HLA-DR and TdT; negative for CD3 (surface and cytoplasmic), CD19 and myeloperoxidase
- Karyotype: 48, XY, t(6;8)(p12;q24.2), add(7)(p11.2), del(13)(q12q22), +16, add(20)(q11.2), +21[10]/46, XY[10]
- Mutation profile: ASXL1, TET2, NRAS
- Acute myeloid leukemia / myeloid sarcoma:
- Mature plasmacytoid dendritic cell proliferation:
- B lymphoblastic lymphoma:
- T lymphoblastic lymphoma:
- Neuroendocrine tumor (including Merkel cell carcinoma):
- Chromogranin+, synaptophysin+, cytokeratin 20+, TCF4 / CD123- dual color
- CD56
- CD123
- CD3
- CD4
- CD45
Comment Here
Reference: Blastic plasmacytoid dendritic cell neoplasm
- CD56 and CD4
- CD123 and TCF4
- CD123 and CD19
- TCF4 and MPO
- TCL1 and CD3
Comment Here
Reference: Blastic plasmacytoid dendritic cell neoplasm
- Chronic eosinophilic leukemia (CEL) is an autonomous, clonal proliferation of eosinophils that leads to a persistent increase in eosinophils in peripheral blood, bone marrow and sometimes tissue
- Absolute eosinophil count is ≥ 1.5 x 109/L on at least 2 occasions for 4 weeks (notable change from 6 months in the 4th edition of WHO)
- Bone marrow shows abnormal morphology (e.g., dysmegakaryopoiesis, eosinophilic infiltrate associated overt fibrosis)
- Clonal
- Clonal myeloid or lymphoid neoplasms and reactive causes of eosinophilia should be excluded
- Rare myeloproliferative neoplasm characterized by a gradual increase in circulating eosinophils with cytologic atypia, left shifted granulocytic maturation and often hepatomegaly or splenomegaly
- Organ / tissue infiltration by eosinophils and eosinophilic microabscesses are frequently present, leading to tissue damage and organ dysfunction
- Clinical course is variable from indolent to life threatening; transformation to acute myeloid leukemia can occur and prognosis is generally poor
- Fatal cardiac damage has been documented secondary to the release of cytokines produced by eosinophils
- Diagnosis should be made after exclusion of reactive causes of eosinophilia and myeloid and lymphoid neoplasms associated with PDGFRA, PDGFRB, FGFR1, FLT3 or JAK2 rearrangements with evidence of clonality (see updated full WHO criteria) (Am J Hematol 2017;92:1243)
- Cell of origin: pluripotent hematopoietic stem cell
- Due to difficulty in distinguishing CEL from nonclonal hypereosinophilic syndrome, the true incidence is unknown
- A recent study indicates that only 1.2% of cases with peripheral eosinophilia met criteria for CEL (Am J Hematol 2020;95:E172)
- The Surveillance, Epidemiology and End Results (SEER) database from 2004 - 2015 estimates the incidence of hypereosinophilic syndrome (HES) including CEL, not otherwise specified (NOS) is 0.4 cases per 1,000,000 (Am J Hematol 2022;97:129)
- Some reports show a predilection for men in the seventh decade (Mod Pathol 2016;29:854, Am J Hematol 2012;87:643)
- Peripheral blood and bone marrow are always involved
- Splenic and hepatic involvement are also common
- Other frequent sites of involvement include the heart, lungs, central nervous system (CNS), skin and gastrointestinal (GI) tract
- Natural history varies considerably between individuals
- Patients may be asymptomatic with longstanding eosinophilia (2 episodes over a 4 week interval), without specific etiology (i.e., no allergy, asthma, drug reaction, parasitic infection or connective disease)
- Absolute eosinophil count can vary from 1.5 to 400 x 109/L
- Eosinophils account for 10% of white blood cells (WBCs)
- Can have leukocytosis (20 - 30 x 109/L), anemia, thrombocytopenia or thrombocytosis
- Symptoms: weight loss, night sweats, fever (12%), fatigue (26%), myalgias or angioedema (14%), cough (24%), dyspnea (16%), rhinitis (10%), pruritus and diarrhea
- Skin manifestations are the most common, followed by lung (44%) and GI tract (38%)
- Cardiovascular
- Endomyocarditis, endomyocardial fibrosis followed by restrictive cardiomegaly
- Hypertension, atherosclerosis and heart failure
- Scarring of mitral and tricuspid valves leading to regurgitation, thrombi formation and embolization in end organs
- Peripheral neuropathy, CNS dysfunction and rheumatologic findings (Am J Hematol 2012;87:643)
- World Health Organization (WHO) classification (5th edition)
- Essential
- Persistent eosinophilia (≥ 1.5 x 109/L) on at least 2 occasions over a minimum of 4 weeks in which reactive and genetically defined causes have been excluded
- Evidence of clonality (see note 2)
- Abnormal bone marrow morphology
- Does not meet WHO diagnostic criteria for other myeloid or lymphoid neoplasms (see note 3)
- Blasts of < 20% in peripheral blood and bone marrow cells and does not have the following cytogenetic aberrations
- Tyrosine kinase gene fusions including PDGFRA, PDGFRB or FGFR1 rearrangements
- FLT3 rearrangements (Leukemia 2022;36:1703)
- ETV6::ABL1 fusion (Leukemia 2022;36:1703)
- JAK2 rearrangements: t(8;9)(p22;p24.1) / PCM1::JAK2, t(9;12)(p24.1;p13.2) / ETV6::JAK2 or t(9;22)(p24.1;q11.2) / BCR::JAK2 fusions
- AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) / CBFB::MYH11; t(15;17)(q22;q11-12) / PML::RARA or t(8;21)(q22;q22.1) / RUNX1::RUNX1T1
- CML with t(9;22)(q24;q31) / BCR::ABL1
- Blasts of < 20% in peripheral blood and bone marrow cells and does not have the following cytogenetic aberrations
- Notes
- Note 1
- Note 2: possibility of clonal hematopoiesis of indeterminate potential (CHIP) should be considered
- Certain gene mutations (e.g., DNMT3A, TET2, ASXL1 with variant allele frequency ≥ 2%) can be seen in a subpopulation of elderly patients who do not have hematologic disorders, otherwise known as CHIP
- Diagnostic interpretation should be done with caution (N Engl J Med 2014;371:2477, Nat Med 2014;20:1472)
- Mutations in other genes with variant allele frequency (VAF) > 10% in combination with other mutations is a better indicator of clonality
- Note 3: including myeloproliferative neoplasms (MPN), myelodysplastic syndrome (MDS), MDS / MPN, myeloid / lymphoid neoplasms with eosinophilia (MLN-eo), mastocytosis and acute myeloid leukemia (AML)
- Essential
- International Consensus Classification (ICC) criteria; requires all 6 criteria
- Hypereosinophilia (≥ 1.5 x 109/L) with eosinophils comprising ≥ 10% of white blood cells
- Does not meet criteria for AML; blasts < 20% in the peripheral blood and bone marrow, excluding AML with recurrent genetic abnormalities
- AML with recurrent genetic aberrations with < 20% is also excluded
- No tyrosine kinase fusion genes including BCR::ABL1, other ABL, PDGFRA, PDGFRB, FGFR1, JAK2 or FLT3 fusions
- Does not meet criteria for MPN, chronic myelomonocytic leukemia (CMML) or systemic mastocytosis (SM)
- Of note, although eosinophilia can be seen with SM, true CEL, NOS may also occur as SM with an associated myeloid neoplasm (Blood 2022;140:1200)
- Marrow shows increased cellularity with dysplastic megakaryocytes with or without dysplasia in other cell lineages; often with significant myelofibrosis, associated eosinophilic infiltrate or increased blasts ≥ 5% in bone marrow or ≥ 2% in peripheral blood
- Clonal cytogenetic abnormality or somatic mutation identified
- If no clonality / somatic mutation demonstrated or no increased blasts, persistent eosinophilia may suffice provided the bone marrow findings are supportive and other causes of eosinophilia have been ruled out
- WHO classification (5th edition)
- Variable survival; median survival is 22.2 months secondary to infection, bleeding and age related comorbidity
- Transformation to acute leukemia can occur
- Response to imatinib therapy is uncommon except for some CEL with PDGFRA mutations (Am J Hematol 2012;87:643, Blood 2011;117:2935)
- STAT5B p. N642H with or without SF3B1 mutations demonstrated better overall survival (Leukemia 2019;33:415)
- 4 cases showed somatic activating KIT M541L mutation that was responsive to low dose imatinib therapy (Oncotarget 2014;5:4665)
- Unfavorable prognostic findings include marked splenomegaly, bone marrow fibrosis, megakaryocytic atypia and thrombocytopenia (Am J Hematol 2020;95:E172)
- 52 year old man with t(5;12)(q31;p13) / ETV6::ACSL6 gene fusion, a novel variant of myeloid proliferative neoplasm with eosinophilia (Hum Pathol (N Y) 2016;5:6)
- 68 year old woman with liver infiltration resembling Budd-Chiari syndrome (Rinsho Ketsueki 2007;48:505)
- 72 year old man with autoimmune hemolytic anemia and erythrophagocytosis by eosinophils (Am J Hematol 2006;81:458)
- Bone marrow
- Hypercellular due to eosinophilic proliferation; however, maturation is orderly without disproportionate increase in myeloblasts
- Charcot-Leyden crystals often present
- Usually normal erythropoiesis and megakaryocytopoiesis; however, abnormal dysplastic megakaryocytes can be seen with or without dysplasia in other lineage (Blood 2022;140:1200)
- Subpopulation of patients (27%) show morphologic features resembling BCR::ABL1 negative neoplasms, myelodysplastic syndromes or myelodysplastic / myeloproliferative neoplasms (Mod Pathol 2016;29:854, Haematologica 2017;102:1352)
- Increased myeloblasts (commonly 5 - 19% in bone marrow)
- ~33% of cases show myelofibrosis; often significant fibrosis associated with eosinophilic infiltrate (Blood 2022;140:1200)
- Tissue
- Eosinophilic infiltration or microabscesses
- Charcot-Leyden crystals
- Fibrosis (caused by the degranulation and release of eosinophilic basic and cationic proteins)
Contributed by Lynh Nguyen, M.D. and AFIP

Numerous eosinophilic precursors
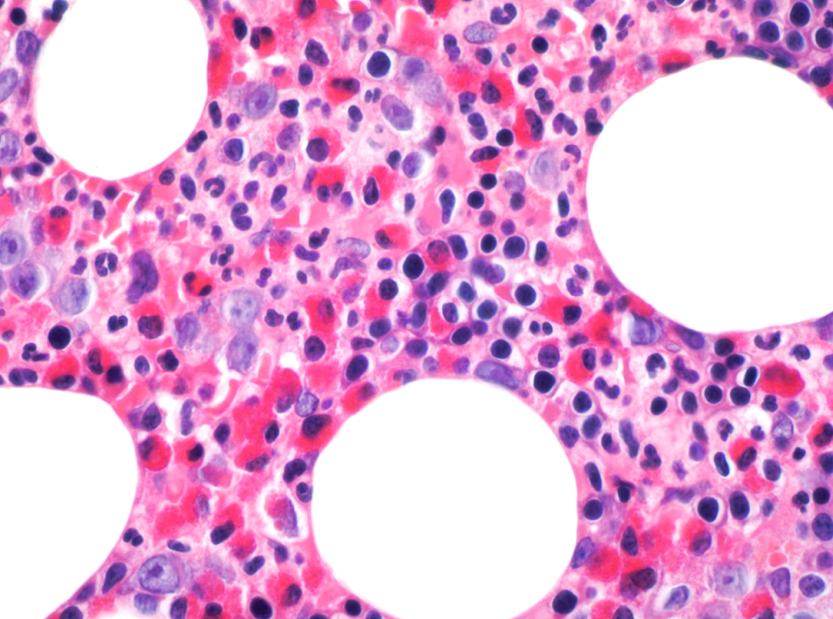
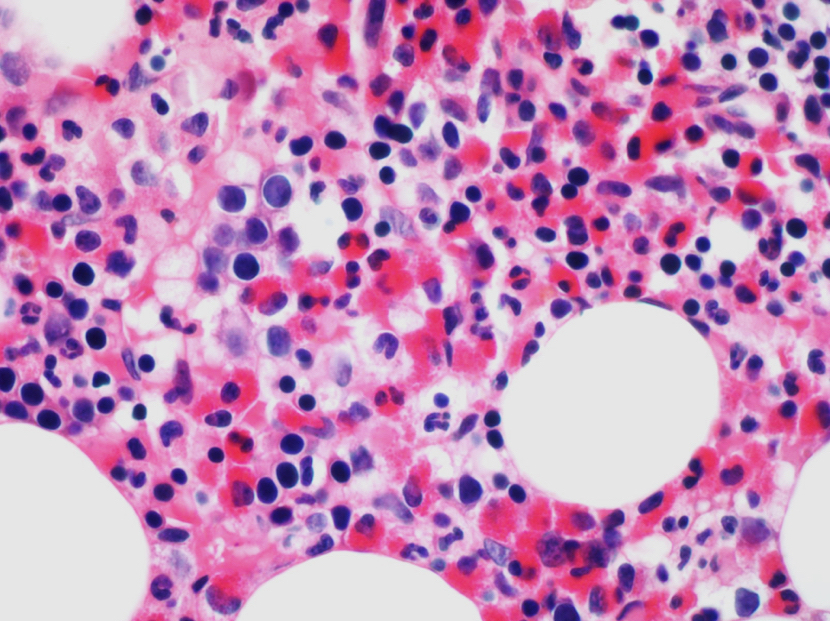
Myeloid preponderance

Marked hypercellularity
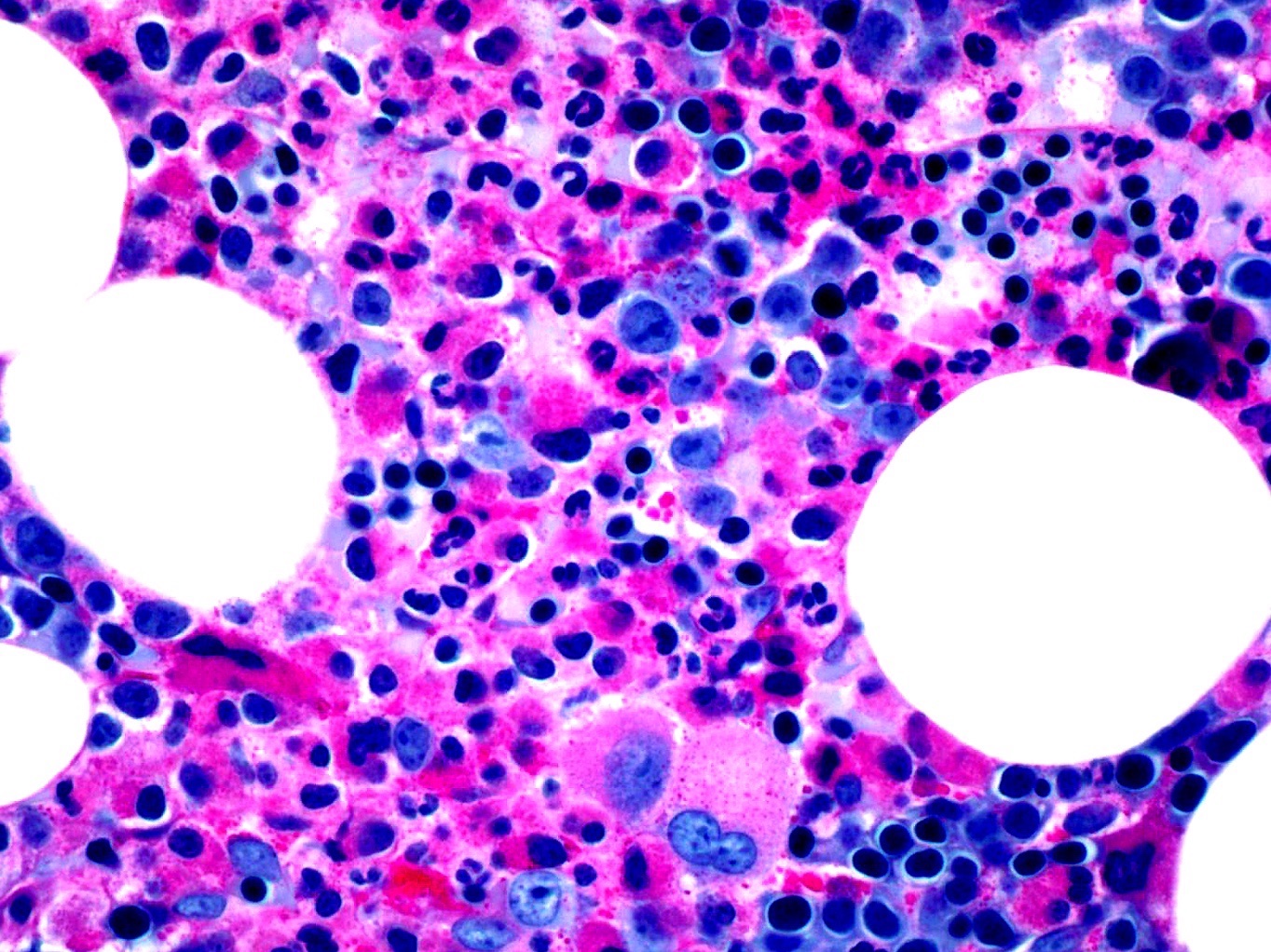
Granulocytic hyperplasia
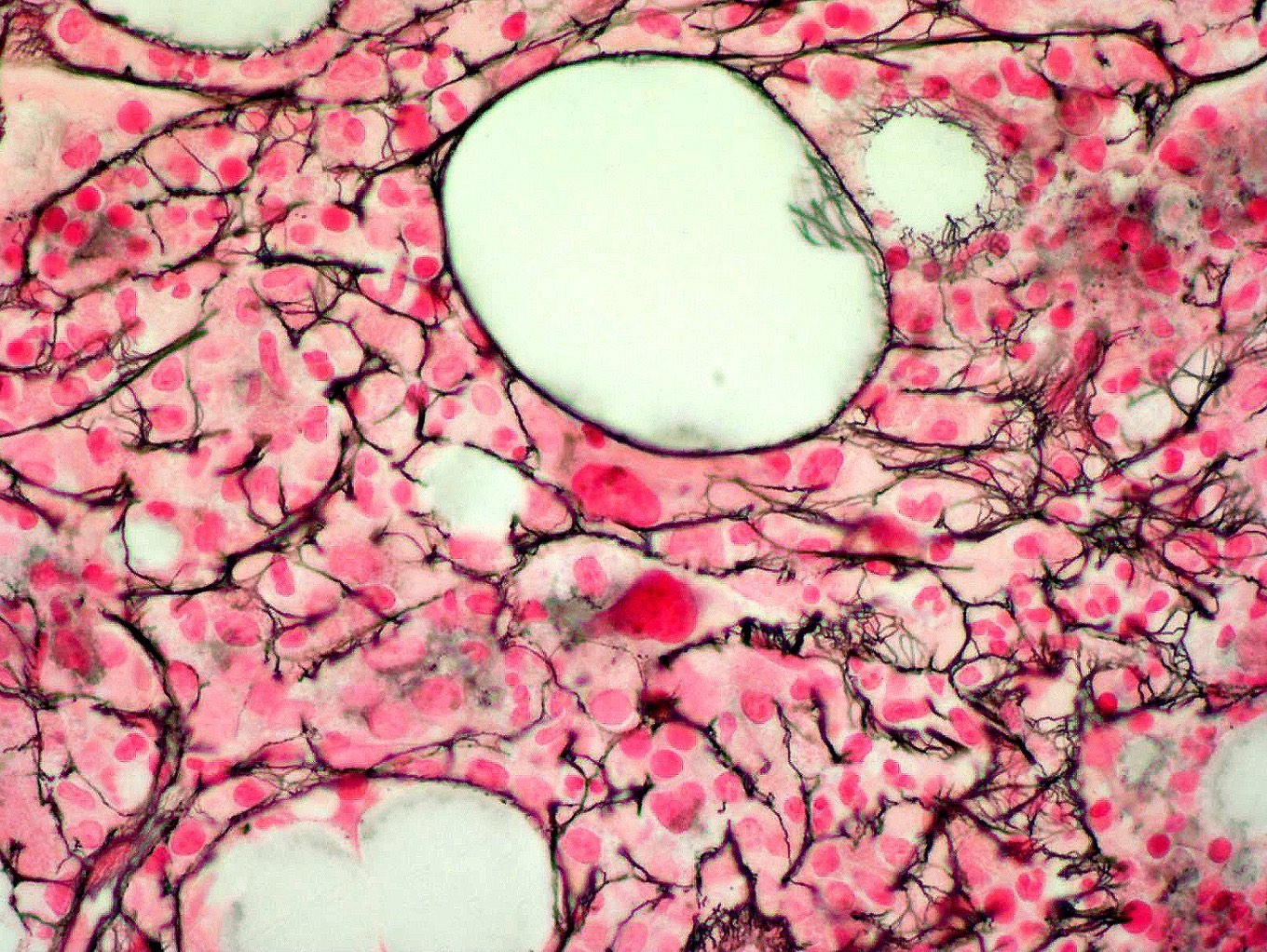
Reticulin fibrosis
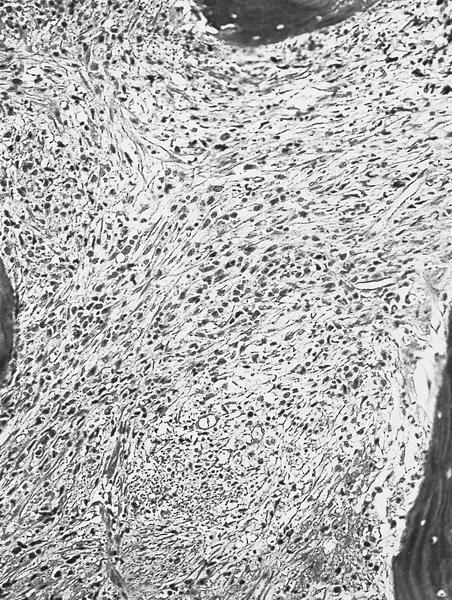
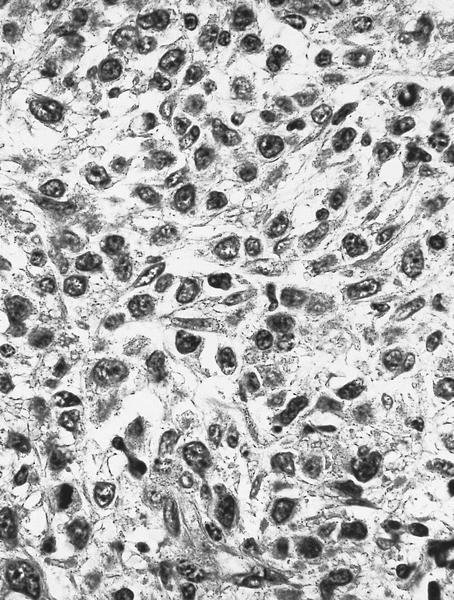
Markedly fibrotic marrow

Markedly hypercellular
- Peripheral blood
- Striking eosinophilia (≥ 1.5 x 109/L) mainly composed of mature eosinophils and occasional immature eosinophilic precursors
- Circulating blasts can be seen but comprise < 20%
- Spectrum of nonspecific eosinophil abnormalities: sparse granulation, cytoplasmic vacuolation, nuclear hyper / hyposegmentation or increased size
- Often accompanied by neutrophilia, some with mild monocytosis (> 1 x 109/L) and a few with mild basophilia
- Striking eosinophilia (≥ 1.5 x 109/L) mainly composed of mature eosinophils and occasional immature eosinophilic precursors
Contributed by Lynh Nguyen, M.D.
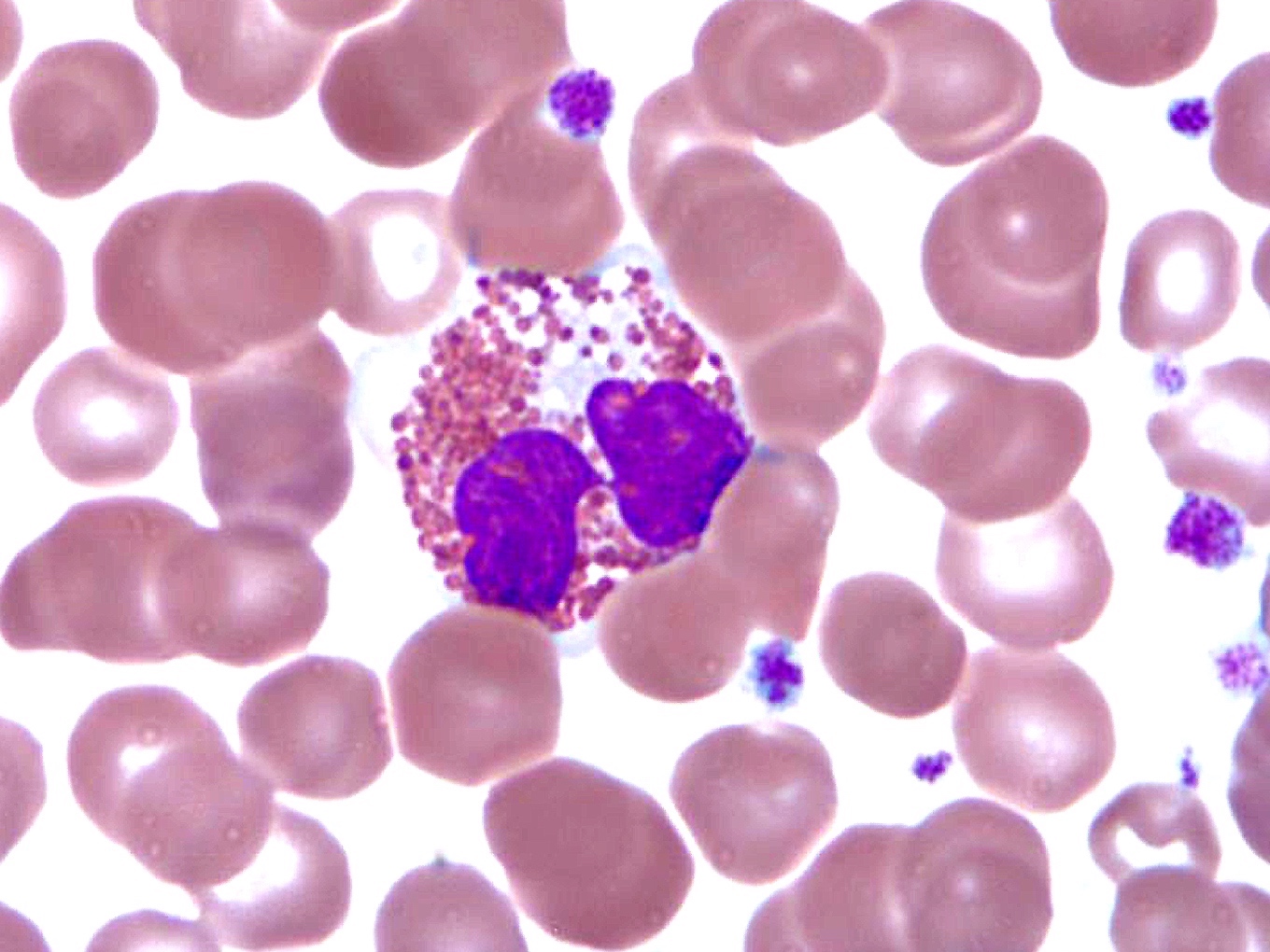
Moderately degranulated eosinophils
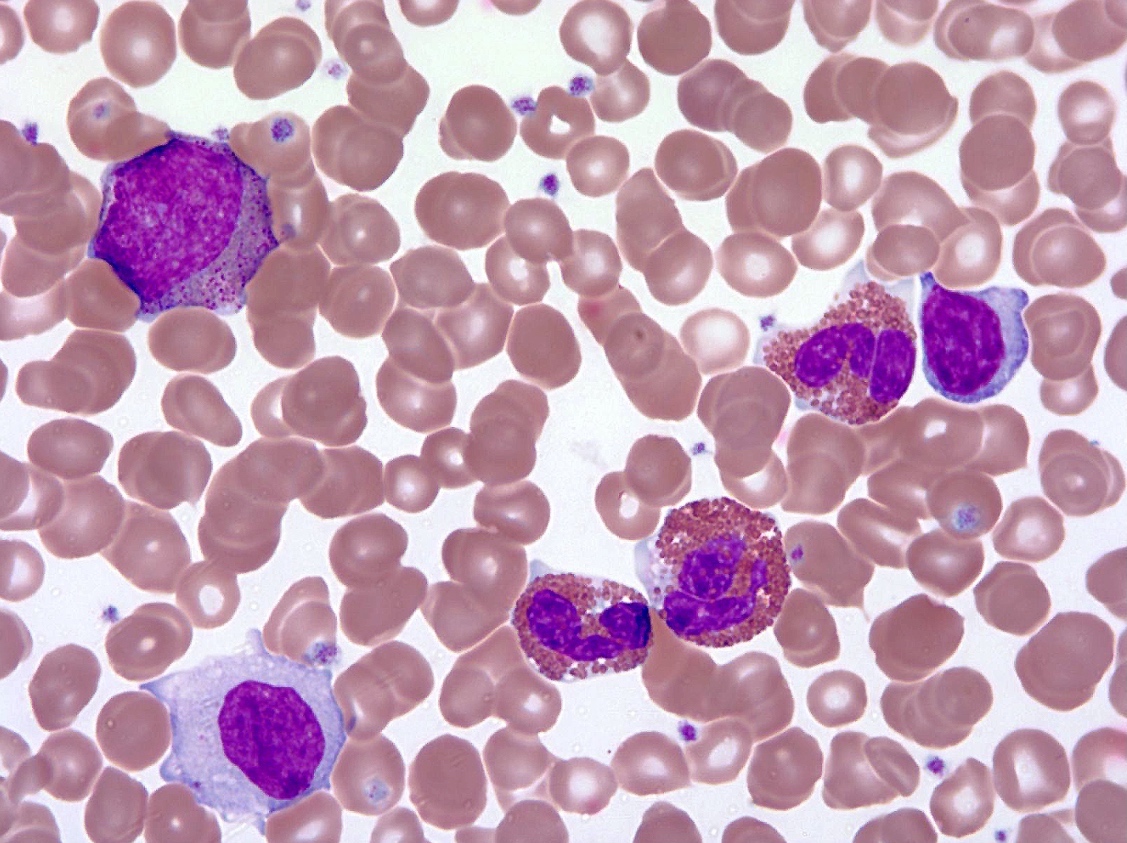
Patient with longstanding hypereosinophilia
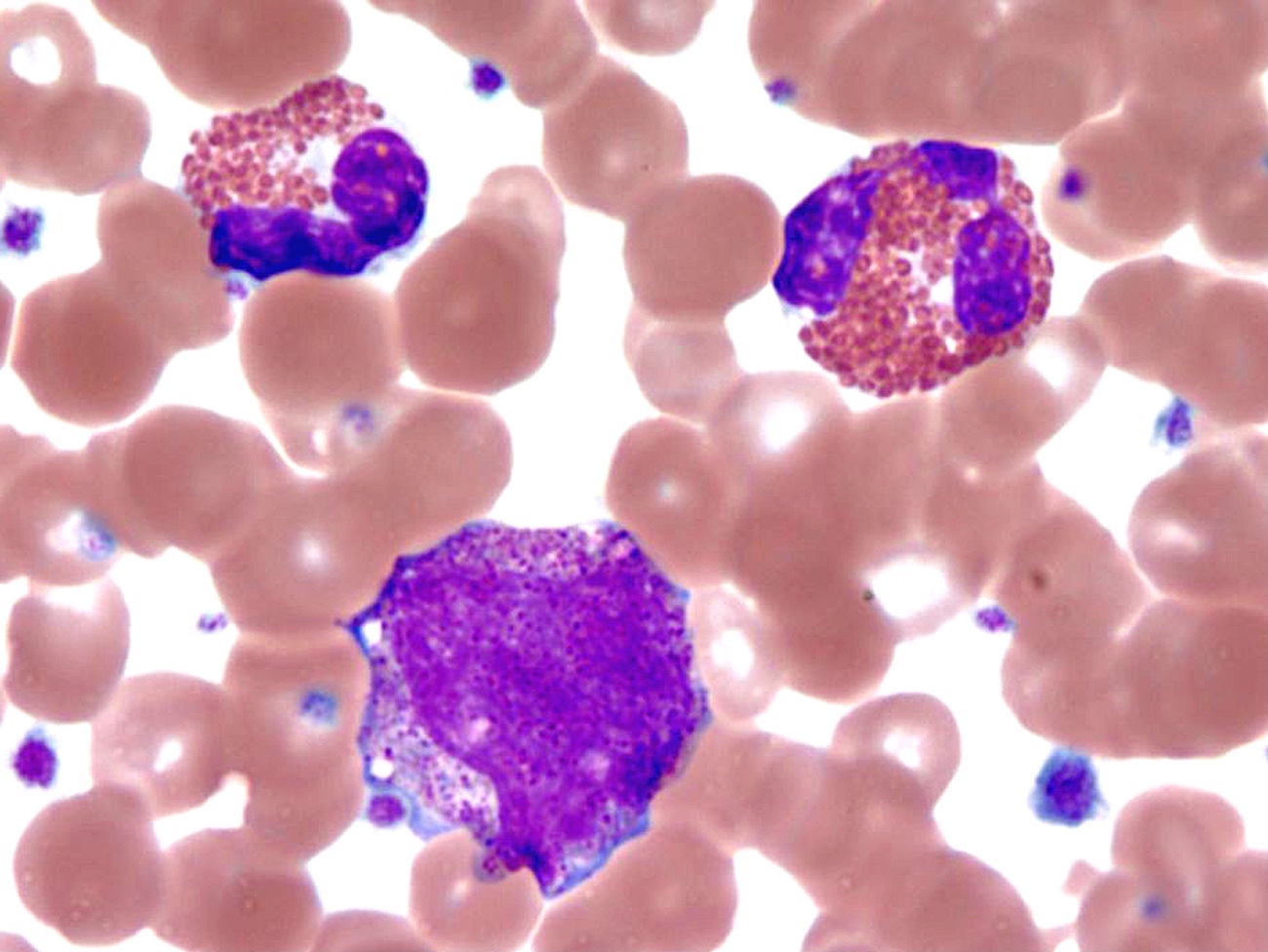
Circulating abnormal eosinophils
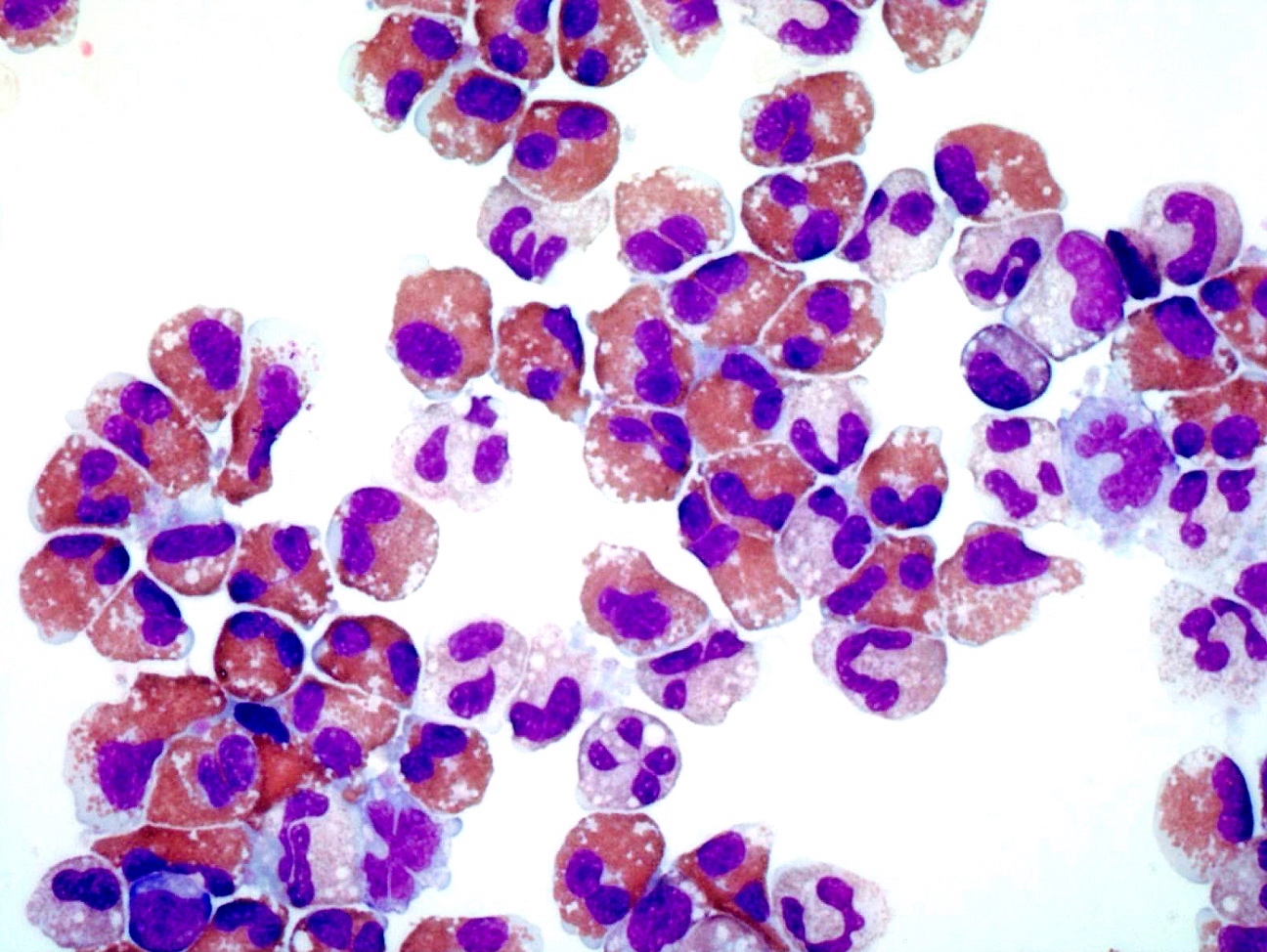
Markedly increased eosinophils
- No specific immunophenotype has been associated with CEL, NOS; however, immunophenotyping is important to exclude T lymphocyte driven eosinophilia or acute leukemia
AFIP images
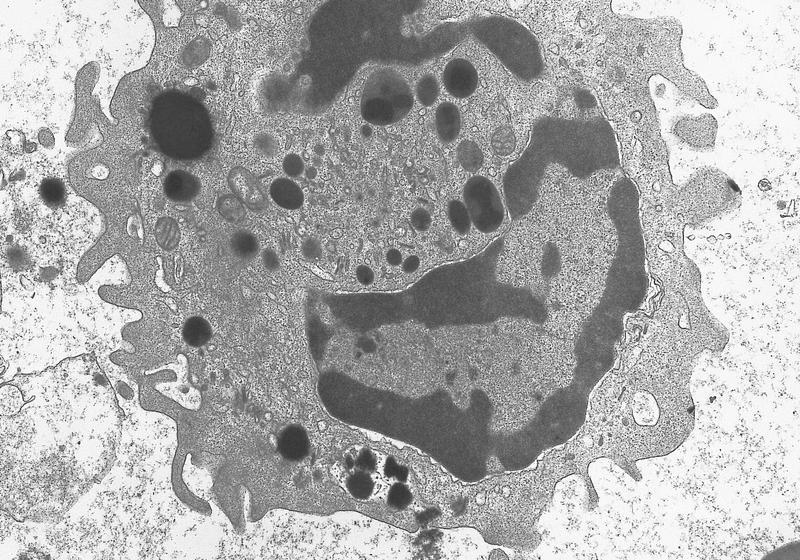
Abnormal eosinophil with decreased granules
- No specific cytogenetic or molecular genetic abnormality identified
- Must exclude neoplasms with rearrangements of BCR::ABL1, PDGFRA, PDGFRB, FGFR1, FLT3 or PCM1::JAK2, ETV6::JAK2 or BCR::JAK2 fusions
- Presence of recurrent translocations and karyotypic abnormalities that are seen in myeloid disorders [-8, -7, i(17q)] also support the diagnosis (Br J Haematol 1996;95:2, Br J Haematol 1986;62:659)
- Next generation sequencing (NGS) is helpful in establishing clonality
- Common mutations in ASXL1, TET2 and EZH2; occasional JAK2 mutation (Blood 2005;106:2162, Mod Pathol 2016;29:854)
- Mutations in TET2, ASXL1 and DNMT3A can be detected in the elderly population without neoplasms (clonal hematopoiesis of indeterminate potential) (N Engl J Med 2014;371:2477, Nat Med 2014;20:1472)
- Peripheral blood, peripheral blood smear:
- Mild normocytic anemia
- Leukocytosis with marked eosinophilia
- No circulating blasts identified
- Bone marrow, aspirate, clot section and biopsy:
- Markedly hypercellular marrow with myeloid preponderance, marked eosinophilic hyperplasia and scattered blasts (2%) (see comment)
- Moderate myelofibrosis (MF2)
- Comment: Examination of the peripheral blood and bone marrow aspirate smears show a marked increase in eosinophils with abnormal morphology including sparse granulation and nuclear hyper / hyposegmentation. There is no peripheral monocytosis or basophilia. Manual differential counts performed on the submitted bone marrow aspirate demonstrate approximately 2% myeloblasts. Megakaryocytes are present and slightly increased in number. Erythroid precursors show essentially normoblastic maturation. The M:E ratio is increased, estimated 5:1. The bone marrow core biopsy demonstrates normal bony trabeculae surrounded marrow space with overt myeloid predominance, increased eosinophils and their precursors. Occasional blasts are identified. Megakaryocytes are evenly distributed without syncytial clustering. Erythroid precursors are present and relatively low in proportion. Occasional small loose lymphoid aggregates composed of small mature forms are focally noted. No granulomata are evident. Flow cytometry performed on the bone marrow aspirate with adequate cells showed no immunophenotypic evidence of clonal B cell or aberrant T cell population. Rare CD34 positive blasts (1 - 2%) are identified.
- Karyotyping showed normal karyotype, 46,XX[20]. Fluorescence in situ hybridization is negative for BCR::ABL1, PDGFRA, PDGFRB or FGFR1 rearrangements. Next generation sequencing identified multiple mutations involving ASXL1, TET2, EZH2, SETBP1, CBL and NOTCH genes. According to WHO classification, the patient showed typical clinical presentation, along with morphologic and molecular findings supporting a diagnosis of chronic eosinophilic leukemia. Clinically, all other neoplastic and reactive causes of eosinophilia have been excluded.
- Markedly hypercellular marrow with myeloid preponderance, marked eosinophilic hyperplasia and scattered blasts (2%) (see comment)
- Acute myeloid leukemia with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22); CBFB::MYH11:
- Presence of inv(16)(p13.1;q22) or t(16;16)(p13.1;q22)
- Most cases show myelomonocytic differentiation and presence of abnormal eosinophils with immature basoeosinophilic granules in addition to increased myeloblasts
- Atypical chronic myeloid leukemia (aCML); BCR::ABL1 negative:
- Myelodysplastic / myeloproliferative neoplasm with neutrophilia according to the 5th edition of WHO
- Besides features of myeloproliferative neoplasms and eosinophilia, dysplasia is present in > 10% of neutrophilic precursors but there is no monocytosis
- Myeloproliferative chronic myelomonocytic leukemia (MP-CMML) (5th edition of WHO):
- Sustained monocytosis ≥ 1 x 109/L for > 6 months with bone marrow morphologic findings compatible with CMML
- Nonclonal hypereosinophilic syndrome:
- Diagnosis of exclusion given when
- Eosinophil count ≥ 1.5 x 109/L for ≥ 6 months
- Exclude reactive causes of eosinophilia
- Exclude acute myeloid leukemia, myeloproliferative neoplasms, myelodysplastic syndromes, myelodysplastic / myeloproliferative neoplasms and systemic mastocytosis and familial causes of eosinophilia
- Exclude cytokine producing, immunophenotypically aberrant T cell population
- Tissue damage present due to hypereosinophilia
- Diagnosis of exclusion given when
- Idiopathic hypereosinophilia (hypereosinophilia of uncertain significance):
- When criteria 1 - 4 listed above for idiopathic hypereosinophilic syndrome are met but there is no end organ damage
- Presence of clonal genetic aberration favors chronic eosinophilic leukemia, not otherwise specified
- Lymphocytic variant of hypereosinophilic syndrome:
- Abnormal cytokine release by immunophenotypically abnormal or clonal T cells
- Aberrant CD3- / CD4+ T cell population which can be detected by flow cytometry can be used to differentiate; absolute CD3- / CD4+ T cell count of 0.01 - 28.3 k/L with median of 0.35 k/L
- TCRγδ rearrangements were frequently identified (76%) (Medicine (Baltimore) 2014;93:255)
- Other phenotypic changes (e.g., CD3+ / CD4+ / CD7- and CD3+ / CD4- / CD8- TCRαβ+) have been reported (Immunol Allergy Clin North Am 2007;27:389, N Engl J Med 1999;341:1112)
- Medication related:
- Administration of cytokines IL3, IL5 or GMCSF
- Myeloid neoplasms with rearrangement of PDGFRA, PDGFRB, FGFR1 or FLT3 or PCM1::JAK2, ETV6::JAK2 or BCR::JAK2 fusion (Blood 2017;129:704):
- Presence of these genetic abnormalities show morphologic findings similar to other myeloid and lymphoid neoplasms
- PDGFRA rearrangement: can present as CEL, AML or T ALL with prominent eosinophilia
- PDGFRB rearrangement: most often presents as CMML but have been characterized as atypical CML, BCR::ABL negative, CEL or MPN with eosinophilia; atypical CML is now called myelodysplastic / myeloproliferative neoplasm with neutrophilia
- FGFR1 rearrangement: presents as a AML, T or B ALL or mixed phenotype acute leukemia with prominent eosinophilia
- PCM1::JAK2: CEL, PMF or atypical CML, BCR::ABL negative
- ETV6::JAK2: B and T ALL but can also present as MDS
- BCR::JAK2: most commonly as atypical CML, BCR::ABL negative
- Paraneoplastic eosinophilia:
- T cell lymphoma, Hodgkin lymphoma, systemic mastocytosis, acute lymphoblastic leukemia t(5;14) / IgH::IL3, myeloproliferative neoplasms or certain solid tumors (e.g., lung cancer, metastatic renal cell carcinoma, etc.) associated with abnormal release of IL2, IL3, IL5 or GMCSF (Arch Pathol Lab Med 2013;137:259, Rinsho Ketsueki 1991;32:874, Acta Clin Belg 2011;66:293, Am J Hematol 2007;82:234)
- Reactive eosinophilia:
- Parasitic or fungal infections, allergies, pulmonary disease (Löffler syndrome), cyclical eosinophilia, skin diseases (angiolymphoid hyperplasia), collagen vascular diseases such as Churg-Strauss syndrome and Kimura disease
- Systemic mastocytosis:
- Clonal proliferation of atypical mast cells that meet WHO diagnostic criteria
- Eosinophils are present or increased, which could be a secondary reaction or derived from a neoplastic clone
- Abnormal T cell population (e.g., CD3- / CD4+)
- Elevated IL5 level
- Eosinophilia ≥ 1.5 k/L
- Eosinophilic tissue infiltrate
- Skin rush and pruritus
Comment Here
Reference: Chronic eosinophilic leukemia
Which of following morphologic or molecular genetic findings is frequently identified in chronic eosinophilic leukemia, not otherwise specified?
- Dysplastic noneosinophilic granulocytes
- ETV6::JAK2 fusion
- Hypercellular marrow with markedly increased eosinophils and left shifted myelopoiesis
- KIT D816V gene mutation
- Marked monocytosis
Comment Here
Reference: Chronic eosinophilic leukemia
- Chronic myeloid leukemia (CML), BCR-ABL1 positive, is a myeloproliferative neoplasm (MPN) characterized by clonal granulocytic proliferation
- Arises in a pluripotent stem cell with t(9;22)(q34.1;q11.2) chromosomal translocation and formation of the Philadelphia (Ph) chromosome, containing the BCR-ABL1 fusion gene (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 2017)
- Occurs in 3 different phases (Am J Hematol 2018;93:442)
- Chronic phase (CP)
- Accelerated phase (AP)
- Blast phase (BP)
- Natural progress: most cases progress from chronic phase to blast phase in 3 - 5 years
- Relatively rare in pediatric patients
- Children and young adults tend to have a more aggressive clinical presentation than older adults, more frequent in accelerated or blast phase (Blood 2012;119:1821)
- Chronic myelogenous leukemia, BCR-ABL1 positive
- Chronic granulocytic leukemia, BCR-ABL1 positive
- Chronic myelogenous leukemia, Philadelphia chromosome positive (Ph+)
- Chronic myelogenous leukemia, t(9;22)(q34;q11)
- Chronic granulocytic leukemia, Philadelphia chromosome positive (Ph+)
- Chronic granulocytic leukemia, (9;22)(q34;q11)
- Chronic granulocytic leukemia, BCR-ABL1
- ICD-10: C92.1 - Chronic myeloid leukemia, BCR-ABL1 positive
- Incidence: 10 - 20 cases per million/year
- M:F = 1.2 ~ 1.7:1 (Ann Hematol 2015;94:S241)
- Median age: about 65 years; pediatric cases rare
- Chronic phase: blood, bone marrow, spleen and liver
- Blast phase: spleen, liver, lymph nodes, skin and soft tissue are the most common extramedullary sites of involvement
- Reciprocal translocation t(9;22)(q34;q11) leads to the formation of the chimeric fusion gene BCR-ABL1, which is responsible for production of the oncoprotein BCR-ABL
- BCR-ABL protein has uncontrolled tyrosine kinase activity, making abnormal pluripotent hematopoietic progenitor cell initiate excessive production of all myeloid lineage cells (Am J Hematol 2020;95:691)
- Underlying causes are largely unknown
- Radiation exposure has been indicated (Lancet Haematol 2018;5:e346)
- Most patients are diagnosed in the chronic phase
- Almost 50% of patients diagnosed in the United States are asymptomatic and diagnosed during a routine examination (Mayo Clin Proc 2015;90:1440)
- Insidious onset with leukocytosis, anemia, fatigue, weight loss, night sweats
- 50% of patients have splenomegaly with dragging sensation in left upper quadrant
- Rare patients present with marked thrombocytosis without leukocytosis, mimicking essential thrombocythemia
- 5% of patients present with accelerated or blast phase
- Accelerated or blast phase: fever, bone pain, bleeding
- Chronic phase (CP) (Mayo Clin Proc 2015;90:1440):
- Leukocytosis (usually 12 - 1,000 x 10⁹/L, median: ~ 80 x 10⁹/L)
- < 2% blasts in blood
- < 5% blasts in bone marrow
- BCR-ABL1 positive by cytogenetics or molecular study
- Does not meet any of the following diagnostic criteria for accelerated or blast phase
- Accelerated phase (AP) is defined by the presence of ≥ 1 of the following criteria:
- 10 - 19% blasts in blood or marrow (Drug Des Devel Ther 2019;13:825)
- Persistent or increasing WBC > 10 x 10⁹/L, unresponsive to therapy
- Persistent platelet count > 1,000 x 10⁹/L, unresponsive to therapy
- Persistent platelet count < 100 x 10⁹/L, unrelated to therapy
- ≥ 20% basophils in blood
- Persistent or increasing splenomegaly, unresponsive to therapy
- Additional clonal chromosomal abnormalities in Philadelphia chromosome positive cells at diagnosis, including so called major route abnormalities (a second Ph chromosome, trisomy 8, isochromosome 17q, trisomy 19), complex karyotype and abnormalities of 3q26.2
- Large clusters or sheets of small, abnormal megakaryocytes associated with marked reticulin or collagen fibrosis may be considered presumptive evidence of accelerated phase, although these findings are usually associated with one or more of the criteria listed above
- Blast phase (BP):
- ≥ 20% blasts in marrow or blood or the presence of an extramedullary proliferation of blasts (Front Oncol 2019;9:1132)
- Approximately 70% have myeloid blasts (may include neutrophilic, monocytic, megakaryocytic, basophilic, eosinophilic or erythroid blasts), approximately 30% have lymphoid blasts (usually B lymphoblasts but T and NK blasts have been reported) (J Clin Invest 2010;120:2254)
- Chronic phase:
- Leukocytosis with predominantly neutrophils and myelocytes
- Basophilia and eosinophilia
- t(9;22)(q34;q11.2) by FISH, conventional cytogenetics or PCR
- Alkaline phosphatase decreased
- Reference: Mayo Clin Proc 2015;90:1440
- Splenomegaly
Contributed by Yang Shi, M.D., Ph.D.
Splenomegaly
- Most important prognostic indicator is response to tyrosine kinase inhibitors at the hematological, cytogenetic and molecular level (Cancer 2017;123:4391)
- 5 year progression free and overall survival rates: 80 - 95%
- Long term overall survival approaching that of aged matched controls
- 11 year old boy with chronic myeloid leukemia possessing the unique combination of T lymphoblastic transformation and a subclone harboring inv(3)(q21q26.2) at diagnosis (Biomark Res 2016;4:14)
- 39 year old asymptomatic man with hematologically typical chronic phase chronic myeloid leukemia showed an unusual and complex translocation involving chromosomes 9, 11 and 22 (Arch Pathol Lab Med 2001;125:437)
- 52 year old woman with occurrence of chronic myeloid leukemia followed by CLL (Int J Hematol Oncol Stem Cell Res 2014;8:49)
- 68 year old woman with p190 BCR-ABL+ chronic myeloid leukemia and persistent moderate monocytosis (J Hematol 2018;7:120)
- 68 year old man with JAK2 V617F positive PV, evolving to BCR-ABL positive chronic myeloid leukemia (AJHO 2017;13:24)
- Chronic phase: tyrosine kinase inhibitor (TKI), acting by competitive inhibition at the ATP binding site of BCR-ABL1 oncoprotein, resulting in inhibition of phosphorylation of proteins involved in cell signal transduction (Ulster Med J 2007;76:8)
- Tyrosine kinase inhibitor agents help achieve long term control of chronic myeloid leukemia in majority of patients (Leukemia 2020;34:966)
- Imatinib is the prototype drug and first approved tyrosine kinase inhibitor (N Engl J Med 2017;376:917)
- Before tyrosine kinase inhibitor era, interferon alpha and hydroxyurea were used to treat chronic myeloid leukemia (Leukemia 2013;27:803)
- Chronic myeloid leukemia resistant to imatinib can be treated by second generation tyrosine kinase inhibitor agents such as nilotinib, dasatinib and bosutinib (Clin Lymphoma Myeloma Leuk 2015;15:323)
- Complete hematological response (Blood Rev 2011;25:139):
- WBC < 10 x 10⁹/L
- Platelet count < 450 x 10⁹/L
- No immature granulocytes in the differential
- Spleen not palpable
- Treatment failure occurs most commonly due to compliance issues followed by BCR-ABL mutations altering drug binding
- Hematopoietic stem cell transplant is reserved for those rare patients who (Semin Hematol 2010;47:354):
- Present with blast phase
- Are resistant / intolerant to all tyrosine kinase inhibitors
- Progress to accelerated or blast phase while on tyrosine kinase inhibitor therapy
- Bone marrow (chronic phase):
- Hypercellular marrow with absolute myeloid hyperplasia and myelocyte bulge
- Myeloid to erythroid ratio ≥ 10:1
- Layer of immature granulocytes (5 - 10 cells in thickness) around the bone trabeculae (2 - 3 cells layer in normal marrow)
- No significant dysplasia
- Increased basophils and eosinophils (Intern Med 2011;50:501)
- Blasts < 5%
- Normal / slightly decreased megakaryocytes; 40 - 50% show moderate to marked megakaryocytes proliferation
- Megakaryocytes often smaller and hyposegmented with dwarf morphology
- Pseudo-Gaucher cells are common (Leukemia 1998;12:233)
- Bone marrow (accelerated phase):
- 10 - 19% blasts (Am Soc Clin Oncol Educ Book 2015;e381)
- Large clusters or sheets of small, abnormal megakaryocytes associated with marked reticulin or collagen fibrosis may be considered presumptive evidence of accelerated phase
- Bone marrow (blast phase):
- ≥ 20% blasts in marrow or blood or the presence of an extramedullary proliferation of blasts (Leuk Lymphoma 2014;55:1451)
- Approximately 70% have myeloid blasts (may include neutrophilic, monocytic, megakaryocytic, basophilic, eosinophilic or erythroid blasts) and approximately 30% have lymphoid blasts (usually B lymphoblasts but T and NK blasts have been reported) (J Clin Invest 2010;120:2254)
Contributed by Yang Shi, M.D., Ph.D.
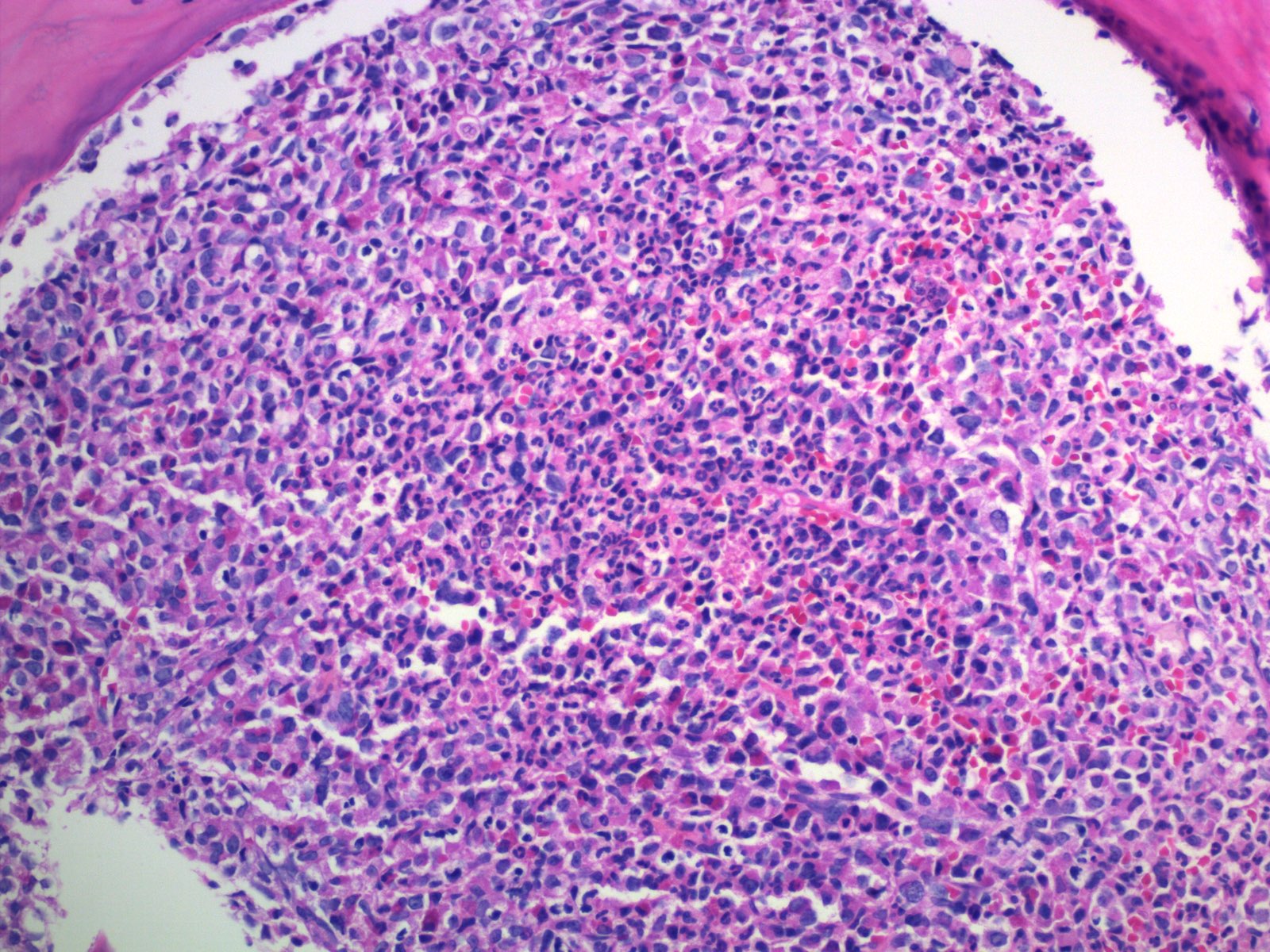
Chronic phase marrow
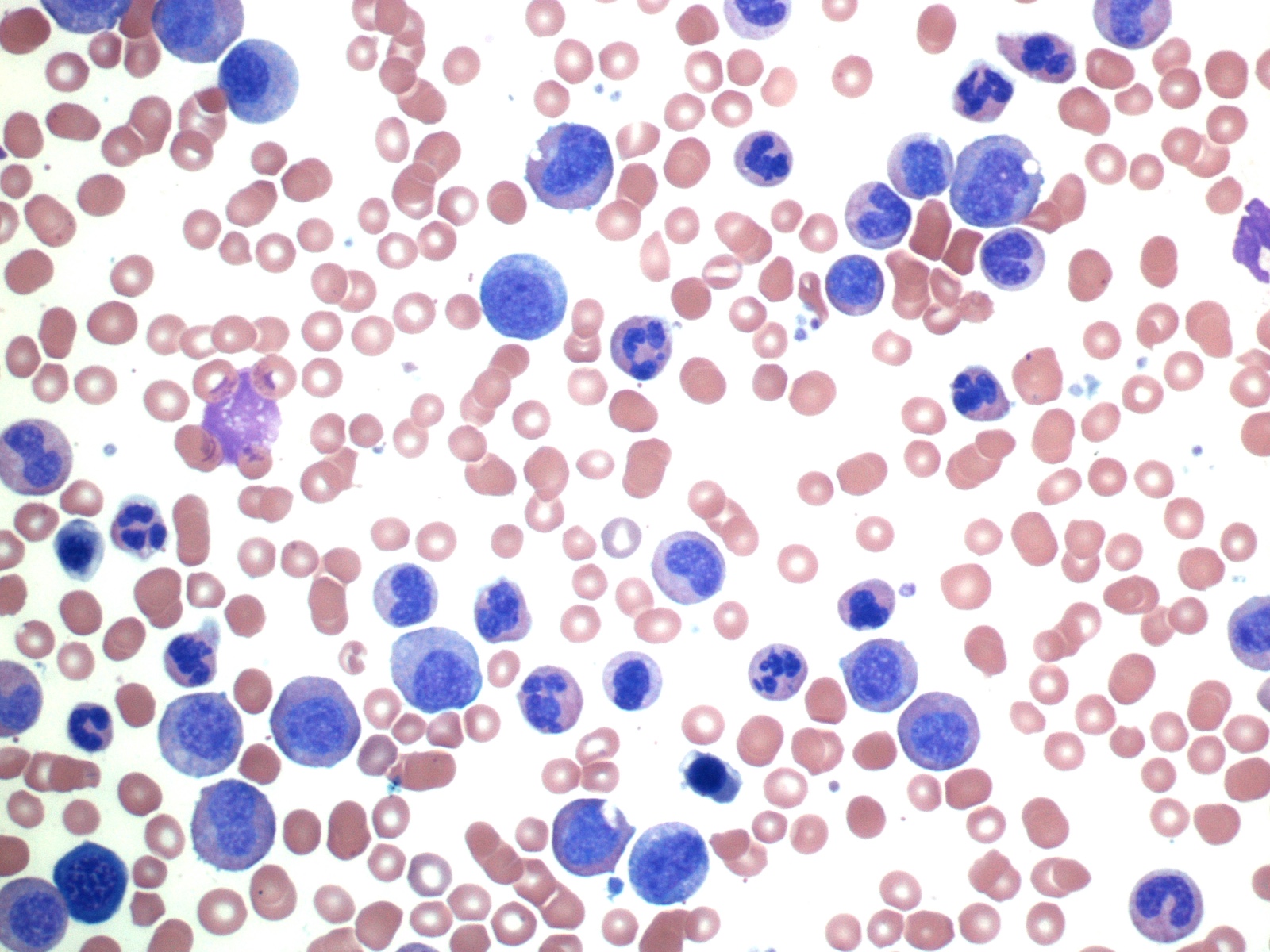
Chronic phase marrow smear

Accelerated phase marrow

Blast phase B-ALL
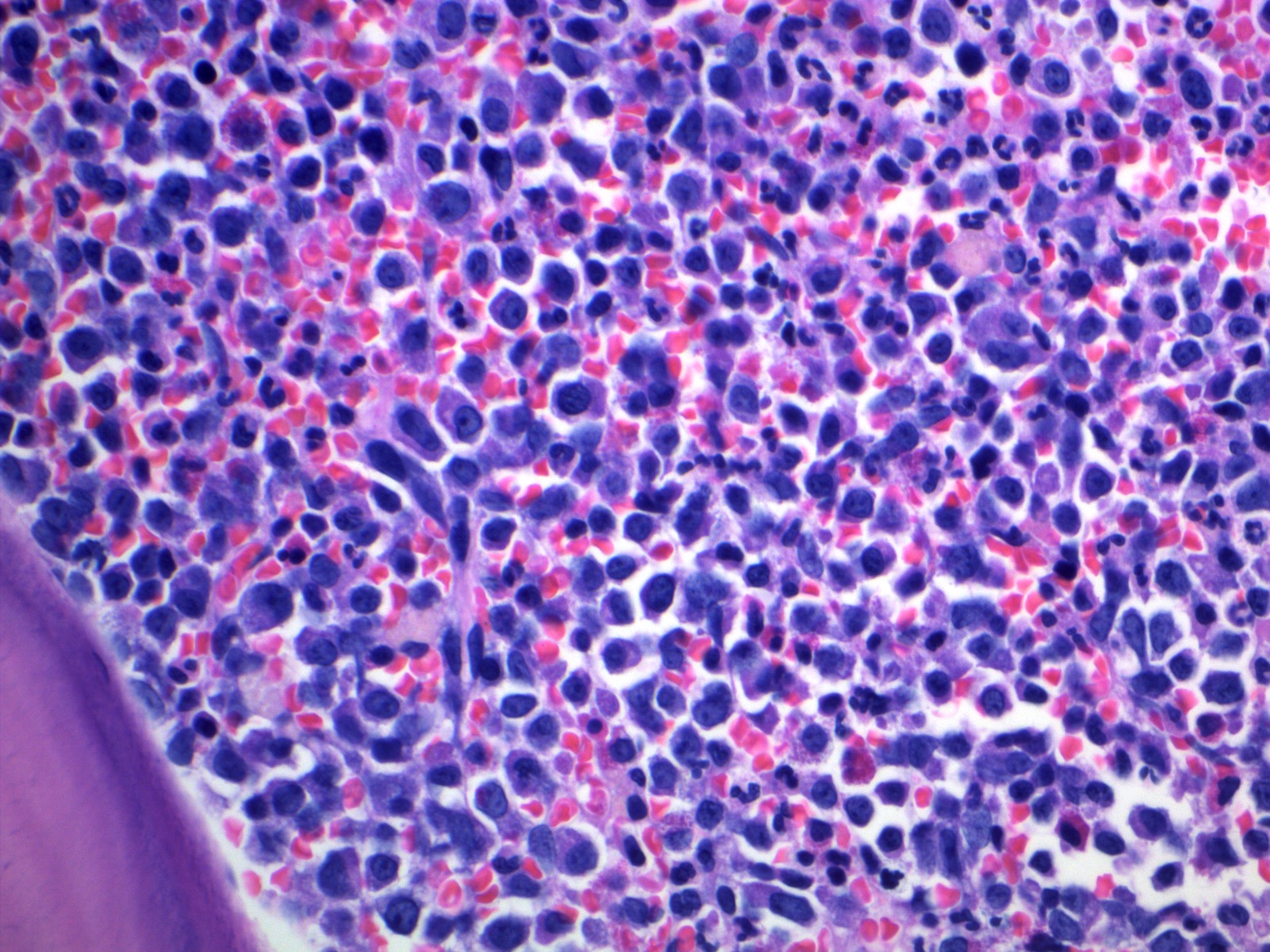
Bone marrow biopsy
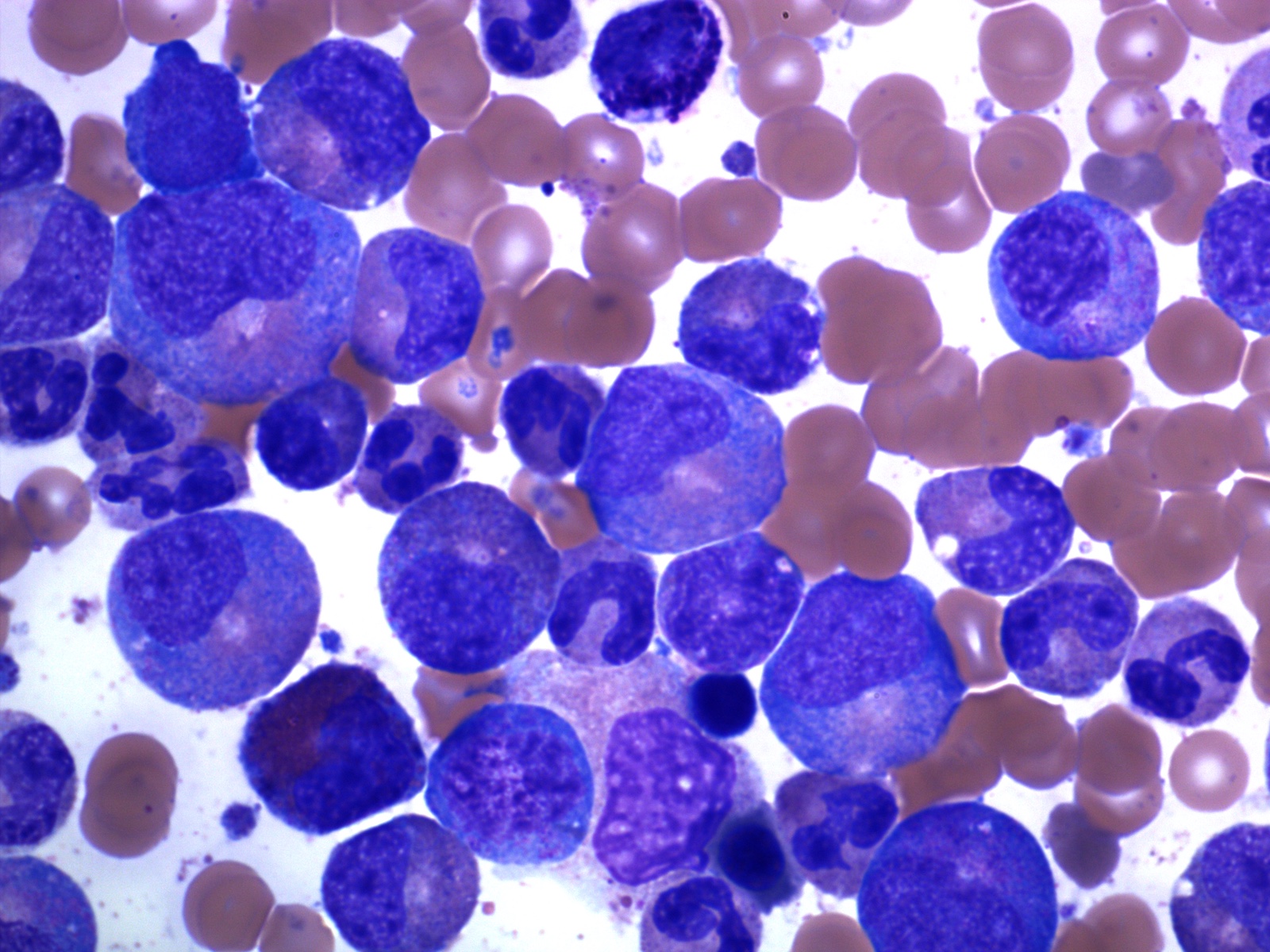
Bone marrow aspirate smear
- Chronic phase:
- Absolute leukocytosis (12 - 1,000 x 10⁹/L, median: ~80 x 10⁹/L)
- Left shifted with 2 peaks: myelocytes and segmented neutrophils
- Blasts < 2%
- No significant granulocytic dysplasia
- Absolute monocytosis may be present but are < 3%
- Absolute basophilia (100%) and eosinophilia (80 - 90%)
- Platelet counts are normal or thrombocytosis ≥ 1,000 x 10⁹/L
- Reference: Mayo Clin Proc 2015;90:1440
Contributed by Yang Shi, M.D., Ph.D.
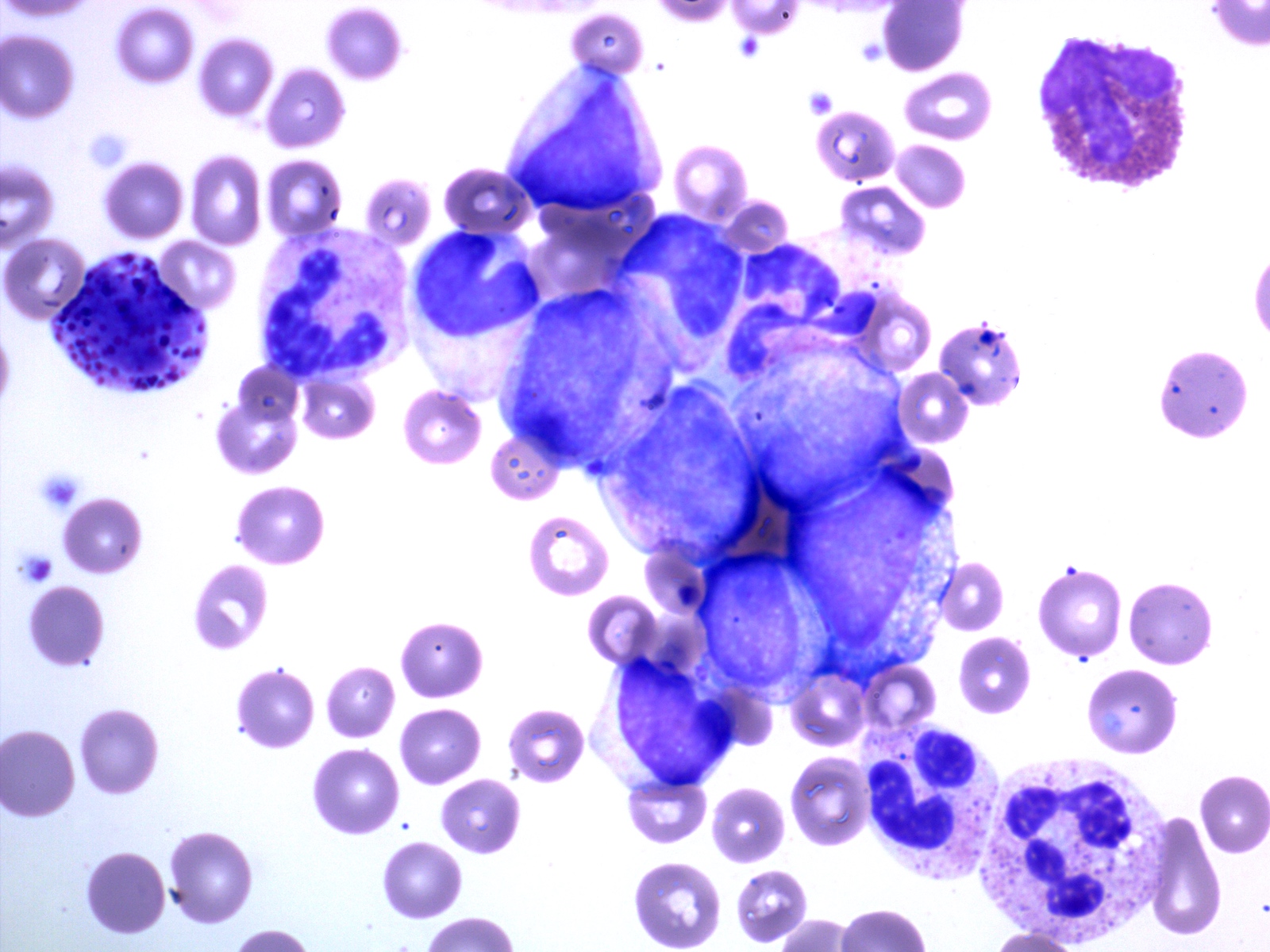
Chronic phase blood smear
- Chronic phase:
- Myeloperoxidase, specific esterase (naphthol AS-D chloroacetate esterase)
- BB1 (basogranulin) stains basophils (Am J Clin Pathol 2006;125:273)
- Blast phase / accelerated phase:
- Peripheral blood (chronic phase) (Mayo Clin Proc 2015;90:1440):
- Left shifted leukocytosis
- No significant increase in blasts: < 2%
- Relative lymphopenia
- Myeloid lineage shows 2 peaks: correspond to myelocytes and segmented neutrophils respectively
- Accelerated phase: 10 - 19% blasts in blood or marrow
- Blast phase: ≥ 20% blasts in marrow or blood or the presence of an extramedullary proliferation of blasts (Front Oncol 2019;9:1132)
- Bona fide lymphoblasts in the peripheral blood or bone marrow (even if < 10%) should prompt immediate investigation for lymphoblastic transformation
Contributed by Yang Shi, M.D., Ph.D.
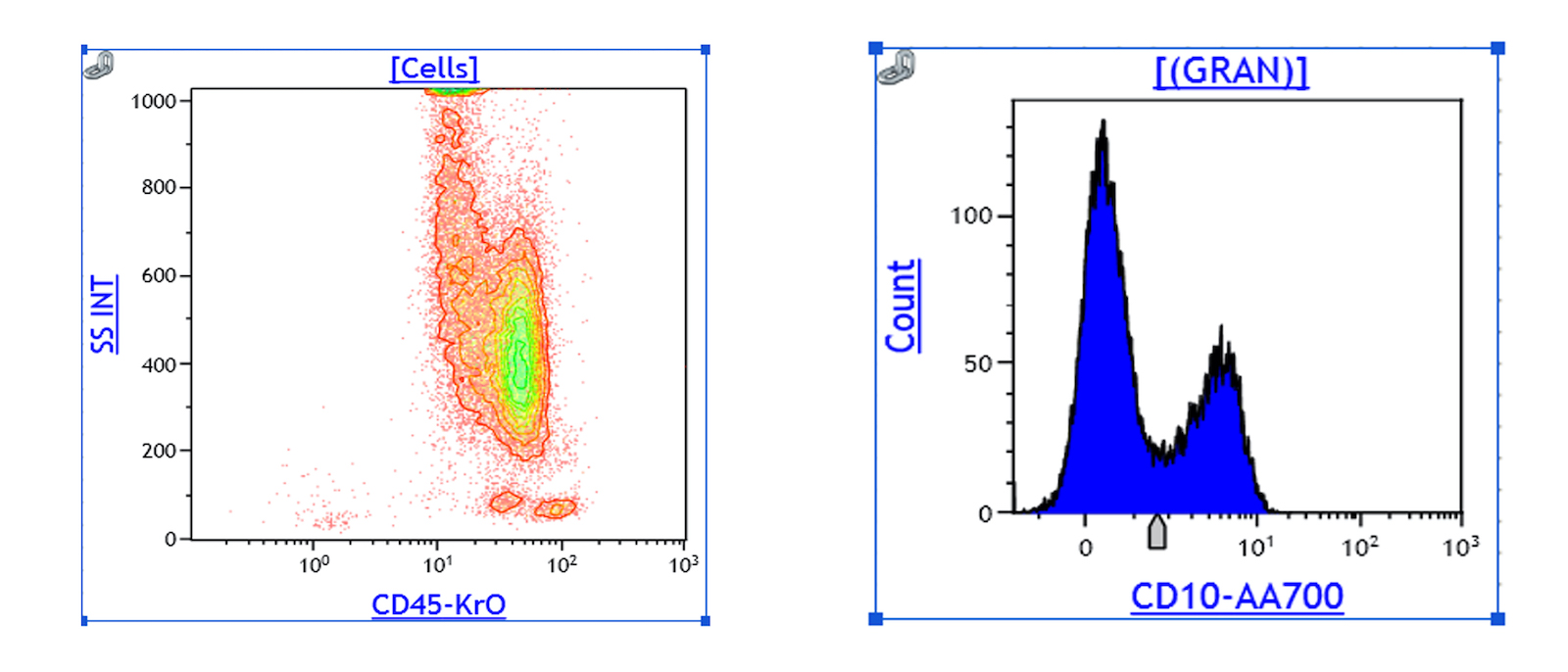
Peripheral blood chronic phase
- 90 - 95% of chronic myeloid leukemia is characterized by t(9;22)(q34.1;q11.2) reciprocal translocation that results in the Ph chromosome, der(22)t(9;22) (Haematologica 2016;101:541)
- Translocation fuses the BCR gene on chromosome 22 with regions of ABL1 on chromosome 9 and generates the BCR-ABL1 fusion gene that characterizes leukemic cells (Am J Hematol 2018;93:442)
- 5 - 10% cases have either variant translocations that involve a third or even a fourth chromosome in addition to chromosomes 9 and 22 or a cryptic translocation of 9q34.1 and 22q11.2 that cannot be identified by routine cytogenetic analysis (Oncotarget 2017;8:29906)
- In these cases, the presence of BCR-ABL1 fusion gene can be detected by FISH or RT-PCR
- In most chronic myeloid leukemia cases the breakpoint cluster region is almost always in the major BCR (M-BCR), spanning exons 12 - 16 and form an abnormal fusion protein p210
- Rarely, the breakpoint in the BCR gene occurs in the p-BCR region, spanning exons 17 - 20 and form a larger fusion protein p230 (Haematologica 2001;86:320)
- Small amounts of P190 transcript with breaks in the minor breakpoint region, m-BCR (exons 1 - 2) can be detected in > 90% of patients with chronic myeloid leukemia due to alternative splicing of the BCR gene
- Very rare chronic myeloid leukemia cases are associated with p190 only and frequently show monocytosis, mimicking chronic myelomonocytic leukemia (J Hematol 2018;7:120)
Contributed by K.H. Ramesh, Ph.D.
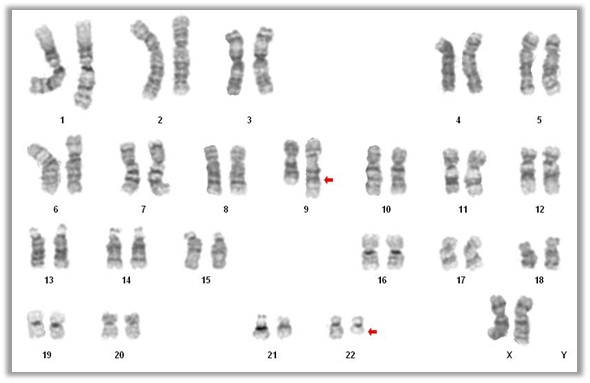
Conventional cytogenetics chronic myeloid leukemia
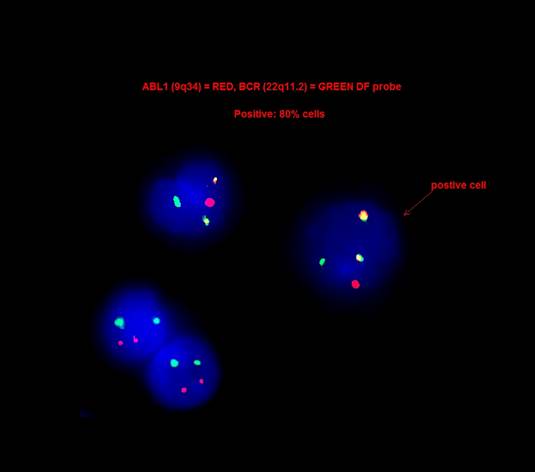
BCR-ABL+ chronic myeloid leukemia FISH
Brief introduction of chronic myeloid leukemia
Chronic myeloid leukemia: pathogenesis, symptoms and treatment
Blast crisis in chronic myeloid leukemia
Chronic myeloid leukemia from Khan Academy
Treatment of chronic myeloid leukemia
Peripheral blood smear of chronic myeloid leukemia
- Bone marrow, left posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Hypercellular (90%) bone marrow with absolute myeloid hyperplasia and < 5% blasts (see comment)
- Comment: Flow cytometric analysis demonstrates 2.5% blasts. There are < 5% blasts by CD34 and CD117 immunohistochemical stains on the core biopsy. A CBC obtained on 03/29/2020 revealed leukocytosis (30.2 x 10⁹/L) and thrombocytosis (625 x 10⁹/L). FISH analysis from peripheral blood (03/29/2020): BCR-ABL1 dual color, dual fusion translocation DNA probe revealed a fusion of 9q34.1 (ABL1) and 22q11.2 (BCR) regions in 62 out of 100 cells (62%) interphase nuclei.
- Peripheral smear: Manual review of the peripheral blood shows left shifted leukocytosis with two bulges: myelocytes and segmented neutrophils. No definite morphologic dysplasia is seen. Rare (~1%) circulating blasts are noted. RBCs: mild normochromic, normocytic anemia. Platelets: moderate thrombocytosis with rare large platelets.
- Bone marrow biopsy: Quality: adequate. Cellularity: > 90%. Hematopoiesis: trilineage maturation with absolute myeloid hyperplasia and relative erythroid hypoplasia. Megakaryocytes are mildly to moderately increased. Most megakaryocytes are small and hyposegmented. Infiltrate: no apparent increase in blasts. Special stains: reticulin: mild diffuse increase in reticulin fibrosis. Trichrome: negative for collagen deposition.
- Bone marrow clot section: Quality: adequate. Cellularity: > 90% morphologic features are similar to those observed in the core biopsy.
- Bone marrow aspirate: Quality: adequate. Granulocytes: markedly increased with significant expansion of myelocytes. No definite morphologic dysplasia. Erythrocytes: relative erythroid hypoplasia with progressive maturation and no definite dysplasia. Megakaryocytes: mildly to moderately increased; most of them are small and hyposegmented. No sheets of megakaryocytes are present. Blasts: < 5% of nucleated cells. Iron: storage iron is adequate (2+ on a scale of 0 - 4). No ring sideroblasts are present.
- Leukemoid reaction (Am J Hematol 2018;93:442):
- Underlying infection / inflammation
- Myelocyte bulge is absent
- Basophilia is absent
- Toxic granulation and cytoplasmic vacuoles might be present
- Alkaline phosphatase increased
- Chronic neutrophilic leukemia:
- Peripheral blood:
- Segmented neutrophils + band neutrophils ≥ 80%
- Neutrophil precursors (promyelocytes, myelocytes and metamyelocytes) < 10%
- Neutrophils might show toxic granulation and Dohle bodies
- No dysplasia
- Strong association with CSF3R mutation (Blood 2017;129:715)
- Peripheral blood:
- Atypical chronic myeloid leukemia, BCR-ABL1 negative (Hematology Am Soc Hematol Educ Program 2015;2015:264):
- Dysplastic neutrophils and their precursors
- Multilineage dysplasia is common
- No or minimal absolute basophilia, basophils < 2% of WBC
- No or minimal monocytosis, monocytes < 10% of WBC
- SETBP1 and ETNK1 mutations are common
- Chronic myelomonocytic leukemia (Am J Hematol 2018;93:824):
- Monocytes ≥ 10% of WBC
- Dysplasia involving ≥ 1 myeloid lineages
- Common mutations: TET2, SRSF2, ASXL1, SETBP1
- Chronic eosinophilic leukemia (Blood Cancer J 2018;8:15):
- Eosinophil count ≥ 1.5 x 10⁹/L
- Blasts < 20%
- A clonal cytogenetics or molecular genetic abnormality is present or blasts ≥ 2% in blood or blasts ≥ 5% in marrow
- Absence of BCR-ABL1 translocation
- Absence of rearrangement of PDGFRA, PDGFRB or FGFR1 and no PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusion
- Other myeloproliferative neoplasms such as polycythemia vera, essential thrombocythemia, primary myelofibrosis, chronic neutrophilic leukemia, chronic myelomonocytic leukemia and BCR-ABL1 negative atypical chronic myeloid leukemia are excluded
- AML with inv(16)(p13.1q22), t(16;16)(p13.1;q22), t(8;21)(q22;q22.1) are excluded
- Essential thrombocythemia (Mayo Clin Proc 2015;90:1440):
- Platelet ≥ 450 x 10⁹/L
- Bone marrow biopsy shows significant megakaryocytic proliferation
- Megakaryocytes are large, hyperlobated
- No significant increase or left shifted granulopoiesis or erythropoiesis
- No basophilia
- JAK2, CALR or MPL mutation (+)
- 5 - 10% blasts in bone marrow or peripheral blood
- Persistent platelet count < 100,000 related to therapy
- ≥ 10% basophils in blood
- Persistent or increasing splenomegaly
- Additional clonal chromosomal abnormalities in Philadelphia chromosome positive cells at diagnosis, including so called major route abnormalities (a second Ph chromosome, trisomy 8, isochromosome 17q, trisomy 19), complex karyotype and abnormalities of 3q26.2
Comment Here
Reference: Chronic myeloid leukemia (CML)
A 15 year old boy with no significant medical history presented with worsening knee pain for 5 days. CBC: WBC: 229.4 x 10⁹/L, hemoglobin 12.1 g/dl, platelet 161 x 10⁹/L. Bone marrow biopsy showed > 90% cellular marrow with absolute myeloid hyperplasia without significant increase in blasts. FISH study showed BCR-ABL1 translocation. Which of the following statements is right?
- CML is very common in pediatric patients
- Children and young adults with CML tend to have a less aggressive clinical presentation than older adults
- Children and young adults with CML present with accelerated or blast phase more frequently
- Children and young adults tend to have higher complete cytogenetic response and molecular response rates compared with older adults
Comment Here
Reference: Chronic myeloid leukemia (CML)
- Monocytosis rules out the possibility of chronic myeloid leukemia
- Thrombocytosis rules out the possibility of chronic myeloid leukemia
- Eosinophilia with basophilia is common in chronic myeloid leukemia
- Toxic granulation and Dohle bodies are common in chronic myeloid leukemia
- Alkaline phosphate is increased in chronic myeloid leukemia
Comment Here
Reference: Chronic myeloid leukemia (CML)
- Clonal myeloid neoplasm defined by persistent relative (> 10%) and absolute monocytosis (≥ 0.5 x 109/L) in peripheral blood
- Features of both a myeloproliferative neoplasm and a myelodysplastic syndrome
- Inherent risk for transformation to acute myeloid leukemia
- No evidence of BCR::ABL1, PDGFRA, PDGFRB, FGFR1 or JAK2 rearrangement
- Persistent relative (> 10%) and absolute monocytosis (≥ 0.5 x 109/L) in peripheral blood
- < 20% blasts (including myeloblasts, monoblasts and promonocytes) in peripheral blood and bone marrow
- Morphologic dysplasia involving at least 1 myeloid lineage
- Acquired clonal cytogenetic or molecular abnormality
- Chronic myelomonocytic leukemia type I: ≤ 5% blasts in the blood and ≤ 10% blasts in the bone marrow and no Auer rods
- Chronic myelomonocytic leukemia type II: 6 - 19% blasts in the blood, 10 - 19% blasts in the bone marrow or Auer rods are present
- Myelodysplastic CMML (MD-CMML): WBC count < 13 × 109/L
- Myeloproliferative CMML (MP-CMML): WBC count ≥ 13 × 109/L
- ICD-10: C93.1 - chronic myelomonocytic leukemia
- Annual incidence rate of ~0.4 cases per 100,000 persons with the highest incidence in patients older than 80 (Leuk Res 2015;39:177, Blood 1984;63:634)
- M:F = ~2.5:1
- Reported median age at diagnosis ranges from 65 to 75 years
- Blood and bone marrow are invariably involved
- Spleen, liver, skin and lymph nodes are the most common extramedullary sites of involvement
- Similar to other myelodysplastic syndromes, chronic myelomonocytic leukemia (CMML) often results in ineffective hematopoiesis and cytopenia
- Acquired cytogenetic and molecular changes involving hematopoietic stem cells
- Heterogeneous clinical manifestations (Curr Hematol Malig Rep 2015;10:292)
- Constitutional symptoms
- Cytopenia
- Splenomegaly
- Cutaneous lesions
- Infection
- Bleeding
- Various inflammatory or autoimmune processes (Leuk Lymphoma 2017;58:1488)
- Persistent absolute (≥ 0.5 x 109/L) and relative (> 10%) peripheral blood monocytosis
- Does not meet WHO criteria for chronic myeloid leukemia, primary myelofibrosis, polycythemia vera or essential thrombocythemia
- No evidence of BCR::ABL1, PDGFRA, PDGFRB, FGFR1 or JAK2 rearrangement
- < 20% blasts (including myeloblasts, monoblasts and promonocytes) in peripheral blood and bone marrow
- Morphologic dysplasia involving at least 1 myeloid lineage
- Myelodysplasia may be absent if all of the above are present and there is evidence of an acquired clonal cytogenetic or molecular abnormality or if monocytosis has persisted ≥ 3 months and all reactive causes of monocytosis have been excluded
- Acquired clonal cytogenetic or molecular abnormality
- Reference: Leukemia 2022;36:1703
- Leukocytosis or leukopenia with relative and absolute monocytosis and neutrophilia or neutropenia
- Anemia
- Thrombocytopenia or rarely thrombocytosis
- Increased serum lactate dehydrogenase (Am J Hematol 2016;91:631)
- Hepatosplenomegaly
- Fluorodeoxyglucose (FDG) avid extramedullary lesions may be seen (Clin Nucl Med 2014;39:811)
Contributed by Sanam Loghavi, M.D.
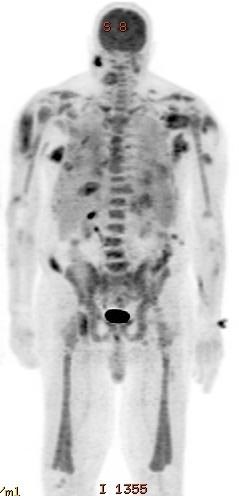
FDG PET / CT
- Prognosis is poor, with a median overall survival of 2 - 3 years (Blood 2002;99:840)
- 15 - 30% risk of transformation to acute myeloid leukemia
- Associated with worse outcome
- Older age
- Higher white blood cell (WBC) count
- Low platelet count
- ASXL1 (frameshift or nonsense mutations)
- Certain cytogenetic alterations including trisomy 8, alterations of chromosome 7 and a complex karyotype (Haematologica 2011;96:375, J Clin Oncol 2013;31:2428)
- Associated with worse outcome
- Several risk stratification systems have been proposed; these risk stratification models incorporate
a variety of clinical, laboratory, cytogenetic or molecular parameters (see table below) (Curr Hematol Malig Rep 2018;13:446)
- MDAPS: MD Anderson Prognostic System (Blood 2002;99:840)
- Global MDAPS (Cancer 2008;113:1351)
- CPSS: CMML Specific Prognostic Scoring System (Blood 2013;121:3005)
- CPSS-Mol: Molecular CPSS (Blood 2016;128:1408)
- Groupe Francophone des Myelodysplasies (J Clin Oncol 2013;31:2428)
- Mayo Prognostic Model (Leukemia 2013;27:1504)
- Mayo Molecular Model (Blood Cancer J 2015;5:e333)
Chronic myelomonocytic leukemia prognostic models
- 30 year old woman with therapy related disease (Int J Hematol 2009;89:699)
- 64 year old man with progression to acute myeloid leukemia FIP1L1::PDGFRA responsive to imatinib (J Hematol Oncol 2014;7:26)
- 64 year old man with blastic plasmacytoid dendritic cell neoplasm associated with chronic myelomonocytic leukemia (Blood 2016;128:1664)
- 67 year old man with a 3 year history of chronic myelomonocytic leukemia (J Cutan Pathol 2017;44:1075)
- 87 year old woman with enlarging neck mass due to involvement of thyroid gland (Br J Haematol 2003;120:548)
- Allogeneic hematopoietic stem cell transplantation remains the only curative treatment option
- Other therapeutic means are also used to alleviate the symptoms related to the associated cytopenia and cytoses; these include the use of erythropoiesis stimulating agents, cytoreductive chemotherapy and hypomethylating agents (Blood 2017;130:126)
- Wait and watch approach may be used in patients with low risk and clinically stable disease
- Ruxolitinib may be considered in order to abrogate the effects of aberrant RAS signaling pathway activation (Clin Cancer Res 2016;22:3707)
Contributed by Michael Tetzlaff, M.D., Ph.D.
Hemorrhagic papules
- Bone marrow is often hypercellular and shows a myelomonocytic proliferation resulting in increased myeloid:erythroid (M:E) ratio but an increase in erythroid precursors may also be seen in some cases
- Monocytic proliferation is always present but may be difficult to appreciate on routine H&E stained sections
- Dysgranulopoiesis is almost always present
- Dyserythropoiesis is also commonly observed but is less striking
- Dysmegakaryopoiesis is seen in up to 80% of cases
- Mild to moderate reticulin fibrosis may be seen
- Neoplastic proliferation of mature plasmacytoid dendritic cells (CD123+) is seen in a subset of cases (Am J Hematol 2010;85:893)
- Splenic red pulp is usually infiltrated by chronic myelomonocytic leukemia
Contributed by Sanam Loghavi, M.D.

Deep dermis
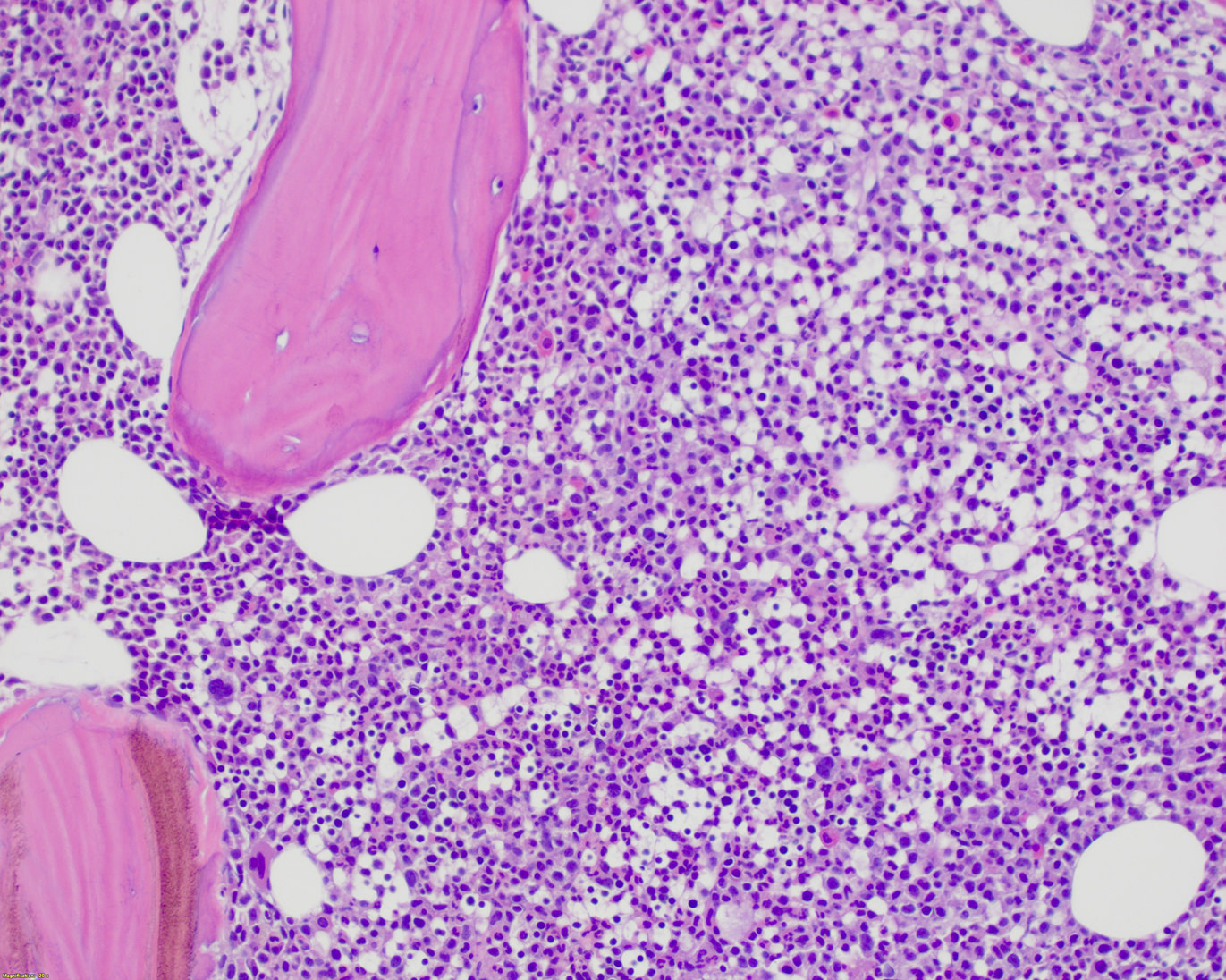
Bone marrow
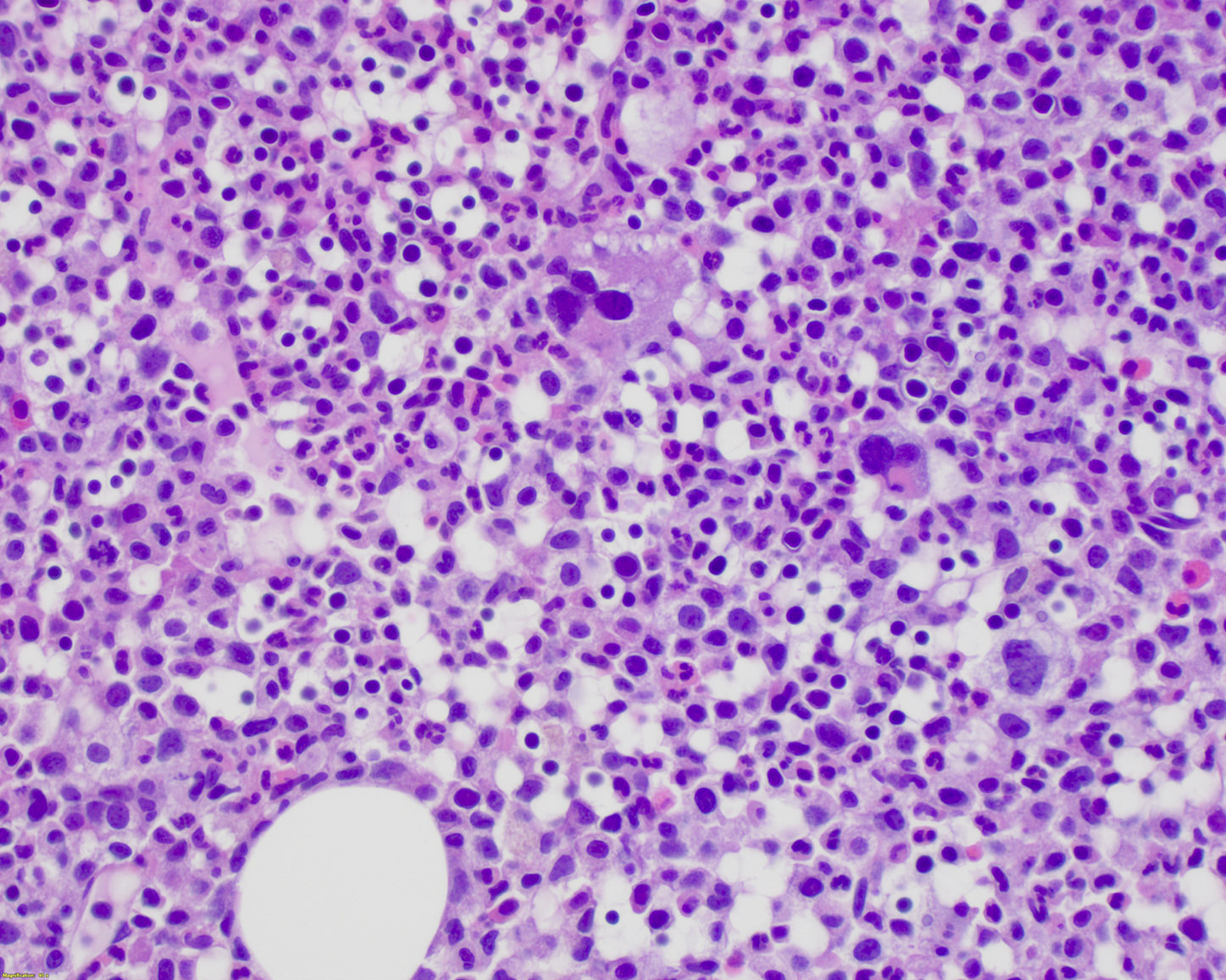
Myelomonocytic hyperplasia in bone marrow

Monocytosis and morphologic dysplasia
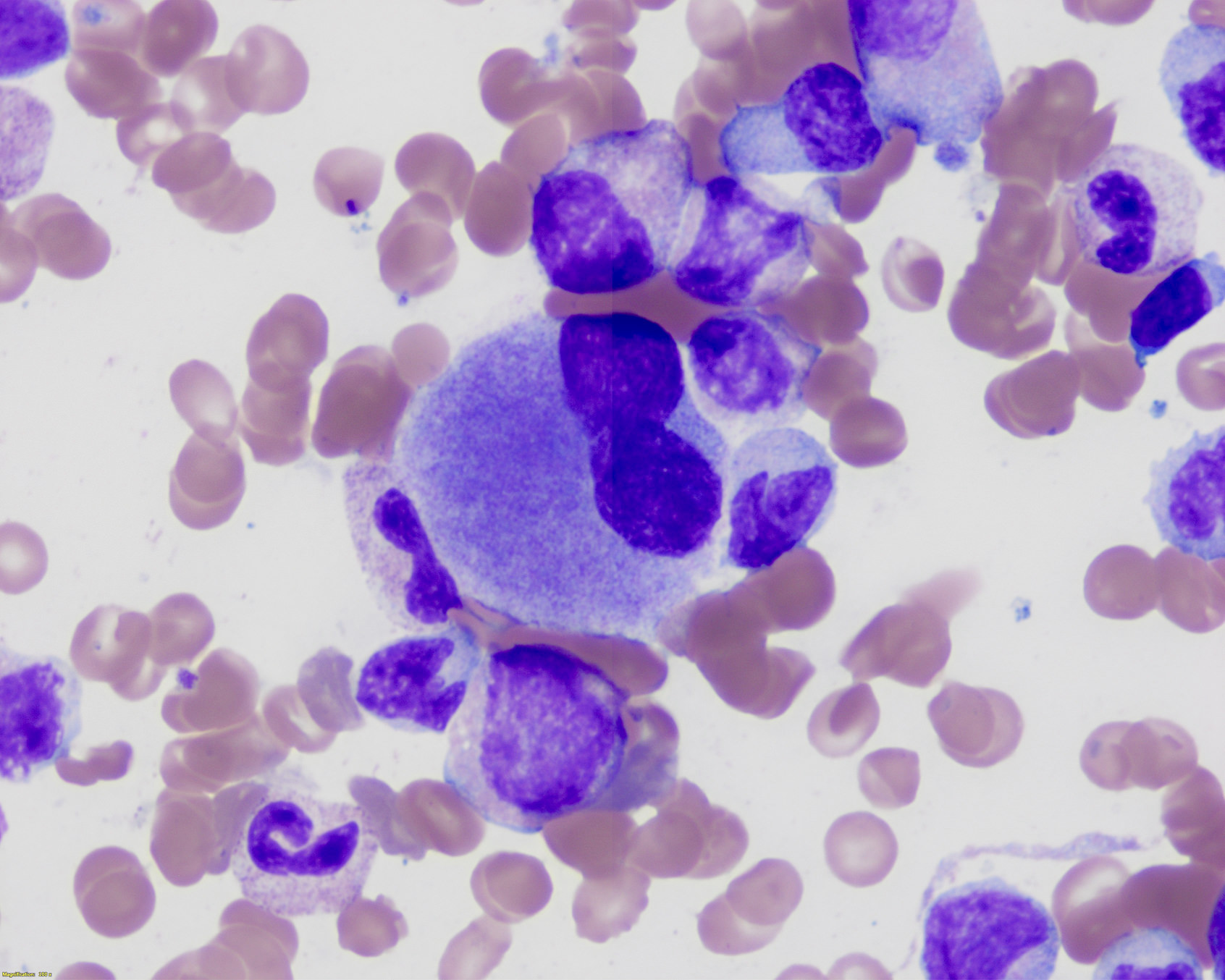
Morphologic dysplasia
- Monocytes may be comprised of mature or immature forms (monoblasts, promonocytes)
- Immature forms in this context are regarded as blast equivalents
- There is typically background dysplasia in one or more of the myeloid lineages including myeloid, erythroid or megakaryocytic lineages
- Relative and absolute monocytosis, variable increased immature monocytes (promonocytes, monoblasts)
- May show neutrophilia or neutropenia with variable dysplastic neutrophils (hypogranular, hypolobated)
- Left shifted granulocytes may be seen but usually account for < 10% of leukocytes and blasts are < 20% by definition
- Variable anemia, with normocytic or macrocytic red blood cells
- Variable thrombocytopenia
- Reference: Leukemia 2022;36:1703
- Butyrate esterase (cytochemistry) highlights increased monocytes on bone marrow and peripheral blood smears
- CD11c, CD14, CD68 and CD163 and lysozyme (immunohistochemistry): highlights increased myelomonocytic cells in bone marrow biopsy and clot
- Reference: Leukemia 2022;36:1703
- Myeloperoxidase may be weakly positive or negative
- Flow cytometry can differentiate reactive monocytosis from chronic myelomonocytic leukemia but findings are not specific and may be seen in other myeloid neoplasms (Eur J Haematol 2015;95:168)
- CD34+ myeloid blasts show antigenic aberrancies (median of 6 aberrancies per case) including increased intensity of CD117, CD123, CD13 and CD34 expression and decreased intensity of CD38 expression as well as altered pattern of CD45 / side scatter and aberrant expression of lineage infidelity markers such as CD2, CD5, CD7, CD19 and CD56, among others
- Monocytic and granulocytic cells show frequent immunophenotypic aberrancies (96% and 83%, respectively)
- Granulocytes tend to show altered patterns of maturation markers CD11b / CD13 / CD16
- Monocytes show decreased expression of CD45 and HLA-DR, altered (increased or decreased) expression of CD64 and CD14 and aberrantly increased expression of CD56 (positive in ≥ 25% of monocytes) and CD2
- Hematogones are completely absent in ~93% of cases
- Classical monocyte (CD14+ / CD16-) population is expanded (Blood 2015;125:3618)
- Expansion of classical monocyte subsets (≥ 94%) has also been shown to be able to discriminate from other myelodysplastic syndrome subtypes with relatively good sensitivity (72%) and specificity (86%)
- Flow cytometry immunophenotyping can distinguish from other myeloproliferative with monocytosis using a classical monocyte fraction cutoff of 92% with a sensitivity of 93% and specificity of 100% (Blood Cancer J 2017;7:e584)
- Clonal cytogenetic abnormalities are seen in 20 - 30%
- Most frequent chromosomal alterations include +8 (23%), -Y (20%), -7/del 7q (14%), +21 (8%) and del20q (8%) and der(3q) (8%) (Haematologica 2011;96:375)
- Cytogenetic changes have important prognostic implications and are included in all major risk stratification models (Am J Hematol 2014;89:813)
- Clones have up to 15 mutations per kilobase of coding DNA
- Most commonly mutated genes including epigenetic and transcriptional regulation
- TET2 (60%)
- ASXL1 (40%)
- EZH2 (< 5%; more common in myeloproliferative chronic myelomonocytic leukemia), DNMT3A (~5%), IDH1 (< 2%) and IDH2 (~5%), mutations in the spliceosome machinery including SRSF2 (50%), U2AF1 (7%), SF3B1 (5 - 10%), ZRSR2 (3%), PRPF8 (Nat Commun 2016;7:10767)
- Epigenetic and splicing gene mutations frequently co-occur with mutations involving genes in the cell signaling pathways (~30%) including JAK2 (5%), KRAS (5 - 10%), NRAS (10 - 20%), CBL (13%), BRAF (7%), PTPN11 (< 5%), FLT3 (< 5%), NPM1 (< 3%) (J Clin Oncol 2013;31:2428, Am J Hematol 2014;89:499)
- Signaling pathway mutations are more frequent in myeloproliferative chronic myelomonocytic leukemia
- Mutations in the BRAF kinase domain in ~7%
- Co-occurrence of SRSF2 and TET2 mutations is highly specific (~98%) to myeloid neoplasms with a CMML-like phenotype (Blood 2014;124:1513)
- Worse survival
- Truncating ASXL1 mutations (frameshift and nonsense) (Leukemia 2014;28:2206)
- ASXL1 / EZH2 comutations
- DNMT3A mutations (Blood Cancer J 2016;6:e385)
- Favorable outcome
- Clonal TET2 mutations with wild type ASXL1 (Blood Cancer J 2016;6:e385)
- RUNX1 mutations
- Associated with more severe cytopenia and progression to acute myeloid leukemia
- No impact on survival (Leukemia 2009;23:1426)
- TP53 mutations
- Uncommon (~4%, more common in myeloproliferative chronic myelomonocytic leukemia)
- May be clonal or subclonal
- Mostly detected at initial diagnosis
- In contrast to other cases with TP53 mutations, usually have a noncomplex karyotype
- Prognostic significance is unclear (Leuk Res 2018;70:97)
- Bone marrow, right posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Chronic myelomonocytic leukemia (CMML 1) (see comment)
- Hypercellular (90%) bone marrow showing myelomonocytic and megakaryocytic hyperplasia, trilineage dysplasia, monocytosis and 5% blasts
- Comment: Flow cytometric analysis demonstrates changes consistent with myelodysplastic syndrome or related stem cell disorder. CD34+ myeloblasts are ~1.2% (note that hemodilution and other technical factors may limit the precision of blast counts by flow cytometry). They have an altered immunophenotype (CD34+, CD117+, CD13+, CD33+, CD123+, CD38 decreased). Hematogones are absent. Granulocytes show decreased side scatter, in keeping with aberrant hypogranularity. Monocytes are increased (~12%) show increased expression of CD2. No monotypic B cells or immunophenotypically abnormal T cells are detected. Fluorescence in situ hybridization is negative for BCR::ABL1. The monocyte count as indicated by the accompanying CBC / differential is noted; however, a manual 500 cell differential count of the peripheral blood smear (10/2/2020) reveals 11% monocytes resulting in an absolute monocyte count of (1.02 K/uL). A CBC obtained on 04/29/2022 also revealed relative (19%) and absolute (1.4 K/uL) monocytosis. Additional cytogenetic and molecular studies are in progress and will be reported separately.
- Peripheral smear: Manual review of the peripheral blood shows normochromic, normocytic anemia, thrombocytopenia, relative (11%) and absolute (1.02 K/uL) monocytosis and rare circulating blasts. RBCs: mild normochromic, normocytic anemia with minimal anisopoikilocytosis. WBCs: overall adequate in number with relative neutrophilia and relative and absolute monocytosis. A manual 500 cell differential count reveals 11% monocytes, resulting in an absolute monocyte count of 1.02 K/uL). Granulocytes show left shifted maturation and are dysplastic (increased hypogranular or hyposegmented forms). Monocytes include atypical forms. Rare (~1%) circulating blasts are noted. Platelets: thrombocytopenia with occasional large, hypogranular platelets.
- Bone marrow biopsy: Quality: adequate. Cellularity: 90%. Hematopoiesis: trilineage maturation with myelomonocytic hyperplasia and relative erythroid hypoplasia. Megakaryocytes are increased (15/high power field on average) and are dysplastic (pleomorphic increased small or hypolobated forms, many with hyperchromatic nuclei, rarely with separation of nuclear lobes. Also identified are large forms with abnormal nuclear lobulation). Infiltrate: no apparent increase in blasts. Special stains: loose network of reticulin without significant intersections (minimal reticulin fibrosis); trichrome is negative for collagen deposition.
- Bone marrow clot section: Quality: adequate. Cellularity: 90% morphologic features are similar to those observed in the core biopsy.
- Bone marrow aspirate: Quality: adequate. Granulocytes: increased; left shifted maturation; dysplastic (hyposegmented nuclei or hypogranular cytoplasm). Erythrocytes: progressive maturation, mild dysplasia (slight nuclear irregularities, basophilic stippling of cytoplasm or N:C maturation asynchrony). Megakaryocytes: increased; dysplastic (variable in size, increased hypolobated forms, frequently with nuclear hyperchromasia). Blasts: overall ~5% of nucleated cells, including monocytic precursors. Iron: storage iron is adequate (2+ on a scale of 0 - 4). Occasional (9%) ring sideroblasts are present.
- Myeloid neoplasms with PDGFRA, PDGFRB, FGFR1 or JAK2 rearrangement:
- Typically show eosinophilia
- Specifically exclude these entities using FISH in cases of monocytosis with prominent eosinophilia
- Chronic myeloid leukemia:
- May present with monocytosis
- Presence of BCR::ABL1
- Reactive monocytosis:
- Lacks clonal cytogenetic and molecular alterations
- Bone marrow morphology is unremarkable, dysplasia is absent
- Monocyte partitioning studies may be helpful
- Primary myelofibrosis with monocytosis:
- Megakaryocytes are large, atypical and clustered in primary myelofibrosis
- Primary myelofibrosis with monocytosis is reported to have a higher JAK2 V617F allelic burden (median: 43%; range: 20 - 62%) compared with CMML cases in which the reported JAK2 V617F allelic burden is lower (median: 17%; range: 5 - 36%) (Hum Pathol 2019;85:290)
- SRSF2 and RAS mutations
- Rare gray zone cases exist with hybrid features (Am J Surg Pathol 2018;42:799, Hum Pathol 2019;85:290)
- < 10% blasts (including monoblasts and promonocytes) in the peripheral blood and bone marrow
- Presence of ASXL1 mutation of BCR::ABL1, PDGFRA, PDGFRB or FGFR1 rearrangement or PCM1::JAK2
- Persistent absolute (≥ 0.5 x 109/L) and relative (> 10%) peripheral blood monocytosis
- Persistent absolute (≥ 1 x 109/L) and relative (> 10%) peripheral blood monocytosis
- Presence of SRSF2 mutation
1. Persistent absolute (≥ 0.5 × 109/L) and relative (≥ 10%) peripheral blood monocytosis.
2. Blasts constitute < 20% of the cells in the peripheral blood and bone marrow.
3. Not meeting diagnostic criteria of chronic myeloid leukemia or other myeloproliferative neoplasms.
4. Not meeting diagnostic criteria of myeloid/lymphoid neoplasms with eosinophilia and defining gene rearrangements (e.g. PDGFRA, PDGFRB, FGFR1, or JAK2).
Answer A is incorrect because up to 19% blasts are permitted for the diagnosis. Answer B is incorrect because ASXL1 mutation is not a required criterion and BCR::ABL1, PDGFRA, PDGFRB or FGFR1 rearrangement or PCM1::JAK2. Exclude the diagnosis of CMML. Answer D is incorrect because the minimum absolute monocyte count has been lowered to (≥ 0.5 × 109/L) in the WHO 5th edition. Answer E is incorrect because SRSF2 mutation is not a required criterion.
Comment here
Reference: Chronic myelomonocytic leukemia
A bone marrow aspirate shows the above image. A diagnosis of CMML is established. What is the most likely mutated gene in this case?
- DNMT3A
- EZH2
- IDH2
- TET2
Comment here
Reference: Chronic myelomonocytic leukemia
- Rare myeloproliferative neoplasm characterized by persistent peripheral blood neutrophilia, bone marrow hypercellularity and hepatosplenomegaly
- Requires exclusion of reactive neutrophilia as well as other myeloproliferative neoplasms such as chronic myeloid leukemia (including typical, atypical and neutrophilic variants), polycythemia vera, primary myelofibrosis or myelodysplastic / myeloproliferative neoplasm
- Persistent blood neutrophilia (≥ 25 x 109/L); with neutrophil ≥ 80% and myeloid precursors (promyelocytes, myelocytes and metamyelocytes) comprising < 10% of the white blood cells in peripheral blood
- Hypercellular bone marrow with predominance of neutrophilic granulocytes and blasts < 5%
- No dysgranulopoiesis
- CSF3R activating mutations are common
- Lacks BCR-ABL1 gene rearrangement, which distinguishes CNL from the neutrophilic variant of chronic myeloid leukemia
- Must be distinguished from reactive leukemoid reactions that can be seen in association with infectious or inflammatory processes or as a paraneoplastic phenomenon secondary to plasma cell neoplasms, lymphomas and some carcinomas
- ICD-10: D47.1 - Chronic myeloproliferative disease; also applicable to myeloproliferative disease, unspecified
- Predominantly seen in older patients, with median age 66.5 years at diagnosis (range: 15 - 86) (Leukemia 2005;19:313)
- Slight male predominance (M:F = 1.6:1) (Br J Haematol 2015;171:400)
- Peripheral blood
- Bone marrow
- Spleen
- Liver
- CSF3R activating mutations identified in 89% of cases (N Engl J Med 2013;368:1781)
- Constitutional CSF3R T618I mutation in familial CNL (Blood 2016;128:2097, Br J Haematol 1999;105:428)
- Unknown
- Some cases are associated with cytotoxic chemotherapy (Br J Haematol 2005;128:275)
- Substantial number of reported cases are associated with plasma cell neoplasm; these cases likely represent a neutrophilic leukemoid reaction secondary to production of G-CSF by neoplastic plasma cells (Br J Haematol 2015;171:400)
- Most consistent clinical feature is splenomegaly, which may be symptomatic (Cureus 2021;13:e15433, Clin Adv Hematol Oncol 2021;19:450)
- Other findings include hepatomegaly, fatigue, pruritus, easy bruising, purpura and bone pain
- World Health Organization (WHO) 2016 revised diagnostic criteria:
- Peripheral blood white blood cell count ≥ 25 x 109/L
- Segmented neutrophils plus bands neutrophils constitute ≥ 80% of the white blood cells
- Neutrophil precursors (promyelocytes, myelocytes and metamyelocytes) constitute < 10% of the white blood cells
- Myeloblasts rarely if ever observed
- Monocyte count < 1 x 109/L
- No dysgranulopoiesis
- Hypercellular bone marrow
- Neutrophilic granulocytes increased in percentage and number
- Neutrophil maturation appears normal
- Myeloblasts constitute < 5% of the nucleated cells
- Not meeting WHO criteria for BCR-ABL1 positive chronic myeloid leukemia, polycythemia vera, essential thrombocythemia or primary myelofibrosis
- No rearrangement of PDGFRA, PDGFRB or FGFR1 and no PCM1-JAK2 fusion
- CSF3R T618I or another activating CSF3R mutation or persistent neutrophilia (≥ 3 months), splenomegaly and no identifiable cause of reactive neutrophilia including absence of a plasma cell neoplasm or if a plasma cell neoplasm is present, demonstration of clonality of myeloid cells by cytogenetic or molecular studies (Pathology 2021;53:339)
- Peripheral blood white blood cell count ≥ 25 x 109/L
- Persistent mature neutrophilia
- Absence of monocytosis, basophilia, eosinophilia or increased immature myeloid cells
- Majority of patients present with mild anemia (Leukemia 2005;19:313)
- Elevated lactate dehydrogenase (LDH) and leukocyte alkaline phosphatase (LAP)
- Elevated vitamin B12 and uric acid
- Median overall survival after diagnosis, approximately 1.8 years (Curr Treat Options Oncol 2021;22:59)
- Transformation to acute myeloid leukemia can occur (Curr Hematol Rep 2004;3:210, Ann Hematol 2021;100:1459, Blood Adv 2020;4:5389)
- CSF3R T618I mutated CNL has worse prognosis compared with CNL with other CSF3R activating mutations (Blood Cancer J 2018;8:21)
- Coexistence of CSF3R and ASXL1 mutations is associated with a worse prognosis (Am J Hematol 2015;90:653)
- Risk model for CSF3R mutated CNL (Blood Cancer J 2018;8:21)
- Low risk (0 - 1 point) versus high risk (2 - 4 points) groups, based on the following criteria:
- Platelets < 160 x 109/L (2 points)
- Leukocytes > 60 x 109/L (1 point)
- ASXL1 mutation (1 point)
- Low risk (0 - 1 point) versus high risk (2 - 4 points) groups, based on the following criteria:
- 18 year old man with abdominal pain, splenomegaly and unexplained leukocytosis (Blood 2019;134:2414)
- 65 year old woman patient who presented with leukocytosis (BMC Cancer 2018;18:343)
- 70 year old woman presented with pain in the right great toe and ankle (Indian J Cancer 2020;57:201)
- 78 year old man presented with leukocytosis (Oncol Lett 2015;9:2208)
- 80 year old man with 1 month of fatigue (World J Clin Cases 2020;8:6337)
- Hydroxyurea: most commonly used agent, with most patients showing an initial response (Leukemia 2005;19:313)
- Most patients will require second or third line agents (Blood Cancer J 2018;8:21):
- Interferon alpha
- Ruxolitinib
- Thalidomide
- Cladribine
- Imatinib
- Hematopoietic stem cell transplantation is the only potentially curative therapy (Curr Treat Options Oncol 2021;22:59)
- Hypercellular bone marrow with increased myeloid to erythroid ratio; up to 20:1
- Increase in neutrophilic granulocytes
- Myeloblasts and promyelocytes are not increased
- No dysplasia in erythroid and myeloid series or megakaryocytes
- Spleen with leukemic neutrophilic infiltrate in the red pulp
- Liver with mature leukemic granulocytes in sinusoids or portal spaces
Contributed by Jingwei Li, M.D., Ph.D., Vignesh Shanmugam, M.D. and Roberto N. Miranda, M.D.

Hypercellular bone marrow with myeloid predominance
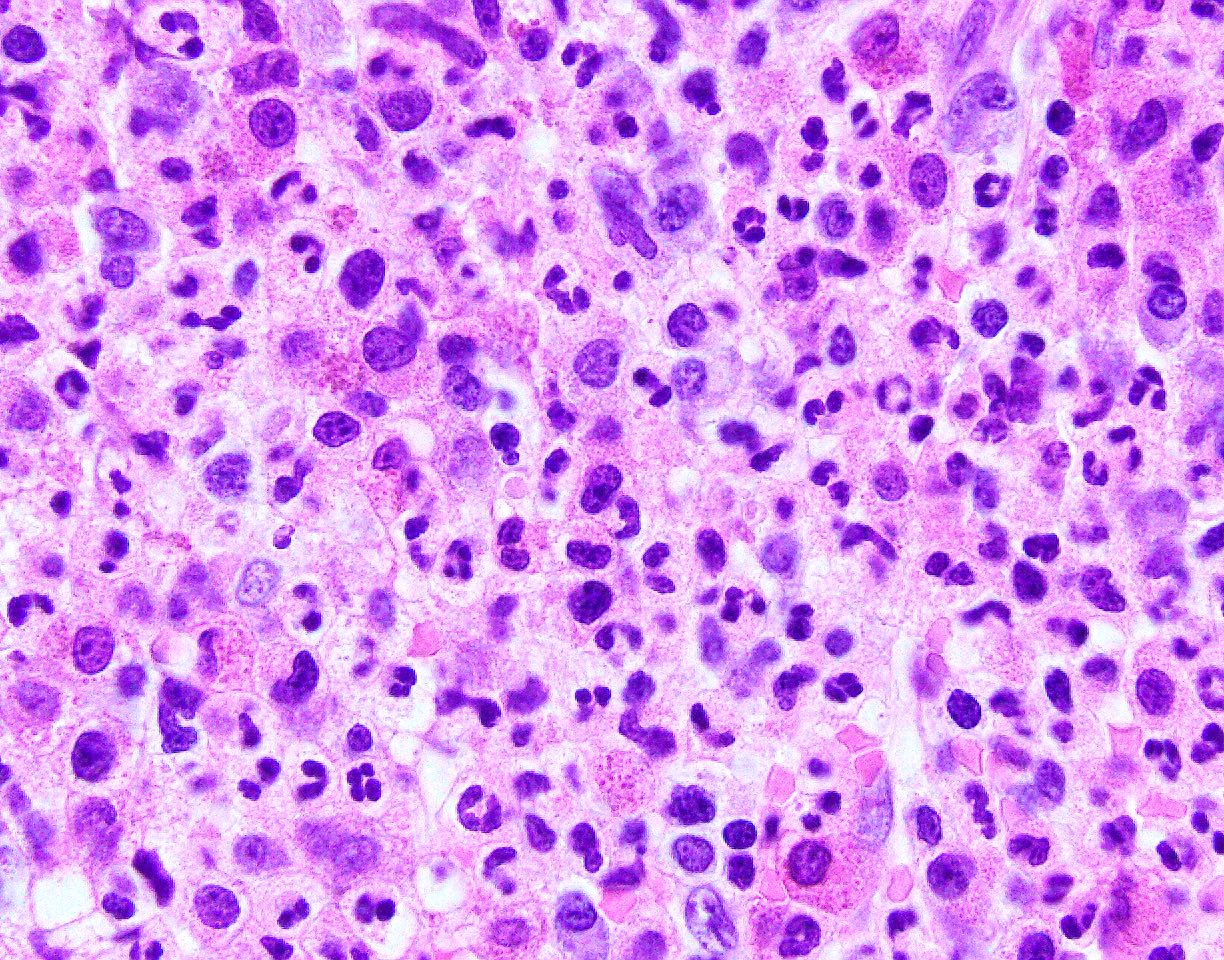
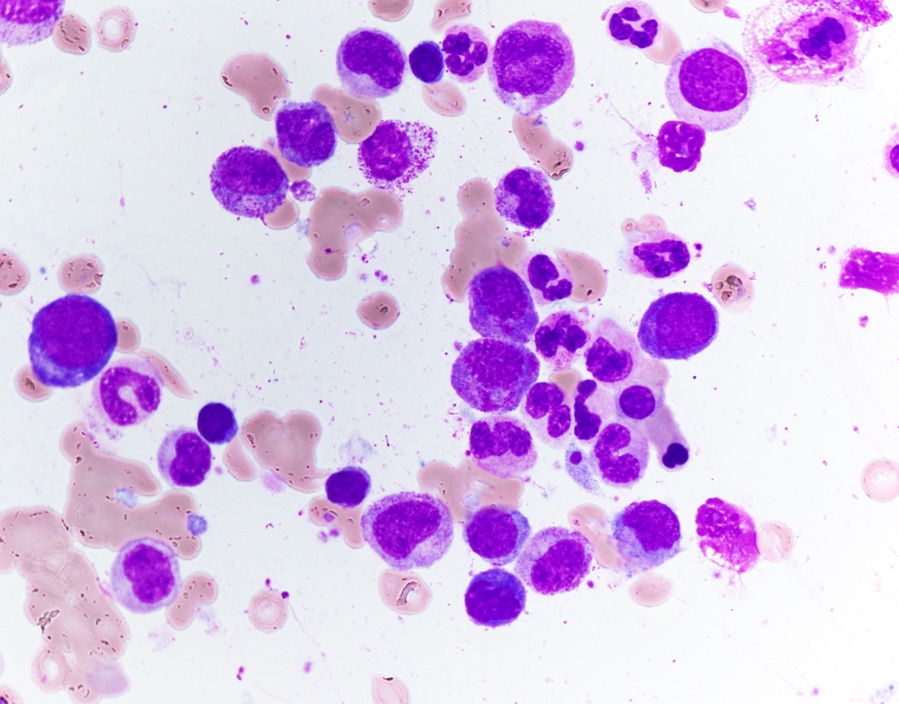
Increased neutrophilic granulocytes
Toxic granulation and Döhle bodies

Myeloid cells
- Peripheral blood with neutrophilia, including mature neutrophils and bands
- Toxic granulation and Döhle bodies in neutrophils
Contributed by Jingwei Li, M.D., Ph.D., Vignesh Shanmugam, M.D. and Roberto N. Miranda, M.D.
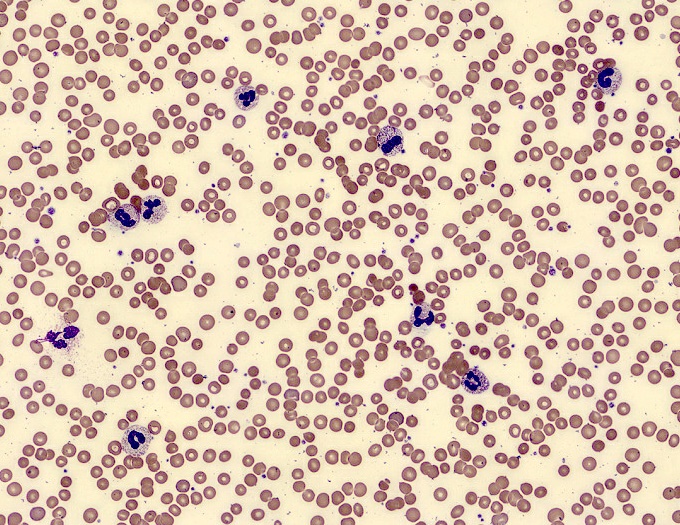
Neutrophilic leukocytosis
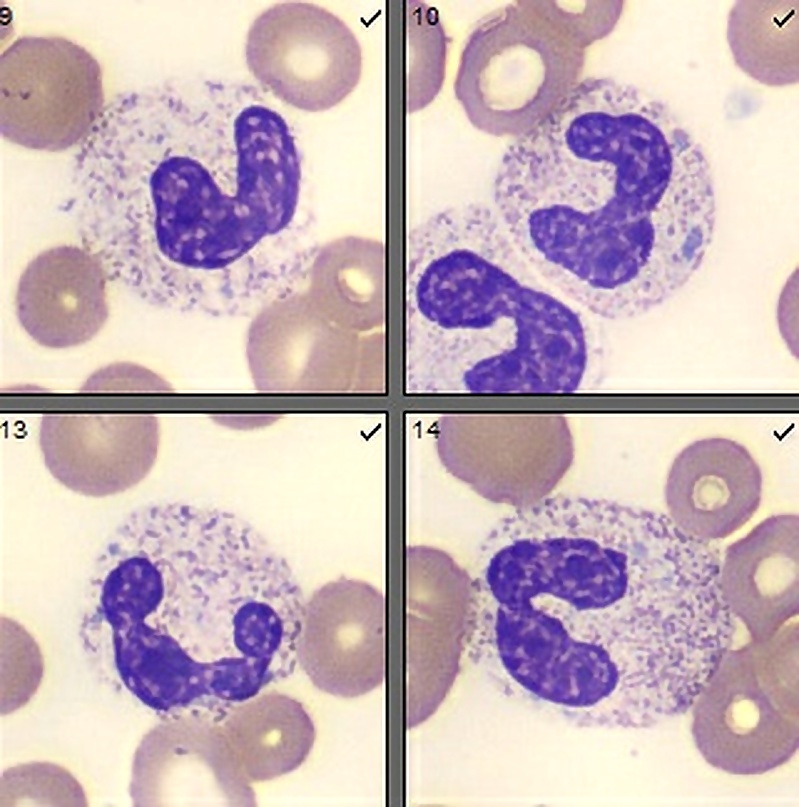
Toxic granulation and Döhle bodies
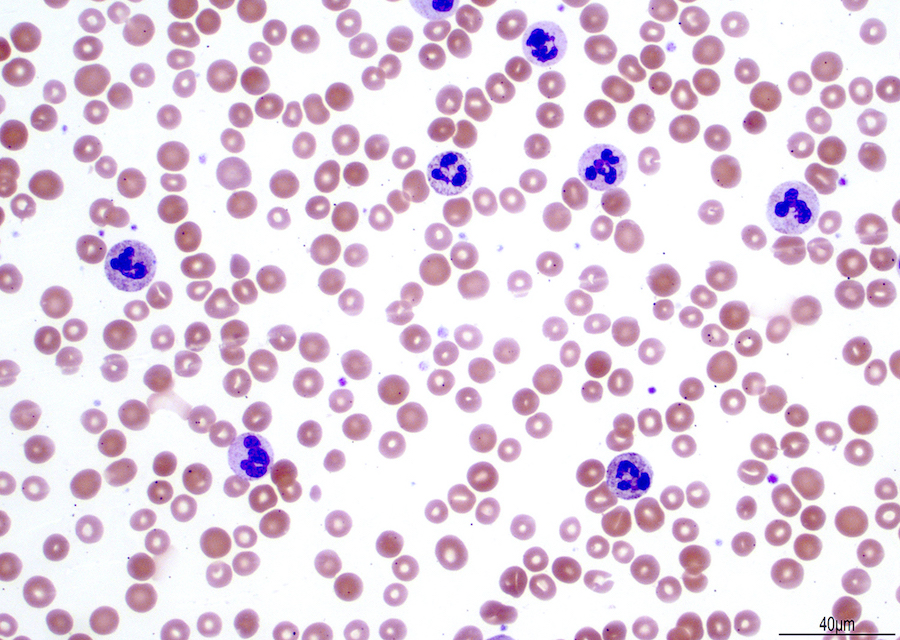
Leukocytosis
- CSF3R mutations, up to 89% in a case series (N Engl J Med 2013;368:1781)
- Can associate with ASXL1 or SETBP1 mutations
- JAK2 V617F can also be observed
- Cytogenetic studies are normal in 90% of cases
- Remaining cases show:
- Gains of chromosomes 8, 9 and 21
- del(7q), del(11q), del(12p) and del(20q)
- Nullisomy 17
- Absence of BCR-ABL1 rearrangement
- Absence of rearrangements in PDGFRA, PDGFRB, FGRF1 or PCM1-JAK2
- Bone marrow core biopsy and clot specimen, aspirate smears and peripheral blood:
- Chronic neutrophilic leukemia, 1% blasts (see comment)
- Comment:
- The bone marrow core biopsy is adequate in size and quality. The marrow is markedly cellular (> 95% cellular). Erythroid precursors are decreased and exhibit adequate maturation. Myeloid cells are markedly increased and exhibit maturation. Megakaryocytes are present in normal number and morphologically are unremarkable. Scattered mast cells are seen and do not form aggregates. Scattered lymphocytes and plasma cells are seen. Lymphoid aggregates are not identified. Bone trabeculae exhibit focal osteoblastic activity with no osteosclerosis.
- Reticulin stain shows no significant reticulin fibrosis. Trichrome stain shows no abnormal collagen fibrosis.
- Immunohistochemistry for CD34 reveals that less than 5% of marrow cells are positive. In situ hybridization for kappa and lambda immunoglobulin light chains demonstrate polytypic expression in the plasma cells.
- Bone marrow aspirate smears show a cell differential with blasts 1%, promyelocytes 2%, myelocytes 2%, metamyelocytes 2%, and granulocytes and bands aggregate to 82%; lymphocytes 3%, plasma cells 2% and erythroid precursors 4%. The myeloid to erythroid ratio is 23 (91:4).
- FISH assays did not show rearrangements of PDGFRA, PDGFRB, FGFR1 or BCR-ABL1.
- Next generation sequencing showed the following findings: Pathogenic single nucleotide variants and small insertions / deletions:
- ASXL1 NM_015338 c.1534C>T p.Q512* - in 46.6% of 1104 reads
- CSF3R NM_156039 c.1853C>T p.T618I - in 41.2% of 699 reads
- Read count analysis shows gain of JAK2 (on 9p).
- FLT3 ITD is not detected.
- Comment: The findings are of a hypercellular marrow with myeloid hyperplasia with no significant fibrosis or increase in blasts. No significant dysplasia is noted on the aspirate smear. Next generation sequencing using probes for 81 gene mutations associated with myeloid neoplasms demonstrated a canonical CSF3R (T618I) mutation along with pathogenic ASXL1 mutations. FISH analysis did not show rearrangements involving PDGFRA, PDGFRB, FGFR1 or BCR-ABL1.
- The diagnosis of chronic neutrophilic leukemia is rendered based on the presence of persistent neutrophilia (69.78 x 109/L neutrophils, immature myeloid cells < 10%) and CSF3R mutation.
- Chronic myeloid leukemia (CML):
- Associated with BCR-ABL1 fusion gene, frequent basophilia, thrombocytosis or eosinophilia
- Neutrophilic chronic myeloid leukemia (N-CML):
- Uncommon BCR-ABL1 fusion that results in a 230 kD fusion protein (p230)
- Atypical CML:
- Prominent granulocytic dysplasia and left shift
- Chronic myelomonocytic leukemia (CMML):
- Chronic monocytosis (> 1 x 109/L) and dysplasia
- Leukemoid reaction / infection:
- Rule out underlying infection or other malignancies (plasma cell neoplasms and others)
- Lacks CSF3R mutations or other cytogenetic abnormalities seen in CNL listed above in the molecular / cytogenetics section
A 53 year old man presents with abdominal pain. Abdominal imaging shows splenomegaly and a CBC shows leukocytosis (70 x 109/L) with 95% neutrophils. A representative image of the bone marrow biopsy is shown below. An extensive laboratory work up is negative for infection or other causes that could explain these findings. Molecular studies do not reveal a BCR-ABL rearrangement. What is the gene that is most commonly mutated in this condition?
- CSF3R
- JAK2
- NOTCH2
- RUNX1
- TET2
Comment Here
Reference: Chronic neutrophilic leukemia
- Lack of basophilia in CNL
- Lack of dysgranulopoiesis in CNL
- Lack of monocytosis in CNL
- Presence of ASXL1 mutation in CNL
- WBC count > 50 x 109/L
Comment Here
Reference: Chronic neutrophilic leukemia
- See also topic in Kidney nontumor chapter
- Cryoglobulinemia is a clinical disorder defined as the presence of immunoglobulins that precipitate below 37°C but redissolve upon warming
- Cryoglobulinemia is a clinical entity characterized by the presence of cryoglobulins in the serum
- Cryoglobulins precipitate below normal body temperature (37°C) and dissolve on rewarming
- Underlying causes of cryoglobulinemia include B cell / plasma cell malignances, infections such as hepatitis C virus and collagen vascular disorders such as Sjögren syndrome
- Clinical features of cryoglobulinemia include the triad of purpura, arthralgia and weakness
- ICD-10: D89.1 - cryoglobulinemia
- Overall prevalence is unknown (Lancet 2012;379:348)
- Cryoglobulinemia is associated with infection (most frequently hepatitis C), autoimmune disease (e.g. Sjögren syndrome) and malignancy (B cell / plasma cell hematopoietic neoplasms)
- 12 - 56% of hepatitis C virus patients have circulating cryoglobulins
- Prevalence in patients coinfected with HIV and hepatitis C virus is 35 - 64%
- Cold exposure leads to precipitation of cryoglobulins (typically in distal extremities) (Lancet 2012;379:348)
- Internal organ involvement may occur
- 3 types exist: type I (monoclonal IgM or monoclonal IgG), type II (mixture of monoclonal IgM and polyclonal IgG) and type III (polyclonal IgM and polyclonal IgG) (Blood 2017;129:289)
- Type I cryoglobulinemia is characterized by hyperviscosity and occlusion of small blood vessels
- In type II and III cryoglobulinemia, immune complexes are formed between monoclonal / polyclonal IgM and polyclonal IgG with RF formation and complement fixation
- In hepatitis C virus (HCV) associated cryoglobulinemia, HCV drives a clonal B cell expansion producing monoclonal IgM that binds to anti-HCV IgG, forming immune complexes that promote vasculitis
- Underlying causes of type I cryoglobulinemia include plasma cell myeloma, monoclonal gammopathy of undetermined significance (MGUS) and Waldenström macroglobulinemia (Am J Hematol 2011;86:500)
- Causes of the mixed cryoglobulinemias (type II and III) include hepatitis C, HIV, Sjögren syndrome, rheumatoid arthritis, systemic lupus erythematosus and essential (idiopathic) cryoglobulinemia
- Purpura, arthralgia and weakness are noted in 80% of patients (Lancet 2012;379:348)
- Type I cryoglobulinemia patients may present with hyperviscosity syndrome
- Flares of immune complex mediated vasculitis can be seen
- Joint pain may be present without inflammation
- 20% of patients present with nephropathy
- 17 - 60% present with peripheral neuropathy
- Less common manifestations include gastrointestinal involvement, pulmonary involvement and myocardial vasculitis
- Cryoglobulins are identified by collecting blood (transported at 37°C) in a red top tube and dividing the serum into 3 tubes (Am J Hematol 2011;86:500)
- First tube is used to quantify the amount of cryoprecipitate (cryocrit) by cooling to 4°C for up to 72 hours
- Second tube is used to identify and analyze the cryoprecipitate
- Third tube is used to demonstrate solubility of the cryoprecipitate on rewarming to 37°C
- Diagnosis of underlying conditions is made on clinical, laboratory and histopathologic findings
- Monoclonal gammopathy in types I and II (Lancet 2012;379:348)
- Hyperviscosity
- Unexplained low complement (C4)
- Unexplained high rheumatoid factor
- Raised serum creatinine
- Microscopic hematuria and red blood cell casts
- Worse outcome with older age, renal insufficiency, pulmonary involvement and gastrointestinal involvement
- 47 year old man with persistent mixed cryoglobulinemia despite successful treatment of hepatitis C, aggressive B cell directed therapies and long term plasma exchanges (Kidney Int Rep 2019;4:1194)
- 55 year old woman with IgG4 related sialadenitis complicated with type III mixed cryoglobulinemia (Medicine (Baltimore) 2019;98:e16571)
- 56 year old man with type I immunoglobulin M cryoglobulinemic vasculitis with chronic lymphocytic leukemia and a history of hepatitis C virus (Cureus 2019;11:e4729)
- 58 year old woman with severe cutaneous necrosis associated with type I cryoglobulinemia (JAAD Case Rep 2019;5:736)
- Treatment is based on underlying pathogenesis (Lancet 2012;379:348)
- Conventional immunosuppression: similar to treatment of other vasculitides
- Apheresis: removes cryoglobulin from circulation
- Antiviral therapy: for hepatitis C virus related cryoglobulinemia
- Biologics: rituximab driven B cell depletion
- Precipitated cryoglobulin appear as hyaline thrombi that occlude blood vessels (Lancet 2012;379:348)
- In the kidney, glomeruli may show a membranoproliferative pattern of injury with PAS positive intracapillary deposits and double contour basement membrane
- Vasculitis may involve small and medium sized blood vessels; skin, kidney and nerves are most commonly affected
Contributed by Maria M. Picken, M.D., Ph.D.
Cryoglobulinemic nephropathy
- Cryoglobulins (seen as pale staining material), rouleaux formation, pseudothrombocytosis
Contributed by Anas Nasir, M.Sc.
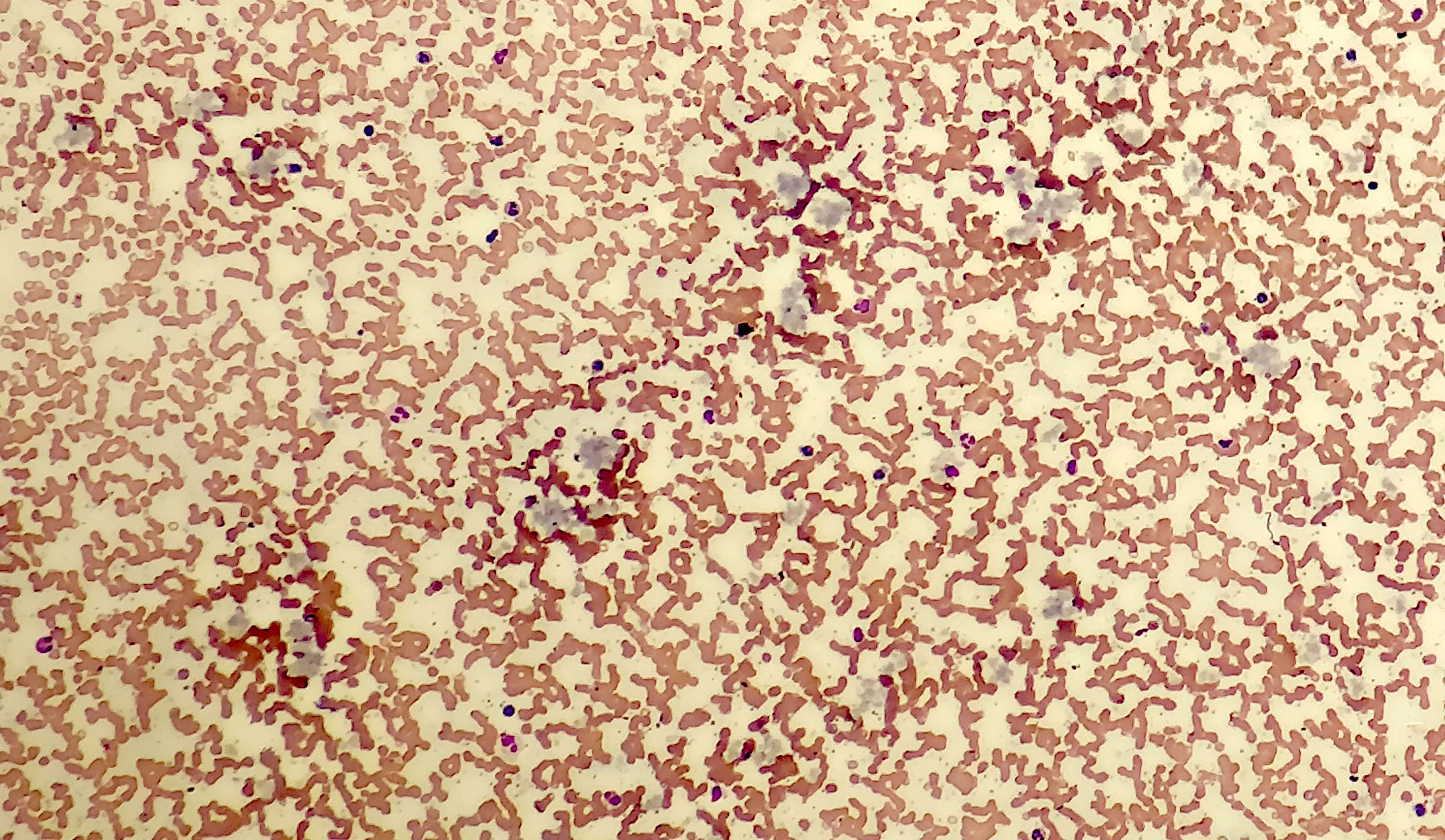

Circulating cryoglobulins
Managing HCV cryoglobulinemia
Cryoglobulinemia and vasculitis
Cryoglobulinemia, lymphoproliferation and hepatitis C
- Other vasculitides (hypersensitivity vasculitis, IgA vasculitis, ANCA associated vasculitis and infection related vasculitis):
- Cryoglobulins are not detected in these entities
A 62 year old woman has increasing weight loss, fatigue and lack of appetite over the past 7 months. She is now complaining of intermittent headaches, dizziness and decreased vision bilaterally. Her hands get cold easily and she gets frequent nosebleeds. On physical exam, she has generalized lymphadenopathy. Laboratory studies show presence of cryoglobulin. Serum electrophoresis / immunofixation is pictured above (ELP = serum electrophoresis, G = IgG, A = IgA, M = IgM, K = kappa, L = lambda). Which underlying condition is most likely in this patient?
- Henoch-Schönlein purpura
- Hepatitis C infection
- Sjögren syndrome
- Systemic lupus erythematosus
- Waldenström macroglobulinemia
- Anti centromere antibody
- Anti desmosome antibody
- Anti glomerular basement antibody
- Anti hemidesmosome antibody
- Anti hepatitis C antibody
- Crescent moon shape on light microscopy
- Mesangial expansion with eosinophilic nodular glomerulosclerosis on light microscopy
- Normal renal biopsy
- PAS positive intracapillary deposits with double contour basement membrane
- Podocyte hypertrophy, capillary wall thickening and diffuse glomerular sclerosis
Comment Here
Reference: Cryoglobulinemia
- Aggressive T cell neoplasm with immunophenotypic markers of early T cell differentiation, including stem cell markers or myeloid differentiation markers
- Histologic morphology of lymphoblastic leukemia / lymphoma
- Early T cell precursor (ETP) blasts have the following immunophenotype: positive for cytoplasmic CD3, absent (< 5% positive blasts) CD1a and CD8 expression, absent or dim (< 75% positive blasts) CD5 expression, positive (≥ 25% positive blasts) for ≥ 1 myeloid (CD11b, CD13, CD33, CD65, CD117) or stem cell (CD34, HLA-DR) marker and negative (< 3%) myeloperoxidase (Lancet Oncol 2009;10:147)
- 12 - 17% of pediatric acute lymphoblastic leukemia (ALL) (Lancet Oncol 2009;10:147)
- 22 - 40% of adult ALL (Lancet Haematol 2016;3:e80, Lancet Oncol 2009;10:147)
- M:F = 4:1
- Incidence of 3 cases per million person years; does not vary by ethnicity
- Can involve the bone marrow, thymus, lymph nodes or extranodal sites
- Physiologically, early T cell precursors characteristically migrate from bone marrow to thymus
- They express lymphoid, myeloid and stem cell markers and are characterized by multilineage differentiation potential (Nature 2008;452:764, Nature 2008;452:768)
- Single cell RNA sequencing in ETP ALL demonstrates leukemic cell plasticity, with both stem cell and T cell signatures and can differentiate to stimulate an immunosuppressive microenvironment through expression of the LGALS9 checkpoint molecule and support of dysfunctional CD8 positive T cells (Blood 2021;137:2463)
- ETP ALL shows increased expression of MEF2C and decreased expression of BCL11B and GATA3 (Cancer Cell 2011;19:484, Blood 2021;138:773, Br J Haematol 2021;194:1034, J Exp Med 2009;206:2987, PLoS One 2013;8:e53190)
- Concurrent EZH2 and RUNX1 inactivating mutations in a recent murine molecular pathogenesis model were hypothesized to be the initiating events that lead to expansion of early thymic progenitors with an ETP ALL gene expression signature
- Leukemic potential is further stimulated by acquisition of mutations (e.g., FLT3), resulting in constitutive activation of the RAS signaling pathway (Cancer Cell 2018;33:274)
- Pathogenesis in this model is similar to that of myeloid neoplasms, by harboring activating mutations in genes regulating cytokine receptor and RAS signaling (FLT3, NRAS, KRAS, IL7R, JAK3, JAK1, SH2B3 and BRAF), hematopoietic development (GATA3, ETV6, RUNX1, IKZF1) and histone modifications (EZH2, EED, SUZ12, SETD2, EP300) (Nature 2012;481:157, Blood 2013;121:4749, PLoS One 2013;8:e53190)
- NOTCH1 mutations are uncommon in ETP ALL, in contrast to other T lymphoblastic lymphoma / leukemia (T ALL) (PLoS One 2013;8:e53190)
- Frequency of CDKN2A / CDKN2B deletion is low in patients with ETP ALL (Haematologica 2020;105:e294, Am J Hematol 2021;96:312, Blood Adv 2021;5:2890)
- Nonrearranged TRG gene rearrangement is present in most ETP ALL patients; they may lack more frequent biallelic deletion of the TRG locus than non-ETP ALL, confirming early maturation stage (PLoS One 2013;8:e53190, Haematologica 2014;99:94)
- Unknown etiology in most cases
- A study has demonstrated an association between germline RUNX1 variants and ETP ALL; it has been observed that mice with RUNX1 variants and mutated JAK3 induce ETP ALL (J Clin Invest 2021;131:e147898)
- Cervical, supraclavicular and axillary lymphadenopathy (50%)
- Mediastinal mass (50 - 75%) with associated compressive superior vena cava syndrome, tracheal obstruction and pericardial effusion (Blood Cancer J 2012;2:e55)
- CNS involvement (18%) (Blood Cancer J 2012;2:e55)
- Features related to extramedullary hematopoiesis (such as hepatosplenomegaly)
- Morphologic features of lymphoblastic leukemia / lymphoma (Lancet Oncol 2009;10:147)
- Blasts with expression of ETP immunophenotype: positive for cytoplasmic CD3, absent (< 5% positive blasts) CD1a and CD8 expression, absent or dim (< 75% positive blasts) CD5 expression, positive (≥ 25% positive blasts) for ≥ 1 myeloid (CD11b, CD13, CD33, CD65, CD117) or stem cell (CD34, HLA-DR) marker and negative (< 3%) myeloperoxidase (Lancet Oncol 2009;10:147, Blood 2016;127:1863)
- White blood count (WBC) may or may not be elevated and in some studies, ETP ALL patients tend to have lower WBC in comparison to those with non-ETP ALL (Pediatr Blood Cancer 2021;68:e28719)
- Anemia and thrombocytopenia are secondary to bone marrow involvement
- Several studies showed that long term outcomes for pediatric and adult ETP ALL patients were similar to those of other T ALL patients (Br J Haematol 2014;166:421)
- Favorable outcomes were reported despite higher rates of minimal residual disease (MRD) positive disease postinduction among ETP ALL patients; this can be because of treatment intensification for MRD positive ETP ALL patients and the increased rate of allogeneic stem cell transplantation after first remission (Pediatr Blood Cancer 2021;68:e28719, Blood Cancer Discov 2021;2:326)
- Because of BCL2 overexpression in ETP ALL, clinical trials are studying BCL2 inhibition as a potential treatment (Haematologica 2020;105:e294, Cancer Discov 2014;4:1074, Cancer Discov 2021;11:1440)
- 42 year old woman presented with mild thrombocytopenia and generalized lymphadenopathy in the absence of constitutional symptoms (Hemasphere 2020;4:e379)
- 43 year old woman presented with fatigue, pancytopenia and increased blasts in the peripheral blood smear (Onco Targets Ther 2021;14:3795)
- 117 newly diagnosed children with T ALL with varying disease free survival rates based on treatment regimen (J Cancer Res Clin Oncol 2021;147:2775)
- Myeloid oriented chemotherapy (as frontline induction treatment) along with gene targeting inhibitors, followed by allogeneic hematopoietic stem cell transplant
- Early T cell precursor acute lymphoblastic leukemia: diagnosis, updates in molecular pathogenesis, management and novel therapies (Front Oncol 2021;11:750789, Onco Targets Ther 2021;14:3795)
- As seen in patients with other ALL, blasts from patients with ETP ALL are small to medium sized with scant cytoplasm and inconspicuous nucleoli
Contributed by Kamran M. Mirza, M.D., Ph.D. and Yazan Alhalaseh, M.D.
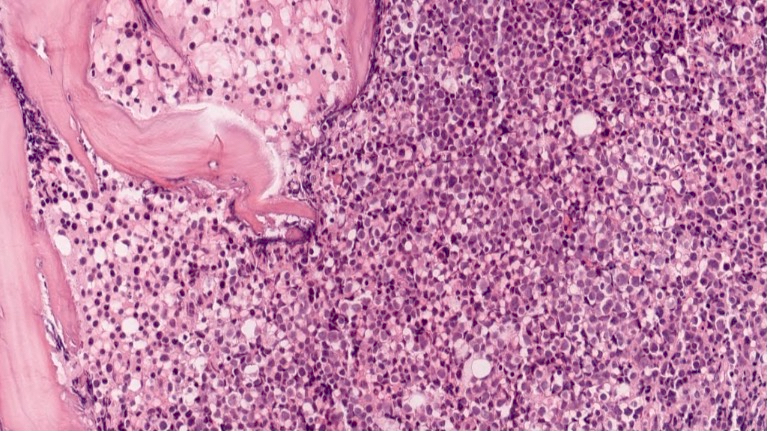
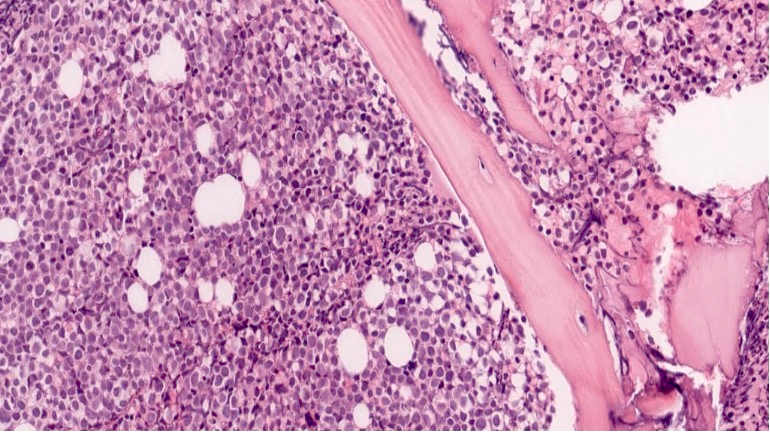
Hypercellular bone marrow core biopsy
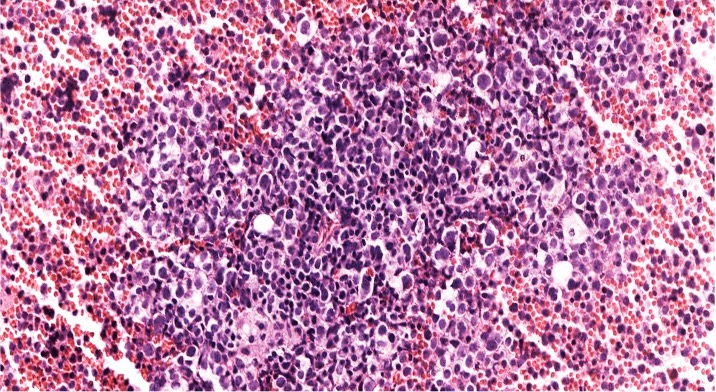
Hypercellular bone marrow clot section
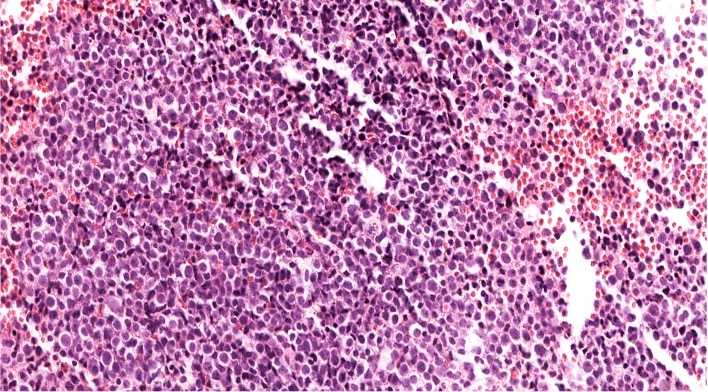
Hypercellular bone marrow
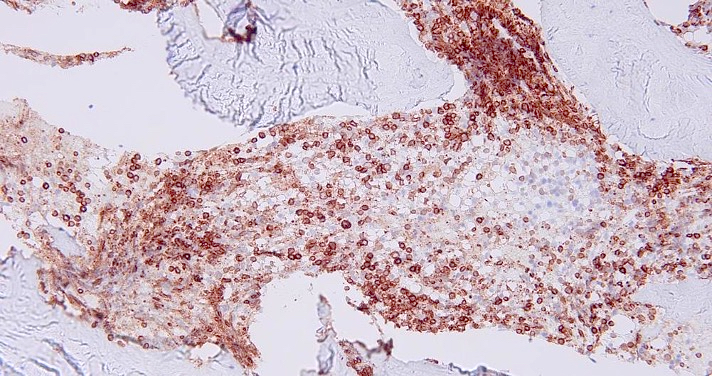
CD3 IHC
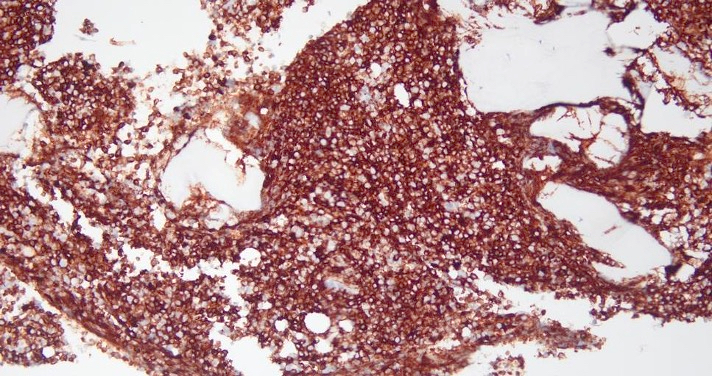
CD34 IHC
- Peripheral blood smear reveals increase in circulating blasts with high N:C ratio
Contributed by Kamran M. Mirza, M.D., Ph.D. and Yazan Alhalaseh, M.D.
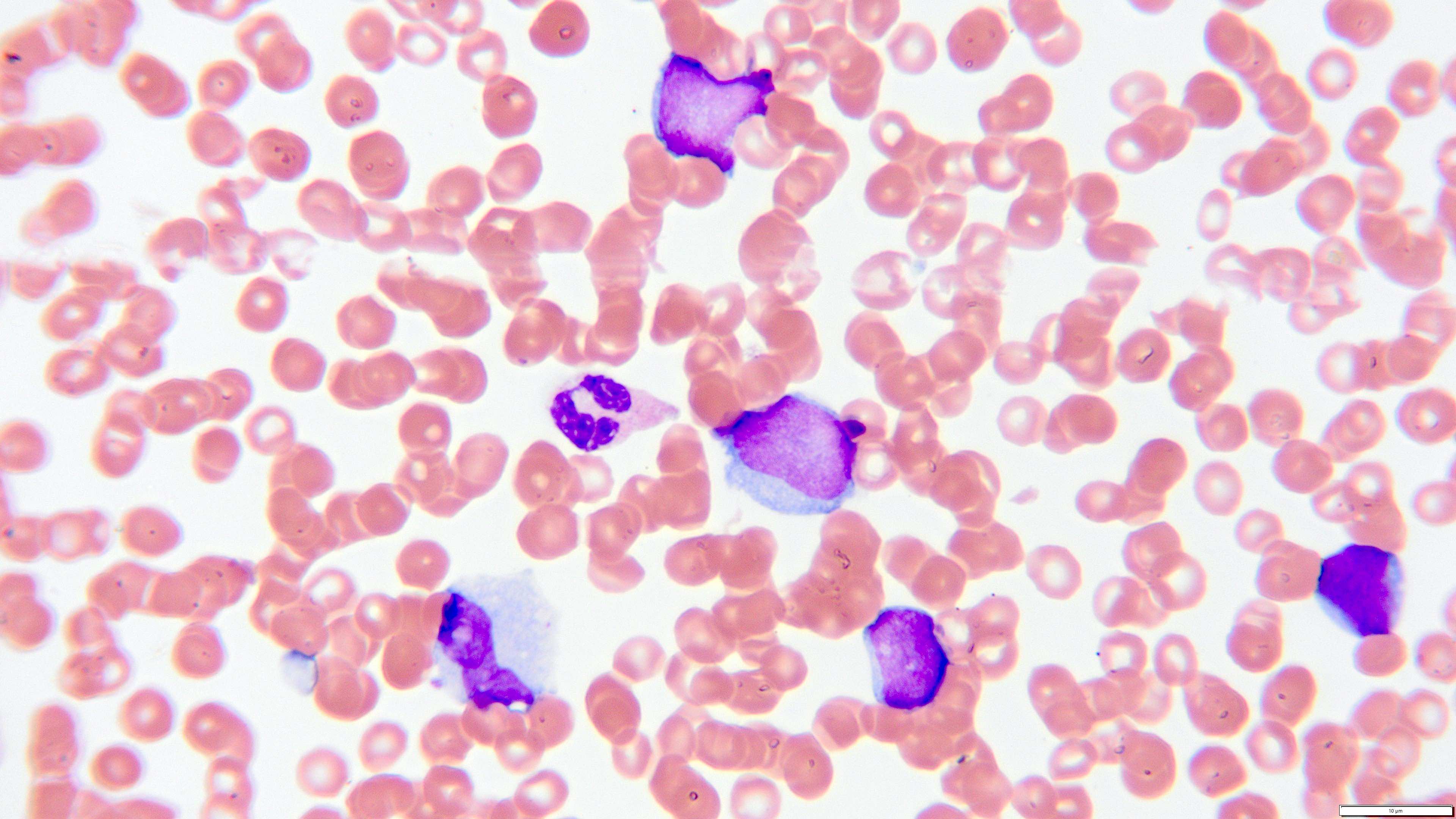
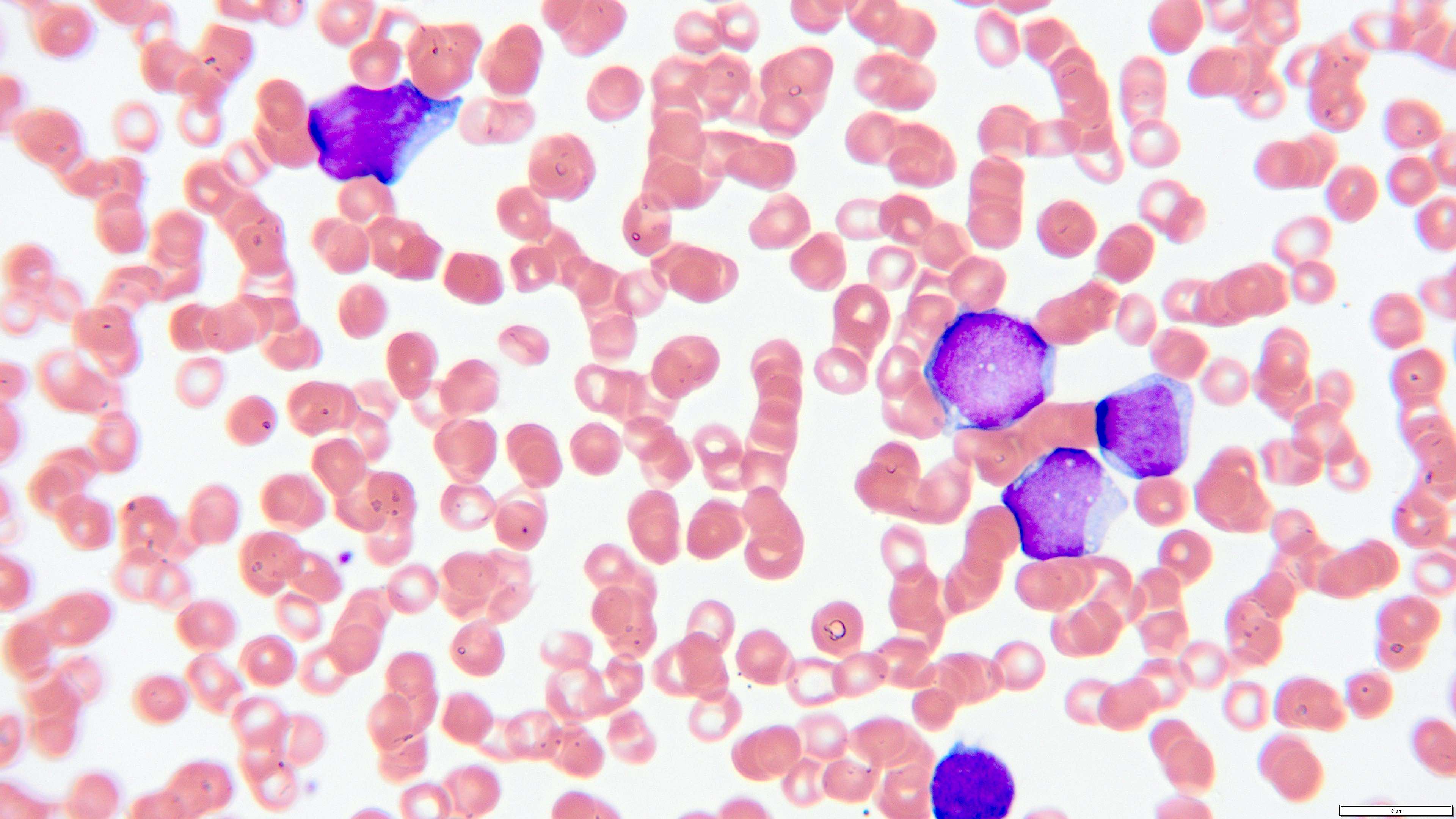
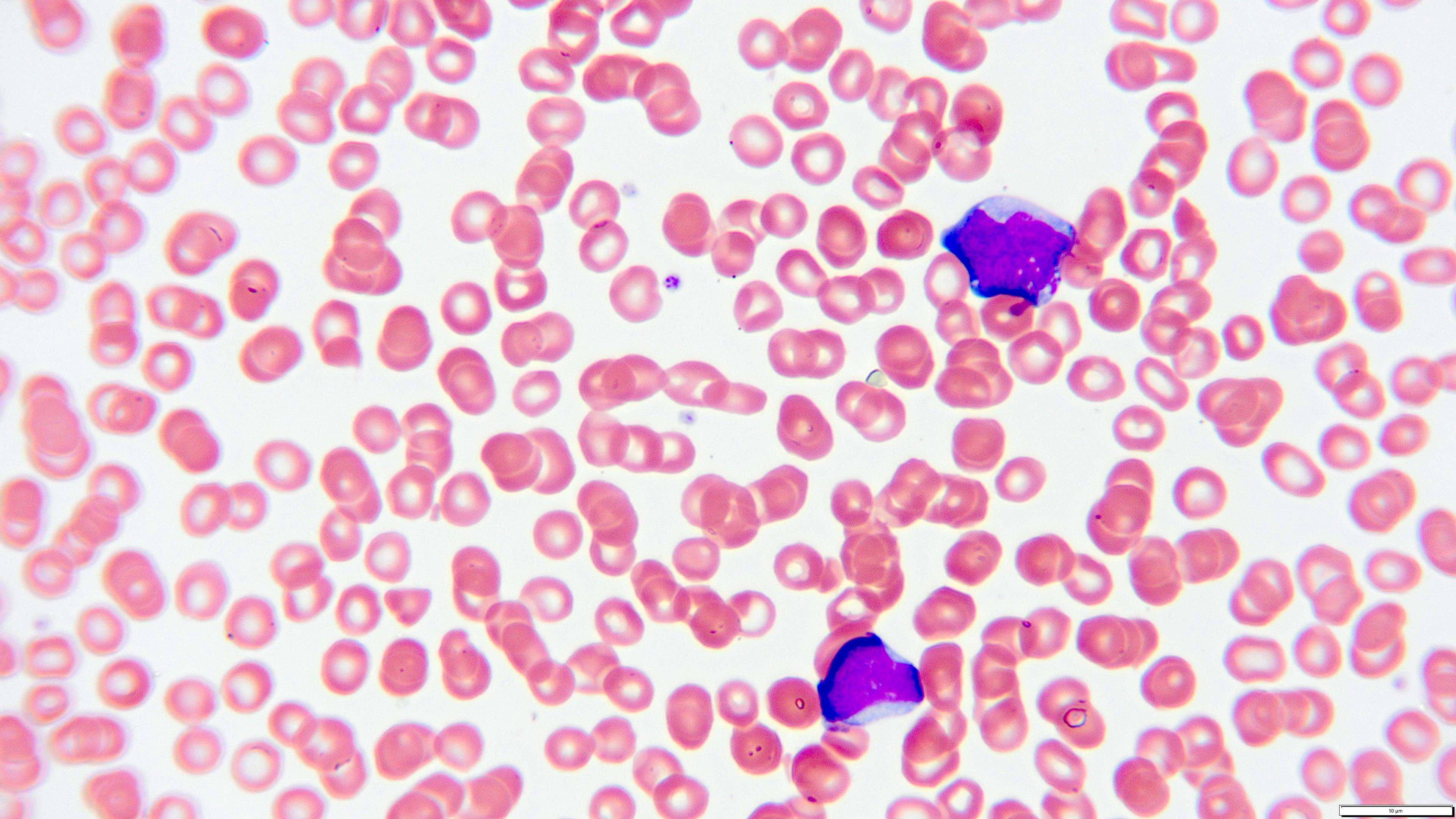
Atypical circulating blasts
- Positive for cytoplasmic CD3, absent (< 5% positive blasts) CD1a and CD8 expression (Lancet Oncol 2009;10:147, Blood 2016;127:1863)
- Positive (≥ 25% positive blasts) for ≥ 1 myeloid (CD11b, CD13, CD33, CD65, CD117) or stem cell (CD34, HLA-DR) markers (Lancet Oncol 2009;10:147, Blood 2016;127:1863)
- Absent or dim (< 75% positive blasts), CD5 expression (Lancet Oncol 2009;10:147, Blood 2016;127:1863)
- Negative (< 3%) myeloperoxidase (MPO) (Lancet Oncol 2009;10:147, Blood 2016;127:1863)
- Flow cytometry phenotyping of the blasts (dim CD45 gate) feature (Lancet Oncol 2009;10:147)
Contributed by Kamran M. Mirza, M.D., Ph.D. and Yazan Alhalaseh, M.D.
Side scatter versus CD45 panel
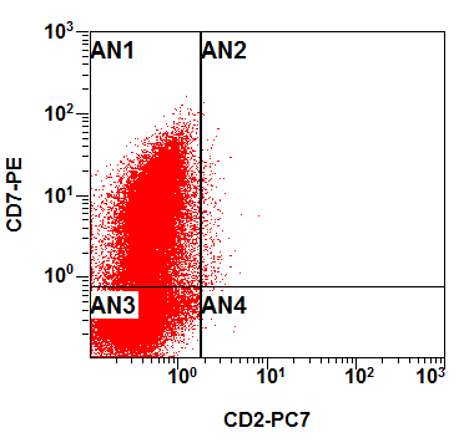
CD7 versus CD2 panel
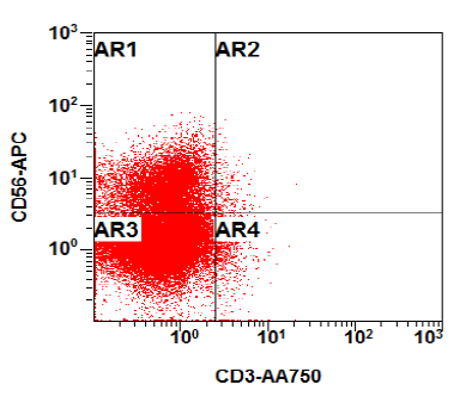
CD56 versus CD3 panel
- Not relevant for diagnostic evaluation
ETP ALL case
- Right posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprints:
- T lymphoblastic leukemia / lymphoma (T ALL phenotypically compatible with ETP ALL) (see comment)
- Hypercellular marrow with 87% blasts and markedly diminished trilineage hematopoiesis
- Correlation with cytogenetics is recommended for complete assessment
- Eosinophils (2%), lymphocytes (2%), plasma cells (1%) (500 cell count on aspirate smear)
- Comment: The peripheral blood film reveals 76% circulating blasts with high N:C ratio. The most recent hemogram shows leukocytosis, anemia and thrombocytopenia.
- The bone marrow core is fragmented and reveals sheets of blasts replacing normal bone marrow elements. The clot shows similar findings. Immunohistochemical stains (CD34, TdT, CD3) on the core and clot reveal blast positivity for CD34 and CD3. Stains for MPO and TdT are negative. Cytochemical analysis for MPO and alpha naphthyl acetate esterase (ANAE) stains are performed. The blasts are negative for MPO and ANAE by cytochemical as well.
- The marrow aspirate smears reveal markedly diminished normal trilineage hematopoiesis and blasts for 87% of a 500 cell differential count. The blasts have high N:C ratio, smudged chromatin with irregular nuclear membranes and occasional cytoplasmic vacuoles. Flow cytometry phenotyping of the blasts (dim CD45 gate) reveals positivity for cytoplasmic CD3, CD34, dim CD4, CD56, CD38, CD33, CD13 and CD117. The blasts are negative for MPO, surface CD3, CD5, CD8, CD22, CD10, CD20, HLA-DR, CD11c, CD64, CD14, TdT and cytoplasmic CD22. Gating on the mature lymphocytes reveals no monotypic B cell population or T cell abnormality based on markers assayed.
- Overall, the presence of cytoplasmic CD3 expression by flow cytometry and CD3 positivity by immunohistochemistry with concurrent lack of MPO (flow cytometry, immunohistochemistry and cytochemistry) monocytic markers and B cell antigens is diagnostic of T lymphoblastic leukemia / lymphoma. Please correlate with corresponding cytogenetics and FISH findings for complete assessment. The phenotype (CD3, CD4, CD7, CD33, CD13 positive, with negativity for MPO, CD8 and CD5) is compatible with ETP ALL (Nature 2012;481:157).
- Mixed phenotype acute leukemia (MPAL), T / myeloid:
- MPAL blasts express both T and myeloid lineage markers
- Cytoplasmic CD3, CD7 are expressed in the majority of cases
- CD2, CD4, CD5 and CD8 may also be present (Curr Oncol Rep 2021;23:22, Hum Pathol 2021;114:66)
- Surface CD3 expression is uncommon but may occur in an isolated population of T lineage blasts (Curr Oncol Rep 2021;23:22)
- Myeloid lineage markers: myeloperoxidase, CD13, CD15, CD33, CD65 and CD117
- Some cases meet the criteria for monocytic differentiation and express monocytic markers such as CD11c, CD14, CD36 and CD64 (Blood 2011;117:3163, Cytometry B Clin Cytom 2019;96:183, Curr Oncol Rep 2021;23:22)
- Bilineage (2 populations) or biphenotypic (single population) lineage expression patterns are identified in MPAL, including MPAL T / M
- A subset of early T precursor lymphoblastic leukemia / lymphoma (ETP ALL) may have immunophenotypic profiles that overlap with MPAL T / M
- In the initial definition of ETP ALL, a threshold of < 3% to define negative myeloperoxidase expression was adopted and may be a helpful distinctive feature (Lancet Oncol 2009;10:147)
- MPAL blasts express both T and myeloid lineage markers
- Chronic myeloid leukemia
- Early T cell precursor lymphoblastic leukemia
- Myelodysplastic neoplasm
- Natural killer cell (NK) large granular lymphocytic leukemia
Answer C (myelodysplastic neoplasm) is incorrect because the patient's blasts are significantly increased in the peripheral blood and bone marrow biopsy. Answer A (chronic myeloid leukemia) is incorrect because it can have increased blasts in the blast phase; however, the absence of BCR::ABL1 translocation rules out this diagnosis. Answer D (natural killer cell [NK] large granular lymphocytic leukemia) is incorrect because it is not characterized by increased CD34+ blasts.
Comment Here
Reference: Early T cell precursor lymphoblastic leukemia
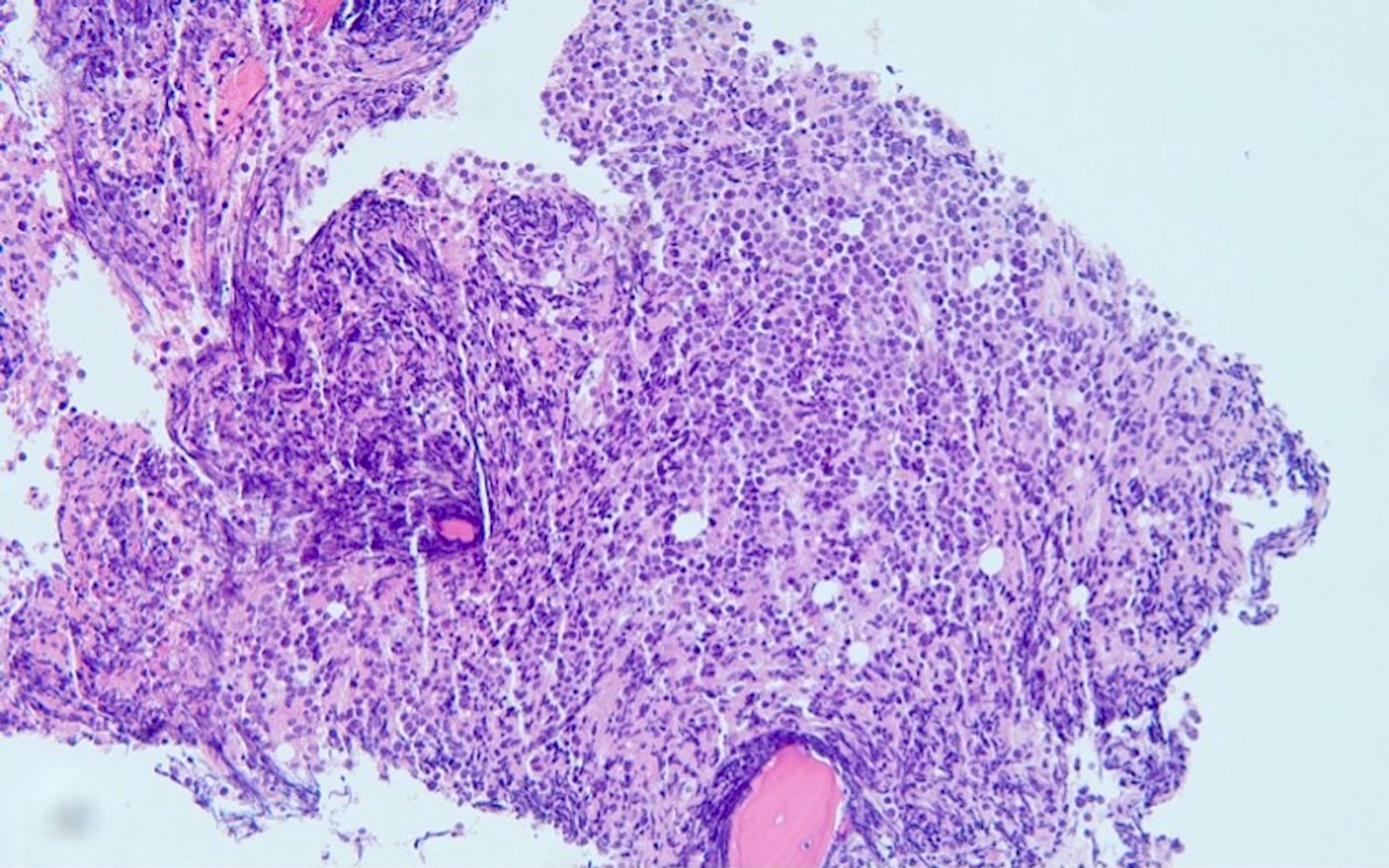
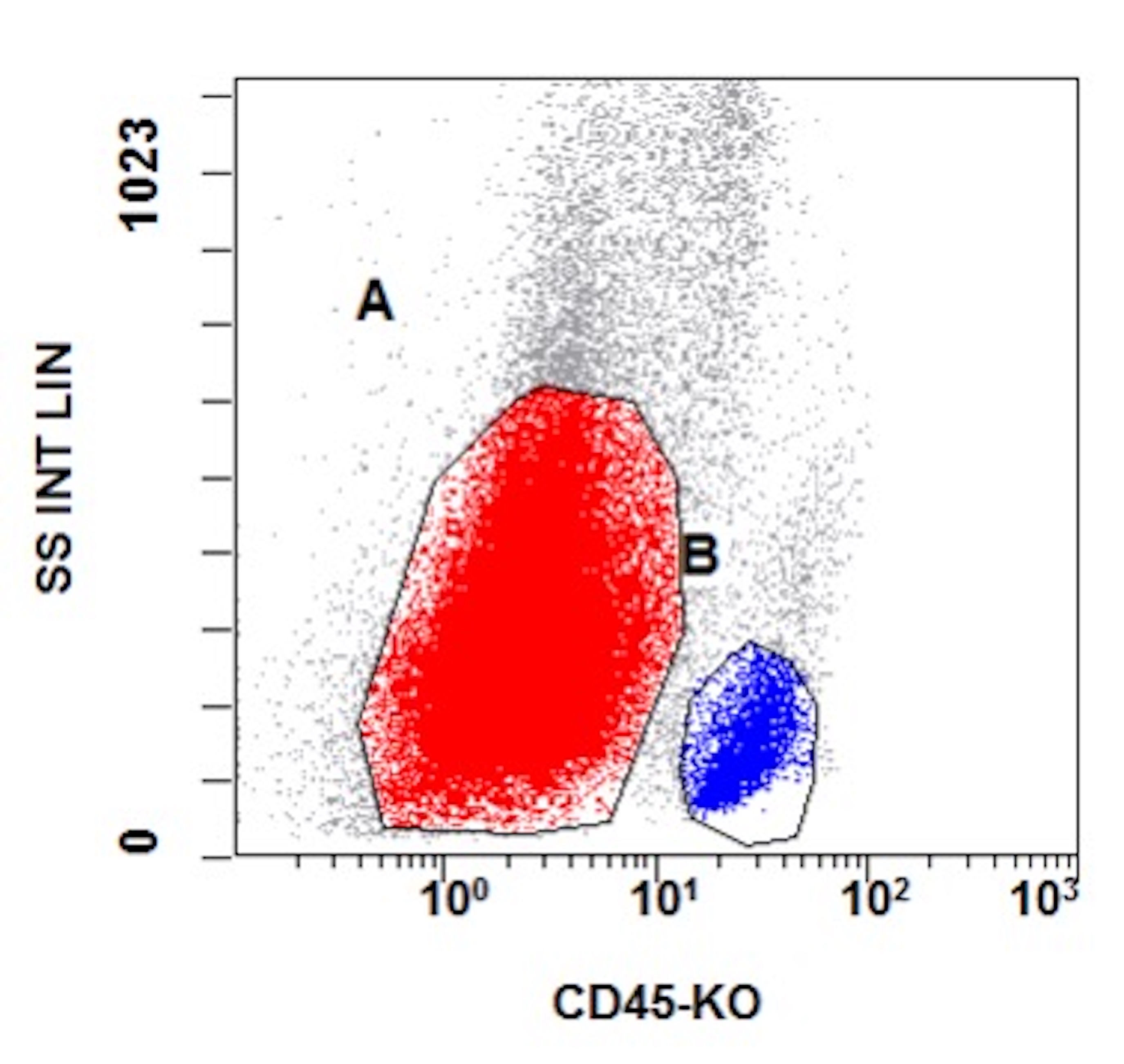
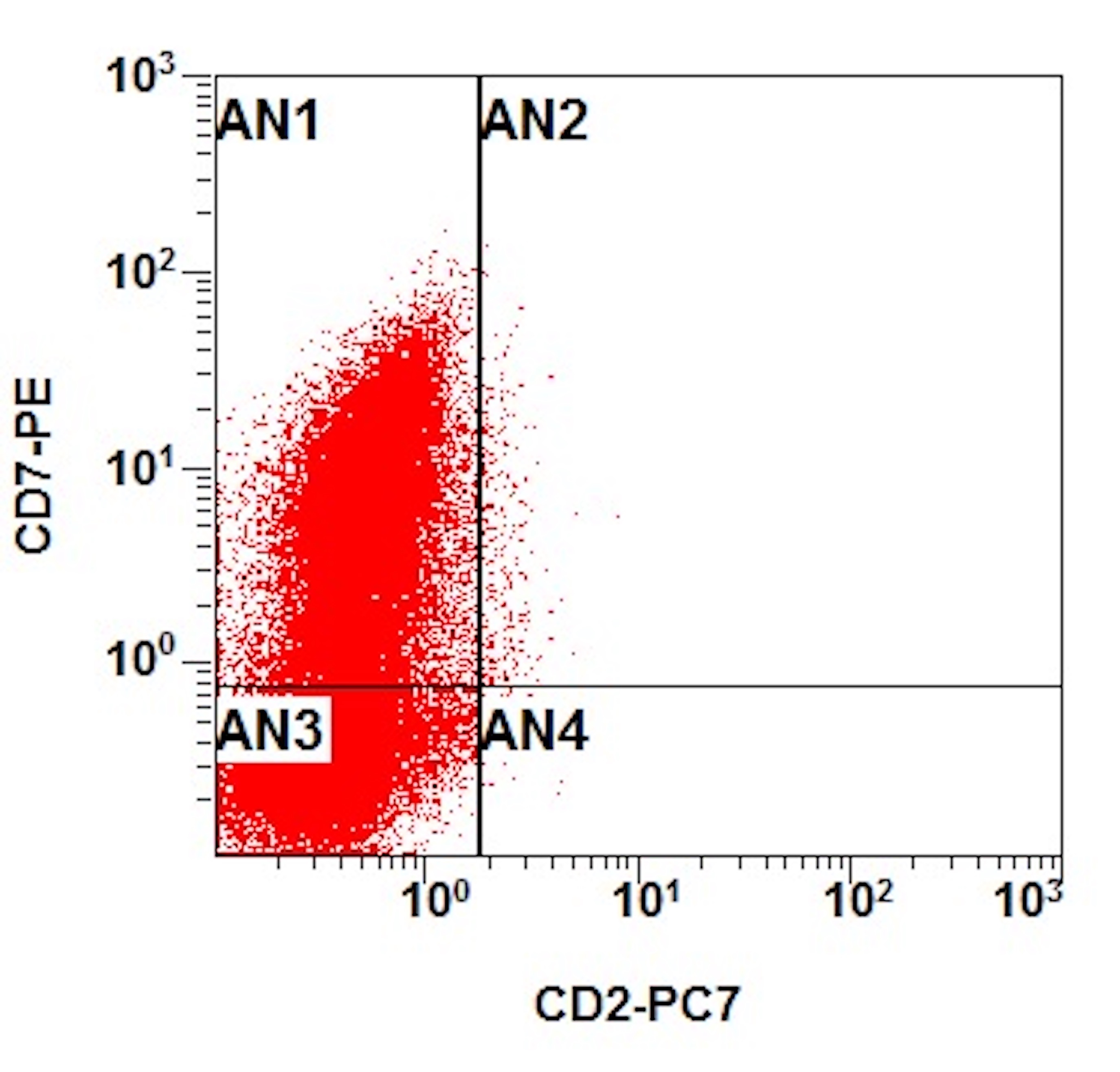
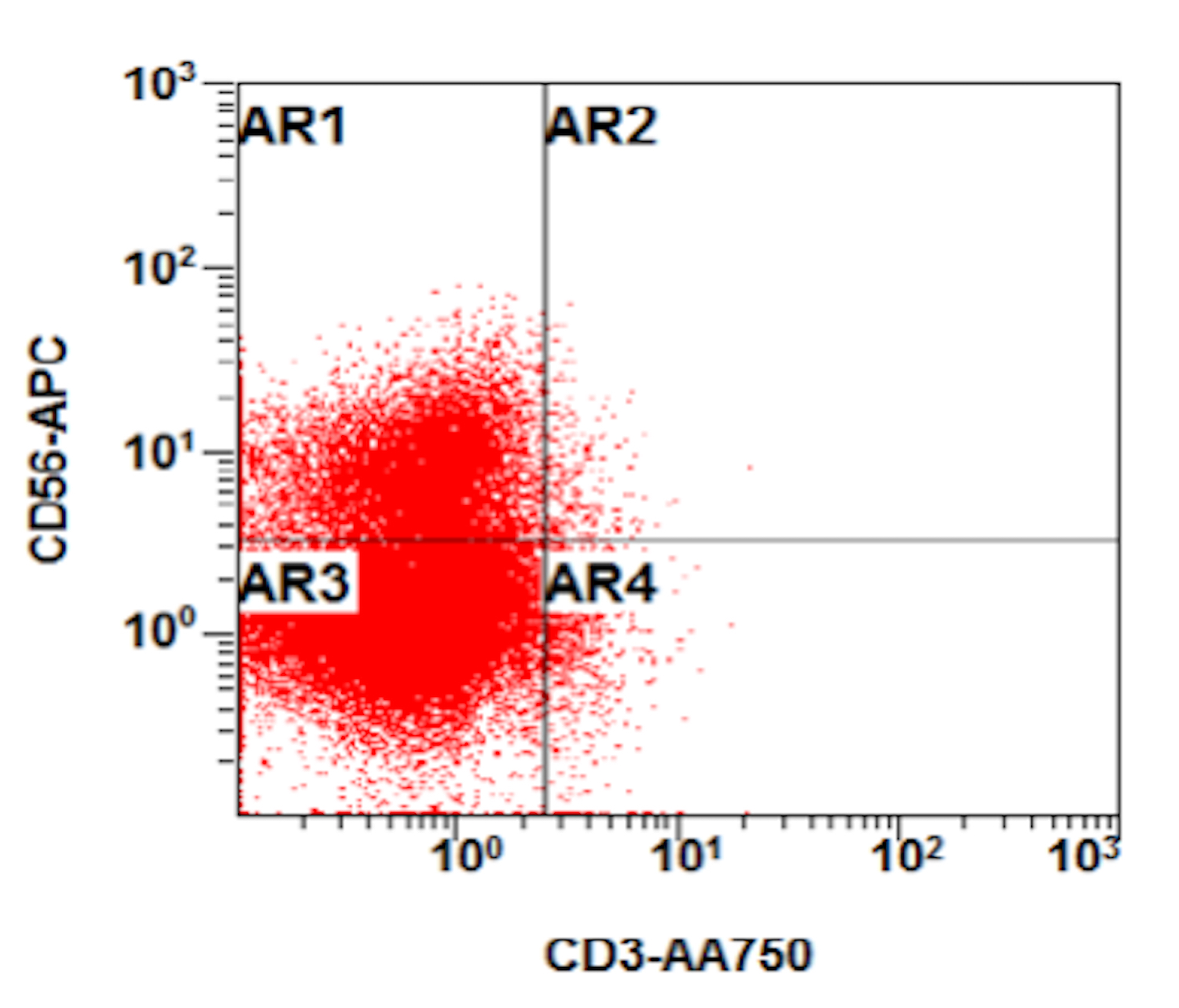
A 48 year old woman presents for evaluation of shortness of breath and fatigue over a 4 month period. She notes swelling in her neck and axillary region. Physical examination shows generalized lymphadenopathy and hepatosplenomegaly. Her complete blood count shows a white blood cell (WBC) count of 22,000, hemoglobin of 8.0 and platelet count of 100,000. A peripheral blood smear and flow cytometry on the peripheral blood are shown above. A bone marrow biopsy is performed as shown above and there is a neoplastic lymphoid population that is positive for cytoplasmic CD3, CD5 and CD34 and negative for CD1a, CD8 and myeloperoxidase. Which of the following is the most likely diagnosis?
- Adult T cell leukemia
- Early T cell precursor lymphoblastic leukemia
- Mixed phenotype acute leukemia (MPAL), T / myeloid
- Natural killer cell (NK) large granular lymphocytic leukemia
Answer C (mixed phenotype acute leukemia [MPAL], T / myeloid) is incorrect because the tumor cells in this case lack the expression of CD8 and myeloperoxidase and the immunoprofile meets criteria for early T cell precursor acute lymphoblastic leukemia. Answer D (natural killer cell [NK] large granular lymphocytic leukemia) is incorrect because it is not characterized by increased CD34 positive blasts. Answer A (adult T cell leukemia) is incorrect because the tumor cells of this entity do not express stem cell markers such as CD34.
Comment Here
Reference: Early T cell precursor lymphoblastic leukemia
- Essential thrombocythemia is a chronic myeloproliferative neoplasm that primarily involves the megakaryocytic lineage
- Characterized by sustained thrombocytosis (platelet count > 450 x 109/L) in the peripheral blood and increased numbers of large, atypical megakaryocytes in the bone marrow; clinically by the occurrence of thrombosis or hemorrhage
- Idiopathic thrombocythemia / thrombocytosis; essential hemorrhagic thrombocythemia; idiopathic hemorrhagic thrombocythemia; idiopathic thrombocythemia
- Has also been called essential thrombocytosis and primary thrombocytosis
- ICD-10: D47.3 - essential (hemorrhagic) thrombocythemia
- In the U.S. and Europe the incidence is 0.2 - 2.3 cases/100,000
- Median age of 60 years at diagnosis but may occur at any age
- Higher incidence among African Americans
- More common in women (F:M = 2:1) (Br J Haematol 2016;174:382)
- Rare in children (must rule out hereditary thrombocytosis)
- Clonal stem cell disorder characterized by increased platelet production
- Serum thrombopoietin levels can be normal or elevated
- Harbor a phenotypic driver mutation in JAK2 (present in 50 - 60% of cases), CALR (30%) or MPL (3%); ~12% of cases are triple negative for these mutations (Leuk Lymphoma 2017;58:2786)
- JAK2, CALR or MPL mutations result in the upregulation of JAK-STAT pathway
- JAK2 is a nonreceptor tyrosine kinase and has been found to play a crucial role in hematopoiesis
- JAK2 mutations leads to a gain of function leading to the activation of intracellular signaling pathways associated with the receptors of hematopoietic cytokines
- CALR is involved in cellular proliferation, differentiation and apoptosis
- JAK2, CALR or MPL mutations result in the upregulation of JAK-STAT pathway
- Etiology unknown
- Most cases are sporadic
- Some mutations of JAK2 and GSN genes have been found in families with hereditary thrombocytosis
- Patient can either be asymptomatic or symptomatic
- Asymptomatic
- More than half of cases
- Detected on routine peripheral blood count
- Less than half present with symptoms related to thrombosis or hemorrhage
- Long asymptomatic periods alternating with thrombotic or hemorrhagic crises
- May rarely progress to myelofibrosis
- Symptoms are due to qualitative and quantitative abnormalities in platelets and include
- Bleeding tendency
- Microvascular occlusion
- Thrombosis of major arteries leading to symptoms such as stroke and transient ischemic attacks
- Vasomotor manifestations such as headache, transient visual disturbances and syncope
- Mild splenomegaly
- Hepatomegaly and lymphadenopathy are uncommon
- Unexplained abortion (Blood 2007;110:485)
- Closely related to polycythemia vera but no increase in red blood cell mass
- Increased proliferation is usually confined to megakaryocytes with platelet count > 450 x 109/L
- Diagnostic criteria for early / prefibrotic essential thrombocythemia: requires either all major criteria or the first 3 major criteria plus the minor criterion (Blood 2022;140:1200, Leukemia 2022;36:1703) (WHO 2022 and ICC 2022)
- Major
- Criterion #1: sustained platelet count ≥ 450 x 109/L
- Criterion #2: megakaryocyte proliferation, large and mature with hyperlobulated nuclei (atypical), little to no granulocyte or erythroid proliferation; very rarely a minor (grade P) increase in reticulin fibers
- Criterion #3: does not meet WHO criteria for chronic myeloid leukemia, polycythemia vera, primary myelofibrosis, myelodysplastic syndrome or other myeloid neoplasm
- Criterion #4: demonstration of JAK2 or CALR or MPL mutation
- Minor
- Presence of a clonal marker
- Absence of evidence of reactive thrombocytosis
- Major
- Diagnostic criteria for post essential thrombocythemia myelofibrosis (Blood 2022;140:1200, Leukemia 2022;36:1703)
- 2 major criteria and at least 2 minor criteria (WHO 2022 and ICC 2022)
- Required criteria
- Established diagnosis of essential thrombocythemia
- Bone marrow fibrosis grade 2 - 3 (on 0 - 3 scale)
- Additional criteria (2 required)
- Anemia or decrease in hemoglobin by 2 gm/dL from the baseline level
- Leukoerythroblastic peripheral blood
- Splenomegaly
- Rise in lactate dehydrogenase (LDH) levels
- Constitutional symptoms (at least 2 or all 3): unexplained fever (> 37.5 °C), night sweats, > 10% weight loss in 6 months
- Required criteria
- Bone marrow examination is necessary to exclude other myeloproliferative neoplasms, myelodysplastic syndromes associated with isolated del(5q) and myelodysplastic / myeloproliferative neoplasm
- 2 major criteria and at least 2 minor criteria (WHO 2022 and ICC 2022)
- Essential thrombocythemia is generally an indolent disease with a good prognosis
- Initial management is largely dictated by the risk of thrombotic complication based on age, JAK2 mutation status and the history of thrombosis, and is stratified as follows (Blood Cancer J 2015;5:e369)
- High risk: history of thrombosis at any age or age > 60 with a JAK2 V617F mutation
- Intermediate risk: age > 60, no JAK2 mutation detected and no history of thrombosis
- Low risk: age ≤ 60 with JAK2 mutation and no history of thrombosis
- Very low risk: age ≤ 60, no JAK2 mutation detected and no history of thrombosis
- Variable rate of transformation to acute myeloid leukemia or myelodysplastic syndrome; generally these patients have a poor prognosis
- Can undergo delayed disease progression into a fibrotic state called post essential thrombocythemia myelofibrosis; < 5% conversion rate after 10 - 20 years
- Median survival is 10 - 15 years, hence life expectancy is almost normal (Am J Med 2004;117:755)
- CALR mutations are associated with a favorable prognosis in essential thrombocythemia compared to other genetic changes (Semin Hematol 2018;55:215)
- 35 year old woman with calreticulin mutation presenting with postpartum hemorrhage (Blood Coagul Fibrinolysis 2016;27:727)
- 37 year old woman presenting with acute renal infarction (Kidney Int 2017;92:1292)
- 50 year old man presenting with coronary artery dissection (WMJ 2015;114:26)
- 61 year old woman with abdominal surgery (Medicine (Baltimore) 2017;96:e8856)
- 3 patients with essential thrombocythemia presenting as acute coronary syndrome (J Thromb Thrombolysis 2017;44:57)
- Goal of management is to control symptoms arising from thrombotic and hemorrhagic complications
- Aspirin is given to the majority of patients, to reduce the odds of vascular events and vasomotor symptoms
- Low risk and very low risk patients (Br J Haematol 2000;110:577)
- Should undergo an annual or semi annual visits with aspirin treatment
- No randomized trials comparing low risk and very low risk treatment options
- Intermediate risk and high risk patients (Br J Haematol 2000;110:577)
- Should be treated with cytoreductive therapy (preferably hydroxyurea followed by anagrelide) or systemic anticoagulation (such as low dose aspirin)
- Bone marrow
- Normal / mildly hypercellular
- Atypical megakaryocytes (Blood 2016;127:2391)
- Increased number and large atypical forms with deeply lobulated or hyperlobulated nuclei (staghorn nuclear appearance)
- Dispersed throughout the marrow with occasional loose clusters; dense clusters are uncommon
- No highly bizarre / dysplastic forms, no predominance of small megakaryocytes with monolobulated nuclei
- Delicate reticulin fibers (myelofibrosis absent / minimal) but no overt fibrosis (usually grade 1)
- Usually normal erythroid and granulocytic proliferation
- Usually no dyserythropoiesis, dysgranulopoiesis, macrocytosis or monocytosis
- Features in bone marrow aspirate / biopsy suggesting an alternative diagnosis (Blood 2016;127:2391)
- Megakaryocytes with highly atypical morphology
- Increased myeloblasts
- Myelodysplastic features
- Significant (> grade 1) reticulin fibrosis or collagen fibrosis
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
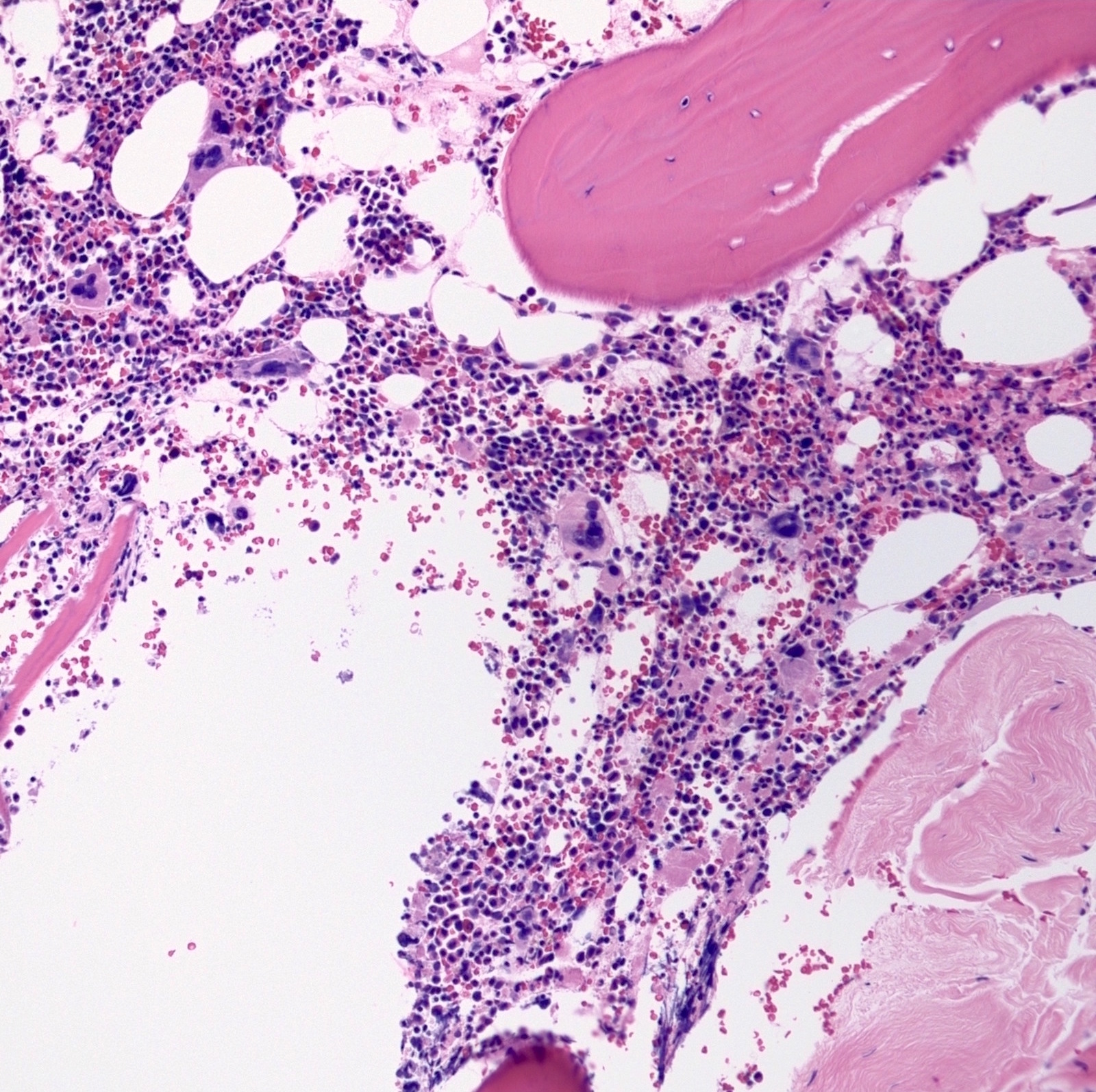
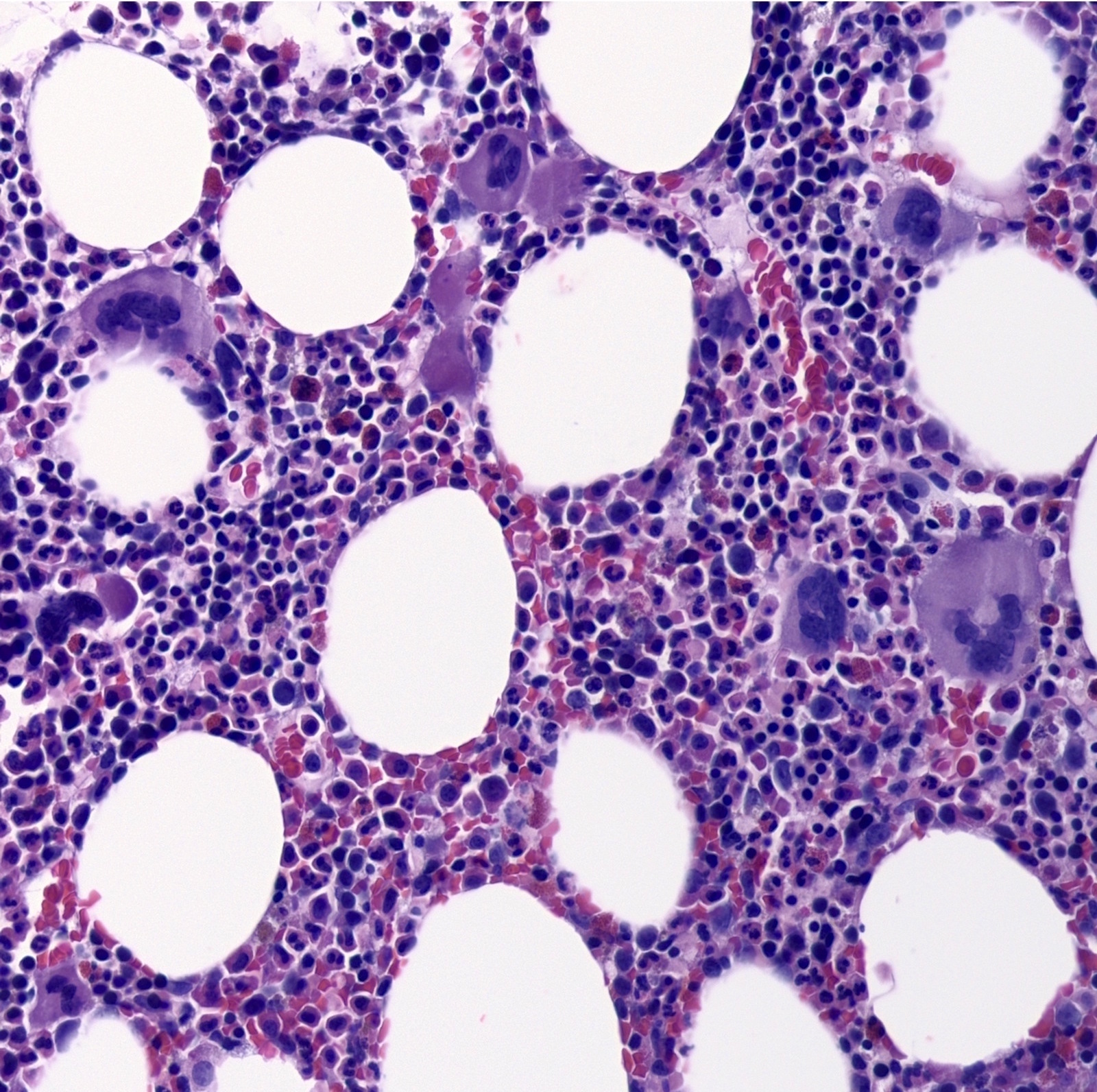
Bone marrow biopsy with atypical megakaryocytes
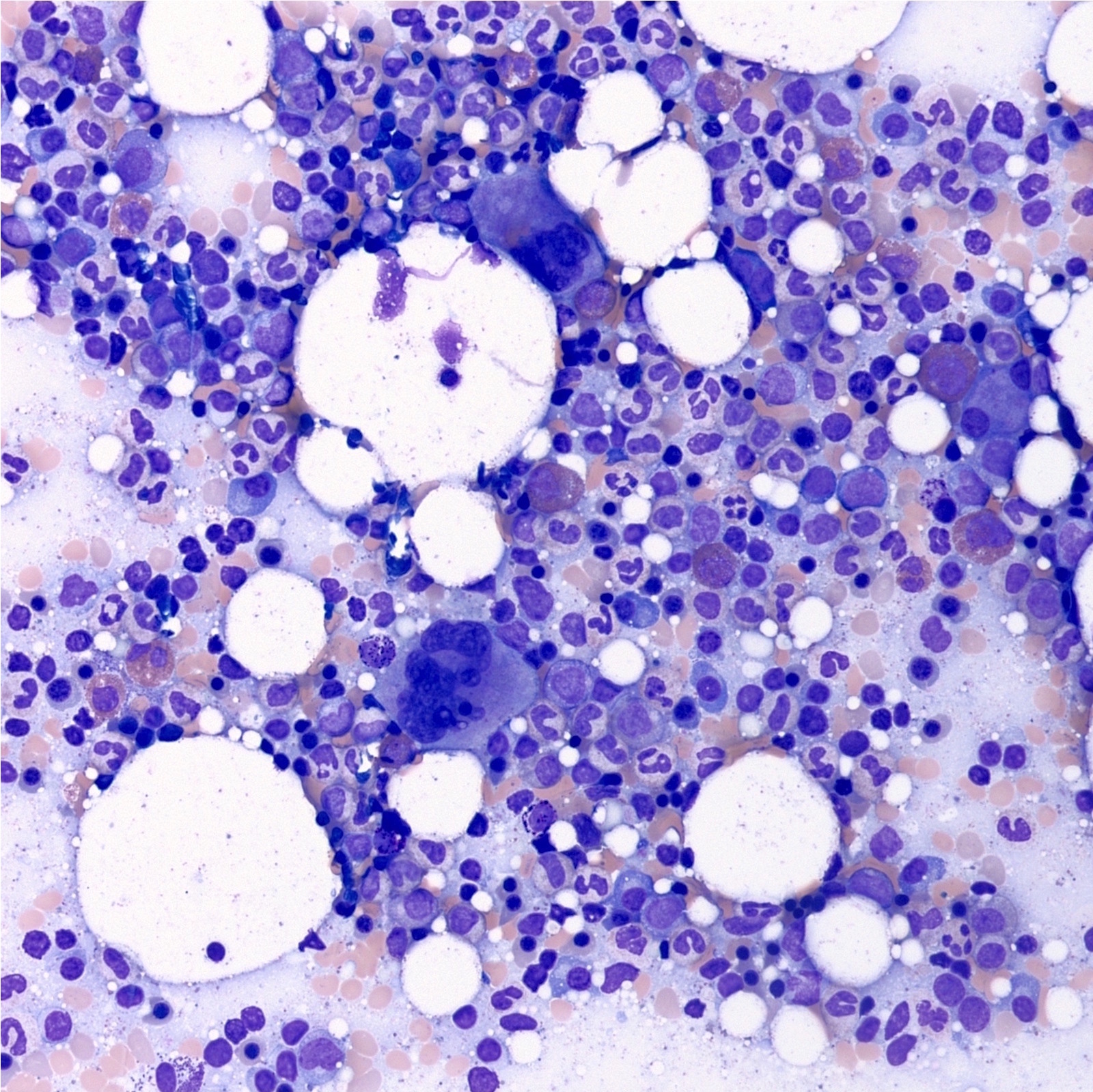
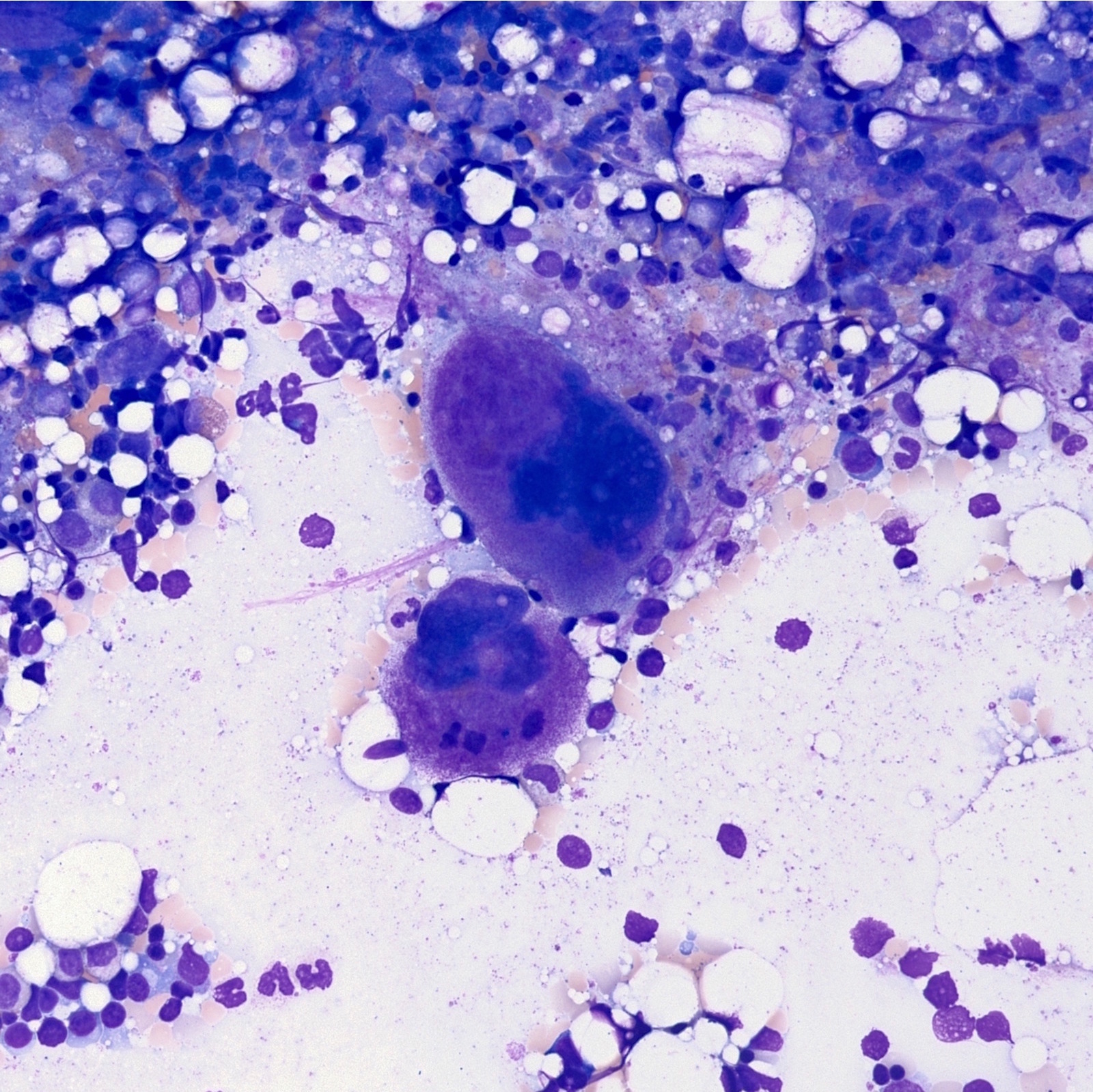
Bone marrow aspirate with atypical megakaryocytes
- Normal / mild increase in the white blood cell count
- Moderate to marked thrombocytosis
- Platelet anisocytosis, abnormally large platelets and increased platelets; bizarre shapes, pseudopods and agranular forms
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
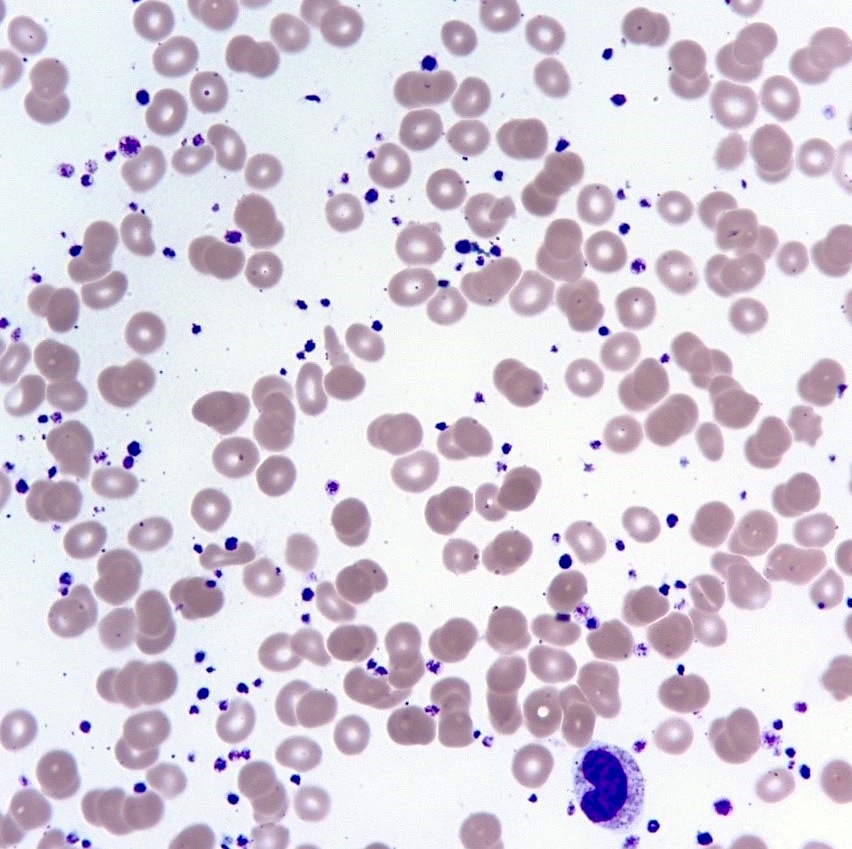
Anisocytosis and bizarre forms
- Normal number and phenotype myeloblasts with normal myeloid scatter by CD45 / SSC
- Normal CD10 / CD13 / CD16 / CD11b myeloid maturation pattern and all other myeloid markers are normally expressed; hence, there is no immunophenotypic evidence of myelodysplasia
- No evidence for monoclonal B cell lymphoproliferative disease
- Although no unusual phenotype T cells are identified, surface markers will not routinely detect monoclonal T lymphocytes
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
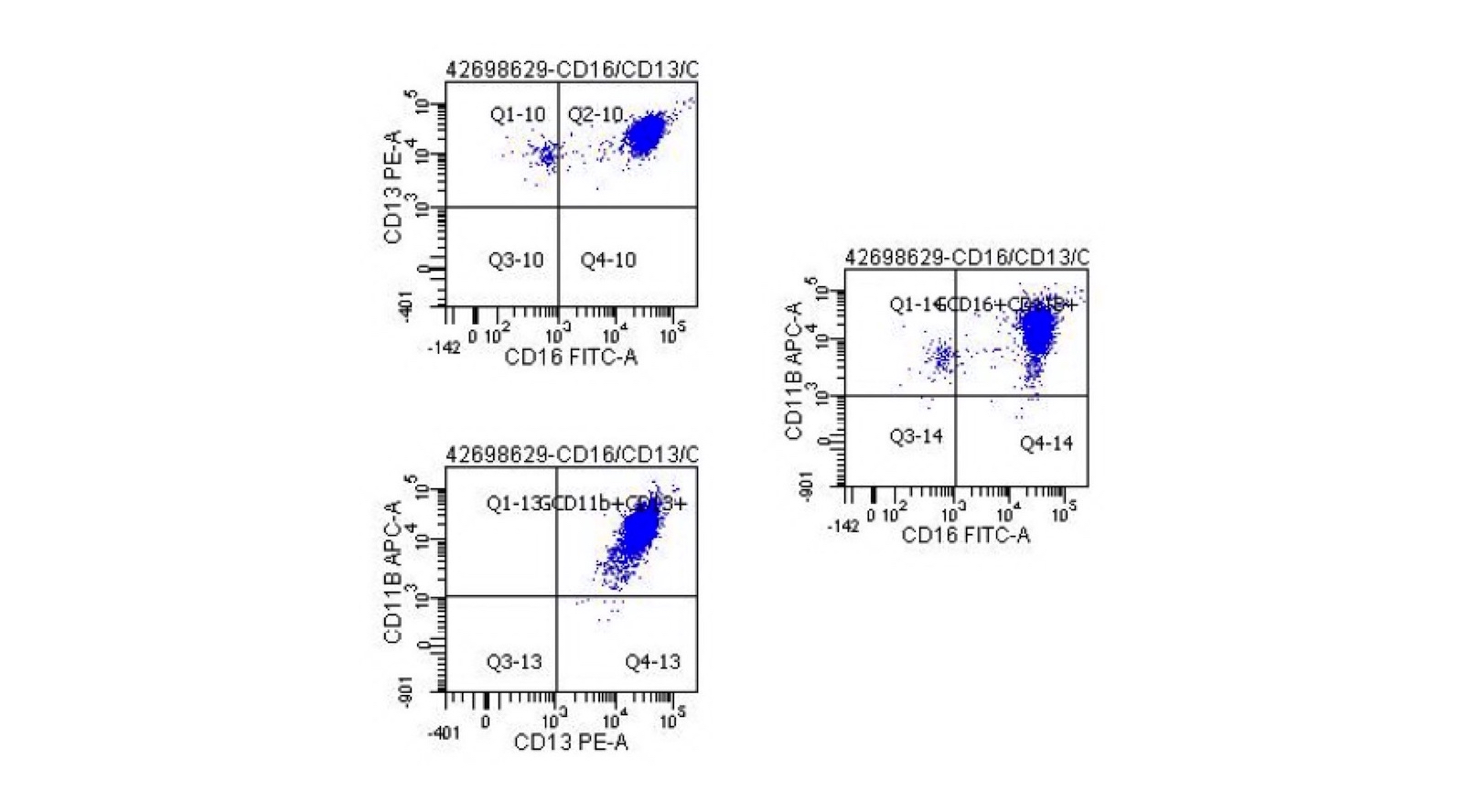
No myelodysplasia
- Phenotypic driver mutation
- JAK2 (present in 50 - 60% of cases), CALR (30%) or MPL (3%), ~12% of cases are triple negative for these mutations
- Mutations are specific for essential thrombocythemia but their presence excludes reactive thrombocytosis
- Abnormal karyotype is found in 5 - 10% of cases
- No consistent abnormality
- Reported abnormalities include gain of chromosome 8, abnormalities in 9q and del(20q) and unbalanced translocations between 1q and 7p
- JAK2 mutant allele burden contributes to clinical phenotype (Haematologica 2009;94:144)
- Subset of triple negative cases have been found to have
- Gain of function mutations (e.g., MPL S204P and MPL Y591N) through whole exome sequencing or other molecular techniques
- W515L and W515K mutations in thrombopoietin receptor c-Mpl (J Transl Med 2006;4:41)
- Must rule out BCR::ABL1 fusion gene status
- Children usually do not have JAK2 mutation
- Bone marrow, biopsy and aspirate:
- Myeloproliferative neoplasm (see comment)
- Comment: The patient's positive JAK2 mutation is noted. The morphologic findings are compatible with a myeloproliferative neoplasm and in conjunction with the thrombocytosis, essential thrombocythemia is favored. Correlation with clinical, cytogenetic and laboratory findings is recommended.
- CBC (4/29/2019): HGB: 12.7 g/dL; MCV: 89.7 fL; WBC: 9.7 K/uL; PLT: 735 K/uL
- Bone marrow biopsy microscopic description:
- Slightly hypercellular marrow for age (30% cellular). Megakaryocytes are increased in number, occasionally in clusters and show frequent atypical forms, including large and hyperlobated forms. The myeloid:erythroid (M:E) ratio is normal. Erythroid elements exhibit maturation. Myeloid elements exhibit maturation. Prominent lymphoid aggregates are not seen. Granulomas are not seen. Reticulin stain reveals mild and patchy reticulin fibrosis. Trabecular bone is unremarkable. The clot section is mainly blood and shows small fragments with findings similar to the core biopsy. CD34 highlights blasts comprising < 5% of the marrow cellularity.
- Bone marrow aspirate microscopic description:
- The marrow aspirate smears are spicular and cellular. Megakaryocytes are increased. The M:E ratio is approximately 1:1. Erythroid maturation is seen. Myeloid maturation is seen. Prussian blue iron stain shows decreased iron storage and no significant increase in ring sideroblasts.
- Aspirate cell count: A 226 cell count reveals 2% blasts, 9% promyelocytes / myelocytes, 34% maturing granulocyte forms, 38% erythroid forms, 8% lymphocytes, 5% eosinophils, 2% plasma cells, 0% basophils / mast cells and 0% monocytes.
- Other myeloproliferative neoplasms:
- Chronic myeloid leukemia:
- BCR::ABL translocation is diagnostic
- Polycythemia vera:
- Most cases harbor a clonal JAK2 mutation
- Increase in erythroid / granulocytic population should raise suspicion of polycythemia vera; polycythemia vera patients also present with decreased erythropoietin (EPO)
- Polycythemia vera patients usually present with some symptoms related to hyperviscosity such as headaches or visual disturbance; some patients may present with pruritus exacerbated by hot water
- Primary myelofibrosis and prefibrotic myelofibrosis:
- Increased bone marrow reticulin fibrosis, markedly bizarre megakaryocytes and a leukoerythroblastic peripheral blood smear favor myelofibrosis
- Prefibrotic myelofibrosis:
- Will show features of primary myelofibrosis
- The main feature that can help differentiate this entity from essential thrombocythemia is the presence of reticulin fibrosis in pre-PMF
- Myelodysplastic syndrome:
- Dysplastic features and genetics such as 5q loss
- Familial essential thrombocythemia:
- Autosomal dominant disease
- Genetic analysis will show mutations in thrombopoietin, TPO receptor (c-MPL) or others related to thrombopoiesis
- Reactive thrombocythemia:
- Causes include exercise, allergic reaction, reaction to medications, inflammatory disorders, asplenism, infection, connective tissue disorders, metastatic cancer, lymphoproliferative disorders and iron deficiency
- Spurious thrombocytosis:
- Due to the miscount of structures that are not platelets (e.g., cryoglobulin crystals, cytoplasmic fragments of circulating leukemic cells and bacteria) as platelets leading to thrombocytosis (Thrombosis 2011;2011:536062)
- Chronic myeloid leukemia:
What is the prevalence of genetic mutation in essential thrombocythemia?
- CAL > JAK2 > MPL > triple negative
- JAK2 > CAL > MPL > triple negative (JAK2, CAL and RET)
- JAK2 > CAL > triple negative > MPL
- JAK2 > MPL > CAL > triple negative
Comment Here
Reference: Essential thrombocythemia
- Abnormal karyotype
- JAK2 mutation
- Leukoerythroblastic reaction
- Positive BCR::ABL translocation
Comment Here
Reference: Essential thrombocythemia
- Pluripotent, hematopoietic, stem cell driven malignancy that may present as myeloproliferative neoplasm (MPN), acute myeloid leukemia (AML), T or B lymphoblastic lymphoma leukemia (T-ALL, B-ALL) or mixed phenotypic acute leukemia (MPAL)
- MPN or myelodysplastic syndrome / myeloproliferative neoplasm (MDS / MPN) with prominent eosinophilia, with or without lymphocytosis or neutrophilia
- Alternatively, AML or T-ALL or B-ALL or MPAL with eosinophilia in peripheral blood or bone marrow
- Presence of t(8:13)(p11.2;q12) or a variant of translocation leading to FGFR rearrangement in a myeloid progenitor, a lymphoblast or both
- Hematopoietic stem cell neoplasm with FGFR1 abnormalities
- Myeloid and lymphoid neoplasm with FGFR1 abnormalities
- 8p11 stem cell leukemia / lymphoma syndrome
- 8p11 myeloproliferative syndrome
- 8p11 stem cell syndrome
- ICD-10: D47.7 - other specified neoplasms of uncertain or unknown behavior of lymphoid, hematopoietic and related tissue
- M:F = 1.5:1 (Arkh Patol 2019;81:59)
- Age range of 3 - 84 years; median age of 32 years (Arkh Patol 2019;81:59)
- Bone marrow, peripheral blood, liver, spleen and lymph nodes
- FGFR1 gene rearrangement affects the pluripotent stem cells, which could differentiate to myeloid or lymphoid neoplasm
- Most cases present with chronic phase then may transform to acute blast phase; however, some cases may represent as de novo acute proliferation
- Etiologic factors have not been identified
Cytogenetics and molecular genetics reported in myeloid / lymphoid neoplasms with FGFR1 rearrangement
| t(8;13)(p11.2;q12.1) | ZMYM2-FGFR1, previously ZNF198-FGFR1 |
| t(8;9)(p11.2;q33.2) | CNTRL-FGFR1, previously CEP110-FGFR1 |
| t(6;8)(q27;p11.2) | FGFR10P-FGFR1 |
| t(8;22)(p11.2;q11.2) | BCR-FGFR1 |
| t(7;8)(q33;p11.2) | TRIM24-FGFR1 |
| t(8;17)(p11.2;q11.2) | MY018A-FGFR1 |
| t(8;19)(p11.2;q13.3) | HERVK-FGFR1 |
| ins(12;8)(p11.2;p11.2;p22) | FGFR10P2-FGFR1 |
| t(1;8)(q31.1;p11.2) | TPR-FGFR1 |
| t(2;8)(q13;p11.2) | RANBP2-FGFR1 |
| t(2;8)(q37.3;p11.2) | LRRFIP1-FGFR1 |
| t(7;8)(q22.1;p11.2) | CUX1-FGFR1 |
| t(8;12)(p11.2;q15) | CPSF6-FGFR1 |
- FGFR1 rearrangement has also been found in association with t(8;12)(p11.2;q15) and t(8;17)(p11.2;q25) but suspected involvement of FGFR1 in t(8;11)(p11.1-p11.2;p15) was not confirmed
- Additional cytogenetic abnormalities have been reported, of which trisomy 21 is the most common secondary chromosomal abnormality
- Lymphadenopathy, mediastinal mass or organomegaly
- Myeloproliferative features
- AML-like features or myeloid sarcoma
- Systemic symptoms (e.g., fever, weight loss and night sweats)
- Cases usually present as chronic eosinophilic leukemia (CEL), atypical chronic myeloid leukemia (aCML) or chronic myelomonocytic leukemia (CMML), the latter are usually with eosinophilia
- Overall, ~90% of chronic phase patients have peripheral blood or bone marrow clonal eosinophilia (including those with acute transformation), neutrophilia and occasionally monocytosis
- De novo AML or de novo T-ALL with or without eosinophilia
- Some cases show subsequent transformation to T-ALL, B-ALL, MPAL or AML (including myeloid sarcoma)
- Basophilia is not typical but may be seen in cases with BCR-FGFR1 fusion, t(1;8)(q31.1;p11.2) and TPR-FGFR, t(1;8)(q25;p11.2)
- Polycythemia vera has been reported in 5 cases with t(6;8) / FGFR10P-FGFR1 fusion (Hematol J 2004;5:534)
- Some patients primarily present as B-ALL or MPAL with t(8;13)
- T-ALL characteristically shows eosinophilic infiltration within the lymphoma, which can point toward this diagnosis
- Leukocytosis and sometimes anemia or thrombocytopenia
- Often presenting with eosinophilia but may show normal eosinophil count
- Poor prognosis, even for patients presenting in the chronic phase due to the high incidence of transformation
- 34 year old woman with no subjective symptoms diagnosed with T lymphoblastic lymphoma (LBL) with ZNF198-FGFR1 or ZMYM2-FGFR1; bone marrow (BM) aspirate and biopsy showed hypercellularity with hypereosinophilia (Hematology 2018;23:470)
- 48 year old Japanese man with peripheral leukocytosis, diagnosed with B-ALL with BCR-FGFR1 gene rearrangement (Bone Marrow Transplant 2019;54:326)
- 66 year old man had clinical features that resembled CML; however, bone marrow cytogenetic and fluorescent in situ hybridization (FISH) studies showed t(8;22)(p11;q11) and BCR-FGFR1 fusion gene (Case Rep Hematol 2018;2018:5724960)
- In several cases, interferon has induced a cytogenetic response
- Intensive chemotherapy with regimens such as hyper-CVAD for T or B cell lymphoblastic lymphoma, followed by early allogeneic transplantation, is recommended
- Hematopoietic stem cell transplantation should be considered, even for patients who present in the chronic phase
- Optional targeted therapy:
- Midostaurin (PKC412) for a patient with ZNF198 / ZMYM2-FGFR1 fusion (Curr Hematol Malig Rep 2015;10:351, Haematologica 2010;95:696)
- Ponatinib has partial activity against the FGFR1 tyrosine kinase, which is reported effective in a patient with BCR-FGFR1 positive trilineage T / B / myeloid mixed phenotype acute leukemia (Med Res Rev 2014;34:280)
- B-ALL with FGFR1 rearrangement:
- Peripheral blood smear (PBS): shows left shifted granulocytosis with eosinophilia
- Bone marrow biopsy:
- Low power: hypercellular marrow (75%) with involvement by myeloid / lymphoid neoplasm
- High power: abnormal immature B cell proliferation indicates B lymphoblastic leukemia / lymphoma
- Aspirate smear:
- Granulocytic hyperplasia with increased eosinophils (10%)
- Immature lymphoid cells are increased, approximately 15% (hemodiluted aspirate)
- Megakaryocytes are present and show normal morphology
- Erythroid precursors show normoblastic maturation
- AML with FGFR1 rearrangement:
- PBS: leukocytosis with increased blast count (> 20%), with eosinophilia (20%) and normocytic anemia with thrombocytopenia
- Bone marrow biopsy:
- Hypercellular marrow (80%) with involvement by sheets of blasts > 40%, which stains positive for CD34 and MPO
- Reticulin fibrosis is mildly increased (1/3)
- Aspirate smear: hypercellularity and diffuse infiltration by myeloblasts
- Myeloproliferative neoplasms (MPN) with FGFR1 rearrangement:
- PBS: leukocytosis with eosinophilia and normocytic anemia with thrombocytopenia
- Bone marrow biopsy: hypercellular marrow (80%) with left shifted maturation of the granulocyte lineage with increased eosinophils (which may be seen in clusters)
- Aspirate smear: hypercellularity with left shifted maturation of the granulocytes and eosinophilia
Contributed by Ling Zhang, M.D.

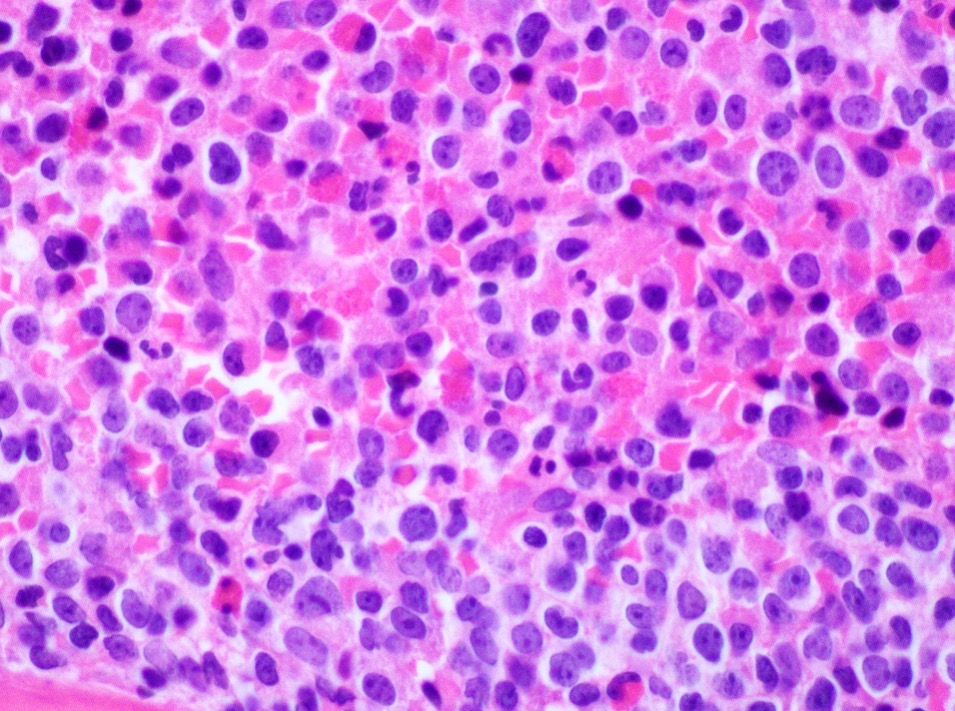
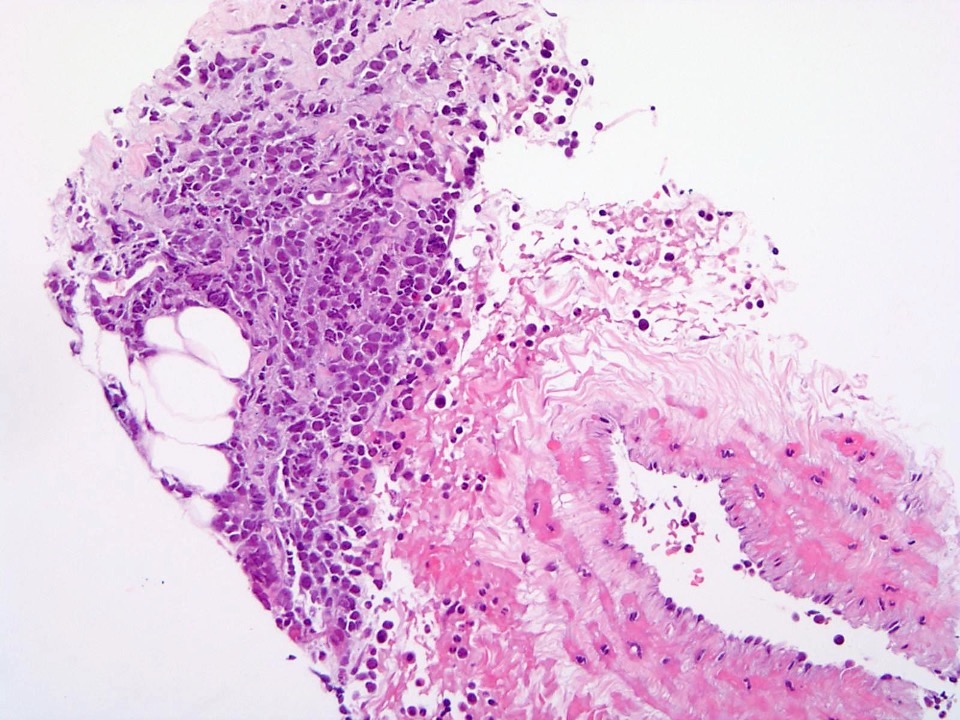
AML with FGFR1 rearrangement
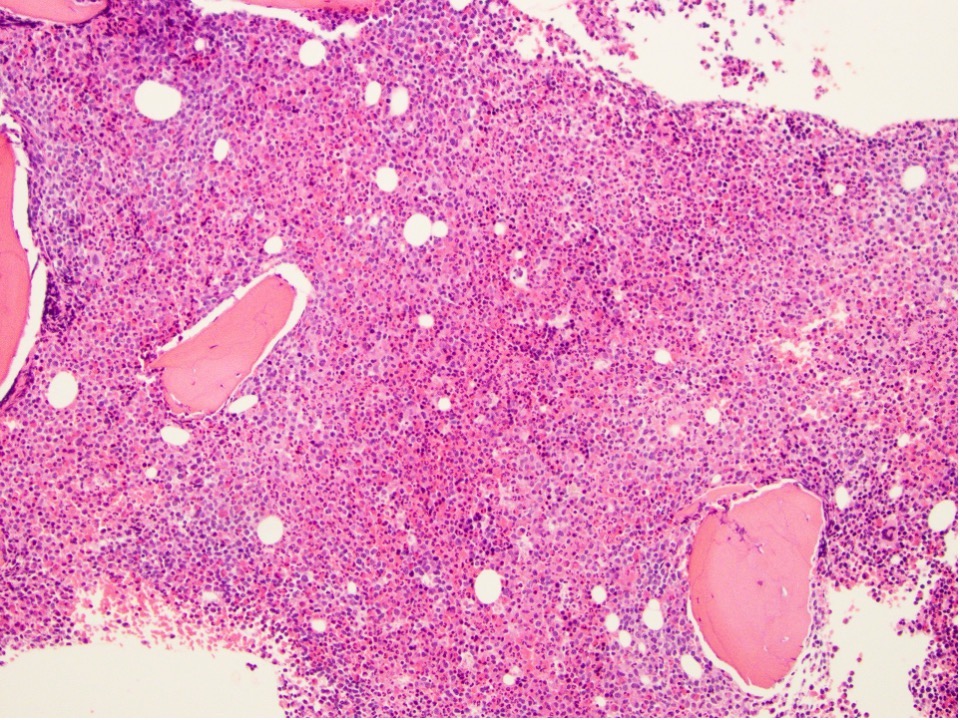
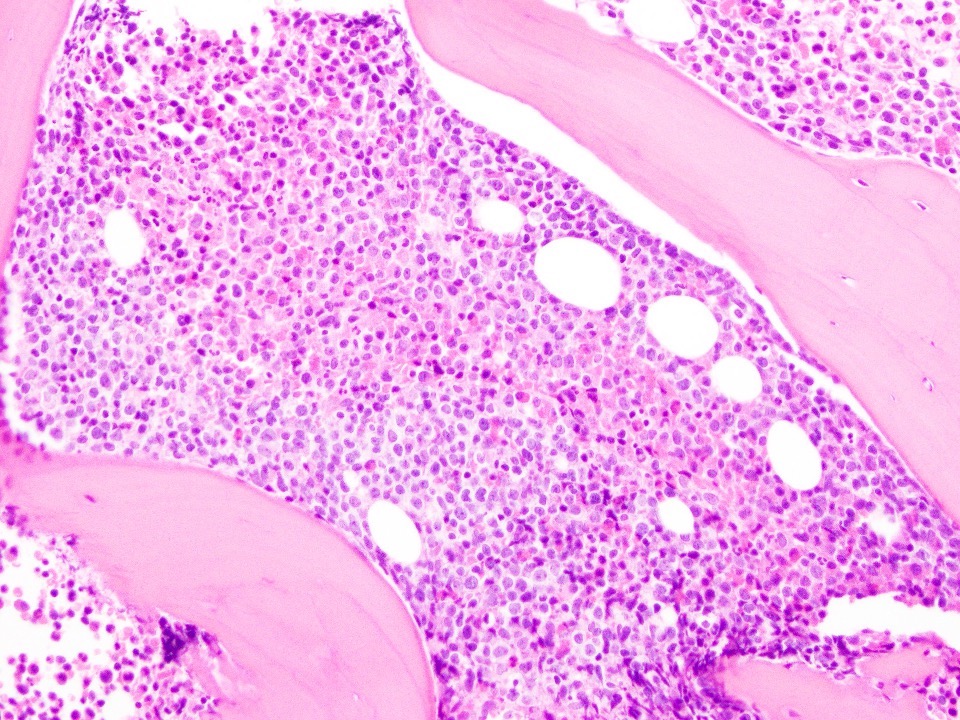
B-ALL with FGFR1 rearrangement
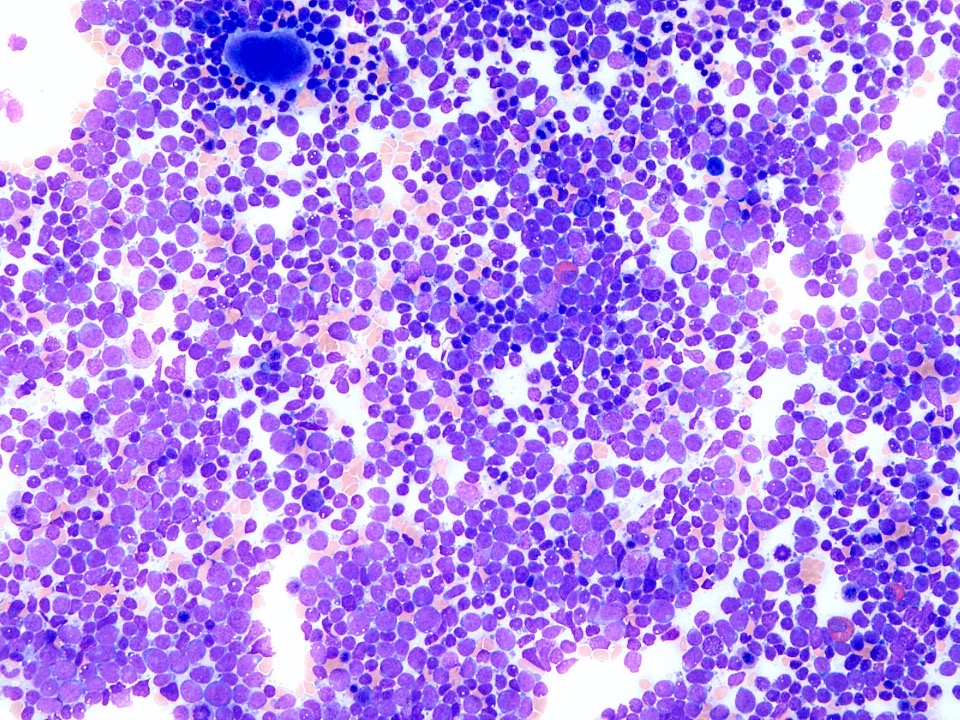

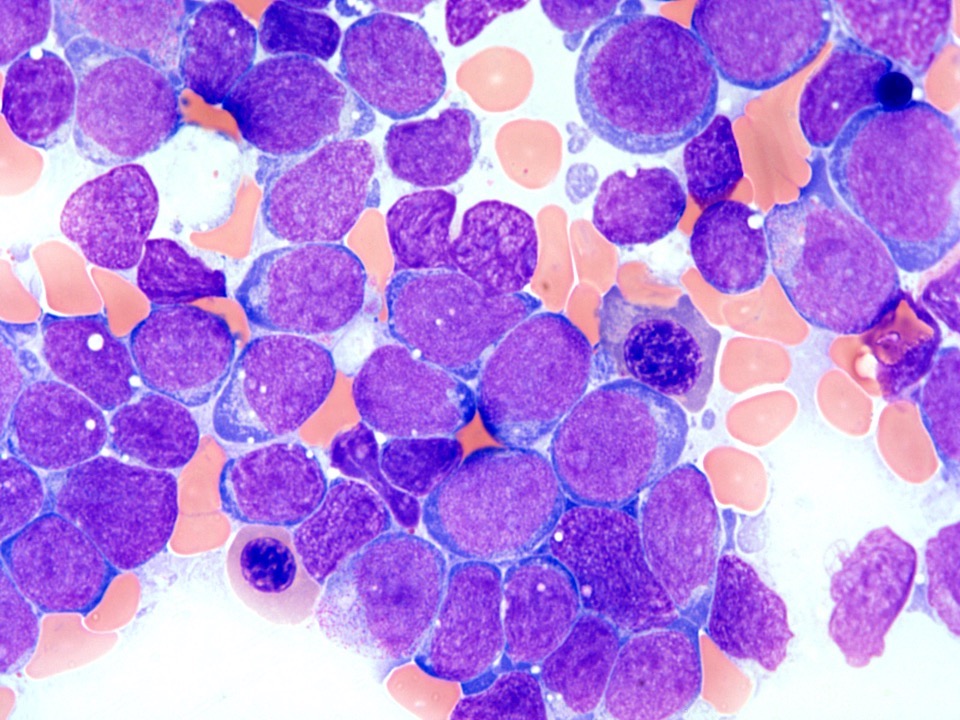
AML with FGFR1 rearrangement
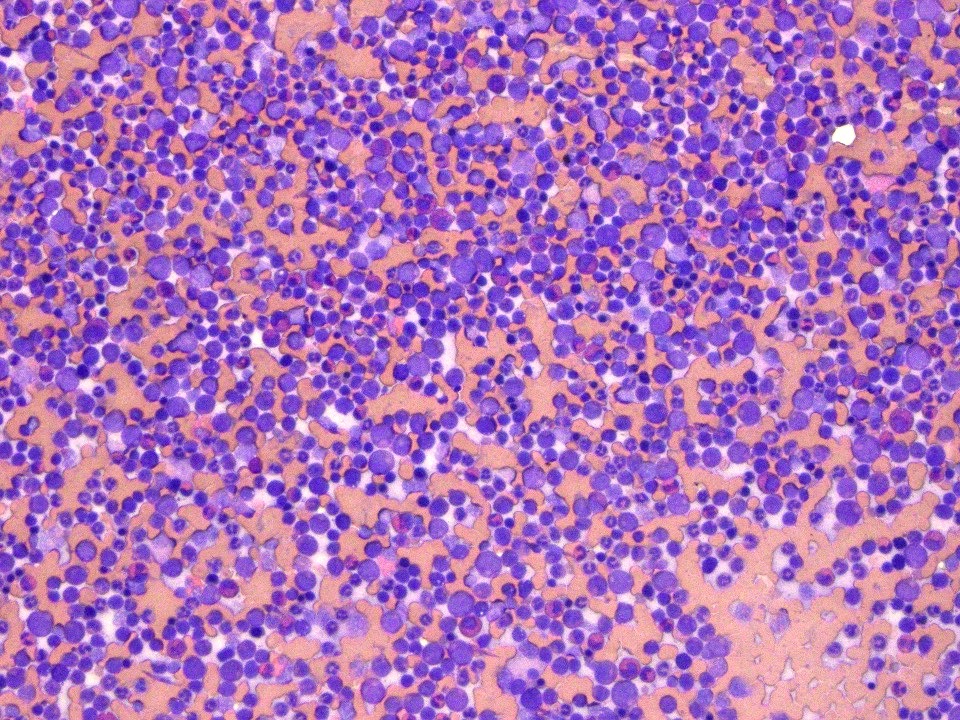
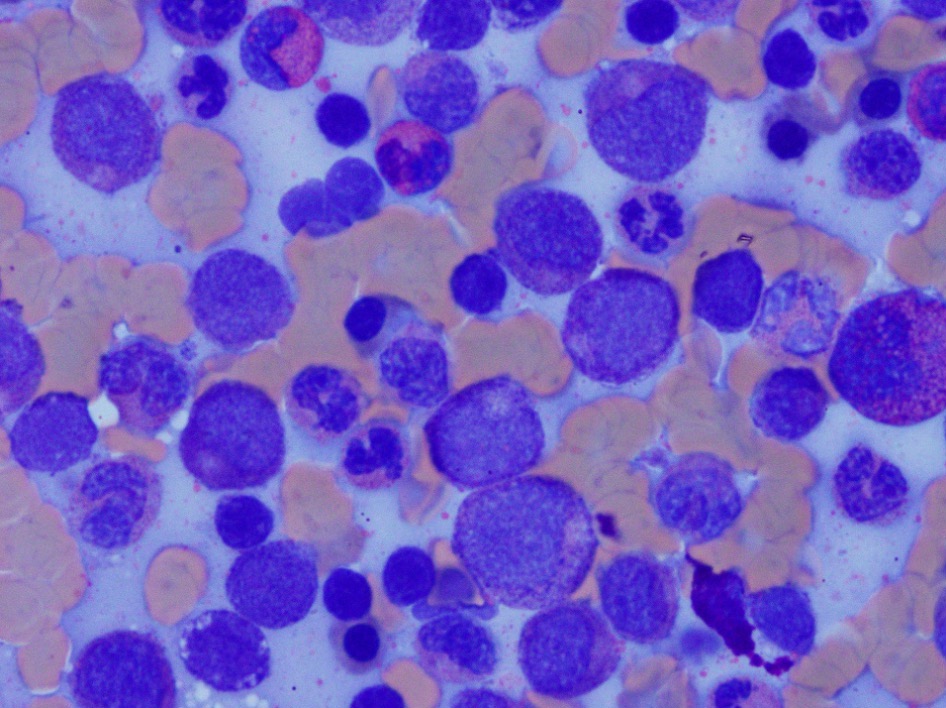
B-ALL with FGFR1 rearrangement
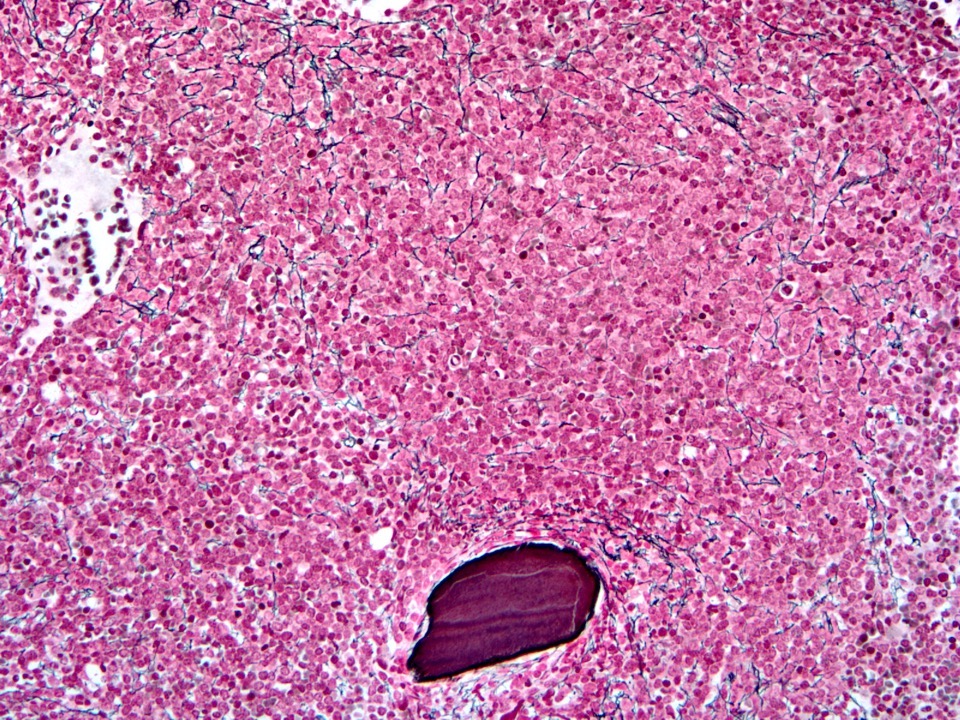
Reticulin stain
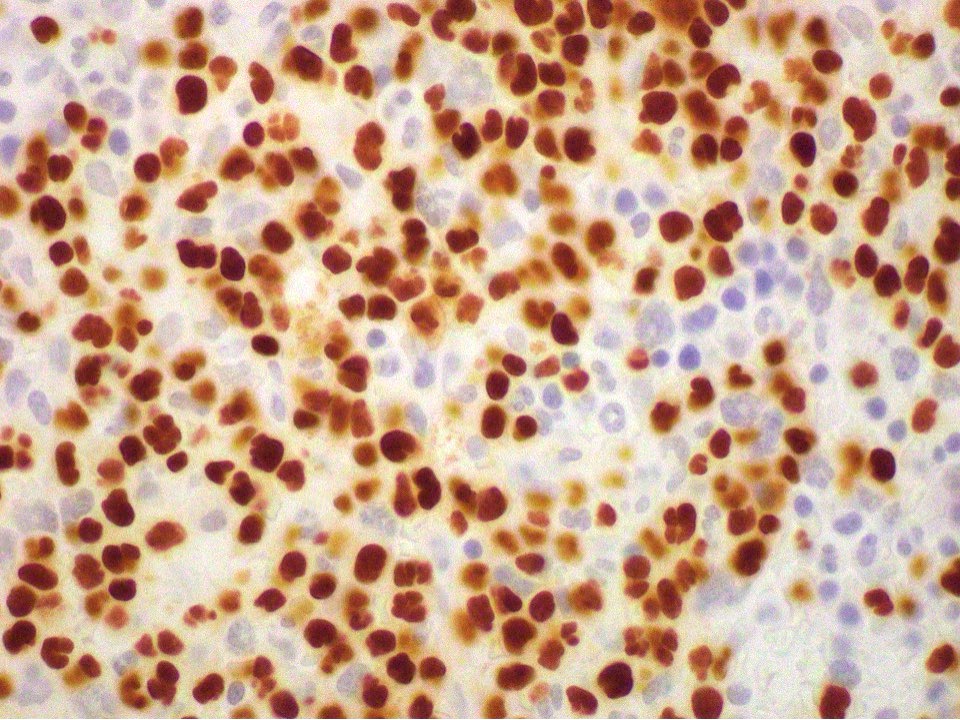
TdT positive stain
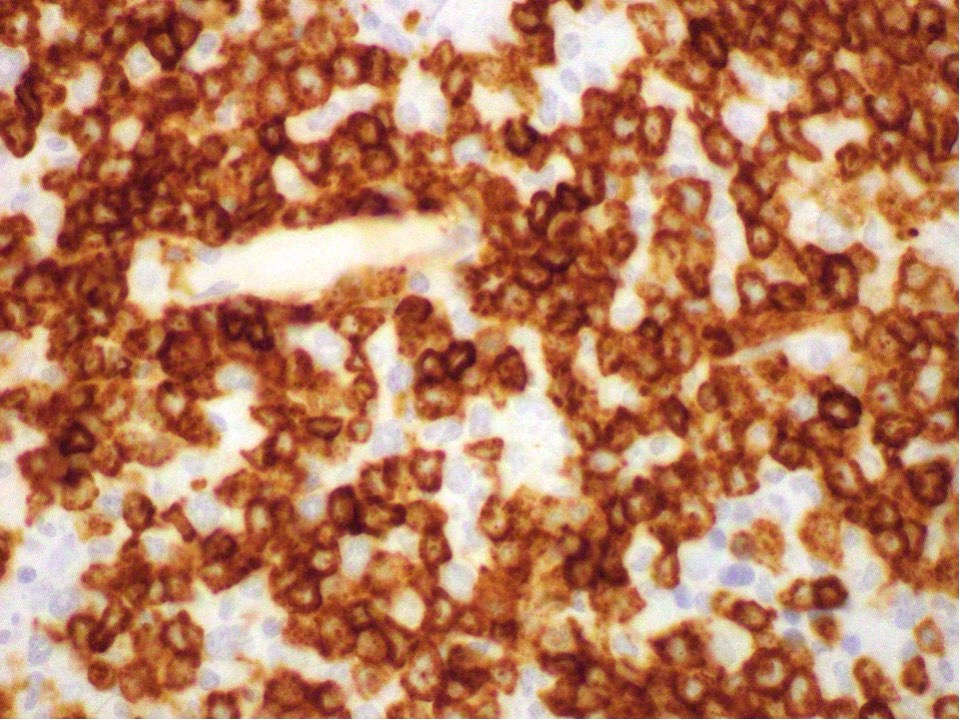
CD34 positive stain
- B-ALL with FGFR1 rearrangement:
- Left shifted leukocytosis with eosinophilia; normocytic normochromic red cell morphology and adequate reticulocytosis
- Anemia or thrombocytopenia is present, occasional monocytosis
- AML with FGFR1 rearrangement:
- Normocytic normochromic red cell morphology with adequate reticulocytosis; left shifted leukocytosis with increased blast cells and eosinophilia
Contributed by Ling Zhang, M.D.
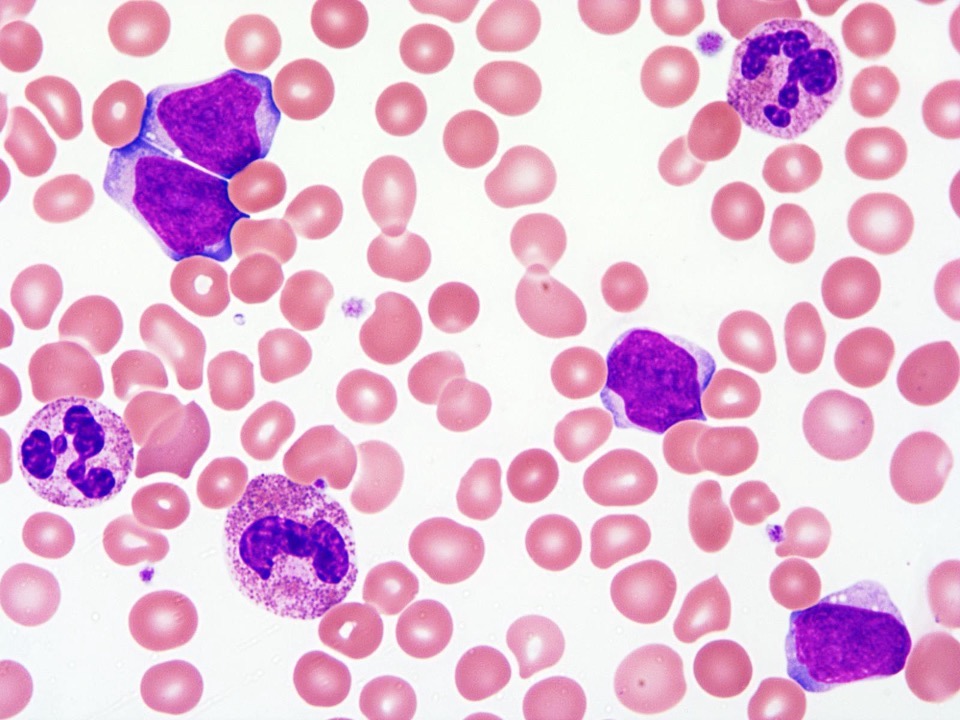
Myeloblast with eosinophils
- FISH test with FGFR1 break apart probe: 1 red, 1 green and 1 fusion signal pattern indicates positive FGFR1 gene rearrangement detected in 72.5% of cells
Contributed by Ling Zhang, M.D.

FISH for FGFR1 rearrangement
Images hosted on other servers:

FISH assay of FIM (also called ZNF198) and FGFR1
- Peripheral blood:
- Left shifted granulocytosis with eosinophilia
- Bone marrow, right posterior iliac crest, aspirate and biopsy:
- Hypercellular marrow (75%) with involvement by myeloid / lymphoid neoplasm with FGFR1 rearrangement (see comment)
- Decreased stainable storage iron
- Comment: The concurrent flow cytometry identified immature abnormal B cell population. Immunohistochemical stains are performed on the current bone marrow core biopsy shows 30 - 40% PAX5+, TdT+, CD34+ subset immature B cells. FISH study reveals 64.5% cells with FGFR1 rearrangement. The patient's previous outside bone marrow biopsy showed abnormal karyotype with t(8:13)(p11.2;q12.1).
- The abnormal immature B cell proliferation indicates B lymphoblastic leukemia / lymphoma. With the clinical presentation of left shifted leukocytosis, monocytosis, eosinophilia and FGFR1 rearrangement, the findings are consistent with a myeloid / lymphoid neoplasm with FGFR1 rearrangement. Please correlate with pending FoundationOne Heme analysis, reported separately.
- Microscopic description:
- Peripheral blood smear:
- Cytopenias: not present
- Red cell morphology: normocytic normochromic
- Reticulocytosis: adequate
- Other findings: left shifted leukocytosis with monocytosis and eosinophilia
- Aspirate smear:
- Adequacy: adequate
- Significant findings: There is granulocytic hyperplasia with increased eosinophils (20%). Immature lymphoid cells are increased, approximately 15%. Megakaryocytes show normal morphology. Erythroid precursors show normal maturation.
- Clot section and core biopsy:
- The clot section (A) and the core biopsy (B) are flaccid in B plus fix (BBC) fixative. The core biopsy (B) is decalcified and formical (formic acid, formalin diet and EDTA). On level of the clot section (A) and core biopsy (B) are stained with PAS to evaluate myeloid maturation and more accurately estimate and M:E ratio. One level of the clot section (A) and core biopsy (B) is each stained with Prussian blue to evaluate iron stores and the average value reported. One level of the colon biopsy (B) is stained with reticulin stain to evaluate marrow fibrosis.
- Bony trabeculae: unremarkable
- Cellularity: 75%
- Megakaryocytes: adequate in number with normal morphology
- Estimated M:E ratio: 3 - 4:1
- Iron stores: decreased
- Reticulin fibrosis: normal
- Significant findings: hypercellular marrow with increased eosinophils and increased immature mononuclear cells
- Immunohistochemical stains are performed on the core biopsy with adequate controls. PAX5 and TdT highlight markedly increased to scattered and aggregates of immature B cells, overall 30 - 40%. Majority of this immature B cells are also positive for CD34. Spectrin stains mildly decreased erythroid precursor. CD61 highlights megakaryocytes.
- Peripheral blood smear:
- Hypereosinophilic syndrome:
- Sustained eosinophilia (> 1.5 x 109/L) for at least 6 months with other reactive processes excluded with induced tissue damage, other reactive processes excluded
- Exclude cytokine related lymphoid variant hypereosinophilia
- Increased eosinophils with either no dysplastic features or no increase in blast cells in the bone marrow
- No lymphoid or myeloid neoplasms (e.g., MDS / MPNs, MPNs, systemic mastocytosis or AML)
- No cytogenetic or molecular alteration is identified
- No FGFR, PDGFRA, PDGFRB rearrangement, PCM1-JAK2, BCR-ABL1, ETV6-JAK2 or BCR-JAK2 fusions
- No inv(16)(p13.1q22), t(16;16)(p13.1q22) or t(8;21)(q22;q22.1)
- Reactive eosinophilia:
- Occurs secondary to allergies, drugs, infection, skin diseases, asthma, adrenal insufficiency, gastrointestinal eosinophilia
- Peripheral blood shows increased eosinophils (may be with abnormal forms)
- No evidence of FGFR1 rearrangement or active mutation or other cytogenetic / molecular abnormalities
- Systemic macrocytosis with eosinophilia:
- Major diagnostic criteria: multifocal infiltrates of mast cells (≥ 15 mast cells in aggregates) + 1 minor criterion or 4 minor criteria
- Minor criteria:
- Bone marrow shows eosinophilia with variable dysplastic features
- No FGFR1 rearrangement
- Chronic eosinophilic leukemia, NOS:
- Sustained eosinophilia (> 1.5 x 109/L) with other reactive processes excluded
- < 20% blasts in peripheral blood or bone marrow
- No inv(16)(p13.1q22), t(16;16)(p13.1q22), t(8;21)(q22;q22.1)
- Bone marrow aspirate is hypercellular with abundant eosinophils and eosinophilic precursors with dysplastic features
- Clonal cytogenetic (e.g., +8, -20q) or molecular genetic abnormalities (e.g., TET2, ASXL1 and DNMT3A); if no clonal abnormalities are found, then blast count must be > 2% in peripheral blood or > 5% in bone marrow aspirate
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR-ABL1, PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusions
- No lymphoid or myeloid neoplasms (e.g., MDS / MPN, MPNs, systemic mastocytosis or AML) are identified
- Idiopathic hypereosinophilia:
- Sustained eosinophilia (> 1.5 x 109/L) for at least 6 months with no tissue damages
- No reactive causes, lymphoid or myeloid neoplasms are identified
- Increased eosinophils with no dysplastic features or no increase in blasts in the bone marrow
- No molecular alteration is identified and no PDGFRA, PDGFRB, FGFR rearrangement, BCR-ABL1, PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusions
- BCR
- CNTRL
- FGFR10P
- ZMYM2
- BCR-FGFR1 fusion, t(1;8)(q31.1;p11.2)
- CPSF6-FGFR1 fusion, t(8;12)(p11.2;q15)
- CUX1-FGFR1 fusion, t(7;8)(q22.1;p11.2)
- FGFR10P-FGFR1 fusion, t(6;8)
- Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by mutation of the GBA1 gene that codes for glucocerebrosidase (GCase)
- Impaired activity of GCase causes its substrate, glucocerebroside, to accumulate in lysosomes of reticuloendothelial system (liver, spleen, bone marrow)
- Macrophages (Gaucher cells) become laden with lipid and dysfunctional
- One of the most common lysosomal storage disorders with a predisposition among those of Ashkenazi Jewish descent
- Caused by mutation in GBA1 gene on chromosome 1q that codes for GCase enzyme
- Impaired enzymatic activity leads to accumulation of Gaucher cells in reticuloendothelial system (liver, spleen, bone marrow), which leads to hepatosplenomegaly and bone marrow infiltration with cytopenia and bone pain
- Diagnosis is made by assessing GCase activity
- Treatment includes enzyme replacement therapy (GCase infusions) and substrate reduction therapy
- Gaucher disease (pronounced as GO-SHEY)
- ICD-10: E75.22 - Gaucher disease
- First described in 1882 in a patient with massive splenomegaly (Int J Mol Sci 2017;18:441)
- Prevalence is highest among the Ashkenazi Jewish population (Eastern European) with a frequency of 6% compared to 0.7 - 0.8% of all other ethnic groups (Hematology 2017;22:65)
- Liver
- Hepatomegaly with or without elevated liver enzymes (alkaline phosphatase, aminotransferase) due to intracellular accumulation of Gaucher cells and secondary inflammatory response
- Concurrent chronic liver diseases such as chronic viral hepatitis, steatohepatitis and vascular diseases are often seen
- Liver fibrosis (20 - 50% of cases) and cirrhosis with portal hypertension due to accumulation of Gaucher cells inciting an inflammatory response with fibrosis causing ischemia, infarction and progression of liver fibrosis (Mol Genet Metab Rep 2020;22:100564)
- Focal liver lesions also known as Gaucheroma (benign cluster of Gaucher cells associated with inflammation, fibrosis and iron accumulation)
- Increased risk of chronic liver disease and hepatocellular carcinoma due to cirrhosis
- Hypergammaglobulinemia and hyperferritinemia with probable underlying factor include chronic inflammation, impaired macrophage function and dysregulation in hepcidin - ferroportin axis
- Spleen
- Splenomegaly with focal splenic lesions characterized by clusters of Gaucher cells involving mostly the splenic cords in the red pulp
- Extramedullary hematopoiesis
- Bone marrow
- Infiltration of bone marrow by Gaucher cells causes reduced hematopoiesis and cytopenia
- Reduced proportion of marrow fat
- Bone
- Skeletal remodeling is also affected by replacement of marrow adipocytes with Gaucher cells; this causes fractures, remodeling defects, loss of bone mineral density and bone infarction
- Renal involvement is rare but proteinuria with or without renal insufficiency can occur
- Reference: Am J Hematol 2015;90:S6
- Mutation in GBA1 gene causes decrease in GCase activity with accumulation of glucocerebroside within activated macrophages (Gaucher cells) that infiltrate liver, spleen, bone marrow and other organs, leading to organomegaly
- Gaucher cells release cytokines and chemokines (IL1 beta, IL6, TNF alpha, IL10, IL18 and CCL18)
- Accumulated glucocerebroside is metabolized into sphingosine that can cause direct damage to hepatocytes, hematopoietic cells and the CNS
- GGase deficiency causes reduced osteoblast differentiation and function, reduced bone mineral deposition, defective bone remodeling caused by decreased bone formation leading to bone weakening, cortical thinning and trabecular resorption
- Infiltration of marrow by Gaucher cells leads to change in medullary microenvironment and compression of intraosseous blood vessels with potential bone infarction and necrosis
- References: Int J Mol Sci 2017;18:441, Am J Hematol 2015;90:S6
- Autosomal recessive disease caused by mutation in gene GBA1 that codes for GCase causing impaired function and excessive accumulation of glucocerebroside in the reticuloendothelial system (liver, spleen, bone marrow)
- GD patients inherit 2 copies of mutated GBA1 (homozygous) from parents who are carriers
- Reference: Am J Hematol 2015;90:S6
Images hosted on other servers:

Glucosylceramide metabolic pathway
- Splenomegaly is often the initial presentation
- Primary hypersplenism with anemia and thrombocytopenia, leukopenia also occurs frequently
- Hepatomegaly occurs late in the disease course with an increase in liver enzymes
- Bone marrow involvement / infiltration leading to anemia, thrombocytopenia, plasma cell dyscrasia
- Skeletal involvement in the form of osteopenia, osteoporosis, defective bone remodeling, osteonecrosis
- Pulmonary involvement in the form of interstitial lung disease
- Central nervous system (CNS) involvement
- Impaired cognition (type I GD)
- Convulsions, intellectual disability, apnea (type II GD)
- Myoclonus, seizures, dementia (type III GD)
- Categorized into 3 main types
- Type I: nonneuronopathic; most common type (90 - 95% of GD); variable presentation from asymptomatic patients discovered incidentally to early childhood presentation (OMIM: Gaucher Disease, Type I; GD1 [Accessed 1 May 2023])
- Type II: acute and severe neuronopathic type; affects CNS in infants, usually death before third year of life (OMIM: Gaucher Disease, Type II; GD2 [Accessed 1 May 2023])
- Can cause hydrops fetalis
- Perinatal lethal / collodion type: most severe form of type II with clinical manifestations presenting before birth and causing neonatal death
- Type III: subacute or juvenile neuronopathic, with systemic and CNS involvement, including oculomotor symptoms (OMIM: Gaucher Disease, Type III; GD3 [Accessed 1 May 2023])
- Intermediate in severity
- Associated with increased risk of multiple myeloma (relative risk: 5.9) (Blood 2005;105:4569)
- References: Expert Rev Endocrinol Metab 2018;13:107, Hematology 2017;22:65, J Bone Miner Res 2019;34:996
- Diagnosis is made by assessing GCase activity in circulating lymphocytes or fibroblast culture; < 10 - 15% of mean normal activity is diagnostic
- Disease activity or response to therapy is evaluated by measuring biochemical markers in activated macrophages (e.g., chitotriosidase, angiotensin converting enzyme, ferritin, chemokine CCL18 / PARC)
- Genotype testing in Ashkenazi Jewish patients for the most common alleles (L444P, N370S, IVS2+1g>a, V394L and R496H)
- References: Int J Mol Sci 2017;18:441, Am J Hematol 2015;90:S6
- Anemia
- Thrombocytopenia due to hypersplenism
- Hyperferritinemia
- Hypergammaglobulinemia
- GCase enzyme activity assay
- Serum protein electrophoresis
- Bone marrow
- Large (30 - 100 micron) macrophages with fibrillary pale blue to grey cytoplasm
- Liver biopsy
- Performed for hepatomegaly and shows clusters of Gaucher cells (macrophages with amphophilic cytoplasm resembling wrinkled tissue paper)
- Reference: Mol Genet Metab Rep 2020;22:100564
- Radiographic / Xray findings
- Erlenmeyer flask deformity (enlargement of metaphysis and absence of the typical concave diametaphyseal curve) in distal femur
- Osteonecrosis, proximal femur
- Osteopenia
- Lytic lesions
- MRI performed to assess skeletal involvement
- Osteonecrosis
- Gaucher cell infiltration produce low signal on T1 - T2 weighted images
- Degree of bone marrow infiltration by Gaucher cells
- CT / ultrasound findings
- Hepatosplenomegaly
- Bone scan
- Decreased uptake
- EEG and swallowing studies for neuronopathic GD
- Reference: J Bone Miner Res 2019;34:996
Images hosted on other servers:
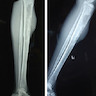
Gaucher lytic lesions

Erlenmeyer flask deformity
- Variable prognosis depending on clinical type of disease, ranging from asymptomatic to fatal outcome
- Type I: normal life span
- Type II: patients die within first 3 years of life
- Type III: intermediate between type I and type II
- Early diagnosis and treatment can have a good outcome
- Reference: Mol Genet Metab 2021;132:49
- 4 year old Albanian boy diagnosed with GD type 1 through newborn screening presented with multiple osteonecrosis in femur on follow up (JIMD Rep 2022;63:414)
- 15 year old boy who presented with fever, pulmonary interstitial fibrosis and hepatomegaly and was ultimately diagnosed with GD (J Med Case Rep 2018;12:306)
- 18 year old man diagnosed with neuroblastoma, also found to have GD (Blood Cells Mol Dis 2018;68:106)
- 24 year old woman diagnosed as carrier of GD when she presented with gestational thrombocytopenia and anemia that did not respond to any medications (J Med Case Rep 2022;16:203)
- 38 year old man who presented with solitary swelling of proximal tibia, mimicking a musculoskeletal tumor (J Orthop Case Rep 2022;12:64)
- Enzyme replacement therapy, which includes glucocerebrosidase IV infusions
- FDA approved Cerezyme (imiglucerase), VPRIV (velaglucerase alfa) and Elelyso (taliglucerase alfa) effective against hematologic, visceral and bone symptoms
- Not known to be effective against neurologic symptoms
- Substrate reduction therapy (oral medication) to reduce the accumulation of toxic substrate
- Cerdelga (eliglustat), a glucosylceramide synthase inhibitor, is indicated only for type 1 GD
- Zavesca (miglustat) for type 1 GD; not known to be effective against neurological symptoms (Ann Neurol 2008;64:514)
- Symptomatic treatment
- Bone remodeling: targeted treatments and vitamin D
- Splenectomy for hypersplenism
- Analgesics for bone pain and joint pain
- Bone marrow transplant only in cases of neuronopathic GD
- Somatic cell gene therapy under investigation
- References: Expert Rev Endocrinol Metab 2018;13:107, Mol Genet Metab 2021;132:49, Eur J Pharmacol 2022;926:175023
Images hosted on other servers:
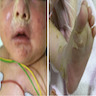
Perinatal collodion Gaucher (type 2)
- Bone
- Mass-like lesions that are soft, hemorrhagic and irregular, resulting in extensive localized bone loss
- Osteonecrosis of bone appears as pale yellow zones
- Spleen
- Splenomegaly with pale homogenous cut surface with reddish yellow nodules of infarction
- References: Am J Hematol 2016;91:736, J Orthop Case Rep 2022;12:64
Images hosted on other servers:

Splenomegaly in Gaucher disease
- Bone marrow, liver, spleen
- Infiltration by Gaucher cells containing abundant cytoplasm with fine, fibrillary, amphophilic characteristics resembling wrinkled tissue paper
- May have increased reticulin fibers and reduced fat in bone marrow biopsy
- Liver often demonstrates infiltration in the zone 3 region by Gaucher cells
- Small focal accumulations (Gaucheroma) or diffuse replacement by large, ovoid histiocytes (30 - 100 microns) with abundant, fibrillary eosinophilic granular cytoplasm resembling wrinkled tissue paper
- Small, bland nucleus that may be centrally or eccentrically located
- Cytoplasm has periodic acid Schiff diastase (PASD) resistant granules
- Reference: Mol Genet Metab Rep 2020;22:100564
Contributed by Patricia Tsang, M.D. and Mowafak Hamodat, M.B.Ch.B., M.Sc. (Case #164)

Gaucher in marrow biopsy
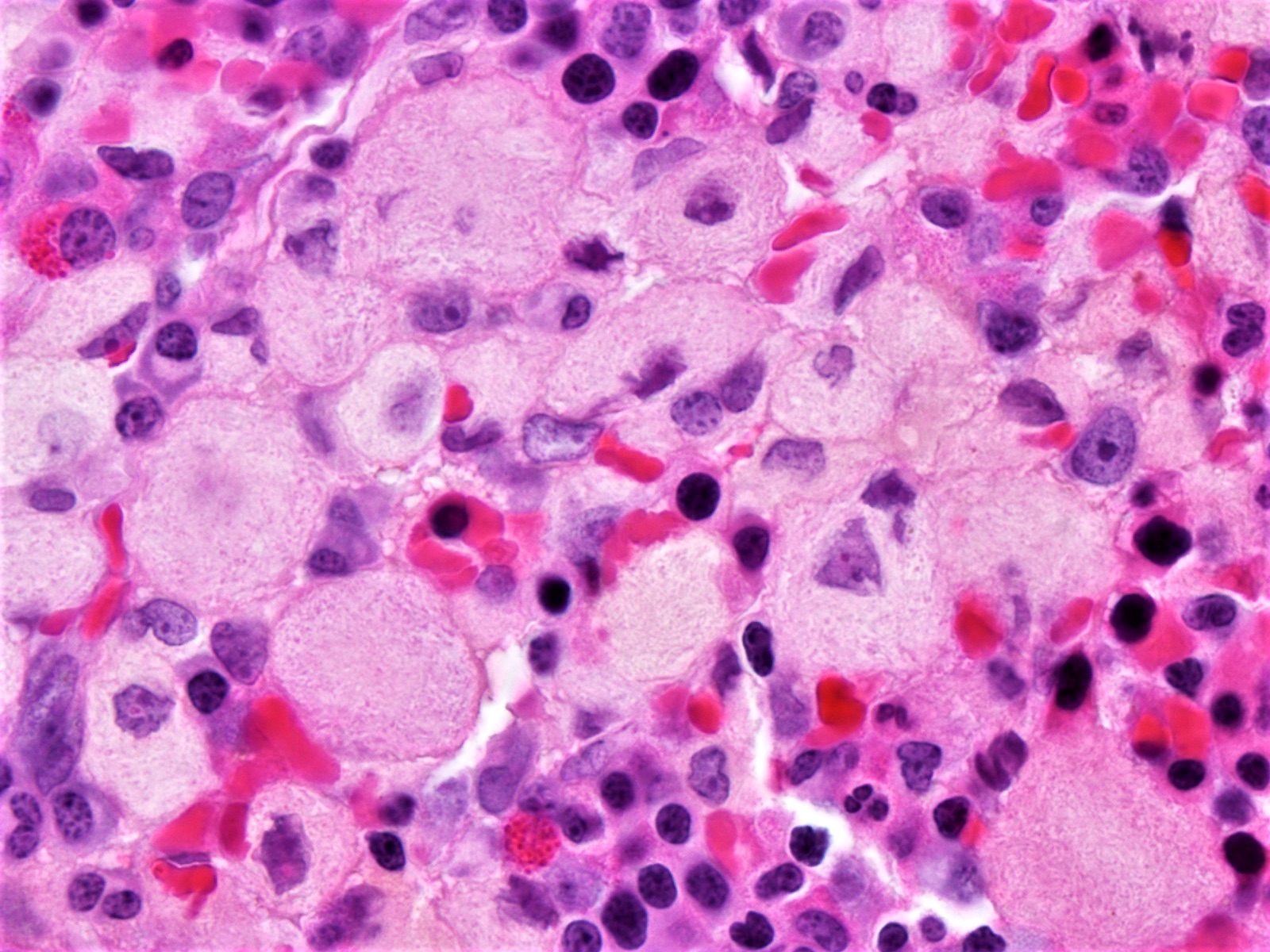
Fibrillary Gaucher cell cytoplasm

Gaucher cells marrow clot
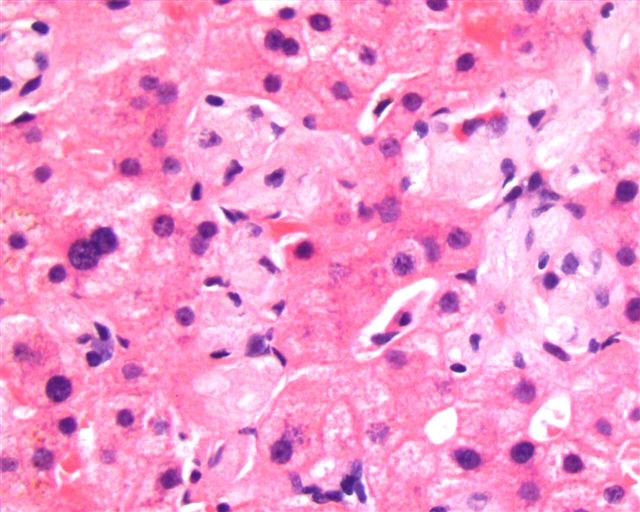
Liver biopsy

PASD positive Gaucher cells
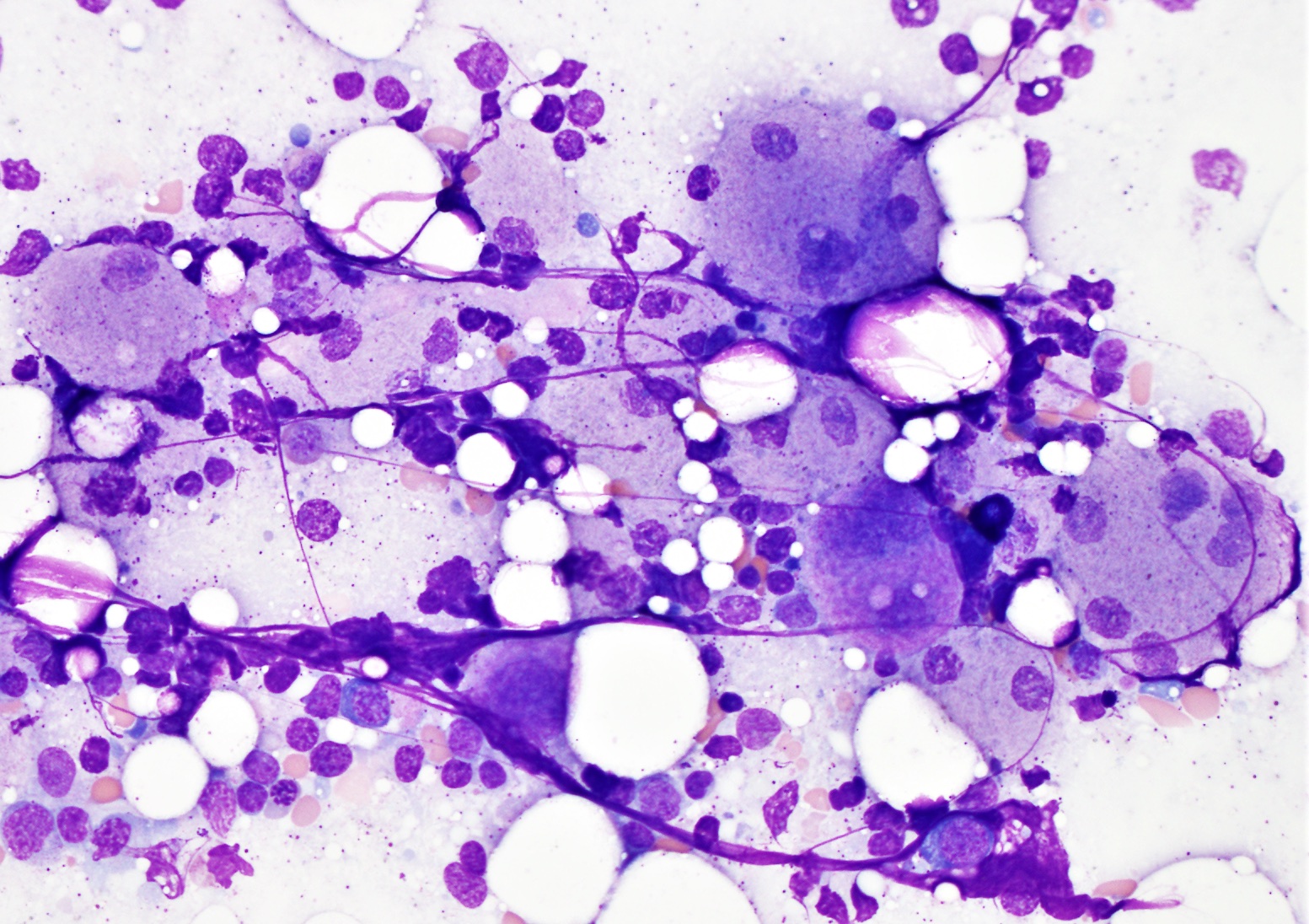
Marrow aspirate
Images hosted on other servers:
Granular, fibrillary blue gray cytoplasm
- Wright-Giemsa stained bone marrow aspirate shows Gaucher cell containing abundant granular or fibrillary, blue-gray cytoplasm with wrinkled tissue paper-like appearance
- PAS and PASD are positive in Gaucher cells (diastase resistant)
- Iron
- CD68
- Reference: Arch Pathol Lab Med 2008;132:851
- Phospholipid stains, acid fast stains, CD11b, CD40
- Elongated / rod shaped lysosomes filled with tubular structures
Images hosted on other servers:

Gaucher cell cytoplasmic fibrils
- Gene encoding glucocerebrosidase (Gcase) GBA1 is located on the long arm of chromosome 1 (1q21) and genetic testing is performed using saliva or blood samples
- DNA result is obtained by whole gene sequencing or selective sequencing within families
- More than 400 different mutations have been described in GBA1 gene; most prevalent are N370s, L444P
- c.1226A>G (N370S) mutation is common in the Ashkenazi Jewish population and most of these patients are asymptomatic
- L444P mutation seen in GD is at high risk for developing CNS complications
- Homozygous (D409H) mutation presents with cardiac complications
- References: Int J Mol Sci 2017;18:441, Mol Genet Metab 2021;132:49
Gaucher disease involving bone
- Liver, right lobe, needle core biopsy:
- Gaucher disease involving liver (see comment)
- Comment: Sections of the liver biopsy show clusters of Gaucher cells (pale staining histiocytes with striated cytoplasm, PASD positive) involving mainly the perivenular zone with atrophy of hepatocyte plates.
- Pseudo-Gaucher cells:
- Mimic Gaucher cells on light microscopy but iron stain negative and lack the typical electron microscopy findings seen in GD (Turk J Haematol 2014;31:428)
- Seen in conditions that cause rapid cell turnover (e.g., myeloproliferative and myelodysplastic neoplasms, myeloma)
- Mycobacterial infection (AFB stain+)
- Sea blue histiocytosis (Arch Pathol Lab Med 2008;132:851):
- Seen in various conditions with increased marrow cell turnover (e.g., myeloproliferative or myelodysplastic neoplasms)
- Macrophage contains ceroid (insoluble lipid pigment); inclusions are globular (not fibrillary)
- Stains more intensely blue with Wright-Giemsa than Gaucher cells
- Niemann-Pick disease (Humpath.com: Niemann-Pick Diseases [Accessed 6 June 2023]):
- Lipid storage disorder caused by sphingomyelinase deficiency, which leads to accumulation of lipids in histiocytes that appear foamy
- Electron microscopy shows lamellar inclusions in lysosomes
A 5 year old boy presents to the clinic for evaluation of peripheral pancytopenia. His parents are of Ashkenazi Jewish descent. On physical examination, he has hepatosplenomegaly. Bone marrow aspirate smears show large cells with fibrillary cytoplasm that are positive for periodic acid Schiff (PASD) stain. Which of the following lysosomal enzymes are most likely deficient?
- Alpha galactosidase
- Glucocerebrosidase
- Hexosaminidase A
- Sphingomyelinase
Comment Here
Reference: Gaucher disease
- Gastric ulcers
- Hydrops fetalis
- Normal expected life span
- Oculomotor symptoms
Comment Here
Reference: Gaucher disease
- Rare B cell neoplasm characterized by production of monoclonal heavy chain unable to bind light chain (truncated or mutated)
- Alpha heavy chain disease is the most common; it is also known as immunoproliferative small intestinal disease (IPSID), a variant of extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT lymphoma)
- 3 main types: alpha (IgA), gamma (IgG) and mu (IgM)
- Resultant lymphoma resembles other lymphoma types with each heavy chain disease associated with distinct features
- Must demonstrate free heavy chain by protein electrophoresis / immunofixation
- Alpha heavy chain disease is the most common, presents in younger patients, marginal zone phenotype
- Gamma is often associated with autoimmune disease, may be polymorphic
- Mu may resemble chronic lymphocytic leukemia (CLL) with mix of lymphocytes and characteristic vacuolated plasma cells
- Alpha heavy chain is also known as immunoproliferative small intestinal disease (IPSID)
- ICD-10: C88.2 - heavy chain disease
- Alpha: younger age, peak incidence in 20s and 30s, equal prevalence in men and women, more common in areas neighboring Mediterranean Sea, most prevalent in Africa and the Middle East, associated with factors linked to low socioeconomic status (poor hygiene, malnutrition, frequent intestinal infections)
- Gamma: rare, ~150 cases reported, median age 60, female predominance
- Mu: extremely rare, 30 - 40 cases reported, median age 60, equal prevalence in men and women
- Alpha: mainly small intestine, mesenteric lymph nodes; gastric and colonic mucosa may be involved, rarely respiratory tract and thyroid; usually not bone marrow or other organs
- Gamma: lymph nodes or extranodal, Waldeyer ring, gastrointestinal tract, bone marrow, liver, spleen, peripheral blood, skin, subcutaneous tissue
- Mu: spleen, liver, bone marrow, blood, usually not lymphadenopathy
- All: deletions in heavy chain gene result in defective heavy chain protein that cannot bind to light chain to form complete Ig molecule
- Alpha: chronic intestinal infection (e.g. Campylobacter jejuni) may underlie development of the disease
- Alpha: typically presents with malabsorption, diarrhea, hypocalcemia, abdominal pain, wasting, fever; patients may die from malnutrition, intestinal obstruction, sepsis or other complications due to massive bowel involvement
- Gamma: typically associated with systemic symptoms (anorexia, weakness, fever, weight loss, bacterial infections), concurrent autoimmune disease, generalized disease (hepatomegaly, splenomegaly, lymphadenopathy); rarely transforms to large cell lymphoma; disease patterns range from indolent to aggressive
- Mu: may resemble chronic lymphocytic leukemia (CLL) but differs in the high frequency of hepatosplenomegaly and absence of peripheral lymphadenopathy; has mixture of plasma cells and B lymphocytes with monoclonal cytoplasmic mu heavy chain; similar slow progression
- Combination of histopathologic features and detection of serum heavy chain by immunofixation
- Variably sized heavy chains may be produced, which may NOT produce characteristic monoclonal peak on serum protein electrophoresis
- Immunofixation may be critical for detection
- Gamma: may detect IgG without light chain in blood or urine
- Mu: mu heavy chain not detected in urine but Bence Jones light chains may still be detected (especially kappa) (Am J Hematol 1992;40:56)
- Alpha: early stages may respond to antibiotic therapy; more advanced disease requires multiagent chemotherapy
- 48 year old woman with gamma heavy chain disease and a history of seronegative rheumatoid arthritis (Biochem Med (Zagreb) 2012;22:373)
- 48 year old woman with Hodgkin lymphoma and gamma heavy chain disease (Arch Pathol Lab Med 2001;125:803)
- 59 year old woman with immunotactoid glomerulopathy, follicular lymphoma and gamma heavy chain disease (Arch Pathol Lab Med 2004;128:689)
- 64 year old patient with mu heavy chain disease (Blood 2017;130:558)
- Man with 30 year course of immunoproliferative small intestinal disease (IPSID) without development of lymphoma (Am J Gastroenterol 2001;96:2769)
- Alpha: early disease may respond to antibiotic therapy; for more advanced immunoproliferative small intestinal disease, anthracycline containing regimens have been reported to result in remission and longterm survival
- Alpha: lamina propria of bowel mucosa heavily infiltrated with plasma cells and admixed lymphocytes which separate the crypts; may have marginal zone B cells, lymphoepithelial lesions and villous atrophy; progression to diffuse large B cell lymphoma may be characterized by destructive sheets of large plasmacytoid cells, immunoblasts and ulceration
- Gamma: circulating plasma cells, may resemble splenic marginal zone or MALT lymphoma; polymorphous proliferation with lymphocytes, plasma cells, immunoblasts, histiocytes, eosinophils
- Mu: bone marrow contains vacuolated plasma cells admixed with small lymphocytes
Contributed by Genevieve M. Crane, M.D., Ph.D.
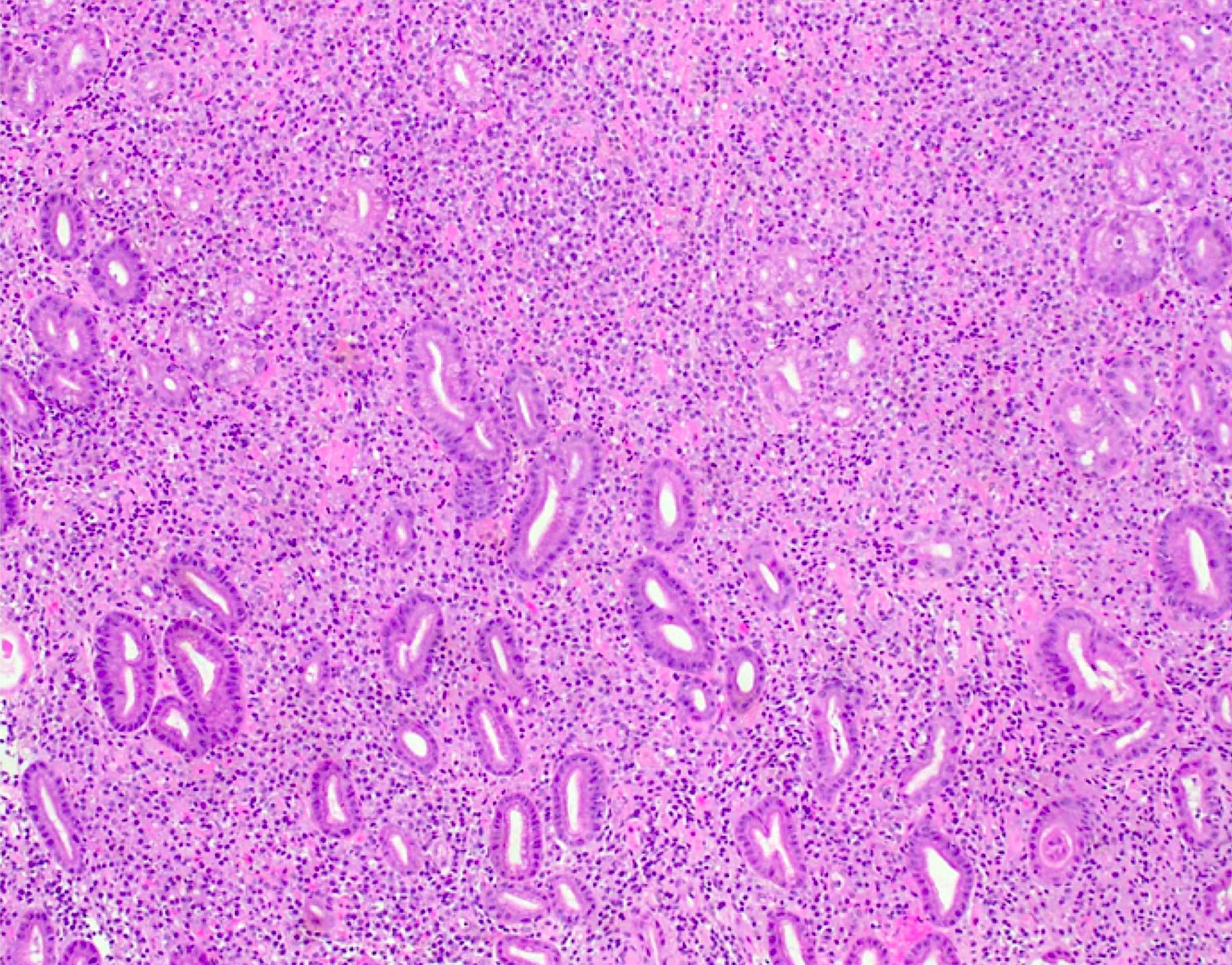

Lamina propria is diffusely expanded by a plasma cell rich infiltrate
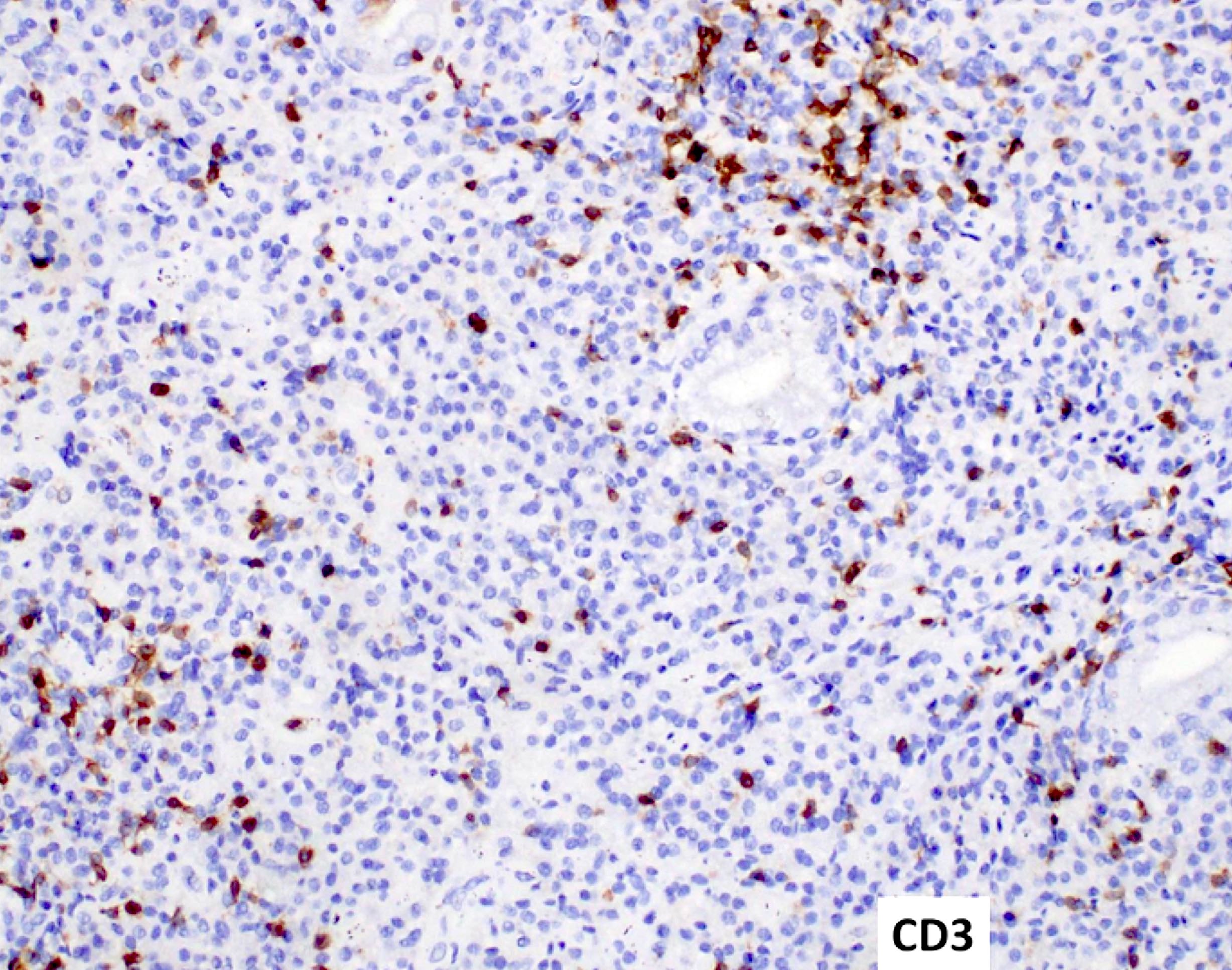
CD3 highlights scattered infiltrating T cells
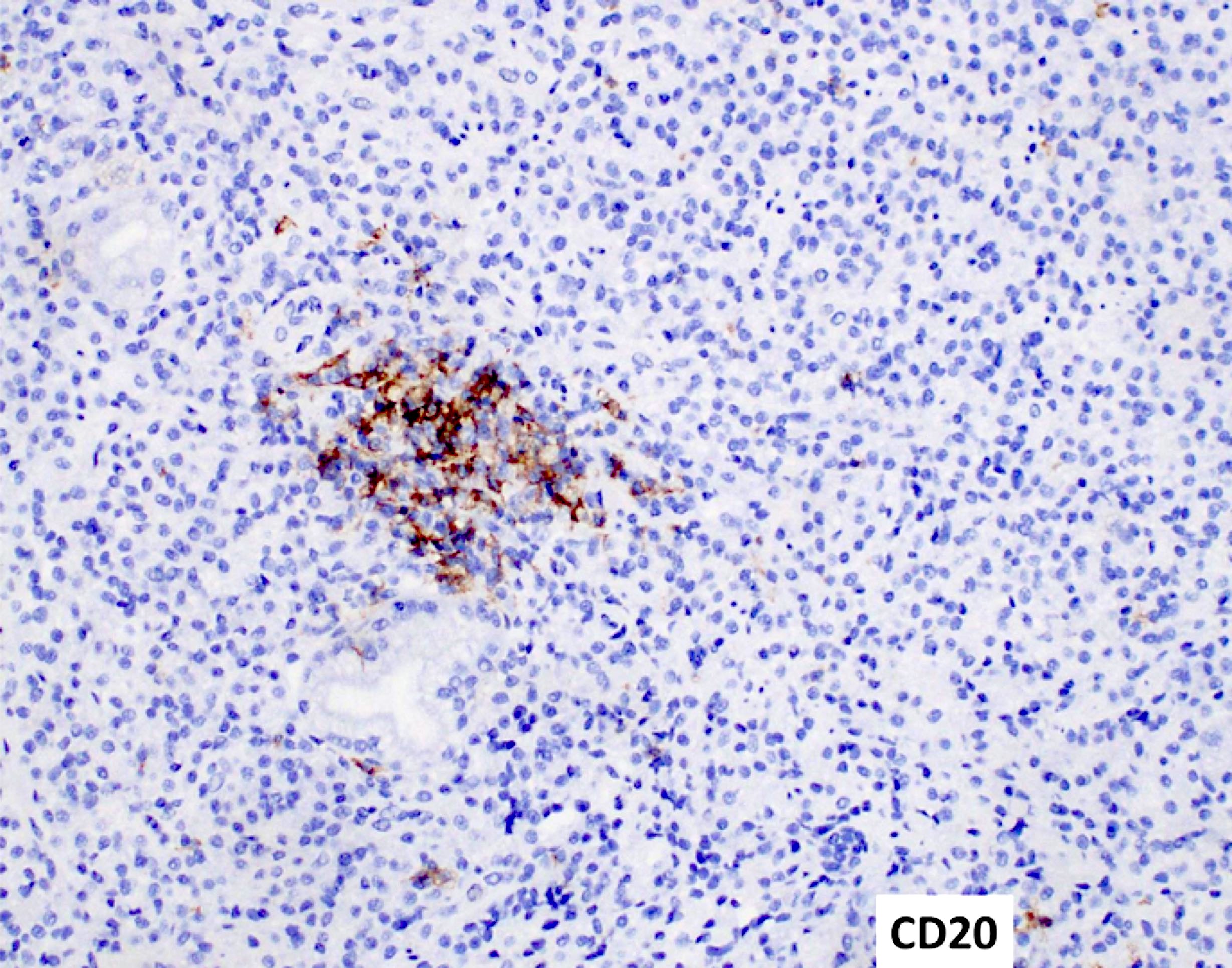
CD20 highlights rare B cell nodules

CD79a stains the majority of the infiltrate

CD138 highlights plasma cells and epithelium
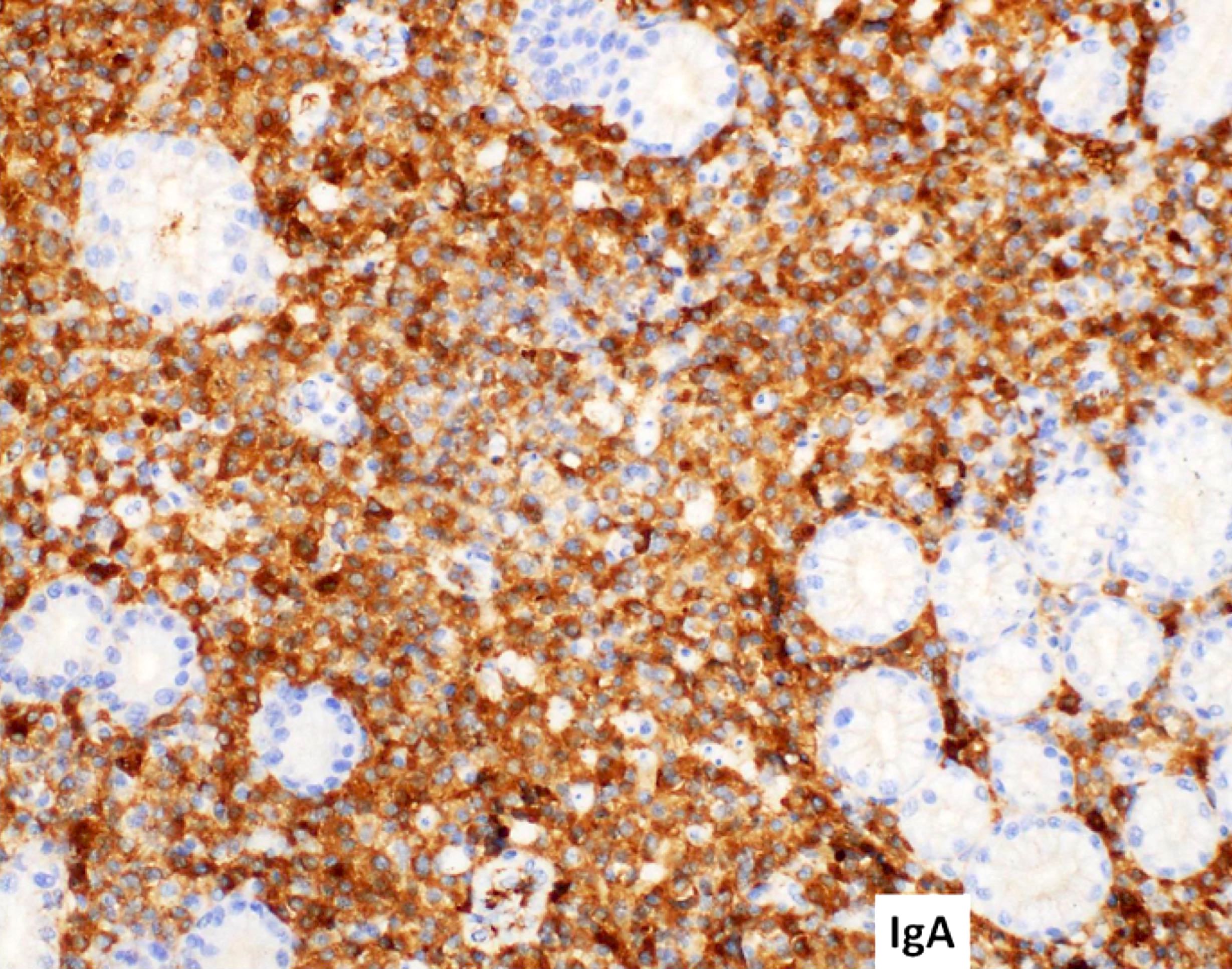
Plasma cells are predominantly positive for cytoplasmic IgA
Images hosted on other servers:

Characteristic
intracytoplasmic
vacuoles
- All have clonally rearranged IGH
- Deletions / insertions in the IGHV or CH1 domain result in defective or truncated heavy chain that cannot bind light chain to form a complete immunoglobulin molecule
- Alpha: case reports of cytogenetic abnormalities but not the t(11;18)(q21;q21) (BIRC3 / MALT1) translocation associated with gastric and pulmonary MALT lymphoma
- Gamma: abnormal karyotype in 50% of cases, MYD88 mutation is absent
- Mu: immunoglobulin genes show high levels of somatic mutation
- Alpha: a variant of MALT lymphoma, suspicion based on location demographics, clinical features
- Gamma:
- If more monomorphic, may resemble:
- If polymorphous, may resemble:
- Mu: typically resembles chronic lymphocytic leukemia (CLL)
- Lymphoplasmacytic lymphoma
- MALT variant immunoproliferative small intestinal disease
- Mu heavy chain disease
- Myeloma with a t(11;14) translocation
- Splenic marginal zone lymphoma
Comment Here
Reference: Heavy chain disease
- Alpha heavy chain disease
- Gamma heavy chain disease
- Mu heavy chain disease
- None of the above
- Idiopathic hypereosinophilic syndrome is a disorder defined by peripheral blood eosinophilia (absolute eosinophil count ≥ 1.5 x 109/L) for at least 6 months with organ damage / dysfunction attributable to tissue hypereosinophilic infiltrate per biopsy and no discernible underlying etiology
- Diagnosis of exclusion
- Hypereosinophilia (HE) is defined by an increase in eosinophils in the peripheral blood (≥ 1.5 x 109/L); however, if eosinophilia is sustained for ≥ 6 months and there is associated tissue damage, this should be classified as hypereosinophilic syndrome (HES)
- Clinically, if treatment is necessary to minimize tissue / organ damage, the criteria of 6 months might not be enforced and an eosinophil count ≥ 1.5 x 109/L on 2 occasions for ≥ 1 month apart may be sufficient (Pathobiology 2019;86:39)
- Idiopathic hypereosinophilia diagnostic criteria
- Eosinophil count ≥ 1.5 x 109/L for ≥ 6 months
- Tissue or organ damage associated with eosinophilic infiltration
- Cardiac and cutaneous manifestations are common but liver, CNS, muscle, pulmonary and nasal sinus involvement may occur as well
- No discernible underlying cause
- Exclude reactive causes of eosinophilia (allergy, infection including parasites, drug related, connective tissue disorder, etc.)
- Exclude acute myeloid leukemia, myeloproliferative neoplasms, myelodysplastic syndromes, myelodysplastic / myeloproliferative neoplasms, systemic mastocytosis and myeloid / lymphoid neoplasms with eosinophilia and PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2 rearrangement
- Exclude cytokine producing, immunophenotypically aberrant T cell population
- No cytogenetic or molecular abnormalities, however, must exclude clonal hematopoiesis (CHIP) mutations
- Idiopathic HES
- ICD-10: D72.110 - idiopathic hypereosinophilic syndrome (IHES)
- Typically occurs in adults age 20 - 50 years (Blood 1994;83:2759, Pediatr Hematol Oncol 2009;26:129, Br J Haematol 2010;151:440)
- Estimated prevalence is 0.36 - 6.3 per 100,000 (J Allergy Clin Immunol 2010;126:179)
- Previously thought to have male predominance until cases with FIP1L1-PDGFRA fusion were excluded
- Skin and cardiac involvement common
- Minority of patients present with splenomegaly and lymphadenopathy
- Liver, CNS, muscle, pulmonary and nasal sinus involvement occur more commonly than in eosinophilic leukemia (Haematologica 2014;99:e148)
- Cause is unknown
- Onset of symptoms is often insidious with eosinophilia being detected incidentally; however, some patients initially present with severe and life threatening problems due to the rapid progression of cardiovascular and neurologic complications
- Retrospective study of 188 patients with hypereosinophilia reported the frequency of symptoms at presentation (J Allergy Clin Immunol 2009;124:1319):
- Dermatologic symptoms were most common, followed by pulmonary, gastrointestinal, cardiac and lastly neurologic
- 6% of patients presented initially with incidentally detected asymptomatic hypereosinophilia
- Dermatologic: angioedema, dermographism, eczema, erythroderma, pruritis, urticaria
- Pulmonary: cough, dyspnea, wheezing
- Gastrointestinal: weight loss, abdominal pain, vomiting, diarrhea, hepatitis, cholangitis, Budd-Chiari syndrome
- Cardiac: cardiac damage, valvular fibrosis, thromboembolism
- Neurologic: behavioral changes, confusion, ataxia, memory loss, peripheral neuropathy
- Unexplained eosinophilia of ≥ 1.5 x 109/L for at least 6 months that leads to tissue damage
- Exclude reactive causes of eosinophilia (parasites, drug related, allergy, etc.)
- Exclude acute myeloid leukemia, myeloproliferative neoplasms, myelodysplastic syndromes, myelodysplastic / myeloproliferative neoplasms and systemic mastocytosis
- Exclude cytokine producing, immunophenotypically aberrant T cell population
- Tissue damage present due to hypereosinophilia
- Bone marrow with increased eosinophilic precursors; proposed criteria of ≥ 20% marrow cellularity with or without peripheral blood eosinophilia (Pathobiology 2019;86:39)
- Affected organs may have increased eosinophils with eosinophil degranulation (Pathobiology 2019;86:39)
- Serum IgE level is elevated in about half of patients
- Serum tryptase is occasionally elevated (Blood 2003;101:4660)
- Serum B12 and platelet counts are typically normal (Haematologica 2014;99:e148)
- Slowly progressive clinical course with death occurring due to cardiac damage in some cases
- 26 year old man with ascites (JRSM Open 2020;11:2054270419894826)
- 36 year old man with erythroderma (An Bras Dermatol 2018;93:451)
- 43 year old man with pulmonary hypertension (Pulm Circ 2019;9:2045894018793999)
- 83 year old woman with dyspnea (Am J Case Rep 2019;20:381)
- Patients are typically observed during the initial stages of the disease
- As HES progresses, initial treatment is often systemic glucocorticoid therapy with second line treatments including imatinib, interferon alfa and hydroxyurea
- Recent study showed that mepolizumab could be effective at treating flares in HES (J Allergy Clin Immunol 2020;146:1397)
- Bone marrow
- Normocellular or hypercellular marrow with an increase in eosinophils and eosinophilic precursors with orderly maturation
- Bone marrow eosinophilia proposed criteria: ≥ 20% marrow cellularity with or without peripheral blood eosinophilia (Pathobiology 2019;86:39)
- Normal differential counts
- No increase in blasts (< 5%)
- No neoplastic process
- Tissue / organ
- Eosinophils are typically scattered or absent in normal tissue; affected organs may have increased eosinophils with eosinophil degranulation (Pathobiology 2019;86:39)
- Possible eosinophilic microabscesses
- Organs affected include gastrointestinal tract, lung, lymph nodes, spleen and thymus
AFIP images
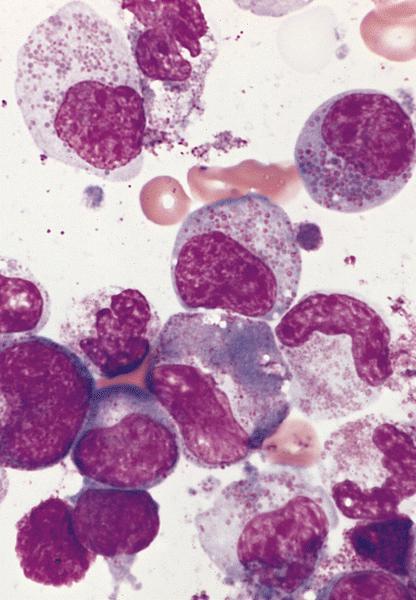
Bone marrow aspirate
- Marked eosinophilia (≥ 1.5 x 109/L) with normal eosinophil morphology (Foucar) (J Allergy Clin Immunol 2012;130:607)
- No increase in blasts (< 2%)
AFIP images
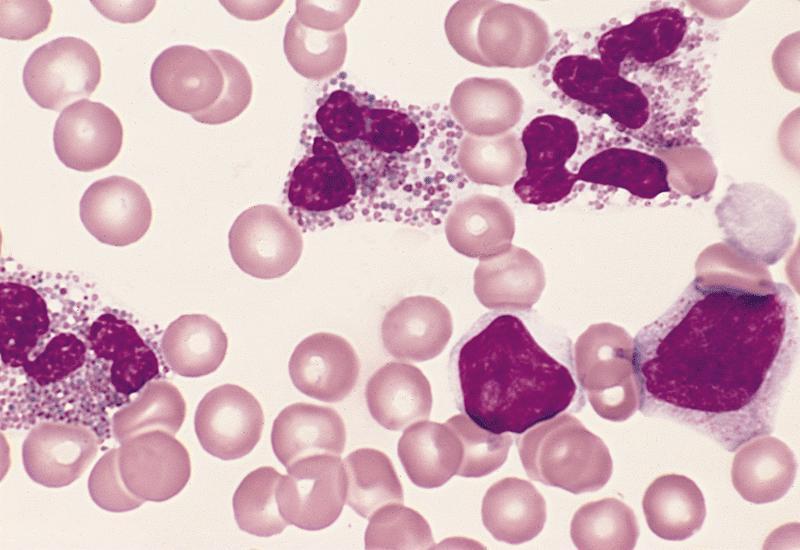
40% eosinophils with normal morphology
- Flow cytometry on blood, bone marrow or other tissue specimen may be important for excluding a potential associated lymphoid, myeloid or mast cell neoplasm
- No clonal myeloid or lymphoid cells
- If an aberrant T cell population is identified, lymphocytic variant of hypereosinophilic syndrome should be considered (Arch Pathol Lab Med 2016;140:1060, J Allergy Clin Immunol 2012;130:607, Immunol Allergy Clin North Am 2007;27:389, Medicine (Baltimore) 2014;93:255)
- Generally, not useful when examining eosinophils
- No cytogenetic or molecular genetic abnormalities identified by definition (Jaffe: Hematopathology, 2nd Edition, 2016)
- Must exclude neoplasms with rearrangements of BCR-ABL1, PDGFRA, PDGRB or FGFR1 or PCM1-JAK2, ETV6-JAK2, BCR-JAK2 or FLT3 fusions as well as activating PDGFRA gene mutation
- Bone marrow, posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Hypereosinophilic syndrome (see comment)
- Hypercellular (80 - 90%) bone marrow with myeloid hyperplasia with markedly increased eosinophils and no overt increase in blasts (< 5%)
- No evidence of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2 gene rearrangements
- No evidence of BCR-ABL1, CBFB-MYH11 / inv(16) gene rearrangements
- No gene mutations identified per next generation sequencing (NGS) panel
- Comment: There is no morphologic evidence of a myeloid neoplasm associated with eosinophilia. Clinically there are no identifiable etiologies of reactive eosinophilia such as allergic reactions, parasitic infections, autoimmune diseases and medications. The patient presents with a persistent eosinophilia for over 6 months (ranging from 1600 - 5500/µL). Electrocardiogram and echocardiograms were reportedly abnormal. Imaging studies showed subendothelial fibrosis within the ventricles. The corresponding tissue biopsy showed eosinophilic infiltration (20 - 40 eosinophils/high power field) with degranulation within the myocardium.
- Flow cytometric analysis demonstrates occasional CD34+ myeloblasts which comprise about 1.0% of total events. Hematogones are absent. No clonal B cell population or immunophenotypically abnormal T cells (e.g. CD3- / CD4+) were noted to suggest lymphoid variant of hypereosinophilic syndrome or T cell lymphoma. No clonal plasma cell population is identified.
- Additional immunohistochemical stains performed on the core biopsy show no increase in CD34 or CD117 positive immature myeloid precursors. CD117 also highlights occasional normal appearing mast cells which do not form aggregates. CD3 and CD20 highlight scattered T and B cells respectively. CD138 stains plasma cells, which comprise < 1% of the marrow cellularity.
- Peripheral smear: Manual review of the peripheral blood shows normochromic, normocytic anemia, mild leukocytosis, absolute eosinophilia and essentially normal platelets. There is no absolute monocytosis or circulating blasts. Morphologically, RBCs show normochromic, normocytic anemia with mild anisopoikilocytosis. WBCs: slightly increased in number (13000/µL) without neutrophilia, blastosis or immature monocytosis. Occasional circulating immature granulocytic precursors, e.g. metamyelocytes and myelocytes, are noted. A manual 500 cell differential count reveals 42.0% of eosinophils, resulting in an absolute eosinophilic count of 5500/µL and 3.0% monocytes, resulting in an absolute monocyte count of 390/µL. No dysplastic granulocytes such as hypogranular or hyposegmented forms are identified. Platelets: normal by count with occasional large platelets.
- Bone marrow biopsy: Quality: adequate. Cellularity: 80 - 90%. Hematopoiesis: trilineage maturation with mild myeloid hyperplasia and relatively normal erythropoiesis. Megakaryocytes are identified and normally distributed. There is no apparent increase in blasts or abnormal localization of immature myeloid precursors. Mature eosinophils are increased, making up approximately 30 - 40% of marrow cellularity. Special stains: reticulin: loose network of reticulin without significant intersections (minimal reticulin fibrosis). Small loose lymphoid aggregates composed of small mature lymphocytes are focally noted, overall < 5% of marrow cellularity.
- Bone marrow clot section: Quality: adequate. Cellularity: 80 - 90%, morphologic features are similar to those observed in the core biopsy.
- Bone marrow aspirate: Quality: adequate. Granulocytes: mildly increased; mild left shifted maturation without morphologic dysplasia. Eosinophilic precursors, predominantly mature forms, are markedly increased comprising approximately 40 - 50% of cellularity. Erythrocytes exhibit normal normoblastic maturation. Megakaryocytes appear normal by morphology and adequate in number. Myeloblasts: overall ~3% of nucleated cells. There is no monocytosis or increase promonocytes. Iron: storage iron is adequate (2+ on a scale of 0 - 4). No ring sideroblasts are present. Scattered mature mast cells are present (2%).
- Acute myeloid leukemia with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) / CBFB / MYH11:
- Immature eosinophils containing rough basoeosinophilic granules
- Oligoblastic myelogenous leukemia or increased blasts, not necessarily ≥ 20%
- FISH or cytogenetic evidence of inv(16) or t(16;16) / CBFB-MYH11
- Atypical chronic myeloid leukemia (aCML), BCR-ABL1 negative:
- Besides features of myeloproliferative neoplasms and eosinophilia
- Leukocytosis, often > 25 x 109/µL
- Dysplasia is present in > 10% of neutrophilic precursors
- No absolute monocytosis
- < 20% blasts
- NGS study with frequent mutations involving SETBP1
- Besides features of myeloproliferative neoplasms and eosinophilia
- Chronic myelomonocytic leukemia (CMML) with eosinophilia:
- Normal or leukocytosis
- Sustained monocytosis ≥ 1 x 109/L, > 10% of WBCs for > 6 months
- < 20% blasts
- Bone marrow morphologic findings: hyper or normocellular, myeloproliferative or myelodysplastic predominance with or without marrow monocytosis
- Negative for t(5;12) or PDGFRB gene rearrangement
- Negative for BCR-ABL1 gene rearrangement
- NGS study with frequent mutations involving TET2, SRSF2, ASXL1 and SETBP1
- Chronic eosinophilic leukemia, NOS:
- Persistent eosinophilia (≥ 1.5 x 109/L) in which reactive causes have been excluded
- Does not meet diagnostic criteria for chronic myeloid leukemia with t(9;22)(q34.1;q11.2) / BCR-ABL1 gene fusion, polycythemia vera, essential thrombocythemia, primary myelofibrosis, chronic neutrophilic leukemia, chronic myelomonocytic leukemia, atypical chronic myeloid leukemia (BCR-ABL1 gene fusion negative) or systemic mastocytosis
- Blasts < 20% of peripheral blood and bone marrow cells and does not have the following cytogenetic aberrations:
- PDGFRA, PDGFRB or FGFR1 rearrangements
- t(8;9)(p22;p24.1) / PCM1-JAK2, t(9;12)(p24.1;p13.2) / ETV6-JAK2 or t(9;22)(p24.1;q11.2) / BCR-JAK2 fusions
- inv(16)(p13.1;q22) or t(16;16)(p13.1;q22) / CBFB / MYH11, t(15;17)(q22;q11-12) / PML-RARA or t(8;21)(q22;q22.1) / RUNX1 / RUNX1T1
- t(9;22)(q24;q31) / BCR-ABL1 fusions
- Presence of clonal cytogenetic or molecular genetic abnormality(ies) or a blast count ≥ 2% in the peripheral blood or ≥ 5% in the bone marrow
- Systemic mastocytosis with eosinophilia:
- Clonal proliferation of atypical mast cells
- Major diagnostic criteria: aggregates of ≥ 15 mast cells in the bone marrow or extracutaneous organ + 1 minor criterion or
- Minor diagnostic criteria (≥ 3 of the following 4 present)
- Morphology in BM or extracutaneous organ
- Infiltrate: > 25% mast cells are spindled or have abnormal morphology or
- BM aspirate: > 25% of mast cells are immature or atypical
- Molecular: KIT D816V point mutation, negative for FIP1L1-PDGFRA gene rearrangement
- Flow cytometry / immunohistochemistry: mast cells express aberrant CD25 +/- CD2 in addition to normal mast cell markers
- Laboratory findings: serum tryptase > 20 ng/ml persistently
- Morphology in BM or extracutaneous organ
- Bone marrow: eosinophils are present or increased, which could be a secondary reaction or derived from a neoplastic clone
- Idiopathic hypereosinophilia:
- When criteria 1 - 4 are met but there is no end organ damage
- Eosinophil count ≥ 1.5 x 109/L for ≥ 6 months
- Exclude reactive causes of eosinophilia
- Exclude acute myeloid leukemia, myeloproliferative neoplasms, myelodysplastic syndromes, myelodysplastic / myeloproliferative neoplasms and systemic mastocytosis
- Exclude cytokine producing, immunophenotypically aberrant T cell population
- Subset of patients harbor certain gene mutations, such as ASXL1 (43%), TET2 (36%), EZH2 (29%), SETBP1 (22%), CBL (14%) and NOTCH1 (14%) (Mod Pathol 2016;29:854)
- Presence of clonal genetic aberration favors chronic eosinophilic leukemia, not otherwise specified
- Lymphocytic variant of hypereosinophilic syndrome (L-HES):
- Abnormal cytokine release by immunophenotypically abnormal or clonal T cells
- Most commonly aberrant CD3- / CD4+ T cell population which can be detected by flow cytometry; absolute CD3- / CD4+ T cell count 0.01 - 28.3 k/L with median 0.35 k/L
- TCRγδ rearrangements were frequently identified (76%) (Medicine (Baltimore) 2014;93:255)
- High frequency of cutaneous manifestations, soft tissue involvement and lymphadenopathy (Medicine (Baltimore) 2014;93:255)
- Other phenotypic changes, e.g. CD3+ / CD4+ / CD7- and CD3+ / CD4- / CD8- TCRαβ+ have been reported (Immunol Allergy Clin North Am 2007;27:389, N Engl J Med 1999;341:1112)
- Elevated serum IgE and polyclonal hypergammaglobulinemia produced by aberrant T cells via T helper 2 cytokines (i.e. IL5, IL4, IL13, GM-CSF) (Br J Haematol 2000;109:540)
- 33% of cases have elevated absolute lymphocyte counts (Medicine (Baltimore) 2014;93:255)
- T cell clone infected by EBV can also produce cytokines that stimulate eosinophil production (Blood 2013;121:2364)
- Unknown molecular mechanism; however, STAT3 gain of function mutation, upregulation of STAT3 pathways and STAT5B gain of function mutations have been described independently (Blood 2016;127:948, Blood 2017;129:650)
- Medication related eosinophilia:
- Administration of cytokines IL2, IL3, IL5 or GMCSF
- Myeloid neoplasms with rearrangement of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusion (Blood 2017;129:704)
- Paraneoplastic eosinophilia:
- T cell lymphoma, Hodgkin lymphoma, systemic mastocytosis, acute lymphoblastic leukemia, myeloproliferative neoplasms or certain solid tumors (e.g. lung cancer, metastatic renal cell carcinoma, etc.) associated with abnormal release of IL2, IL3, IL5 or GMCSF (Arch Pathol Lab Med 2013;137:259, Rinsho Ketsueki 1991;32:874, Acta Clin Belg 2011;66:293, Am J Hematol 2007;82:234, N Z Med J 1990;103:537)
- Reactive eosinophilia:
- Parasitic or fungal infections, allergies, pulmonary disease (Löffler syndrome), cyclical eosinophilia, skin diseases (angiolymphoid hyperplasia), collagen vascular diseases such as Churg-Strauss syndrome and Kimura disease
- Myeloproliferative variants of HES (M-HES):
- HES with features of myeloproliferative disorder:
- Anemia, thrombocytopenia, circulating leukocyte precursors
- Hepatosplenomegaly
- Increased serum vitamin B12
- Chromosomal abnormalities
- Myeloid / lymphoid with following abnormalities
- FIP1L1-PDGFRA fusion
- PDGFRB rearrangement
- FGFR1 rearrangement
- PCM1-JAK2 fusion
- PDGFRA fusion with other partner genes have been described (Leukemia 2006;20:827, Br J Haematol 2007;138:77, Blood 2011;117:2935)
- KIT mutated systemic mastocytosis
- Myeloid / lymphoid with following abnormalities
- Often refractory to glucocorticoid therapy but may respond to tyrosine kinase inhibitors depending on mutation profile
- HES with features of myeloproliferative disorder:
- Familial HES:
- Autosomal dominant inheritance
- Family history of eosinophilia with or without endomyocardial fibrosis
- Aberrant gene was mapped to chromosome 5q21-33 in one affected family (Am J Hum Genet 1998;63:1086)
- May present as single organ damage, e.g. eosinophilic esophagitis and eosinophilic fasciitis (Ann Rheum Dis 1994;53:281, Clin Gastroenterol Hepatol 2008;6:621, Arthritis Rheum 1989;32:96)
- Organ restricted hypereosinophilic conditions:
- Disorder with single organ involvement together with eosinophilia ≥ 1.5 x 109/L or lower in the setting of clear single organ involvement
- When eosinophilia is ≥ 1.5 x 109/L, can be difficult to distinguish from IHES
- Can overlap with single organ restricted eosinophilic disorders, such as eosinophilic gastrointestinal disorders, chronic eosinophilic pneumonia and Wells syndrome
- Specific / defined syndromes associated with hypereosinophilia:
- Associated with immune dysregulation
- Autoimmune lymphoproliferative syndrome (ALPS) (Am J Hematol 2007;82:615, Blood Cells Mol Dis 1999;25:227)
- CARD9 deficiency collagen vascular diseases (N Engl J Med 2013;369:1704)
- HIV infection (Arch Dermatol 1994;130:119, Allergy 1997;52:110)
- Hyperimmunoglobulin E syndromes
- IgG4 related disease (Eur J Haematol 2017;98:378)
- Inflammatory bowel disease (i.e Crohn's, ulcerative colitis) (Cardiology 2013;125:258, Postgrad Med J 1986;62:1101)
- Sarcoidosis (Mayo Clin Proc 2000;75:586)
- Etiology of eosinophilia (≥ 1.5 x 109/L) is unknown but correlates with the severity of the disease
- Associated with immune dysregulation
- 1.0 x 109/L for at least 3 months
- 1.0 x 109/L for at least 6 months
- 1.0 x 109/L for at least 12 months
- 1.5 x 109/L for at least 6 months
- 1.5 x 109/L for at least 12 months
- Aberrant T cell population must be present
- Can accompany systemic mastocytosis
- Carries an autosomal dominant inheritance
- End organ damage is absent
- No cytogenetic or molecular abnormalities are present
Comment Here
Reference: Hypereosinophilic syndrome
- PCM1-JAK2 fusion is a rare genomic abnormality resulting from t(8;9)(p22;p24.1), which fuses the Janus activated kinase 2 gene (JAK2) with the human autoantigen pericentriolar material gene 1 (PCM1), resulting in a constitutively activated tyrosine kinase
- Observed in both myeloid and lymphoid neoplasms and can present as myeloproliferative neoplasm (MPN), myelodysplastic / myeloproliferative neoplasm (MDS / MPN), acute leukemia (B or T lymphoid, myeloid or mixed phenotype), lymphoblastic lymphoma or blast stage of a chronic myeloid neoplasm
- Presence of t(8;9)(p22;p24.1) PCM1/JAK2
- Chronic myeloid leukemia and rearrangements of PDGFRA, PDGFRB and FGFR1 must be excluded
- Variable peripheral blood and bone marrow eosinophilia
- Bone marrow morphology; usually like an MPN, MDS / MPN or acute leukemia - both myeloid and lymphoid
- Proliferation of early erythroblasts in the bone marrow
- Dyserythropoiesis may be present
- Marrow reticulin fibrosis
- Neoplasms with t(8;9)(p22;p24.1) / PCM1-JAK2 are included as a provisional entity in the 2016 revision of the WHO classification in the category of myeloid / lymphoid neoplasm with eosinophilia and specific gene rearrangements
- Rare cases with other fusion partners of JAK2, such as t(9;12)(p24.1;p13) / ETV6-JAK2 and t(9;22)(p24.1;q11.2) / BCR-JAK2, are considered as variants of this entity
- ICD-O: 9968/3 - Myeloid / lymphoid neoplasms with PCM1-JAK2
- M:F = 27:5 showing distinct male preponderance
- Wide age range (12 - 75), with median age of 47 years
- Reference: Br J Haematol 2014;166:809
- Peripheral blood and bone marrow
- Rarely lymph nodes and extramedullary locations
- Due to the presentation as both myeloid and lymphoid neoplasms, the cell of origin is purported to be the pluripotential hematopoietic stem cell (Cancer Res 2005;65:2662)
- PCM1-JAK2 fusion mediates an oligomerization that brings together the linked JAK2 domains, resulting in a constitutively active tyrosine kinase domain of JAK2, which activates multiple downstream signal transducers via the JAK-STAT pathway to promote cell proliferation and differentiation
- Underlying pathobiology does not depend on the fusion partner of JAK2
- As far as is known, the translocation partners do not affect the clinical picture, which is determined by the activation of the JAK2 tyrosine kinase
Images hosted on other servers:

Structure of PCM1-JAK2 translocation
- Clinical features are variable depending on the presentation as a chronic myeloid neoplasm, acute leukemia or a lymphoblastic lymphoma (Leukemia 2005;19:1692)
- Common presenting symptoms are fatigue, pallor, weight loss, abdominal discomfort, splenomegaly and lymph node enlargement (Leukemia 2005;19:1692)
- May represent the blastic phase of an underlying chronic neoplasm
- Reference: Ann Lab Med 2016;36:79
- Review of the CBC and peripheral smear evaluation; if eosinophilia is present, all secondary causes of eosinophilia should be excluded clinically and by appropriate investigation (of note, eosinophilia may not be a prominent feature)
- Flow cytometry to exclude the presence of an immunophenotypically aberrant T cell population as cause of eosinophilia (also known as lymphocyte variant eosinophilia)
- Bone marrow core biopsy and aspirate morphology
- Conventional cytogenetics or FISH to exclude BCR-ABL1 and FISH to detect rearrangements of PDGFRA, PDGFRB and FGFR1
- Identification of the t(8;9)(p22;p24.1) PCM1-JAK2 translocation by conventional cytogenetics and confirmation by FISH or RT-PCR for the fusion gene product
- References: Blood 2017;129:704, Am J Hematol 2019;94:1149
- Peripheral blood evaluation may show anemia or pancytopenia, normal, mildly or markedly increased WBC, leukoerythroblastosis, mild or striking eosinophilia or increased blasts if presentation is as an acute leukemia
- Bone marrow shows myeloid proliferation with maturation; there may be dysplasia in one or more lineages, variable eosinophilia and may show increase in mast cells, usually as loose clusters
- Frequent finding is presence of paratrabecular clusters of early erythroid precursors
- At least some myelofibrosis is frequently present, which is highlighted by the reticulin stain (Eur J Haematol 2011;86:87)
- Metaphase cytogenetics will reveal the characteristic translocation
- In the variant forms of the PCM1-JAK2 translocation involving other fusion partners with JAK2, a JAK2 break apart FISH probe identifying a rearrangement as a first step may be a clue for further FISH studies to identify the specific chromosomal partner
- Fusion is confirmed by PCM1-JAK2 dual color, dual fusion probe or by RT-PCR for the fusion gene product
- In the variant forms of the PCM1-JAK2 translocation involving other fusion partners with JAK2, or in rare cases of crytic rearrangement, a JAK2 break apart FISH probe identifying a rearrangement as a first step may be a clue for further FISH studies to identify the specific chromosomal partner
- Next generation sequencing usually does not reveal additional somatic mutations (Am J Clin Pathol 2021;155:160)
- Aggressive behavior; usually does not respond to imatinib
- Good response has been seen with JAK1/2 inhibitor ruxolinitib, although eventually allo-SCT is required
- Prognosis is quite variable (from few weeks to > 5 years) or longer if patient undergoes allogenic bone marrow transplant
- Reference: Blood 2012;120:1529
- 35 year old man presented with a history of abdominal discomfort, early satiety, low grade fever, night sweats and weight loss (Blood 2013;122:861)
- 42 year old man with left inguinal and multiple cervical lymphadenopathy; a CT scan identified splenomegaly (Ann Lab Med 2016;36:79)
- 51 year old woman was diagnosed as having acute myeloid leukemia at a tertiary hospital (Ann Lab Med 2018;38:492)
- 57 year old man presented with fatigue, weight loss and dyspnea for 3 months; clinical examination only revealed a splenomegaly (Eur J Haematol 2011;86:87)
- Neoplasms resulting from PCM1-JAK2 mutation do not respond to imatinib; the JAK1/2 inhibitor ruxolitinib has shown good response, although eventually these patients require allogenic stem cell transplantation
- Classic triad features of bone marrow biopsy are: hypercellularity with eosinophilic infilterate, aggregates of immature erythroblasts and frequent marrow fibrosis; this triad may not be seen in variants with fusion of ETV6-JAK2 and BCR-JAK2 (Am J Clin Pathol 2021;155:160)
- Variable morphology: may be of a chronic myeloid neoplasm or of acute leukemia; the latter may develop as a blast crisis of the chronic myeloid neoplasm
- Some cases may not have eosinophilia
- Megakaryocytes may be increased or decreased but are often dysplastic
- Variable grades of reticulin fibrosis; usually present
- Reference: Eur J Haematol 2011;86:87
Contributed by Zeba N. Singh, M.B.B.S., M.D.

Markedly hypercellular bone marrow

Megakaryocytic atypia
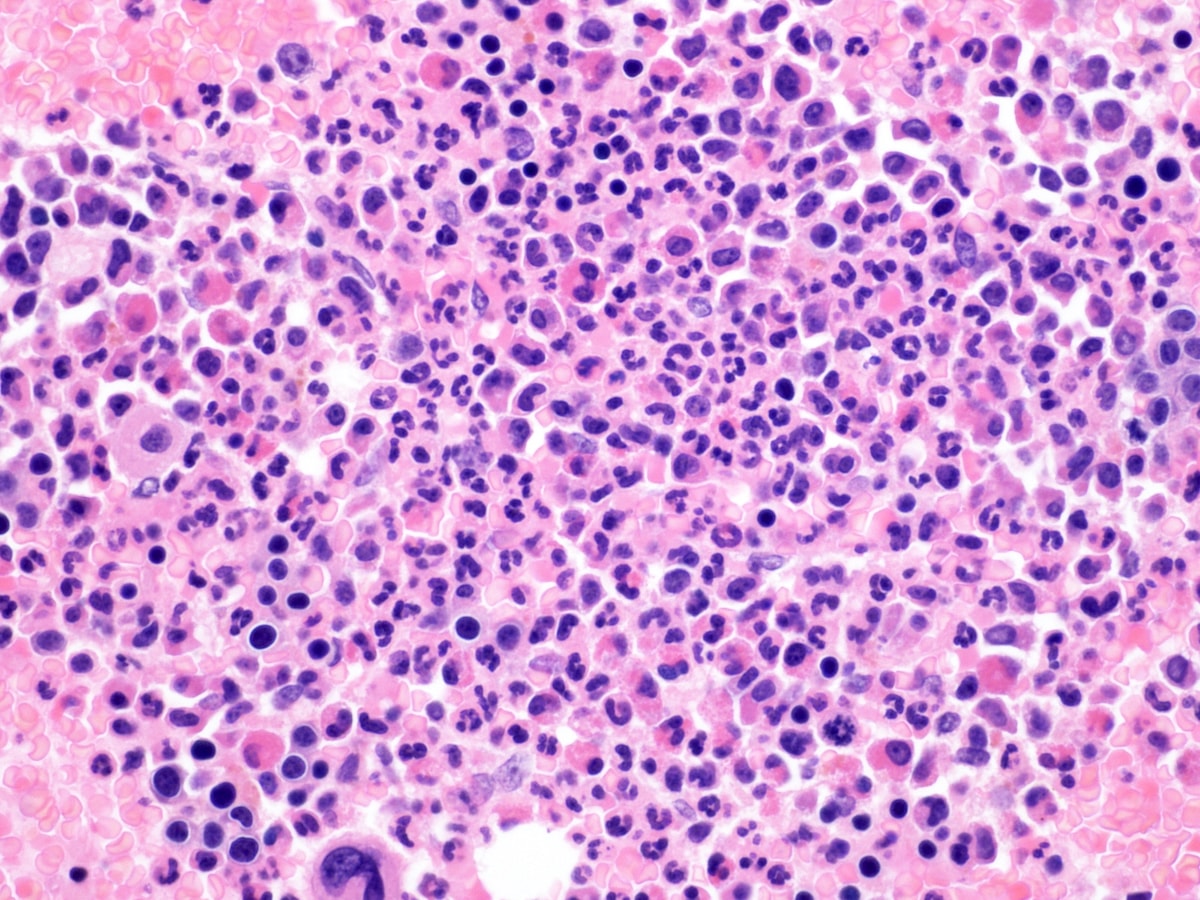
Bone marrow eosinophilia
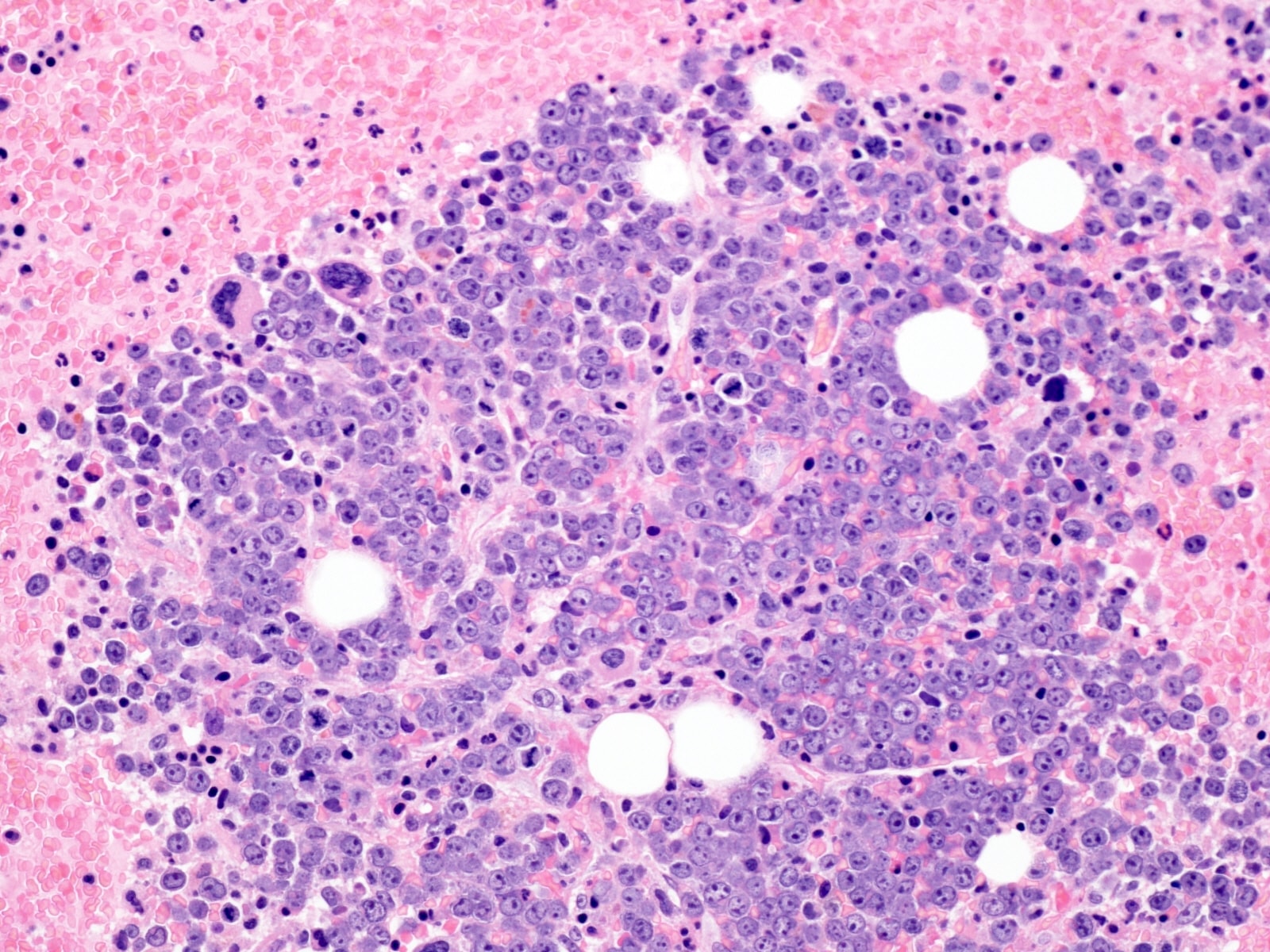
Large aggregate of early erythroid precursors
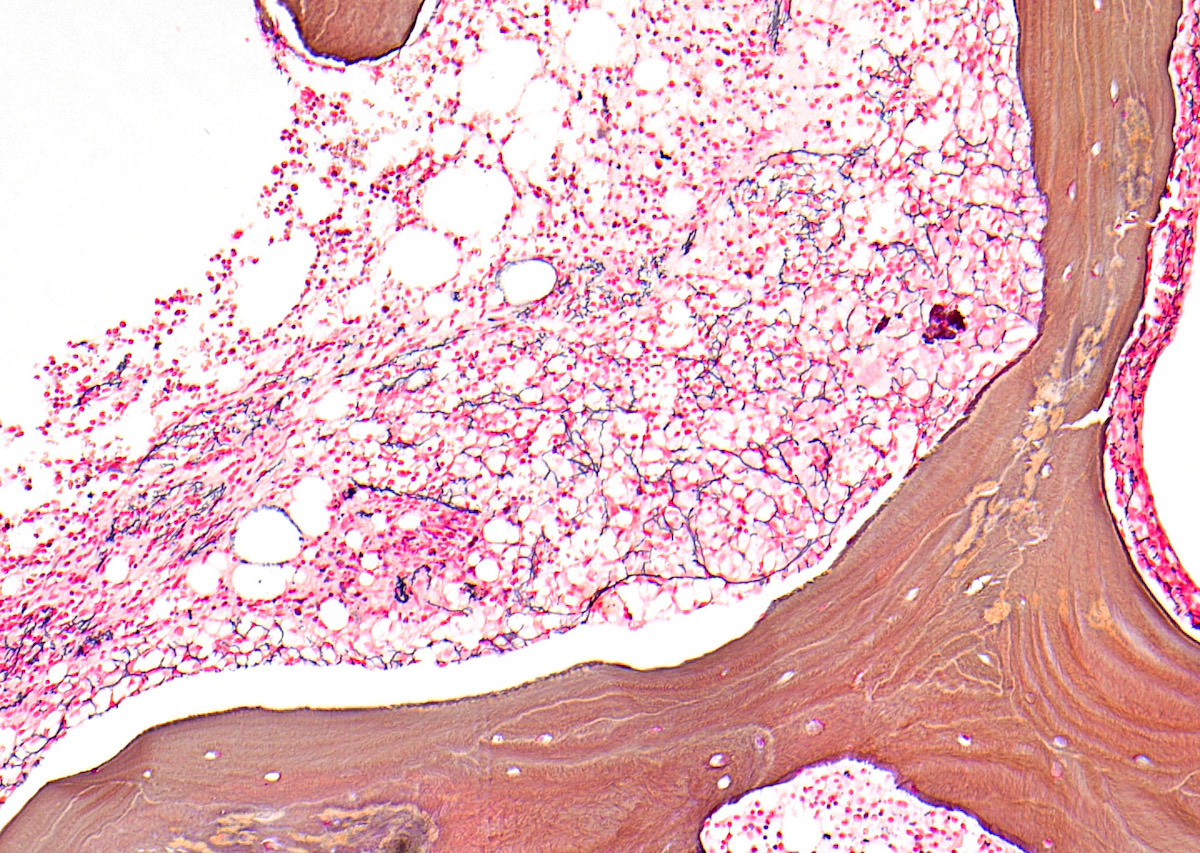
Reticulin fibrosis

CD34 immunostain
- Normochromic, normocytic anemia
- Leukocytosis, leukopenia or leukoerythroblastic picture
- Eosinophilia often but not always
- In cases that present as acute leukemia, circulating blasts are present
- Reference: Am J Hematol 2019;94:1149
Contributed by Zeba N. Singh, M.B.B.S., M.D.
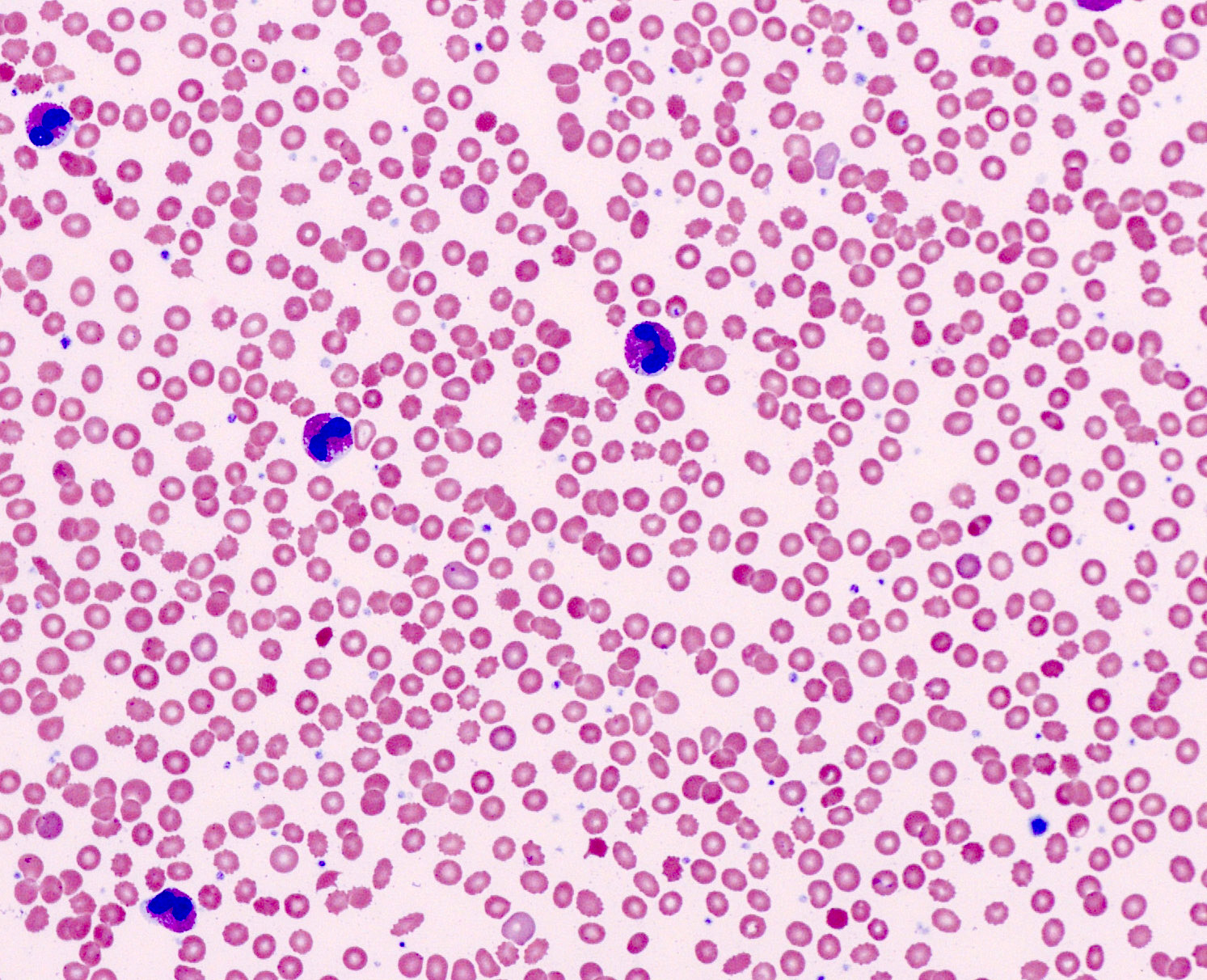
Peripheral blood eosinophilia
- PCM1-JAK2 fusion mediates an oligomerization that brings together the linked JAK2 domains resulting in a constitutively active tyrosine kinase
- JAK2 is a cytoplasmic tyrosine kinase; it activates multiple downstream signal transducers via the JAK-STAT pathway to promote cell proliferation and differentiation
- BCR-JAK2 fusion protein contains the coiled coil dimerization domain of BCR and the protein tyrosine kinase domain (JH1) of JAK2
- t(8;9)(p22;p24.1) resulting in PCM1-JAK2 fusion
- t(9;12)(p24.1;p13.2) resulting in ETV6-JAK2
- t(9;22)(p24.1;q11.2) resulting in fusion of BCR-JAK2
- Reference: Cancer Res 2005;65:2662
Images hosted on other servers:
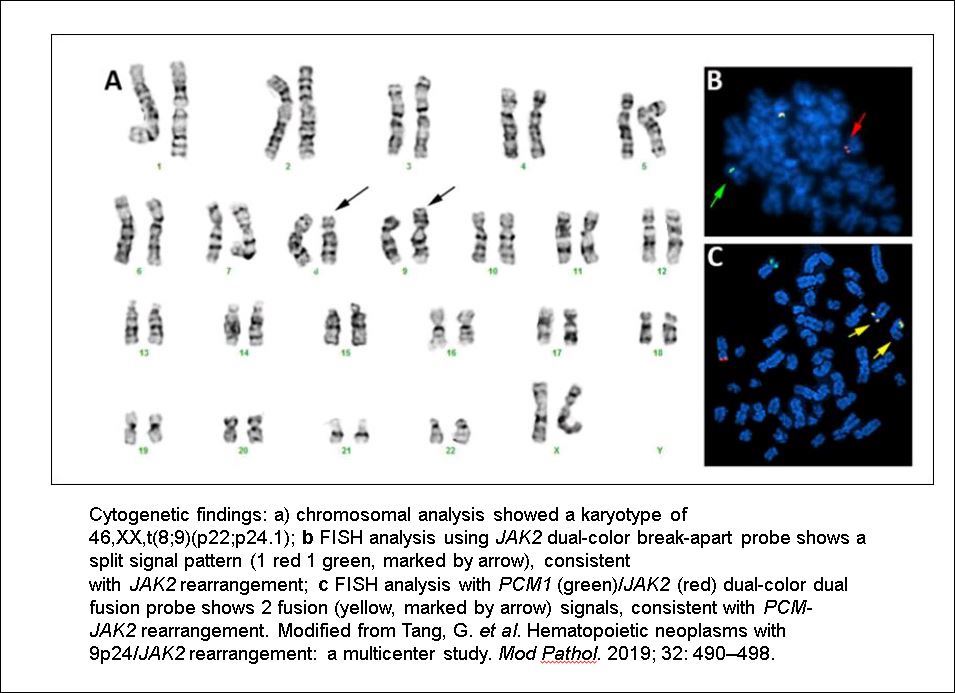
Karyogram and FISH analysis
- Bone marrow, right posterior iliac crest, core biopsy and clot section:
- Chronic myeloid neoplasm with t(8;9)(p22;p24.1) PCM1-JAK2 fusion (see comment)
- Hypercellular (90%) bone marrow showing myeloid hyperplasia, dysmegakaryopoiesis, eosinophilia and reticulin fibrosis (grade 1/3); blasts less than 5%.
- Comment: The morphological features of the bone marrow core biopsy and clot section are consistent with a myelodysplastic / myeloproliferative neoplasm, unclassifiable (MDS / MPN-U). There is peripheral blood eosinophilia and an increase in marrow eosinophils. Cytogenetic analysis identified t(8;9)(p22;p24.1) abnormality in 20 / 20 metaphases. RT-PCR for the chimeric BCR-ABL1; FISH for PDGFR alpha, PDGFR beta, FGFR1 rearrangements and JAK2 V617F mutation were negative. The patient had hepatosplenomegaly, pulmonary, cardiac and bone involvement.
- The t(8;9)(p22;p24.1) abnormality fuses the PCM1 gene with the JAK2 gene. Based on the WHO classification (2016 revision), the neoplasm is classified under the group of myeloid / lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, FGFR1 rearrangements or t(8;9) PCM1-JAK2 fusion. The neoplasm is aggressive in behavior. Response to ruxolitinib has been demonstrated (Blood 2012;120:1529).
- Peripheral smear: Manual review of the peripheral blood shows normochromic, normocytic anemia, thrombocytopenia, relative (11%) and absolute (1.65 K/uL) eosinophilia. RBCs: Mild normochromic, normocytic anemia with minimal anisopoikilocytosis. WBCs: There is mild leukocytosis with absolute eosinophilia. The granulocytes show normal morphology. There is a mild left shift with few metamyelocytes and myelocytes. Blasts are not present. The lymphocytes are mature. There is no increase in large granular lymphocytes. Platelets are mildly reduced (100 K/uL) and include some large forms.
- Bone marrow biopsy: Quality: adequate. Cellularity: 90%. Hematopoiesis: full spectrum trilineage hematopoiesis. There is myeloid hyperplasia. There is a prominence of eosinophils and their precursors. Large, paratrabecular aggregates of early erythroid precursors are identified. There is no overt increase in blasts. Megakaryocytes are normal in number but have dysplastic morphology including nuclear hypolobation, hyperchromasia, loose clustering and paratrabecular localization. There is no increase in lymphocytes or plasma cells. Special stains: reticulin; loose network of reticulin without significant intersections (grade 1 fibrosis on a scale of 0 - 3). Trichrome: negative for collagen deposition.
- Bone marrow clot section: Quality: adequate. Cellularity: 90% morphologic features are similar to the core biopsy. Bone marrow aspirate: Quality: adequate. Spicules present. Myeloid hyperplasia. M:E ratio increased (5:1). Full maturation in myeloid and erythroid series. No dysgranulopoiesis or increase in blasts. Eosinophils and eosinophil precursors are increased. Clusters of early erythroid precursors present. Dysmegakaryopoiesis with hypolobated nuclei.
- Immunohistochemistry: Immunohistochemical stains were performed on paraffin fixed and decalcified bone core biopsy sections using appropriate controls. CD34 marks mostly vascular endothelium. Occasional blasts (less than 5%) are identified. CD117 marks the blasts, promyelocytes, early erythroid precursors and mast cells (15%). E-cadherin marks the large aggregates of early erythroid precursors.
- Reactive eosinophilia:
- Nonclonal, secondary cause for eosinophilia (e.g. allergic conditions, parasitic infections, drugs, etc.)
- Detailed clinical history should be taken for exclusion of an underlying cause of eosinophilia
- Chronic myeloid leukemia:
- Uncommonly CML may have absolute eosinophilia
- Absence of BCR-ABL1 fusion excludes CML
- Chronic myelomonocytic leukemia (CMML):
- Absolute monocytosis of 1000/uL or more and absence of genetic abnormalities defining CM, or neoplasms with rearrangements of PDGFRA, PDGFRB and FGFR1; both of these entities are excluded by absence of specific genetic abnormality
- Myeloid neoplasms with PDGFRA, PDGFRB or FGFR1:
- These neoplasms share many morphological features but are distinguished by the specific genetic abnormality
- Chronic eosinophilic leukemia, NOS:
- Persistent eosinophilia, clonal in etiology but not one of the chromosomal translocations included in the entities listed above
- Systemic mastocytosis:
- May be associated with peripheral blood, bone marrow and tissue eosinophilia; bone marrow shows more multifocal aggregates of mast cells with abnormal immunophenotype (CD25+, CD2 variable)
- May be associated with non mast cell clonal hematological disorders
- Lack the specific abnormalities (PDGFRA, PDGFRB, FGFR1 rearrangements or PCM1-JAK2 translocation)
- Idiopathic hypereosinophilic syndrome:
- Peripheral blood eosinophil ≥ 1,500/uL associated with tissue damage, after exclusion of secondary causes of eosinophilia and morphological, flow cytometric, cytogenetic and molecular evaluation for clonal evidence for an acute or chronic myeloid or lymphoproliferative disorder
- Other entities to consider: atypical chronic myeloid leukemia, T and B acute lymphoblastic leukemia and acute myeloid leukemia; all of these entities can be excluded by specific genetic abnormality
- Lymphocyte variant eosinophilia:
- Flow cytometry shows aberrant T cell population
A 60 year old man with leukocytosis, mild thrombocytopenia and a differential leukocyte count (showing neutrophils 55%, bands 4%, lymphocytes 22%, monocytes 7%, eosinophils 12%, basophils 1%, metamyelocytes 2%, myelocytes 2% and blasts 1%) undergoes a bone marrow evaluation. Images from the bone marrow core biopsy are provided. Which set of investigations is the most appropriate?
- FISH for inv(16)(13.1;q22), 11q23 / MLL and t(8;21)
- Metaphase cytogenetics, comparative genomic hybridization and next generation sequencing for myeloid mutations
- Metaphase cytogenetics, FISH for PDGFRA, PDGFRB and FGFR1 rearrangement
- qRT-PCR for BCR-ABL1, FISH for NPM1-ALK, FLT3 ITD
Comment Here
Reference: PCM1-JAK2
- Clonal proliferation of (predominantly) granulocytic and monocytic precursors in children, with < 20% blasts in blood and bone marrow
- Mutations involving RAS pathway are characteristic
- Myelodysplastic / myeloproliferative disorder in childhood, can occur in children from 1 month of age to adolescence
- Approximately 75% of cases occur in children less than 3 years old
- Greater than 75% of cases have mutations in the RAS pathways: PTPN11, KRAS, NRAS, CBL and NF1
- About 15% of cases are diagnosed in patients with Noonan syndrome (or Noonan-like syndrome) and ~10% occur in conjunction with neurofibromatosis type 1
- Patients present with hepatosplenomegaly and sustained increased monocyte counts
- Stem cell transplant improves survival
- Juvenile chronic myelomonocytic leukemia
- About 1.2 per million people per year
- 75% of cases occur in children < 3 years old, more frequent in males (~2:1)
- Median age of 2 years
- 10% of cases are associated with neurofibromatosis 1 and deletion of NF1 gene
- 15% of cases in infants with Noonan syndrome (CBL gene mutation)
- Rare association with EBV infection (Leuk Res 2008;32:181)
- Primarily involves peripheral blood and bone marrow
- Nearly all cases involve the liver and spleen
- Other common sites of involvement include lymph nodes, skin and respiratory tract
- Reference: Blood 1998;91:365
- Due to stem cell defect causing deranged hematopoiesis (Blood 2019;133:1060)
- In vitro hypersensitivity to GM-CSF, considered to be a hallmark of the disease (and previously used for diagnosis)
- Multiple genes affecting the RAS / MAPK signaling pathways have been implicated
- Gain of function mutations in more than 75% of patients
- Somatic mutations in NRAS or KRAS identified in ~30% of cases, which can lead to accumulation of activated protein
- ~30% of cases have PTPN11 mutations, which can be somatic or germline as seen in Noonan syndrome
- Loss of function mutation in NF1, which can be somatic or germline
- ~15 - 20% of cases can have NF1 mutation or deletion without neurofibromatosis
- CBL mutation, which can be somatic or germline (Noonan-like syndrome)
- ~10 - 15% of cases can have this loss of function mutation
- Gain of function mutations in more than 75% of patients
- Leads to reduced degradation of tyrosine kinase receptors and therefore increased RAS pathway signaling
- Varied clinical presentation may include failure to thrive, malaise, fever, bleeding, pallor, infection, with or without lymphadenopathy; rarely CNS involvement (J Pediatr Hematol Oncol 2007;29:770)
- Splenomegaly: found in all cases and prerequisite for diagnosis of JMML but may be initially absent at presentation
- Skin lesions: can be seen in patients with germline mutations (e.g., café au lait spots in NF1 and CBL mutations)
- Gastrointestinal and pulmonary symptoms can also be observed
- Most patients die of disease, although it may wax and wane
- Multiple diagnostic criteria, according to WHO
- Genetic criteria (at least 1):
- Somatic mutations in PTNP11, KRAS or NRAS
- If present, germline mutations or transient abnormal myelopoiesis of Noonan syndrome have to be excluded
- NF1 mutation or clinical diagnosis of neurofibromatosis type 1
- CBL mutation (germline) or loss of heterozygosity
- Somatic mutations in PTNP11, KRAS or NRAS
- Clinical and hematologic criteria (must fulfill all 4):
- Sustained peripheral blood absolute monocyte count ≥ 1 x 109/L
- < 20% blasts + promonocytes in blood and bone marrow
- No Philadelphia chromosome or BCR::ABL1 fusion
- Splenomegaly
- If no genetic criteria are present, then the following criteria should be used (along with the clinical and hematologic criteria):
- Monosomy 7 or other chromosomal abnormality
- Or 2 or more of the following criteria:
- Increased hemoglobin F (for age)
- Myeloid / erythroid precursors on peripheral blood smear
- In vitro hypersensitivity to granulocyte macrophage colony stimulating factor (GM-CSF or CSF2)
- Hyperphosphorylation of STAT5
- Typically, WBC count is 20 - 30 x 109/L with granulocytes and monocytes and occasional dysplasia (which may not be prominent) (Blood 2019;133:1060)
- Peripheral blood monocytosis ≥ 1 x 109/L
- Abnormally increased hemoglobin F for patient's age
- Peripheral smear showing leukocytosis with myeloid left shift, in addition to thrombocytopenia and often anemia
- Nucleated RBCs are often seen
- Reference: Blood 1998;91:365
- According to WHO, low platelet counts, age at diagnosis greater than 2 years and high HgF levels are clinical predictors of shorter survival
- PTPN11 mutated JMML or in patients with NF1 is fatal without rapid treatment
- Aggressive JMML with increased risk of progression to AML seen in patients with more than 1 RAS activating mutation (Leukemia 2020;34:1658)
- BMP4, CALC4, CDKN2B, RARB, RASA4 isoform 2: other genes known to be hypermethylated in JMML
- Methylation pattern in JMML divided into 3 categories with different prognosis:
- Favorable prognosis: low methylation, underlying somatic mutations in NRAS and CBL
- Poor prognosis: high methylation, somatic PTPN11 mutations
- Intermediate category: somatic KRAS mutations and monosomy 7
- Reference: Blood Adv 2021;5:5507
- 22 month old boy with concurrent neurofibromatosis type 1 and juvenile xanthogranuloma diagnosed with JMML (Pediatr Dermatol 2017;34:114)
- 2 year old boy with JMML with t(3;5), Auer rods and marked myelodysplasia (Pathol Res Pract 2018;214:919)
- 2 year old girl who presented with MDS / MPN driven by a cryptic NUP98::NSD1 fusion (J Pediatr Hematol Oncol 2021;43:e808)
- Stem cell transplantation
- Recommended early in disease in patients with PTPN11, KRAS or NF1 mutations
- Deferred in patients with CBL or NRAS (germline) mutations (mainly due to spontaneous regression of JMML)
- Reference: Haematologica 2015;100:17
- Bone marrow: hypercellular with granulocytic hyperplasia, 5 - 30% monocytes, decreased megakaryocytes, blasts + promonocytes < 20%, dysplasia not prominent, reticulin fibrosis is rare (Blood 1997;89:3534)
- Skin: common presentation, with increased myelomonocytic infiltrates in the papillary and reticular dermis
- Liver: leukemic infiltrates in the portal regions and sinusoids
- Lung: leukemic infiltrates spread from the capillaries within the alveolar septa into alveoli
- Spleen: infiltrates in the red pulp with a preference for trabecular and central arteries
Contributed by Alexa J. Siddon, M.D. and AFIP
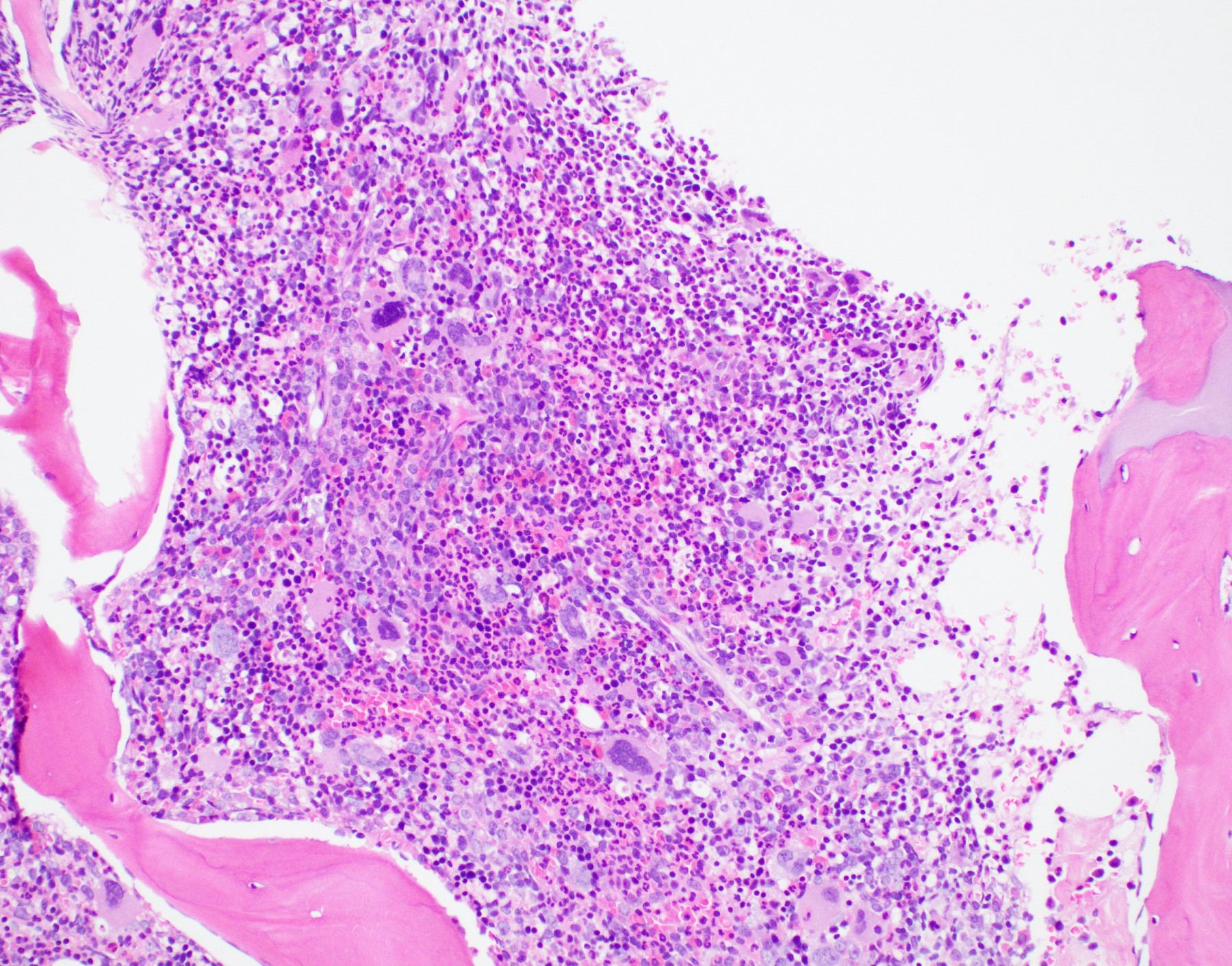
Hypercellular marrow
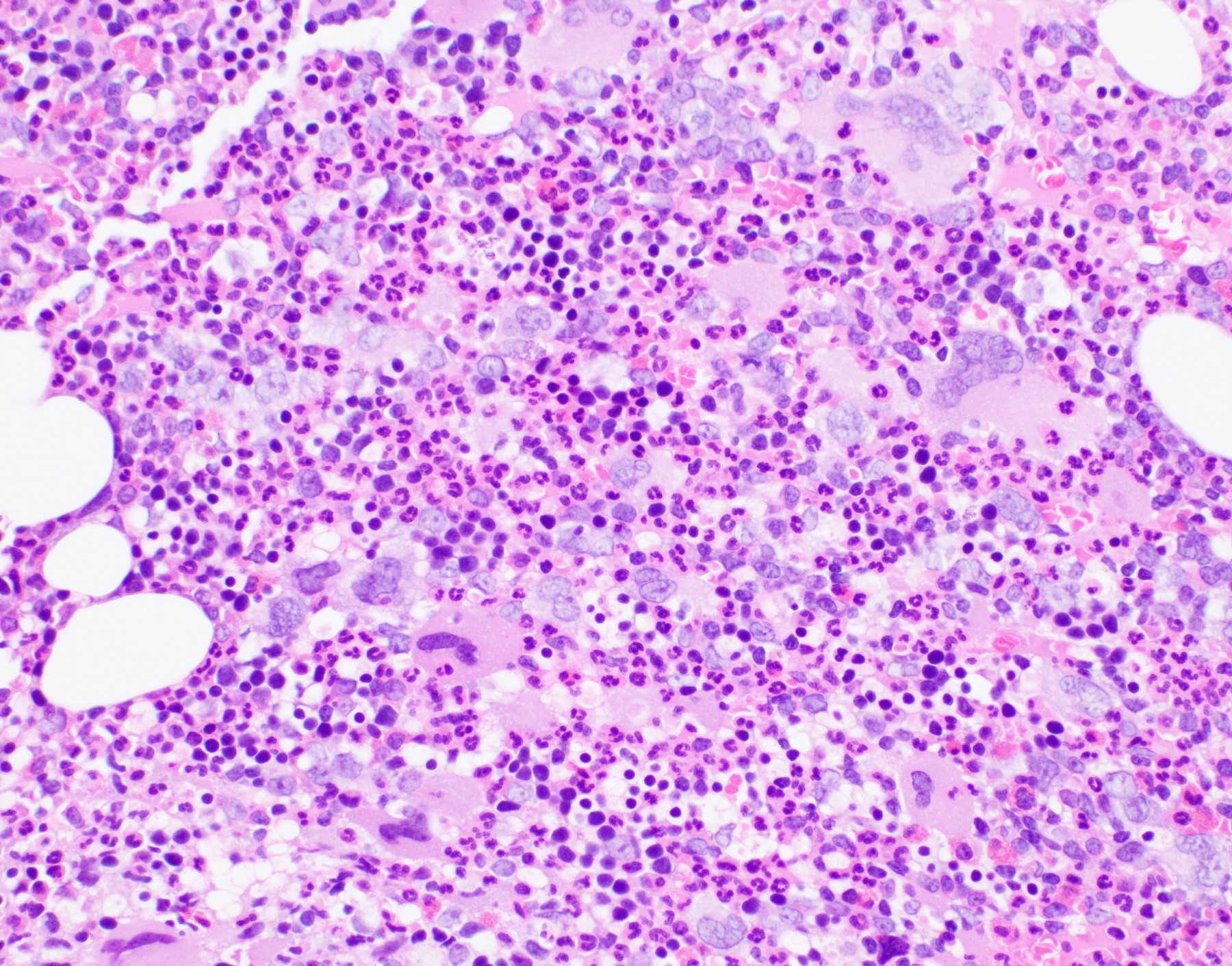
Myeloid predominant marrow
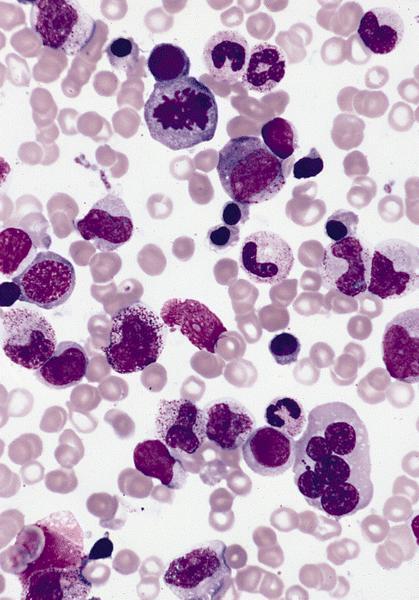
Aspirate with monocytes and promonocytes
- Important specimen for diagnosis
- Leukocytosis with neutrophilia with immature forms and monocytosis, WBC count is 20 - 30 x 109/L with granulocytes and monocytes and occasional dysplasia (which may not be prominent) (Blood 2019;133:1060)
- Anemia: most commonly normochromic and nucleated red blood cells are often identified
- Macrocytosis seen in cases with monosomy 7
- Thrombocytopenia
- Blasts and blast equivalents are usually less than 5% (no more than 20%)
AFIP images
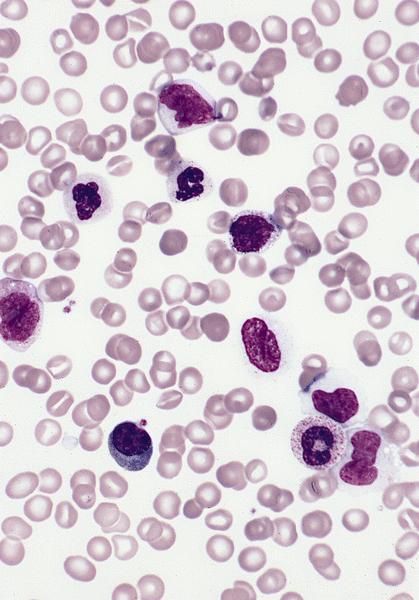
Blood shows mature monocytes and neutrophils
- CD68 and lysozyme helpful to identify the monocytic component
- Cytochemical stains for alpha naphthyl acetate esterase / butyrase helpful to identify monocytic cells
- Blasts, if present: CD34 (should be < 20%)
- Can see increased monocytes
- PCR used for KRAS, NRAS, NF1, PTNT11 or CBL mutations
- RAS pathway mutations can occur in about 90% cases
- Next generation sequencing (NGS) has identified other mutations in patients with JMML
- SETBP1: found at disease initiation and progression, also considered a potential marker for how aggressive the disease is
- JAK3, ALK, ROS1
- EZH2, ASXL1 and DNMT3A: in about 15% of cases
- 35% of cases can have abnormal karyotype, most commonly monosomy 7
- References: Am J Blood Res 2021;11:1, Pediatr Int 2016;58:681
- Left posterior iliac crest, core biopsy and aspirate smear:
- Hypercellular marrow with granulocytic proliferation and (< 20%) blasts, consistent with juvenile myelomonocytic leukemia (see comment)
- Comment: The bone core biopsy demonstrates hypercellularity and myeloid predominance, with no increase in blasts. In conjunction with peripheral blood monocytosis, splenomegaly, negative BCR::ABL1 studies and a clinical diagnosis of neurofibromatosis type 1, the findings are consistent with juvenile myelomonocytic leukemia.
- Childhood myelodysplastic syndrome:
- More likely to have karyotype abnormalities than gene mutations
- May be hypocellular for age
- Myeloproliferative neoplasms such as chronic myeloid leukemia:
- Must assess for BCR::ABL1 to exclude CML
- Viral infections:
- EBV, CMV or HHV6 also have increase in granulocytic and monocytic precursors, hypersensitivity to GM-CSF / CSF2 and NRAS mutation in proliferating B cells (J Pediatr Hematol Oncol 2002;24:136, Leuk Res 2008;32:181)
- Wiskott-Aldrich syndrome:
- Atopic dermatitis-like eczema, no intracellular WASP expression, WASP gene mutation (J Pediatr Hematol Oncol 2007;29:836)
- Infantile malignant osteopetrosis:
- Radiologic evidence of increased bone density
- Familial hemophagocytic lymphohistiocytosis (HLH):
- Splenomegaly and extensive specific clinical and genetic criteria
- Acute leukemia with KMT2A (MLL) rearrangement:
- Patients with massive hepatosplenomegaly and low blast count (Oman Med J 2019;34:553)
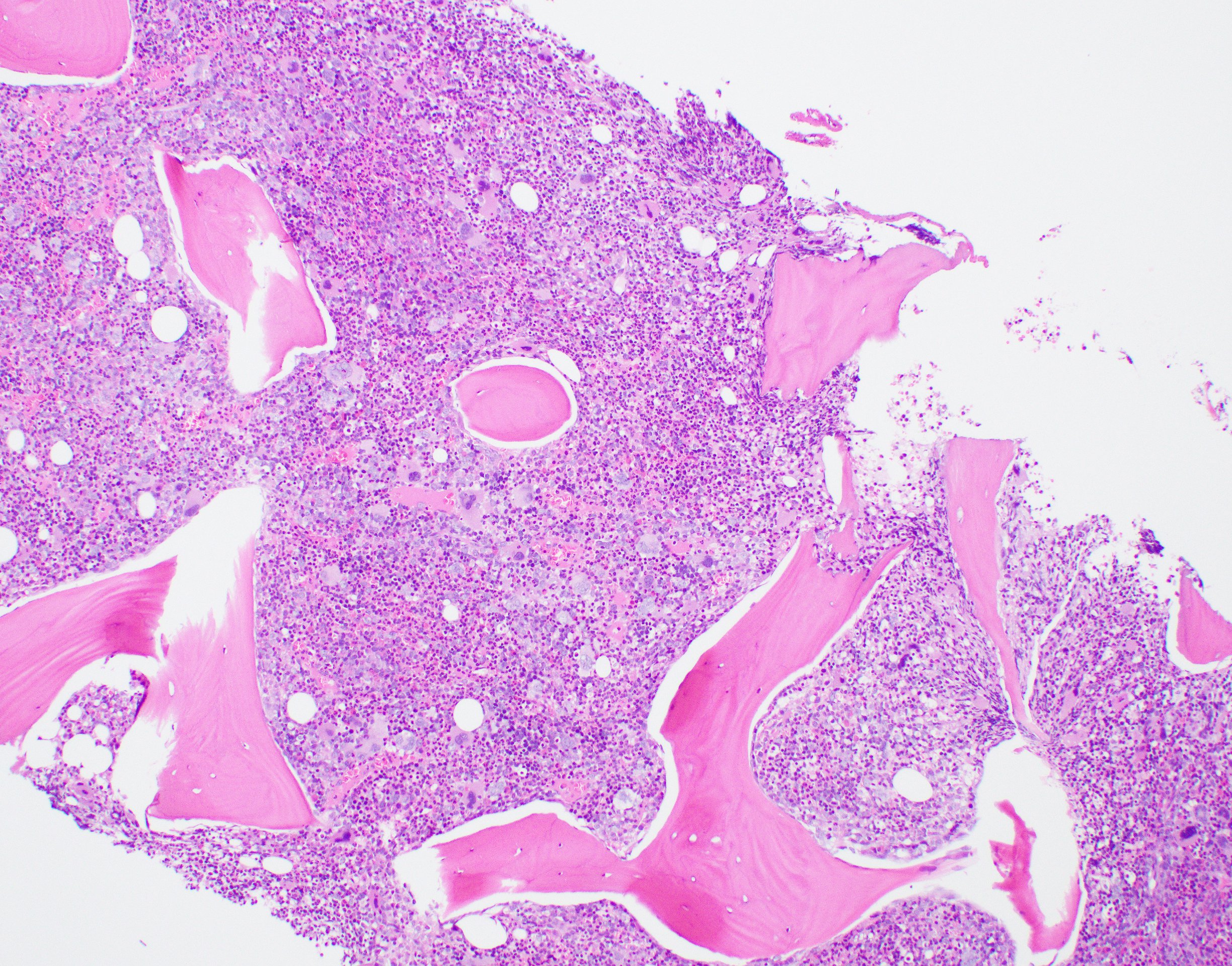
A 9 year old boy with relevant prior history was noted to have enlarged tonsils and cough. His CBC showed leukocytosis with monocytosis. His physical exam was notable for lymphadenopathy and splenomegaly. Flow cytometry of blood showed no increase in blasts or evidence of acute leukemia. A bone marrow biopsy was pursued, which showed a myeloid predominant marrow with no increase in blasts and little granulocytic dysplasia. A pathogenic mutation in which gene is most supportive of a diagnosis of juvenile myelomonocytic leukemia?
- IDH2
- KRAS
- NPM1
- SF3B1
Comment Here
Reference: Juvenile myelomonocytic leukemia
- Average age of newly diagnosed patients is 50
- Commonly associated with neurofibromatosis type 2
- There is often increased hemoglobin F for age
- Usually driven by Epstein-Barr virus
Comment Here
Reference: Juvenile myelomonocytic leukemia
- Acute monoblastic leukemia (M5a)
- 5 - 8% of AML
- Children and young adults
- 80%+ of monocyte lineage cells are monoblasts
- 66 year old man with erythropoietin dependent transformation of refractory anemia with ringed sideroblasts into acute monoblastic leukemia (Blood 2001;98:3492)
- 73 year old woman with coexisting mantle cell lymphoma (Leuk Lymphoma 2005;46:1813)
- 82 year old man with acute monoblastic leukemia following granular lymphocyte proliferative disorder (Rinsho Ketsueki 2011;52:1870)
- Hypercellular marrow with large number of monoblasts
- Monoblasts are large with moderately abundant intensely basophilic cytoplasm, variably basophilic and delicate azurophilic granules but no / rare Auer rods
- May have pseudopods or vacuoles
- Have round nuclei and lacy chromatin with one or more prominent nucleoli but no folds
- Promonocytes have abundant less basophilic cytoplasm with obvious azurophilic granules and nuclei have delicate folds
AFIP images

Abundant cytoplasm with azurophilic granules
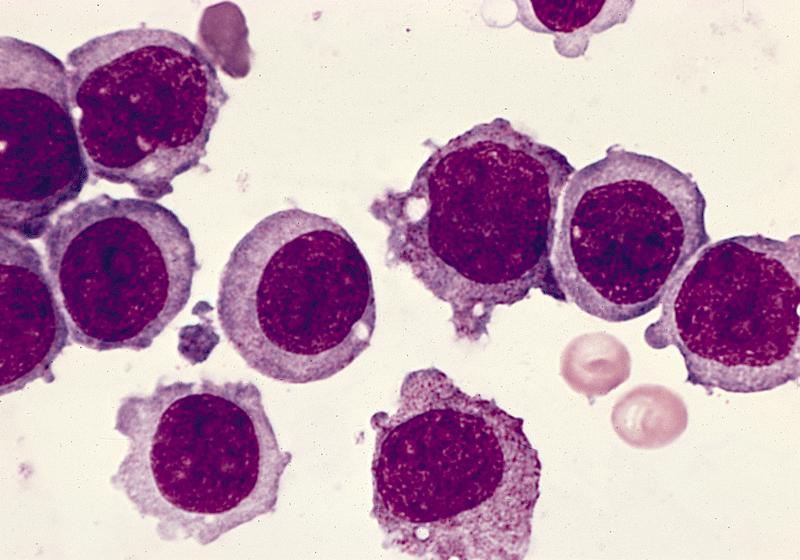
Monoblasts are large with abundant cytoplasm
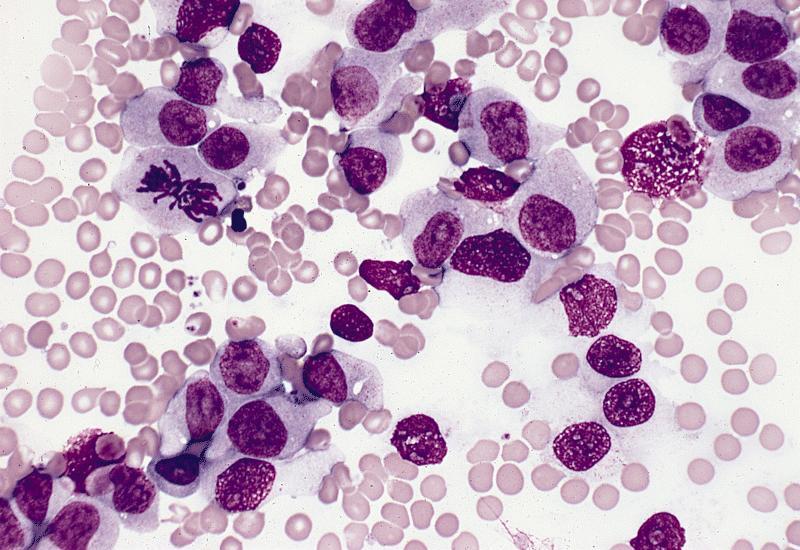
Some monoblasts also show pseudopods
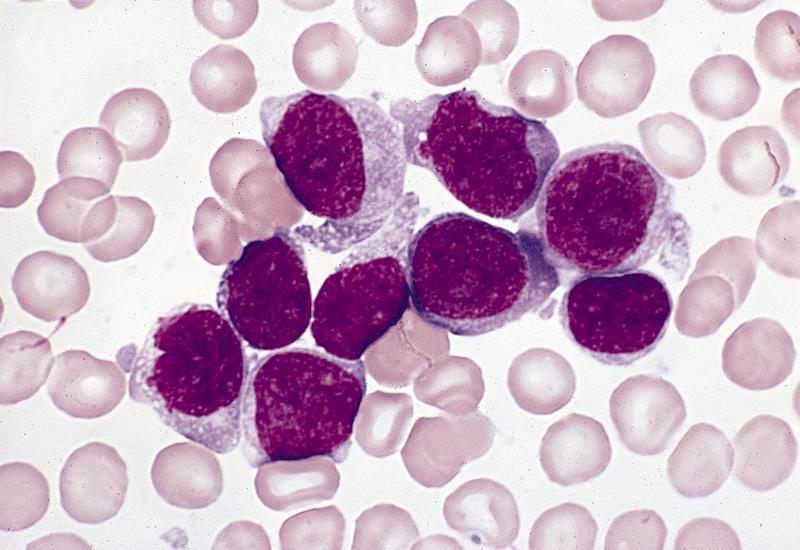
Monoblasts have variable cytoplasm
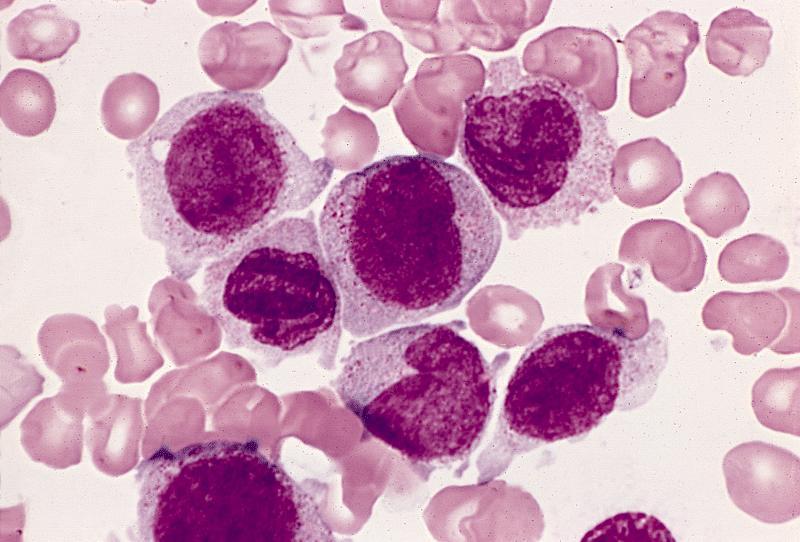
Monoblasts and promonocytes
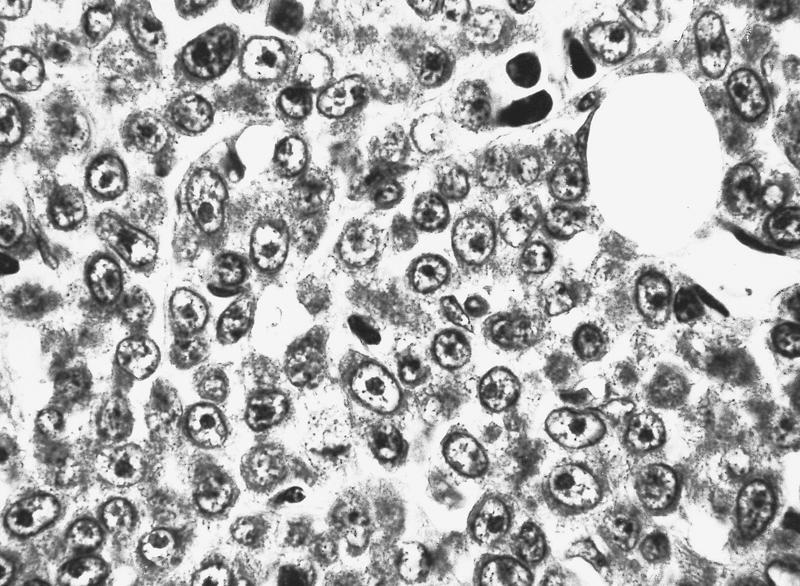
Marrow completely replaced by monoblasts
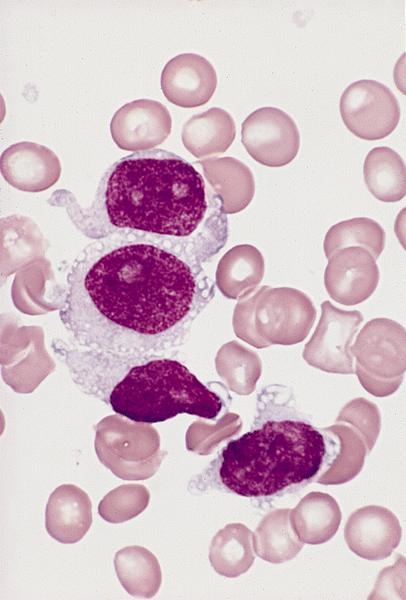
Monoblasts are large with abundant pale cytoplasm
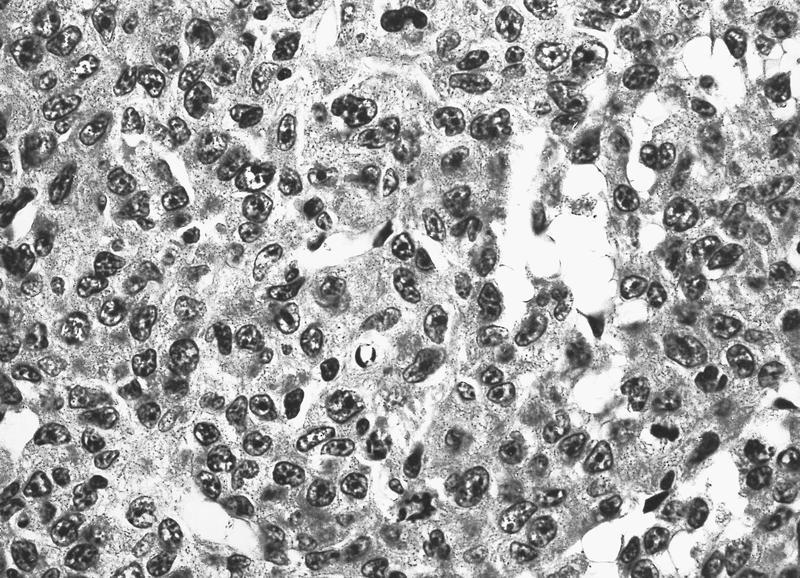
Large monoblasts with abundant cytoplasm
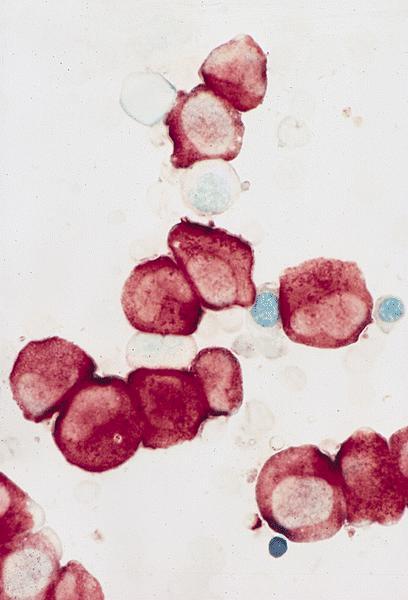
Nonspecific esterase positive
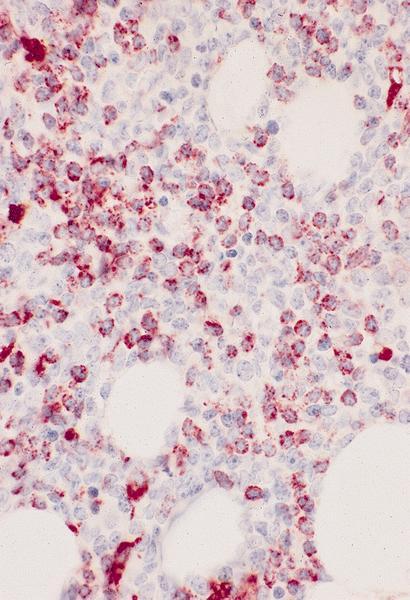
CD68 #1 (KP-1) positive
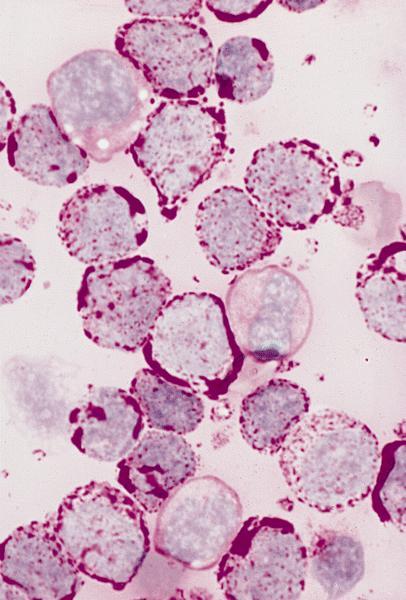
PAS positive
AFIP images
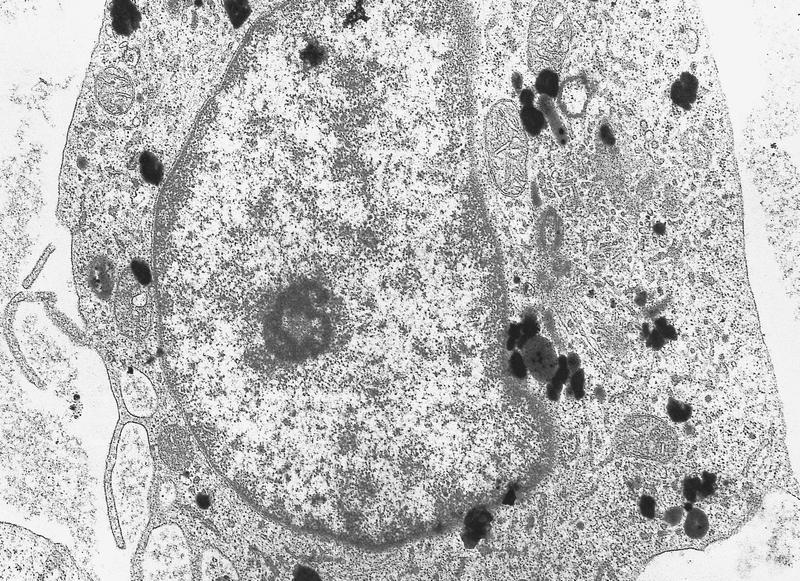
Scattered electron dense deposits
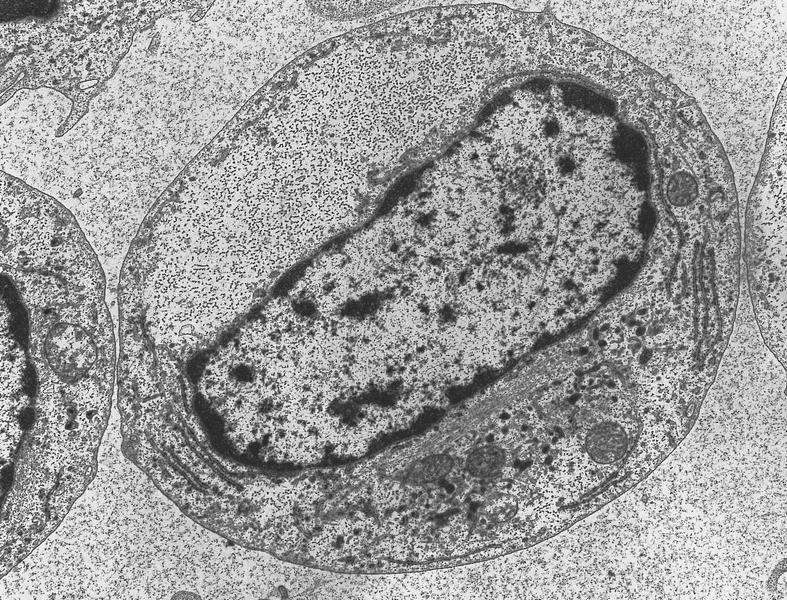
Cytoplasm contains focal area of glycogen deposition
- 75% have cytogenetics abnormalities, including 11q23 in 30% (these cases should be classified as a recurrent genetic abnormality)
- FLT3 mutations in 7%
- Acute monocytic leukemia (M5b)
- 3 - 6% of AML
- Affects all ages
- Mature monocytes or promonocytes predominate in peripheral blood (< 80% of monocyte lineage cells are monoblasts, usually < 20%)
- Treatment may cause tumor lysis syndrome, DIC and falsely elevated platelet counts (Arch Pathol Lab Med 1999;123:1111)
- 47 year old man with spontaneous remission after infection (Int J Lab Hematol 2007;29:386)
- 50 year old man with cutaneous disease (Arch Pathol Lab Med 2005;129:425)
- 63 year old man with preceding aleukemic leukemia cutis (Case Rep Oncol 2011;4:547)
- 74 year old man with mycosis fungoides now with increased circulating immature mononuclear cells (Univ Pittsburgh case #460)
- 77 year old man with coexisting myeloma (Arch Pathol Lab Med 2003;127:1506)
- With mononucleosis syndrome due to varicella zoster virus (Eur J Haematol 2002;68:236)
- Leukemic cells are often promonocytes with less basophilic cytoplasm and more azurophilic granules than monoblasts
- Have folded or cerebriform nuclei with fine chromatin
- Erythrophagocytosis is common
AFIP images
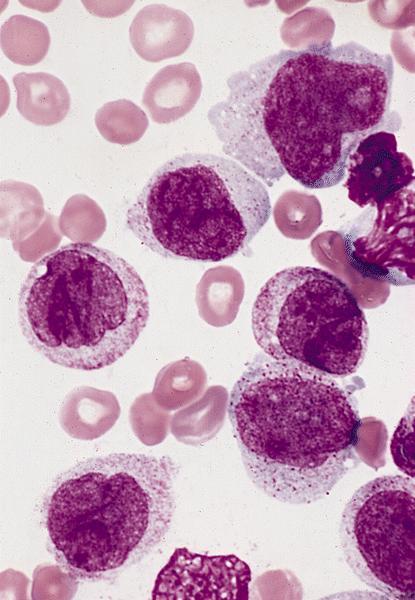
Monocytic cells have range of differentiation
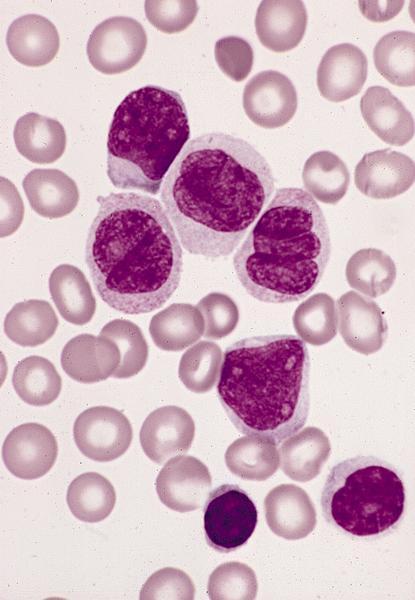
2 myeloblasts and 3 promonocytes
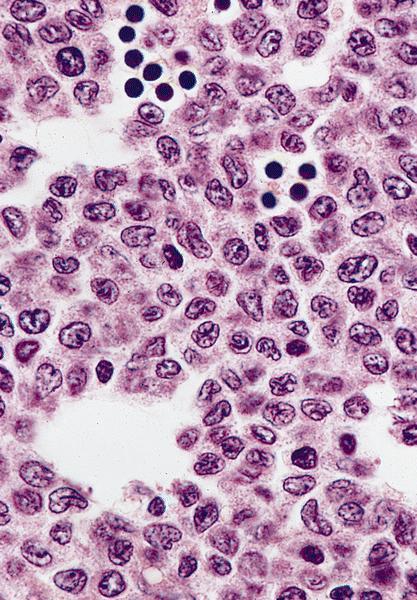
Moderate granular cytoplasm
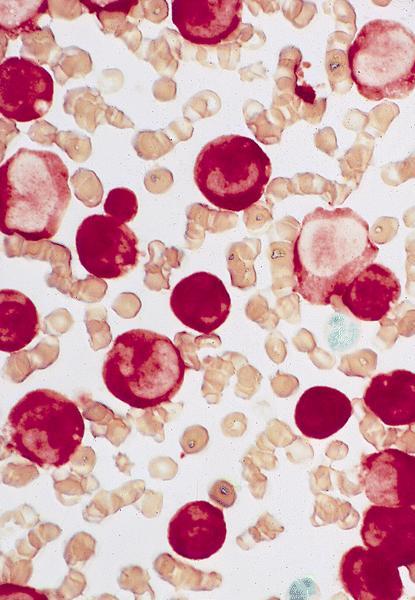
Nonspecific esterase positive
AFIP images
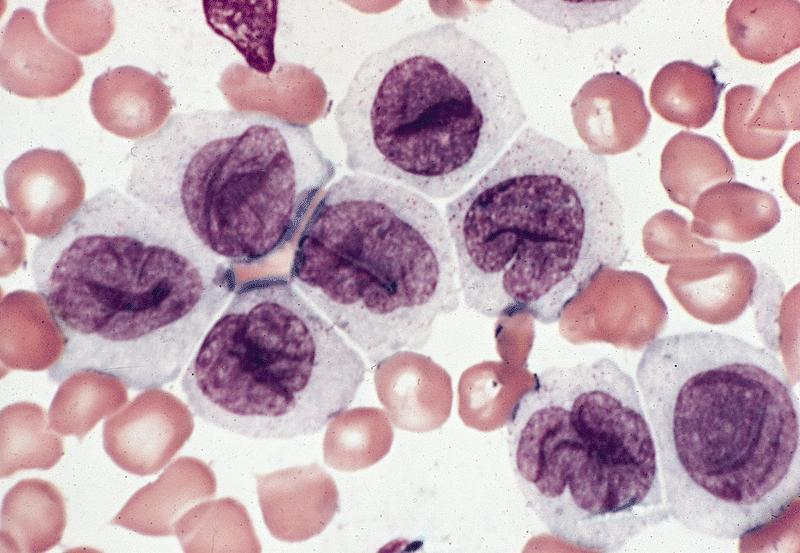
Promonocytes have abundant cytoplasm
AFIP images
Promonocyte has cytoplasm with numerous small cisterna
- 30% have cytogenetics abnormalities, including 11q23 in 12% (these cases should be classified as a recurrent genetic abnormality)
- FLT3 mutations in 30%
- t(8;16)(p11;p13) fuses MOZ gene at 8p11 with CBP gene at 16p13 and is associated with erythrophagocytosis and coagulopathy
- AML M4
- Malignant histiocytic disorders
- Microgranular acute promyelocytic leukemia
- Myelodysplastic syndrome: to distinguish, count promonoblasts in M5 with monoblasts
- This is a provisional myelodysplasia / myeloproliferative disorder, with JAK2 V617F mutation present in 48 - 67% (Haematologica 2008;93:34, Blood 2006;108:2173)
- Diagnostic criteria: features of one of the myelodysplasia / myeloproliferative categories but no history of MDS/MPN, no history of growth factor or cytotoxic therapy, no BCR-ABL1 gene fusion, no rearrangement of PDGFRA, PDGFRB or FGFR1, no del(5q), no inv(3)(q21;q26) OR de novo disease with MPN and MDS features which cannot be assigned to any specific category of MDS/MPN
- Includes refractory anemia with ringed sideroblasts (RARS) with dyserythropoiesis and marked thrombocytosis (RARS-T) with megakaryocytes similar to those in ET, PV or primary myelofibrosis
- Essential thrombocythemia with ringed sideroblasts (Arch Pathol Lab Med 2004;128:815)
- Prominent erythroid dysplasia and t(8;9) progressing to acute erythroid leukemia (Hum Pathol 2005;36:1148)
- Unclassified disorder with thrombocytosis and splenomegaly (Ann Hematol 2002;81:308)
- Clonal myeloid disorder with mixed myelodysplastic and myeloproliferative features
- Ring sideroblasts ≥ 15% of erythroblasts (Cancer Biol Med 2016;13:360)
- Persistent thrombocytosis ≥ 450 x 109/L (Blood Cancer J 2018;8:15)
- No increase in blast count, < 1% in blood and < 5% in bone marrow (Blood Cancer J 2018;8:15)
- Lacking BCR-ABL1, isolated del(5q), t(3;3)(q21.3;q26.2) and inv(3)(q21.3;q26.2) (Cancer Biol Med 2016;13:360)
- No previous history of myelodysplastic syndrome (MDS) or myeloproliferative neoplasm (MPN), except for MDS with ring sideroblasts
- New distinct entity in 2016 WHO classification; related to 2008 WHO provisional entity, refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T) (Blood Cancer J 2018;8:15, Haematologica 2015;100:1117)
- Mixed myelodysplastic and myeloproliferative features with ring sideroblasts (≥ 15% of marrow erythroblasts), persistent thrombocytosis (≥ 450 x 109/L), anemia and normal blast count (Blood Cancer J 2018;8:15)
- Strong association with SF3B1 mutation; if absent, there should be no history of recent cytotoxic or growth factor therapy that could explain the MDS / MPN overlap features
- Often seen in conjunction with JAK2 V617F (and less often CALR or MPL gene mutations), which support but are not required for diagnosis (Haematologica 2015;100:1117, Blood 2016;127:2391)
- In contrast to MDS with ring sideroblasts, at least 15% ring sideroblasts are required even if an SF3B1 mutation is present (Blood 2016;127:2391)
- Myelodysplastic and myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS / MPN-RS-T)
- Previously, refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T)
- ICD-10: D46.9 - myelodysplastic syndrome, unspecified
- Median age mid 70s, range 18 - 95 years (Cancer Biol Med 2016;13:360)
- About equal gender distribution (Cancer Biol Med 2016;13:360)
- Myeloid cells primarily in bone marrow and blood
- Splenomegaly 40%
- Occasional hepatomegaly
- Probable multistep genetic and epigenetic changes, leading to signaling deregulation, impaired DNA damage response and other cellular abnormalities (Int J Hematol 2015;101:229)
- Unclear
- Anemia due to ineffective erythropoiesis
- Thrombosis more likely than MDS with ring sideroblasts (Cancer Biol Med 2016;13:360)
- Anemia with dyserythropoietic changes (Haematologica 2015;100:1117)
- Ring sideroblasts ≥ 15% of erythroblasts (Blood Cancer J 2018;8:15)
- Persistent thrombocytosis ≥ 450 x 109/L (Blood Cancer J 2018;8:15)
- Blast count < 1% in blood and < 5% in bone marrow (Blood Cancer J 2018;8:15)
- SF3B1 mutation or in absence of SF3B1 mutation, no history of recent cytotoxic or growth factor therapy (Haematologica 2015;100:1117)
- No BCR-ABL1 fusion; no rearrangement of PDGFRA, PDGFRB or FGFR1; no PCM1-JAK2, t(3;3)(q21.3;q26.2), inv(3)(q21.3;q26.2) or del(5q) (Cancer Biol Med 2016;13:360)
- No history of MPN, MDS (except for MDS with ring sideroblasts) or MDS / MPN neoplasm (Blood Cancer J 2018;8:15)
- Macrocytic or normocytic anemia
- Thrombocytosis, ≥ 450 x 109/L
- White blood cell (WBC) absolute and differential counts usually normal but mild leukocytosis can occur
- Median survival 76 - 128 months (Am J Hematol 2016;91:492)
- Worse outcome than essential thrombocythemia (Am J Hematol 2019;94:475)
- Low risk of acute leukemic transformation (Am J Hematol 2017;92:297, Am J Hematol 2016;91:492)
- SF3B1 mutation longer survival (6.9 years) than wild type (3.3 years) (Leukemia 2013;27:1826)
- JAK2 V617F mutation more favorable than wild type (Leukemia 2013;27:1826, Am J Hematol 2016;91:492)
- ASXL1 or SETBP1 mutation shorter survival (Am J Hematol 2017;92:297, Am J Hematol 2016;91:492)
- Hemoglobin < 10 g/dL or abnormal karyotype shorter survival (Am J Hematol 2016;91:492)
- 58 year old man with transfusion dependent anemia, markedly elevated platelet count and marrow changes (Blood Res 2017;52:8)
- 69 year old man with progressive thrombocytosis and suspicion of essential thrombocythemia (Blood 2013;121:4256)
- 75 year old woman presenting with chronic anemia (Blood 2014;123:314)
- 75 year old man with prefibrotic / early primary myelofibrosis (Blood 2017;129:657)
- 80 year old man with history of radiotherapy treated prostate cancer (Rev Bras Hematol Hemoter 2014;36:69)
- No specific therapy, only general treatment for lower risk MDS and MPN (Haematologica 2015;100:1117)
- Aspirin may be used (Am J Hematol 2017;92:297)
- Luspatercept may improve anemia (Am J Hematol 2019;94:475)
- Anecdotal reports of response with single agent lenalidomide (Blood 2010;116:180, Am J Hematol 2018;93:E27)
- Hypercellularity of bone marrow with < 5% blasts
- Erythroid hyperplasia with dyserythropoiesis, including megaloblastic change, common in marrow
- Ring sideroblasts ≥ 15% of normoblasts on iron stain (Blood Cancer J 2018;8:15)
- Multilineage dysplasia possible
- Atypical megakaryocytic hyperplasia and cluster formation with large, hyperlobated megakaryocytes, reminiscent of BCR-ABL negative MPN (Haematologica 2015;100:1117)
- Marrow fibrosis can occur
Contributed by Patricia Tsang, M.D., M.B.A.
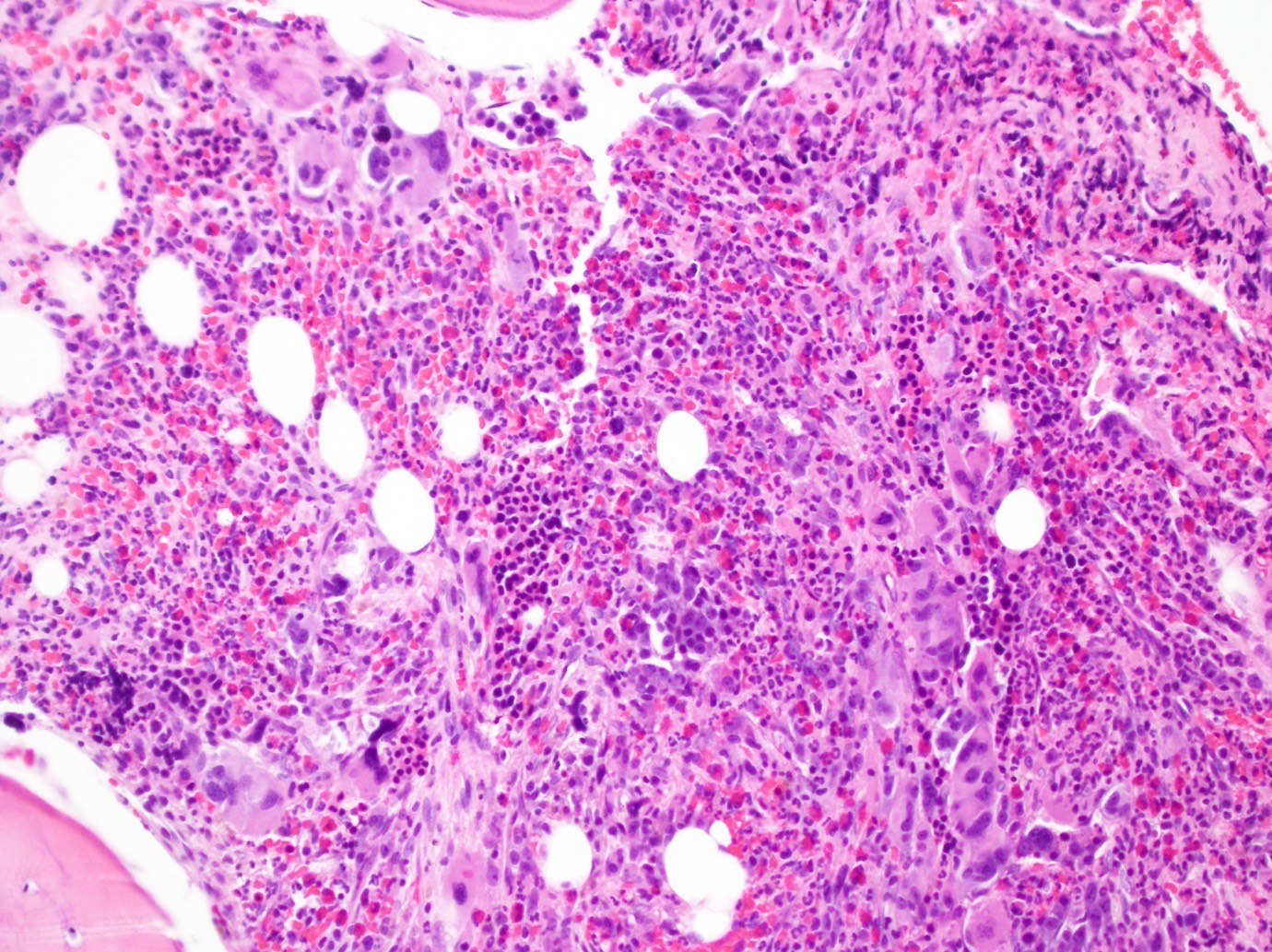
Hypercellular marrow
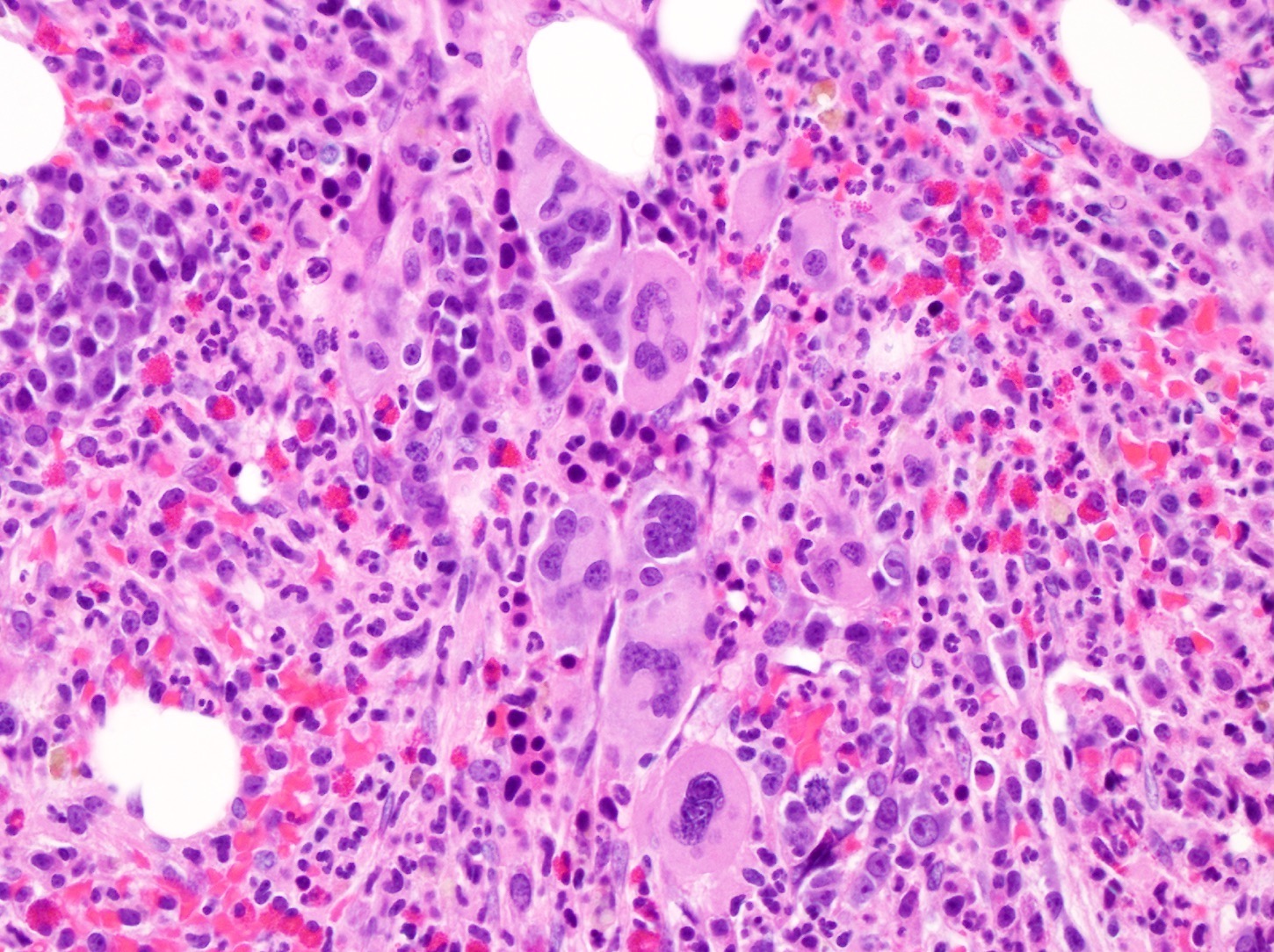
Megakaryocyte proliferation

Iron stain with ring sideroblast
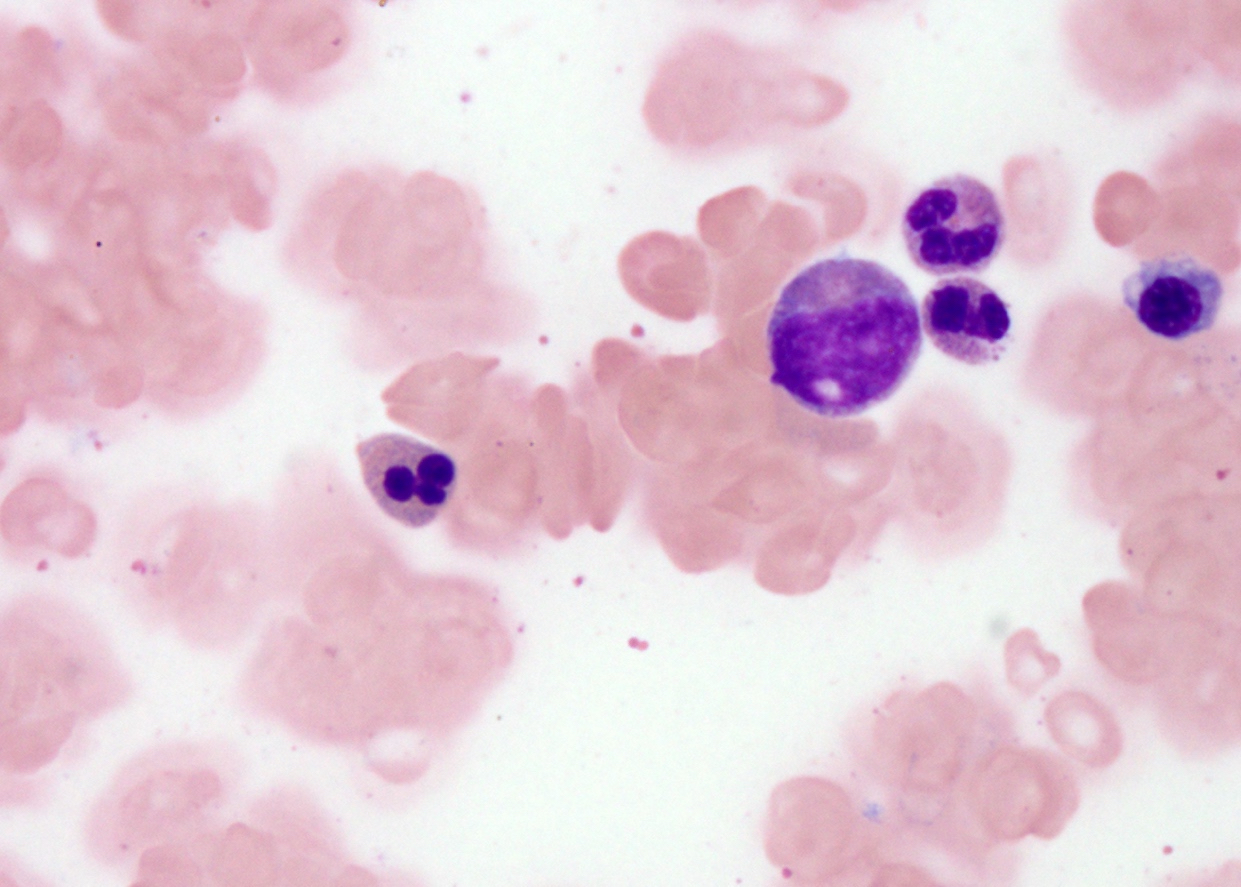

Dyserythropoiesis
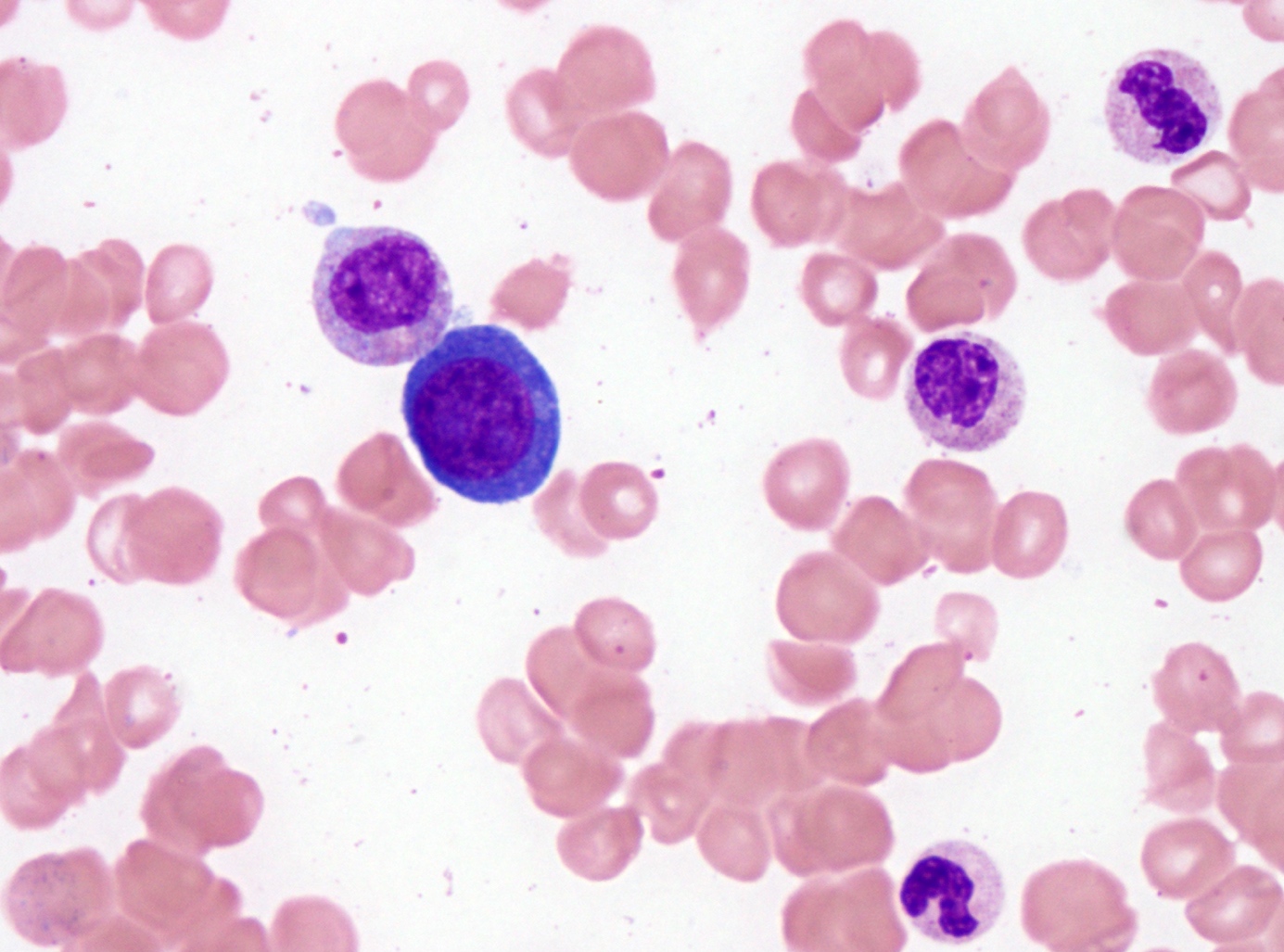
Normoblast with megaloblastic change
- Normocytic to macrocytic, normochromic red blood cells
- Thrombocytosis, sometimes with large, giant or atypical platelets
- No or rare blasts, < 1% (Blood Cancer J 2018;8:15)
Contributed by Patricia Tsang, M.D., M.B.A.
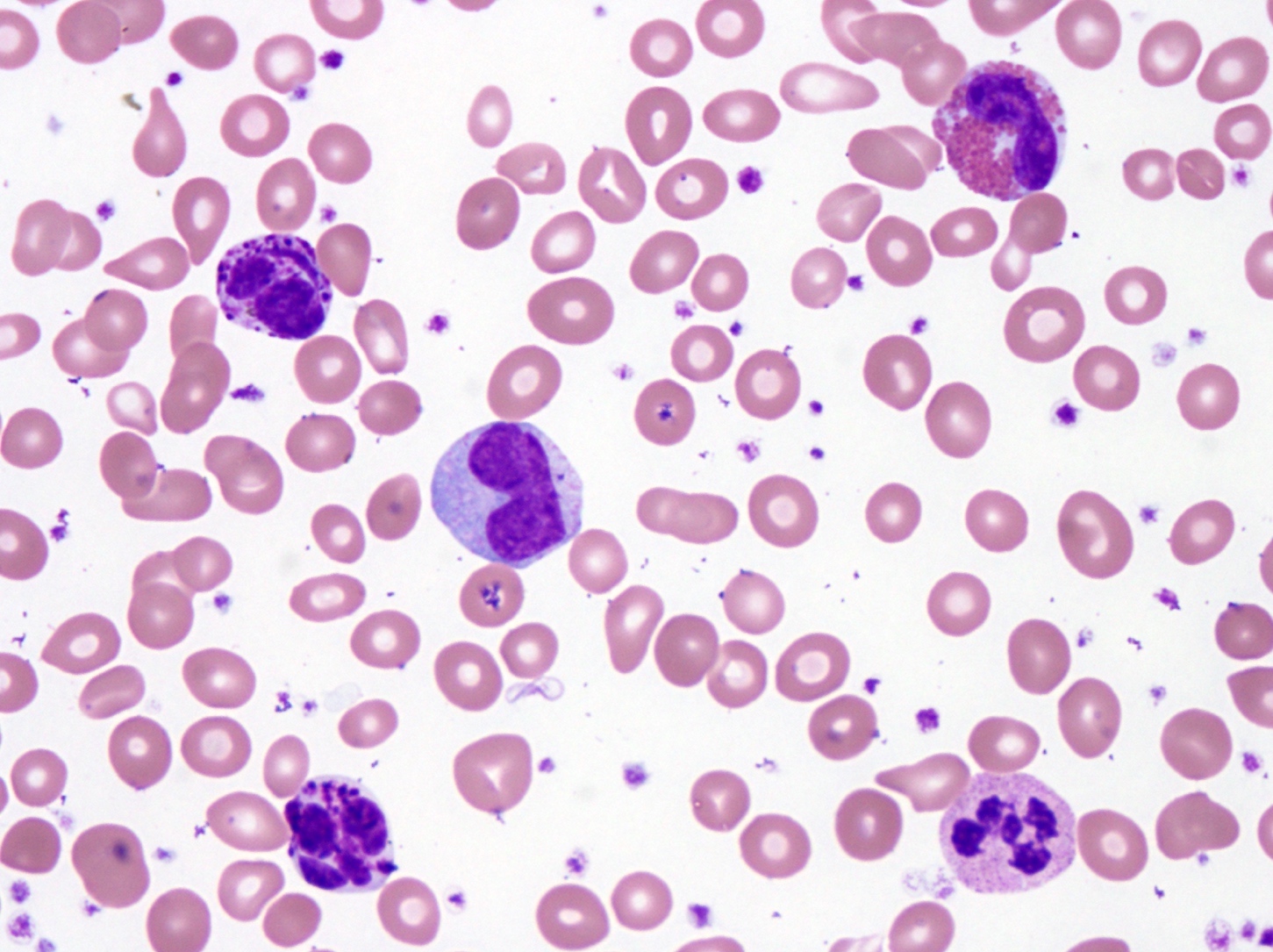
Thrombocytosis
- Iron stain on aspirate smear shows ring sideroblasts
- Immunostains generally not needed for diagnosis
- No increase in blasts detected
- Aberrant CD56 on myeloid cells sometimes seen
- SF3B1 mutation (60 - 90%), often in conjunction with JAK2 V617F mutation (> 60%), infrequently with MPL or CALR gene mutation (< 10%) (Am J Hematol 2018;93:E27, Am J Hematol 2016;91:492)
- ASXL1 (20%), DNMT3A (15%), SETBP1 (10%) and TET2 (25%) mutations (Am J Hematol 2017;92:297, Am J Hematol 2018;93:E27)
- Abnormal cytogenetics (~20%) (Am J Hematol 2018;93:E27)
- Bone marrow biopsy and aspirate smears, left posterior iliac crest:
- Myelodysplastic / myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, formerly refractory anemia with ring sideroblasts and thrombocytosis
- Cytogenetics, next generation sequencing for myeloid disorders, reticulin and trichrome stains are pending for further evaluation
- Peripheral blood smear:
- Macrocytic anemia with occasional teardrop RBCs
- Mild neutrophilia and marked thrombocytosis with some giant platelets
- Microscopic description:
- The peripheral blood smear shows leukocytosis with mild neutrophilia (8,400/mcL) and no circulating blasts. Anisopoikilocytosis with macrocytic RBCs and occasional teardrop cells are seen. Platelet count is elevated (610,000/mcL).
- The bone marrow aspirate (dry tap) is aspicular and hypocellular with evidence of maturing granulopoiesis and erythropoiesis. Some of the normoblasts show megaloblastic change and nuclear lobation, consistent with dyserythropoietic changes. The iron stain on aspirate reveals adequate storage iron. Ring sideroblasts constitute about 35% of the erythroid precursors.
- The bone marrow biopsy is hypercellular (about 80%) with trilineage hyperplasia. There is focal crush cell artifact, suggestive of marrow fibrosis. Myeloid:erythroid ratio appears unremarkable. No increase in myeloblasts is seen histologically. Proliferation of atypical megakaryocytes with cluster formation is present. Megakaryocytes are characteristically enlarged and hyperlobated. No abnormal lymphoid or plasma cell infiltrate is noted.
- Myelodysplastic syndrome with ring sideroblasts (MDS-RS): lower platelet count, lacking myeloproliferative morphologic features, lacking JAK2 / CALR / MPL gene mutation
- Essential thrombocythemia: typically higher white blood cell count, higher hemoglobin levels, lacking ring sideroblasts, lacking dysplastic features, lacking SF3B1 mutation
- Myelodysplastic syndrome with isolated del(5q): unilobated or hypolobated megakaryocytes, del(5q) occurring singly or with one other cytogenetic abnormality, often del(7q) or monosomy 7
- ABL1 and CALR
- ASXL1 and KRAS
- SETBP1 and TP53
- SF3B1 and JAK2
- TET2 and FLT3
Comment Here
Reference: Myelodysplastic / myeloproliferative neoplasm with ring sideroblasts and thrombocytosis
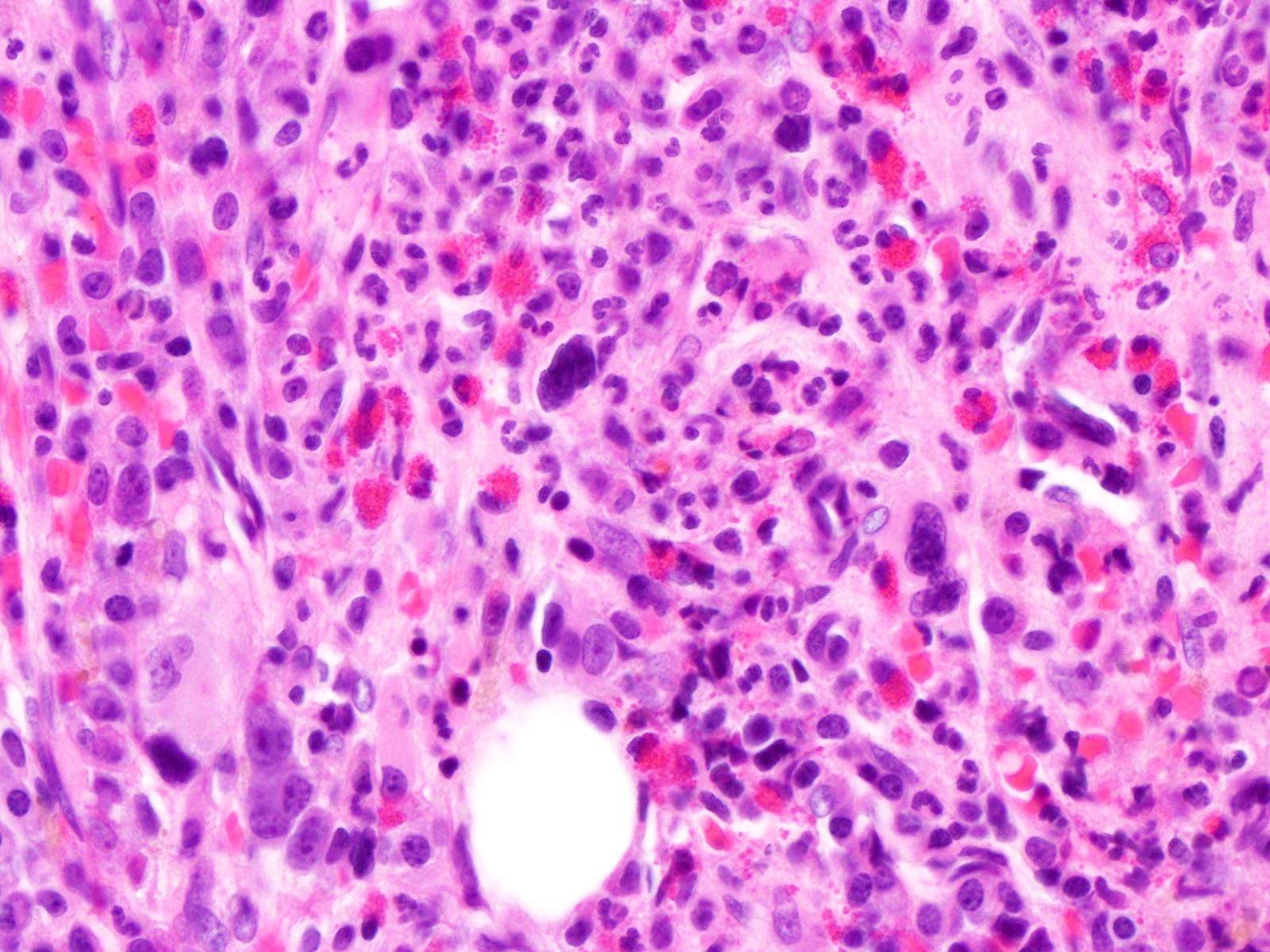
- Blasts < 1% in the blood and < 5% in the bone marrow
- Circulating ring sideroblasts > 15% in the peripheral blood
- Deletion 5q or monosomy 5
- Persistent monocytosis with no secondary etiology
- Proliferation of hypolobated megakaryocytes in the bone marrow
Comment Here
Reference: Myelodysplastic / myeloproliferative neoplasm with ring sideroblasts and thrombocytosis
- 3 - 9% of pediatric hematologic malignancies
- Estimated annual incidence in U.S. is 0.5 to 4 cases per million for children compared to 20 - 40 cases for adults
- Usually diagnosed at ages 6 - 8 years, although juvenile myelomonocytic leukemia is usually diagnosed at age 2 years
- Difficult to diagnose because:
- Less dysplasia at younger ages
- Dysplasia may be confused with treatment effects of G-CSF
- Bone marrow often hypocellular, resembling aplastic anemia
- Resembles HHV6 (Pediatr Blood Cancer 2006;47:543) or parvovirus infections (Rinsho Ketsueki 2001;42:1096, Pediatr Hematol Oncol 2000;17:475)
- Modified WHO classification for childhood MDS
- Diagnosis requires:
- Sustained and unexplained cytopenia
- At least bilineage dysplasia
- A clonal cytogenetic abnormality
- At least 5% blasts (Leukemia 2003;17:277); may be most successful system for classification (Arch Pathol Lab Med 2007;131:1110)
- Diagnosis requires:
- 69% idiopathic, 24% associated with constitutional / inherited disorders (Down syndrome, neurofibromatosis, Bloom syndrome, Fanconi anemia, Atlas of Genetics and Cytogenetics), 7% therapy related (Arch Pathol Lab Med 2007;131:1110)
- Similar prognosis as AML M6 and M7, with poor induction success rate (Pediatr Blood Cancer 2007;49:17)
- Monosomy 7 in 30%
- Myelodysplastic neoplasms (MDS) are a heterogeneous group of diseases with variable outcomes depending on multiple parameters
- International Prognostic Scoring System for MDS is the most commonly used tool to predict long term outcomes
- Outcomes of MDS are variable and depend on many different parameters including cytogenetics, blast percentage, hemoglobin levels, absolute neutophil count and platelet count; the International Prognostic Scoring System for MDS is the most commonly used tool to predict long term outcomes
- Patients in the low risk group will often have more prolonged survival and oncological goals include improving their cytopenias (neutropenia, anemia and thrombocytopenia) and reducing iron overload
- Patients in the high risk group with increased blasts and cytogenetic abnormalities often have shorter survival and are treated more aggressively with 5-azacitidine to delay the transformation to acute myeloid leukemia
- Myelodysplastic syndrome / myelodysplastic neoplasm (MDS)
- International Prognostic Scoring System (IPSS)
- Revised International Prognostic Scoring System (IPSS-R)
- ICD-10: D46.9 - myelodysplastic syndrome, unspecified
- Median age at the diagnosis of MDS: 77 years (Br J Haematol 1992;82:358)
- Incidence of MDS below age 40 is extremely low (0.1 cases per 100,000) (Blood Rev 2019;34:1)
- Early age of onset (< 50 years) is associated with MDS with genetic predisposition due to inherited genetic mutations (Blood Rev 2019;34:1)
- Younger age of onset is more common amongst the Asian population (Leuk Res 1998;22:453)
- Early onset can also be caused by exposure to chemicals (e.g., benzene) or radiation (J Natl Cancer Inst 2012;104:1724)
- Men are more commonly affected compared to women, except in MDS with low blasts and 5q deletion (MDS 5q), where women are the predominate population affected (Leuk Res 2011;35:1591)
- There is a higher rate of MDS among the Caucasian population (Blood Rev 2019;34:1)
- Bone marrow
- Genetic defects play a major role in the pathogenesis of MDS, including cytogenetic abnormalities, genetic mutations and alterations, abnormal gene expression and epigenetic dysregulation (Int J Clin Oncol 2019;24:885)
- Chromosomal abnormalities have been seen in at least 50% of MDS patients, including the abnormalities of chromosome 5, 7, 8 and 20; complex karyotypes are also identified (Blood 2008;111:1534)
- Genetic abnormalities have been identified in over 50 genes in at least 80% of MDS patients, including SF3B1, SRSF2, U2AF1, ZRSR2, LUC7L2, DDX41, ASXL1, EZH2, TET2, DNMT3A, IDH1 / IDH2, CSNK1A1, RUNX1, BCOR, TP53 and CUX1 (Eur J Haematol 2015;95:3)
- Old age is considered a significant risk factor and the risk increases with advancing age due to an increased propensity for genetic mutations (Blood 2013;122:2943)
- Exposure to certain environmental toxins and certain chemicals (e.g., benzene), has been linked to an increased risk of developing MDS (Leuk Lymphoma 1999;35:269)
- Previous exposure to chemotherapy or radiation therapy for the treatment of another malignancy is also known risk factor for developing secondary MDS (Cancer Genet Cytogenet 2007;176:1, Eur J Haematol 1991;47:17)

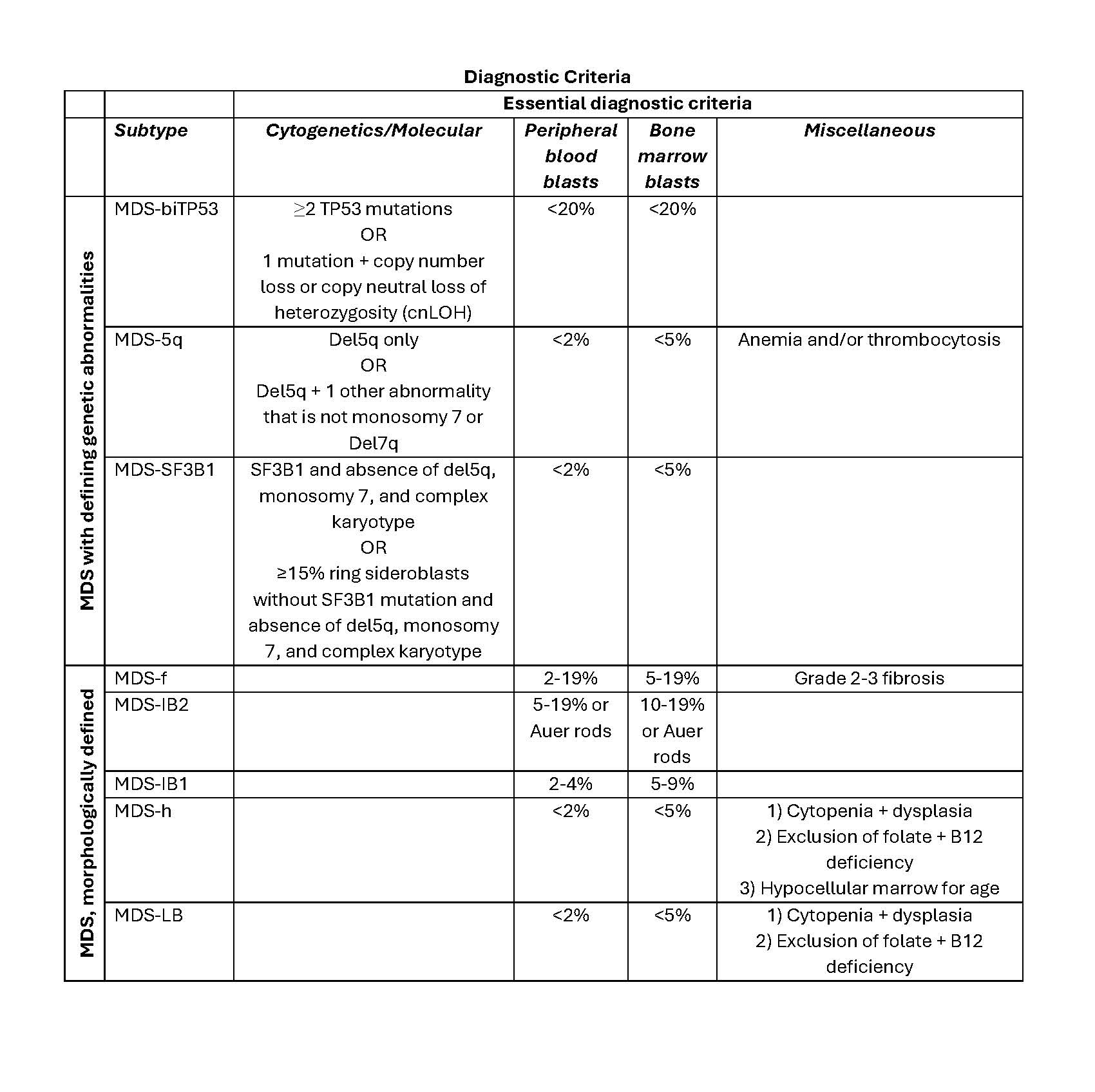
- MDS patients typically present with at least 1 cytopenia: neutropenia, anemia or thrombocytopenia (Leukemia 2022;36:1720)
- Neutropenia can lead to increased susceptibility to infections (Clin Hematol Int 2021;3:41)
- Anemia can contribute to persistent fatigue, weakness and shortness of breath (Ann N Y Acad Sci 2019;1450:15)
- Thrombocytopenia can lead to easy bruising, petechiae and an increased risk of bleeding (Cureus 2022;14:e27718)
- Please see diagnostic criteria table
- Please see features of dysplasia table for the entities that require dysplasia as an essential diagnostic criteria; dysplasia must be seen in at least 10% of the total lineage on examation of the bone marrow aspirate smears, touch preparations, bone marrow clot section and bone marrow biopsy sections (Leukemia 2022;36:1720)
- Cytopenia in at least 1 hematopoietic lineage is usually seen in most cases of MDS
- Anemia: Hb < 13 g/dL in men and < 12 g/dL in women
- Leukopenia: absolute neutrophil count < 1.8 x 109/L
- Thrombocytopenia: platelets < 150 x 109/L
- Chromosome analysis or conventional karyotyping, fluorescence in situ hybridization (FISH) and next generation sequencing (NGS) for mutation analysis are required for some diagnoses
- Revised IPSS (IPSS-R) utilizes cytogenetics, blast percentage in the bone marrow, hemoglobin levels, platelet levels and the absolute neutrophil count to create the prognostic risk scores and classify patients under the risk categories
- Please see IPSS-R table above (Blood 2012;120:2454)
- Molecular IPSS (IPSS-M) utilizes molecular data for risk prognostication using a list of 31 focused genes: TP53, MLL, FLT3, ASXL1, CBL, DNMT3A, ETV6, EZH2, IDH2, KRAS, NPM1, NRAS, RUNX1, SF3B1, SRSF2, U2AF1, BCOR, BCORL1, CEBPA, ETNK1, GATA2, GNB1, IDH1, NF1, PFH6, PPM1D, PRPF8, PTPN11, SETBP1, STAG2, WT1 (NEJM Evid 2022;1:EVIDoa2200008)
- 8 year old girl with childhood MDS and transformation to acute lymphoblastic leukemia (Ann Lab Med 2013;33:130)
- 53 year old woman with fibromyalgia and MDS (Case Rep Hematol 2011;2011:560106)
- 71 year old man with idiopathic cytopenia of undetermined significance (ICUS) progressing to MDS and AML (Blood 2024;143:473)
- 72 year old woman with interstitial lung disease and MDS (Front Med (Lausanne) 2021;8:673573)
- Treatment approach can be divided based on the risk group; for those with low risk MDS, it depends on the transfusion needs
- Transfusion independent patients are followed with observation only
- Patients with severe anemia with increasing red blood cell transfusion needs can be managed with erythroid stimulating agents (ESA) (Am J Hematol 2023;98:1307)
- Patients with severe neutropenia with increased infection risk can be managed with granulocyte colony stimulating factors (GCSF) (Am J Hematol 2023;98:1307)
- Lenalidomide is the standard of care for low risk MDS with del(5q), anemia and increasing transfusion needs; it is contraindicated if there is thrombocytopenia
- 5-azacitidine and decitabine are used for transfusion dependent, low risk MDS cases that have failed or are not candidates for growth factors or lenalidomide (Am J Hematol 2023;98:1307)
- For low risk MDS that have failed multiple therapies, allogeneic stem cell transplant (allo-SCT) must be considered (Am J Hematol 2023;98:1307)
- 5-azacitidine is the standard of care for high risk MDS; furthermore, allo-SCT should also be considered in high risk MDS (Am J Hematol 2023;98:1307)
- Myelodysplastic neoplasms with defining genetic abnormalities
- Myelodysplastic neoplasm with biallelic TP53 inactivation (MDS biTP53)
- In general, MDS biTP53 has high risk morphologic findings, such as multilineage dysplasia, higher blast count and fibrosis in the bone marrow
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Bone marrow is typically hypercellular but can be normocellular or hypocellular
- Blast percentage < 20% in the bone marrow and < 20% in the peripheral blood
- Myelodysplastic neoplasm with low blasts and 5q deletion (MDS 5q)
- Complete blood count and peripheral blood smear evaluation can show thrombocytosis in the absence of a cytopenia
- Megakaryocytes are usually dysplastic with small and hypolobated forms (micromegakaryocytes)
- Dysplasia in the erythroid and granulocytic lineages are less prominent; however, dysplastic changes can still overlap with those of other MDS types (please see features of dysplasia table)
- Ring sideroblasts (or SF3B1 mutation) can be present and do not exclude the diagnosis of MDS 5q, if the other criteria are met
- Bone marrow is typically hypercellular or normocellular and can have megakaryocytic hyperplasia and erythroid hypoplasia
- Blast percentage < 5% in the bone marrow and < 2% in the peripheral blood
- Myelodysplastic neoplasm with low blasts and SF3B1 mutation (MDS SF3B1)
- Complete blood count and peripheral blood smear evaluation can show normocytic to macrocytic anemia
- Bone marrow is typically hypercellular with left shifted erythroid predominance and dyserythropoiesis
- Ring sideroblasts are increased; ring sideroblasts are defined as having ≥ 5 iron granules or granules encircling at least one - third or more of the nucleus
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Blast percentage < 5% in the bone marrow and < 2% in the peripheral blood
- Myelodysplastic neoplasm with biallelic TP53 inactivation (MDS biTP53)
- Myelodysplastic neoplasms morphologically defined
- Myelodysplastic neoplasm with increased blasts
- Bone marrow is usually hypercellular
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Subtypes
- MDS with increased blasts 1 (MDS IB1): blast percentage 5 - 9% in the bone marrow or 2 - 4% in the peripheral blood without significant fibrosis
- MDS with increased blasts 2 (MDS IB2): blast percentage 10 - 19% in the bone marrow or 5 - 19% in the peripheral blood or Auer rods without significant fibrosis
- MDS with increased blasts and fibrosis (MDS-f): Blast percentage 5 - 19% in the bone marrow or 2 - 19% in the peripheral blood with fibrosis grade 2 to 3 out of 3
- Myelodysplastic neoplasm, hypoplastic (MDS-h)
- Bone marrow is hypocellular; the hypocellularity can be diffuse or patchy
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Blast percentage < 5% in the bone marrow and < 2% in the peripheral blood
- Myelodysplastic neoplasm with low blasts (MDS LB)
- Bone marrow is usually hypercellular
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Blast percentage < 5% in the bone marrow and < 2% in the peripheral blood
- Myelodysplastic neoplasm with increased blasts
- Myelodysplastic neoplasms of childhood
- Childhood myelodysplastic neoplasm with increased blasts (cMDS IB)
- Complete blood count and peripheral blood smear evaluation shows at least 1 cytopenia
- Bone marrow is usually hypercellular
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Blast percentage 5 - 19% in the bone marrow and 2 - 19% in the peripheral blood
- Childhood myelodysplastic neoplasm with low blasts (cMDS LB)
- Complete blood count and peripheral blood smear evaluation shows at least 1 cytopenia
- Bone marrow is usually hypercellular
- Dysplastic changes overlap with those of other MDS types; please see features of dysplasia table
- Blast percentage < 5% in the bone marrow and < 2% in the peripheral blood
- Childhood myelodysplastic neoplasm with increased blasts (cMDS IB)
Contributed by Brenda Mai, M.D.
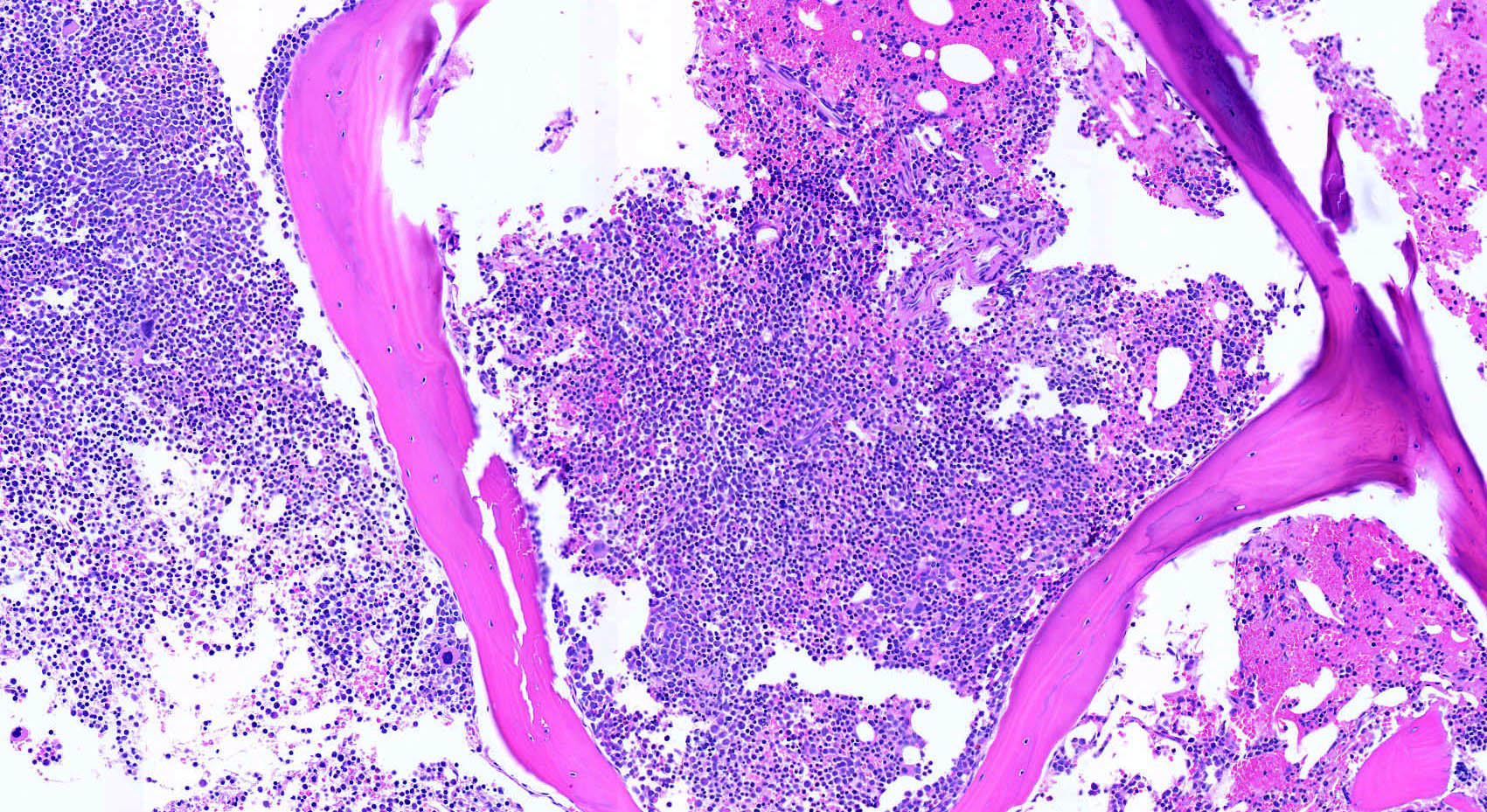
Hypercellular marrow for age

Osteosclerosis
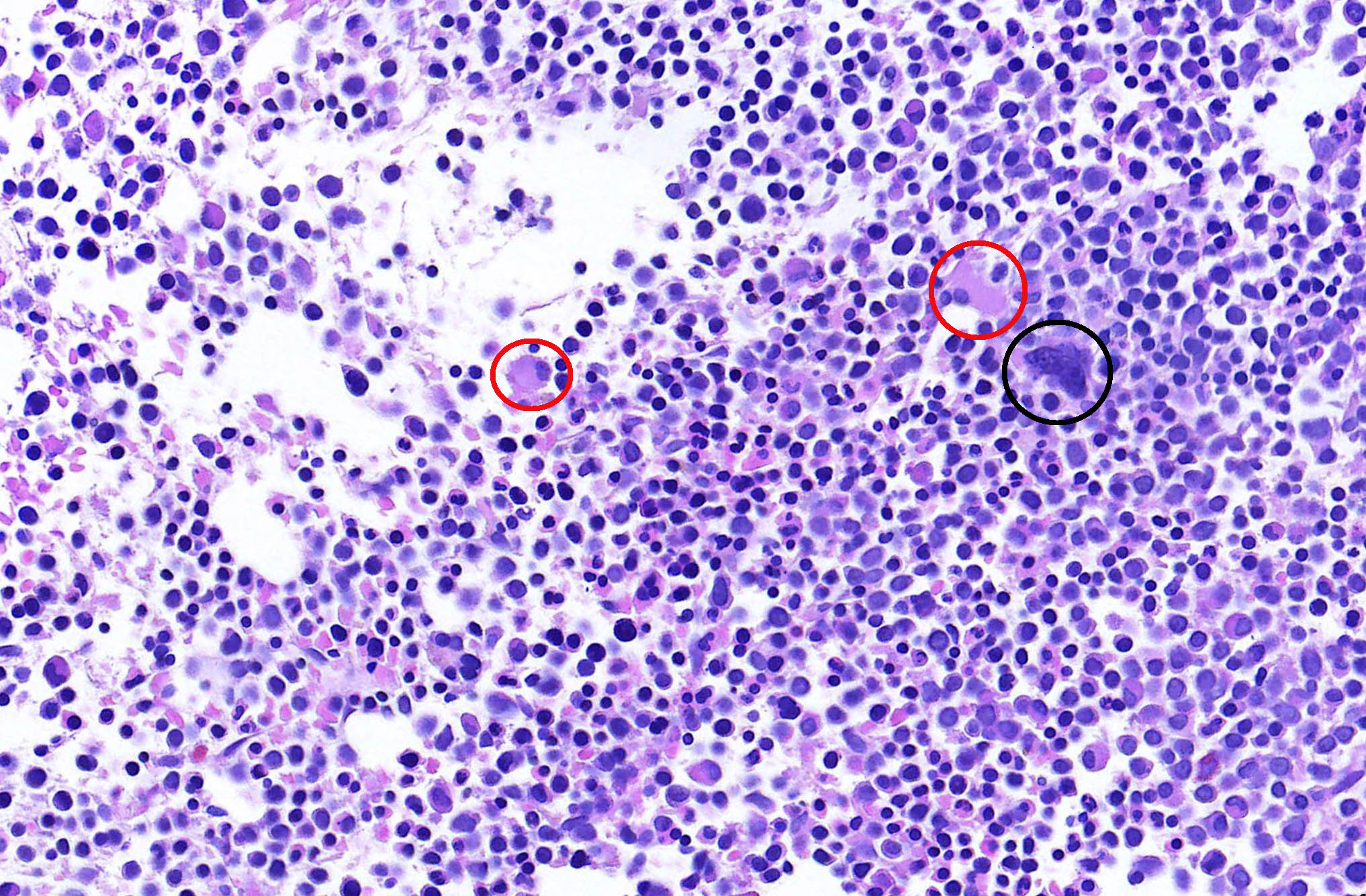
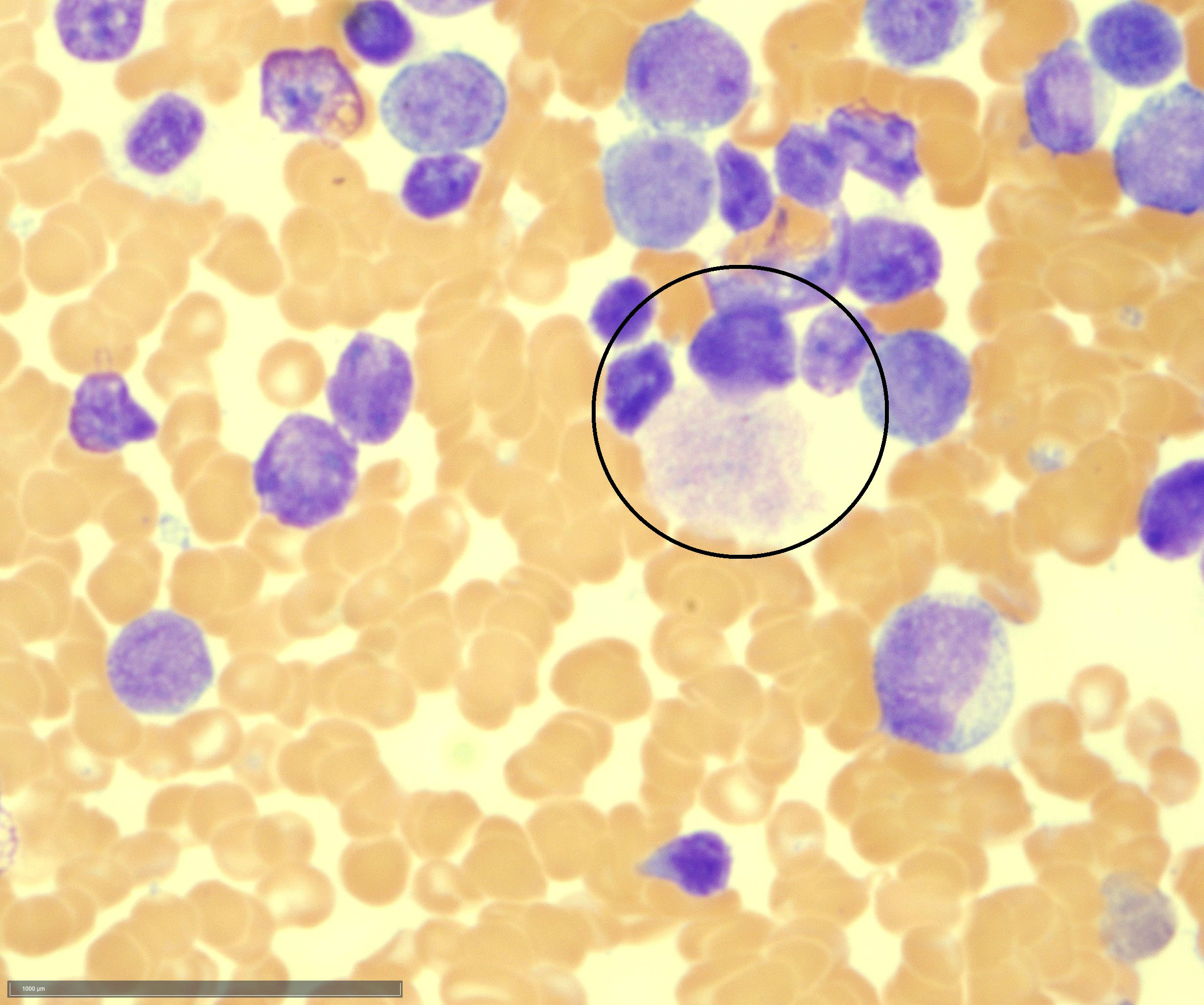
Micromegakaryocyte

Paratrabecular megakaryocyte
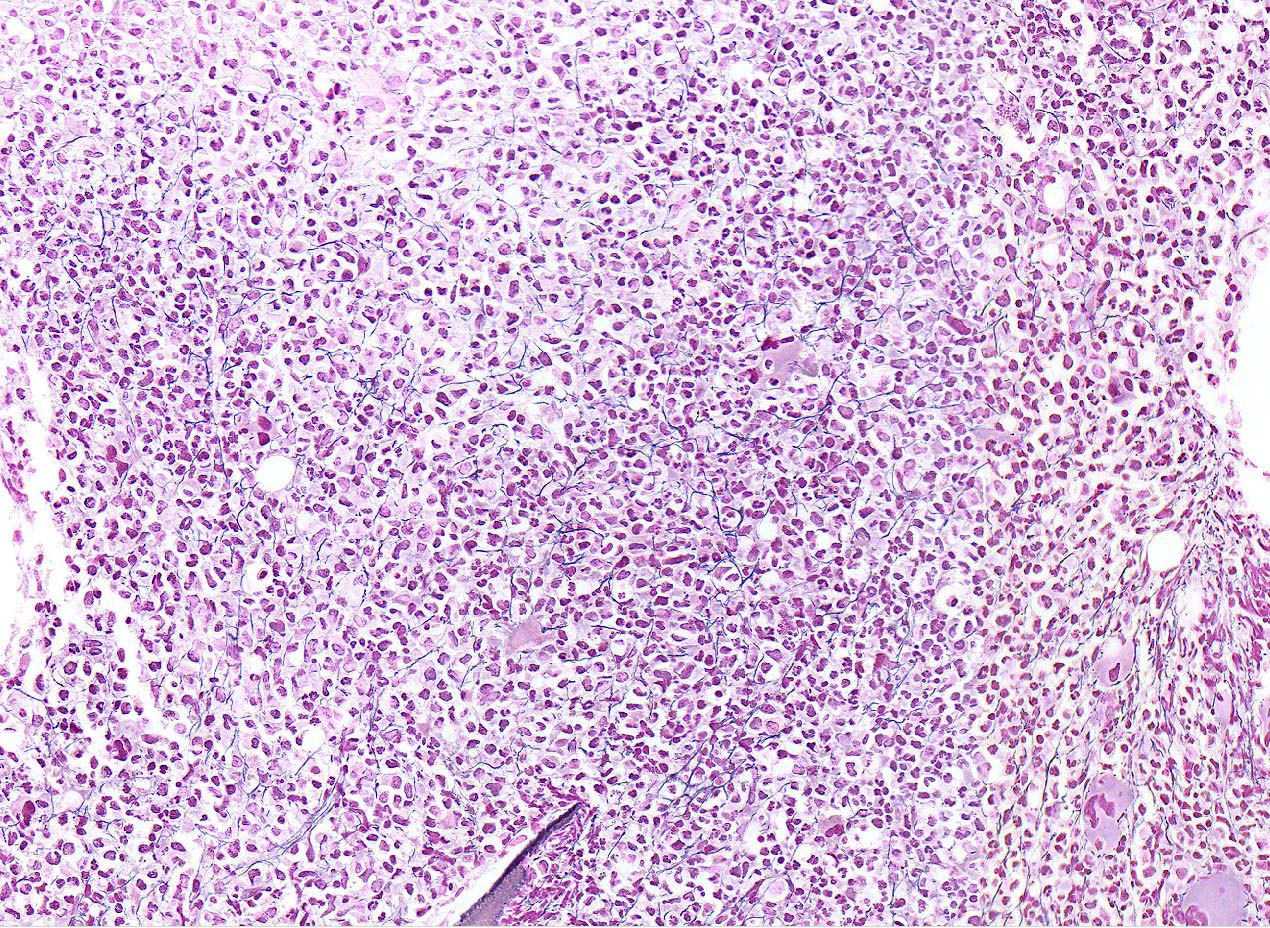
Grade 1 reticulin fibrosis
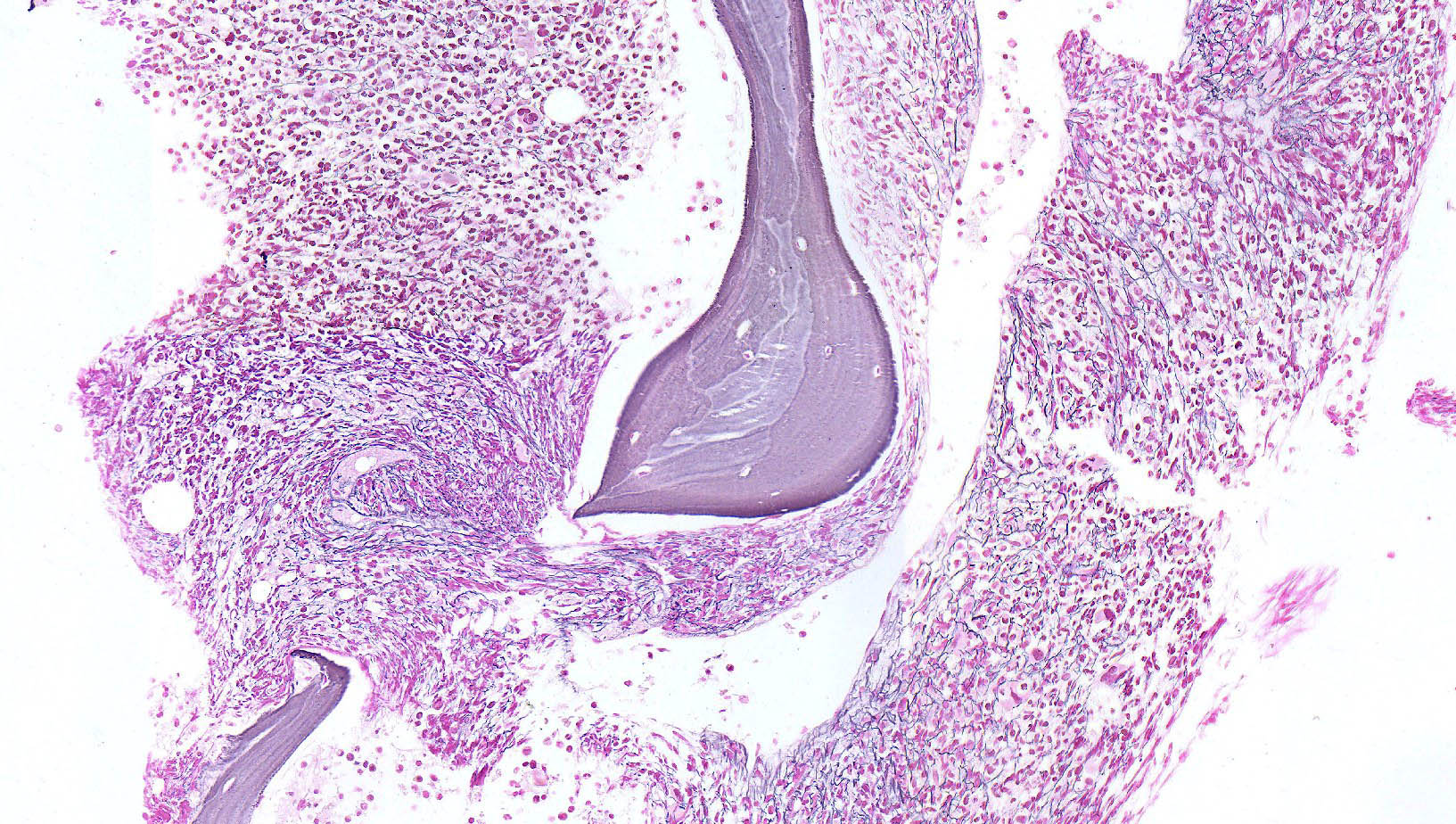
Grade 2 reticulin fibrosis
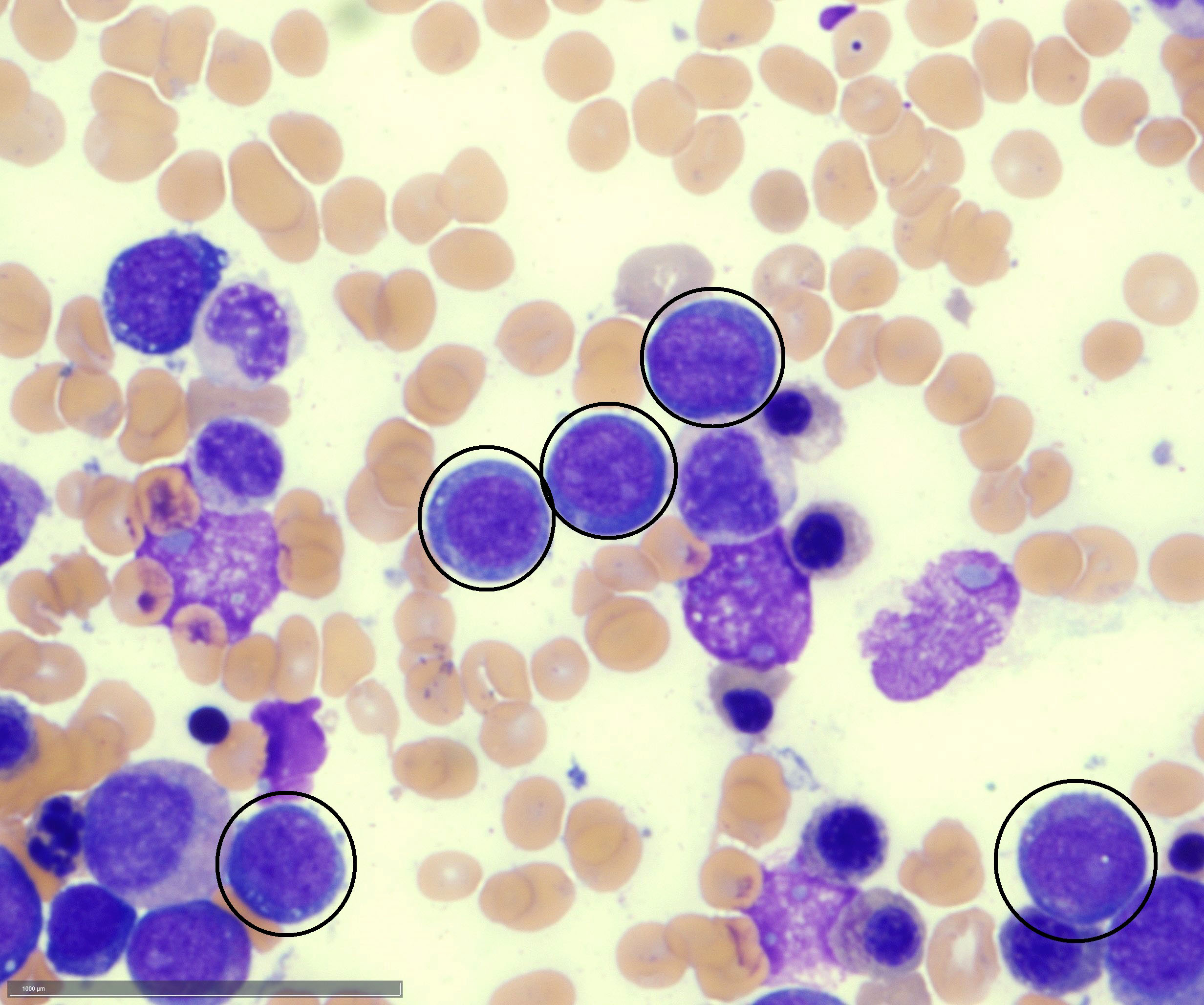
Increased blasts
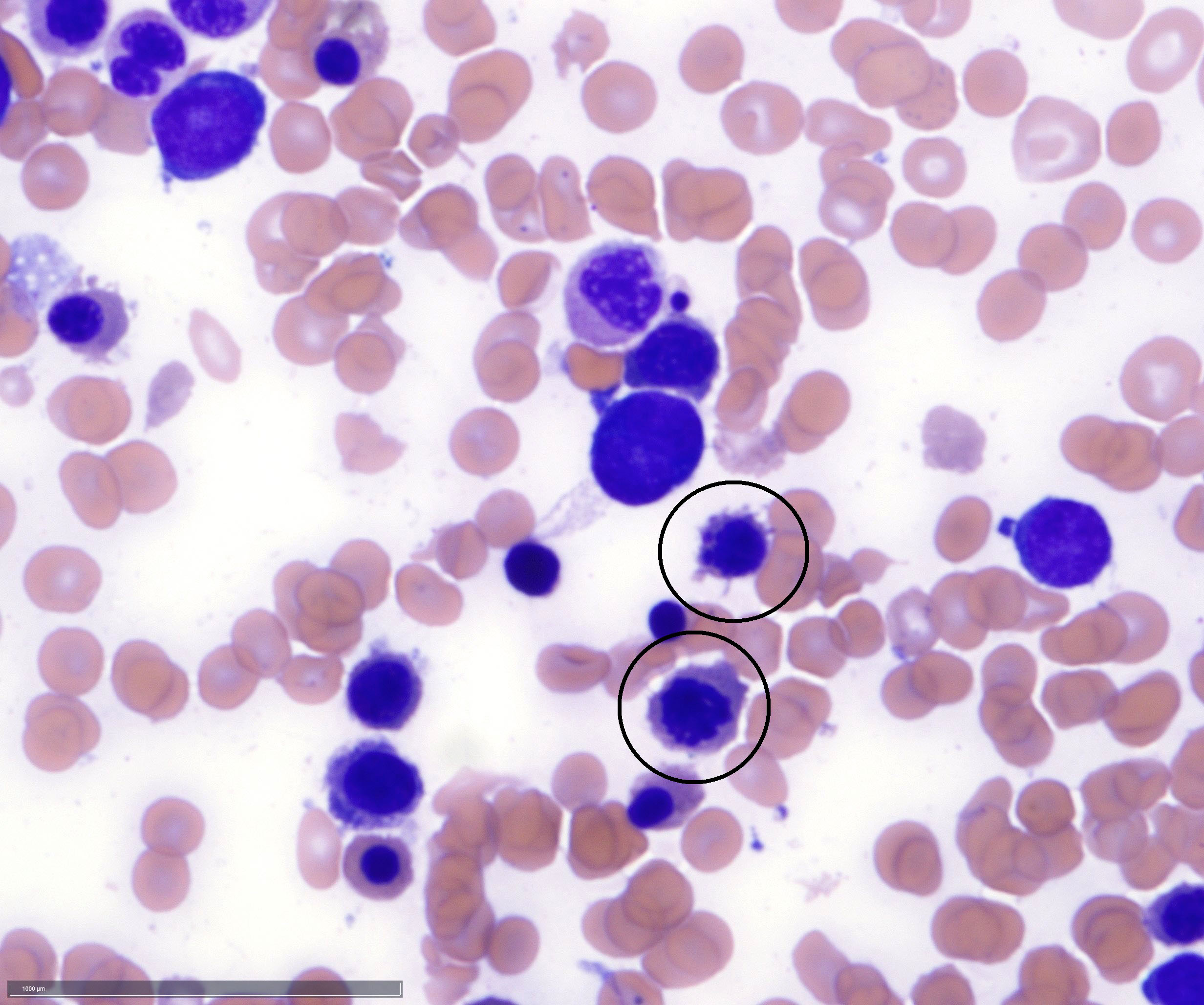
Irregular erythroids

Erythroid binucleation
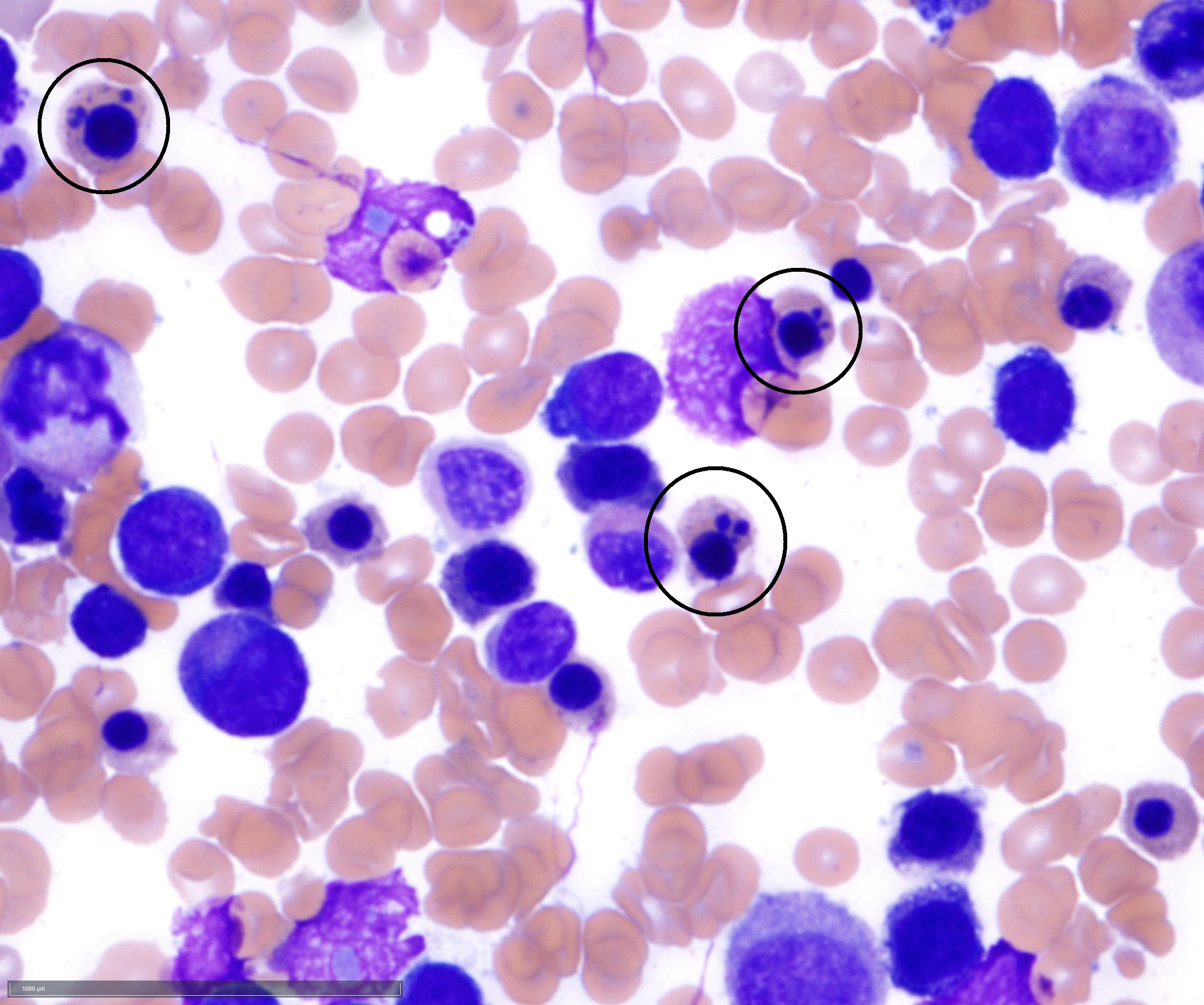
Erythroid budding and multinucleation

Erythroid karryorhexis and budding / binucleation

Erythroid vacuolization

Megaloblastoid change

Hypolobated megakaryocyte
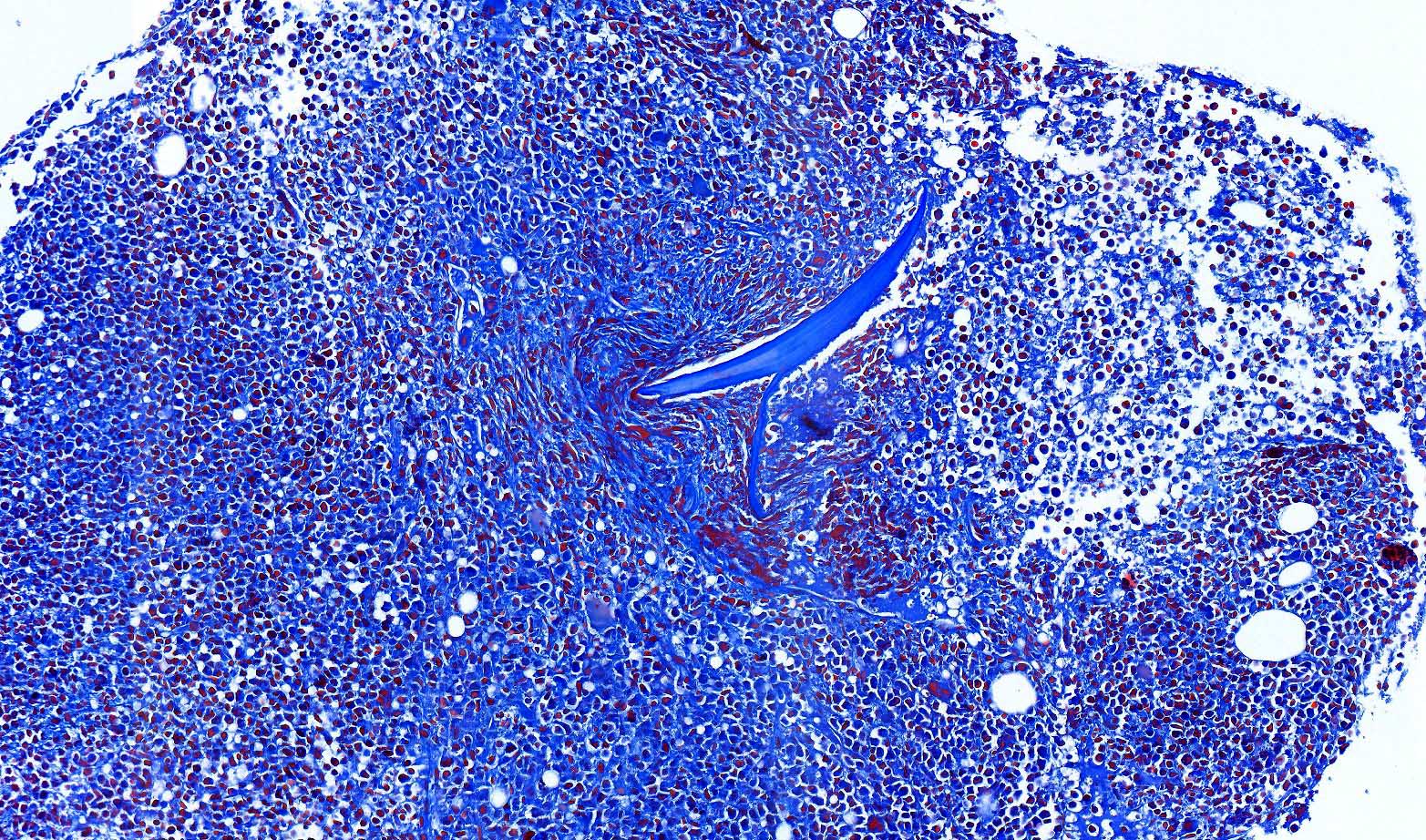
Trichrome fibrosis
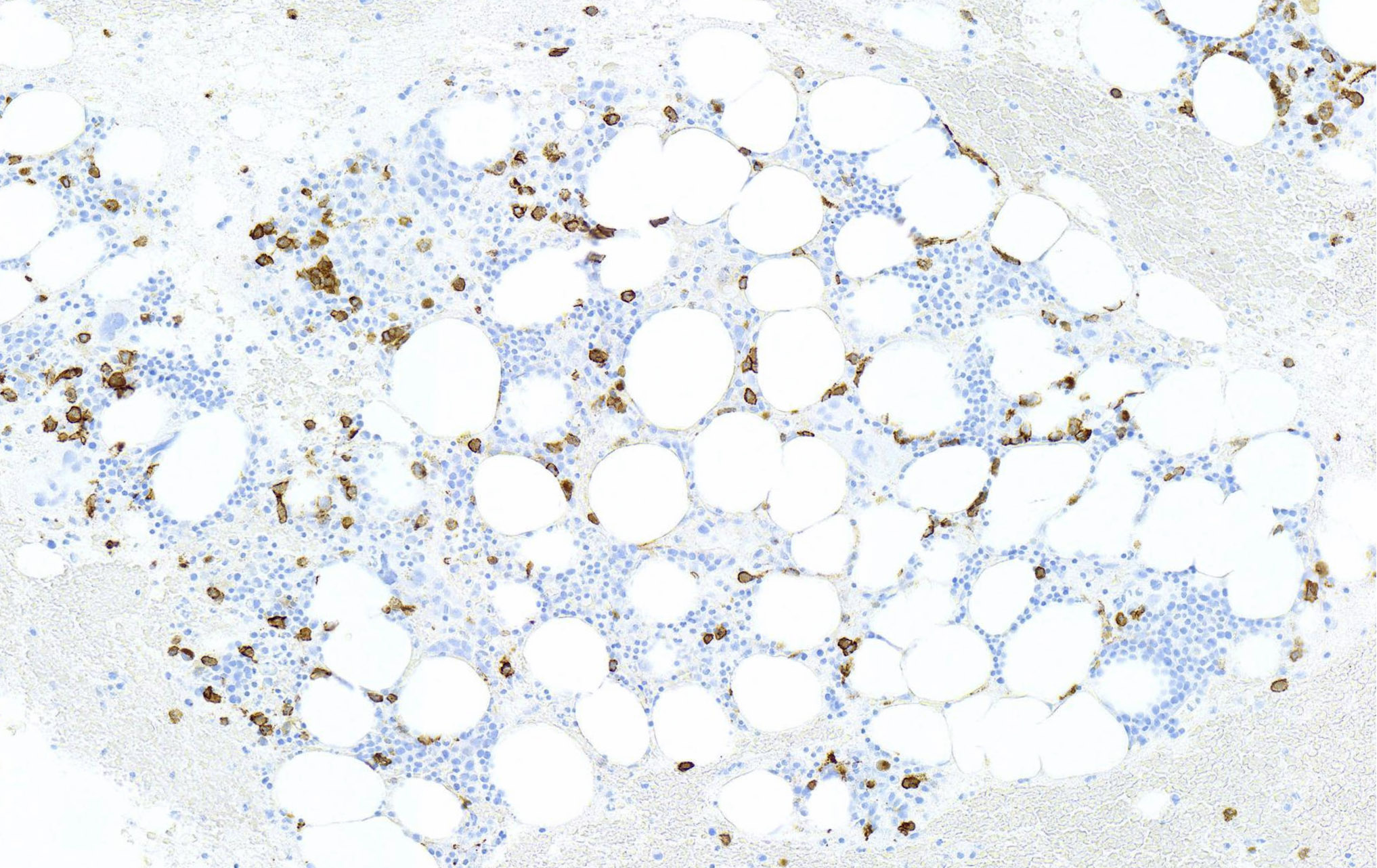
CD34 with increased blasts
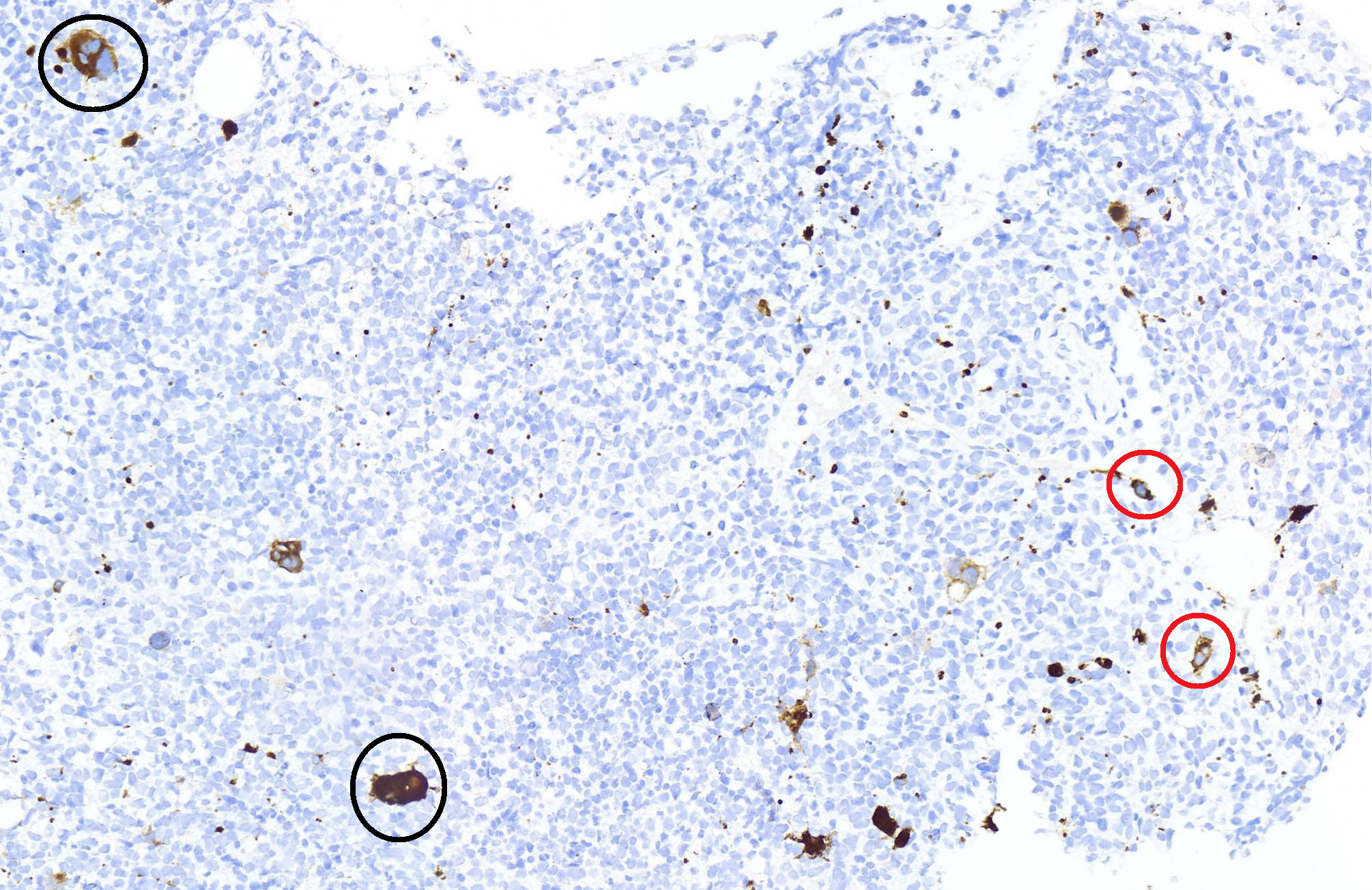
CD61 with micro-
megakaryocytes
- 500 cell count is recommended for morphologic evaluation in bone marrow aspirate smears and touch preparations
- Limitations occur when there is a dry tap or the aspirate smears are aspicular and suboptimal for evaluation
- Cytologic dysplastic changes are listed in dysplastic features table, especially the top table, features of dysplasia seen in aspirate smears
- Cytopenia in at least 1 hematopoietic lineage is usually seen in most cases of MDS
- Anemia: Hb < 13 g/dL in men and < 12 g/dL in women
- Leukopenia: absolute neutrophil count < 1.8 x 109/L
- Thrombocytopenia: platelets < 150 x 109/L
- Only granulocytic dysplasia and increased blasts are usually identified in the peripheral blood; erythroid dysplasia and megakaryocytic dysplasia are not seen
- Cytologic dysplastic changes are listed in dysplastic features table, especially the top table, especially the top table, features of dysplasia seen in aspirate smears, under dysgranulopoiesis
Contributed by Brenda Mai, M.D.

Hyposegmented neutrophil
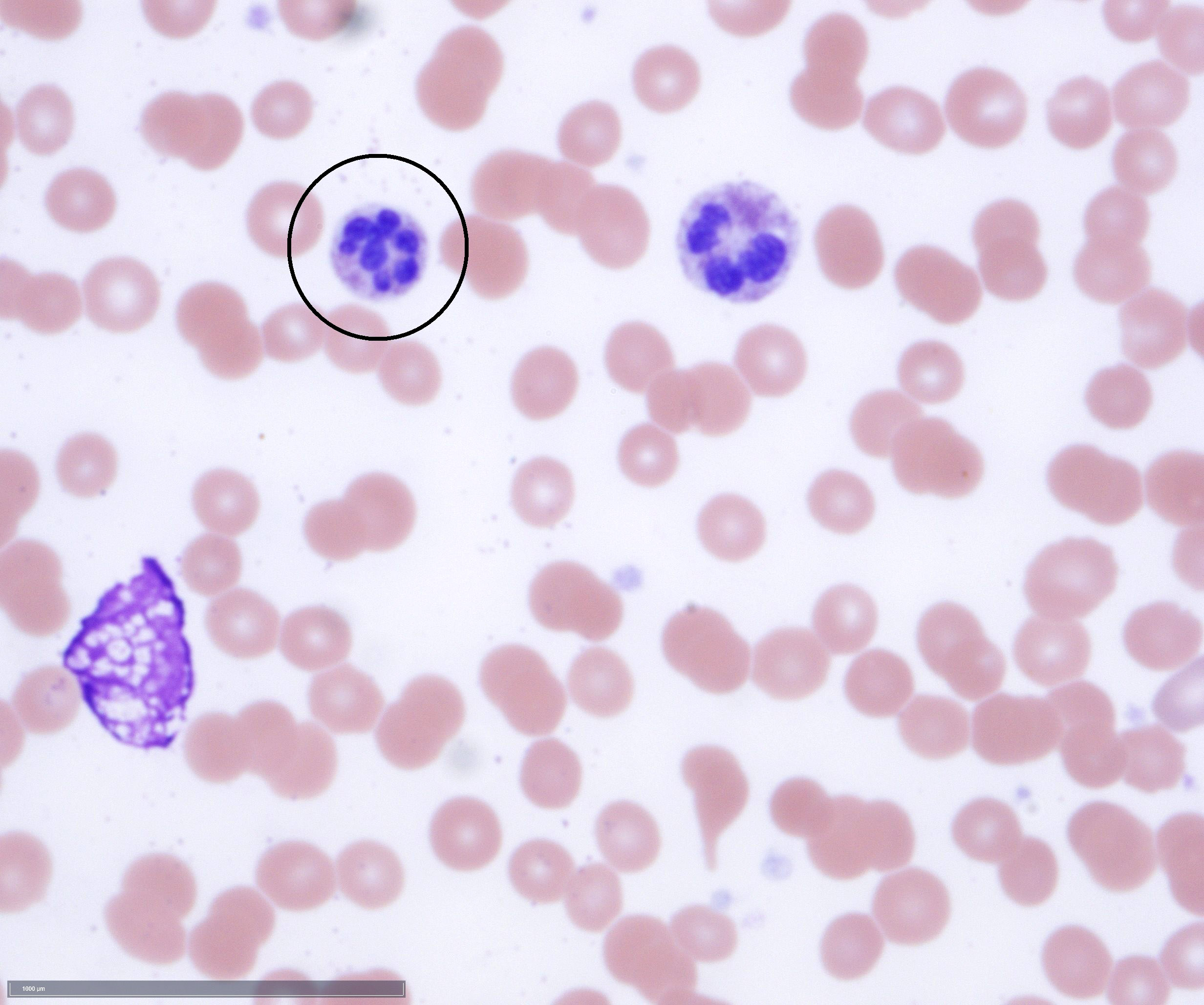
Hypersegmented neutrophil

Hypogranular neutrophil
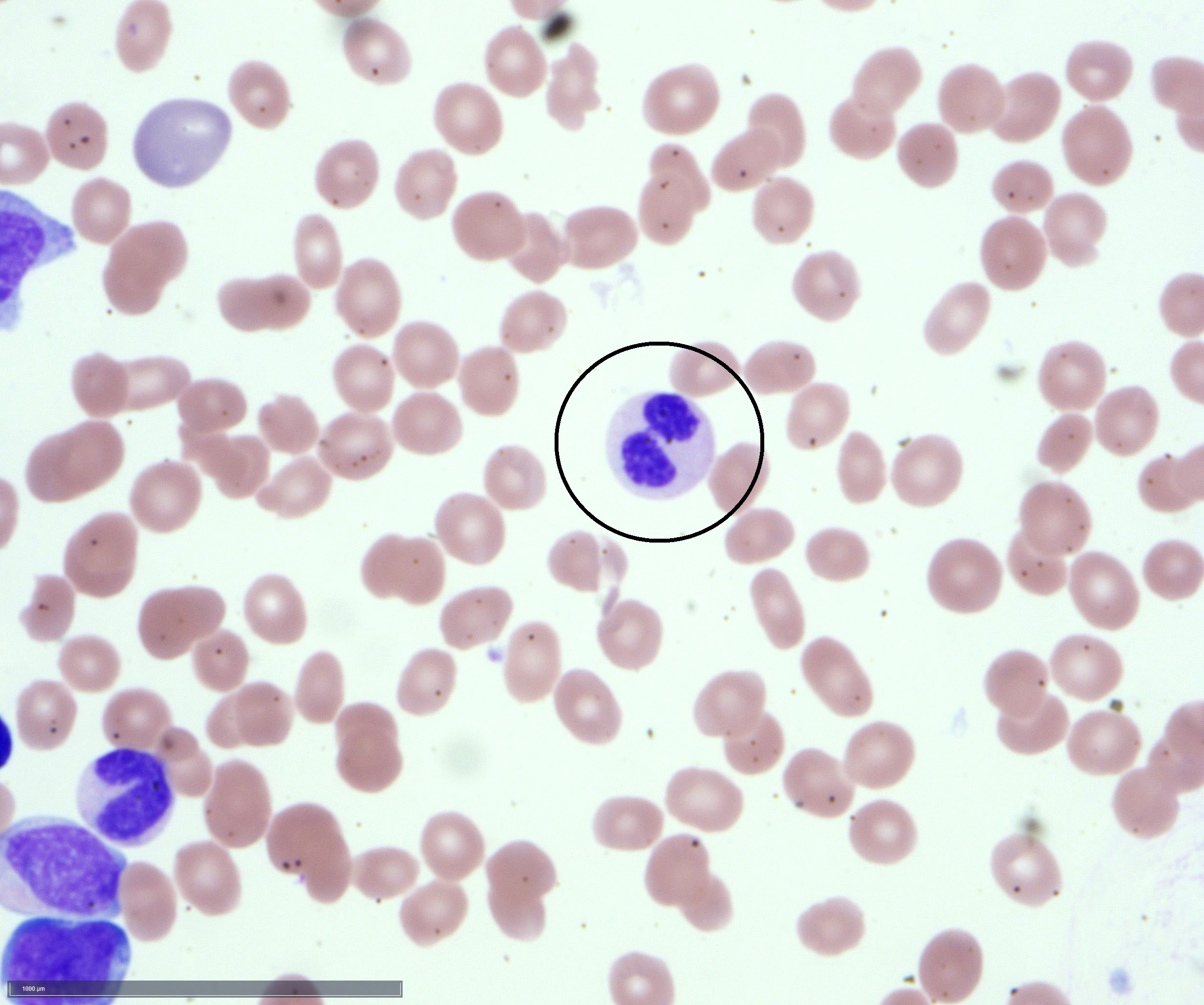
Hypogranular and hyposegmented neutrophil
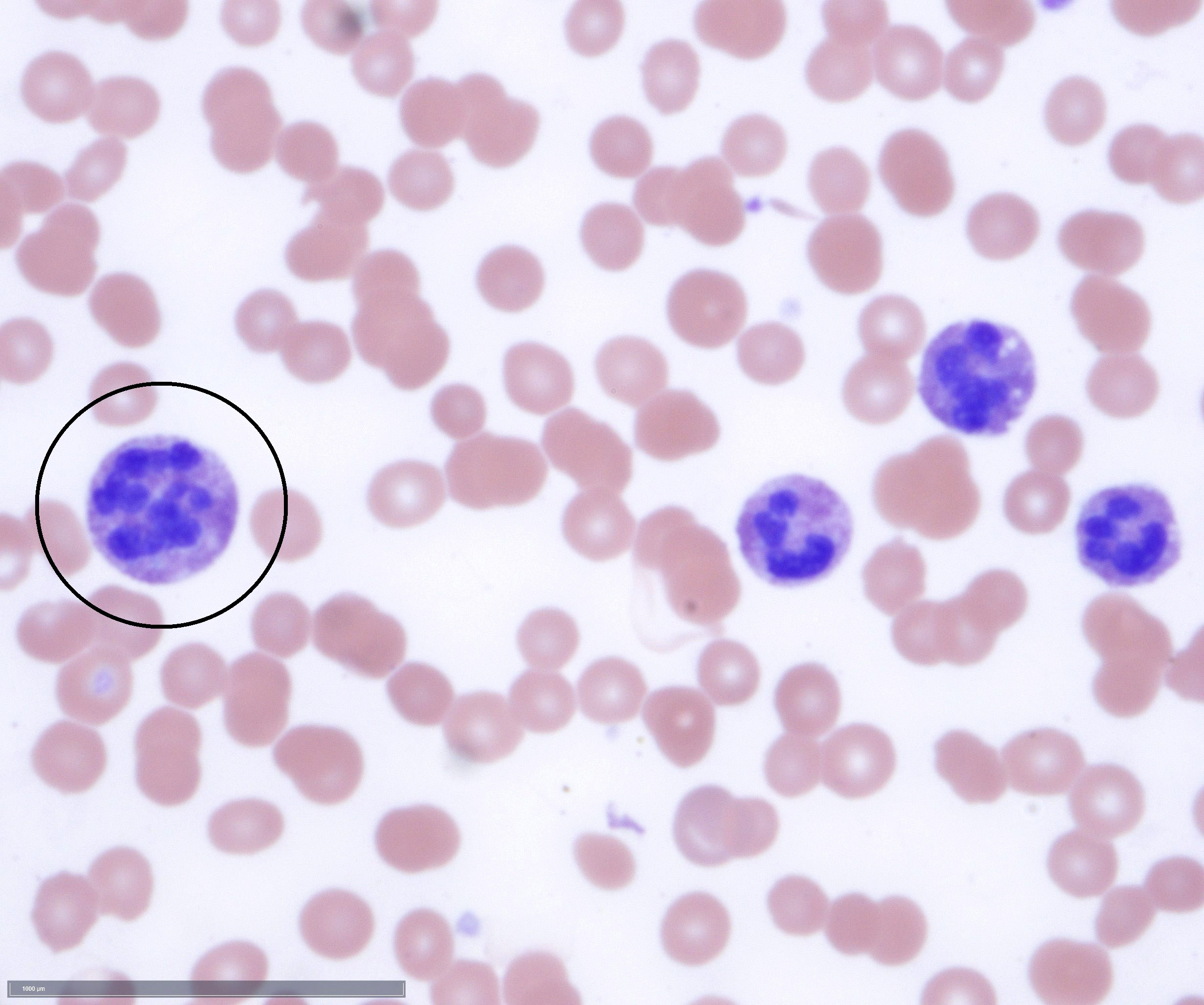
Neutrophil that is larger in size
- CD34 and CD117 can be used to enumerate blasts in the tissue sections
- CD42b, CD61 and factor VIII can be used to evaluate for small megakaryocytes and atypical clustering in the tissue sections
- Reticulin and trichrome are used to evaluate for fibrosis
- PAS positivity in the erythroids is a feature of dysplasia
- Prussian blue iron stain is used to visualize ring sideroblasts
- There are no definitive diagnostic flow cytometry features that are seen in MDS
- Decreased side scatter in the granulocytes are suggestive of hypogranularity, which is a feature of granulocytic dysplasia (Front Oncol 2016;6:161)
- CD34 and CD117 can be used to enumerate cases with increased blasts; the blasts can have aberrant expression of lymphoid antigens (e.g., CD2, CD56, etc.) (Oman Med J 2021;36:e218)
- Granulocytes can have dyssynchronous expression patterns of immature and maturate antigens, particularly on neutrophils (CD10, CD11c, CD13, CD15, CD33) and monocytes (CD4, CD11b, CD11c, CD14, CD16, CD64) (Front Oncol 2017;7:270)
Contributed by Brenda Mai, M.D.
Decreased side scatter
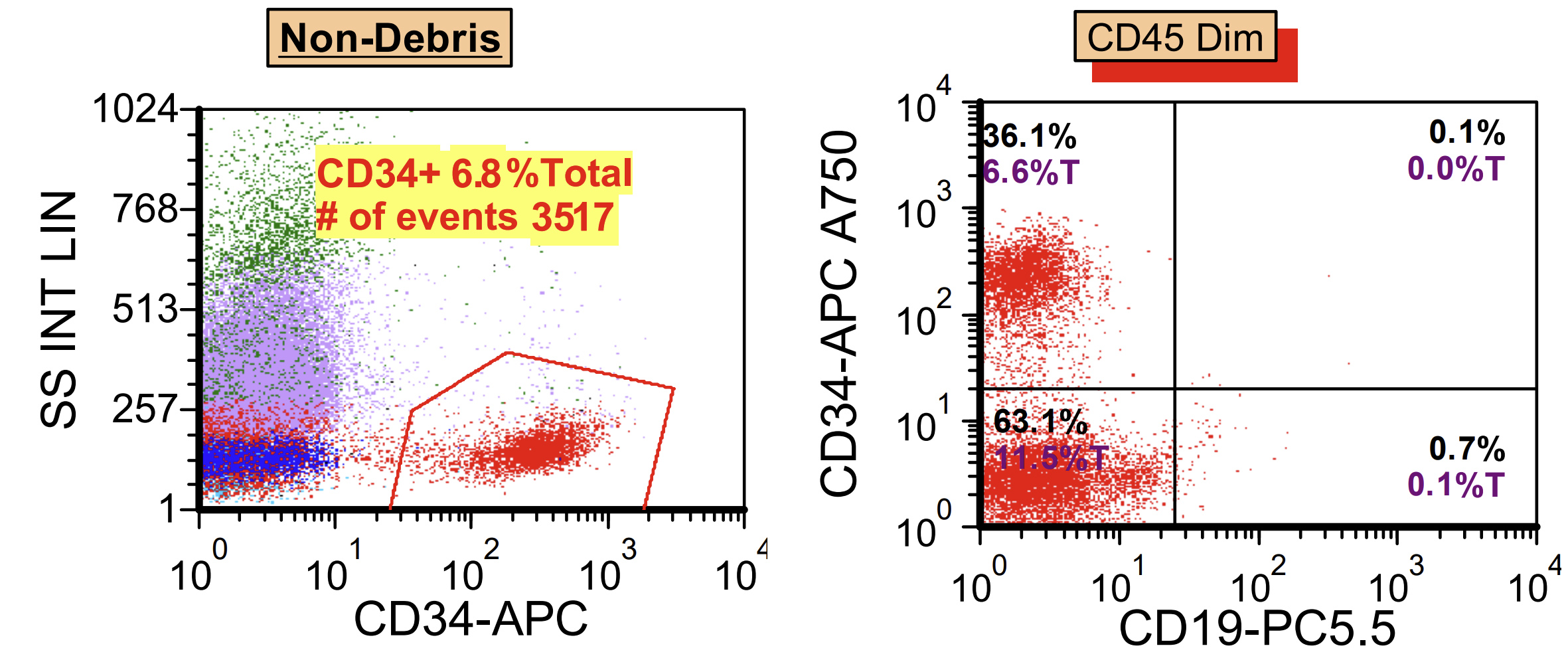
Increased blasts
- Chromosomal abnormalities have been seen in at least 50% of MDS patients, including the abnormalities of chromosome 5, 7, 8 and 20; complex karyotypes are also identified (Blood 2008;111:1534)
- Genetic abnormalities have been identified in over 50 genes in at least 80% of MDS patients, including SF3B1, SRSF2, U2AF1, ZRSR2, LUC7L2, DDX41, ASXL1, EZH2, TET2, DNMT3A, IDH1 / IDH2, CSNK1A1, NRAS, STAG2, RUNX1, BCOR, TP53 and CUX1 (Eur J Haematol 2015;95:3)
- Cases of childhood MDS can been associated with SETBP1, GATA2, PTPN11, RAS and SAMD mutations (Nat Commun 2017;8:1557)
Contributed by Brenda Mai, M.D.

Complex karyotype

Karyotype with deletion 5q
FISH with deletion 5q
Updates in myelodysplastic syndromes
- Bone marrow, biopsy:
- Hypercellular (80%) bone marrow with dyserythropoiesis and ~12% blasts (see comment)
- Comment: Bone marrow aspirate smears show erythroid dysplasia and a mild increase in blasts, constituting ~10% of the total nucleated cells on the aspirate smear. The bone marrow core biopsy and clot section are hypercellular for age with increased scattered blasts highlighted by CD34 immunostain. Flow cytometric analysis shows increased abnormal myeloblasts that constitute about 11.3% of total events with a normal immunophenotype (CD13+, CD33+, CD34+, CD38+, CD117+, MPO+). These findings are compatible with a myelodysplastic neoplasm with increased blasts (MDS IB2); however, correlation with karyotype, FISH and NGS studies is required for complete evaluation.
- Vitamin B12, folate and copper deficiency:
- Can lead to ineffective hematopoiesis and may mimic some features of MDS
- Correlation with laboratory values, cytogenetics and molecular studies are required (Proc (Bayl Univ Med Cent) 2019;32:589, Case Rep Hematol 2021;2021:9661765)
- Recent chemotherapy or radiation therapy exposure:
- Can cause bone marrow suppression, cytopenias and dysplastic changes, which may resemble MDS
- Close examination of the patient's recent medication and radiation exposure is required (Leukemia 2021;35:835)
- Acute myeloid leukemias:
- Can have accompanying dysplasia; however, they tend to have a higher blast count (> 20%) than MDS
- In some cases of AML with defining genetic abnormalities (e.g., PML::RARA, RUNX1::RUNX1T1, NPM1, etc.), there may be less than 20% blasts and the identification of the defining genetic abnormality by karyotype, FISH or molecular studies precludes diagnosis of MDS (Blood Res 2020;55:S1)
- Myeloproliferative neoplasms (e.g., primary myelofibrosis) and myelodysplastic / myeloproliferative neoplasms (e.g., chronic myelomonocytic leukemia [CMML]):
- Can have atypical or dysplastic findings, resembling MDS
- These cases usually have a cytosis instead of a cytopenia (Am J Hematol 2022;97:352)
- Autoimmune disorders, such as systemic lupus erythematosus (SLE) or rheumatoid arthritis:
- Can cause autoimmune mediated bone marrow suppression, leading to cytopenias and dysplastic changes (Ann Hematol 2018;97:2015)
- Aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) or Fanconi anemia:
- Can mimic MDS-h
- Flow cytometry can be used to rule out PNH
- Molecular testing is required to differentiate between MDS-h, aplastic anemia and Fanconi anemia (Cancers (Basel) 2021;13:3380)

A 72 year old man presents with fatigue and easy bruising. Complete blood count reveals
- Hemoglobin: 8.5 g/dL
- Absolute neutrophil count: 3,500/μL
- Platelets: 60,000/μL
Peripheral blood smear shows dysplastic neutrophils and macrocytic red blood cells with occasional circulating nucleated RBCs; circulating blasts are not identified. The bone marrow aspirate smears show erythroid dysplasia with 4% blasts on differential. The marrow cellularity is 60% without evidence of fibrosis. A Prussian blue iron stain is shown above. Which of the following is the most likely MDS subtype given the provided information?
- MDS-f
- MDS-h
- MDS IB2
- MDS SF3B1
Comment Here
Reference: MDS prognostic scoring
- White blood cells: 1,200/μL (neutrophils 500/μL)
- Hemoglobin: 11 g/dL
- Platelets: 150,000/μL
The peripheral blood smear shows 3% blasts. The bone marrow aspirate smears show dysplastic erythroid precursors and 8% blasts. Reticulin shows grade 1 fibrosis. Karyotype analysis shows a normal female. Which of the following is the most likely MDS subtype given the provided information?
- MDS 5q
- MDS biTP53
- MDS-f
- MDS IB1
Comment Here
Reference: MDS prognostic scoring
- Unequivocal dysplasia < 10% in one or more lineage, with cytogenetic abnormalities presumptive of MDS, and < 1% blasts in peripheral blood, 5% blasts in bone marrow
- Refractory anemia or refractory cytopenia with multilineage dysplasia with 1% blasts in peripheral blood
- Unilineage dysplasia with pancytopenia
- Cases can be reclassified into specific subtypes later if characteristic features develop
- Some cases are associated with prior aplastic anemia and monosomy 7 (Am J Clin Pathol 2006;126:925)
- Myelofibrosis: when present, often is difficult to obtain bone marrow aspirate; patients often have pancytopenia with dysplasia in 3 lineages; CD34 staining helps identify blast population
- 82 year old man with moderate thrombocytopenia (University of Pittsburgh Case #520)
AFIP images
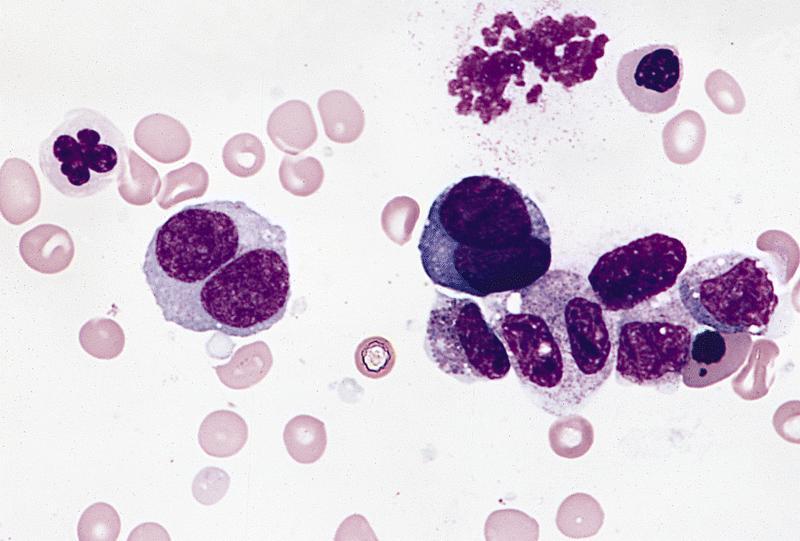
Pancytopenia, abnormally bilobed nuclei in granulocytes

Marked increase in megakaryocytes
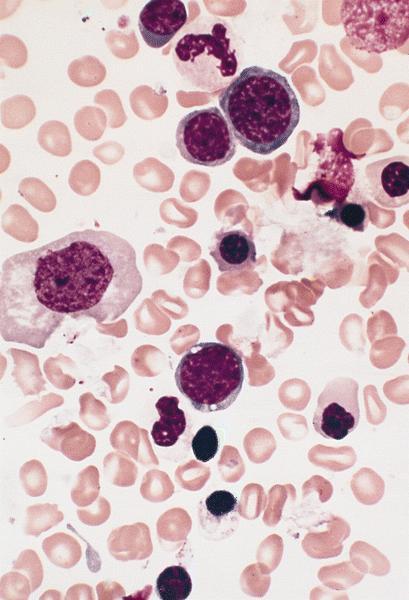
Erythroid hyperplasia
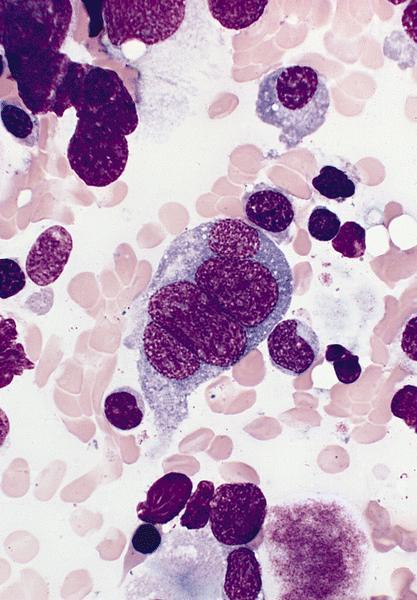
Large erythroid precursors
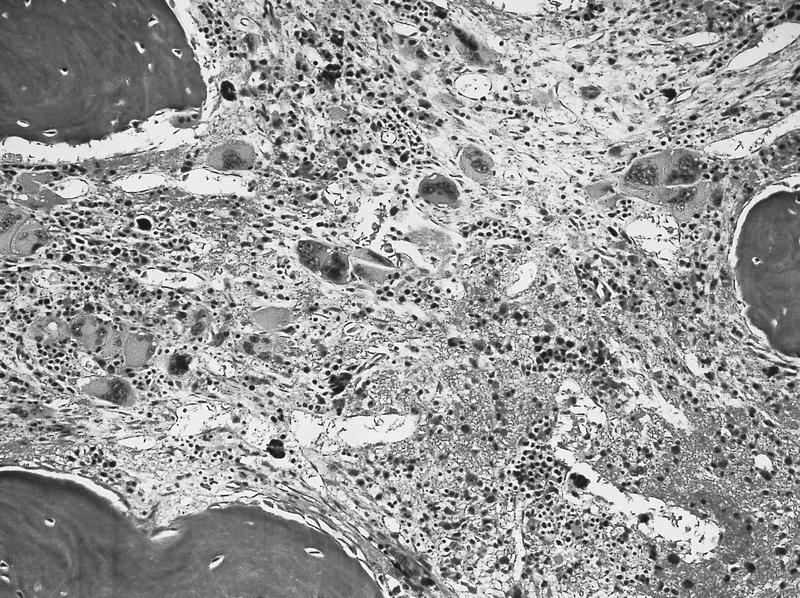
Hypercellular marrow with marked fibrosis
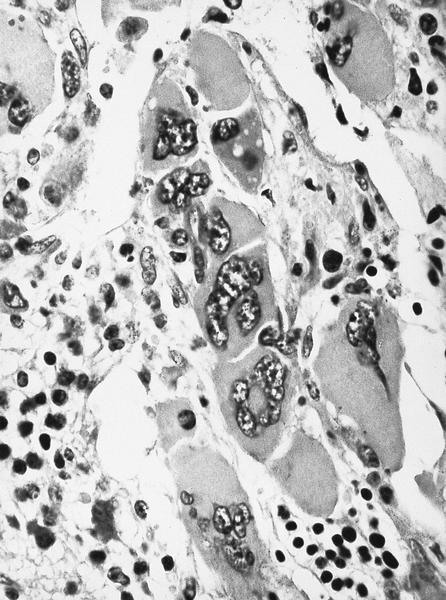
Cluster of large megakaryocytes
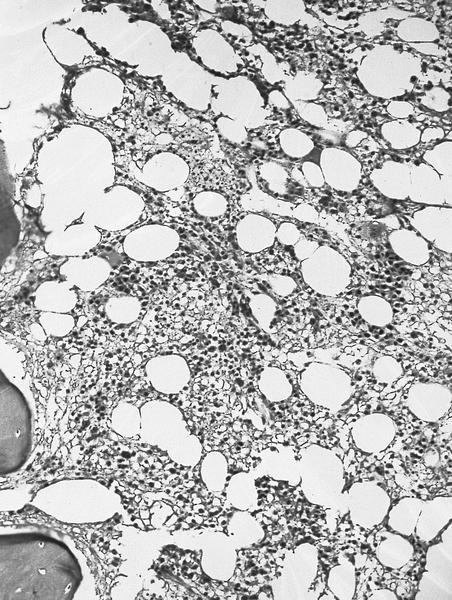
Marked reduction in megakaryocytes
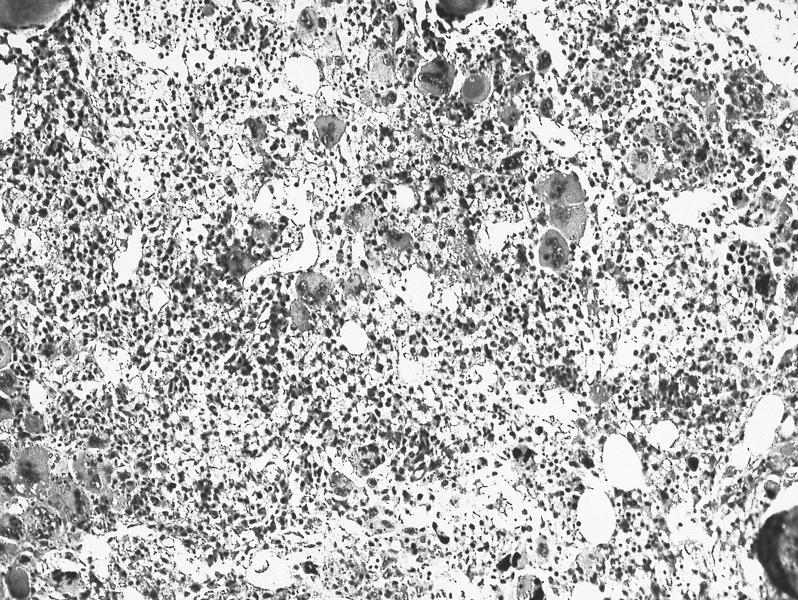
Recurrence with increased marrow cellularity
Images hosted on other servers:

Megakaryocyte clustering and hyperlobulated forms (figs 2 / 3)
- Acute panmyelosis with myelofibrosis (20% or more blasts),
- AML-M7 (20% or more blasts),
- Chronic idiopathic myelofibrosis (usually marked splenomegaly, no dysplasia)
- Myelodysplastic neoplasms (MDS), previously called myelodysplastic syndromes, are clonal hematopoietic stem cell neoplasms characterized by ineffective marrow hematopoiesis that results in morphologic dysplasia and peripheral cytopenia; there is increased risk of transformation to acute myeloid leukemia (AML)
- New MDS subtype per WHO 5th edition and international consensus classification (ICC) 2022
- MDS biTP53 (MDS with biallelic TP53 inactivation) is a new subtype per WHO 5th edition, 2022
- Detection of ≥ 1 TP53 mutation, meeting molecular diagnostic criteria of biallelic TP53 mutations (Nat Med 2020;26:1549)
- 33% of TP53 mutated MDS patients had monoallelic mutations whereas 67% had multiple hits (multihit) consistent with biallelic targeting (Nat Med 2020;26:1549)
- MDS biTP53 is characterized by increased blast counts, higher risk of leukemic transformation and higher risk of mortality
- TP53 mutated myeloid neoplasms (MN) represent a unique entity with very poor prognosis, irrespective of whether the blast percentage indicates MDS or AML or whether the disease is therapy related or de novo in its ontogeny (Blood Adv 2022;6:2847)
- Per the 5th edition of WHO published in 2022, the terminology of myelodysplastic neoplasms has been introduced to replace the previous term of myelodysplastic syndromes, in parallel to myeloproliferative neoplasm and to emphasize the neoplastic course of the disease; however, the abbreviation of MDS is kept for myelodysplastic neoplasm
- ICD-10: D46 - myelodysplastic syndromes
- In the U.S., the age adjusted incidence of MDS is estimated to be 4 in 100,000 individuals (Blood Rev 2019;34:1)
- Incidence notably increases with advancing age, male gender, smoking, obesity and history of radiotherapy or chemotherapy (Blood Rev 2019;34:1)
- Estimated incidence of MDS biTP53 is ~0.03/100,000/year
- ~10% of MDS patients exhibit TP53 alteration, with 67% of these cases involving biallelic alterations (Leukemia 2019;33:1747)
- TP53 mutated MDS has lower response to treatment compared to non-TP53 mutated MDS and an overall survival (OS) of 5 - 10 months with prognosis worsening in biallelic TP53 mutations (Cancer Discov 2022;12:2516, Nat Med 2020;26:1549)
- It is important to note that TP53 has been found to play an important role in both de novo and therapy related MDS; interestingly TP53 mutations were found to be the most common genetic mutation in treatment related MDS (t-MDS) (Nature 2015;518:552)
- History of cytotoxic treatment is a qualifier that moves the diagnosis to myeloid neoplasm postcytotoxic therapy
- Defining characteristic of MDS is bone marrow failure, which occurs as hematopoietic stem cells acquire mutations, resulting in ineffective hematopoiesis, morphological dysplasia and peripheral blood cytopenias (Blood 2019;133:1086)
- TP53 suppressor gene is mutated in about half of human tumors (Nat Med 2020;26:1549)
- TP53 gene encodes for the p53 protein, which plays a crucial role in regulating various cellular processes, such as the cell cycle and cell death, which is usually triggered by oxidative stress (WHO 5); thus, the inactivation of p53 allows tumor initiation and its evasion from cell death (Nat Rev Cancer 2018;18:89)
- It is hypothesized that monoallelic TP53 inactivation may contain biallelic TP53 inactivation subclones that can negatively affect disease outcomes, as patients with single TP53 and VAF of > 23% have similar outcomes to biallelic TP53 patients
- In addition, biallelic inactivation can be also formed from subclonal TP53 mosaicism (Res Sq 2023 Mar 9 [Preprint])
- Patients with TP53 mutations were found to have fewer comutated genes; however, they more commonly exhibit del(5q) chromosomal abnormality, monosomal karyotype and complex karyotype (Leukemia 2019;33:1747)
- Neurofibromin 1 (NF1), which is a negative regulator of Ras signaling, and del(12p), which includes ETV6, were found to be prevalent in TP53 inactivation AML / MDS cases (Blood Adv 2023;7:4586)
- TP53 mutated MN represents a unique entity with very poor prognosis, irrespective of whether the blast percentage indicates MDS or AML or whether the disease is therapy related or de novo in its ontogeny (Blood Adv 2022;6:2847)
- Current assignment of these cases into MDS, AML with myelodysplasia related changes (AML MRC) or therapy related myeloid neoplasms (t-MN) appears to divide a biologically similar disease into different diagnostic groups
- TP53 mutations play an important role in the pathogenesis of many cases of t-AML / t-MDS (Nature 2015;518:552)
- Loss of TP53 provides a selective advantage for neoplastic growth; TP53 mutations are resistant to chemotherapy and expand preferentially after treatment
- Variable and includes
- Idiopathic
- Exposure to certain DNA damaging agents such as chemotherapy or radiation
- TP53 mutations were found to be more common in t-MN, which was also a poor diagnostic factor (Leukemia 2019;33:2842)
- Chemotherapy leads to selective expansion of the progenitor cells with TP53 mutations that are resistant to treatment (Nature 2015;518:552)
- Inherited genetic disorders
- E.g., Li-Fraumeni syndrome or inherited bone marrow failure syndromes
| WHO 4R | WHO 5th | ICC classification |
|
|
|
| ICC classification of MDS with mutated TP53 | |
| MDS with mutated TP53 | AML / MDS with mutated TP53 |
|
|
| WHO 5th edition, 2022 | ICC 2022 | |
| Morphologically defined | ||
| Lineage | Subclassification using dysplastic lineages removed, replaced with MDS LB (MDS with low blasts) | MDS, NOS SLD MDS, NOS MLD |
| Excessive blast counts in bone marrow (5 - 9%) or in peripheral blood (2 - 4%) | MDS IB1 | MDS EB (blast count 5 - 9% in bone marrow, 2 - 4% in peripheral blood) |
| Excessive blast counts in bone marrow (10 - 19%) or in peripheral blood (5 - 19%) | MDS IB2 | MDS AML (blast count 10 - 19% in bone marrow and 2 - 4% in peripheral blood) |
| Others | cMDS (MDS of childhood) MDS-h (hypoplastic MDS) MDS-f (MDS with fibrosis, blasts 5 - 19% in bone marrow, 2 - 19% in peripheral blood) | MDS, NOS (without dysplasia) |
| Defining genetic abnormalities | ||
| SF3B1 mutation | MDS SF3B1 (MDS with low blasts and SF3B1 mutation or MDS with ring sideroblasts if SF3B1 wild type) | MDS SF3B1 (MDS with mutated SF3B1) or MDS, NOS (with ring sideroblasts and SF3B1 wild type) |
| del(5q) | MDS 5q (MDS with low blasts and 5q deletion) [5q deletion alone or with 1 other genetic aberration other than del(7q) / -7] | MDS with del(5q) [del(5q) is isolated or with up 1 genetic aberration except del(7q) / -7 or multihit TP53] |
| TP53 | MDS biTP53 (MDS with biallelic TP53 inactivation) | MDS with mutated TP53 Multihit TP53 Includes MDS, MDS / AML, AML |
- Multihit TP53 is approximated by (Blood Adv 2022;6:2847, Nat Med 2020;26:1549)
- Next generation sequencing analysis or Sanger sequencing covering at least exons 4 - 11 of TP53 gene is required for detection of biallelic TP53 alterations, coupled with a technique to detect copy number status, usually fluorescence in situ hybridization (FISH) with a probe set specific for the TP53 locus on 17p13.1 (Leukemia 2014;28:241, Blood 2013;122:3616)
- Detection of ≥ 2 TP53 mutations, usually affecting both alleles that can be considered multihit status (Nat Med 2020;26:1549)
- After exclusion of constitutional changes, a TP53 VAF > 49% may be regarded as presumptive (not definitive) of copy loss on the trans allele or copy neutral LOH
- In the presence of one TP53 mutation, evidence of TP53 copy loss or copy neutral LOH is required as concurrent 17p LOH, suggestive of biallelic TP53 alterations (Nat Med 2021;27:1239, Nat Commun 2020;11:4980)
- TP53 mutations are typically associated with complex karyotype; these 2 features are associated with unfavorable risk (N Engl J Med 2016;375:2023, Nat Med 2020;26:1549)
- Up to 55% of patients with complex karyotype (CK) (> 3 cytogenetic abnormalities) harbor a TP53 mutation (Leukemia 2019;33:1747, Blood Adv 2022;6:2847)
- Complex karotype TP53 mutated MDS has been shown to predict poor prognosis irrespective of blast percentage and other TP53 molecular characteristics (Blood Adv 2022;6:2847)
- Presence of TP53 mutation is an independent impact on prognosis that is as significant as severe anemia and more significant than having a bone marrow blast proportion of 10 - 29% (Leukemia 2019;33:1747)
- To assess patients with TP53 mutations, research has shown that relying on VAF estimates is not sufficient for precise assessment of TP53 allelic state and recommends other approaches for confirmation (Nat Med 2020;26:1549)
- Factors associated with significant decrease in survival in MDS patients with TP53 mutations include (Leukemia 2019;33:1747)
- Severe anemia (hemoglobin < 8.0 g/dL), NRAS mutation, SF3B1 mutation, TP53 mutation, elevated blast percentage (> 10%), abnormal 3q, abnormal 9 and monosomy 7 as having the greatest survival risk
- Monosomal karyotype, high complexity and TP53 mutation are individually associated with shorter overall survival (Leukemia 2019;33:1747)
- Type and abundance of TP53 mutation (VAF) in question may further refine its prognostic impact (Leukemia 2019;33:1747)
- TP53 mutant CK MDS patients also had significantly higher bone marrow blast proportion and lower platelet counts, 2 factors strongly associated with elevated prognostic risk considered by clinical scoring systems like the IPSS-R (Leukemia 2019;33:1747)
- Some studies have shown that the effect of TP53 on survival is significant in both TP53 monoallelic and TP53 multihit (Blood Adv 2022;6:2847)
- No TP53 mutation (median: 33.9 months)
- TP53 monoallelic (median: 12.5 months)
- TP53 multihit (median: 9.4 months)
- No significant influence of TP53 VAF as a continuous variable or using VAF cutoffs (Blood Adv 2022;6:2847)
- Some studies have shown that only MDS with TP53 multihit shows poor outcomes and response to therapy compared to MDS with TP53 monoallelic mutations (Nat Med 2020;26:1549)
- Median OS in MDS TP53 multihit state was 8.7 months
- Median OS in MDS TP53 monoallelic patients was 2.5 years
- Median OS wild type patients was 3.5 years
- Patients with monoallelic TP53 mutations and VAF > 22% (n = 38) had increased risk of death compared to wild type patients; monoallelic TP53 mutations and VAF ≤ 22% (n = 87) had OS similar to wild type patients
- 7 year old boy, 44 year old woman and 47 year old woman with Li-Fraumeni syndrome and MDS (Arch Pathol Lab Med 2010;134:1010)
- 65 year old woman with TP53 germline mutation developed Bowen disease and myelodysplastic syndrome (Intern Med 2005;44:490)
- 76 year old man presented with refractory anemia with ring sideroblasts, i(17q) and TP53 mutation (Cancer Genet Cytogenet 2002;136:86)
- 9 patients with TP53 mutant myelodysplastic syndromes managed by allogeneic hematopoietic stem cell transplantation and decitabine containing preconditioning regimen (Front Oncol 2022;12:928324)
- Risk stratification is essential in the clinical care of patients with myelodysplastic syndromes (Blood 2012;120:2454)
- Types of chromosomal abnormalities present, co-occurring somatic mutations and clinical features all contribute to the actual risk in patients (Leukemia 2019;33:1747)
- Revised International Prognostic Scoring System (IPSS-R) (Blood 2012;120:2454)
- MDS are usually classified according to the IPSS-R into 5 prognostic groups
- Molecular International Prognostic Scoring System (IPSS-M) (NEJM Evid 2022;1:EVIDoa2200008)
- MDS is classified into 6 IPSS-M risk categories with prognostic differences
- Compared with the IPSS-R, the IPSS-M shows better prognostic discrimination
- Patients with TP53 mutations who receive conventional chemotherapy, such as anthracycline based or cytarabine based induction chemotherapy, have poor outcomes; decitabine has been shown to improve overall survival (N Engl J Med 2016;375:2023, Leukemia 2019;33:1747)
- Possible explanation is that standard chemotherapy leads to outgrowth of preexisting TP53 mutated clones rather than creating TP53 mutations (Nature 2015;518:552)
- Blast count must be < 19% in bone marrow
- Bone marrow cellularity is typically hyper / normocellular
- Significant dysplasia in 1 or more of the myeloid lineages
- Dysmegakaryopoiesis
- Dyserythropoiesis
- Dysgranulopoiesis
- Reference: Leukemia 2022;36:1703
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
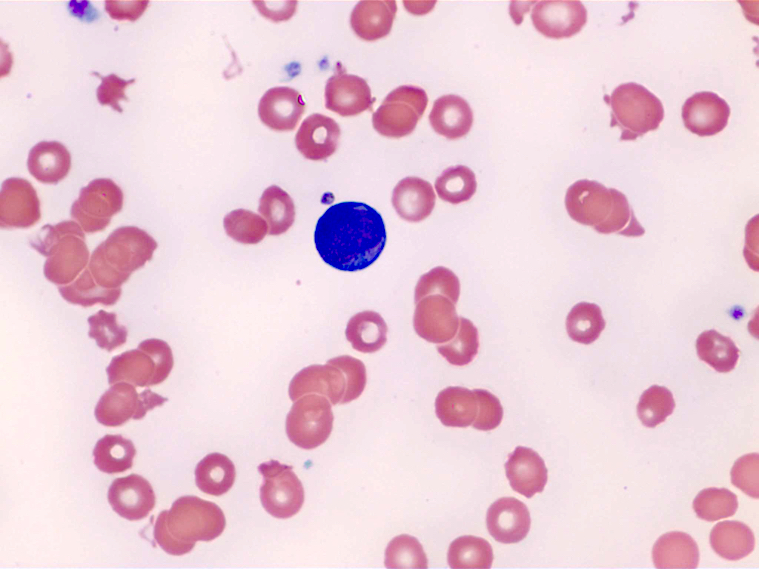
Blast

Bone marrow with increased blasts

Dyserythropoiesis
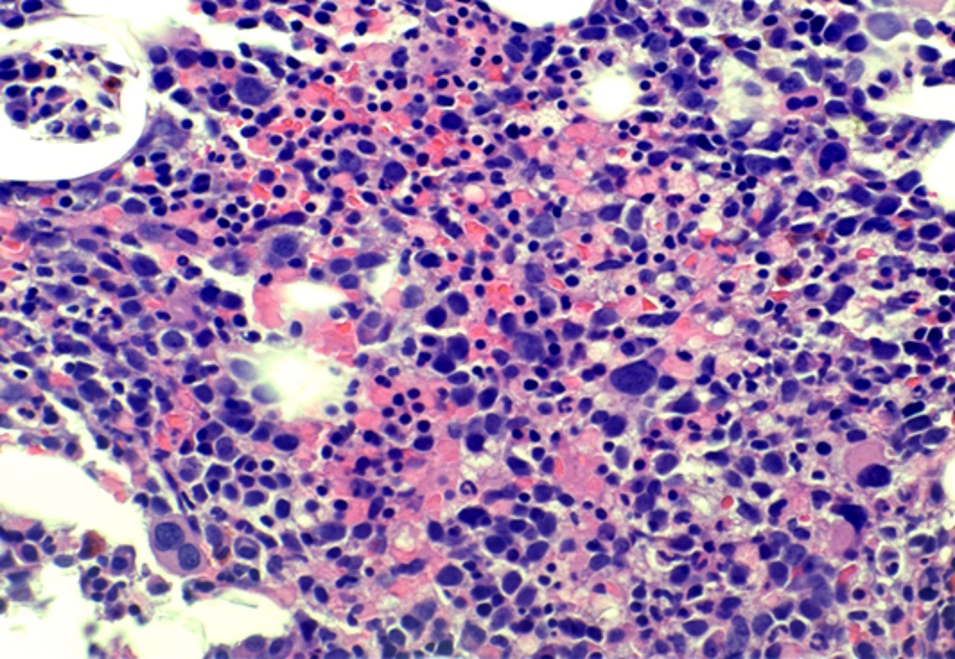
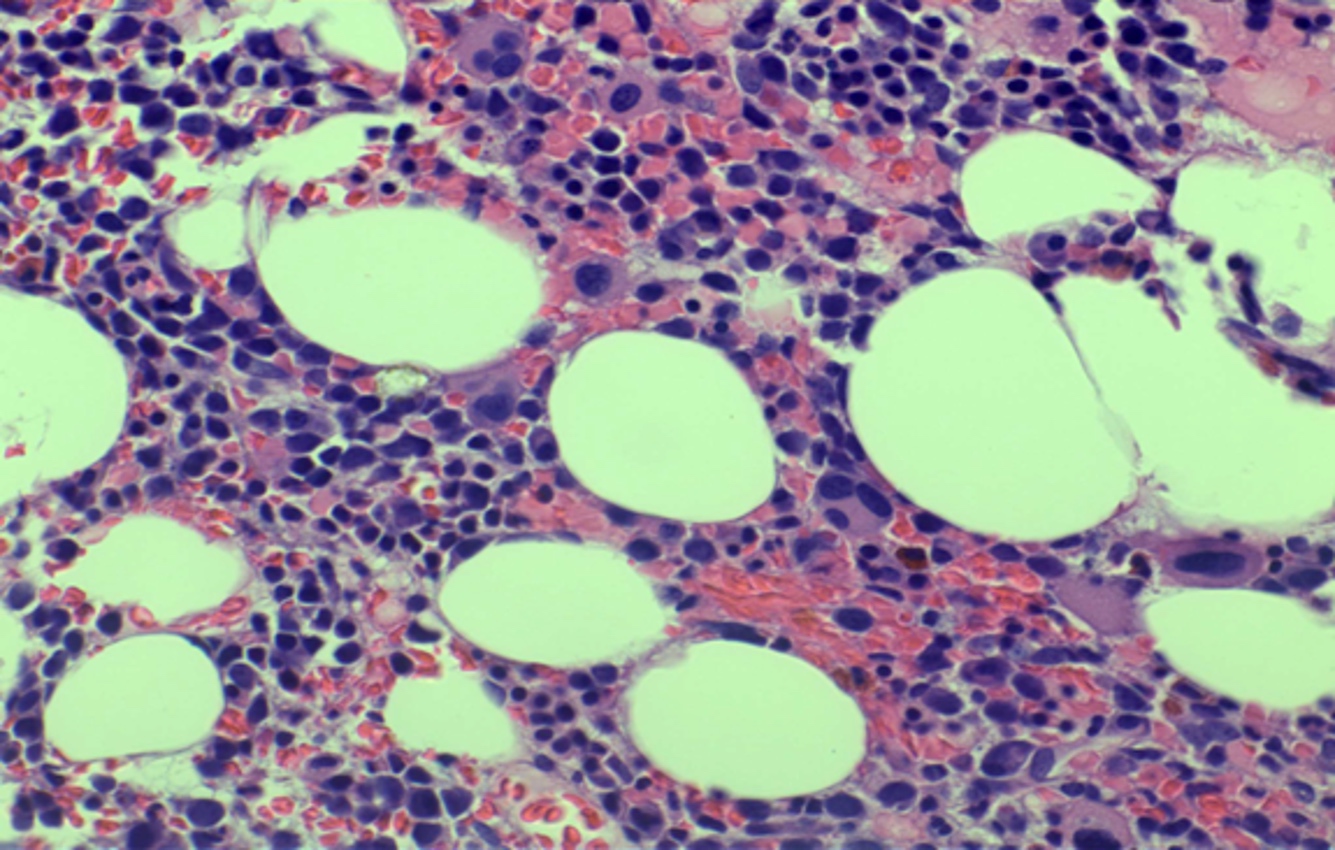
Hypercellular bone marrow with dysplasia

Hypercellular bone marrow with dysplasia
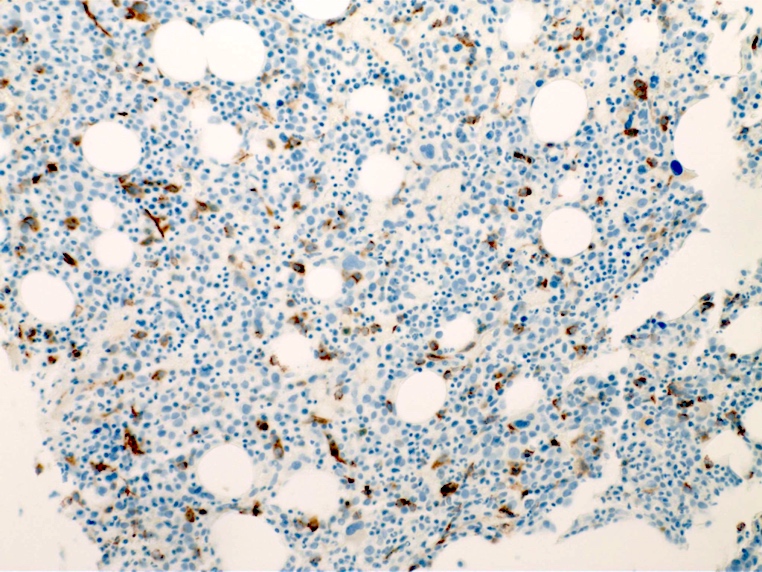
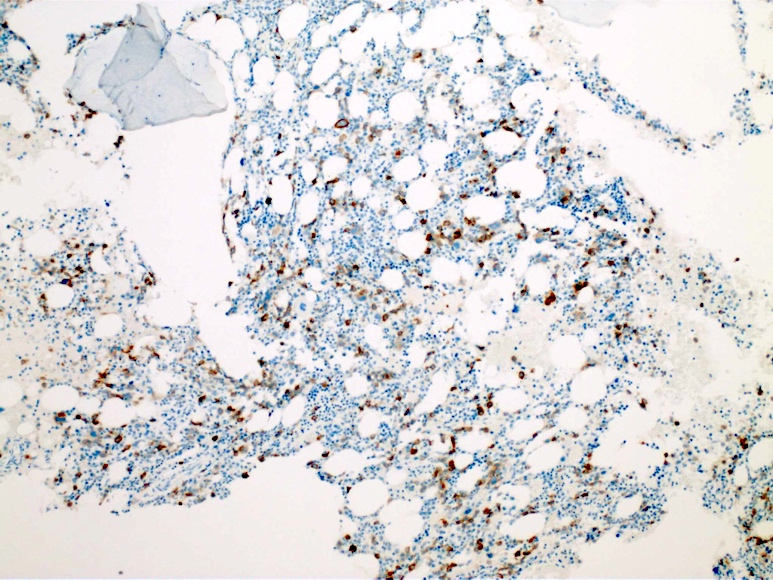
Increased blasts in bone marrow
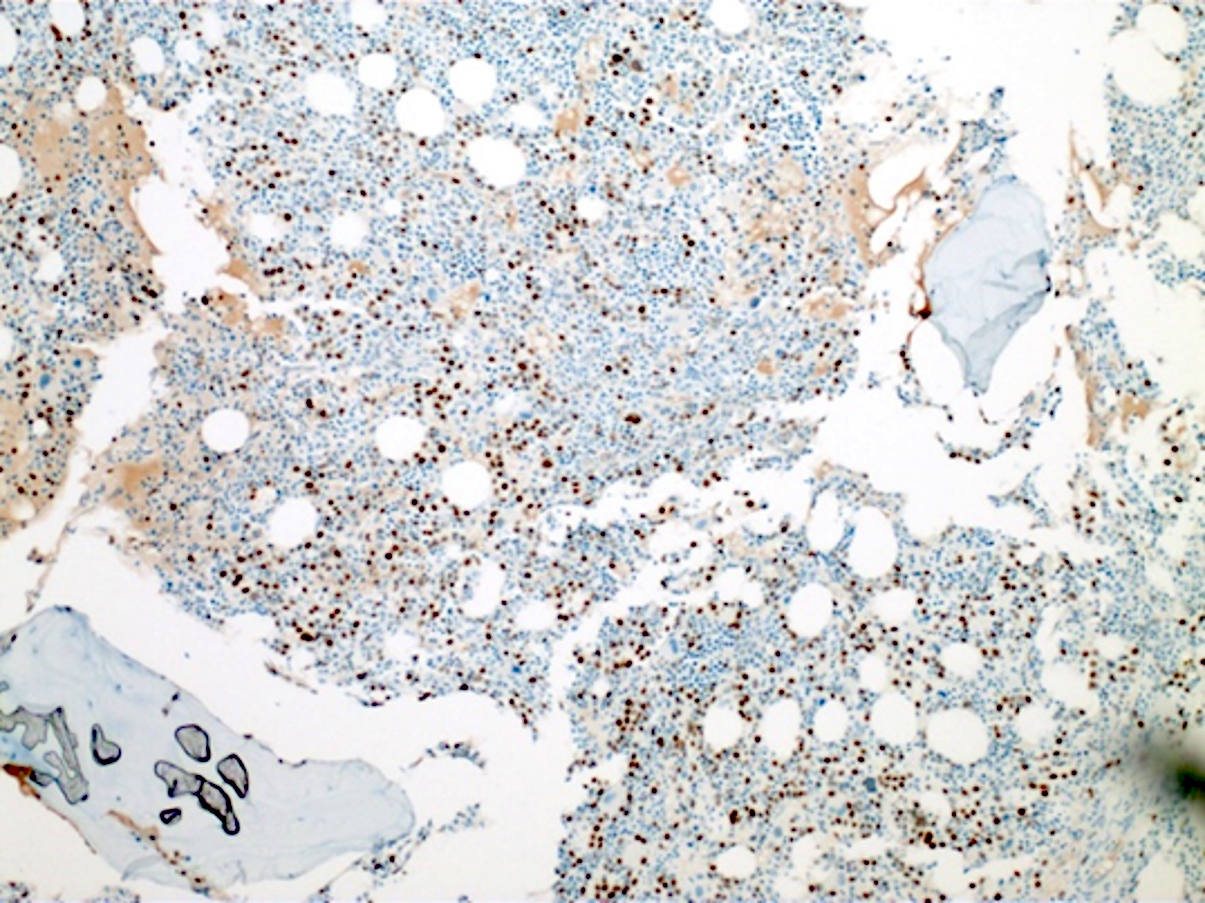
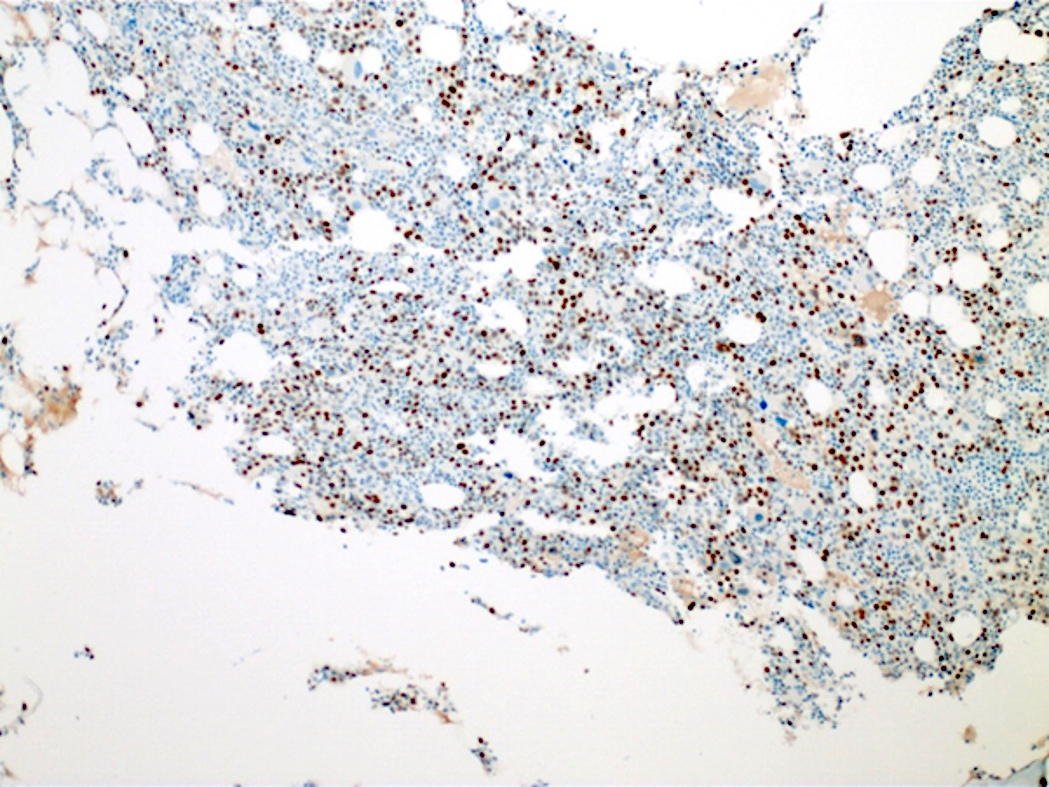
TP53 IHC
- No findings specific to this entity
- Myeloblasts with aberrant phenotype may be detected
- Multihit TP53 is approximated by (Blood Adv 2022;6:2847, Nat Med 2020;26:1549)
- Detection of ≥ 2 TP53 mutations, usually affecting both alleles that can be considered multihit status (Nat Med 2020;26:1549)
- After exclusion of constitutional changes, a TP53 VAF > 49% may be regarded as presumptive (not definitive) of copy loss on the trans allele or copy neutral LOH
- In the presence of one TP53 mutation, evidence of TP53 copy loss or copy neutral LOH is required as concurrent 17p LOH, suggestive of biallelic TP53 alterations (Nat Med 2021;27:1239, Nat Commun 2020;11:4980)
- Bone marrow aspirate, core biopsy, clot section and peripheral blood smear:
- Myeloid neoplasm with multihit TP53 (see comment)
- Comment: This neoplasm meets criteria for MDS with increased blasts 2 (MDS IB2) (WHO 5). The presence of TP53 in MDS supersedes other MDS types in classification; therefore, in conjunction with the reported TP53 mutation (VAF: 87%), it is compatible with myelodysplastic syndrome with biallelic TP53 inactivation (MDS biTP53) (WHO 5) and MDS / AML with mutated TP53 (ICC).
- Associated with 5q loss
- Association with SF3B1 mutation is common
- Favorable outcome among MDS types
- Rarely transforms to acute myeloid leukemia
- TP53 VAF > 49% supports the diagnosis
Comment Here
Reference: MDS with biallelic TP53 inactivation
Which of the following is correct regarding myelodysplastic neoplasm (MDS) with TP53 inactivation / multihit?
- Blast count must be < 10% in bone marrow
- Disease shows specific flow cytometry findings
- Dysmegakaryopoiesis is the most common dysplastic finding
- MDS TP53 monoallelic patients typically show similar prognosis to MDS TP53 multihit patients
- Usually associated with complex karyotype
Comment Here
Reference: MDS with biallelic TP53 inactivation
- Myeloblasts are 5 - 19% of bone marrow differential
- Usually cytopenias in 2 or 3 lineages
- Considered high risk MDS
- Type 1: 5 - 9% blasts in bone marrow or 2 - 4% blasts in peripheral blood, no Auer rods, < 1 billion/L monocytes
- Type 2: 10 - 19% blasts in bone marrow or 5 - 19% blasts in peripheral blood or Auer rods in any MDS, < 1 billion/L monocytes; more aggressive, greater tendency to progress to AML (Am J Clin Pathol 2005;124:191)
- Refractory anemia with excess blasts in transformation (RAEB-T): classified as AML under WHO classification
- Median survival of 16 months for RAEB-1 and 9 months for RAEB-2 (Br J Haematol 2006;132:162)
- Anemia (normochromic, normocytic or macrocytic), usually neutropenia and thrombocytopenia
- 31 year old man with cutaneous involvement (Cutis 2007;80:223)
- 64 year old woman with extensive myocardial infiltration (BMC Blood Disord 2006;6:4)
- Peripheral blood: nucleated red blood cells, immature granulocytes, neutrophilic hyposegmentation, pseudo-Pelger-Huet cells and hypogranulation, myeloblasts 2 - 4% (RAEB-1) or 5 - 19% (RAEB-2), occasional micromegakaryocytes
- Bone marrow: normocellular or hypercellular; hyperplasia of granulocytes or erythrocytes; myeloblasts comprise 5 - 9% (RAEB-1) or 10 -19% (RAEB-2) of white blood cells; Auer rods often seen; severe dysplastic changes in all 3 lineages, more severe than other MDS; abnormal localization of immature precursors (ALIP / clusters or aggregates of blasts located away from bone trabeculae and vascular structures); may have increased reticulin fibers
AFIP images

Blast in center with adjacent neutrophils
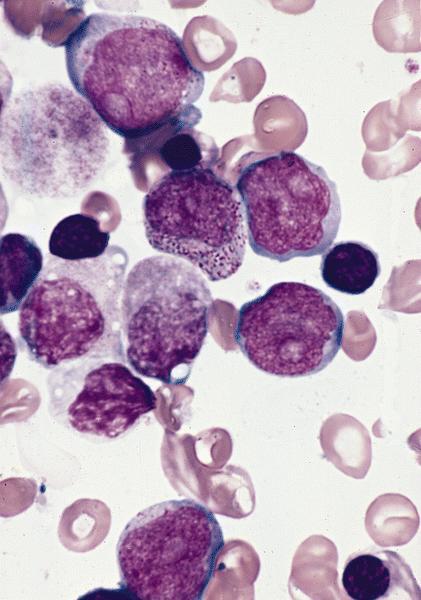
Promyelocytes and myelocytes
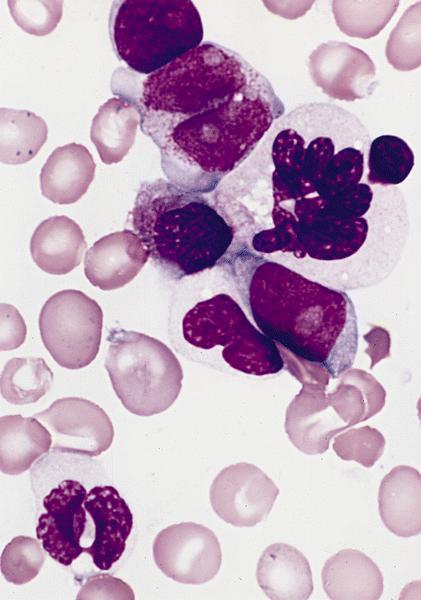
Markedly hypogranular cytoplasm

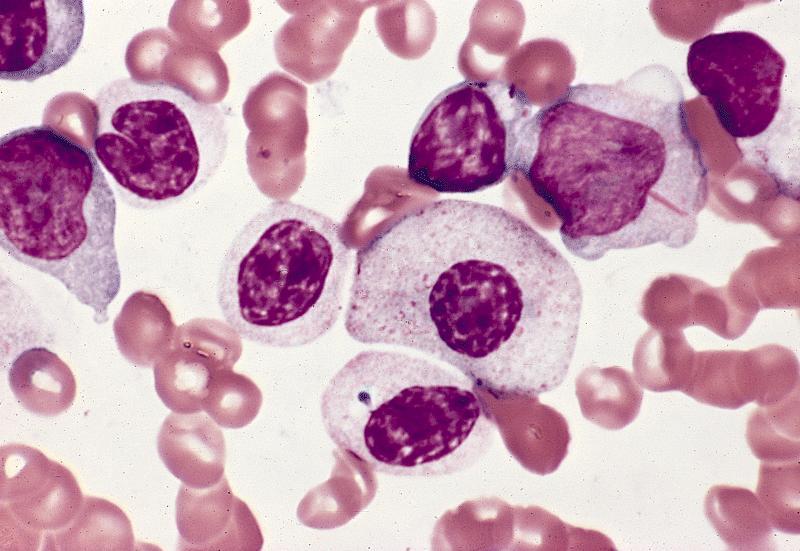
Auer rod present in blast; neutrophils show nuclear hypolobulation
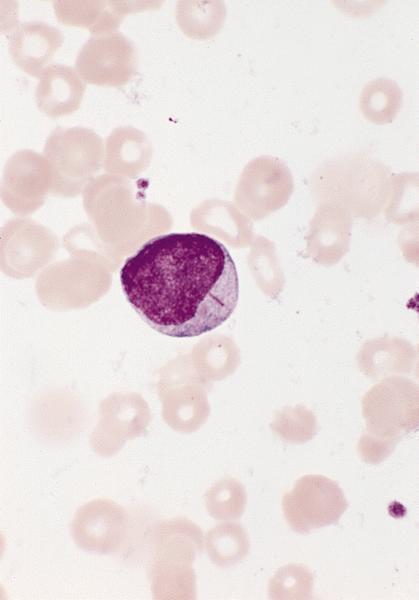
Bone marrow aspirate

Normocellular biopsy
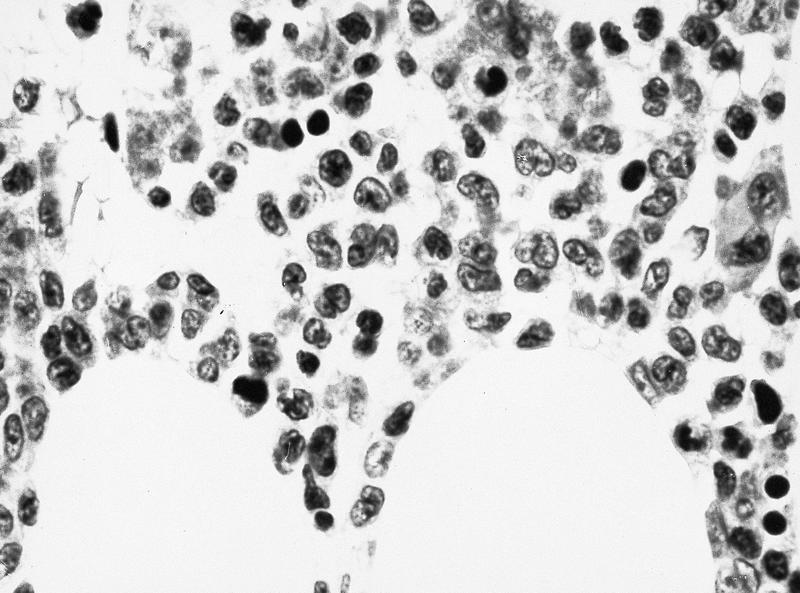
Normal erythroid precursors
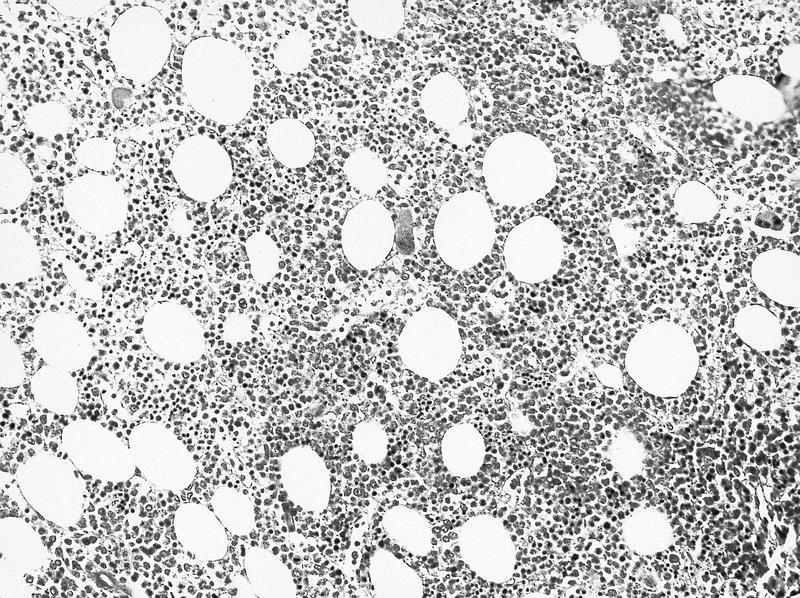
Slightly more cellular marrow
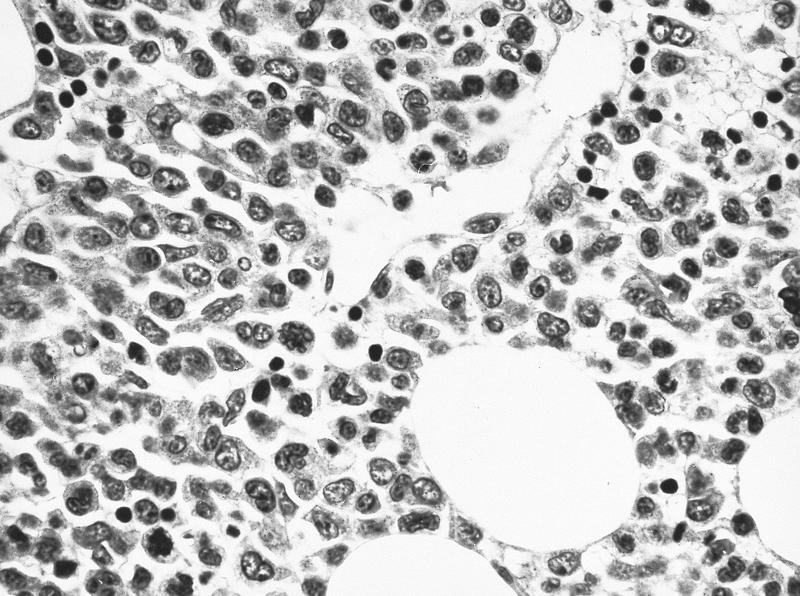
More immature granulocytes

Marked fibrosis with streaming effect
Images hosted on other servers:
RAEB-1
AFIP images

Neutrophil has hypogranular cytoplasm

Hypogranular cytoplasm
Images hosted on other servers:

RAEB-1
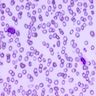
RAEB-2: blood, marrow and myocardial infiltration
- No specific abnormality (Atlas of Genetics and Cytogenetics)
- Copper deficiency: reduced copper levels (Leuk Res 2008;32:495)
- Myelodysplastic syndrome (MDS) with multilineage dysplasia is characterized by 1 or more cytopenias and dysplastic changes in 2 or more of the myeloid lineages (erythroid, granulocytic and megakaryocytic)
- Myelodysplastic syndrome with ring sideroblasts and multilineage dysplasia (MDS RS MLD) if ring sideroblasts are > 15% (> 5% with SF3B1 mutation)
- Dysplastic changes in 2 or more of the myeloid lineages
- Blast counts < 1% in the peripheral blood and < 5% in the bone marrow
- Assess ring sideroblasts (< 15%) and SF3B1 mutation status (if sideroblasts comprise 5 - 15% of erythroid elements) to categorize as MDS MLD or MDS RS MLD
- Monocytes < 1 x 109/L
- Exclusion of other causes of dysplasia
- References: Leukemia 2015;29:66, Leukemia 2007;21:668
- Refractory cytopenia with multilineage dysplasia
- Myelodysplastic syndrome with ring sideroblasts and multilineage dysplasia (MDS RS MLD)
- Median age: 67 - 70 years (M = 70 - 74, F = 75 - 79) (Leuk Res 2012;36:727)
- M > F (Leuk Res 2012;36:727)
- 30% of all cases of MDS (Leuk Res 2013;37:64)
- Bone marrow
- Cell of origin is a hematopoietic stem cell
- Complex interplay between genetic and epigenetic alterations and the bone marrow microenvironment lead to dysregulated hematopoietic differentiation, resulting in impaired differentiation, morphological dysplasia and cytopenia
- Age associated risk attributed to hematopoietic stem cell mutations
- Exposure to toxins (e.g., benzene, cigarette smoking, agricultural chemicals or solvents)
- Family history of hematopoietic neoplasms
- Acquired aplastic anemia
- References: N Engl J Med 2014;371:2477, N Engl J Med 2014;371:2488
Images hosted on other servers:

Pathogenesis of myelodyspastic syndromes
- Unicytopenia, bicytopenia (common)
- Pancytopenia, milder cytopenia (less common)
- Peripheral blood analysis
- Bone marrow examination
- Hemoglobin concentration < 10 g/dL (recommended by 2016 WHO)
- Platelet count < 100 x 109/L (recommended by 2016 WHO)
- Standard hematologic values for cytopenia (hemoglobin < 13 g/dL [males], < 12 g/dL [females] and platelets < 150 109/L) are also used by some authors
- Absolute neutrophil count < 1.8 x 109/L
- Monocytes < 1 x 109/L
- Blasts < 1% in the peripheral blood
- Reference: Blood 2016;128:2096
- Median overall survival 36 months
- Evolution of acute leukemia is ~15% at 2 years and 28% at 5 years
- Revised International Prognostic Scoring System (IPSS-R) scores
- Abnormal findings in flow cytometry analysis (see Flow cytometry description)
- Karyotype
- References: Leuk Res 2013;37:64, J Clin Oncol 2005;23:7594
- 17 year old girl with cri du chat syndrome (Int J Hematol 2020;112:728)
- 64 year old woman with longstanding anemia and fatigue (Intern Med 2017;56:1213)
- 70 year old man with a 6 - 8 year history of anemia and thrombocytopenia (Case Rep Hematol 2017;2017:3625946)
- 70 year old woman with pancytopenia (Case Rep Hematol 2012;2012:167653)
- Transfusion
- Iron chelation
- Erythropoiesis stimulating factors
- Immunosuppressive treatment
- Lenalidomide for lower risk del(5q) MDS
- Luspatercept for MDS with ring sideroblasts
- Azacytidine
- Decitabine
- Induction chemotherapy
- Stem cell transplantation
- Reference: Blood 2013;122:2943
- Bone marrow findings (Leuk Res 2013;37:64)
- General
- Cellularity: majority of cases are normocellular or hypercellular; a subset of MDS-MLD show age adjusted hypocellularity
- Erythroid hyperplasia
- Myeloid hyperplasia with left shift
- Myeloblasts must be < 5%
- Occasional fibrosis (16% of cases)
- Erythroid lineage dysplasia
- Nuclear: nuclear budding, internuclear bridging, karyorrhexis, multinuclearity, megaloblastoid changes
- Cytoplasmic: ring sideroblasts (< 15%, < 5% with SF3B1 mutation)
- Vacuolization
- Myeloid lineage dysplasia
- Small or unusually large size
- Nuclear cytoplasmic asynchrony
- Nuclear hyposegmentation (pseudo Pelger-Huët anomaly)
- Nuclear hypersegmentation
- Decreased granules; agranularity
- Pseudo Chédiak-Higashi granules
- Döhle bodies
- Megakaryocyte dysplasia
- Micromegakaryocytes (most reliable and reproducible)
- Nuclear hypolobation or nonlobation
- Binucleation or multinucleation
- General
Contributed by Julia T. Geyer, M.D. and Shajo Kunnath-Velayudhan, M.B.B.S., M.M.S.T.

Hypercellular bone marrow
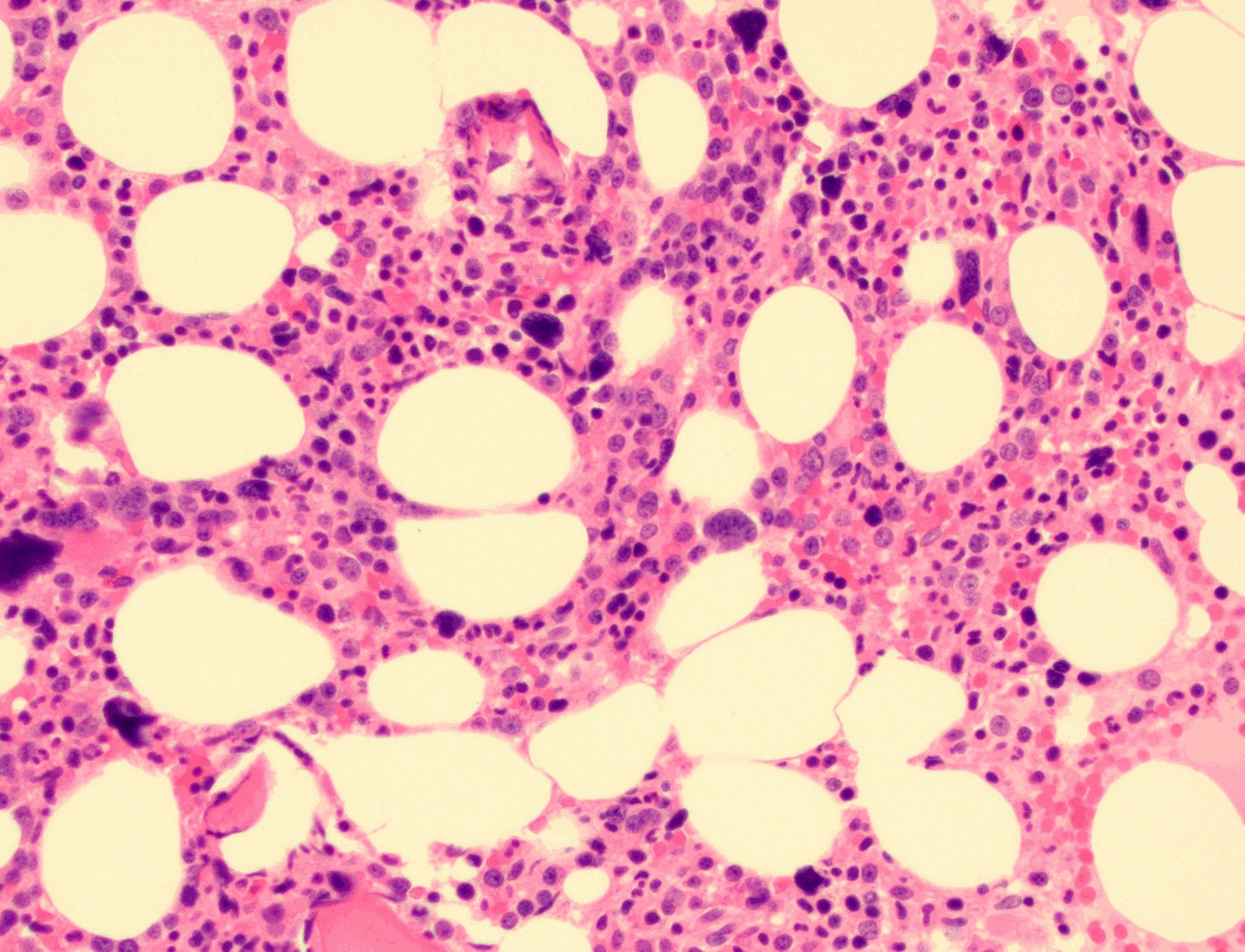
Dysplastic megakaryocytes
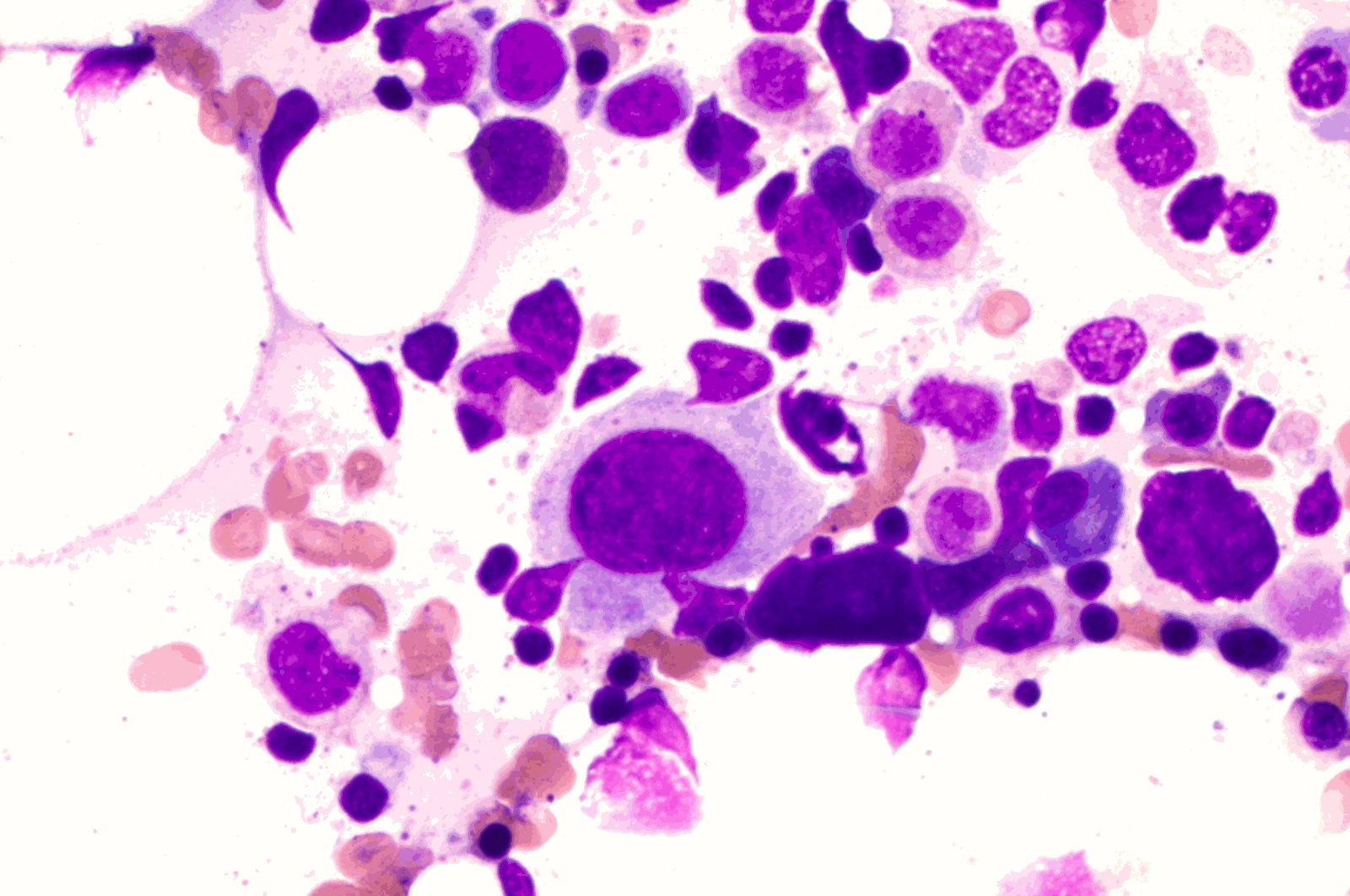
Dysmegakaryopoiesis
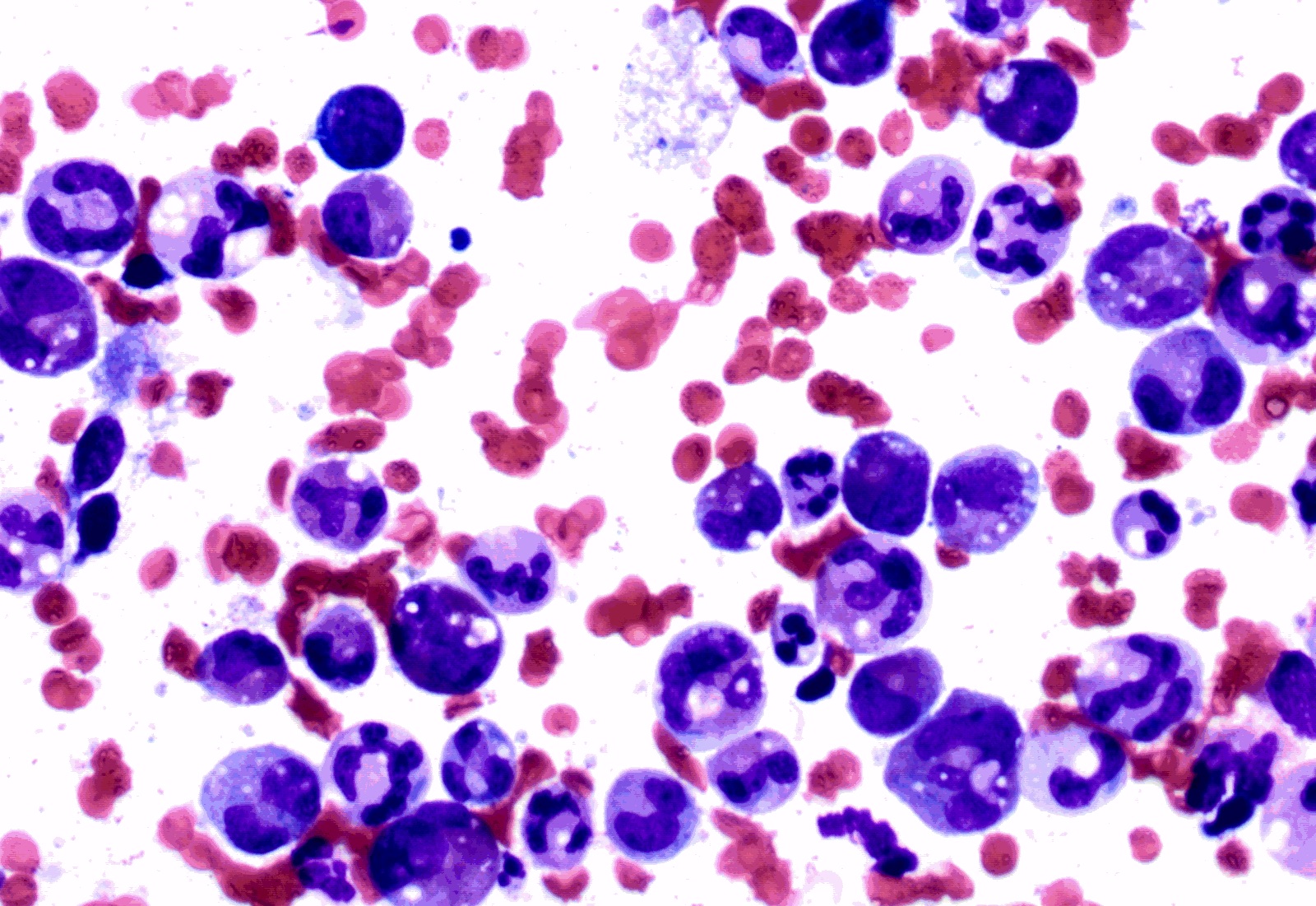
Dysgranulopoiesis
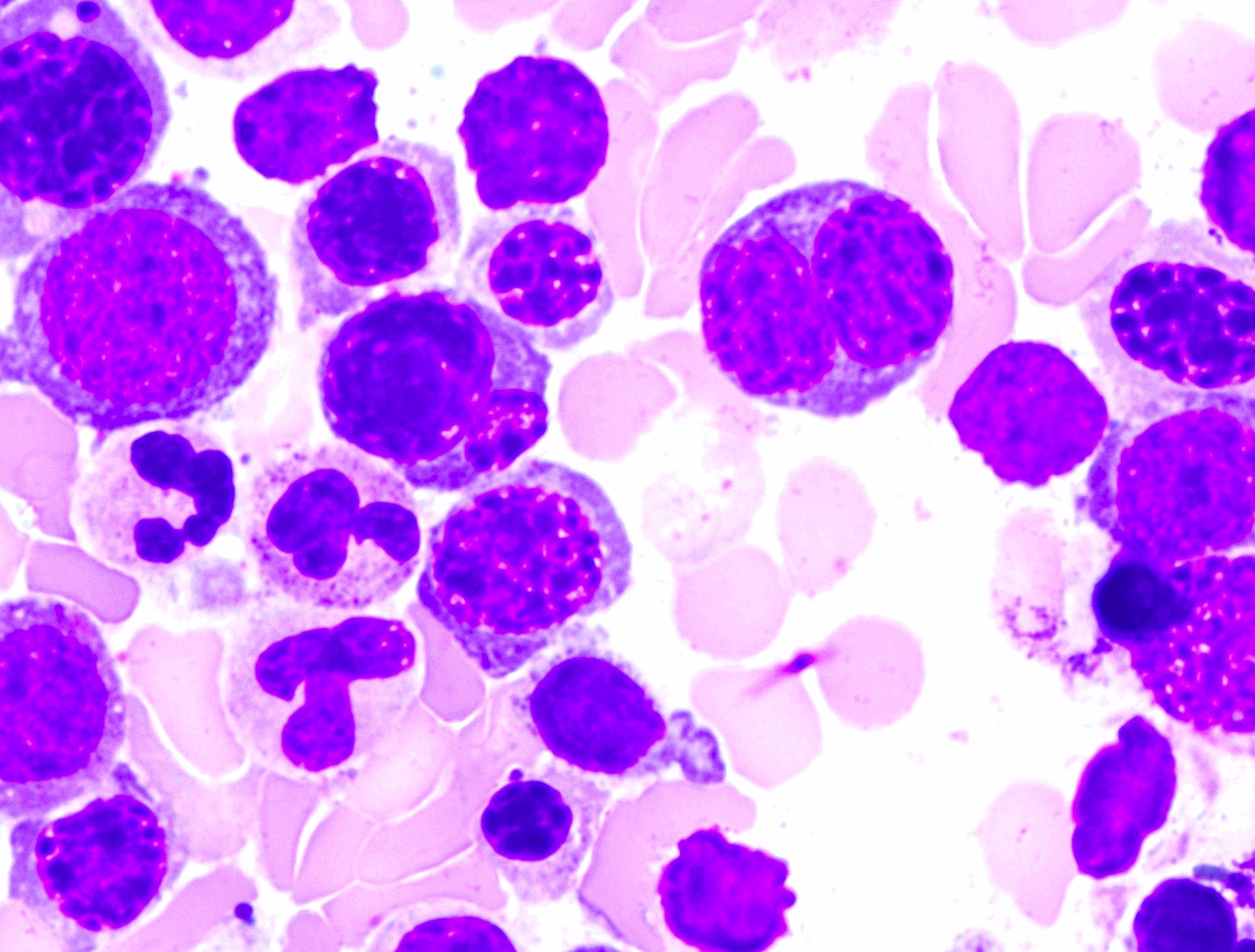
Dyserythropoiesis
- Red blood cells: anisopoikilocytosis with macrocytosis of red blood cells
- White blood cells
- Nuclear clumping
- Hyposegmentation
- Lack of lobation (pseudo Pelger-Huët anomaly)
- Cytoplasmic hypogranularity or agranularity
- Abnormal granular clumping (similar to Chédiak-Higashi syndrome): infrequent
- No increase in blasts (must be < 1%)
- Reference: Haematologica 2013;98:568
Contributed by Genevieve M. Crane, M.D., Ph.D.
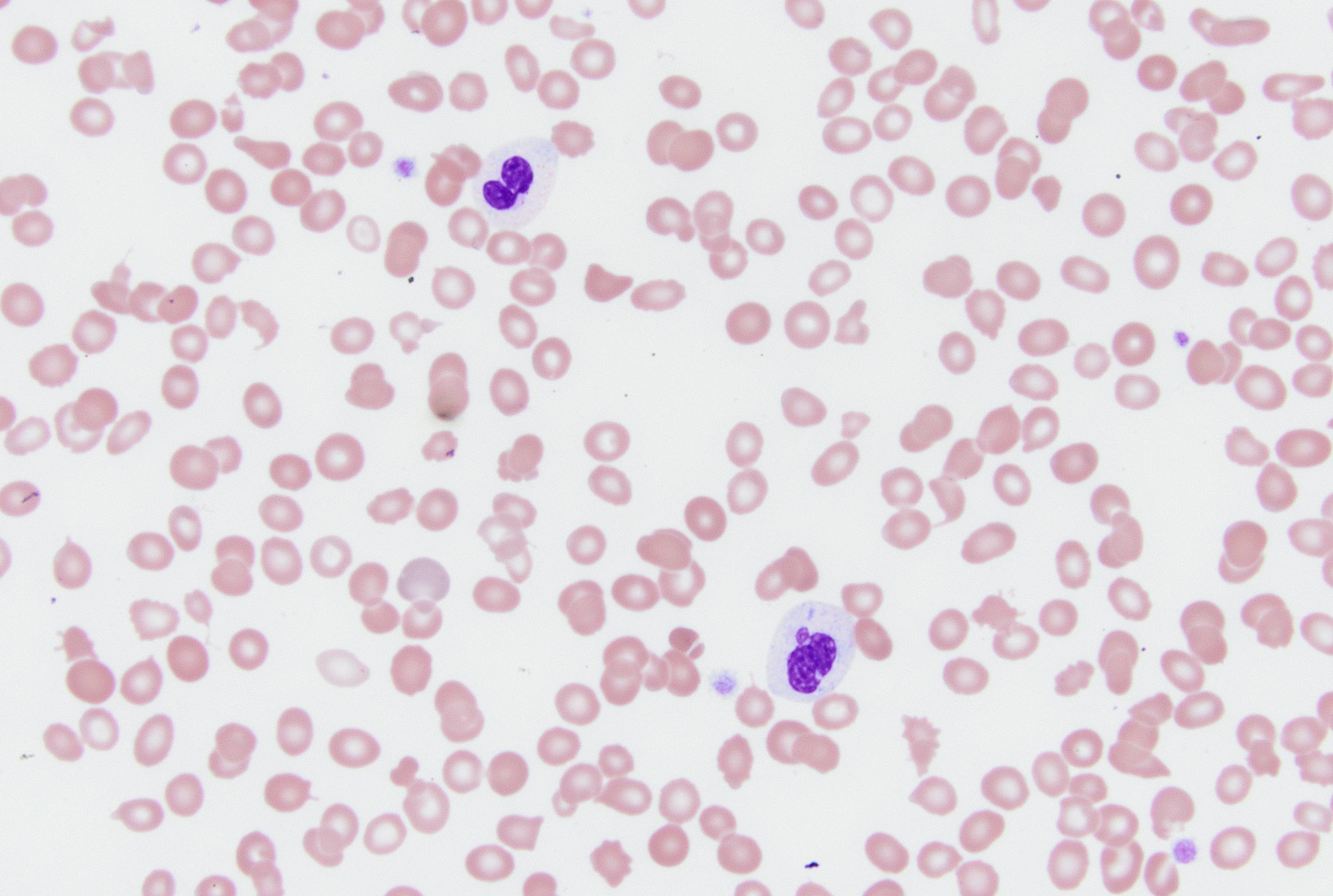
Dysplastic neutrophils
- Flow cytometry findings alone are not diagnostic
- Decreased side scatter of granulocytes
- Erythroid: increased coefficient of variation and decreased intensity of CD71 or CD36
- Aberrant expression of CD56 or CD7 on progenitors, granulocytes or monocytes
- Asynchrony of CD15 and CD16 on granulocytes
- Altered expression of CD13 in relation to CD11b or CD16
- Reference: Leukemia 2014;28:1793
- Cytogenetics
- Trisomy 8
- Monosomy 7
- del(7q)
- Monosomy 5
- del(5q)
- del(20q)
- Complex karyotypes
- Molecular aberrations
- Cohesion family (STAG2)
- Chromatin modifiers (ASXL1)
- Spliceosome genes (SRSF2)
- Transcription factors (RUNX1)
- Signaling molecules (CBL)
- Tumor suppressors (TP53)
- DNA modifiers (TET2)
- SF3B1 (in setting of MDS RS MLD with at least 5% ring sideroblasts)
- Reference: Leuk Res 2013;37:64
What is multilineage dysplasia?
- Bone marrow, posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Myelodysplastic syndrome with trilineage dysplasia
- Flow cytometry: abnormal myeloid blasts present
- Peripheral blood: anemia, moderate, macrocytic
- Cytogenetics: 46,XY,+8[16]/46,XY[4]
- Next generation sequencing:
- SRSF2 P95R with mutant allele frequency of 31%
- ASXL1 E635%fs*15 with mutant allele frequency of 28%
- Secondary causes of dysplasia:
- Nutritional deficiencies, toxic exposures, drugs and biologic agents, infection, congenital disorders and sideroblastic anemias
- History and physical examination
- Appropriate laboratory investigations
- Myelodysplastic syndrome with single lineage dysplasia:
- Only 1 hematopoietic lineage shows dysplasia
- Myelodysplastic syndrome with excess blasts:
- 5 - 19% myeloblasts in the bone marrow or 2 - 19% blasts in the peripheral blood
- Presence of Auer rods in blasts
- Myelodysplastic syndrome, unclassifiable:
- MDS (MDS with excess blasts excluded) associated with 1% blasts in the peripheral blood on 2 separate occasions
- MDS (MDS with multilineage dysplasia excluded) associated with pancytopenia
- Persistent cytopenia with < 2% blasts in the blood and < 5% in the bone marrow and no significant dysplasia in any myeloid lineage and the presence of cytogenetic abnormalities considered presumptive evidence of MDS
A 60 year old man with anemia underwent bone marrow core biopsy (an H&E image is shown). Which of the following is essential to make the diagnosis of myelodysplastic syndrome?
- Absence of secondary causes of dysplasia
- Presence of dysplasia in the erythroid lineage
- Presence of dysplasia in the megakaryocyte lineage
- Presence of dysplasia in the myeloid lineage
- Myelodysplastic syndrome, unclassifiable
- Myelodysplastic syndrome with multilineage dysplasia
- Myelodysplastic syndrome with ring sideroblasts
- Myelodysplastic syndrome with ring sideroblasts and multilineage dysplasia
Comment Here
Reference: MDS with multilineage dysplasia
- Hyperchromatic nuclei
- Micromegakaryocytes
- Multinucleation
- Nuclear hypolobation
- Myelodysplastic syndrome (MDS) characterized by morphological dysplasia with presence of ring sideroblasts (RS); may or may not have an SF3B1 mutation
- Characterized by cytopenias, morphological dysplasia and ring sideroblasts, often with concomitant SF3B1 mutation
- In the absence of SF3B1 mutation, ≥ 15% ring sideroblasts are needed, whereas ≥ 5% ring sideroblasts are required for diagnosis in the presence of SF3B1 mutation
- Ring sideroblasts are erythroid precursors with ≥ 5 iron granules encircling a third or more of the nucleus
- Lacks Auer rods or del 5q as an isolated cytogenetic abnormality; myeloid blasts are not increased in the bone marrow or peripheral blood
- Formerly called refractory anemia with ring sideroblasts, refractory cytopenia with multilineage dysplasia and ring sideroblasts
- Current nomenclature: MDS RS SLD (single lineage dysplasia) and MDS RS MLD (multilineage dysplasia)
- ICD-10: D46.1 - refractory anemia with ring sideroblasts
- 3 - 11% of all MDS cases
- Median age: 60 - 73 years
- M = F
- Myelodysplastic syndrome with ring sideroblasts, multilineage dysplasia (MDS RS MLD) is more common than myelodysplastic syndrome with ring sideroblasts, single lineage dysplasia (MDS RS SLD)
- Reference: Am J Med 2012;125:S2
- SF3B1 gene encodes a component of U2 small nuclear ribonucleoproteins (snRNP) spliceosome
- Mutations in the SF3B1 gene lead to altered splicing of mitochondrial iron transporter gene and other metabolic genes, which in turn leads to the ineffective erythropoiesis and formation of ring sideroblasts
- Ring sideroblasts are erythroid precursors with abnormal iron accumulation within the mitochondria
- These changes lead to morphologic dysplasia, ineffective erythropoiesis and iron overload
- References: Blood 2016;128:462, Blood 2012;120:3173
- Anemia
- Thrombocytopenia and neutropenia in a minority
- Hepatosplenomegaly due to iron overload in some
- Reference: Am J Hematol 2021;96:379
- Refractory anemia or bicytopenia / pancytopenia on complete blood count; 1 - 2 cytopenias MDS RS SLD, 1 - 3 cytopenias MDS RS MLD
- Bone marrow aspiration and biopsy, < 5% bone marrow blasts, < 1% peripheral blood blasts
- Molecular studies for diagnosis and prognosis
- Cytogenetic studies: any abnormality unless it fulfills criteria for MDS with isolated del(5q)
- References: Am J Hematol 2021;96:379, Blood 2016;127:2391
- Normochromic macrocytic or normochromic normocytic anemia (hemoglobin usually is < 10 g/dL), usually no other cytopenias but rarely may have thrombocytosis (correlate with JAK2 mutation status and diagnosis of MDS / myeloproliferative neoplasm with ring sideroblasts and thrombocytosis [MPN RS T]) (Am J Hematol 2021;96:379)
- Concurrent iron deficiency can mask the presence of MDS RS (Blood 2011;117:5793)
- May demonstrate lab values of progressive iron overload - e.g. elevated serum ferritin and transferrin saturation (Hemasphere 2020;4:e357)
- 1 - 2% cases evolve into acute myeloid leukemia (AML)
- Age > 70 years, hemoglobin < 8 g/dL in women and < 9 g/dL in men and platelet count < 75 x 109/L are poor prognostic factors
- Absence of SF3B1 mutation, presence of high risk cytogenetics, thrombocytopenia and multilineage dysplasia are poor prognostic factors
- Monosomal karyotype (MK), non-MK other than single or double del(5q) abnormalities, presence of RUNX1 and ASXL1 mutations (Am J Hematol 2019;94:475)
- More recently, SF3B1 mutant MDS proposed as a distinct subtype with relatively good prognosis (Blood 2020;136:157, Am J Clin Pathol 2021 May 12 [Epub ahead of print])
- Reference: Am J Hematol 2021;96:379
- 16 year old girl with hypochromic microcytic anemia (Arch Pathol Lab Med 2005;129:e199)
- 59 year old man with beta thalassemia trait and MDS RS (NAJMS 2017;10:32)
- 73 year old woman with MDS with ring sideroblasts (ASH: Reference cases - MDS with ring sideroblasts [Accessed 13 July 2021])
- Recombinant erythropoietin (EPO) for anemia
- Iron chelation therapy for iron overload
- Lusatercept and sotatercept: soluble fusion proteins that increase hemoglobin levels by a different mechanism than EPO (Am J Hematol 2019;94:475)
- Microscopic criteria
- Characterized by cytopenias (1 - 2 MDS RS SLD, 1 - 3 MDS RS MLD)
- Ring sideroblasts and morphological dysplasia (1 lineage [erythroid] MDS RS SLD, 2 - 3 lineages MDS RS MLD), often with concomitant SF3B1 mutations
- In the absence of SF3B1 mutations, ≥ 15% ring sideroblasts are needed, whereas > 5% ring sideroblasts are required for diagnosis in the presence of SF3B1 mutations (Blood 2012;119:5674)
- Ring sideroblasts are erythroid precursors with ≥ 5 iron granules encircling a third or more of the nucleus (Haematologica 2008;93:1712)
- Lacks Auer rods, < 5% bone marrow blasts, < 1% peripheral blood blasts
- Does not meet criteria for MDS with isolated del (5q); other cytogenetic abnormalities allowed
- Bone marrow histology
- Bone marrow general:
- Hypercellular or normocellular marrow with erythroid hyperplasia
- Bone marrow erythroid:
- Erythroid hyperplasia (may be mild to moderate dysplasia, including irregular nuclear contours, nuclear budding, megaloblastic change), markedly increased iron stores
- Bone marrow myeloid:
- Myeloblasts < 5% (or call MDS with excess blasts)
- Bone marrow megakaryocytes:
- Normal numbers and morphology (in SLD)
- In addition to presence of ring sideroblasts and erythroid hyperplasia and dysplasia, > 10% dysplastic cells within the myeloid or megakaryocytic series in multilineage dysplasia subtype
- Bone marrow general:
- Reference: Mediterr J Hematol Infect Dis 2015;7:e2015035, Blood 2016;127:2391
Contributed by Cecilia C.S. Yeung, M.D.
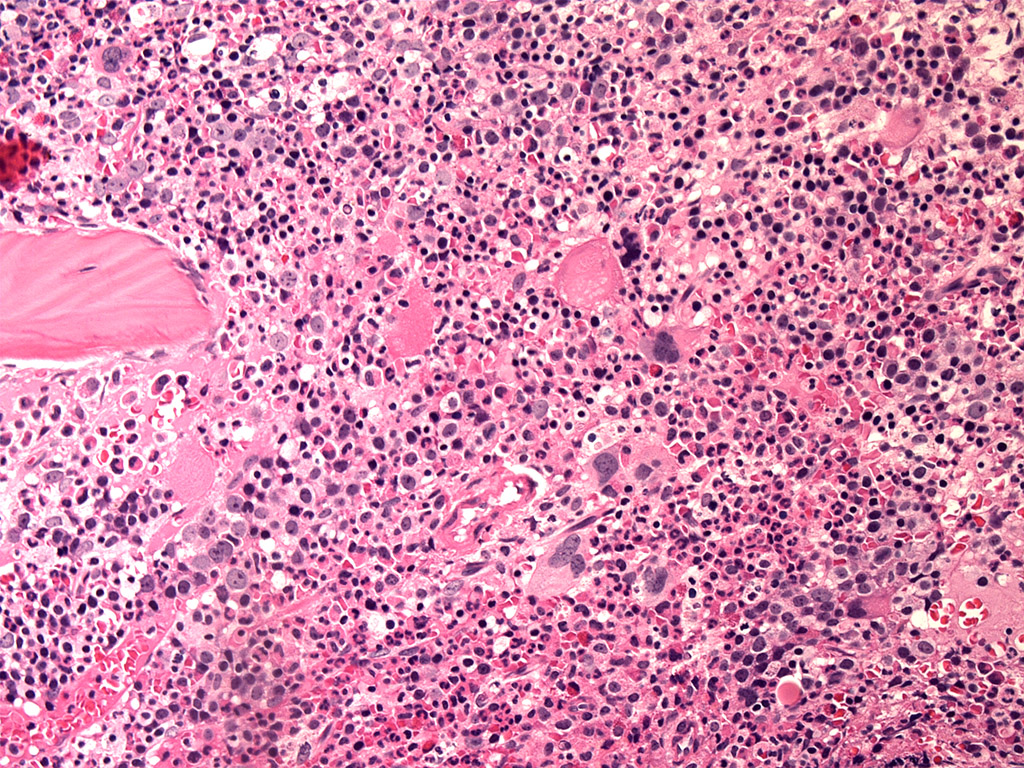
Erythroid predominance
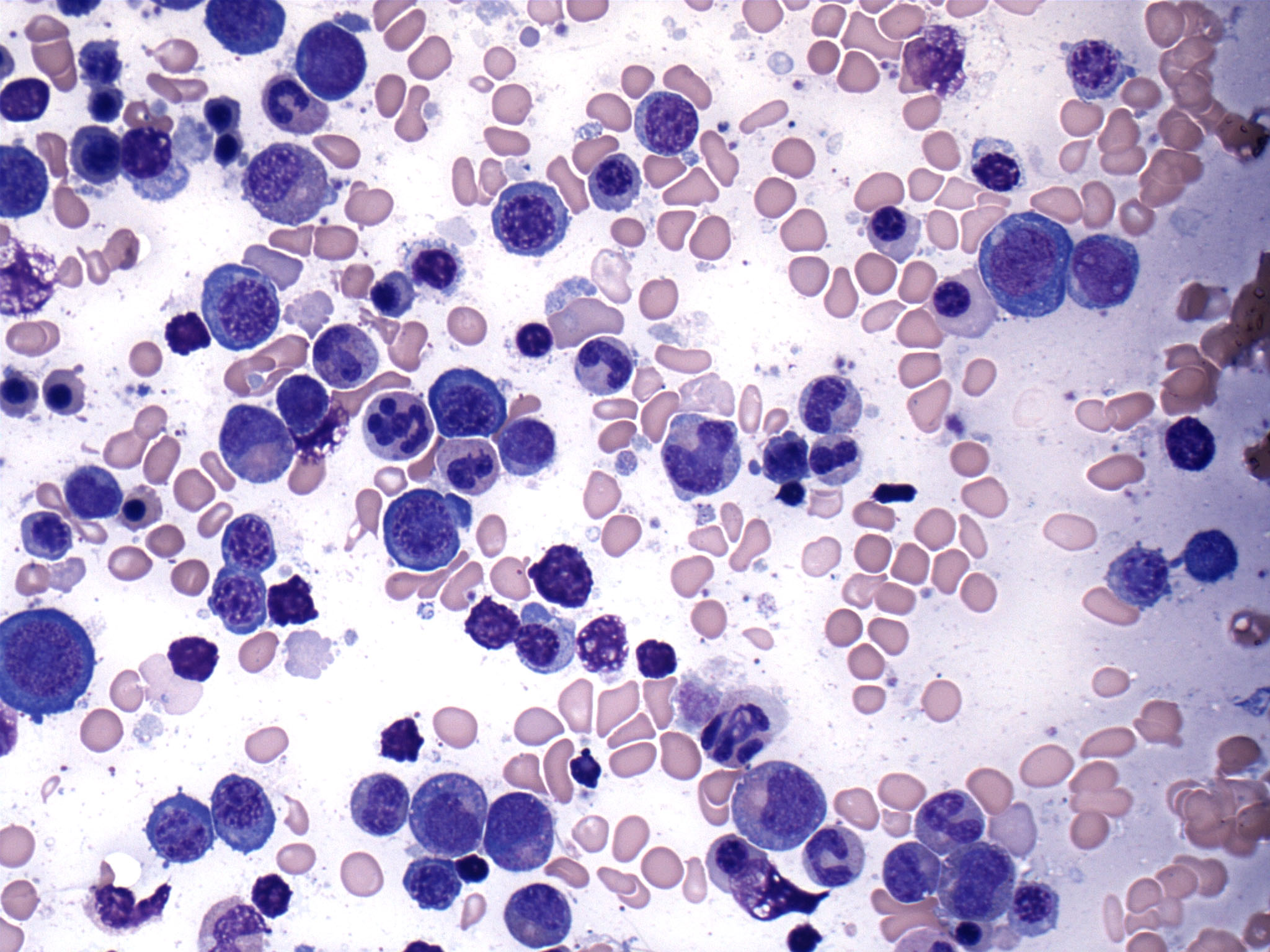
Erythroid hyperplasia and dyserythropoiesis
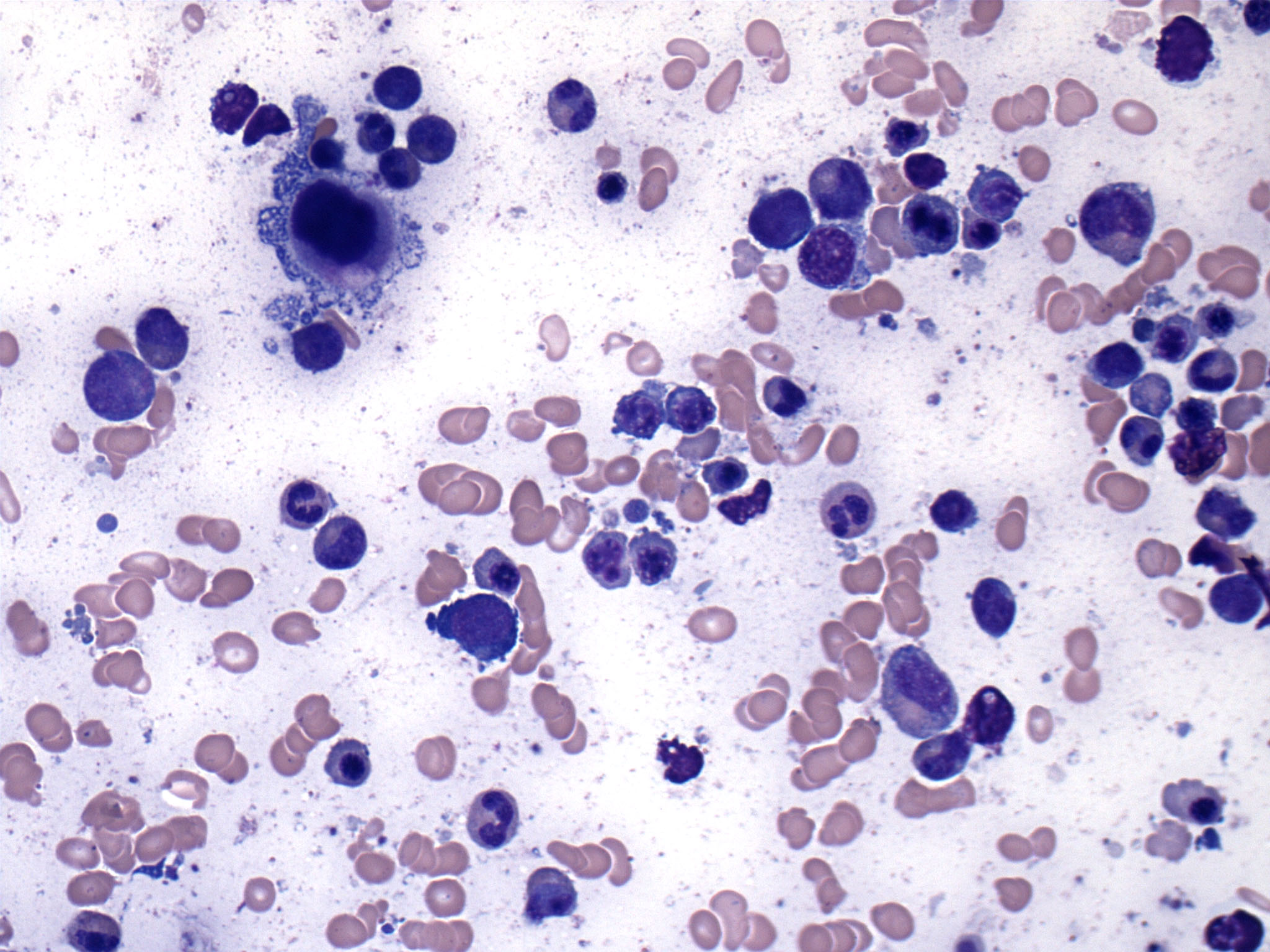
Multilineage dysplasia
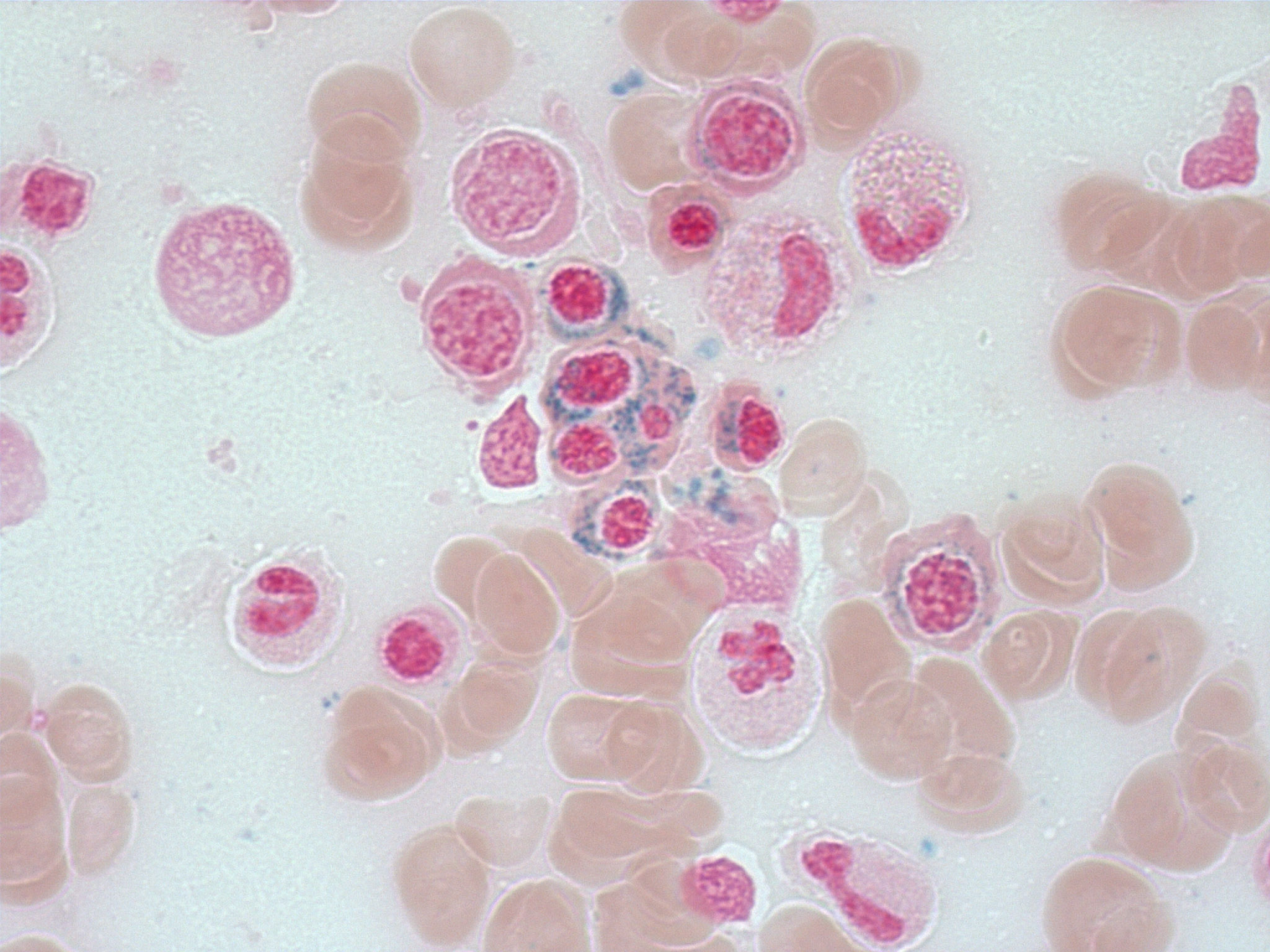
Ring sideroblasts
- Peripheral blood:
- Dimorphic red blood cell populations normal and microcytic hypochromic
- Occasional coarse basophilic stippling and Pappenheimer bodies
- < 1% myeloblasts
- Evaluate for cytopenias and granulocytic dysplasia to subclassify
AFIP images
Peripheral blood
- Iron stain highlights the ring sideroblasts
- Reticulin stain is used to assess the degree of fibrosis in the bone marrow biopsy
- CD34 is used to highlight the blasts in the bone marrow biopsy
- Reference: Hematol Transfus Cell Ther 2019;41:279
- Transmission electron microscopy analysis of ring sideroblasts reveals features of apoptosis with condensed nuclear chromatin and fragmentation of the erythroid cells because of iron deposition in mitochondria (Oncogene 2006;25:4757)
AFIP images
Erythroid precursor shows iron deposits
- Mutation in the spliceosome gene SF3B1 is found in 90% cases of MDS RS SLD and in 30 - 70% of MDS RS MLD
- Clonal cytogenetic abnormalities involve a single chromosome in 50% of MDS RS MLD cases, including high risk loss of chromosome 7
- Other MDS related gene mutations are identified in more than half of cases of MDS MLD, including mutations in STAG2, ASXL1, SRSF2, RUNZ1, CBL, TP53 and TET2
- Bone marrow, aspiration and biopsy:
- Final diagnosis
- Myelodysplastic syndrome with ring sideroblasts with multilineage dysplasia (see comment)
- Negative for increased blasts
- Positive for SF3B1 mutation
- Peripheral blood with normocytic normochromic anemia and pancytopenia
- Comment: The bone marrow aspirates are cellular and demonstrate trilineage dysplasia with presence of 40% ring sideroblasts and without significant increase in blast count (3% by morphology and flow cytometry). SF3B1 mutations are identified by next generation sequencing. Overall, the morphologic, flow cytometric and molecular features are diagnostic of myelodysplasia with ring sideroblasts with multilineage dysplasia. Cytogenetic studies reveal loss of chromosome 7, which is associated with high risk of leukemic transformation.
- Cytogenetics
- Loss of chromosome 7
- Molecular studies
- SF3B1 mutation
- Flow cytometry
- 3% myeloid blasts (see comment)
- Comment: Bone marrow flow cytometry reveals presence of 3% myeloid blasts expressing CD33, CD13, CD117 and MPO. There is no abnormal B cell clone identified and B cells constitute approximately 4% of all nucleated cells. Plasma cells comprise 2% of all nucleated cells and normally express CD138 and CD38.
- 3% myeloid blasts (see comment)
- Peripheral blood
- Differential:
- Complete blood count: 3,500/cumm
- Neutrophils: 60%
- Lymphocytes: 30%
- Eosinophils: 4%
- Monocytes: 4%
- Basophils: 2%
- Blasts: 0%
- Morphology:
- Red blood cells: decreased numbers, normocytic normochromic with mild anisopoikilocytosis and polychromasia
- White blood cells: leukopenia, no blasts, normal morphology
- Platelets: mild thrombocytopenia, few giant platelets seen
- Differential:
- Bone marrow aspirate
- Differential:
- Erythroid: 65%
- Myeloid: 32%
- Lymphocytes: 3%
- Plasma cells: 2%
- Blasts: 3%
- M:E ratio: 1:2
- Morphology:
- Erythroid: erythroid hyperplasia with features of dyserythropoeisis - binucleation, nuclear budding, nucleocytoplasmic asynchrony
- Myeloid: normal maturation with mild dysgranulopoeisis
- Megakaryocytes: normal in numbers with hypolobated and monolobated forms
- Plasma cells: normal in number and morphology
- Lymphocytes: normal in number and morphology
- Iron stain: 40% ring sideroblasts are identified with presence of perinuclear iron granules
- Differential:
- Bone marrow biopsy
- Morphology:
- Hypercellular (70% cellularity) bone marrow with erythroid hyperplasia, dyserythropoiesis, dysgranulopoeisis and dysmegakaryopoeisis
- Reticulin stain shows grade I fibrosis
- Morphology:
- MDS / MPN with ring sideroblasts and thrombocytosis:
- Clonal disorder with diagnostic features of MDS RS SLD
- Platelet count of ≥ 450 × 109/L (persistent)
- Large, atypical megakaryocytes
- Various nonclonal conditions can present with ring sideroblasts in the bone marrow; thorough clinical history and absence of diagnostic criteria for clonal MDS RS (> 15% RS or SF3B1 mutation with > 5% RS) are essential for differential diagnosis
- Genetically inherited congenital sideroblastic anemia (Br J Haematol 2016;174:847):
- Characterized by mutations in 1 of 3 mitochondrial pathways: heme synthesis, iron sulfur cluster biogenesis and protein synthesis
- X linked sideroblastic anemia is the most common form attributed to mutation in aminolevulinic acid (ALA) synthase
- Microcytic hypochromic anemia and iron overload
- Acquired sideroblastic anemia due to chronic alcohol use (Postgrad Med 1992;92:147):
- History of chronic alcoholism
- Elevated serum aspartate aminotransferase (AST):alanine aminotransferase (ALT) ratio
- Macrocytic anemia
- < 15% ring sideroblasts
- Drug induced sideroblastic anemia - chloramphenicol, penicillamine, linezolid, isoniazid (Haematologica 2013;98:e138):
- History of medication use
- May have pure red cell aplasia
- Reversible after discontinuation of drug
- Lead poisoning:
- High serum lead levels, lead lines (Burton lines) on teeth and gingiva
- < 15% ring sideroblasts
- Basophilic stippling in red cells on peripheral smear, microcytic and hemolytic anemia
- Copper deficiency / zinc toxicity (Clin Case Rep 2015;3:325):
- Low serum copper levels or high serum zinc levels
- < 15% ring sideroblasts
- Cytoplasmic vacuoles in erythroid and myeloid cells
- Genetically inherited congenital sideroblastic anemia (Br J Haematol 2016;174:847):
- Reference: Am J Hematol 2021;96:379
Bone marrow aspirate with iron stain is shown in the image above. Which molecular alteration is most commonly observed in patients who have myelodysplastic syndrome (MDS) with this histologic feature?
- Isolated del 5q
- JAK 517F mutation
- Monosomy 7
- SF3B1 mutation
- If no SF3B1 mutation is seen, only 5% ring sideroblasts are needed to make the diagnosis of MDS with ring sideroblasts
- If SF3B1 mutation is seen, only 5% ring sideroblasts are needed to make the diagnosis of MDS with ring sideroblasts
- If SF3B1 mutation is seen, only 10% ring sideroblasts are needed to make the diagnosis of MDS with ring sideroblasts
- There is no impact; 15% ring sideroblasts are required for the diagnosis of MDS with ring sideroblasts
Comment Here
Reference: MDS with ring sideroblasts
- Myelodysplastic syndrome with mutated SF3B1 (MDS SF3B1) is a myeloid neoplasm with cytopenia and dysplasia characterized by SF3B1 mutation and often ring sideroblasts
- Acceptable definition: MDS with low blasts and ring sideroblasts
- ICC 2022 Nomenclature: MDS with mutated SF3B1
- Blood counts: characterized by cytopenia of one or more lineages, without thrombocytosis
- Morphology: erythroid lineage dysplasia, ring sideroblasts are present in over 90% cases but not required for diagnosis in the presence of SF3B1 mutation, blasts < 5% in the bone marrow and < 2% in the peripheral blood
- Cytogenetics: absence of 5q deletion, monosomy 7/7q deletion or complex karyotype
- Molecular genetics: SF3B1 mutation at a variant allele frequency (VAF) of > 10% (per International Consensus Classification [ICC]), no multihit TP53 or RUNX1 mutation
- In the absence of SF3B1 mutation, ≥ 15% ring sideroblasts are needed
- Reference: Blood 2020;136:157
- WHO 4th edition equivalents: MDS RS SLD (myelodysplastic syndrome with ring sideroblasts and single lineage dysplasia) and MDS RS MLD (multilineage dysplasia)
- Formerly called refractory anemia with ring sideroblasts, refractory cytopenia with multilineage dysplasia and ring sideroblasts
- 17% of all MDS cases (Leuk Res 2017;57:78)
- Annual incidence: 0.84/100,000 persons (Leuk Res 2011;35:1591)
- Median age: 70 - 75 years (Leuk Res 2017;57:78)
- Slight male preponderance
- Patients with low blasts and ≥ 15% ring sideroblasts without SF3B1 mutation account for 3 - 4% of all MDS cases
- Reference: Blood 2020;136:157
- SF3B1 gene encodes a component of the U2 small nuclear ribonucleoproteins (snRNP) spliceosome
- Mutations in the SF3B1 gene lead to altered splicing of mitochondrial iron transporter gene and other metabolic genes, which in turn leads to the ineffective erythropoiesis and formation of ring sideroblasts
- Ring sideroblasts are erythroid precursors with abnormal iron accumulation within the mitochondria
- These changes lead to morphologic dysplasia, ineffective erythropoiesis and iron overload
- References: Blood 2016;128:462, Blood 2012;120:3173
- Driver mutations in SF3B1 gene in hematopoietic stem cells
- Anemia
- Thrombocytopenia and neutropenia in a minority
- Hepatosplenomegaly due to iron overload in some
- Reference: Am J Hematol 2021;96:379
- Refractory anemia or bicytopenia / pancytopenia on complete blood count
- Bone marrow aspiration and biopsy, < 5% bone marrow blasts, < 2% peripheral blood blasts
- Molecular studies: next generation sequencing (NGS) panel including SF3B1 and associated myeloid neoplasm genes for diagnosis and prognosis; absence of biallelic TP53 inactivation
- Cytogenetic studies: absence of del(5q), monosomy 7 or 7q deletion, complex karyotype
- Does not fulfill criteria for other MDS / myeloproliferative neoplasm (MPN) entities
- References: Am J Hematol 2021;96:379, Blood 2016;127:2391
- Normochromic macrocytic or normochromic normocytic anemia (hemoglobin usually is < 10 g/dL); usually no other cytopenias
- Thrombocytosis should be absent as these are classified separately as MDS / MPN with SF3B1 mutation and thrombocytosis; these usually have concurrent JAK2 V617F, CARR or MPL mutations
- Concurrent iron deficiency can mask the presence of MDS RS (Blood 2011;117:5793)
- May demonstrate lab values of progressive iron overload (e.g., elevated serum ferritin and transferrin saturation) (Hemasphere 2020;4:e357)
- Among other WHO 5th edition and ICC 2022 described MDS subtypes, MDS SF3B1 has the best outcomes (Leukemia 2022;36:1703, Blood 2022;140:1200)
- When SF3B1 mutations are seen, the presence of multilineage dysplasia has not demonstrated significant prognostic impact
- Transfusion dependence is associated with worse outcomes and higher risk of transformation to acute myeloid leukemia (AML)
- In comparison to patients with MDS with low blasts and SF3B1 mutations, MDS with low blasts, ring sideroblasts and wild type SF3B1 has a less favorable overall survival and leukemia free survival (Crit Rev Oncol Hematol 2019;133:74)
- Presence of excess blasts has a worse prognosis even in the presence of SF3B1 mutation
- Patients with this diagnosis who have additional mutations in TP53, RUNX1, EZH2 mutations have been associated with a worse outcome as compared to patients with SF3B1 mutation alone (Blood 2015;126:233)
- 16 year old girl with hypochromic microcytic anemia (Arch Pathol Lab Med 2005;129:e199)
- 59 year old man with beta thalassemia trait, MDS with ring sideroblasts and SF3B1 and TET2 mutations (NAJMS 2017;10:32)
- 73 year old woman with MDS with ring sideroblasts (ASH: MDS with Ring Sideroblasts [Accessed 9 April 2024])
- Recombinant erythropoietin (EPO) for anemia
- Iron chelation therapy for iron overload
- Lusatercept and sotatercept: soluble fusion proteins that increase hemoglobin levels by a different mechanism than EPO (Am J Hematol 2019;94:475)
- Microscopic criteria
- Characterized by cytopenias (1 - 2 MDS RS SLD, 1 - 3 MDS RS MLD)
- Ring sideroblasts and morphological dysplasia (1 lineage [erythroid] or 2 - 3 lineages), often with concomitant SF3B1 mutations
- In the absence of SF3B1 mutations, ≥ 15% ring sideroblasts are needed, whereas > 5% ring sideroblasts are required for diagnosis in the presence of SF3B1 mutations
- Ring sideroblasts are erythroid precursors with ≥ 5 iron granules encircling a third or more of the nucleus (Haematologica 2008;93:1712)
- Lacks Auer rods, < 5% bone marrow blasts, < 2% peripheral blood blasts
- Does not meet criteria for MDS with isolated del (5q); other cytogenetic abnormalities allowed
- Bone marrow histology
- Bone marrow general
- Hypercellular or normocellular marrow with erythroid hyperplasia
- Bone marrow erythroid
- Erythroid hyperplasia (may be mild to moderate dysplasia, including irregular nuclear contours, nuclear budding, megaloblastic change), markedly increased iron stores
- Bone marrow myeloid
- Myeloblasts < 5% (or call MDS with excess blasts)
- Bone marrow megakaryocytes
- Normal numbers and morphology (in SLD)
- In addition to presence of ring sideroblasts and erythroid hyperplasia and dysplasia, > 10% dysplastic cells within the myeloid or megakaryocytic series in multilineage dysplasia subtype
- Bone marrow general
- References: Mediterr J Hematol Infect Dis 2015;7:e2015035, Blood 2016;127:2391
Contributed by Cecilia C.S. Yeung, M.D.
Erythroid predominance
Erythroid hyperplasia and dyserythropoiesis
Multilineage dysplasia
Ring sideroblasts

Bone marrow aspirate, iron stain
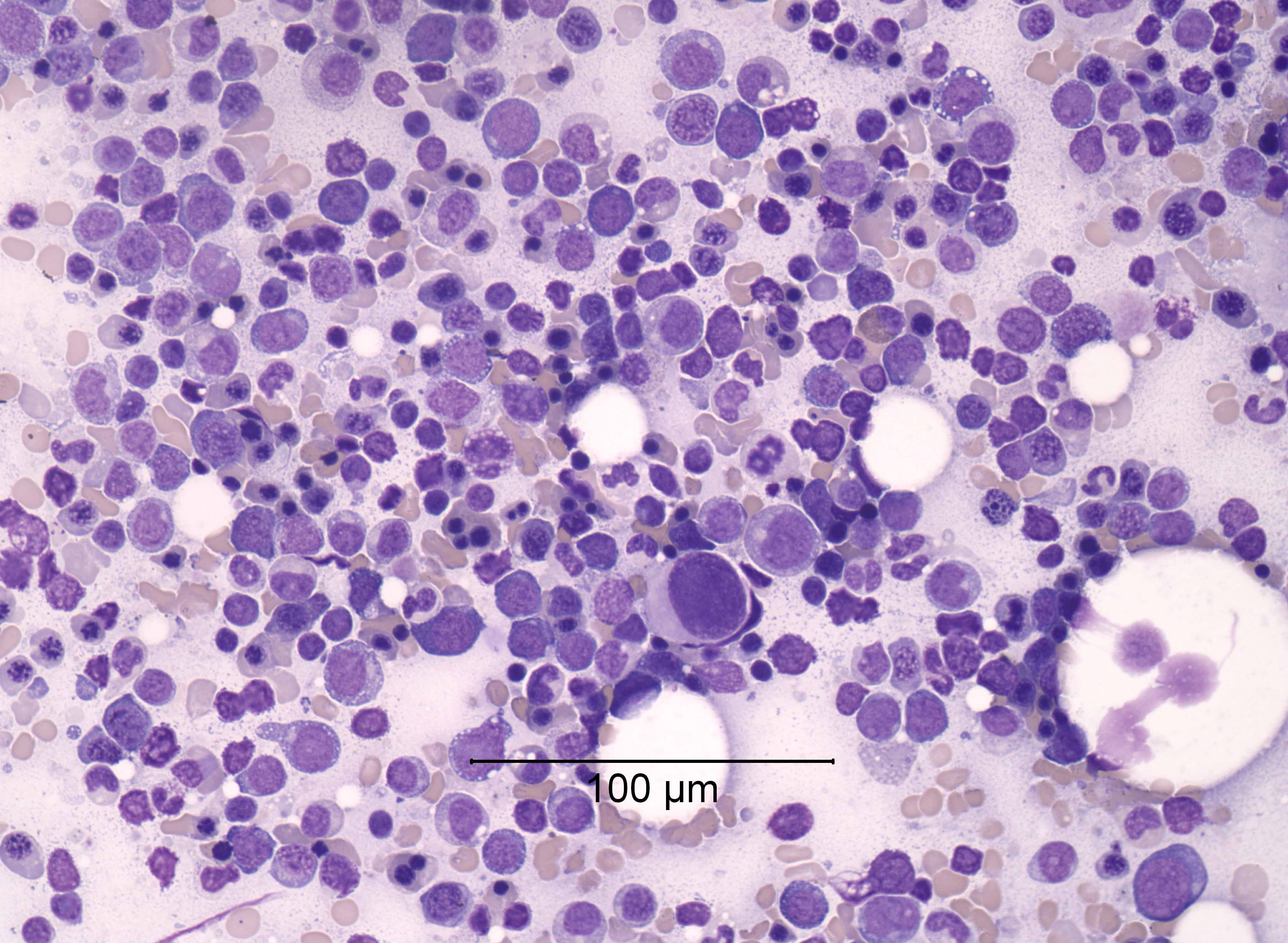

Bone marrow aspirate, Wright-Giemsa stain
- Peripheral blood
- Dimorphic red blood cell populations, normal and microcytic hypochromic
- Occasional coarse basophilic stippling and Pappenheimer bodies
- < 1% myeloblasts
- Evaluate for cytopenias and granulocytic dysplasia to subclassify
- Reference: Blood 2020;136:157
AFIP images
Dimorphic red blood cells
- Iron stain highlights the ring sideroblasts
- Reticulin stain is used to assess the degree of fibrosis in the bone marrow biopsy
- CD34 is used to highlight the blasts in the bone marrow biopsy
- Reference: Hematol Transfus Cell Ther 2019;41:279
- Transmission electron microscopy analysis of ring sideroblasts reveals features of apoptosis with condensed nuclear chromatin and fragmentation of the erythroid cells due to iron deposition in mitochondria (Oncogene 2006;25:4757)
AFIP images
Erythroid precursor shows iron deposits
- Mutation in the spliceosome gene SF3B1 is found in 90% of cases, usually at a high VAF (35 - 43%)
- 50% of SF3B1 mutations affect codon 700, with other hotspots at codons 666, 662, 622, 625
- Presence of comutations involving BCOR, BCORL1, NRAS, RUNX1, SRSF2 or STAG2 are associated with significantly different outcomes in comparison to mutation in SF3B1 alone (NEJM Evidence 2022;1:EVIDoa2200008)
- Most cases of MDS SF3B1 have a normal karyotype or abnormalities involving a single chromosome
- Cases with 5q deletion, monosomy 7/7q deletion or complex karyotype are excluded
- Bone marrow, right posterior iliac crest, aspirate smears, core biopsy, particle prep and peripheral blood smear:
- Hypercellular marrow with megakaryocytic and erythroid dysplasia, 1+ reticulin deposition and a normal blast count (see comment)
- Increased ring sideroblasts are present
- Anemia with no circulating blasts
- Comment: The sum of the morphological findings (notably, the presence of ringed sideroblasts and a dominant SF3B1 mutation [VAF 44%]) is diagnostic of a myeloid neoplasm and compatible with myelodysplastic neoplasm with low blasts and SF3B1 mutation (MDS SF3B1) under the WHO classification system. Under the International Consensus classification system, this would be called myelodysplastic syndrome with mutated SF3B1 (also called MDS SF3B1). Myelodysplastic / myeloproliferative neoplasm with SF3B1 mutation and thrombocytosis (MDS / MPN SF3B1 T) was a consideration but the lack of thrombocytosis argues against this entity.
- Cytogenetics
- Normal male, 46, XY
- Molecular studies
- Positive for clinically significant variants: SF3B1 (p.N626S, NM_012433.3:c.1877A>G, VAF 44%)
- Flow cytometry
- 3% myeloid blasts (see comment)
- Comment: Bone marrow flow cytometry reveals presence of 3% myeloid blasts expressing CD33, CD13, CD117 and MPO. There is no abnormal B cell clone identified and B cells constitute approximately 4% of all nucleated cells. Plasma cells comprise 2% of all nucleated cells and normally express CD138 and CD38.
- Peripheral blood
- Differential
- Complete blood count: 3,500/mm³
- Neutrophils: 60%
- Lymphocytes: 30%
- Eosinophils: 4%
- Monocytes: 4%
- Basophils: 2%
- Blasts: 0%
- Morphology
- Red blood cells: macrocytic anemia with anisopoikilocytosis
- White blood cells: leukopenia, no circulating blasts, normal morphology
- Platelets: mild thrombocytopenia, few giant platelets seen
- Differential
- Bone marrow aspirate
- Differential
- Erythroid: 65%
- Myeloid: 32%
- Lymphocytes: 3%
- Plasma cells: 2%
- Blasts: 3%
- M:E ratio = 1:2
- Morphology
- Erythroid: erythroid hyperplasia with features of dyserythropoiesis - binucleation, nuclear budding, nucleocytoplasmic asynchrony
- Myeloid: normal maturation with mild dysgranulopoiesis
- Megakaryocytes: normal in numbers with hypolobated and monolobated forms
- Plasma cells: normal in number and morphology
- Lymphocytes: normal in number and morphology
- Iron stain: 40% ring sideroblasts are identified with presence of perinuclear iron granules
- Differential
- Bone marrow biopsy
- Morphology
- Hypercellular (70% cellularity) bone marrow with erythroid hyperplasia, dyserythropoiesis, dysgranulopoiesis and dysmegakaryopoiesis
- Reticulin stain shows grade I fibrosis
- Morphology
- MDS / MPN with SF3B1 mutation and thrombocytosis:
- Anemia associated with dysplastic erythropoiesis and ≥ 15% ring sideroblasts
- Persistent thrombocytosis with platelet count ≥ 450 x 109/L
- Detection of SF3B1 mutation concurrently with JAK2 p.V617F
- Various nonclonal conditions can present with ring sideroblasts in the bone marrow; thorough clinical history and absence of diagnostic criteria for clonal MDS RS (> 15% RS or SF3B1 mutation) are essential for differential diagnosis
- Genetically inherited congenital sideroblastic anemia (Br J Haematol 2016;174:847):
- Characterized by mutations in 1 of 3 mitochondrial pathways: heme synthesis, iron sulfur cluster biogenesis and protein synthesis
- X linked sideroblastic anemia is the most common form attributed to mutation in aminolevulinic acid (ALA) synthase
- Microcytic hypochromic anemia and iron overload
- Acquired sideroblastic anemia due to chronic alcohol use (Postgrad Med 1992;92:147):
- History of chronic alcoholism
- Elevated serum aspartate aminotransferase (AST):alanine aminotransferase (ALT) ratio
- Macrocytic anemia
- < 15% ring sideroblasts
- Drug induced sideroblastic anemia - chloramphenicol, penicillamine, linezolid, isoniazid (Haematologica 2013;98:e138):
- History of medication use
- May have pure red cell aplasia
- Reversible after discontinuation of drug
- Lead poisoning:
- High serum lead levels, lead lines (Burton lines) on teeth and gingiva
- < 15% ring sideroblasts
- Basophilic stippling in red cells on peripheral smear, microcytic and hemolytic anemia
- Copper deficiency / zinc toxicity (Clin Case Rep 2015;3:325):
- Low serum copper levels or high serum zinc levels
- < 15% ring sideroblasts
- Cytoplasmic vacuoles in erythroid and myeloid cells
- Genetically inherited congenital sideroblastic anemia (Br J Haematol 2016;174:847):
- Reference: Am J Hematol 2021;96:379
Bone marrow aspirate with iron stain is shown in the image above. Which molecular alteration is most commonly observed in patients who have myelodysplastic syndrome (MDS) with this histologic feature?
- Isolated del 5q
- JAK 617F mutation
- Monosomy 7
- SF3B1 mutation
Comment Here
Reference: MDS with ring sideroblasts
- MDS with mutated SF3B1 has poor disease outcomes as compared to other subtypes of MDS
- Presence of excess blasts has a worse prognosis, even in the presence of SF3B1 mutation
- Presence of ring sideroblasts is an essential diagnostic requirement for MDS with mutated SF3B1
- Ring sideroblasts as well as SF3B1 mutation are not detected in other MDS types or other myeloid neoplasms
Comment Here
Reference: MDS with ring sideroblasts
- Myelodysplastic syndrome (MDS) with mutated TP53 is characterized by ≥ 1 TP53 mutation or mutation with loss of TP53 function resulting in ≥ 1 cytopenia and either blasts in the blood or bone marrow (International Consensus Classification ([ICC]) or single / multilineage dysplasia (WHO 5th edition [WHO HAEM5])
- Other MDS subtypes should not be diagnosed in patients with multihit TP53 (ICC)
- Myelodysplastic neoplasm (MDS) with multihit TP53 mutation is associated with aggressive behavior with a rapid transformation to acute myeloid leukemia (AML)
- Poor prognosis, median overall survival of 0.7 years
- Genomic testing is essential to make the diagnosis
- No morphologic features specific to MDS with mutated TP53
- Reference: Cancer Discov 2022;12:2954
- Myelodysplastic neoplasm (MDS) (WHO 5th edition [WHO HAEM5])
- Myelodysplastic syndrome (MDS) (International Consensus Classification ([ICC])
- MDS with mutated TP53 (MDS-TP53) (ICC)
- MDS with biallelic TP53 inactivation (MDS-biTP53) (WHO HAEM5)
- ICD-10: D46.9 - myelodysplastic syndrome, unspecified
- Median age: 69.7 years
- Increased frequency of TP53 mutation in patients following cytotoxic chemotherapy or radiation therapy
- ~10% of all cytopenias
- Reference: Blood Adv 2022;6:2847
- Bone marrow and peripheral blood
- Mutations affecting TP53 function, a tumor suppressor gene that plays a critical role in cellular apoptosis (Blood Rev 2023;60:101055)
- DNA damaging agents
- Chemotherapy and radiation
- Hereditary
- Li-Fraumeni syndrome (germline TP53 mutations) (Cold Spring Harb Perspect Med 2017;7:a026187)
- Symptoms associated with cytopenias
- Anemia
- Fatigue, shortness of breath, pallor
- Thrombocytopenia
- Purpura, bleeding, easy bruising
- Neutropenia
- Infections
- Anemia
- Reference: Blood 2023;142:2247
- WHO HAEM5
- Myelodysplastic neoplasms with defining genetic abnormalities
- MDS with biallelic TP53 inactivation
- < 20% blasts in peripheral blood and bone marrow, usually complex cytogenetics, ≥ 2 TP53 mutations or 1 mutation with TP53 copy number loss or copy neutral loss of heterozygosity
- MDS with biallelic TP53 inactivation
- Myelodysplastic neoplasms with defining genetic abnormalities
- ICC
- MDS cases with defining cytogenetic or molecular criteria along with blast percentage and cytopenias, no requirement for dysplasia
- MDS with mutated TP53
- 0 - 9% peripheral blood / bone marrow blasts, multihit TP53 mutations or TP53 mutation with variant allele frequency (VAF) > 10% and complex karyotype and often with loss of 17p
- MDS / AML with mutated TP53
- 10 - 19% bone marrow or peripheral blood blasts with any somatic TP53 mutation with VAF ≥ 10%
- MDS with mutated TP53
- MDS cases with defining cytogenetic or molecular criteria along with blast percentage and cytopenias, no requirement for dysplasia
- Reference: Virchows Arch 2023;482:39, Ann Lab Med 2023;43:503
- ≥ 1 cytopenia
- Reticulocytopenia is commonly found in MDS
- TP53 mutation is the strongest prognostic factor in MDS (Cancer Discov 2022;12:2954)
- Associated with treatment resistant disease, rapid disease progression and low overall survival
- 3 year transformation to AML rate: 40%
- 51 year old man with dysphagia (Oncol Lett 2020;20:266)
- 62 year old woman with pancytopenia and dyspnea (Mol Cytogenet 2022;15:51)
- 70 year old woman presenting with palpitations (Intern Med 2005;44:490)
- Stem cell transplant associated with increased overall survival (OS = 20.6 months) (N Engl J Med 2017;376:536)
- Median relapse free survival of 14.5 months
- Multilineage dysplasia in most cases
- Increased erythroid precursors common
- Occasional bone marrow fibrosis
- Reference: Blood 2023;142:2247
Contributed by Hamzah Rehan, D.O.
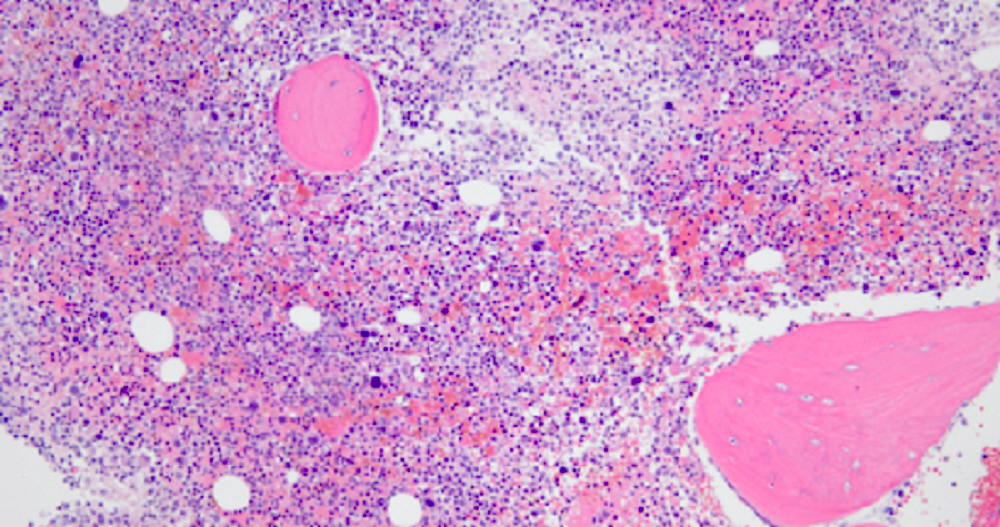
Hypercellular bone marrow

Dysplastic megakaryocytes

Clot section

CD34
- Multilineage dysplasia
Contributed by Hamzah Rehan, D.O.

Aspirate smear
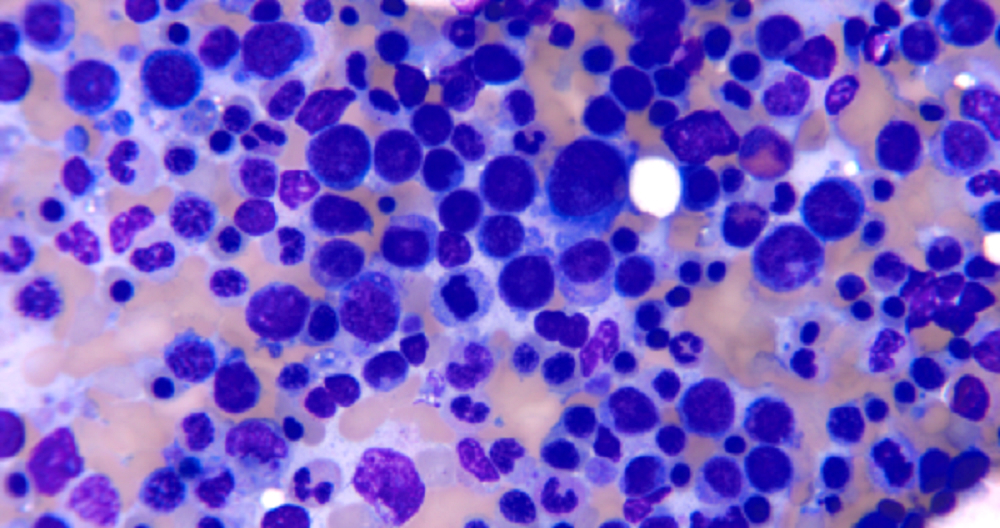
Erythroid budding
- Red blood cells
- Anisopoikilocytosis with macrocytosis of red blood cells
- White blood cells
- Hyposegmentation, lack of lobation, nuclear clumping, cytoplasmic hypogranularity or agranularity
- Reference: Blood 2023;142:2247
- p53 IHC staining of > 2% is associated with TP53 mutation (Leuk Res 2016;41:21)
- Flow cytometry alone is not diagnostic
- Decreased side scatter of granulocytes
- Erythroid: increased coefficient of variation and decreased intensity of CD71 or CD36
- Aberrant expression of CD56 or CD7 on progenitors, granulocytes or monocytes
- Asynchrony of CD15 and CD16 on granulocytes
- Altered expression of CD13 in relation to CD11b or CD16
- Reference: Ann Hematol 2023;102:3015
- Cytogenetics
- Usually accompanied by complex karyotype
- del(5q), 17p loss of heterozygosity (LOH), -7/del(7q), -18
- Other mutations as well
- Usually accompanied by complex karyotype
- Molecular genetics
- TP53 mutations
- Commonly in DNA binding domain, inactivating
- Copy number status can be detected by FISH probe
- TP53 mutations
- Reference: Blood 2022;139:2347
- Bone marrow, left posterior iliac crest; core biopsy, clot section, aspirate smears and touch imprint:
- Myelodysplastic neoplasm with low blasts
- Hypercellular marrow with trilineage hematopoiesis showing multilineage dysplasia
- Aspirate smear: blasts 3%, lymphocytes 11%
- Cytogenetics: -7/del(7q)
- Next generation sequencing: multiallelic TP53 mutation, deletion 5q and 20q
- Myelodysplastic neoplasm with low blasts
- Myelodysplastic neoplasm / syndrome, other subtypes:
- Does not have TP53 mutation or multihit loss of TP53
- MDS / AML with mutated TP53 (ICC):
- Blasts 10 - 19% in bone marrow or peripheral blood
A 71 year old man presenting with anemia and thrombocytopenia underwent a bone marrow biopsy (H&E image shown above) of his left posterior iliac crest. Which of the following mutations is associated with the worst prognosis?
- 5q deletion
- JAK2 V617F
- Multihit TP53 inactivation
- SF3B1
Comment Here
Reference: MDS with mutated TP53
- 30 pack year smoking history
- Brother with 3 prior basal cell carcinomas and a prior meningioma
- Deceased donor kidney transplant
- HIV infection
Comment Here
Reference: MDS with mutated TP53
- Myelodysplastic syndrome with single lineage dysplasia (MDS SLD) is defined as persistent unexplained cytopenias (anemia [most common], neutropenia or thrombocytopenia) or bicytopenia with ≥ 10% dysplastic cells in 1 myeloid lineage
- Per 2016 revised WHO classification, MDS SLD includes:
- Peripheral blood with anemia (Hb < 10 g/dL) or neutropenia (< 1.8 x 109/L) or thrombocytopenia (< 100 x 109/L) or bicytopenia with < 1% blasts
- Bone marrow (BM) with ≥ 10% dysplastic cells in one myeloid lineage
- MDS with erythroid dysplasia only and 5 - 14% ring sideroblasts in absence of SF3B1 mutation
- Reference: Blood 2016;127:2391
- According to 2008 WHO classification, it was known as refractory cytopenia with unilineage dysplasia (refractory anemia, refractory neutropenia or refractory thrombocytopenia)
- Incidence: 7 - 20% of all MDS cases
- Median age of onset: 65 - 70 years
- M = F
- Peripheral blood and bone marrow
- Risk factors:
- Benzene exposure (J Natl Cancer Inst 2012;104:1724)
- Cigarette smoking (also in part due to benzene in cigarette smoke)
- Agricultural solvents or chemicals
- Family history of hematopoietic neoplasms
- Inherited hematological disorders, such as Fanconi anemia, dyskeratosis congenita, Shwachman-Diamond syndrome (SDS) and Diamond-Blackfan anemia
- These cases should also be separately designated as myeloid neoplasms with germline predisposition (Curr Opin Hematol 2021;28:86)
- Acquired aplastic anemia (Blood 2002;100:786)
Contributed by Karan Saluja, M.D. and Beenu Thakral, M.D.
Classification algorithm for MDS SLD
- Clinical presentation may include fatigue, infection or petechiae related to the type and degree of cytopenia(s)
- MDS is a comprehensive diagnosis made by review of complete clinical history, complete blood count, peripheral blood smear findings, bone marrow morphology assessment, flow cytometry immunophenotyping (if available), cytogenetics and molecular findings
- Review of:
- Complete clinical history (personal and family history), history of chemotherapy or radiation exposure
- Medications that can contribute to cytopenia or dysplasia
- Peripheral blood smear for blasts percentage, absolute monocyte count, monocyte percentage, degree of dysplasia and number of lineages involved
- Bone marrow morphology should include cellularity, blast percentage, degree of dysplasia, ring sideroblasts percentage (if any), presence or absence of fibrosis
- Flow cytometry to evaluate for aberrancies in CD34 positive myeloblast population and abnormal myelomonocytic maturation; in addition, any monoclonal B cell population or immunophenotypically abnormal T cells or NK cell population, if present, can also be assessed
- Cytogenetic abnormality in the absence of morphologic criteria, such as gain of chromosome 8, del 20q and loss of Y chromosome, is not considered definitive evidence of MDS, according to the WHO 2016 revised classification; in the setting of persistent cytopenia of undetermined origin, other MDS defining cytogenetic abnormalities are considered presumptive evidence of MDS, even in the absence of definitive morphologic features
- MDS associated somatic mutations identified alone in the absence of morphologic features of dysplasia are not considered diagnostic evidence of MDS, according to the current WHO 2016 revised classification
- References: Hematology Am Soc Hematol Educ Program 2015;2015:294, Pathobiology 2019;86:7
- Peripheral blood cytopenias with anemia (Hb < 10 g/dL) or neutropenia (< 1.8 x 109/L) or thrombocytopenia (< 100 x 109/L) or bicytopenia and < 1% blasts
- Prognosis in MDS is determined using IPSS-R diagnostic scoring system, which takes into account cytogenetic findings, bone marrow blast percentage, hemoglobin, platelet count and absolute neutrophil count (ANC)
- 5 risk groups:
- Very low risk: ≤ 1.5
- Low: > 1.5 - 3.0
- Intermediate: > 3.0 - 4.5
- High: > 4.5 - 6.0
- Very high: > 6.0
- References: Blood 2012;120:2454, J Natl Compr Canc Netw 2015;13:261
IPPS-R scoring for MDS
| Prognostic variables | Score value | ||||||
| 0 | 0.5 | 1.0 | 1.5 | 2.0 | 3.0 | 4.0 | |
| Karyotype | Very good | - | Good | - | Intermediate | Poor | Very poor |
| Percentage of bone marrow blasts | ≤ 2% | - | > 2% to < 5% | - | 5 - 10% | > 10% | - |
| Hemoglobin (Hb) (g/dL) | ≥ 10 | - | 8 to < 10 | < 8 | - | - | - |
| Platelets (x109/L) | ≥ 100 | 50 to < 100 | < 50 | - | - | - | - |
| Absolute neutrophil count (ANC) (x109/L) | ≥ 0.8 | < 0.8 | - | - | - | - | - |
Cytogenetic prognostic group:
| |||||||
- 46 year old man and his daughter presented with a novel germline frameshift GATA2 mutation presenting as congenital sensorineural hearing loss and familial MDS (Int J Hematol 2021;114:286)
- 53 year old man with history of shortness of breath, dizziness and fatigue (pernicious anemia with spuriously normal vitamin B12 level might be misdiagnosed as myelodysplastic syndrome) (Clin Lymphoma Myeloma Leuk 2014;14:e141)
- 55 year old man with history of fatigue and generalized weakness (zinc induced copper deficiency: a diagnostic pitfall of myelodysplastic syndrome) (Pathology 2014;46:246)
- Low grade MDS can be observed if the patient is asymptomatic with mild cytopenias
- ESA (erythropoietin stimulating agent) to treat anemia
- Hypomethylating agents (azacitidine or decitabine) if the patient is symptomatic, with or without moderate to severe cytopenias
- Reference: Cancers (Basel) 2021;13:784
- Adequate morphologic assessment for MDS requires high quality, good stained aspirate smears prepared from fresh aspirate specimen (< 2 hours); bone marrow core biopsy should be of adequate length (≥ 1.5 cm) and good quality peripheral blood smear
- Morphologic features of dysplasia in erythroid, myeloid or megakaryocytic lineages
| Dyserythropoiesis | Dysgranulopoiesis | Dysmegakaryopoiesis |
| Nuclear budding or multinucleation | Small or unusually large size granulocytes | Micromegakaryocytes |
| Internuclear bridging | Nuclear hyposegmentation (pseudo Pelger-Huët) | Nuclear hypolobation |
| Karyorrhexis | Nuclear hypersegmentation | Multinucleation or widely separated nuclear lobes |
| Megaloblastoid changes | Hypogranularity; agranularity | |
| Presence of ring sideroblasts | Pseudo Chediak-Higashi granules | |
| Cytoplasmic vacoulization | Auer rod |
Contributed by Beenu Thakral, M.D.
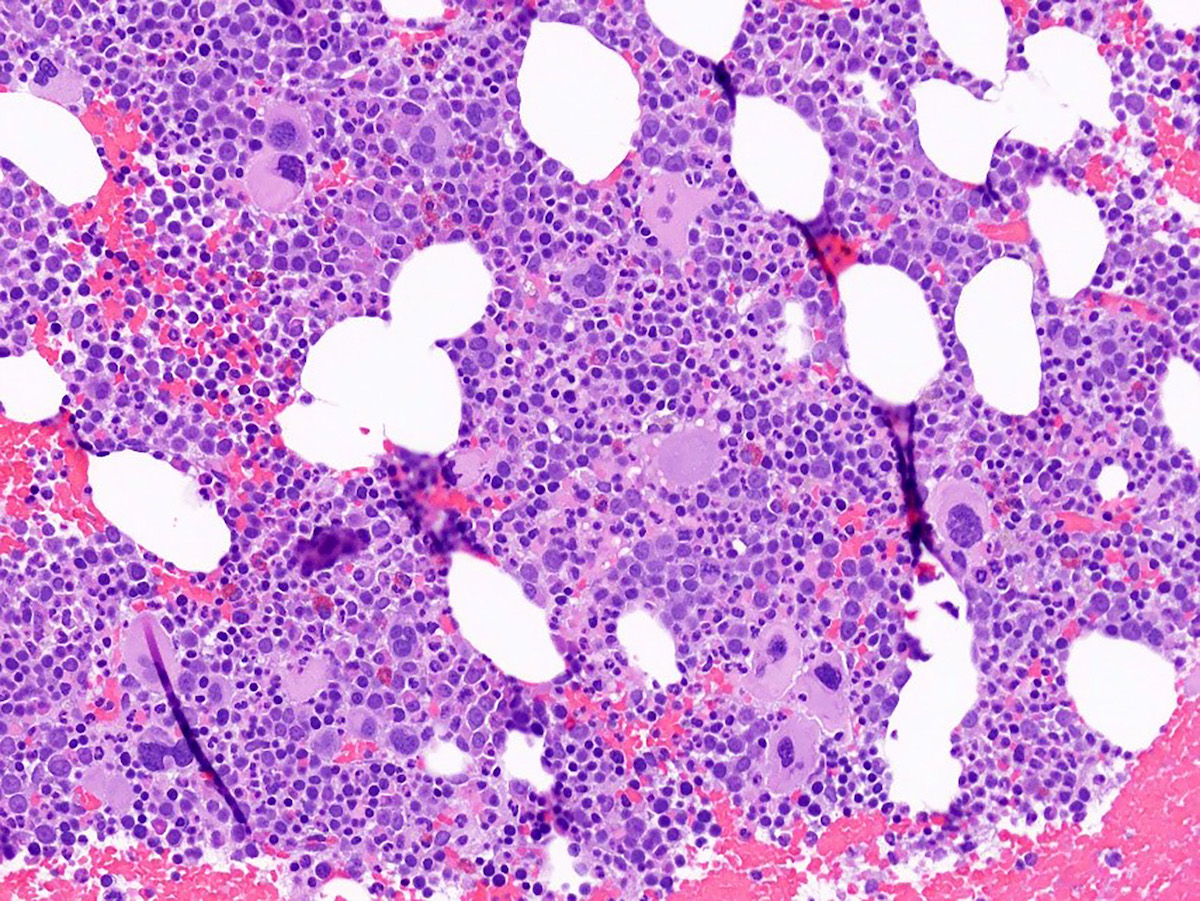
Hypercellular BM and megakaryocytic dysplasia
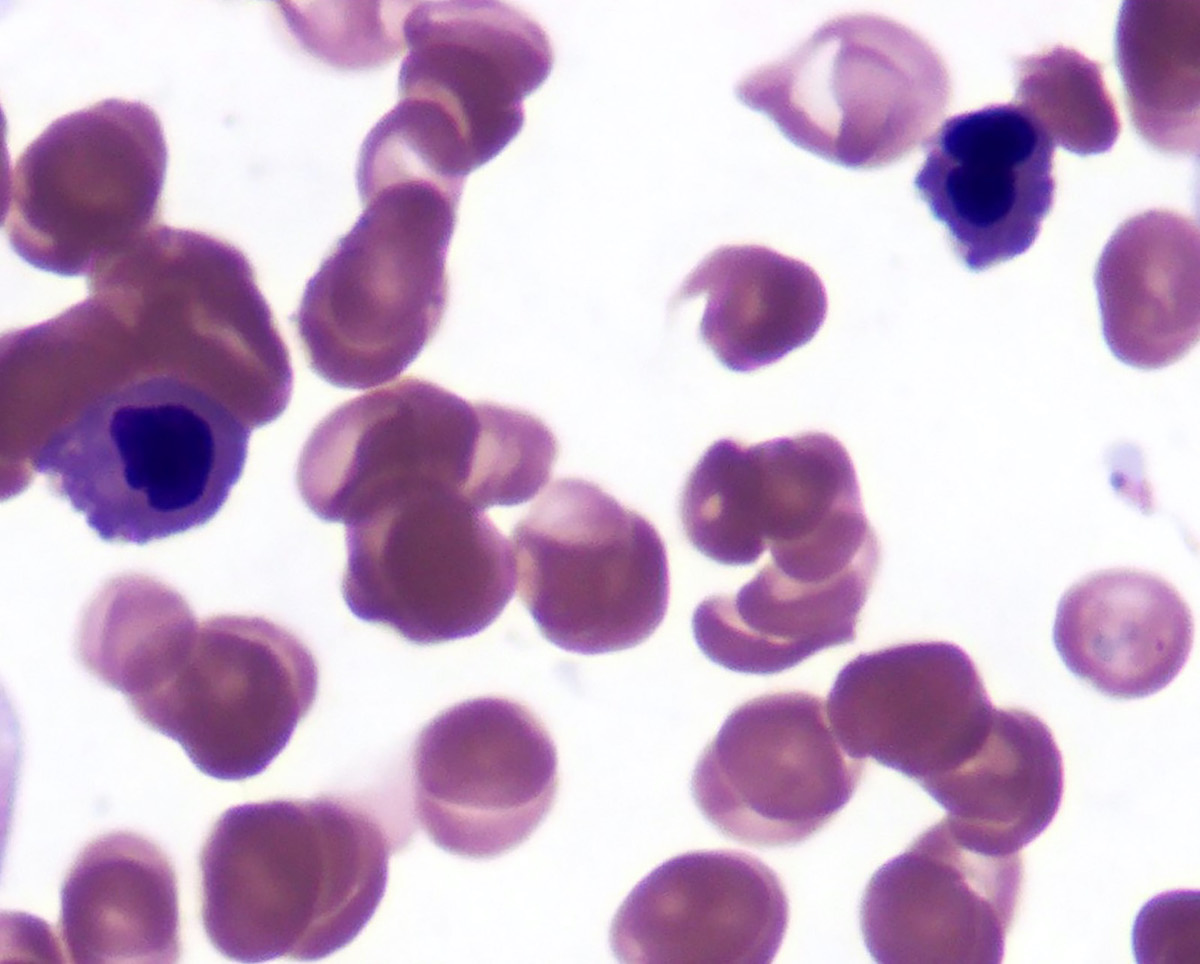

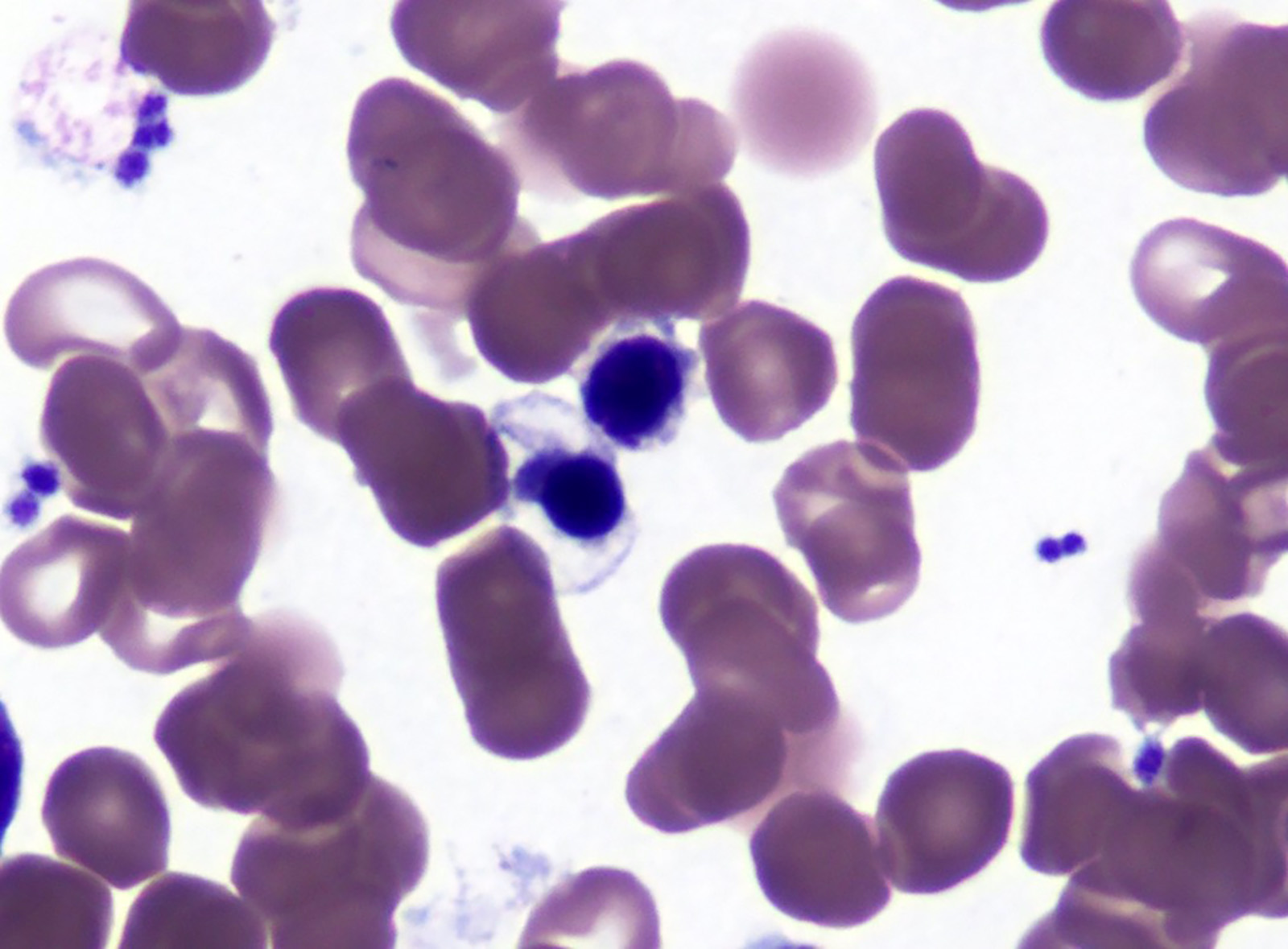
Erythroid dysplasia

Ring sideroblasts (Pearl stain)
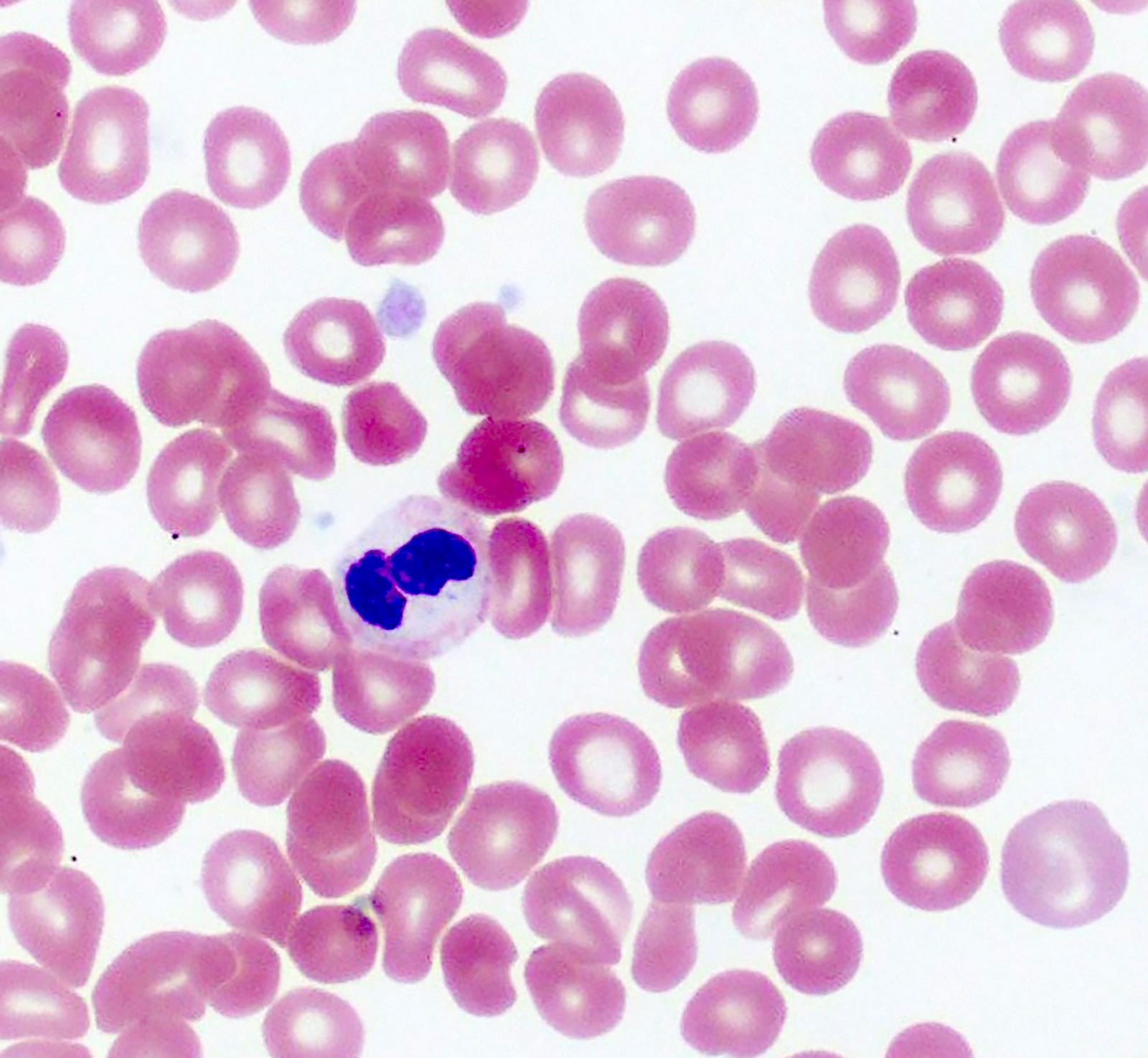
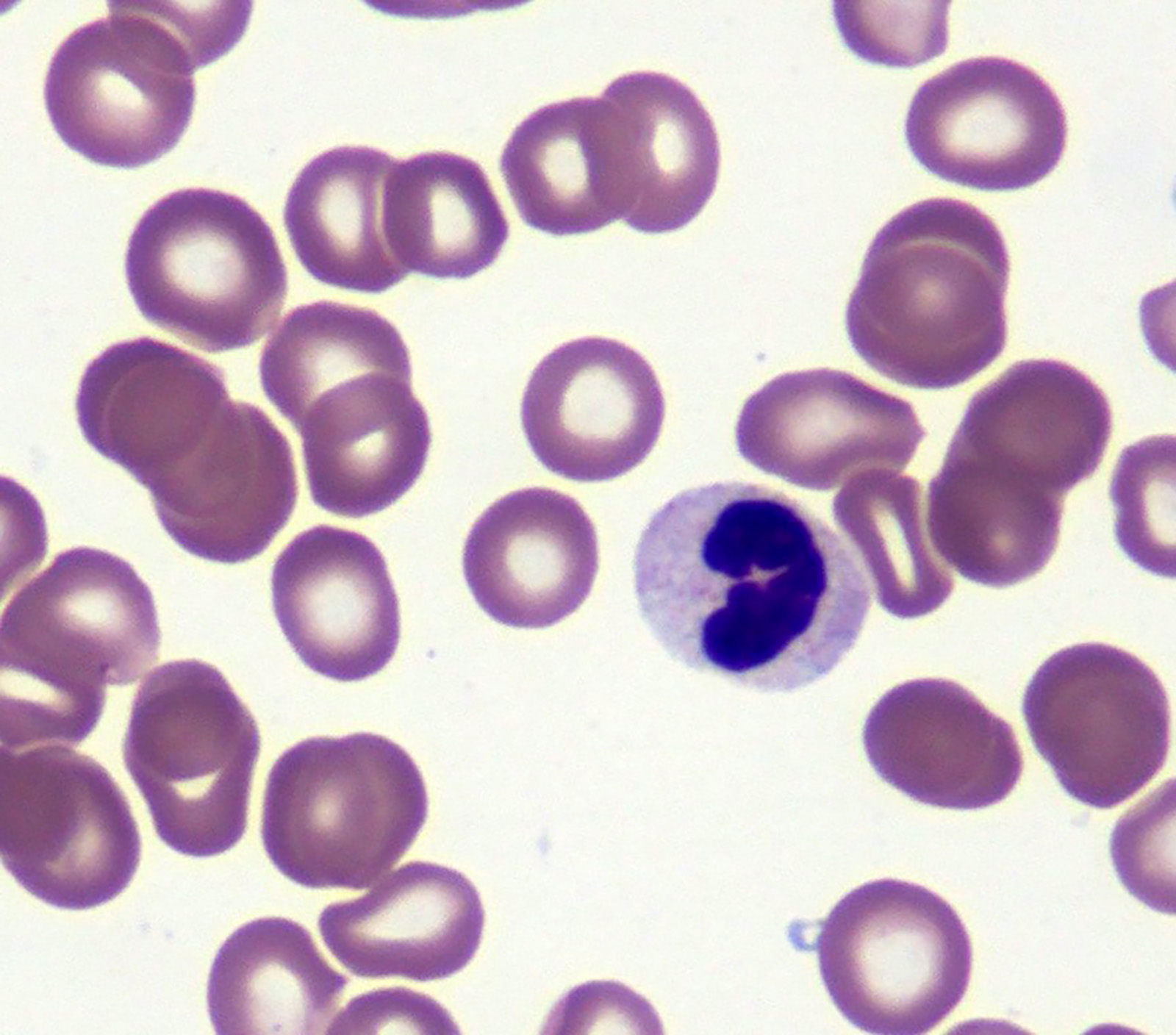
Myeloid dysplasia
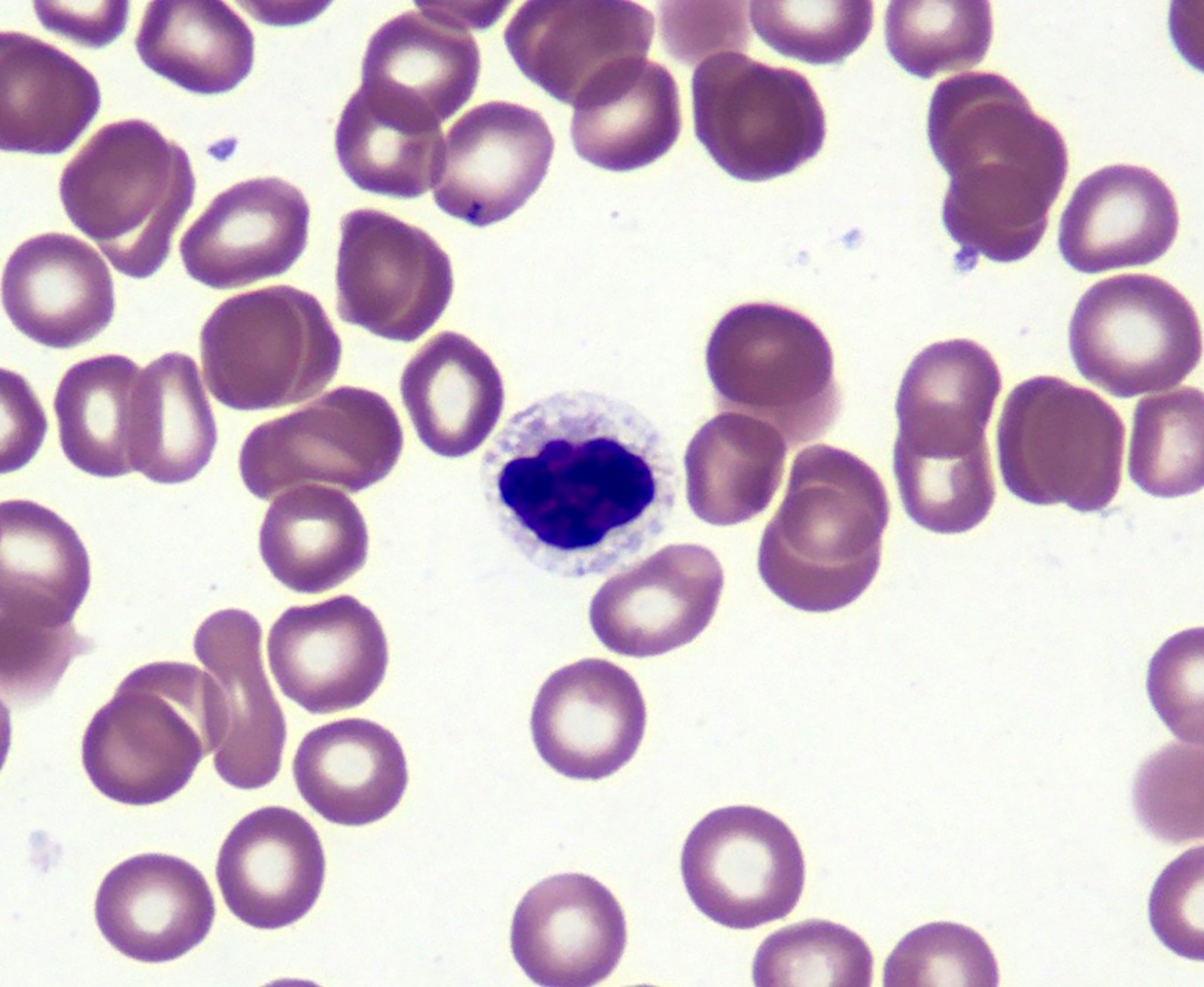
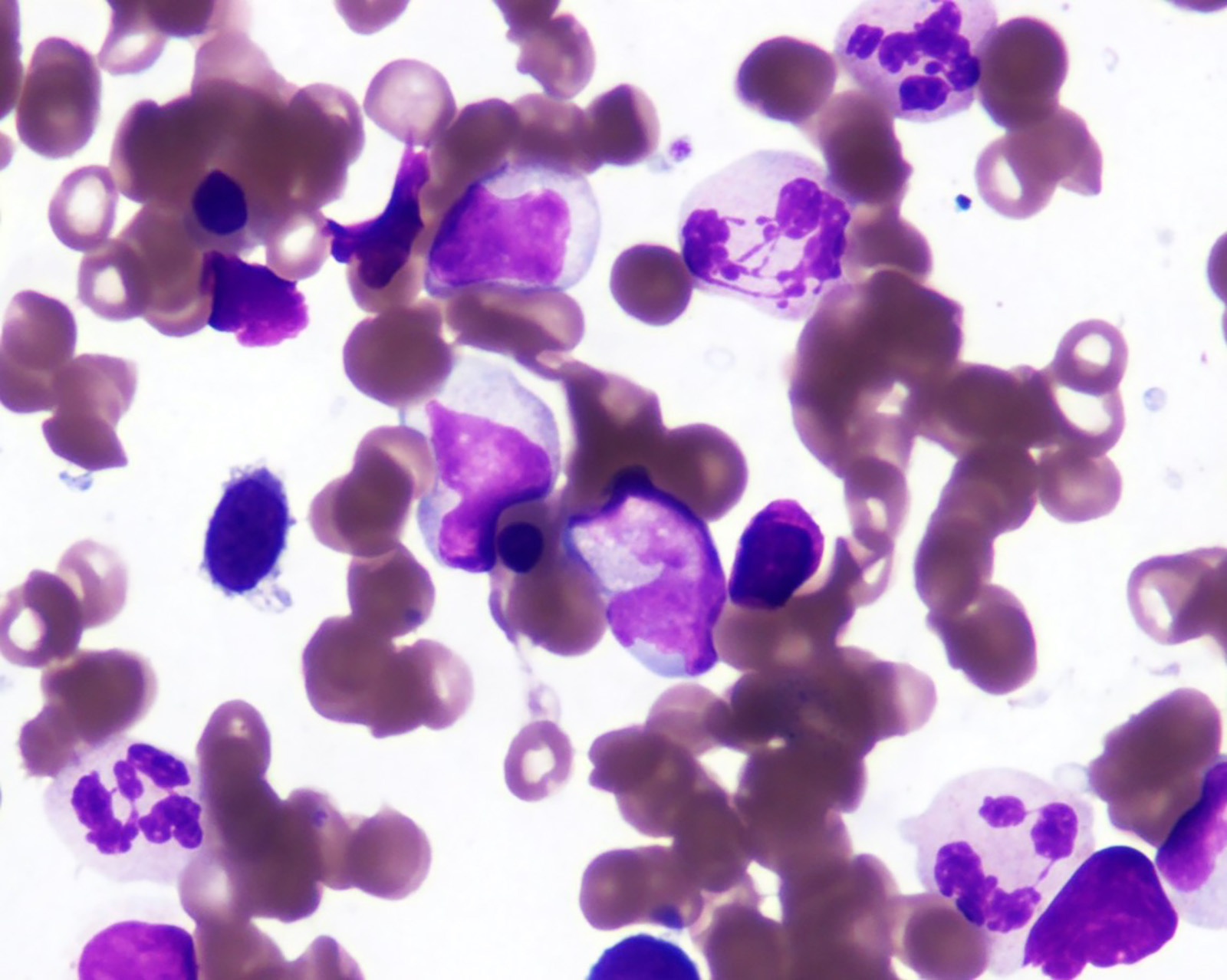
Myeloid dysplasia

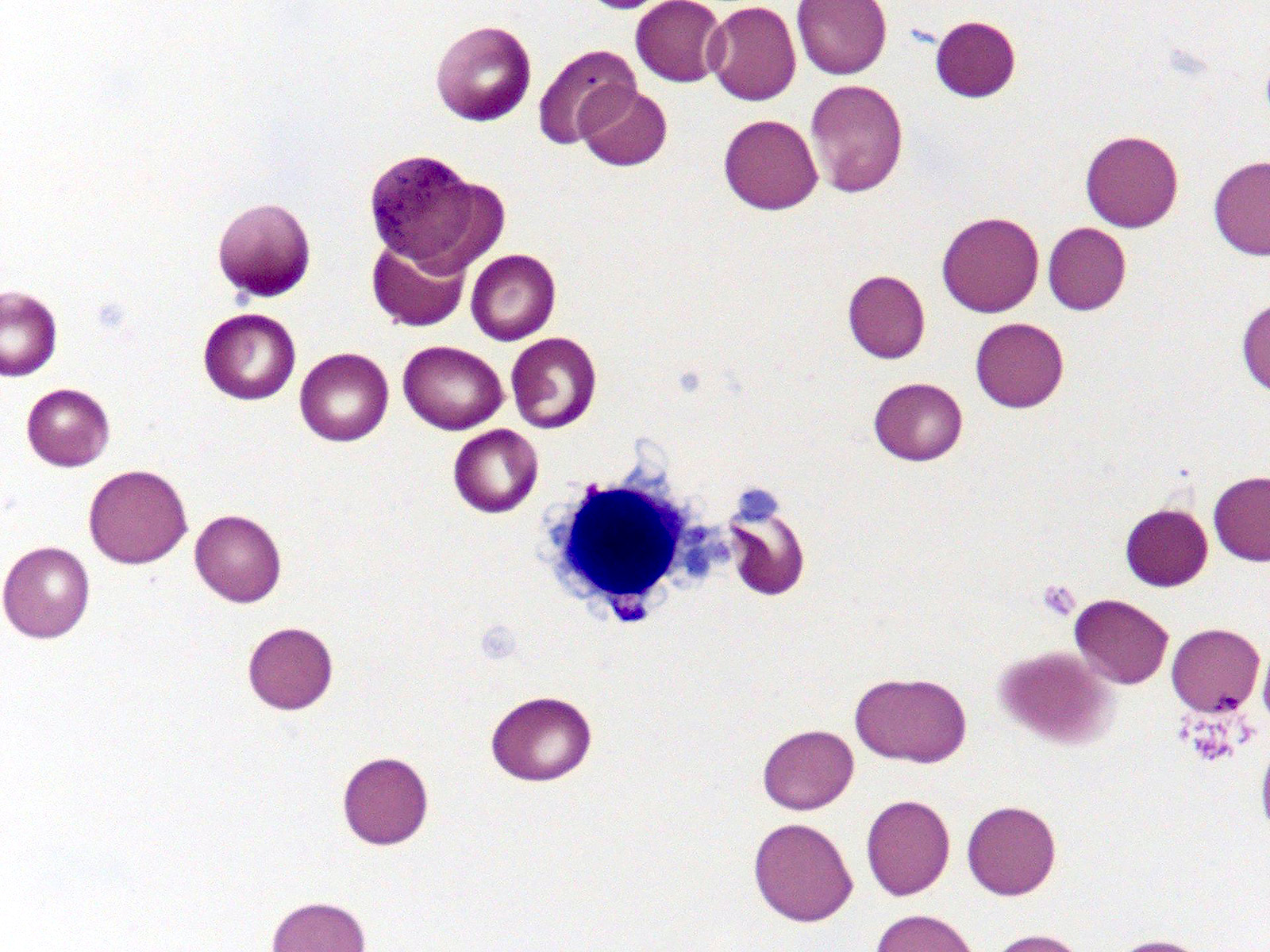
Megakaryocytic dysplasia
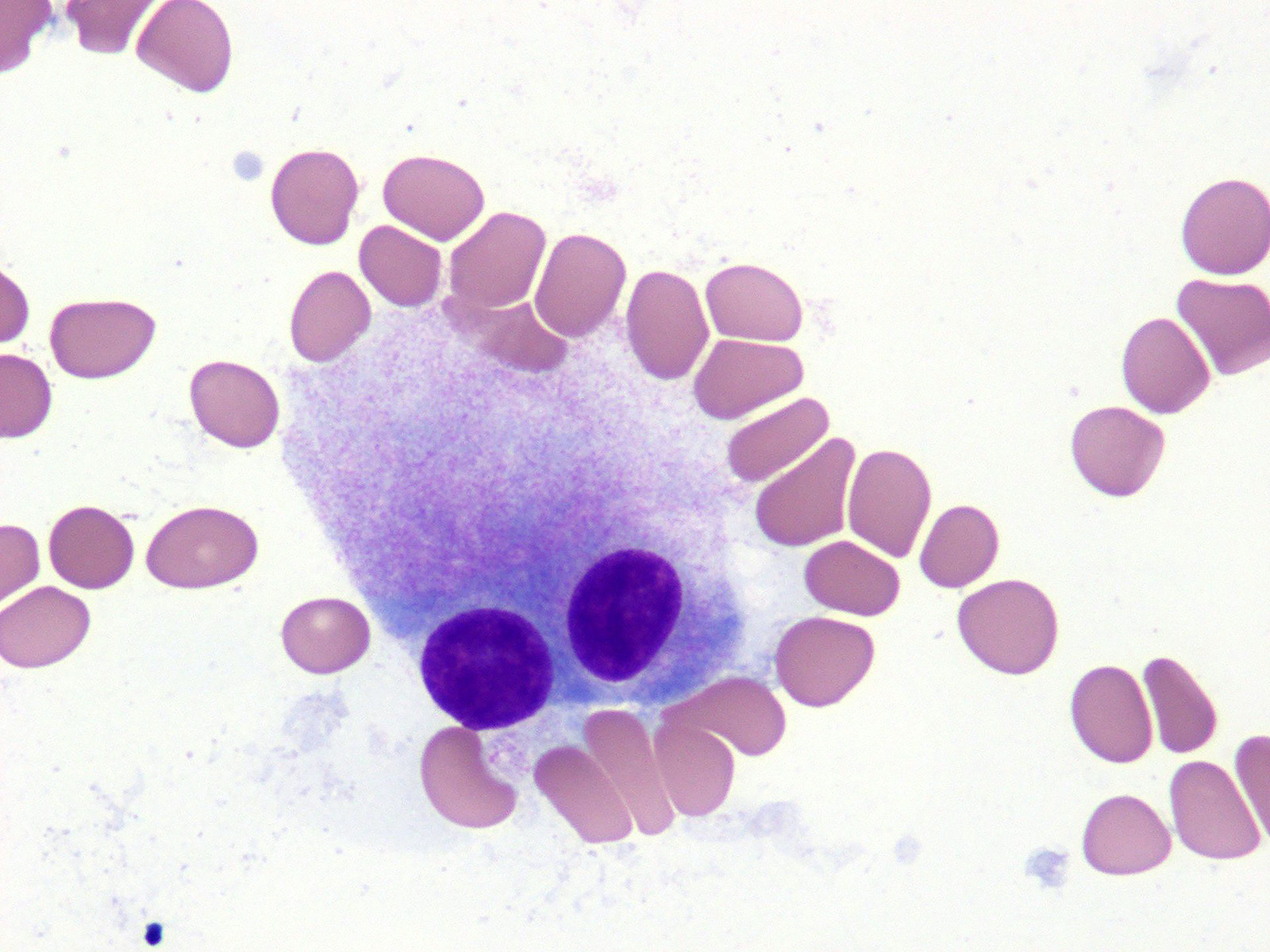
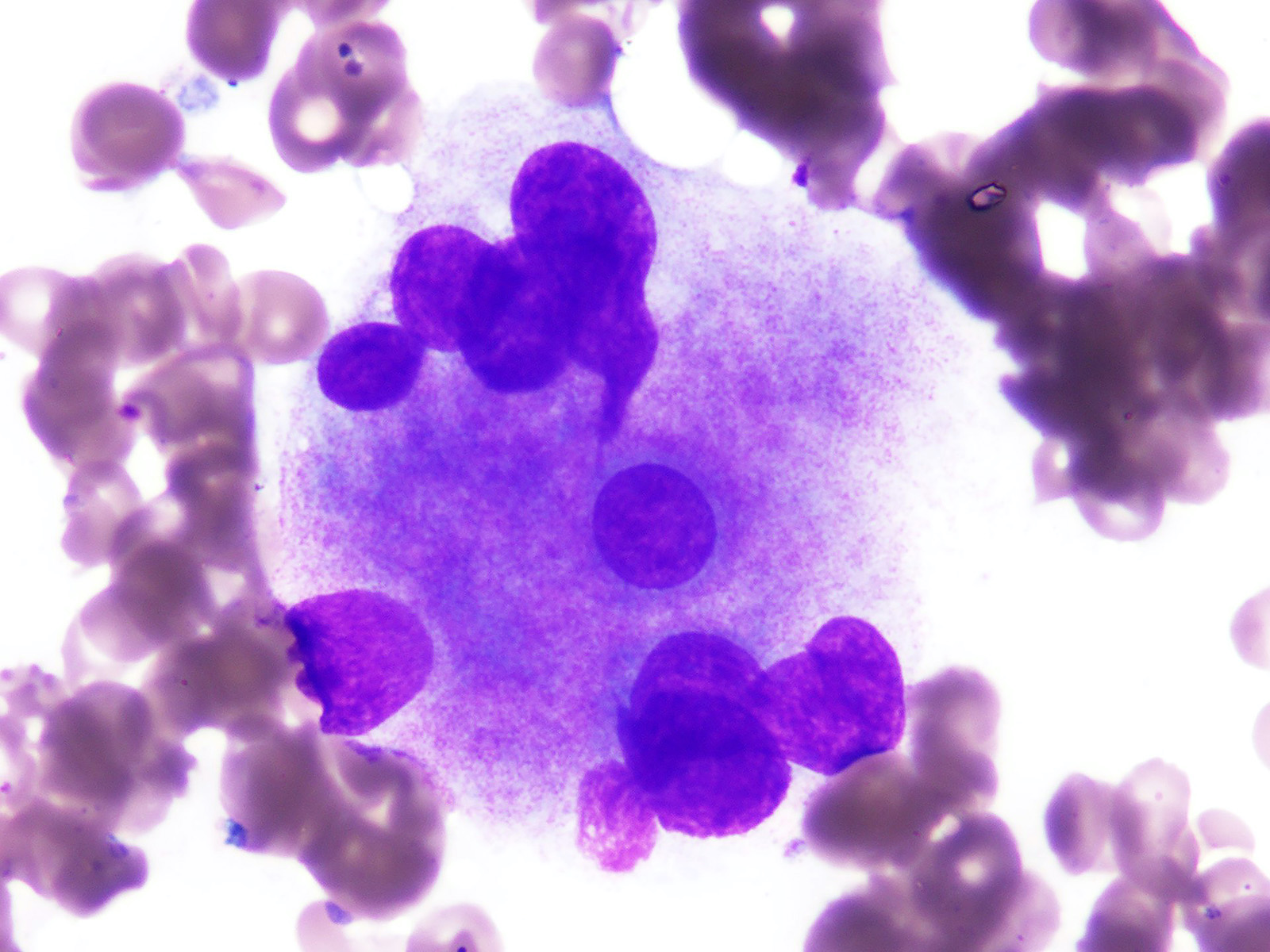
Megakaryocytic dysplasia
- Red blood cells are usually normocytic or macrocytic and normochromic with anisopoikilocytsois
- Granulocytes may show hypogranularity, uneven distribution of cytoplasmic granules, pseudo Pelger-Huët anomaly
- Blasts are usually < 1%
- Subset of platelets may be hypogranular
- Flow cytometry abnormalities in MDS (Oncotarget 2017;8:73483, Leukemia 2012;26:1730, Haematologica 2009;94:1066, Haematologica 2013;98:201):
- Detection of an aberrant myeloblast population by flow cytometry helps in distinguishing reactive / nonclonal causes of dysplasia from clonal causes of MDS
- Abnormal CD34 positive myeloblast population: altered CD45 and side scatter (CD45 / side scatter), increased blasts (> 2%), increased intensity of CD13, CD34, CD117, CD123, decreased intensity of CD38 or HLA-DR, substantial increase in cells with CD4, CD15, CD64 or CD65 expression, aberrant expression of lymphoid antigens (CD2, CD5 or CD7) and decreased hematogones
- Abnormal myelomonocytic maturation:
- Myeloid cells: decreased side scatter of granulocytes, abnormal maturation patterns of CD11b, CD13 and CD16 expression, lack of CD10 expression on neutrophils
- Monocytes: decreased CD45 and side scatter, decreased expression of HLA-DR, CD13, CD14 and CD36; increased expression of CD15 or CD16, aberrant expression of CD56 (bright) and CD2
- Detection of an aberrant myeloblast population by flow cytometry helps in distinguishing reactive / nonclonal causes of dysplasia from clonal causes of MDS
Contributed by Beenu Thakral, M.D.
CD45 versus side scatter
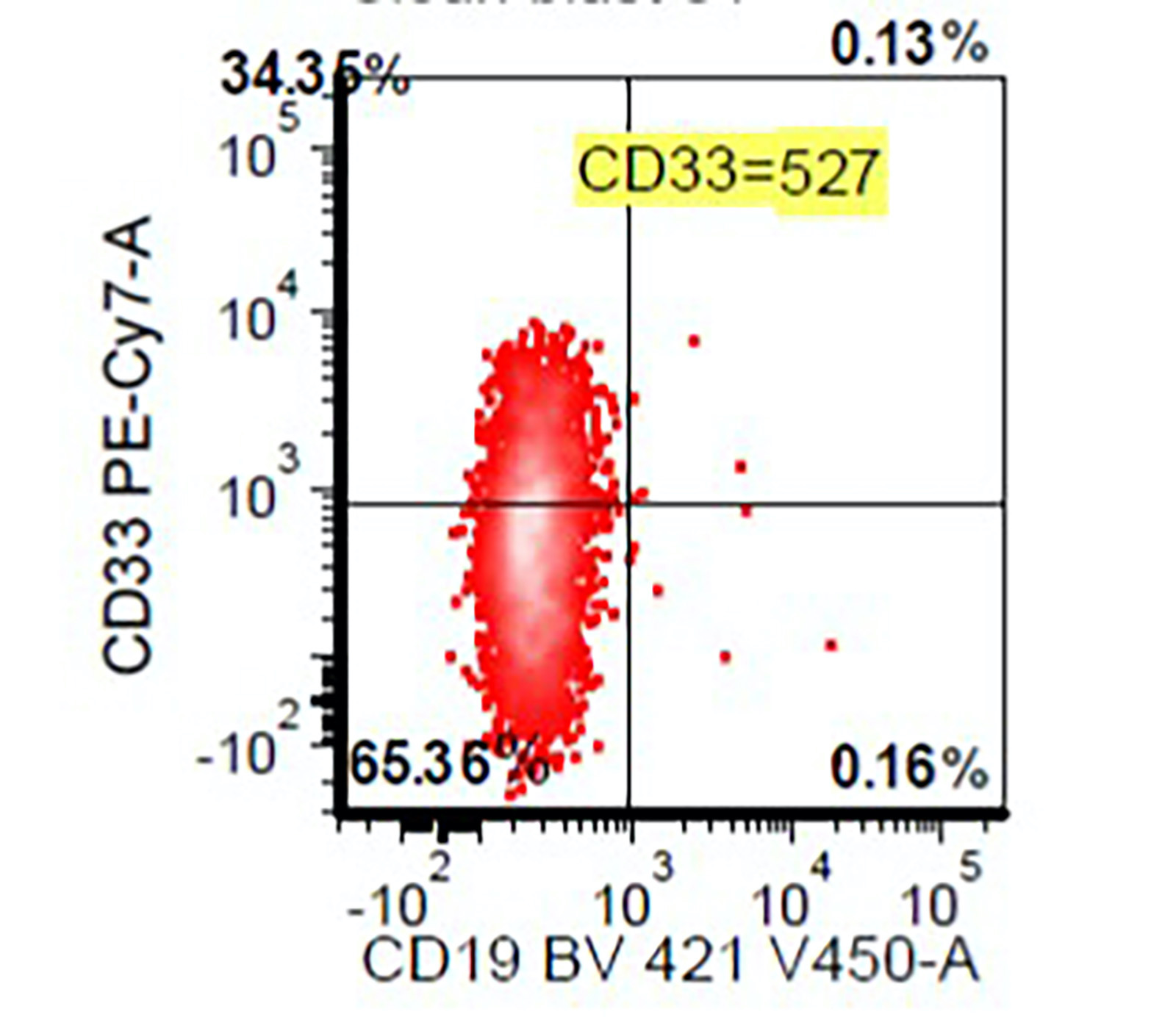
CD19 versus CD33
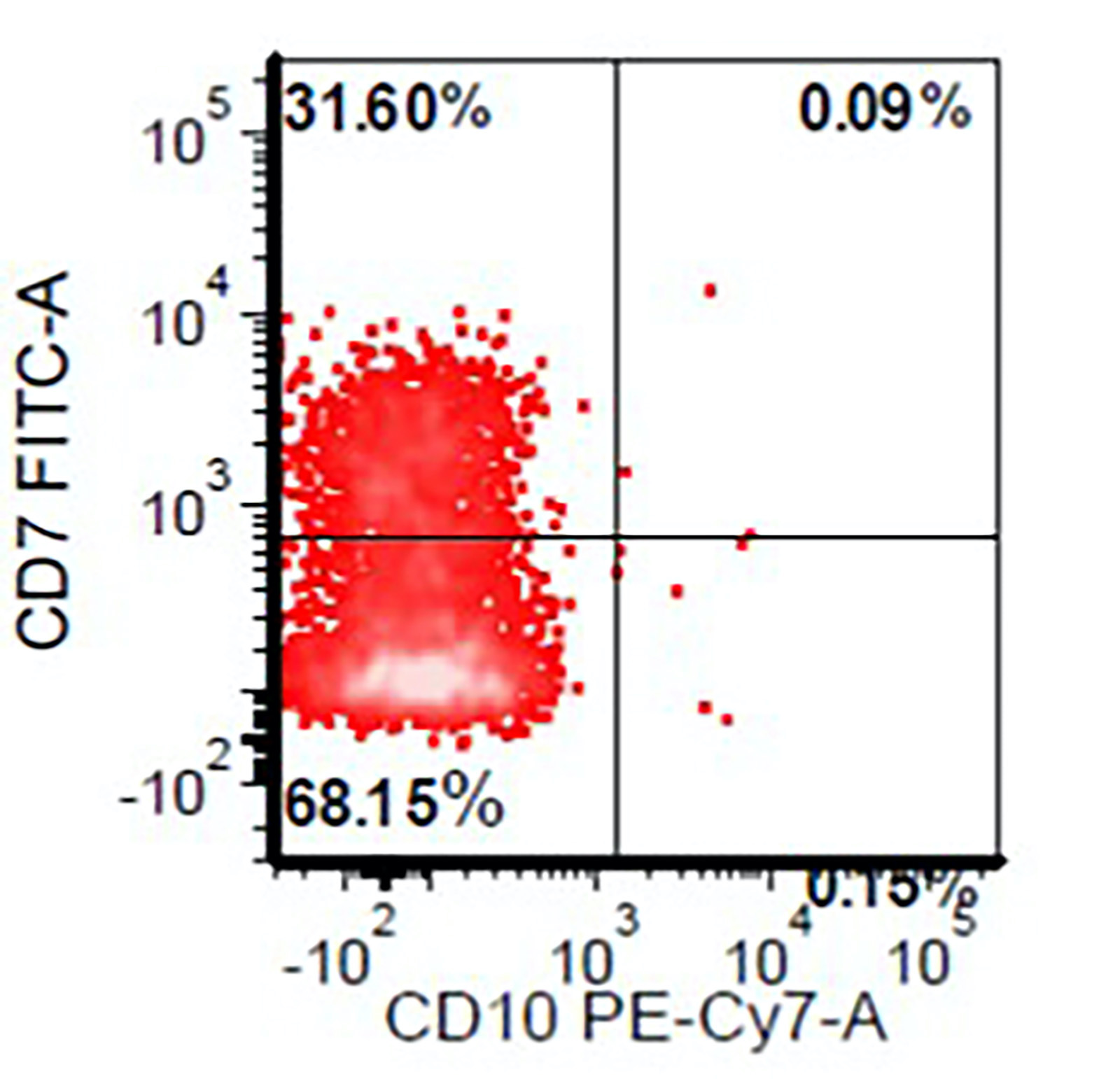
CD7 versus CD10
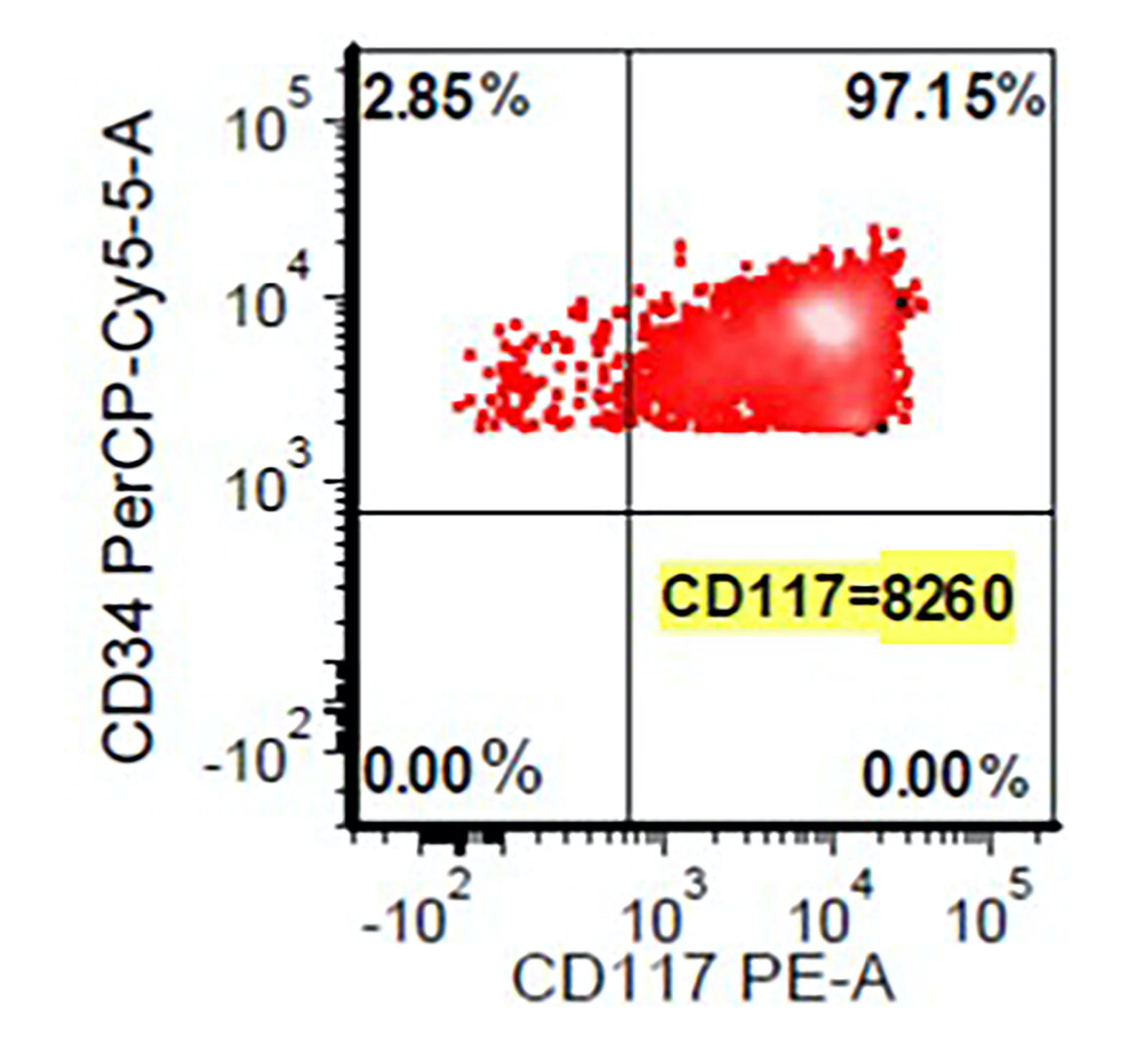
CD117 versus CD34
- Cytogenetics findings (J Clin Oncol 2012;30:820):
- Conventional karyotyping should be performed in all MDS cases at diagnosis, as it defines prognosis
- Cytogenetic abnormalities are seen in 50 - 60% of MDS; these abnormalities usually include del20q, gain in chromosome 8 or abnormalities in chromosome 5 and 7
- MDS SLD with isolated thrombocytopenia, the presence of cytogenetic or molecular abnormality is helpful in distinguishing MDS from immune mediated thrombocytopenia
MDS defining cytogenetic abnormalities
Unbalanced cytogenetic abnormalities
|
Balanced cytogenetic abnormalities
|
- Molecular findings (Int J Lab Hematol 2020;42:671, Blood 2012;119:3578, Leuk Res 2015;39:6):
- Recurrent somatic mutations are identified in 80 - 90% of MDS cases
- Most commonly mutated genes in MDS include RNA splicing (SF3B1, SRSF2, U2AF1 and ZRSR2) or epigenetic regulation of gene expression via DNA methylation (TET2, DNMT3A, IDH1 and IDH2) or genes involved in histone modification (ASXL1 and EZH2)
- Specific mutations associated with morphologic features and MDS include SF3B1 mutation with ring sideroblasts and mutations in ASXL1, RUNX1, TP53 and SRSF2 are associated with severe granulocytic dysplasia
- Acquired clonal mutations identical to those seen in MDS (TET2, DNMT3A, SF3B1, ASXL1 and JAK2) can also occur in hematopoietic cells of apparently healthy older individuals without MDS; thus, the presence of MDS associated somatic mutations alone are not considered diagnostic of MDS even in patients with unexplained cytopenia
Genes commonly mutated in MDS
| SF3B1 | RNA splicing | 20 - 30% | Favorable |
| TET2 | DNA methylation | 20 - 30% | Neutral |
| ASXL1 | Histone modification | 15 - 20% | Adverse |
| SRSF2 | RNA splicing | 15% | Adverse |
| DNMT3A | DNA methylation | 10% | Adverse |
| RUNX1 | Transcription factor | 10% | Adverse |
| U2AF1 | RNA splicing | 5 - 10% | Adverse |
| TP53 | Tumor suppressor | 5 - 10% | Adverse |
| EZH2 | Histone modification | 5 - 10% | Adverse |
| ZRSF2 | RNA splicing | 5 - 10% | Neutral |
| STAG2 | Cohesion complex | 5 - 7% | Adverse |
| IDH1 / IDH2 | DNA methylation | 5% | Neutral |
| CBL | Signaling | 5% | Adverse |
| NRAS | Transcription factor | 5% | Adverse |
| BCOR | Transcription factor | 5% | Adverse |
- Bone marrow, right posterior iliac crest, core biopsy, clot section, aspirate smears, touch imprint and peripheral blood smear:
- Myelodysplastic syndrome with single lineage dysplasia, 1% blast (see comment)
- Comment: Flow cytometry immunophenotyping performed on bone marrow aspirate shows an aberrant myeloblast population (0.8% of total events) with the following immunophenotype: CD7 partial, CD13+, CD33+, CD34+, CD117 inc and CD38 dec. Hematogones are absent. No monoclonal B cells or immunophenotypically abnormal T cells are identified.
- Review of peripheral blood smear shows moderate macrocytic anemia with slight anisopoikilocytosis and no circulating blast.
- Cytogenetic study shows a normal female karyotype: 46,XX[20]
- Next generation sequencing identified mutations in U2AF1 and TET2 genes.
- The current bone marrow specimen shows hypercellular (80%) bone marrow with erythroid predominant trilineage hematopoiesis, dyserythropoiesis, 3% ring sideroblasts and no increase in blasts; this is consistent with myelodysplastic syndrome with single lineage dysplasia (WHO 2016 revised classification).
Characteristics of ICUS, CHIP / ARCH, CCUS and MDS
(ICUS) | (CHIP / ARCH) | (CCUS) | (MDS) | |
| Clonality | ||||
| Cytopenia | ||||
| Dysplasia | ||||
| Bone marrow blasts | ||||
| Cytogenetic abnormality | ||||
| Typical number of mutated genes | ||||
| Commonly mutated genes | NA | |||
| Typical VAF (variants with allele frequency) | ||||
| Risk of developing MDS or acute myeloid leukemia | Very low | Low | High | NA |
- MDS with ring sideroblasts and single lineage dysplasia:
- Characterized by unilineage dysplasia and distinguished from MDS SLD by presence of ≥ 15% ring sideroblasts or ≥ 5% ring sideroblasts, if SF3B1 mutation is present
- MDS unclassifiable:
- Cases with single lineage dysplasia in presence of 1% blasts in peripheral blood on 2 separate occasions or by presence of pancytopenia
- Nonclonal causes of dysplasia:
- Drugs, medications and toxin exposure
- Growth factor therapy
- Viral infection
- Immunological disorders
- Vitamin deficiencies: B12, folate (Clin Lymphoma Myeloma Leuk 2014;14:e141)
- Copper deficiency or zinc excess (Pathology 2014;46:246)
- References: Am Soc Clin Oncol Educ Book 2019;39:400, Am J Clin Pathol 2019;152:347, Eur J Haematol 2021;106:500, Blood Adv 2021;5:2272
- Myelodysplastic syndrome, unclassifiable
- Myelodysplastic syndrome with del5q
- Myelodysplastic syndrome with ring sideroblasts
- Myelodysplastic syndrome with single lineage dysplasia
Comment Here
Reference: MDS with single lineage dysplasia
- ASXL1
- RUNX1
- SF3B1
- STAG2
- TP53
- Myelodysplastic neoplasms (MDS), previously called myelodysplastic syndromes, are clonal hematopoietic stem cell neoplasms characterized by ineffective marrow hematopoiesis that results in morphologic dysplasia and peripheral cytopenia; there is increased risk of transformation to acute myeloid leukemia (AML)
- This topic discusses myelodysplastic syndromes / neoplasms based on the WHO 2022 classification; differences between WHO 2022 and the ICS classification system are noted in Diagrams / tables below
- The definition of cytopenia comes from clonal cytopenia of undetermined significance (CCUS), MDS and myelodysplastic / myeloproliferative neoplasm (MDS / MPN)
- 2 main categories are accepted for subclassification of MDS
- MDS with defining genetic abnormalities, which is encompassed by MDS-5q, MDS-SF3B1 and MDS with biallelic TP53
- MDS, morphologically defined, consisting of MDS with low blasts, MDS with increased blasts and MDS, hypoplastic
- There is also a third category named MDS of childhood
- The previous subclassification of MDS-U has been eliminated from the new version of WHO classification
- Clarifications for the 2022 WHO MDS diagnoses
- Criteria for cytopenia
- Anemia: hemoglobin < 13 g/dL in men, < 12 g/dL in women
- Leukopenia: absolute neutrophil count < 1.8 x 109/L
- Thrombocytopenia: low platelet count < 150 x 109/L
- Threshold for dysplasia remains at 10% dysplastic cells
- Criteria for cytopenia
- Reference: Blood 2016;127:2391
- Per the fifth edition of WHO published in 2022, the terminology of myelodysplastic neoplasms has been introduced to replace the previous term of myelodysplastic syndromes (MDS), in parallel to myeloproliferative neoplasm and to emphasize the neoplastic course of the disease; however, the abbreviation of MDS is kept for myelodysplastic neoplasm
- Increased blasts is a term that has been adopted to replace excess blasts as previously used in the fourth edition of WHO classification
- ICD-10: D46 - myelodysplastic syndromes
- Myelodysplastic neoplasms with defining genetic abnormalities
- Myelodysplastic neoplasm with low blasts and 5q deletion (MDS-5q)
- Anemia, commonly macrocytic, transfusion dependent, with or without other cytopenias; thrombocytopenia is uncommon, instead, 33% of patients present with thrombocytosis (platelet count ≥ 450 x 109)
- Blasts < 5% in bone marrow and < 2% in peripheral blood
- Normo or hypercellular marrow
- Dysplasia involving megakaryocytes with characteristic features (megakaryocytes, intermediate in size, with single lobation), with or without dysplasia involving other lineages (see Microscopic (histologic) images)
- Often associated with erythroid hypoplasia
- Ring sideroblasts can be seen
- Detection of 5q deletion, isolated or with one other cytogenetic aberration but not including monosomy 7 or 7q deletion; not fulfilling diagnostic criteria of AML, MDS with biallelic TP53 inactivation, MDS with increased blasts or MDS / MPN
- MDS-5q with SF3B1 mutation (20% of the cases) are included in this category, as the SF3B1 mutation is likely a secondary event in this context
- Myelodysplastic neoplasm with low blasts and SF3B1 mutation (MDS-SF3B1)
- Detection of SF3B1 mutation with variant allele frequency (VAF) ≥ 5%; lower than the threshold is not considered diagnostic
- Cytopenia involving ≥ 1 lineages, most commonly anemia, without thrombocytosis (presence of thrombocytosis and SF3B1 without del(5q) belongs to MDS / MPN with SF3B1 mutation)
- Blasts < 5% in bone marrow and < 2% in peripheral blood
- Erythroid preponderance, mild left shifted, with megaloblastoid maturation or dysplasia; single or multilineage dysplasia can be observed in the subcategory, without impact on diagnosis or prognosis (Blood 2015;126:233, Blood 2020;136:157)
- Ring sideroblasts are often identified; however, an absence of ring sideroblasts in the setting does not exclude the diagnosis
- If SF3B1 mutation analysis is not available, demonstration of ring sideroblasts comprising ≥ 15% of erythroid precursors can be used as a surrogate
- Absence of 5q deletion, monosomy 7 / 7q deletion or complex karyotype
- Not fulfilling diagnostic criteria of AML, MDS with low blasts and 5q deletion, MDS with biallelic TP53 inactivation, MDS with increased blasts or any MDS / MPN type
- Myelodysplastic neoplasm with biallelic TP53 inactivation (MDS-biTP53)
- Detection of ≥ 1 TP53 mutations, meeting molecular diagnostic criteria of biallelic TP53 mutations (see Diagnosis)
- Cytopenia, dysplasia and < 20% blasts or 30% erythroblasts in bone marrow
- Next generation sequencing analysis or Sanger sequencing covering at least exons 4 - 11 of TP53 gene is required for detection of biallelic TP53 alterations, coupled with a technique to detect copy number status, usually fluorescence in situ hybridization (FISH) with a probe set specific for the TP53 locus on 17p13.1 (Leukemia 2014;28:241, Blood 2013;122:3616)
- Detection of ≥ 2 TP53 mutations, usually affecting both alleles that can be considered multihit status (Nat Med 2020;26:1549)
- After exclusion of constitutional changes, a TP53 VAF > 49% may be regarded as presumptive (not definitive) of copy loss on the trans allele or copy neutral loss of heterozygosity (LOH)
- In the presence of one TP53 mutation, evidence of TP53 copy loss or copy neutral LOH is required as concurrent 17p LOH, suggestive of biallelic TP53 alterations (Nat Med 2021;27:1239, Nat Commun 2020;11:4980)
- Characterized by increased blast counts, higher risk of leukemic transformation and higher risk of mortality
- Myelodysplastic neoplasm with low blasts and 5q deletion (MDS-5q)
- Myelodysplastic neoplasms, morphologically defined
- Myelodysplastic neoplasm with low blasts (MDS-LB)
- Low blasts are defined as < 5% in bone marrow and < 2% in peripheral blood
- Cytopenia involving ≥ 1 lineages
- Dysplastic changes in ≥ 1 lineages, involving at least 10% of cells
- Blasts < 5% in bone marrow and < 2% in peripheral blood
- Need to exclude folate and vitamin B12 deficiency
- No fulfilling diagnostic criteria of MDS with defining genetic alterations or hypoplastic MDS
- Myelodysplastic neoplasm, hypoplastic (h-MDS)
- Hypocellular bone marrow (adjusted for age of the patient); drug / toxin exposure or pertinent nutritional deficiency need to be excluded
- Cytopenia involving ≥ 1 lineages
- Dysplasia involving myeloid or megakaryocytic lineages
- Blasts < 5% in bone marrow and < 2% in peripheral blood
- Not meeting criteria for MDS with defining genetic abnormalities or MDS with increased blasts
- Myelodysplastic neoplasm with increased blasts (MDS-IB)
- Cytopenia involving ≥ 1 lineages
- Dysplastic changes in ≥ 1 lineages, involving at least 10% of cells
- Blasts ≥ 5% in bone marrow or ≥ 2% in peripheral blood
- No fulfilling diagnostic criteria of MDS with biallelic TP53 inactivation or AML
- Subclassification
- MDS with increased blasts 1 (MDS-IB1): 5 - 9% blasts in bone marrow or 2 - 4% blasts in peripheral blood, without significant reticulin fibrosis
- MDS with increased blasts 2 (MDS-IB2): 10 - 19% blasts in bone marrow or 5 - 19% blasts in peripheral blood, without significant reticulin fibrosis; or with presence of Auer rods
- MDS with increased blasts and fibrosis (MDS-F): 5 - 19% blasts in bone marrow or 2 - 19% blasts in peripheral blood, with significant reticulin fibrosis (defined as grade 2 or 3)
- Myelodysplastic neoplasm with low blasts (MDS-LB)
- Myelodysplastic neoplasms of childhood
- Childhood myelodysplastic neoplasm with low blasts (cMDS-LB)
- Myeloid neoplasm with cytopenia and dysplasia arising in children and adolescents (< 18 years of age)
- Cytopenia involving ≥ 1 lineage
- Dysplastic changes in ≥ 1 lineage, involving at least 10% of cells
- Blasts < 5% in bone marrow and < 2% in peripheral blood
- Meeting at least 1 of the following criteria
- Detection of clonal cytogenetic or molecular abnormality; monosomy 7 is the most common cytogenetic abnormality
- Need to exclude other causes of cytopenia (nonneoplastic and some germline mutations)
- Subclassification
- cMDS-LB, hypocellular
- cMDS-LB, not otherwise specified
- Childhood myelodysplastic neoplasm with increased blasts (cMDS-IB)
- Myeloid neoplasm with cytopenia and dysplasia arising in children and adolescents (< 18 years of age)
- Cytopenia involving ≥ 1 lineages
- Dysplastic changes in ≥ 1 lineages, involving at least 10% of cells
- Blasts 5 - 19% in bone marrow or 2 - 19% in peripheral blood
- Need to exclude Down syndrome, juvenile myelomonocytic leukemia and AML with defining genetic abnormalities
- Childhood myelodysplastic neoplasm with low blasts (cMDS-LB)
- See differences from WHO (2022) in Diagrams / tables
- MDS with single lineage dysplasia (MDS-SLD)
- MDS with ring sideroblasts
- MDS with ring sideroblasts and single lineage dysplasia (MDS-RS-SLD)
- MDS with ring sideroblasts and multiple lineage dysplasia (MDS-RS-MLD)
- MDS with multilineage dysplasia (MDS-MLD)
- MDS with excess blasts, type I and II (MDS-EB-I and MDS-EB-II)
- MDS with isolated del(5q) [MDS del(5q)]
- MDS, unclassifiable (MDS-U)
- Provisional: refractory cytopenia of childhood (RCC)
- Median age of MDS is 77 years at diagnosis; < 10% are < 50 years old
- Incidence of MDS increases progressively with age from 60 to 90 years of age (from 2.2 to 56.8 cases per 100,000) (Blood Rev 2019;34:1)
- M > F except in MDS with low blasts and 5q deletion (MDS-5q) where F > M (Am J Med 2012;125:S2)
| Comparison of guidelines | WHO 4th edition, 2016 | WHO 5th edition, 2022 | ICC 2022 | |
| Name of the entity | Myelodysplastic syndrome (MDS) | Myelodysplastic neoplasms (MDS) | Myelodysplastic syndromes (MDS) | |
| Morphologically defined | ||||
| Lineage | MDS-SLD MDS-MLD | Subclassification using dysplastic lineages removed, replaced with MDS-LB (MDS with low blasts) | MDS, NOS-SLD MDS, NOS-MLD | |
| Excessive blast counts in bone marrow (5 - 9%) or in peripheral blood (2 - 4%) | MDS-EB-I# | MDS-IB1 | MDS-EB* | |
| Excessive blast counts in bone marrow (10 - 19%) or in peripheral blood (5 - 19%) | MDS-EB-II# | MDS-IB2 | MDS-AML** | |
| Others | MDS-U | cMDS (MDS of childhood) MDS-h (hypoplastic MDS) MDS-f (MDS with fibrosis, blasts 5 - 19% in bone marrow, 2 - 19% in peripheral blood) | MDS, NOS (without dysplasia) | |
| Defining genetic abnormalities | ||||
| SF3B1 mutation | MDS-RS-SLD MDS-RS-MLD (5% ring sideroblasts if SF3B1 mutated, 15% ring sideroblasts if SF3B1 wild type) | MDS-SF3B1 (MDS with low blasts and SF3B1 mutation or MDS with ring sideroblasts if SF3B1 wild type) | MDS-SF3B1 (MDS with mutated SF3B1) or MDS, NOS (with ring sideroblasts and SF3B1 wild type) | |
| del(5q) | MDS with isolated del(5q) | MDS-5q (MDS with low blasts and 5q deletion) [5q deletion alone or with 1 other genetic aberration other than del(7q) / -7] | MDS with del(5q) [del(5q) is isolated or with up 1 genetic aberration except del(7q) / -7 or multihit TP53] | |
| TP53 | Not included in classification | MDS-biTP53 (MDS with biallelic TP53 inactivation) | MDS with mutated TP53 Multihit TP53 Includes MDS, MDS / AML, AML | |
- WHO 4th edition, 2016
- MDS-SLD: MDS with single lineage dysplasia
- MDS-MLD: MDS with multilineage dysplasia
- MDS-RS-SLD: MDS with ring sideroblasts and single lineage dysplasia
- MDS-RS-MLD: MDS with ring sideroblasts and multilineage dysplasia
- MDS-EB-I: MDS with excess blast I
- MDS-EB-II: MDS with excess blast II
- MDS del(5q): MDS with isolated del(5q)
- MDS-U: MDS, unclassifiable
- RCC: refractory cytopenia of childhood
- WHO 5th edition, 2022
- MDS-LB: MDS with low blasts
- MDS-IB1: MDS with increased blasts I
- MDS-IB2: MDS with increased blasts II
- cMDS: MDS of childhood
- MDS-h: hypoplastic MDS
- MDS-f: MDS with fibrosis
- MDS-SF3B1: MDS with low blasts and SF3B1 mutation or MDS with RS if SF3B1 wild type
- MDS-5q: MDS with low blasts and 5q deletion
- MDS-biTP53: MDS with biallelic TP53 inactivation
- ICC 2022
- MDS, NOS-SLD: MDS, not otherwise specified with single lineage dysplasia
- MDS, NOS-MLD: MDS, not otherwise specified with multilineage dysplasia
- MDS-EB: MDS with excess blasts
- MDS-AML: myelodysplastic syndrome / acute myeloid leukemia
- MDS-SF3B1: MDS with mutated SF3B1
- MDS, NOS: MDS, not otherwise specified
- * ICC MDS-EB, MDS with excess blasts (blast count 5 - 9% in bone marrow, 2 - 4% in peripheral blood)
- ** ICC MDS-AML: myelodysplastic syndrome / acute myeloid leukemia (blast count 10 - 19% in bone marrow and 2 - 4% in peripheral blood)
- # WHO 4th edition, 2016: MDS-EB including MDS-EB and erythroid preponderance and MDS-EB and fibrosis
- If a patient presented with sustained cytopenia and suspected MDS, an extensive clinical and laboratory workup is necessary to exclude other etiologies that could clinically or morphologically mimic MDS (e.g., nutritional deficiency [e.g., folate acid, vitamin B12, copper deficiency], infection [e.g., HIV], medication induced marrow suppression [e.g., cytoreduction therapy], autoimmune disorders [e.g., rheumatoid arthritis, SLE])
- Imaging study
- To detect hepatomegaly or splenomegaly, which is usually not present in MDS patients but often seen with primary myelofibrosis or chronic myelomonocytic leukemia
- Cytogenetic consulting
- For the patients with germline gene mutations (e.g., DDX41, GATA2, RUNX1) or family history of MDS or other bone marrow failure syndromes
- Reference: Blood 2016;127:2391
- A series of laboratory tests should be ordered
- Routine complete blood count (CBC) data with differential count
- Reticulocyte percentage: reticulocytopenia is commonly found in bone marrow failure, including MDS
- Serum vitamin B12, folate level: within normal limits
- Erythropoietin (EPO) levels are usually lower than normal serum ferritin; elevated in the presence of inflammatory conditions such as rheumatoid arthritis
- Serum iron levels and total iron binding capacity, along with serum ferritin, may be helpful to exclude iron deficiency anemia
- HIV screening
- Screening for paroxysmal nocturnal hemoglobinuria (PNH) and T cell large granular lymphocytic leukemia
- Microscopic examination of bone marrow and peripheral blood smears to identify morphologic dysplasia and enumerate blasts
- Chromosome analysis, e.g., conventional karyotyping, fluorescence in situ hybridization using specific probes, most commonly including del(5q) / -5, del(7q) / -7, +8, del(17p) / TP53 and del(20q)
- Mutation analysis (e.g., next generation sequencing myeloid targeted mutation profiling certain gene mutations are often seen in MDS cells including DNMT3A, TET2, ASXL1, TP53, RUNX1, SRSF2 and SF3B1)
- Optional flow cytometric analysis for immunophenotyping for monocyte subset, blasts or altered myeloid maturation (Blood 2014;123:3675)
- Immunohistochemical staining for assessment of blasts, abnormal localization of immature precursors, dysplastic megakaryocytes, p53 expression and altered trilineage distribution
- Myelodysplastic neoplasm with low blasts and 5q deletion (MDS-5q)
- Good prognosis cytogenetic subgroup in the Revised International Prognostic Scoring System (Blood 2012;120:2454)
- Overexpressed p53 by IHC (≥ 1%, strong single) is associated with higher risk for AML transformation (Haematologica 2014;99:1041)
- Concurrent TP53, RUNX1, TET2 or single SF3B1 mutations may predict worse outcomes in MDS-5q (Leukemia 2016;30:1956, Blood 2020;136:157)
- Myelodysplastic neoplasm with low blasts and SF3B1 mutation (MDS-SF3B1)
- Favorable outcome among MDS types (Blood 2020;136:157)
- Concurrent mutations with TP53, RUNX1, EZH2 and FLT3 are associated with worse outcome (Blood 2020;136:157, Blood 2021;138:989)
- Presence of other molecular mutations (e.g., BCOR, BCORL1, NRAS, RUNX1, SRSF2 or STAG2) also alter clinical outcome when compared with single SF3B1 mutation
- Favorable outcome among MDS types (Blood 2020;136:157)
- Myelodysplastic neoplasm with biallelic TP53 inactivation (MDS-biTP53)
- Higher risk of leukemic transformation and higher risk of mortality, independent of Revised International Prognostic Scoring System risk categorization and independent of treatment (Nat Med 2020;26:1549)
- Similar adverse clinical outcomes are observed in MDS patients with mutated TP53 (VAF ≥ 40%) or complex cytogenetics
- Myelodysplastic neoplasm with low blasts (MDS-LB)
- Prognosis is usually linked to the degree of cytopenia (Blood 2012;120:2454)
- Myelodysplastic neoplasm, hypoplastic (h-MDS)
- Prognosis is better in this subtype of MDS (Leukemia 2022;36:1947)
- Myelodysplastic neoplasm with increased blasts (MDS-IB)
- Presence of fibrosis and CD7 expression were linked with worse outcome (Mod Pathol 2014;27:681, Blood 2002;100:3887)
- Childhood myelodysplastic neoplasm with low blasts (cMDS-LB)
- Patients with monosomy 7, 7q deletion or complex karyotype usually progress to AML and typically treated with hematopoietic stem cell transplantation (Blood 2018;131:1406)
- Normal karyotype or trisomy 8 are usually associated with a stable disease (Br J Haematol 2011;154:185)
- Patients with monosomy 7, 7q deletion or complex karyotype usually progress to AML and typically treated with hematopoietic stem cell transplantation (Blood 2018;131:1406)
- Childhood myelodysplastic neoplasm with increased blasts (cMDS-IB)
- Complex karyotype is associated with worse outcome (Nat Med 2021;27:1806)
Contributed by Ling Zhang, M.D.

Dysplastic erythroid precursors

Dysplastic myeloid
and megaloblastic
erythroid precursors
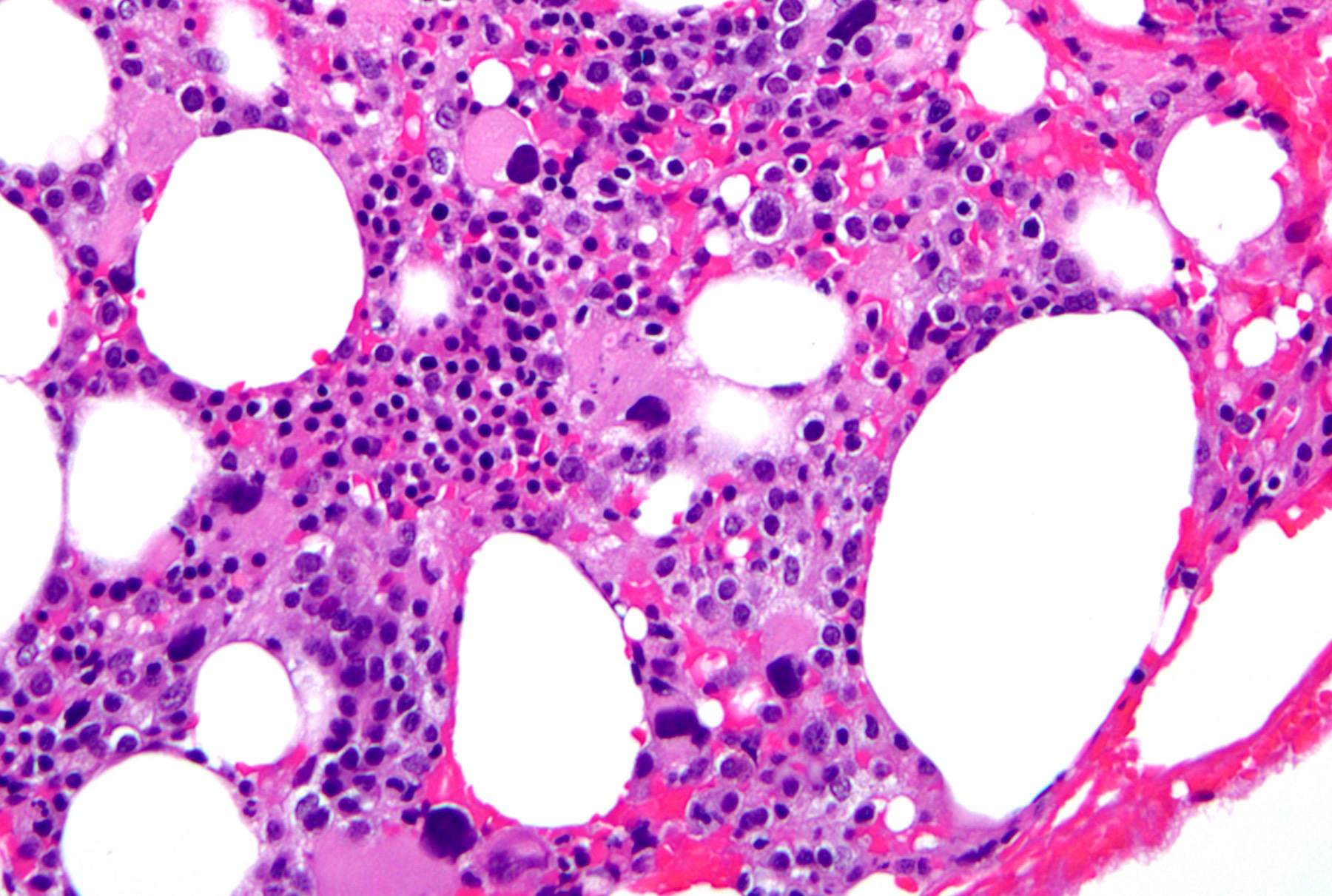
Bone marrow core biopsy, MDS with del(5q)


Aspirate smear, MDS with del(5q)

MDS, SF3B1 mutated with increased ring sideroblasts
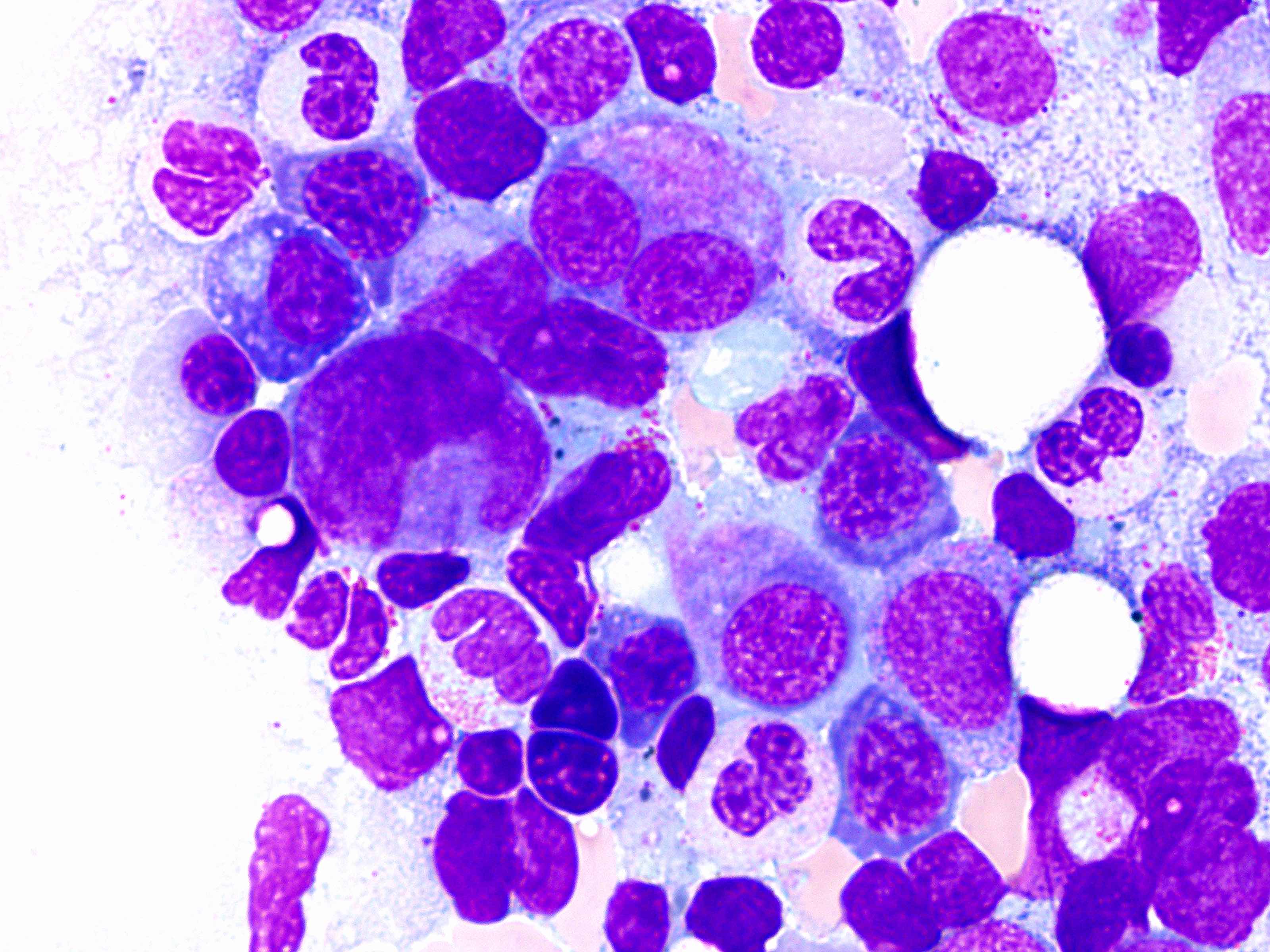
Dysplastic
megakaryocytes
and granulocytes
with biallelic TP53

MDS transformation to AML-MR

ALIP in bone marrow biopsy

ALIP CD34 immunostaining
Contributed by Ling Zhang, M.D.
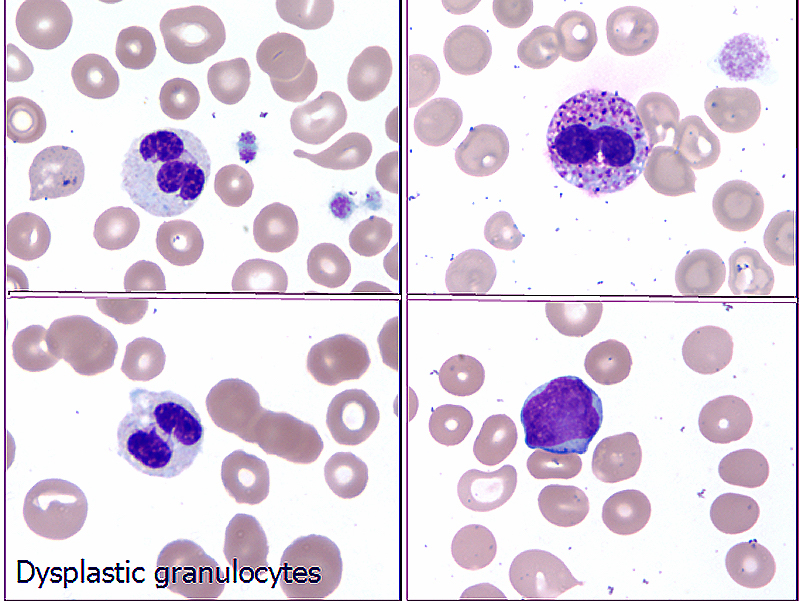
Dysplastic granulocytes and peripheral blood blast
- Bone marrow, right posterior iliac crest, biopsy and aspiration:
- Hypercellular marrow (90%) with trilineage dyspoiesis and increased blasts (7%) consistent with myelodysplastic syndrome with increased blasts (MDS-IB) (see comment)
- Comment: Immunochemistry with CD34 and CD117 confirms the presence of increased proportion of blasts. Mild diffuse reticulin fibrosis is noted. FISH MDS panel shows both del(5q) and del(7q). No BCR::ABL1 rearrangement is identified. Karyotyping shows 46,XY,del(5q)(27;q33),del(7q)(q22),+21[10]/46,XY[10] and NGS study reveals several tier 1 pathologic mutations involving ASXL1 (VAF of 27%), DNMTA (VAF of 33%) and RUNX1 (VAF of 6.5%). There are no JAK2, NPM1 or TP53 mutations detected.
- Absence of ring sideroblasts excludes the diagnosis
- Blasts are > 5% in bone marrow
- Detection of SF3B1 mutation with any percentage of variant allele frequency is considered diagnostic
- Favorable outcome among MDS types
- If SF3B1 mutation analysis is not available, demonstration of ring sideroblasts comprising ≥ 5% of erythroid precursors can be used as a surrogate
Comment Here
Reference: MDS-general
- Associated with low risk disease
- Presence of biallelic TP53 mutations is required for diagnosis
- Rarely transforms to acute myeloid leukemia
- Sensitive to conventional therapies
Comment Here
Reference: MDS-general
- The term monoclonal gammopathy of undetermined significance (MGUS) was introduced in 1978 by Robert Kyle to describe benign asymptomatic patients having an M protein that may progress to other malignancies
- By definition, no evidence of multiple myeloma, other B cell lymphoproliferative disorders or other diseases known to produce M protein
- 2 distinct types of MGUS: non-IgM MGUS (the focus of this topic) and IgM MGUS
- Non-IgM MGUS and IgM MGUS (lymphoplasmacytic lymphoma / Waldenström macroglobulinemia) are considered separate entities by the WHO / ICC classification due to the differences in cell of origin, clinical picture and disease progression (Lancet Oncol 2014;15:e538, Leukemia 2022;36:1720, Blood 2022;140:1229)
- Non-IgM MGUS (IgG, IgA, IgD or light chain MGUS) accounts for most MGUS cases and is characterized by a monoclonal plasma cell proliferation
- Light chain MGUS (a subset) has only monoclonal light chains with complete loss of heavy chain expression
- Non-IgM MGUS is a benign premalignant plasma cell disorder that is characterized by the presence of serum M protein < 30 g/L, bone marrow clonal plasma cells < 10%, absence of plasma cell myeloma related end organ damage and amyloidosis
- Non-IgM MGUS is usually asymptomatic; conditions with symptoms should be categorized as monoclonal gammopathy of renal significance (MGRS) or monoclonal gammopathy of clinical significance (MGCS) based on symptoms (Blood 2022;140:1229)
- Rarely, plasma cell MGUS produces an IgM M protein (1% of cases)
- Non-IgM MGUS is a benign, asymptomatic premalignant plasma cell disorder that is characterized by the presence of serum M protein < 30 g/L, bone marrow clonal plasma cells < 10% and absence of plasma cell myeloma related end organ damage or amyloidosis
- Non-IgM MGUS may progress to a malignant plasma cell neoplasm (rate of 1% per year)
- ICD-10: D47.2 - monoclonal gammopathy of undetermined significance (MGUS)
- Most common plasma cell neoplasm (Blood 2009;113:5412)
- Incidence increases with age and is found in ~2 - 3% of adults over age 50 and 5% of adults over age 70 (N Engl J Med 2018;378:241)
- More common than multiple myeloma (1 million versus 13,000 cases/year in the U.S.)
- More prevalent in African Americans than in Caucasians (2 - 3 fold) (Leukemia 2014;28:1537)
- More common in men than women (1.5:1)
- Median age at diagnosis: 70 years
- Non-IgM MGUS represents up to 85% of MGUS cases
- Originates from clonal mature plasma cells residing in the bone marrow that harbor somatic hypermutation errors or IgH class switched variable regions; these events occur during antigen selection in germinal centers
- Most non-IgM MGUS cases are IgG, followed by IgA, biclonal and IgD
- Random secondary genetic alterations have been associated with increased risk of progression to plasma cell myeloma
- Reference: Clin Lymphoma Myeloma Leuk 2023;23:e195
- No specific cause has been identified; however, a gene - environment interaction is possible (Hematol Oncol Clin North Am 2014;28:775)
- Associated with
- Connective tissue disorders, peripheral neuropathies, dermatological diseases (such as acquired C1 esterase inhibitor deficiency [angioedema]), endocrine diseases and liver infections (such as hepatitis C and HIV liver disease)
- Radiation, pesticides and obesity (Blood 2009;113:6386, Blood 2009;113:1639, Blood 2010;116:1056)
- First degree relatives have higher incidence (Blood 2012;119:4608)
- Risk increased by polymorphisms at 2p23.3, 3p22.1, 3q26.2, 6p21.33, 7p15.3, 17p11.2 and 22q13.1 (Blood 2014;123:2513)
- Most patients are asymptomatic
- Usually diagnosed as an incidental finding on protein electrophoresis performed to evaluate peripheral neuropathy, vasculitis, hemolytic anemia, skin rashes, hypercalcemia or elevated erythrocyte sedimentation rate
- Diagnosis should be established only if the disease persists and is not transient (CMAJ 2013;185:1345)
- Paraprotein incidence: 70% IgG, 15% IgM, 12% IgA, 3% biclonal, 1% IgD, 1% IgE; monoclonal light chain in urine in 33% of cases; 20% with only light chain (N Engl J Med 2006;354:1362)
- No myeloma related organ or tissue impairment, such as hypercalcemia (> 1 mg/dL higher than the upper limit of normal or > 11 mg/dL), renal insufficiency (Cr clearance < 40 mL/min or serum Cr > 2 mg/dL), anemia (Hb > 20 g/L below lower limit of normal or > 100 g/L) or bone lesions (≥ 1 by skeletal radiography, CT or PET / CT) (= CRAB features of myeloma)
- 3 diagnostic criteria
- Serum monoclonal protein < 30 g/L
- < 10% clonal plasma cells in the bone marrow
- Absence of end organ damage attributable to the plasma cell disorder (CRAB features) and absence of amyloidosis
- Light chain only MGUS diagnostic criteria: abnormal light chain ratio (< 0.26 or > 1.65), urinary light chain must be < 0.5 g/24 h, < 10% plasma cells, end organ damage or amyloidosis
- Need thorough investigation to exclude other plasma cell neoplasms
- Complete blood count / peripheral smear: usually normal but some cases show rouleaux formation
- Plasma cells are associated with high risk MGUS
- Bone marrow with < 10% clonal plasma cells
- Plasma cells are usually mature with an interstitial distribution
- Serum calcium and creatinine: important in differentiating non-lgM MGUS from plasma cell myeloma
- Serum protein electrophoresis and immunofixation, serum free light chains, urine protein electrophoresis and immunofixation assay, quantitation of IgM
- Complete blood count / peripheral smear: usually normal but some cases show rouleaux formation
- Quantification of M protein is useful to monitor disease course (Hematol Oncol Clin North Am 2014;28:775)
- Serum protein electrophoresis can be performed on agarose gel or capillary zone electrophoresis
- Serum free light chains: measures κ and λ light immunoglobulin chains that circulate unbound to heavy chains in the serum; the normal ratio for κ/λ is 0.26 - 1.65
- Light chain MGUS: no immunoglobulin heavy chain on immunofixation electrophoresis
- Bone survey of skull, spine and pelvis (Xray, CT, MRI and PET with differing sensitivities)
- Important in differentiating non-lgM MGUS from other plasma cell neoplasm entities and other B cell lymphoproliferative disorders
- Osteolytic bone destruction (≥ 5 mm in size) on imaging is considered positive for bone involvement of plasma cell myeloma or plasmacytoma (Lancet Oncol 2014;15:e538)
- Considered a preneoplastic condition with an annual risk of progression of ~1%
- May progress to multiple myeloma, amyloidosis, Waldenström macroglobulinemia, solitary plasmacytoma or other lymphoproliferative disorder (Lancet Oncol 2014;15:e538)
- Risk of progression of non-IgM MGUS is lower than IgM MGUS (N Engl J Med 2018;378:241)
- Risk assessment based on serum M protein < 1.5 g/dL, IgG subtype, normal FLC ratio (0.26 - 1.65) (N Engl J Med 2002;346:564)
- Light chain MGUS: lower rate of progression (0.3% per year)
- 49 year old man with polycythemia and renal failure (Nephrol Dial Transplant 2012;27:448)
- 61 year old man with dystrophic plasma cells (Eur J Haematol 2012;88:461)
- 70 year old woman with Good syndrome (BMJ Case Rep 2012;2012:bcr2012007601)
- 74 year old woman with leukocyte cytoplasmic inclusions associated with cryoglobulinemia (Eur J Haematol 2011;86:550)
- 81 year old woman with crystalline keratopathy (Br J Haematol 2012;159:258)
- 82 year old man with von Willebrand disease and IgG MGUS (Blood Coagul Fibrinolysis 2014;25:631)
- 86 year old woman with blue finger syndrome (BMJ Case Rep 2013;2013:bcr2012007966)
- Management requires an understanding of the risk of progression
- Follow up of MGUS patients depends on the risk assessment
- Risk stratification (Blood 2005;106:812)
- Serum monoclonal protein level ≥ 15 g/L
- Non-IgG MGUS (i.e., IgA, IgM and IgD MGUS)
- Abnormal serum free light chain ratio
- Patients are categorized by number of risk factors present
- 3 risk factors: high risk MGUS
- 2 risk factors: high intermediate risk MGUS
- 1 risk factor: low intermediate risk MGUS
- 0 risk factors: low risk MGUS
- Despite close observation, can progress abruptly to plasma cell myeloma
- Follow up
- Low risk MGUS: every 2 - 3 years without laboratory testing (CMAJ 2013;185:1345)
- Intermediate and high risk MGUS: clinical and laboratory follow up every 6 months (Hematol Oncol Clin North Am 2014;28:775)
- < 10% clonal plasma cells in bone marrow aspirate / biopsy
- Plasma cells evenly scattered or in occasional small clusters
- Plasma cells lack nucleoli
- Reference: Clin Lymphoma Myeloma Leuk 2023;23:e195
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
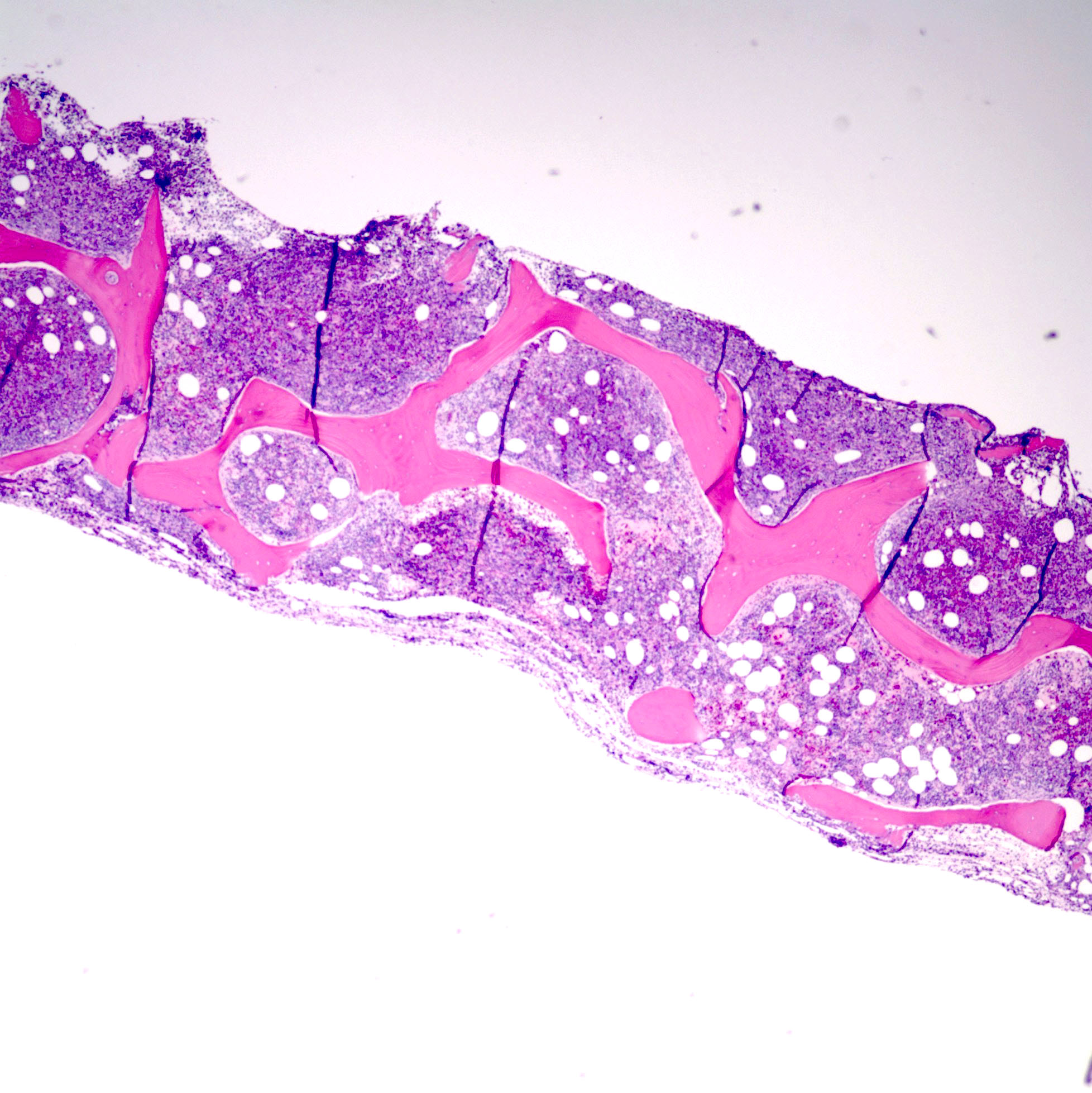
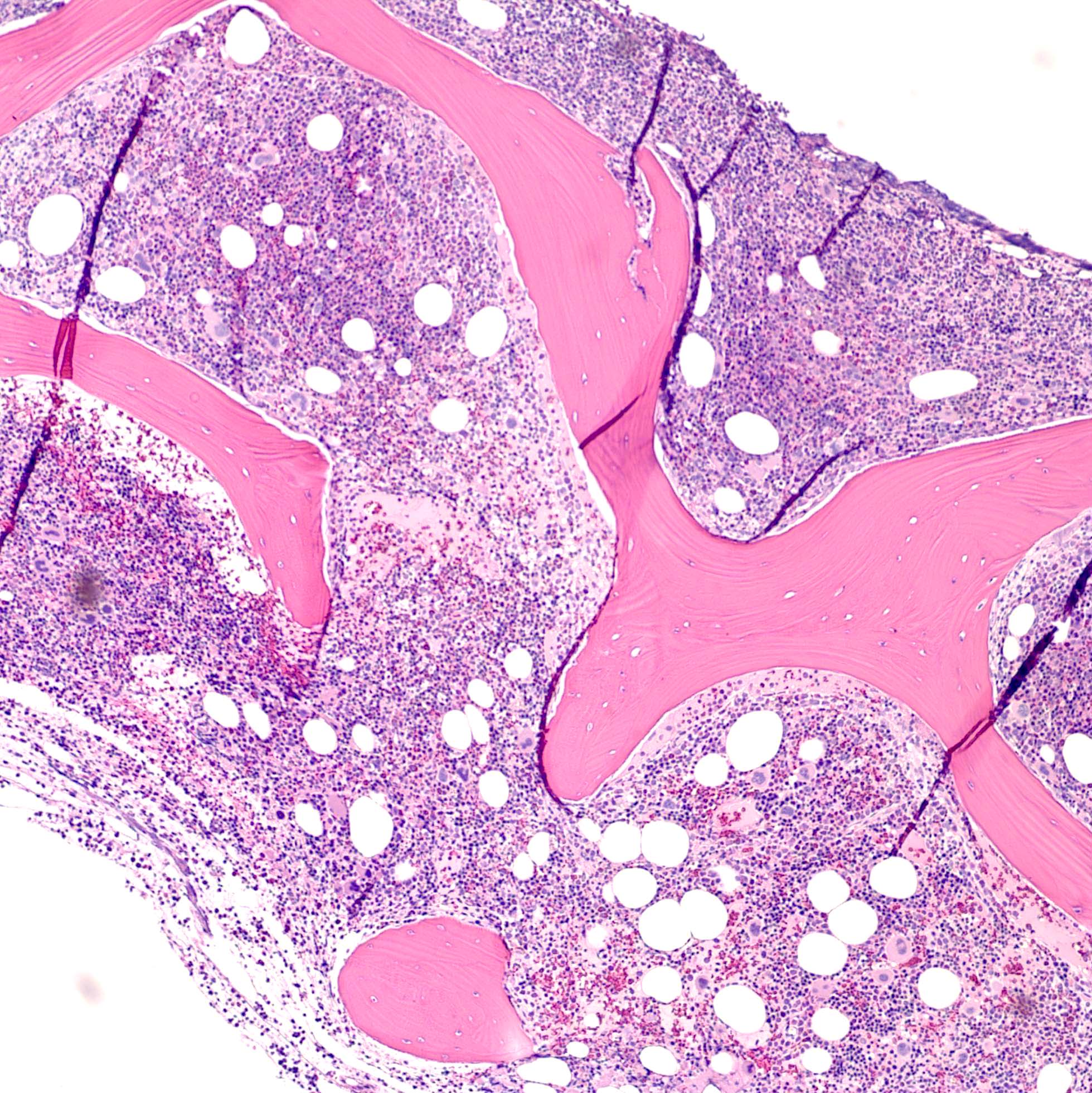
Bone biopsy
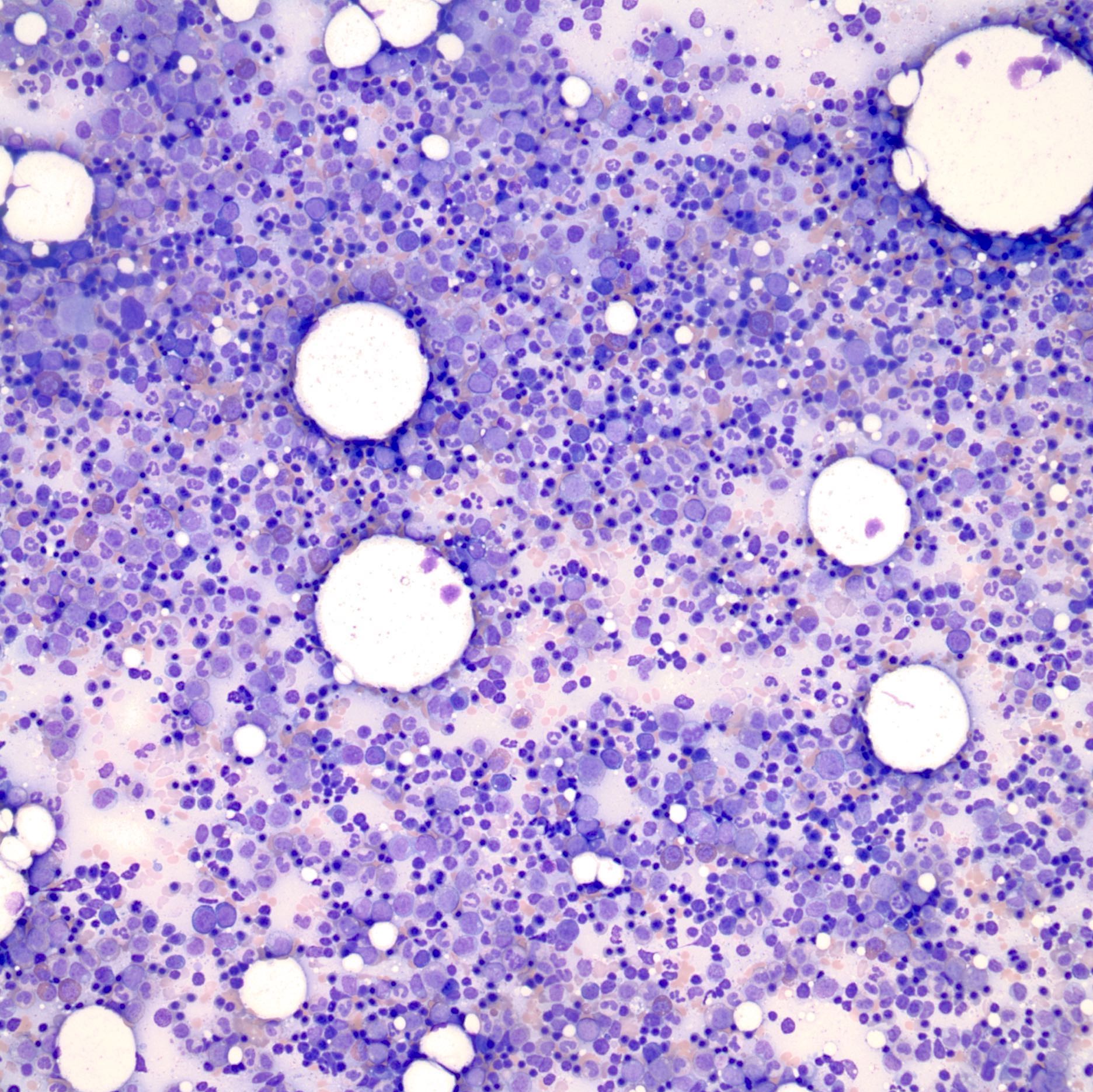
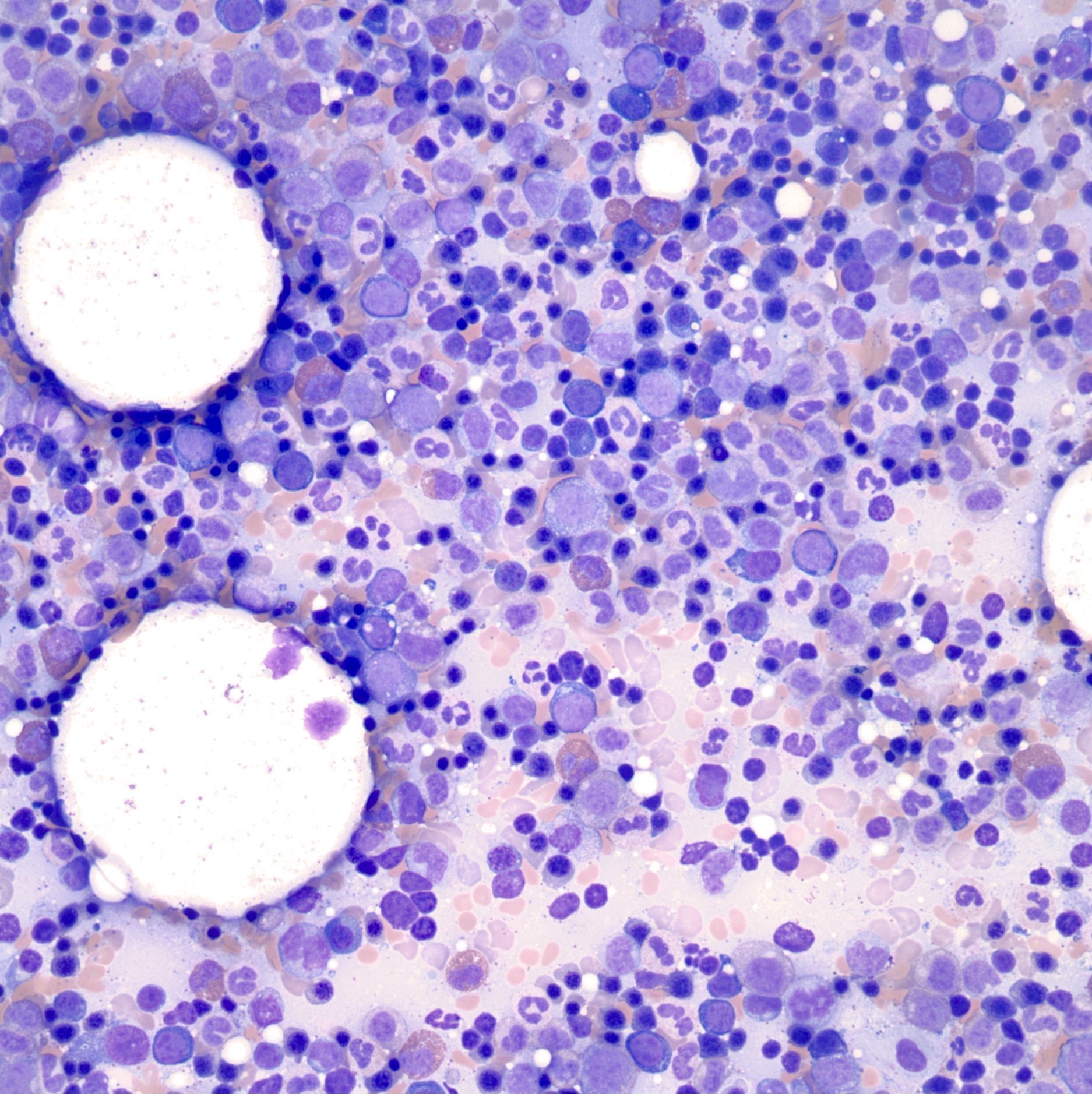
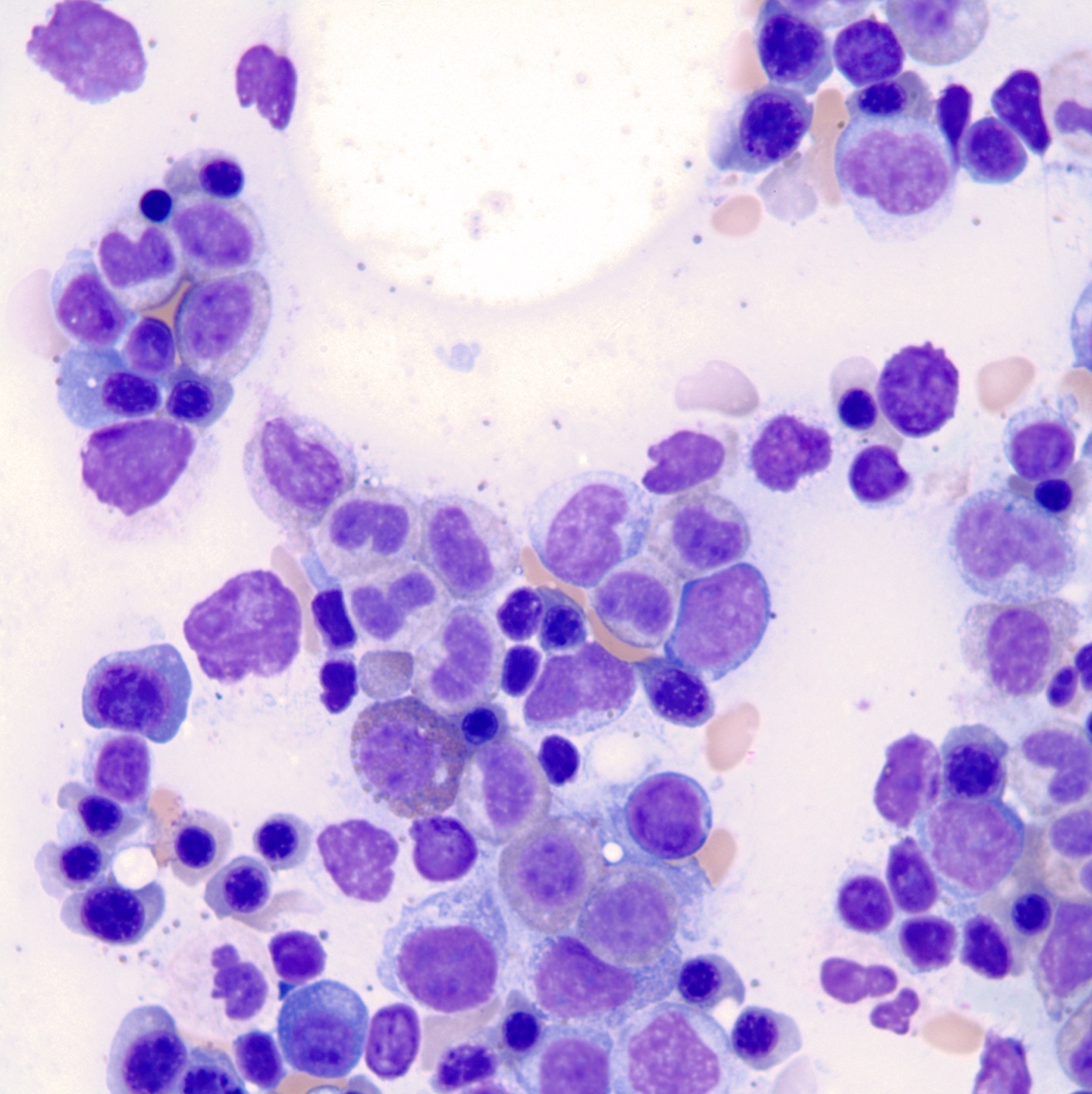
Interspersed plasma cells
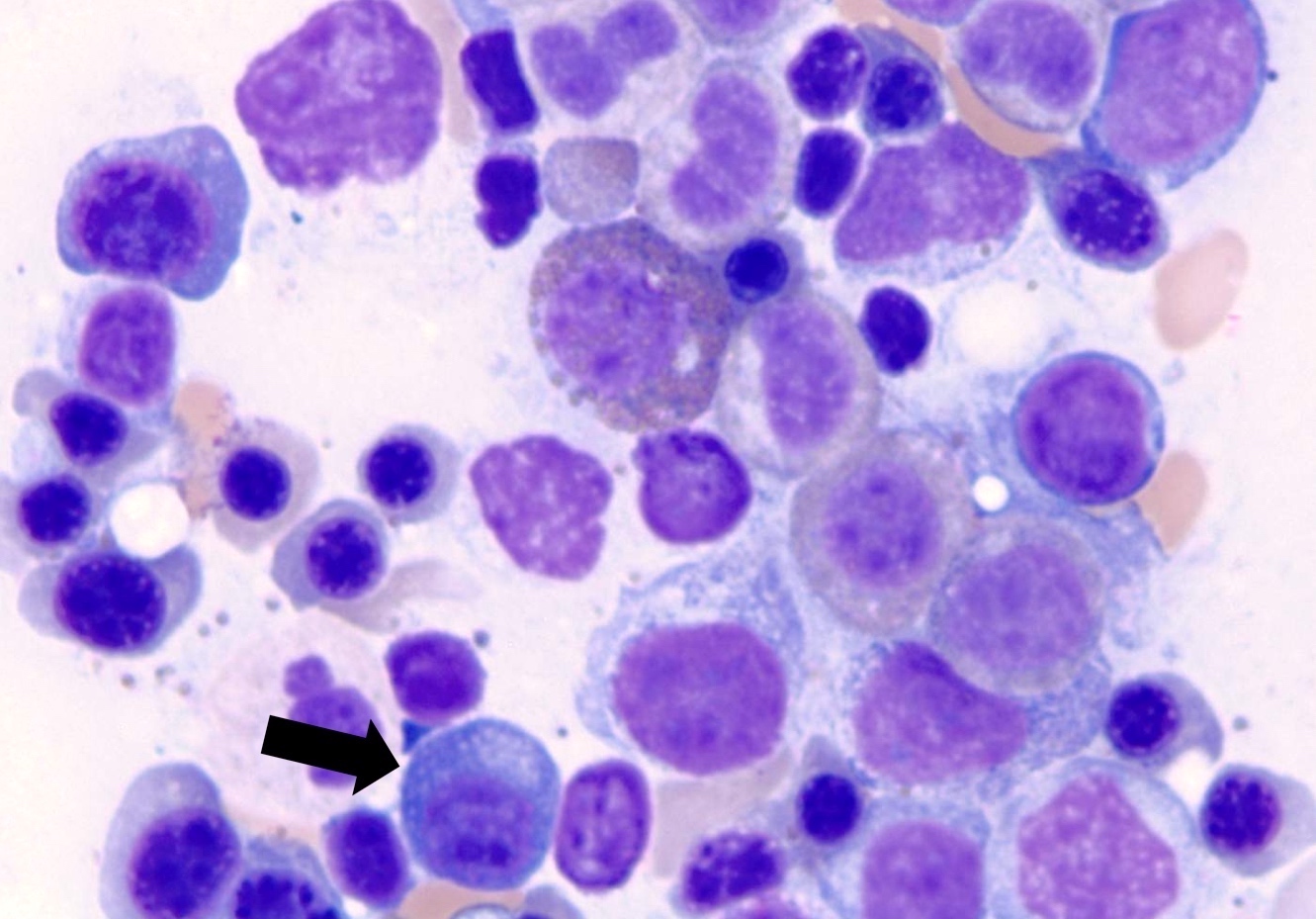
Few mature plasma cells
- CD19 (in contrast to normal plasma cells)
- Usually shows a normal karyotype because of the relatively small number of plasma cells
- Possible chromosomal alterations include t(11;14), t(4;14), t(14;16), deletions of 13q and hyperdiploidy; no clinical correlation for these genetic alterations have been found in non-IgM MGUS
- Reference: Clin Lymphoma Myeloma Leuk 2023;23:e195
- Bone marrow, biopsy and aspirate:
- Plasma cell neoplasm with 5% plasma cells (see comment)
- Comment: This is a 70 year old man with a medical history of MGUS, currently presenting with congestive heart failure and anemia. The bone marrow shows increased plasma cells (5%); these cells show monotypic kappa light chain restriction by immunohistochemistry. Flow cytometry studies show 0.1% of the total events consistent with kappa restricted plasma cells. Further classification of plasma cell neoplasms requires correlation with clinical, laboratory and radiologic findings.
- Smoldering plasma cell myeloma:
- Serum M protein ≥ 3 g/dL, bone marrow plasma cells ≥ 10% (but < 60%) and absence of CRAB features
- Waldenström macroglobulinemia (smoldering or symptomatic):
- IgM on immunofixation
- Light chain monoclonal gammopathy of undetermined significance (LC MGUS):
- Monoclonal protein that lacks the immunoglobulin heavy chain component
- Monoclonal gammopathy of renal significance:
- Meets diagnostic criteria for MGUS as well as renal insufficiency and monoclonal immunoglobulin deposits in the kidney by immunofluorescence (Blood 2012;120:4292)
- Light chain smoldering multiple myeloma (idiopathic Bence Jones proteinuria):
- Presence of monoclonal light chains in the urine (Bence Jones proteinuria) and no immunoglobulin heavy chain expression in the serum or urine
- Primary (amyloid light chain) amyloidosis and light chain deposition disease:
- Plasma cell neoplasm associated with the pathologic deposition of monoclonal light chains in tissue
- Presence of amyloid in tissue and evidence of plasma cell neoplasm
- Other B cell lymphoproliferative disorders:
- Diagnosis of the specific entity is based on biopsy, IHC, cytogenetics and molecular studies
- 0.5% per year
- 1% per year
- 1.5% per year
- 2% per year
Which of the following factors is associated with increased risk of progression in non-IgM monoclonal gammopathy of uncertain significance (MGUS)?
- Generalized lymphadenopathy
- Lambda restriction of plasma cells in bone marrow aspirate
- M protein of 5 g/L
- M protein of 15 g/L
Comment Here
Reference: MGUS - non-IgM
- IgM monoclonal gammopathy of undetermined significance (MGUS) is characterized by < 3 g/dL of monoclonal IgM protein, < 10% clonal lymphoplasmacytic or clonal plasma cells, with no anemia, constitutional symptoms, hyperviscosity, lymphadenopathy or hepatosplenomegaly
- Serum IgM monoclonal protein < 3 g/dL
- Bone marrow clonal lymphoplasmacytic cells or clonal plasma cells < 10% of cellular elements
- No anemia, constitutional symptoms, hyperviscosity, lymphadenopathy or hepatosplenomegaly caused by the underlying disorder
- Can progress to lymphoplasmacytic lymphoma / Waldenström macroglobulinemia, IgM multiple myeloma or AL amyloidosis
- Prevalence of MGUS is 2 - 3% of adults over 50 years (F1000Res 2017;6:2142)
- IgM MGUS comprises 15% of MGUS, compared with 70% for IgG MGUS and 12% for IgA MGUS (Blood 2003;102:3759)
- Monoclonal IgM kappa and lambda proteins are found in the serum or urine
- Clonal plasma cells or lymphocytes can be seen in the bone marrow
- Initial step is an abnormal immune response to an antigenic stimulus (F1000Res 2017;6:2142)
- MYD88 (42 - 54%) and CXCR4 (7%) mutations are seen in IgM MGUS
- Additional steps (such as the 6q deletion) may be involved in progression to Waldenström macroglobulinemia (F1000Res 2017;6:2142)
- Noncoding RNAs may drive development of Waldenström macroglobulinemia (Cancers (Basel) 2020;12:320)
- IgM MGUS patients with t(11:14) may progress to IgM plasma cell myeloma
- Unknown
- Patients with IgM MGUS do not have constitutional symptoms (Clin Lymphoma Myeloma 2009;9:17)
- Progression among IgM MGUS patients is 2% per year in the first 10 years and 1% per year thereafter (N Engl J Med 2018;378:241)
- Risk of progression is higher than non-IgM MGUS
- Some patients have associated peripheral neuropathy
- Diagnosis of IgM MGUS is based on the presence of a serum IgM monoclonal protein < 3 g/dL, fewer than 10% clonal lymphoplasmacytic or plasma cells in the bone marrow and lack of evidence of progression to lymphoplasmacytic lymphoma / Waldenström macroglobulinemia, IgM multiple myeloma or AL amyloidosis (Blood 2018;131:163)
- Serum electrophoresis shows a restricted band (< 3 g/dL)
- Serum immunofixation shows an IgM kappa or IgM lambda monoclonal protein
- Skeletal survey does not show lytic lesions
- CT / PET scans are negative for lymphoma
- Risk factors for progression are initial concentration of the monoclonal protein (≥ 1.5 g/dL) and abnormal serum free light chain ratio (N Engl J Med 2018;378:241)
- Overall survival rate at 30 years is 4% for patients with IgM MGUS and 7% for non-IgM MGUS, compared with 12% in an age and sex matched control population (N Engl J Med 2018;378:241)
- 62 year old immunocompetent woman was admitted with cytomegalovirus (CMV) infection, pulmonary embolism, splenic vein thrombosis and monoclonal gammopathy of undetermined significance (MGUS) (BMJ Case Rep 2019;12:e226448)
- 70 and 84 year old Caucasian men showed an M spike by protein electrophoresis (J Med Case Rep 2020;14:75)
- 72 year old Japanese man with diabetes mellitus and hypertension presented with an acutely elevated serum creatinine level (CEN Case Rep 2020;9:109)
- 72 year old Japanese woman was hospitalized because of massive proteinuria (Front Med (Lausanne) 2021;8:608741)
- 75 year old woman presented with maculopapular purpura and erythematous (inflammatory) retiform purpura on her lower leg (JAAD Case Rep 2019;5:288)
- Treatment is not recommended for asymptomatic patients with IgM MGUS (F1000Res 2017;6:2142)
- "Wait and watch" approach is usually followed
- Patients with peripheral neuropathy are treated with IVIG, rituximab, steroids or plasma exchange
- Bone marrow biopsy is usually normocellular with normal trilineage hematopoiesis
- Plasma cells and CD20+ B cells may be slightly increased but do not form prominent aggregates
Contributed by Ameet R. Kini, M.D., Ph.D. and Maryam F. Raouf, M.D.
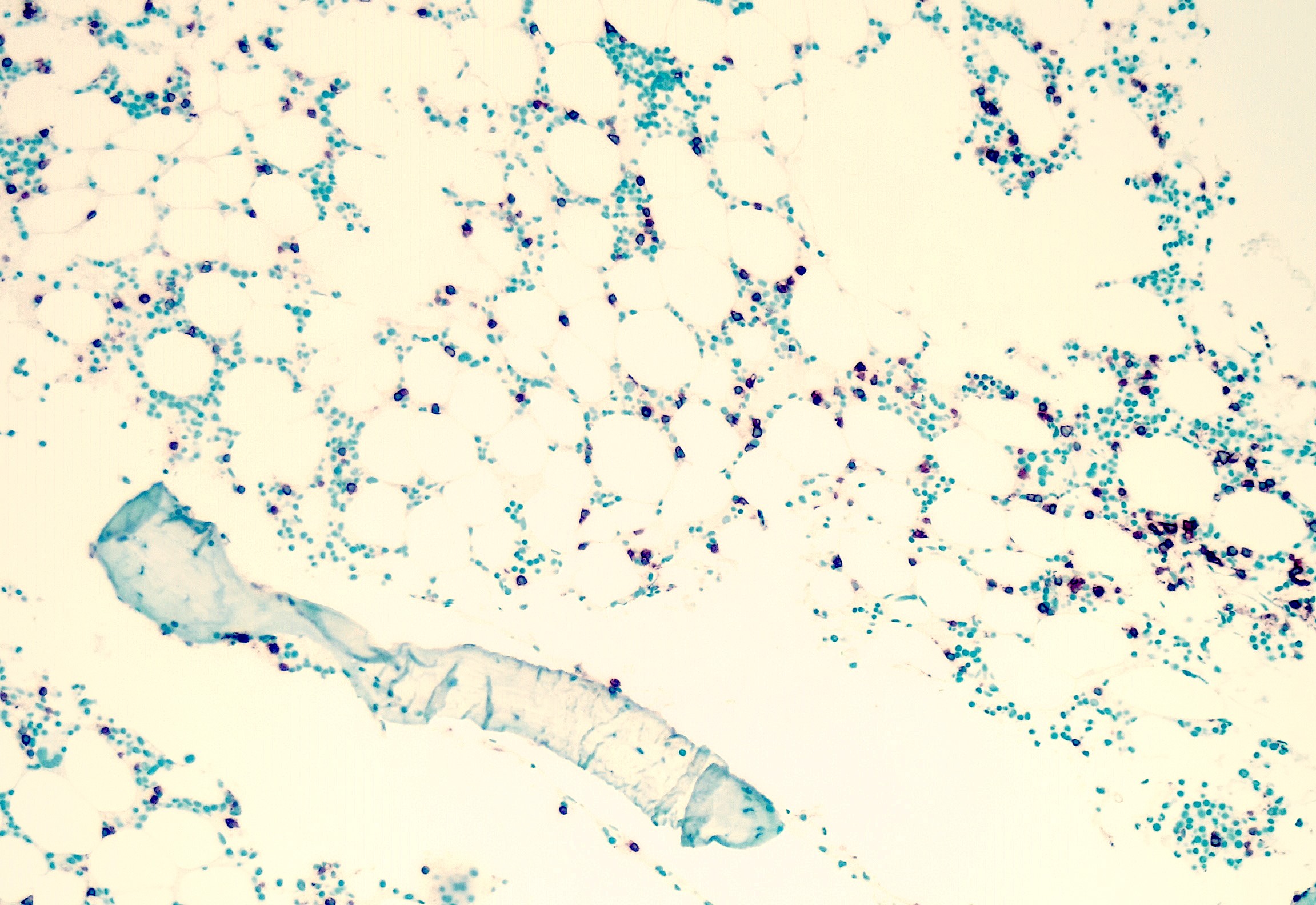
Increased CD20+ B cells
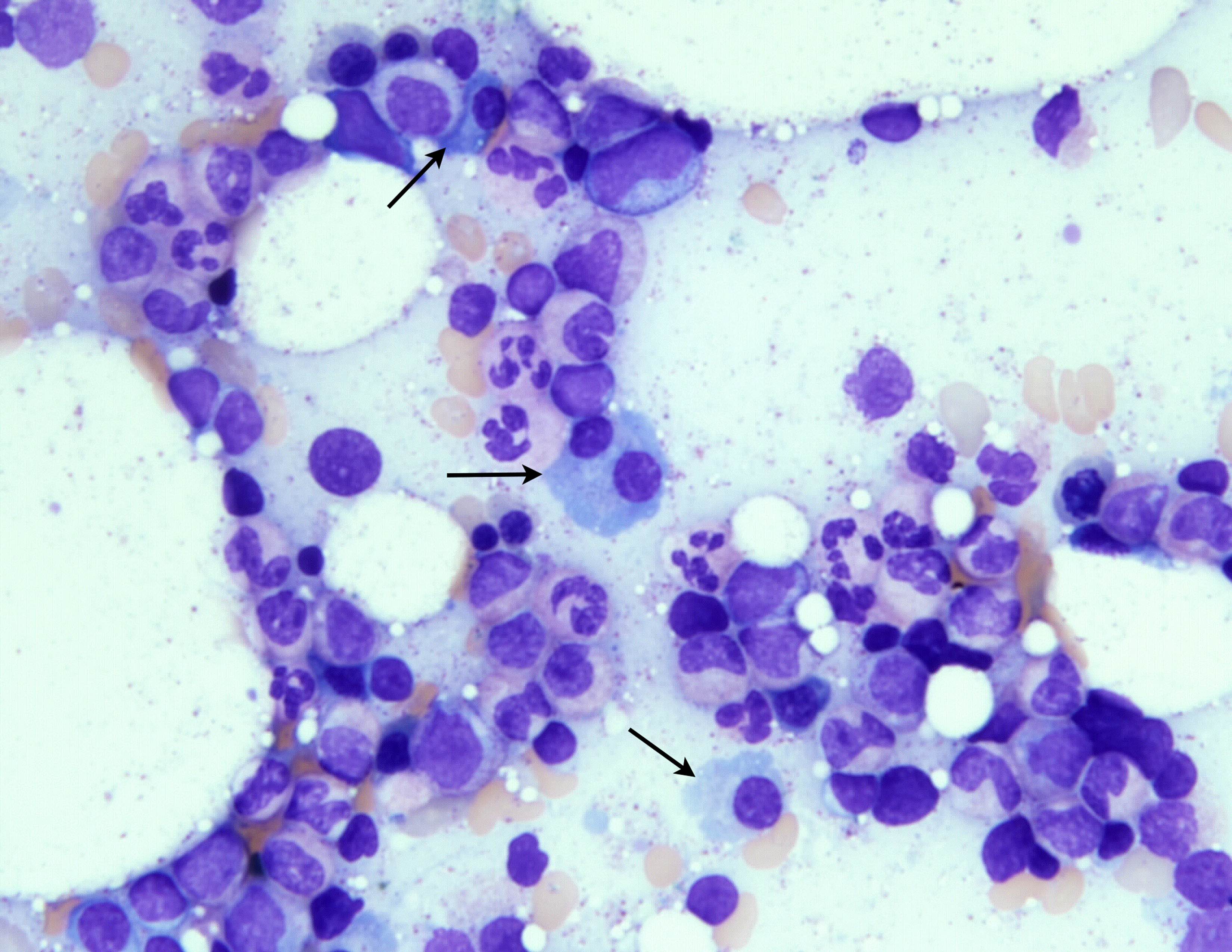
Increased plasma cells
- Bone marrow aspirate smears show normal trilineage hematopoiesis
- Plasma cells and lymphocytes may be slightly increased but are < 10% of cells
- Peripheral blood smear shows no abnormal findings
- CD20 detects B cells that may be slightly increased
- CD138 detects plasma cells
- Plasma cells may show aberrant expression of CD56 or CD117
- Plasma cells show monoclonal expression of either kappa or lambda light chain
- Negative for T cell markers (such as CD2, CD3 and CD5), myeloid markers (including CD33 and myeloperoxidase) and nonhematopoietic markers (such as pancytokeratin)
Contributed by Ameet R. Kini, M.D., Ph.D. and Maryam F. Raouf, M.D.
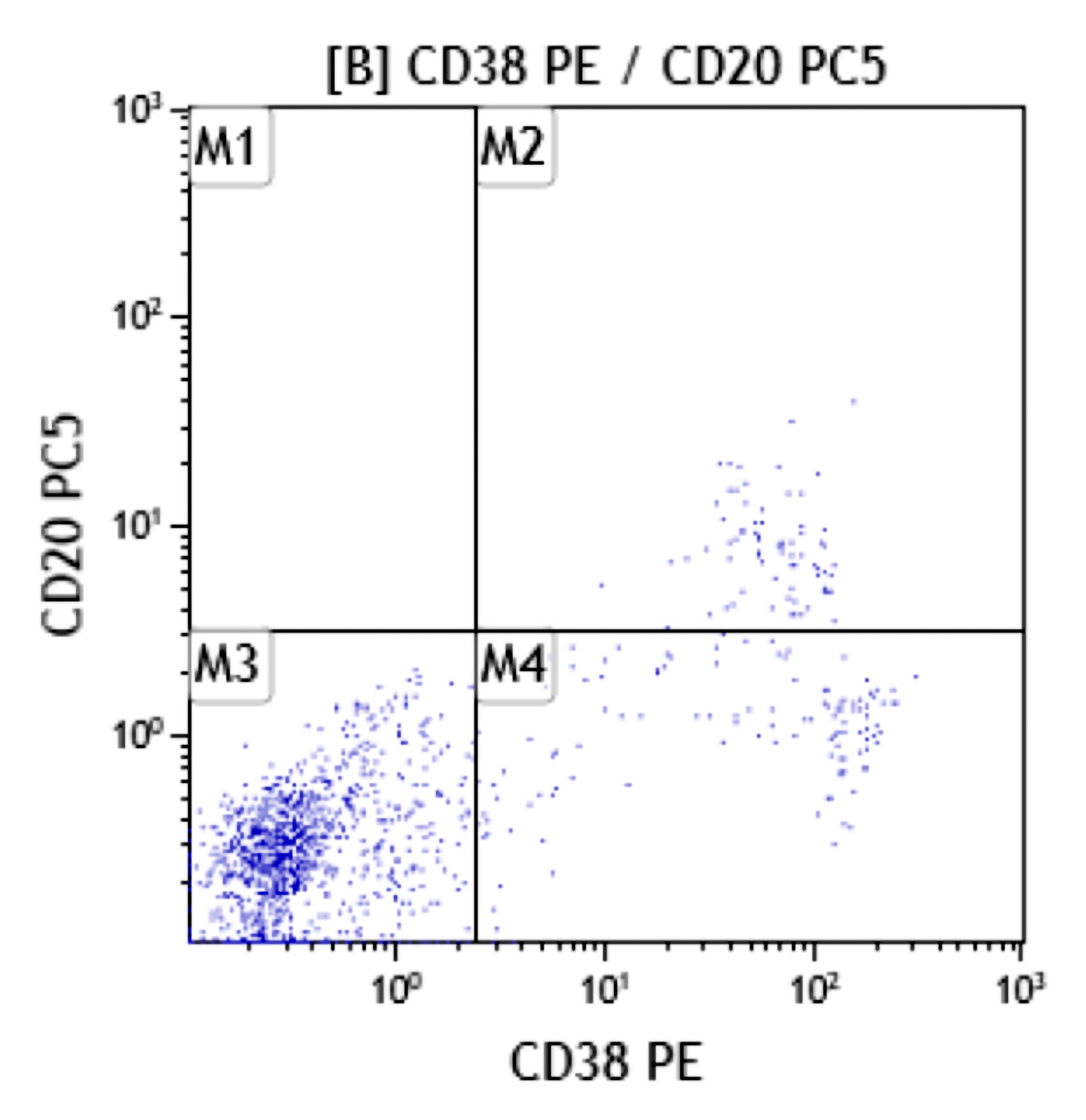
Abnormal plasma cells
- FISH analysis may detect presence of t(11;14), CCND1-IGH in plasma cells
- MYD88 mutation may be detected (Blood 2013;121:2051)
Contributed by Ameet R. Kini, M.D., Ph.D. and Maryam F. Raouf, M.D.
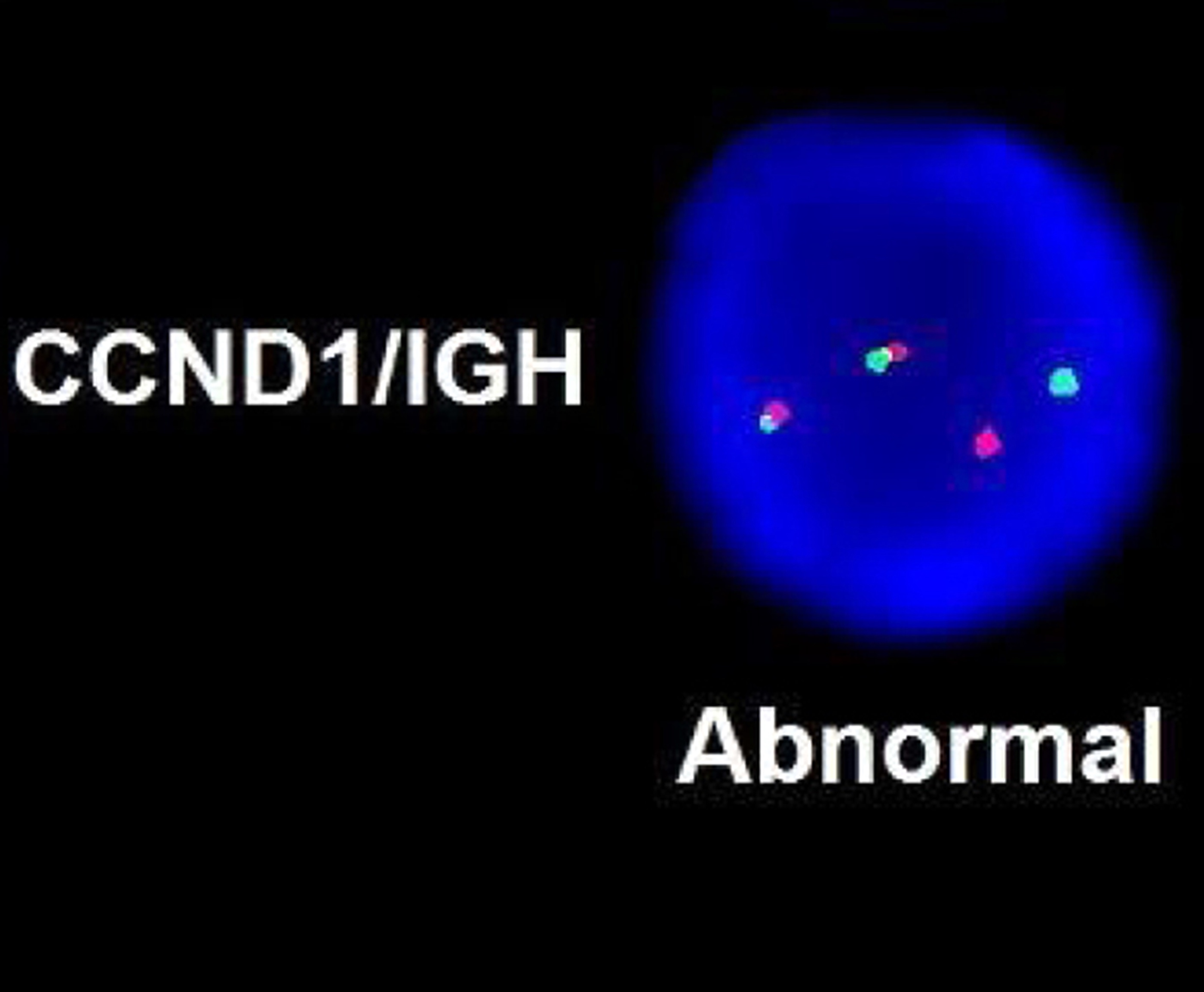
CCND1-IGH fusion
Introduction to MGUS
MGUS diagnosis
MGUS management
MGUS associated diseases
IgM MGUS pathophysiology
- Bone marrow, right posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Normocellular bone marrow (50%) with trilineage hematopoiesis and slightly increased plasma cells (4%) (see comment)
- Comment: Review of laboratory data shows a monoclonal protein in the serum (IgM lambda, 1.2 g/dL). The CBC is within normal limits. Imaging studies reveal bone marrow lytic lesions and no lymphadenopathy or organomegaly.
- The bone marrow core biopsy is normocellular for age (50%). Trilineage hematopoiesis is present. Megakaryocytes are adequate and appear normal in morphology.
- The aspirate clot section is adequate and shows features similar to the core biopsy.
- The bone marrow aspirate smear is adequate. The myeloid and erythroid precursors show progressive and orderly maturation. Megakaryocytes are adequate in number and show normal morphology. Blasts are not increased. Plasma cells are slightly increased (4% plasma cells based on a 500 cell differential count performed on the bone marrow aspirate smear).
- The touch preparation findings are similar to the aspirate smear.
- Flow cytometry shows a small abnormal plasma cell population (CD45 negative, bright CD38 positive) with expression of CD20, CD56 and CD117. These plasma cells show lambda light chain restriction. Gating on the lymphocytes shows no evidence of a monoclonal B cell population or T cell abnormality based on the markers assayed. There is no increase in blasts.
- FISH analysis performed on an enriched plasma cell population reveals presence of t(11;14). Molecular analysis for the MYD88 mutation is negative.
- The findings are consistent with IgM MGUS. IgM MGUS patients can progress to lymphoplasmacytic lymphoma / Waldenström macroglobulinemia, IgM multiple myeloma or AL amyloidosis.
- IgM myeloma:
- IgM protein monoclonal ≥ 3 g/dL or ≥ 10% clonal plasma cells
- Lytic bone lesions may be seen
- Lymphoplasmacytic lymphoma:
- IgM protein monoclonal ≥ 3 g/dL or ≥ 10% clonal lymphoplasmacytic cells
- Hyperviscosity and lymphadenopathy may be seen
- AL amyloidosis:
- Presence of amyloid deposits that display apple green birefringence under polarized microscopy on Congo red stain
A 58 year old woman with a past medical history significant for type 2 diabetes and hypertension presents to her PCP office for routine care. The patient's diabetes and hypertension are well controlled and she reports no significant constitutional symptoms. Physical exam shows no lymphadenopathy or organomegaly. Laboratory studies show her CBC within normal limits. Serum electrophoresis shows a restricted band at 1.5 g/dL. A serum immunofixation is shown above (ELP = serum electrophoresis, G = IgG, A = IgA, M = IgM, K = kappa, L = lambda). This patient's most likely diagnosis is
- IgG MGUS
- IgM MGUS
- Polyclonal gammopathy
- Smoldering myeloma
- Waldenström macroglobulinemia
- CALR
- FLT3
- JAK2
- MYD88
- SF3B1
- Has clinical phenotype of myeloproliferative neoplasms (MPN) but does not meet diagnostic criteria for any classic or nonclassic myeloproliferative neoplasms
- May have JAK2 617F mutation (Mod Pathol 2007;20:929)
- De novo acute leukemia containing separate populations of blasts of more than one lineage (bilineal or bilineage), or a single population of blasts co-expressing antigens of more than one lineage (biphenotypic)
Excludes:
- Acute myeloid leukemia (AML) with recurrent translocations t(8;21), t(15;17) or inv(16)
- Leukemias with FGFR1 mutations
- Chronic myelogenous leukemia (CML) in blast crisis
- Myelodysplastic syndrome (MDS) related AML and therapy related AML, even if they have MPAL immunophenotype
Criteria for biphenotypic leukemia:
- Score of 2 or more for each of two separate lineages (shown below)
Contributed by Daniela Mihova, M.D.
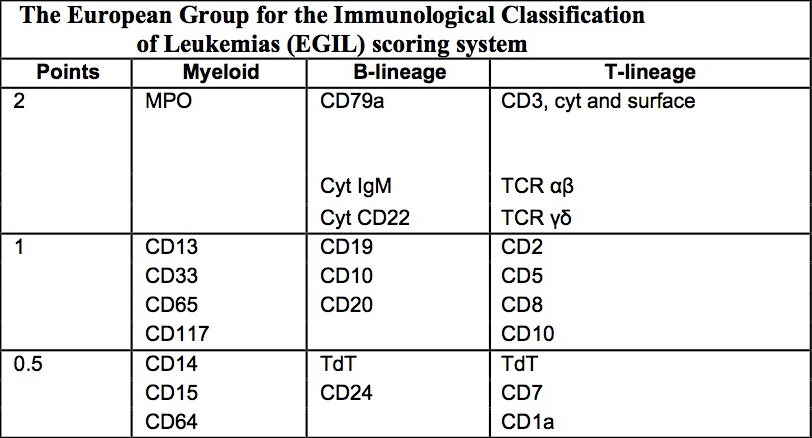
The European Group
for the Immunological
Classification of Leukemias
(EGIL) scoring system
2008 WHO
classification of
acute leukemias of
ambiguous lineage
- Poor, overall survival of 18 months
- Young age, normal karyotype and ALL induction therapy are associated with favorable survival
- Ph+ is a predictor for poor prognosis
- Bone marrow transplantation should be considered in first remission
- 20% of all MPAL
- Blasts with t(9;22)(q34;q11.2) translocation or BCR-ABL1 rearrangement (Ph+) without history of CML
- Majority in adults
- High WBC counts
- Most of the cases B/myeloid phenotype
- Rare T/myeloid, B and T lineage, or trilineage leukemias
Morphology:
- Many cases show a dimorphic blast population, one resembling myeloblasts and the other lymphoblasts
Cytogenetic abnormalities:
- Conventional karyotyping for t(9;22), FISH or PCR for BCR-ABL1 translocation
- Additional complex karyotypes
- Ph+ is a poor prognostic factor for MPAL with a reported median survival of 8 months
- Worse than patients of all other types of MPAL
- Meeting the diagnostic criteria for MPAL with blasts bearing a translocation involving the 11q23 breakpoint (MLL gene)
- MPAL with MLL rearranged rare
- More often seen in children and relatively common in infancy
- High WBC counts
- Poor prognosis
- Dimorphic blast population with one resembling monoblasts and the other resembling lymphoblasts
- Lymphoblast population often shows a CD19+, CD10- B precursor immunophenotype, frequently CD15+
- Expression of other B markers usually weak
- Translocations involving MLL gene include t(4;11)(q21;q23), t(11;19)(q23;p13) and t(9;11)(p22;q23)
- Cases with chromosome 11q23 deletion should not be classified in this category
AFIP images
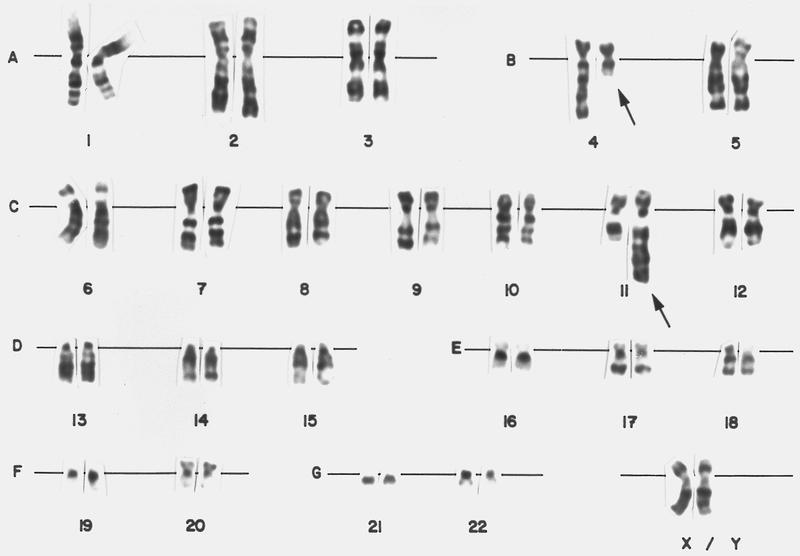
t(4;11)(q21;23) in bilineal leukemia
- Meets the diagnostic criteria for MPAL with both B and myeloid lineages without Ph+ and MLL rearrangement
- B/myeloid acute leukemia accounts for 1% of all leukemias
- More common in adults but also seen in children
Morphology:
- Dimorphic populations, resembling lymphoblasts and myeloblasts, or a single population resembling ALL
Genetics:
- Multiple different cytogenetic changes have been demonstrated, however none is proven to be specific in this subtype
- Meets the diagnostic criteria for MPAL with both T and myeloid lineages without Ph+ and MLL rearrangement
- More often in children, though in adults as well
- Dimorphic populations, resembling lymphoblasts and myeloblasts, or a single population resembling ALL
- Other commonly expressed T-lineage markers include CD2, CD7
- Myeloid markers MPO, CD117, TdT, CD13 and CD33
- T cell plus myeloid cases may have 2p13 translocations or other unrelated anomalies (Leukemia 2007;21:2264)
- Myeloid proliferations seen in individuals with Down syndrome (DS) include:
- Transient abnormal myelopoiesis (TAM) is a hematopoietic stem cell disorder of newborns with Down syndrome which presents with clinical and morphological findings of acute myeloid leukemia but is self limiting; this entity is discussed separately
- Myeloid leukemia associated with Down syndrome
- Myeloid proliferations associated with Down syndrome include transient abnormal myelopoiesis which is self limiting and acute myeloid leukemia, specifically, acute megakaryoblastic leukemia
- Trisomy 21 and GATA1 mutations are implicated in pathogenesis
- Prognosis is better than in cases with other pediatric acute leukemias
- Individuals with Down syndrome have 10 - 100 times the risk of acute leukemia as compared with the general population (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised Edition, 2017)
- 1 - 2% of children with Down syndrome will develop acute myeloid leukemia before 5 years of age
- If patient is a neonate, consider the diagnosis transient abnormal myelopoiesis associated with Down syndrome
- 20 - 30% of children with history of transient abnormal myelopoiesis get acute myeloid leukemia within 1 - 3 years following transient abnormal myelopoiesis
- Primary sites: peripheral blood and bone marrow
- Extramedullary: spleen and liver, usually present
- Trisomy 21 in fetal hematopoietic precursors followed by acquired GATA1 mutations is implicated in the development of myeloid proliferations associated with Down syndrome
- Secondary epigenetic and genetic events lead to acute myeloid leukemia in individuals with transient abnormal myelopoiesis
- Trisomy 21 affects fetal hematopoietic stem cell biology and leads to defective megakaryocytic, erythroid and B lymphoid lineage development
- N terminal truncating mutations in the key megakaryocyte erythroid transcription factor, GATA1, are commonly seen in association with trisomy 21; mutated GATA1 gene affects megakaryocytic lineage in the fetal liver
- Some of the secondary events that lead to acute leukemia in patients with transient abnormal myelopoiesis are gain of function mutation in myeloid cytokine receptor CSF2RB and tyrosine kinases such as JAK1, JAK2 and JAK3; loss of function mutations in genes encoding cohesion core subunits - CTCF, NIPBL, JAK1-3, SRSF2 and SF3B (Cancer Cell 2019;36:123)
- Often a prolonged preleukemic or myelodysplastic phase may be present when patient presents with refractory cytopenias
- Individuals with Down syndrome and GATA1 mutated acute myeloid leukemia have a better prognosis than those with other pediatric acute myeloid leukemias (must use Down specific treatment protocols)
- Older age at diagnosis is associated with poor prognosis
- Monozygotic twins with Down syndrome and distinct GATA1 point mutations (Am J Clin Pathol 2016;146:753)
- 19 month old girl with Down syndrome and acute megakaryoblastic leukemia with pentasomy of chromosome 21 (Ann Lab Med 2015;35:373)
- 1 year old boy with myeloid leukemia associated with Down syndrome harboring complex chromosomal abnormalities (Mol Cytogenet 2017;10:35)
- 2 year old girl with Down syndrome diagnosed with acute erythroleukemia (J Cancer Res Ther 2017;13:381)
- 3 year old boy with Cornelia de Lange syndrome and Down syndrome-like acute megakaryoblastic leukemia (Haematologica 2018;103:e274)
- Preleukemic phase has morphologic features of refractory cytopenia of childhood; no increase in blasts
- Acute megakaryoblastic leukemia accounts for ≥ 50% of cases
- Bone marrow:
- Variable blast count
- Leukemic blasts have round to slightly irregular nuclei and a moderate amount of basophilic cytoplasm, basophilic granules; cytoplasmic blebs (megakaryoblastic features)
- Erythroid, granulocytic and megakaryocytic dysplasia
- Variable degree of reticulin fibrosis
- Reference: Arch Pathol Lab Med 2020;144:466
Contributed by Alexa J. Siddon, M.D.

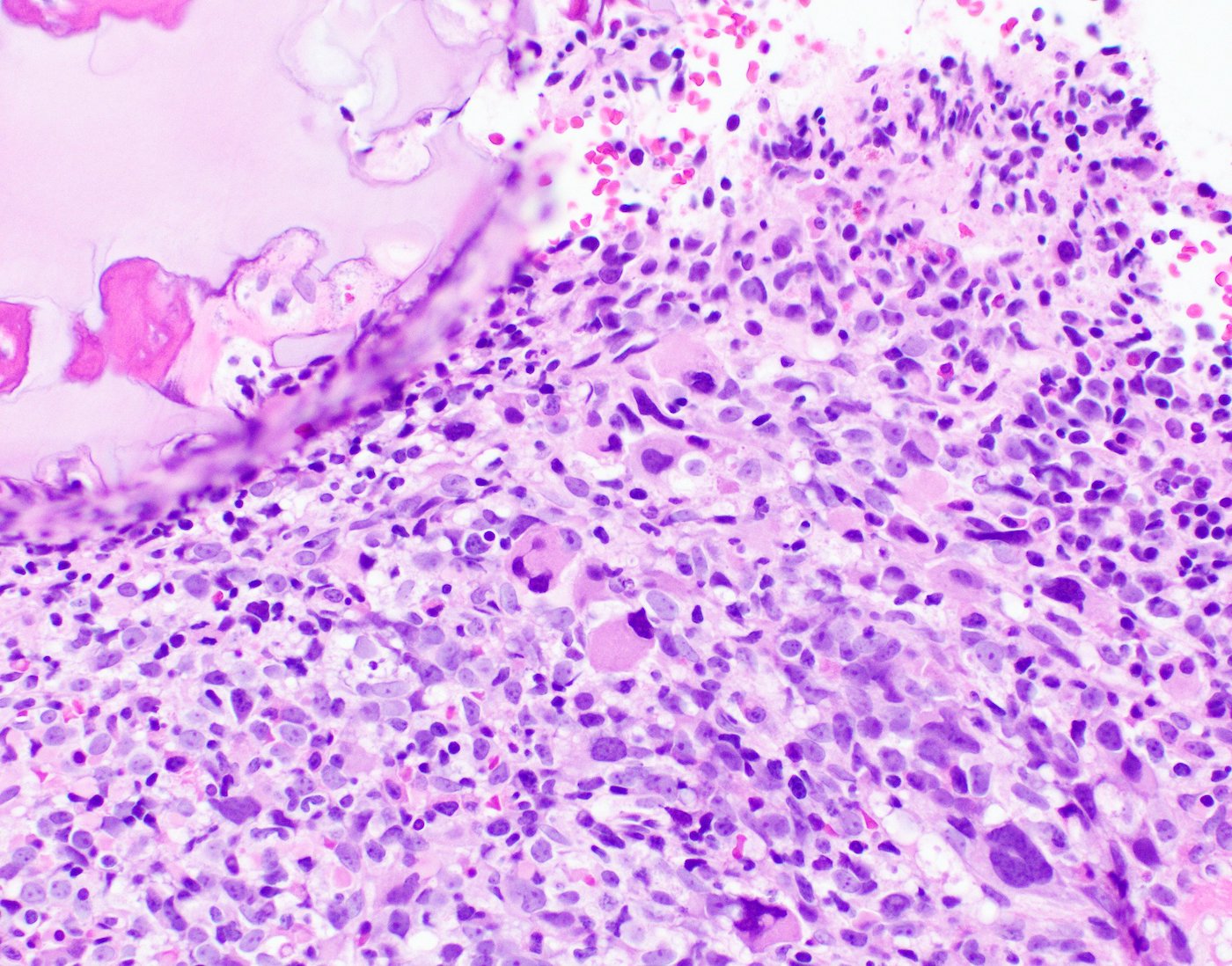
Hypercellular bone marrow
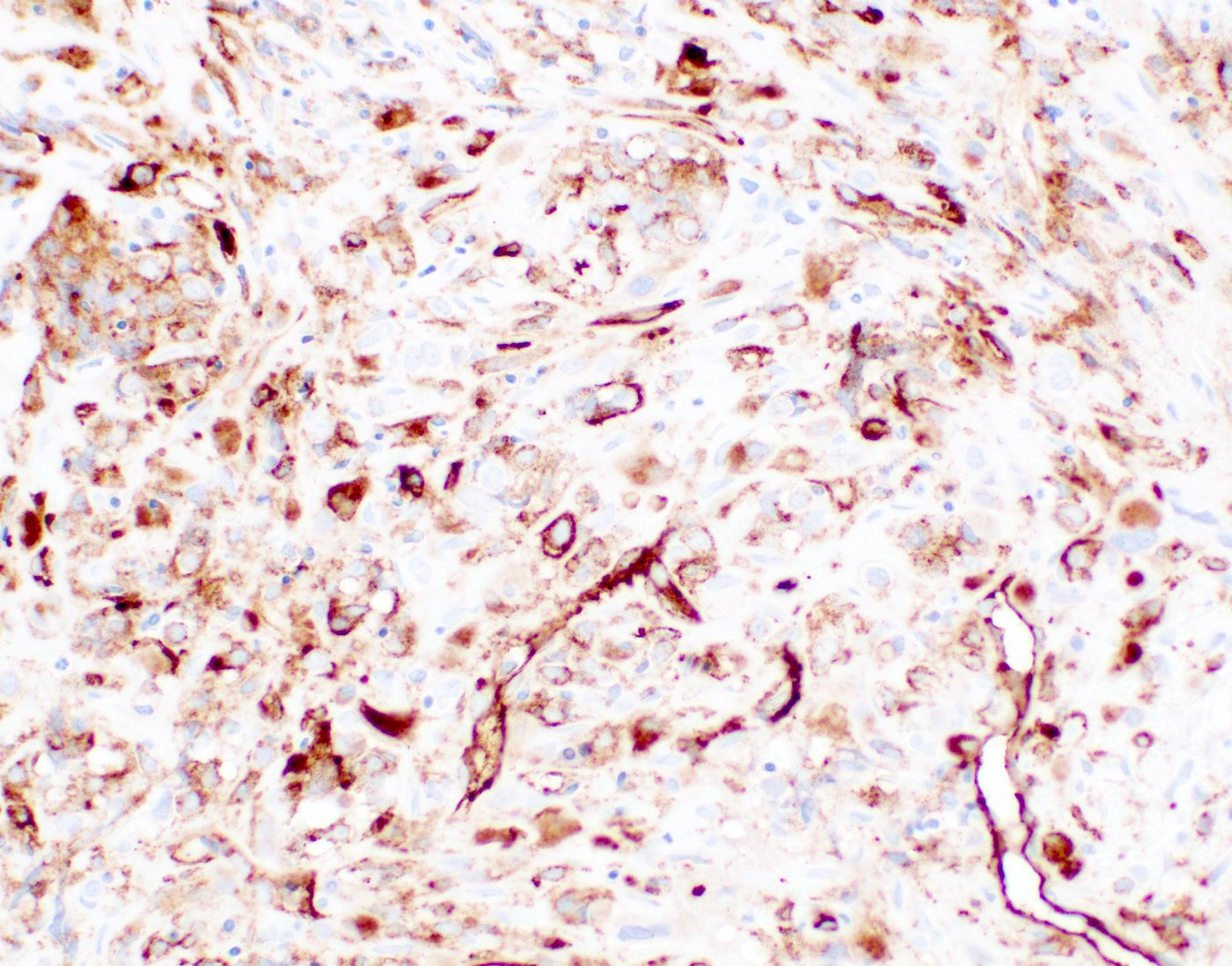
Bone marrow with CD34+ blasts

Bone marrow with CD61+ megakaryocytes and blasts
- Circulating blasts are typically present in blood; may have cytoplasmic blebs
- Macrocytic red cells with anisopoikilocytosis including dacrocytes
- Thrombocytopenia with giant platelets
Contributed by Alexa J. Siddon, M.D.
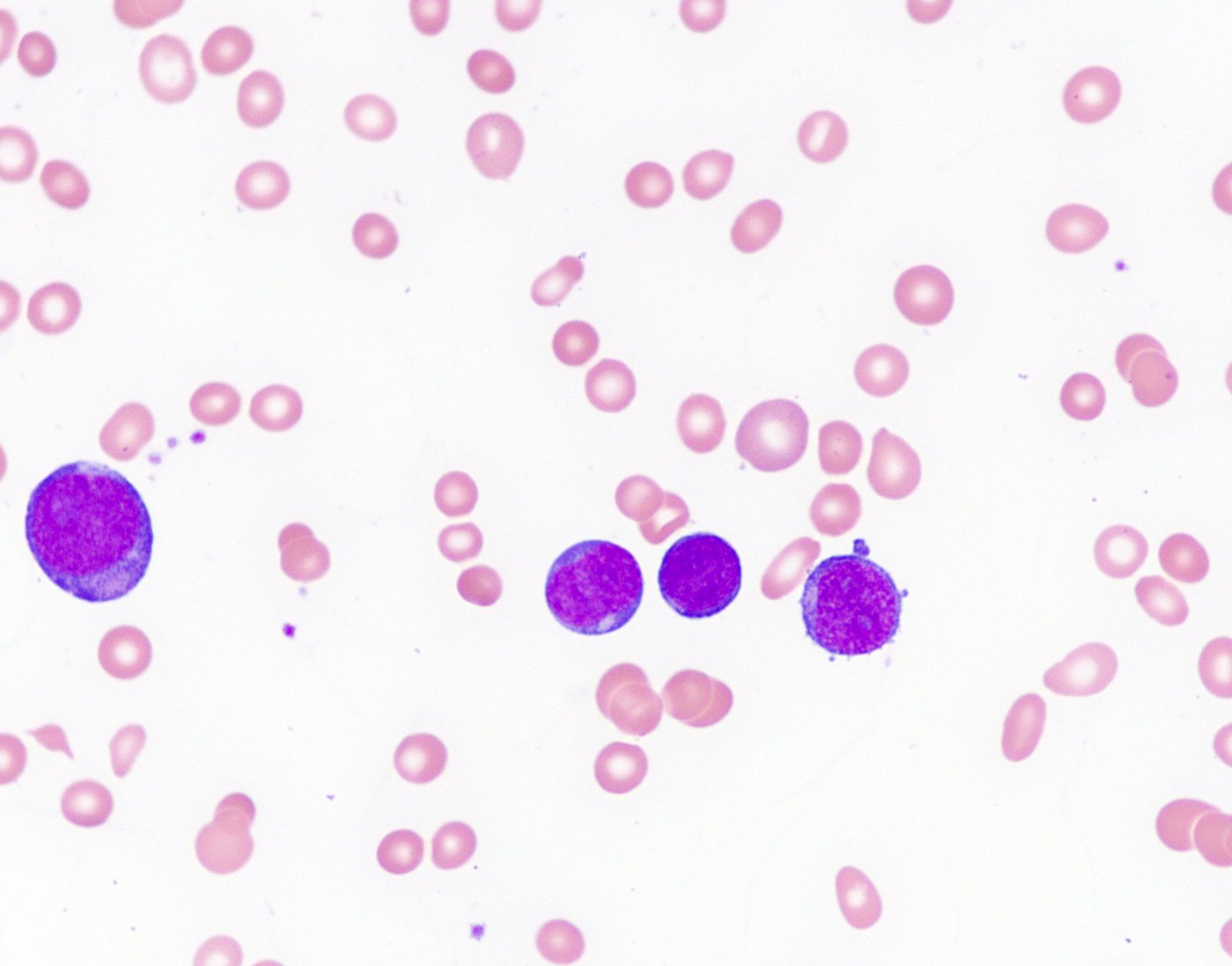
Megakaryoblasts in peripheral blood
- MPO, glycophorin A, CD15, CD41 (30%), CD56 (30%)
- Similar to stains
- Can be helpful to show that the blasts are positive for both CD34 and platelet markers
- Trisomy 21, trisomy 8
- GATA1 mutations are identified in nearly all DS-AMKL (Blood Cells Mol Dis 2003;31:351)
- Secondary mutations in CTCF, EZH2, KANSL1, JAK2, JAK3, MPL, SH2B3 and RAS pathway genes
- Unlikely to have monosomy 7
- Reference: Cancer Cell 2019;36:123
- Bone marrow, biopsy, aspirate and peripheral smear:
- Acute megakaryoblastic leukemia (see comment)
- Comment: The overall features, including smear morphology, immunophenotype of the blast population and detection of a GATA1 mutation are most consistent with myeloid leukemia associated with Down syndrome in this 3 year old child with trisomy 21.
- Peripheral smear:
- The peripheral blood shows anemia and scattered intermediate sized blasts with basophilic cytoplasm, a subset of which has cytoplasmic blebs.
- Bone marrow biopsy:
- Cellular marrow for age (100% cellular). The marrow shows marked fibrosis, clusters of immature cells and frequent dysplastic megakaryocytes which are small with hypolobated nuclei. Residual trilineage hematopoiesis is markedly decreased. A reticulin stain reveals that reticulin is markedly increased (MF 3 of 3). Trabecular bone is unremarkable and there are no lymphoid aggregates. The clot section consists predominantly of clotted blood admixed with rare marrow elements.
- Immunostains:
- CD34 highlights abundant blasts, accounting for 50% of cellularity, which are negative for myeloperoxidase. CD61 highlights abundant dysplastic megakaryocytes as well as blasts.
- Bone marrow aspirate:
- The aspirate smear is aparticulate hemodilute; a formal differential count was not performed. Frequent blasts similar to those in blood are noted. They are medium in size and have a high N/C ratio. The nuclei are round or irregular with moderately delicate chromatin and inconspicuous nucleolus. The cytoplasm is scant and blue staining with occasional cytoplasmic blebs. Prussian blue iron stain on diluted smear is inconclusive for storage iron assessment; no increase in ring sideroblasts is seen.
- Flow cytometry:
- Flow cytometric analysis reveals approximately 44% of analyzed cells are blasts with a megakaryoblastic immunophenotype: CD45dim+ CD34+ CD117+ CD7dim+ CD41a(var)+ CD42+ CD61+ CD14- CD64- CD13dim+ CD33dim CD4- HLADR- CD19- CD10- CD79a- TDT- surfaceCD3- CD56(var) CD235a- MPO- cytoCD3-. The differential based on immunophenotype alone includes acute megakaryoblastic leukemia (AMKL) and transient abnormal myelopoiesis of Down syndrome. Blasts cannot be reliably differentiated by flow cytometry for these two entities but age is more consistent with AMKL. Please correlate with core biopsy analysis and molecular/cytogenetic results as available.
- Cytogenetics:
- Karyotype: 47,XY,+21[20]
- Refractory cytopenias of childhood:
- Can affect children of any age
- Blasts are < 5% and MPO+, monosomy 7 is common
- GATA2 mutations are seen in ~ 5% of cases
- Acute myeloid leukemia (megakaryoblastic) with t(1k;22)(p13.3;q13.1); RBM1S-MKL1:
- Not associated with Down syndrome
- Blasts are usually negative for CD34
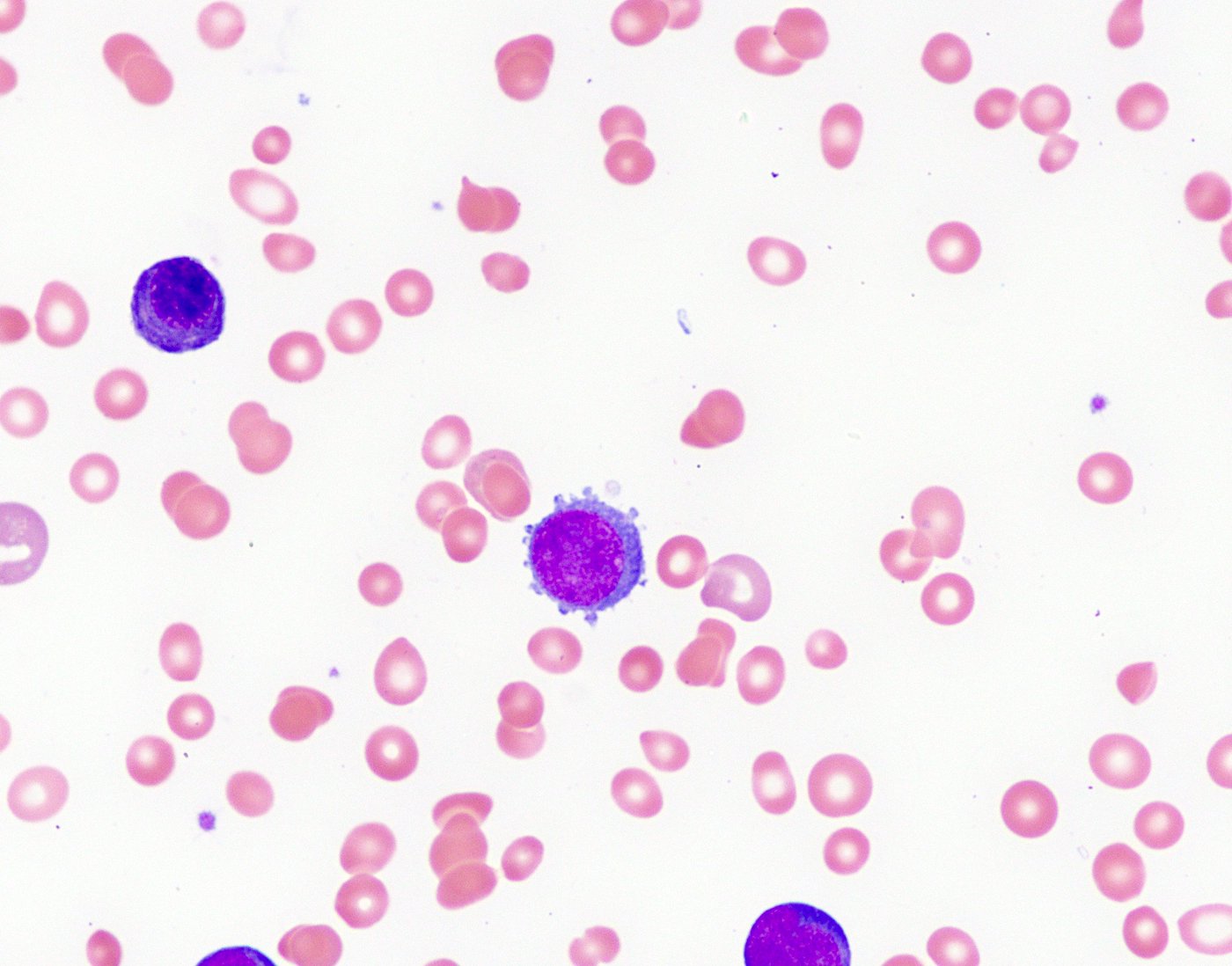
A 29 month old child with Down syndrome presents to the clinic with easy bruising. Peripheral blood smear is significant for thrombocytopenia, anisopoikilocytosis and blasts (~ 30%). A flow cytometry analysis of peripheral blood specimen is pending. What is the most likely immunophenotype of the blasts?
- CD34-, CD117+, HLADR-, MPO++, CD33,
- CD34+, CD33+, CD11b+, CD41+, CD61+, MPO-, CD71-
- CD34+, CD117+, CD33+, HLA-DR+, CD41-, CD61-
- CD34-, CD4+, CD7+, CD33+, CD123+, CD56+
- CD34+, CD19+, CD20-, CD10+, TdT+
Comment Here
Reference: Myeloid leukemia associated with Down syndrome
- Monosomy 7
- t(1;22)
- t(1;19)
- t(2;5)
- GATA1 mutation
- Classified as one of the myeloid neoplasms with germline predisposition and other organ dysfunction in the revised 2016 WHO classification (Blood 2016;127:2391)
- Now known to underlie several syndromes: MonoMAC syndrome, dendritic cell, monocyte, B and NK lymphoid (DCML) deficiency and Emberger syndrome
- Counts for the cause of 7 - 15% of primary myelodysplastic syndrome cases in children and adolescents and for approximately 37% of myeloid neoplasms with monosomy 7 (-7) or other forms of 7q- / -7 in adolescents and young adults
- Differentiating somatic from germline GATA2 mutations is necessary
- Genetic counseling of family members and timely screening of donors for presence of predisposition germline mutations are necessary prior to hematopoietic stem cell transplant for optimal patient outcomes
- Skin fibroblasts are the gold standard for obtaining germline DNA in patients with myeloid neoplasms
- Myeloid neoplasms with germline GATA2 mutation
- Familial myeloid neoplasms
- Familial myelodysplastic syndrome (MDS) / acute myeloid leukemia (AML)
- Prevalence in general population is unknown
- Approximately 550 myeloid neoplasm cases with germline GATA2 mutations reported in literature (Best Pract Res Clin Haematol 2020;33:101197)
- In children, adolescents and young adults, 7 - 15% of primary myelodysplastic syndrome cases and up to 37% of patients with monosomy 7 (-7) are associated with germline GATA2 mutation (Blood 2016;127:1387, Best Pract Res Clin Haematol 2020;33:101197)
- Myeloid neoplasms mainly involve the peripheral blood and bone marrow
- Many other organs other than bone marrow (e.g. ear, lung, heart, joints, skin, blood vessels and immune system) can also be involved, especially if a germline GATA2 mutation associated syndrome exists (Blood 2014;123:809)
- GATA2 is a member of the GATA2 family of zinc finger transcription factors that regulate transcription of many genes
- Plays an essential role in genesis and function of hematopoietic stem and progenitor cells and thus all subsequent blood cell lineages
- Also involved in autoimmunity, inflammation and developmental processes
- Germline GATA2 mutations may result in loss of function of the mutated allele, which leads to GATA2 haploinsufficiency and then affecting function of multiple cell lineages (Hematol Oncol Clin North Am 2018;32:713)
- 4 types of germline GATA2 mutations are categorized:
- Missense mutations, including all mutations resulting in a mutant protein
- Null mutations, including all nonsense, frameshift mutations and large deletions that result in no protein product
- Regulatory mutations, including all mutations within the enhancer region of intron 5
- Uniallelic mutations, including cases with phenotypical GATA2 deficiency but without a defined gene mutation
- According to Spinner et al, null mutations are closely associated with severe viral infections and earlier onset of those infections, while lymphedema was only observed in patients with null or regulatory mutations (Blood 2014;123:809)
- Large deletions are associated with extramedullary developmental defects
- Somatic ASXL1 mutations are commonly observed in patients with germline GATA2 mutations and are considered as an important second hit for myeloid transformation observed in these patients (Haematologica 2014;99:276)
- Inheritance: spontaneously arising but transmitted mainly as autosomal dominant (approximately 80%) (Blood 2014;123:809, Best Pract Res Clin Haematol 2020;33:101197)
- Penetrance was estimated at 90% by the age of 60 years; cases with sporadic germline GATA2 mutations have been reported as well (Br J Haematol 2013;161:701)
- Onset of myeloid neoplasm in patients with germline GATA mutation(s) is between ages of 5 months and 78 years (median 20 years) (Blood 2014;123:809, Best Pract Res Clin Haematol 2020;33:101197)
- Spectrum of the diseases include pediatric MDS or familial MDS / AML, chronic myelomonocytic leukemia (CMML) and myeloproliferative neoplasms (MPNs); especially, germline GATA2 mutations count for approximately 7% of all primary MDS cases in children but are absent in children with secondary MDS
- Initial presentations in these patients are heterogeneous, ranging from asymptomatic, mild or moderate cytopenia (e.g. refractory cytopenia of childhood, RCC) to mild to severe immunodeficiency (e.g. lymphedema, pulmonary diseases, vascular problems, virus or bacteria infections, skin lesions or congenital deafness) (Best Pract Res Clin Haematol 2020;33:101197)
- Can present or be diagnosed as a standalone disease / syndrome prior to myeloid neoplasm as well as coexist with myeloid neoplasm
- The following conditions can be indications to start a workup for potential germline GATA2 mutations that are associated with existing or preexisting myeloid neoplasm:
- Idiopathic cytopenia (especially neutropenia, CD4 lymphocytopenia, depletion of monocytes, B cells, NK cells and dendritic cells), idiopathic bone marrow failure, susceptibility to refractory virus or other pathogen infection (e.g. human papillomavirus [HPV], herpes virus, nontuberculous mycobacteria [NTM] and Histoplasma capsulatum, etc.), pulmonary alveolar proteinosis (PAP) and ventilatory defects, skin lesions (e.g. flat warts), idiopathic lymphedema, solid tumor, especially those being associated with HPV, EBV infections and so on
- Diagnostic criteria for each type of germline GATA2 mutation associated myeloid neoplasm (e.g. MDS, MPN or AML) are the same as that for cases without germline GATA2 mutation; the prevalence of MDL, AML and CMML in a study of 57 patients with GATA2 deficiency were 84%, 14% and 8% respectively and all the AML cases were evolved from MDS (Blood 2016;127:1387)
- Different from de novo MDS cases with a bone marrow feature of hypercellularity, germline GATA2 mutation associated MDS patients mostly present with hypocellular bone marrow for their ages, also frequently with increased fibrosis and dysplastic magakryocytes (Blood 2016;127:1387)
- In general, the status of germline GATA2 mutation does not influence overall survival and outcome of hematopoietic stem cell transplantation (HSCT) (Blood 2014;123:809, Blood 2016;127:2391, Semin Hematol 2017;54:81)
- However, children and adolescence with germline GATA2 mutation(s) and monosomy 7 had a shorter overall survival than their controls with wild type GATA2 (Blood 2016;127:1387, Semin Hematol 2017;54:81)
- Acquired somatic mutations, such as monosomy 7, SETBP1 and ASXL1 mutations that are more frequently detected in adolescents and adults usually indicate a disease progression (Blood 2016;127:2391, Blood 2016;127:1387)
- 16 year old boy with acute myeloid leukemia and severe bilateral warts on hands and fingers since childhood (Leuk Lymphoma 2020;61:3010)
- 22 year old woman with acute monoblastic leukemia diagnosed at age 13 (Br J Haematol 2020;188:768)
- 24 year old man with pancytopenia and recurrent extensive warts on bilateral hands and recurrent panniculitis (JCO Precis Oncol 2019;3:PO.18.00301)
- 27 year old man with myelodysplastic syndrome diagnosed at age 9 and a history of recurrent viral and bacterial infections and congenital deafness (Br J Haematol 2020;188:768)
- 36 year old woman with prolonged pancytopenia, chronic active Epstein-Barr virus infection and later onset of acute myeloid leukemia (JCO Precis Oncol 2019;3:PO.18.00301)
- Intensive chemotherapy is not suitable for patients with germline GATA2 mutation associated myeloid neoplasm due to the underlying immunodeficiency and stem cell defects; therefore, timely HSCT is necessary for patients with disease progression or high risk karyotypes (Blood 2014;123:809, JCO Precis Oncol 2019;3:PO.18.00301, Best Pract Res Clin Haematol 2020;33:101197, Blood 2016;127:2391)
- However, genetic screening of related donors to avoid reintroduction of germline mutation with predisposition is necessary
Images hosted on other servers:

Recalcitrant periungual warts
Contributed by Wei Wang, M.D., Ph.D.
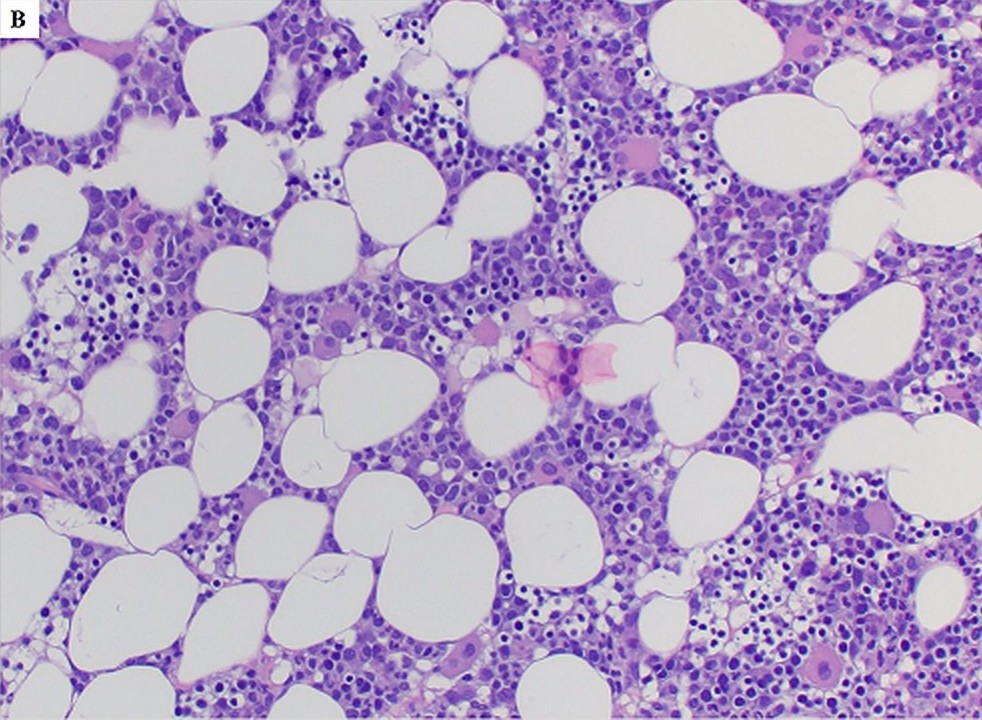
Megakaryocytic dysplasia in MDS
Contributed by Wei Wang, M.D., Ph.D.

Pancytopenia in MDS
- Variable, depending on the type and stages of myeloid neoplasm but not specifically associated with the underlying germline GATA2 mutation
- Variable, depending on the type and stages of myeloid neoplasm but not specifically associated with the underlying germline GATA2 mutation
- Variable, depending on the type and stages of myeloid neoplasm but not specifically associated with the underlying germline GATA2 mutation; for example, in childhood myelodysplastic syndrome with germline GATA2 mutation, characteristic immunophenotyping changes include but are not limited to granulocytopenia, monocytopenia, decreased B cells and NK cells, presence of CD56+ plasma cells, etc. (Haematologica 2011;96:1221, Hematol Oncol Clin North Am 2018;32:713)
Images hosted on other servers:

Flow cytometry study of peripheral blood
- Molecular findings:
- Mutation of various types (missense, nonsense, indels, etc.), involving exon(s) or intron(s) of GATA2 gene; differentiating somatic from germline GATA2 mutations is necessary (see below)
- Cytogenetic findings:
- In the study by Wlodarski et al, the frequent chromosomal abnormalities were monosomy 7 (68%), trisomy 8 (9%), der(1;7)(q10;p10) resulting loss of 1p and 7q (7%); the rest of the cases had a normal karyotype (Blood 2016;127:2391)
- Other chromosomal abnormalities, such as trisomy 21 and t(2;12)(p21;p13), have also reported (JCO Precis Oncol 2019;3:PO.18.00301, Best Pract Res Clin Haematol 2020;33:101197)
- Next generation sequencing (NGS): in 10 - 15% cases, large deletions or mutations in intronic region of GATA2 gene may be involved and they can be missed with standard gene panel based NGS; therefore, NGS based whole exome (WES) or even whole genome (WGS) sequencing can be applicable to identify rare mutation(s) or those located within an intron (Leuk Lymphoma 2020;61:3010, JCO Precis Oncol 2019;3:PO.18.00301)
- Approximately 20% of all cases are de novo mutations without a family history (IUBMB Life 2020;72:142)
- Sanger sequencing, especially for known mutation(s)
- Conventional cytogenetic analysis or FISH, with focus on monosomy 7 or 7q abnormality, trisomy 8 and trisomy 21
- Note: somatic GATA2 mutations reportedly coexist with other germline mutations (e.g. ASXL1, CEBPA) or other acquired drive mutations in myeloid neoplasm cases; differentiating somatic from germline GATA2 mutations is necessary
- Additional reference: Blood 2014;123:809
Contributed by Zhenya Tang, M.D., Ph.D.

Abnormal karyotype with -7

FISH analysis
- Bone marrow biopsy, right posterior iliac crest:
- Gross measurement: 1.2 cm in aggregate. Quality: limited, subcortical. Cellularity: near empty marrow with only 2 small foci of cellular areas with erythroid predominant. Megakaryocytes: present. Infiltrate: no increase in immature cells.
- Bone marrow clot:
- Quality: adequate, 10 - 20% cellularity, morphology similar to biopsy.
- Bone marrow smear / touch preparations:
- Quality: adequate. Granulocytes: decreased with maturation and dysplasia. Erythrocytes: relatively increased with maturation and occasional dyserythropoiesis. Megakaryocytes: rare. Lymphocytes: not increased. Plasma cells: not increased. Blasts: not increased.
- Stains of bone marrow smears, clots and biopsy:
- Iron: slightly increased, no ringed sideroblasts. Reticulin / trichrome: inadequate for reticulin and trichrome stain evaluation. MPO: positive in scattered cells, decreased. CD71 / glycophorin A: highlight increased erythroids. CD61: highlights few scattered megakaryocytes.
- Bone marrow diagnosis:
- Hypocellular marrow with erythroid predominance and granulocytic dysplasia, 2% blasts, morphologically and immunophenotypically consistent with myelodysplastic syndrome (MDS). An underlying congenital condition should be considered (see comment).
- Comment: 23 year old man with a history of recurrent skin infections and warts, CBC with mild pancytopenia and marrow biopsy showed hypocellularity. Concurrent flow cytometry analysis demonstrated aberrant myeloblasts, lack of hematogones, virtual absence of monocytes, minimal NK cells and only 1.1% of B cells. In addition, a tiny atypical T cells population is also detected, CD2+, CD3+, CD5 dim, CD4-, CD8 partial, CD56+, CD57 bright. The overall picture is highly suspicious for a congenital GATA2 mutation. Correlation with pending NGS result will be helpful for confirmation. Correlation with pending ancillary study results is recommended for complete evaluation.
- Germline versus somatic GATA2 mutation
- Syndromic versus nonsyndromic germline GATA2 mutation
- 1 case with somatic GATA2 mutation but mimicking clinical presentation of GATA2 deficiency of germline GATA2 mutations has been reported (Blood Adv 2018;2:904)
Table 1. Differentials of Germline and Somatic GATA2 mutations
| ~ 150 | ~ 50 | |
| Whole GATA2 gene, ZF2 especially | ZF1 | |
| Multiple (see pathophysiology) | Mostly point mutations | |
| GATA2 deficiency of loss of function of affected copy | Gain of function | |
| Syndromic and nonsyndromic with or without myeloid neoplasm; myeloid neoplasm as standalone, especially MDS and AML in children and young adults | Mainly MDS, AML, CML blast phase in all age groups | |
| Other germline mutation, somatic ASXL1, etc. | Biallelic CEBPA mutations; other germline mutations (e.g. RUNX1) | |
| Both disease affected and nonaffected tissues; especially skin biopsy | Disease affected tissue only (e.g. bone marrow and skin biopsy will be mostly negative) |
- Bone marrow biopsy and cell culture
- Buccal swabs and saliva
- Nails or hair
- Peripheral blood and cell culture
- Skin biopsy and cell culture
Comment Here
Reference: Myeloid neoplasms with germline GATA2 mutation
- Myeloid or lymphoid neoplasms with germline RUNX1 variant
- Confirmation of germline origin of RUNX1 variant in skin fibroblasts or persistent presence of RUNX1 variant at ~50% allelic frequency in all complete remission samples
- Hematologic neoplasms with RUNX1 germline variant
- Up to 10% of acute myeloid leukemia (AML) cases with mutated RUNX1 at diagnosis have the RUNX1 mutation confirmed as germline change (Blood 2021;137:1428, Blood Adv 2018;2:146)
- Carriers of familial RUNX1 variants are at ~30 - 40% lifetime risk of having a myeloid neoplasm (Leukemia 2020;34:2519)
- Incidence of development of T cell or B cell lymphoid neoplasm is unknown but recognized (see Case reports) (Pediatr Blood Cancer 2023;70:e30184, Blood Adv 2021;5:3199)
- Variable tissues, including skin, peripheral blood and bone marrow
- Frequency of tissue and germline mosaicism is unknown
- RUNX1 encodes a transcription factor, a binding partner for core binding factor beta (CBFB) and plays a crucial role in thrombocyte differentiation
- Germline pathogenic or likely pathogenic (P / LP) RUNX1 variants are common in familial platelet disorder with lifelong thrombocytopenia and a risk of progression to myeloid neoplasms and less frequently, T cell lymphoblastic leukemia / lymphoma and B cell neoplasms (OMIM: Runt related transcription factor 1; RUNX1 [Accessed 11 January 2024], OMIM: Platelet disorder, familial, with associated myeloid malignancy; FPDMM [Accessed 11 January 2024], Blood Adv 2019;3:2962, Haematologica 2020;105:870)
- Germline P / LP RUNX1 variants in patients with familial platelet disorder demonstrate a wide spectrum of RUNX1 mutations: missense, stop codon, frameshift, splice site and intragenic or whole gene deletions, which result in loss of function of mutated RUNX1 allele (Blood Adv 2019;3:2962, Blood Adv 2020;4:1131, Leukemia 2016;30:999)
- Majority of germline variants in the RUNX1 cluster are in the Runt domain and result in loss of binding activity
- Germline RUNX1 P / LP variants in the transactivation domain have also been noted
- Stop codon, splice site and frameshift mutations, as well as gene deletions, result in loss of function of one allele and haploinsufficiency
- Patients with germline RUNX1 variants experiencing malignant transformation acquire additional mutation(s), affecting either the second RUNX1 allele or other gene (DNMT3A, FLT3, GATA2, PHF6, BCOR, WT1, TET2, ASXL1) (Blood Adv 2020;4:1131, Leukemia 2016;30:999)
- History of thrombocytopenia and progression to hematologic neoplasm in individuals with germline RUNX1 P / LP variant can occur at any age from early infancy to late adulthood (Blood Adv 2020;4:1131, Pediatr Blood Cancer 2023;70:e30184, Blood Adv 2021;5:3199, Leukemia 2020;34:2519)
- Easy bruising and prolonged bleeding following trauma or dental procedures (Blood Adv 2020;4:1131)
- Thrombocytopenia (Blood Adv 2020;4:1131)
- Eczema, psoriasis, arthritis (Blood Adv 2020;4:1131)
- Detection of heterozygous germline P / LP variant in RUNX1 by molecular genetics test
- Mild to moderate thrombocytopenia (50 - 150 x 109/L)
- Qualitative platelet defect with impaired platelet aggregation and dense granule deficiency
- 10 year old boy with history of thrombocytopenia, germline RUNX1 variant and new diagnosis of T cell lymphoblastic lymphoma (Pediatr Blood Cancer 2023;70:e30184)
- 12 year old girl with revertant mosaicism for the familial RUNX1 mutation (Haematologica 2020;105:e535)
- 16 year old girl and 16 year old boy (unrelated patients) with history of mild thrombocytopenia, germline RUNX1 variants and new diagnosis of B cell acute lymphoblastic leukemia (Blood Adv 2021;5:3199)
- Best practice consensus guidelines from the UK Cancer Genetics Group, CanGene-CanVar and the NHS England Hematological Oncology Working group recommend CBC every 3 - 4 months and, if abnormal, thorough clinical and bone marrow examination for all carriers of familial RUNX1 P / LP variants (Br J Haematol 2023;201:25)
- For bleeding: recommendation to use clotting promoters during procedures with bleeding risk
- For early onset of hematologic malignancy: consideration for allogeneic stem cell transplantation from related donor who does not carry RUNX1 germline variant or matched unrelated donor
- For skin manifestations: topical steroids
- Genetic counseling to identify unaffected relatives at risk of being a carrier of germline RUNX1 variant for further surveillance
- Hypocellular or normocellular bone marrow for patient's age (Pediatr Dev Pathol 2019;22:315, Haematologica 2017;102:1661)
- Atypical but not dysplastic small megakaryocytes with hypolobated nuclei (Pediatr Dev Pathol 2019;22:315, Haematologica 2017;102:1661)
- Eosinophilia (Haematologica 2017;102:1661)
Contributed by Barina Aqil, M.D.

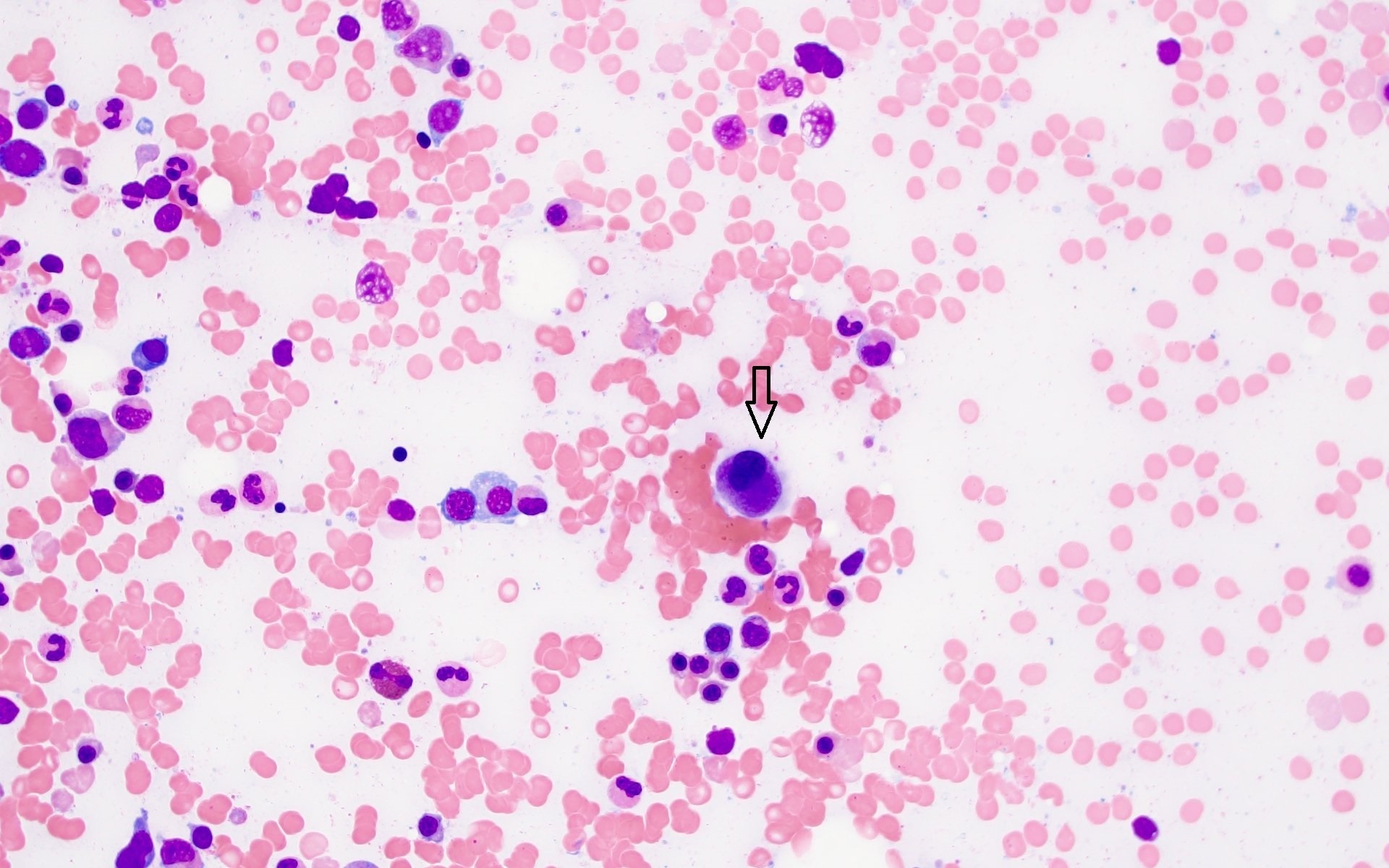

Small hypolobated megakaryocyte

Erythroid dysplasia

Scattered blasts

CD61 highlights small megakaryocytes
- CD34 highlights blasts
- MPO highlights myeloid precursors
- CD61 highlights megakaryocytes including small hypolobated / monolobated forms (Haematologica 2017;102:1661)
- CD71 highlights erythroid precursors
- Reticulin does not show increased fibrosis
- Prussian blue shows decreased iron storage with no ring sideroblasts identified
- Sequence analysis (single gene sequencing or next generation sequencing [NGS] of gene panel) detects heterozygous P / LP RUNX1 variant in ~80% of cases (Blood Adv 2020;4:1131, Blood Adv 2019;3:2962, Eur J Hum Genet 2008;16:1014)
- Copy number analysis (chromosomal microarray, multiplex ligation dependent probe amplification (MLPA), quantitative PCR, fluorescence in situ hybridization) detects a whole RUNX1 gene deletion or intragenic deletion in ~20% of cases (Cancers (Basel) 2022;14:3431, Am J Med Genet A 2016;170:2580, Blood Adv 2019;3:2962, Eur J Hum Genet 2008;16:1014)
- In some patients, analysis of hematopoietic tissue can be negative for mutation or deletion of RUNX1 due to somatic loss of heterozygosity event with a loss of chromosome 21 with P / LP RUNX1 variant; thus, genetic testing of cultured skin fibroblasts is a preferred method of confirmation of the presence of P / LP germline RUNX1 variant, if possible (see Case reports) (Haematologica 2020;105:e535)
Contributed by Madina Sukhanova, Ph.D.
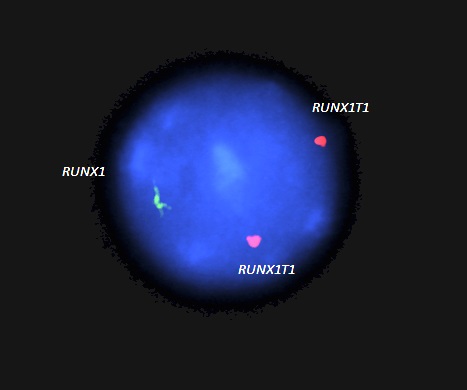
RUNX1 whole gene deletion
Images hosted on other servers:

RUNX1 germline mutations

Reverse mosaicism due to LOH
- Peripheral blood, bone marrow aspirate and bone marrow core biopsy:
- Myelodysplastic syndrome involving a variably cellular bone marrow (see comment)
- Comment: There is dysplasia in the granulocyte, erythroid and megakaryocyte lineages. Immunochemical stain for CD34 highlights 3 - 4% blasts. No ring sideroblasts are identified. Myeloid NGS detected DNMT3A (Q656*, VAF 47%) and TET2 (Q1828*, VAF 46%) mutations. The overall findings are consistent with myelodysplastic syndrome with low blasts (MDS LB) per the WHO, 5th edition, 2022, online beta version and myelodysplastic syndrome, not otherwise specified (MDS, NOS), with multilineage dysplasia per ICC (Blood 2023;141:437)
- Peripheral blood smear
- Peripheral blood smear shows mild normocytic anemia
- Red blood cells show anisopoikilocytosis including occasional teardrop forms, ovalocytes and acanthocytes
- Absolute neutropenia with shift to immaturity
- Thrombocytopenia with unremarkable morphology
- Bone marrow aspirate smear / touch preparation
- Bone marrow aspirate smear slides show several cellular spicules for evaluation
- Myeloid precursors show shift to immaturity with many hypolobated / monolobated neutrophils
- Erythroid precursors show progressive maturations with megaloblastic changes and occasional nuclear budding and irregular nuclear contour
- Megakaryocytes are decreased but occasional small monolobated megakaryocytes are present
- Touch preparations are cellular with similar findings to the bone marrow aspirates
- Small hypolobated / monolobated megakaryocytes are present
- Bone marrow core biopsy / particle clot
- Bone marrow core biopsy is fragmented and variable cellularity, overall normocellular
- Myeloid precursors show progressive maturation with unremarkable morphology
- Small megakaryocytes are present
- Particle clot sections show many bone marrow particles for evaluation
- Marrow is hypercellular for age (~80% cellular)
- Myeloid precursors show progressive maturation with many hypolobated / monolobated neutrophils
- Erythroid precursors are relatively decreased and show progressive maturation with unremarkable morphology
- Megakaryocytes are present with small hypolobated / monolobated forms
- Bone marrow core biopsy is fragmented and variable cellularity, overall normocellular
- Presentation with pre-existing platelet disorder and risk of hematologic neoplasms
- Myeloid neoplasms with germline predisposition and pre-existing platelet disorder due to germline ETV6 P / LP variant (familial platelet disorder with associated myeloid malignancy):
- Presentation with red cell macrocytosis and neutropenia
- Higher likelihood of progression to lymphoid neoplasms
- Thrombocytopenia 2 due to germline ANKRD26 P / LP variant:
- Presentation with erythrocytosis and leukocytosis
- Myeloid neoplasms with germline predisposition and pre-existing platelet disorder due to germline ETV6 P / LP variant (familial platelet disorder with associated myeloid malignancy):
- Presentation without pre-existing platelet disorder and risk of hematologic neoplasms
- Acute myeloid leukemia with germline CEBPA P / LP variant (aka CEBPA associated familial AML):
- Presentation with anemia and leukopenia
- Myeloid neoplasms with germline DDX41 P / LP variant:
- Bone marrow examination reveals megakaryocytic and erythroid dysplasia in some cases
- Myeloid neoplasms with germline TP53 P / LP variant (Li-Fraumeni syndrome):
- Patients with Li-Fraumeni syndromes are at high risk of not only hematologic neoplasms but also soft tissue sarcoma, osteosarcoma and breast cancer
- Myeloid neoplasms with germline GATA2 P / LP variant (GATA2 deficiency):
- Presentation with immunodeficiency
- Acute myeloid leukemia with germline CEBPA P / LP variant (aka CEBPA associated familial AML):
Which of the following is a characteristic phenotype of megakaryocytes in bone marrow biopsy of an asymptomatic individual with familial RUNX1 mutation (shown in the image above)?
- Atypical megakaryocytes with separated nuclear lobes
- Dysplastic megakaryocytes with nonlobated or bilobed nuclei
- Numerous micromegakaryocytes
- Small hypolobated / monolobated megakaryocytes
Comment Here
Reference: Myeloid or lymphoid neoplasms with germline RUNX1 mutation
- ASXL1 mutation
- DNMT3A mutation
- RUNX1 intragenic deletion
- TET2 mutation
Comment Here
Reference: Myeloid or lymphoid neoplasms with germline RUNX1 mutation
- Tumor mass consisting of myeloblasts occurring at an anatomical site other than the bone marrow and distorts the normal tissue architecture
- Although commonly associated with concurrent acute myeloid leukemia, myeloid sarcoma can occur in isolation
- Diagnosis of myeloid sarcoma should be considered an equivalent to a diagnosis of acute myeloid leukemia (AML)
- Based on the WHO classification, myeloid sarcoma must present as a tumor mass with tissue architectural effacement
- Important distinction
- Leukemia cutis (LC) is a somewhat nonspecific term defined as a cutaneous infiltration by neoplastic leukocytes (myeloid or lymphoid; mature or immature) (Am J Clin Pathol 2008;129:130)
- Leukemia cutis is not an equivalent of myeloid sarcoma without fulfilling the myeloid sarcoma diagnostic criteria by WHO classification
- Leukemia cutis could be seen in non-AML patients with myeloproliferative neoplasm (MPN), myelodysplastic syndrome (MDS) or myelodysplastic / myeloproliferative neoplasm (MDS / MPN), such as chronic myelomonocytic leukemia (CMML)
- Can present as de novo lesions, as therapy related myeloid neoplasms or as disease progression of MPN, MDS or MDS / MPN
- Granulocytic sarcoma
- Chloroma
- Extramedullary myeloid tumor
- ICD-10: C92.3 - myeloid sarcoma
- Can occur at any age
- Can involve any site; the most common sites of involvement are lymph nodes, skin, soft tissue, GI tract, bone and testicles
- Exact mechanism is not fully understood
- Chemokine / chemokine receptor interactions are implicated in the migration of leukemia cells migration to skin (Pediatr Blood Cancer 2010;55:344)
- Matrix metalloproteinases (MMPs) and integrin are implicated in the leukemia cells migration and the blood brain barrier disruption (Blood 2009;114:3008, PLoS One 2011;6:e20599)
- CXCR4, CXCL12 and CD56 have been previously reported to be associated with specific sites of myeloid sarcoma, which was not confirmed by a more recent study (Am J Surg Pathol 2016;40:1473)
- Variable clinical presentation depending on the location of tumor (Clin Lymphoma Myeloma Leuk 2017;17:263)
- Primary myeloid sarcoma (de novo): no pre-existing diagnosis of myeloid neoplasm or AML
- Secondary myeloid sarcoma: concurrent or pre-existing diagnosis of myeloid neoplasm or AML
- Isolated myeloid sarcoma: primary or secondary myeloid sarcoma with no bone marrow involvement
- Broad panel of immunohistochemical stains required to avoid misdiagnosis (Am J Clin Pathol 2015;144:219)
- Cases without concurrent bone marrow involvement may have no fresh material for karyotyping and flow cytometric studies; fluorescence in situ hybridization (FISH) studies and mutation analysis should still be attempted on formalin fixed, paraffin embedded (FFPE) samples
- Findings vary, depending on the location of the tumor and the bone marrow involvement status
- Limited data on the prognostic significance of myeloid sarcoma
- Survival likely is similar to that for AML (Acta Haematol 2009;122:238, Leukemia 2003;17:1100)
- Tumor site may play a role in the prognosis
- 47 year old woman with 2 large vulvar ulcers (Diagn Pathol 2019;14:126)
- 54 year old man with repeated symptoms of intestinal obstruction (Surg Case Rep 2020;6:2)
- 58 year old woman with right breast mass and history of essential thrombocythemia (ET) (Hematol Oncol Stem Cell Ther 2018;11:178)
- 84 year old man with superior mediastinal mass causing airway obstruction (Proc (Bayl Univ Med Cent) 2017;30:195)
- No universal consensus on treatment (Leukemia 2021;35:1193)
- Treatment strategies may depend on the disease presentation (initial or relapse) and vary from institution to institution
- Treatment includes systemic chemotherapy, local therapy (surgery and radiation therapy), bone marrow transplant and target therapy
- At least partial effacement of the architecture
- Neoplastic cells can be seen forming cohesive nests, mimicking carcinoma or lymphoma or infiltrating as single files
- Granulocytic, monocytic or rarely, erythroblastic or megakaryoblastic differentiation can be seen
- Neoplastic cells usually demonstrate dispersed chromatin with high N/C ratio
- Mitotic figures and apoptosis are commonly identified indicating a high proliferation rate
Contributed by Yen-Chun Liu, M.D., Ph.D.
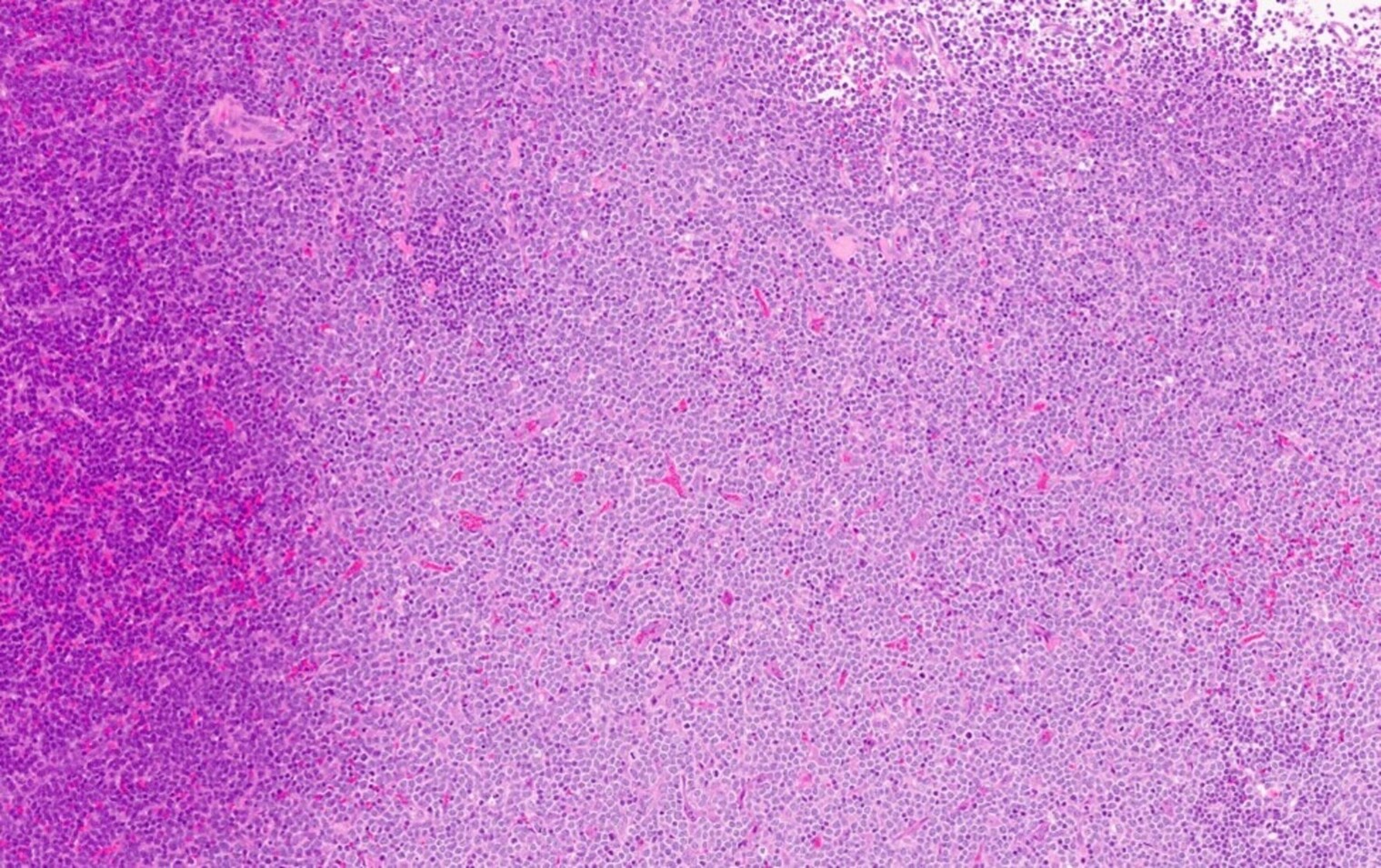
Architecture effaced by neoplasm
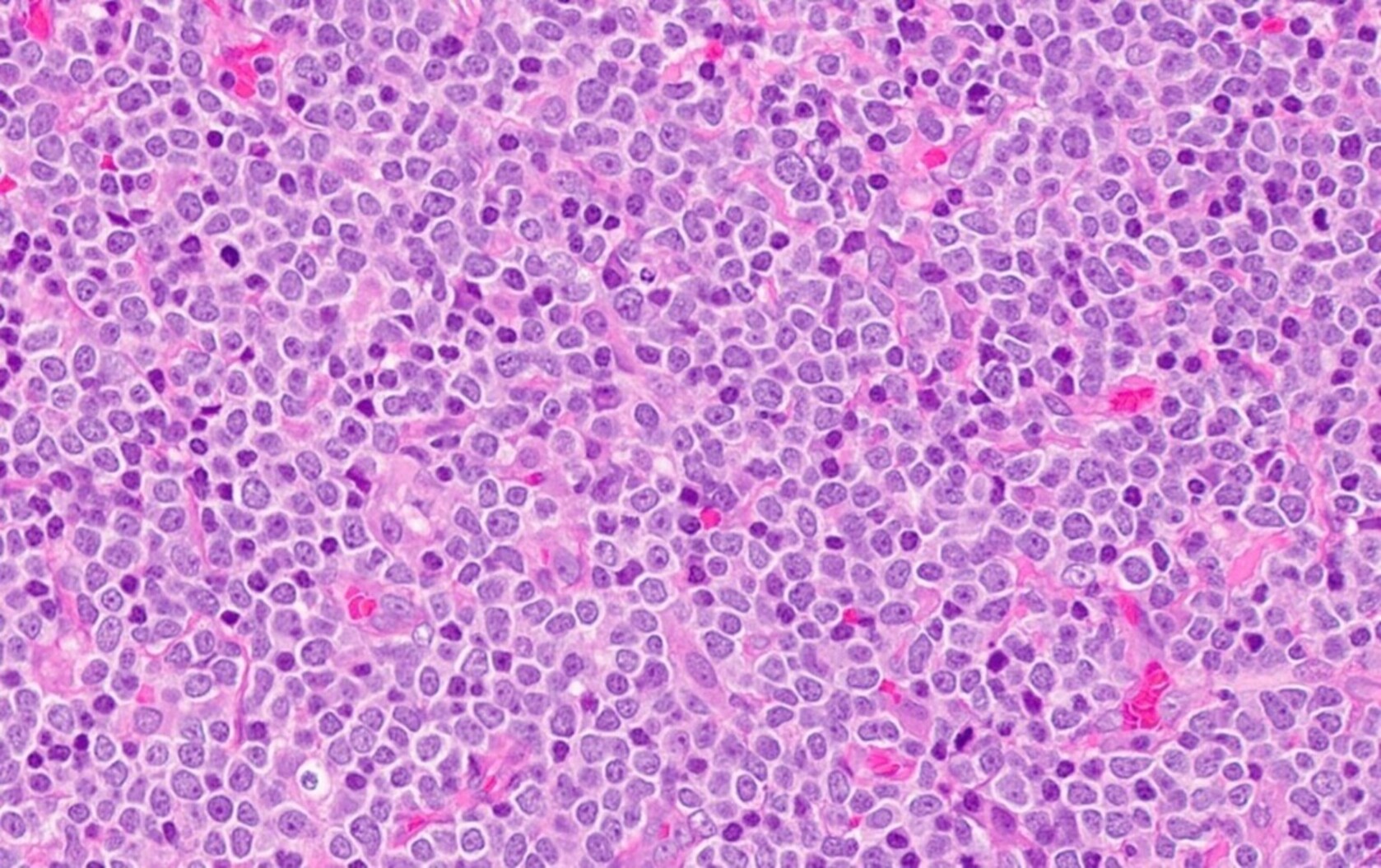
Sheets of myeloid blasts
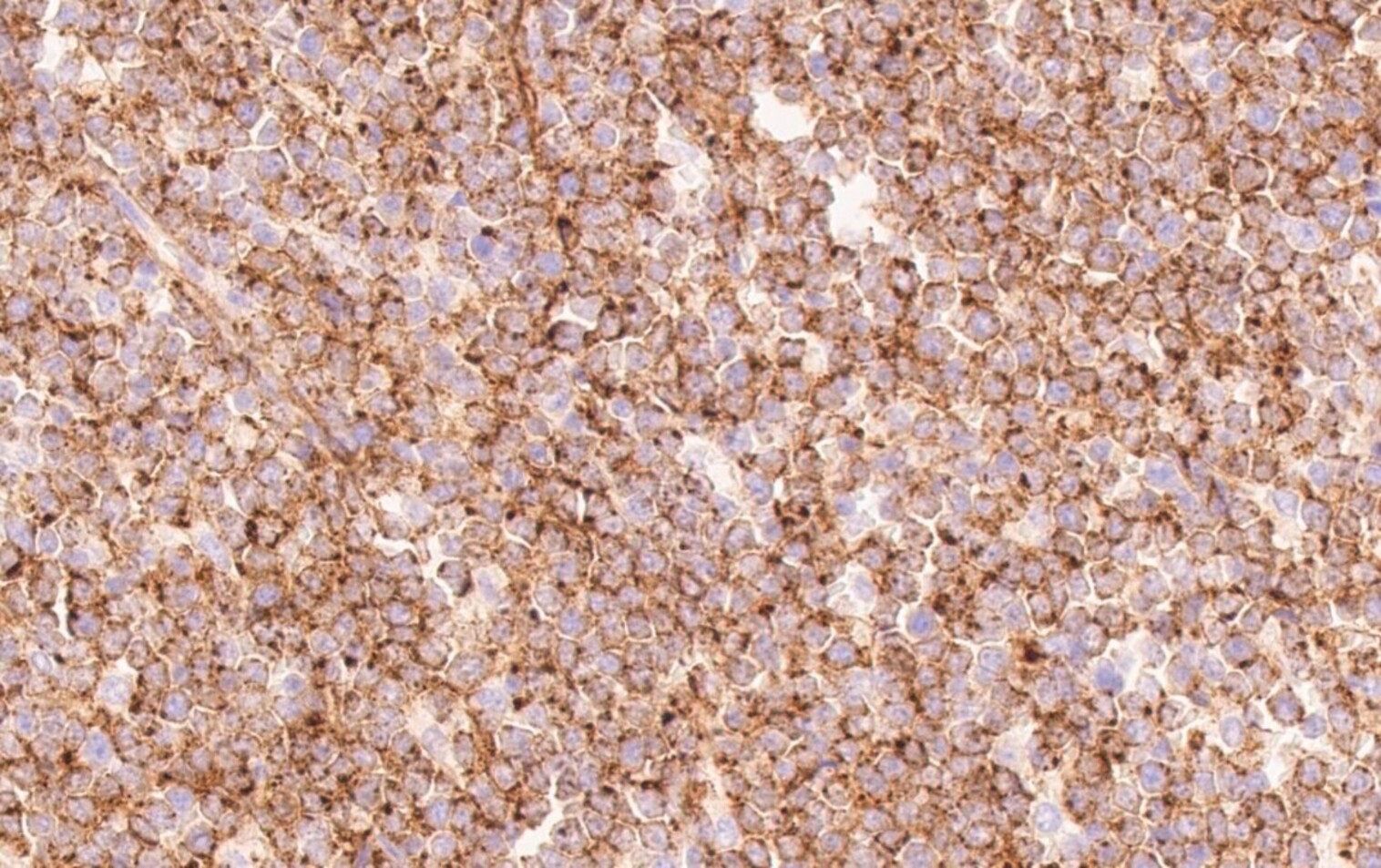
Blasts positive for CD34
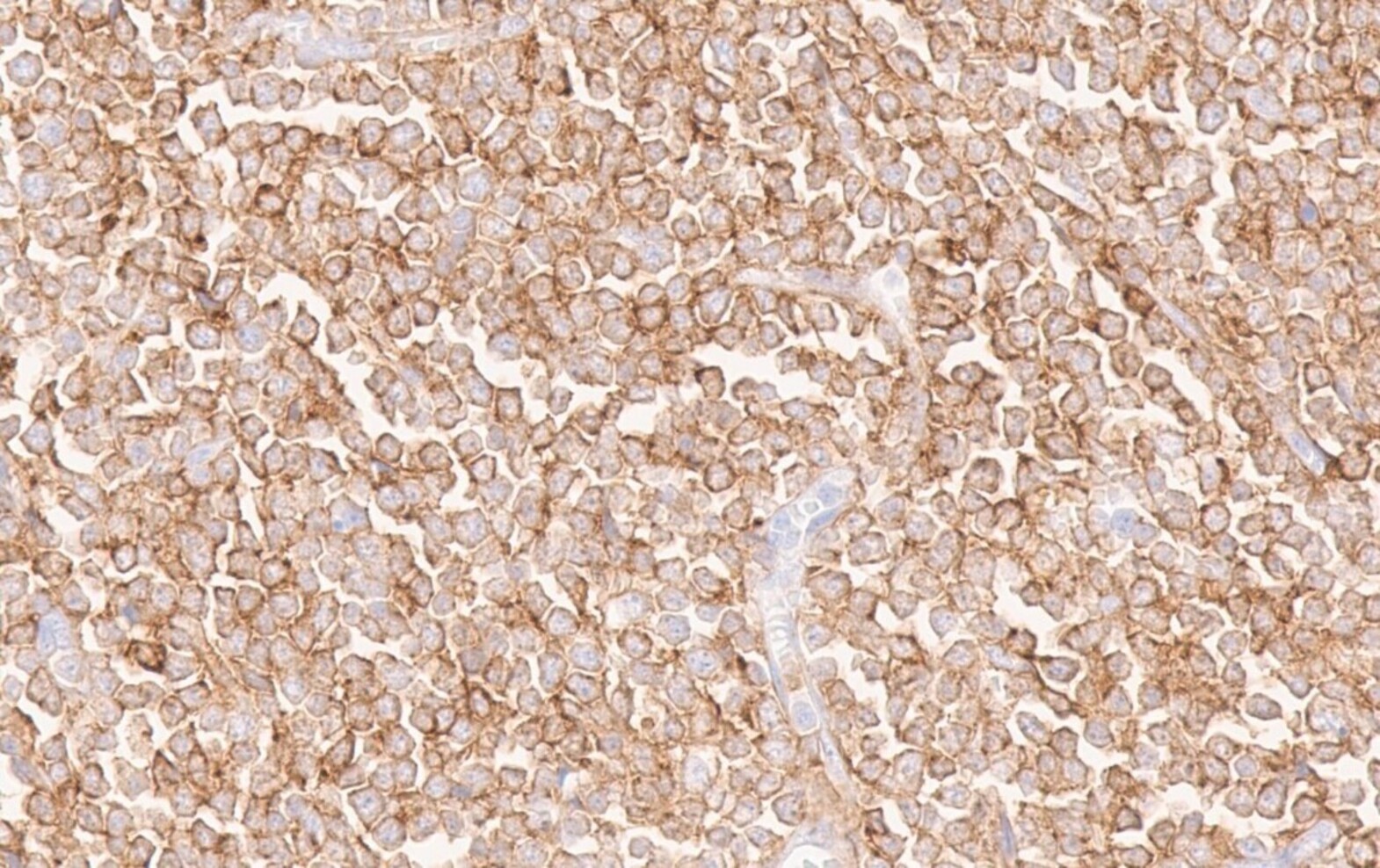
Blasts positive for CD33
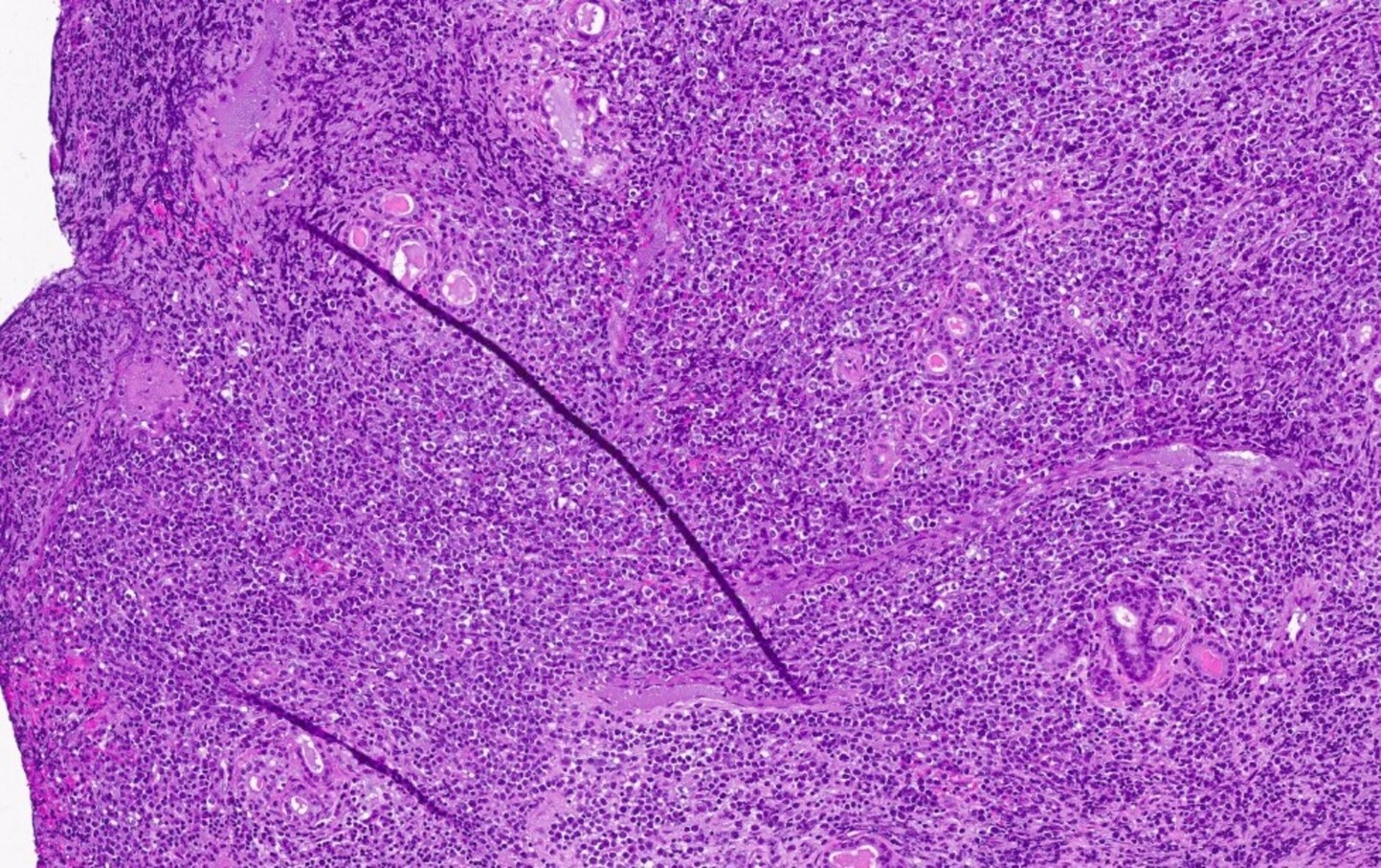
Nasopharyngeal mass
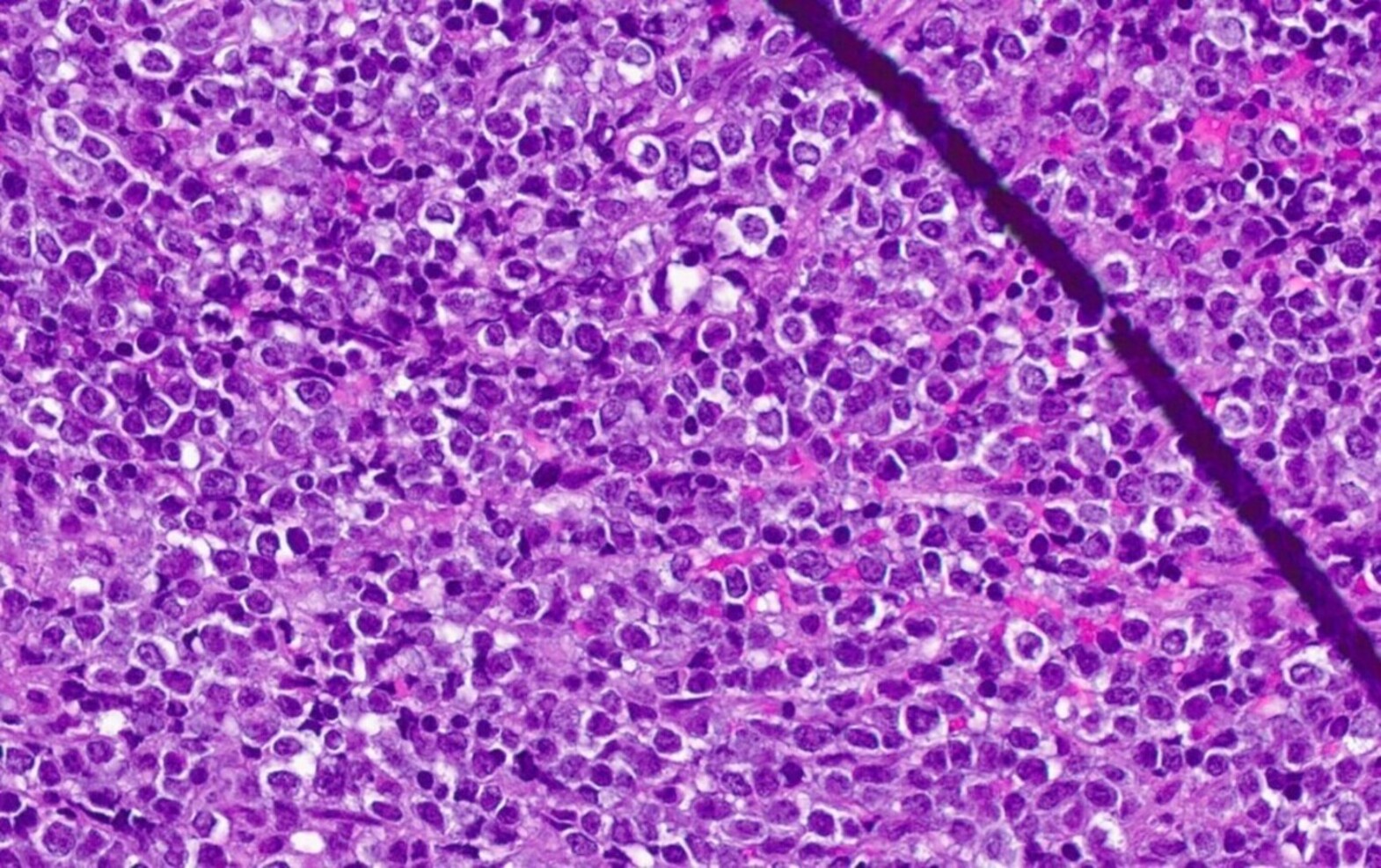
Mitosis and apoptosis identified

Blasts positive for CD117
Blasts positive for MPO
- Positivity may depend on the patterns of differentiation (Diagn Pathol 2007;2:42)
- CD68 / KP1, lysozyme, CD33, CD43: positive in high percentage of cases
- CD45, MPO and CD117 are also commonly seen but are not present in all cases (likely in > 50% of cases)
- CD56, CD34 and TdT: positive in only subset of cases (likely in < 50% of cases)
- CD71 and glycophorin for lesions with erythroblastic differentiation; CD42b and CD61 for lesions with megakaryoblastic differentiation
- Negative for lymphoid lineage markers, such as CD20 and CD3
- Usually negative for CD19 and PAX5 but association with t(8;21)(q22;q22.1) RUNX1-RUNX1T1 can show positive expression (Blood 1992;80:470)
- Workup similar to that for acute leukemia but fresh material is usually lacking
- Roughly half of cases show cytogenetic abnormalities
- Rare cases may show discrepancies from the karyotype obtained from the corresponding bone marrow, which may suggest clonal evolution
- Cytogenetic and molecular abnormalities seen in myeloid sarcoma are similar to those seen in AML
- Common translocations include t(8;21)(q22;q22) RUNX1-RUNX1T1 and inv(16)(p13.1q22) CBFB-MYH11
- Complex karyotype can be seen, usually in secondary myeloid sarcoma (Eur J Haematol 2018;100:603)
- Common mutations include NRAS, NPM1, FLT3-ITD, KRAS and IDH2 (Blood Cancer J 2018;8:43)
- JAK2 V617F mutation can be seen, especially in cases with pre-existing MPN
- BCR-ABL1 fusion can be seen and may represent disease progression of chronic myeloid leukemia (CML)
- Nasopharyngeal mass, biopsy:
- Myeloid sarcoma (see comment)
- Comment: The histologic sections demonstrate sheets of immature mononuclear cells. The immature cells are positive for CD34, CD33, CD117, MPO and negative for CD19 and CD3 by immunohistochemical stains. The morphologic and immunohistologic findings in the current sample support a diagnosis of myeloid sarcoma. A bone marrow examination to investigate for potential involvement by a myeloid neoplasm / acute myeloid leukemia will be of interest if clinically indicated. If no fresh diagnostic material can be retrieved from additional tissue / bone marrow examination, fluorescence in situ hybridization studies and molecular studies can be requested on the current sample for further characterization.
- Lymphoblastic lymphoma:
- Can demonstrate similar blast-like morphology
- Relies on immunophenotyping to differentiate
- Positive for T cell or B cell lineage markers; commonly positive for TdT
- Burkitt lymphoma:
- Usually has a prominent starry sky appearance, though the starry sky appearance is not entirely specific
- Positive for B cell lineage markers, not myeloid markers
- MYC rearrangement present
- Diffuse large B cell lymphoma:
- Predominately large cells; cytologic features are less likely blastoid
- Positive for B cell lineage markers, not myeloid markers
- Histiocytic sarcoma:
- Can be a very challenging differential diagnosis due to significant overlapping immunophenotypic features
- Less likely to demonstrate bone marrow involvement
- Nonhematopoietic tumor:
- Relies on immunophenotyping in some scenarios
- Commonly used markers for screening include cytokeratin, CD45 and S100
- Extramedullary hematopoiesis:
- More likely occurs in patients with MPN
- Trilineage hematopoiesis with maturation is usually present
- Blastic plasmacytoid dendritic cell neoplasm (BPDCN):
- Always presents as a solitary mass
- Can occur with a concurrent myeloid disorder
- CD43 is not a good marker for the diagnosis
- Only occurs in the GI tract
Comment Here
Reference: Myeloid sarcoma
Which of the following statements is true about myeloid sarcoma?
- Always shows megakaryoblastic differentiation
- Can only be identified with concurrent acute myeloid leukemia in bone marrow
- Never shows cytogenetic abnormalities
- NPM1 mutation can be found in myeloid sarcoma and lesions with NPM1 mutations tend to demonstrate myelomonocytic morphology
Comment Here
Reference: Myeloid sarcoma
- Myeloid, lymphoid or combined myeloid / lymphoid neoplasms often accompanied by eosinophilia and associated with FIP1L1-PDGFRA fusion, other PDGFRA gene rearrangements or an activating mutation of the PDGFRA gene
- Myeloid or lymphoid neoplasm, often accompanied by prominent eosinophilia
- Involved PDGFRA gene, cytogenetic location 4q12 (N Engl J Med 2003;348:1201)
- Rearrangement of PDGFRA must be defined by FISH or reverse transcription PCR
- Multiple fusion partners are identified
- Can have different bone marrow pictures, most commonly chronic eosinophilic leukemia (CEL) or other diseases (e.g., acute transformation to acute myeloid leukemia or T cell acute lymphoblastic leukemia [T ALL])
- Very sensitive to imatinib therapy
- Associated mast cell hyperplasia
- Myeloid and lymphoid neoplasms with PDGFRA rearrangement
- Myeloid and lymphoid neoplasms associated with PDGFRA rearrangement
- ICD-11: 2A50 - myeloid neoplasm associated with PDGFRA rearrangement
- M:F = 17:1
- Age range 7 - 77; mostly between 25 - 55
- Reference: Immunol Allergy Clin North Am 2007;27:377
- Usually, peripheral blood and bone marrow
- Most common translocation partner is FIP1L1; FIP1L1-PDGFRA is generated from a submicroscopic, 800 kb interstitial deletion on chromosome 4, del(4)(q12q12), a cryptic deletion of CHIC2
- Other fusion partners have been identified:
- t(4;10)(q12;p11) KIF5B:PDGFRA (Leukemia 2006;20:827)
- ins(9;4)(q33;q12q25) CDK5RAP2:PDGFRA (Genes Chromosomes Cancer 2006;45:950)
- t(4;12)(q12;p13) PDGFRA:ETV6 (Br J Haematol 2007;138:77)
- t(4;22)(q12;q11) BCR:PDGFRA
- t(4;2) (q12;p24) STRN:PDGFRA
- t(4;10)(q12;q23) PDGFRA:TNKS2
- t(4;22)(q12;q11) BCR-PDGFRA translocation is a neoplasm with features intermediate between chronic myeloid leukemia and chronic eosinophilic leukemia (CEL) (Immunol Allergy Clin North Am 2007;27:377)
- Extramedullary myeloid tumor can harbor PDGFRA rearrangement despite the bone marrow not displaying the rearrangement (Cancer Genet 2018;220:13)
- Activating mutations of PDGFRA can play a role in tumorigenesis (e.g., Y288C mutation) (Nat Commun 2018;9:4583)
- Unknown
- Some cases occurred after cytotoxic chemotherapy (Br J Haematol 2006;134:547)
- Always involves peripheral blood and bone marrow; however, other organs can be affected (e.g., lungs, CNS, skin and GI tract)
- Usually presents as chronic eosinophilic leukemia (CEL); can present as acute myeloid leukemia, T acute lymphoblastic leukemia or myeloid sarcoma (Leukemia 2007;21:1183, Head Neck Pathol 2021;15:1399)
- Symptoms range from asymptomatic to generalized fatigue, respiratory (dyspnea, cough) or GI symptoms (Leuk Lymphoma 2013;54:897)
- Can present with cardiac symptoms, such as endomyocardial fibrosis, Loeffler endocarditis and valvular regurgitation (Korean J Intern Med 2018;33:642)
- Splenomegaly is more common than hepatomegaly
- Can present as arterial or venous thrombosis or thrombotic thrombocytopenic purpura (TTP) (Case Rep Hematol 2019;2019:2820954)
- May present with anemia and thrombocytopenia
- Peripheral blood and bone marrow examination both usually show eosinophilia (sometimes normal)
- FISH is the most used method for diagnosis (also known as CHIC2 deletion) and chromosomal microarray; however, reverse transcription PCR is more sensitive (Oncol Lett 2020;19:3587)
- Phosphoflow cytometry can be used for detection of PDGFRA rearrangement; however, it is not very sensitive in patients on treatment (Cytometry A 2021;99:784)
- Next generation sequencing can show additional mutations associated with this rearrangement (e.g., somatic KIT and CSF3R mutations) (Haematologica 2018;103:e348, Am J Hematol 2016;91:E10)
- Diagnostic criteria for myeloid / lymphoid neoplasms with eosinophilia associated with FIP1L1-PDGFRA or a variant fusion gene (A):
- Myeloid or lymphoid neoplasm, usually with prominent eosinophilia and presence of FIP1L1-PDGFRA fusion gene or a variant fusion gene with rearrangement of PDGFRA or an activating mutation of PDGFRA (B):
- A) Cases presenting as a myeloproliferative neoplasm, acute myeloid leukemia or lymphoblastic leukemia / lymphoma with eosinophilia and FIP1L1-PDGFRA gene fusion are assigned to this category
- B) If appropriate molecular analysis is not possible, this diagnosis should be suspected if there is a myeloproliferative neoplasm with no Philadelphia chromosome and with the hematological features of chronic eosinophilic leukemia associated with splenomegaly, marked elevation of serum vitamin, elevation of serum tryptase (> 12 ng/dL and < 20 ng/dL) and an increased number of bone marrow mast cells
- Myeloid or lymphoid neoplasm, usually with prominent eosinophilia and presence of FIP1L1-PDGFRA fusion gene or a variant fusion gene with rearrangement of PDGFRA or an activating mutation of PDGFRA (B):
- Complete blood count usually shows eosinophilia, usually > 1,500/uL; however, normal counts can be found
- Increased vitamin B12 levels > 1,900 ng/L in all cases with FIP1L1-PDFGRA due to increased levels of haptocorrins (Am J Hematol 2019;94:1149)
- Increased tryptase levels > 12 ng/dL and < 20 ng/dL (minor criteria for systemic mastocytosis)
- Good prognosis with absence of cardiac involvement
- 51 year old woman with cutaneous T cell lymphoma with PDGFRA rearrangement and absence of eosinophilia (Am J Dermatopathol 2018;40:610)
- 60 year old man with chronic eosinophilic leukemia with PDGFRA rearrangement associated with tumor lysis syndrome as a complication of imatinib therapy (J Glob Oncol 2018;4:1)
- 70 year old man presenting with myeloid sarcoma in the retromolar area with PDGFRA rearrangement (Head Neck Pathol 2021;15:1399)
- Usually sensitive to imatinib; alternative tyrosine kinase inhibitors (TKIs; e.g., midostaurin or sorafenib) may be effective
- Treatment with imatinib even in cases presenting with acute myeloid leukemia or T cell acute lymphoblastic leukemia
- However, T674I mutation within the adenosine triphosphate (ATP) binding domains of PDGFRA is resistant to imatinib and second generation tyrosine kinase inhibitors; a panresistant D842 variant was also identified (Hemasphere 2019;3:e182, Leukemia 2009;23:845)
- Third generation TKI ponatinib is active against both FIP1L1-PDGFRα p.T674I and p.D842 V (Hemasphere 2019;3:e182)
- Midostaurin and sorafenib are alternative regimens
- Some activating mutations of PDGFRA (e.g., Y288C mutations) are resistant to PDGFRA inhibitors; however, they are sensitive to PI3K / mTOR inhibitors (Nat Commun 2018;9:4583)
- Cases of acute myeloid leukemia with PDGFRA rearrangement and another mutation can achieve complete remission with tyrosine kinase inhibitors (Oncol Lett 2020;19:3587)
- End point of treatment is unclear; in general, imatinib can effectively suppress but not eliminate the FIP1L1-PDGFRA clone in most patients (Am J Hematol 2019;94:1149)
- Allogeneic hematopoietic stem cell transplantation would be the option for those resistant / refractory cases (Blood 2017;129:704)
- Monocytes and basophil counts are normal
- Hypercellular marrow with increased eosinophils and its precursors (see Microscopic [histologic] images); clusters or scattered, increased spindle shaped CD25+ or CD2- mast cells present (Blood 2003;101:4660)
- Reticulin fibrosis is identified; sometimes can be normal (Blood 2004;103:473)
- Increased blasts identified in accelerated / blastic phase of myeloproliferative neoplasms or de novo acute myeloid leukemia or T cell acute lymphoblastic leukemia (see Microscopic [histologic] images)
- Tissue eosinophilic infiltrate forming eosinophilic microabscesses or identifiable Charcot-Leyden crystals
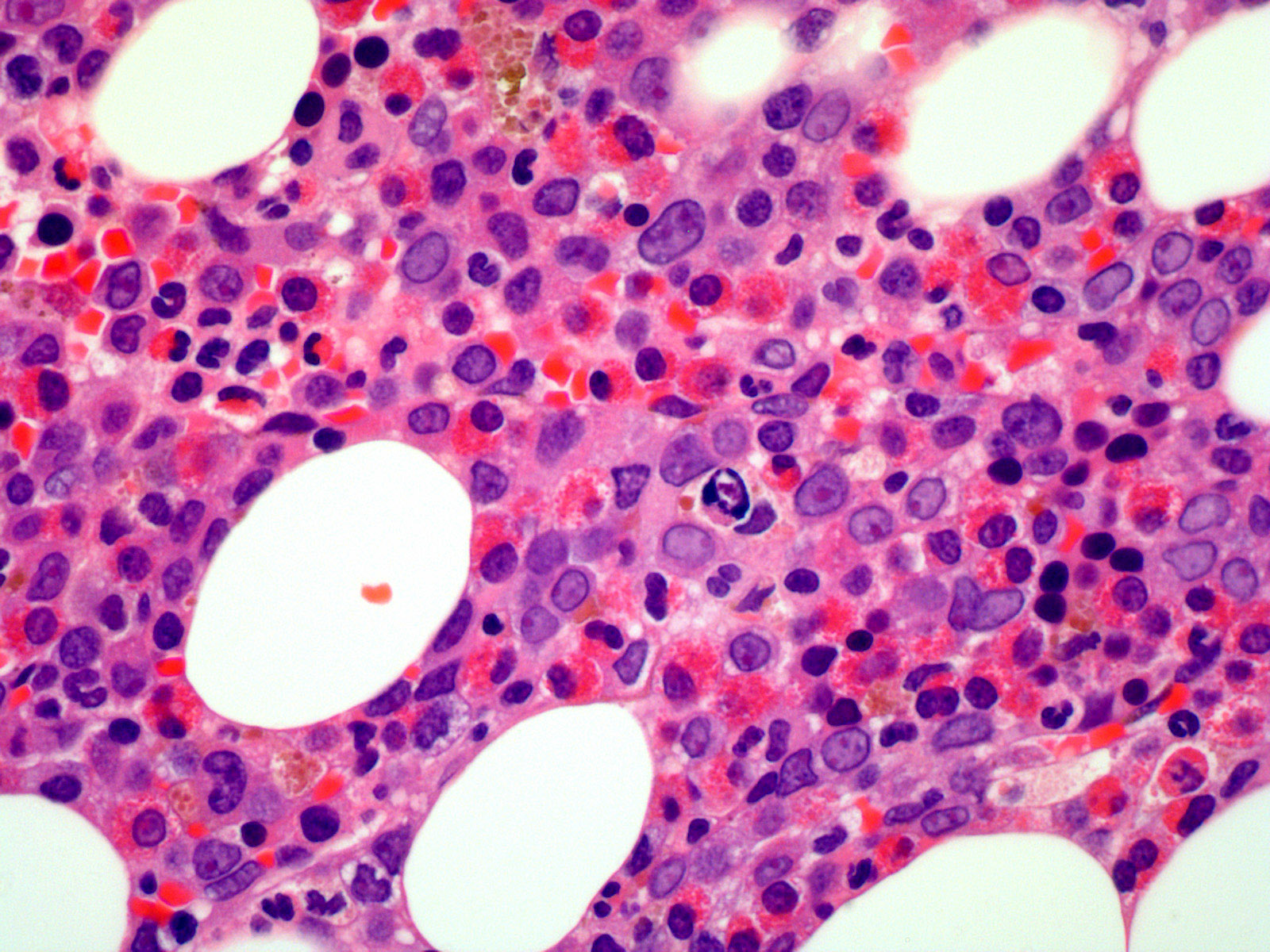
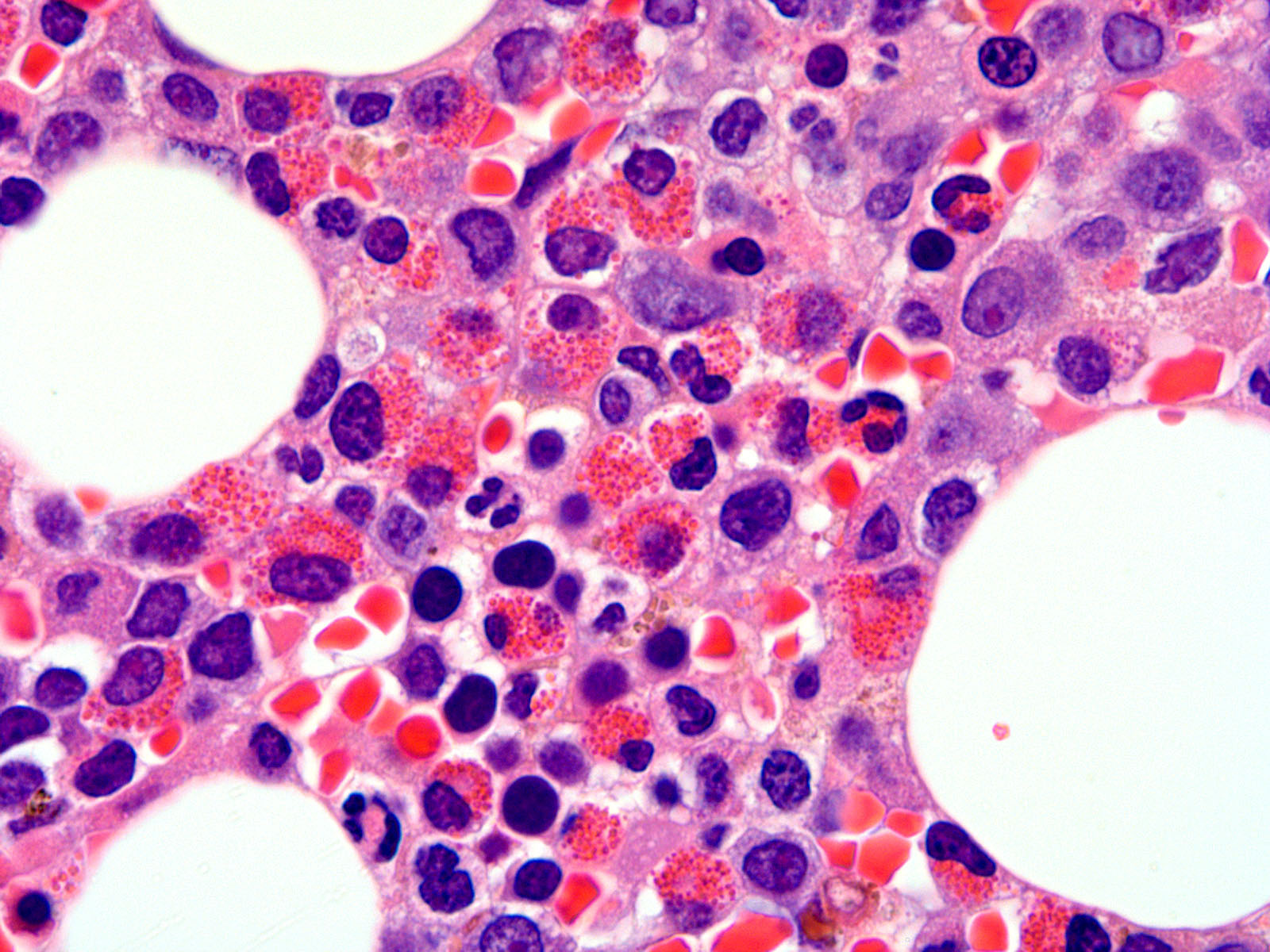
- Peripheral blood eosinophilia, occasionally neutrophilia; eosinophils are usually mature
- Variable morphologic abnormalities can be present in the eosinophils, including cytoplasmic vacuolation, nuclear hypersegmentation / hyposegmentation; morphologic changes are not unique as reactive conditions can have these changes as well (Foucar: Bone Marrow Pathology, 4th Edition, 2019)
- Some cases will present with increased blasts if the patient has acute lymphocytic leukemia or acute myeloid leukemia
Contributed by Ismail Elbaz Younes, M.D. and Ling Zhang, M.D.
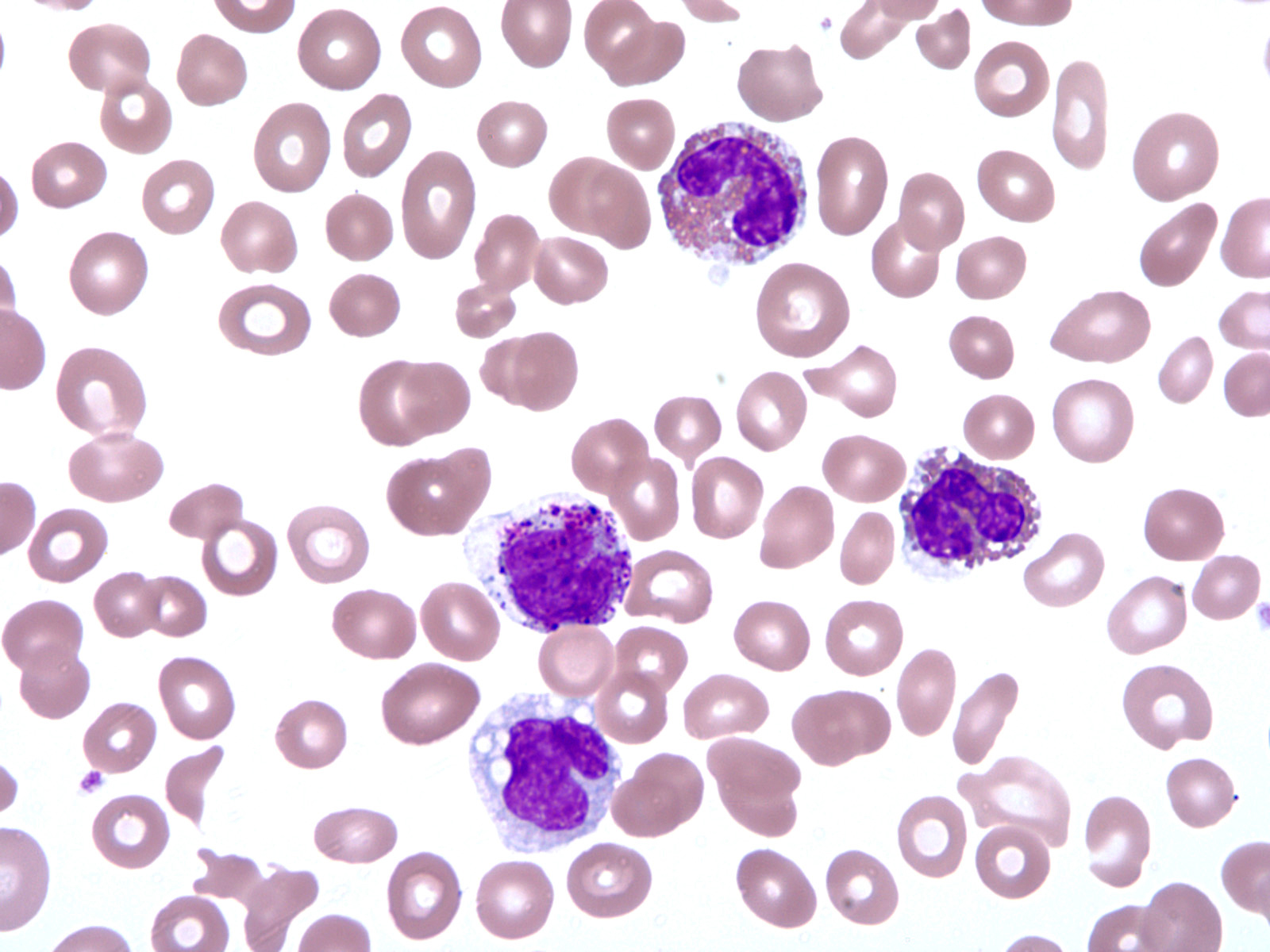
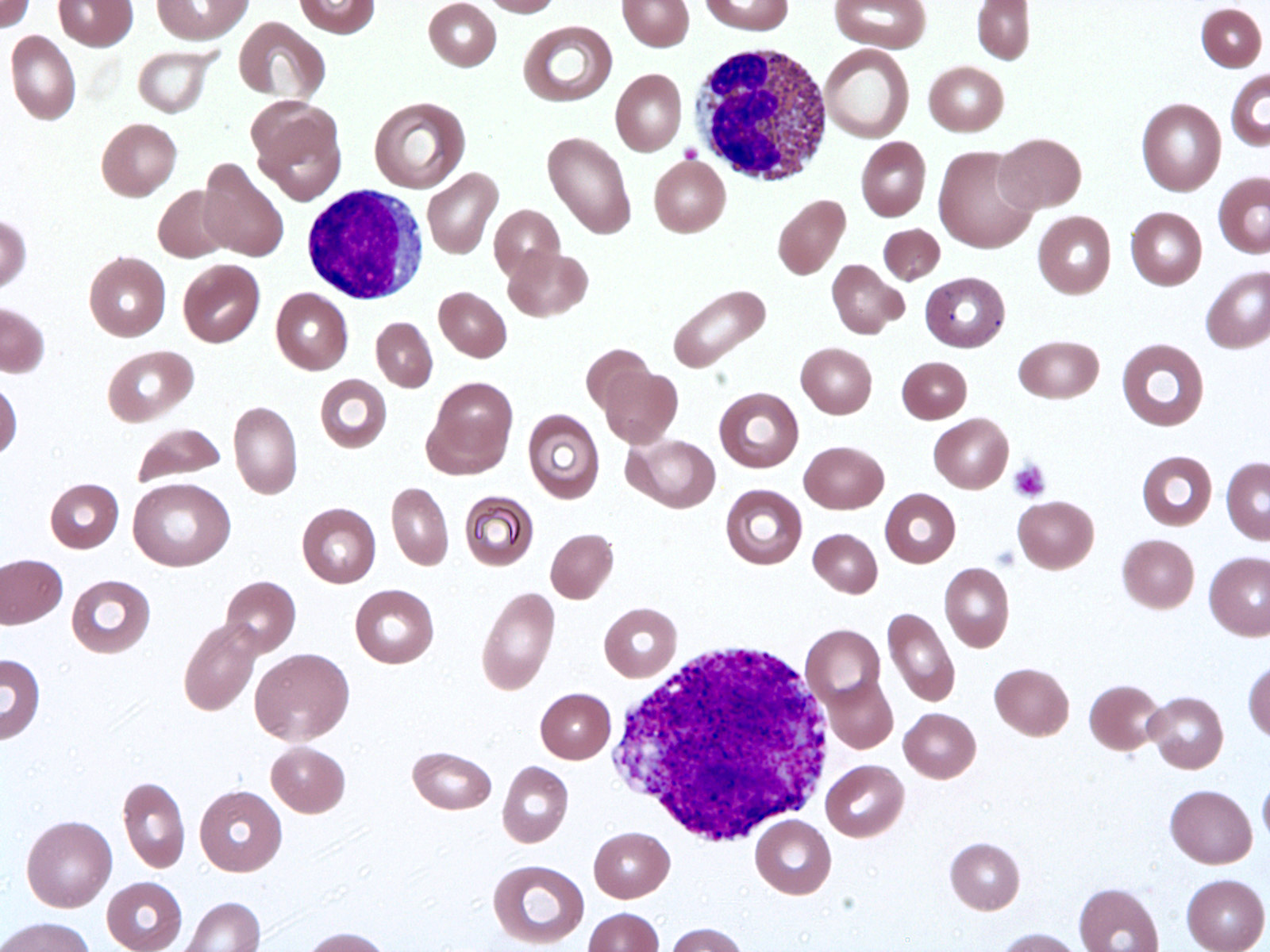
Increased eosinophils with
abnormal granulation
- FISH shows loss of CHIC2 gene
Contributed by Ismail Elbaz Younes, M.D. and Ling Zhang, M.D.
Deletion of CHIC2
- Peripheral blood:
- Microcytosis with mild anisopoikilocytosis
- Moderate neutropenia with marked eosinophilia
- Moderate thrombocytopenia
- Bone marrow, left posterior iliac crest, aspirate and biopsy:
- Hypercellular marrow with marked hypereosinophilia and FIP1L1-PDGFRA rearrangement (see comment)
- Marked reticulin and diffuse collagen fibrosis
- Absent iron stores
- Comment: Cytogenetics confirms a normal male karyotype (46,XY). FISH identifies a microdeletion of the CHIC2 gene on chromosome 4q12, resulting in fusion of FIP1L1-PDGFRA. No abnormality of PDGFRB (5q33) is identified.
- Microscopic description:
- Peripheral smear: The hemoglobin is normal, with microcytic indices and mild anisopoikilocytosis. The reticulocyte count is decreased for the degree of anemia. The total white cell count is moderately increased, with a moderately severe neutropenia and a marked eosinophilia composed of relatively mature appearing eosinophils with normal granulation. Circulating blasts are not seen. The platelet count is moderately decreased, with relatively normal platelet morphology.
- Aspirate smear: The Wright stained aspirate smears are hypercellular with several poorly spread spicules, adequate for evaluation. Megakaryocytes are noted in moderately increased numbers, including many large hyperlobulated forms and few hypolobulated forms with increased cytoplasm. Myeloid and erythroid precursors are evenly distributed, with an M:E ratio calculated at 2.6. Myeloid precursors show adequate maturation and relatively normal morphology. There is no increase in blast cells. There is a marked eosinophilia, with predominantly mature segmented eosinophils showing normal granulation. Erythroid precursors are admixed, with adequate maturation and mild megaloblastic change. There is no significant increase in mast cells.
- Clot section and core biopsy: The clot section (A) and core biopsy (B) are fixed in B-Plus Fix (BBC) fixative. The core biopsy (B) is decalcified in Nitrical (5% nitric acid). 1 level of the clot section (A) and 1 level of the core biopsy (B) are stained with PAS to evaluate myeloid maturation and more accurately estimate an M:E ratio. 1 level of the clot section (A) and 1 level of the core biopsy (B) are stained with Prussian blue to evaluate iron stores and the average value reported. 1 level of the core biopsy (B) is stained with reticulin stain to evaluate marrow fibrosis.
- Thickened bony trabeculae show extensive evidence of bony remodeling and surround a marrow that is variably densely fibrotic and variably hypercellular, with prominent dilatation of the marrow sinuses. The overall cellularity approaches 100%. An occasional interstitial benign lymphoid aggregate is noted. Megakaryocytes showing the aforementioned abnormalities are present, forming distinct clusters and few small aggregates. Large erythroid islands are noted, with interspersed marrow parenchyma containing predominantly mature eosinophils. Admixed neutrophils are also seen. An M:E ratio is estimated at 10:1. Stainable iron is absent. There is a diffuse marked reticulin fibrosis associated with the aforementioned collagen fibrosis. Collagen accounts for approximately 20% of the total marrow space.
- Chronic eosinophilic leukemia, NOS:
- Sustained eosinophilia (> 1.5 x 109/L) with other reactive processes excluded
- < 20% blasts in peripheral blood or bone marrow, no inv(16)(p13.1q22), t(16;16)(p13.1q22), t(8;21)(q22;q22.1)
- Bone marrow aspirate is hypercellular with abundant eosinophils and eosinophilic precursors with dysplastic features
- Clonal cytogenetic (e.g., +8, -20q) or molecular genetic abnormalities (e.g., TET2, ASXL1 and DNMT3A); if no clonal abnormalities are found, then blast count must be > 2% in peripheral blood or > 5% in bone marrow aspirate
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR-ABL1, PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusions
- No lymphoid or myeloid neoplasms (e.g., myelodysplastic syndrome, myeloproliferative neoplasms, systemic mastocytosis or acute myeloid leukemia) are identified
- Hypereosinophilic syndrome:
- Sustained eosinophilia (> 1.5 x 109/L) for at least 6 months with other reactive processes excluded
- Tissue damage must be present
- Exclude cytokine related lymphoid variant hypereosinophilia
- Bone marrow shows increased eosinophils with no dysplastic features and no increase in blast cells
- No cytogenetic or molecular alteration is identified
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR-ABL1, PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusions; no inv(16)(p13.1q22), t(16;16)(p13.1q22) or t(8;21)(q22;q22.1)
- No lymphoid or myeloid neoplasms (e.g., myelodysplastic syndrome, myeloproliferative neoplasms, systemic mastocytosis or acute myeloid leukemia) are identified
- Reactive eosinophilia:
- Can present with the increased eosinophils and can have abnormal eosinophils in the peripheral blood
- Secondary causes (infection, allergies, drugs, skin diseases, asthma, adrenal insufficiency, gastrointestinal eosinophilia) are present
- No evidence of PDGFRA rearrangement or active mutation or other cytogenetic / molecular abnormalities
- Idiopathic hypereosinophilia:
- Sustained eosinophilia (> 1.5 x 109/L) for at least 6 months
- No tissue damage
- No reactive causes, lymphoid or myeloid neoplasms are identified
- Bone marrow shows increased eosinophils with no dysplastic features and no increase in blast cells
- No molecular alteration is identified
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR-ABL1, PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusions
- Systemic mastocytosis with eosinophilia:
- Major diagnostic criteria: 4 minor criteria or multifocal infiltrates of mast cells (≥ 15 mast cells in aggregates) + 1 minor criterion
- Minor criteria:
- Bone marrow shows eosinophilia with variable dysplastic features
- No PDGFRA rearrangement
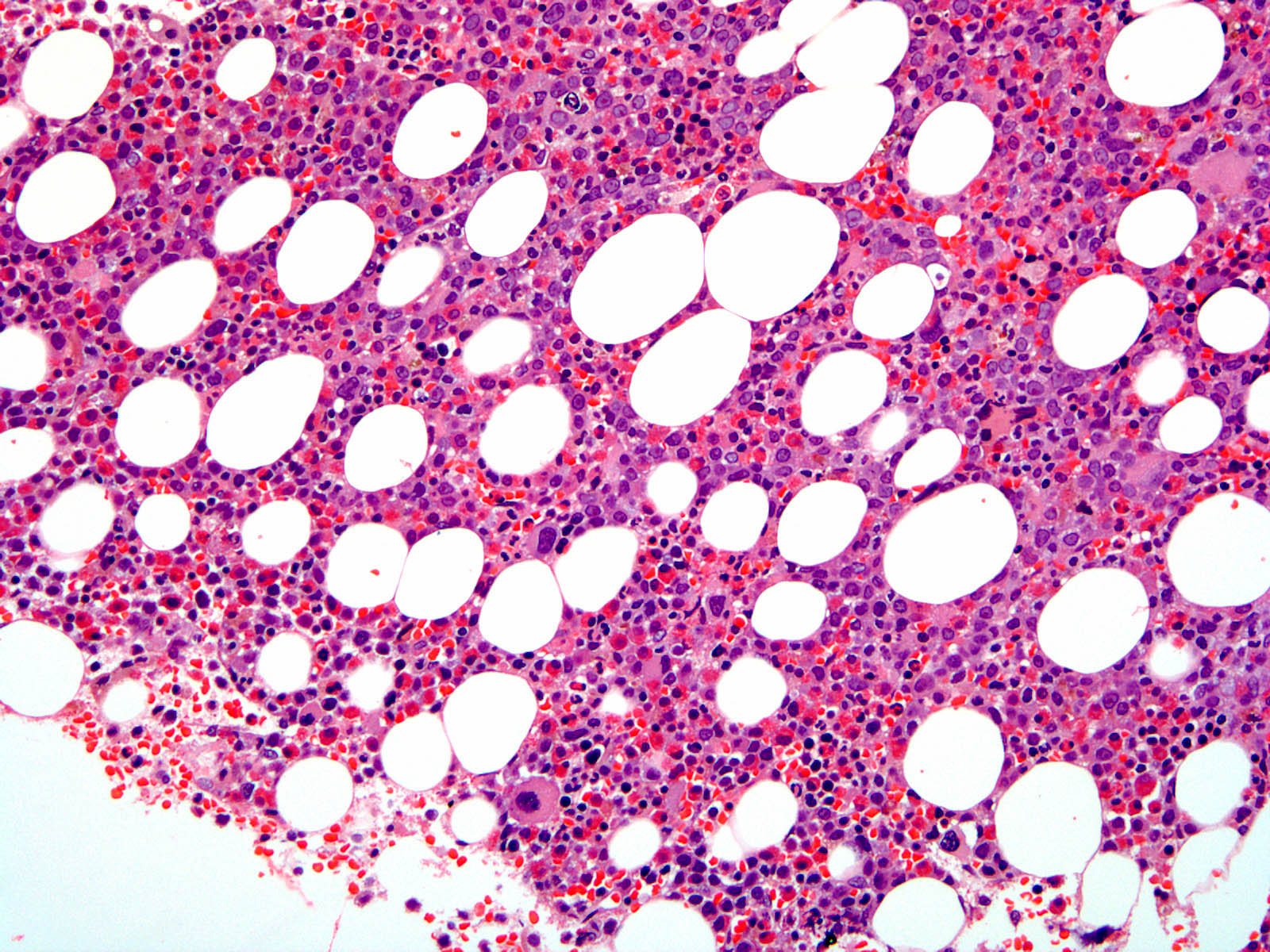
What is the most common translocation partner in myeloid and lymphoid neoplasms with PDGFRA rearrangement?
- BCR
- ETV6
- FIP1L1
- KIF5B
- STRN
- ABL
- BCR
- CHIC2
- FGFR
- TNK2
- Myeloid or lymphoid neoplasm with frequent eosinophilia, sometimes neutrophilia or monocytosis; associated with rearrangement of PDGFRB, at chromosome 5, long arm (5q32)
- Discovered in 1994 by Golub TR, Barker GF, Lovett M, Gilliland DG (Cell 1994;77:307)
- To date, more than 40 fusion partners identified
- Can have different presentation; e.g., myeloproliferative neoplasm (MPN), myelodysplastic syndrome / myeloproliferative neoplasm (MDS / MPN), acute myeloid leukemia (AML), with MDS / MPN overlap syndrome being more common
- Conventional karyotyping is the most common used modality for diagnosis, confirmation by FISH PDGFRB (break apart probe) or RT-PCR is recommended due to multiple other genes being located at 5q31-33 (e.g., IL3, IL5, GM-CSF)
- Chronic myelomonocytic leukemia with eosinophilia associated with t(5;12); myeloid neoplasms with PDGFRB rearrangement
- Myeloid neoplasms associated with PDGFRB rearrangement
- ICD-11: 2A51 - myeloid neoplasm associated with PDGFRB rearrangement
- M:F = 2:1
- Age range is 8 - 72 years with peak incidence at late 40s (Acta Haematol 2002;107:113)
- Peripheral blood and bone marrow is always involved
- Spleen; other organs can be involved as well
- Most common translocation t(5;12)(q32;p13.2) ETV6::PDGFRB fusion gene
- More than 40 different partners are identified (Histopathology 2020;76:1042)
- t(1;3;5)(p36;p21;q33) WDR48::PDGFRB
- t(1;5)(q21;q33) TPM3::PDGFRB
- t(1;5)(q23;q33) PDE4DIP::PDGFRB
- t(5;7)(q33;q11.2) HIP1::PDGFRB
- t(5;10)(q33;q21) CCDC6 (H4/D10S170)::PDGFRB
- Some fusions are typically associated with BCR::ABL1-like B cell acute lymphoblastic leukemia (B ALL) (e.g., PDGFRB fused with EBF1, SSBP2, TNIP1, ZEB2 and ATF7IP)
- Those fusions are not diagnosed as PDGFRB rearranged entities but rather as BCR::ABL1-like B ALL (N Engl J Med 2014;371:1005)
- Unknown
- Can present with MPN, MDS / MPN (e.g., atypical chronic myeloid leukemia [CML], chronic myelomonocytic leukemia [CMML] or juvenile myelomonocytic leukemia [JMML])
- Rarely myeloid blast phase / secondary AML or de novo B ALL (Histopathology 2020;76:1042, Blood 2017;129:704)
- Can present with B symptoms
- Hepatosplenomegaly
- Cardiac and skin damage
- Good response to imatinib or other tyrosine kinase inhibitor (TKI) therapy
- Peripheral blood and bone marrow examination both usually show eosinophilia (sometimes normal) and other features representative of MPN, MDS / MPN or transformed AML
- Conventional karyotyping
- FISH PDGFRB for confirmation, especially cryptic cases
- RT-PCR has limited utility due to multiple fusion partners (Foucar: Bone Marrow Pathology, 4th Edition, 2019)
- Next generation sequencing (NGS) can detect cases missed by FISH (Cancer Genet 2020;244:55)
- According to WHO, diagnostic criteria for myeloid / lymphoid neoplasms associated with ETV6::PDGFRB or other rearrangement of PDGFRB:
- Myeloid or lymphoid neoplasm often with prominent eosinophilia and sometimes with neutrophilia or monocytosis and presence of t(5;12)(q32;p13.2) or a variant translocationa,b or demonstration of ETV6::PDGFRB fusion gene or other rearrangement of PDGFRBb
- Cases with fusion genes typically associated only with BCR::ABL1-like B lymphoblastic leukemia are specifically excluded
- Because t(5;12)(q32;p13.2) does not always result in ETV6::PDGFRB fusion, molecular confirmation is highly desirable; if molecular analysis is not possible, this diagnosis should be suspected if there is a myeloproliferative neoplasm associated with eosinophilia, with no Philadelphia (Ph) chromosome and with a translocation with a 5q32 breakpoint
- Myeloid or lymphoid neoplasm often with prominent eosinophilia and sometimes with neutrophilia or monocytosis and presence of t(5;12)(q32;p13.2) or a variant translocationa,b or demonstration of ETV6::PDGFRB fusion gene or other rearrangement of PDGFRBb
- Complete blood count (CBC) usually shows eosinophilia; however, normal counts can be found
- Increased tryptase levels (> 12 ng/dL); however, < 20 ng/dL (Expert Rev Hematol 2014;7:683)
- Vitamin B12 levels may be increased
- Median overall survival is < 2 years before introduction of tyrosine kinase inhibitor; a case series of 26 cases treated with imatinib showed a longer survival (up to 6.6 years)
- 4 month old girl with refractory juvenile xanthogranuloma with a novel MRC1::PDGFRB fusion and a dramatic response induced by dasatinib (Blood Adv 2020;4:2991)
- 22 year old woman with relapsed BCR::ABL1-like ALL with CCDC88C::PDGFRB fusion and complete remission after treatment with tyrosine kinase inhibitor (Int J Hematol 2021;113:285)
- 22 year old man with a cryptic imatinib sensitive G3BP1::PDGFRB rearrangement in a myeloid neoplasm with eosinophilia (Blood Adv 2020;4:445)
- 70 year old man with asymptomatic leukocytosis, thrombocytosis with PDGFRB rearrangement and absence of eosinophilia (Leukemia 2016;30:1402)
- 76 year old man with myeloid / lymphoid neoplasms associated with eosinophilia, with negative FISH and positive next generation sequencing for PDGFRB rearrangement (Cancer Genet 2020;244:55)
- Sensitive to tyrosine kinase inhibitor (TKI) (e.g., imatinib)
- Study showed a response rate to imatinib treatment of 96% with a 10 year overall survival rate of 90% (Blood 2014;123:3574, Blood 2017;129:704)
- Some mutations confer resistance to all TKIs (e.g., PDGFRB C843G and EBF1::PDGFRB Ph-like ALL with gatekeeper mutation T681I) (Blood 2018;131:2256, Haematologica 2021;106:2242)
- Peripheral blood shows leukocytosis including eosinophilia, neutrophilia or monocytosis; sometimes circulating immature myeloid precursors (e.g., metamyelocytes, myelocytes or promyelocytes and occasional blasts / blast equivalents)
- Increased blasts up to 20% seen in blastic phage
- Anemia and thrombocytopenia can be seen
- Bone marrow is hypercellular with myeloid preponderance including many eosinophils and neutrophils
- Rarely there might be increase in basophils
- Can show increase in mast cells with identified abnormal spindle shaped forms and expression of CD2 and CD25 (Am J Hematol 2007;82:77, Haematologica 2007;92:163)
- Reticulin fibrosis (Blood 2008;111:1855)
Contributed by Ismail Elbaz Younes, M.D. and Ling Zhang, M.D.
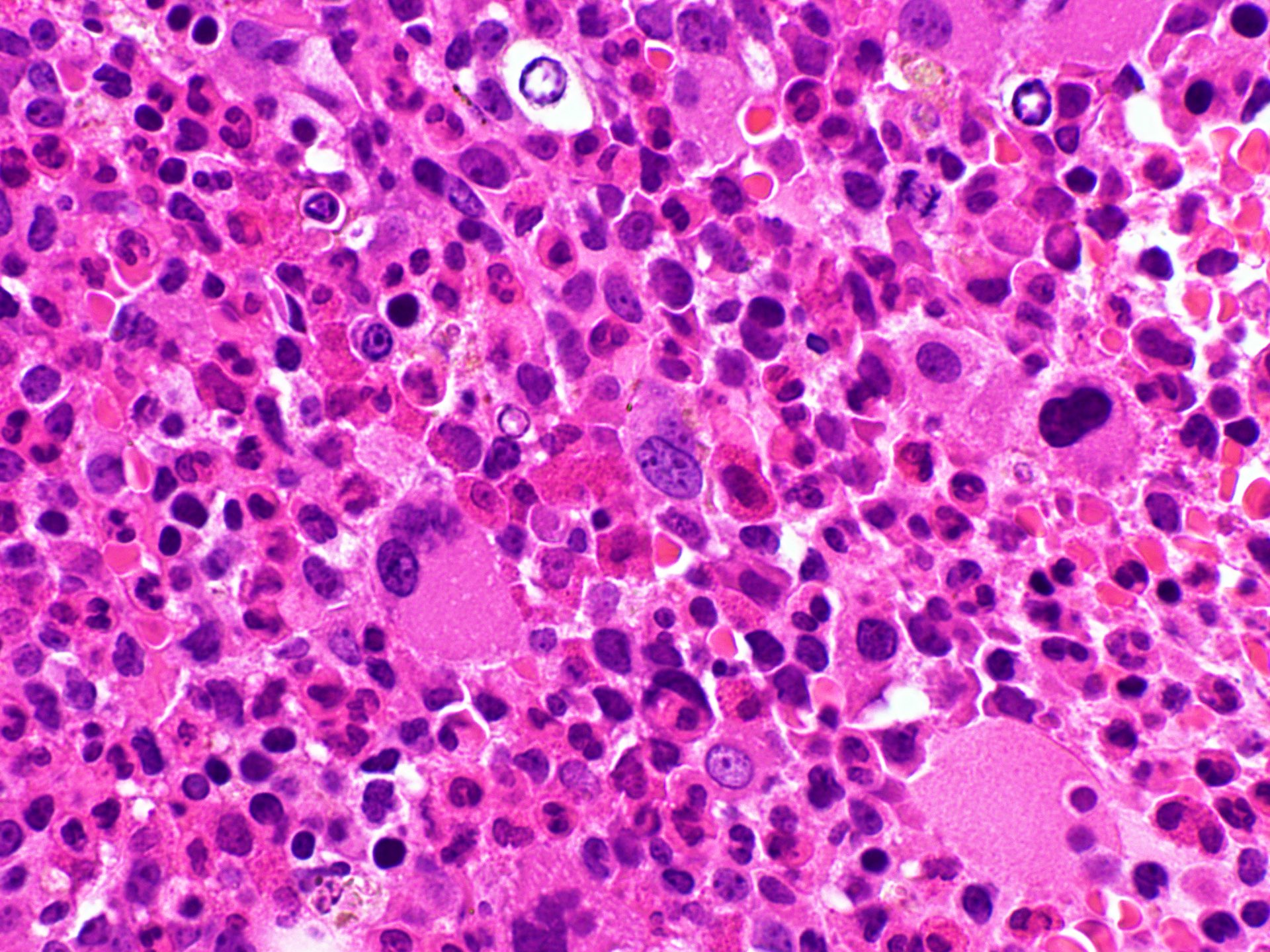
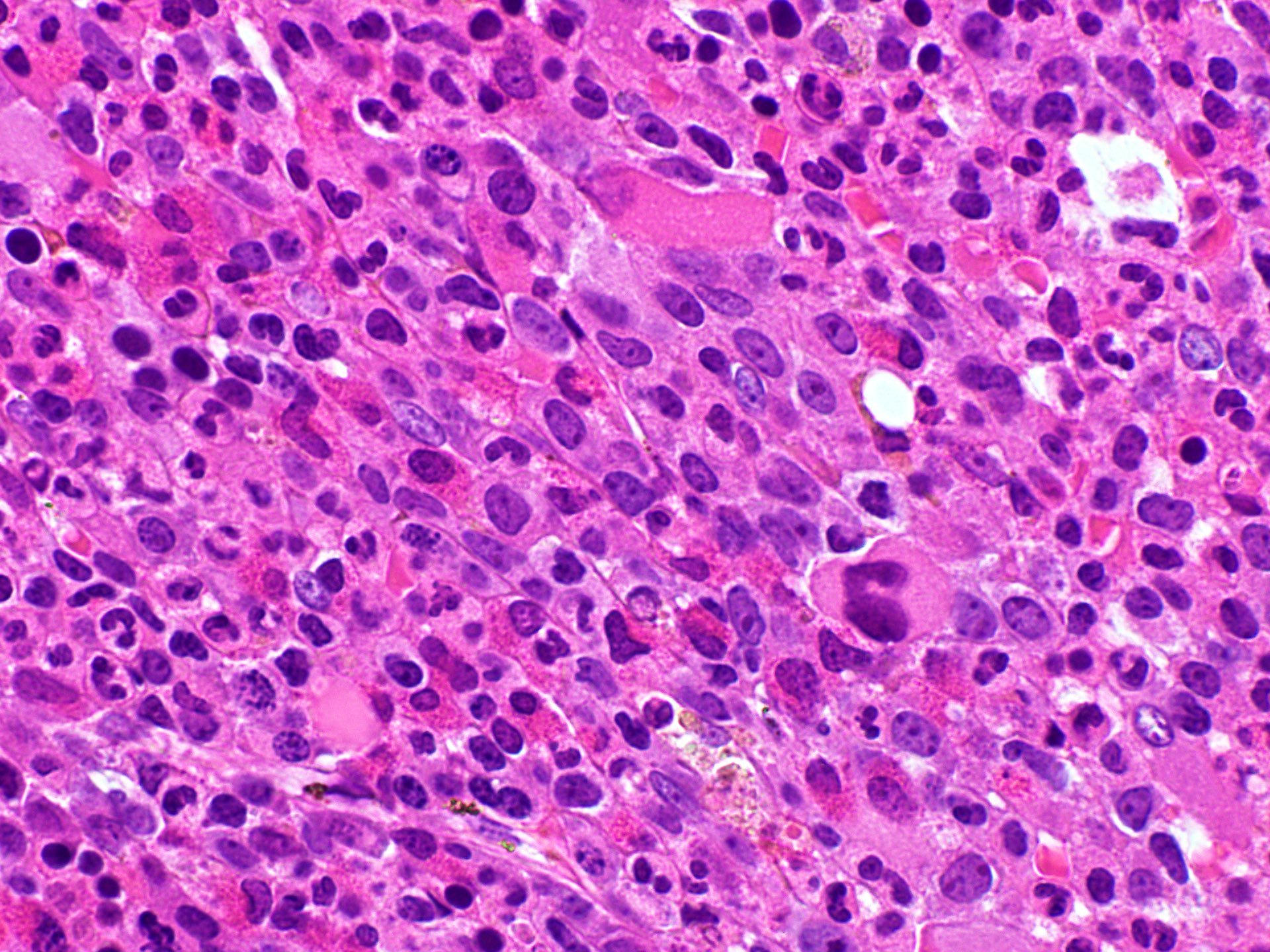
Increased blasts in bone marrow core biopsy
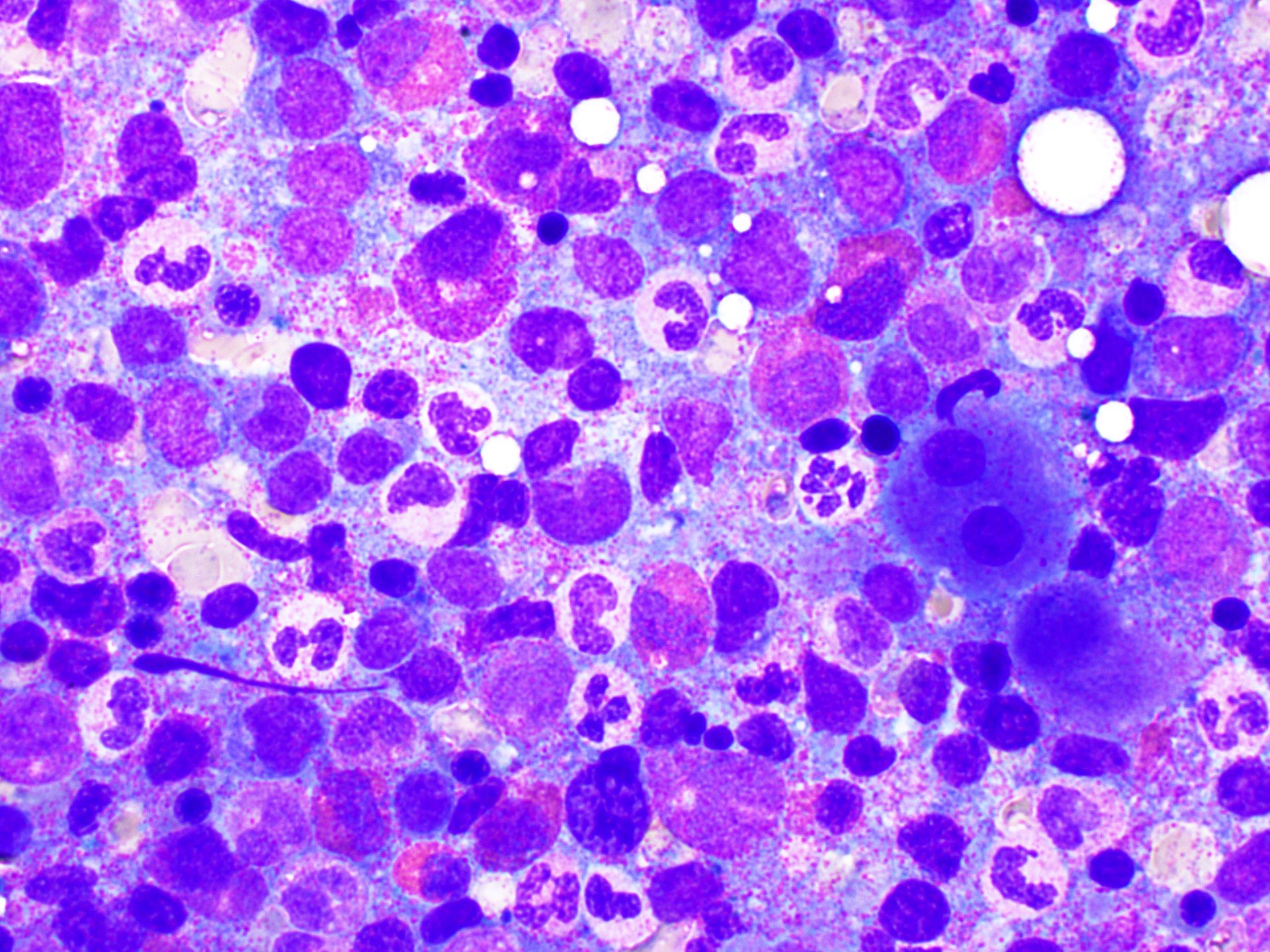
Dysplastic megakaryocytes and neutrophils
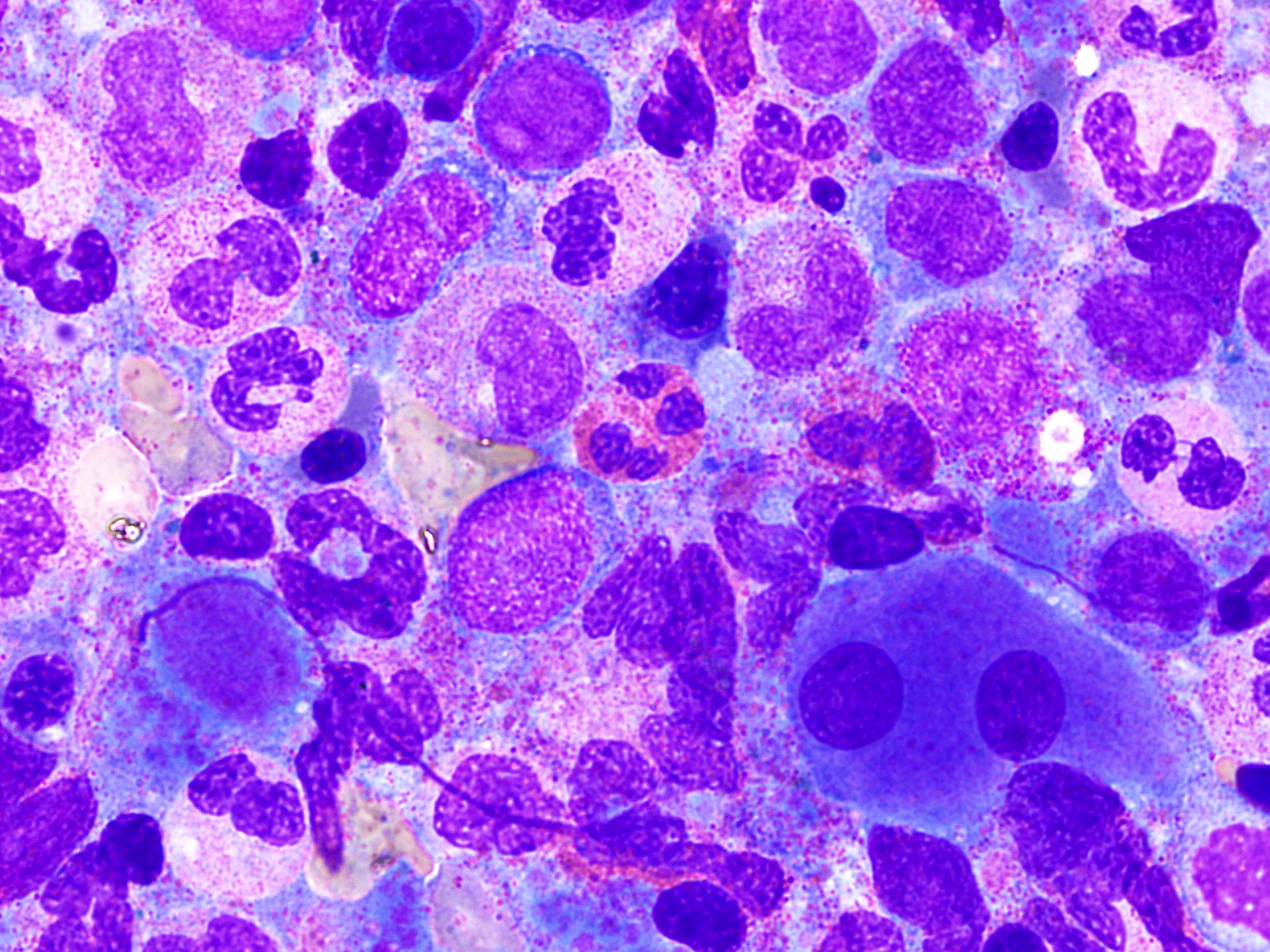
Increased blasts
- Peripheral blood eosinophilia and occasionally neutrophilia, eosinophils are usually mature
- Variable morphologic abnormalities can be present in the eosinophils, including cytoplasmic vacuolation, nuclear hyper / hyposegmentation
- Morphologic changes are not unique as reactive conditions can have these changes as well (Foucar: Bone Marrow Pathology, 4th Edition, 2019)
Contributed by Ismail Elbaz Younes, M.D. and Ling Zhang, M.D.
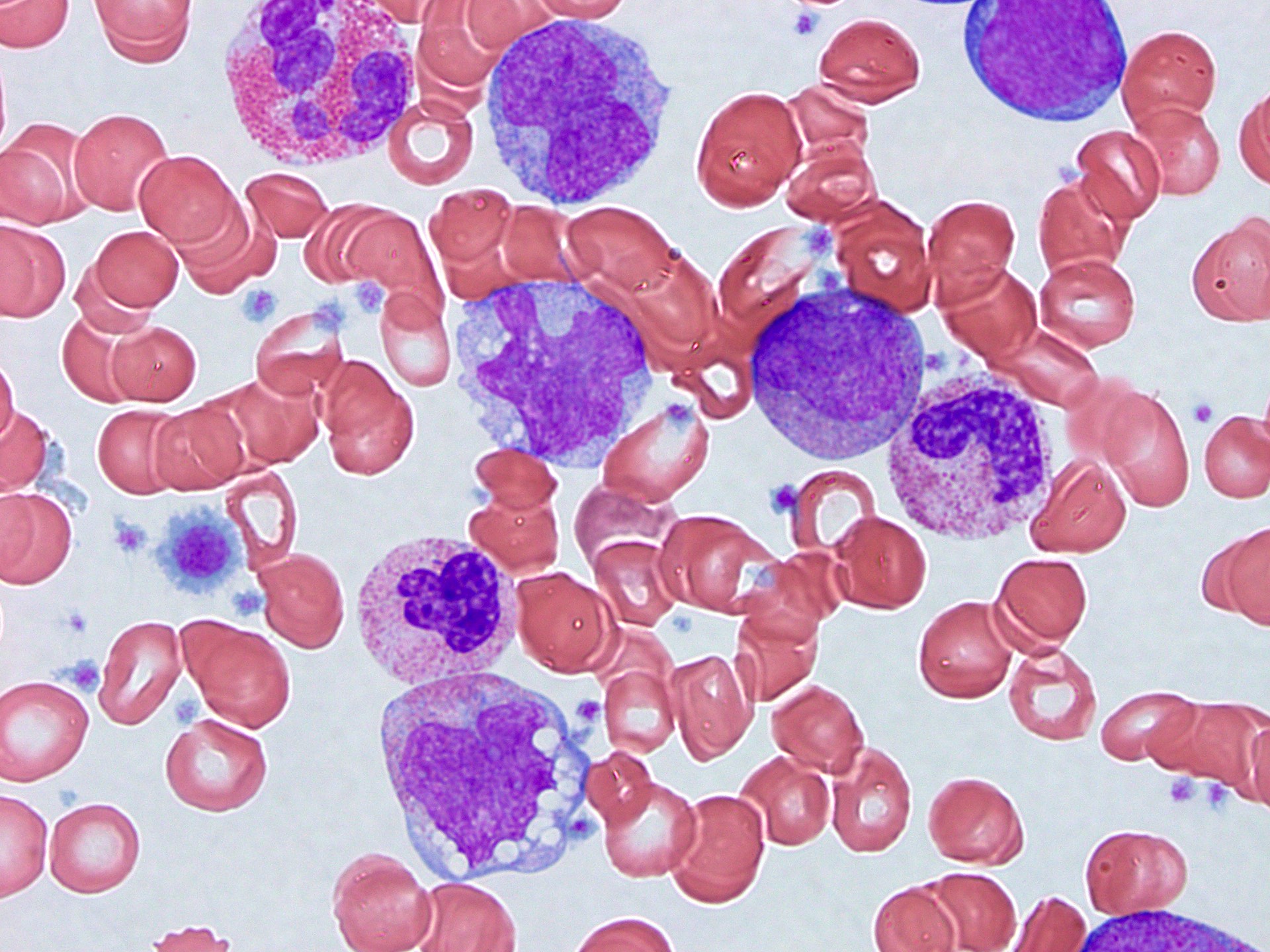
Increased myeloid blasts
- FISH rearrangement with 5q31~33 breakpoint, typically t(5;12)(q31~33;p12) creating ETV6::PDGFRB
Contributed by Ismail Elbaz Younes, M.D. and Ling Zhang, M.D.

Rearrangement with 5q31~33 breakpoint
Images hosted on other servers:


FISH assays indicate rearrangement of PDGFRB
- Peripheral blood:
- Mild macrocytic anemia
- Neutrophilia with left shifted granulocyte maturation
- Monocytosis
- Mild thrombocytopenia
- Bone marrow, left posterior iliac crest, aspirate and biopsy:
- Hypercellular marrow with myeloid hyperplasia and monocytosis, consistent with a myeloid neoplasm with PDGFRB rearrangement (see comment)
- Mildly increased reticulin fibers
- Comment: Cytogenetics reported 46,XY,t(5;14)(q33;q32)[18]/46,XY[2]. FISH was reported positive for PDGFRB gene rearrangement. An MDS FISH panel and FISH for BCR::ABL were reported negative. Flow cytometry reported atypical myeloid maturation with decreased CD10, atypical maturation of CD64 / CD14 and increased HLA-DR expression. Given the cytogenetic findings, this myeloid neoplasm with PDGFRB rearrangement is associated with a hematologic diagnosis of chronic myelomonocytic leukemia with eosinophilia. The morphology is compatible with chronic myelomonocytic leukemia and there are focal areas of slightly increased eosinophils seen on the core biopsy.
- Microscopic description:
- Peripheral smear: There is a mild macrocytic anemia with mild anisopoikilocytosis. The total white blood cell count is elevated, with a neutrophilia and monocytosis. Left shifted granulocytes are seen, including an occasional circulating blast. The monocytes are maturing. The granulocytes have variable cytoplasmic granulation. Platelets are mildly decreased.
- Aspirate smear: The Wright stained aspirate smear is cellular, with spicules available for evaluation. Megakaryocytes are seen in adequate number, with an occasional hypolobated form. Myeloid and erythroid precursors are seen. The myeloid precursors show slight left shifted maturation, with approximately 4% blasts. The myeloid precursors have variable toxic granulation and include a few precursors with mild nuclear:cytoplasmic asynchrony. Monocytes comprise approximately 25% of the differential and are unevenly distributed. The monocytes include approximately 5% promonocytes and many immature monocytes. Eosinophils include forms with some large basophilic granules. Erythroid precursors show maturation. Lymphocytes and scattered plasma cells are seen in the background.
- Aspirate iron: An aspirate smear is stained for iron by the contributor. Stainable iron stores are present. A rare ring sideroblast is seen.
- Clot section and core biopsy: Subcortical bone and normal bony trabeculae surround the marrow space with a cellularity approaching 100%. Megakaryocytes are seen in slightly decreased numbers, with occasional hypolobated forms. Small erythroid islands with admixed myeloid precursors are seen, with an M:E ratio estimated at greater than 5:1. The myeloid precursors have focal areas of left shifted maturation. No clusters of blasts are seen on the H&E stained section. Focal areas of slightly increased eosinophils are noted. Stainable iron stores are adequate. Reticulin fibers are mildly increased.
- Chronic eosinophilic leukemia, NOS:
- Sustained eosinophilia (> 1.5 x 109/L) with other reactive processes excluded
- < 20% blasts in peripheral blood or bone marrow, no inv(16)(p13.1q22), t(16;16)(p13.1q22), t(8;21)(q22;q22.1)
- Bone marrow aspirate is hypercellular with abundant eosinophils and eosinophilic precursors with dysplastic features
- Clonal cytogenetic (e.g., +8, -20q) or molecular genetic abnormalities (e.g., TET2, ASXL1 and DNMT3A)
- If no clonal abnormalities are found, then blast count must be > 2% in peripheral blood or > 5% in bone marrow aspirate
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR::ABL1, PCM1::JAK2, ETV6::JAK2 or BCR::JAK2 fusions
- No lymphoid or myeloid neoplasms (e.g., MDS, MPNs, systemic mastocytosis [SM] or AML) are identified
- Hypereosinophilic syndrome:
- Sustained eosinophilia (> 1.5 x 109/L) for at least 6 months with other reactive processes excluded
- Tissue damage must be present
- Exclude cytokine related lymphoid variant hypereosinophilia
- Bone marrow shows increased eosinophils with no dysplastic features and no increase in blast cells
- No cytogenetic or molecular alteration is identified
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR::ABL1, PCM1::JAK2, ETV6::JAK2 or BCR::JAK2 fusions
- No inv(16)(p13.1q22), t(16;16)(p13.1q22) or t(8;21)(q22;q22.1)
- No lymphoid or myeloid neoplasms (e.g., MDS, MPNs, SM or AML) are identified
- Reactive eosinophilia:
- Can present with increased eosinophils and can have abnormal eosinophils in the peripheral blood
- Secondary causes are present: infection (e.g., parasites; the most common cause), connective tissue disorders, certain cutaneous lesions (e.g., dermatitis herpetiform), paraneoplastic (e.g., Hodgkin lymphoma, T cell lymphoma, colon carcinoma), certain lung diseases (e.g., Churg-Strauss syndrome, aspergillosis), autoimmune disorder or on IL2 therapy (Hematology Am Soc Hematol Educ Program 2018;2018:326)
- No evidence of PDGFRB rearrangement
- Idiopathic hypereosinophilia:
- Sustained eosinophilia (> 1.5 x 109/L) for at least 6 months
- No tissue damage
- No reactive causes, lymphoid or myeloid neoplasms are identified
- Bone marrow shows increased eosinophils with no dysplastic features and no increase in blast cells
- No molecular alteration is identified
- No PDGFRA, PDGFRB, FGFR rearrangement, BCR::ABL1, PCM1::JAK2, ETV6::JAK2 or BCR::JAK2 fusions
- Systemic mastocytosis with eosinophilia:
- Major diagnostic criteria: 4 minor criteria or multifocal infiltrates of mast cells (≥ 15 mast cells in aggregates) + 1 minor criterion
- Minor criteria
- Bone marrow shows eosinophilia with variable dysplastic features
- No PDGFRB rearrangement
What is the most common translocation in myeloid / lymphoid neoplasms with eosinophilia and PDGFRB rearrangement?
- t(1;3;5)(p36;p21;q33) WDR48::PDGFRB
- t(1;5)(q21;q33) TPM3::PDGFRB
- t(1;5)(q23;q33) PDE4DIP::PDGFRB
- t(5;7)(q33;q11.2) HIP1::PDGFRB
- t(5;12)(q32;p13.2) ETV6::PDGFRB
- FISH
- Karyotyping
- RT-PCR
- Snap array
- Paraneoplastic syndrome due to an underlying plasma cell neoplasm
- POEMS = polyneuropathy, organomegaly, endocrinopathy, M protein spike and skin changes
- But the acronym does not include several of the common clinical manifestations
- Major criteria for diagnosis are:
- Polyradiculopathy
- Clonal plasma cell disorder
- Sclerotic bone lesions
- Elevated vascular endothelial growth factor
- Castleman disease (also known as angiofollicular hyperplasia, 11 - 30% of cases, most commonly multicentric Castleman disease)
- POEMS acronym coined in 1980 by Bardwick et al. (Medicine (Baltimore) 1980;59:311)
- Other names: osteosclerotic myeloma, Takatsuki syndrome, Crow-Fukase syndrome
- ICD-10: D47.7 - other neoplasms of uncertain behavior of lymphoid, hematopoietic and related tissue
- See also WHO coding
- Prevalence of approximately 0.3 per 100,000 reported in Japan but occurs worldwide
- VEGF levels are high and correlate with disease activity but may not drive the process
- IL12 also correlates with disease activity; IL6 produced by plasma cells may play a role
- In addition to essential features above, other clinical features include:
- Organomegaly
- Endocrinopathy
- Characteristic skin changes
- Papilledema
- Extravascular volume overload / anasarca
- Thrombocytosis, with propensity for thrombosis
- Abnormal pulmonary function tests, pulmonary fibrosis, pulmonary hypertension
- Diarrhea, calciphylaxis (rare)
- Generally no bone pain
- Diagnosis is made by fulfilling at least 3 major criteria, 2 of which must include polyradiculoneuropathy and a clonal plasma cell disorder, along with at least 1 minor criterion (Am J Hematol 2017;92:814)
- Mandatory criteria (must have both): polyneuropathy and monoclonal plasma cell disorder
- Other major criteria (at least 1 present): Castleman disease, sclerotic bone lesions, increased VEGF levels
- Minor criteria (at least 1 present):
- Organomegaly
- Extravascular volume overload (edema, pleural effusion, ascites),
- Endocrinopathy (adrenal, thyroid, pituitary, gonadal, parathyroid, pancreatic); hypogonadism most common
- Skin changes (hyperpigmentation, hypertrichosis, glomeruloid angiomata, plethora, acrocyanosis, white nails, sclerodermoid changes, clubbing)
- Papilledema
- Thrombocytosis / polycythemia
- Monoclonal light chain (especially lambda)
- VEGF levels are high
- Thrombocytosis or erythrocytosis, absence of cytopenias
- Bone lesions
- Signs of volume overload (e.g. pleural effusion, ascites)
- Risk associated with the extent of the plasma cell disorder
- Bone marrow plasmacytosis increases risk of cerebrovascular event
- Number of clinical criteria met is not prognostic
- Generally better overall survival than myeloma, older data shows median survival of 14 years
- 47 year old man with POEMS syndrome with a plasmacytoma causing craniocervical instability (J Clin Neurosci 2017 Oct 31 [Epub ahead of print])
- 48 year old woman with 2 week history of peripheral neuropathy (Blood 2013;122:159)
- 2 patients with plasma cell dyscrasias, original description by Bardwick et al. (Medicine (Baltimore) 1980;59:311)
- If no bone marrow involvement by plasma cell clone, local radiation therapy may be adequate (e.g. sclerotic plasmacytoma)
- If disseminated disease or progression following local radiation, systemic therapy advised
- Corticosterioids as a temporizing measure
- Alkylating agents a mainstay, including high dose with stem cell transplant
- Thalidomide, bortezomib can be considered but may worsen peripheral neuropathy
- Prompt treatment recommended
Images hosted on other servers:

Classic findings of POEMS syndrome
- Bone marrow biopsy may show megakaryocyte hyperplasia and clustering (no JAK2 mutation)
- Lymphoid aggregates (50% of cases) with clonal plasma cell rimming
Contributed by Genevieve M. Crane, M.D., Ph.D. and Sarah Elsoukkary, M.D.
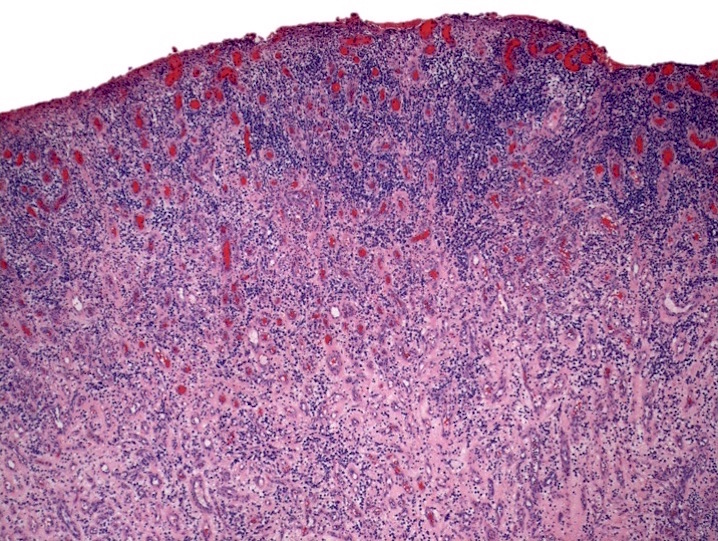
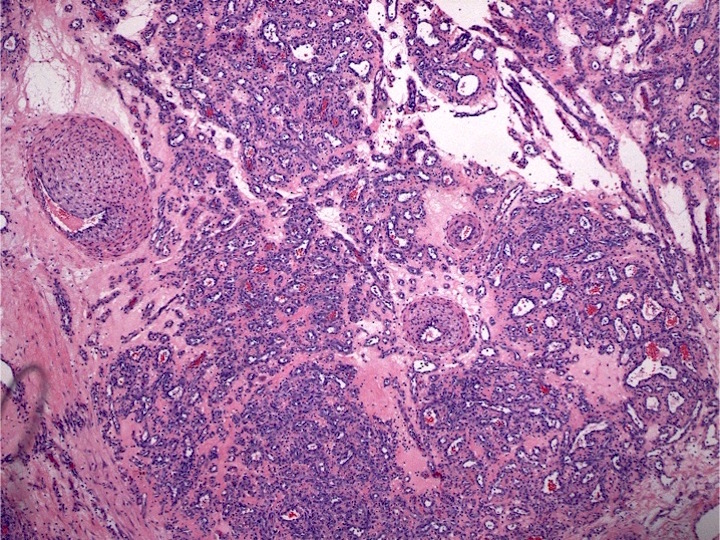
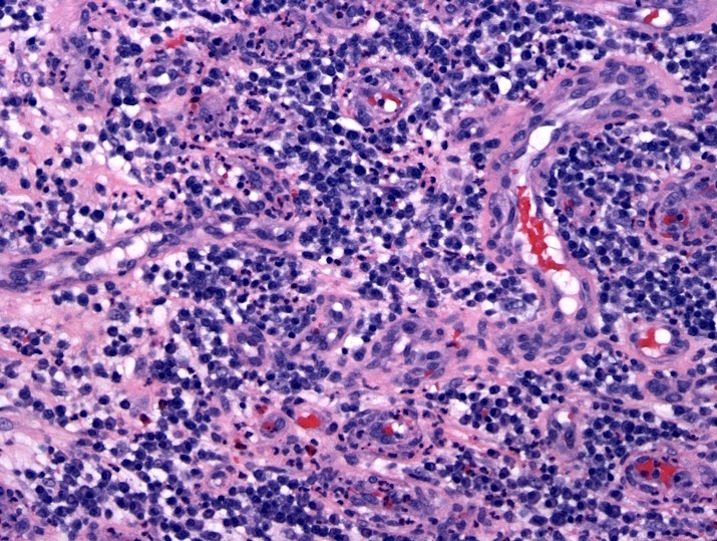
A pyogenic granuloma arising in a patient with POEMS syndrome
Biopsy of sclerotic bone lesions in a patient with POEMS syndrome
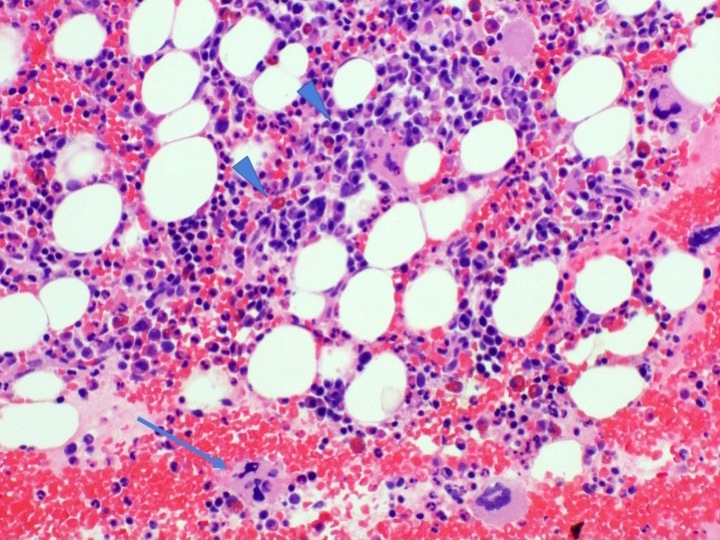
Bone marrow clot section
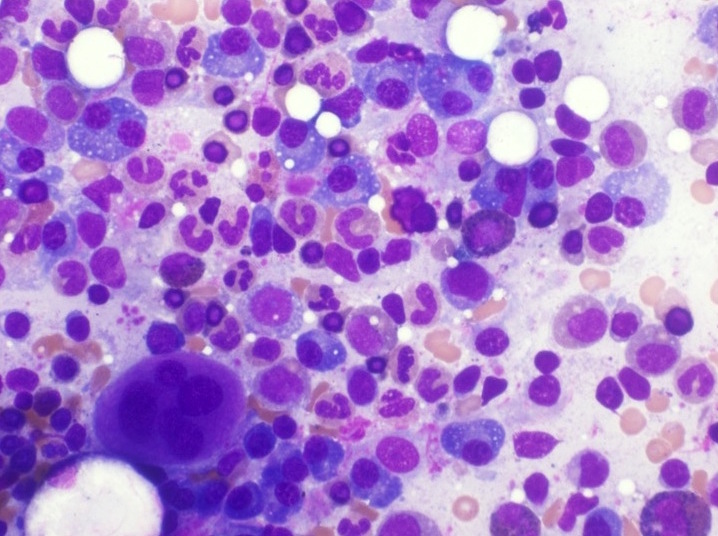
Bone marrow aspirate smear
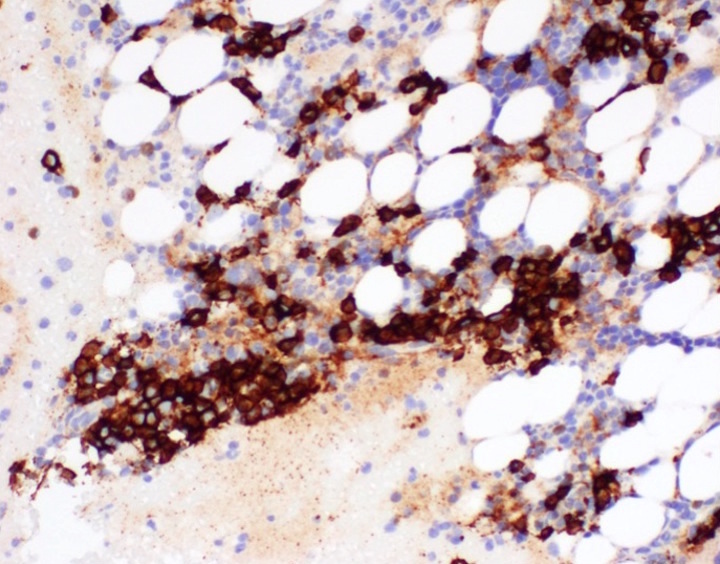
CD138

CD138 / Ki67
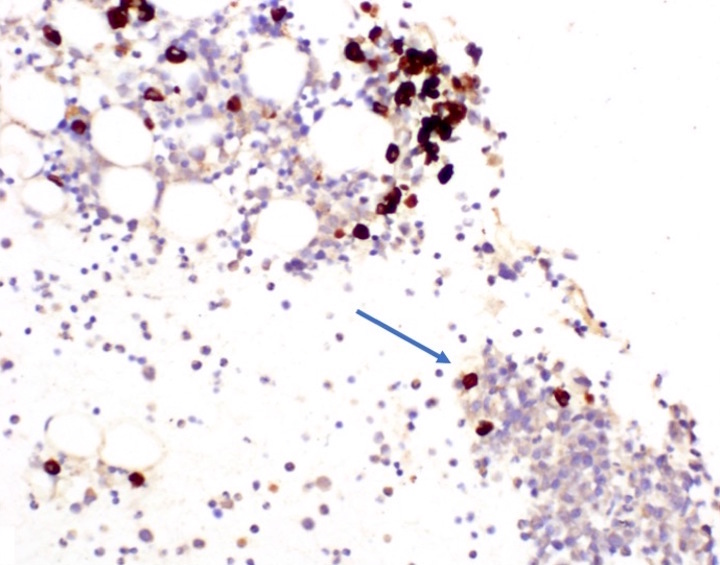
CD56+ clone

Lambda
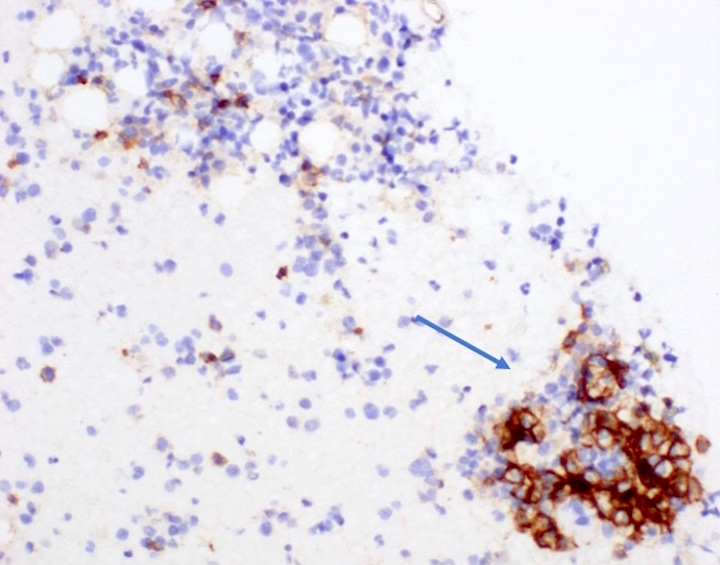
IHC for kappa and lambda of the bone marrow clot
- Lambda clones predominate (95% of cases)
- Other features are as for clonal plasma cell populations
- No features of macrophage associated demyelination in contrast to chronic inflammatory demyelinating polyradiculopathy
- Tend to have restricted immunoglobulin variable chain usage (IGLV1)
- Translocations and deletion of chromosome 13 have been described
- POEMS disease is rare and can be mistaken for other processes:
- Castleman disease variant of POEMS syndrome: no associated clonal plasma cell process and little to no peripheral neuropathy
- Chronic inflammatory demyelinating polyradiculopathy: a predominantly neurologic disorder (Acta Neuropathol Commun 2016;4:116)
- Monoclonal gammopathy of undetermined significance (MGUS), solitary plasmacytoma
- Osteosclerotic lesion differential by radiology: aneurysmal bone cysts, nonossifying fibromas, fibrous dysplasia
- Plasma cell myeloma: POEMS symptoms of neuropathy, endocrine dysfunction, volume overload not associated with myeloma; myeloma symptoms of bone pain, renal failure less common in POEMS
- Bone pain
- Demonstration of clonal plasma cells
- Neuropathy
- Pleural effusion
- Renal failure
Comment Here
Reference: POEMS syndrome (osteosclerotic myeloma)
- Rare subtype of multiple myeloma characterized by the presence of clonal plasma cells in the peripheral blood and an aggressive clinical course
- Current definition: ≥ 5% circulating plasma cells, detected by manual differential of peripheral blood (recently updated from prior cutoff of ≥ 20% or ≥ 2.0 x 109/L) (Blood Cancer J 2021;11:192)
- Detection of ≥ 5% circulating plasma cells by manual differential of peripheral blood, in a patient with newly diagnosed or relapsed / refractory multiple myeloma
- Aggressive clinical course marked by high tumor burden and shortened overall survival
- Primary plasma cell leukemia (pPCL): ≥ 5% circulating plasma cells by manual differential of peripheral blood, in a patient with newly diagnosed multiple myeloma
- Secondary plasma cell leukemia (sPCL): ≥ 5% circulating plasma cells by manual differential of peripheral blood, in a patient with relapsed / refractory multiple myeloma
- Incidence increases with age
- Incidence of pPCL (using updated criteria): 4 - 7% of patients with multiple myeloma (Leuk Lymphoma 2022;63:2955, J Clin Oncol 2023;41:1342)
- pPCL is more common than sPCL (60 - 70% versus 30 - 40%) (Leukemia 2013;27:780)
- Incidence of sPCL: 1 - 2% of patients with advanced or refractory multiple myeloma (Blood Cancer J 2021;11:192)
- Median time from diagnosis of multiple myeloma to development of sPCL is ~20 - 22 months (Leuk Lymphoma 2017;58:1538)
- Patients with pPCL are younger (occasionally diagnosed in second to fourth decade of life)
- Median age of presentation is ~10 years younger than for multiple myeloma (fifth to sixth decade versus sixth to seventh decade) (Leuk Lymphoma 2017;58:1538, Leukemia 2013;27:780)
- Incidence increases with age
- Peripheral blood: by definition, plasma cells are ≥ 5% of circulating white blood cells
- Bone marrow: variable, often extensive involvement by sheets of plasma cells with varying degrees of cytologic atypia
- Extramedullary sites: liver, spleen, lymph nodes and CNS are common sites of extramedullary involvement (Curr Oncol Rep 2019;21:8, Leuk Lymphoma 2022;63:2955)
- Pathophysiology is incompletely understood
- Alterations in the bone marrow microenvironment and downregulated expression of cell adhesion molecules may decrease neoplastic cell dependence on the bone marrow and promote extramedullary involvement (Leuk Lymphoma 2017;58:1538, Leukemia 2013;27:780)
- Loss of CD56 / NCAM expression disrupts adhesion of plasma cells to bone marrow stroma (Int J Hematol Oncol 2022;11:IJH39)
- Decreased expression of chemokine receptors such as CXCR4 may disrupt plasma cell localization to bone marrow stroma
- Expression of CD27 is associated with antiapoptotic processes via the nuclear factor Κβ pathway (which can be targeted with antiproteosome agents such as bortezomib) (Blood 2014;123:3770)
- Exact etiology of primary and secondary PCL remains under investigation
- Altered expression of adhesion molecules and extracellular matrix play an incompletely understood role in pathogenesis (Blood Cancer J 2020;10:70, Leukemia 2020;34:1866)
- pPCL is characterized by an aggressive clinical presentation and high tumor burden
- High tumor burden is reflected by increased serum lactate dehydrogenase (LDH) and beta 2 microglobulin (Blood Cancer J 2021;11:23)
- Compared to multiple myeloma, patients with pPCL are more likely to show the following at presentation (Blood Cancer J 2021;11:23)
- High levels of bone marrow involvement
- Elevated levels of serum LDH and beta 2 microglobulin
- Hypercalcemia
- Cytopenias (anemia and thrombocytopenia)
- Renal dysfunction
- Extramedullary disease
- Rare report of pPCL presenting with tumor lysis syndrome (Clin Case Rep 2022;10:e05933)
- Of patients with detectable monoclonal gammopathy, IgG is most common, followed by IgA, IgD and IgE
- ~35 - 40% of patients show increased light chains only or are nonsecretory (2 - 8%) (Curr Oncol Rep 2019;21:8)
- Poor overall survival (OS) (5 - 30 months) (Curr Oncol Rep 2019;21:8, Leuk Lymphoma 2022;63:2955)
- References: Leuk Lymphoma 2017;58:1538, Leuk Lymphoma 2022;63:2955, J Clin Oncol 2023;41:1342, Leukemia 2013;27:780
- pPCL is defined by detection of ≥ 5% circulating plasma cells, identified by manual differential of the peripheral blood, in a patient with a new diagnosis of multiple myeloma (Blood Cancer J 2021;11:192)
- sPCL is defined by detection of ≥ 5% circulating plasma cells, identified by manual differential of the peripheral blood, in a patient with relapsed or refractory multiple myeloma
- Detection of ≥ 2% circulating plasma cells by flow cytometry in patients with multiple myeloma portends a pPCL-like clinicopathologic course but is not yet recognized as pPCL (J Clin Oncol 2023;41:1383)
- When present, the percentage of circulating plasma cells and the method used for detection (e.g., manual differential, flow) should be included in the pathology report
- Monoclonal gammopathy by serum or urine protein electrophoresis
- IgG is most common, followed by IgA, IgD, light chain only and nonsecretory
- Serum and urine immunofixation to characterize the monoclonal gammopathy
- Elevated levels of serum or urine free light chains
- Abnormal kappa:lambda free light chain ratio
- Anemia and thrombocytopenia
- Renal dysfunction and elevated creatinine
- Elevated serum LDH and beta 2 microglobulin
- Bone marrow with variable, often extensive involvement by plasma cells
- ≥ 5% circulating plasma cells by morphologic examination of concurrent peripheral blood
- Flow cytometry of peripheral blood and bone marrow shows aberrant plasma cells with monotypic, cytoplasmic light chain expression
- Background B lymphocytes are polytypic and lack abnormalities
- Hypodiploidy and high risk cytogenetic abnormalities, by fluorescence in situ hybridization
- Mutation of TP53 is frequently observed in both pPCL and sPCL
- Frequent detection of t(11;14) and cyclin D1 expression observed in both pPCL and sPCL
- References: Leuk Lymphoma 2017;58:1538, Leuk Lymphoma 2022;63:2955, Leukemia 2013;27:780, J Clin Oncol 2023;41:1342
- pPCL has an unfavorable prognosis with decreased overall survival as compared to multiple myeloma
- Overall survival (using > 20% circulating plasma cells as cutoff): ~6.8 - 12.6 months (Leukemia 2013;27:780)
- Overall and progression free survival is approximately doubled by use of lower cutoffs, novel therapeutic agents and autologous stem cell transplant
- Overall survival rates of 16 - 30 months have been reported (Leuk Lymphoma 2022;63:2955, Leuk Lymphoma 2017;58:1538)
- Presence of any amount of circulating plasma cells in patients with multiple myeloma negatively affects prognosis and survival rates progressively decline with increasing amounts of circulating cells (J Clin Oncol 2023;41:1342)
- A recent study using flow cytometry based identification and quantitation of circulating plasma cells reported that achievement of minimal residual disease (MRD) negativity improved progression free survival in patients with multiple myeloma and circulating tumor cells (J Clin Oncol 2022;40:3120)
- Majority of patients with pPCL have high (II or III) Revised International Staging System (R-ISS) scores due to elevated beta 2 microglobulin and LDH, indicating a high tumor burden and poor prognosis (Leuk Lymphoma 2022;63:2955, Curr Oncol Rep 2019;21:8, J Clin Oncol 2015;33:2863)
- Prognostic influence of various cytogenetic abnormalities, molecular genetic abnormalities and prognosis remains under investigation
- 60 year old woman with secondary plasma cell leukemia, successfully treated with chimeric antigen receptor T cell therapy targeting B cell maturation antigen (BCMA; CD269) (Front Oncol 2022;12:901266)
- 62 year old man presenting with hypercalcemia, tumor lysis syndrome and primary plasma cell leukemia (Clin Case Rep 2022;10:e05933)
- Combination of immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs) may be the optimal induction therapy for pPCL (Leuk Lymphoma 2017;58:1538, Curr Oncol Rep 2019;21:8, Leukemia 2013;27:780)
- Autologous stem cell transplant (ASCT) after induction therapy and maintenance therapy reduced early relapse and improved survival in eligible patients (Expert Rev Hematol 2019;12:245)
- Venetoclax (BCL2 inhibitor) as a single agent or in combination with daratumumab, dexamethasone and bortezomib can be considered in patients with complex genomic characteristics and t(11;14) (Haematologica 2023;108:941, Eur J Haematol 2018;100:215, Blood 2022;139:2666)
- Chimeric antigen receptor (CAR) T cell therapy, other immunotherapies (mAbs, bispecific T cell engagers, antibody drug conjugates) and small molecule inhibitors are under more investigation (Haematologica 2023;108:941)
- A therapeutic focus on achieving minimal residual disease (MRD) negativity, rather than complete remission, may improve a patient’s outcome (Clin Lab Med 2017;37:821, J Clin Oncol 2023;41:1342, J Clin Oncol 2022;40:3120)
- Peripheral blood
- White blood cells may be decreased or markedly increased with ≥ 5.0% plasma cells detected by manual differential
- Plasma cells are usually larger than background lymphocytes with eccentric nuclei, condensed chromatin with peripherally located chromatin clumps imparting a clock face-like appearance, abundant, basophilic cytoplasm and a perinuclear hof of clear cytoplasm
- Anemia and thrombocytopenia are often present
- Rouleaux formation is often present
- Bone marrow aspirate
- Aspirate contains increased plasma cells
- Background hematopoietic elements may be markedly reduced
- Bone marrow core biopsy and clot section
- Marrow usually shows infiltration by sheets and clusters of plasma cells
- CD138 immunohistochemistry is used to estimate the percentage of bone marrow involvement and to confirm diagnosis in cases with atypical morphology
- Bone lesions and extramedullary lesions
- Sheets and clusters of plasma cells (morphologic features of osseous and nonosseous plasmacytoma, respectively)
- Reference: Semin Diagn Pathol 2003;20:211
Contributed by Jesse Manuel Jaso, M.D.

Sheets of plasma cells
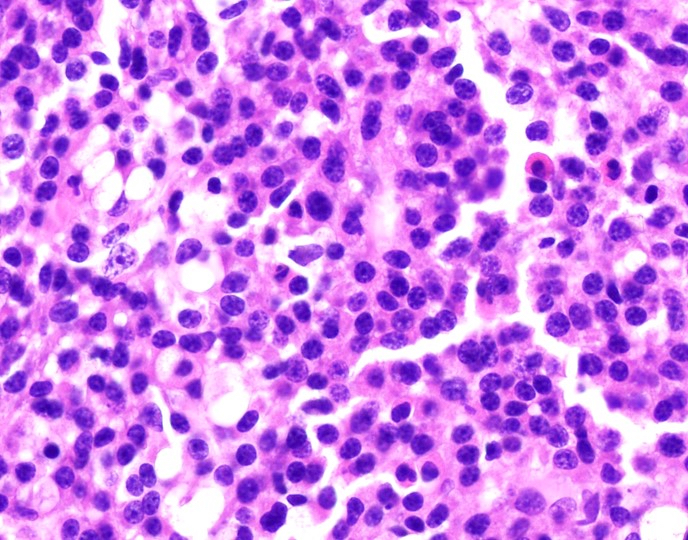
Bone marrow core
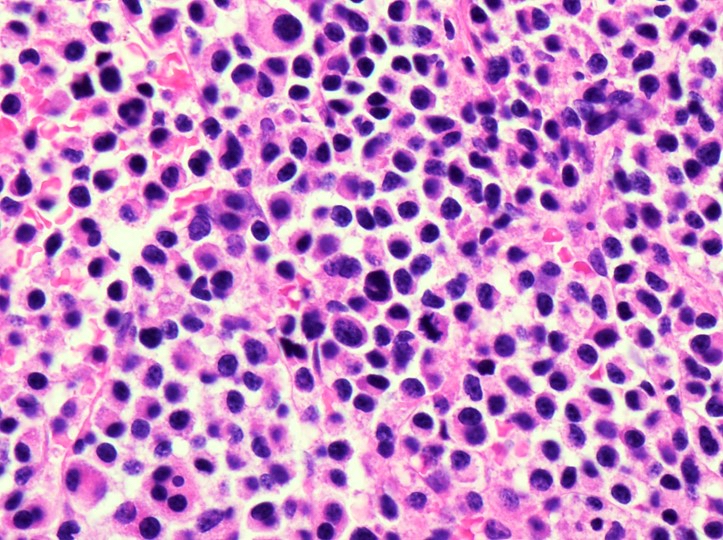
Pleomorphic plasma cells
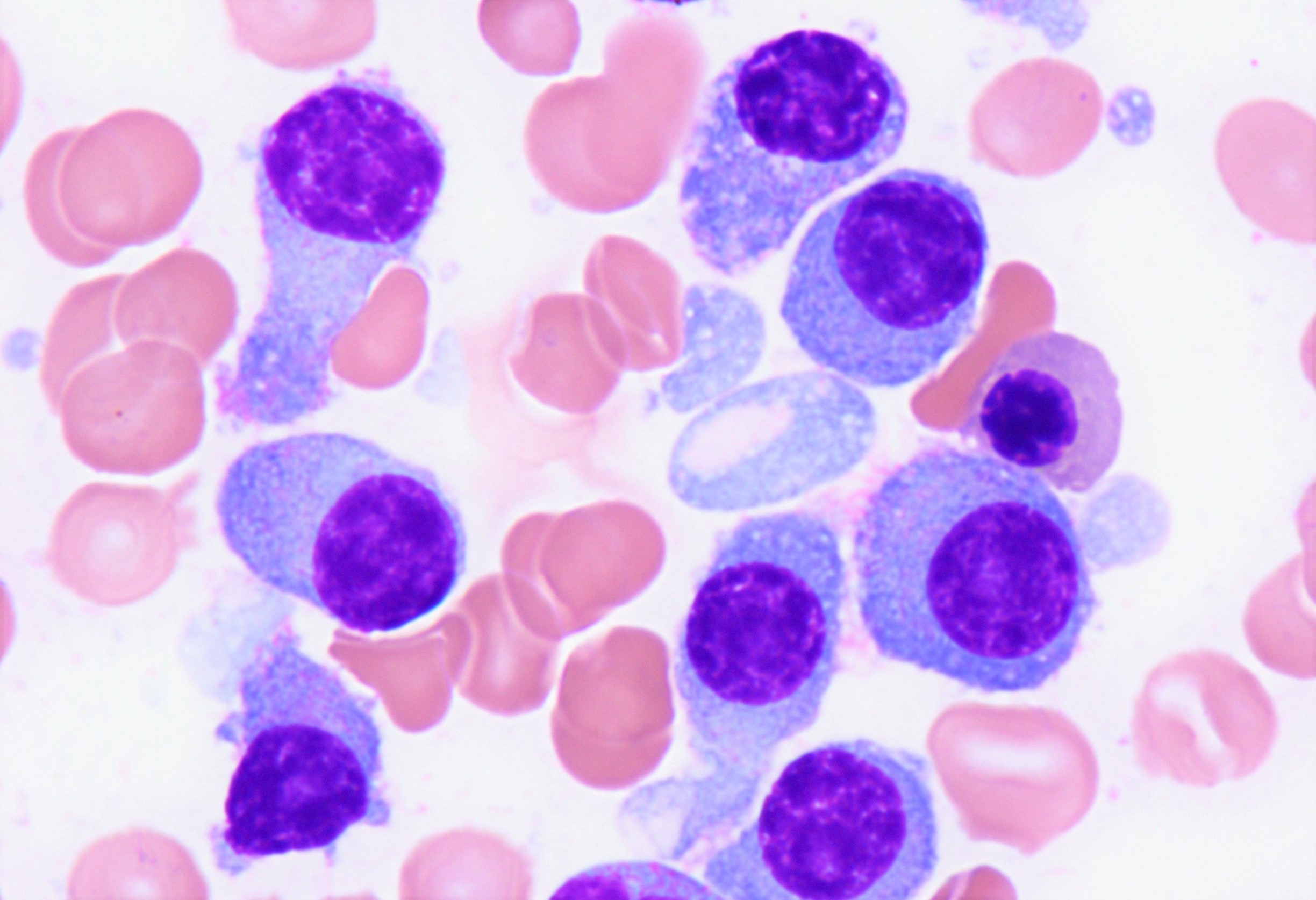
Plasma cells
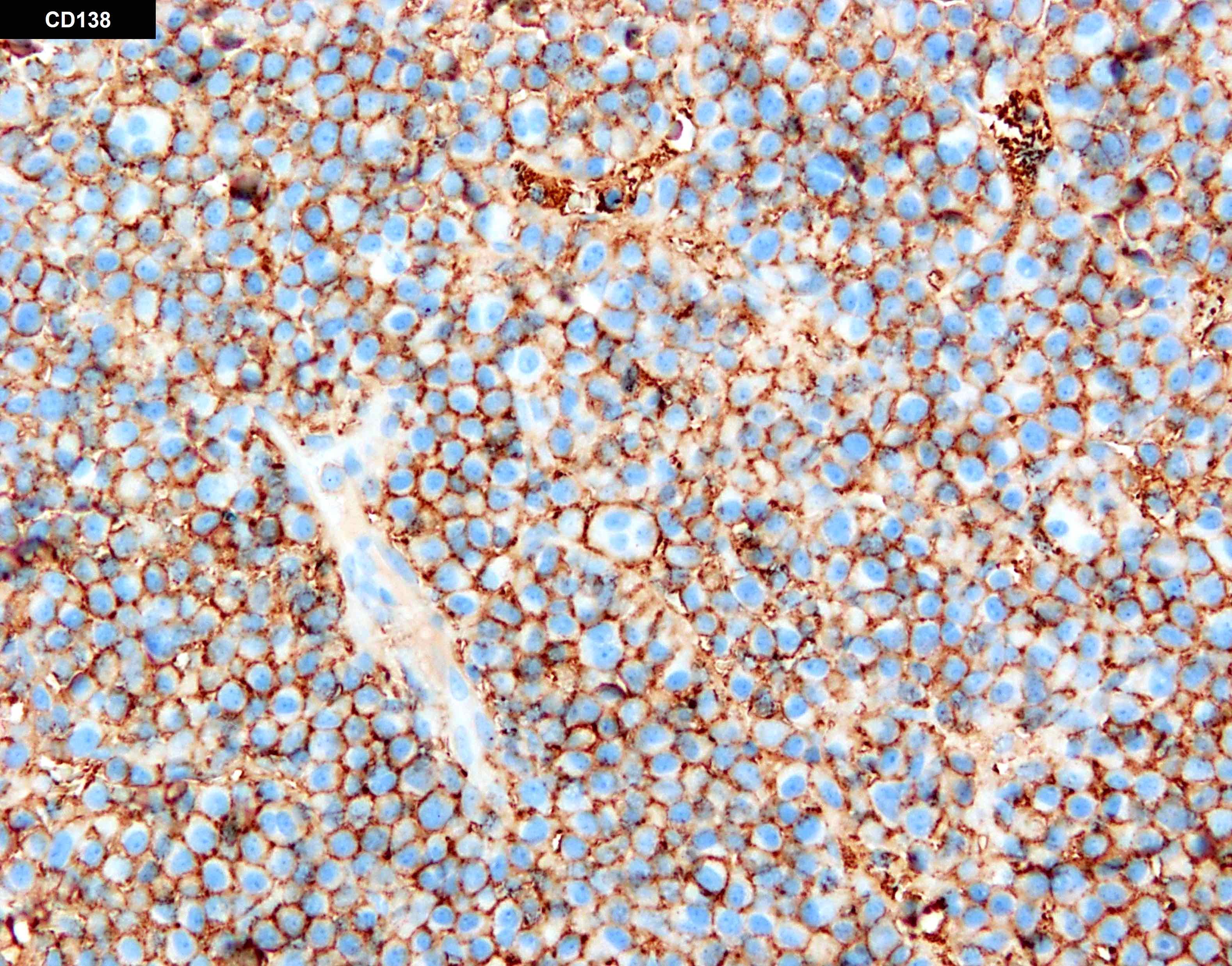
CD138
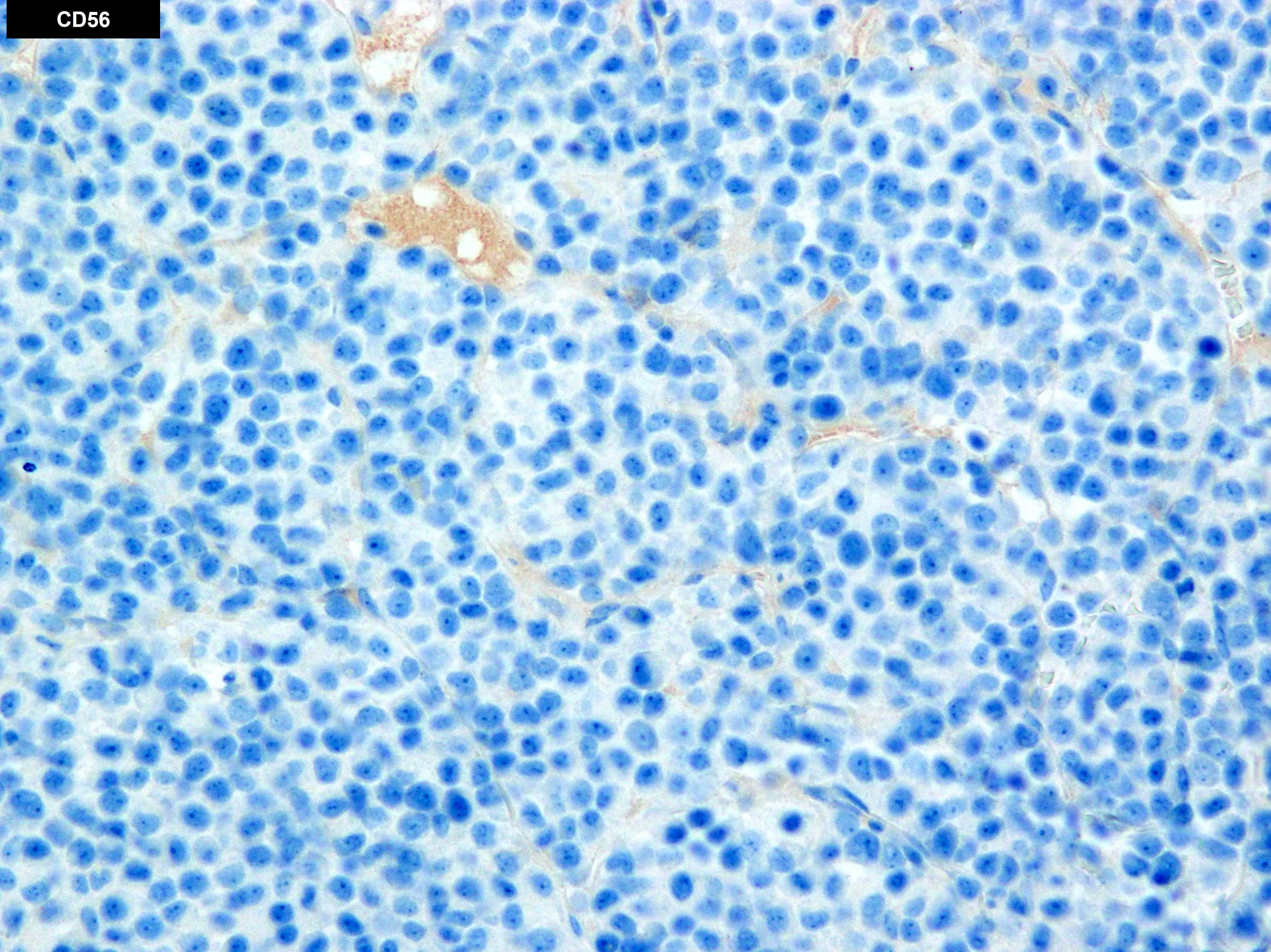
CD56 negative plasma cells

Cyclin D1 expression in plasma cells
- Medium sized cells with eccentric nuclei, condensed, clock face or spoke wheel-like chromatin, abundant basophilic cytoplasm and a perinuclear clearing
- Cells may show variable amounts of morphologic atypia
- Intranuclear (Dutcher bodies), cytoplasmic (Russell bodies) or extracellular immunoglobulin may be present
- Peripheral blood
- White blood cells may be decreased or markedly increased with ≥ 5.0% plasma cells detected by manual differential
- Plasma cells are usually larger than background lymphocytes with eccentric nuclei, condensed chromatin with peripherally located chromatin clumps imparting a clock face-like appearance, abundant, basophilic cytoplasm and a perinuclear hof of clear cytoplasm
- Reference: Semin Diagn Pathol 2003;20:211
Contributed by Jesse Manuel Jaso, M.D.
Circulating plasma cells
- Flow cytometry: neoplastic plasma cells in primary and secondary PCL are variably positive for CD38 and CD138
- Monotypic expression of cytoplasmic kappa or lambda light chains can be assessed via flow cytometry, immunohistochemistry or in situ hybridization
- Neoplastic plasma cells express variable amounts of CD20, CD23, CD44 and CD45
- CD20 expression may be associated with the presence of t(11;14) CCND1::IGH (Leuk Res 2013;37:1251, Front Oncol 2022;12:1061438)
- CD23 expression is highly associated with the presence of t(11;14) CCND1::IGH (Br J Haematol 2010;150:724, Br J Haematol 2010;149:292, Leukemia 2013;27:780)
- CD28 is more frequently expressed in sPCL versus pPCL; its expression has been linked with proliferation / progression and chemotherapeutic resistance in multiple myeloma (Leuk Lymphoma 2017;58:1538, Blood 2014;123:3770)
- CD27 is more frequently expressed in pPCL than sPCL and is associated with antiapoptotic pathway activation (Leukemia 2013;27:780)
- Immunohistochemistry: positive for CD138 with more frequent expression of CD20, CD45 and cyclin D1 in primary and secondary PCL
- Neoplastic cells express CD38 and CD138 and typically lack expression of CD19 and CD45 with monotypic cytoplasmic light chain expression (Leuk Res 2011;35:169)
- Compared to MM, the neoplastic cells in PCL less frequently express CD56 (Br J Haematol 2021;195:95)
- Variable expression of CD20, CD23, CD45 and CD200 may be seen (Blood Cancer J 2021;11:23, Leukemia 2013;27:780)
- CD23 expression is often associated with presence of t(11;14) and cyclin D1 expression (Leukemia 2013;27:780)
Contributed by Franklin Fuda, D.O.

Increased forward and side scatter
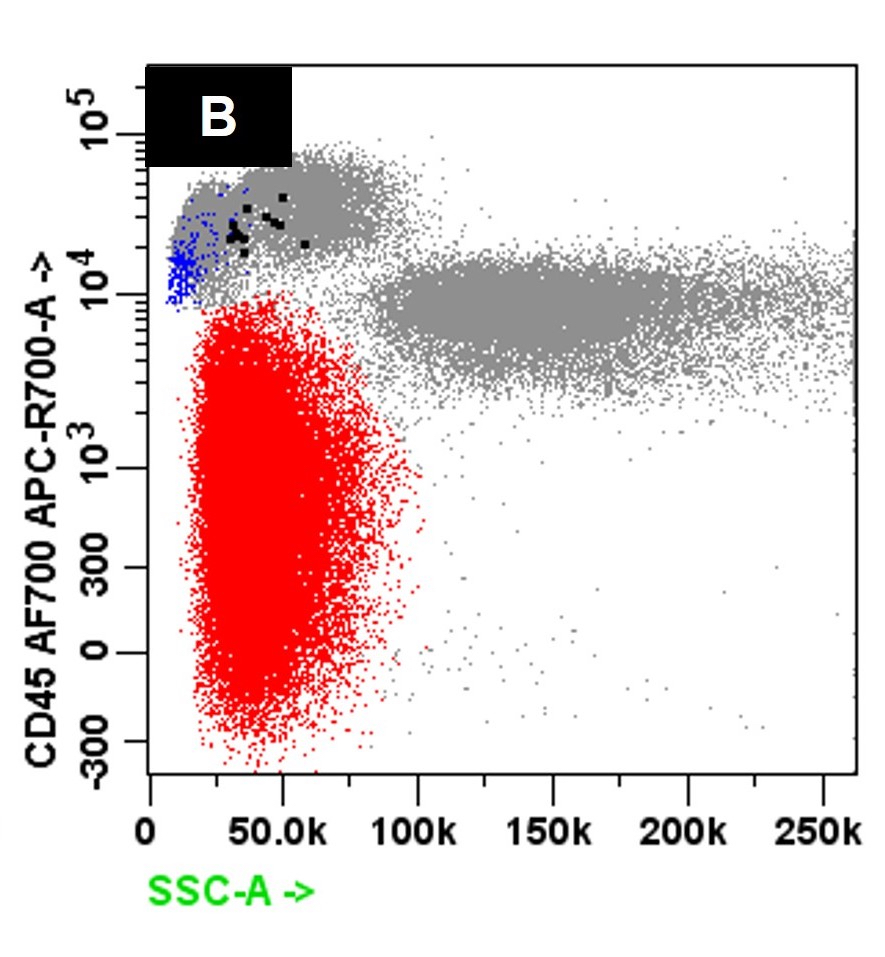
Heterogeneous CD45 expression
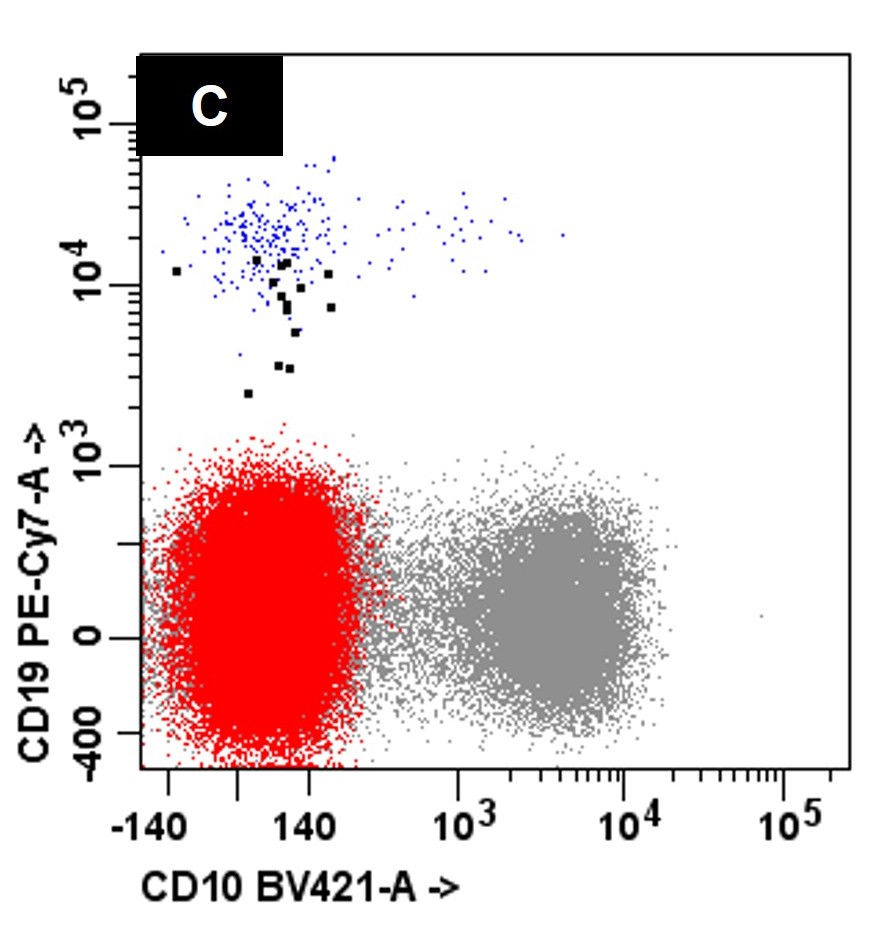
Negative for CD19 and CD10

Negative for CD19 and CD5
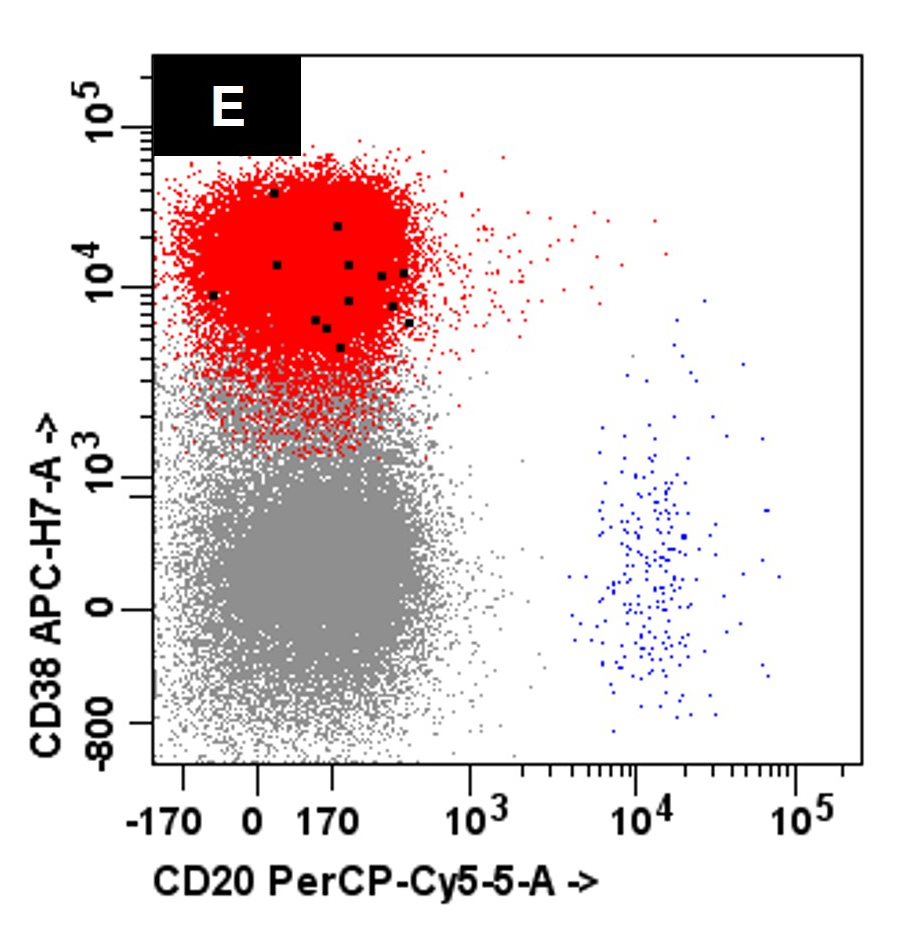
Bright expression of CD38
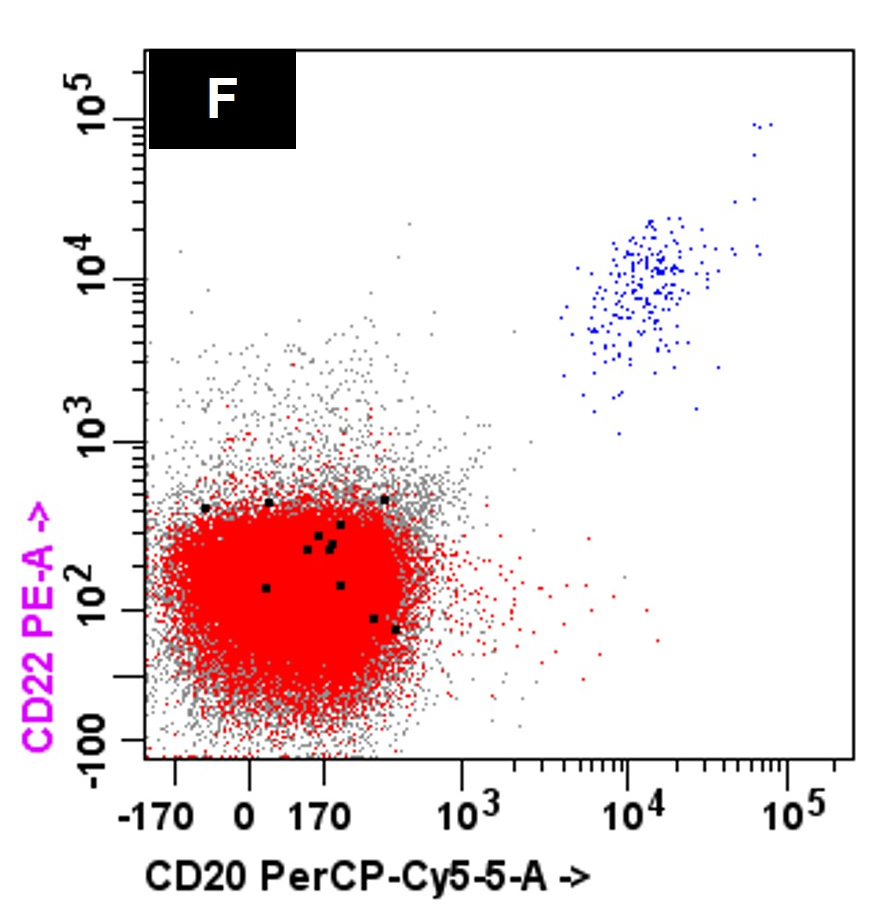
Negative for CD22
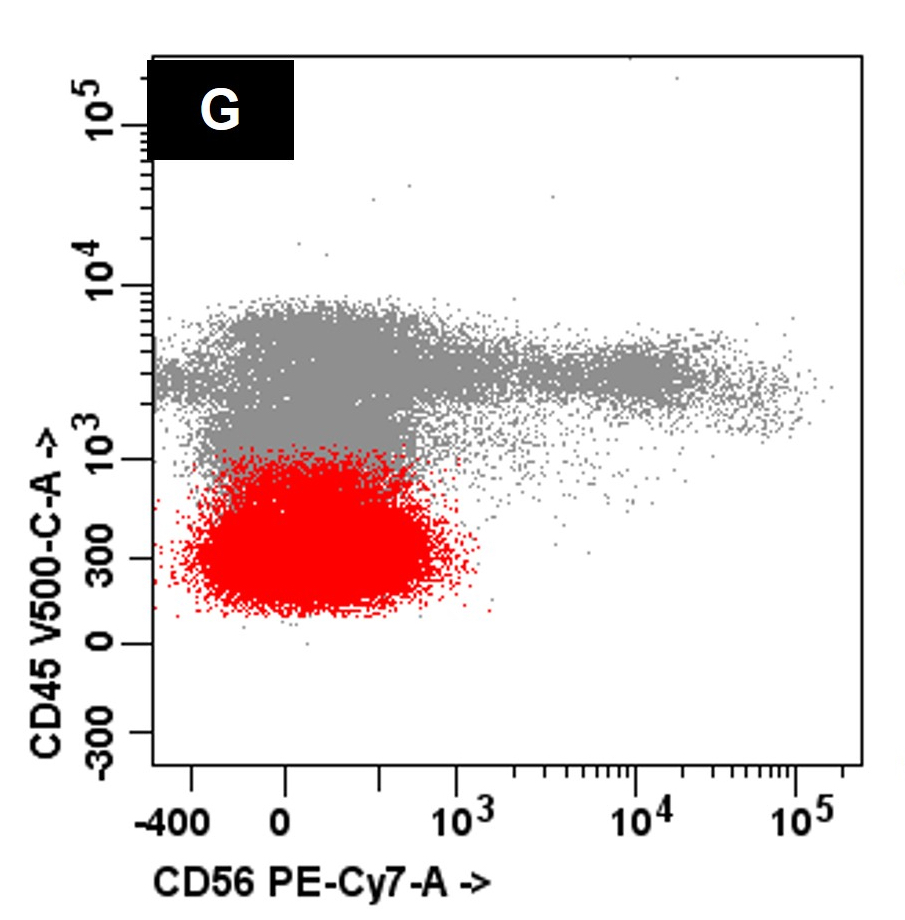
Lack of CD56 expression
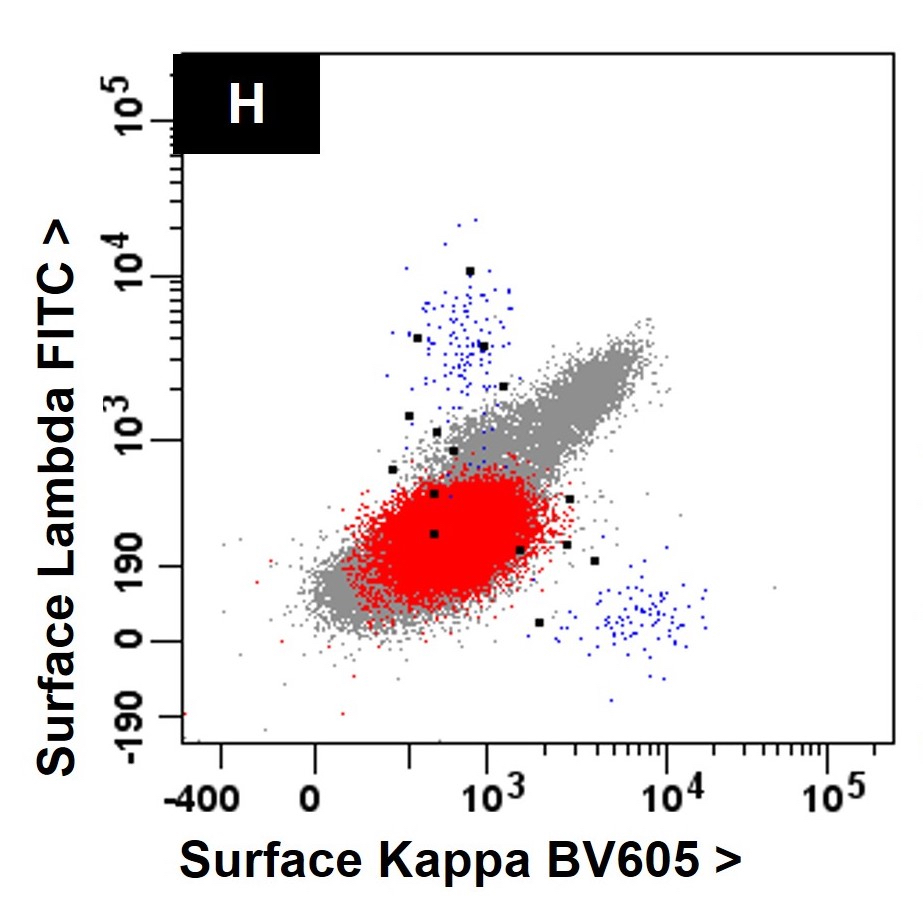
Lack of surface light chain expression

Cytoplasmic kappa expression
- Complex cytogenetic and molecular genetic abnormalities are more frequent in PCL than in multiple myeloma; the most commonly encountered findings are
- Abnormal karyotype
- High risk cytogenetics: t(4;14) or t(14;16) or del(17p13) or t(8q24) (Biomedicines 2022;10:209)
- Hypodiploidy (Leukemia 2013;27:780)
- Loss of chromosome 1p
- Gain of chromosome 1q
- Del 13q (Cancers (Basel) 2022;14:1594)
- Loss of chromosome 16 and 7 (80%, 11%) (Cancers (Basel) 2022;14:1594)
- Trisomy 8: 43% (Cancers (Basel) 2022;14:1594)
- Translocations involving the IGH locus (14q32)
- t(11;14) CCND1::IGH (cyclin D1) with atypical breakpoint in almost half of pPCL (Biomedicines 2022;10:209)
- t(4;14) IGH::FGFR3, t(14;16) IGH::MAF: 13 - 25% may be seen (Cancers (Basel) 2022;14:1594, Biomedicines 2022;10:209)
- Commonly encountered molecular genetic abnormalities
- High prevalence of TP53 mutation or deletion 17p by FISH (Blood 2022;139:2666)
- In patients with t(11;14), commonly associated with a deletion 17p
- In non-t(11;14) commonly associated to biallelic inactivation (Oncotarget 2015;6:17543, Curr Oncol Rep 2019;21:8, Blood Cancer J 2020;10:70)
- High prevalence of MYC abnormalities (loss, rearrangement, amplification) (Biomedicines 2022;10:209, Genes Chromosomes Cancer 2009;48:624)
- Overexpression of MYC by RT-PCR (Leuk Lymphoma 2017;58:1538)
- High prevalence of TP53 mutation or deletion 17p by FISH (Blood 2022;139:2666)
- Bone marrow, aspirate, clot, core biopsy and peripheral blood:
- Primary plasma cell leukemia, 25% circulating plasma cells (see comment)
- Plasma cell neoplasm, involving > 95% of bone marrow
- Comment:
- The peripheral blood shows leukocytosis with numerous circulating plasma cells (25%). The bone marrow is hypercellular (95 - 100%) and shows near total replacement by sheets of atypical, pleomorphic plasma cells (67% by manual differential; > 95% by immunohistochemistry for CD138).
- Concurrent flow cytometry shows an aberrant plasma cell population with monotypic, cytoplasmic light chain expression.
- Review of the electronic medical record indicates that the patient does not have a prior diagnosis of plasma cell neoplasm or other malignancy.
- The combined findings are diagnostic of primary plasma cell leukemia. Clinical correlation and correlation with pending ancillary and staging studies is recommended.
- Bone marrow, aspirate, clot, core biopsy and peripheral blood:
- Secondary plasma cell leukemia, 35% circulating plasma cells (see comment)
- Relapsed plasma cell myeloma, involving 80 - 90% of bone marrow
- Comment:
- The peripheral blood shows leukocytosis with numerous circulating plasma cells (35%). The bone marrow is hypercellular (95 - 100%) and shows near total replacement by sheets of atypical, pleomorphic plasma cells (45% by manual differential; 80 - 90% by immunohistochemistry for CD138).
- Concurrent flow cytometry shows an aberrant plasma cell population with monotypic, cytoplasmic light chain expression.
- The combined findings are diagnostic of relapse of the patient’s previously diagnosed plasma cell myeloma with development of secondary leukemic transformation (secondary plasma cell leukemia). Clinical correlation and correlation with pending ancillary and staging studies is recommended.
- Florid polytypic plasmacytosis:
- In rare cases, patients with autoimmune disease, HIV and other infectious / inflammatory disorders may have a florid increase in reactive bone marrow plasma cells
- Plasma cells will lack immunophenotypic aberrancies by flow cytometry or immunohistochemistry
- Plasma cells will show polytypic, cytoplasmic kappa and lambda light chain expression by flow cytometry or immunohistochemistry
- B cell lymphoma with plasmacytic differentiation:
- B cell non-Hodgkin lymphoma shows a variable propensity for plasmacytic differentiation (e.g., marginal zone lymphoma and lymphoplasmacytic lymphoma)
- Flow cytometry will demonstrate the presence of 2 monoclonal cell populations: an aberrant B cell and plasma cell population with monotypic expression of the same light chain (assessed on the cell surface for B cells and within cytoplasm / intracellularly for plasma cells)
- Neoplastic B cells will express CD19, CD20, CD22 and monotypic, surface light chain expression
- Neoplastic plasma cells will show monotypic, cytoplasmic expression of the same light chain as the B cells
- Neoplastic plasma cells often express CD19, CD20 (partial) and CD45 and lack expression of CD56
- Neoplastic plasmacytic cells often show monotypic, surface light chain expression similar to the neoplastic B lymphocytes
- B cell non-Hodgkin lymphoma shows a variable propensity for plasmacytic differentiation (e.g., marginal zone lymphoma and lymphoplasmacytic lymphoma)
A 45 year old patient presents with weight loss, leukocytosis, anemia and thrombocytopenia. Serum protein electrophoresis shows a monoclonal IgG kappa protein. Bone marrow examination shows ~90% involvement by sheets of atypical plasma cells with monotypic, intracytoplasmic kappa light chain expression. Review of a concurrent peripheral blood smear shows ~6% atypical cells (see images above). Which of the following statements regarding these cells is most correct?
- Neoplastic cells have monotypic, surface light chain expression by flow cytometry
- Neoplastic cells typically lack expression of CD38 by flow cytometry
- Neoplastic cells typically lack expression of CD56 by flow cytometry
- Neoplastic cells with bright expression of CD19 by flow cytometry
- Neoplastic cells with bright expression of CD45 by flow cytometry
Comment Here
Reference: Plasma cell leukemia
- Serum beta 2 microglobulin
- Serum calcium
- Serum creatinine
- Urine immunofixation electrophoresis
- White blood cell count
Comment Here
Reference: Plasma cell leukemia
- Bone marrow based, multifocal plasma cell neoplasm usually associated with a monoclonal immunoglobulin (M protein) in serum or urine and evidence of organ damage related to the plasma cell neoplasm (J Natl Compr Canc Netw 2019;17:1154)
- Diagnosis requires synthesis of clinical, laboratory, radiologic and histologic findings
- Assessment of serum or urine M protein, clonal plasma cells in bone marrow and presence of end organ damage related to the plasma cell neoplasm critical for diagnosis
- Multiple myeloma
- Plasma cell myeloma
- Myeloma
- Medullary plasmacytoma
- Myelomatosis
- Kahler disease (no longer used) (Recent Results Cancer Res 2011;183:3)
- Smoldering (asymptomatic) plasma cell myeloma
- Nonsecretory myeloma
- Plasma cell leukemia
- 1.8% of malignant tumor diagnoses in the U.S. annually, 19% of hematopoietic neoplasms, 21% of deaths from hematopoietic malignancies (CA Cancer J Clin 2021;71:7)
- M:F = 1.2:1
- Twice as frequent in African Americans compared to Caucasians
- Not seen in children, rarely seen in adults under 30; median age of diagnosis is ~70 years (Semin Oncol 2016;43:676, CA Cancer J Clin 2021;71:7)
- Potential genetic predisposition, relative risk of 2.1 to develop multiple myeloma among those with an affected first degree relative (Int J Cancer 2009;125:2147, Leukemia 2020;34:697)
- Bone marrow site of origin for nearly all cases
- Interactions between bone marrow stroma and neoplastic plasma cells directly influences disease with a potential key role of IL6 to support survival and expansion of myeloma cells (Leukemia 2014;28:1647, Cancer 2003;97:2440, Cancers (Basel) 2021;13:216)
- Role of IL6 and other cytokines in promoting osteoclastic activity and lytic bone lesions
- Multiple myeloma cells suppress the differentiation and proliferation of osteoblasts while inducing osteoclast differentiation and hyperfunction (Cancers (Basel) 2021;13:216)
- Chronic antigen stimulation, exposure to radiation or toxins may result in increased risk; however, most patients do not have these associated factors (Cancer 1993;72:2148, Occup Environ Med 2011;68:391)
- Association with radiation may be weak and not identified in some studies (PLoS One 2016;11:e0162710)
- Almost all cases appear to arise in patients with precursor monoclonal gammopathy of undetermined significance (MGUS) (Blood 2009;113:5418)
- Bone disease is the most frequent disease defining clinical feature (Am Soc Clin Oncol Educ Book 2018;38:638)
- Often presents with bone pain due to lytic bone lesions (thoracic vertebrae most common, also ribs, skull, shoulders, pelvis and long bones); spinal cord compression or peripheral neuropathy are less common presenting symptoms
- Renal failure / elevated creatinine / hyperuricemia (monoclonal light chain proteinuria results in renal tubular damage); hypercalcemia; hypoalbuminemia
- Recurrent infections due to impaired humoral immunity (immunoglobulin production, often < 50% normal)
- Anemia (bone marrow infiltration often in areas of most active hematopoiesis and renal failure causing loss of erythropoietin)
- Extramedullary involvement generally associated with advanced disease
- Historical fact: Bence Jones proteins were the first tumor marker (Clin Kidney J 2012;5:478)
- Smoldering (asymptomatic):
- More likely to progress to symptomatic myeloma than monoclonal gammopathy of uncertain significance (MGUS)
- Both show gammopathy without myeloma defining events (hypercalcemia, anemia, bone lesions and renal insufficiency)
- Risk of progression 10% per year for the first 5 years
- Lower risk if no progression in the first 5 years after diagnosis (N Engl J Med 2007;356:2582)
- Nonsecretory myeloma (~1% of cases):
- Serum protein electrophoresis (SPE) / immunofixation electrophoresis (IFE) negative, 85% with impaired secretion and have cytoplasmic immunoglobulin (Ig) by IHC (Am J Clin Pathol 2011;136:168)
- 15% nonproducers, serum free light chain may still be detected
- Lower incidence of renal insufficiency, hypercalcemia and depression of normal IgG
- Must be distinguished from the rare IgD and IgE myelomas
- Plasma cell leukemia:
- Typically aggressive with short survival < 1 year
- > 2 x 109/L or 20% of the leukocyte count on differential are monoclonal plasma cells
- Primary plasma cell leukemia (0.6% of myeloma) or develop as a late stage transformation (secondary) (Curr Oncol Rep 2019;21:8)
- Usually lack CD56 (80% of PCLs), more frequent high risk genetic findings
- Bone pain and osteolytic lesions less common
- Typically associated with extramedullary lesions (e.g., body cavity effusions, lymphadenopathy and organomegaly)
- Division into these categories will guide plan for therapy (Am Soc Clin Oncol Educ Book 2016;35:e418):
- Multiple (symptomatic) myeloma (J Natl Compr Canc Netw 2019;17:1154):
- Clonal bone marrow plasma cells ≥ 10% or biopsy proven bony or extramedullary plasmacytoma
- And ≥ 1 of the following myeloma defining events:
- Calcium > 1 mg/dL above upper limit of normal or > 11 mg/dL
- Renal insufficiency*; creatinine > 2 mg/dL or creatinine clearance < 40 mL/min (preferred)
- Hemoglobin < 10 g/dL or > 2 g/dL below the lower limit of normal
- 1 or more osteolytic bone lesions on skeletal radiography, CT or FDG PET / CT
- Clonal bone marrow plasma cells ≥ 60% of bone marrow cellularity
- Involved / uninvolved serum free light chain (FLC) ratio ≥ 100 and involved free light chain concentration 10 mg/dL or higher
- > 1 focal lesion on MRI studies ≥ 5 mm
- These include traditional CRAB features associated with end organ damage (hypercalcemia, renal failure, anemia and osteolytic bone lesions) and biomarkers associated with ~80% risk of progression to end organ damage (≥ 60% clonal plasma cells in bone marrow, serum FLC ≥ 100 with FLC level ≥ 10 mg/dL or > 1 focal lesion by MRI) (Am Soc Clin Oncol Educ Book 2016;35:e418)
- *Only suspected or proven light chain cast nephropathy is considered a multiple myeloma defining event, consider renal biopsy to clarify if FLC < 500 mg/L (Am Soc Clin Oncol Educ Book 2016;35:e418)
- Smoldering (asymptomatic) myeloma (J Natl Compr Canc Netw 2019;17:1154):
- M protein in serum (IgG or IgA) at ≥ 3 g/dL
- Or Bence Jones protein ≥ 500 mg/24 h urine
- Or 10 - 59% clonal plasma cells in bone marrow
- And no related tissue damage / myeloma defining event or amyloidosis; if bone survey negative, bone disease should be assessed with whole body MRI, FDG PET / CT or low dose CT scan
- Multiple (symptomatic) myeloma (J Natl Compr Canc Netw 2019;17:1154):
- 97% have an M protein in serum or urine, 3% are nonsecretory
- IgG (50%), IgA (20%), light chain (20%), others < 10% (IgD, IgE, IgM and biclonal)
- SPEP: serum proteins normally separate into 5 major fractions based on electric charge and size (Am Fam Physician 2005;71:105):
- Albumin
- Alpha 1 globulins
- Alpha 2 globulins
- Beta 1 and beta 2 globulins
- Gamma globulins
- Gamma globulins include polyclonal antibodies and light chains, with a normal gamma zone appearing as a symmetrical smear; myeloma may appear as a spike in this region
- Urine protein electrophoresis (UPEP): monoclonal light chains in urine = Bence Jones protein
- IFE: used to characterize the M spike, by reacting with specific antisera to heavy chains IgG, IgA, IgM, IgD, IgE and kappa and lambda light chains
- Serum free light chain assay (SFLCA) (Freelite): more sensitive for monitoring light chain disease and nonsecretory myeloma (Blood Cancer J 2020;10:2)
- Mass spectrometry may have equivalent performance to IFE and better differentiate M protein from therapeutic antibodies (Blood Cancer J 2021;11:24)
Images hosted on other servers:
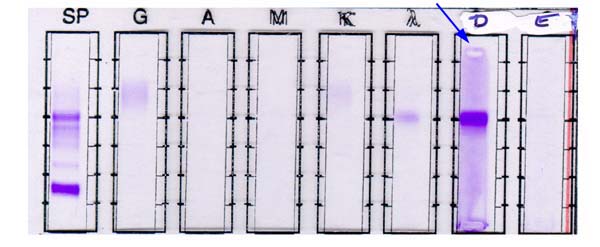
Serum protein immunofixation shows IgD lambda myeloma
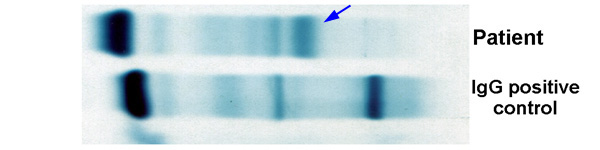

IgD lambda myeloma

Another IgD myeloma
- Evidence of 1 or more sites of osteolytic bone destruction (at least 5 mm in size)
- Advanced methods include low dose whole body CT, MRI and (18F) fluorodeoxyglucose PET (FDG PET), and FDG PET with PET / CT (Am Soc Clin Oncol Educ Book 2016;35:e418)
- With greater sensitivity than radiographic bone survey and recommended prior to a diagnosis of smoldering multiple myeloma or solitary plasmacytoma (Am Soc Clin Oncol Educ Book 2016;35:e418)
- Increased uptake on PET / CT alone is not adequate without evidence of underlying osteolytic bone destruction; bone biopsy recommended if any doubt
Contributed by Mark R. Wick, M.D. and AFIP images
%20skull%20xray.jpg)
%20skull%20xray2.jpg)
Skull Xray
%20tibia%20xray.jpg)
Tibia Xray

Solitary plasma cell myeloma

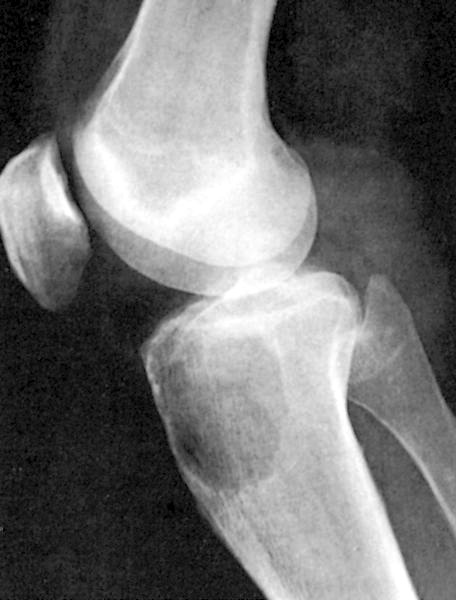
Plasma cell myeloma
Images hosted on other servers:
Prominent skull defect
- Usually incurable with median survival of ~5.5 years with a 5 year survival rate of 54% (ASCO: Multiple Myeloma - Statistics [Accessed 26 May 2022])
- However, improvements in treatment are leading to longer survival times (Semin Oncol 2016;43:676)
- Higher risk:
- Elevated beta 2 microglobulin, lactate dehydrogenase, C reactive protein, serum soluble receptor for IL6 and plasma cell proliferation or bone marrow infiltration
- Reduced polyclonal (uninvolved) serum immunoglobulins
- Plasmablastic morphology
- Abnormalities by conventional cytogenetics
- Active myeloma can be staged using the International Staging System (ISS) or revised International Staging System (R-ISS) (J Natl Compr Canc Netw 2019;17:1154)
- Revised International Staging System:
- Stage I: serum beta 2 microglobulin < 3.5 mg/L, serum albumin ≥ 3.5 g/dL, standard risk chromosomal abnormalities by FISH [absence of del(17p), t(4:14) or t(14;16)] and normal serum LDH
- Stage II: not R-ISS stage I or III
- Stage III: serum beta 2 microglobulin ≥ 5.5 mg/L or either high risk chromosomal abnormalities [del(17p), t(4:14) or t(14;16)] and serum LDH > upper limit of normal
- Additional prognostic chromosomal abnormalities:
- Worse: t(4;14), MAF translocations t(14;16) and t(14;20), del(17p), del 13, aneuploidy, hypodiploidy
- Better: hyperdiploidy, t(11;14), t(6;14), cyclin D1 or D3 positive
- References: analysis of prognostic value of most frequent chromosomal changes in a large series of patients with newly diagnosed symptomatic myeloma (Blood 2007;109:3489)
- Model for identifying patients with increased risk of progression of smoldering myeloma (Mayo 2018 criteria or 20/2/20 criteria) with 3 independent risk factors:
- Serum monoclonal protein > 2 g/dL
- Involved to uninvolved serum free light chain ratio > 20
- Bone marrow plasma cells > 20%
- Low (0 factor), intermediate (1 factor) and high risk (2 - 3 factors) shown to have 2 year rates of progression to multiple myeloma of 5%, 17% and 46%, respectively (Am Soc Clin Oncol Educ Book 2020;40:1)
- 49 year old woman with a history of severe, chronic, active sarcoidosis (Arch Pathol Lab Med 2002;126:365)
- 55 year old man with multiple myeloma and prognosis of undetermined significance (American Society of Hematology: Case Study [Accessed 26 May 2022])
- 56 year old woman diagnosed with multiple myeloma (type IgG kappa, 59 g/L) (J Hematother Stem Cell Res 2001;10:657)
- 61 year old woman with EBV+ multiple myeloma (Int J Clin Exp Pathol 2015;8:2090)
- 64 year old man with IgM multiple myeloma (Cancer Control 2018;25:1073274817744448)
- 67 year old man with acute kidney injury (J Hematol 2016;5:76)
- 77 year old woman with plasma cell leukemia with t(11;14)(q13;q32) simulating lymphoplasmacytic lymphoma (Rev Bras Hematol Hemoter 2017;39:66)
- Original description of Bence Jones proteinuria in 1850 (Med Chir Trans 1850;33:211)
- Updated diagnostic criteria (see Diagnosis) from the International Myeloma Working Group to initiate therapy, including sensitive imaging, may help identify patients who would benefit from treatment before end organ damage occurs (CRAB features) (J Natl Compr Canc Netw 2019;17:1154, Lancet Oncol 2016;17:e328, Am Soc Clin Oncol Educ Book 2020;40:1)
- Treatment regimens are discussed in the National Comprehensive Cancer Network (NCCN) guidelines (J Natl Compr Canc Netw 2019;17:1154)
- May include proteasome inhibitors; immunomodulatory drugs, steroids, antibody based therapy (including elotuzumab [anti-SLAM7], daratumumab and isatuximab [CD38] and new BCMA targeting agents), traditional chemotherapeutic agents, radiation therapy (Multiple Myeloma Treatment Foundation: Standard Treatments [Accessed 26 May 2022])
- Autologous bone marrow transplant, particularly for young patients with newly diagnosed myeloma (Blood Cancer J 2019;9:44)
- Sensitive measure of response to therapy to guide treatment decisions
- Flow cytometry (Blood 2013;122:1088)
- Molecular methods monitoring immunoglobulin rearrangement by NGS (Blood Adv 2020;4:4573)
- Clonotypic peptides by mass spectrometry by liquid chromatography with tandem mass spectrometry (LC-MS / MS) (Clin Chem 2016;62:243)
- MRD negativity associated with better progression free survival (Blood Adv 2020;4:4573)
- Bone defects are filled with a soft, gelatinous "fish flesh" hemorrhagic tissue
Images hosted on other servers:

Vertebrae with myeloma lesions

Skull lesions
- Osseous or extraosseous plasmacytomas, particularly if the patient is not known to have a history of plasma cell myeloma, may be sent for frozen section evaluation (Borczuk: Frozen Section Pathology, 1st Edition, 2021)
- Sites can include: mucosa of the upper respiratory tract, lymph nodes, thyroid, testes, breast, salivary gland and CNS (Int J Otolaryngol 2010;2010:302656)
- Morphology may range from monotonous plasma cells to more irregular, multinucleated or pleomorphic forms in more advanced myeloma
Contributed by Genevieve M. Crane, M.D., Ph.D.
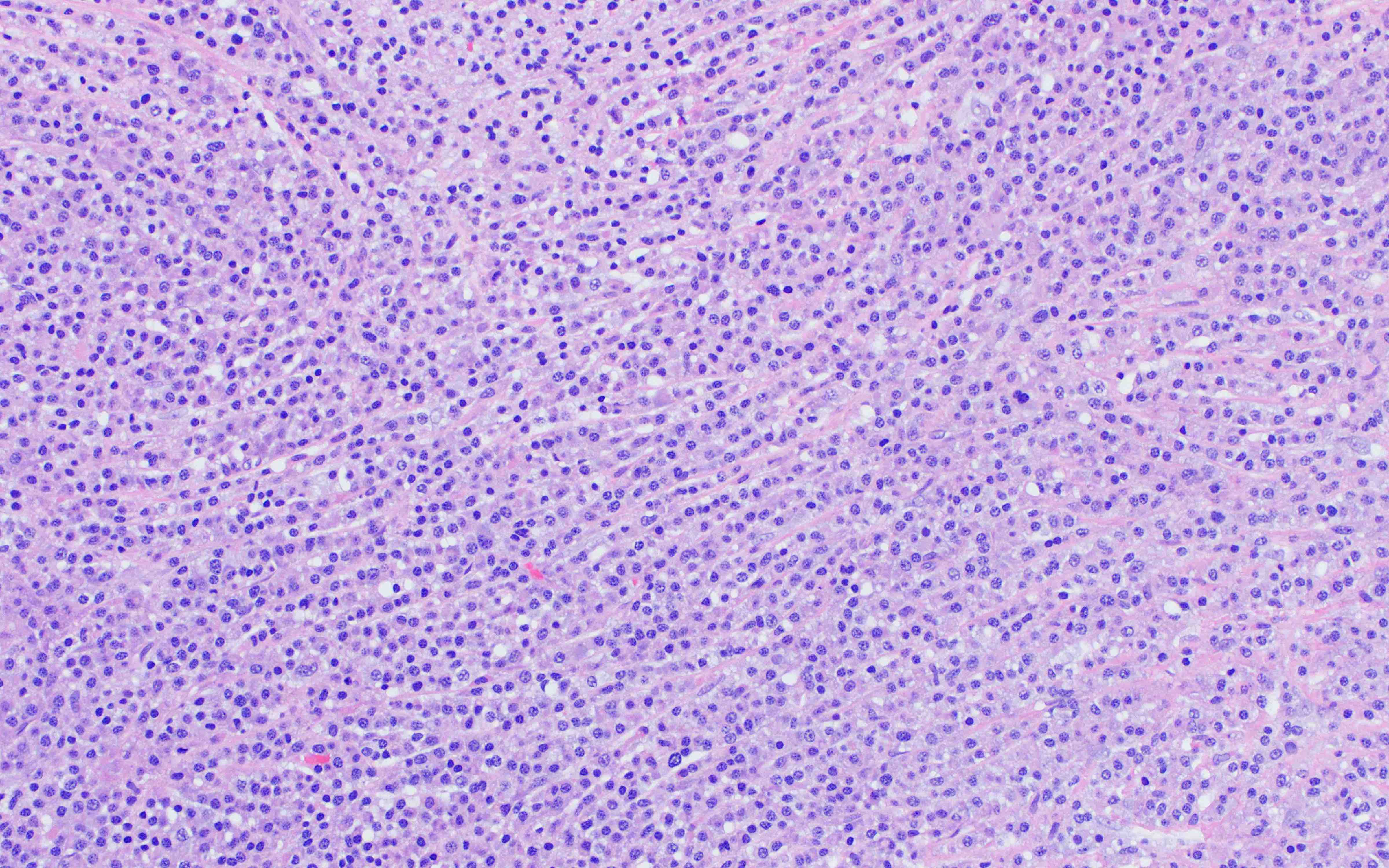
Touch preparation plasmacytoma
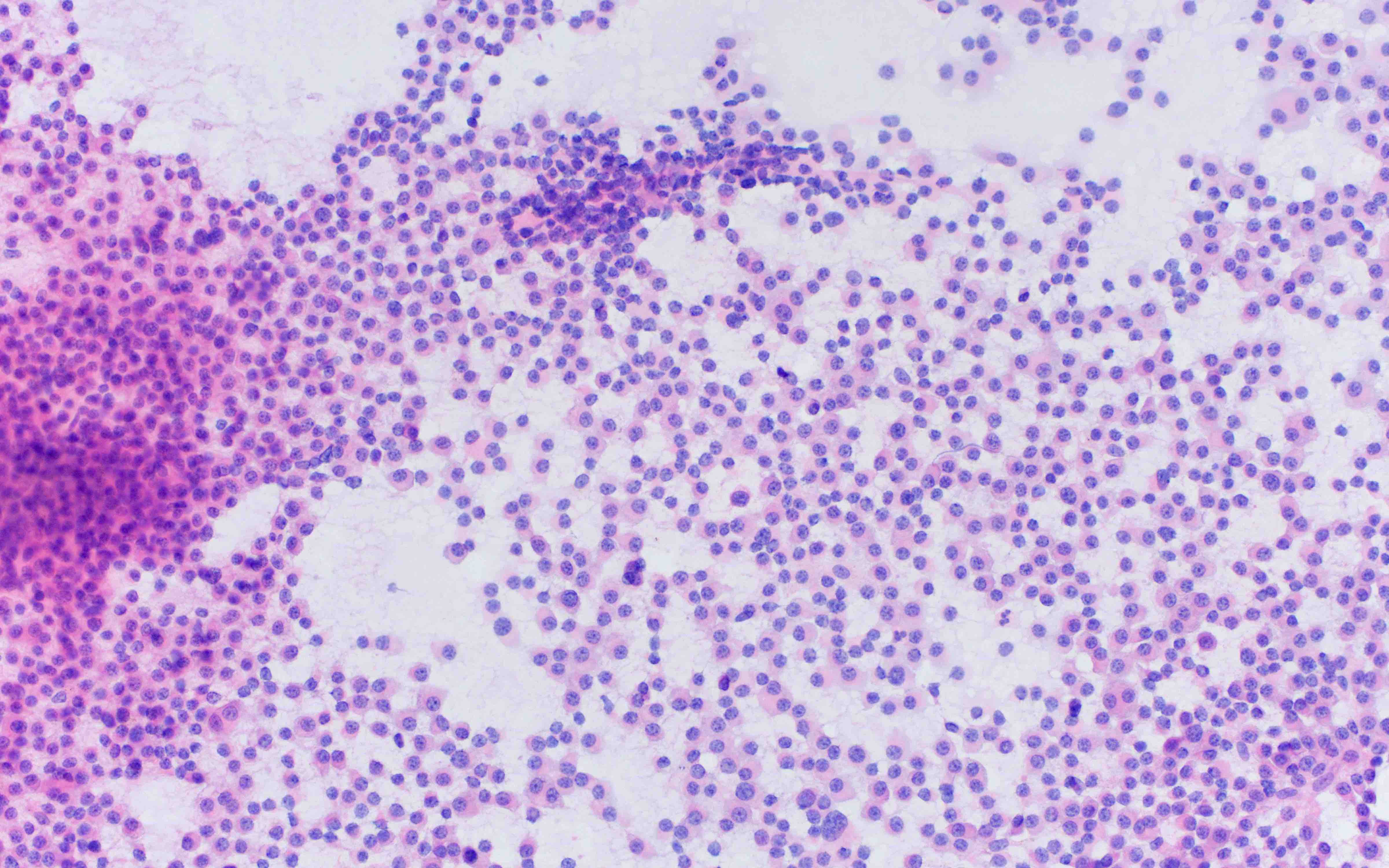
Frozen section plasmacytoma
- Core biopsy (Am J Clin Path 1987;87:342):
- Interstitial clusters, nodules or sheets of plasma cells
- Areas of bone marrow may be spared with preserved hematopoiesis, other cases may have diffuse involvement and markedly suppressed hematopoiesis
- Prominent osteoclastic activity may be seen
- IHC to quantify plasma cells (CD138), stains for Ig kappa and lambda to establish clonality
Contributed by Genevieve M. Crane, M.D., Ph.D. and Tapan Bhavsar, M.D., Ph.D.
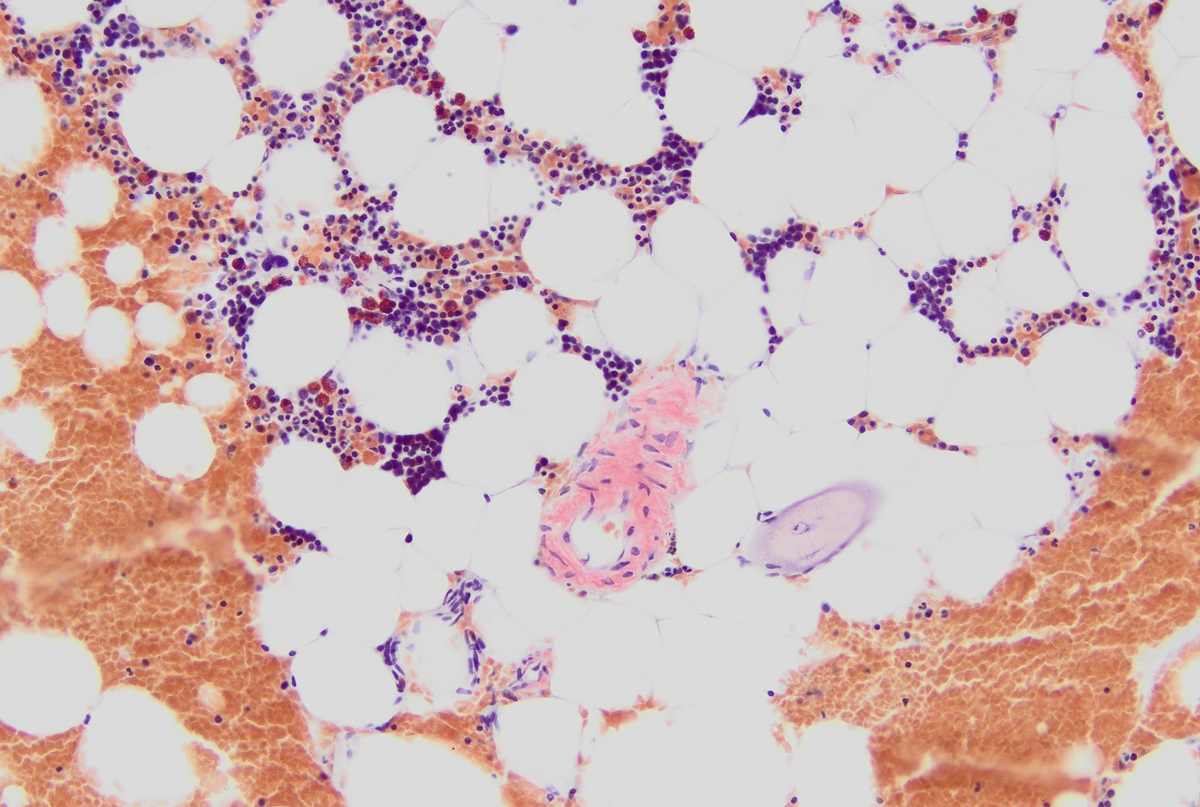
Amyloid deposition (Congo red stain)
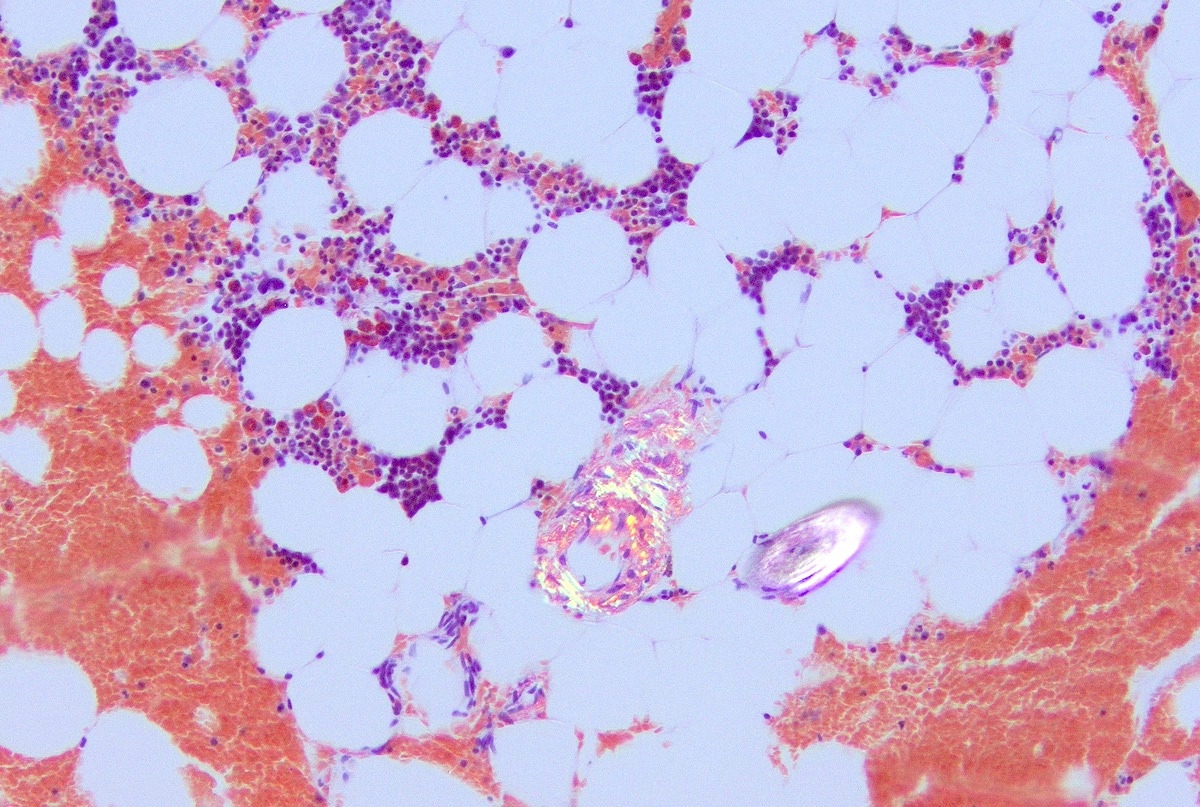
Amyloid deposition
(Congo red stain
with birefringence)
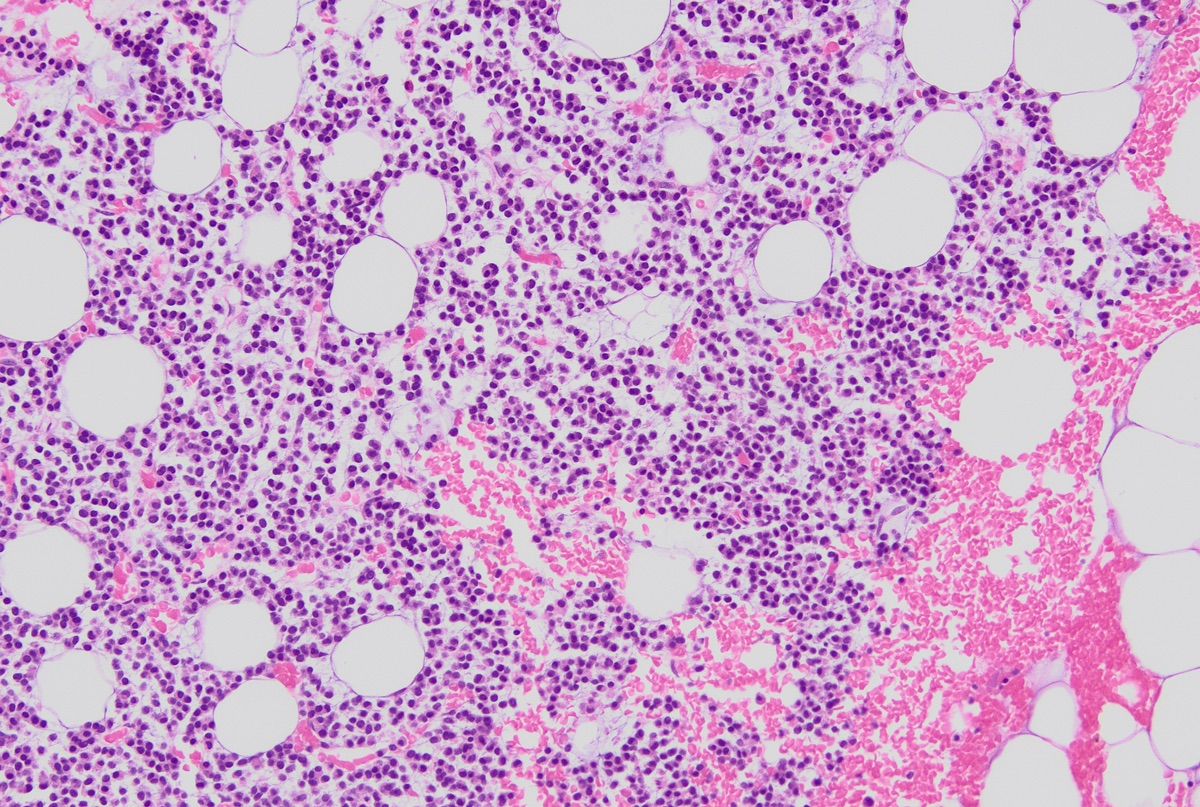
Focal sheets of plasma cells

Extensive amyloid, bone marrow
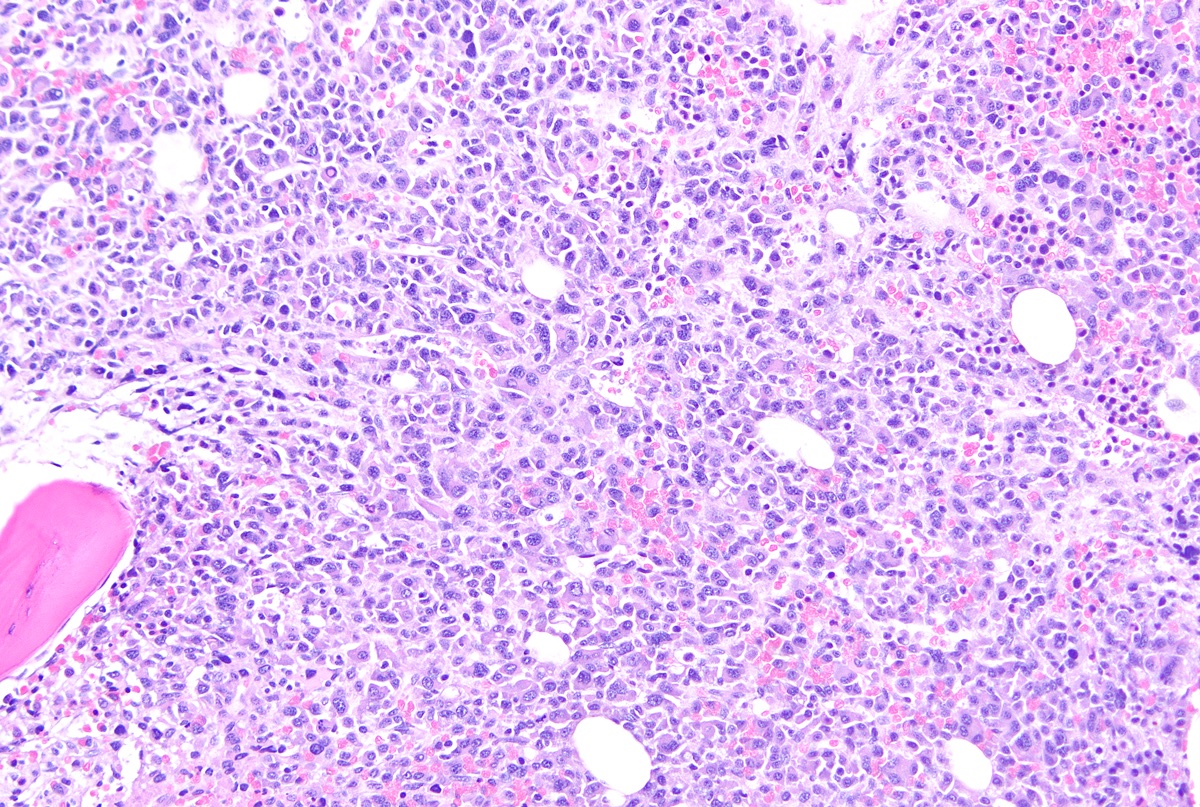
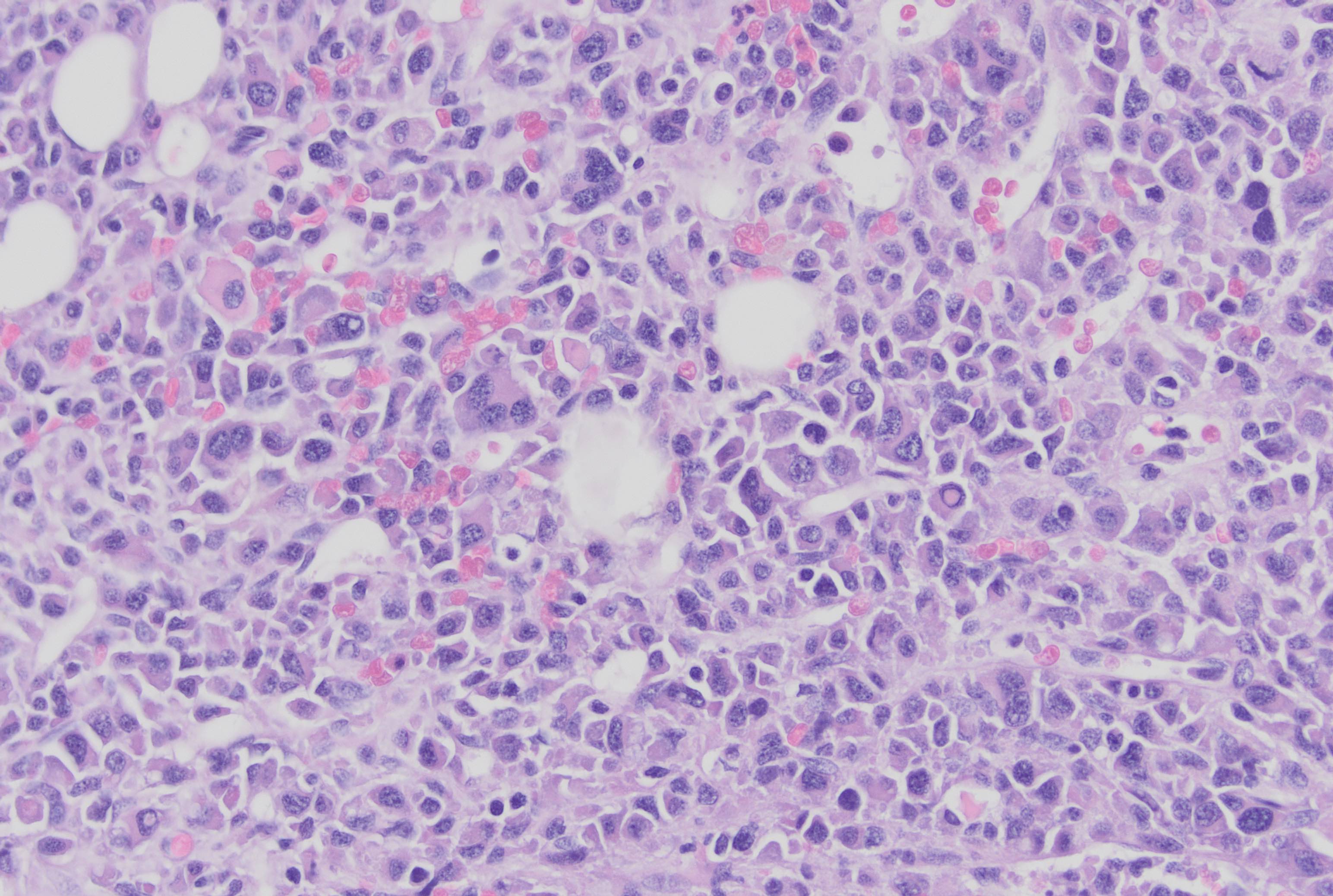
Extensive bone marrow involvement by myeloma
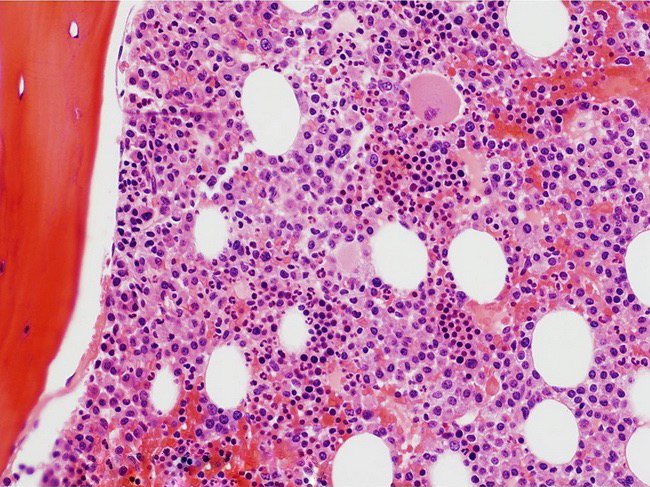
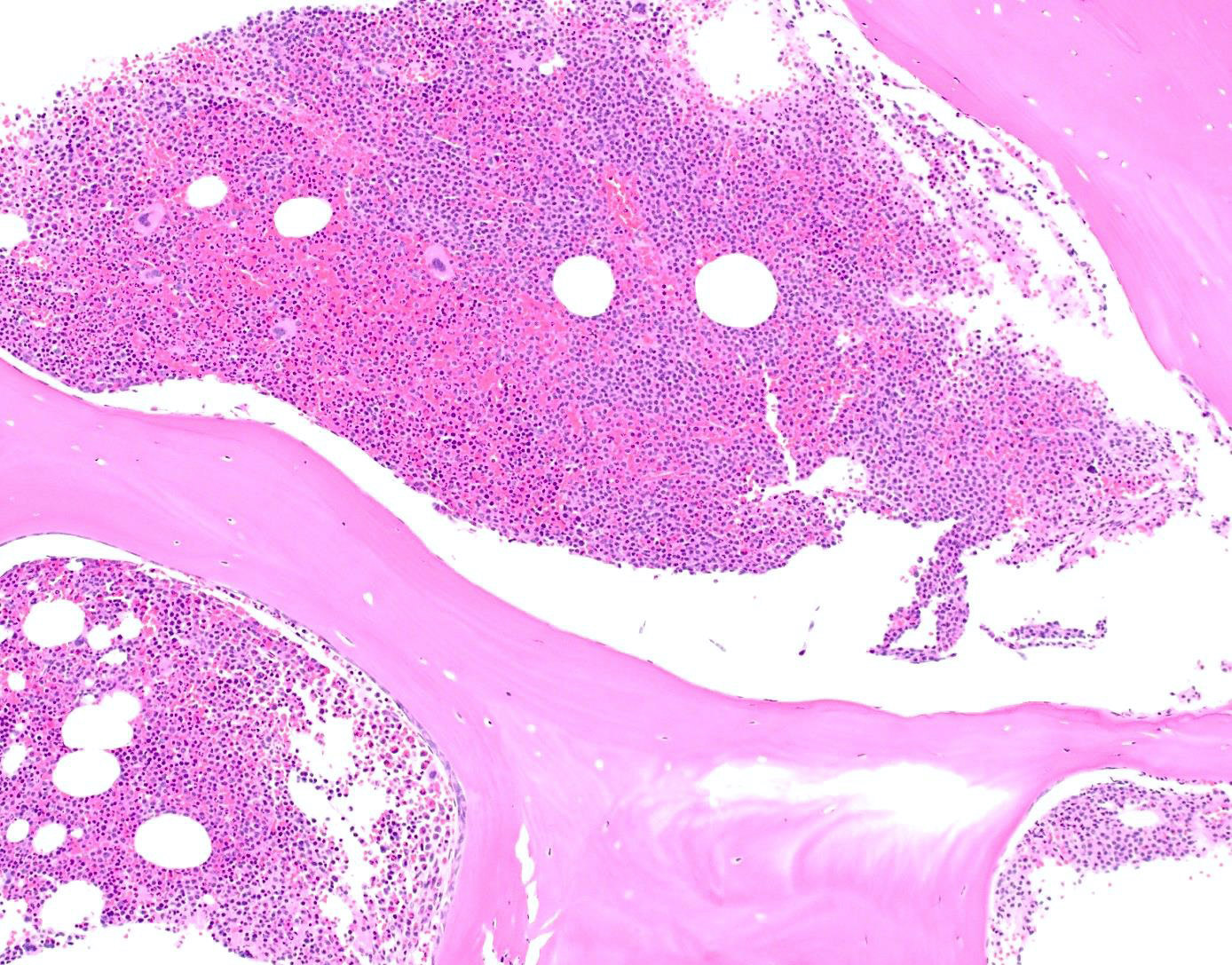
Trephine biopsy
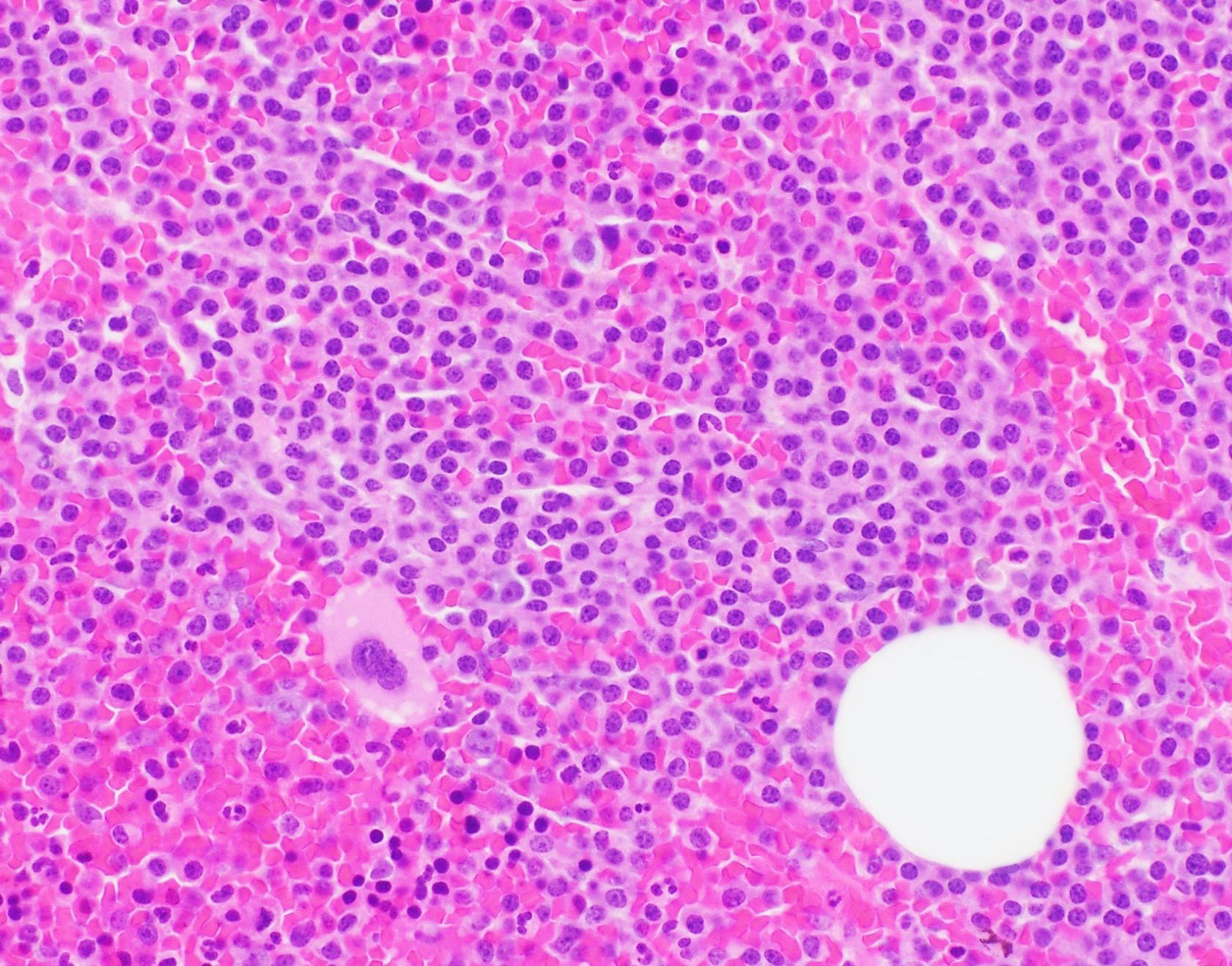
Trephine biopsy
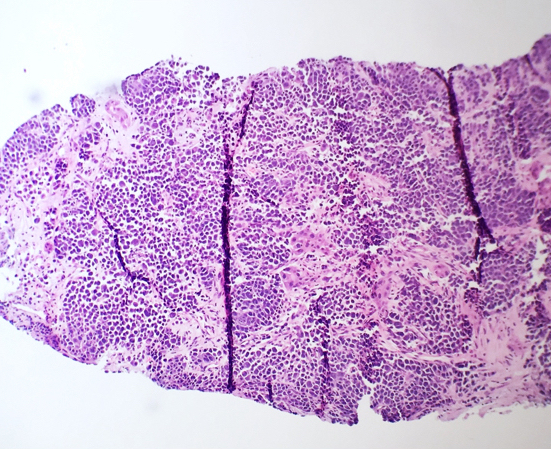
Myeloma with plasmablastic transformation
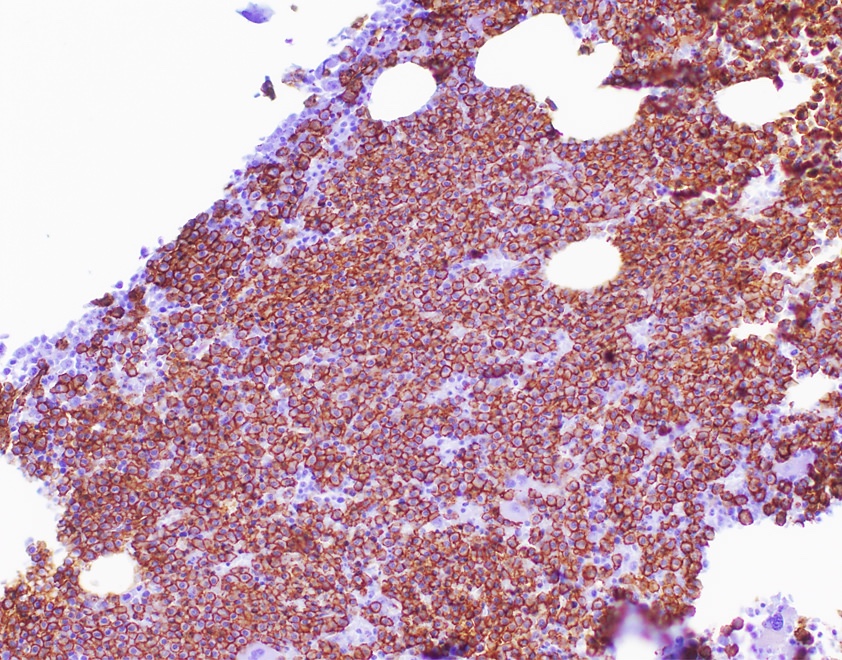
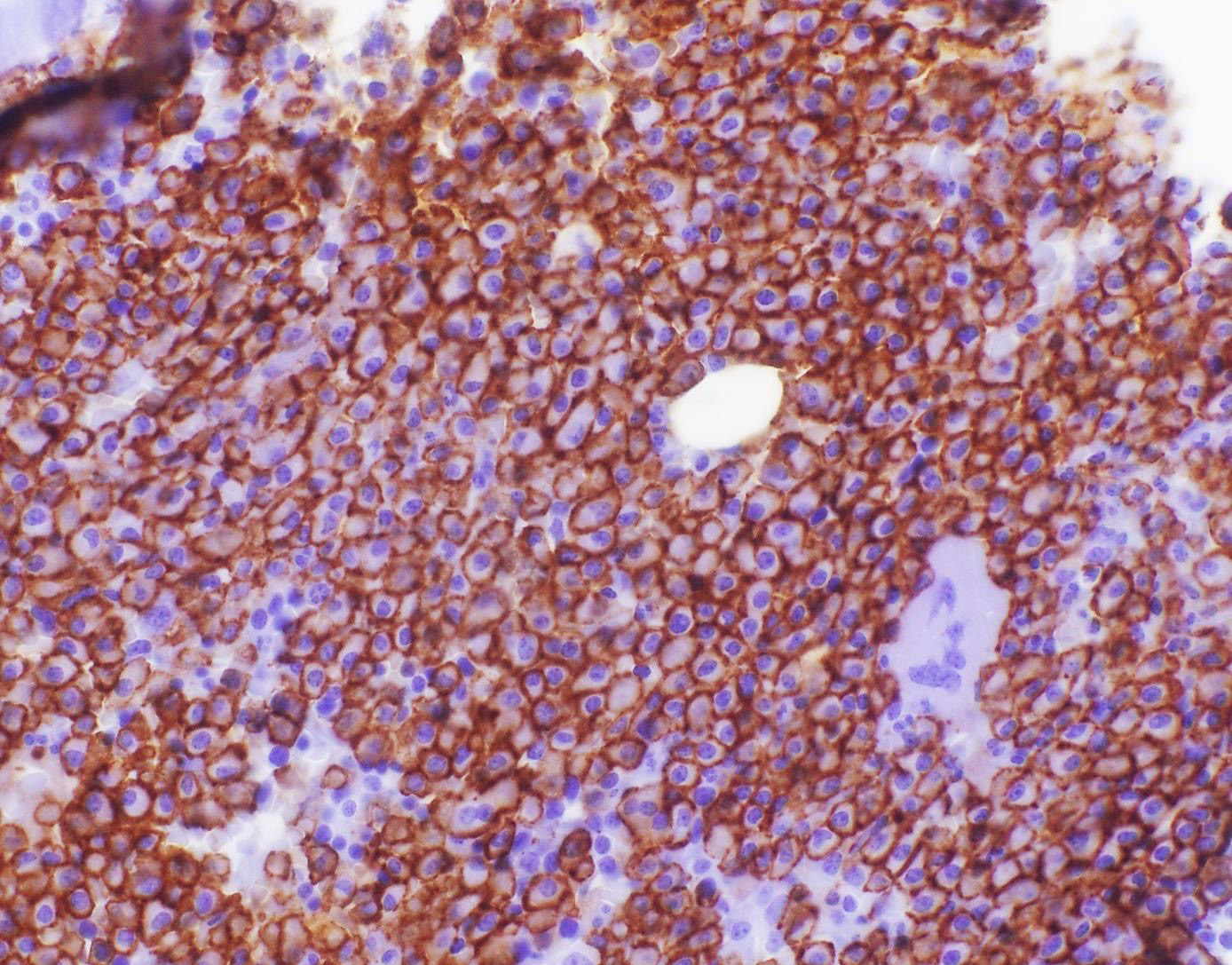
CD138
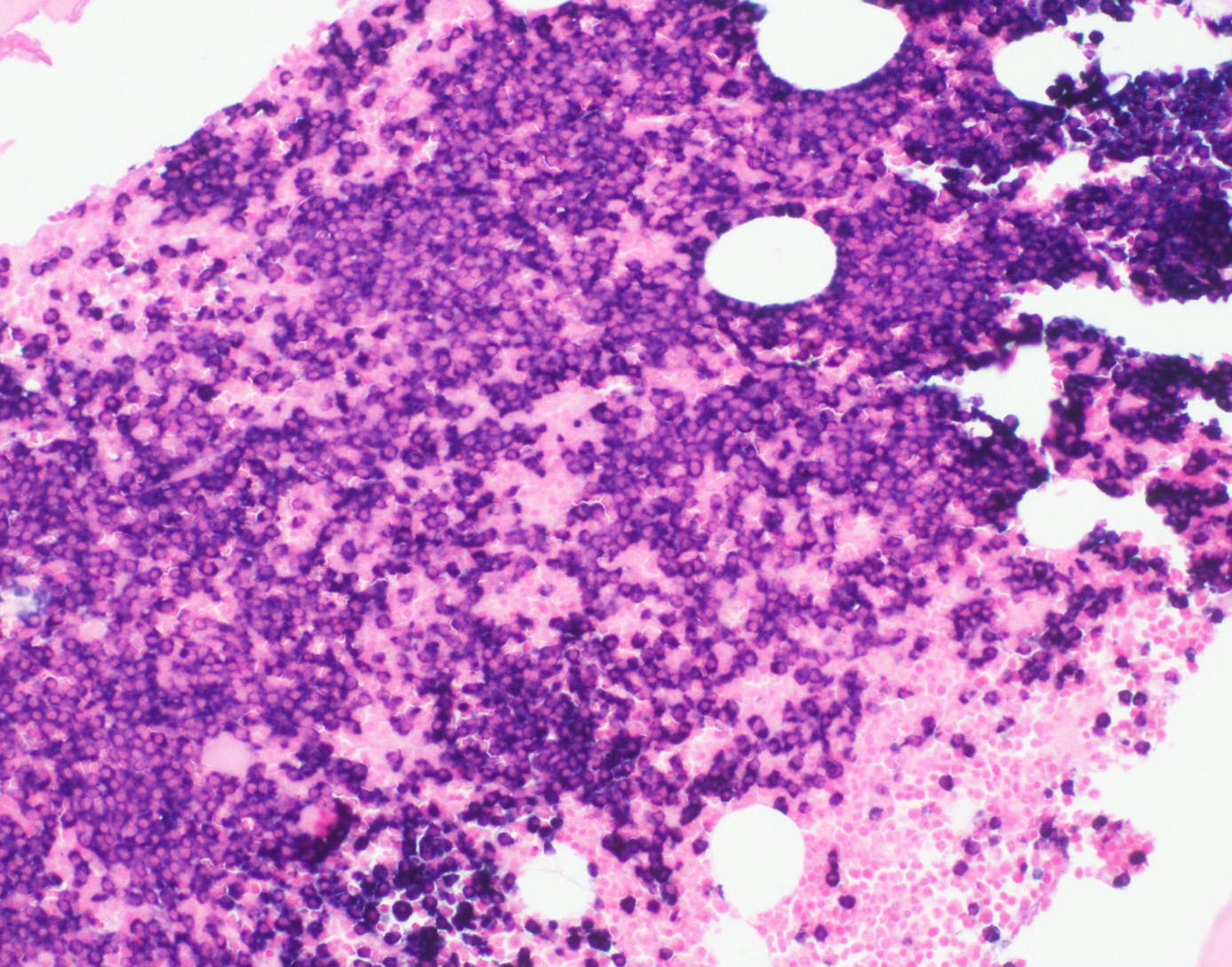
In situ hybridization for kappa
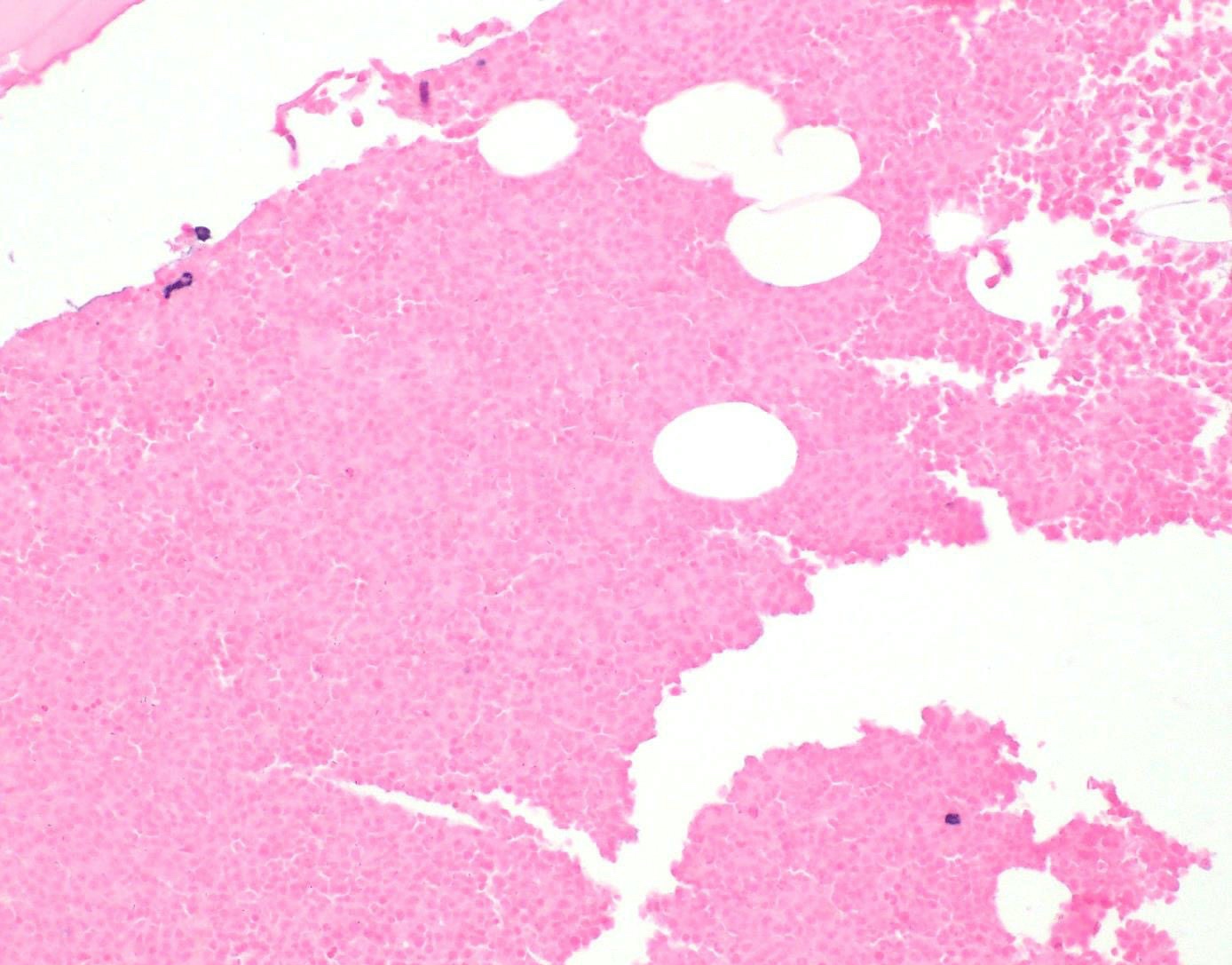
In situ hybridization for lambda
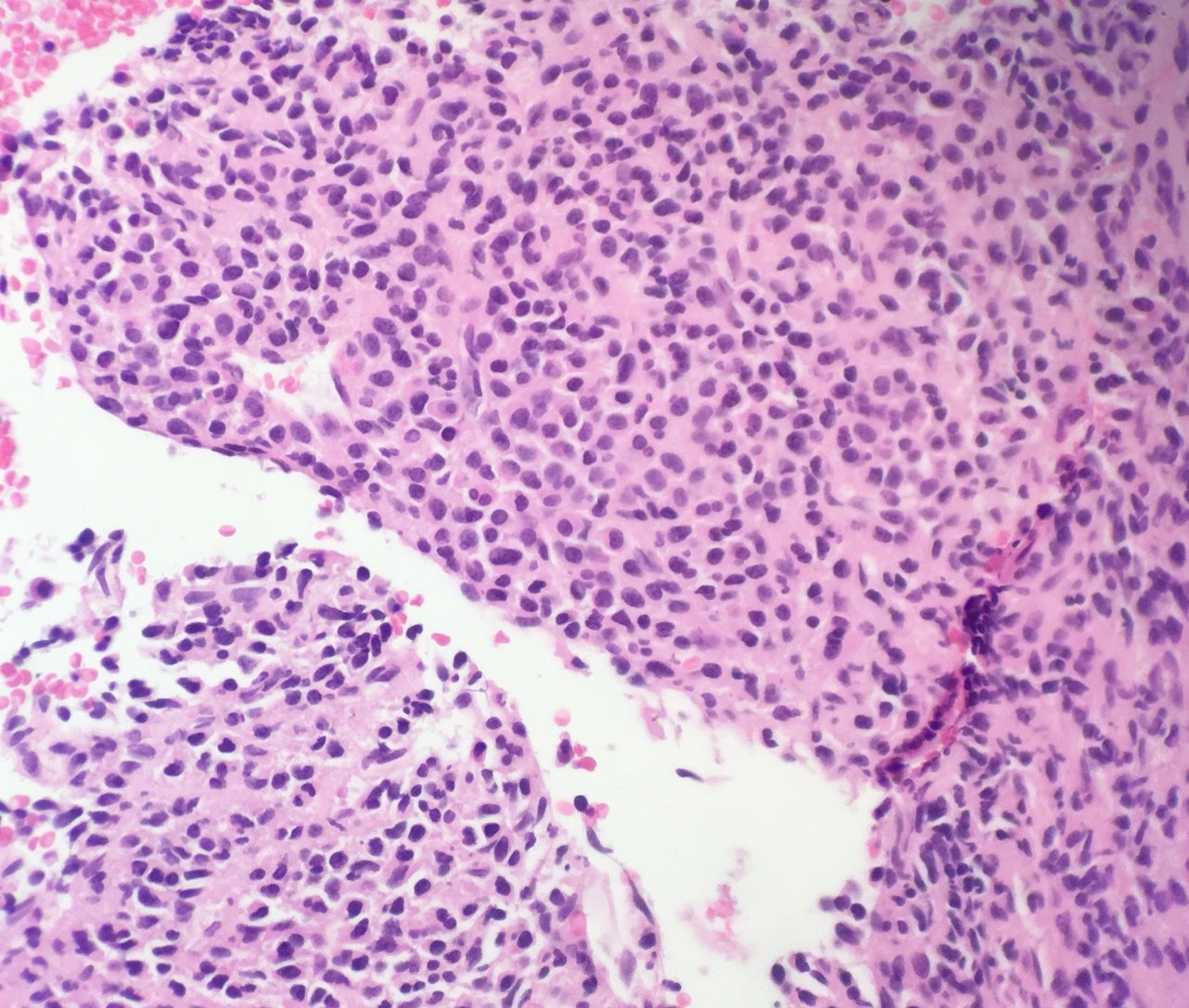
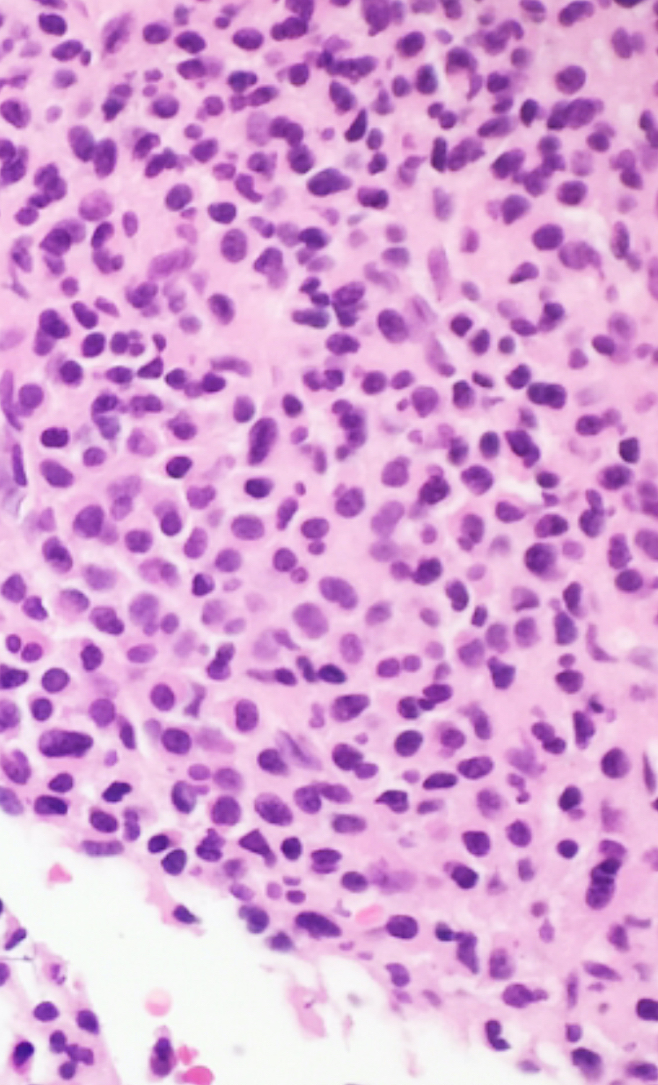
Myeloma involving bone marrow
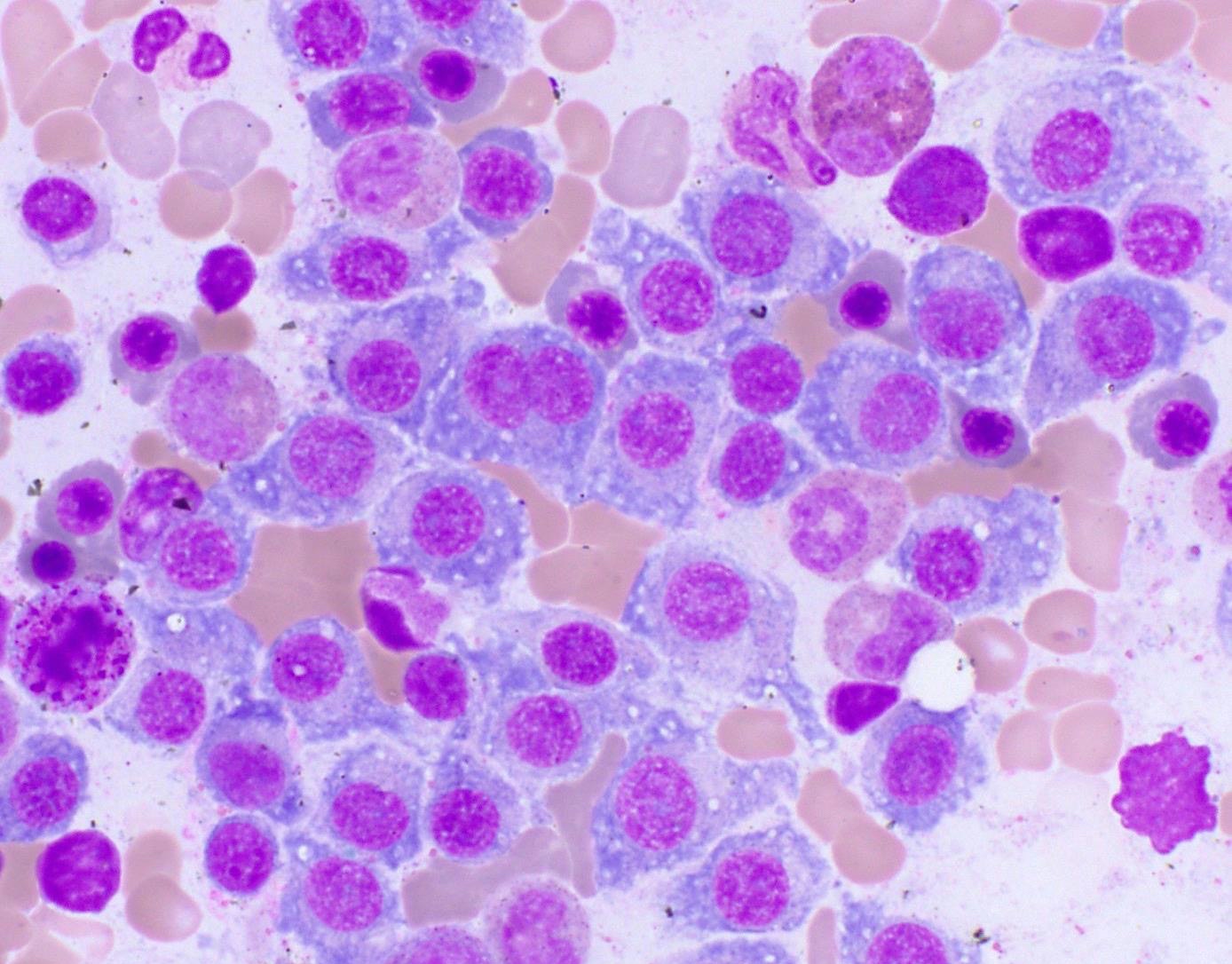
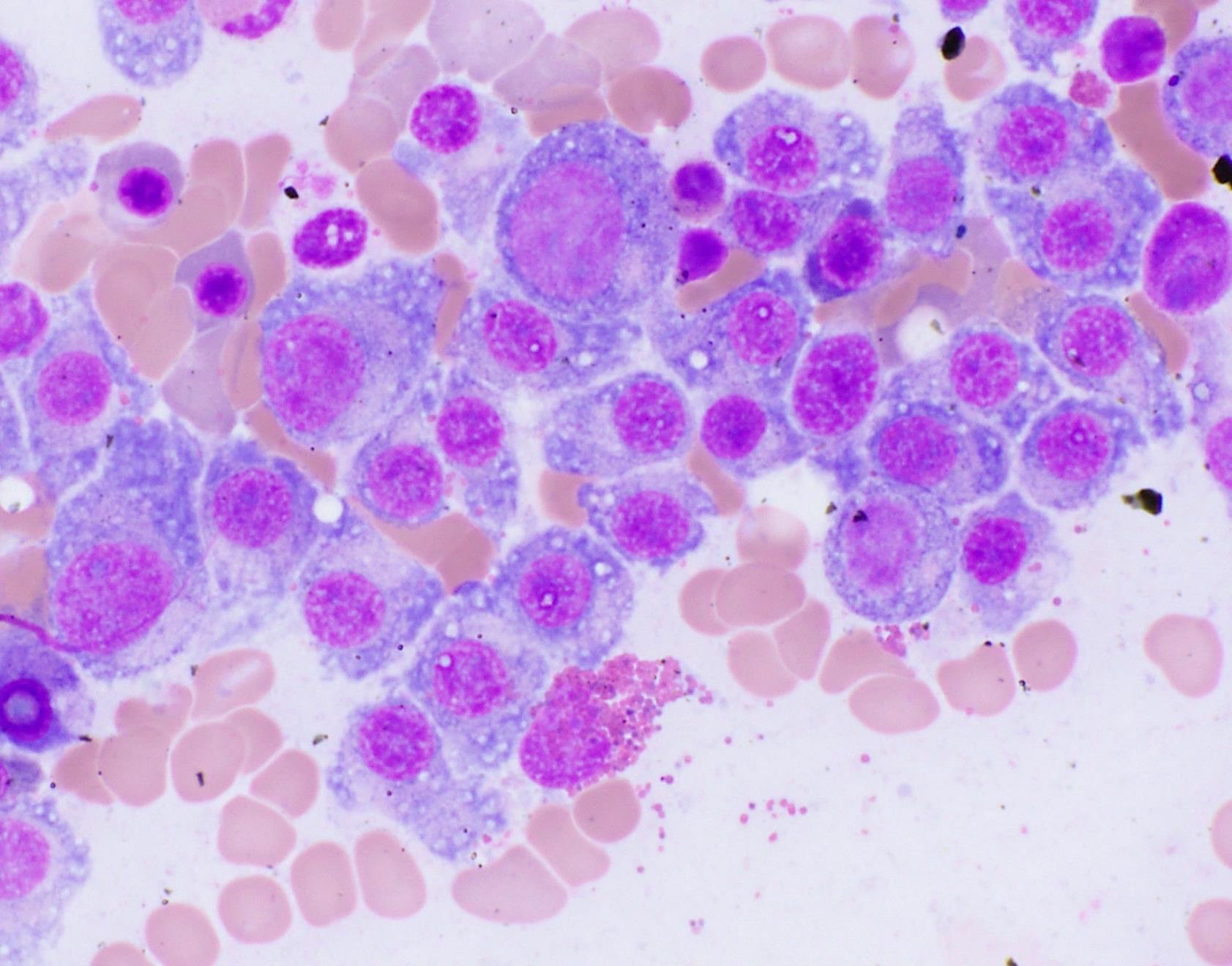
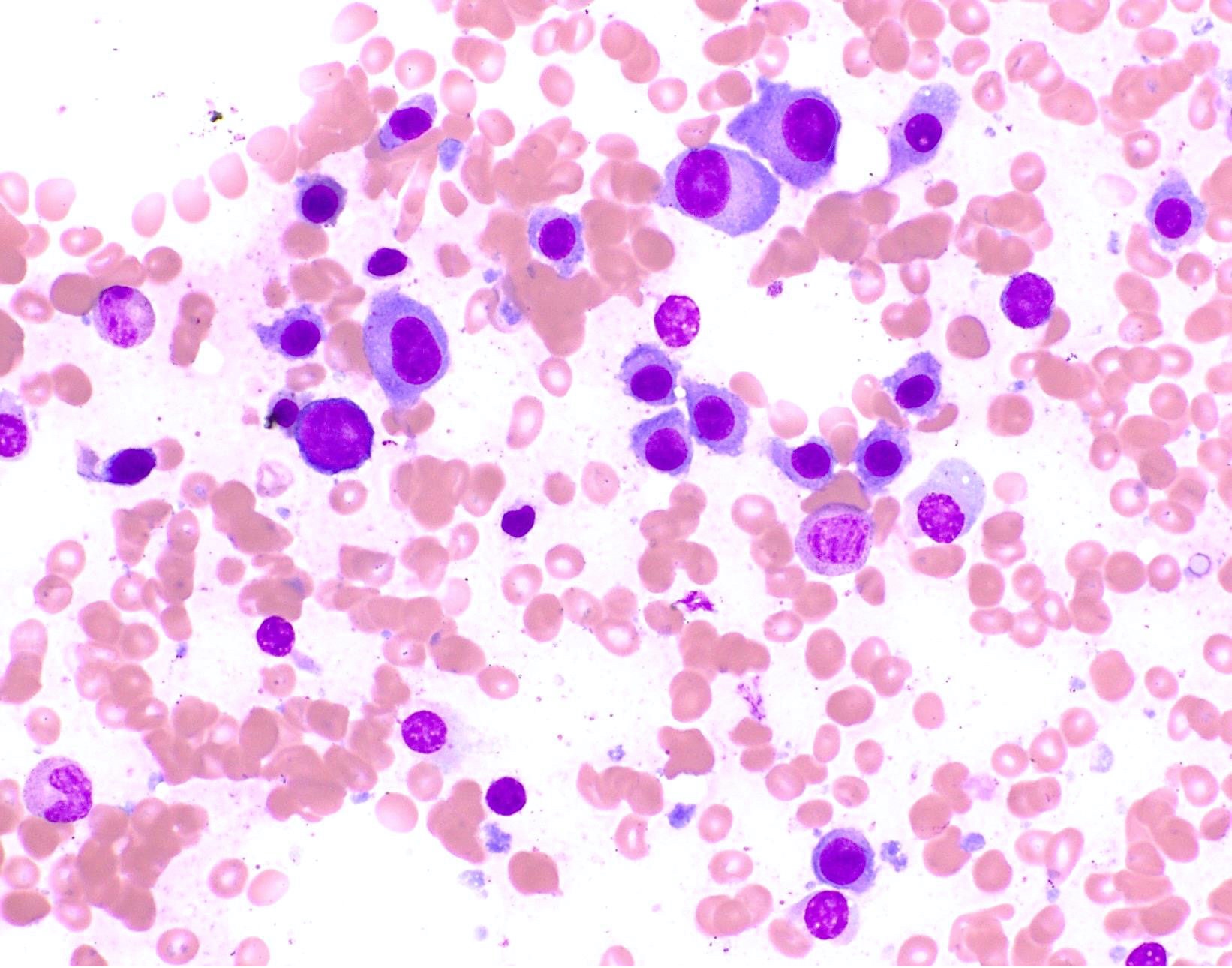
Bone marrow touch preparation
Images hosted on other servers:

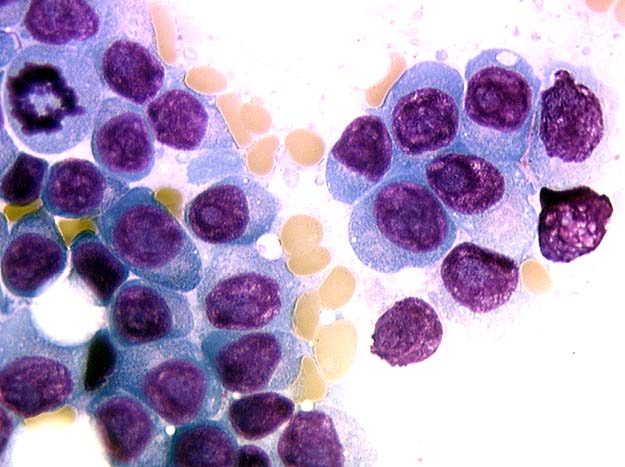
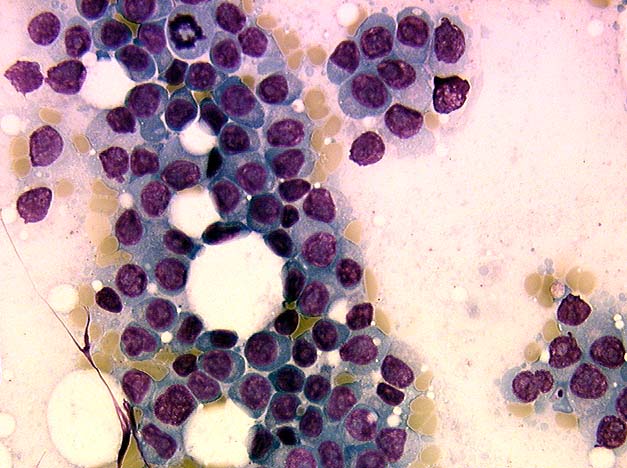
Bone marrow aspirate has plasmacytoid cells
Images hosted on other servers:

Plasmacytoma involving thyroid

Myeloma with pleomorphic features involving lymph node

Plasmacytoma
Myeloma with
pleomorphic features
diffusely involving
bone marrow
- Cytology can assess plasma cell morphology (e.g., mature, immature, plasmablastic) but number of plasma cells present may vary substantially from the core biopsy
- Mature plasma cells: oval with abundant basophilic cytoplasm, perinuclear hof, round eccentric nuclei, clock face chromatin and indiscernible nucleoli
- Immature plasma cells: higher nuclear to cytoplasmic ratio, more abundant cytoplasm and hof region compared to plasmablastic, more dispersed chromatin, often prominent nucleoli
- Plasmablastic: less abundant cytoplasm with little or no hof region, fine reticular chromatin, large nucleus (> 10 microns) or large nucleolus (> 2 microns) (Blood 1998;91:2501)
- Pleomorphic: multinucleated, polylobated
- Rare cases may have small, lymphoid appearing plasma cells or plasma cells with marked nuclear lobation
- Immature or pleomorphic features are rare in reactive plasma cell proliferations
- Morphologic features:
- Mott cells / morula cells: multiple grape-like cytoplasmic inclusions comprised of crystalized immunoglobulin
- Russell bodies: hyaline intracytoplasmic inclusions
- Flame cells: vermillion staining glycogen rich IgA in cytoplasmic projections (American Society of Hematology: Flame Cells in Multiple Myeloma [Accessed 26 May 2022])
- Pseudo-Gaucher cells / thesaurocytes: overstuffed fibrils (J Clin Pathol 1976;29:916)
- Cytoplasmic crystals: occasional in myeloma, common in adult Fanconi syndrome (Am J Clin Pathol 1983;80:224)
- Dutcher body: pale staining immunoglobulin filled cytoplasm invaginating into the nucleus and appearing as an intranuclear inclusion, single and usually large, more common in IgA myeloma
Contributed by Genevieve M. Crane, M.D., Ph.D.
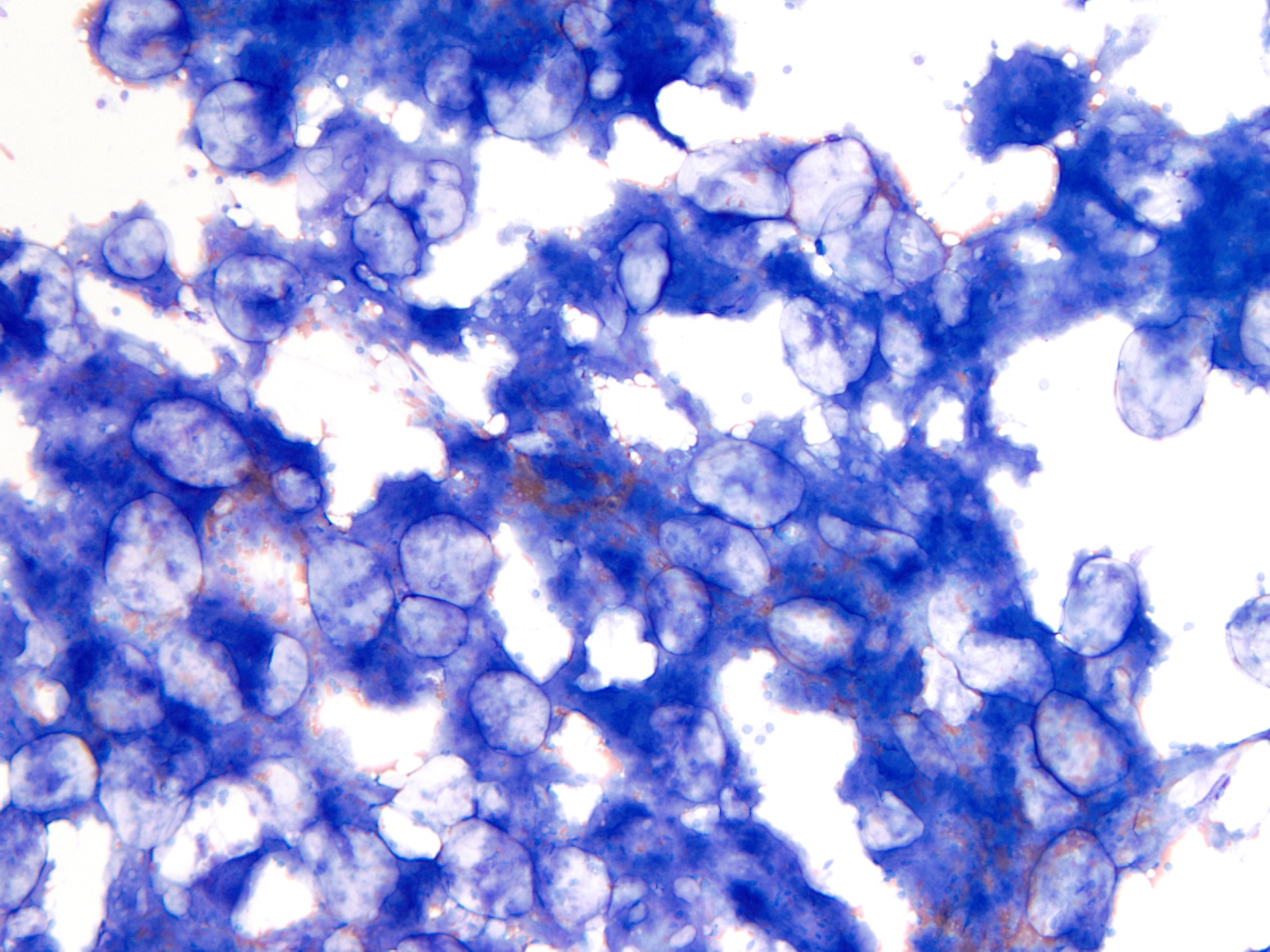
Extensive amyloid, aspirate
- Rouleaux formation: erythrocytes resemble stacked coins; related to quantity and type of M protein, not specific and may be caused by alterations in other plasma proteins (Biophys J 2000;78:2470, American Society of Hematology: Rouleaux Formation [Accessed 26 May 2022])
- Leukoerythroblastic reaction can occur with extensive marrow involvement
- Circulating plasma cells can be seen in ~15% of cases, usually small numbers not meeting criteria for plasma cell leukemia (> 2 x 109/L or 20% of the leukocyte count)
Contributed by Genevieve M. Crane, M.D., Ph.D.
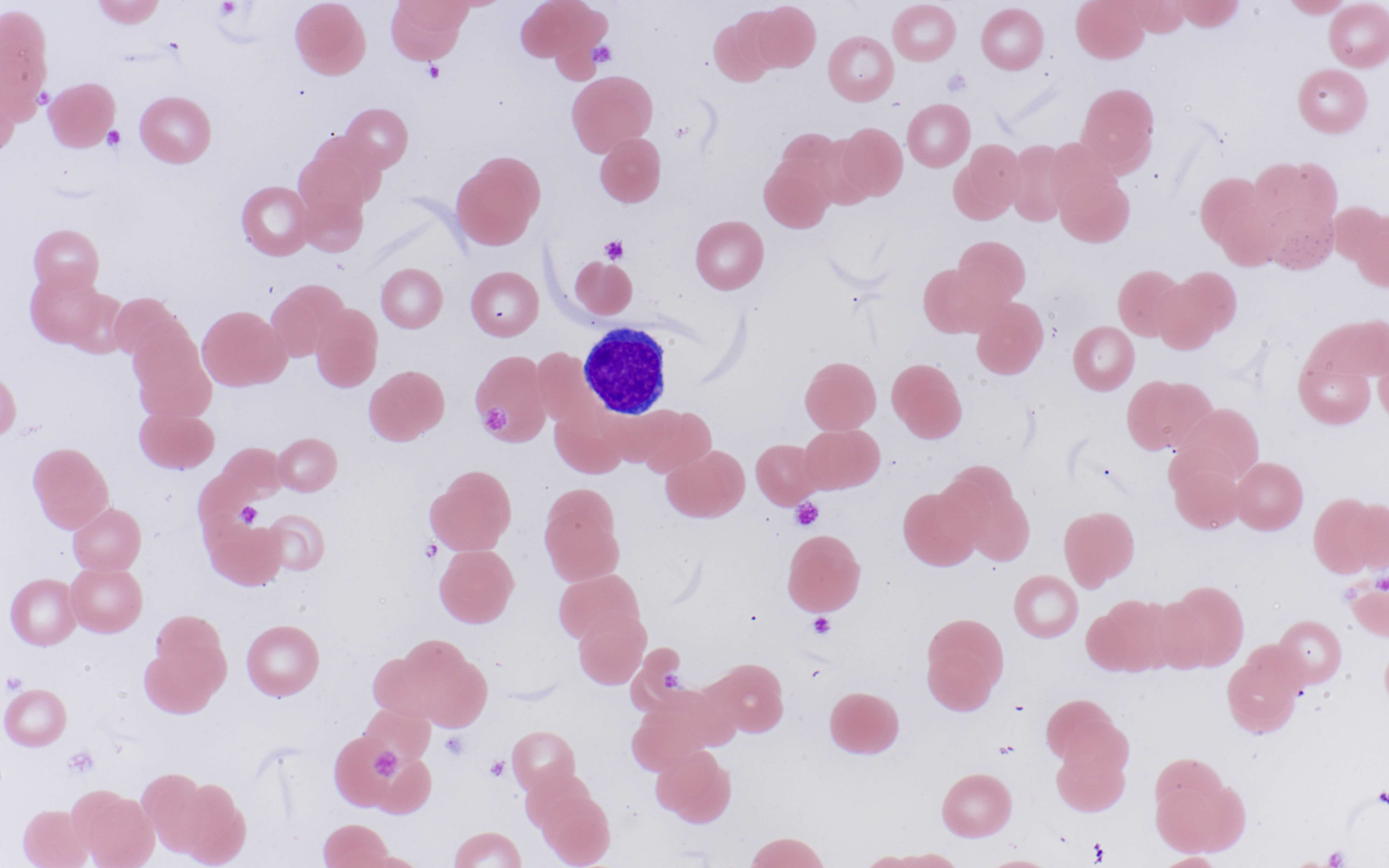
Peripheral blood rouleaux

Rouleaux formation
Images hosted on other servers:


Peripheral blood shows plasma cells (right: with blastic features)
- CD38, CD138, VS38c
- MUM1, EMA, CD79a (variable)
- May have aberrant expression of CD56 (75 - 80%), CD200 (60 - 75%), CD28 (~40%), CD117 / KIT (20 - 35%), CD20 (10 - 20%), CD52 (8 - 14%), CD10 and occasional myeloid or monocytic markers; may correlate with cytogenetics in some cases (Leuk Lymphoma 2015;56:426, Cytometry B Clin Cytom 2016;90:61)
- Monoclonal light chain
- Cyclin D1 positive in presence of t(11;14)(q13;q32), variable expression levels with hyperdiploidy or 11q13 amplification (Blood 2004;104:1120)
- t(11;14) translocation associated with expression of B cell markers on the clonal plasma cells including CD19, CD20, CD79a (Leuk Res 2013;37:1251)
- Increased MYC expression may be seen and potentially distinguish from MGUS (Am J Surg Pathol 2014;38:776)
- Monotypic cytoplasmic Ig and usually lack surface light chain
- Express CD38, CD138, often CD56+ or CD117+; may have partial CD45, usually negative for CD20, CD19 and CD10 (Cytometry B Clin Cytom 2016;90:61)
- Exception in myeloma with t(11:14) where plasma cells more often express B cell markers including CD19 and CD20
Contributed by Genevieve M. Crane, M.D., Ph.D.
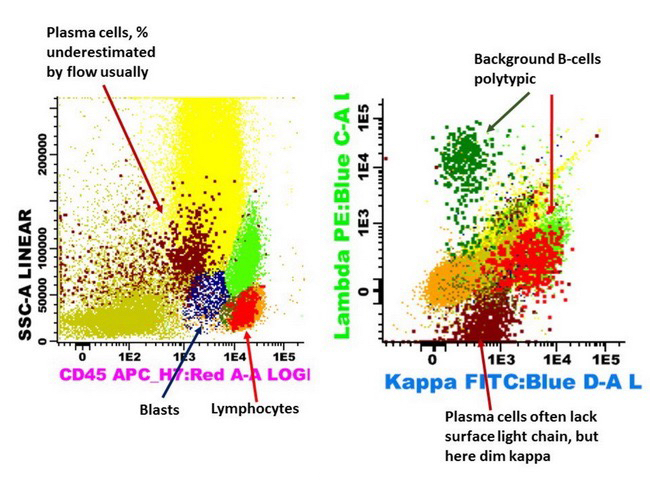
Dim kappa
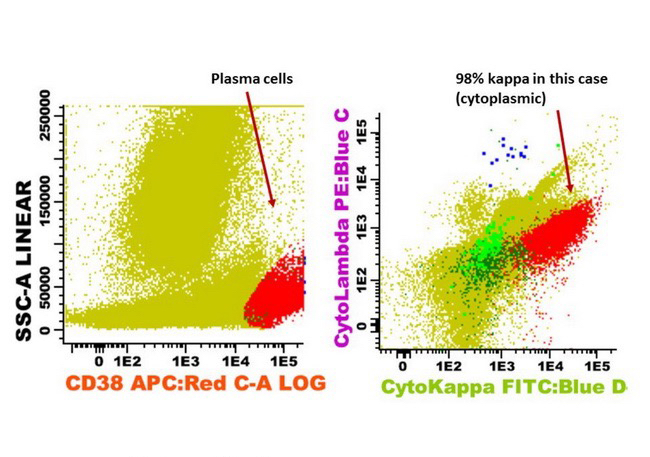
Strong cytoplasmic expression
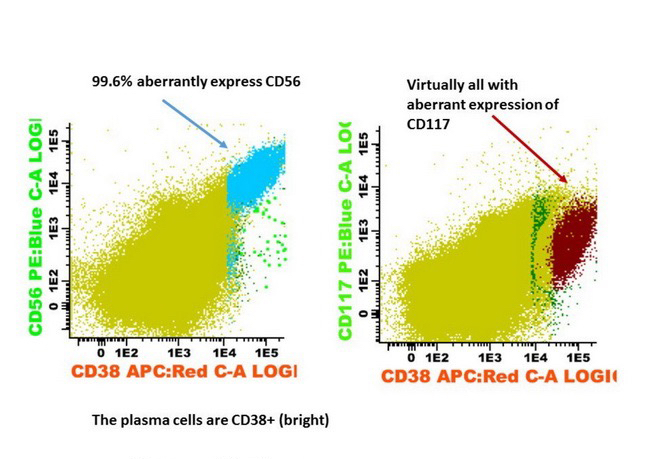
Neoplastic plasma cells
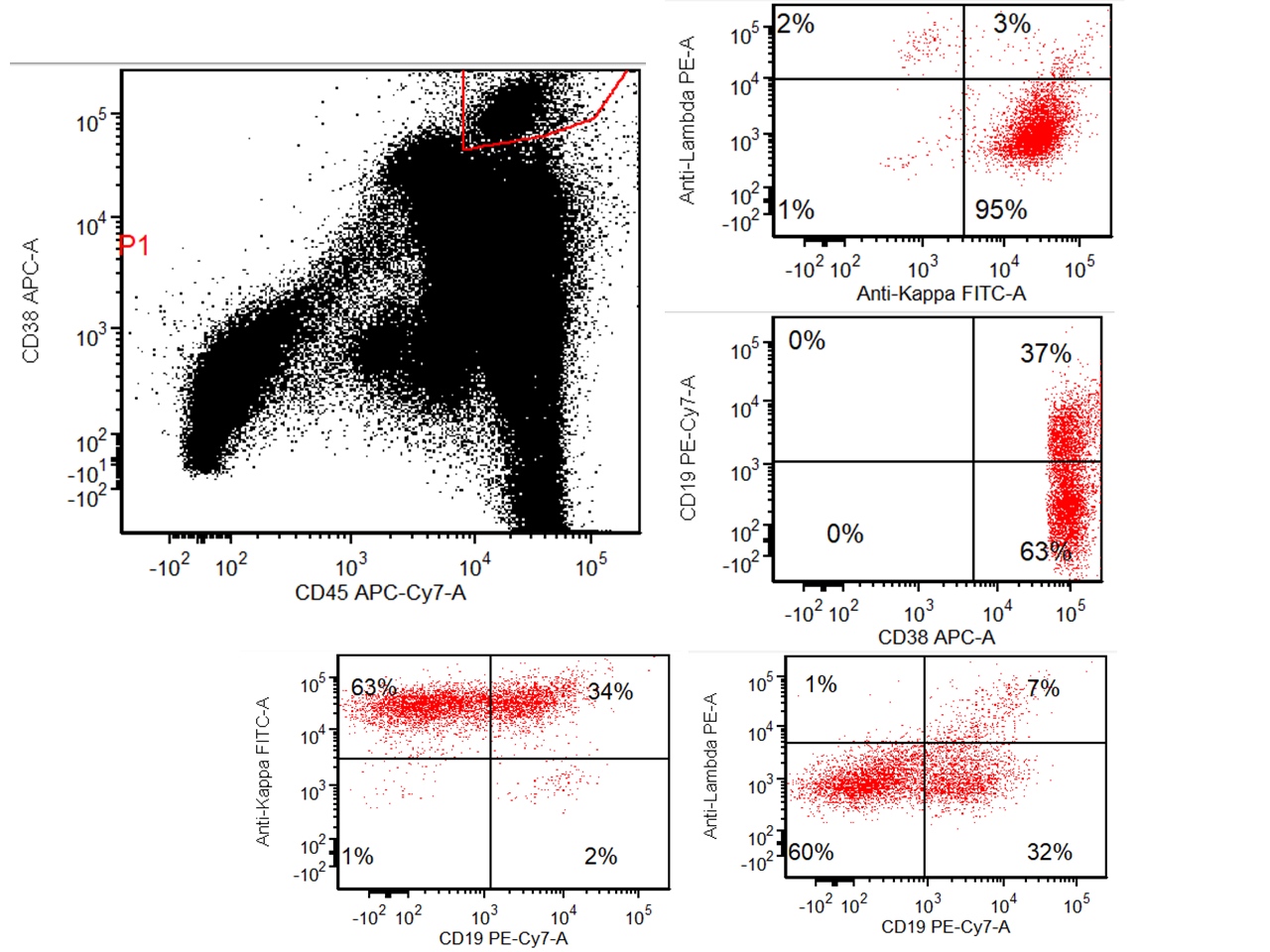
Myeloma with t(11;14) and partial CD19
Images hosted on other servers:

Prominent monotypic
pattern (lambda,
with minimal kappa)
- Prominent rough endoplasmic reticulum, often with Russell bodies budding off of it (Acta Haematol 1977;58:173)
- Immunoglobulin heavy and light chain genes are clonally rearranged with a high load of IGHV gene somatic hypermutation; abnormalities in IGH rearrangement may be seen with light chain only disease (Blood 2004;103:3869)
- Cytogenetics: abnormalities by conventional cytogenetics seen in 33% of cases but identified by FISH in > 90%, including trisomies, deletions and translocations (Blood Cancer J 2015;5:e365)
- FISH studies often performed by enriching the plasma cell fraction using magnetic cell sorting (MACS) and fluorescence activated cell sorting (FACS) to increase detection of cytogenetic abnormalities in interphase cells (Am J Clin Pathol 2011;136:712)
- IGH (14q32) translocations in 40% of tumors, recurrent oncogenes:
- t(4;14) FGFR3 / MMSET (NSD2) (4p16.3); 15%
- t(6;14) cyclin D3 (6p21); 4%
- t(11;14) cyclin D1 (11q13); 20%
- t(14;16) MAF (16q23); 4%
- t(14;20) MAFB (20q11); 1%
- MAFA (8q24)
- Cyclin D2 (12p13)
- Tumors without 1 of the frequent IGH are more often hyperdiploid with gains of odd numbered chromosomes (3, 5, 7, 9, 11, 15, 19, 21); 50%
- Monosomy or partial deletion of chromosome 13 (13q14) found in nearly 50% by FISH
- MYC rearrangements in nearly half of tumors
- Activating mutations of KRAS, NRAS or BRAF in 40% of tumors
- Other: TP53 deletion or mutation, gain of 1q, loss of 1p, NF kappa B pathway activation, inactivation of CDKN2C, RB1, FAM46C, DIS3 and DNA methylation changes
Video on myeloma MRD

Plasma cell neoplasms

Podcast on SPEP

Introduction to bone marrow interpretation
- Bone marrow aspirate smears, touch imprints, core biopsy and clot section with peripheral smear:
- Plasma cell myeloma (see comment)
- Normocytic anemia, rouleaux formation
- Comment: The patient is a 65 year old man who recently presented with back pain, anemia and hypercalcemia. Xrays demonstrated evidence of lytic bone lesions and serum protein analysis demonstrated an IgG kappa M protein. The bone marrow shows sheets of atypical plasma cells in a hypercellular bone marrow overall comprising 80% of the intertrabecular space. Flow cytometry as reported separately, demonstrates a CD56+, CD19- kappa restricted plasma cell population. Please correlate with forthcoming cytogenetics and myeloma FISH panel, which will be reported as an addendum.
- Mature B cell lymphoma with extensive plasmacytic differentiation (marginal zone or lymphoplasmacytic lymphoma):
- Monoclonal gammopathy of undetermined significance (MGUS):
- No myeloma defining events, < 10% bone marrow plasma cells and < 3 g/dL M protein
- Plasmablastic lymphoma:
- Differential consideration from myeloma with plasmablastic features with presence of associated lymphadenopathy, oral mass in absence of myeloma defining signs helpful to distinguish plasmablastic lymphoma
- EBER more often positive in PBL but can be seen in myeloma or plasmacytoma (Histopathology 2015;67:225, J Clin Pathol 2017;70:775)
- Primary effusion lymphoma:
- May express plasma cell markers, particularly MUM1 but universally HHV8 positive, majority coexpress EBER (J Cutan Pathol 2014;41:928)
- Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS) syndrome:
- Rare disorder, VEGF correlates with disease activity, predominantly lambda, if other symptoms are not recognized may be more likely to be diagnosed as MGUS (Am J Hematol 2019;94:812)
- Reactive plasmacytosis:
- Rare cases may reach up to 30% plasma cells, will be polytypic
- Solitary plasmacytoma of bone:
- Focal bone symptoms, single lesion on imaging, may have M spike, no bone marrow plasmacytosis (< 10% clonal plasma cells in bone marrow), no CRAB (hypercalcemia, renal failure / insufficiency, anemia, lytic bone lesions)
- Systemic AL amyloidosis:
- Evidence of a clonal plasma cell disorder, demonstration of amyloid (Congo red) and that the amyloid is light chain related, amyloid can occur in association with myeloma
- Telangiectasias, elevated erythropoietin and erythrocytosis, monoclonal gammopathy, perinephric fluid collection and intrapulmonary shunting (TEMPI) syndrome:
- Very rare, identified by constellation of symptoms, responds to plasma cell directed therapy (Blood 2020;135:1199)
- Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th Edition, 2017, Leukemia 2021;35:18, Hematology Am Soc Hematol Educ Program 2020;2020:264, Leukemia 2020;34:985, Curr Hematol Malig Rep 2016;11:111, Nat Rev Dis Primers 2017;3:17046, Nat Rev Clin Oncol 2021;18:71, Acta Med Acad 2019;48:57, Morphologie 2015;99:38, Nat Rev Cancer 2012;12:335, Lancet Oncol 2019;20:e302
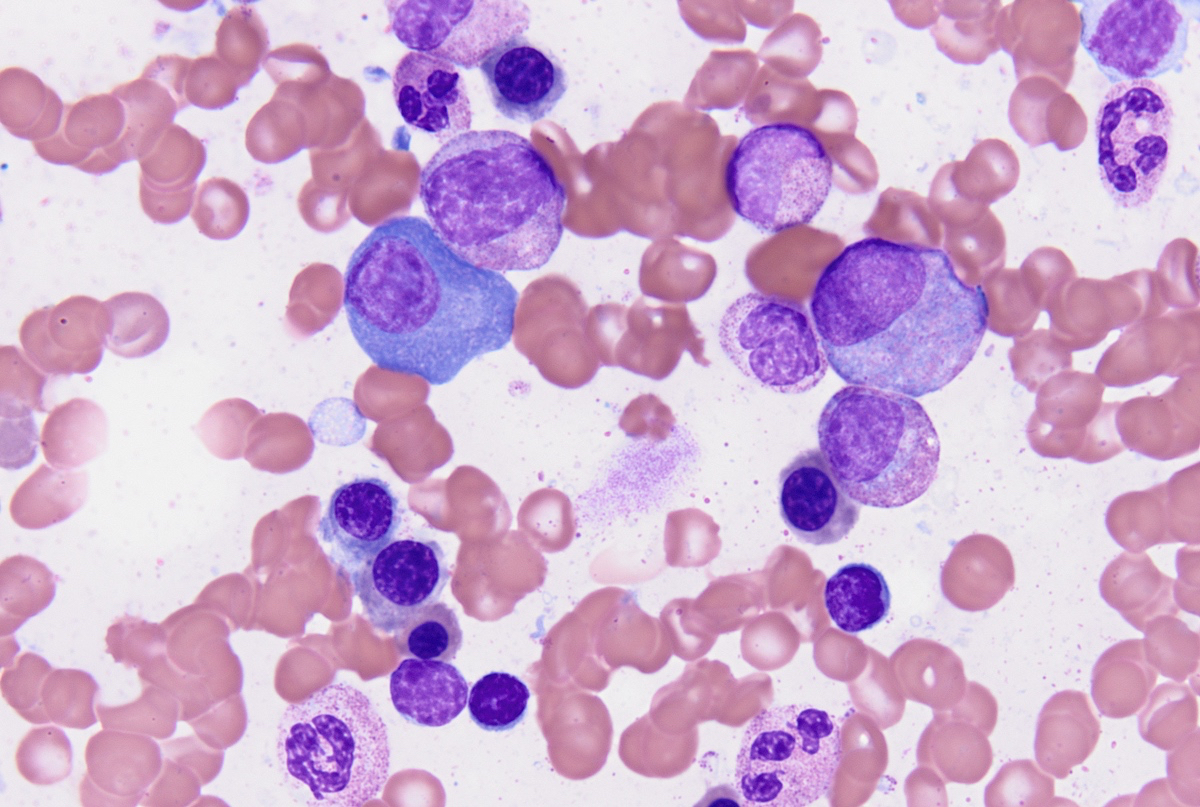
Which of the following findings would be compatible with a diagnosis of smoldering myeloma?
- 70% plasma cells in bone marrow
- Free light chain ratio is 0.01
- IgG lambda M protein in serum of 3.5 g/dL
- 1 osteolytic lesion on skeletal radiography
- Serum calcium of 11.2 mg/dL
Comment Here
Reference: Plasma cell myeloma (multiple myeloma)
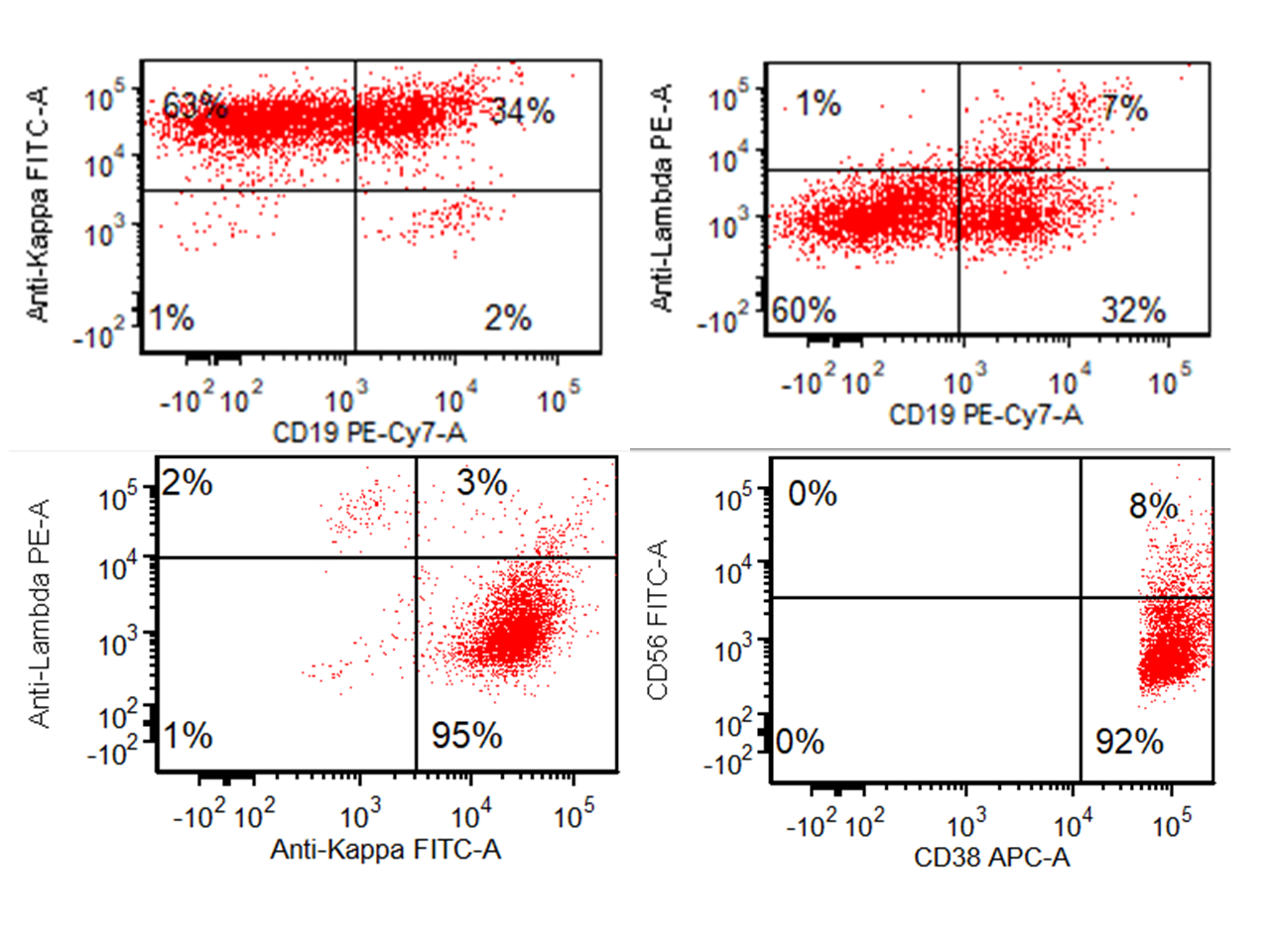
Clonal plasma cells in the setting of multiple myeloma are typically negative for CD19. What cytogenetic abnormality is typically associated with CD19 or CD20 positive plasma cells in the setting of a plasma cell neoplasm?
- Deletion 13
- Hypodiploidy
- t(4:14)
- t(11;14)
- t(14:16)
Comment Here
Reference: Plasma cell myeloma (multiple myeloma)

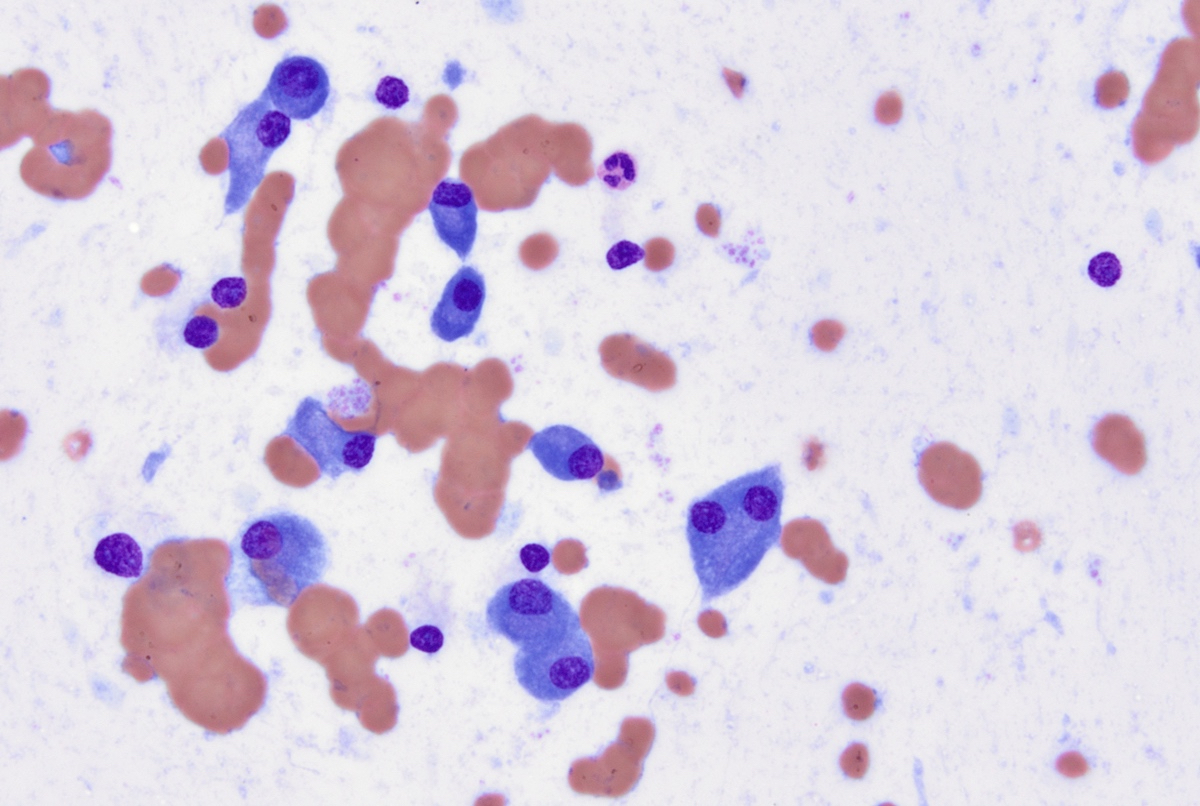
Which of the following findings would most strongly favor the presence of a neoplastic plasma cell process rather than a reactive plasma cell proliferation?
- Interstitial plasma cells comprising 10% of cellularity
- Plasma cells with prominent clock face chromatin
- Russell bodies
- Scattered immature plasma cells
- Scattered Mott cells with grape-like inclusions
Comment Here
Reference: Plasma cell myeloma (multiple myeloma)
- Solitary lesion of clonal plasma cells that are cytologically, immunophenotypically and genetically similar to plasma cell myeloma
- Solitary plasmacytoma of bone: localized bone tumor consisting of monoclonal plasma cells with no other radiographic lesions and no evidence of other bone marrow involvement
- Extraosseous (extramedullary) plasmacytoma: localized tumor of plasma cells (not in bone)
- Monoclonal plasma cells, no associated clonal B cell population, isolated lesion without evidence of additional bone marrow plasmacytosis
- Solitary plasmacytoma of bone
- Extraosseous (extramedullary) plasmacytoma
- Bone and extramedullary tumors each comprise 3 - 5% of plasma cell neoplasms
- Both are more common in men (65%), median age 55 years
- Extraosseous: 80% in upper respiratory tract (15% spread to cervical lymph nodes)
- Other: GI, lymph nodes (Am J Clin Pathol 2001;115:119), bladder, CNS, breast, thyroid, testis, parotid, skin
- Bone:
- Areas of active hematopoiesis
- In order of decreasing frequency: vertebrae, ribs, skull, pelvis, femur, clavicle, scapula, rare in distal long bones
- Most present with bone pain
- Vertebral lesions may cause cord compression
- Palpable mass due to soft tissue extension
- Up to 2/3 progress to myeloma or additional plasmacytomas; 1/3 remain disease free for > 10 years following local (radiation) control
- 5% may have multiple or recurrent plasmacytomas but no evidence of myeloma
Extraosseous (clinical features depend on site):
- Rhinorrhea, epistaxis, nasal obstruction
- Approximately 15% progress to myeloma, 70% are disease free at 10 years
- 25% have local recurrence, may spread to regional lymph nodes or metastasize to distant sites
- Monoclonal plasma cells, less frequently plasmablastic or anaplastic morphology
- Histologic findings are combined with laboratory and radiographic findings (below) showing absence of additional disease
- M protein in serum or urine (absent or lower than in myeloma):
- Bone: approximately 50%, usually IgG
- Extraosseous: 20%, often small, IgA
- No CRAB (hypercalcemia, renal failure, anemia, additional bone lesions)
- Unlike myeloma, normal levels of uninvolved immunoglobulins
- Monitor free light chain to measure progression
- MRI may be preferred method to exclude additional bone lesions
- Solitary bone lesions are usually purely lytic with a narrow zone of transition to normal bone
- Abnormalities may persist after successful treatment
- Older patients
- Plasmacytoma > 5 cm
- Persistence of M protein following radiotherapy
- Low polyclonal immunoglobulins (also raises concern for myeloma)
- Osteopenia
- 52 year old man with plasmacytoma involving a lymph node that evolved to plasma cell myeloma (N Engl J Med 1950;243:335)
- 67 year old African American man with extramedullary plasmacytoma associated with amyloidoma (Arch Pathol Lab Med 2002;126:969)
- Local radiation therapy
- Similar to myeloma, may contain mature, immature, plasmablastic or anaplastic plasma cells
- Amyloid deposits may appear in extraosseous tumors as pink amorphous material with scattered multinucleated giant cells
Contributed by Genevieve M. Crane, M.D., Ph.D. and Kelly Magliocca, D.D.S., M.P.H.

Plasmacytoma
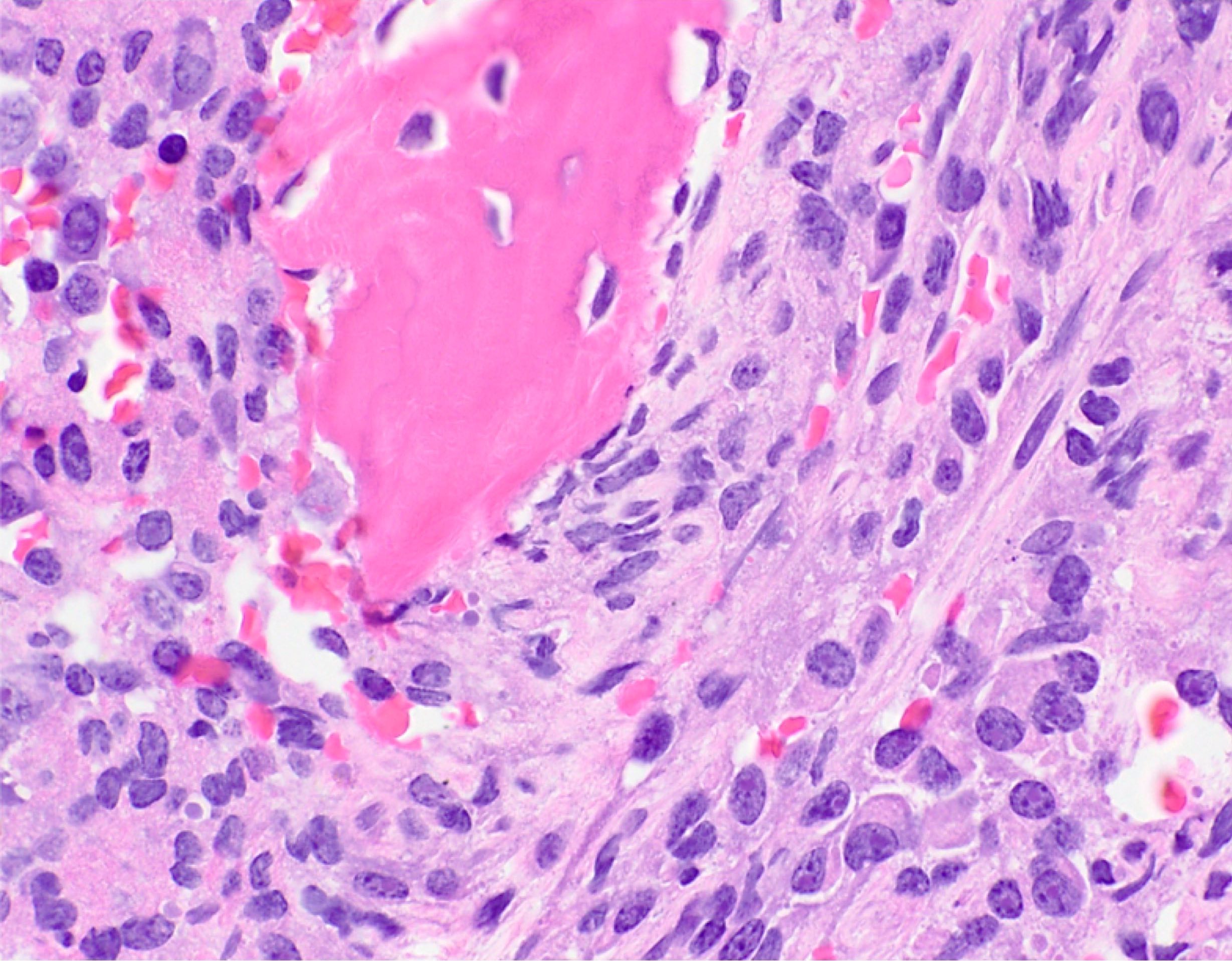
Plasmacytoma of bone with osteolytic destruction
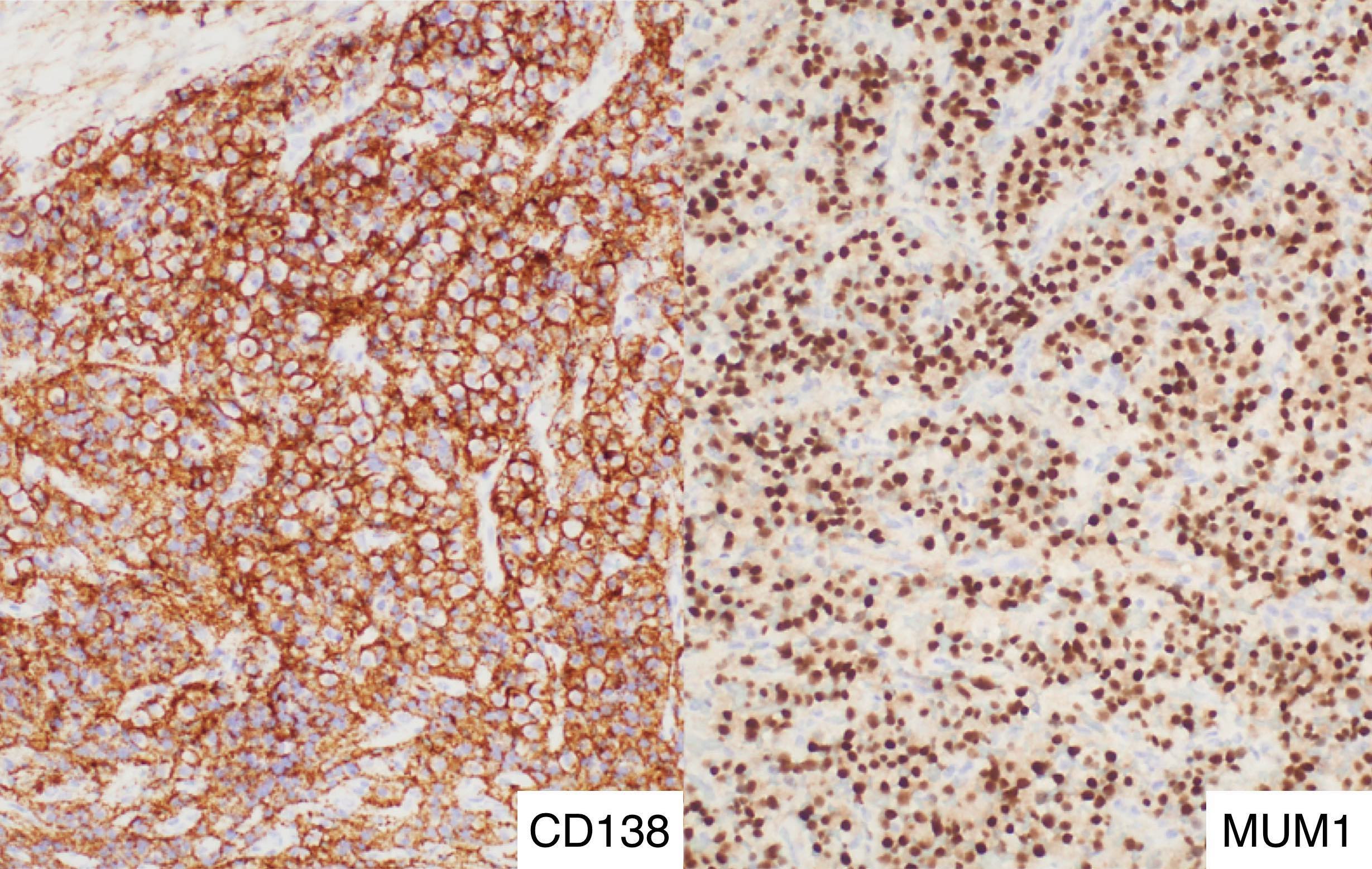
CD138 positive and MUM1 positive

CD79 variable and CD20 negative

Core biopsy section

Core biopsy, kappa and lambda
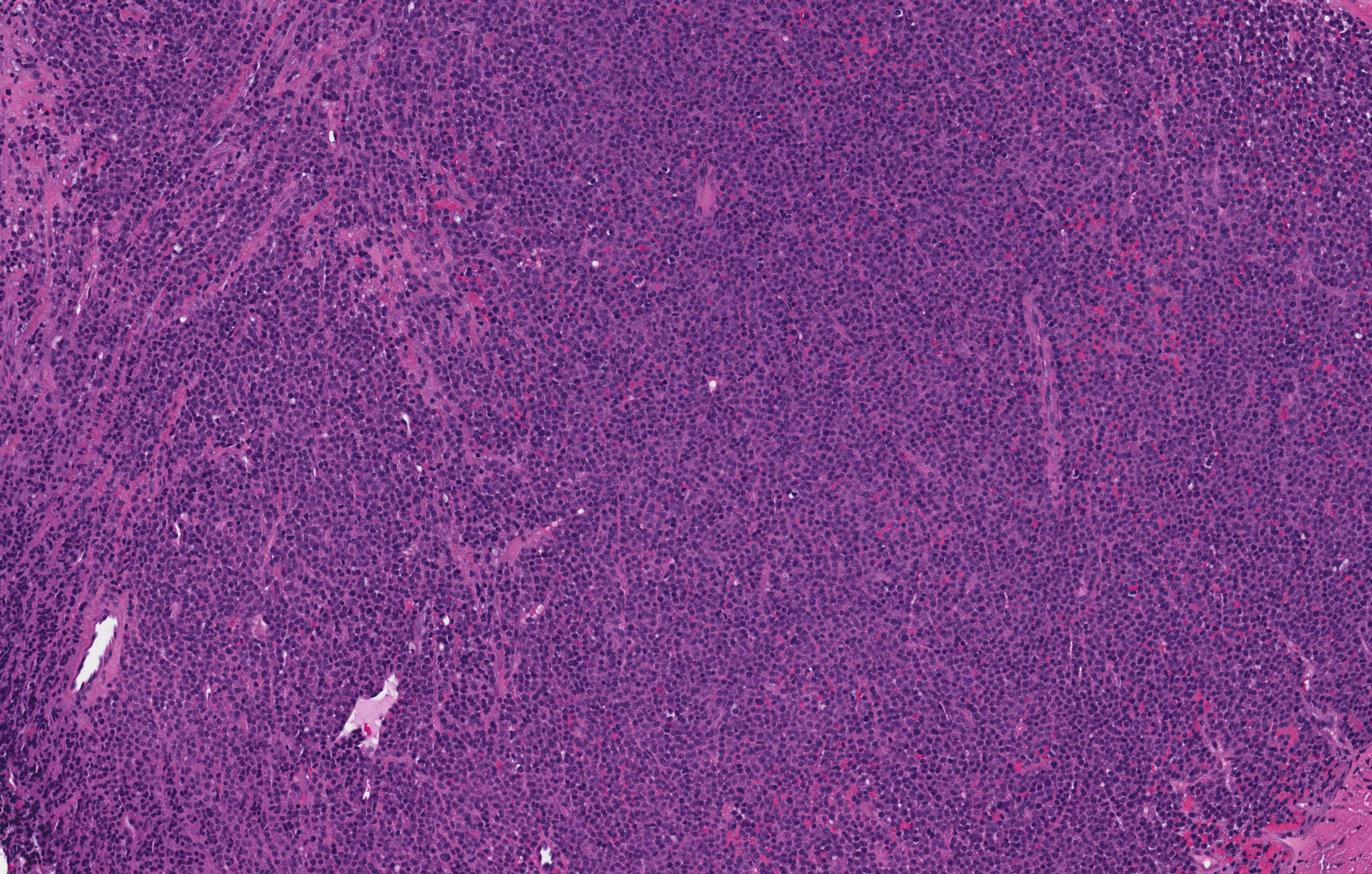
Solitary plasmacytoma of bone
Contributed by Genevieve M. Crane, M.D., Ph.D.

EBV+ tumor
- Similar to myeloma
- Mature plasma cells: oval with abundant basophilic cytoplasm, perinuclear hof, round eccentric nuclei, "clock face" chromatin and indiscernible nucleoli
- Immature plasma cells: higher nuclear / cytoplasmic ratio, more abundant cytoplasm and hof region compared to plasmablastic, more dispersed chromatin, often prominent nucleoli
- Plasmablastic: less abundant cytoplasm with little or no hof region, fine reticular chromatin, large nucleus ( > 10 microns) or large nucleolus ( > 2 microns) (Blood 1998;91:2501)
- Pleomorphic: multinucleated, polylobated
- Rare cases may have small, lymphoid appearing plasma cells or plasma cells with marked nuclear lobation
- Morphologic features:
- Mott cells / morula cells: multiple grape-like cytoplasmic inclusions comprised of crystalized Ig
- Russell bodies: hyaline intracytoplasmic and intranuclear inclusions
- Flame cells: vermillion staining glycogen rich IgA in cytoplasmic projections
- Gaucher-like cells / thesaurocytes: overstuffed fibrils
- Cytoplasmic crystals: occasional in myeloma, common in adult Fanconi syndrome
- Dutcher bodies: pale staining nuclear inclusions, single and usually large, more common in IgA myeloma
Contributed by Genevieve M. Crane, M.D., Ph.D.
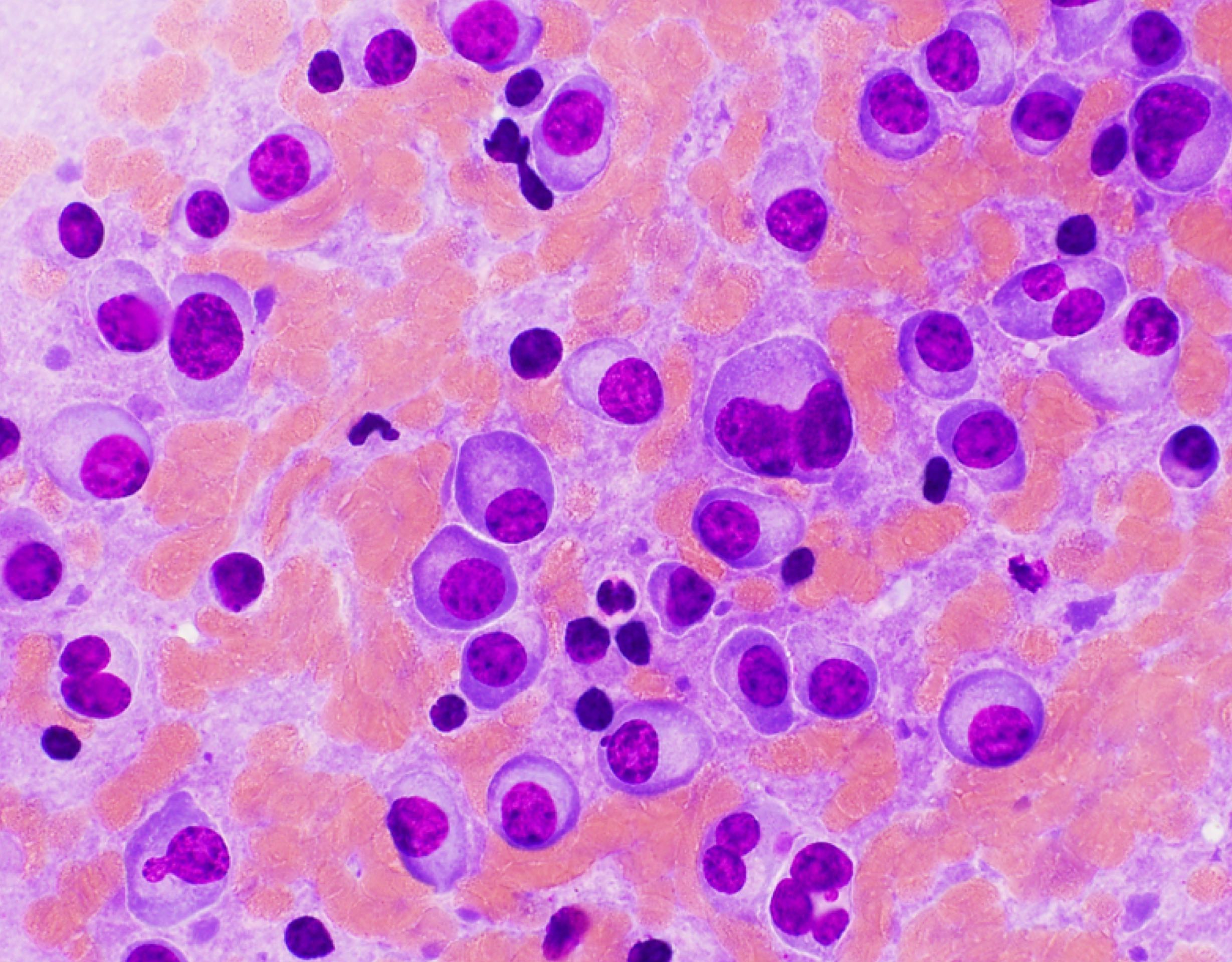
Plasmacytoma touch preparation
- Similar to plasma cell myeloma
- In bone, genetics similar to plasma cell myeloma, extraosseous not extensively studied
- Cutaneous or GI plasmacytoma:
- Particularly difficult to differentiate from marginal zone lymphoma, may not be possible
- Large cell lymphoma:
- With immunoblastic or plasmablastic features
- Marginal zone lymphoma, lymphoplasmacytic lymphoma:
- May have plasmacytoid features but more extensive sampling may reveal B cell component
- CD20 expression by lymphocytes or plasmacytoid cells
- Clonal B cell population by flow; molecular or cytogenetic features of marginal zone or lymphoplasmacytic lymphoma
- Plasma cell myeloma:
- Must be excluded by radiographs, bone marrow biopsy
- Radiographically, may mimic metastatic tumor, Langerhans cell histiocytosis
- Reactive plasma cell infiltrate:
- CD19 expression
- Cytogenetics demonstrating a t(11;14) translocation
- Plasmablastic features
- Radiographs
- Lymph node involvement by myeloma
- Nodal marginal zone lymphoma with extensive plasmacytic differentiation
- Potential spread from the upper respiratory tract
- All of the above
- BCR::ABL1 negative myeloproliferative neoplasm (MPN) characterized by peripheral erythrocytosis frequently accompanied by leukocytosis or thrombocytosis
- Relatively common MPN characterized by increased red blood cell (RBC) production with elevated red blood cell count and hemoglobin level, which is not proportional to erythropoietin (EPO) production (usually has markedly decreased EPO level)
- Diagnosis requires all 3 major criteria or 2 major plus the minor criterion (see 2016 and 2022 WHO classification in Diagnosis below)
- JAK2 V617F (exon 14, valine to phenylalanine) and JAK2 exon 12 mutations are identified in > 95% and ~5% of patients, respectively (Blood 2006;108:1865, Hum Pathol 2006;37:1458, N Engl J Med 2007;356:459)
- Occurs in 3 phases
- Prodromal / prepolycythemic
- Overt polycythemic
- Postpolycythemic myelofibrosis / spent
- < 20% undergo blastic transformation within 15 years
- Polycythemia vera (PV)
- Not accepted: Vaquez disease, Osler-Vaquez disease, polycythemia rubra vera, primary polycythemia, polycythemia
- Annual incidence increases with advanced age, varies from 0.7 to 2.6 per 100,000 people in Europe and North America (Semin Thromb Hemost 2006;32:171)
- Slight male predominance; M:F = 2:1 (Leukemia 2013;27:1874)
- Median age at diagnosis is 61 years
- Peripheral blood and bone marrow are major sites of involvement
- Spleen and liver are common sites of extramedullary hematopoiesis in later stages (Blood 2016;128:5491)
- Clonal hematopoietic stem cell disorder (Blood 2003;102:3793)
- Point mutation in Janus kinase 2 (JAK2) gene at 9p24 results in constitutional activation of transcription factors of the STAT family, which promote growth factor independent proliferation and survival (Blood 2006;108:1865)
- Mechanisms of thrombosis and bleeding are due to increased circulating red blood cells and elevated platelets (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- 3 phases
- Prodromal / prepolycythemic: characterized by borderline to mild erythrocytosis
- Overt polycythemic: associated with significantly increased red cell mass
- Postpolycythemic myelofibrosis / spent: cytopenias, including anemia, are associated with ineffective hematopoiesis, bone marrow fibrosis, extramedullary hematopoiesis and hypersplenism
- Underlying cause is unknown
- Presence of JAK2 V617F mutation is detected in over 95% of patients with PV (Blood 2008;111:1686)
- Abnormal karyotype in hematopoietic progenitor cells has been found in ~34% of patients with PV (J Clin Oncol 2011;29:761)
- Major symptoms are related to hypertension or vascular abnormalities caused by increased red cell mass
- Plethora can occur due to increased RBC mass
- Common symptoms include fatigue, headache, dizziness, tinnitus, vision changes, insomnia, claudication, pruritus, gastritis and early satiety (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Thrombotic events and thrombosis have been observed in up to 41% of patients with PV (Ann Intern Med 1995;123:656)
- 66% of thrombosis events tend to be arterial in nature (Rodgers: The Bethesda Handbook of Clinical Hematology, 4th Edition, 2018)
- Pruritis that occurs during or after a hot shower (aquagenic pruritus) is found in 40% of patients (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Gastrointestinal (GI) discomfort and peptic ulcer disease are common (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Early satiety may occur from impaired gastric filling due to splenomegaly (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Median survival of untreated symptomatic PV is 6 - 18 months from diagnosis, 3.5 years for PV treated with phlebotomy and 7 - 12 years for PV treated with cytoreduction (Rodgers: The Bethesda Handbook of Clinical Hematology, 4th Edition, 2018)
- Diagnosis criteria (WHO 5th edition) for polycythemia vera requires meeting either all 3 major criteria or the first 2 major criteria and the minor criterion
- Major criteria
- Hemoglobin > 16.5 g/dL in men, > 16.0 g/dL in women; hematocrit (HCT) > 49% in men, > 48% in women
- Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis), including prominent erythroid, granulocytic and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size)
- Presence of JAK2 V617F or JAK2 exon 12 mutation
- Minor criterion
- Subnormal serum erythropoietin level (normal range of EPO in adults: 4.1 - 19.5 mU/mL)
- Major criterion 2 may not be required in cases with sustained absolute erythrocytosis
- Sustained absolute erythrocytosis is defined as hemoglobin levels greater than 18.5 g/dL in men or 16.5 g/dL in women or hematocrit greater than 55.5% in men and 49.5% in women
- Major criterion 3 (presence of JAK2 V617F or JAK2 exon 12 mutation) plus the minor criterion must also be present
- Accelerated phase: presence of 10 - 19% blasts in bone marrow or peripheral blood (Best Pract Res Clin Haematol 2022;35:101379)
- Blast phase: presence of ≥ 20% blasts in bone marrow or peripheral blood (Best Pract Res Clin Haematol 2022;35:101379)
- Major criteria
- Diagnostic criteria for postpolycythemic myelofibrosis (post-PV MF)
- Criteria based on International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and WHO (Blood 2016;127:2391, Am J Hematol 2017;92:94)
- Required criteria
- Documentation of a previous diagnosis of PV according to 2016 or 2022 WHO criteria
- Presence of grade 2 or 3 bone marrow fibrosis, based on either 2016 or 2022 WHO classification or the European consensus on grading of bone marrow fibrosis (Haematologica 2005;90:1128)
- Additional criteria (2 required)
- Anemia
- Leukoerythroblastosis
- Splenomegaly - either development of newly palpable splenomegaly or increased splenomegaly of > 5 cm from baseline
- Development of any 2 (or all 3) of the following constitutional symptoms
- Night sweats
- > 10% weight loss within past 6 months
- Unexplained fevers greater than 37.5 °C
- Required criteria
- Criteria based on International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and WHO (Blood 2016;127:2391, Am J Hematol 2017;92:94)
- Laboratory studies usually demonstrate erythrocytosis
- May also present with asymptomatic leukocytosis or thrombocytosis
- Bone marrow biopsy may show hypercellularity and panmyelosis
- Iron deficiency can be seen in many cases (Hematology 2023;28:2204621)
- JAK2 V617F mutation is detectable in over 95% of patients with PV (Blood 2008;111:1686)
- JAK2 exon 12 mutations may be seen in 2 - 5% of patients who are negative for the JAK2 V617F mutation (Ann Hematol 2020;99:983)
- Leukocytosis may predict inferior survival and leukemic transformation
- Risk of PV progressing to postpolycythemic myelofibrosis is 4.9% at 10 years and 9.4% at 15 years (Hematol J 2003;4:198)
- 15% develop spent phase with marrow fibrosis and marked cytopenias after 10 years; death in months if no treatment
- JAK2 V617F mutant allele burden of > 50% has been associated with fibrotic transformation (Am J Hematol 2017;92:94)
- Leukemic transformation rates at 20 years are estimated at < 10% for PV (Am J Hematol 2017;92:94)
- Risk factors for transformation include advanced age, leukocytosis and abnormal karyotype
- Leukemic transformation has been associated with treatment exposure to pipobroman or P32 / chlorambucil (Leukemia 2013;27:1874)
- Recent study showed older age, leukocytosis, venous thrombosis and abnormal karyotype are associated with adverse clinical outcomes (Leukemia 2013;27:1874)
- 3 risk groups are proposed, including low risk (0 points), intermediate risk (1 or 2 points) and high risk (3 points)
- Adverse points are assigned to age 67 years and older (5 points), age 57 - 66 years (2 points), leukocyte count 15 x 109/L (1 point) and venous thrombosis (1 point) with median survival of 28 years (low risk), 19 years (intermediate risk) and 11 years (high risk) (Leukemia 2013;27:1874)
- Next generation sequencing (NGS) study indicated that ASXL1, SRSF2 and IDH2 mutations are negative prognostic parameters associated with shorter overall, leukemia free and fibrosis free survival (Am J Hematol 2017;92:94)
- Cytogenetic abnormalities can be detected in up to 20% of patients at the time of initial diagnosis (Haematologica 2017;102:1511)
- Abnormalities may vary in different phases of disease
- Isolated del(20q), +8 and +9 are shown to be the most common abnormalities in polycythemic phase
- Del(20q) and +1q have been shown to be the most common abnormalities in postpolycythemic myelofibrosis
- Complex karyotypes have been shown to be the most common in accelerated and blast phases
- Abnormalities may vary in different phases of disease
- 15 year old boy with cooperation of germline JAK2 mutations E846D and R1063H in hereditary erythrocytosis with megakaryocytic atypia (Blood 2016;128:1418)
- 40 year old woman who presented with subdural hematoma was found to have polycythemia vera (Neurol India 2022;70:1717)
- 51 year old woman with PV treated with intermittent phlebotomy and oral hydroxyurea irregularly for 2 years developed acute promyelocytic leukemia (Medicine (Baltimore) 2022;101:e30064)
- 56 year old woman with progression of primary myelofibrosis to polycythemia vera (Medicine (Baltimore) 2017;96:e7464)
- 62 year old woman with postpolycythemic myelofibrosis with tumor lysis syndrome after the administration of ruxolitinib (Intern Med 2017;56:2335)
- 66 year old man with PV and Erdheim-Chester disease with multiorgan involvement (Medicine (Baltimore) 2016;95:e3697)
- Risk of PV progressing to postpolycythemic myelofibrosis is 4.9% at 10 years and 9.4% at 15 years (Hematol J 2003;4:198)
- Due to increased red blood cell production independent of mechanisms that typically regulate erythropoiesis
- 15% develop spent phase with marrow fibrosis and marked cytopenias after 10 years; death in months if no treatment
- Leukemic transformation rates at 20 years are estimated at < 10% for PV (Am J Hematol 2017;92:94)
- 2% develop acute myeloid leukemia with phlebotomy treatment but 15% with alkylating agents or radioactive phosphorus (no longer used)
- Risk of leukemic transformation (ALL) is rare, except in patients treated with cytotoxic regimens
- Therapeutic goals
- Decrease thrombosis without increasing bleeding
- Prevent progression
- Ameliorate symptoms
- Treatment guidelines (Ann Oncol 2015;26:v85, Blood 2014;124:3212, N Engl J Med 2004;350:114)
- Low / intermediate risk for thrombosis
- First line: phlebotomy
- Low dose aspirin
- Intensive management of cardiovascular risk factors
- High risk for thrombosis
- Cytoreductive therapy (hydroxyurea)
- Adjunct phlebotomy
- Low dose aspirin
- Pregnant women
- First line: phlebotomy
- Low dose aspirin
- Low molecular weight heparin in postpartum period
- Low / intermediate risk for thrombosis
- European collaboration on low dose aspirin in polycythemia vera (ECLAP) trial (2004): multicenter project that investigated the benefits and risks of low dose aspirin in patients with PV
- Study showed that daily aspirin (100 mg) lowered the risk of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke and total mortality
- Treatment also was known to nonsignificantly increase the risk of major bleeding (Am Heart J 2004;148:1068)
- Randomized study of efficacy and safety in polycythemia vera with JAK inhibitor ruxolitinib versus best available care (RESPONSE) trial (2020): longterm efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (Lancet Haematol 2020;7:e226)
- 5 year follow up of a phase 3 trial comparing patients with failed response to hydroxyurea
- Study results showed that ruxolitinib is a safe and effective longterm treatment option for patients with polycythemia vera who are resistant to or intolerant of hydroxyurea
- Bone marrow (prepolycythemic and overt polycythemic phases) (see Microscopic (histologic) images 1, 4 & 7)
- Hypercellular with panmyelosis; notable predominance of erythroid and megakaryocytic lineages
- Complete and progressive maturation of all 3 hematopoietic lineages
- Abnormal megakaryocyte morphology and architecture is a prominent feature
- Mekagaryocytes are variably hyperlobulated and often seen in loose clusters; frequently seen close to bony trabeculae
- Minority of cases (< 20%) may show mild increase in reticulin fibrosis (Blood 2012;119:2239)
- Iron stores are decreased, often absent
- Reactive lymphoid aggregates may be seen in up to 20% of cases (Histol Histopathol 2005;20:317)
- Blasts can be seen during accelerated or blast phase
- Post-PV / spent phase (see Peripheral smear image and Microscopic (histologic) images 2, 5 & 6)
- Leukoerythroblastosis (Leukemia 2021;35:3339)
- Overt bone marrow reticulin and collagen fibrosis; osteosclerosis often prominent (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Dilated sinuses with intrasinusoidal hematopoiesis
- Decline in hematopoietic cells
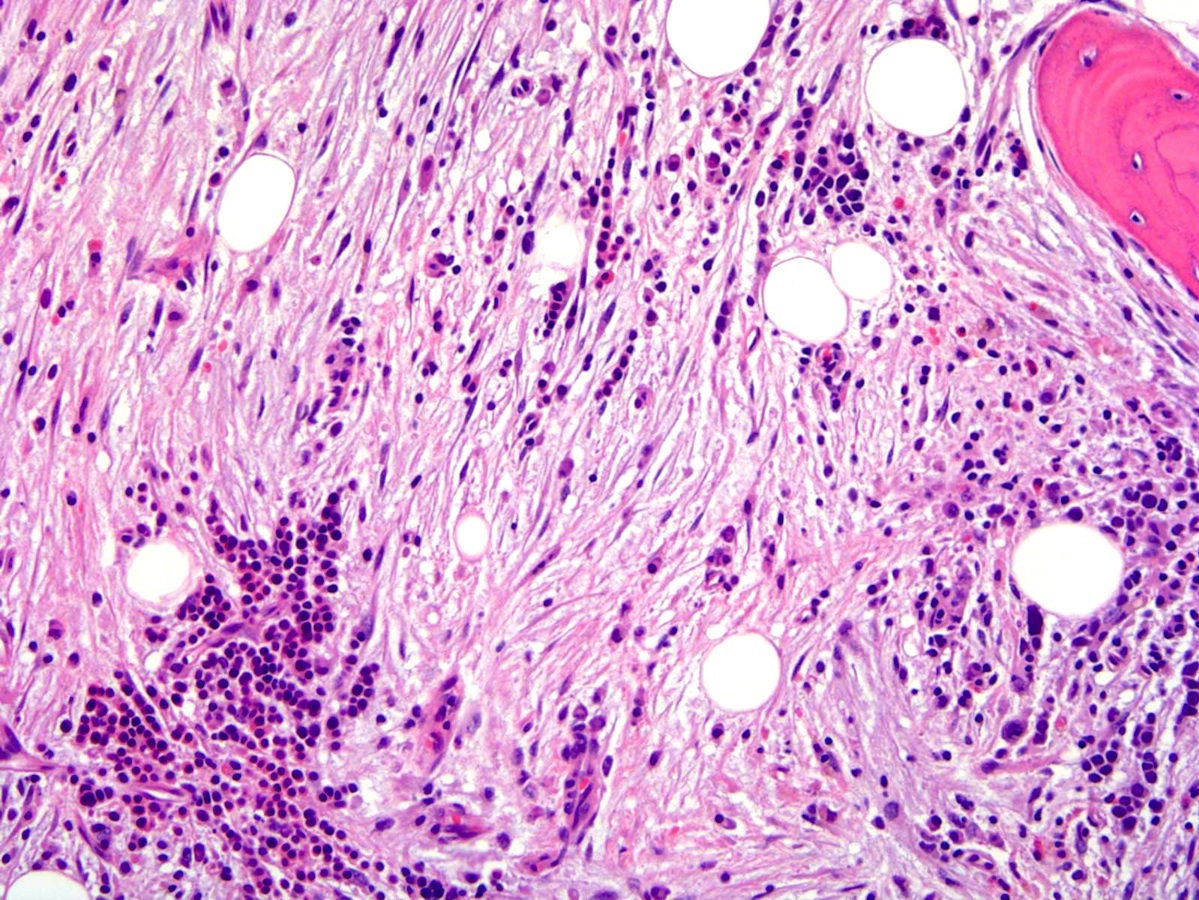
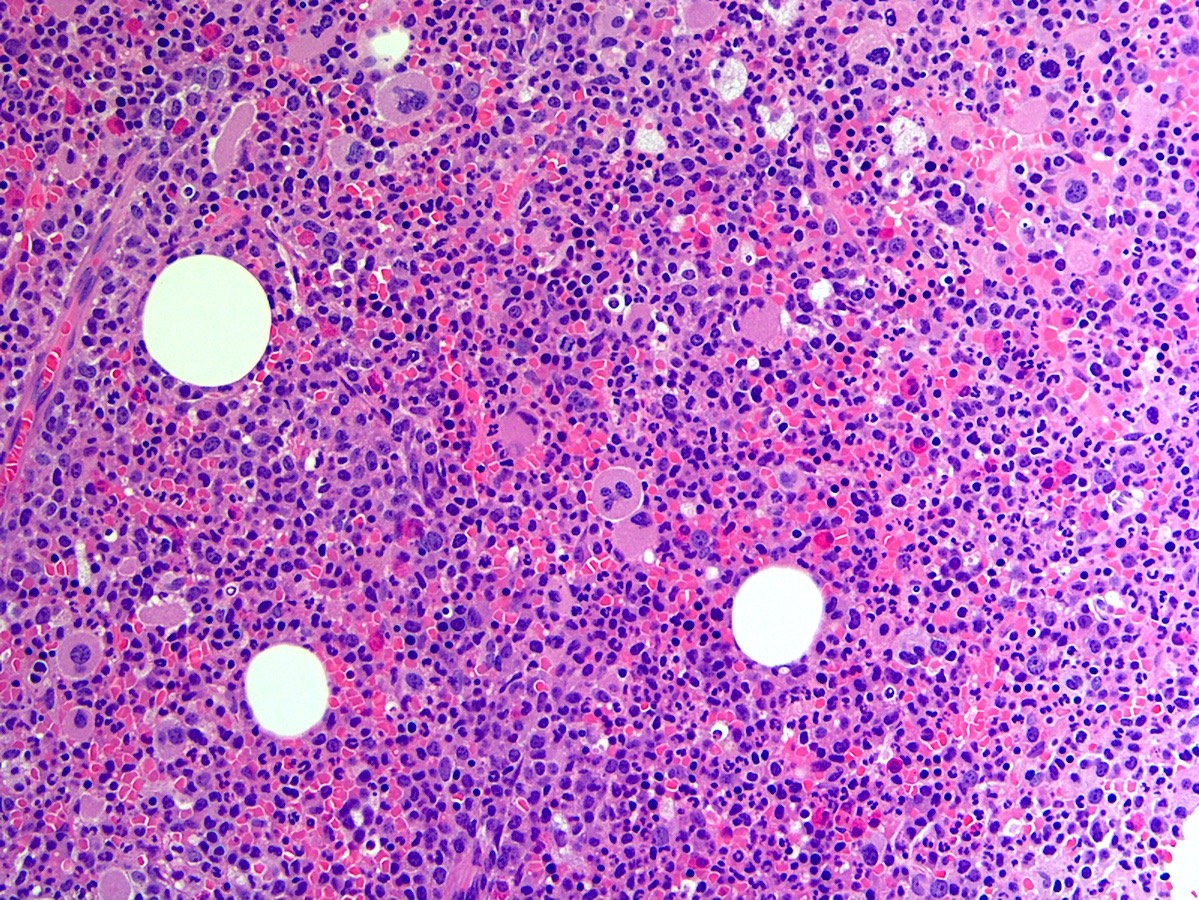

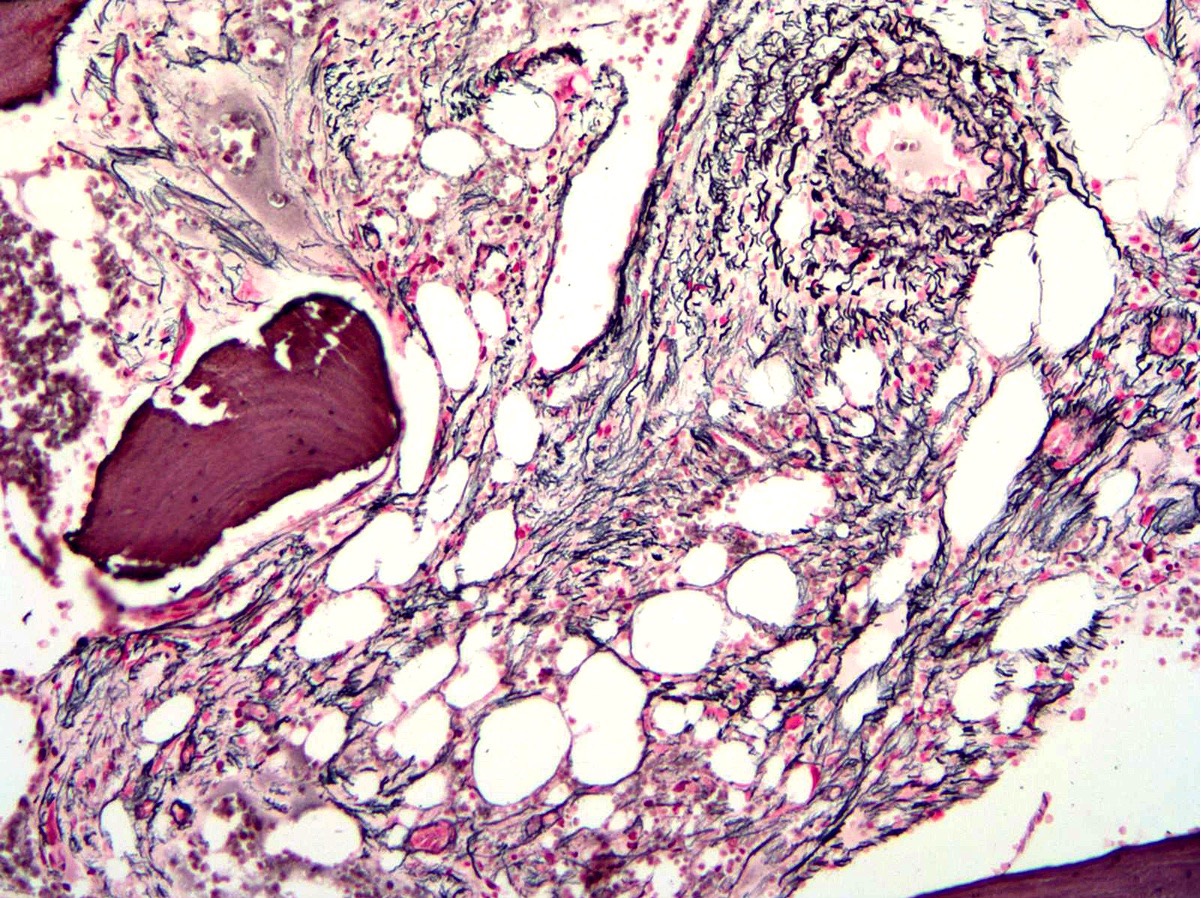
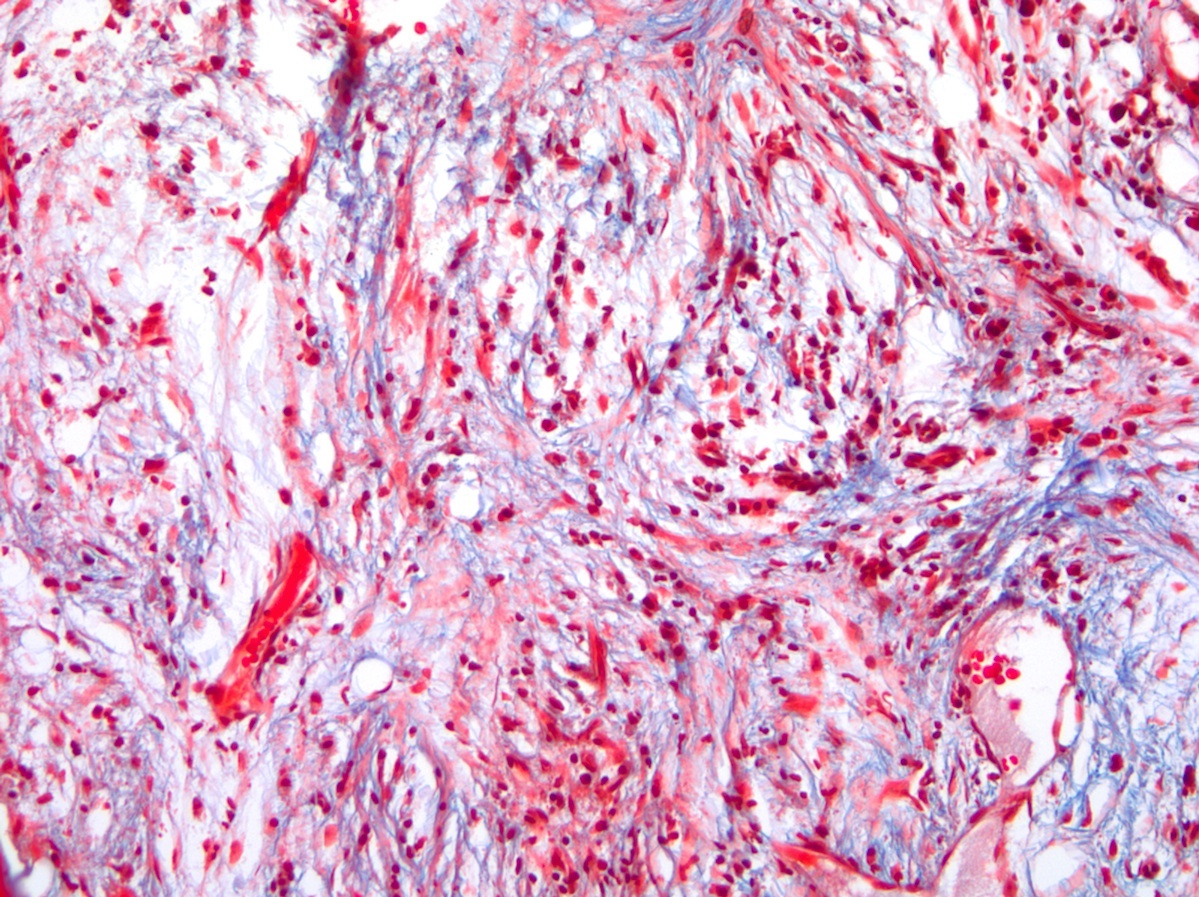
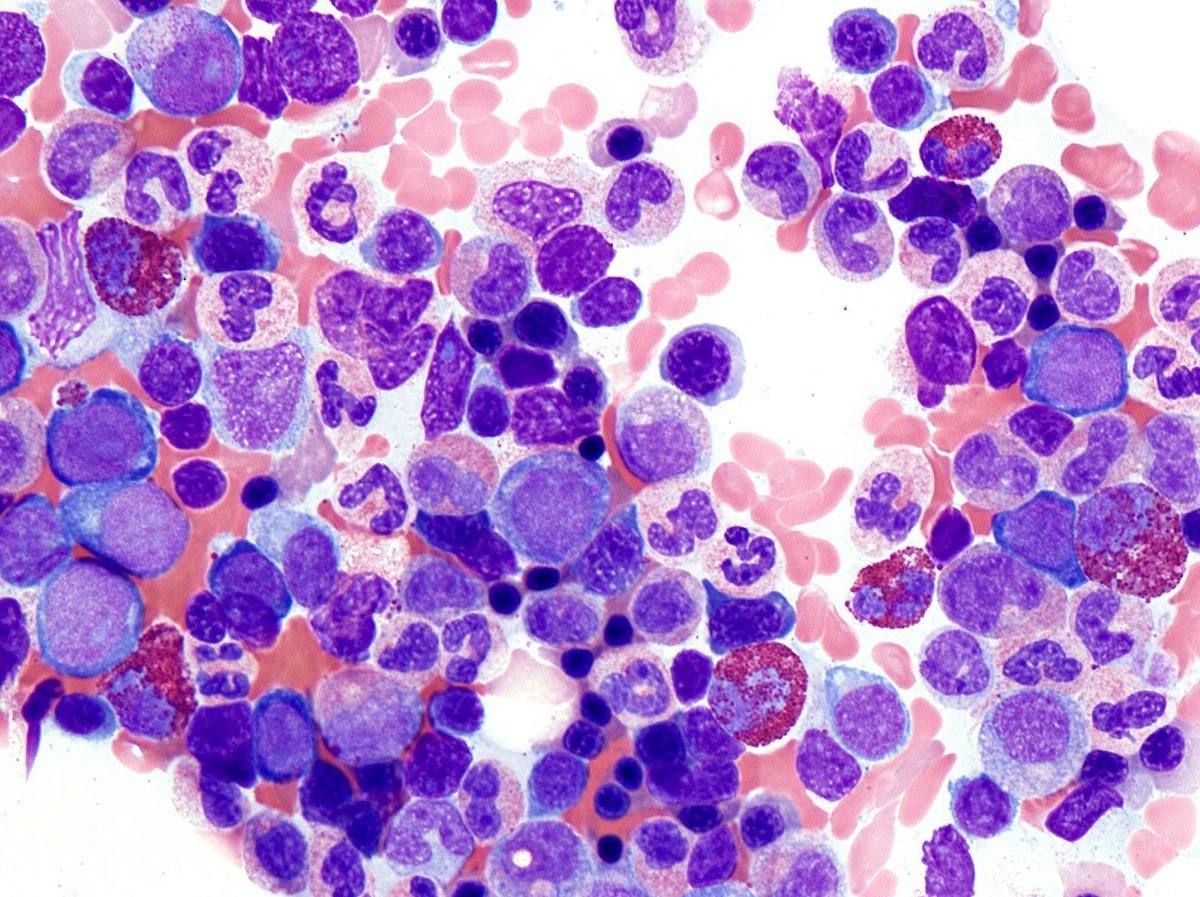
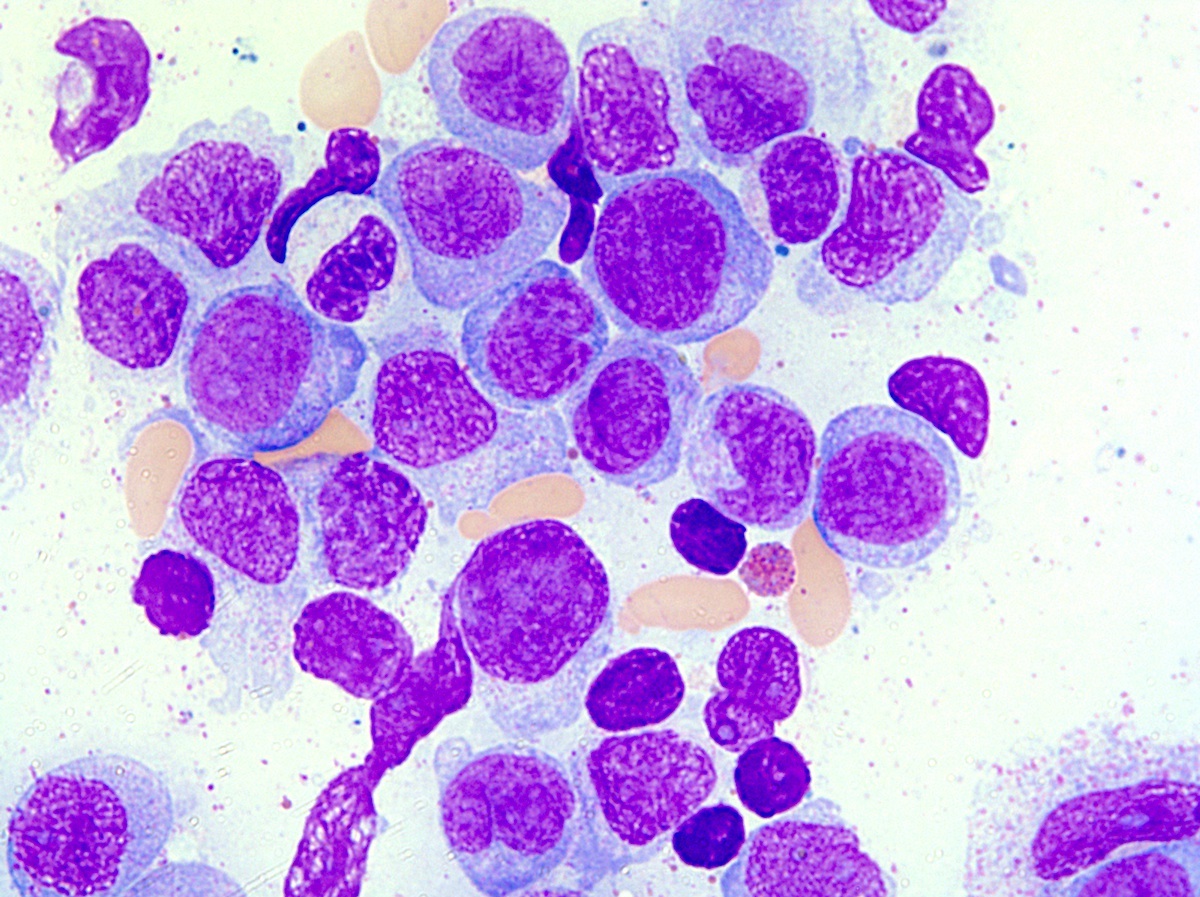
- Peripheral blood (prepolycythemic and overt polycythemic phases)
- Normochromic, normocytic red blood cells or microcytic, hypochromatic when iron deficiency is present
- Erythrocytosis with increased hemoglobin or hematocrit (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Red blood cell morphology is unremarkable except in cases with concomitant iron deficiency
- Deeply basophilic reticulocytes or rare normoblasts may be present
- Mild neutrophilic leukocytosis with rare left shift (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Mild basophilia may be seen
- Thrombocytosis; may be particularly prominent in prodromal phase or exaggerated by concurrent iron deficiency (Am J Hematol 2020;95:1599)
- Post-PV / spent phase: myeloid metaplasia characterized by leukoerythroblastosis (see Peripheral smear image)
- Poikilocytosis with teardrop shaped red blood cells (StatPearls: Polycythemia Vera [Accessed 12 September 2023])
- Findings of > 10% blasts in the peripheral blood or bone marrow or the presence of significant myelodysplasia is unusual and most likely signals transformation to an accelerated phase
- Cases in which 20% or more blasts are found are considered acute myeloid leukemia (AML) (see Microscopic (histologic) images 3 & 8) (J Clin Med 2021;10:436)
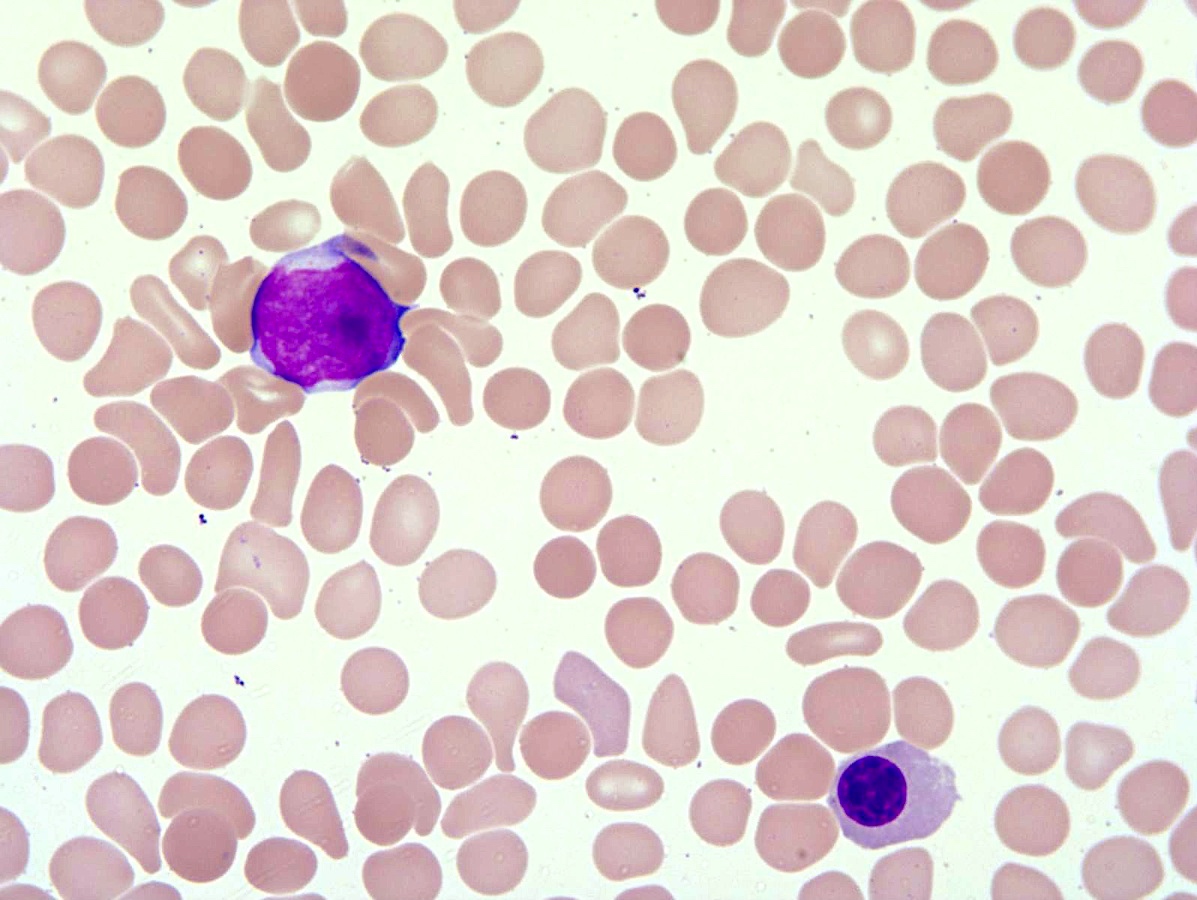
- High expression of VEGF but not used for diagnosis (Am J Clin Pathol 2007;128:966)
- Glycophorin, spectrin or hemoglobin highlight increased number of erythroid precursors
- CD34 highlights increased myeloblasts when transformed
- CD61, CD42b and factor VIII highlight megakaryocytic hyperplasia
- MPL (thrombopoietin receptor) on megakaryocytes is usually moderate to strong in normal controls and secondary erythrocytosis but is not used for diagnosis (Blood 2000;96:771)
- Most frequent somatic activating mutations of JAK2 are located in exon 14 (JAK2 V617F) (> 95%) followed by exon 12 (~5%)
- JAK2 V617F is not specific and is found at a lower frequency in other MPN
- Patients with JAK2 V617F are usually elderly with higher level of hemoglobin, white blood cell (WBC) count and lower level of platelets, while patients with JAK2 exon 12 mutation are frequently younger with erythroid hyperplasia but low erythropoietin production (low EPO level) (Am J Hematol 2017;92:94)
- Cytogenetic abnormalities are detectable in about 20% of patients and most commonly include del(20q), +8, +9 and +1q (Haematologica 2017;102:1511)
- Cytogenetic abnormalities increase in frequency when patients with PV transform to acute myeloid leukemia (AML), myelodysplastic syndrome (MDS) or myelofibrosis (MF) (Leuk Lymphoma 2013;54:2667)
- Next generation sequencing identified several gene mutations in PV in addition to JAK2, which includes most frequently TET2 and ASXL1 followed by SRSF2 and IDH2, with combined prevalence of 15% (Blood Adv 2016;1:21)
- Bone marrow, left posterior iliac crest, aspirate and biopsy:
- Myeloproliferative neoplasm (see comment)
- Comment: The patient has a history of persistent erythrocytosis with a decreased erythropoietin level. A JAK2 V617F mutation was identified in the peripheral blood (VAF: 10%). The combined morphologic and ancillary laboratory findings are diagnostic of polycythemia vera.
- Hypercellular bone marrow (80 - 90%) with panmyelosis
- Microscopic examination
- Peripheral blood
- Erythrocytosis
- Normal WBCs and platelets
- Peripheral smear: normocytic erythrocytosis
- Cytopenias present: none
- Red cell morphology: normocytic erythrocytosis
- Reticulocytosis: not increased
- Other significant findings: none
- Bone marrow aspirate smear
- Adequacy: spicular, cellular
- M:E ratio: 2:1
- Bone marrow differential count: blasts: 2.1 %, promyelocytes: 1.0%, myelocytes: 14.2%, metamyelocytes: 15.8%, band neutrophils: 3.5%, segmented neutrophils: 9.8%, eosinophils: 11.8%, basophils: 1.2%, monocytes: 2.0%, lymphocytes: 10.0%, early normoblasts: 2.5%, late normoblasts: 24.0%
- Significant findings: maturing trilineage hematopoiesis, no overt dysplasia, megakaryocytes are relatively increased and show variable morphology, including small forms and large hyperlobulated forms
- Bone marrow clot section and core biopsy
- Bony trabeculae: unremarkable
- Cellularity: 80 - 90%
- Megakaryocytes: increased, with occasional loose clustering
- Estimated M:E ratio: 3:1
- Iron stores: decreased
- Reticulin fibrosis: none (marrow fibrosis 0/3)
- Significant findings: hypercellular bone marrow with panmyelosis
- Peripheral blood
- Chronic myelogenous leukemia (CML):
- Leukocytosis (WBC ranges from 12 k/L to 1,000 k/L; median 100 k/L), basophilia, thrombocytosis, monocytosis (often accounts for < 2 - 3% of WBC)
- No evidence of true erythrocytosis
- Positive for BCR::ABL1 gene rearrangement
- Negative for JAK2 V617F or JAK2 exon 12 or 13 mutation
- Essential thrombocythemia (ET) and early phase of primary myelofibrosis (PMF):
- Following the adoption of the updated 2016 WHO classification of PV or when a patient presents with thrombocytosis, it is particularly important to differentiate masked / prodromal polycythemia from JAK2 positive ET by careful examination of bone marrow morphology
- In contrast to PV, ET and PMF show distinct clinical and pathologic features (bone marrow biopsy of PV shows panmyelopoiesis with more atypical or pleomorphic appearing mature megakaryocytes) (Am J Hematol 2017;92:1062, Blood 2016;127:2391)
- ET: peripheral thrombocytosis but no evidence of erythrocytosis; bone marrow with essentially normocellular marrow, hyperlobated megakaryocytes
- PMF, early phase: cytosis but no evidence of erythrocytosis; bone marrow with atypical megakaryocytic proliferation, often associated with clustering, accompanied by myeloid hyperplasia
- PMF fibrotic phase: peripheral cytopenia; bone marrow with atypical megakaryopoiesis, syncytial clustering of megakaryocytes, reticulin fibrosis (marrow fibrosis 2 - 3/3)
- Following the adoption of the updated 2016 WHO classification of PV or when a patient presents with thrombocytosis, it is particularly important to differentiate masked / prodromal polycythemia from JAK2 positive ET by careful examination of bone marrow morphology
- Secondary erythrocytosis / polycythemia:
- In contrast to primary polycythemia vera, EPO levels are elevated, usually as a secondary response to chronic hypoxemia
- May be compensatory to various physiologic or environmental processes
- Lung disease, high altitude living, cyanotic heart disease, heavy smoking, carbon monoxide poisoning, body builder or hormone replacement (e.g., testolone)
- May be caused by EPO secreting tumors (i.e., renal cell carcinoma, hepatocellular carcinoma, cerebellar hemangioblastoma)
- May be caused by an inherited defect that stabilizes HIF1A (prolyl hydroxylase mutations, homozygous VHL mutations, etc.)
- Negative JAK2 mutation and normal serum EPO level help to differentiate from PV
- Gaisbock syndrome: normal megakaryocytes, less cellular marrow, no increase in reticulin, more normal iron stores, normal or high erythropoietin levels (Angiology 1978;29:520)
- Grade of marrow fibrosis
- Increased erythroid precursors seen in the bone marrow
- JAK2 V617F mutation status
- Presence of BCR::ABL1 gene fusion
- Red blood cell morphology
Comment Here
Reference: Polycythemia vera
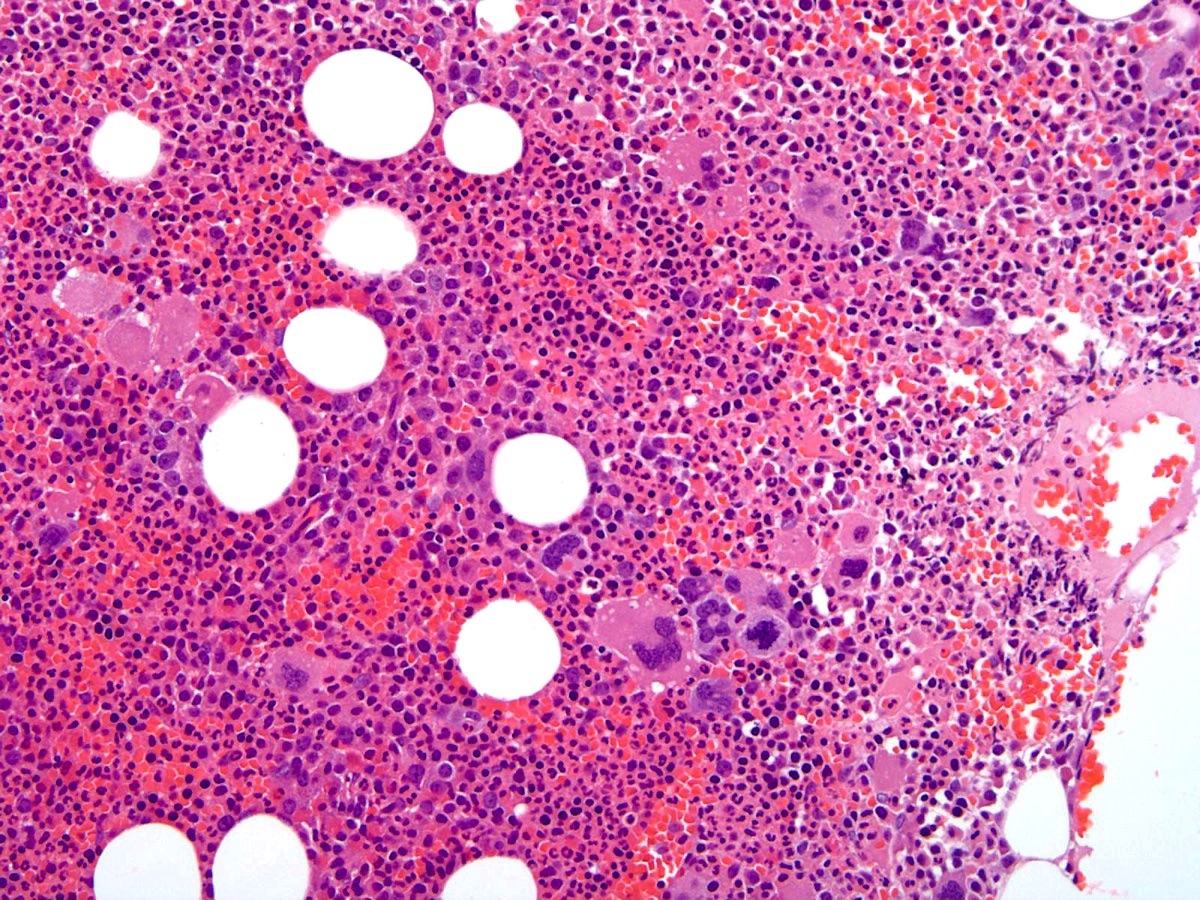
A patient underwent bone marrow biopsy for unexplainable pruritus and erythromelalgia. The bone marrow findings are depicted in the image above. What is a notable microscopic feature of polycythemia vera (PV) that can be seen in the bone marrow during the prepolycythemic and overt polycythemic phases?
- Decreased erythroid precursors
- Hypercellularity with panmyelosis
- Hypocellularity with sparse megakaryocytes
- Increased reticulin fibrosis
- Presence of collagen fibrosis highlighted by trichrome stain
Comment Here
Reference: Polycythemia vera
- Bone marrow hypocellularity for age
- Hemoglobin greater than 16.5 g/dL
- Platelet count less than 100 x 103/μL
- Presence of extramedullary hematopoiesis
Comment Here
Reference: Polycythemia vera
- Light chain (AL) amyloidosis is the most common form of systemic amyloidosis (Clin J Am Soc Nephrol 2006;1:1331)
- Monoclonal immunoglobulin deposition disease with visceral and soft tissue Ig deposition resulting in organ dysfunction
- Underlying plasma cell dyscrasia or (less commonly, 5 - 7%) lymphoplasmacytic neoplasm (Expert Rev Hematol 2018;11:117)
- Fibrils consist of whole or fragments of immunoglobulin light chains
- Form beta pleated sheets (AL amyloid) in tissue that bind Congo red dye and show characteristic apple green birefringence in polarized light (Clin J Am Soc Nephrol 2006;1:1331)
- M protein is found in the serum or urine in > 80% of patients
- Diagnostic criteria for myeloma met in a subset of cases (12 - 15%), typically Ig molecule accumulates before there is a large tumor burden (N Engl J Med 2003;349:583)
- Primary amyloidosis and light chain and heavy chain deposition disease (nonamyloid immunoglobulin deposition in tissues) appear to be chemically different manifestations of similar pathologic processes
- Rare and may present with a broad range of symptoms, some vague, making it challenging to recognize (Clin J Am Soc Nephrol 2006;1:1331)
- Accepted nomenclature for Amyloid is AX, where A stands for Amyloid and X represents the protein in the fibril
- Historically light chain amyloidosis (AL) referred to as primary amyloidosis
- Overall, > 30 different proteins can cause amyloidosis (Blood 2009;114:4957, Clin J Am Soc Nephrol 2010;5:2180)
- Those involving kidney recently grouped into the category of monoclonal gammopathies of renal significance (MGUS) to separate them from MGUS based on need for treatment (Blood 2012;120:4292)
- Incidence: approximately 1 case per 100,000; approximately 3,000 cases in the U.S. per year
- Most common type of systemic amyloidosis in developed countries (75%) (Blood 2016;128:159)
- Median age 64, > 95% are older than 40
- Predominant in men (65 - 70%)
- Protean clinical presentation due to widespread organ involvement, involving all except possibly the brain (Expert Rev Hematol 2018;11:117)
- Survival mostly determined by extent of cardiac involvement (Blood 2016;128:159)
- Abnormal folding of a protein that is normally soluble as a result of a proteolytic event or mutation that renders it unstable / prone to self aggregation (Clin J Am Soc Nephrol 2006;1:1331)
- Aggregates form protofilaments that associate into amyloid fibrils
- Interactions with the extracellular environment may result in proteolytic cleavage and matrix components such as glycosaminoglycans and collagen may serve as scaffolds to facilitate aggregation and fibril buildup (Sci Rep 2016;6:29096)
- Only a subset of Ig light chains are amyloidogenic (12 - 15% of myeloma) (N Engl J Med 2003;349:583)
- Repertoire of variable region germline genes involved in systemic amyloidosis is restricted and certain gene usage may affect organ targeting; e.g. use of IGVL1-44 had 5 fold odds of dominant cardiac involvement (Blood 2012;119:144)
- Amyloid precursor proteins directly impair cardiac function independent of fibril formation, invoking oxidative stress, cellular dysfunction, apoptosis (Proc Natl Acad Sci U S A 2010;107:4188)
- Clonal expansion of plasma cells or low grade lymphoproliferative disorder secreting monoclonal immunoglobulin light chains which misfold, aggregate and deposit as interstitial fibrils of amyloid
- ~30% present with advanced, irreversible organ damage (Blood 2016;128:159)
- General: dyspnea, fatigue, weight loss
- GI: macroglossia (highly suggestive of AL but only present in 10 - 15%), hepatomegaly (25 - 30%), malabsorption (5%)
- Renal: nephrotic syndrome (28%)
- Heart: restrictive cardiomyopathy, congestive heart failure (17%), amyloid precursor proteins with direct cardiotoxic effect (Proc Natl Acad Sci U S A 2010;107:4188)
- Neural: peripheral sensory neuropathy (17%), carpal tunnel syndrome (21%), autonomic dysfunction
- Bone pain (5%), periorbital or facial purpura
- Hemorrhage: can result from factor X binding to amyloid (Blood 2001;97:1885)
- Patients with underlying IgM-secreting lymphoplasmacytic lymphoma are typically older, higher prevalence of neuropathy and lymph node involvement and lower level of cardiac involvement than non IgM AL amyloidosis (Expert Rev Hematol 2018;11:117)
- Tissue diagnosis is the gold standard
- Laser microdissection with analysis by mass spectrometry is the most reliable method of characterizing amyloid type (Blood 2009;114:4957)
- Abdominal fat pad aspirate: most common and easily accessible tissue (positive in more than 80% of patients with primary amyloidosis)
- Rectal biopsies diagnostic in 80% of cases
- Biopsy of kidney, heart, liver
- Bone marrow: 60% of cases show amyloid; detection improved if adequate sized vessels present
- Serum and urine protein electrophoresis and immunoelectrophoresis identifies M spike (99% of cases); IgG most frequent (followed by light chain only, IgA, IgM and IgD); 80% lambda (Expert Rev Hematol 2018;11:117)
- Critical to test for free light chain as some patients lack circulating intact immunoglobulin
- Nephrotic range proteinuria and hypoalbuminemia with renal involvement
- Elevated erythrocyte sedimentation rate (ESR)
- Elevated brain natriuretic peptide (BNP), proBNP and troponin in cardiac involvement
- Cholestasis with elevated alkaline phosphatase with liver involvement
- AL / primary amyloidosis may also present with hypothyroidism / hypoadrenalism or hypopituitarism
- Scintigraphy with radiolabeled serum amyloid P(SAP) can be used to test for amyloid where SAP binds to the amyloid and reveals its presence (Annu Rev Med 2006;57:223)
- Bone lesions restricted to patients with myeloma
- Uncommon to see splenomegaly or lymphadenopathy
- Extent of cardiac involvement a major determinant of outcome and most frequent cause of death (40% of cases)
- Higher risk: monoclonal plasma cells > 10% bone marrow involvement, high serum free light chain, elevated beta-2-microglobulin, multiple organ involvement, elevated uric acid level
- Median survival time of 12 months if untreated
- Multiple staging systems have been proposed based on cardiac markers or renal function (Blood 2016;128:159)
- 18 year old man with primary AL (kappa light chain) amyloidosis manifesting as peripheral neuropathy (Clin Neuropathol 2005;24:118)
- 27 year old woman with primary systemic amyloidosis (Cent Eur J Immunol 2014;39:61)
- 70 year old woman with undiagnosed primary amyloidosis unmasked during surgical treatment (J Endocr Soc 2018;2:112)
- Antiplasma cell chemotherapy to reduce concentration of toxic light chains based on myeloma regimens
- Myeloma treatment regimens are discussed in the NCCN guidelines (J Natl Compr Canc Netw 2016;14:389)
- Not solely a hematologic malignancy with amyloid related organ dysfunction limiting access to aggressive treatment; clinical trials are recommended with close monitoring of response to treatment (Blood 2016;128:159)
- Common regimens include melphalan or dexamethasone; thalidomide and its analogues may also be used
- May have increased sensitivity to proteasome inhibitors such as bortezomib due to presence of misfolded light chain (Blood 2017;129:2132)
- Autologous stem cell transplant may also be considered with caution for transplant related mortality; not solely a hematologic malignancy with amyloid related organ dysfunction limiting access to aggressive treatment (Blood 2016;128:159)
- Targeting amyloid deposits to interfere with organ damage is a newer strategy: the small molecule anthracycline 49-iodo-49-deoxy-doxorubicin and related molecule doxycycline (antibiotic) may disrupt amyloid fibrils and improve clinical status (Proc Natl Acad Sci U S A 1995;92:2959, Blood Cancer J 2017;7:e546) and other approaches as (Blood 2016;128:159)
Images hosted on other servers:

Systemic AL amyloidosis
- Amyloidosis may or may not be apparent on macroscopic examination
- Classic gross appearance porcelain-like or waxy
- Involved organ may appear enlarged, gray, firm and waxy when amyloid accumulates in larger amounts
- Pale pink, extracellular, glassy, hyaline material on H&E staining, mostly deposited in vascular or perivascular location
- Characteristic cracking artifact
- Macrophages and foreign body giant cells may be found around deposits
Contributed by Meenakshi Bansal, M.D.
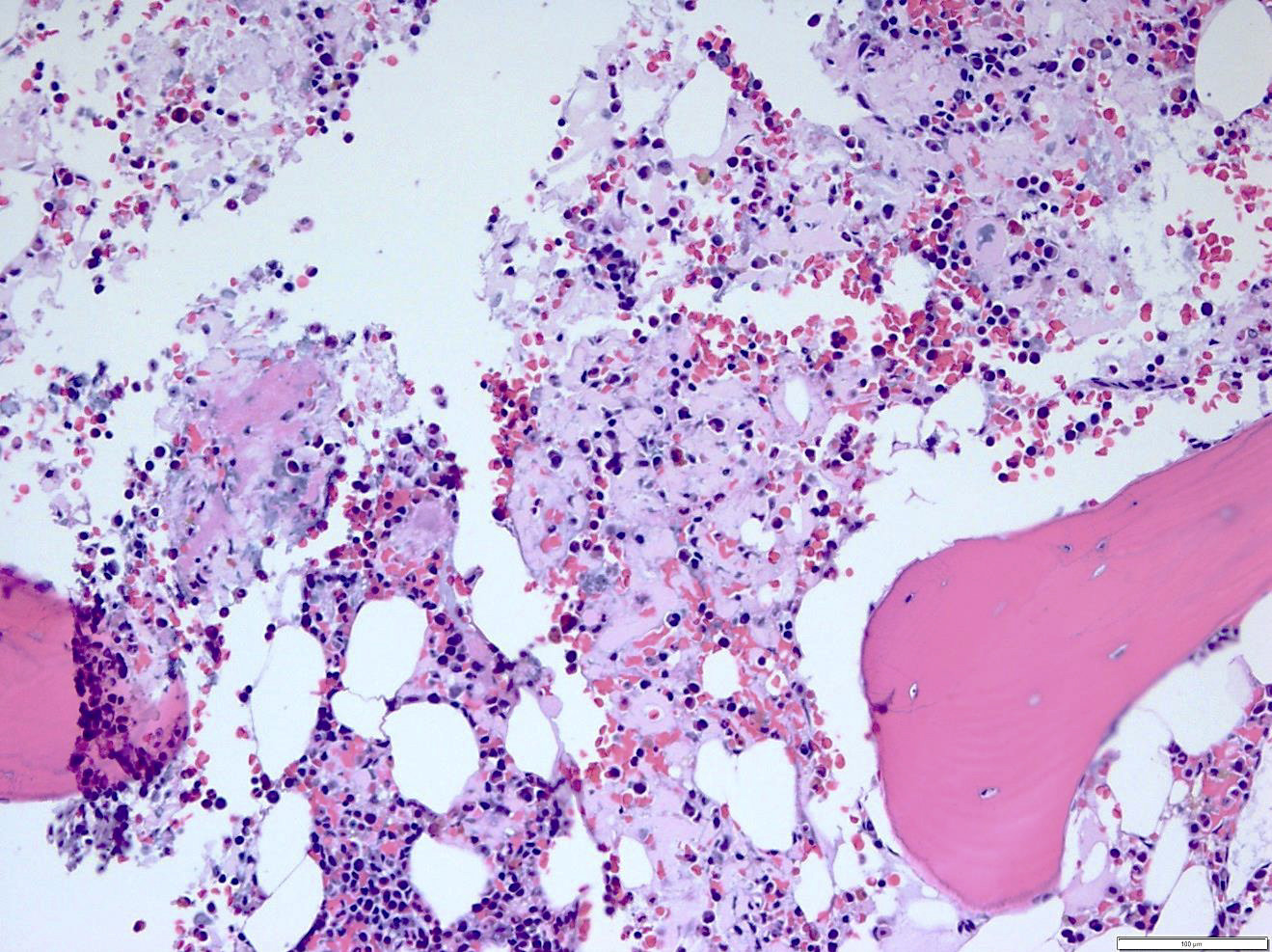
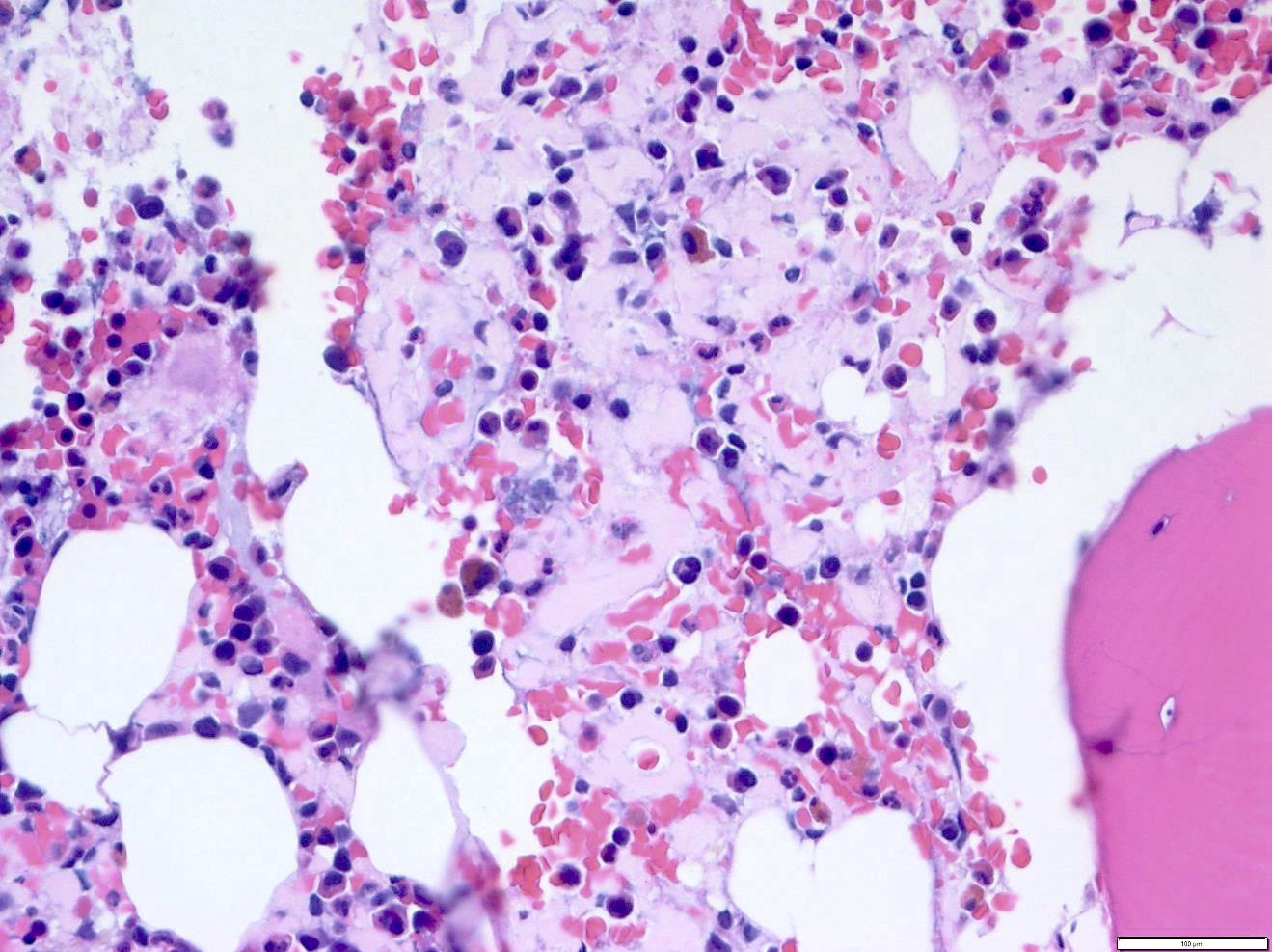
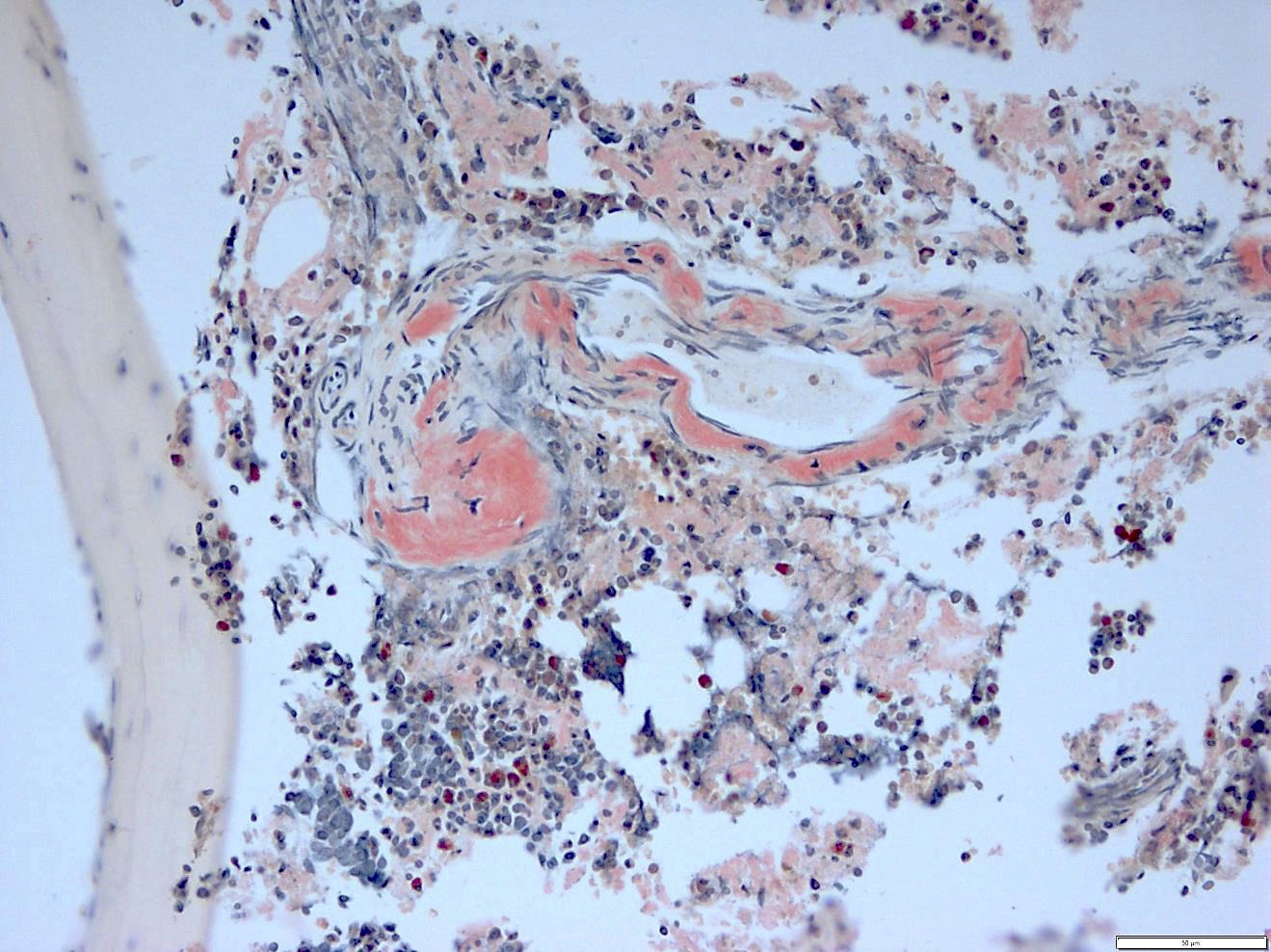
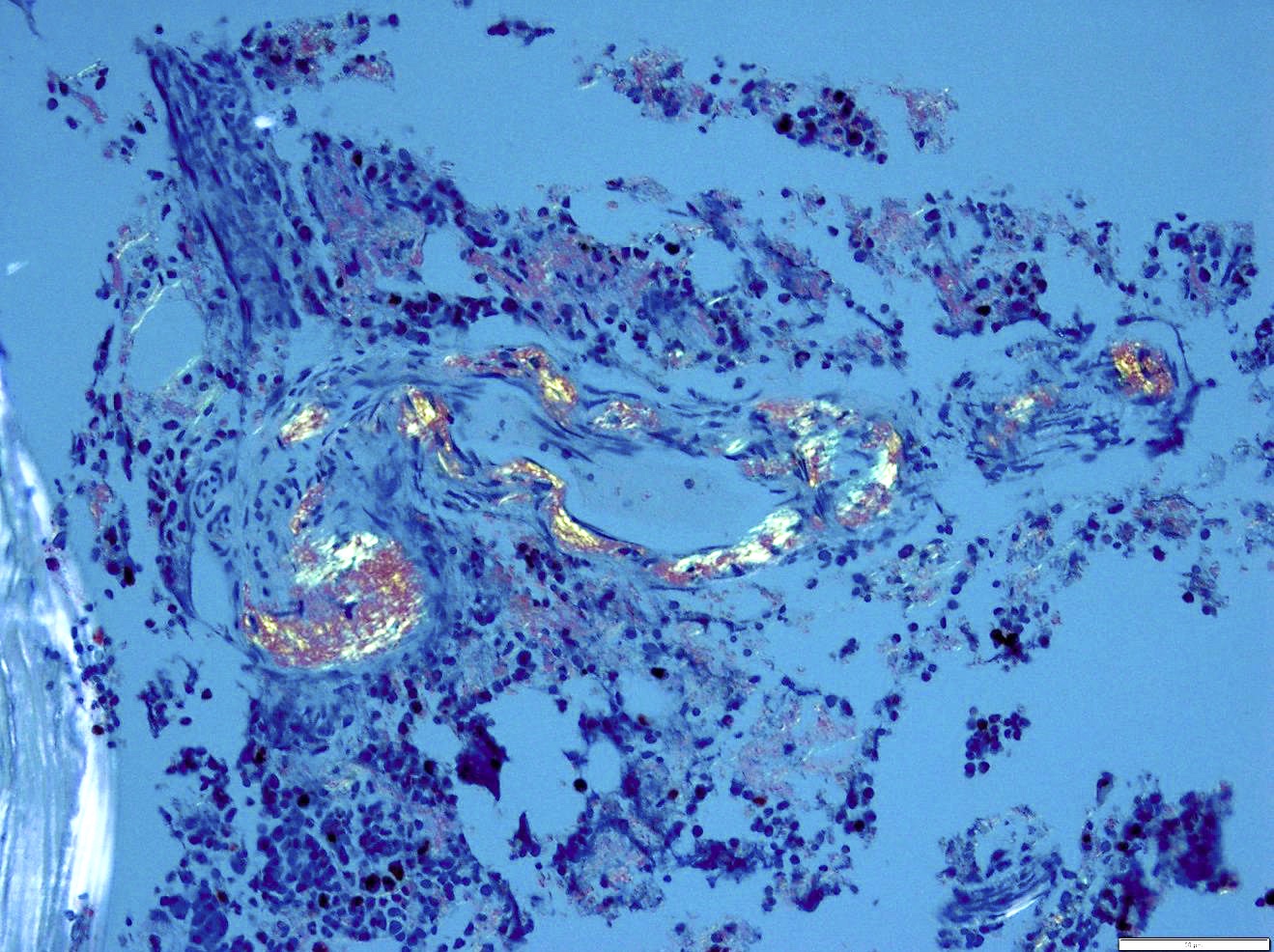
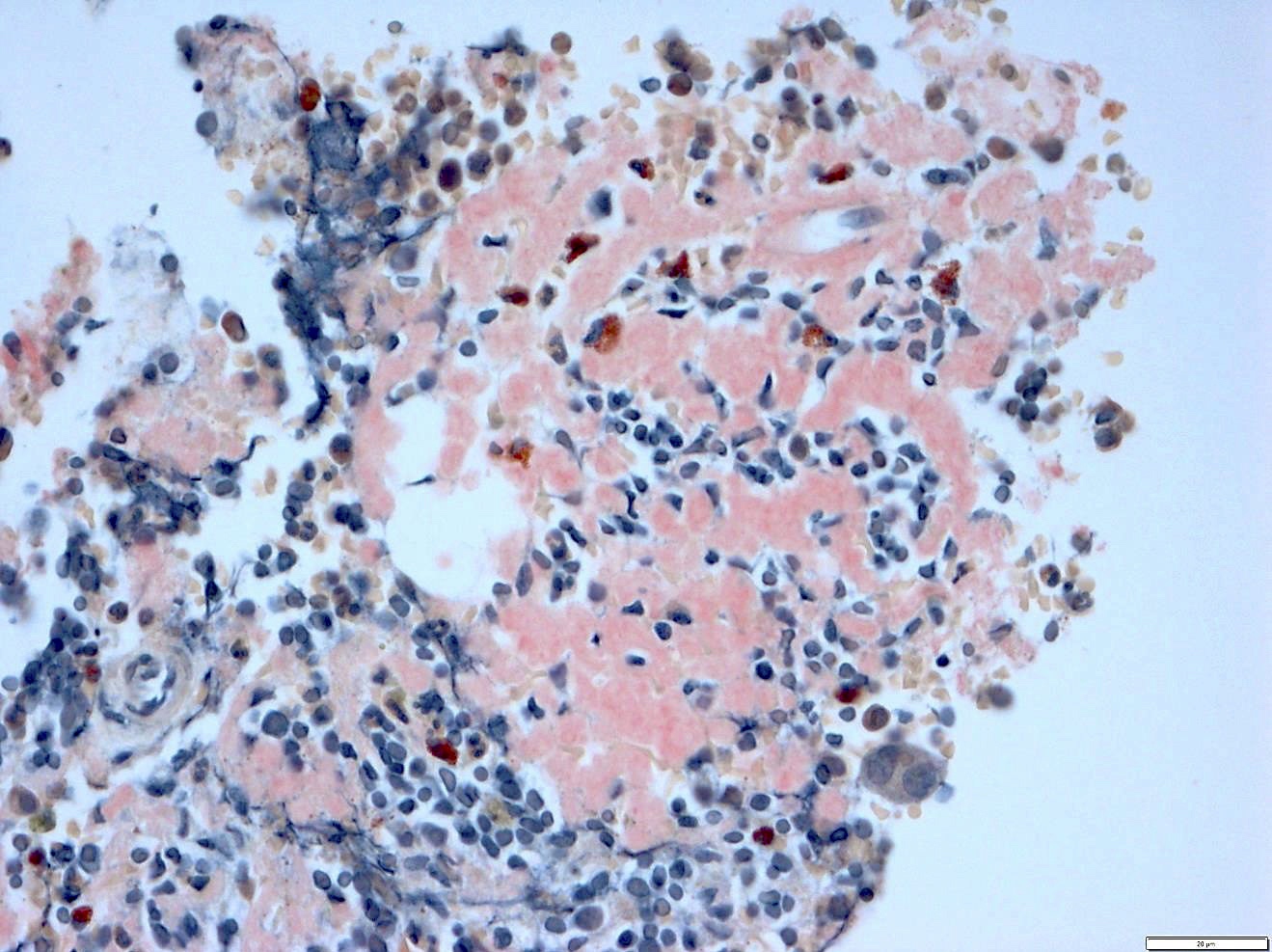
Bone marrow with extensive involvement by amyloid
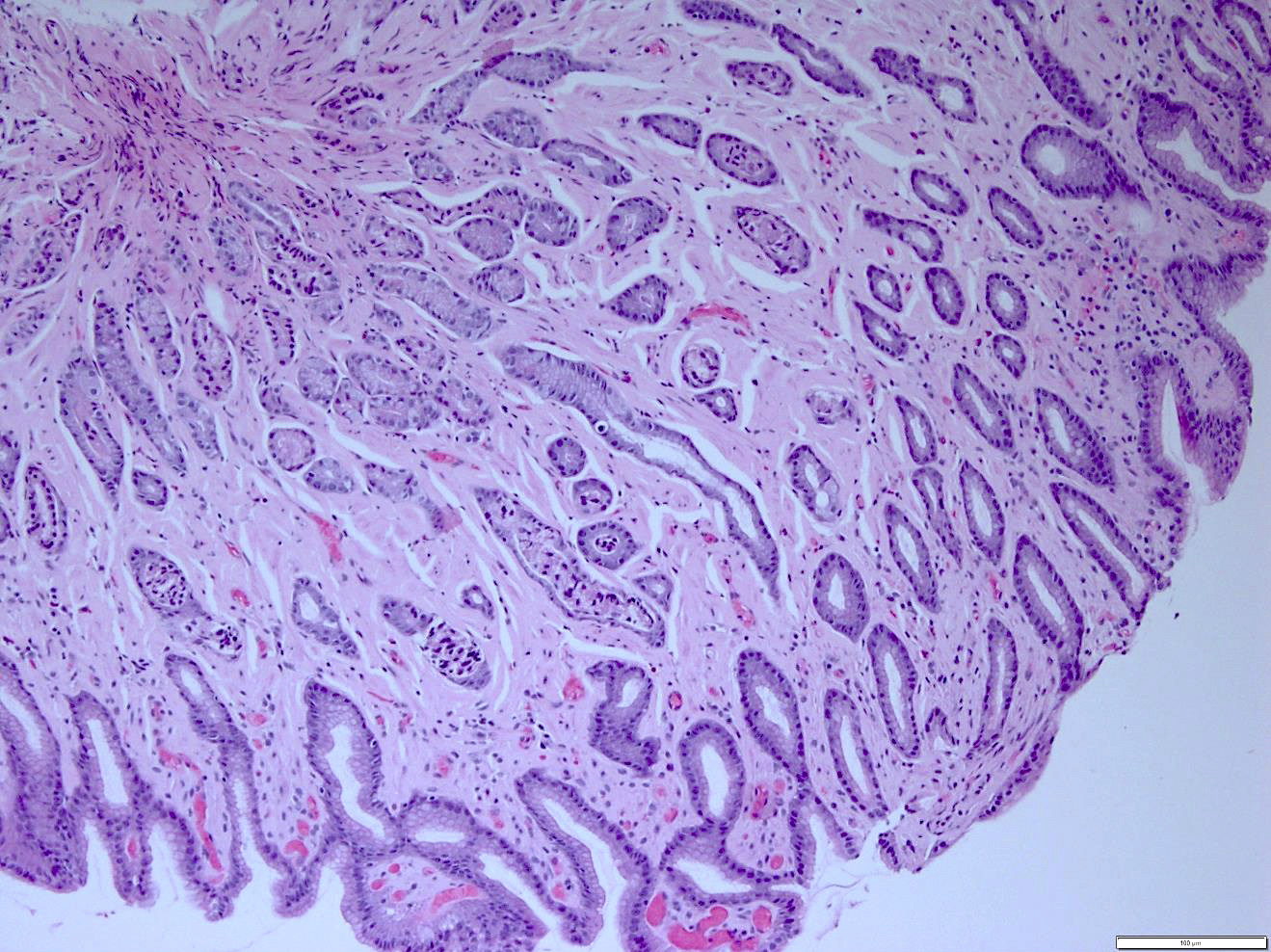

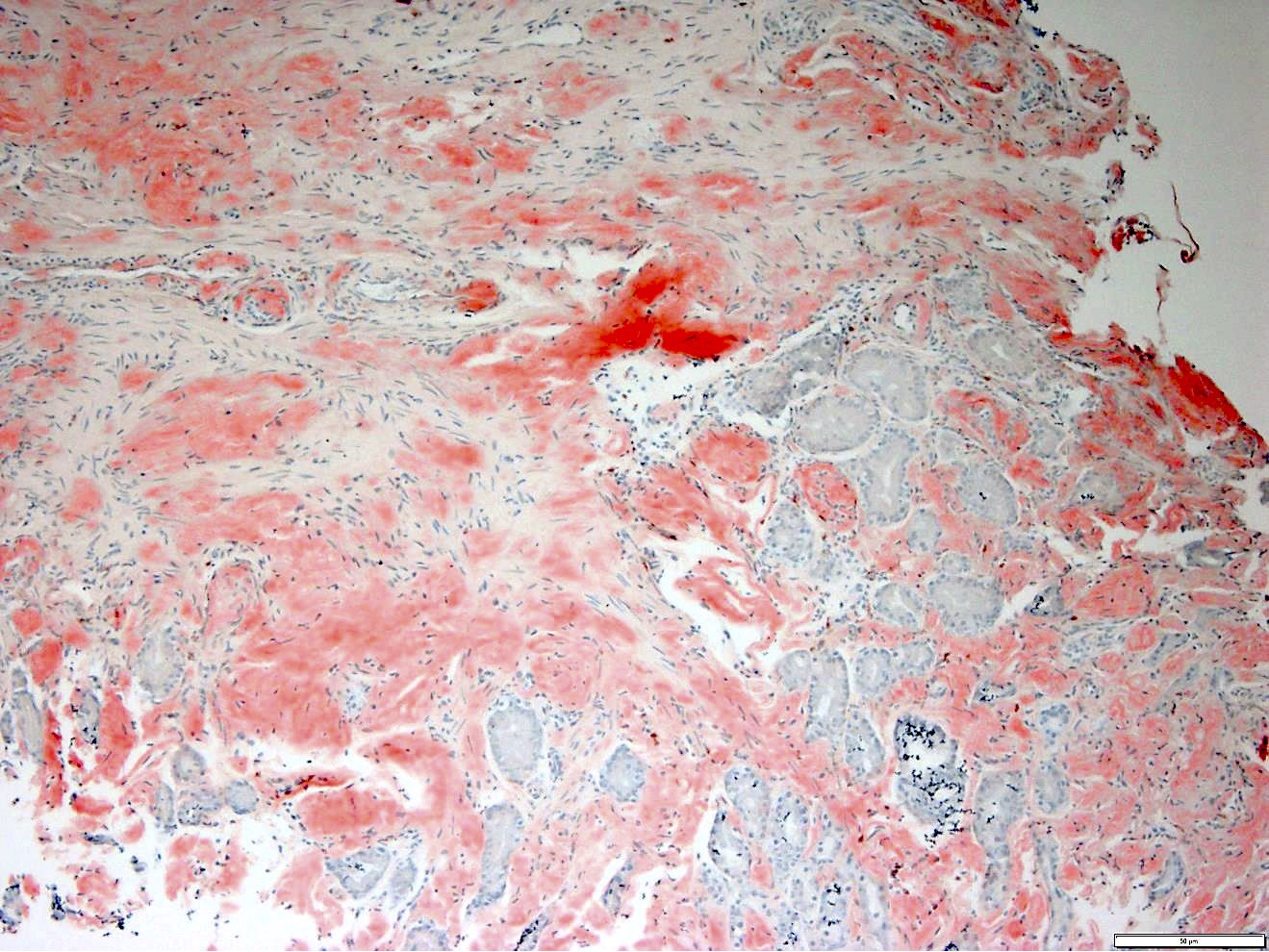

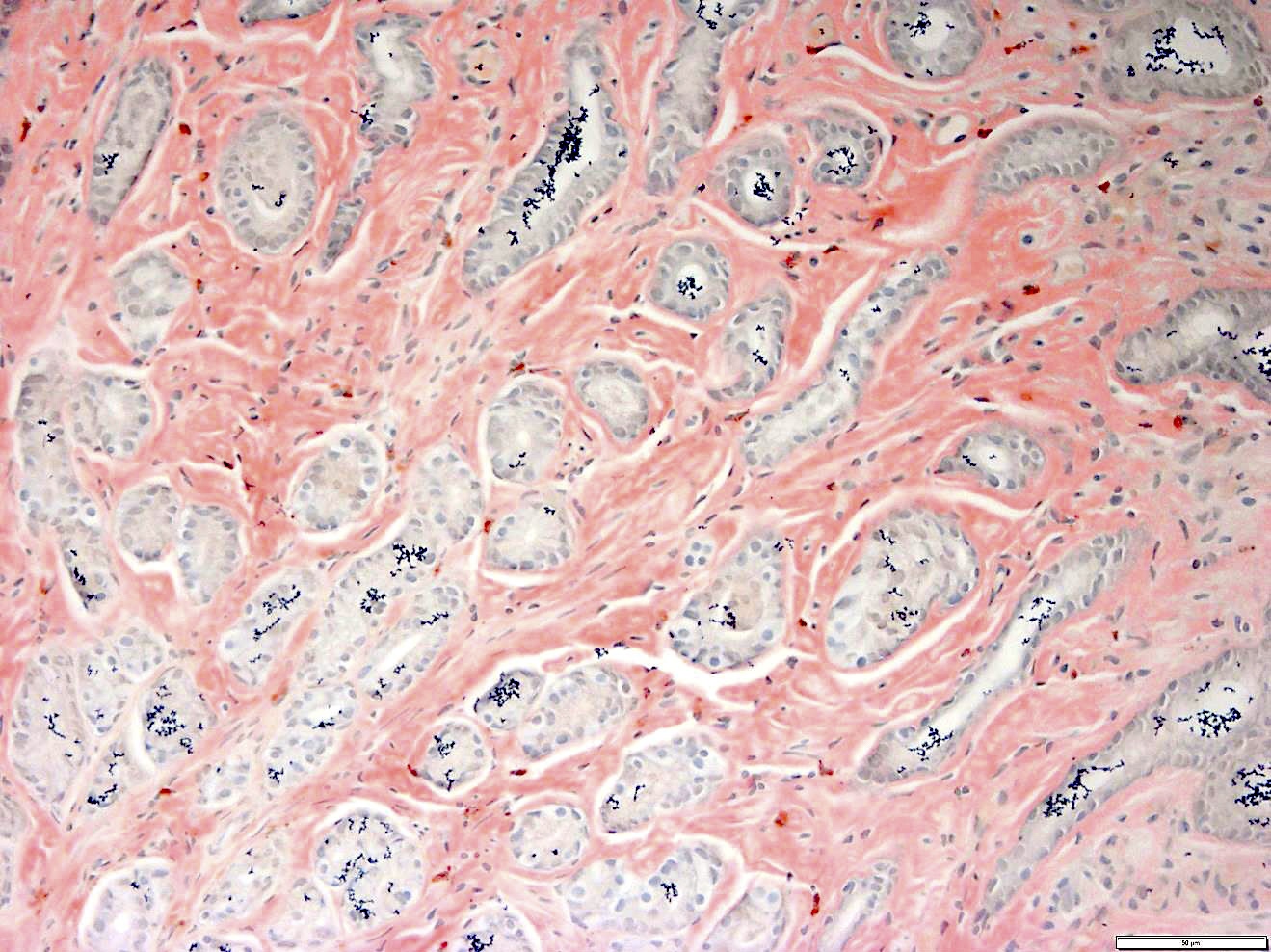
Gastric biopsies from the same patient showing extensive involvement by amyloid
- Marrow aspirate smears typically contain > 10% plasma cells
- Plasma cells may show a range of morphologies including those seen in myeloma, vacuolated as seen in mu heavy chain disease may be seen
- If extensive amyloid deposition, proteinaceous clumps (lightly eosinophilic to basophilic) may be present
- Congo red: ordinary light imparts a pink to red color to amyloid deposits; the beta pleated sheet structure of amyloid produces an apple green birefringence under polarized microscopy when stained by Congo red or sirius red stain
- PAS: moderately positive
- Iodine: stains deep brown but turns blue after treatment with concentrated sulfuric acid ("iodine sulphuric reaction" historical review Histochemistry 1976;49:131)
- Crystal violet and toluidine blue: metachromatic staining
- Thioflavin T and thioflavin S: exhibits fluorescence
- Clonal plasma cells can also be identified
- Trichrome for collagen
- Extracellular, amorphous, straight nonbranching thin fibrils of 4 - 9 nm
Contributed by Karen Vanderbilt and Bruce Goldman, M.D.
Amyloid by electron micrograph
- Genetic abnormalities similar to non-IgM MGUS; e.g. 13q14 deletion, 1q21 gain
- t(11;14) overrepresented (present > 40% of cases) and may be associated with worse survival in contrast to the favorable prognosis in myeloma (Hematology Am Soc Hematol Educ Program 2010;2010:287)
- Not typically seen are t(4;14) and del 17p
- Light chain or heavy chain deposition disease: plasma cell tumors that secrete abnormal light or heavy chain which deposit in tissues and cause organ dysfunction
- Do NOT have amyloid beta pleated sheets or bind Congo red
- Often do have monoclonal gammopathy (75%), most often kappa (80%), variable myeloma
- Secondary or hereditary amyloidosis: laser microdissection and analysis by mass spectrometry most reliable to distinguish; IHC with anti-amyloid fibril antibodies can also be used; presence of MGUS does not exclude concurrent secondary or familial amyloidosis (Engl J Med 2002;346:1786)
- Serous fat atrophy: extensive extravascular deposits may resemble serous fat atrophy (clinical history, Congo red staining will help distinguish)
- Extent of cardiac involvement
- Liver involvement
- Macroglossia
- Nephrotic range proteinuria
- Presence of myeloma
Comment Here
Reference: Primary amyloidosis
- del 17p
- Hyperdiploidy
- Monosomy 13q14
- t(11;14)
- TP53 mutation
While all of these alterations have been described in plasma cell myeloma and may be seen in primary amyloidosis, the t(11;14) translocation is particularly overrepresented in primary amyloidosis (> 40% of cases).13q14 deletion is also common but del 17p is rarely seen.
Comment Here
Reference: Primary amyloidosis
- Primary myelofibrosis is an uncommon myeloproliferative neoplasm characterized by a proliferation of predominantly abnormal megakaryocytes and granulocytes in the bone marrow, which in fully developed disease is associated with reactive deposition of fibrous connective tissue and extramedullary hematopoiesis
- Shows a stepwise evolution from an initial prefibrotic / early stage to overt fibrotic stage
- Is a clonal stem cell defect characterized by:
- Impaired medullary hematopoiesis
- Granulocytic and megakaryocytic proliferation in the bone marrow
- Gradual increase in bone marrow reticulin and collagen fibrosis
- Extramedullary hematopoiesis in the spleen, liver and other organs
- Impaired medullary hematopoiesis
- Chronic idiopathic myelofibrosis, myelofibrosis / sclerosis with myeloid metaplasia, agnogenic myeloid metaplasia, megakaryocytic myelosclerosis, idiopathic myelofibrosis, myelofibrosis with myeloid metaplasia, myelofibrosis as a result of myeloproliferative disease (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Myelofibrosis refers to the increase in the amount and density of reticulin fibers in the bone marrow (can be caused by infections, inflammatory, neoplasms, etc)
- ICD-10: D75.81 - myelofibrosis
- Least common of all myeloproliferative neoplasms
- Estimated annual incidence of overt phase is 0.5 - 1.5 cases per 100,000 population
- Prefibrotic / early phase accounts for 30 - 50% of all cases
- Increasing prevalence due to earlier diagnosis (pre-primary myelofibrosis)
- Equal gender predisposition
- Disease of older adults (mean age 60 years)
- Extremely rare in children; however, some childhood cases may be inherited and associated with other anomalies (Jaffe: Hematopathology, 2nd Edition, 2016)
- Increased incidence in Ashkenazi Jews (Hsi: Hematopathology - Foundations in Diagnostic Pathology, 3rd Edition, 2017)
- Clonal stem cell defect
- Fibrosis is due to neoplastic megakaryocytes releasing platelet derived growth factor, basic fibroblast growth factor, transforming growth factor beta or other cytokines, which causes nonneoplastic fibroblasts in marrow to deposit collagen
- Myelofibrosis and osteosclerosis are secondary changes due to the abnormal release of growth factors and fibrogenic cytokines
- Prominent angiogenesis in the bone marrow and spleen due to increased serum levels of vascular endothelial growth factor
- Teardrop red blood cells and leukoerythroblastosis are caused by extramedullary hematopoiesis
- Presentation
- Asymptomatic (30%)
- Symptomatic (70%)
- Splenomegaly (90%): often considered a hallmark
- Hepatomegaly (50%): portal hypertension can develop as a complication of hepatomegaly and may precede the onset of the disease (Gastroenterology 1988;94:1063)
- Constitutional symptoms such as fatigue, dyspnea, weight loss, night sweats, low grade fever and cachexia
- Fatigue is the most common presenting symptom
- Anemia
- Leukocytosis or thrombocytosis is common; leukopenia or thrombocytopenia is less common
- Gouty arthritis and renal stones
- Marked thrombocytosis may lead to thrombosis or hemorrhagic episodes
- Uncommon symptoms include pruritus and pulmonary hypertension (Am J Hematol 2012;87:136, Leukemia 2008;22:646)
- 2 phases (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017):
- Prefibrotic:
- Usually presents with thrombocytosis
- No blasts
- No / borderline anemia
- No / borderline splenomegaly
- Normal or borderline increased lactate dehydrogenase
- Misdiagnosis of essential thrombocythemia is possible if the bone marrow biopsy is not carefully examined
- Overt:
- Thrombocytopenia
- Leukoerythroblastosis
- Anemia
- Splenomegaly
- Increased lactate dehydrogenase
- Increased myeloblasts can be seen (< 20%)
- Transformation to acute myeloid leukemia occurs in ~5 - 20% at a median of 3 years after the diagnosis of the overt phase (Jaffe: Hematopathology, 2nd Edition, 2016)
- 2 factors at the time of diagnosis are considered predictors of leukemic transformation: circulating blasts ≥ 3% and platelet count < 100,000/μL (Cancer 2008;112:2726)
- Prefibrotic:
- Extramedullary hematopoiesis can form masses
- May coexist with systemic mastocytosis (J Mol Diagn 2008;10:58)
- Survival 3.5 - 5.5 years
- Diagnostic criteria for primary myelofibrosis, prefibrotic / early stage: must meet all 3 major criteria plus at least 2 minor criteria (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Major
- Major criterion 1: megakaryocyte proliferation and atypia in the absence of reticulin fibrosis, accompanied by increased marrow cellularity, granulocytic proliferation and often decreased erythropoiesis
- Major criterion 2: not meeting WHO criteria for other myeloproliferative neoplasms or myelodysplastic syndromes
- Major criterion 3: demonstration of JAK2, CALR or MPL mutation OR other clonal marker OR no evidence of reactive marrow fibrosis
- Minor
- Minor criterion 1: leukoerythroblastosis
- Minor criterion 2: increased serum lactate dehydrogenase
- Minor criterion 3: anemia
- Minor criterion 4: palpable splenomegaly
- Comparing WHO 2017 to WHO 2008, only minor changes to the diagnostic criteria; the change involved the addition of CALR or MPL mutations in addition to the JAK2 mutation
- Major
- Diagnostic criteria for primary myelofibrosis, overt fibrotic stage: all 3 major criteria and at least 1 minor criterion must be confirmed in 2 consecutive determinations (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Major
- Major criterion 1: megakaryocytic proliferation and atypia, accompanied by reticulin or collagen fibrosis grades 2 or 3
- Major criterion 2: not meeting WHO criteria for other myeloproliferative neoplasms or myelodysplastic syndromes
- Major criterion 3: demonstration of JAK2, CALR or MPL mutation OR other clonal marker OR no evidence of reactive marrow fibrosis
- Minor
- Minor criterion 1: anemia not attributed to a comorbid condition
- Minor criterion 2: leukocytosis
- Minor criterion 3: palpable splenomegaly
- Minor criterion 4: increased serum lactate dehydrogenase
- Minor criterion 5: leukoerythroblastosis
- Major
- Complete blood count / peripheral blood smear
- Prefibrotic:
- Initially normal or increased blood counts
- Mild neutrophilia with a left shift
- Thrombocytosis (mild / moderate)
- No / borderline anemia
- No myeloblasts
- No leukoerythroblastosis
- Initially normal or increased blood counts
- Overt:
- Thrombocytopenia with bizarre abnormal large platelets with altered granulation; in addition, fragmented megakaryocytes can be seen on the peripheral smear
- Leukoerythroblastosis and anemia
- Myeloblasts (usually > 5%)
- Prefibrotic:
- Lactate dehydrogenase
- Prefibrotic: normal or borderline increased lactate dehydrogenase
- Overt: increased lactate dehydrogenase
- Alkaline phosphatase, uric acid, leukocyte alkaline phosphatase and vitamin B12 are increased (Leuk Lymphoma 1996;22:303)
- Bone marrow aspirate / biopsy
- Prefibrotic:
- Hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations
- Usually morphologically more variable in size, atypical and bizarre than other myeloproliferative neoplasms
- Bulbous hypolobated nucleus
- Erythropoiesis is reduced
- Absent or only slight reticulin fibrosis
- Hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations
- Overt:
- Hypocellular bone marrow with usually alternating cellular and hypocellular regions
- Atypical megakaryocytes can form clusters or sheets
- More variable in size, atypical and bizarre than other myeloproliferative neoplasms
- Bulbous hypolobated nucleus
- Marked reticulin or collagen fibrosis
- New bone formation and osteosclerosis
- Bone marrow aspirate is often difficult (dry tap)
- Prefibrotic:
- See also Molecular description
- Has the least favorable prognosis among the myeloproliferative neoplasms
- Patients are at risk of death due to disease progression, leukemic transformation, thrombohemorrhagic complications and infections
- Survival
- Prefibrotic: 10 - 15 years
- Overt: 3 - 5 years
- Dynamic International Prognostic Scoring System (DIPSS plus) (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Includes 8 predictors of inferior survival:
- Patient age > 65 years
- Red blood cell transfusion dependency
- Unfavorable karyotype
- Hemoglobin < 10 g/dL
- White blood cells > 25 × 109/L
- Constitutional symptoms (fever, night sweats, weight loss)
- Circulating blasts ≥ 1%
- Platelet count < 100 × 109/L
- Risk status is defined by the number of adverse prognostic factors present: 0 (low risk), 1 (intermediate - 1 risk), 2 or 3 (intermediate - 2 risk) or > 4 (high risk)
- Includes 8 predictors of inferior survival:
- Absolute monocyte count ≥ 1 × 109/L confers an unfavorable outcome
- Unfavorable karyotype
- Complex karyotype, sole abnormality
- 2 abnormalities that include +8, -7 / 7q-, i(17q), inv(3), -5 / del(5q), 12p- or 11q23.3 rearrangement
- CALR mutation confers a better prognosis compared to other mutations
- 51 year old Korean man with tumor lysis syndrome (Am J Case Rep 2019;20:146)
- 54 year old woman with massive hemothorax due to primary extramedullary hematopoiesis of the pleura (Cureus 2018;10:e3675)
- 71 year old patient treated with ruxolitinib (Drugs Context 2019;8:212569)
- 80 year old man with a JAK2 V617F mutation transforming into Philadelphia chromosome positive acute myeloid leukemia (Biomark Res 2018;6:33)
- 86 year old woman presenting with chronic lymphocytic lymphoma (Case Rep Hematol 2018;2018:7426739)
- Management based on the risk stratification by
- GIPSS (Genetically Inspired Prognostic Scoring System): uses karyotyping, driver mutations and HMA mutations
- MIPSS70+ v2.0 (Mutation and Karyotype Enhanced International Prognostic Scoring System) criteria: uses clinical information, karyotyping and mutations
- Higher risk:
- Eligible for transplantation: allogeneic hematopoietic cell transplantation
- Ineligible for transplantation: enrollment in a clinical trial or treatment with ruxolitinib or hydroxyurea for relief of symptoms (N Engl J Med 2012;366:787)
- Lower risk:
- Asymptomatic: observation
- Symptomatic: treatment with ruxolitinib or hydroxyurea
- Bone marrow:
- Prefibrotic stage:
- Hypercellular with large, dysplastic, clustered (loose or tight) megakaryocytes and excess granulocytes
- Increased reticulin is present around clusters of megakaryocytes; megakaryocytes have aberrant nuclear/cytoplasmic ratios and hyperchromatic, bulbous or irregularly folded nuclei
- Often bare megakaryocytic nuclei
- Megakaryocytic features are most useful to distinguish this stage of primary myelofibrosis from essential thrombocythemia
- Fibrotic phase:
- Hypocellular and diffusely fibrotic bone marrow with atypical streaming megakaryocytes
- Marrow osteosclerosis with irregular, broad bony trabeculae
- Markedly dilated sinuses; associated with dry bone marrow taps
- Prefibrotic stage:
- Spleen: red pulp sinuses contain megakaryocytes, granulocyte precursors, nucleated red cells; may be nodules of extramedullary hematopoiesis
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H. and AFIP images

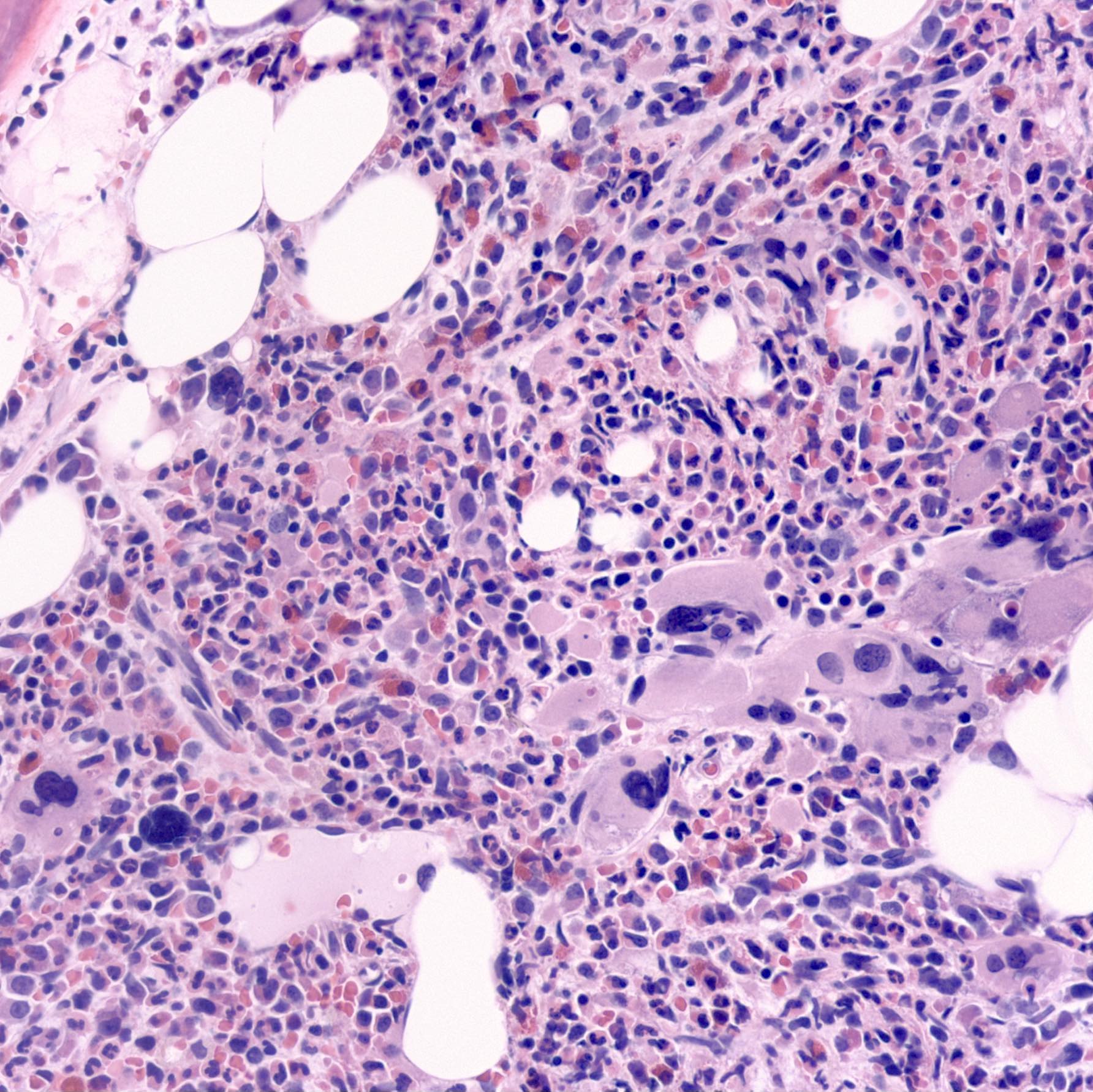
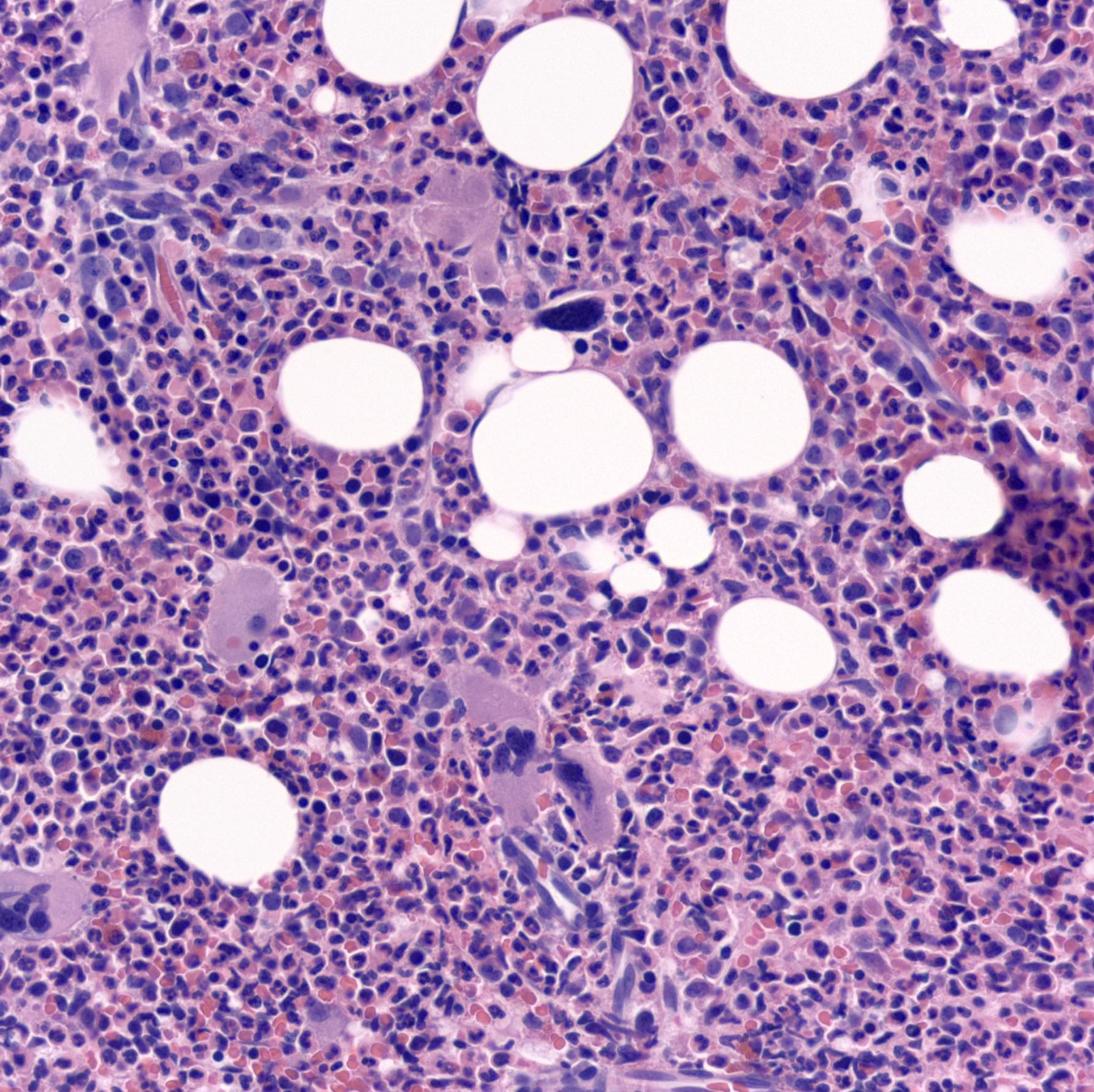
Atypical megakaryocytes
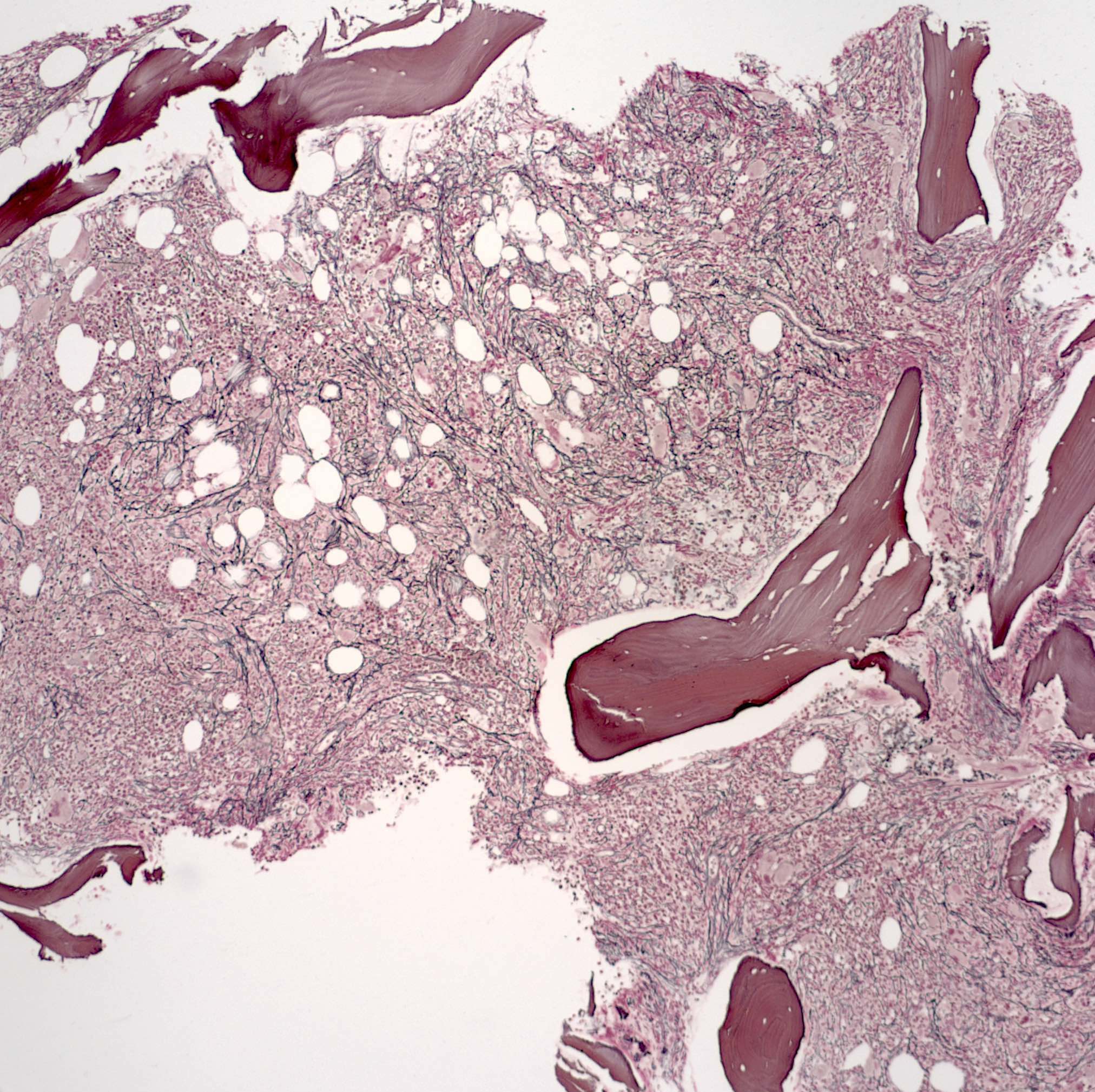
Fibrotic bone marrow
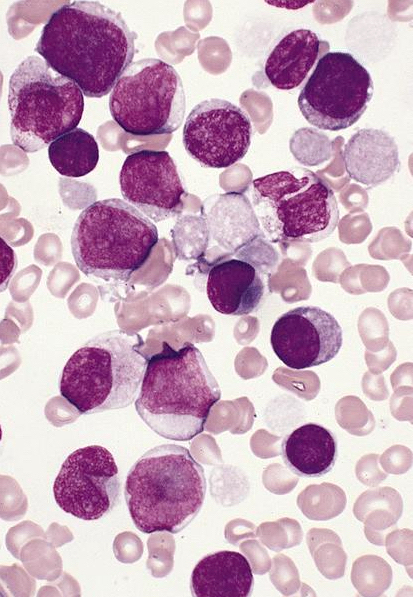
Blast transformation
- Leukoerythroblastosis (immature granulocytes and normoblasts in peripheral blood) is common in later phases; also dacrocytes (teardrop erythrocytes)
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H. and AFIP images
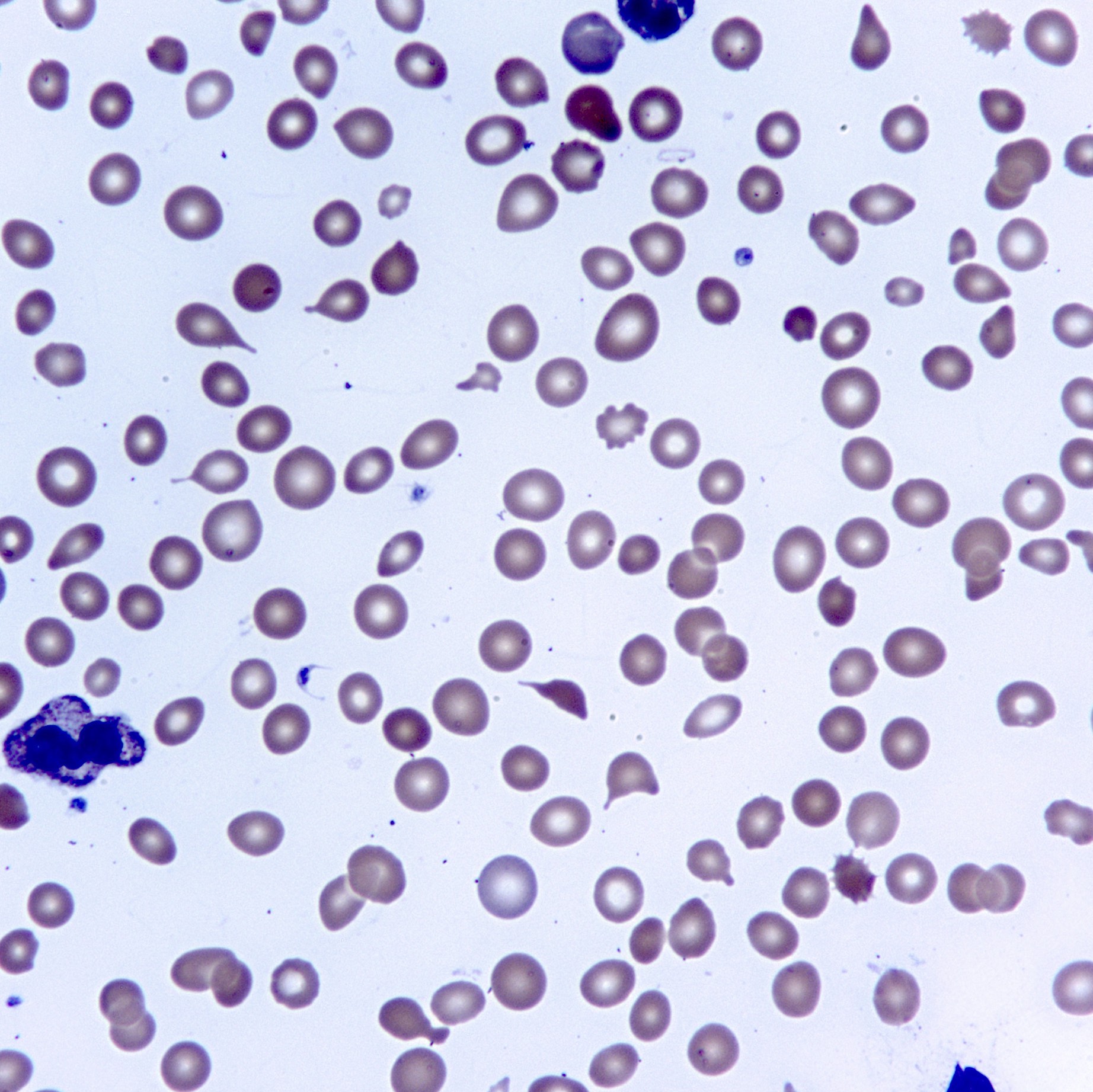
Dacrocytes

Cell intermediate
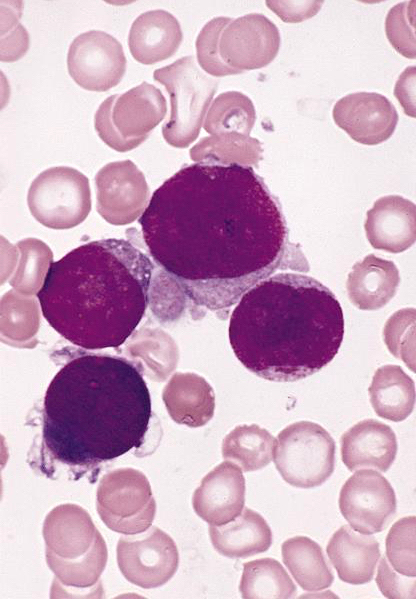
Blast transformation
- VEGF (high expression) (Am J Clin Pathol 2007;128:966)
- CD105 (to assess microvascular density, may have prognostic value) (Mod Pathol 2004;17:1513)
- Chloroacetate esterase helps mark granulocytic proliferation in the prefibrotic phase
- Normal number and phenotype myeloblasts with normal myeloid scatter by CD45 / side scatter
- Normal CD10 / CD13 / CD16 / CD11b myeloid maturation pattern and all other myeloid markers are normally expressed, hence there is no immunophenotypic evidence of myelodysplasia
- No evidence for monoclonal B cell lymphoproliferative disease
- Although no unusual phenotype T cells are identified, surface markers will not routinely detect monoclonal T lymphocytes
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.

Normal myeloid maturation
- No specific genetic marker
- No BCR-ABL fusion gene (or classify as chronic myeloid leukemia); presence of del(13)(q12-22) or der(6)t(1;6)(q21-23;p21.3) is strongly suggestive of primary myelofibrosis
- Shows karyotypic abnormalities in up to 50% of cases
- Frequent abnormalities include del(20q), del(13q), +8, +9 and abnormalities of 1q
- Driver mutations
- JAK2 V617F mutation (50 - 60%)
- CALR mutations (25 - 30%)
- MPL mutations (5 - 10%)
- Triple negative for mutations in JAK2, CALR and MPL (8 - 12%)
- Other gene mutations include ASXL1, EZH2, TET2, IDH1 / IDH2, SRSF2 or SR3B1
- Driver mutational profile is associated with overall survival
- CALR mutated cases have better survival compared to others
- Triple negative cases have the shortest survival compared to others
- Bone marrow, biopsy and aspirate:
- Myeloproliferative neoplasm, favoring prefibrotic primary myelofibrosis (see comment)
- Comment: The marrow biopsy shows morphologic features of a myeloproliferative neoplasm, in conjunction with thrombocytosis. The major differential diagnosis includes essential thrombocythemia and prefibrotic stage of primary myelofibrosis. Overall, we favor a diagnosis of the latter, due to hypercellular marrow, distribution of megakaryocytes (focal clusters) and markedly dysplastic cytology of megakaryocytes. Recommend correlation with clinical findings (presence of splenomegaly and disease progression) and molecular studies (JAK2, CALR and MPL mutational studies).
- Complete blood count (9/17/2018), by report: hemoglobin - 11.4 g/dL, mean corpuscular volume - 100 fL, white blood cells - 21.7 K/uL, platelets - 698 K/uL
- Bone marrow biopsy microscopic description:
- Hypercellular marrow for age (80% cellular). Megakaryocytes are increased in number, forming focal loose clusters and showing abnormal morphology including hypersegmented nuclei with hyperchromasia. The myeloid:erythroid ratio is markedly increased. Erythroid elements exhibit maturation. Myeloid elements exhibit maturation. Myeloblasts appear to be increased. No lymphoid aggregates are identified. Granulomas are absent. Reticulin stain reveals that reticulin is mildly increased (MF1/3). Trabecular bone is normal.
- Bone marrow aspirate microscopic description:
- The marrow aspirate smears are spicular and cellular. Megakaryocytes are increased and show prominent dysmegakaryopoiesis, including hyper and hyposegmented nuclei and increased nuclear/cytoplasmic ratio. The myeloid:erythroid ratio is approximately 4:1. Erythroid maturation is identified. Myeloid maturation is slightly left shifted. Prussian blue iron stain shows # storage iron and no significant increase in ring sideroblasts.
- Aspirate cell count: A 207 cell count reveals 1% blasts, 15% promyelocytes / myelocytes, 57% maturing granulocyte forms, 20% erythroid forms, 3% lymphocytes, 2% eosinophils, 1% plasma cells, 1% basophils / mast cells and 2% monocytes.
- Other causes of leukoerythroblastosis or dry taps:
- Granulomatous marrow disease, metastases to marrow (desmoplasia but no increased reticulin) or lymphoma
- Other myeloid disorders:
- Myeloproliferative neoplasms:
- Chronic myelogenous leukemia: positive BCR / ABL translocation
- Polycythemia vera: dispersed less atypical megakaryocytes and prominent erythroid element
- Essential thrombocythemia: uniformly enlarged megakaryocytes with deeply lobulated (staghorn) nuclei, in loose clusters; normal myeloid:erythroid ratio
- Myelodysplastic / myeloproliferative neoplasms:
- Show hybrid myelodysplastic and myeloproliferative features manifested by at least 1 cytopenia and at least 1 cytosis in blood
- Myeloid maturation is intact and usually mature cells predominate
- Exact subtype should be diagnosed based on the specific criteria of the entity
- Myelodysplastic syndromes are
a heterogeneous group of entities that demonstrate:
- Variable degrees of ineffective hematopoiesis
- < 20% blasts in marrow
- Peripheral blood with unexplained and persistent cytopenia(s), < 20% blasts, < 1.0 x 109/L monocytes and dysplasia in 1 or more myeloid lineages
- Mastocytosis:
- Shows focal, compact aggregates of mast cells in bone marrow core biopsy
- Cells can show the following patterns of infiltration: paratrabecular, perivascular or parafollicular
- Presence of atypical mast cell supports the diagnosis
- Most cases will harbor a KIT D816V mutation
- Myeloproliferative neoplasms:
- Reactive thrombocythemia:
- Causes include exercise, allergic reaction, reaction to medications, inflammatory disorders, asplenism, infection, connective tissue disorders, metastatic cancer, lymphoproliferative disorders and iron deficiency
- Acute myelofibrosis:
- Subtype of acute myeloid leukemia that shows > 20% blasts in peripheral blood or bone marrow
- Patients also present with bone marrow fibrosis, fever and pancytopenia
- Autoimmune myelofibrosis: generally shows diffuse reticulin fibrosis and an associated autoimmune disease such as rheumatoid arthritis
- Primary hyperparathyroidism: rarely causes myelofibrosis and pancytopenia (Int J Lab Hematol 2007;29:464)
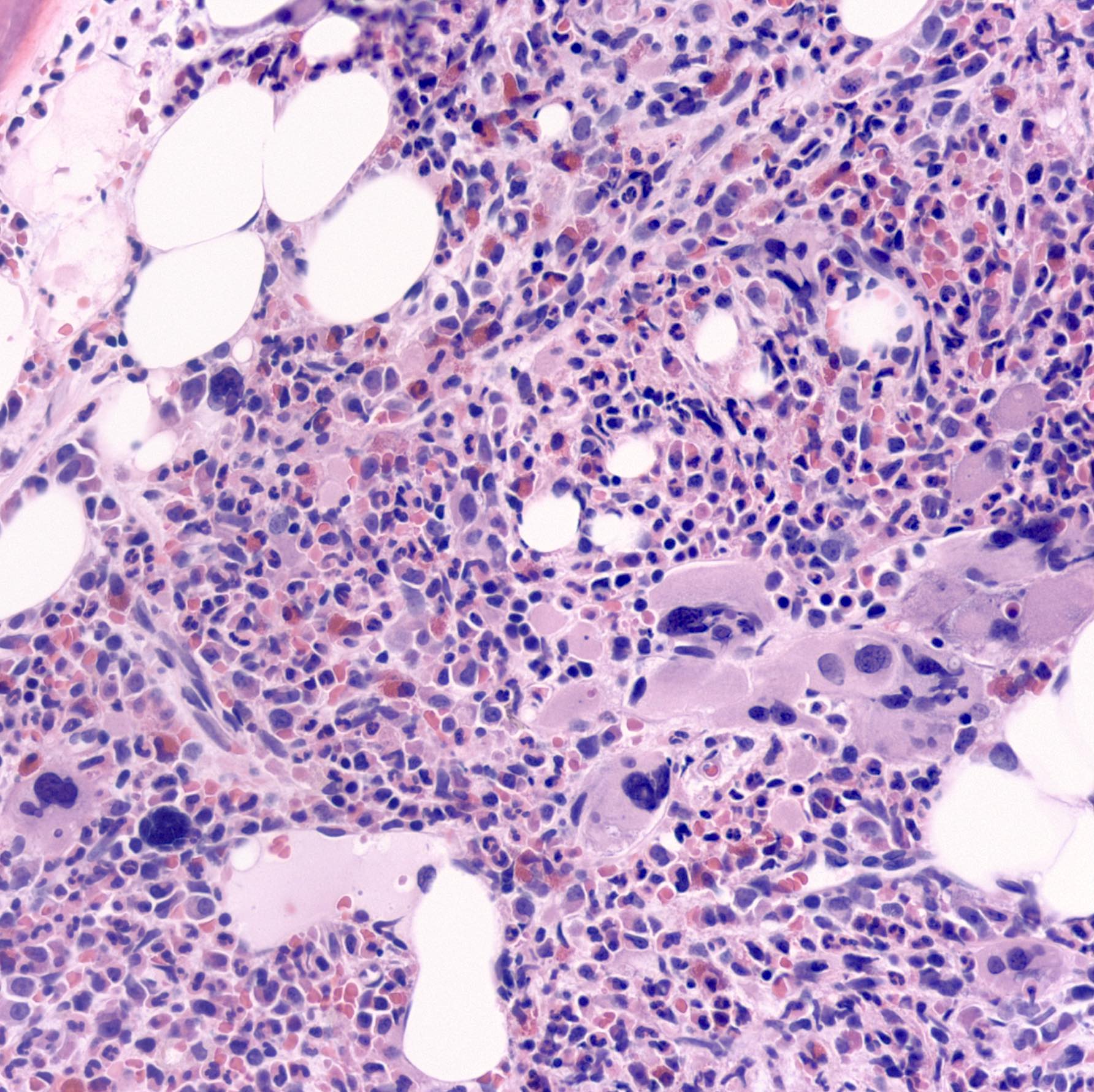
A 57 year old man presented with fatigue, splenomegaly and hepatomegaly. A complete blood count demonstrated anemia and thrombocytosis. A bone marrow aspirate and biopsy were ordered (shown above). What is the most likely diagnosis?
- Acute myeloid leukemia
- Chronic myelogenous leukemia
- Myelodysplastic syndrome
- Primary myelofibrosis
The symptoms and bone marrow biopsy support the diagnosis of primary myelofibrosis. The biopsy above shows hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations.
Comment Here
Reference: Primary myelofibrosis
- ABL
- CALR
- JAK2
- MPL
Primary myelofibrosis cases harbor a phenotypic driver mutation in JAK2 V617F mutation (50 - 60%), CALR mutations (25 - 30%), MPL mutations (5 - 10%), triple negative for mutations in JAK2, CALR and MPL (8 - 12%).
Comment Here
Reference: Primary myelofibrosis
- Primary myelofibrosis is an uncommon myeloproliferative neoplasm characterized by a proliferation of predominantly abnormal megakaryocytes and granulocytes in the bone marrow, which in fully developed disease is associated with reactive deposition of fibrous connective tissue and extramedullary hematopoiesis
- Shows a stepwise evolution from an initial prefibrotic / early stage to overt fibrotic stage
- Is a clonal stem cell defect characterized by
- Impaired medullary hematopoiesis
- Granulocytic and megakaryocytic proliferation in the bone marrow
- Gradual increase in bone marrow reticulin and collagen fibrosis
- Extramedullary hematopoiesis in the spleen, liver and other organs
- Impaired medullary hematopoiesis
- The International Consensus Classification (ICC) 2022 distinguishes prefibrotic from overtly fibrotic PMF (Blood 2022;140:1200)
- Myelofibrosis refers to the increase in the amount and density of reticulin fibers in the bone marrow (can be caused by infections, inflammatory, neoplasms, etc.)
- ICD-10: D75.81 - myelofibrosis
- Estimated annual incidence of overt phase is 0.5 - 1.5 cases per 100,000 population
- Prefibrotic / early phase accounts for 30 - 50% of all cases
- Increasing prevalence due to earlier diagnosis (preprimary myelofibrosis)
- Equal gender predisposition (Am J Hematol 2014;89:915)
- Disease of older adults (mean age: 60 years)
- Extremely rare in children; however, some childhood cases may be inherited and associated with other anomalies (Curr Hematol Malig Rep 2020;15:141, Jaffe: Hematopathology, 2nd Edition, 2016)
- Increased incidence in individuals of Ashkenazi Jewish descent (Leuk Lymphoma 1992;7:251, Hsi: Hematopathology - Foundations in Diagnostic Pathology, 3rd Edition, 2017)
- Mainly blood and bone marrow
- Other variably included sites: liver, spleen
- Clonal stem cell defect
- Fibrosis is due to neoplastic megakaryocytes releasing platelet derived growth factor, basic fibroblast growth factor, transforming growth factor beta or other cytokines, which causes nonneoplastic fibroblasts in marrow to deposit collagen
- Myelofibrosis and osteosclerosis are secondary changes due to the abnormal release of growth factors and fibrogenic cytokines
- Prominent angiogenesis in the bone marrow and spleen due to increased serum levels of vascular endothelial growth factor
- Teardrop red blood cells and leukoerythroblastosis are caused by extramedullary hematopoiesis
- References: Int J Lab Hematol 2023:45:59, Leukemia 2022;36:1703
- Presentation
- Asymptomatic (30%)
- Symptomatic (70%)
- Splenomegaly (90%): often considered a hallmark
- Hepatomegaly (50%): portal hypertension can develop as a complication of hepatomegaly and may precede the onset of the disease (Gastroenterology 1988;94:1063)
- Constitutional symptoms such as fatigue, dyspnea, weight loss, night sweats, low grade fever and cachexia
- Fatigue is the most common presenting symptom
- Anemia
- Leukocytosis or thrombocytosis is common; leukopenia or thrombocytopenia is less common
- Gouty arthritis and renal stones
- Marked thrombocytosis may lead to thrombosis or hemorrhagic episodes
- Uncommon symptoms include pruritus and pulmonary hypertension (Am J Hematol 2012;87:136, Leukemia 2008;22:646)
- 2 phases
- Prefibrotic
- Usually presents with thrombocytosis
- No blasts
- No / borderline anemia
- No / borderline splenomegaly
- Normal or borderline increased lactate dehydrogenase
- Misdiagnosis of essential thrombocythemia is possible if the bone marrow biopsy is not carefully examined
- Overt / fibrotic
- Thrombocytopenia
- Leukoerythroblastosis
- Anemia
- Splenomegaly
- Increased lactate dehydrogenase
- Increased myeloblasts can be seen (< 20%); 10 - 19% blasts indicates progression to accelerated phase
- Transformation to acute myeloid leukemia occurs in ~5 - 20% at a median of 3 years after the diagnosis of the overt phase (Jaffe: Hematopathology, 2nd Edition, 2016)
- 2 factors at the time of diagnosis are considered predictors of leukemic transformation: circulating blasts ≥ 3% and platelet count < 100,000/μL (Cancer 2008;112:2726)
- Prefibrotic
- Extramedullary hematopoiesis can form masses
- Survival: 3.5 - 5.5 years
| Diagnostic criteria for prefibrotic primary myelofibrosis | Diagnostic criteria for primary myelofibrosis, fibrotic stage |
| Diagnosis of prefibrotic primary myelofibrosis requires that all 3 major criteria and at least 1 minor criterion are met | Diagnosis of overt primary myelofibrosis requires that all 3 major criteria and at least 1 minor criterion are met |
| Major criteria | |
|
|
| Minor criteria | |
Presence of at least 1 of the following, confirmed in 2 consecutive determinations
|
Presence of at least 1 of the following, confirmed in 2 consecutive determinations
|
- Complete blood count / peripheral blood smear
- Prefibrotic
- Initially normal or increased blood counts
- Mild neutrophilia with a left shift
- Thrombocytosis (mild / moderate)
- No / borderline anemia
- No myeloblasts
- No leukoerythroblastosis
- Initially normal or increased blood counts
- Overt
- Thrombocytopenia with bizarre abnormal large platelets with altered granulation; in addition, fragmented megakaryocytes can be seen on the peripheral smear
- Leukoerythroblastosis and anemia
- Myeloblasts (usually > 5%)
- Prefibrotic
- Lactate dehydrogenase (LDH)
- Prefibrotic: normal or borderline increased lactate dehydrogenase
- Overt: increased lactate dehydrogenase
- Alkaline phosphatase, uric acid, leukocyte alkaline phosphatase and vitamin B12 are increased (Leuk Lymphoma 1996;22:303)
- Bone marrow aspirate / biopsy
- Prefibrotic
- Hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations
- Usually morphologically more variable in size, atypical and bizarre than other myeloproliferative neoplasms
- Bulbous hypolobated nucleus
- Erythropoiesis is reduced
- Absent or only slight reticulin fibrosis
- Hypercellular bone marrow with granulocytic and atypical megakaryocytic proliferations
- Overt
- Hypocellular bone marrow with usually alternating cellular and hypocellular regions
- Atypical megakaryocytes can form clusters or sheets
- More variable in size, atypical and bizarre than other myeloproliferative neoplasms
- Bulbous hypolobated nucleus
- Marked reticulin (MF2 / 3) or collagen fibrosis
- New bone formation and osteosclerosis
- Bone marrow aspirate is often difficult (dry tap)
- Prefibrotic
- See also Molecular description
- Has the least favorable prognosis among the myeloproliferative neoplasms
- Patients are at risk of death due to disease progression, leukemic transformation, thrombohemorrhagic complications and infections
- Survival
- Prefibrotic: 10 - 15 years
- Overt: 3 - 5 years
- Dynamic International Prognostic Scoring System (DIPSS plus)
- Includes 8 predictors of inferior survival:
- Patient age > 65 years
- Red blood cell transfusion dependency
- Unfavorable karyotype
- Hemoglobin < 10 g/dL
- White blood cells > 25 × 109/L
- Constitutional symptoms (fever, night sweats, weight loss)
- Circulating blasts ≥ 1%
- Platelet count < 100 × 109/L
- Risk status is defined by the number of adverse prognostic factors present
- 0 (low risk), 1 (intermediate - 1 risk), 2 or 3 (intermediate - 2 risk) or > 4 (high risk)
- Includes 8 predictors of inferior survival:
- Genetics inspired International Prognostic Scoring System (Leukemia 2018;32:1631)
- Points are scored as follows
- Very high risk karyotype f (2 points)
- Unfavorable karyotype g (1 point)
- ASXL1 mutation (1 point)
- SRSF2 mutation (1 point)
- U2AF1 Q157 mutation (1 point)
- Type 1-like CALR absent (1 point)
- Risk status is defined by the points
- 0 (low), 1 (intermediate - 1), 2 (intermediate - 2), > 3 (high)
- Points are scored as follows
- Absolute monocyte count ≥ 1 × 109/L confers an unfavorable outcome
- Unfavorable karyotype
- Complex karyotype, sole abnormality
- 2 abnormalities that include +8, -7 / 7q-, i(17q), inv(3), -5 / del(5q), 12p- or 11q23.3 rearrangement
- CALR mutation confers a better prognosis compared to other mutations
- 51 year old Korean man with tumor lysis syndrome (Am J Case Rep 2019;20:146)
- 54 year old woman with massive hemothorax due to primary extramedullary hematopoiesis of the pleura (Cureus 2018;10:e3675)
- 71 year old patient treated with ruxolitinib (Drugs Context 2019;8:212569)
- 80 year old man with a JAK2 V617F mutation transforming into Philadelphia chromosome positive acute myeloid leukemia (Biomark Res 2018;6:33)
- 86 year old woman presented with chronic lymphocytic lymphoma (Case Rep Hematol 2018;2018:7426739)
- Management based on the risk stratification by
- GIPSS (Genetically Inspired Prognostic Scoring System): uses karyotyping, driver mutations and HMA mutations
- MIPSS70+ v2.0 (Mutation and Karyotype Enhanced International Prognostic Scoring System) criteria: uses clinical information, karyotyping and mutations
- Higher risk
- Eligible for transplantation: allogeneic hematopoietic cell transplantation
- Ineligible for transplantation: enrollment in a clinical trial or treatment with ruxolitinib or hydroxyurea for relief of symptoms (N Engl J Med 2012;366:787)
- Lower risk
- Asymptomatic: observation
- Symptomatic: treatment with ruxolitinib or hydroxyurea
- Bone marrow
- Prefibrotic stage
- Hypercellular with large, dysplastic, clustered (loose or tight) megakaryocytes and excess granulocytes
- Increased reticulin is present around clusters of megakaryocytes; megakaryocytes have aberrant N:C ratios and hyperchromatic, bulbous or irregularly folded nuclei
- Often bare megakaryocytic nuclei
- Megakaryocytic features are most useful to distinguish this stage of primary myelofibrosis from essential thrombocythemia
- Fibrotic phase
- Hypocellular and diffusely fibrotic bone marrow with atypical streaming megakaryocytes
- Marrow osteosclerosis with irregular, broad bony trabeculae
- Markedly dilated sinuses; associated with dry bone marrow taps
- Prefibrotic stage
- Spleen: red pulp sinuses contain megakaryocytes, granulocyte precursors, nucleated red cells; may be nodules of extramedullary hematopoiesis
- References: Int J Lab Hematol 2023:45:59, Leukemia 2022;36:1703
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H. and AFIP
Atypical megakaryocytes
Fibrotic bone marrow
Blast transformation
Cell intermediate
- Leukoerythroblastosis (immature granulocytes and normoblasts in peripheral blood) is common in later phases; also dacrocytes (teardrop erythrocytes)
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
Dacrocytes
- VEGF (high expression) (Am J Clin Pathol 2007;128:966)
- CD105 (to assess microvascular density, may have prognostic value) (Mod Pathol 2004;17:1513)
- Chloroacetate esterase helps mark granulocytic proliferation in the prefibrotic phase
- Normal number and phenotype myeloblasts with normal myeloid scatter by CD45 / side scatter
- Normal CD10 / CD13 / CD16 / CD11b myeloid maturation pattern and all other myeloid markers are normally expressed, hence there is no immunophenotypic evidence of myelodysplasia
- No evidence for monoclonal B cell lymphoproliferative disease
- Although no unusual phenotype T cells are identified, surface markers will not routinely detect monoclonal T lymphocytes
- Reference: Clin Lab Med 2023;43:411
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
Normal myeloid maturation
- No specific genetic marker
- No BCR::ABL fusion gene (or classify as chronic myeloid leukemia); presence of del(13)(q12-22) or der(6)t(1;6)(q21-23;p21.3) is strongly suggestive of primary myelofibrosis
- Shows karyotypic abnormalities in up to 50% of cases
- Frequent abnormalities include del(20q), del(13q), +8, +9 and abnormalities of 1q
- Unfavorable karyotype (complex karyotype; > 3 abnormalities) (Virchows Arch 2023;482:53)
- Isolated +8, isolated -7/7q-, i(17q), -5/5q-, 12p-, 11q23 rearrangement or inv(3) and an abnormal karyotype with abnormalities of chromosomes 5, 7, 17 or 12p-
- Mutations (Am J Hematol 2023;98:801)
- Driver mutations
- JAK2 V617F mutation (50 - 60%)
- CALR mutations (25 - 30%)
- MPL mutations (5 - 10%)
- Triple negative for mutations in JAK2, CALR and MPL (8 - 12%)
- Other mutations
- Include ASXL1, EZH2, TET2, IDH1 / IDH2, SRSF2 or SR3B1
- Driver mutations
- Driver mutational profile is associated with overall survival
- CALR mutated cases have better survival compared to others
- Triple negative cases have the shortest survival compared to others
- Bone marrow, biopsy and aspirate:
- Myeloproliferative neoplasm, favoring prefibrotic primary myelofibrosis (see comment)
- Comment: The marrow biopsy shows morphologic features of a myeloproliferative neoplasm, in conjunction with thrombocytosis. The major differential diagnosis includes essential thrombocythemia and prefibrotic stage of primary myelofibrosis. Overall, we favor a diagnosis of the latter, due to hypercellular marrow, distribution of megakaryocytes (focal clusters) and markedly dysplastic cytology of megakaryocytes. Recommend correlation with clinical findings (presence of splenomegaly and disease progression) and molecular studies (JAK2, CALR and MPL mutational studies).
- Complete blood count (9/17/2018), by report: hemoglobin - 11.4 g/dL, mean corpuscular volume - 100 fL, white blood cells - 21.7 K/uL, platelets - 698 K/uL
- Bone marrow biopsy microscopic description:
- Hypercellular marrow for age (80% cellular). Megakaryocytes are increased in number, forming focal loose clusters and showing abnormal morphology including hypersegmented nuclei with hyperchromasia. The myeloid:erythroid ratio is markedly increased. Erythroid elements exhibit maturation. Myeloid elements exhibit maturation. Myeloblasts appear to be increased. No lymphoid aggregates are identified. Granulomas are absent. Reticulin stain reveals that reticulin is mildly increased (MF1/3). Trabecular bone is normal.
- Bone marrow aspirate microscopic description:
- The marrow aspirate smears are spicular and cellular. Megakaryocytes are increased and show prominent dysmegakaryopoiesis, including hyper and hyposegmented nuclei and increased N:C ratio. The myeloid:erythroid ratio is approximately 4:1. Erythroid maturation is identified. Myeloid maturation is slightly left shifted. Prussian blue iron stain shows # storage iron and no significant increase in ring sideroblasts.
- Aspirate cell count: A 207 cell count reveals 1% blasts, 15% promyelocytes / myelocytes, 57% maturing granulocyte forms, 20% erythroid forms, 3% lymphocytes, 2% eosinophils, 1% plasma cells, 1% basophils / mast cells and 2% monocytes.
- Other causes of leukoerythroblastosis or dry taps:
- Granulomatous marrow disease, metastases to marrow (desmoplasia but no increased reticulin) or lymphoma
- Other myeloid disorders:
- Myeloproliferative neoplasms:
- Chronic myelogenous leukemia:
- Positive BCR::ABL translocation
- Polycythemia vera:
- Dispersed less atypical megakaryocytes and prominent erythroid element
- Essential thrombocythemia:
- Uniformly enlarged megakaryocytes with deeply lobulated (staghorn) nuclei, in loose clusters; normal myeloid:erythroid ratio
- Chronic myelogenous leukemia:
- Myelodysplastic / myeloproliferative neoplasms:
- Show hybrid myelodysplastic and myeloproliferative features manifested by at least 1 cytopenia and at least 1 cytosis in blood
- Myeloid maturation is intact and usually mature cells predominate
- Exact subtype should be diagnosed based on the specific criteria of the entity
- Myelodysplastic syndromes are
a heterogeneous group of entities that demonstrate:
- Variable degrees of ineffective hematopoiesis
- < 20% blasts in marrow
- Peripheral blood with unexplained and persistent cytopenia(s), < 20% blasts, < 1.0 x 109/L monocytes and dysplasia in 1 or more myeloid lineages
- Mastocytosis:
- Shows focal, compact aggregates of mast cells in bone marrow core biopsy
- Cells can show the following patterns of infiltration: paratrabecular, perivascular or parafollicular
- Presence of atypical mast cell supports the diagnosis
- Most cases will harbor a KIT D816V mutation
- Myeloproliferative neoplasms:
- Reactive thrombocythemia:
- Causes include exercise, allergic reaction, reaction to medications, inflammatory disorders, asplenism, infection, connective tissue disorders, metastatic cancer, lymphoproliferative disorders and iron deficiency
- Acute myelofibrosis:
- Subtype of acute myeloid leukemia that shows > 20% blasts in peripheral blood or bone marrow
- Patients also present with bone marrow fibrosis, fever and pancytopenia
- Autoimmune myelofibrosis:
- Generally shows diffuse reticulin fibrosis and an associated autoimmune disease such as rheumatoid arthritis
- Primary hyperparathyroidism:
- Rarely causes myelofibrosis and pancytopenia (Int J Lab Hematol 2007;29:464)
A 57 year old man presented with fatigue, splenomegaly and hepatomegaly. A complete blood count demonstrated anemia and thrombocytosis. A bone marrow aspirate and biopsy were ordered (shown above). What is the most likely diagnosis?
- Acute myeloid leukemia
- Chronic myelogenous leukemia
- Myelodysplastic syndrome
- Primary myelofibrosis
Comment Here
Reference: Primary myelofibrosis
- ABL
- CALR
- JAK2
- MPL
Comment Here
Reference: Primary myelofibrosis
- Mastocytoses are a heterogeneous group of disorders characterized by abnormal growth and accumulation of mast cells in 1 or more organ systems
- Associated with activating point mutations in proto-oncogene c-KIT, which codes for tyrosine protein kinase KIT / CD117, driving mast cell survival and growth (Int J Biochem Cell Biol 2007;39:1995)
- Mast cells are cytologically and immunophenotypically abnormal in neoplastic mast cell proliferations
- Systemic mastocytosis is a neoplastic condition in which histomorphologically atypical mast cells involve tissue sites including bone marrow, skin and other tissues
- Occurs primarily in adults and is strongly associated with c-KIT mutations with the majority being KIT D816V
- Also associated with myeloid neoplasms
- Major or minor criteria are required for diagnosis and include identification of atypical spindled mast cell clusters, aberrant expression of CD25, CD2 or CD30, the presence of a KIT mutation or elevated serum tryptase
- Categories of disease are based upon sites of involvement, related symptoms and laboratory findings
- According to the 2017 WHO classification, mastocytosis is classified into the following categories
- Cutaneous mastocytosis
- Urticaria pigmentosa / maculopapular cutaneous mastocytosis
- Diffuse cutaneous mastocytosis
- Mastocytoma of skin
- Systemic mastocytosis
- Indolent systemic mastocytosis
- Bone marrow mastocytosis
- Smoldering systemic mastocytosis
- Systemic mastocytosis with an associated hematological neoplasm
- Aggressive systemic mastocytosis
- Mast cell leukemia
- Diagnosis of these entities requires correlation with burden of disease (B findings) and cytoreduction requiring (C findings), which indicate organ involvement with and without organ dysfunction, respectively (see Clinical features)
- Cutaneous mastocytosis
- Prevalence estimate of 10 - 13 per 100,000 (Transl Res 2016;174:86, Cancers (Basel) 2021;13:6380)
- Indolent / smoldering types present at median age of ~50 years; more aggressive types present at older age with a median of ~70 years
- Pediatric systemic mastocytosis is extremely rare
- Mastocytoses in pediatric patients are predominantly cutaneous limited (not systemic) with a bimodal incidence of birth to 2 years and over 15 years
- ~50% of cases are self limited, resolving after onset of puberty
- M ≈ F; most studies show no significant difference or only slight differences in incidence among men and women (Transl Res 2016;174:86)
- Familial disease is rare, accounting for ~1 - 2% of all cases (Br J Haematol 2021;193:845)
- Bone marrow or bone involvement is present in up to 90% of systemic mastocytosis cases
- Bone marrow and bone
- Skin
- Liver (Cancer 1989;63:532)
- Spleen (Cancer 1992;70:459)
- Other sites including lymph nodes and extracutaneous organs (Histopathology 1992;21:439)
- Sites of involvement affect diagnosis and classification (see Clinical features)
- Clonal expansion of cells with mast cell differentiation having c-KIT mutation
- Mast cell proliferation and degranulation results in destructive lesions
- Generally thought to differentiate from a CD34+, CD117 / KIT+, CD13+ hematopoietic progenitor cells or a mast cell committed precursor
- Proto-oncogene c-KIT (Leukemia 2015;29:1223)
- Chromosome 4
- Codes for tyrosine protein kinase KIT, CD117 or mast / stem cell growth factor receptor (SCFR)
- KIT is dimerized by stem cell growth factor, ultimately inducing cell survival, proliferation and differentiation
- In mastocytosis, c-KIT mutation leads to stem cell growth factor independent activation of KIT
- The most common c-KIT mutation is KIT D816V, while cutaneous mastocytosis has more frequent non-D816V codon 816 mutations
- Mast cell release of mediators including histamine may contribute to increased osteoblast activity, activation of osteoclasts and formation of sclerotic lesions
- Course is variable from benign and self limited to aggressive with a variety of symptoms which differ based on disease subtype (Am J Hematol 2021;96:508)
- Most commonly affected sites are skin (urticaria pigmentosa) and bone marrow
- 4 categories of systemic mastocytosis presenting symptoms
- Constitutional: weight loss, fever and diaphoresis, fatigue
- Skin manifestations: urticaria and pruritus, flushing, dermatographism
- Mediator related systemic events: abdominal pain, headache, hypotension, syncope
- Musculoskeletal symptoms: osteoporosis, osteopenia, bone pain and fracture, arthralgia and myalgia
- Other presentation findings may include organ impairment associated with infiltration, especially in aggressive systemic mastocytosis and mast cell leukemia
- Symptoms related to mast cell degranulation with associated increased serum tryptase may be diagnosed as mast cell activation syndrome
- Mast cell activation syndrome is not considered a subtype of systemic mastocytosis and may be diagnosed in the absence of mastocytosis if criteria are not fulfilled
- In the absence of all required diagnostic criteria, a clonal mast cell population with KIT D816V mutation or aberrant surface CD25 warrants a diagnosis of monoclonal mast cell activation syndrome
- Patients with mediator related symptoms and systemic mastocytosis are designated as having primary (clonal) mast cell activation syndrome, which has diagnostic and therapeutic implications
- Systemic mastocytosis with an associated hematological neoplasm (AHN): associated and often clonally related secondary myeloid neoplasm, most commonly chronic myelomonocytic leukemia
- C (cytoreduction requiring) findings:
- Neoplastic mast cell infiltration impacting bone marrow function is defined as ≤ 1 cytopenia, absolute neutrophil count < 1.0 x 109/L, hemoglobin < 10 g/dL or platelet count < 1.0 x 109/L
- Abnormal liver function with ascites and elevated liver enzymes with or without hepatomegaly, cirrhosis, portal hypertension
- Large osteolytic lesions (≥ 20 mm) with or without bone pain, pathologic fractures (excepting osteoporosis related fractures)
- Palpable splenomegaly with hypersplenism with or without weight loss, hypalbuminemia
- Gastrointestinal mast cell infiltrates causing malabsorption with or without weight loss
- B (burden of disease) findings:
- > 30% of mast cells on bone marrow biopsy and serum total tryptase > 200 ng/mL or KIT D816V mutation with VAF ≥ 10% in bone marrow cells or peripheral blood leukocytes (Leukemia 2022;36:1703)
- International Consensus Classification (ICC): high mast cell burden, > 30% of bone marrow cellularity by mast cell aggregates (assessed on bone marrow biopsy) and serum tryptase > 200 ng/mL)
- Dysplastic changes or myeloproliferation in nonmast cell lineages, without meeting criteria for associated hematological neoplasm, with normal or only slightly abnormal blood counts
- ICC: cytopenia (not meeting criteria for C findings) or cytosis excluding reactive causes and other myeloid neoplasm criteria are not met
- Hepatomegaly without liver dysfunction, palpable splenomegaly without hypersplenism or lymphadenopathy
- Additional provisional B finding proposed for the upcoming WHO fifth edition update
- KIT D816V mutation with variant allele fraction ≥ 10% in bone marrow cells or peripheral blood leukocytes (Leukemia 2022;36:1703)
- > 30% of mast cells on bone marrow biopsy and serum total tryptase > 200 ng/mL or KIT D816V mutation with VAF ≥ 10% in bone marrow cells or peripheral blood leukocytes (Leukemia 2022;36:1703)
- 2017 WHO classification diagnostic criteria for systemic mastocytosis; must demonstrate major criterion and at least 1 minor criterion or ≥ 3 minor criteria
- ICC diagnostic criteria are very similar to the WHO 5th edition criteria; variation from WHO 5th edition are noted
- Major criterion
- Multifocal dense mast cell infiltrates detected in sections of bone marrow or extracutaneous organs; infiltrate is defined as ≥ 15 mast cells in aggregate (ICC specifies that the mast cells must express tryptase or CD117)
- Minor criteria
- > 25% of mast cells in bone marrow biopsy are spindled, have abnormal morphology of bone marrow aspirate / tissue infiltrate mast cells are immature or atypical
- Detection of KIT mutation in blood, bone marrow or other nonskin tissue (activating point mutation at codon 816 or any KIT mutation conferring ligand independent activation (Leukemia 2022;36:1703)
- Expression of one or more of the following antigens in the mast cell population in addition to normal mast cell markers (by flow cytometry or IHC): CD25, CD2, CD30 (Leukemia 2022;36:1703)
- Persistent serum total tryptase > 20 ng/mL (except in context of associated myeloid neoplasm)
- Major criterion
- Diagnostic criteria for variants of systemic mastocytosis (in addition to general criteria above)
- Indolent systemic mastocytosis
- No C findings indicative of organ involvement
- No evidence of associated hematological neoplasm
- Low mast cell burden
- Skin lesions are almost always present
- Bone marrow mastocytosis
- Similar criteria as indolent variant with bone marrow involvement
- No skin lesions
- Proposed fifth edition WHO criteria refinement includes tryptase < 125 ng/mL (Leukemia 2022;36:1703)
- Smoldering systemic mastocytosis
- ≥ 2 B findings in absence of C findings
- No evidence of associated hematological neoplasm
- High mast cell burden
- Does not meet mast cell leukemia criteria
- Systemic mastocytosis with an associated hematological neoplasm
- Associated hematological neoplasm including myelodysplastic syndrome, myeloproliferative neoplasm, acute myeloid leukemia, lymphoma or other WHO classified distinct hematological neoplasm
- Aggressive systemic mastocytosis
- ≥ 1 C finding
- Does not meet mast cell leukemia criteria
- Skin lesions are usually absent
- Mast cell leukemia
- Diffuse infiltration of bone marrow by dense aggregates of atypical, immature mast cells
- ≥ 20% mast cells in bone marrow aspirate
- Classic: ≥ 10% white blood cell count is mast cells in peripheral blood
- Aleukemic: < 10% peripheral blood mast cells (more common than classic)
- Skin lesions are usually absent
- Indolent systemic mastocytosis
- Additional morphologic pattern proposed for the upcoming WHO fifth edition update:
- Well differentiated systemic mastocytosis (Leukemia 2022;36:1703)
- May occur in any subtype with mast cells lacking atypical histomorphologic characteristics, may not have expression of CD25, CD2 or CD30, predominantly lacking KIT D816V mutation (J Allergy Clin Immunol 2016;137:168)
- High percentage of bone marrow mast cells
- Predominantly in women, childhood onset and familial aggregation
- Well differentiated systemic mastocytosis (Leukemia 2022;36:1703)
- Complete blood count (CBC) may demonstrate anemia, thrombocytopenia, thrombocytosis, leukocytosis or eosinophilia
- Serum tryptase > 20 ng/mL (fluoro immunoenzymatic assay [Pharmacia, Uppsala, Sweden]) is a minor diagnostic criterion (Arch Pathol Lab Med 2007;131:784)
- Elevated histamine metabolites in 24 hour urine sample may be present in patients with cutaneous lesions
- Skeletal involvement reported in 70 - 90% of patients (Annu Rev Med 2004;55:419)
- Osteoporosis reported in up to 40% of patients; dual energy Xray absorptiometry (DEXA) of hip and spine should be performed (Cancers (Basel) 2021;13:6380)
- Most common skeletal findings:
- Multiple focal sclerotic axial and appendicular bone lesions
- Diffuse, circumscribed, sclerotic foci with alternating zones having normal or reduced density in axial skeleton, ribs, humerus, femur
- Prognosis is linked to disease subclassification (Am J Hematol 2021;96:508)
- Favorable prognosis
- Indolent / smoldering systemic mastocytosis
- 3% progression to more aggressive forms in study with median follow up of > 10 years; life expectancy is approximately normal
- Indolent / smoldering systemic mastocytosis
- Poor prognosis
- Aggressive systemic mastocytosis
- Overall median survival of ~40 months
- Systemic mastocytosis with an associated hematological neoplasm
- Overall median survival of ~24 months (longer in patients with myeloproliferative neoplasms, shorter in those with chronic myelomonocytic leukemia, myelodysplastic syndrome or acute leukemia)
- Mast cell leukemia
- ~2 year overall median survival
- Aggressive systemic mastocytosis
- Favorable prognosis
- Prognosis is linked to independent risk factors
- Age: > 60 years
- Platelet count: < 150 x 109/L
- Serum alkaline phosphatase above reference interval
- Presence of adverse mutation in ASXL1, RUNX1, SRSF2 or NRAS
- Increased plasma IL2Rɑ / CD25 confers worse prognosis in indolent SM
- 33 year old man with abdominal pain and blood per rectum (Ann Gastroenterol 2019;32:208)
- 50 year old man with foamy mastocytosis who presented with flushing, fever, intestinal disorder, skin lesions and splenomegaly (Blood 2018;131:586)
- 62 year old man with systemic mastocytosis and essential thrombocythemia who presented with thrombocytosis and hepatosplenomegaly with cutaneous papules (Medicina (Kaunas) 2019;55:528)
- 83 year old man with systemic mastocytosis and multiple myeloma who presented with hematuria and bone pain (Blood 2018;132:1545)
- Indolent disease may not require treatment or treatment may be aimed at symptomatic management, including antihistamines, leukotrienes, mast cell stabilizers, proton pump inhibitors, calcium vitamin D, bisphosphonates; epinephrine may be needed for patients having risk of anaphylaxis (Am J Hematol 2021;96:508)
- Avoidance of exposures that aggravate mast cell degranulation including temperature extremes, mechanical irritation, medications and alcohol
- Aggressive disease requiring cytoreductive therapy may be treated with the following agents
- Interferon alpha is often paired with prednisone; may have severe side effects including myelosuppression
- 2-chlorodeoxyadenosine issued as a second line therapy for interferon alpha refractory disease; may have severe side effects including myelosuppression, lymphopenia and risk of opportunistic infection
- Cladribine purine analog with mechanism independent of tyrosine kinase inhibition
- Tyrosine kinase inhibitors
- Avapritinib targets PDFRɑ and KIT (Int J Mol Sci 2021;22:2983)
- Midostaurin targets FLT3, KIT D816V mutation (Blood 2020;135:1365)
- Imatinib targets BCR::ABL, wild type KIT
- Nilotinib targets BCR::ABL, wild type KIT
- Dasatinib dual SRC / ABL inhibitor
- Chemotherapy regimens employed in acute myeloid leukemia
- Allogeneic stem cell transplantation in rare cases
- Neoplastic mast cells may be spindled to round / oval (similar to normal mast cells) with variable cytoplasmic granularity
- Spindled mast cell aggregates may invoke a streaming-like appearance
- Bone marrow aspirate sampling of mast cells may be limited due to associated reticulin or collagen fibrosis
- Characteristically, mast cells have a light blue-gray cytoplasm; granules may be inconspicuous on H&E sections but are more prominent with regional distribution on Wright-Giemsa stained bone marrow aspirate and peripheral blood smears
- Basophilic to amphipathic cytoplasmic granules may obscure mast cell nuclei on Wright-Giemsa stain
- On H&E sections, round mast cells with central nuclei may give a fried egg-like appearance at low power, raising a differential that includes hairy cell leukemia (Arch Pathol Lab Med 2007;131:784)
- Mast cell nuclei may have 1 to a few prominent nucleoli and relatively smooth nuclear chromatin
- Mast cell aggregates may be intermixed with lymphohistiocytic proliferations, often with interspersed eosinophils and plasma cells
- Atypical mast cell features include spindle morphology, hypogranulation and atypical nuclear borders
- Bone may demonstrate osteosclerosis, increased trabeculation volume, increased cortical thickness and narrowing of marrow spaces
Contributed by Mark Girton, M.D. and AFIP
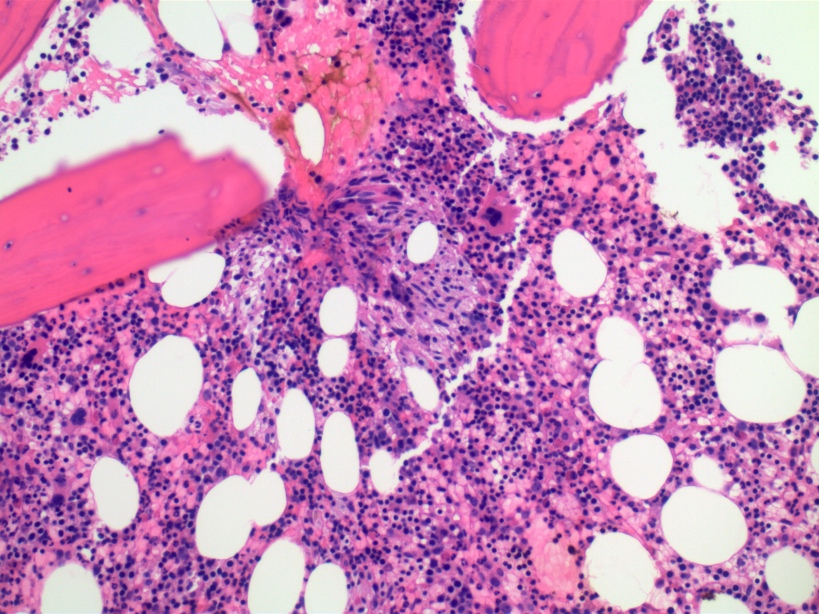
Marrow mast cell cluster
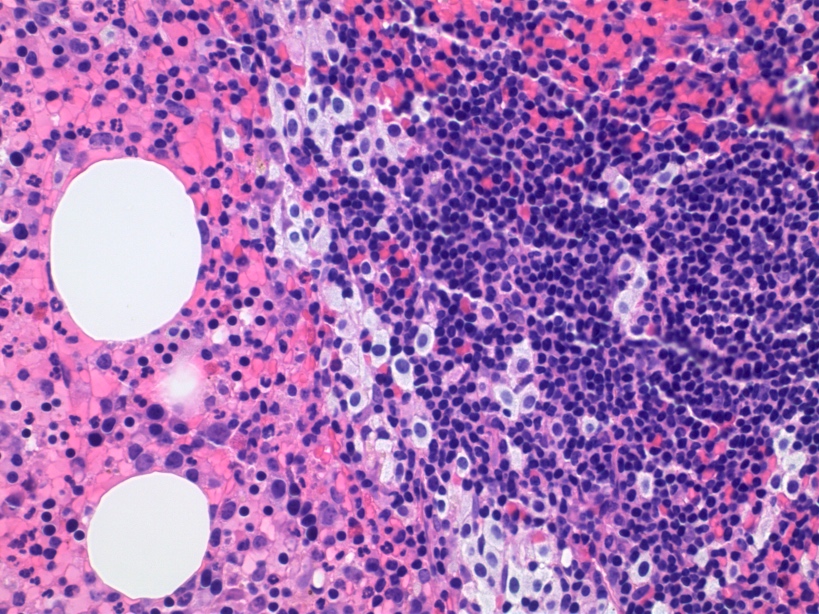
Intermixed with lymphoid aggregate
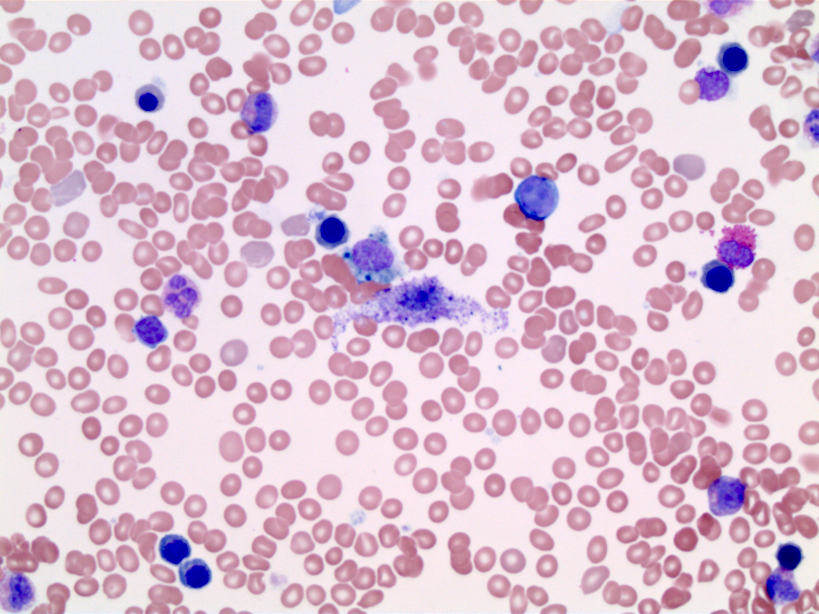
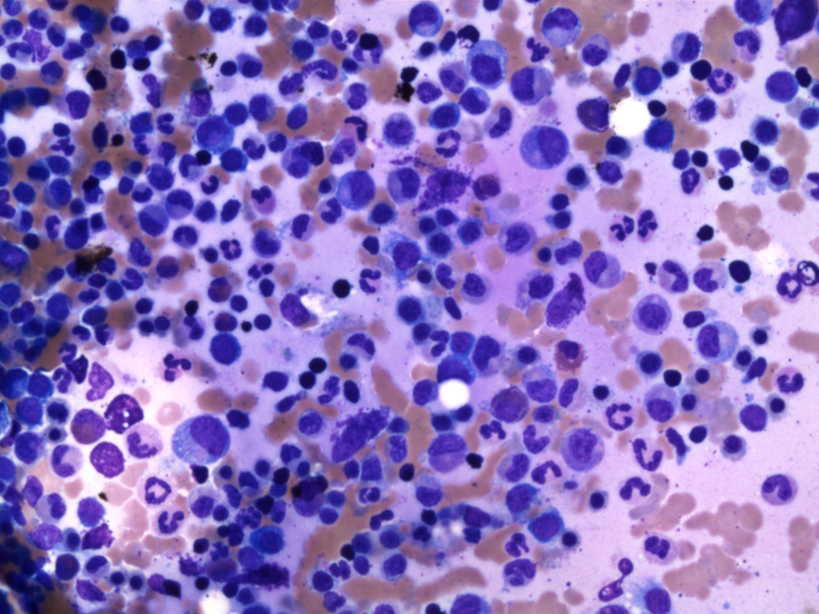
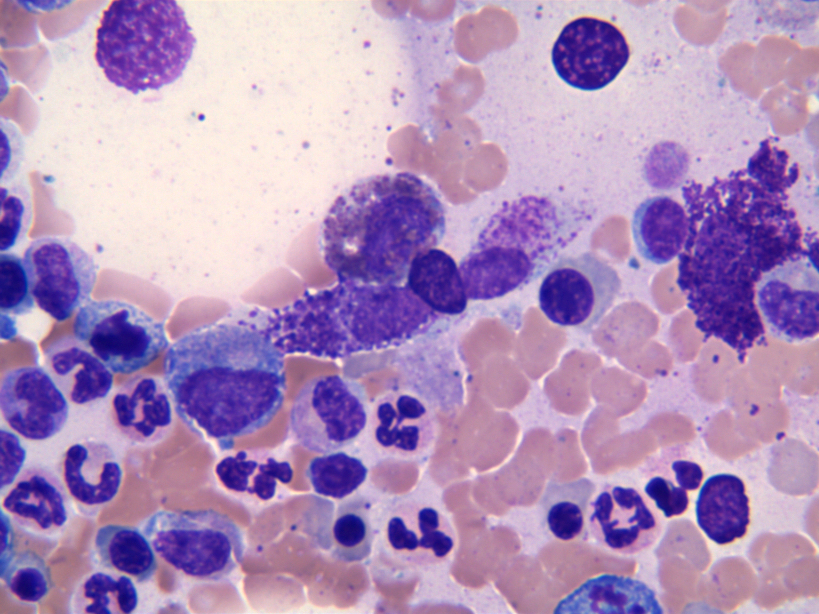
Aspirate with mast cells
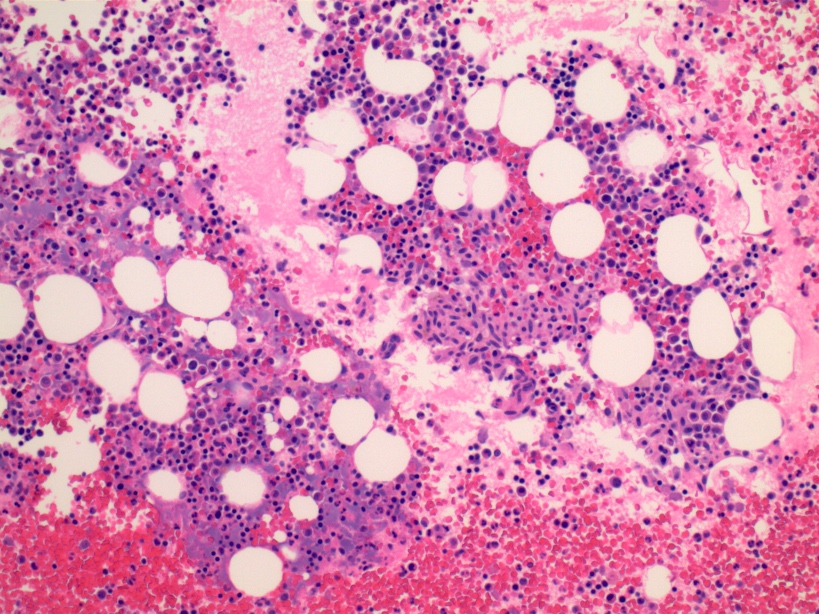
Mast cell cluster
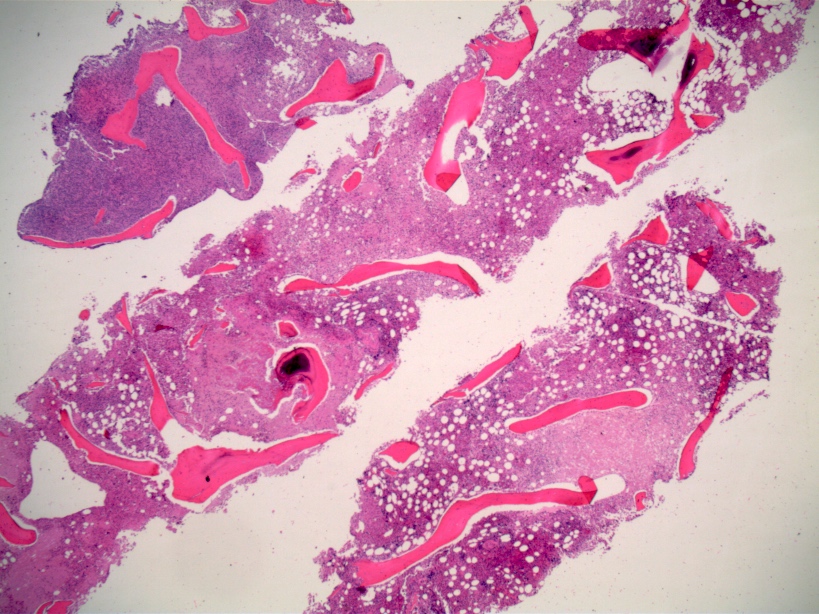
Hypercellular bone marrow biopsy
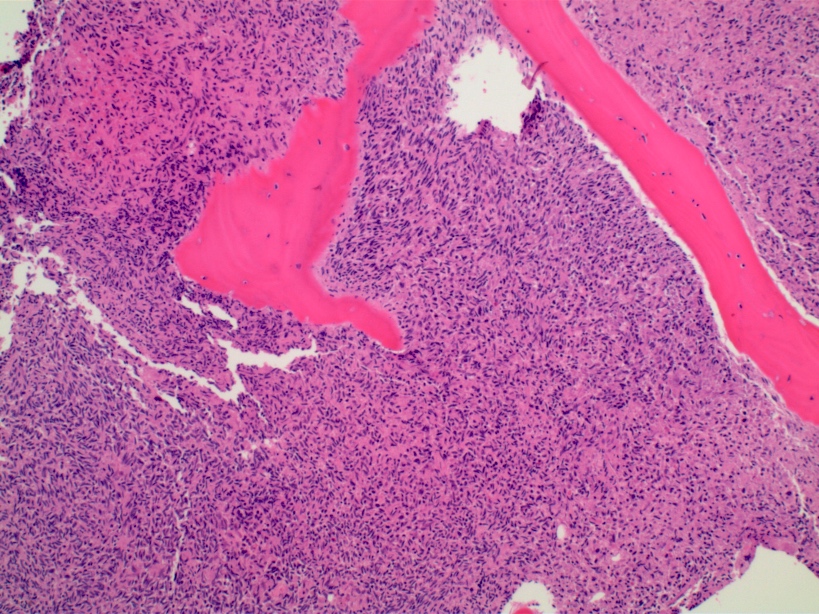
Bone marrow biopsy with marrow replacement
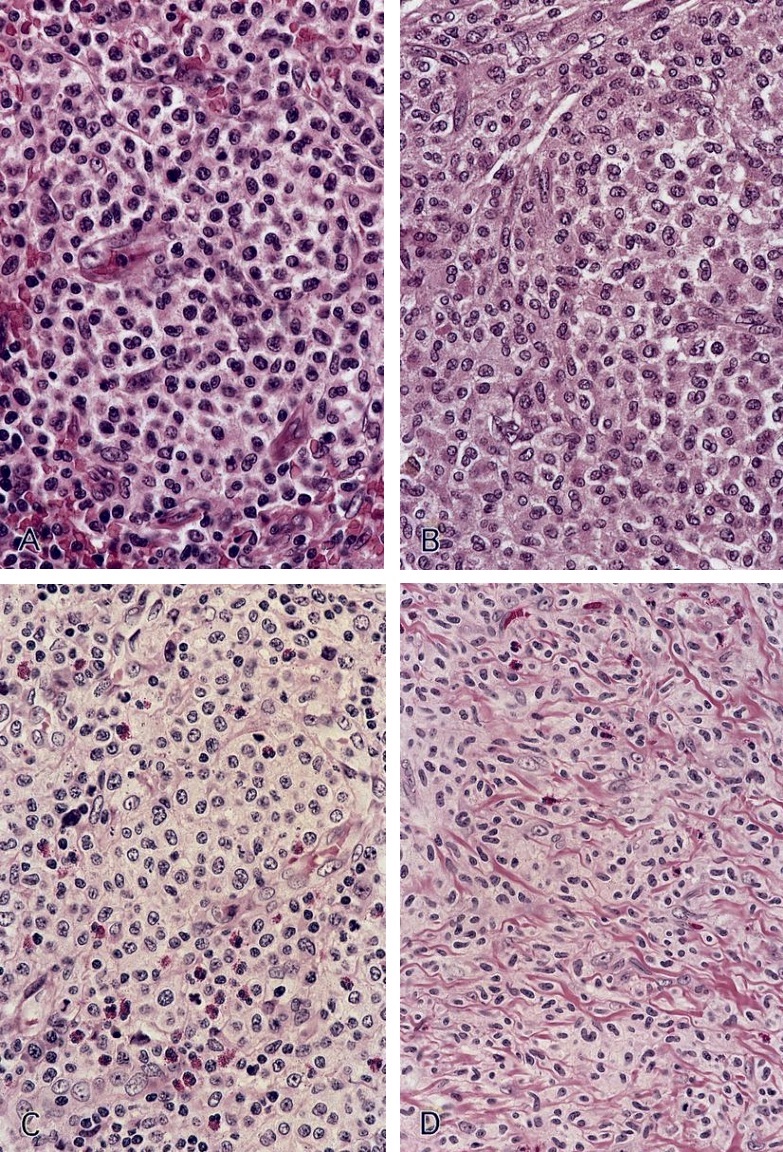
Cytologic spectrum
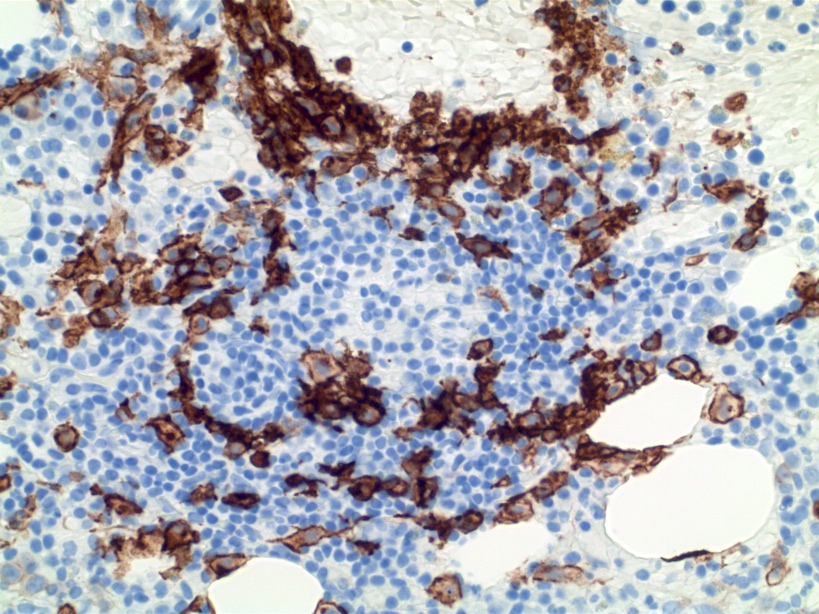
CD117 positive mast cells
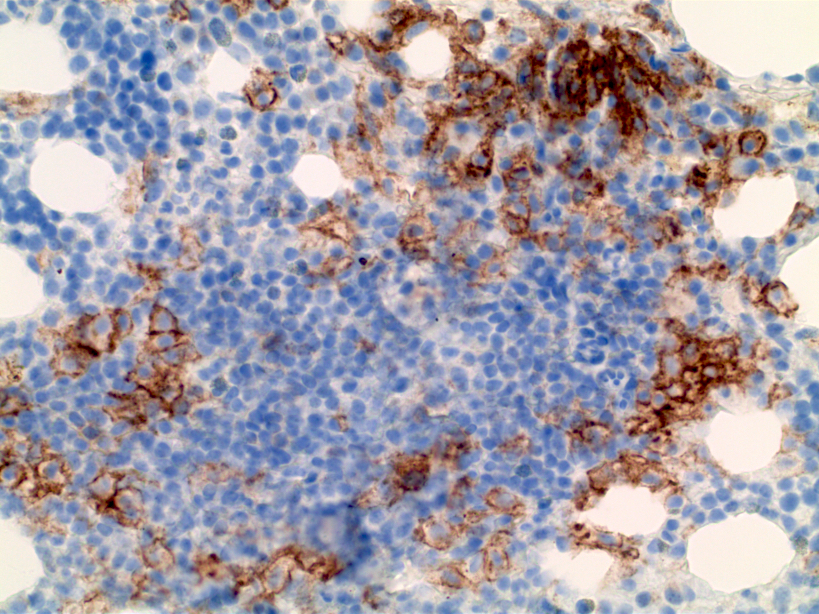
CD25 positive mast cells
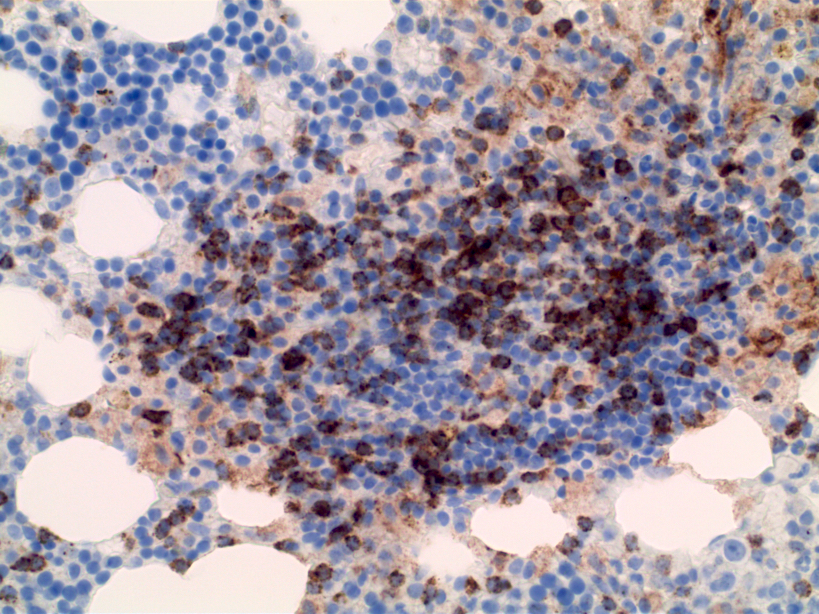
CD2 positive mast cells
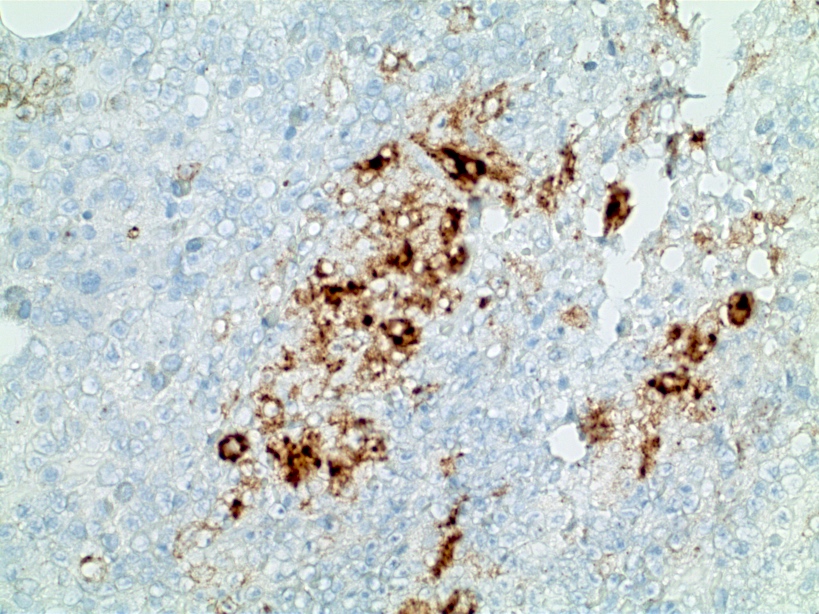
CD30 positive mast cells
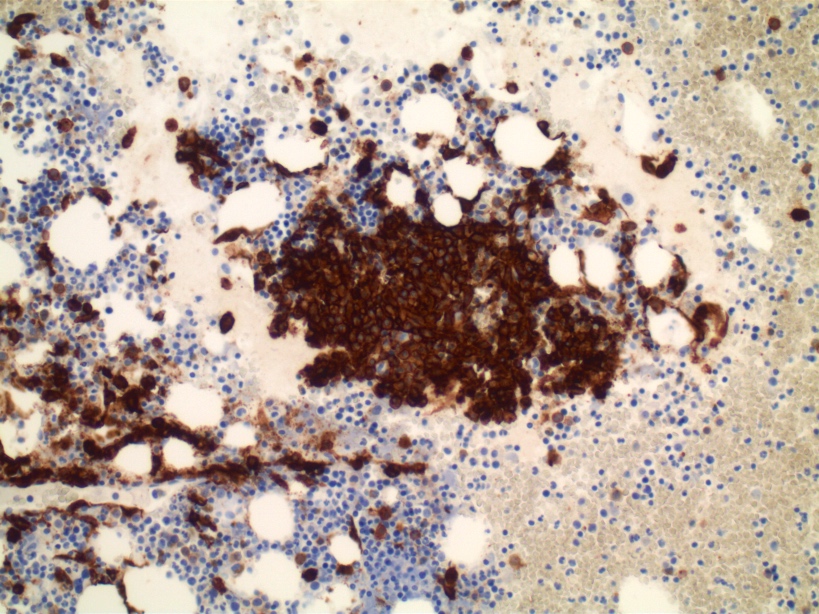
CD117 positive mast cells
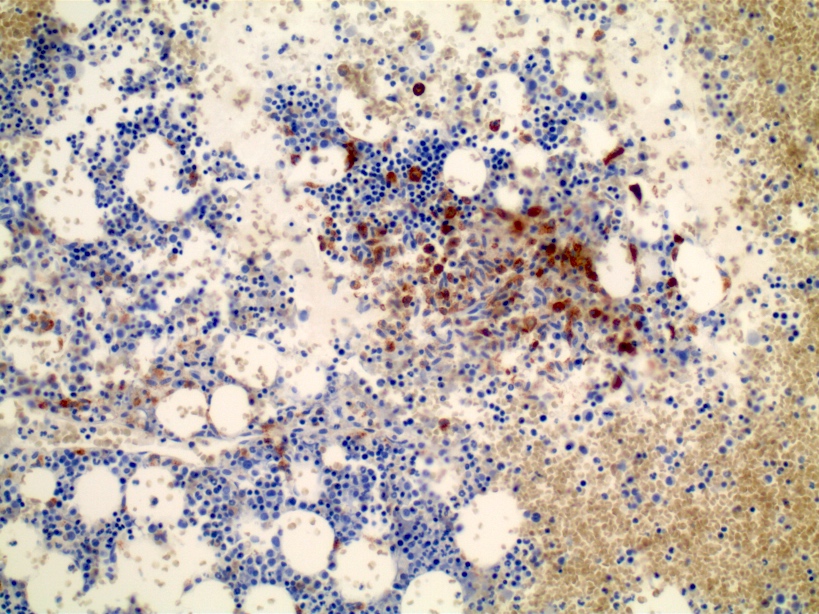
Mast cell tryptase positive
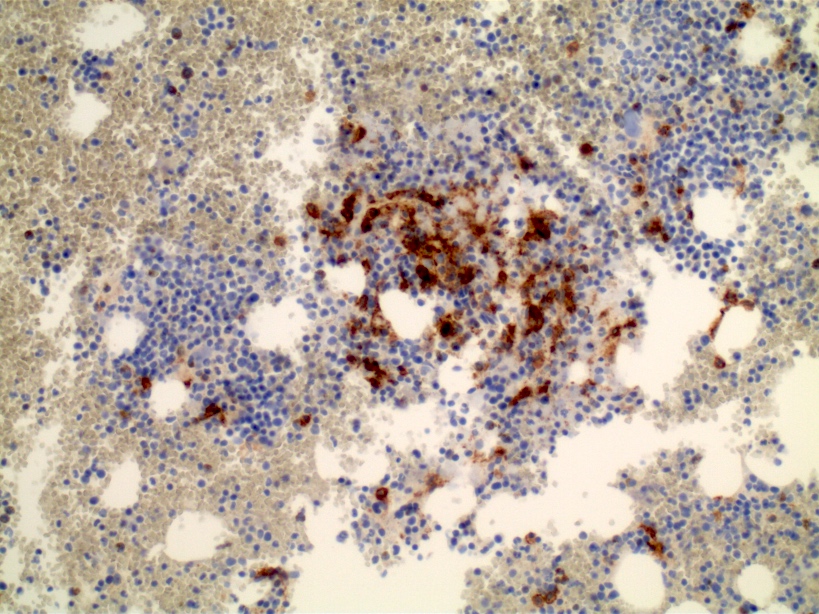
CD25 positive mast cells
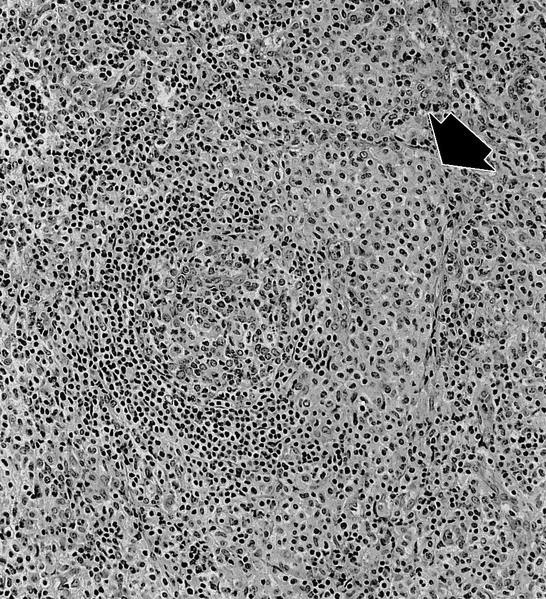
Perifollicular distribution

Characteristic sclerosis
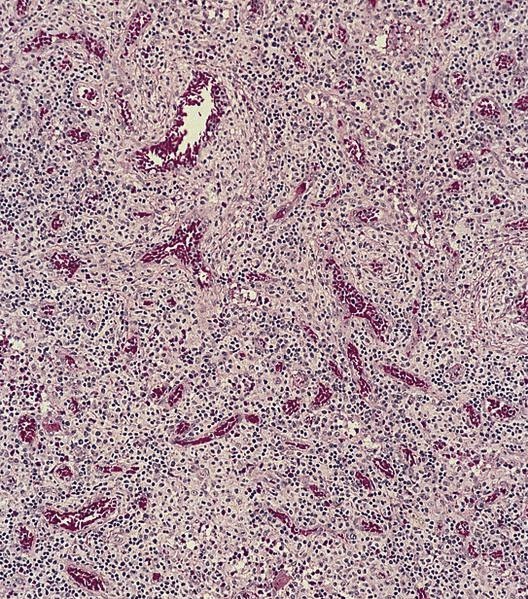
Mast cell infiltrate
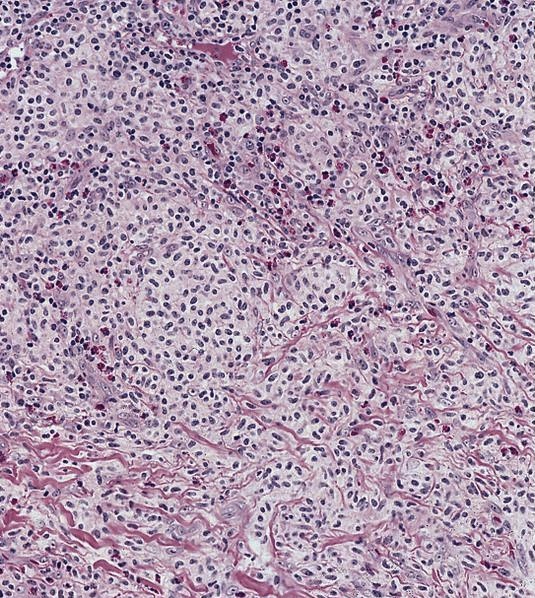
Delicate collagen fibrils and eosinophils
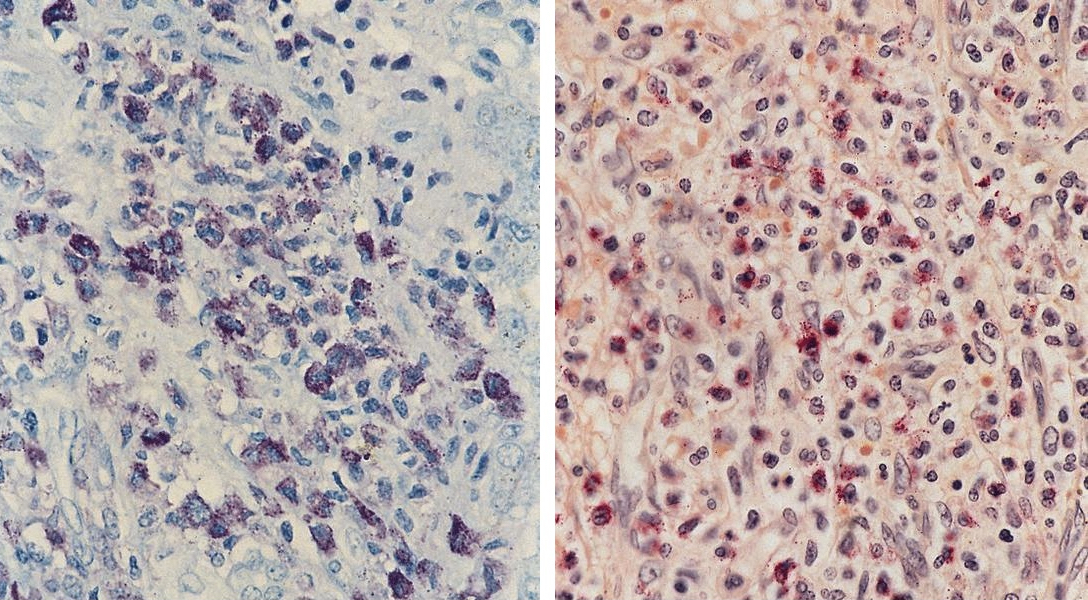
Mast cell staining
Interfollicular pattern
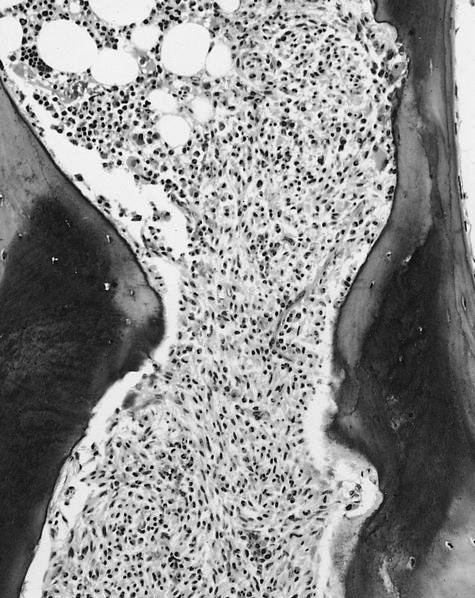
Diffuse involvement
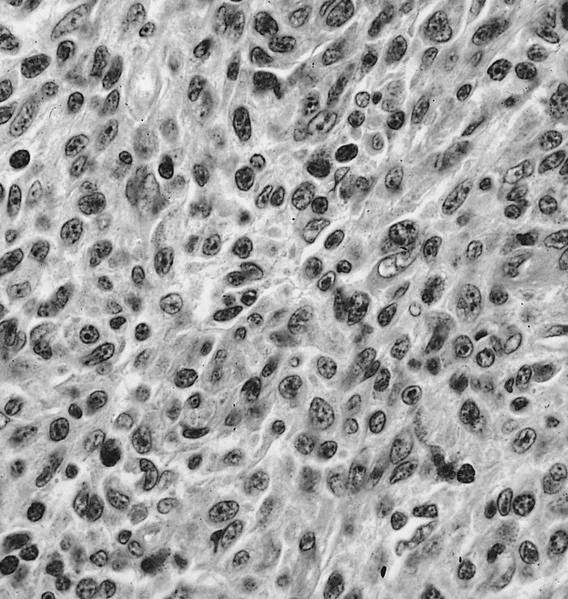
Ovoid nuclei with eosinophilic cytoplasm
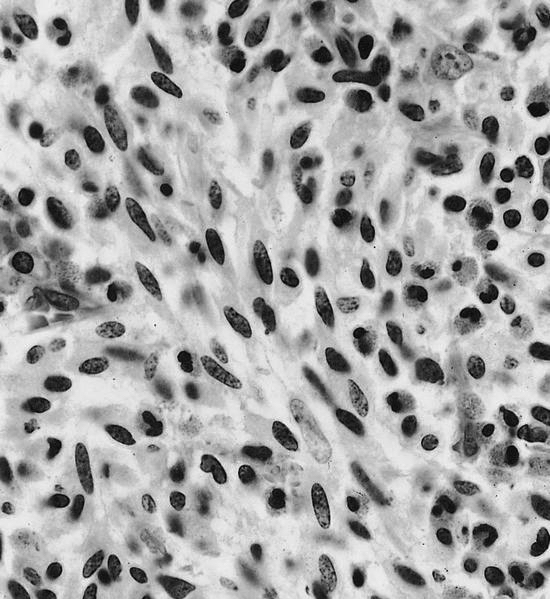
Spindle shaped mast cells
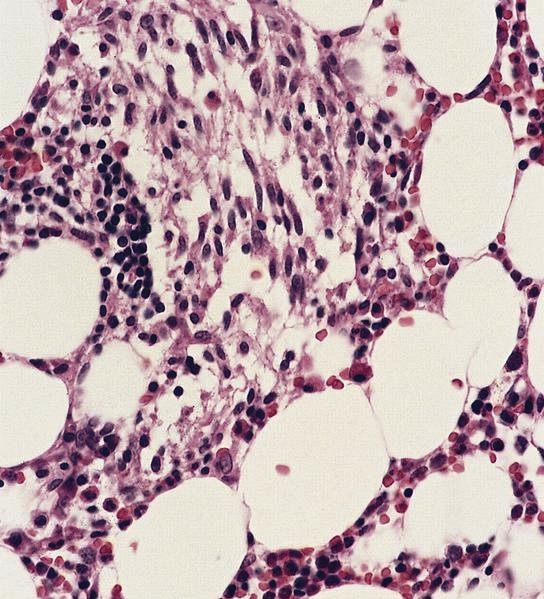
Spindly configuration
Contributed by Mark Girton, M.D.
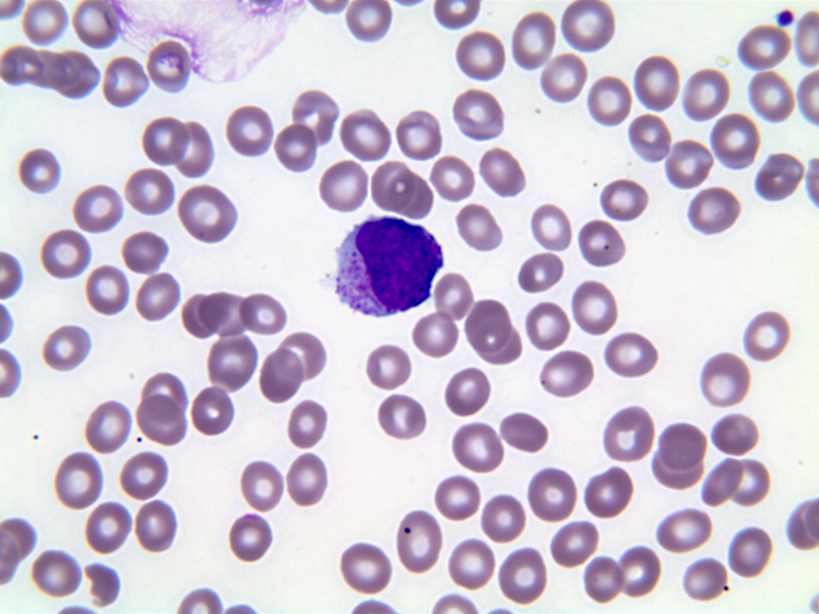
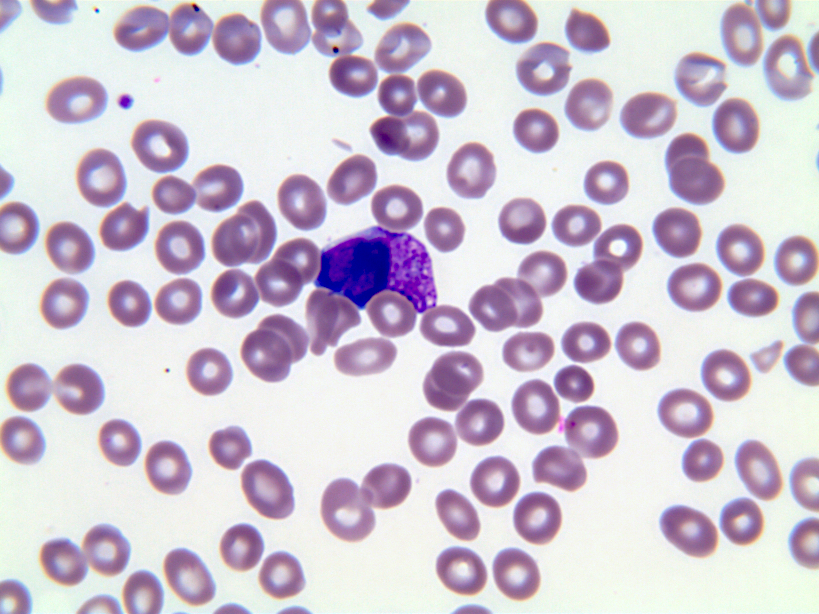
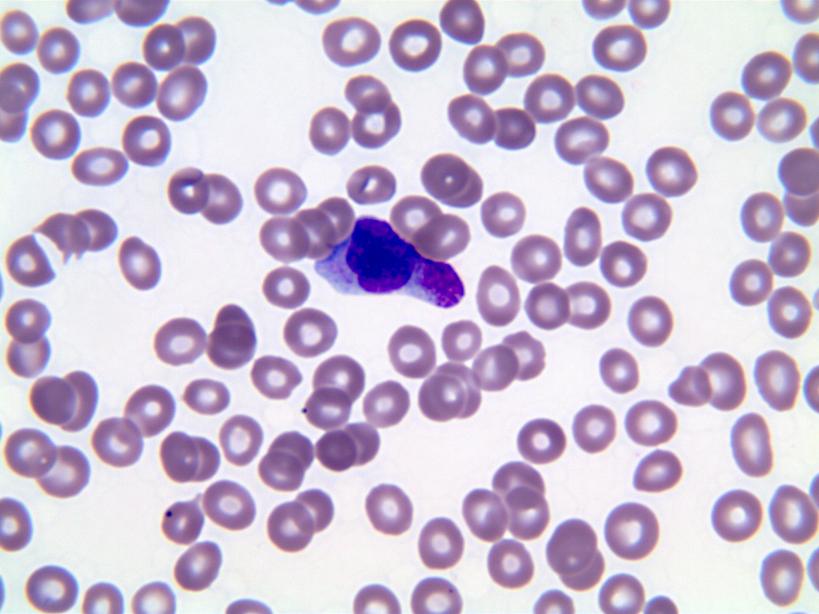
Peripheral blood mast cell
- Mast cell tryptase: positive in mast cells; granular, intracytoplasmic staining
- CD117: membranous staining, differentiates from basophils
- CD25: positive in neoplastic mast cells
- CD2: may demonstrate weak to moderate positivity in neoplastic mast cells, stronger in surrounding reactive T lymphocytes
- Others of less diagnostic utility are: CD68 (may help differentiate from basophils), CD30 (~50%), CD45, HLA-DR, CD43, CD123, Giemsa, toluidine blue, polychrome methylene blue, lysozyme (muramidase), chloroacetate esterase (Leder), CD9, CD33 (Histopathology 2013;63:780, Mod Pathol 2011;24:585)
- Neoplastic mast cell populations may be difficult to sample because of associated reticulin or collagen fibrosis; therefore, the population of interest may be underrepresented in flow cytometry assays
- Antibodies against CD45, CD13, CD33, CD117, CD25 and CD2 are typically positive in neoplastic mast cells, while CD34 is generally expected to be negative, except in mast cell precursors
- CD123, HLA-DR and CD163 may be expressed in less mature neoplastic mast cell populations with reduced expression of CD117, FcεRI (IgE) and cytoplasmic proteases (Leukemia 2016;30:914, Pathobiology 2020;87:2)
- Nonneoplastic mast cells lack expression of CD25 and CD2
- CD38 expression may be seen in normal mast cell precursors
- KIT D816V mutation in exon 17 present in vast majority (> 90%) of cases
- Other KIT point mutations usually involve exon 17, with other mutations reported in exons 8, 9, 10 and 11 (Am J Hematol 2021;96:508, Oncotarget 2016;8:68950)
- Blood, peripheral smear:
- Unremarkable peripheral blood smear (see comment)
- Comment: Microscopic examination of the peripheral blood smear demonstrates a normal red blood cell count with erythrocytes, which are morphologically unremarkable. The white blood cell count is normal and leukocytes are morphologically unremarkable. Platelets are normal in number and morphology.
- Bone marrow, left, aspirate smear:
- Trilineage hematopoiesis with occasional mast cells present (see comment)
- Comment: Microscopic examination of the bone marrow aspirate demonstrates a spicular and cellular specimen which is adequate for evaluation. There is trilineage hematopoiesis. Erythroid precursors demonstrate normoblastic maturation. Myeloid precursors demonstrate the full spectrum of maturation. The myeloid to erythroid (M:E) ratio is normal at 2.3:1. Megakaryocytes are present and appear normal in number and morphology. There are occasional mast cells present, some of which demonstrate a spindled morphology. There is no increase in blasts. An iron stain demonstrates the presence of storage iron and no significant increase in ring sideroblasts.
- Bone marrow, left, core biopsy:
- Limited subcortical specimen showing trilineage hematopoiesis with involvement by systemic mastocytosis (see comment)
- Comment: Microscopic examination of the core biopsy demonstrates a subcortical bone marrow specimen limited sampling, which precludes evaluation of the cellularity. There is trilineage hematopoiesis and small, ill defined lymphoid aggregates. Immunohistochemical staining for CD117 and mast cell tryptase demonstrate clusters of spindle shaped mast cells. Staining for CD25 is positive and staining for CD2 is negative. The lymphoid aggregates are composed of a mixture of B cells and T cells as demonstrated by CD20 and CD3 positivity, respectively. The myeloid to erythroid ratio appears normal. Megakaryocytes are present and appear normal in number and morphology. A PAS stain highlights the myeloid elements and megakaryocytes. An iron stain shows the presence of storage iron. A reticulin stain is not evaluable due to the limited specimen size.
- Bone marrow, left, clot section:
- Normocellular bone marrow with trilineage hematopoiesis and involvement by systemic mastocytosis (see comment)
- Comment: Microscopic examination of the clot preparation demonstrates a cellular specimen that is adequate for evaluation. The clot section appears normocellular. There is trilineage hematopoiesis. The myeloid to erythroid ratio appears normal. Megakaryocytes are present and appear normal in number with unremarkable morphology. There are multiple lymphoid aggregates present with mast cell clusters having focal areas of scattered eosinophils. Immunohistochemical staining for CD117 and mast cell tryptase demonstrate clusters of spindle shaped mast cells. Staining for CD25 is positive and staining for CD2 is negative. The lymphoid aggregates are a mixture of B cells and T cells as demonstrated by CD20 and CD3 positivity, respectively. There is no increase in blasts. An iron stain shows the presence of storage iron and no ring sideroblasts.
- In summary, the patient's specimen demonstrates an overall normocellular bone marrow with trilineage hematopoiesis and involvement by systemic mastocytosis. Correlation with molecular and genetic studies is recommended.
- Mast cell hyperplasia:
- Associated with solid tumors, may be massive but no compact bone marrow infiltration, diffuse interstitial pattern in bone marrow
- CD25 negative mast cells without atypical histomorphology, molecular studies negative for characteristic KIT mutation
- Myeloid tumors with mast cell differentiation:
- Generally lack systemic mastocytosis diagnostic criteria
- Chronic myeloid leukemia with myelomastocytic blast crisis (Leuk Res Rep 2020;14:100222):
- May have significant mast cell differentiation by immunohistochemical profile
- Positive for BCR::ABL1 fusion
- Negative for KIT D816V mutation
- Tryptase positive acute myeloid leukemia (AML) (Blood 2001;98:2200, Expert Rev Hematol 2014;7:431, Expert Rev Hematol 2014;7:431):
- Also known as myelomastocytic leukemia
- Lacks CD25 expression and is negative for KIT D816V mutation
- Acute basophilic leukemia (Leukemia 2017;31:788):
- CD117 expression differentiates between mast cell and basophil differentiation
- International Consensus Classification (ICC) notes that the presence of eosinophilia with a mast cell proliferation should raise a differential including myeloid / lymphoid neoplasms with eosinophilia; this diagnosis, with an associated tyrosine kinase gene fusion involving PDGFRA, would exclude systemic mastocytosis
- Acute myeloid leukemia (AML)
- Chronic myelomonocytic leukemia (CMML)
- Chronic myelocytic leukemia (CML)
- Plasma cell myeloma
- Polycythemia vera (PV)
Comment Here
Reference: Systemic mastocytosis
- Identification of a BRAF V600E mutation in blood, bone marrow aspirate or nonskin tissue
- Mast cell expression of CD25, with or without CD2, in combination with normal mast cell markers
- Mast cells expressing CD117 or tryptase by immunohistochemical staining
- Multiple dense mast cell groups as defined by ≥ 15 mast cells per aggregate in bone marrow or other extracutaneous organ
Comment Here
Reference: Systemic mastocytosis
- Neoplasm of T lineage lymphoblasts which may form lymphomatous masses, involve blood and bone marrow (Stanford School of Medicine: Precursor T Lymphoblastic Leukemia / Lymphoma [Accessed 13 April 2018])
- Also called pre T cell acute lymphocytic leukemia / lymphoma (pre-T ALL), T lymphoblastic leukemia / lymphoma (T LBL)
- Teens and young men (older than B ALL)
- Most cases begin after birth (Blood 2007;110:3036)
- T ALL versus T LBL: T ALL has more immature phenotype, CD47 expression, no 11q23 rearrangement, different gene expression profile and may derive from T cell progenitor of bone marrow; T LBL is derived from thymocytes (Pediatr Blood Cancer 2006;47:130, Leuk Lymphoma 2007;48:1745)
- T ALL constitutes 15% of childhood and 20 - 25% of adult ALL cases
- T LBL constitutes 85 - 95% of LBL, usually presents as mediastinal mass with no / minimal marrow involvement
- CNS involvement if untreated
- T LBL frequently presents with mass in anterior mediastinum, rapid growth, respiratory emergency, pleural effusion
- Younger (age 16 - 60 years) patients compared to older (61+ years) patients have more hepatosplenomegaly, present with mediastinal mass and lymphadenopathy; myeloid antigens and lineage inappropriate gene rearrangements are less common (Am J Clin Pathol 2002;117:252)
Images hosted on other servers:
Rearrangements involving T cell receptor genes
- T ALL if lymphoblasts are 25% or more of marrow cells; T LBL otherwise
- Good prognostic factors: HOX11 overexpression in adults (Am J Clin Pathol 2007;127:528)
- Poor prognostic factors: expression of CFLAR, NOTCH2 and BTG3 genes, 3+ methylated genes, residual disease after treatment (even if minimal) (Br J Haematol 2007;137:319, J Clin Oncol 2005;23:7043)
- Pediatric patients with T ALL: TLX1 (HOX11)+ was associated with favorable outcome and TAL1+ and LYL1+ were associated with unfavorable outcome (Atlas of Genetics and Cytogenetics: T Lineage Acute Lymphoblastic Leukemia (T-ALL) [Accessed 13 April 2018])
- Higher risk than B ALL due to presence of high risk features but T ALL without high risk clinical features has survival comparable to B ALL
- Chemotherapy cures 60%
- Patients have earlier relapse, induction failure and isolated CNS relapse compared to pre-B ALL
- Similar to B cell disease; scant cytoplasm, delicate chromatin, indistinct nucleoli, convoluted nuclear membrane and grooves
- Frequent mitotic figures; starry sky pattern produced by interspersed benign macrophages
- Usually features of FAB L1 or L2; pattern in marrow is usually interstitial
- Lymph nodes: complete architectural effacement or partial involvement with paracortical infiltrate with germinal center sparing
- Thymus: replacement of normal parenchyma
- Occasionally eosinophilia and myeloid hyperplasia with variable t(8;13)(p11.2;q11-22) involving FGFR1 gene; some develop myeloid malignancy (MDS, AML or myeloid sarcoma)
AFIP images
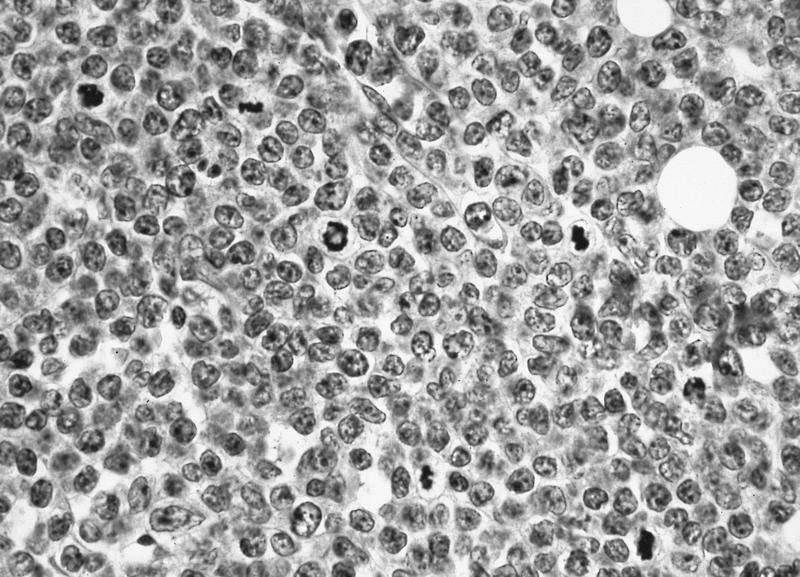
Bone marrow biopsy
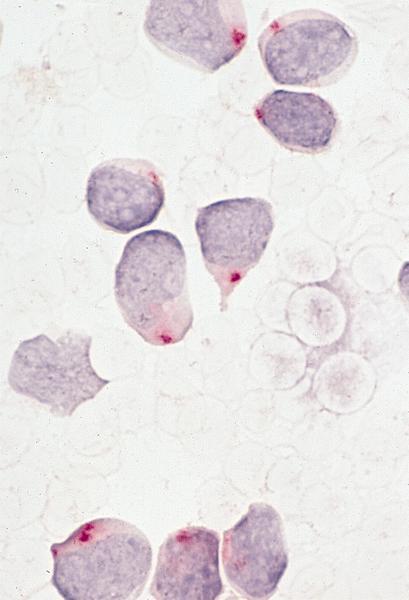
Focal paranuclear acid phosphatase staining
AFIP images
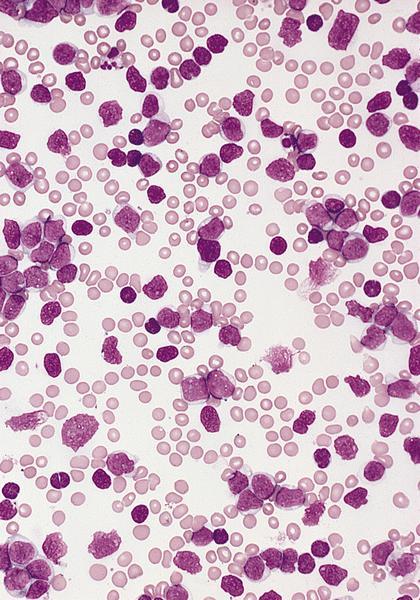
Markedly elevated leukocyte count
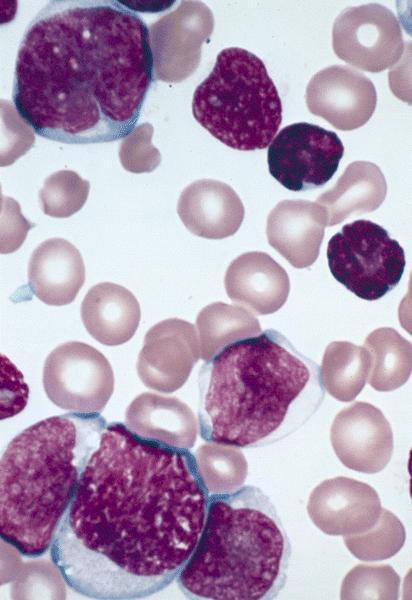
L2 type (blood smears)
- CD1a, CD2 (78%), CD3 (cytoplasmic, not surface in 100%), CD5 (100%), CD7 (100%), TdT (73%)
- CD4 and CD8 both positive in 22%
- Variable CD10 (47%), CD13 (6%), CD16, CD33 (12%), CD56, CD57, CD79a+ (40 - 60%), CD117 (12%) (Haematologica 2009;94:160, Am J Clin Pathol 2000;113:823, Exp Mol Pathol 2006;81:162)
- CD117 associated with activating mutations of FLT3, CD4 (10%) and CD8 (< 10%)
- Different cytogenetic abnormalities than B ALL, often are cryptic and identified only by FISH or PCR (Atlas of Genetics and Cytogenetics: T Lineage Acute Lymphoblastic Leukemia (T-ALL) [Accessed 13 April 2018])
- t(1;14)(p32;q11) involving SCL (TAL1) and T cell receptor delta / alpha in 15 - 30%
- t(10;14)(q24;q11) involving HOX11 (TLX1) and T cell receptor delta / alpha in 7%
- Activating mutations of NOTCH1 in 50% (Science 2004;306:269)
- CDKN2A (INK4A) deletions in up to 80% (Blood 1995;85:2321)
- TCR: may have rearrangement but is not lineage specific
- TCR loci (1/3 of T ALL)
- 14q11.2 (alpha, delta)
- 7q35 (beta)
- 7p(14-15) (gamma)
Genes:
- MYC (8q24.1)
- TAL1 (1p32)
- RBTN1 (LMO1) (11p15)
- RBTN2 (LMO2) (11p13)
- HOX11 (TLX1) (10q24)
- HOX11L2 (TLX3) (5q35)
- LYL1 (19p13)
- LCK (1p34.3-35)
Images hosted on other servers:

FISH: t(5;14)(q35;q32)
- Burkitt leukemia:
- B cell phenotype
- Granulocytic sarcoma:
- Positive for myeloid markers
- Mantle cell lymphoma-blastoid:
- B cell phenotype
- Thymoma (Am J Clin Pathol 2004;121:268)
- B lymphoblastic leukemia / lymphoma arising after cytotoxic or radiation therapy for a prior malignancy
Notes:
- Therapy related T lymphoblastic leukemia is not discussed in this topic; rare reports of such cases have been published but understanding of this entity remains limited
- Secondary B lymphoblastic leukemia / lymphoma is not discussed in this topic
- The term secondary B lymphoblastic leukemia / lymphoma is preferred when the patient has a history of prior malignancy but has not received cytotoxic or radiation therapy
- B lymphoblastic leukemia / lymphoma arising after cytotoxic or radiation therapy for a prior malignancy
- Most common prior malignancy is breast cancer, followed by multiple myeloma and lymphoma
- No distinct morphologic or immunophenotypic features but is commonly associated with KMT2A rearrangement, del(5 / 5q), del(7 / 7q), hypodiploidy, TP53 mutation and BCR::ABL1 rearrangement
- Therapy related B lymphoblastic leukemia / lymphoma (t-B ALL)
- ICD-10: C91.00 - acute lymphoblastic leukemia not having achieved remission
- Estimated to occur in 1 - 10% of patients who receive cytotoxic / radiation therapy (alkylating agents or topoisomerase inhibitors) as primary therapy or in preparation for hematopoietic stem cell transplant (e.g., melphalan)
- Recently discovered association with use of immunomodulatory agents (e.g., lenalidomide, thalidomide) for primary therapy or maintenance therapy (Blood Rev 2019;37:100584, Int J Clin Exp Pathol 2011;4:322, Medicine (Baltimore) 2019;98:e14011)
- More common in older patients; incidence increases with age
- Median age at presentation: fifth and sixth decade (Blood Rev 2019;37:100584, Br J Haematol 2022;196:963)
- Occasionally seen in young adults (second to third decade) and rarely in pediatric patients
- Germline mutations (e.g., TP53 mutation) are frequently present in this age group and should be excluded before a diagnosis of t-B ALL is considered (Leuk Lymphoma 2001;41:255, Blood Rev 2019;37:100584)
- Most common prior malignancy is breast cancer, treated by alkylating agents and topoisomerase inhibitors (Leuk Lymphoma 2001;41:255, Blood Rev 2019;37:100584, Br J Haematol 2022;196:963)
- Second most common are multiple myeloma and lymphoma
- Other prior malignancies include myelodysplastic syndrome with isolated del(5q) (treated with lenalidomide) and a variety of carcinomas and sarcomas
- Interval between diagnosis of primary malignancy and development of t-B ALL is approximately 2 - 7 years but can range from a few months to several decades (Blood Rev 2019;37:100584, Case Rep Hematol 2018;2018:9052314)
- Bone marrow and peripheral blood
- Central nervous system (CNS) involvement can be seen, especially in KMT2A rearranged disease (Curr Hematol Malig Rep 2020;15:83)
- Shows overlapping pathophysiology with WHO defined therapy related myeloid neoplasms (therapy related myelodysplastic syndrome and therapy related acute myeloid leukemia) and may represent a morphologic variant of those diseases (Blood Rev 2019;37:100584)
- Underlying mechanism is exposure to mutagenic agents as treatment for a prior malignancy (Leuk Lymphoma 2001;41:255, Blood Rev 2019;37:100584, Br J Haematol 2022;196:963)
- Accumulating exposure results in DNA damage, likely inducing mitotic instability (Br J Haematol 2022;196:963)
- DNA instability may result in hypodiploidy, including monosomy of chromosomes (5 / 5q), (7 / 7q), (17) and others
- Primarily associated with exposure to alkylating agents, topoisomerase inhibitors and radiation
- Recently associated with use of immunomodulatory agents (e.g., lenalidomide) as maintenance therapy after autologous stem cell transplant for plasma cell myeloma (Lancet Oncol 2014;15:333, Ann Oncol 2017;28:228, Case Rep Hematol 2018;2018:9052314, Am J Clin Pathol 2020;154:816)
- Cases of t-B ALL arising after lenalidomide therapy for myelodysplastic syndrome with isolated del(5q) have been reported (Int J Clin Exp Pathol 2011;4:322, Medicine (Baltimore) 2019;98:e14011)
- History of cytotoxic or radiation for prior malignancy
- Older age at diagnosis (fifth - sixth decade)
- Patients may present with cytopenia(s), fatigue, weight loss and fever or may be asymptomatic
- Examination of blood or bone marrow
- Complete blood cell count (CBC):
- Patients present with lower white blood cell (WBC) counts than patients with de novo disease (Br J Haematol 2022;196:963)
- WBC count ranges widely (may be decreased, normal or increased)
- Anemia, thrombocytopenia or pancytopenia are frequently encountered
- Patients present with lower white blood cell (WBC) counts than patients with de novo disease (Br J Haematol 2022;196:963)
- Previously mentioned cytogenetic abnormalities, as well as prior exposure to mutagenic agents, confer a worse prognosis when compared to de novo disease
- KMT2A rearrangement is associated with faster time to development of t-B ALL, as well as worse prognosis
- Autologous hematopoietic stem cell transplant may improve survival (Blood Rev 2019;37:100584, Br J Haematol 2022;196:963)
- 43 and 64 year old men with B ALL arising during lenalidomide maintenance (Am J Clin Pathol 2020;154:816)
- 53 and 69 year old women with B ALL arising in the context of lenalidomide maintenance (Case Rep Hematol 2018;2018:9052314)
- 60 year old woman with B ALL, 3 years after treatment for multiple myeloma with cytotoxic agents and autologous stem cell transplant with high dose melphalan conditioning (Leukemia 2005;19:299)
- 68 year old Caucasian man and 83 year old woman, the first case series to report development of t-B ALL during lenalidomide therapy for myelodysplastic syndrome with isolated del(5q) (Int J Clin Exp Pathol 2011;4:322)
- 69 year old man with B ALL with mutated EZH2, arising 27 months after initiation of lenalidomide therapy for myelodysplastic syndrome with isolated del(5q) (Medicine (Baltimore) 2019;98:e14011)
- 72 year old man with B ALL, 6 years after treatment with cytotoxic agents and lenalidomide maintenance therapy for multiple myeloma (Ann Clin Lab Sci 2013;43:176)
- 74 year old woman with B ALL, 56 months after treatment for AL amyloidosis with lenalidomide (Int J Clin Exp Pathol 2014;7:2683)
- 80 year old man with B ALL, > 20 years after treatment with alkylating agent (chlorambucil) for Waldenström macroglobulinemia (Leukemia 2004;18:1433)
- Early case series of B ALL arising in the context of lenalidomide maintenance (Hematol Oncol 2017;35:130)
- Intensive chemotherapy induction regimens, such as R hyper CVAD (rituximab, cyclophosphamide, vincristine, doxorubicin and dexamethasone)
- Allogeneic hematopoietic stem cell transplant
- Peripheral blood:
- Circulating blasts are present and can range from rare to numerous
- Blasts have irregular nuclei, variably dispersed chromatin, inconspicuous nucleoli and basophilic vacuolated cytoplasm
- Bone marrow:
- Aspirate smears and touch preparations contain a variable percentage of blasts (rare to numerous)
- Core biopsy and clot sections show variable cellularity (hypocellular, normocellular or hypercellular)
- Blasts are increased and are present in sheets / clusters or as heterogeneously distributed, clusters of immature cells in the interstitium (Am J Clin Pathol 2020;154:816)
- Background trilineage hematopoiesis is variably diminished
- References: Am J Clin Pathol 2020;154:816, Jaffe: Hematopathology, 2nd Edition, 2016
Contributed by Jesse Manuel Jaso, M.D.
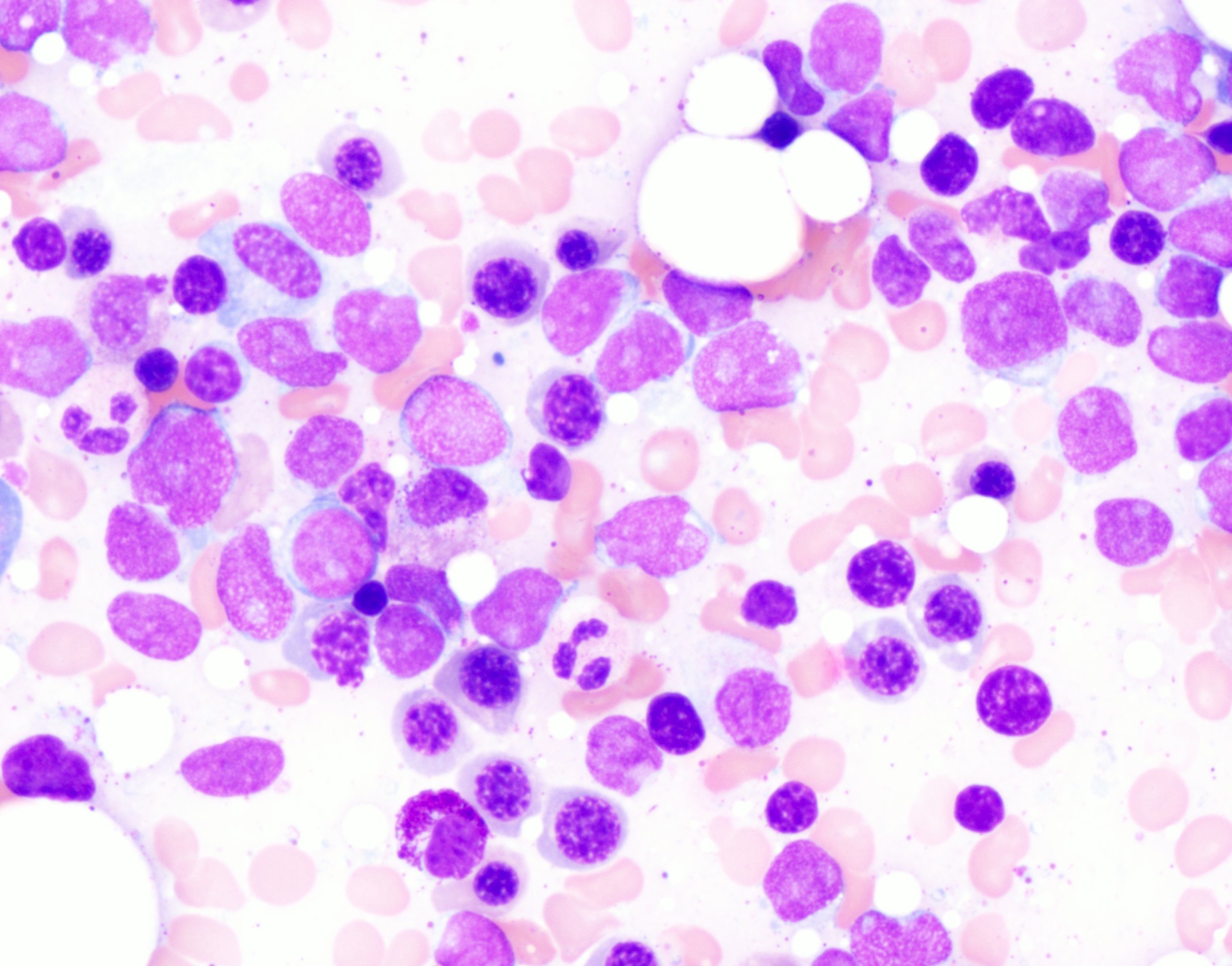
Bone marrow with increased blasts
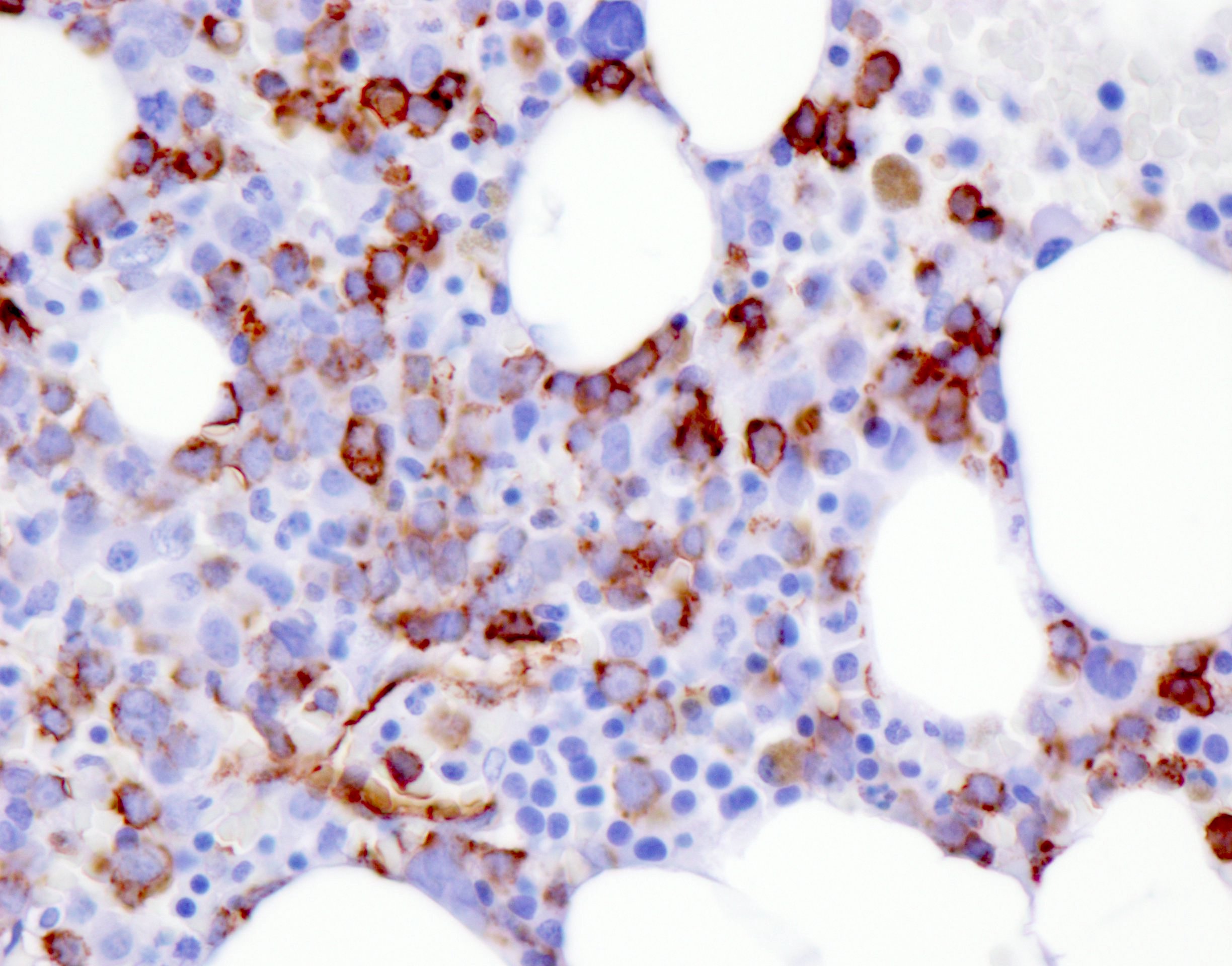
CD34 highlighting clusters of blasts
- CD34, TdT, CD79a, PAX5
- Assist with identification of clusters of blasts in cases with low level involvement
- References: Am J Clin Pathol 2020;154:816, Jaffe: Hematopathology, 2nd Edition, 2016
- Shows several genetic abnormalities that are also seen in therapy related myeloid neoplasms, suggesting a similar pathogenesis (Blood Rev 2019;37:100584)
- KMT2A (formerly MLL) rearrangement:
- Most frequently identified genetic abnormality
- Associated with patients treated for early stage breast cancer with alkylating agents or topoisomerase inhibitors
- Shorter latency interval than other forms of t-B ALL (Blood Rev 2019;37:100584)
- Hypodiploidy:
- Hypodiploidy is a common finding, often with deletion of chromosomes 5 / 5q, 7 / 7q, 13 / 13q, 17 and others
- Low hypodiploid, near triploid karyotype may be seen in t-B ALL (Br J Haematol 2022;196:963)
- Previously described in pediatric patients and associated with aggressive disease (Genes Chromosomes Cancer 2014;53:524)
- Subset of B ALL with low hypodiploidy (31 - 39 chromosomes) may undergo aberrant endoduplication, resulting in a seemingly high hyperdiploid (51 - 65) or near triploid (66 - 79) karyotype on conventional cytogenetic analysis
- Copy number analysis (CNA) can identify this masked hypodiploidy, which is also associated with P53 mutation and loss of chromosome (7p12) (IKZF1 locus), often concurrently (Genes Chromosomes Cancer 2014;53:524)
- IKZF1 encodes Ikaros, a transcription factor involved in B cell development, among other functions
- Relationship between these cytogenetic and molecular findings and t-B ALL pathogenesis remains incompletely understood
- Pediatric patients with this karyotype should also be screened for Li-Fraumeni and other inherited disorders
- BCR::ABL1 rearrangement:
- Seen in a subset of cases
- Incidence and role in t-B ALL pathogenesis is incompletely understood
- Bone marrow, aspirate, clot, core biopsy and peripheral blood:
- Therapy related B lymphoblastic leukemia / lymphoma (see comment)
- Comment: The bone marrow shows increased blasts (***% by manual differential; *** by immunohistochemistry for CD34 / TdT) with atypical morphology and an aberrant B lymphoblast immunophenotype by flow cytometry.
- The findings, in combination with the history of cytotoxic / radiation therapy for prior malignancy (list malignancy) are consistent with therapy related B lymphoblastic leukemia / lymphoma. Correlation with cytogenetic studies is recommended. If clinically indicated, molecular testing for germline mutations should be considered.
- Secondary B lymphoblastic leukemia / lymphoma:
- Diagnosed when patient has a prior malignancy but has not received cytotoxic or radiotherapy
- Inherited disorder (e.g., Li-Fraumeni) predisposing to multiple malignancies:
- Testing for germline mutations should be considered, especially in younger patients
- Hematogone hyperplasia:
- Potential for presentation with low blast percentages may prompt consideration of hematogone hyperplasia / bone marrow regeneration
- Flow cytometry with immunohistochemistry as needed will assist with identification (Leuk Lymphoma 2010;51:10, Am J Clin Pathol 2020;154:816)
- BCR::ABL1 rearrangement
- CREBBP mutation
- ETV6::RUNX1 mutation
- IKZF1 mutation
- KMT2A rearrangement
Comment Here
Reference: Therapy related B ALL
A 20 month old patient presents with lethargy and fever. CBC shows pancytopenia. A bone marrow examination is performed (see image 1 for bone marrow aspirate and image 2 for bone marrow core biopsy with CD34 immunostain). Flow cytometry shows an aberrant B lymphoblast population (15% of total events). Molecular genetic studies identify a mutation in TP53. Which of the following statements is the most correct?
- A concurrent mutation in KMT2A is likely to be identified
- Conventional karyotype and copy number analysis studies are not indicated in these cases
- Germline molecular testing and geneticist consultation should be considered
- The disease is associated with in utero exposure to alkylating agents
- The molecular findings imply a good prognosis
Comment Here
Reference: Therapy related B ALL
- Therapy related myeloid neoplasm refers to cases of acute myeloid leukemia (AML), myelodysplastic syndrome (MDS) and myelodysplastic / myeloproliferative neoplasms (MDS / MPN) occurring as a late complication of cytotoxic chemotherapy or radiation therapy
- Established cases of MPN, MDS or MDS / MPN progressing into AML should not be regarded as therapy related but rather as secondary AML
- Occurs as a late complication of cytotoxic therapy or radiation therapy administered for neoplastic and nonneoplastic conditions
- Can present as either a chronic phase characterized by progressive bone marrow failure (i.e. t-MDS or t-MDS / MPN) or overt leukemic phase (i.e. t-AML)
- Most common etiologies are alkylating agents, topoisomerase II inhibitors and ionizing radiation therapy; other agents are less commonly implicated
- Diagnosed and classified using analogous criteria to the corresponding de novo diseases
- Prognosis is generally poor, compared to the corresponding de novo malignancies
- Therapy related myelodysplastic syndrome (t-MDS); therapy related myelodysplastic / myeloproliferative neoplasm (t-MDS / MPN); therapy related acute myeloid leukemia, alkylating agent related; therapy related acute myeloid leukemia (t-AML)
- ICD-O: 9920/3 - therapy related myeloid neoplasm
- Accounts for around 10 - 20% of all cases of MDS, MDS / MPN and AML (NIH: Therapy Related Myeloid Neoplasms [Accessed 11 May 2020], J Clin Oncol 2015;33:3641, Blood 2011;117:2137)
- Majority of cases arise in patients treated for a previous malignancy; of these, about 70% had a prior solid tumor and 30% a hematological malignancy
- Breast cancer and non-Hodgkin lymphoma account for the largest number of cases (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017, Am J Hematol 2015;90:E80)
- Up to 20% occur following therapy for a nonneoplastic disease (Am J Hematol 2015;90:E80)
- Any age can be affected; however, incidence increases with age proportionally to the higher incidence of cancer in adults
- Risk after exposure to alkylating agents or radiation therapy increases with age; the risk is similar across all ages with topoisomerase inhibitors (Blood 2003;102:43)
- No distinctive gender predilection or ethnic association
- t-MDS, t-MDS / MPN and t-AML occur as the consequence of acquired somatic alterations induced by cytotoxic or radiation therapy
- Alterations occur within hematopoietic stem cells and progenitor cells and confer a proliferative or survival advantage
- Alkylation produces inaccurate base pairing during replication and breaks in the DNA double helix as the alkylated bases are repaired (Mutat Res 1996;355:41)
- Topoisomerase II inhibitors cause chromosomal breakages which favor the development of chromosomal translocations (Chem Res Toxicol 1993;6:585)
- As only a minority of patients treated eventually develop a therapy related myeloid neoplasm, other factors may contribute to the pathogenesis
- Mutations in DNA damage sensing or repair genes (e.g. BRCA1/2 or TP53) or polymorphisms in genes affecting drug metabolism or DNA repair mechanisms are the most common secondary events (Proc Natl Acad Sci U S A 2001;98:11592, Cancer 2016;122:304, Leukemia 2007;21:1413, JAMA 2011;305:1568)
- t-MDS, t-MDS / MPN and t-AML develop mostly following the exposure to (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017):
- Alkylating agents: melphalan, cyclophosphamide, nitrogen mustard, chlorambucil, busulfan, carboplatin, cisplatin, dacarbazine, procarbazine, carmustine, mitomycin C, thiotepa, lomustine
- Topoisomerase II inhibitors: etoposide, teniposide, doxorubicin, daunorubicin, mitoxantrone, actinomycin
- Large field ionizing radiation therapy
- Antimetabolites: thiopurines, mycophenolate mofetil, fludarabine
- Antitubulin agents (often in combination with other agents): vincristine, vinblastine, paclitaxel, docetaxel
- Other agents (hydroxyurea, radioisotopes, L-asparaginase, purine analogues, mycophenolate mofetil) possibly involved but their primary role is unknown
- Majority of t-MDS, t-MDS / MPN and t-AML develop within 10 years after the initial exposure
- Cases related to alkylating agents:
- Represent the majority of cases
- Occur 5 - 10 years after initial exposure
- Are characterized by a chronic phase of bone marrow failure and progressive cytopenia(s) which correspond to t-MDS
- Cases related to topoisomerase II inhibitors:
- Represent a minority of the cases
- Occur after a short latent period (< 5 years)
- Present as overt t-AML, without a preceding chronic phase
- If latency period over 10 years, the hematological malignancy may not be related to therapy (J Clin Oncol 2015;33:3641)
- Signs and symptoms are related to the presence and severity of cytopenia(s) (i.e. anemia, thrombocytopenia, leukopenia)
- Complete blood count with differentials and morphologic review of blood smears to assess for cytopenia(s), dysplasia and blast count
- Bone marrow aspirate and biopsy with features of either MDS, MDS / MPN or AML
- Cytogenetic tests (i.e. conventional karyotyping, FISH) and molecular analysis (i.e. next generation sequencing) for diagnosis, clinical management and risk stratification
- Reference: Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017
- Complete blood counts with differentials:
- Variable cytopenia(s) following exposure to alkylating agents
- Leukocytosis more common after exposure to topoisomerase II inhibitors
- Additional tests to exclude other underlying causes of cytopenia(s), as clinically appropriate (i.e. vitamin B12, folate and iron, Coombs test, etc.)
- Reference: Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017
- Advanced age, poor performance status and low hemoglobin are unfavorable prognostic factors (Am J Hematol 2015;90:E80)
- Cytogenetic findings are key prognostic factors
(Blood 1995;86:3542)
- Abnormalities on chromosome 5 or 7, TP53 mutation or loss and complex karyotype predict poor prognosis with median survival time < 1 year from diagnosis
- Balanced translocations including t(15;17), inv(16) or t(16;16) generally harbor better prognosis
- Therapy related t(15;17), inv(16) or t(16;16) have shorter median survival compared with their de novo counterparts
- 10 month old girl treated with etoposide including regimen for hemophagocytic lymphohistiocytosis (BMC Pediatr 2016;16:116)
- 27 year old woman with systemic lupus erythematous treated with cyclophosphamide and methotrexate (Fukushima J Med Sci 2010;56:121)
- 47 year old woman developing t-MDS after chemoradiation therapy for lobular breast carcinoma (Indian J Pathol Microbiol 2011;54:371)
- 55 year old woman with complete remission after a diagnosis of HTLV1 positive adult T cell leukemia / lymphoma (J Clin Exp Hematop 2015;55:29)
- 56 year old Chinese woman with an erythematous skin rash 18 months after being treated with chemotherapy for ductal breast carcinoma (Acta Derm Venereol 2010;90:649)
- 58 year old woman with 20 year history of rheumatoid arthritis on infliximab and methotrexate therapy (J Clin Exp Hematop 2014;54:137)
- 66 year old man developing pure erythroid leukemia after chemotherapy for pharyngeal cancer (Intern Med 2011;50:3031)
- t-MDS, t-MDS / MPN and t-AML are relatively resistant to conventional therapies
- Allogeneic stem cell transplant can be considered in selective patients
- Therapy related acute promyelocytic leukemia with PML-RARA should be managed as its de novo counterpart (Genes Chromosomes Cancer 2002;33:395, Mediterr J Hematol Infect Dis 2011;3:e2011032)
- Enrollment in clinical trials may be considered
- t-MDS, t-MDS / MPN and t-AML due to alkylating agents
(Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017):
- Increased cellularity
- Multilineage dysplasia, commonly striking
- Increased blast count
- Variable fibrosis
- t-MDS, t-MDS / MPN and t-AML due to topoisomerase II inhibitors
(Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017):
- Increased cellularity
- Frequent features of acute monoblastic, acute monocytic or acute myelomonocytic leukemia
- Common rearrangements involving 11q23
- Other AML with recurrent cytogenetic alterations
- Acute promyelocytic leukemia with t(15;17)(q24.1;q21.2)
- Acute myeloid leukemia with t(8;21)(q22;q22.1)
- Acute myeloid leukemia with inv16(p13.1q22)
- Myeloblasts, monoblasts and megakaryoblasts are included in the blast count
- Promonocytes are considered blasts equivalent in the setting of acute monoblastic, acute monocytic and chronic / acute myelomonocytic leukemia
- Blast count on bone marrow samples (alongside peripheral smears) used to drive final diagnosis (i.e. t-MDS and t-MDS / MPN: < 20%; t-AML: ≥ 20%)
- Primary neoplasm may be manifest (i.e. plasma cell neoplasms, lymphomas): typically form discrete lesions
Contributed by Valentina Sangiorgio, M.D. and AFIP
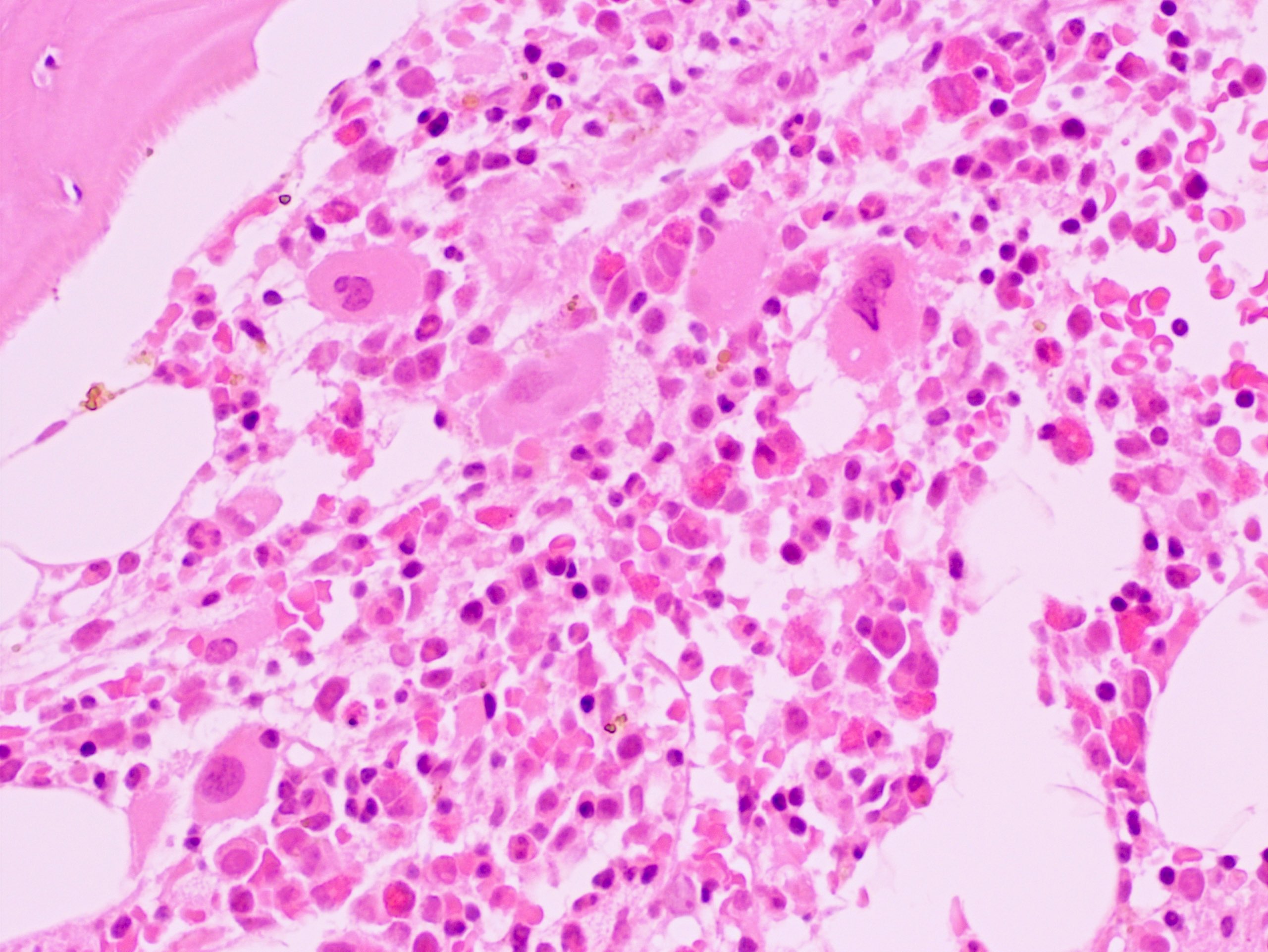
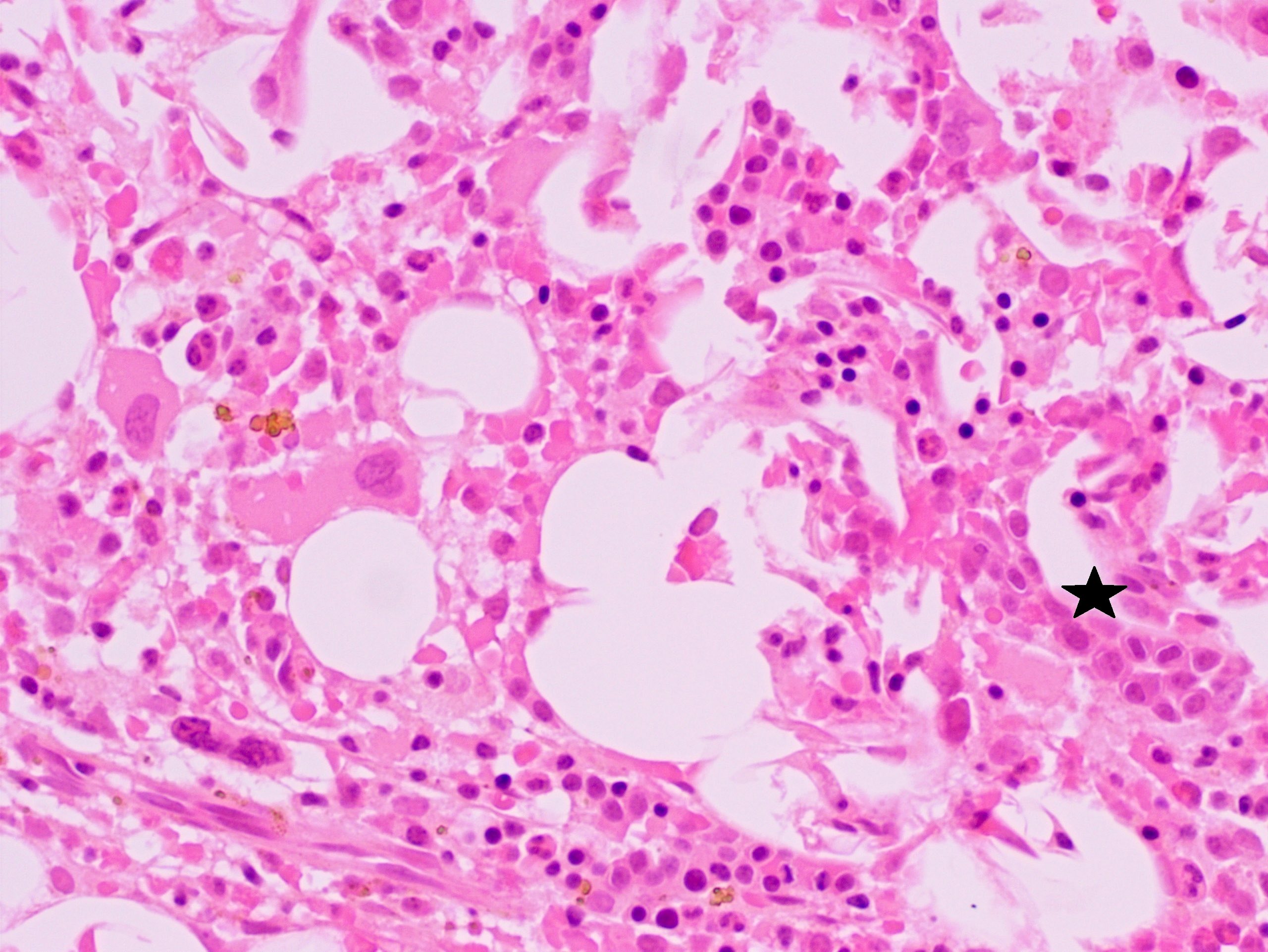
t-MDS: hypercellular bone marrow
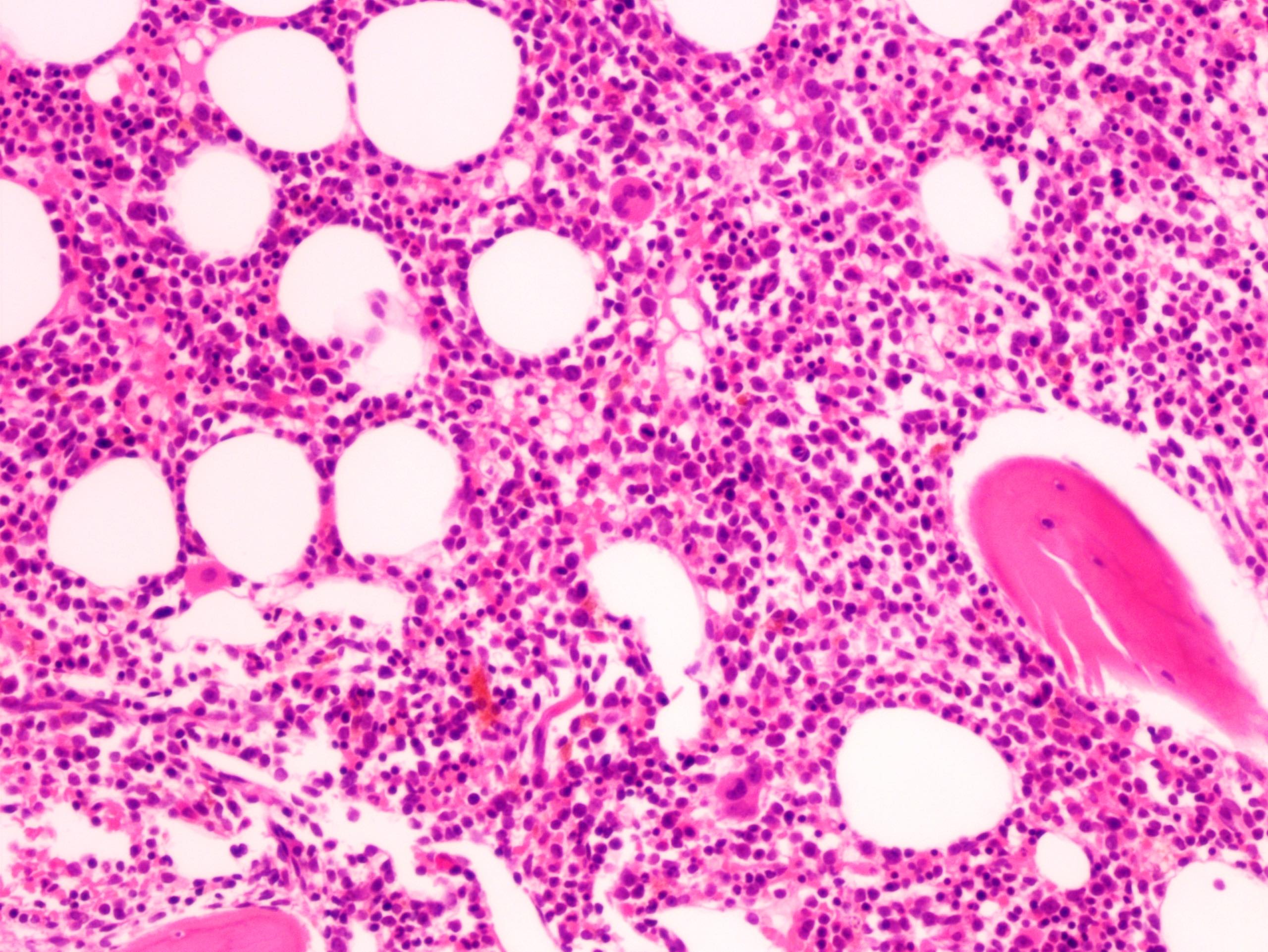
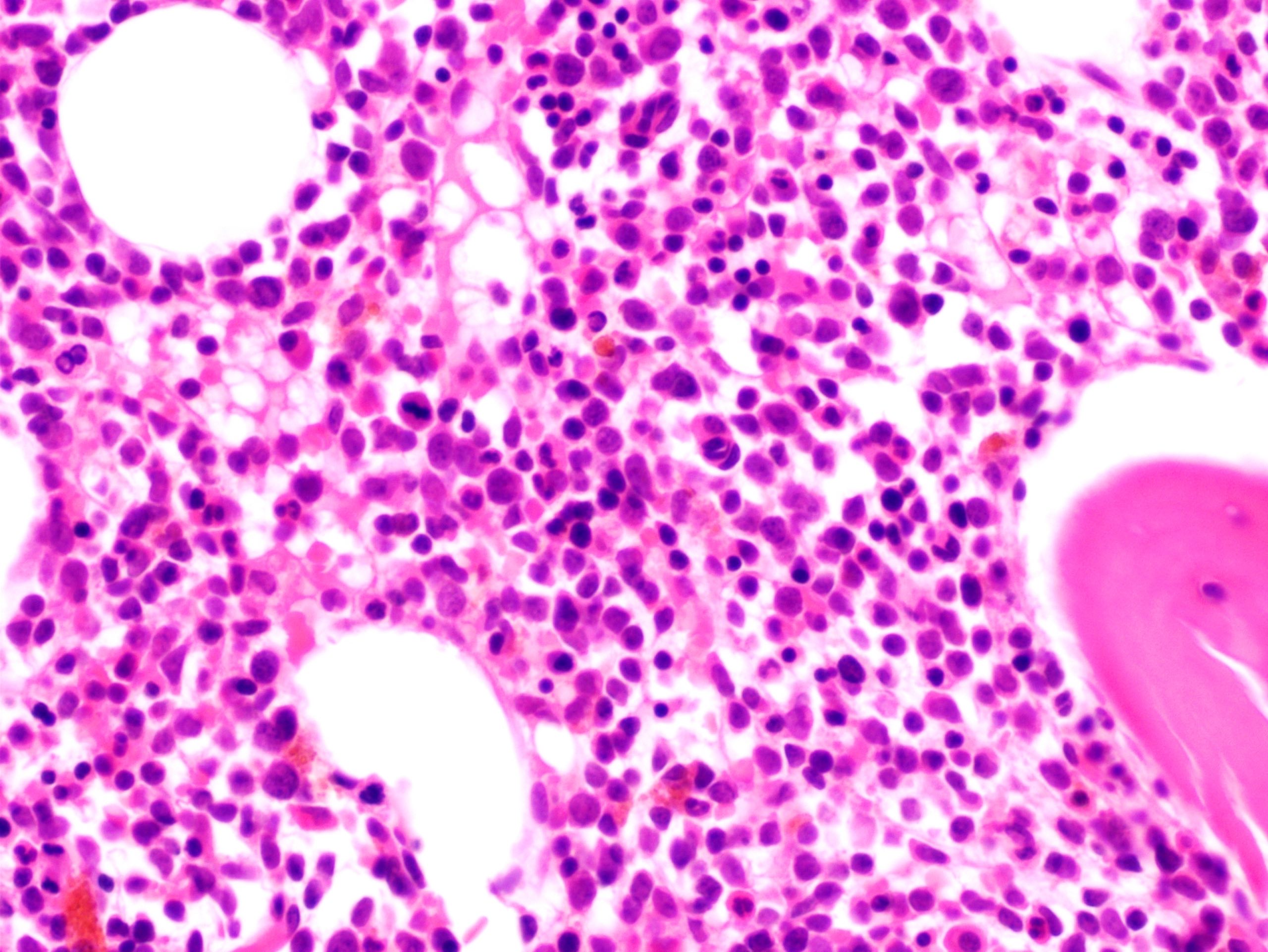
t-AML: hypercellular bone marrow with impaired maturation
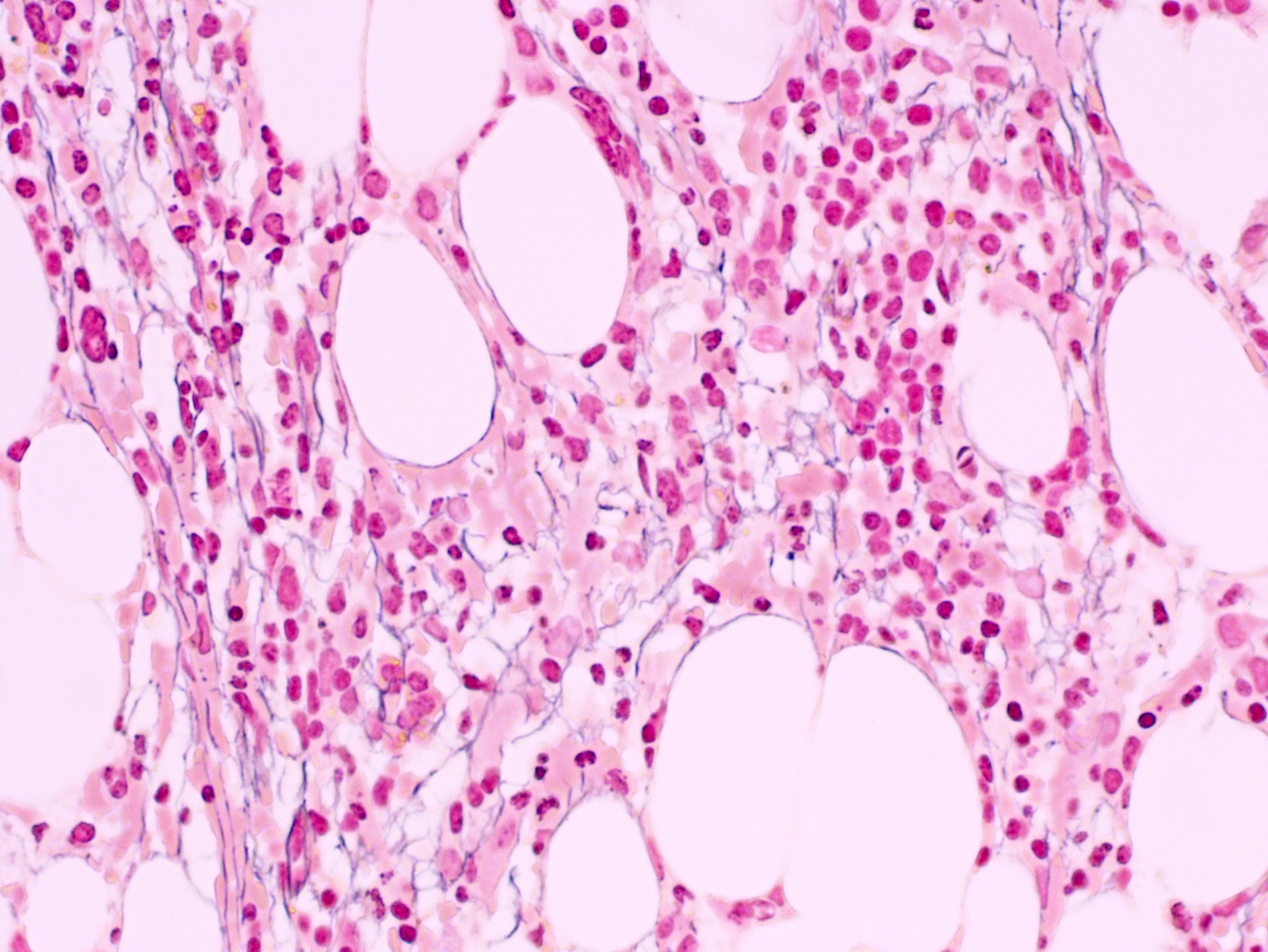
t-MDS: bland reticulin fibrosis
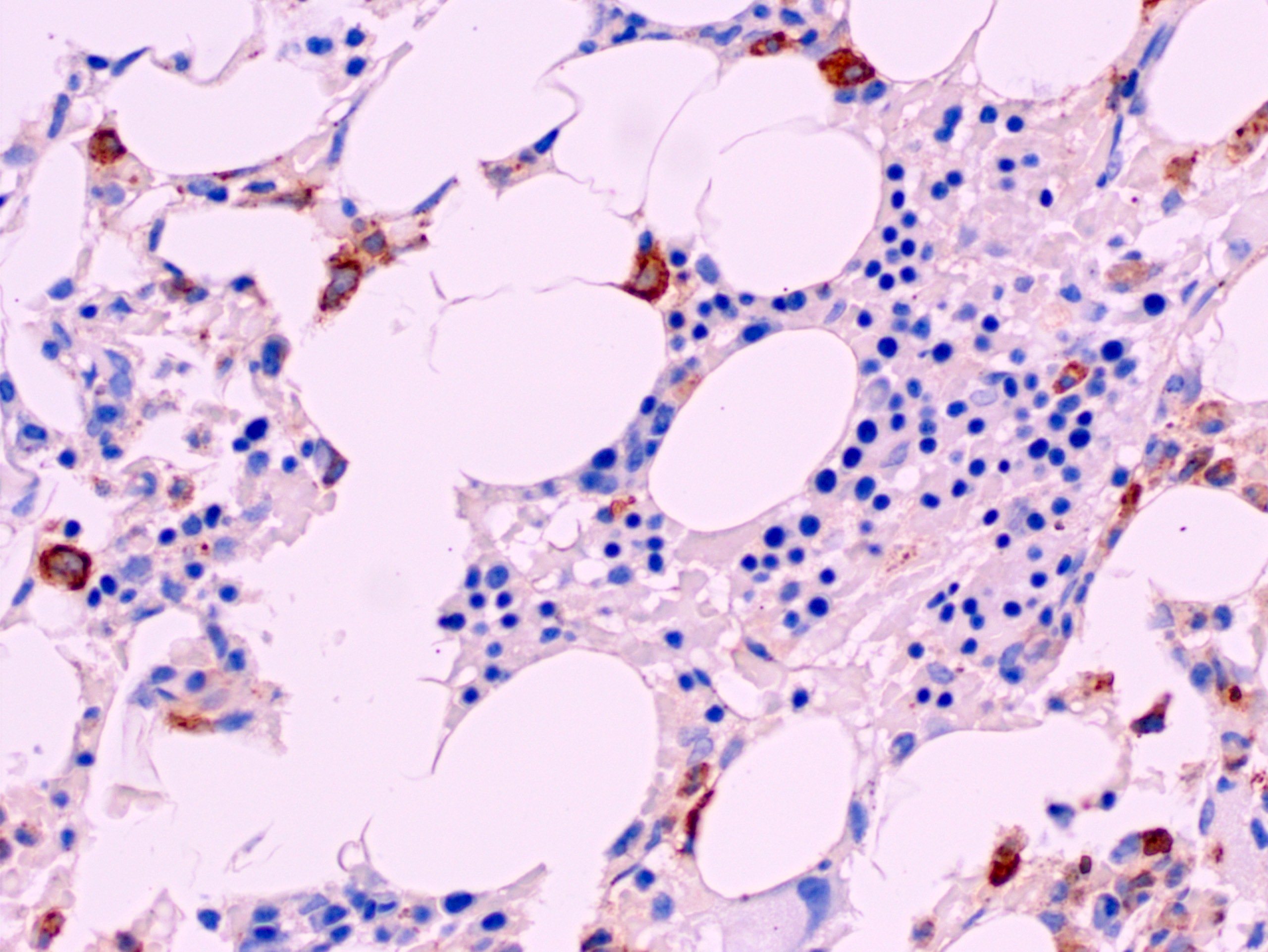
t-MDS: myeloid hypoplasia with left shift
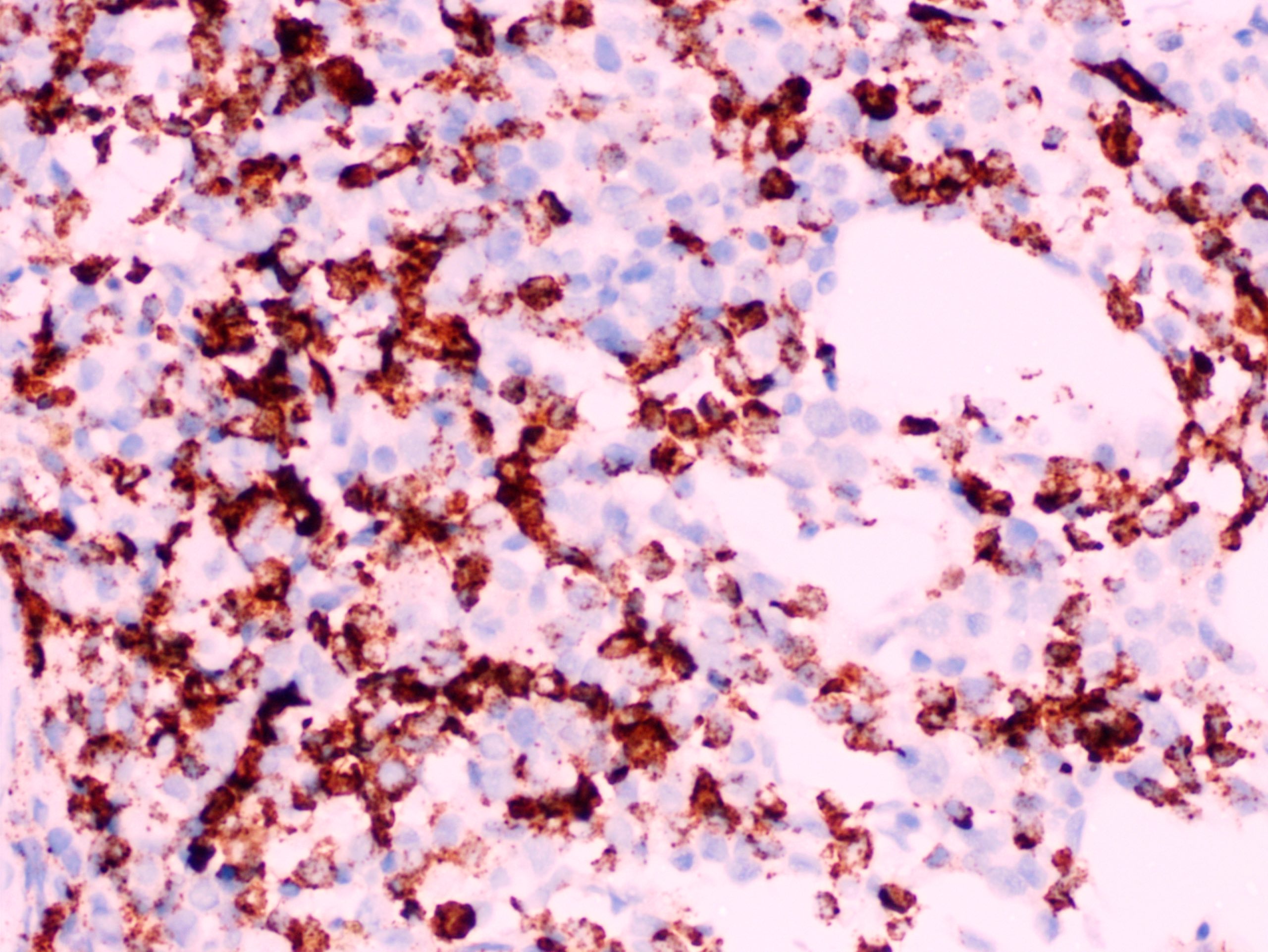
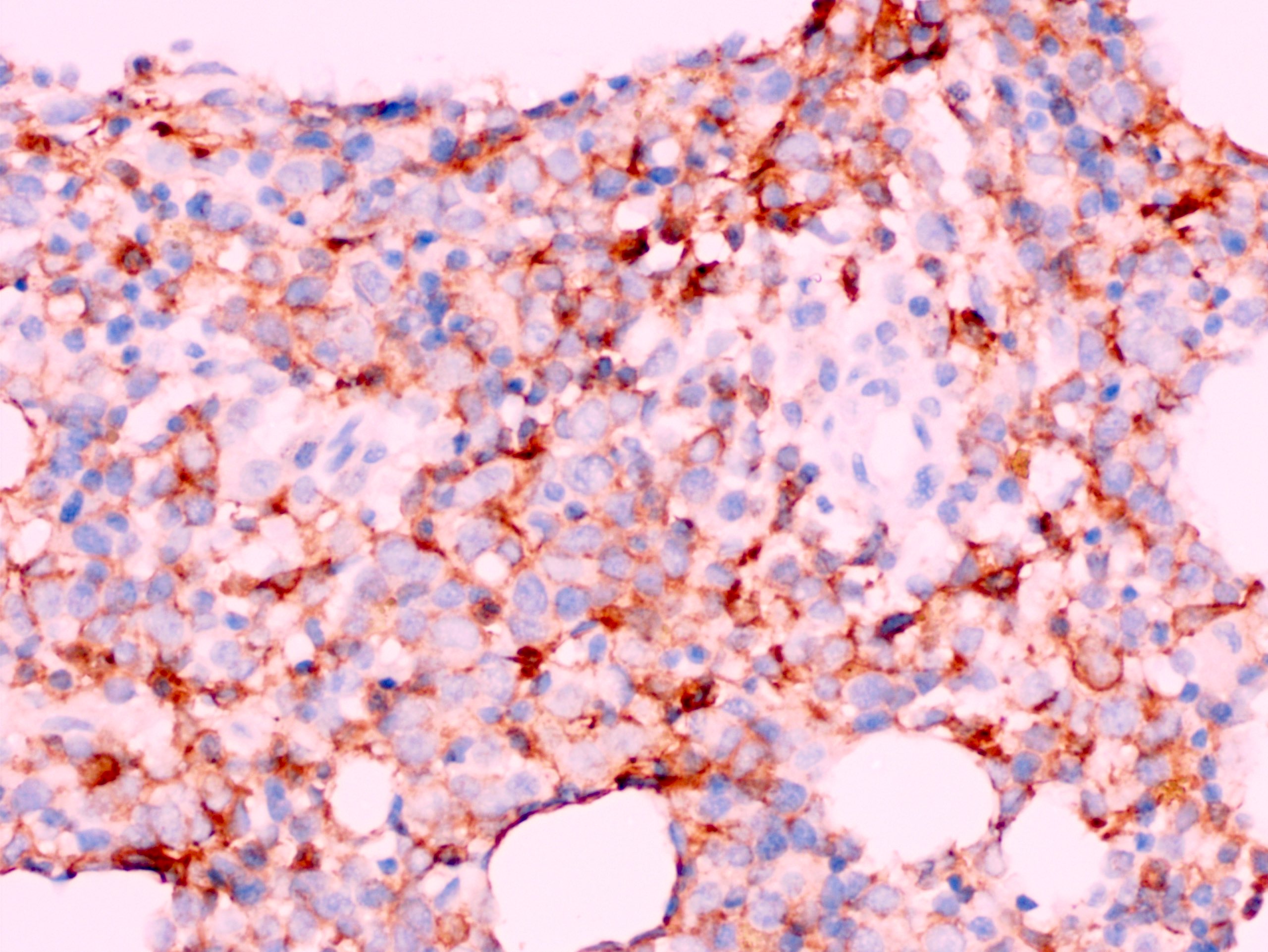
t-AML: clusters of blasts
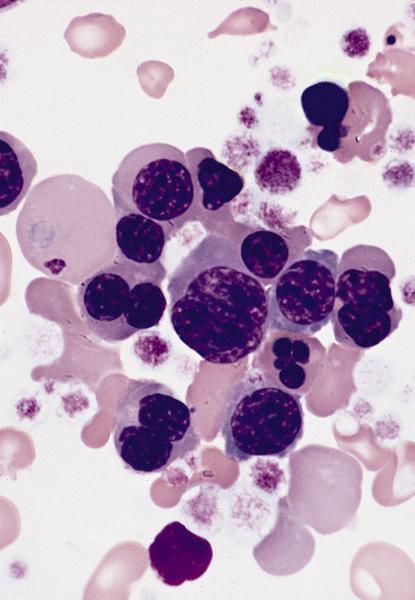
Large erythroid precursors with lobulated nuclei and karyorrhexis

PAS+ erythroid precursors in bone marrow smear
- Cytopenia(s) (i.e. anemia, thrombocytopenia, neutropenia) invariably present
- Unilineage to multilineage dysplasia
- Increased number of circulating blasts
- Leukocytosis more common in topoisomerase II related subtypes (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Monoblastic, acute monocytic or acute myelomonocytic leukemia more common in topoisomerase II related cases (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
AFIP images
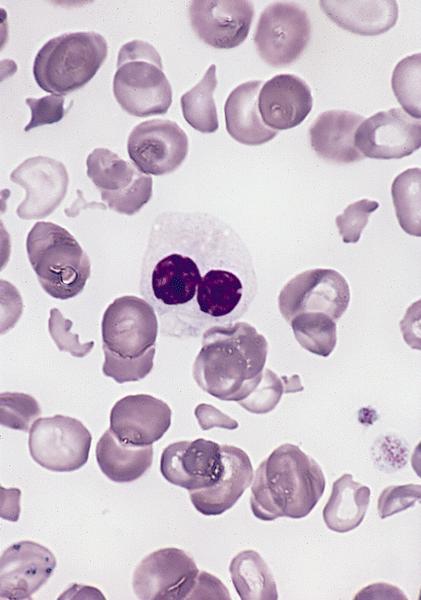
Postradiochemotherapy
and splenectomy for
Hodgkin disease
- CD34 to assess number and distribution of hematopoietic precursors
- CD34 can highlight abnormal localization of immature precursors (ALIP)
- Not all myeloblasts express CD34 alongside monoblasts
- CD117 can be used alternatively
- For diagnostic purposes, blast count should be performed on bone marrow aspirate smears and peripheral smears
- Megakaryocytic markers (CD61, CD42b, factor VIII) used to highlight number and distribution of megakaryocytes and micromegakaryocytes
- Erythroid markers (hemoglobin A, glycophorin A, CD71, E-cadherin) to assess erythroid lineage and colony formation
- Myeloperoxidase highlights myeloid cells and ALIP, if present (Foucar: Diagnostic Pathology - Blood and Bone Marrow, 2nd Edition, 2017)
- Strong p53 immunostaining (≥ 1% of bone marrow cells) is predictive of a TP53 gene mutation and correlates with the presence of a high risk karyotype (Mod Pathol 2015;28:552)
- There are no specific immunophenotypic findings in t-MDS, t-MDS / MPN and t-AML compared with the de novo counterparts
- Blasts generally express CD34 alongside other myeloid markers (i.e. CD13, CD33, CD117, myeloperoxidase)
Contributed by Valentina Sangiorgio, M.D.
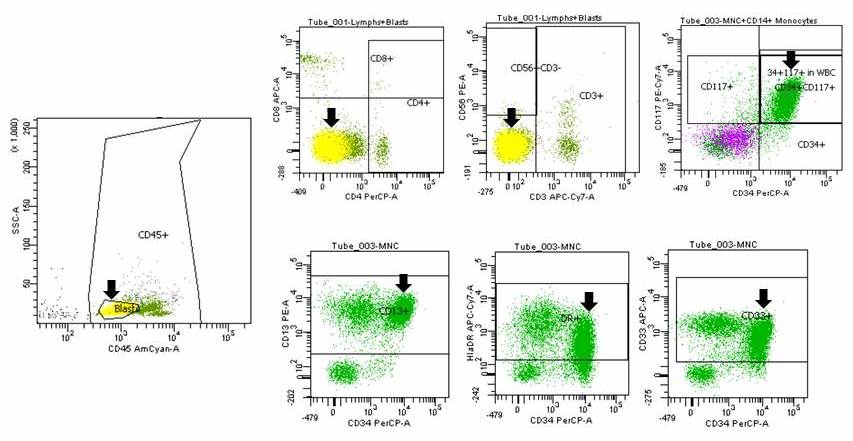
t-AML
- t-MDS, t-MDS / MPN and t-AML show cytogenetic abnormalities in as many as 90% of cases
- Around 70% of cases harbor unbalanced chromosomal aberrations:
- Most common alterations affect chromosome 5 (i.e. -5q or -5) and 7 (i.e. -7q or -7)
- These generally occur in the context of a complex karyotype or co-occur with mutations or loss of TP53
- Such changes correlate with a long latent period, a preceding phase of t-MDS and exposure to alkylating agents or radiation therapy (Leukemia 2008;22:240)
- Around 30% have balanced translocations:
- 11q23 (KMT2A gene) is frequently involved
- Others rearrangements: t(8;21)(q22;q22.1), t(15;17)(q24.1;q21.1) and inv16(p13.1q22)
- These cases develop shortly after exposure to the etiologic agent(s) and present with overt t-AML without a preceding chronic phase
- Common following topoisomerase II inhibitors or radiation therapy alone (J Clin Oncol 1998;16:1897)
- A minority of cases have a normal karyotype
- TP53 is the most commonly mutated gene, occurring in as many as 50% of cases (Haematologica 2013;98:908)
- NPM1 and FLT3 mutations are less common in t-AML compared with de novo AML
- If considering t-AML with normal karyotype, no difference is seen (Leukemia 2008;22:240)
Contributed by Valentina Sangiorgio, M.D.
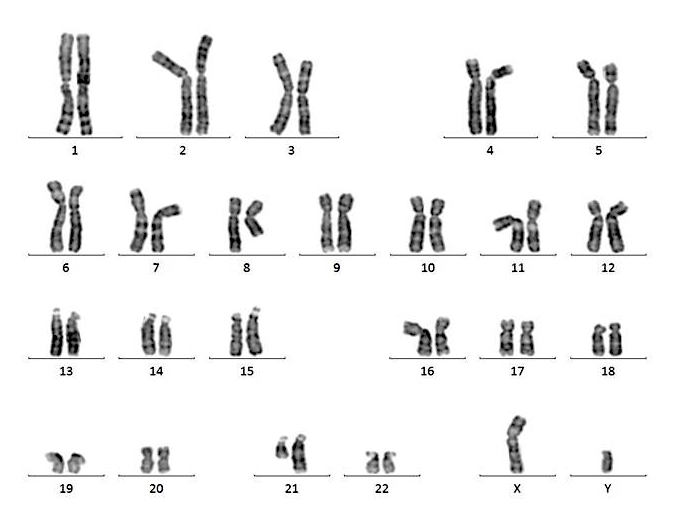
G banding karyotype of
t-AML
Role of bone marrow transplant in therapy related AML and MDS
- Bone marrow, biopsy and aspirate (t-AML):
- Hypercellular for age, with infiltration (around 60%) by acute myeloid leukemia and background of multilineage dysplasia (see comment)
- Comment: In light of the current histologic findings within the bone marrow alongside the detection of a complex karyotype and of the patient's known past medical history (breast carcinoma treated with chemo and radiation therapy), the case is best classified as therapy related acute myeloid leukemia. Clinical correlation is advised.
- Bone marrow aspirate microscopic description (t-AML): Multiparticulate sample showing hypercellular marrow for the patient's age (cellularity around 80%). Erythropoiesis is increased and shows prominent megaloblastoid changes; dysplastic changes in the form of multinucleation and nuclear bridging are seen. Myelopoiesis is increased with left-shifted maturation. Majority of neutrophils shows pseudo-Pelger-Huët changes. Megakaryocytes are seen in adequate number but are overtly dysplastic, the majority being small and hypolobated. There is prominent increase in blasts which are medium in size with scant cytoplasm and open chromatin with variably prominent nucleoli. A cell count (performed on 150 cells) reveals: 39% blasts, 20% promyelocytes / myelocytes, 11% maturing granulocyte forms, 27% erythroid precursors, 1% lymphocytes, 1% plasma cells, 1% monocytes. Prussian blue iron stain shows normal storage iron; no ring sideroblasts are noted.
- Bone marrow biopsy microscopic description (t-AML): The description of bone marrow trephine findings is similar to the aspirate sample (see above). Additional comments should be as follow: "CD34 highlights an increase in hematopoietic precursors which account for around 30% of the total nucleated cells. Aberrant expression of CD34 within the megakaryocytes is also seen (only if seen)." Reticulin stain reveals an increased network of reticulin fibers with many intersections (WHO MF1).
- Peripheral smear microscopic description (t-AML): Pancytopenia with dysplastic neutrophils. 15% of the nucleated cells are blasts.
- Chemotherapy induced dysplasia and bone marrow suppression:
- Recent rather than remote chemoradiation therapy
- Usually resolves after chemotherapy discontinued
- Good response to hematopoietic growth factors (i.e. erythropoietin, G-CSF)
- Coincidental myeloid neoplasms in patients with history of chemotherapy:
- Lacks prototypic morphologic or genetic features of t-MDS, t-MDS / MPN and t-AML (i.e. JAK2 positive MPN unrelated to previous therapy)
- 10% if prior treatment with alkylating agents
- 10% if prior treatment with topoisomerase II inhibitors
- 20%
- 30%
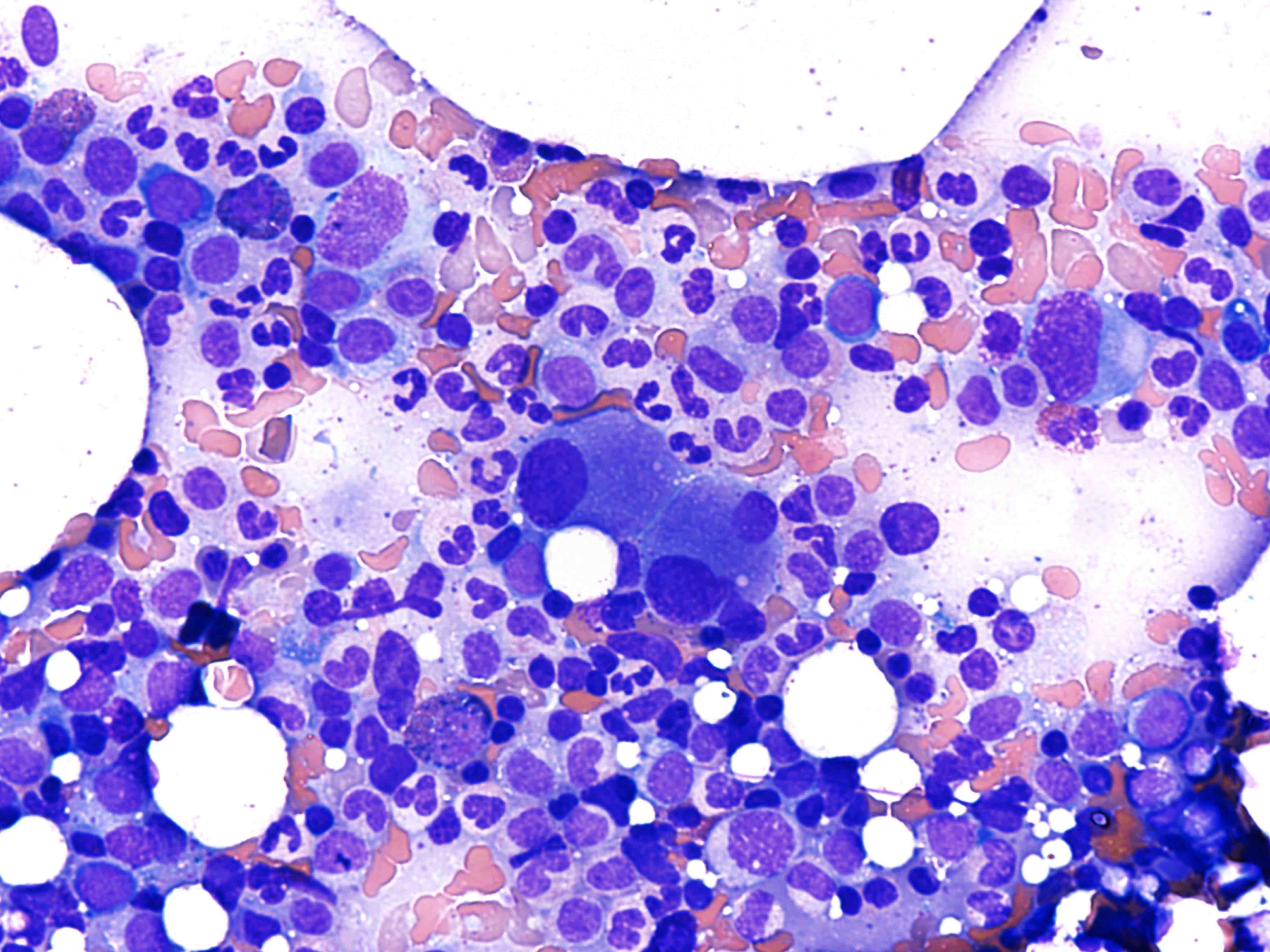
Is the presence of these small and hypolobated megakaryocytes expected in therapy related myeloid neoplasms?
- No; small and hypolobated megakaryocytes are not a common finding in therapy related myeloid neoplasms
- No; small and hypolobated megakaryocytes, especially if many, suggest a myeloproliferative neoplasm
- Yes; small and hypolobated megakaryocytes are a common manifestation of dysplasia and can be seen in therapy related myeloid neoplasms
- Yes; small and hypolobated megakaryocytes are required for t-MDS
Comment Here
Reference: AML therapy related
- Transient disorder of newborns with Down syndrome or phenotypically normal neonates with trisomy 21 mosaicism
- Presents within 3 - 5 days of birth and resolves spontaneously within 3 months
- Proliferation of nonerythroid blasts (commonly megakaryoblasts) in the peripheral blood or organs
- Morphologically indistinguishable from acute myeloid leukemia (AML), specifically acute megakaryoblastic leukemia (AMKL)
- Unique distinguishing clinical, immunophenotypic and molecular genetic features from AML not associated with Down syndrome
- 10% of newborns with Down syndrome or trisomy 21 mosaicism
- Proliferation of nonerythroid blasts in the peripheral blood, bone marrow or organs
- Morphologically indistinguishable from other forms of AML
- Presents within first week of life and resolves within 3 months
- 20 - 30% may progress to nontransient AML (i.e. AMKL) within 1 - 3 years
- Associated with acquired GATA1 mutation
- Transient abnormal myelopoiesis associated with Down syndrome (TAM)
- Transient myeloproliferative disorder (TMD)
- Transient leukemia (TL)
- ICD-O: 9898/1 - Transient abnormal myelopoiesis
- Manifests in approximately 10% of neonates with Down syndrome
- 7 - 16% of TAM is seen in phenotypically normal neonates with trisomy 21 mosaicism
- Peripheral blood
- Common site of blasts, as fetal hematopoiesis occurs predominantly in the liver
- Peripheral blood involvement > bone marrow
- Bone marrow
- Less common site of fetal hematopoiesis
- Other organs
- Liver, spleen, skin, pancreas, kidneys, pleural fluid, pericardial fluid
- 3 step process (Curr Hematol Malig Rep 2016;11:333):
- Perturbation of fetal liver hematopoiesis by trisomy 21
- Acquired or somatic mutation of GATA1 (chromosome X), hematopoietic transcription factor
- 20 - 30% progress to AML with further acquisition of oncogenic mutations
- Risk factors for Down syndrome
- Advanced maternal age
- Most diagnosed at 3 - 5 days of age
- Most patients are asymptomatic but may present with myeloblast organ infiltration:
- Hepatosplenomegaly (common)
- Ascites, pericardial or pleural effusions, hepatic fibrosis, disseminated intravascular coagulopathy (less common)
- Severe organ dysfunction causing renal, hepatic or cardiopulmonary failure (rare)
- If in utero, may present as hydrops fetalis secondary to cardiopulmonary failure and anemia
- References: Curr Hematol Malig Rep 2016;11:333, Silberstein: Hematology - Basic Principles and Practice, 7th Edition, 2017
- No universal diagnostic criteria
- Children's Oncology Group (COG) (Blood 2011;118:6752):
- Detection of nonerythroid blasts in peripheral blood or organs
- Newborns < 90 days old
- Trisomy 21 or mosaicism
- Confirmation with:
- Second blood sample
- > 5% nonerythroid blasts in bone marrow
- Hepatosplenomegaly, lymphadenopathy or pericardial / pleural effusions
- Presence of megakaryoblasts in peripheral blood
- Thrombocytopenia is most common
- Moderate leukocytosis with myeloid left shift
- Basophilia
- Coagulopathy, specifically disseminated intravascular coagulation (DIC) (10%)
- Elevated conjugated bilirubin (common)
- Secondary to liver involvement
- References: Curr Hematol Malig Rep 2016;11:333, Silberstein: Hematology - Basic Principles and Practice, 7th Edition, 2017
- Majority resolve spontaneously over several weeks to 3 months
- Approximately 15 - 23% of patients may die as a result of secondary organ failure:
- Hepatic fibrosis
- Cardiopulmonary failure
- Poor prognostic factors for early death include:
- High WBC count
- Increased bilirubin
- Elevated liver function tests (LFTs)
- Failure to normalize blood counts
- 20 - 30% may develop nontransient AML (usually AMKL) within 1 - 3 years
- Greatest risk factors for AML progression are karyotypic abnormalities in addition to +21 in blast cells
- References: Curr Hematol Malig Rep 2016;11:333, Mycopathologia 2016;181:909
- 7 day old neonate with TAM and severe multiorgan dysfunction (J Pediatr Hematol Oncol 2021;43:e292)
- 8 day old phenotypically normal newborn with TAM associated with Down syndrome mosaicism (Children (Basel) 2020;7:52)
- 14 day old newborn with TAM and pericardial effusion (Clin Case Rep 2019;7:1280)
- 2 neonates with TAM (J Pediatr Genet 2019;8:187)
- Treatment is often supportive due to spontaneous remission
- In severe organ dysfunction, exchange transfusion, leukapheresis or chemotherapy may be necessary
- In TAM progression to AML, cytosine arabinoside is the most commonly used chemotherapeutic agent
- Favorable prognosis
- References: Curr Hematol Malig Rep 2016;11:333, Pediatr Int 2019;61:222
- Blasts are morphologically indistinguishable from those in AMKL associated with Down syndrome
- Features of megakaryoblasts
- Increased nuclear:cytoplasmic (N:C) ratio
- Dispersed nuclear chromatin
- Basophilic cytoplasm
- Coarse basophilic cytoplasmic granules
- Cytoplasmic blebbing
- Erythroid and megakaryocytic dysplasia are also seen
- Dyserythropoiesis with bi and trinucleated forms
- Dysmegakaryopoiesis with dysplastic small forms and micromegakaryocytes
Contributed by Julia T. Geyer, M.D. and Tayler A. van den Akker, M.D.
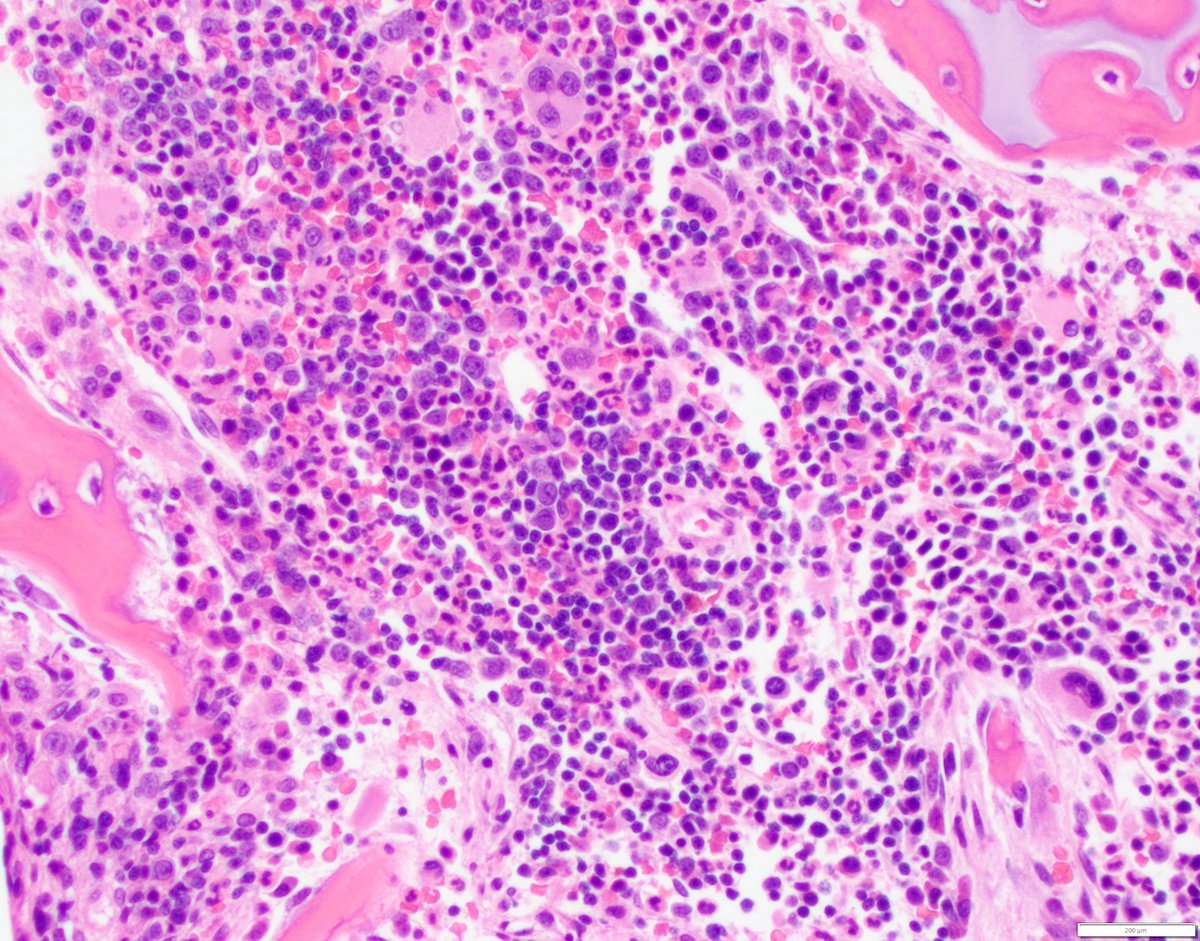
Bone marrow biopsy
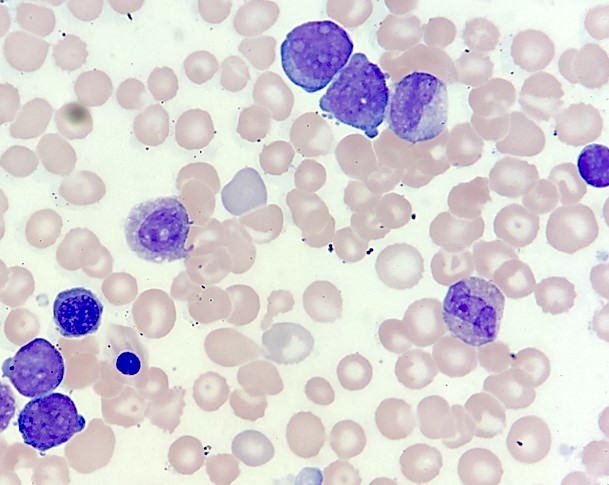
Blasts of TAM-DS
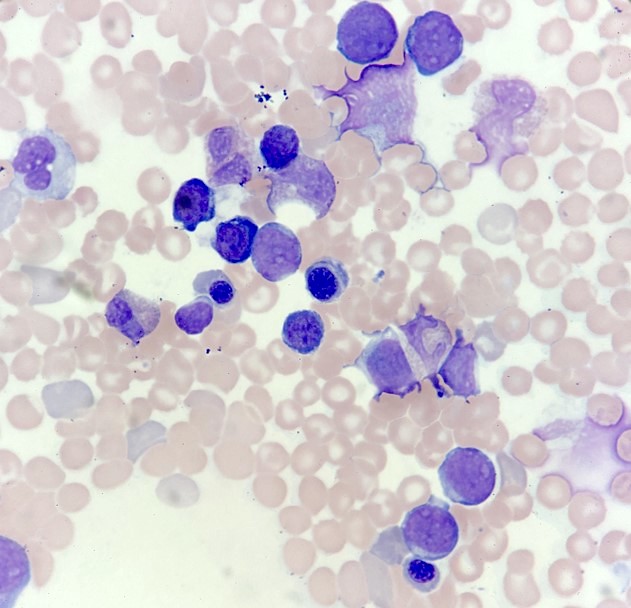
Blasts with hematopoietic elements

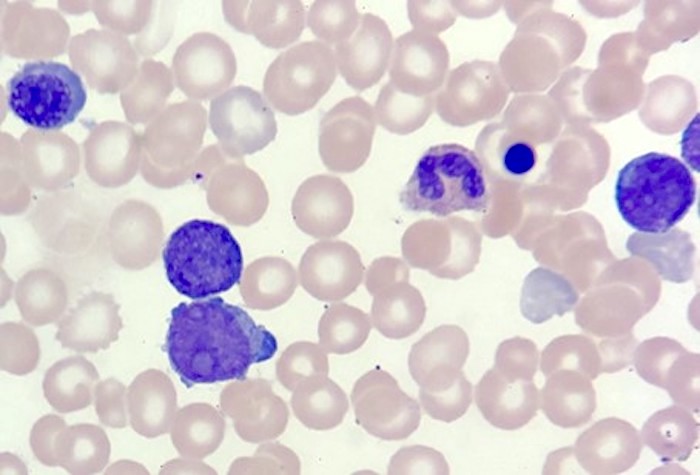
Cytoplasmic blebs
- Features of megakaryoblasts in peripheral blood
- Increased N:C ratio
- Dispersed nuclear chromatin
- Basophilic cytoplasm
- Coarse basophilic cytoplasmic granules
- Cytoplasmic blebbing
- Basophilia
Contributed by Julie Teruya-Feldstein, M.D.

Peripheral blood blasts
- Expanded population of CD45+ myeloid blasts (left shift in myeloid maturation)
- Increased CD34+ myeloid population, may exceed 20% in peripheral blood, expressing CD117, CD33, CD38
- Aberrant expression of CD7 and CD56
- Expression of CD41 and CD61
- Reference: Cherian: Flow Cytometry in Evaluation of Hematopoietic Neoplasms, 1st Edition, 2012
Contributed by Tayler A. van den Akker, M.D.
CD45+ dim population
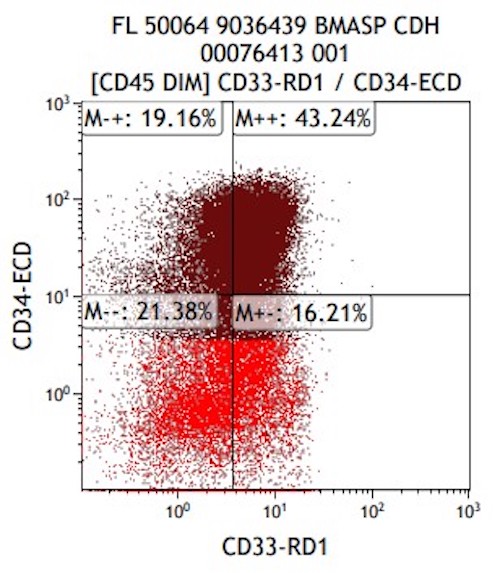
CD34+, CD33+ dim population
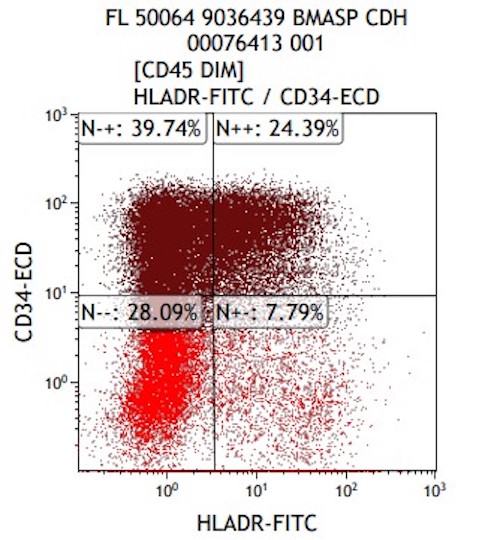
CD34+,
HLA-DR+ partial population
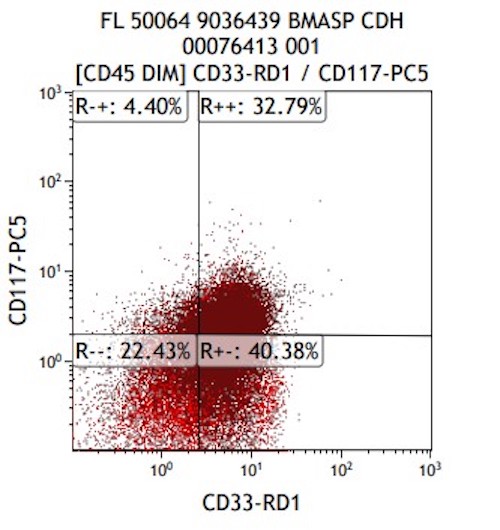
CD33+ dim, CD117+ population
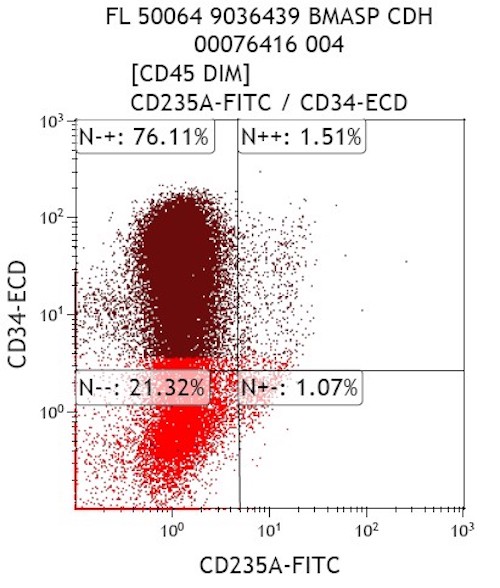
CD34+, CD235A- population
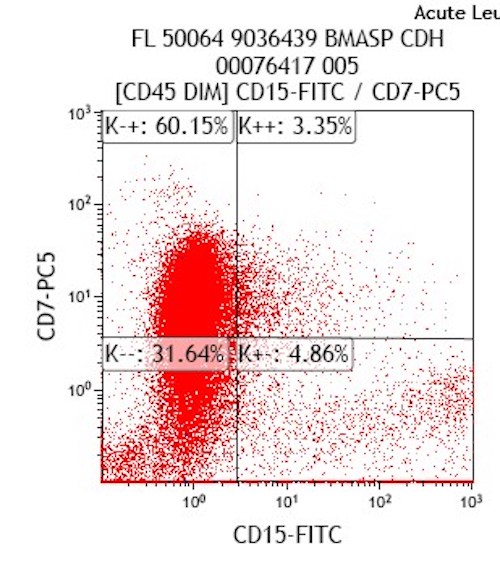
CD7+ heterogeneous, CD15- population
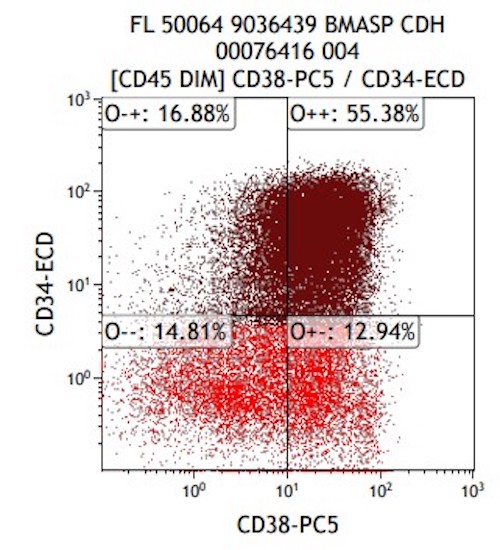
CD34+, CD38+ population
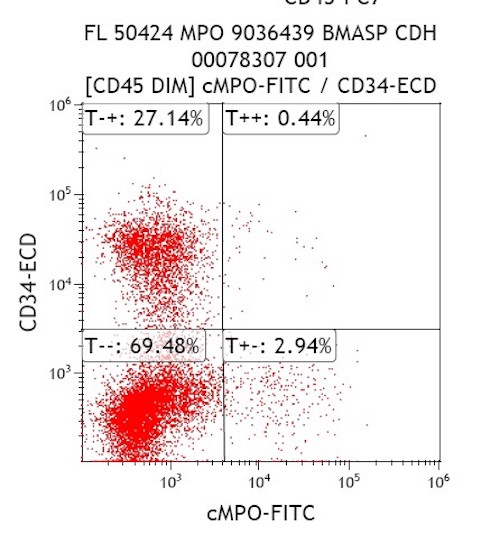
CD34+, MPO- population
- Trisomy 21
- GATA1 mutation in blasts
- Expression of truncated protein, which promotes megakaryocytic and abnormal blast proliferation
- References: Curr Hematol Malig Rep 2016;11:333, Pediatr Int 2019;61:222
- Bone marrow, right posterior iliac crest and aspirate smears:
- Hypercellular marrow (100%), consisting predominantly of left shifted granulocytic precursors with focally interspersed small hypolobated megakaryocytes. Increased blasts (20%) with cytoplasmic blebs and prominent nucleoli. These findings are suspicious for transient abnormal myelopoiesis versus congenital AML (see comment).
- Comment: Flow cytometric analysis demonstrates an aberrant blast population (approximately 35% of total), exhibiting the following immunophenotype: CD45 moderate+, CD34+, CD33 dim+, HLA-DR partial+, CD117+, CD38+. CD7 heterogeneous+, CD4 dim+. These findings do not definitively distinguish between transient abnormal myelopoiesis and acute myeloid leukemia; clinical correlation is required.
- Bone marrow, left posterior iliac crest, trephine biopsy and clot: Quality: adequate. Cellularity: 100%. Normocellular marrow for age (~100%) consisting predominantly of left shifted granulocytic precursors. Megakaryocytes are increased, predominantly hypolobated forms and not seen in dense clusters. No granulomas. The bony trabeculae are unremarkable.
- Bone marrow aspirate smears: Quality: adequate. Blasts are increased, approximately 20% of total by manual differential count. Blasts with cytoplasmic blebs and prominent nucleoli.
- AMKL in Down syndrome:
- Anemia, with preserved WBC count
- Occurs after the first year of life (versus first 3 - 5 days in TAM)
- May have a history of TAM
- Dyserythropoiesis and bone marrow fibrosis more common (versus TAM)
- Frequently negative for CD34, CD56, HLA-DR and positive for CD11b and CD13
- Complex karyotype and other cytogenetic abnormalities (i.e. JAK-STAT pathway) in addition to +21 and GATA1 mutation
- Acute myeloid leukemia not associated with Down syndrome:
- Commonly adults; median age: 65 - 68 years
- Poor prognosis compared with Down syndrome related AML (favorable prognosis)
- AMKL subtype is rare (< 5% AML)
- Absence of trisomy 21
A 4 day old boy with Down syndrome is diagnosed with a transient myeloproliferative condition associated with the cells seen in the above image. Which genetic abnormality would be expected in addition to the additional chromosome 21?
- GATA1 mutation
- NPM1 mutation
- PDGFRA mutation
- t(9;22) with p190 breakpoint
Comment Here
Reference: Transient abnormal myelopoiesis associated with Down syndrome
- CD2+, CD4+, CD8- CD7-, CD10+, CXCL13+, BCL6+
- CD7+, CD3+, MPO-, CD4+, CD8+, TdT+, CD34+
- CD19+, Cd20+dim, CD5+, CD10-, FMC7-
- CD117+, CD34-, CD41+, CD7+, CD56-, CD61+, MPO-
Comment Here
Reference: Transient abnormal myelopoiesis associated with Down syndrome
- 20 - 30% of childhood preB ALL; most common translocation (Chin Med J (Engl) 2003;116:1298) but not infants; also 3% of adults
- 5 different patterns of gene expression involving 14 genes, can detect with gene chip (BMC Genomics 2007;8:385)
- Excellent prognosis due to good response to chemotherapy; 90% remissions; relapses occur later than other ALL
- Persistence of TEL-AML1 transcripts is not necessarily related to relapse (Pediatr Int 2003;45:275)
- No distinct morphology
- Fusion of TEL/ETV6 and AML1/RUNX1/CBFA2 genes
- Detect with FISH or PCR; not found by conventional cytogenetics (i.e. are cryptic) because rearranged segments are too small
- May have no other molecular abnormalities, almost never > 50 chromosomes
Images hosted on other servers:

FISH: t(12;21)(p13;q22)
- 5 - 6% of ALL; commonly detected by conventional cytogenetics in children
- E2A-PBX1 (TCF3-PBX1) is chimeric gene formed by t(1;19)(q23;p13.3) (Mol Cell Biol 1994;14:3938)
- High WBC counts, frequent CNS involvement
- In children (Blood 1984;63:721) and adults (Haematologica 2010;95:241), is no longer a poor prognostic factor due to intensive therapy and bone marrow transplantation
- Child with t(1;19) without fusion transcript by PCR or Southern blot (Rinsho Ketsueki 2005;46:7)
- No distinct morphologic findings
- Produces fusion transcript of PBX1 and E2A; occurs in balanced and unbalanced forms; unbalanced form is der(19)t(1;19)
- The reciprocal product of der(1)t(1;19) is lost and the normal chromosome 1 is duplicated (Leukemia 2001;15:95)
Notes:
- “Unbalanced translocations” means the exchange of chromosomal material is unequal resulting in extra or missing genes
- “der” means derivative chromosome - term is used when only one chromosome from a translocation is present, or when one chromosome has two or more structural abnormalities
- Diagnosis based on immunophenotype and genetic findings, even with low blast count
- Very rare; < 1% of all ALL; both children and adults (Blood 1989;73:2081)
- Presents with variable eosinophilia; may be asymptomatic and lack any blasts (Medicine (Baltimore) 1990;69:232)
- 13 year old boy with cardiac disease (BMJ Case Rep 2012 Jan 20;2012)
- CD10+ acute leukemia with t(5q;14q) translocation and hypereosinophilia (Acta Haematol 1989;82:85)
- CD19+ CD10+ lymphoblasts with circulating reactive eosinophils
- ALL with BCR-ABL1 fusion transcript (Philadelphia chromosome)
- 30% of adults with ALL, 4% of children but 80% of infants
- Excluding infants, older age and higher WBC at presentation than other B ALL
- May have more organomegaly or CNS involvement than other B ALL
- Poor prognosis
- Tyrosine kinase inhibitors (Imatinib) cause some complete responses (Cancer 2007;110:1178), but change in overall survival is minimal (Cancer 2007;109:2068, Hematology Am Soc Hematol Educ Program 2007:435)
- No defining morphology, but large blasts with prominent nucleoli and cytoplasmic granules are more common than other B ALL
AFIP images
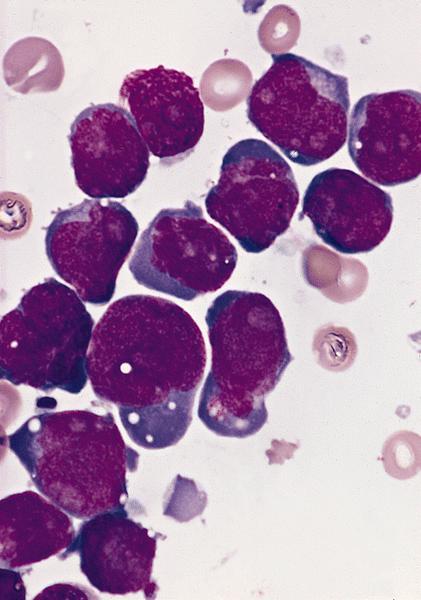
Leukemic cells resemble early erythroblasts
Contributed by Julia Braza, M.D., M.S.
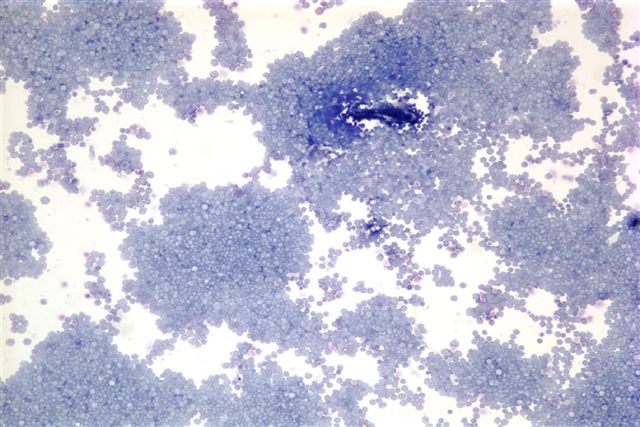
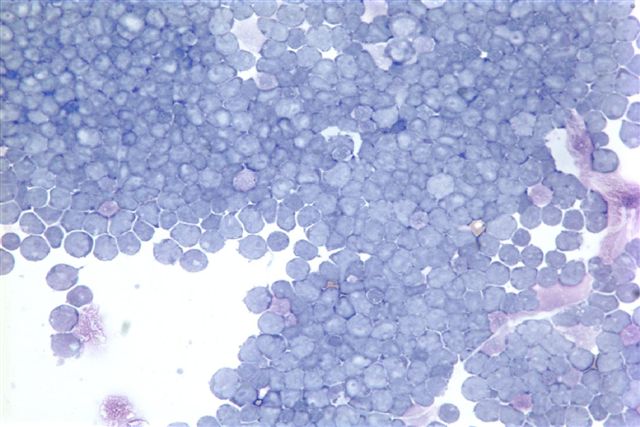

CSF
- Myeloid antigens in 71% (Am J Clin Pathol 1999;111:467)
Images hosted on other servers:

B-ALL associated with t(9;22); lymphoblasts express B-lineage
markers CD19, CD20, CD22, immature markers CD34, TdT, CD10,
and aberrant CD25; they are aberrantly dim for CD38 and CD81;
this pattern is useful in identifying minimal residual disease
- Translocation involves abl on #9q34 (tyrosine kinase) and bcr on #22q11 (breakpoint cluster region)
- Most childhood cases associated with p190 kd BCR-ABL1 fusion protein
- In 50% of adults, has p210 kd fusion protein that is present in CML, the rest is p190 kd; no definitive clinical difference
Contributed by Julia Braza, M.D., M.S. and AFIP
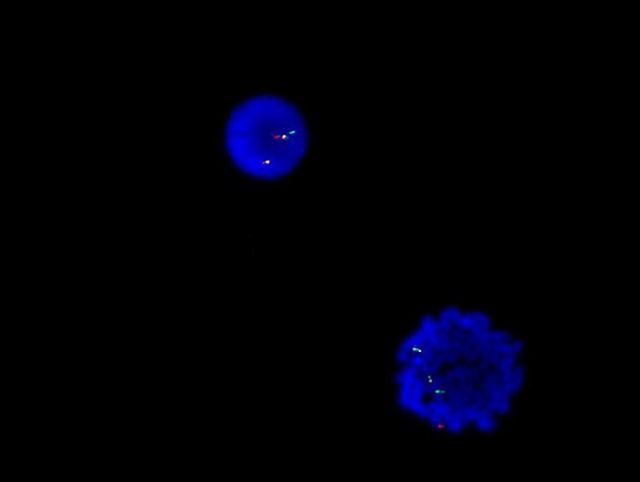
FISH
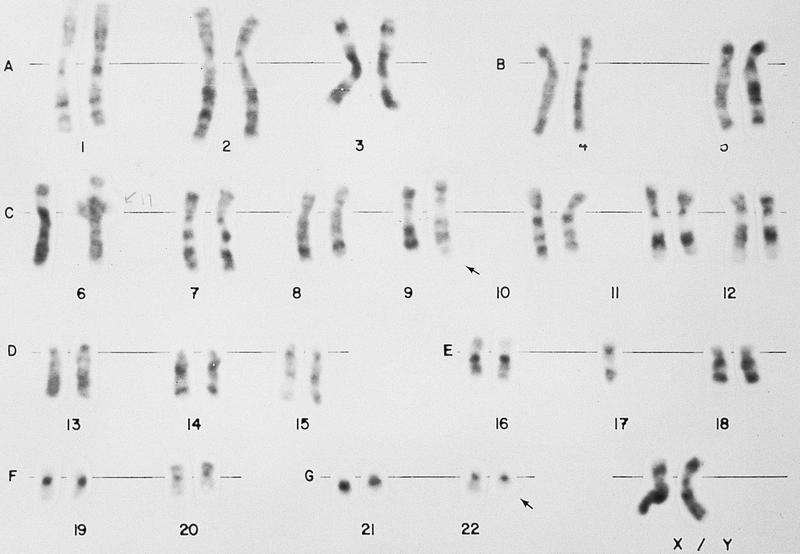
Karyotype
Images hosted on other servers:
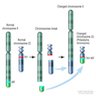
Drawing of translocation
- Rearrangements of MLL gene
- 20% of ALL overall (Anticancer Res 2005;25:1931) including 80% of infants (Leukemia 2007;21:633), 10% of older children and adults
- May have bilineal lymphoblasts with monoblasts and promonocytes
- Usually infants < 1 year with markedly increased WBC (> 100x109/L), CNS involvement; pure lymphomatous involvement not typical
- Poor prognosis (30% event free survival)
- Intensive chemotherapy followed by hematopoietic stem cell transplantation
- No defining morphology
- Over 75 genetic arrangements; most translocations at 11q23 involve MLL (mixed lineage leukemia) gene
- t(4;11)(q21;q23) - MLL-AF4: occurs in 60% of infants, 2% of other children and 3 - 6% of adults
- t(11;19)(q23;p13.3) - MLL-ENL and others
- MLL translocation is associated with FLT3 overexpression
- 11q23 deletions are NOT included in this group because different immunophenotype, clinical and prognostic features
Images hosted on other servers:

ALL1 / MLL duplication
- Autosomal dominant trait caused by pathogenic germline mutational variants in the DDX41 gene, increasing the risk of hematolymphoid neoplasms
- Pathogenic germline mutational variant on chromosome 5q35.3
- Autosomal dominant inheritance
- Familial myeloproliferative / lymphoproliferative neoplasms
- DEAD box helicase 41
- ICD-10: C96.7 - other specified malignant neoplasms of lymphoid, hematopoietic and related tissue
- Germline DDX41 pathogenic variants are the most common cause for genetic predisposition to myeloid neoplasms and are identified in 1 - 4% of patients with myeloid neoplasms (Cancer Cell 2015;27:658, Blood 2019;134:1441, Am J Hematol 2019;94:757)
- Lifelong risk of developing myeloid neoplasms from a pathogenic or likely pathogenic DDX41 variant is ~50%, starting at 40 years of age (Blood 2023;141:534)
- Men with the gene variant have nearly double the risk of developing a myeloid neoplasm compared with female carriers (Blood 2022;140:716)
- Certain DDX41 germline variants are more common in certain populations (e.g., p.Ala500Cysfs*9 in Japanese / Korean and p.D140Gfs*2 in those of Northern European descent) (Blood 2023;141:1544)
- Bone marrow / peripheral blood
- Lymph nodes (less common)
- Precise mechanism by which germline DDX41 variants drive hematopoietic malignancies is still unknown
- DDX41 gene is located on chromosome 5q35.3
- DDX41 is expressed on hematopoietic cells and it encodes an ATP dependent RNA helicase that participates in RNA metabolism (Cancer Cell 2015;27:658)
- Germline DDX41 variants are typically heterozygous and include frameshift, nonsense or splice site mutations whereas somatic mutations are usually missense heterozygous variants that affect the helicase 2 ATP binding domain (Blood 2016;127:1017)
- DDX41 is found both in the cytoplasm and nucleus, where it plays a role in messenger RNA splicing, ribosomal RNA processing or small nucleolar RNA processing (Blood 2023;141:1544)
- Disruption of the DDX41 protein contributes to inflammation and tumorigenesis (Blood 2023;141:1544)
- Germline DDX41 pathogenic mutations are passed on from a parent to child in an autosomal dominant inheritance pattern
- Pathogenic variant is usually heterozygous
Images hosted on other servers:
Proposed mechanisms for DDX41 involvement
- Individuals with a pathogenic DDX41 variant have an increased lifelong risk (~50%) of developing a myeloid neoplasm, including clonal cytopenia of undetermined significance (CCUS), myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), myelodysplastic / myeloproliferative neoplasm (MDS / MPN) and acute myeloid leukemia (AML) starting at age 40 (Blood 2023;141:534)
- Germline DDX41 variants are found in 3 - 5% in MDS, 5 - 9% in secondary AML, 1 - 2% in MDS / MPNs and < 0.5% in MPNs (Blood 2023;141:534)
- Aplastic anemia may occur but is rare
- Some patients may develop a lymphoid neoplasm or a plasma cell neoplasm; however, this is much less common than myeloid neoplasms and includes follicular lymphoma, Hodgkin lymphoma, chronic lymphocytic leukemia, multiple myeloma and acute lymphoblastic leukemia (Blood 2016;127:1017)
- WHO 5th edition diagnostic category: myeloid neoplasms associated with germline predisposition
- WHO 5th edition essential diagnostic criteria: detection of a pathogenic or likely pathogenic DDX41 variant
- WHO 5th edition desirable diagnostic criteria: detection of pathogenic or likely pathogenic germline DDX41 variant at a variant allele frequency consistent with germline status from DNA derived from cultured skin fibroblasts or via segregation in other family members
- Reference: Am J Hematol 2023;98:1780, Am J Hematol 2023;98:1780
- Next generation sequencing based somatic mutation profiling during workup for myeloid neoplasia is diagnostic for the detection of germline DDX41 variants (Front Oncol 2021:10:582213)
- Germline deletion variants are observed but may not be detected by panel based molecular profiling tests and therefore, germline genetic testing should be performed if there is a personal or family history that is worrisome for a germline DDX41 variant (Front Oncol 2021:10:582213)
- Unexplained blood count abnormalities including single or multilineage cytopenias or macrocytosis can be seen on the complete blood count (Front Oncol 2022:12:992340)
- Generally, a normal karyotype is associated with a DDX41 pathogenic variant and chromosome analysis can be done but is not useful in diagnosis (GeneReviews: DDX41-Associated Familial Myelodysplastic Syndrome and Acute Myeloid Leukemia [Accessed 19 October 2023])
- Prognostic impact of germline DDX41 variants is unclear
- Truncating germline DDX41 variants in patients who develop MDS are associated with faster transformation to AML compared with patients with nontruncating germline variants but no difference in overall survival is observed between these groups (Blood 2023;141:534)
- 9 year old boy presented with essential thrombocythemia-like features with a pathogenic germline variant in DDX41, JAK2 and a constitutional inv(7)(q11.2q22) (J Pediatr Hematol Oncol 2023;45:e609)
- 48 year old man with Ph+ B cell lymphoblastic leukemia with biallelic DDX41 mutations (BMC Med Genomics 2022;15:46)
- 62 year old man with myelodysplastic syndrome and DDX41 mutation treated successfully with lenalidomide (Am J Hematol 2020;95:227)
- 70 year old man with an adult onset myeloid neoplasm with excess blasts without dysplasia and germline missense DDX41 variant (Leuk Lymphoma 2019;60:1337)
- Caucasian family with inherited MDS / AML with 2 germline mutations in the DDX41 gene (Haematologica 2016;101:e228)
- No evidence based guidelines on the treatment for DDX41 associated familial MDS / AML have been published and patients are generally treated with standard neoplasm specific therapy
- Patients with MDS associated with germline DDX41 pathogenic variants tend to have better response to hypomethylating agents and better outcomes compared to those with wild type DDX41 (Blood 2023;141:534)
- Allogeneic hematopoietic stem cell transplant evaluation early in the course of hematologic malignancy may be appropriate based on the age of the individual, malignancy and health status
- Hematolymphoid neoplasms arising in individuals with a DDX41 germline variant do not have distinctive morphologic features
- Patients with a DDX41 variant have an increased likelihood of bone marrow hypocellularity and erythroid dysplasia (Am J Hematol 2019;94:757)
- DDX41 mutation can lead to ineffective hematopoiesis resulting in cytopenia and macrocytosis in the peripheral blood (Front Oncol 2022:12:992340)
- It is reported that patients with DDX41 variants can have persistent higher blast count in myelodysplastic syndrome or AML; among MDS subtypes, refractory anemia with excess blasts is the most frequent (Leuk Lymphoma 2019;60:1337)
Contributed by Jacob Armstrong, M.D.

Hypocellular bone marrow core

Hyperchromatic megakaryocytes

Hypercellular bone marrow core
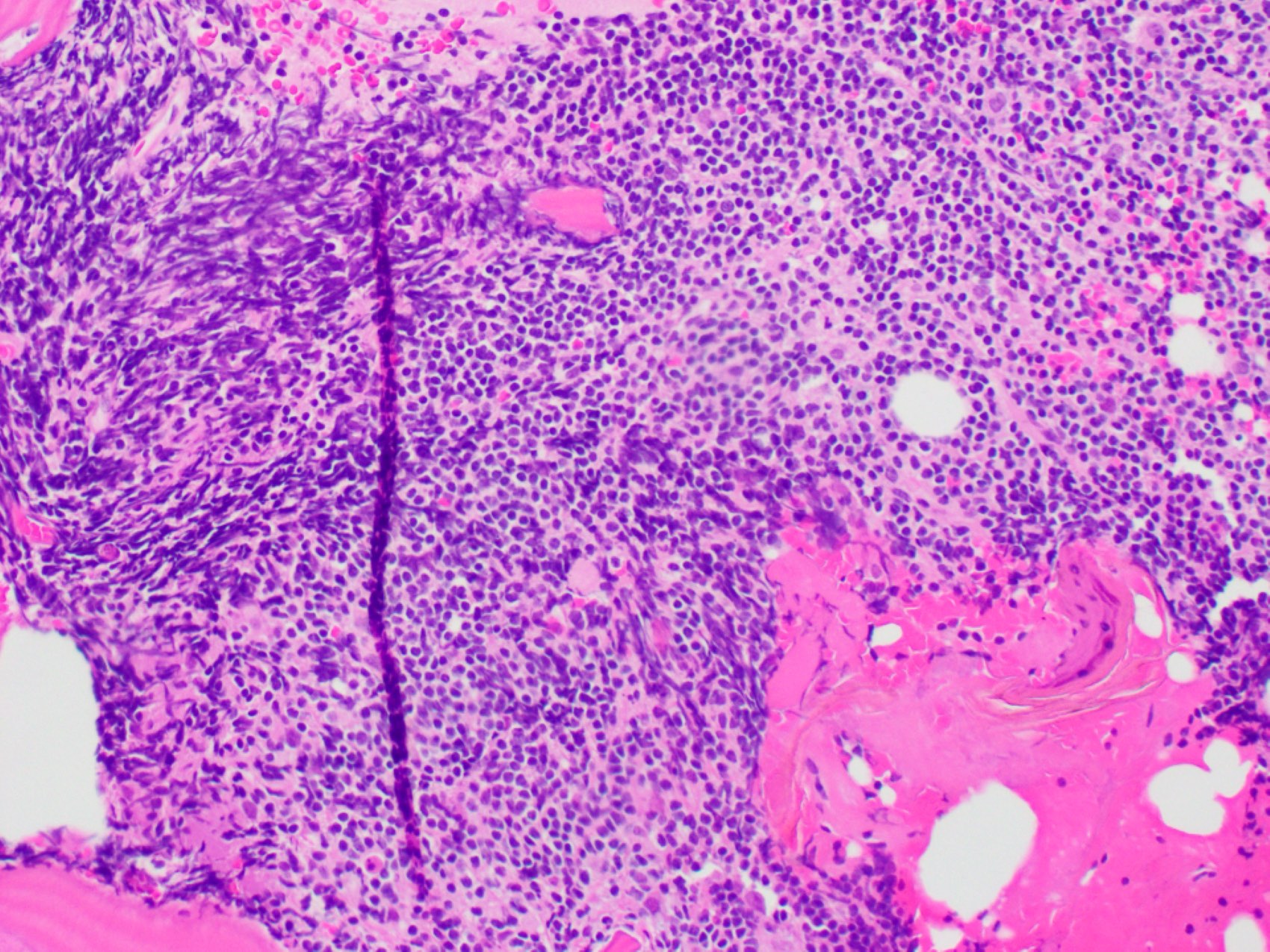
Marrow with AML
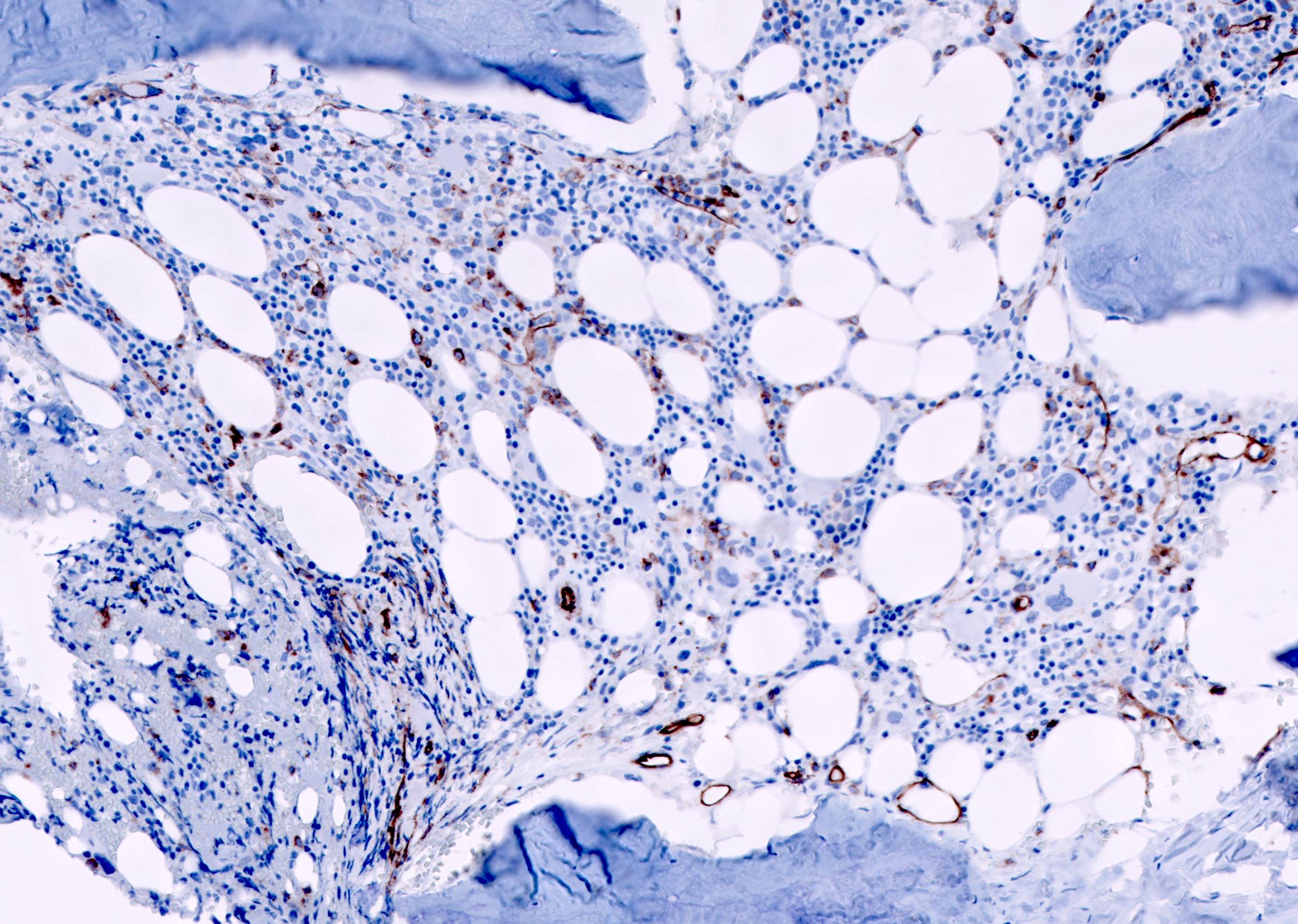
CD34 with ~10% blasts
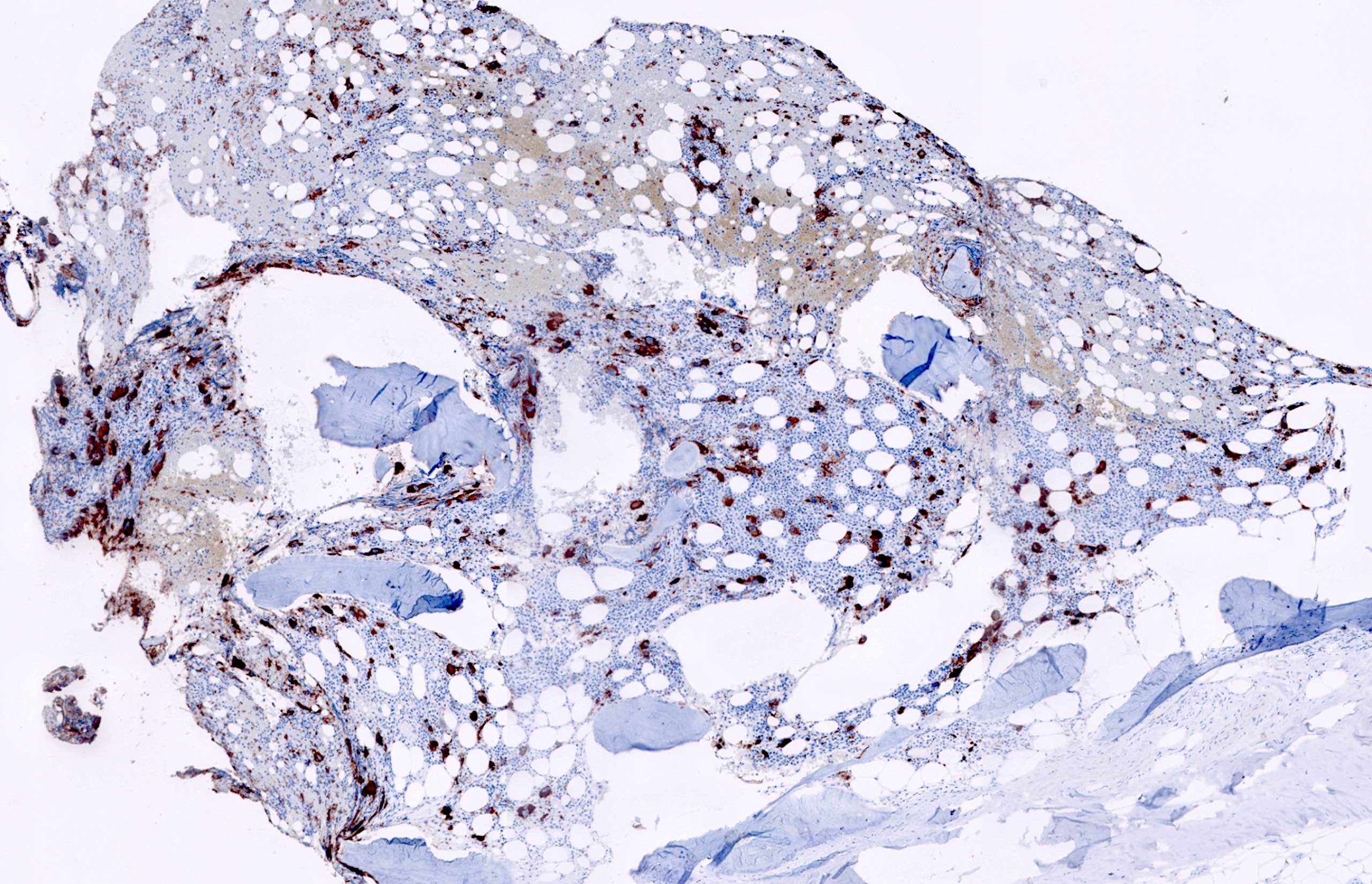
CD61 with megakaryocyte atypia
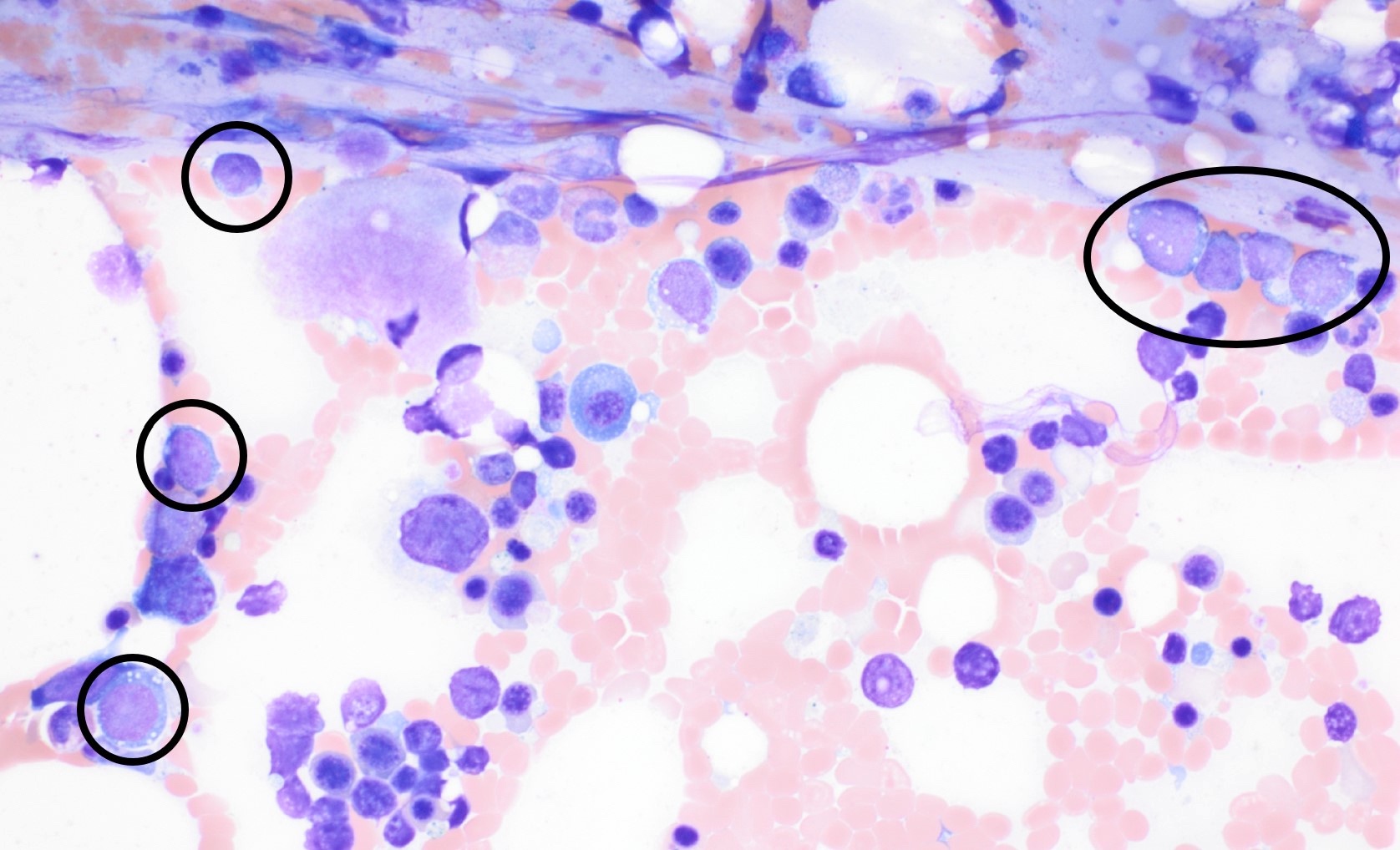
Aspirate with increased blasts
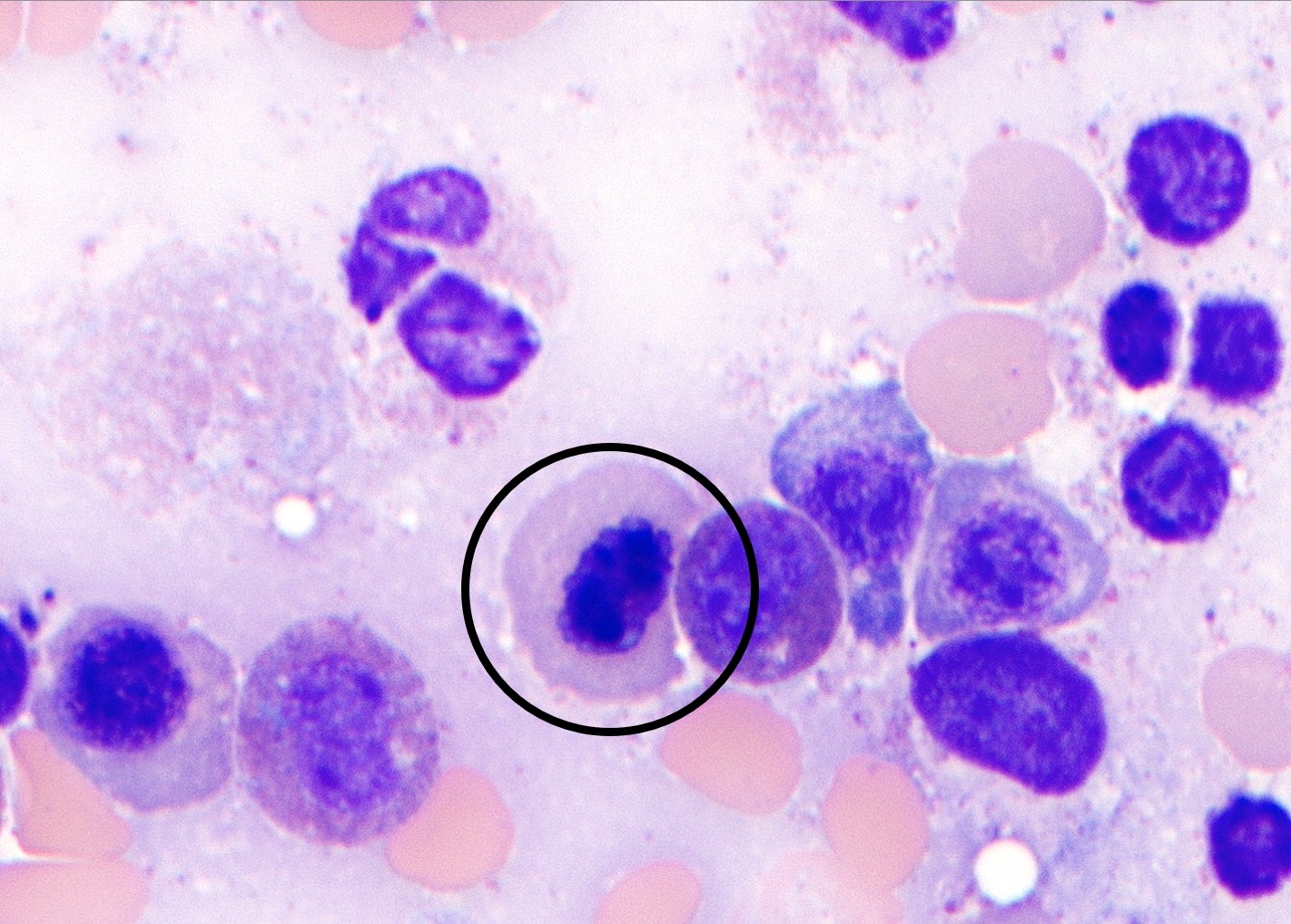
nRBC with irregular nuclei
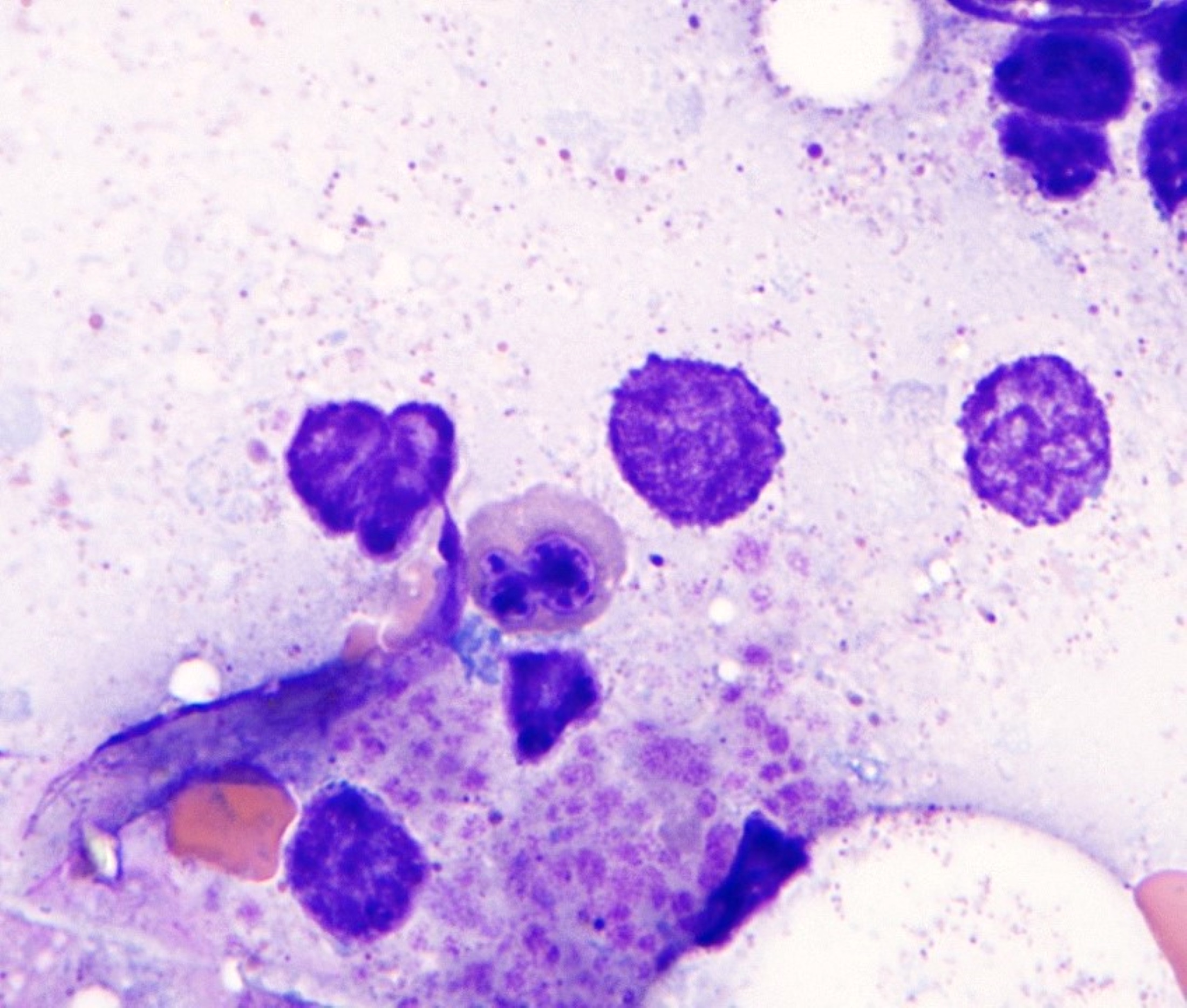
nRBC with nuclear budding

nRBC with multinucleation
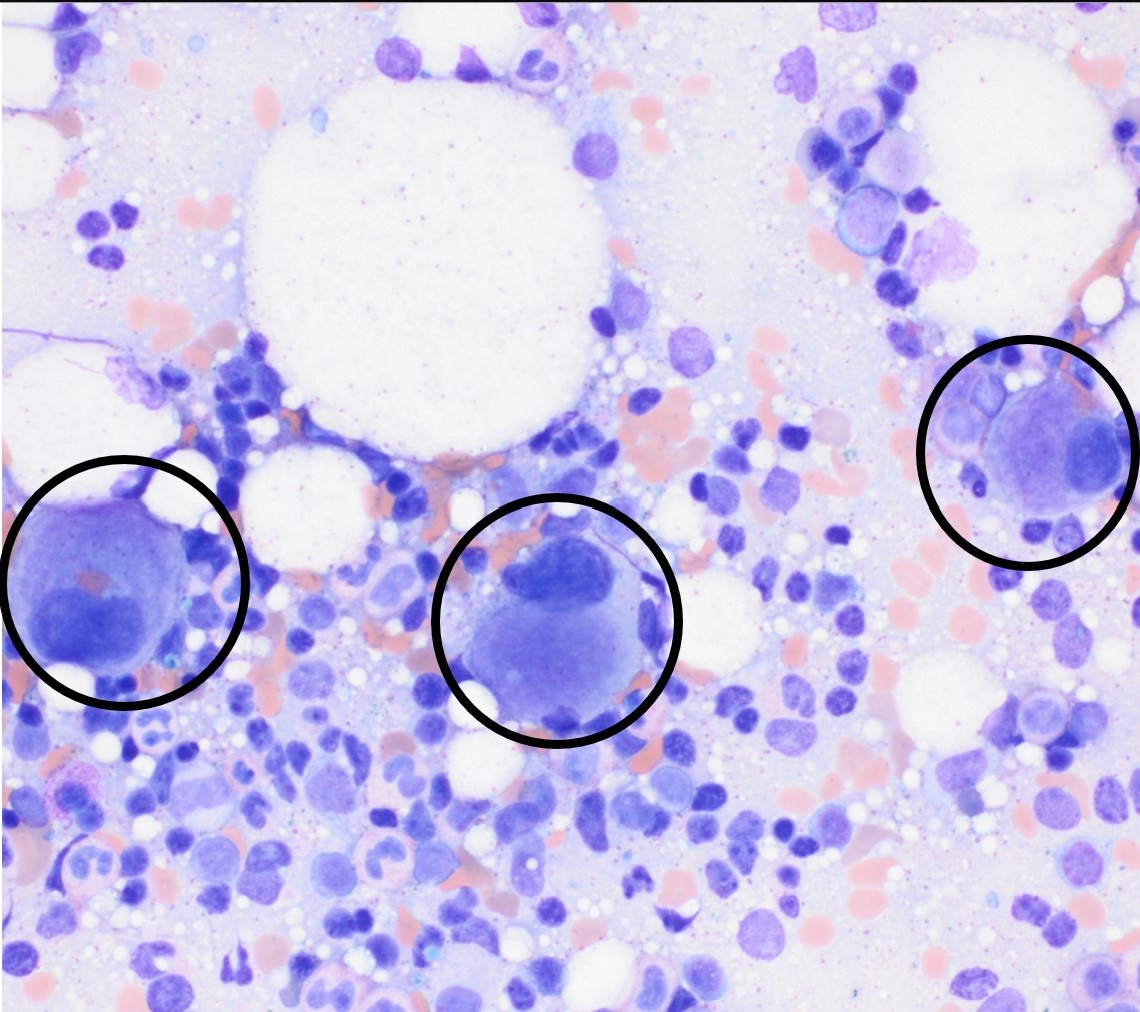
Hypolobated megakaryocytes

Ring sideroblasts
- Granulocytes
- Can show dysplastic features (hypogranular or hyposegmented forms)
- Increased blasts and immature myeloid precursors can be present
- Erythrocytes
- Can have dysplastic features including irregular nuclear contours, nuclear budding, binucleation, multinucleation, nuclear dyssynchrony, megaloblastoid changes and basophilic stippling
- Megakaryocytes
- Can have dysplastic features including large variation in sizes, small forms (micromegakaryocytes), hypolobated nuclei, widely spaced nuclei, hypersegmented (staghorn) nuclei and nuclear hyperchromasia
- Blasts can be increased
- Iron: ring sideroblasts can be present
- Reference: GeneReviews: DDX41-Associated Familial Myelodysplastic Syndrome and Acute Myeloid Leukemia [Accessed 19 October 2023]
- Red blood cells (RBCs)
- Normal RBC count, anemia or erythrocytosis can be present
- Anemia including microcytic, normocytic or macrocytic
- Macrocytosis is the most common finding
- Anisopoikilocytosis, including teardrop cells (if there is myelofibrosis), etc.
- Nucleated red blood cells (nRBC) can be present
- White blood cells (WBCs)
- Normal WBC count, leukocytosis or leukopenia can be present
- Granulocytes can show dysplastic features (hypogranular or hyposegmented forms)
- Circulating blasts and immature myeloid precursors can be present
- Platelets
- Normal platelet count, thrombocytopenia or thrombocytosis can be present
- Morphology of platelets is usually unremarkable
- Reference: GeneReviews: DDX41-Associated Familial Myelodysplastic Syndrome and Acute Myeloid Leukemia [Accessed 19 October 2023]
Contributed by Jacob Armstrong, M.D.
Macrocytic anemia
Circulating blasts and nRBC
- There are no diagnostic stains for DDX41 mutation; helpful stains include
- CD34: enumeration of CD34 positive blasts
- CD117: enumeration of CD117 positive blasts and erythroid precursors
- TdT: positive on some blasts
- CD123: positive on some blasts
- CD42b: highlights megakaryocytes
- CD61: highlights megakaryocytes
- Factor VIII: highlights megakaryocytes
- CD15: highlights granulocytes
- MPO: highlights the myeloid lineage
- E-cadherin: highlights the erythroid lineage
- CD71: highlights the erythroid lineage
- Glycophorin A: highlights the erythroid lineage
- CD4: highlights monocytes and a subset of T cells
- CD68: highlights monocytes
- Reticulin: highlights reticulin fibrosis
- Trichrome: highlights collagen fibrosis
- Reference: Am J Hematol 2023;98:1780
- No reported distinctive flow cytometry immunophenotypic pattern; however, individuals with DDX41 mutation can have an increased blast population or granulocytes with decreased side scatter as a reflection of hypogranularity
Contributed by Zubaidah Al-Jumaili, M.D.


CD45 versus side scatter

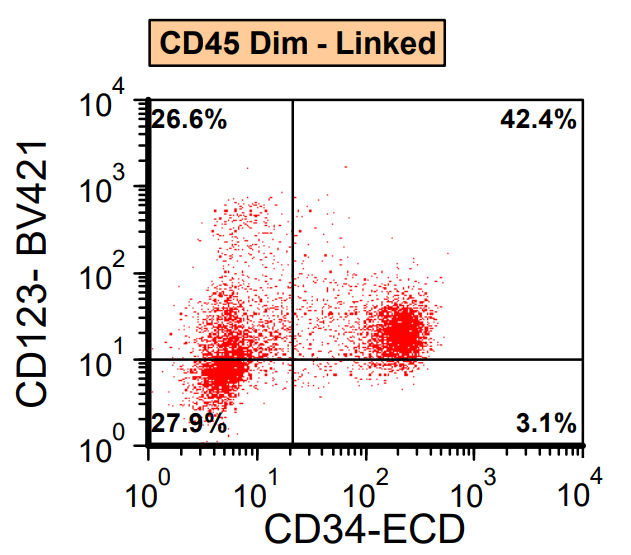
CD34 versus CD123
- Cytogenetics findings
- DDX41 pathogenic variants are not associated with any karyotype abnormalities; while patients with DDX41 variants can have karyotype abnormalities, these changes are independent of the DDX41 mutation (Am J Hematol 2019;94:757)
- Molecular findings
- Deleterious germline DDX41 variants confer increased risk for myeloid neoplasms with an estimated prevalence of 1 - 4% among MNs
- DDX41 can be mutated in the germline or in a somatic fashion
- Germline mutations of DDX41 have been reported to predispose patients to somatic mutations and subsequent MDS or AML
- ~50% of patients with germline mutations also harbor somatic mutations in the other allele of DDX41
- The most frequent DDX41 mutation identified is a frameshift duplication causing a premature stop codon (p.D140Gfs*2); however, missense and splice variants are also described
- The most frequent co-occurring somatic mutations involve ASXL1, CUX1, TP53, DNMT3A and TET2 and among these, CUX1 mutations are preferentially observed in DDX41 mutated cases compared with wild type ones (Am J Hematol 2019;94:757)
Images hosted on other servers:
Distribution of DDX41 mutations along the gene

Germline and somatic DDX41 mutations

Deleterious DDX41 germline variants
What is the clinical impact of DDX41 mutations in myeloid neoplasms?
Germline predisposition to myeloid malignancies
- Bone marrow, biopsy:
- Hypocellular (30%) bone marrow with dyserythropoiesis and ~10% blasts (see comment)
- Comment: Bone marrow aspirate smears show erythroid dysplasia and a mild increase in blasts, comprising ~10% of the total nucleated cells on the aspirate smear. The bone marrow core biopsy and clot section are hypocellular for age with increased scattered blasts marked by CD34 immunostain. Flow cytometric analysis shows increased abnormal myeloblasts that comprise about 10.2% of total events with an atypical immunophenotype (CD2+, CD13+, CD33+, CD34+, CD38+, CD117+, CD123+, MPO+). These findings are compatible with a myelodysplastic neoplasm with increased blasts. NGS studies show a DDX41 mutation. This is consistent with a myeloid neoplasm associated with germline predisposition, more specifically, myelodysplastic neoplasm with increased blasts with DDX41 mutation.
- Myeloid neoplasms associated with a DDX41 pathogenic variant:
- Clonal cytopenias of undetermined significance (CCUS):
- Detection of 1 or more somatic mutations with variant allele frequency (VAF) ≥ 2% (≥ 4% for X linked gene mutations in men) in DNA from blood or bone marrow cells involving genes listed in table #32521 of the clonal hematopoiesis section of the WHO 5th edition or clonal chromosomal abnormalities in myeloid cells
- 1 or more otherwise unexplained cytopenias that persist for > 4 months
- Absence of diagnostic criteria for defined myeloid neoplasms on bone marrow examination
- Myelodysplastic neoplasm (MDS):
- Cytopenia in at least 1 hematopoietic lineage: Hb < 13 g/dL in men and < 12 g/dL in women for anemia, absolute neutrophil count < 1.8 x 109/L for leukopenia and platelets < 150 x 109/L for thrombocytopenia
- Please see diagnostic criteria for MDS with defining genetic abnormalities or morphologically defined MDS
- Defining genetic abnormalities include
- MDS with low blasts and 5q deletion (MDS-5q)
- MDS with low blasts and SF3B1 mutation (MDS-SF3B1)
- MDS with biallelic TP53 inactivation (MDS-biTP53)
- Morphologically defined MDS includes
- MDS with low blasts (MDS-LB)
- MDS, hypoplastic (MDS-h)
- MDS with increased blasts (MDS-IB)
- MDS with fibrosis (MDS-f)
- Defining genetic abnormalities include
- Myelodysplastic neoplasm (MDS) / myeloproliferative neoplasm (MPN):
- Please see diagnostic criteria for MDS / MPN entities
- Chronic myelomonocytic leukemia
- Myelodysplastic / myeloproliferative neoplasm with neutrophilia
- Myelodysplastic / myeloproliferative neoplasm with SF3B1 mutation and thrombocytosis
- Myelodysplastic / myeloproliferative neoplasm, NOS
- Please see diagnostic criteria for MDS / MPN entities
- Myeloproliferative neoplasm (MPN):
- Please see diagnostic criteria for MPN entities
- Chronic myeloid leukemia
- Chronic neutrophilic leukemia
- Chronic eosinophilic leukemia
- Polycythemia vera
- Essential thrombocythemia
- Primary myelofibrosis
- Juvenile myelomonocytic leukemia
- Myeloproliferative neoplasm, NOS
- Please see diagnostic criteria for MPN entities
- Acute myeloid leukemia (AML):
- Blasts comprising at least 20% or AML defining genetic features including t(8;21), t(15;17) or t(16;16)/inv(16)
- Myeloid neoplasm postcytotoxic therapy:
- Myeloid neoplasm meeting diagnostic criteria of any myelodysplastic neoplasm, myelodysplastic / myeloproliferative neoplasm or acute myeloid leukemia
- History of prior exposure to cytotoxic therapy or large field radiation therapy for an unrelated disorder
- Not meeting diagnostic criteria of myeloproliferative neoplasms
- Clonal cytopenias of undetermined significance (CCUS):
- Other germline variants that cause predisposition to myeloid neoplasms:
- Other syndromes that cause predisposition to myeloid neoplasms:
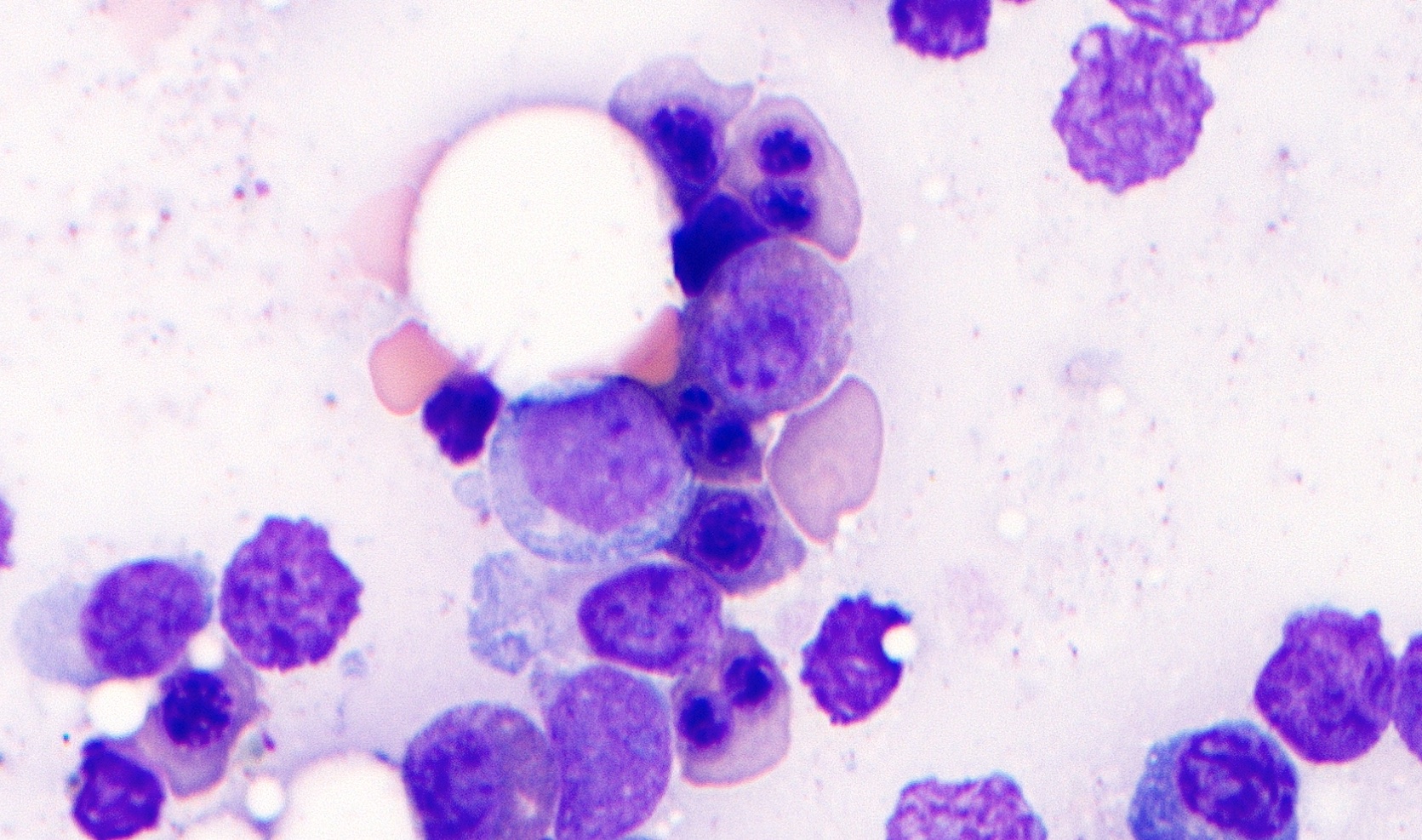
A 46 year old man presents with macrocytic anemia and thrombocytopenia. Bone marrow aspirate smears show the findings above. The bone marrow is hypercellular for age with hyperchromatic megakaryocytes. Cytogenetics findings reveal a 20q deletion and next genome sequencing shows a DDX41 pathogenic variant. The patient has a brother with similar findings. Which of the following is the diagnosis?
- Acute myeloid leukemia
- Chronic myeloid leukemia
- Chronic myelomonocytic leukemia
- Myeloid neoplasm postcytotoxic therapy
- Myeloid neoplasms associated with germline predisposition
Comment Here
Reference: with DDX41 mutation
- Macrocytic anemia
- Microcytic anemia
- Normocytic anemia
- Normocytic normochromic red blood cells
Comment Here
Reference: with DDX41 mutation
- Myelodysplastic syndrome (MDS) characterized by anemia with or without other cytopenias or thrombocytosis in conjunction with chromosome 5q deletion with or without one additional cytogenetic abnormality apart from monosomy 7 or del(7q)
- Myeloblasts represent < 5% of bone marrow cellularity and < 1% of peripheral blood leukocytes
- Subtype of MDS in low risk group with relatively good prognosis
- Requires presence of del(5q) with or without one additional cytogenetic abnormality apart from monosomy 7 or del(7q)
- TP53 mutation is associated with resistance to therapy, increased risk of transformation and decreased overall survival (J Clin Oncol 2011;29:1971, Br J Haematol 2013;160:660, Br J Haematol 2013;162:74, Haematologica 2014;99:1041)
- MDS with 5q deletion
- 5q minus syndrome
- ICD-O: 9986/3 - myelodysplastic syndrome associated with isolated del(5q)
- F > M
- Adults, median age approximately 60 - 70 years
- Blood and bone marrow
- Loss of tumor suppressor genes in deleted region of 5q results in haploinsufficiency of several genes (Blood 2002;99:4638, Nat Med 2007;13:78)
- RPS14
- Encodes a ribosomal structural protein (Nat Med 2010;16:59, Nature 2008;451:335, Nat Med 2016;22:288, Leukemia 2019;33:1759)
- Loss of this gene results in ineffective erythropoiesis
- Thought to play a role in p53 pathway activation (Blood 2011;117:2567)
- CSNK1A1
- Encodes casein kinase 1A1
- Haploinsufficiency leads to WNT / beta catenin pathway dysregulation (Cancer Cell 2014;26:509)
- Important therapeutic target
- miR-145 and miR-146a
- Plays a role in NFκB activation
- Contributes to dysmegakaryopoiesis (Nat Med 2010;16:49)
- Unknown at this time
- Commonly presents with anemia, usually macrocytic, often severe
- 32 - 49% have thrombocytosis at presentation (Leukemia 2004;18:113, Blood 1993;81:1040)
- 17 - 18% have thrombocytopenia (Leukemia 2004;18:113, Blood 1993;81:1040)
- Pancytopenia is very rare and if present, should be categorized as MDS, unclassifiable (Leuk Res 2013;37:64)
- Conventional cytogenetics with del(5q) involving 5q31-5q33 region with or without one additional abnormality apart from monosomy 7 or del (7q) (Leukemia 2012;26:1286, Leukemia 2011;25:110, J Clin Oncol 2012;30:820)
- FISH may be performed to detect del(5q) if conventional cytogenetics is suboptimal but is limited for providing information as to whether or not it is isolated
- TP53 mutation
- Associated with resistance to therapy, increased risk of transformation and decreased overall survival (J Clin Oncol 2011;29:1971, Br J Haematol 2013;160:660, Br J Haematol 2013;162:74, Haematologica 2014;99:1041)
- Detected in approximately 20% of cases
- Can be determined via molecular testing or immunohistochemistry
- Other mutations may be detected with molecular testing, including alterations in JAK2, MPL, SF3B1, RUNX1, WT1, ASXL1 and TET2, among others (Leukemia 2006;20:1319, Leukemia 2010;24:1283, Leuk Res 2010;34:821, Blood 2015;126:233, Blood 2011;118:6239, Am J Hematol 2011;86:393)
- Repetitive with clinical features and diagnosis headings
- Median survival is approximately 5.5 - 12 years (Leukemia 2010;24:1283, Leukemia 2011;25:110, Leukemia 2004;18:113)
- Transformation to AML in < 10% of patients
- Dysgranulopoiesis associated with a poor prognosis (Leukemia 2009;23:796, Hum Pathol 2013;44:346)
- TP53 mutation associated with a poor prognosis (J Clin Oncol 2011;29:1971, Br J Haematol 2013;160:660, Br J Haematol 2013;162:74)
- 50 year old woman with history of autoimmune scleritis treated with immunosuppressive therapy presenting with weakness, pallor and progressive anemia (Case Rep Oncol 2012;5:586)
- 60 year old man with macrocytic anemia, weakness and fatigue (Case Rep Oncol 2014;7:277)
- 66 year old woman presenting with persistent anemia (Blood 2017;129:2333)
- 71 year old woman with history of polycythemia vera presenting with anemia, thrombocytosis and leukocytosis (Mediterr J Hematol Infect Dis 2016;8:e2016050)
- 76 year old woman presenting with dizziness (Blood Res 2018;53:104)
- Lenalidomide, a thalidomide analogue, has been shown to be beneficial (Ann Hematol 2005;84:569, N Engl J Med 2006;355:1456)
- Transfusion independence achieved in approximately 67% of patients
- Blast count must be < 5% in bone marrow
- Bone marrow cellularity typically hyper to normocellular (Leukemia 2004;18:113)
- Megakaryocyte hyperplasia
- Megakaryocytes with nonlobated and hypolobated nuclei
- Erythroid hypoplasia
- Dysgranulopoiesis is uncommon
Contributed by Natasha Iranzad, M.D.

Normocellular bone marrow
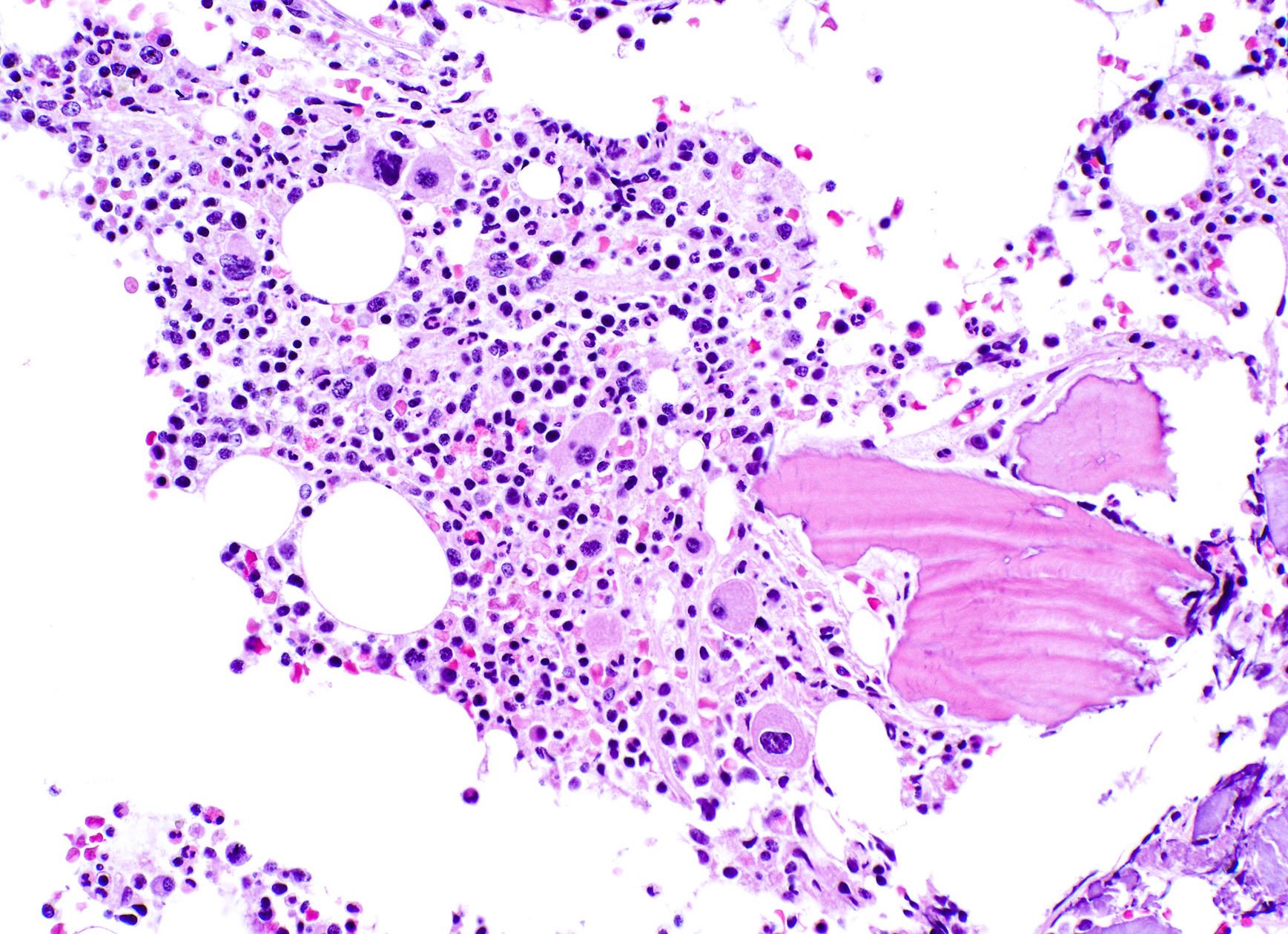
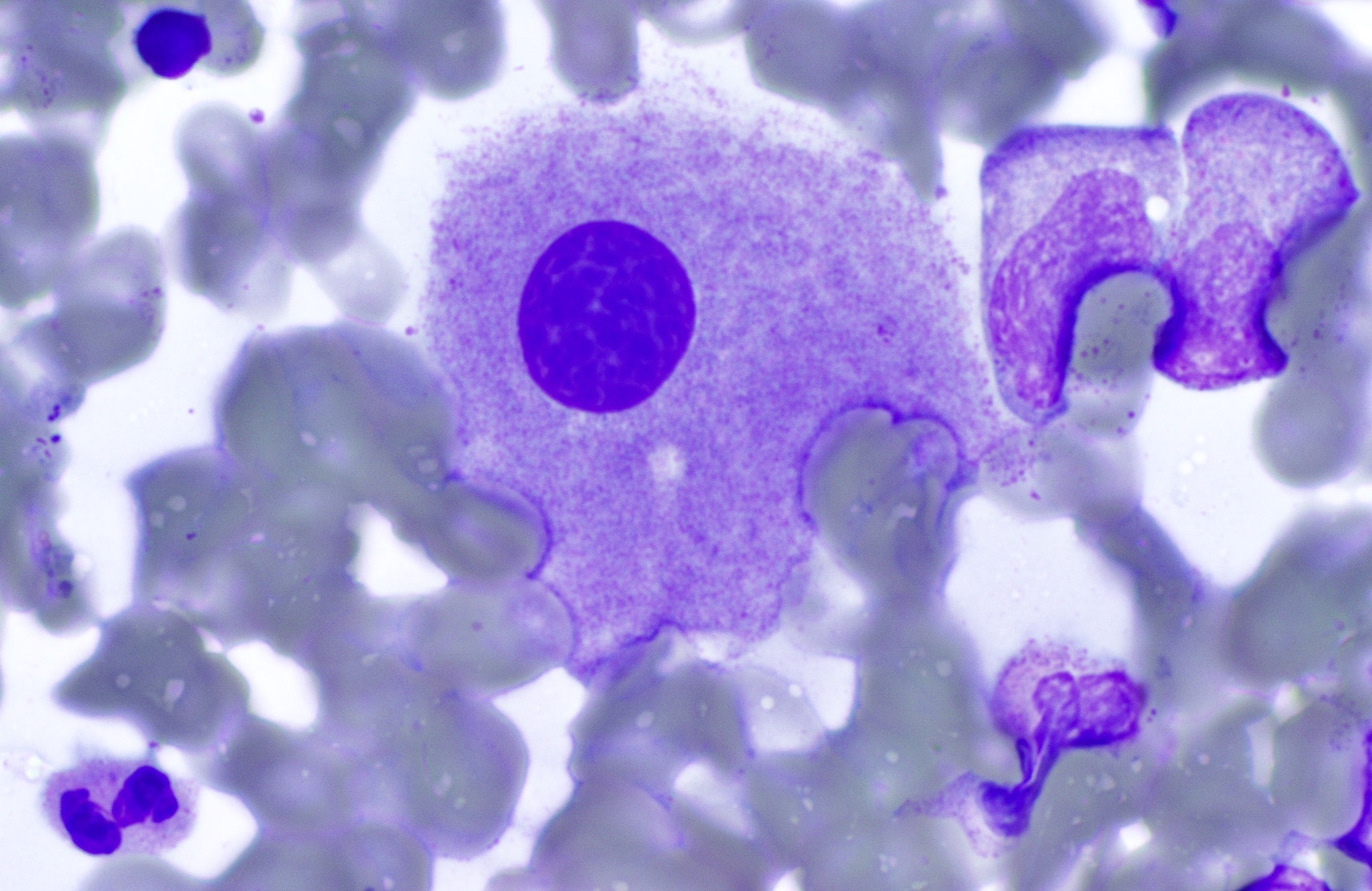
Megakaryocyte dysplasia
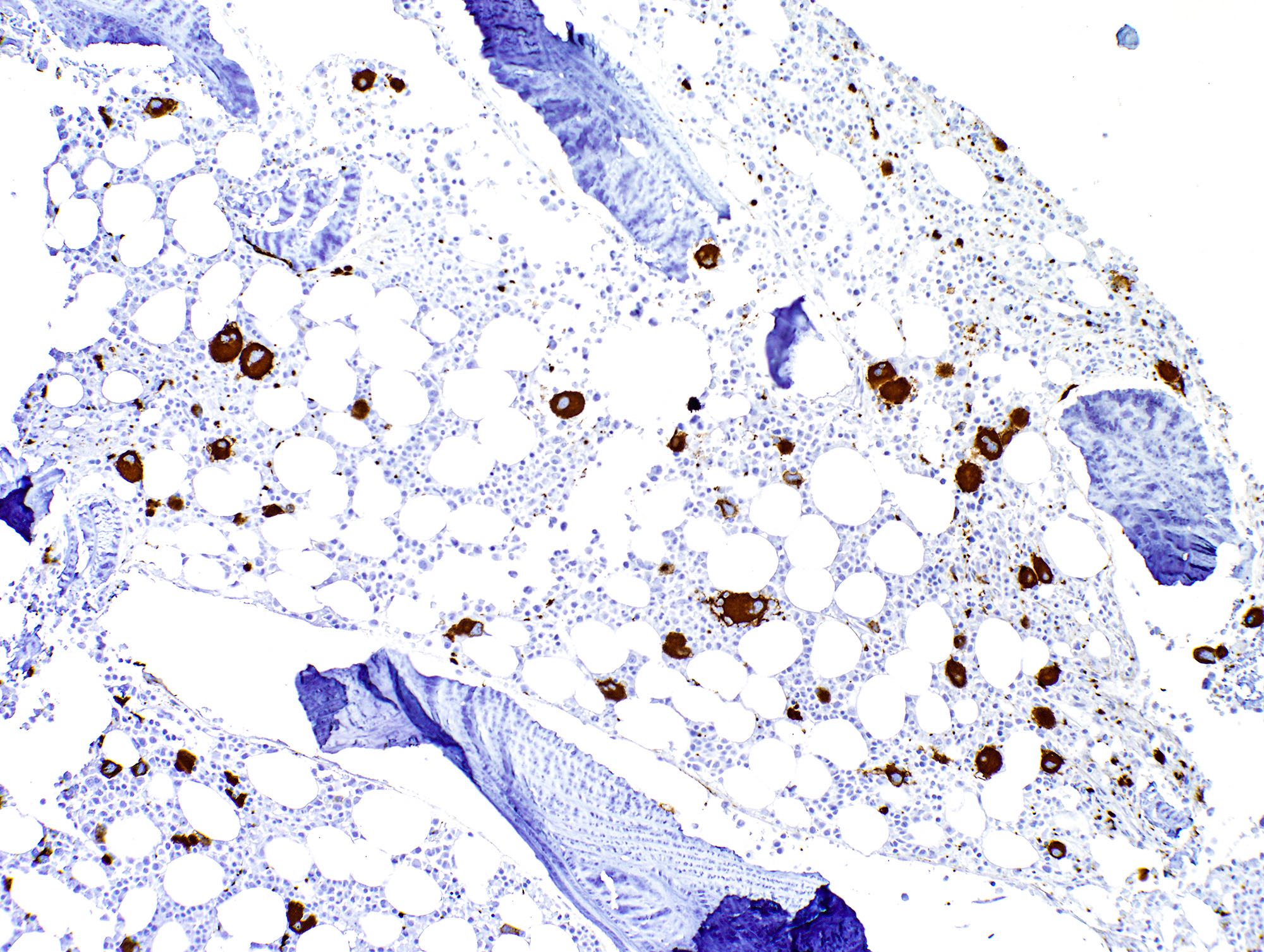
CD61
- Ring sideroblasts may be present
- Auer rods must be absent
- Blast percentage must be < 1%
- Anemia common, often severe and typically macrocytic
- Thrombocytosis in 33% of cases
- Thrombocytopenia less commonly seen
- CD61 highlights increased / dysplastic megakaryocytes
- No findings specific to this entity
- Myeloblasts with aberrant phenotype may be detected
- Repetitive with diagnosis heading
Images hosted on other servers:

Karyogram with del(5q)
- Bone marrow, aspirate smear, touch imprint, clot and core biopsy:
- Normocellular bone marrow (30%) with megakaryocyte dysplasia
- 3% bone marrow blasts (see comment)
- Comment: The presence of dysplastic micromegakaryocytes in this patient with longstanding macrocytic anemia is concerning for myelodysplastic syndrome with single lineage dysplasia (MDS SLD). However, a normocellular marrow would be highly unusual for this diagnosis as MDS typically presents with hypercellularity and rarely with hypocellularity. Thus, a definitive diagnosis of MDS cannot be made at this time without evidence of myeloid clonality, increased blasts or hyper / hypocellularity. Correlation with the pending chromosome analysis and FISH studies are required to establish clonality. In addition, myeloid NGS studies may be diagnostically useful to provide evidence of clonality with additional prognostic or potentially therapeutic information; this test may be performed on the peripheral blood if clinically indicated. The corresponding flow cytometric analysis did not detect any phenotypic abnormalities in myeloid or monocytic lineages or increase in blasts, in keeping with the morphologic findings.
FISH - MDS panel:
- Interpretation
- 18.5% of cells showed deletion of 5q (EGR1) by interphase FISH. No evidence of numeric abnormalities or deletions involving chromosomes 7, 8 or 20 observed by interphase FISH. Correlation with other laboratory and clinical information is recommended.
- nuc ish (D5S721/D5S23x2,EGR1x1)[37/200]
- nuc ish (D7Z1,D7S486)x2[200]
- nuc ish (D8Z2,D20S108)x2[199]
Summary table:
| Locus | Result |
| Monosomy 5/5q- | 18.5% deletion 5q |
| Monosomy 7/7q- | within normal limits |
| Trisomy 8 | within normal limits |
| Deletion of 20q | within normal limits |
Chromosome analysis:
- Interpretation
- 46,XX,del(5)(q12q33)[4]/46,XX[14]
- Abnormal clone with deletion 5q in 4/18 cells, consistent with concurrent interphase FISH analysis showing 18.5% of cells with deletion 5q (EGR1). Please see the Objective Findings section for details. Evaluation for an acquired TP53 mutation is recommended in patients with MDS with isolated del(5q) to help identify an adverse prognostic subgroup in this generally favorable prognosis MDS entity. Correlation with other laboratory and clinical information is recommended. (Blood 2016;127:2391)
- Myelodysplastic syndrome (MDS), unclassifiable (Blood 2016;127:2391):
- Diagnosis of exclusion
- Cases otherwise meeting criteria for MDS with del(5q) but with 1% blasts on peripheral blood smear on ≥ 2 occasions or pancytopenia, defined as:
- Hemoglobin < 10 g/dL
- Absolute neutrophil count < 1.8 x 109/L
- Platelet count < 100 x 10109/L
- MDS with excess blasts (EB):
- As the name suggests, with 2 distinct subcategories depending on the blast count:
- MDS EB1
- 5 - 9% blasts in bone marrow
- 2 - 4% blasts in peripheral blood
- MDS EB2
- 10 - 19% blasts in bone marrow
- 5 - 19% blasts in peripheral blood
- MDS EB1
- As the name suggests, with 2 distinct subcategories depending on the blast count:
- MDS with single / multilineage dysplasia:
- Lacks characteristic 5q deletion
- MDS with ring sideroblasts:
- Lacks characteristic 5q deletion
- Ring sideroblasts can be seen in both
- SF3B1 mutation can be seen in both
A 73 year old woman presents with macrocytic anemia and fatigue. Nutritional deficiency as a cause of her anemia has been ruled out clinically. Bone marrow biopsy shows a normocellular marrow. Aspirate smear shows the findings seen above. On chromosomal analysis, the patient is found to have isolated del(5q).
Which of the following is true about this disease?
- Approximately 40% of patients progress to AML
- Monosomy 7 is commonly seen in this entity
- SF3B1 mutation excludes the diagnosis
- TP53 mutation is associated with shorter overall survival
Comment Here
Reference: Isolated del(5q)
- "High hyperdiploidy" means > 50 chromosomes and < 66 chromosomes without translocations or other structural alternations
- Generally arises by simultaneous gain of all additional chromosomes in a single abnormal mitosis (Genes Chromosomes Cancer 2005;44:113)
- Hyperdiploidy appears to be an early event occurring prenatally (Genes Chromosomes Cancer 2004;40:38, Leukemia 2003;17:2202, Blood 2002;100:347)
- Affects 25 - 33% of children (usually age 3 - 5 years) and 5% of adults with B ALL; not seen in infants
- Favorable overall survival (Am J Hematol 2008;83:34)
- Although outcome in children varies by specific trisomy present (Blood 2003;102:2756)
- Immunophenotype: CD19+, CD10-, CD34+ and CD45-
- No distinct morphology
- Mutations in NRAS (10%), FLT3 (9%), PTPN11 (9%) and KRAS (6%, Genes Chromosomes Cancer 2008;47:26)
- Most common extra copies are 21, X, 14 and 4; least often 1, 2, 3
- Best prognosis of simultaneous trisomies: 4, 10 and 17
- Defined as fewer than 45 or 46 chromosomes
- 5% of B ALL
- 1% in both children and adults
- Most have 45 chromosomes, fewer chromosomes is rare
- Poorer prognosis: near haploid and low hypodiploid groups compared to those with:
- 42 - 45 chromosomes (Br J Haematol 2004;125:552)
- < 44 versus 44 chromosomes (Blood 2007;110:1112)
- < 45 versus 45 chromosomes (Cancer 2003;98:2715)
- Poorest prognosis: near haploid 23 - 29 chromosomes which appear limited to childhood
- Overall 50% survival at 8 years
- No distinct morphology
Images hosted on other servers:

Karyotype of severe hypodiploidy
- B lymphoblastic leukemia / lymphoma (B ALL / LBL) with intrachromosomal amplification of RUNX1 gene at structurally abnormal chromosome 21 (iAMP21)
- B ALL / LBL with iAMP21 is characterized by amplification, which is defined as ≥ 5 copies of RUNX1 per cell and ≥ 3 copies of RUNX1 on a single abnormal chromosome 21 detected by fluorescence in situ hybridization (FISH) study
- Typically occurs in older children with a median age of 9 years
- Pediatric iAMP21 B ALL has a poor outcome with an increased rate of relapse when treated on standard treatment protocols (Hematol Rep 2019;11:7826)
- B ALL / LBL with iAMP21
- ICD-O: 9811/3 - B lymphoblastic leukemia / lymphoma with iAMP21
- ICD-11: 2A70.Y & XH0KD4 - other B lymphoblastic leukemia / lymphoma with recurrent genetic abnormalities & B lymphoblastic leukemia / lymphoma with iAMP21
- Seen in older children with a median age of 9 years (Cancer Genet 2020:243:52)
- 2% of childhood B ALL / LBL and is rarely seen in adults (J Clin Oncol 2013;31:3397, Nature 2014;508:98)
- Peripheral blood and bone marrow are involved
- iAMP21 is considered to be formed through breakage fusion bridge cycles followed by chromothripsis and other complex structural rearrangements of chromosome 21 (Blood 2015;125:1383)
- Genetic studies demonstrated a common 5.1 Mb region that included RUNX1, which is part of the critical region consistently amplified (chr21:32.8 to 37.9 Mb, GRCh37/hg19) (Blood 2011;117:6848)
- Although RUNX1 is present in the amplified region, there is no conclusive evidence which points towards RUNX1 as a critical event in the pathogenesis of the disease given that it is not overexpressed (J Clin Oncol 2012;30:1663, Blood 2011;117:6848)
- Higher risk of B ALL / LBL with iAMP21 occurrence is reported in patients with a constitutional Robertsonian translocation rob(15;21)(q10;q10) and presence of a constitutional ring chromosome 21, r(21)c (Eur J Med Genet 2016;59:162, Nature 2014;508:98)
- Presents with leukopenia and thrombocytopenia (Cancer Genet 2020:243:52)
- Presence of blasts of B cell lineage differentiation
- FISH studies show ≥ 5 copies of RUNX1 per cell with ≥ 3 copies on a single abnormal chromosome 21
- CBC frequently shows leukopenia or thrombocytopenia
- Risk stratification includes older age at the time of diagnosis, initial white blood cell (WBC) count, associated genetic aberrations and minimal residual disease (MRD) (Hematol Rep 2019;11:7826, J Clin Oncol 2013;31:3397)
- Older age (≥ 10 years), WBC count (< 50 K/uL), female sex and detectable MRD at day 29 are considered as poor prognostic factors (J Clin Oncol 2013;31:3397)
- Deletion of SH2B3 is enriched in iAMP21 cases and confers a higher risk of relapse or death (Cancer Genet 2018:226:30, Leukemia 2019;33:1881)
- Pediatric iAMP21 B ALL has a poor outcome with an increased rate of relapse when treated on standard protocols (Hematol Rep 2019;11:7826)
- 4 year event free survival (EFS) and overall survival (OS) have been described as significantly worse for patients with iAMP21 with standard risk B ALL (SR BALL) (J Clin Oncol 2013;31:3397)
- 4 year old girl with Down syndrome who developed B ALL with iAMP21 due to a constitutional isodicentric chromosome 21 (Am J Med Genet A 2022;188:2325)
- 9 year old girl with B ALL with iAMP21 development with constitutional ring chromosome 21, r(21)c (Cytogenet Genome Res 2022;162:231)
- 12 year old girl with homozygous deletion of the SH2B3 gene, chromothripsis of chromosome 21 and a non-Robertsonian somatic t(15;21)(q25.3;q22.1) with NTRK3 gene rearrangement in iAMP21 B ALL (Cancer Genet 2022:262:16)
- Intensive treatment has overcome the higher risk of relapse on standard treatment in these patients, so they are now treated on HR COG ALL protocols (J Clin Oncol 2013;31:3389, J Clin Oncol 2013;31:3397)
- Bone marrow core biopsy is usually hypercellular with extensive marrow replacement by sheets of atypical medium sized blasts, with round to oval nuclear contour, fine chromatin and pinpoint nucleoli
- Lymph nodes show a diffuse or less commonly paracortical blast infiltrate with increased mitotic figures / apoptotic bodies and sometimes interspersed macrophages (starry sky pattern)
Contributed by Barina Aqil, M.D.

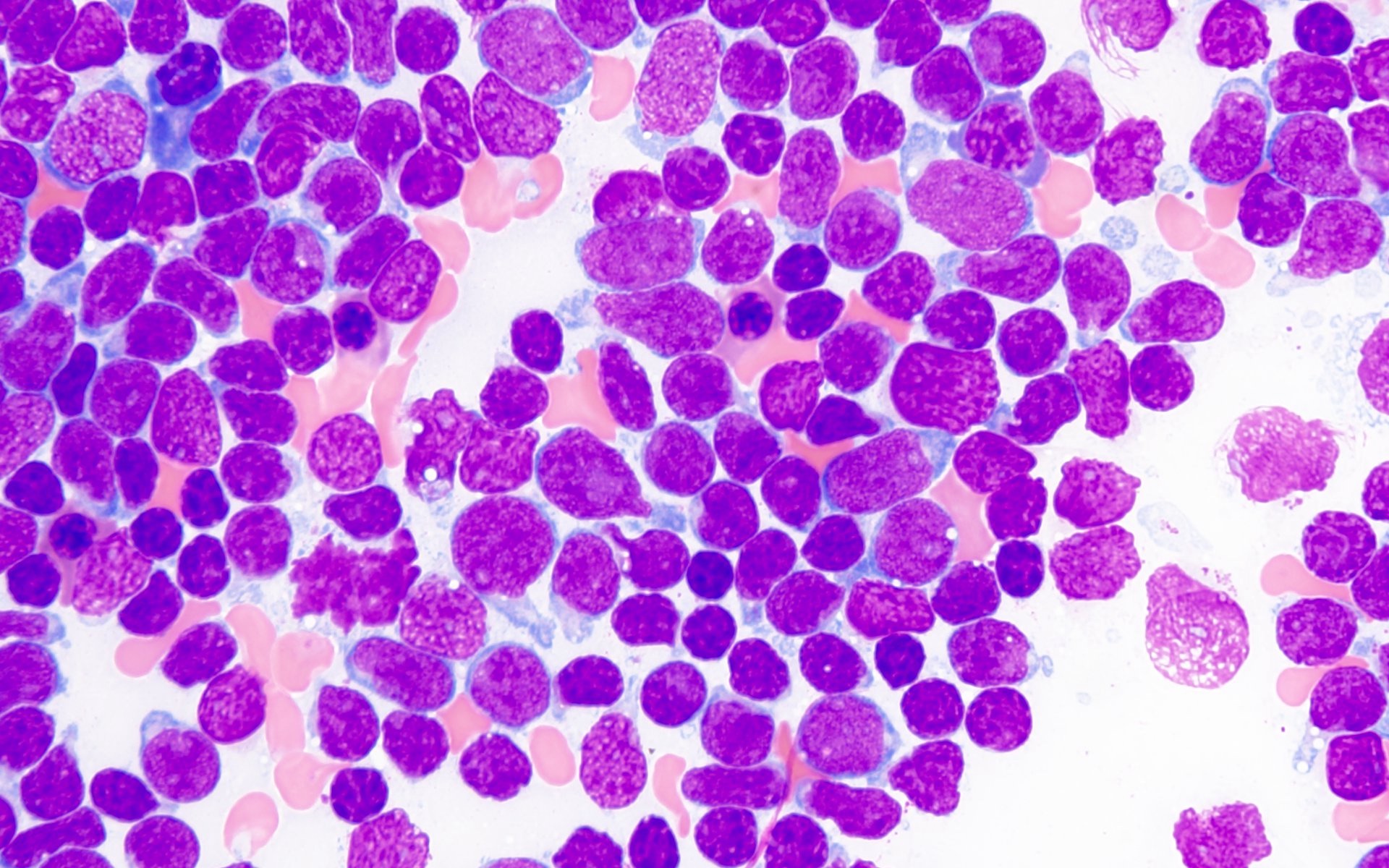
Increased blasts
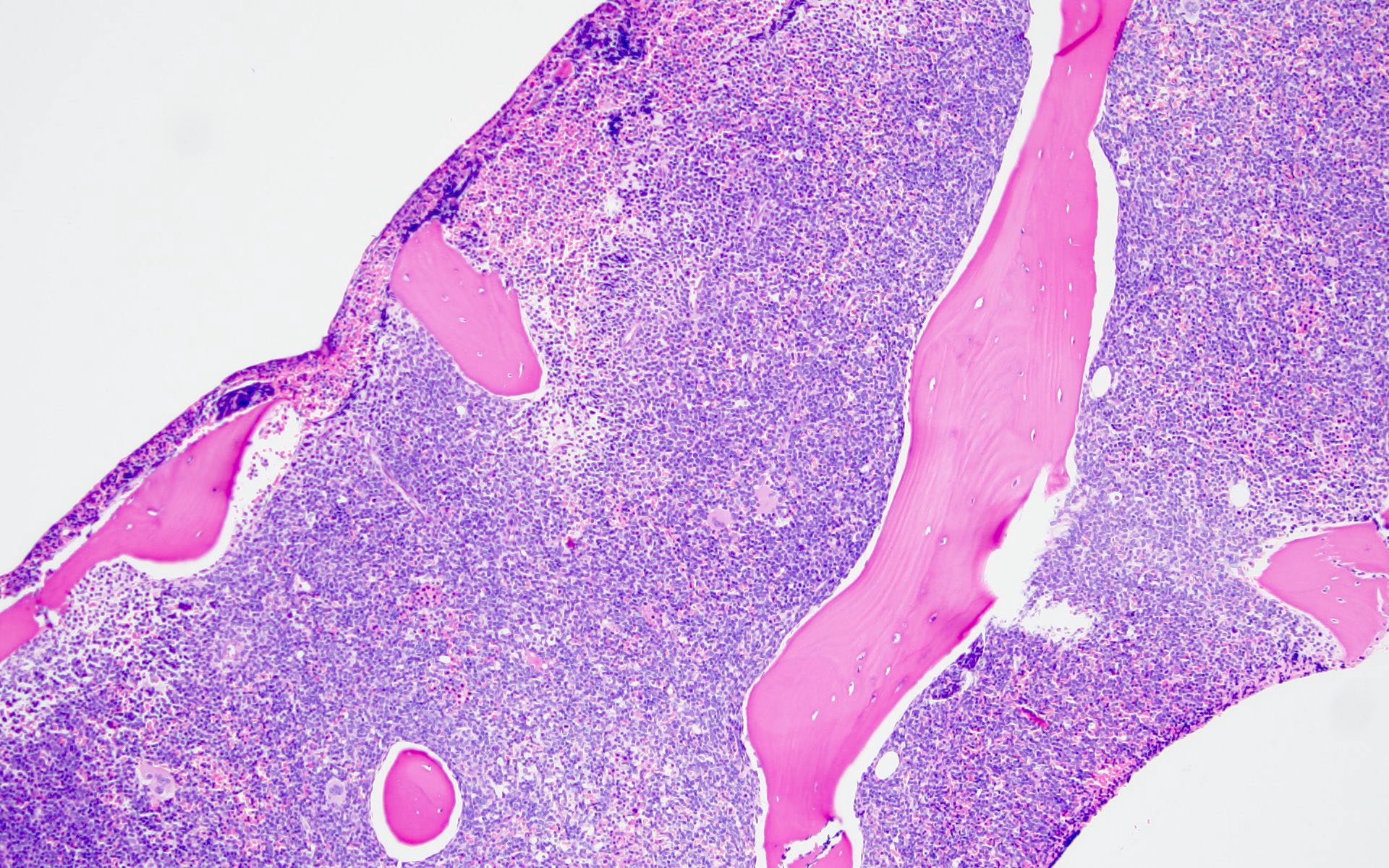
Atypical mononuclear cells

Blast morphology
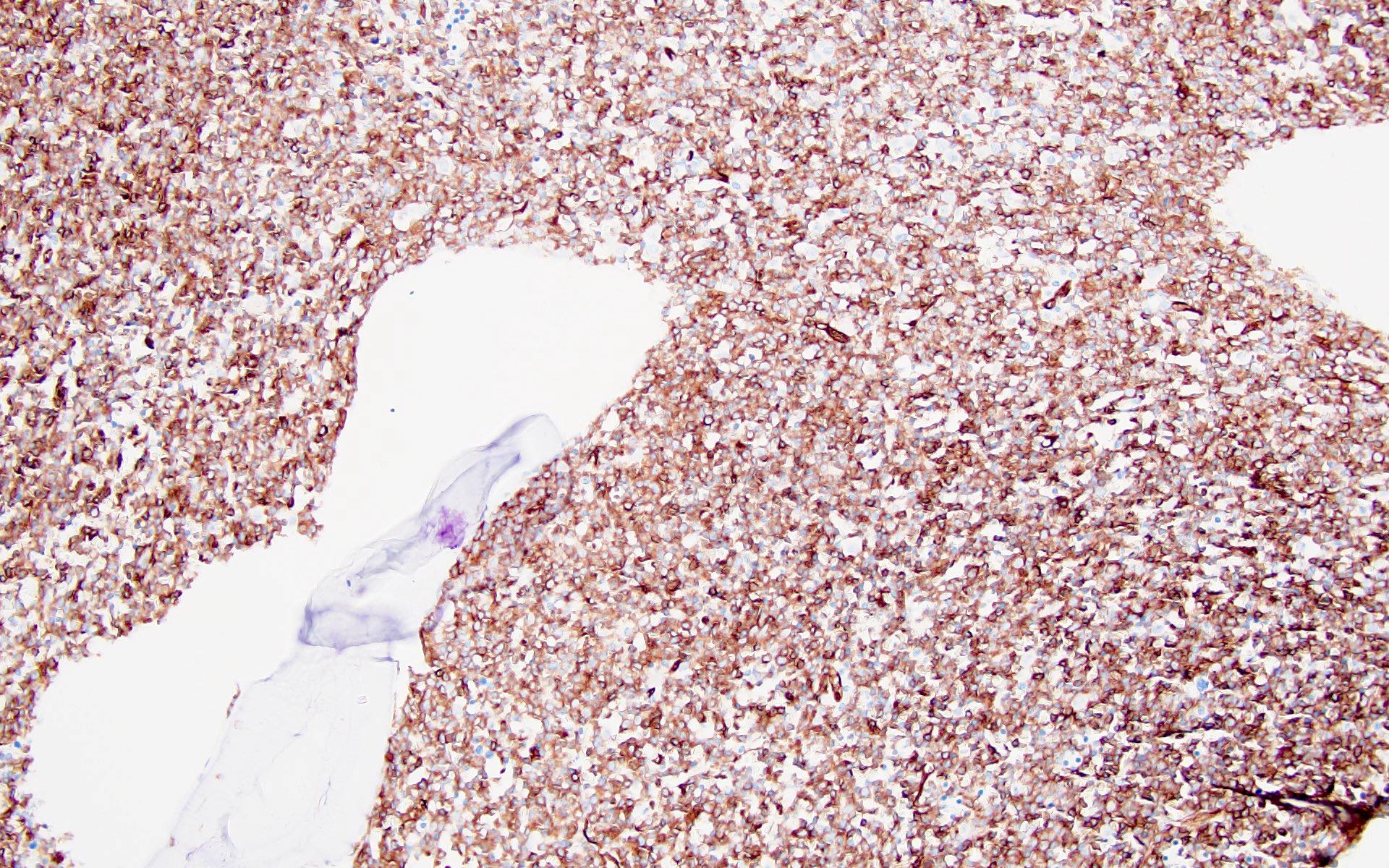
CD34+ blasts

TDT positivity
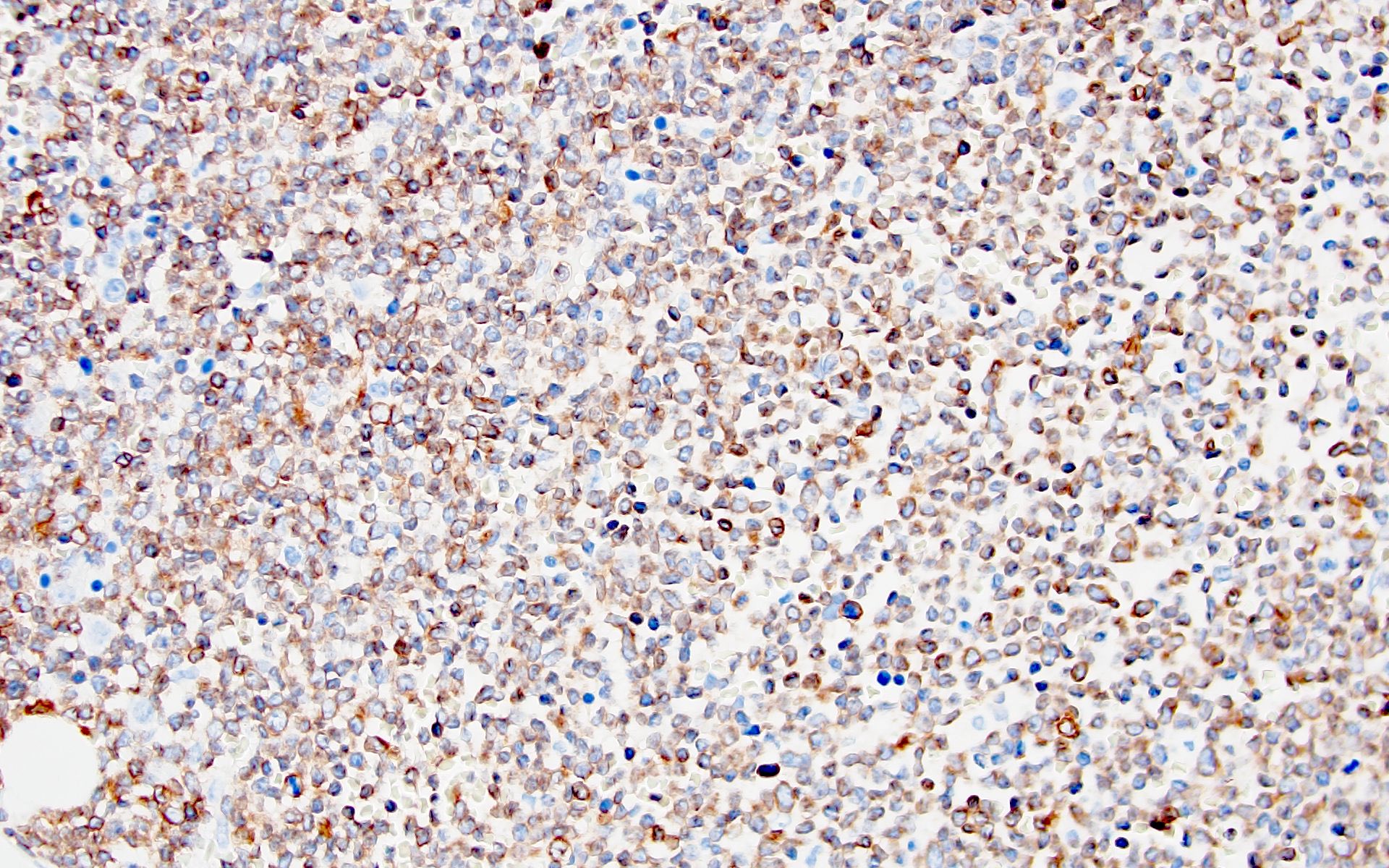
CD79a+ blasts
- Aspirate smears and touch preparations show small to medium sized blasts with high N:C ratio, fine nuclear chromatin and prominent nucleoli
- Some of the blasts show cytoplasmic vacuoles and rarely cytoplasmic granules
- Circulating blasts are seen, which are small to medium sized cells with high N:C ratio, fine nuclear chromatin and prominent nucleoli
Contributed by Barina Aqil, M.D.
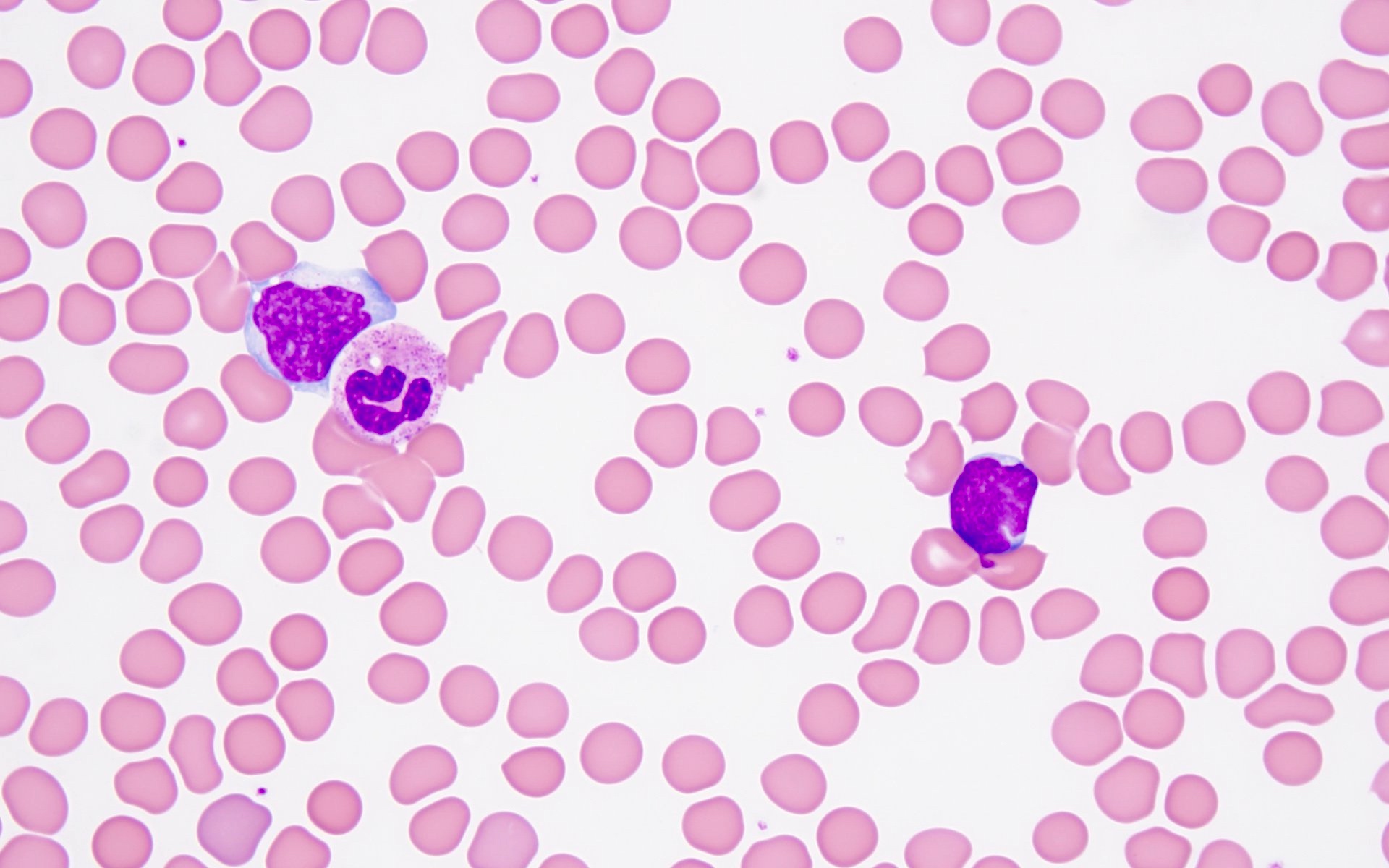
Circulating blasts
- B cell lineage associated antigens CD19, cCD79a and CD22 (cytoplasmic and surface) by flow cytometry
- PAX5, by immunohistochemistry
- Variable CD10 and CD20
- TdT and CD34
- CD38, CD45 (dim) and HLA-DR
- Nonspecific esterase may show multifocal punctate or Golgi staining (inhibited by sodium fluoride)
- Periodic acid-Schiff (PAS) shows coarse chunky cytoplasmic staining
- Myeloperoxidase
- CD3 (surface and cytoplasmic)
- See Positive stains
Contributed by Barina Aqil, M.D.
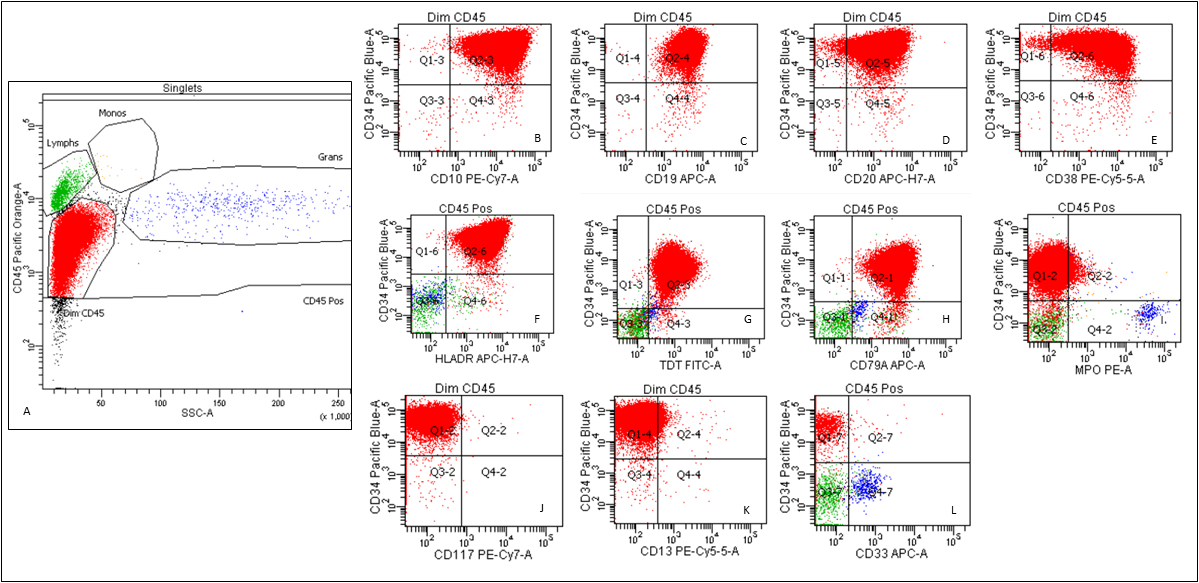
Blast phenotype
- FISH ETV6::RUNX1 fusion probe set is most commonly used to identify iAMP21
- Conventional chromosome analysis further identifies the abnormalities of chromosome 21 along with additional chromosomal aberrations (Blood 2015;125:1383)
- Chromosomal microarray (CMA) or next generation sequencing (NGS) based copy number analysis are being utilized for the identification of iAMP21 (Blood 2015;125:1383)
- Secondary genomic abnormalities that have been reported include gain of chromosomes X, 10 and 14, loss of chromosomes 7/7q and 11q (including KMT2A) and deletions involving IKZF1, CDKN2A/B, PAX5, SH2B3, ETV6 and RB1 as well as rearrangements of P2RY8::CRLF2, ETV6:RUNX1 and BCR::ABL1 (Blood 2011;117:2129, Blood 2011;117:6848, Cancer Genet 2018:226:30, Mol Cytogenet 2016:9:84, Leukemia 2019;33:1881, Leukemia 2014;28:1015)
- Given the unique nature and poor prognosis of the B ALL with iAMP21, cases with genomic lesions that may suggest different subcategory of B ALL / LBL classification should still be included as B ALL / LBL with iAMP21 (Semin Hematol 2019;56:90)
- RAS signaling pathway mutations, including NRAS (45%), KRAS (18%), FLT3 (20%), PTPN11 (11%), BRAF (2%) and NF1 (2%), have been frequently described in cases with iAMP21 (Leukemia 2016;30:1824)
Contributed by Barina Aqil, M.D. and Madina Sukhanova, Ph.D.

Complex karyotype and derivative chromosome 21
- Peripheral blood, bone marrow aspirate and left posterior iliac crest bone marrow core biopsy:
- B lymphoblastic leukemia with iAMP21 involving ~90% of a hypercellular (90% cellular) bone marrow (see comment)
- Comment: The cytogenetic analysis showed complex karyotype including an ider(21) resulting in an amplification of the RUNX1 gene, which was confirmed by the concurrent FISH analysis. FISH study was positive for amplification of RUNX1 (84%).
- Peripheral blood smear: The blood smear shows normocytic anemia with anisopoikilocytosis including ovalocytes, teardrop forms and rare schistocytes. Rare blasts are present, which are small to intermediate sized cells with round to cleaved nuclei, open chromatin, occasional small nucleoli and scant cytoplasm. There is absolute neutropenia. Platelets are decreased with unremarkable morphology.
- Bone marrow aspirate: The bone marrow aspirate smear shows sheets of blasts with similar morphologic features as described above. Few scattered megakaryocytes and rare erythroid precursors are seen.
- Bone marrow core biopsy (decalcified): The unilateral bone marrow core biopsy is hypercellular for age (~90% cellular). There is extensive replacement by sheets of atypical medium sized mononuclear cells, with round to irregular nuclear contour, fine chromatin and pinpoint nucleoli. Background hematopoiesis is markedly decreased. Few scattered megakaryocytes are present.
- B lymphoblastic leukemia / lymphoma with BCR::ABL1 fusion:
- B ALL / LBL defined by BCR::ABL1 fusion due to rearrangement between the BCR gene on chromosome 22q.11.2 and the ABL1 oncogene on chromosome 9q34.1
- Blasts frequently express myeloid markers, such as CD13, CD33 and CD25 (Int J Clin Exp Pathol 2014;7:6225)
- Poor prognosis (Blood 2008;111:1827)
- B lymphoblastic leukemia / lymphoma with KMT2A rearrangement:
Which of the following cytogenetic abnormalities leads to higher risk of development of AML with iAMP21?
- t(9;11)
- t(9;22)
- t(15;17)
- t(15;21)
Comment Here
Reference: with iAMP21
- 5' CRLF2 and 3' CRLF2 (Xp22.3 and Yp11.3), break apart
- 5' PDGFRB and 3' PDGFRB, break apart (5q33)
- ABL1 (9q34) and BCR (22q11.2), fusion
- ETV6 (12p13.2) and RUNX1 (21q22.3)
Comment Here
Reference: with iAMP21
Beck: 2021
Cherry: 2022
Dorfman: 2023
Hsi: 2017
Hudnall: 2019
Husain: 2021
IARC: 2017
Jaffe: 2016
Kantarjian: 2016
Lanzkowsky: 2016
Naeim: 2018
Wang: 2020
Find related Pathology books: hematopathology, immunology / transplant, lab medicine, oncology, pediatric
