
- Aggressive T cell malignancy of mature CD4+ T cells causally associated with human T lymphotropic virus 1 (HTLV-1; also called human T cell leukemia virus) (Blood 2017;129:1071)
- Lymphoma cells show monoclonal integration of HTLV-1 (Blood 2017;129:1071)
- Occurs in regions endemic for HTLV-1 (~ 2.5% in HTLV-1 carriers) (Int J Hematol 2002;76:240)
- Affected individuals are usually exposed to HTLV-1 very early in life (Front Microbiol 2018;9:461)
- Develops after a long latency (therefore occurs in adults)
- Most often composed of highly pleomorphic small to medium sized lymphoid cells
- Widely disseminated and clinically aggressive
- Clinical spectrum of HTLV-1 associated diseases is wide and includes neoplastic and nonneoplastic disorders (Clin Microbiol Rev 2010;23:577)
- Abbreviation: adult T cell leukemia / lymphoma (ATLL)
- Synonyms: adult T cell leukemia, adult T cell lymphoma, adult T cell leukemia / lymphoma (HTLV-1+)
- HTLV-1 virus is endemic in Southwestern Japan, the Caribbean basin, sub-Saharan Africa, South America and parts of the Middle East and Australo-Melanesia (Front Microbiol 2012;3:388)
- Third to ninth decade, mean 58 years
- M:F = 1.5:1
- Transmission: requires the presence of living HTLV-1 infected cells
- Mother to infant (mainly by lymphocytes in breast milk) (J Pediatric Infect Dis Soc 2018;7:350)
- Sexual fluids
- Blood and blood products (not transmitted in fresh frozen plasma)
- Widespread lymph node involvement in most
- Systemic: spleen and extranodal sites including the skin, lungs, liver, gastrointestinal tract and central nervous system
- Main extranodal sites: skin (involved in > 50% of cases) and peripheral blood
- Cell of origin is peripheral CD4+ T regulatory αβ T cell (CD4+ CD25+ FoxP3+ T regulatory cells are the closest normal counterpart)
- ATLL patients suffer from profound immunodeficiency; the concept that the cell of origin of ATLL is a T regulatory T cell provides a biologic basis for disease associated immunodeficiency
- ATLL is uniformly causally associated with HTLV-1 infection
- HTLV was the first human retrovirus to be identified, isolated from a patient derived T cell lymphoma cell line (Proc Natl Acad Sci USA 1980;77:7415)
- HTLV-1 is a type C retrovirus, Deltaretrovirus genus
- Single strand of RNA is converted into double strand of DNA in host via reverse transcriptase enzyme (Viruses 2010;2:2037)
- Monoclonal, randomly integrates into host cell genome (Oncology 2015;89:7)
- Infection alone is not sufficient to result in neoplastic transformation of infected cells
- HTLV-1 can infect immature thymocytes and mature CD4+ T cells
- Enters by cell to cell contact via 3 cellular molecules: neuropilin 1, heparan sulfate proteoglycan (HSPGs) and GLUT1 (J Virol 2006;80:6844)
- Tax protein and HTLV-1 basic leucine zipper factor (HBZ): two major oncoproteins with key role in the development of ATLL in chronically infected individuals
- Tax (p40): activates viral promoter, the cyclic AMP response element binding transcription factor (CREB) and nuclear factor κβ pathways (Retrovirology 2004;1:20)
- HBZ: consistently expressed in all cases of ATLL; contributes to cellular proliferation and survival of the neoplastic clone (Retrovirology 2016;13)
- Epigenetic alterations and hypermethylation are also involved in the development and disease progression (Nat Genet 2015;47:1304, Am J Pathol 2010;176:402)
Contributed by Jennifer Chapman, M.D.
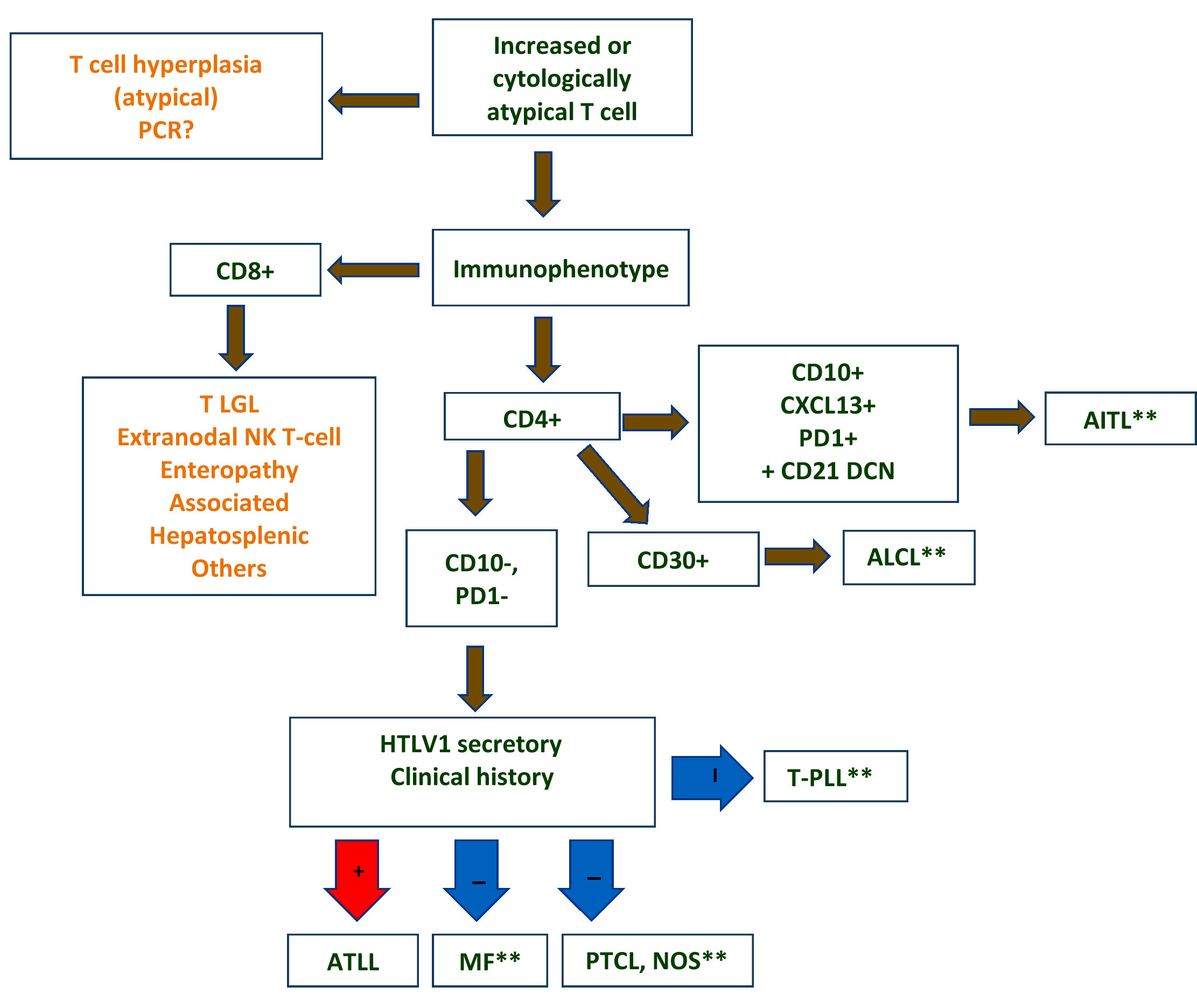
Differential diagnostic algorithm
- 4 clinically defined variants in Shimoyama classification: acute, lymphomatous, chronic and smoldering (Br J Haematol 1991;79:428)
- Acute variant: ~ 55 to 60% of cases, survival usually < 1 year
- Leukocytosis (often greater than 100 x 109/L)
- Peripheral blood involvement with numerous flower cells
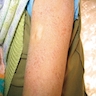
- Skin rash and lymphadenopathy
- Hepatosplenomegaly
- Hypercalcemia
- Elevated serum LDH
- Rapidly progressive course and frequent opportunistic infections
- Lymphomatous variant: ~ 20% of cases, survival usually < 1 year
- Lymphadenopathy
- Skin lesions
- No / minimal peripheral blood involvement
- Hypercalcemia is less frequent (compared with acute variant)
- Chronic variant: ~ 15 to 20% of cases, survival > 2 years but < 5 years due to progression to acute or lymphomatous ATLL
- Lymphocytosis
- Exfoliative skin lesions
- Lymphocyte count is much lower than in the acute variant
- Few atypical lymphocytes in peripheral smear review
- Mild hepatosplenomegaly
- Mild lymphadenopathy
- No hypercalcemia
- Smoldering variant: ~ 5% of cases, survival > 2 years but < 10 years due to progression to
acute ATLL or infectious complications
- Skin or lung lesions
- More than 5% atypical lymphocytes in absence of leukocytosis
- No hepatosplenomegaly, hypercalcemia or lymphadenopathy
- Primary cutaneous tumoral type
- No leukemic phase, lymphadenopathy, hepatosplenomegaly or hypercalcemia
- Proposed as a fifth clinical type in the Shimoyama classification (Int J Dermatol 2010;49:1099)
- Nonneoplastic HTLV-1 associated diseases
- Tropical spastic paraparesis / HTLV-1 associated myelopathy (TSP / HAM) (Rev Neurol (Paris) 2012;168:257)
- HTLV-1-associated infective dermatitis
- Uveitis
- Thyroiditis
- Pneumonitis
- Myositis
| Comparison of clinical forms of adult T cell leukemia / lymphoma | ||||
|---|---|---|---|---|
| Clinical manifestation | Acute | Lymphomatous | Smoldering | Chronic |
| Lymphocytosis | Increased | No | No | Mildly increased |
| Blood abnormal lymphocyte | Increased | No* | > 5% | Mildly increased |
| Increased LDH | Yes | No | No | Minimal |
| Hypercalcemia | Yes | Variable | No | No |
| Skin rash | Variable: > 50% | Variable: > 50% | Erythematous rash | Rash, papules |
| Lymphadenopathy | Usually present | Yes | No | Mild |
| Hepatosplenomegaly | Usually present | Often present | No | Mild |
| Bone marrow infiltration | May be present | No | No | No |
| Median survival | < 1 year | < 1 year | > 2 years | > 2 years |
- Strongest support for a diagnosis of ATLL: demonstration of viral integration in the tumor cells
- Clinical features favoring the diagnosis: appropriate patient demographic (patient derived from HTLV-1 endemic area), hypercalcemia, skin lesions and a leukemic phase
- Histologic features favoring the diagnosis: pleomorphic T cell lymphoma, leukemic phase, CD4+ αβ type with T regulatory immunophenotype, expression of CD25
- Evidence of HTLV-1 or 2 sequences at the molecular level
- Seropositivity for anti-HTLV-1 antibody as surrogate for demonstration of monoclonal integration of virus
- Only useful in areas with low prevalence of HTLV-1 infection
- In areas of high HTLV-1 prevalence, viral integration must be demonstrated
- Expression of CD25 by lymphoma cells
- Other laboratory tests / results:
- CBC: elevated leukocyte count and circulating neoplastic lymphocytes (leukemic phase)
- Elevated serum LDH level reflects disease burden / activity
- Hypercalcemia: more common in patients with acute variant; variably associated with lytic bone lesions
- Eosinophilia and neutrophilia are common
- Elevated soluble IL-2 receptor α chain levels in patients with aggressive ATLL
- May have lytic bone lesions
- Skull, pelvis, spine and long bones can be affected
- Punched out lesions similar to those seen in plasma cell myeloma can be found
- F-18 deoxyglucose PET / CT is usually positive in sites of disease activity
- CT detects sites of nodal and extranodal disease
- Clinical variant, age, performance status, serum calcium and LDH levels are major prognostic factors
- Death often stems from opportunistic infections
- Acute and lymphomatous variants:
- Aggressive clinical course
- Recurrent infection: parasitic (Strongyloides stercoralis) and viral infections (Parasite Immunol 2004;26:487)
- p16 gene deletion and p53 mutation: more aggressive clinical course
- Chronic and smoldering variants:
- More protracted clinical course
- Progression to an acute phase with an aggressive course in ~25%
- p16 gene deletion and chromosomal deletion detected via comparative genomic hybridization (CGH) in the chronic phase is a negative prognostic factor (J Clin Oncol 1997;15:1778)
- 49 year old man with multiple cranial nerve palsies and meningeal lymphoma (Am J Hematol 2017;92:397)
- 50 year old woman with rheumatoid arthritis and panbronchitis-like pulmonary lesions (J UOEH 2017;39:55)
- 58 year old woman with erythematous and itchy plaques (Br J Haematol 2017;177:507)
- 70 year old man with TCL-1 positive lymphoma (Mod Pathol 2018;31:1046)
- 73 year old woman with unilateral conjunctival infiltration (J Clin Exp Hematop 2017;57:143)
- Chronic or smoldering ATLL: observation may be appropriate for patients who are asymptomatic
- ATLL is resistant to most chemotherapy
- No standard chemotherapy regimen
- Intensive high dose combination chemotherapy and bone marrow transplantation have been used in limited numbers of patients (Blood 2010;116:1369)
- Monoclonal antibody based therapies have been attempted directed against:
- IL-2R (anti-Tac)
- CCR4 (mogamulizumab)
- CD52 (alemtuzumab)
- Recent clinical trials use arsenic trioxide, interferon α and zidovudine (Adv Ther 2018;35:135)
Images hosted on other servers:

Nodules
- Skin lesions have been classified as erythema, papules or nodules
- Rare cases show tumor-like lesions or erythroderma as seen in mycosis fungoides / Sézary syndrome
- Enlarged and effaced lymph node
- Bone marrow:
- Degree of bone marrow infiltration is less than expected, given the marked lymphocytosis that is often present
- Pattern of marrow involvement can be diffuse, interstitial or sinusoidal
- Often evidence of bone resorption and osteoclastic activity
- Bone trabeculae: may show remodeling
- Lytic bone lesions can be present even in the absence of tumoral bone infiltration
- Lymph nodes:
- Involves paracortical T cell zones
- Typically show diffuse architectural effacement
- May be subdivided according to cell type and pattern into:
- Pleomorphic small cell
- Pleomorphic medium and large cell type / pattern (most common)
- Anaplastic large cell-like [resembling anaplastic large cell lymphoma (ALCL)]
- Angioimmunoblastic T cell lymphoma-like (AITL)
- Hodgkin lymphoma-like: seen in early phase of some adult T cell lymphoma
- Leukemic pattern of infiltration with preservation or dilation of lymph node sinuses that contain malignant cells
- Size or shape of the neoplastic cells or identification / classification of the above patterns does not impact the clinical course
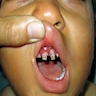
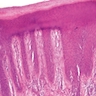
- Skin lesions:
- Epidermal infiltration with Pautrier-like micro-abscesses is common (Mod Pathol 2018;31:1046)
- Hyperparakeratosis is variably present in the overlying epidermis
- Dermal infiltration: mainly perivascular but larger tumor nodules with extension to subcutaneous fat may be observed
- Erythematous lesions: composed of smaller cells in perivascular pattern in dermis
- Papules and nodules: composed of larger cells that replace dermis
- Other sites commonly involved by ATLL include: liver, spleen, lungs and central nervous system
- Grading: there is no formal grading system for ATLL
Note: ATLL should be strongly considered in any T cell lymphoma developing in a patient from an endemic area regardless of histopathologic features and anti-HTLV-1 serology testing should be performed
Contributed by Jennifer Chapman, M.D.
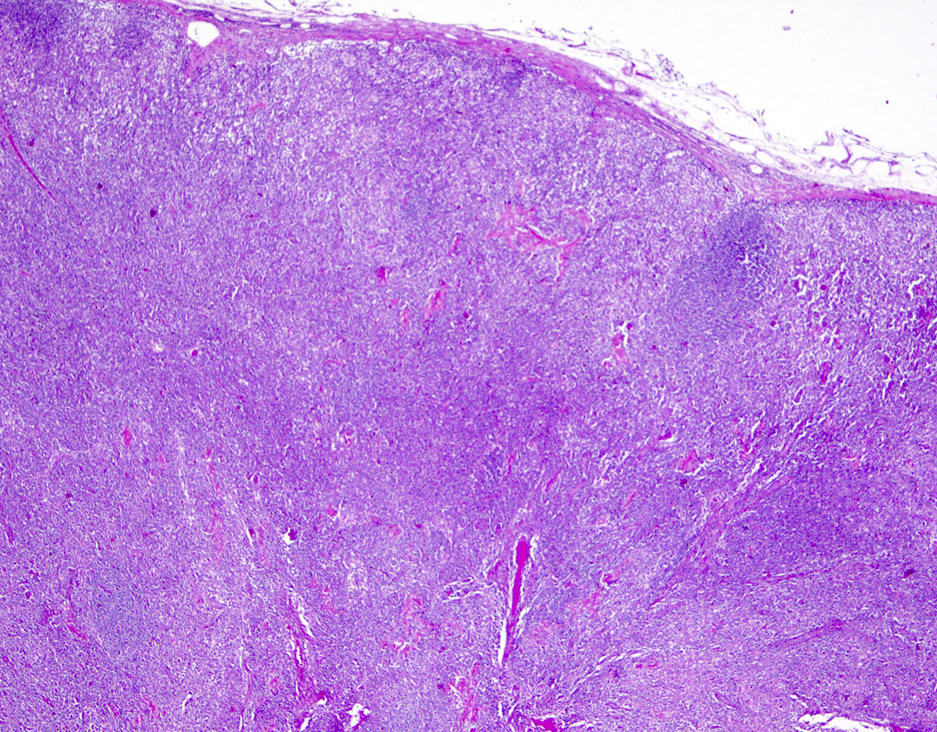
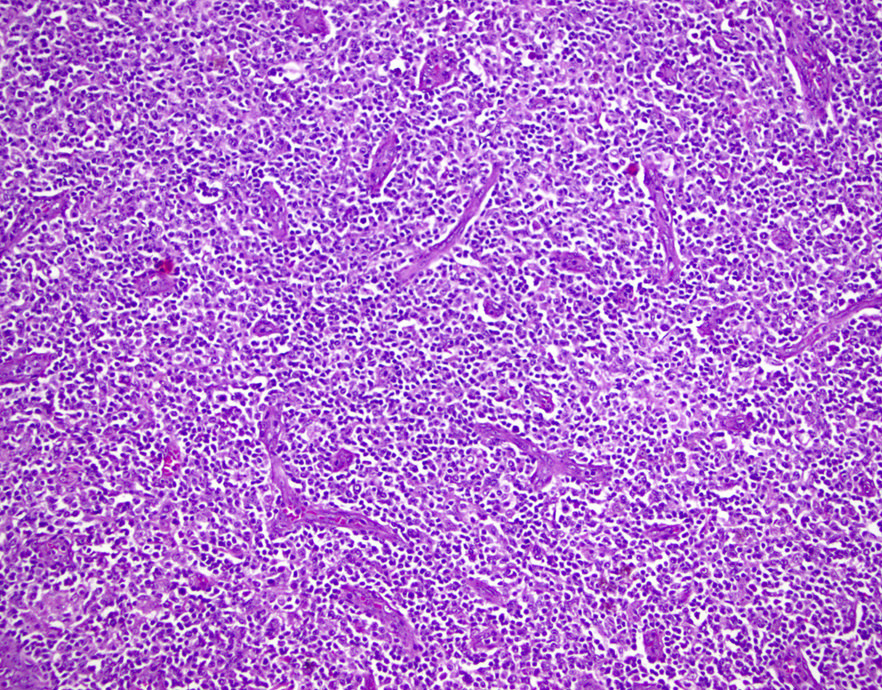
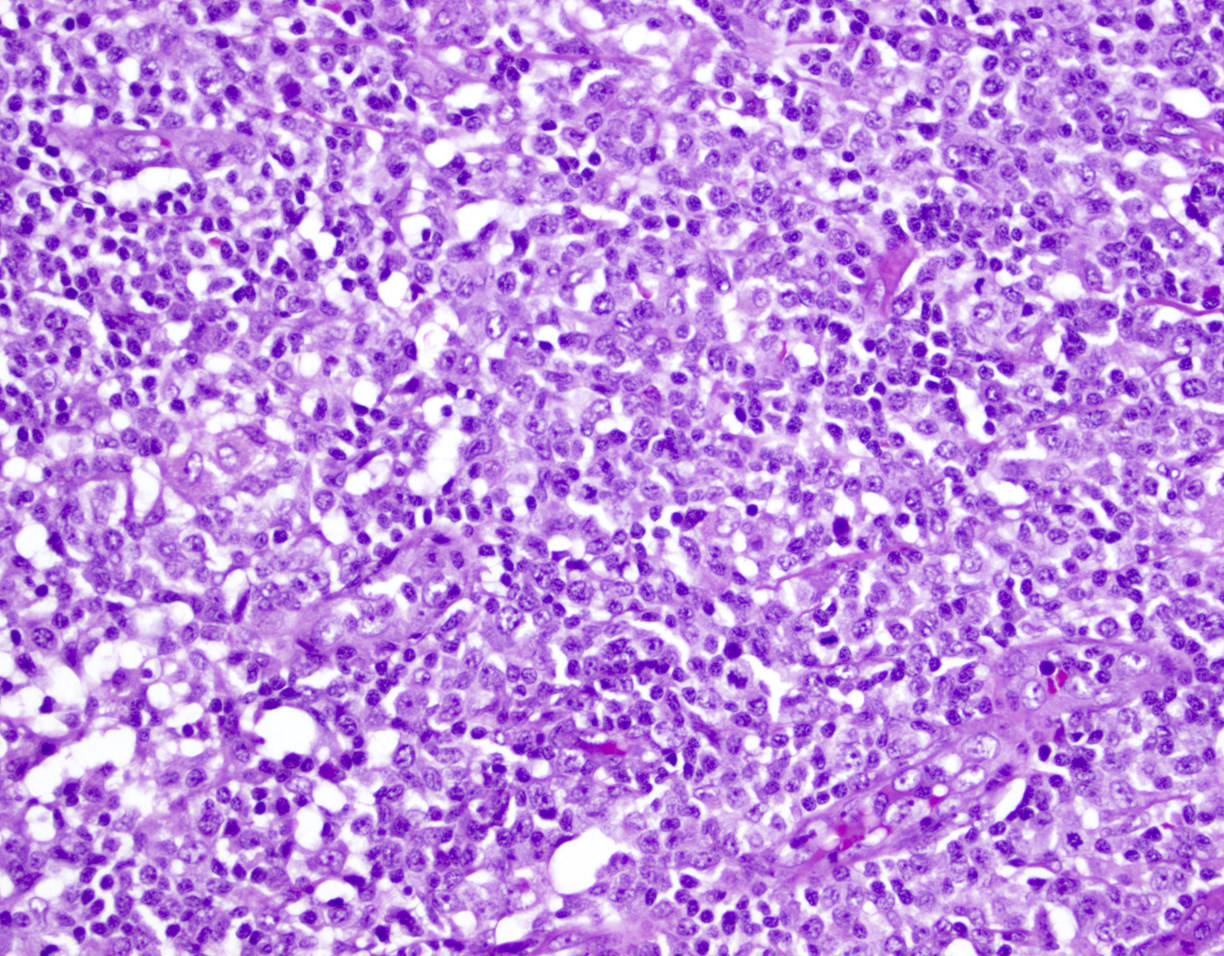
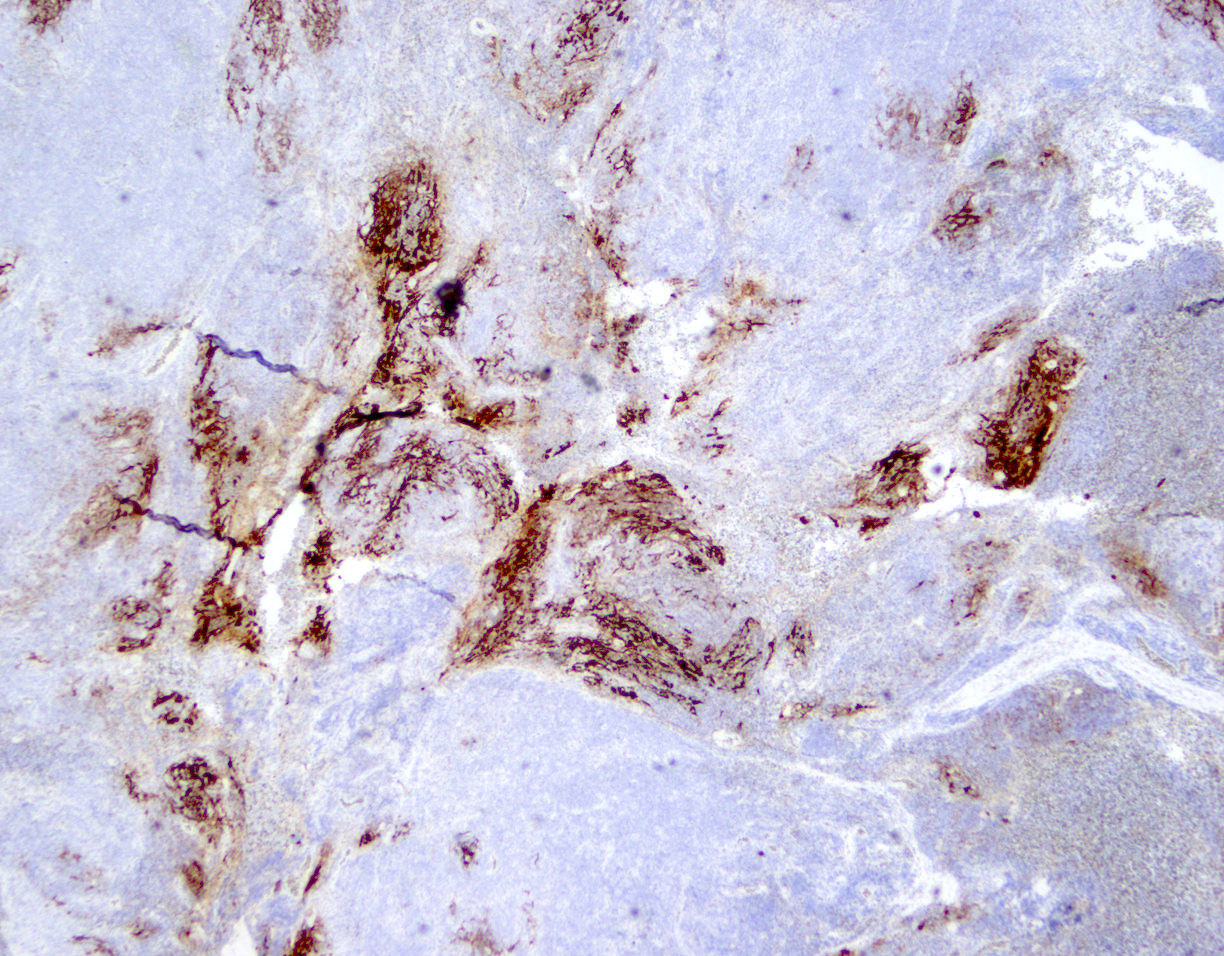
ATLL mimicking angioimmunoblastic T cell lymphoma
ATLL with Hodgkin-like cells mimicking classical Hodgkin lymphoma
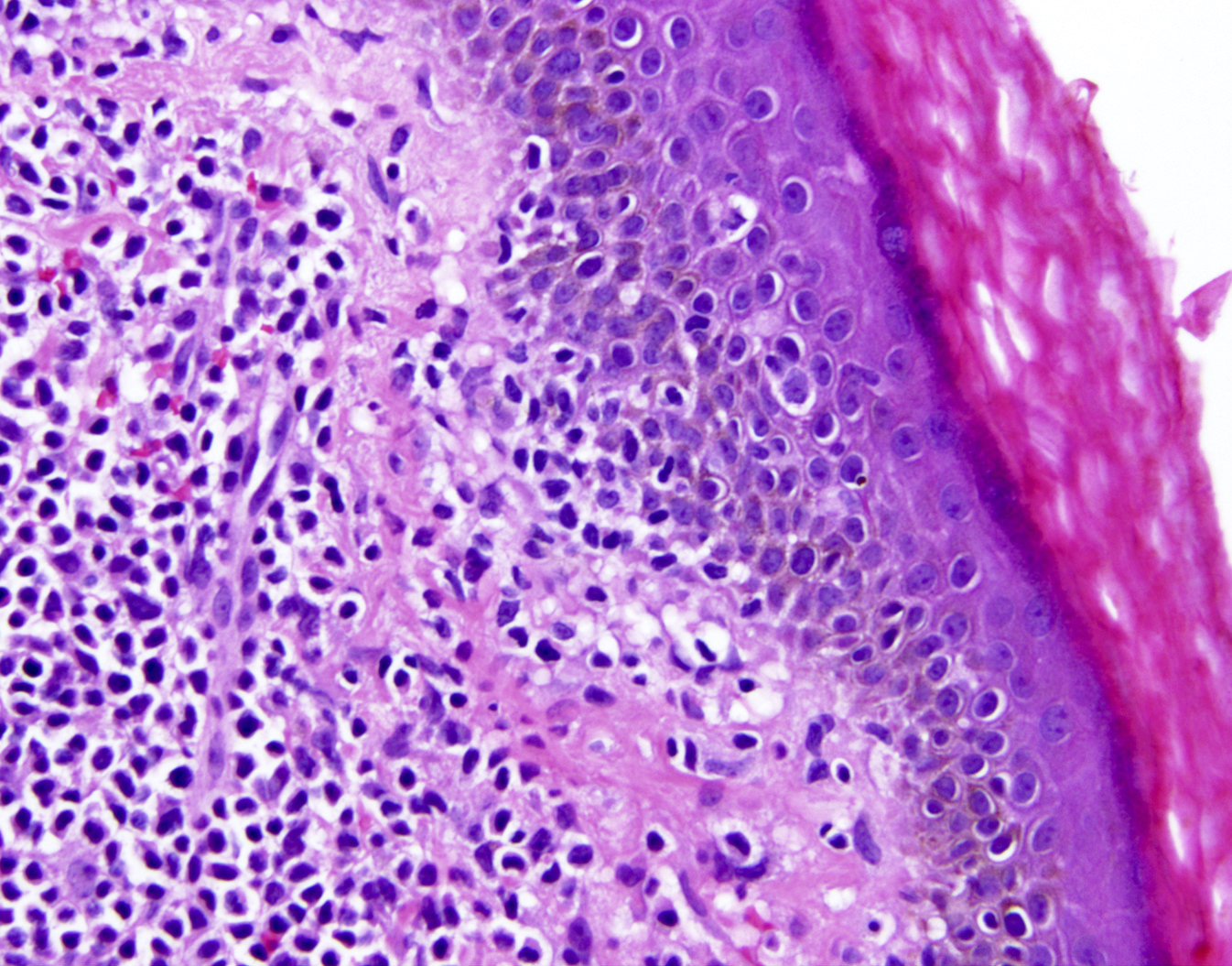
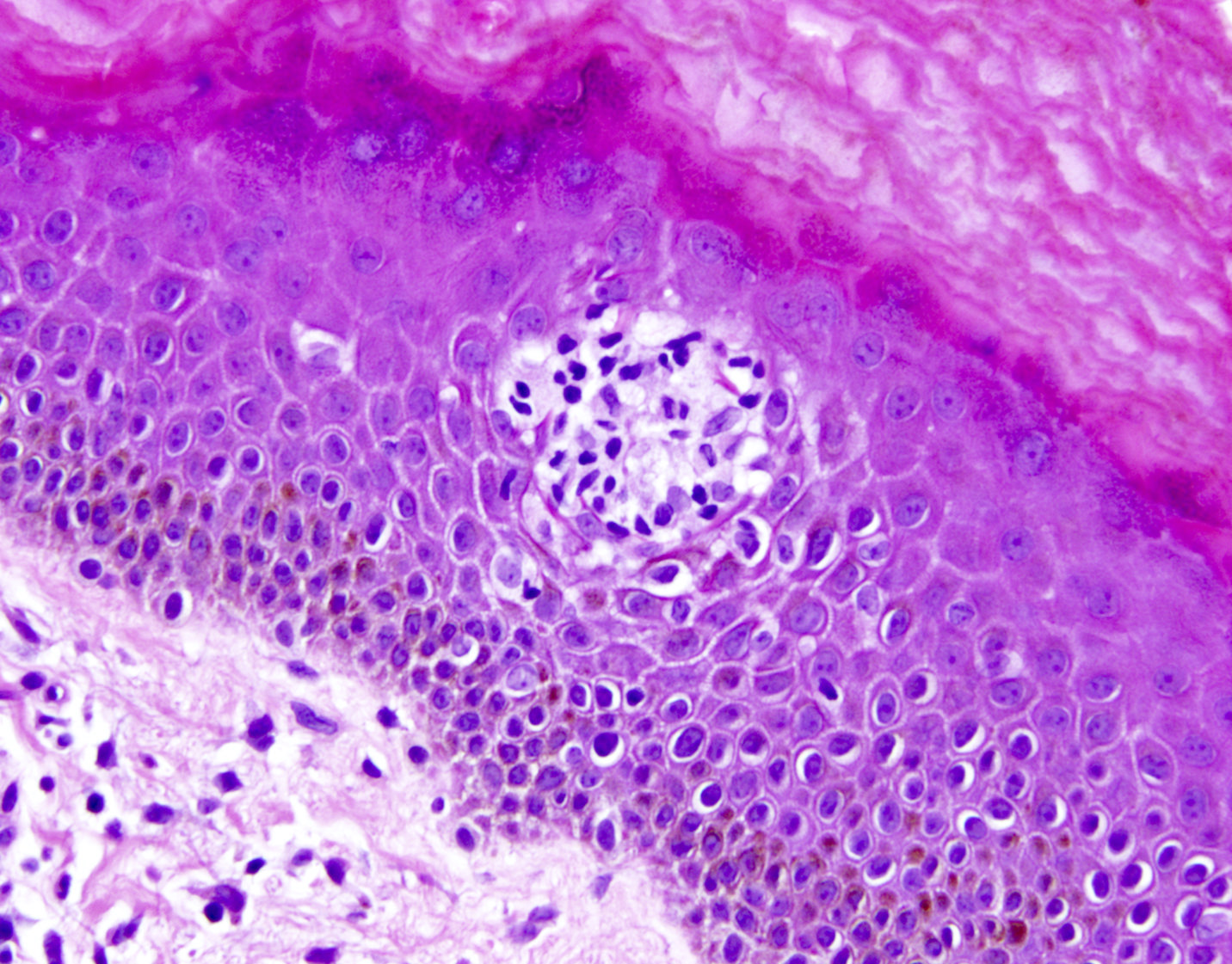
ATLL mimicking mycosis fungoides
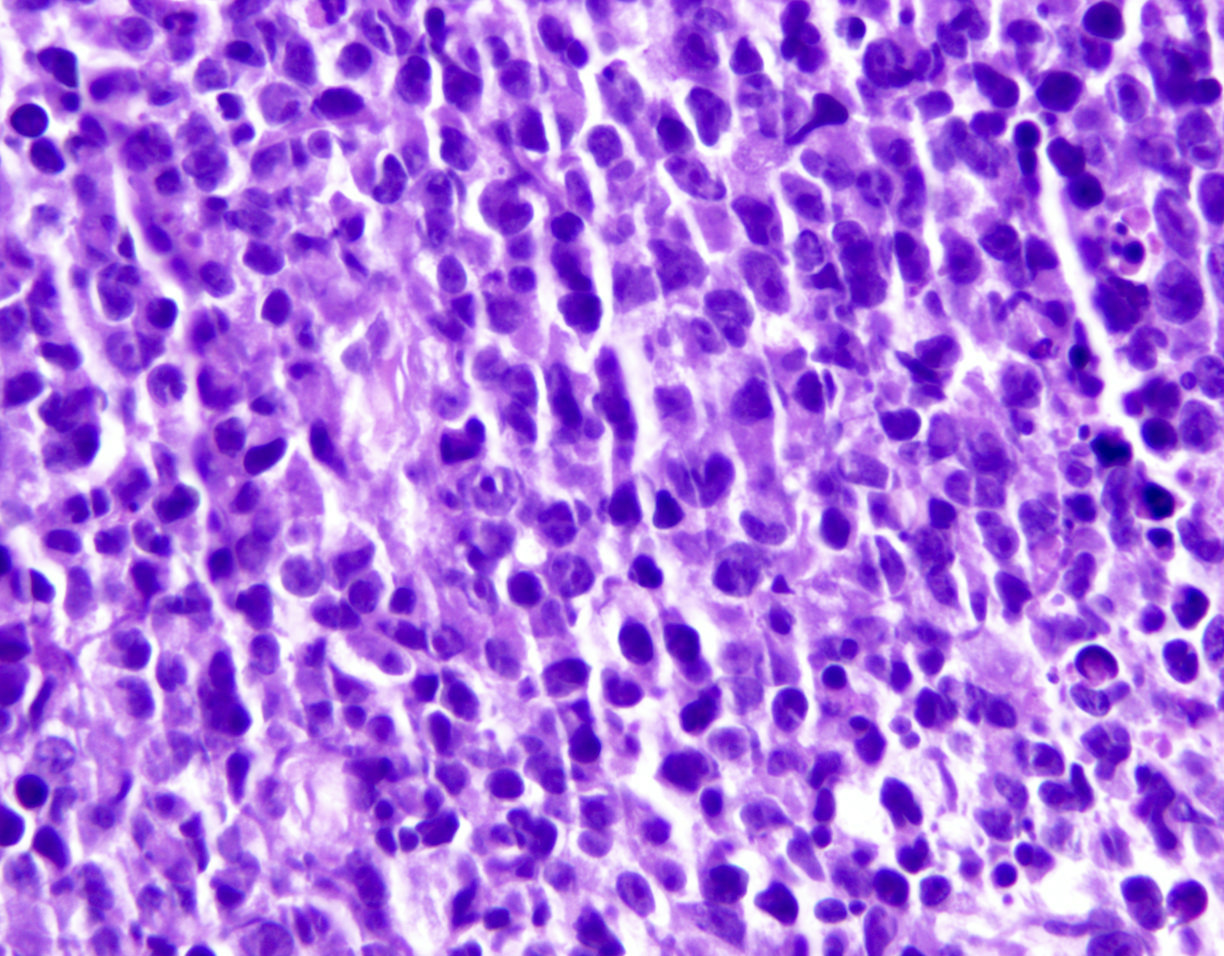

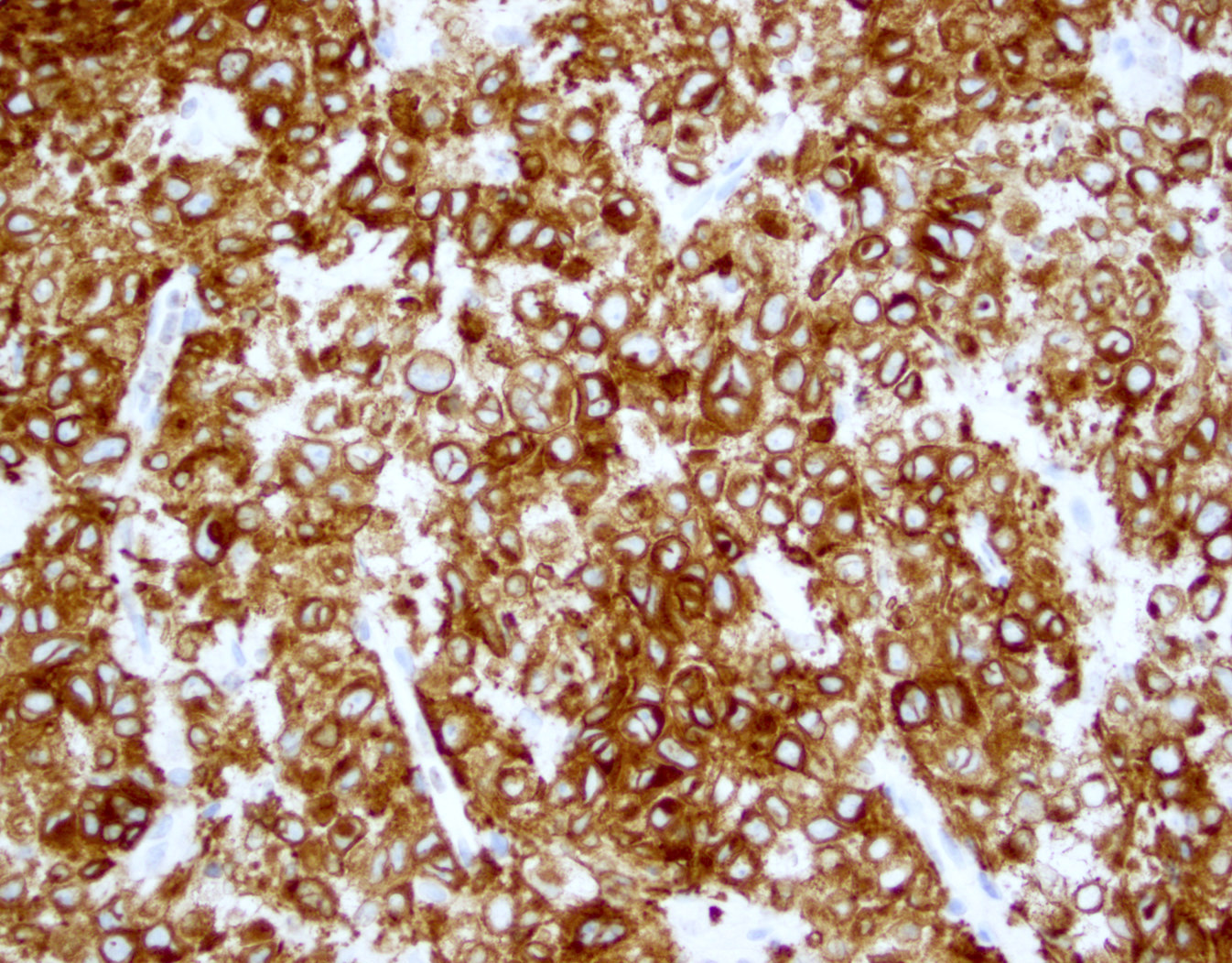
ATLL, lymphomatous type, mimicking anaplastic large cell lymphoma
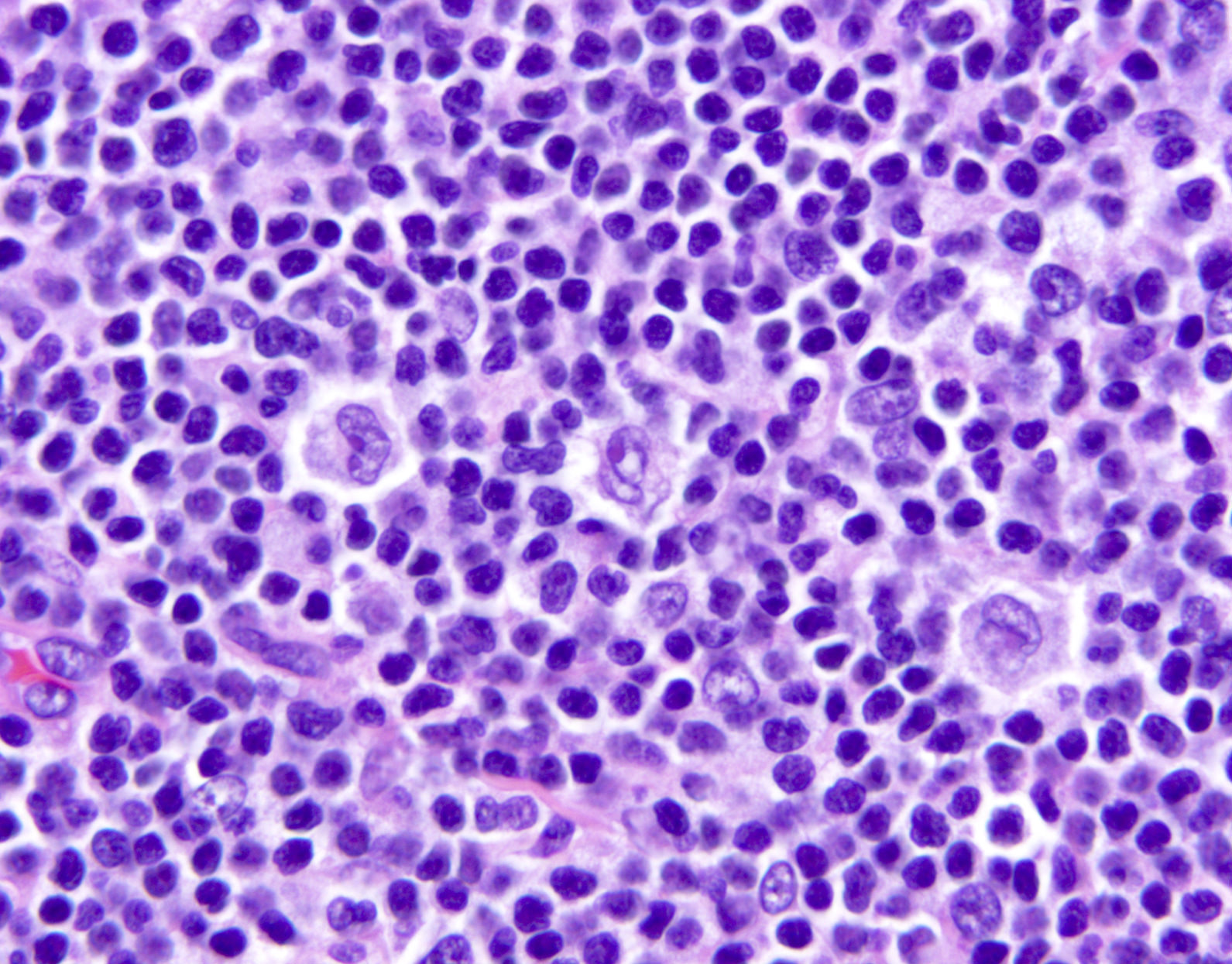
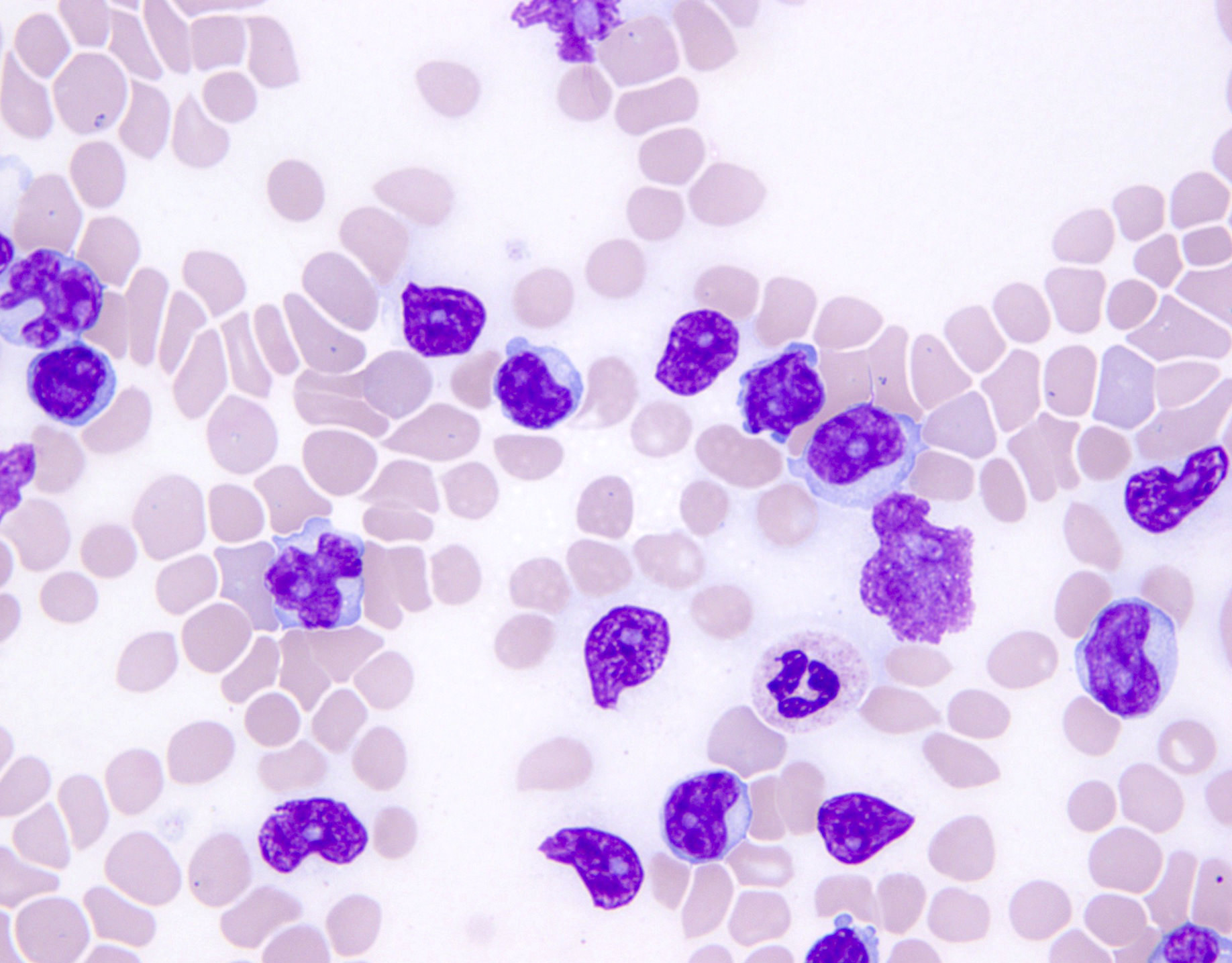
- ATLL cells show variable appearances
- Irregular / polylobulated nuclei, homogeneous, condensed chromatin, small nucleoli
- Agranular basophilic cytoplasm
- Tumor cells in lymph nodes are frequently pleomorphic and most cases have a mixture of small to large pleomorphic cells
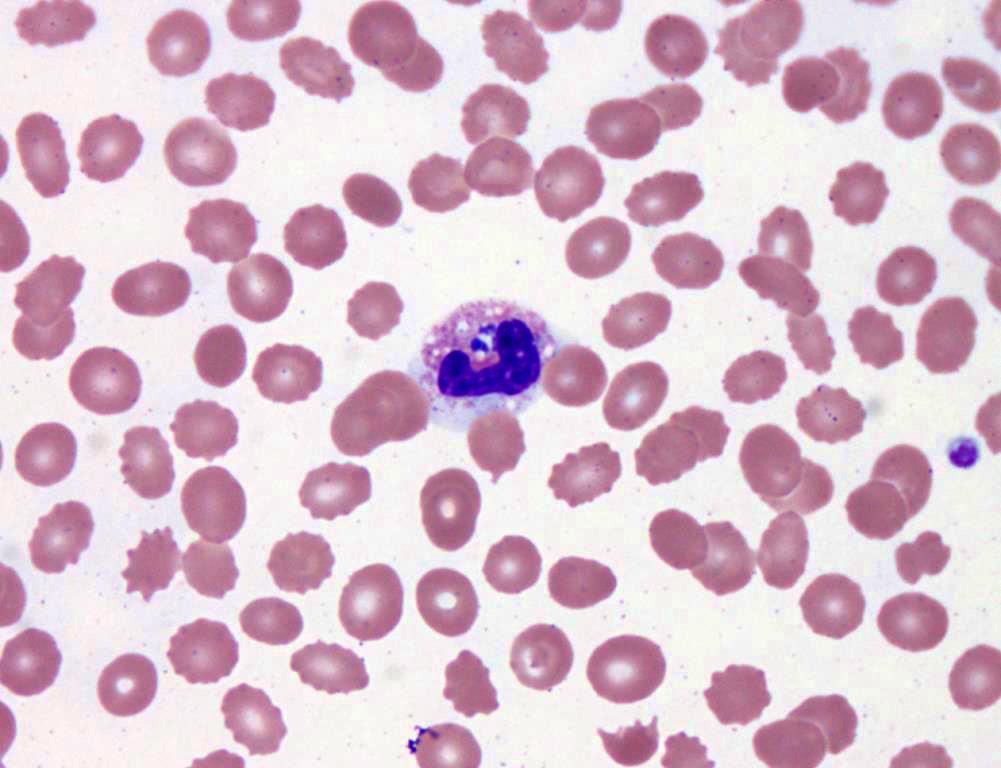
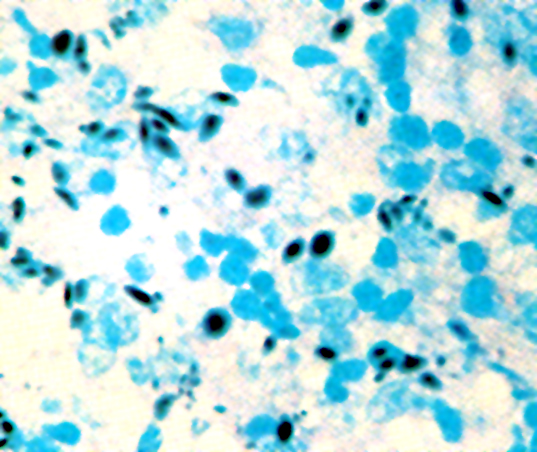
- Medium to large sized lymphocytes with multilobulated nuclei (flower cells) in the peripheral blood of acute (leukemic) ATLL
- Slightly larger than normal peripheral blood lymphocytes with moderately condensed chromatin, absent or small nucleoli, scant slightly basophilic cytoplasm and some with multilobulated nuclei in chronic ATL
Contributed by Jennifer Chapman, M.D.
ATLL, acute variant,
mimicking
T prolymphocytic
lymphoma
Images hosted on other servers:

Flower cells

CLL-like morphology
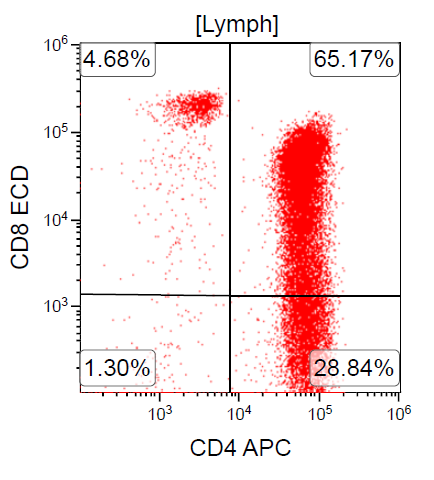
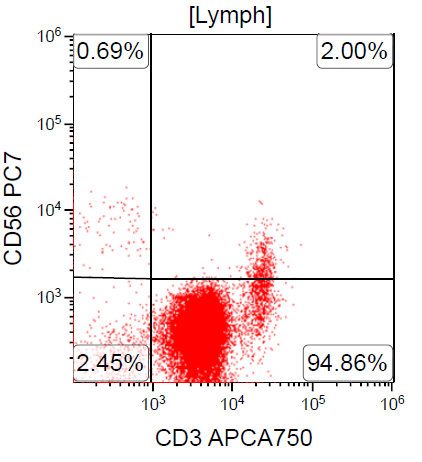
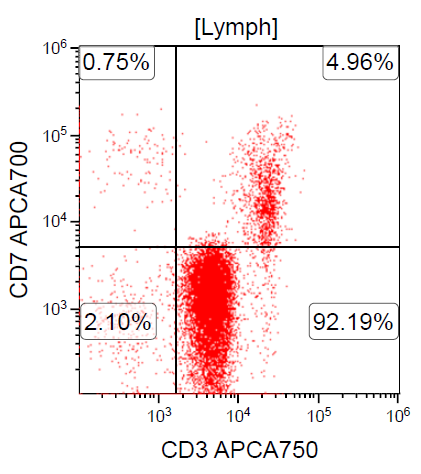
- CD2, CD3, CD4 (most), CD5, CD25 (IL-2R), T cell receptor (TCR) αβ, CD45RO, HLA-DR variable, FOXP3 (68%), CD52 variable, CD62 / selectin L variable and CCR4 (88%) (Leukemia 2005;19:2247, Clin Cancer Res 2003;9:3625)
- Frequent expression of IRF4 / MUM1
- Can be negative for CD30 or can express weak or diffuse and strong CD30 (in large transformed cells)
- EBV+ reactive B cells (immunoblasts) may be present in the background
Contributed by Jennifer Chapman, M.D.
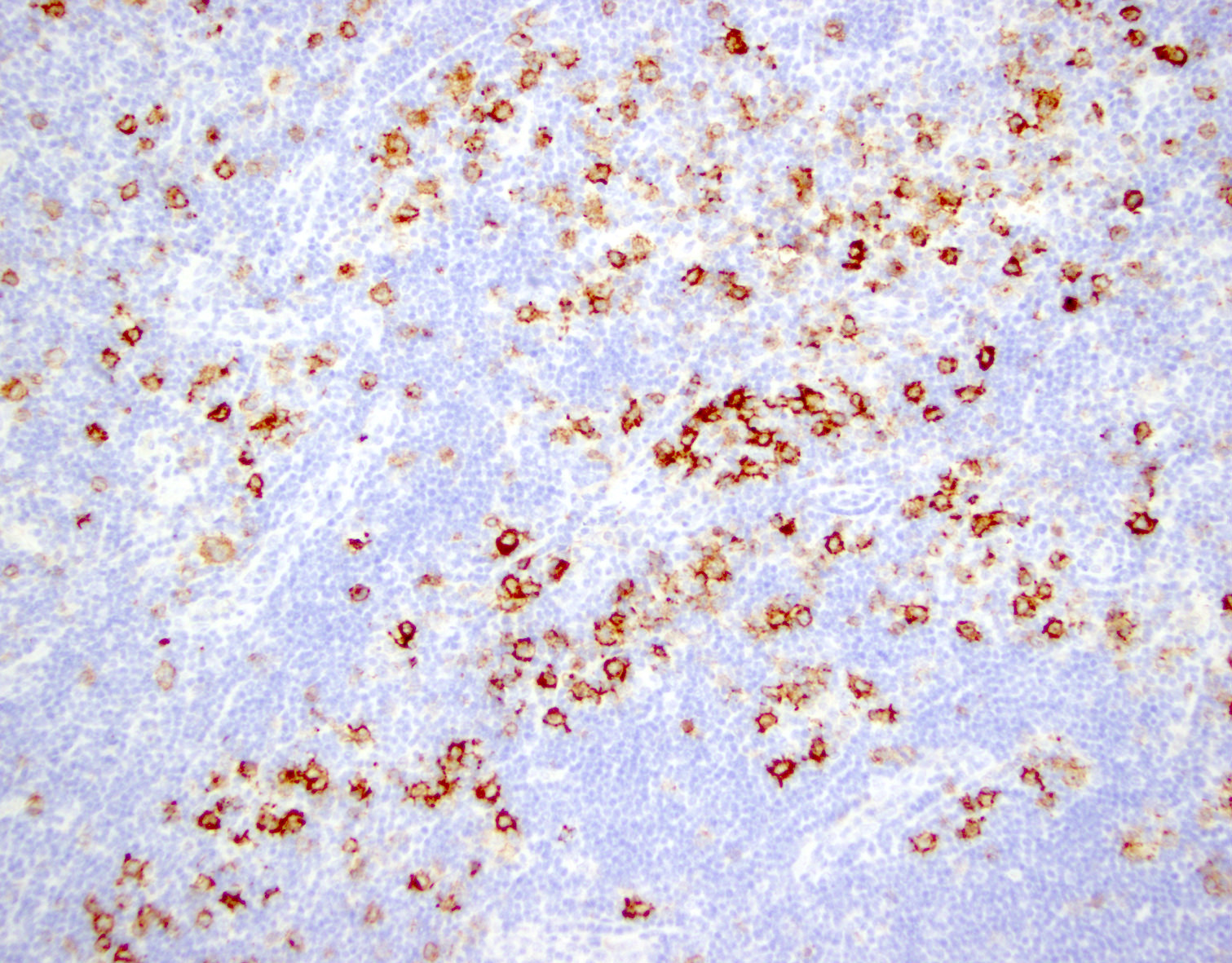
CD45
CD4

CD56
CD7
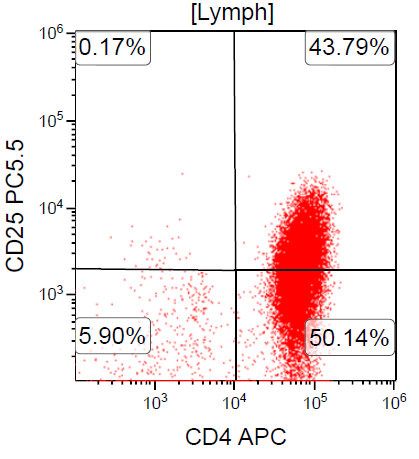
CD25
Images hosted on other servers:

CD3+, CD4+, CD25+, CD7-, CD8-
- Viral particles, 80 to 120 nm, are present in both cytoplasm and extracellular space
Images hosted on other servers:

ATLL cell
- Clonal integration of HTLV-1 genome can be demonstrated
- PCR assays are positive for clonal T cell receptor gene rearrangement
- Quantitative HTLV-1 levels
- CCR4 mutations in ~25% of cases (Nat Genet 2015;47:1304)
- RHOA mutations detected in ~15% of cases (Blood 2016;127:596)
- Tumor suppressor genes are inactivated either by mutation or epigenetic silencing
- Numerous complex chromosomal abnormalities are frequent, especially in acute and lymphomatous variants
- Clonal chromosome abnormalities are frequent but not specific
- Peripheral T cell lymphoma, not otherwise specified (PTCL-NOS)
- Patient from Western hemisphere
- Background of reactive cells, including eosinophils, plasma cells and histiocytes
- Negative serologic studies or lack of molecular evidence of HTLV-1
- Angioimmunoblastic T cell lymphoma (AITL)
- Anaplastic large cell lymphoma (ALCL)
- Sinusoidal distribution in partially involved lymph nodes
- Lymphoma cells are strongly CD30+ (ATLL with strong CD30 expression in 100% of lymphoma cells are reported)
- Expression of cytotoxic granule associated proteins (can be seen in ATLL)
- Translocations involving ALK gene and / or ALK1 protein expression
- ALK- ALCL cases are difficult to differentiate from ATLL without HTLV-1 testing
- Classic Hodgkin lymphoma (HL)
- Background T cells are immunophenotypically normal
- Lacks clonal T cell gene rearrangement
- Lacks malignant cells in peripheral blood, rare reports of skin lesions and hypercalcemia
- Cutaneous T cell lymphoma / mycosis fungoides (CTCL MF / SS)
- T cell prolymphocytic leukemia (T-PLL)
- Involves blood at presentation with ↑ WBC
- Some nuclear irregularity but no flower cell morphology as in ATLL
- Lack of anti-HTLV-1 positive serology
- T lymphoblastic leukemia / lymphoma (T-LBL)
- T cell large granular lymphocytic leukemia (LGL)
- Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue, 4th Edition, 2008, His: Hematopathology - Foundations in Diagnostic Pathology series, 3rd Edition, 2017, Orazi: Knowles Neoplastic Hematopathology, 3rd Edition, 2013, Jaffe: Hematopathology 2nd, Edition, 2016, Medeiros: Diagnostic Pathology - Lymph Nodes and Extranodal Lymphomas, 2nd Edition, 2017
- Central nervous system
- Heart
- Liver
- Skin

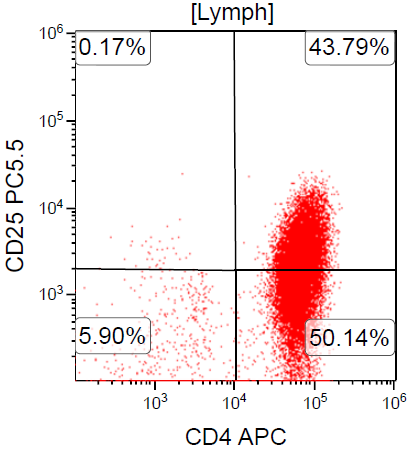
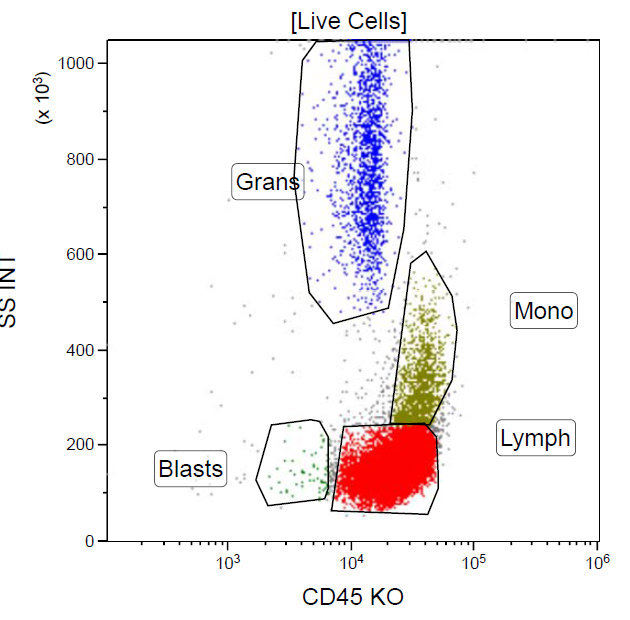
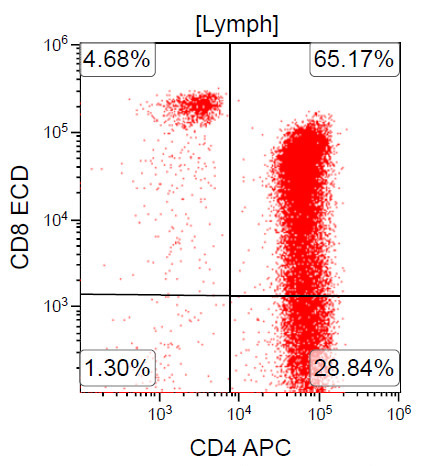
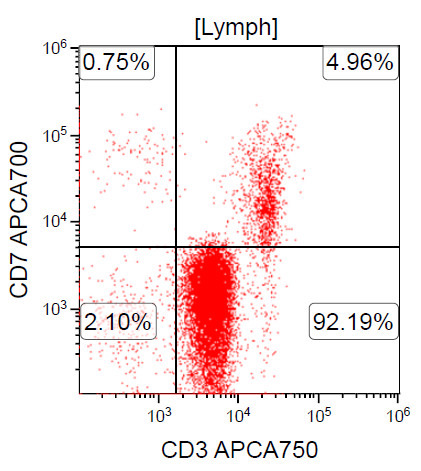
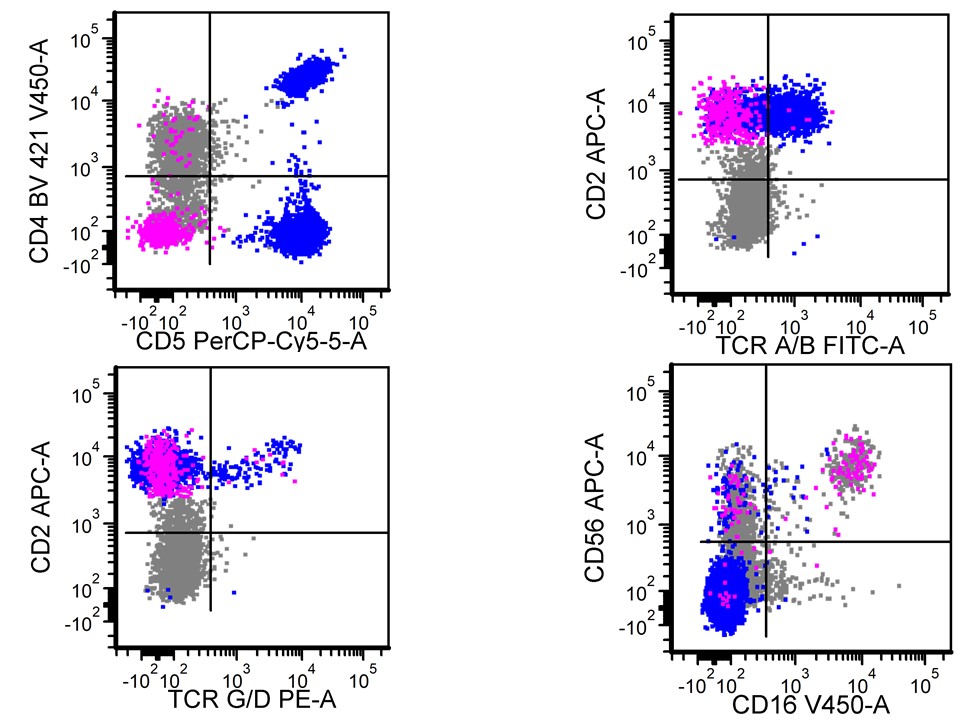
A 67 year old Haitian man with hepatitis B presents with fatigue and weakness. No other symptoms are present. Laboratory work up shows leukocytosis (55 x 109/L) and thrombocytopenia (100 x 109/L). Physical examination and imaging studies show massive splenomegaly with no lymphadenopathy. There was no hypercalcemia or increased in serum LDH. Peripheral smear review showed abnormal lymphocytosis with circulating lymphoma cells having mature nuclear features and highly lobulated nuclei. Flow cytometry performed in bone marrow aspirate is provided above. Serology for anti HTLV-1 antibody is positive. Which of the following is the most accurate statement?
- Dual positivity for CD4 and CD8 is uncommon in adult T cell leukemia / lymphoma
- Expression of CD25 is specific for ATLL among T cell lymphomas / leukemias
- Patient has acute myeloid leukemia with maturation (NK / myeloid type)
- Patient has T cell large granular lymphocytic leukemia (LGL)
Comment here
Reference: ATLL
- Aggressive NK cell leukemia (ANKL) is a rare disease with a dismal prognosis and many diagnostic challenges
- ANKL shares several overlapping features with other NK cell neoplasms
- The clinical presentation and course can help distinguish from other NK cell neoplasms
- ANKL can be subtle to detect morphologically and immunophenotypically
- ANKL overlaps morphologically and genetically with extranodal NK / T cell lymphoma; however, its clinical presentation is acute and outcome is poorer
- As rare EBV negative ANKL have been described, EBV negative status cannot be used as the sole criterion to exclude a diagnosis of ANKL
- Bone marrow involvement by ANKL can be prominent or subtle
- Cytologic atypia can be notable, however, in a subset of cases; neoplastic cells are deceptively bland
- Aggressive NK cell leukemia
- ICD-O: 9948/3 - aggressive NK cell leukemia
- Asian population (mainly in EBV positive ANKL, as EBV negative ANKL have been shown to affect other ethnicities)
- Median age 40 years
- No gender predilection
- Strong association with EBV infection (although Epstein-Barr virus encoded small RNA (EBER) negative ANKL is a well established entity)
- References: Hum Pathol 2020;105:20, Mod Pathol 2017;30:1100, Am J Surg Pathol 2017;41:67, Am J Surg Pathol 2020;44:1235
- Peripheral blood, bone marrow, liver, spleen
- In EBV positive ANKL, EBV encoded small RNAs induce release of high amounts of IL10:
- IL10 activates the JAK / STAT pathway
- Stimulation of STAT3 phosphorylation
- Downstream MYC activation leading to clonal expansion
- In EBV negative ANKL, other unidentified mechanisms lead to
- Stimulation of STAT3 phosphorylation
- Downstream MYC activation leading to clonal expansion
- Reference: Cell Res 2018;28:172
- In EBV positive ANKL: chronic active EBV infection (Jpn J Cancer Res 1997;88:82, Am J Hematol 1997;54:276, J Dermatol 2014;41:29)
- In EBV negative ANKL: unknown
- Fever, B symptoms, hepatosplenomegaly, lymphadenopathy
- Bone marrow biopsy
- Biopsy of organs suspected of involvement by CT scan
- Peripheral blood smear
- Flow cytometry analysis
- Neutropenia, anemia, thrombocytopenia
- Elevated serum lactate dehydrogenase (LDH) levels
- Elevated liver functions tests
- Disseminated intravascular coagulopathy
- Hemophagocytic syndrome
- Reference: Cancers (Basel) 2020;12:2900
- Prognosis is very poor with a median survival of less than 2 months, despite intensive chemotherapy
- 16 year old boy with EBV negative ANKL (J Hematol 2018;7:163)
- 28 year old woman in remission, 1 year post allogeneic hematopoietic stem cell transplant (Intern Med 2010;49:1907)
- 36 year old man with CD56 negative ANKL (Case Rep Hematol 2017;2017:3724017)
- 56 year old man with ANKL diagnosed by cerebrospinal fluid examination (Diagn Cytopathol 2016;44:314)
- 78 year old man with ANKL associated hemophagocytic lymphohistiocytosis (Blood 2016;127:2502)
- Allogeneic hematopoietic stem cell transplantation (SCT) improves outcome in ANKL patients for a limited time
- No consensus chemotherapeutic regimen has been established to manage patients with ANKL; chemotherapy regimens used to manage ANKL include:
- SMILE (dexamethasone, methotrexate, ifosfamide, etoposide and L-asparaginase)
- AspaMetDex (L-asparaginase, methotrexate and dexamethasone)
- VIDL (etoposide, ifosfamide, dexamethasone and L-asparaginase)
- Reference: Cancers (Basel) 2020;12:2900
- Bone marrow involvement by ANKL can be prominent or subtle
- Two main patterns of infiltration: interstitial and sinusoidal
- Neoplastic cells are medium sized, with a moderate amount of cytoplasm, highly irregular nuclei with condensed chromatin and conspicuous nucleoli
- In some cases, atypia is subtle and neoplastic cells can look deceptively bland
- Apoptosis and focal necrosis are common findings
- Geographic necrosis is typically not observed in ANKL, unlike cases of extranodal NK / T cell lymphoma (ENKTL)
- Reference: Cancers (Basel) 2020;12:2900
Contributed by Siba El Hussein, M.D. and Joseph Khoury, M.D.
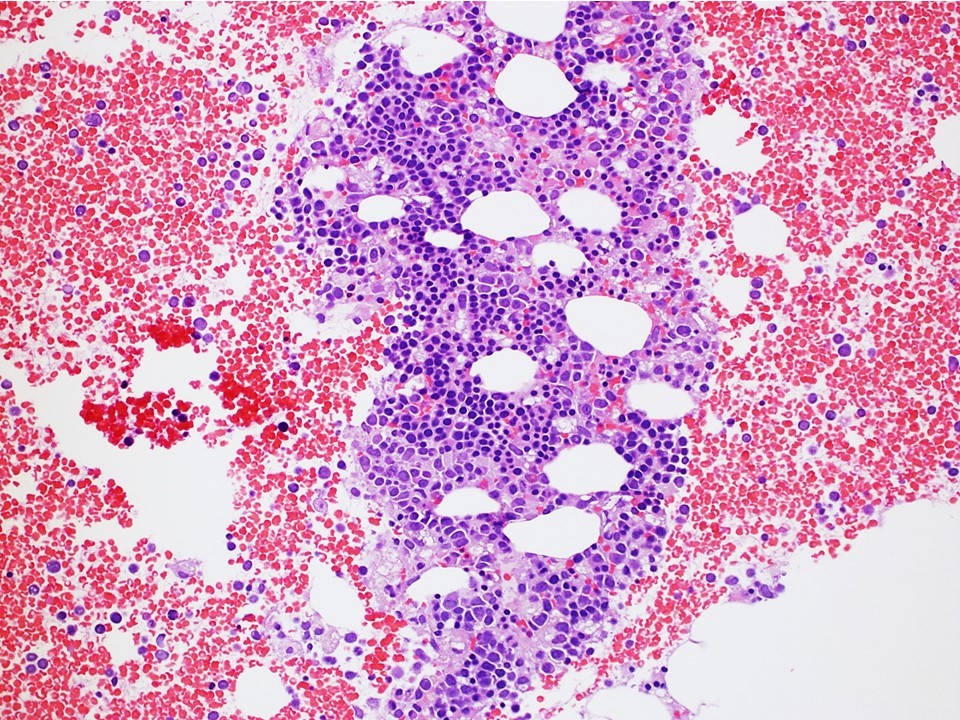
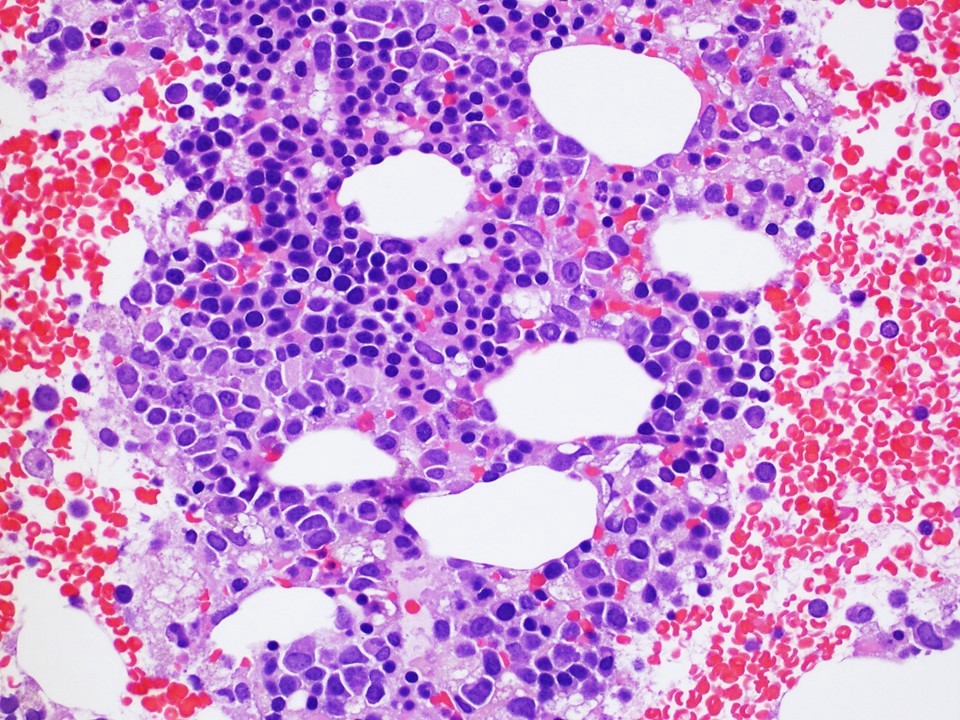
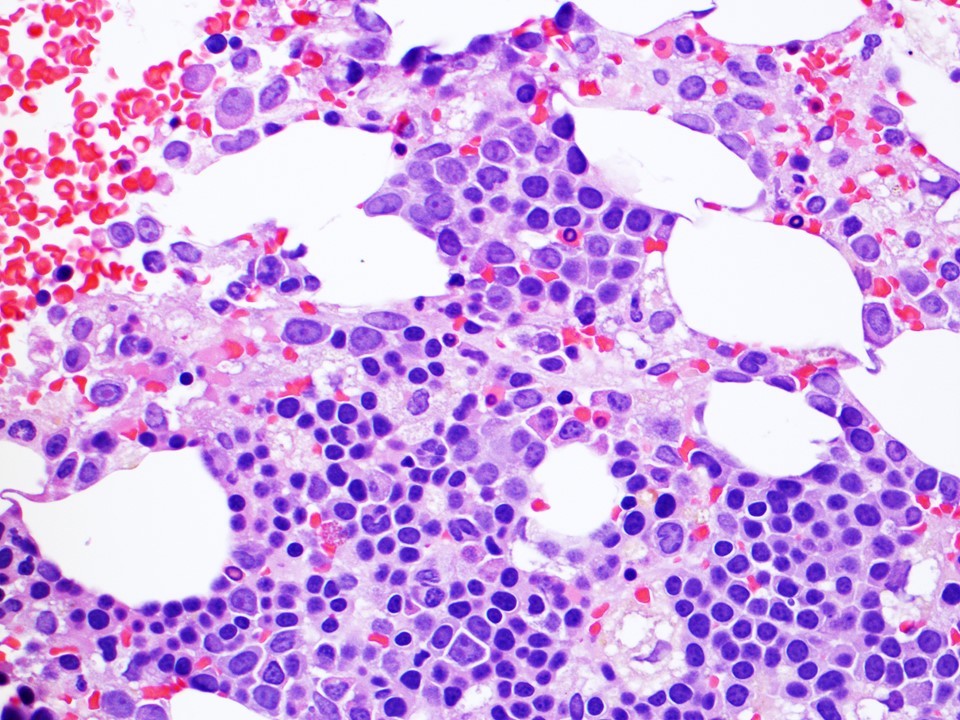
Neoplastic cells involving the bone marrow
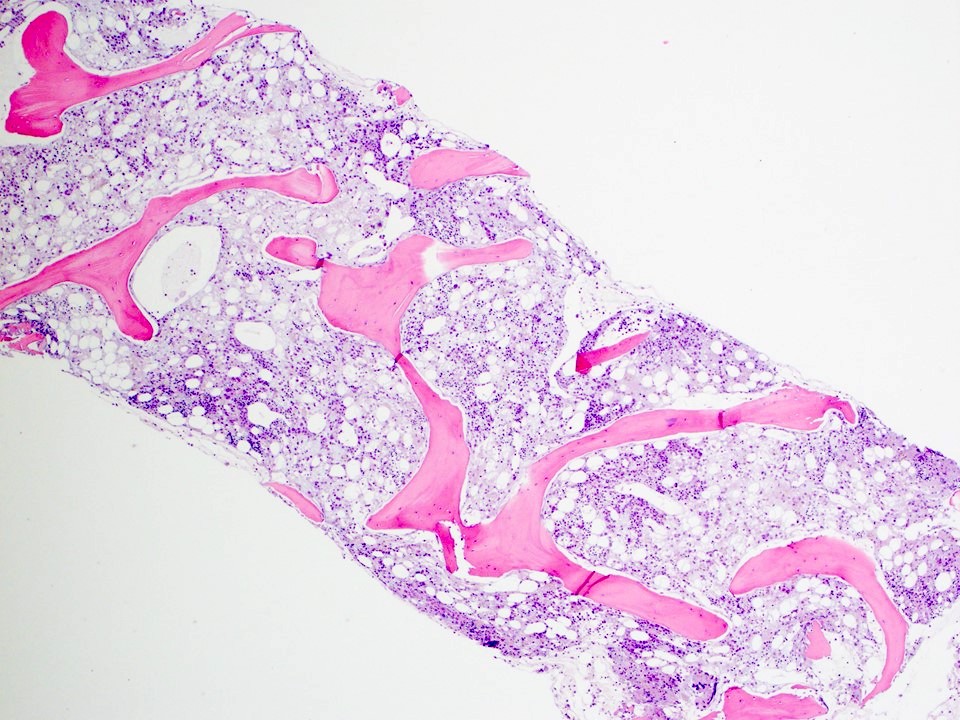
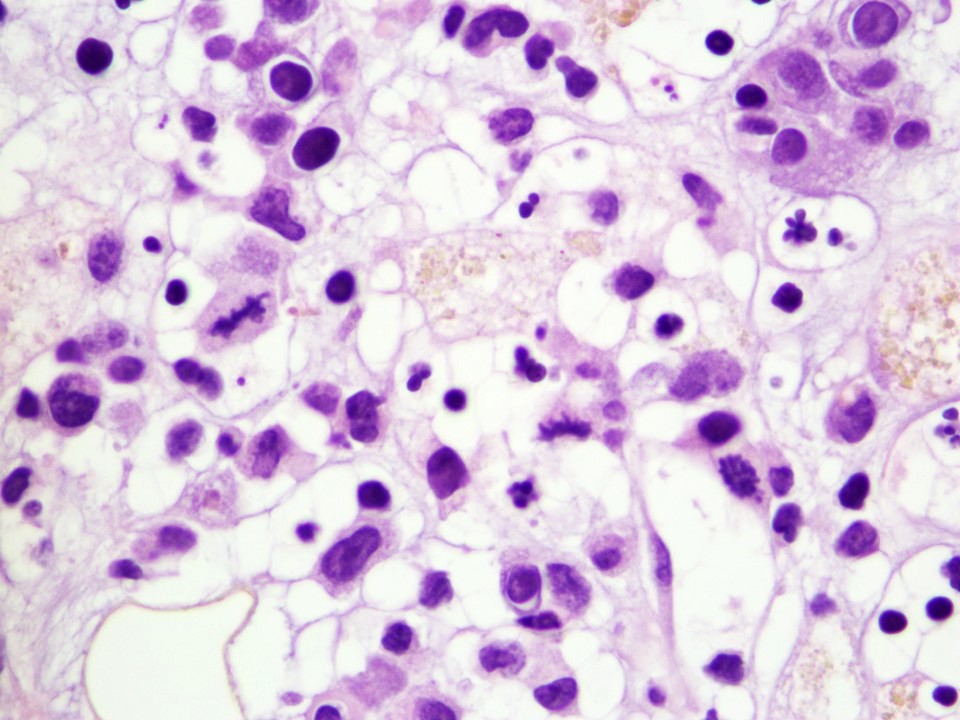
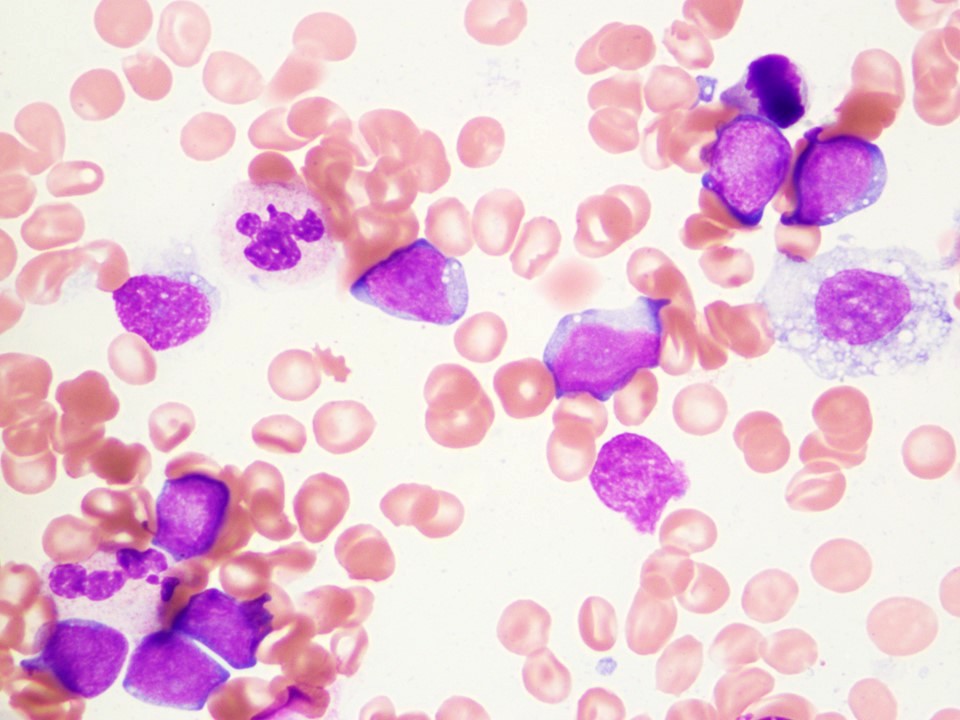
Neoplastic cells involving the bone marrow
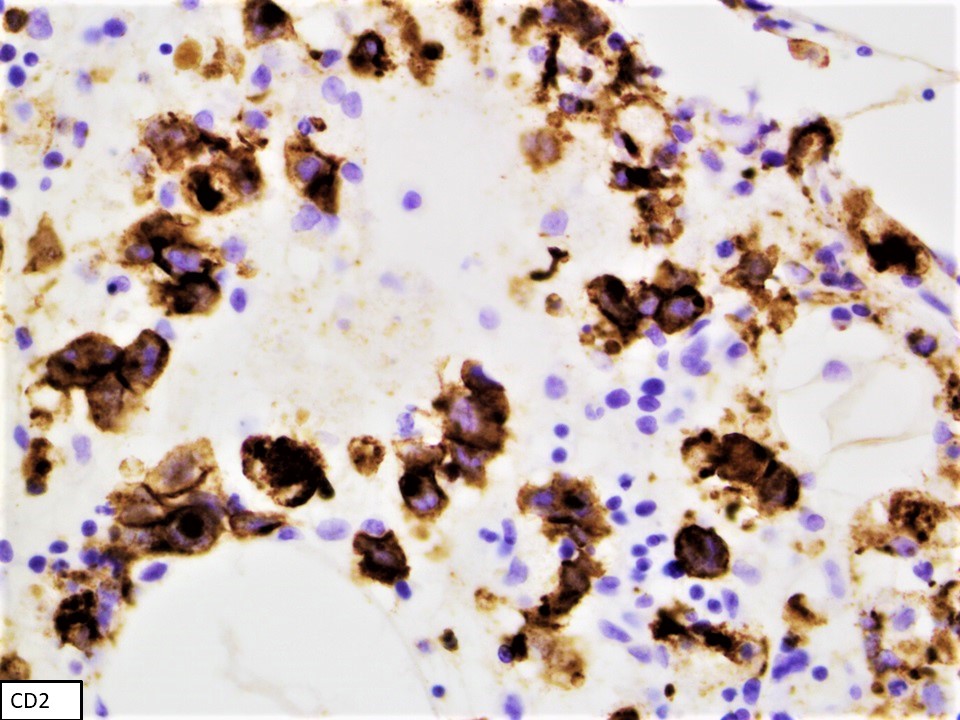
Immunohistochemical stains
Images hosted on other servers:

ANKL bone marrow biopsy
- Moderate amounts of basophilic agranular cytoplasm
- Punched out cytoplasmic vacuoles
- Highly irregular nuclear contours and an open chromatin pattern with prominent nucleoli
- Reference: Cancers (Basel) 2020;12:2900
- Cells are intermediate to large in size
- Moderate amounts of basophilic agranular cytoplasm
- Punched out cytoplasmic vacuoles
- Highly irregular nuclear contours and an open chromatin pattern with prominent nucleoli
- Reference: Cancers (Basel) 2020;12:2900
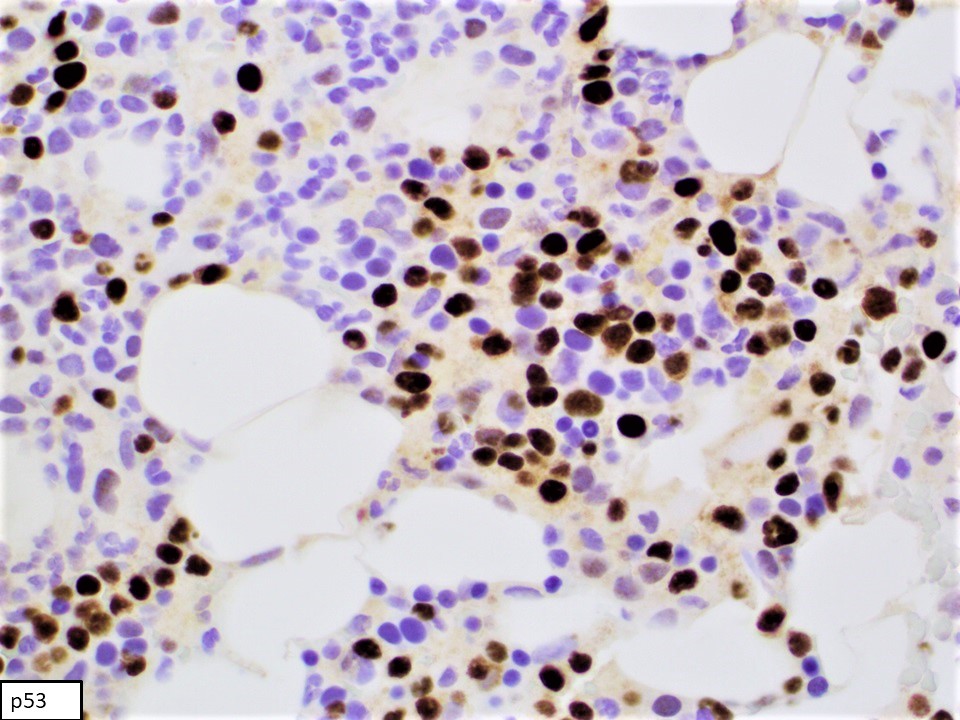
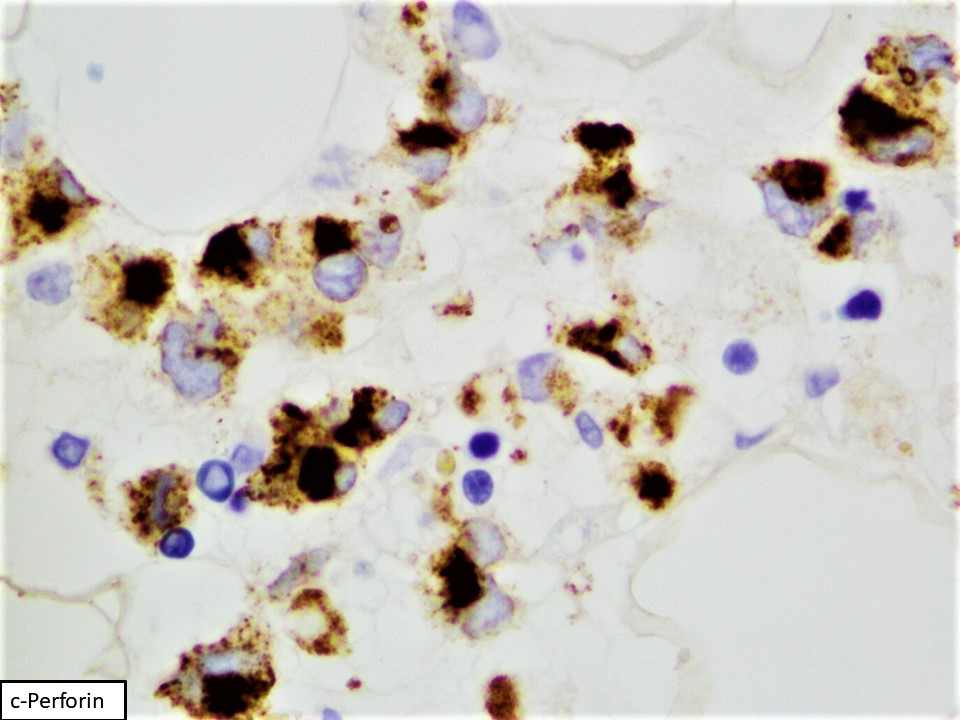
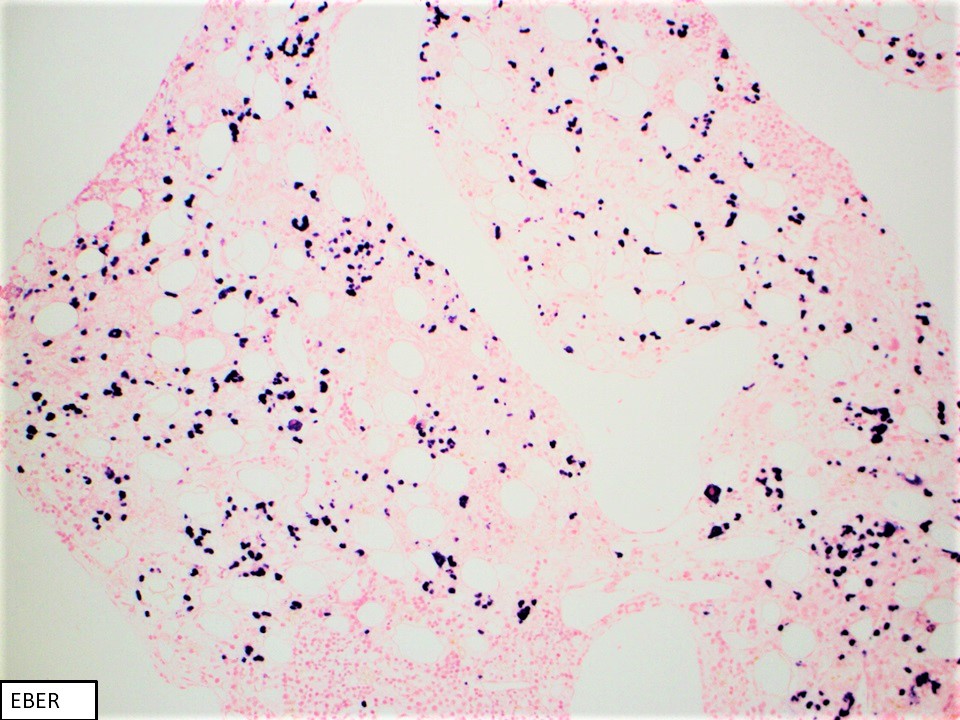
- CD2, cytoplasmic CD3 epsilon protein, CD56, perforin A, granzyme B, TIA, EBER (in most cases), PDL1 (in some cases), p53 (frequent), BCL2, MYC (Am J Surg Pathol 2020;44:1235, Mod Pathol 2017;30:1100)
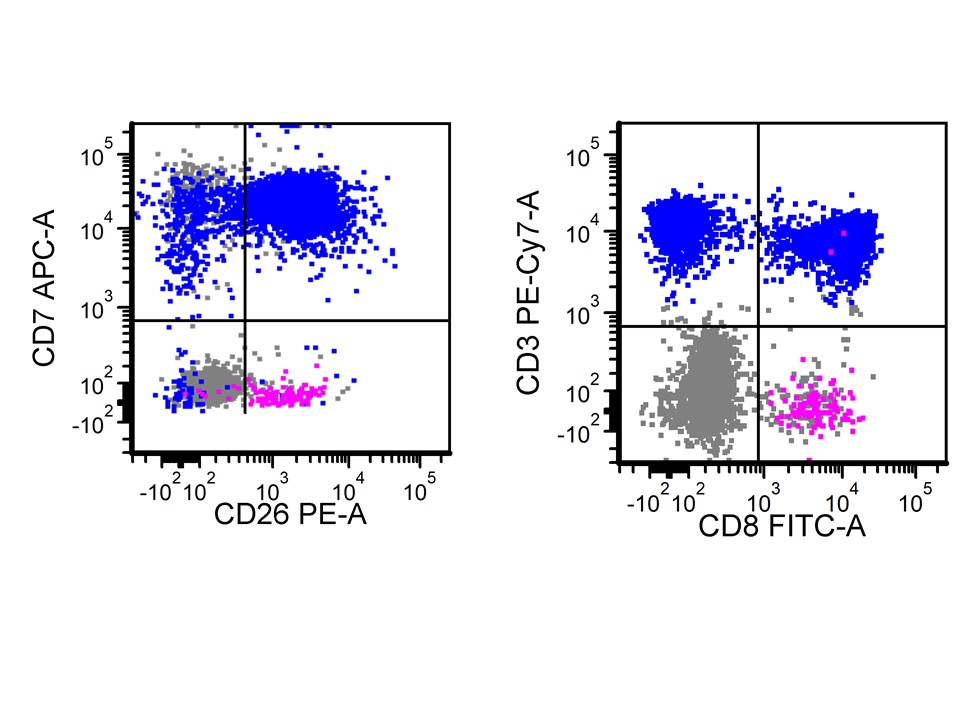
- Prominent forward scatter (FSC) (increased cell size when compared with nonneoplastic background lymphocytes)
- Consistently positive markers: CD2, cCD3 (cytoplasmic CD3), CD16, CD56, CD94
- Frequently positive markers: CD7, CD8
- Frequently negative markers: sCD3 (surface CD3), CD4, CD5, CD57, killer Ig-like receptors (KIR) (CD158a-e), TCRαβ and TCRγδ
- References: Leuk Lymphoma 2015;56:103, Cancers (Basel) 2020;12:2900, Am J Surg Pathol 2020;44:1235
Contributed by Siba El Hussein, M.D. and Joseph Khoury, M.D.
Flow cytometry characteristic
- Array based comparative genomic hybridization (aCGH) analyses can demonstrate nonspecific cytogenetic findings (Genes Chromosomes Cancer 2005;44:247):
- Gains of 1q23.1-q23.2 and 1q31.3-q44
- Losses of 7p15.1-q22.3 and 17p13.1
- Whole genome and exome sequencing and next generation sequencing have shown mutations in the following pathways (Am J Surg Pathol 2020;44:1235, Nat Commun 2018;9:1567, Cell Res 2018;28:172):
- JAK / STAT (STAT3, STAT5B, STAT5A, JAK2, JAK3, STAT6, SOCS1, SOCS3 and PTPN11)
- RAS / MAPK
- Epigenetic modifiers (TET2, CREBBP, KMT2D, BCOR, SET2D, GFI1)
- RNA helicase (DDX3X)
- Cell cycle regulation and DNA damage repair (TP53, ASXL1, ASXL2, BRINP3)
- mRNA splicing (PRPF40B)
- Bone marrow, posterior iliac crest, core biopsy, clot section, aspirate smears and touch imprint:
- Aggressive NK cell leukemia (ANKL) (see comment)
- Comment: Hypocellular bone marrow for age 20 - 30%, with residual trilineage hematopoiesis
- Flow cytometry immunophenotypic studies are performed using bone marrow aspirate material of the specimen. A distinct population of aberrant NK cells is detected. The neoplastic cells account for 49% of all analyzed cells. The neoplastic cells are positive for CD2, cytoplasmic CD3, CD7 (bright), CD45 (bright), CD48 (partial), CD56 and CD158b. The neoplastic cells are negative for CD1a, surface CD3, CD4, CD5, CD10, CD8, CD13+CD33, CD19, CD25, CD30, CD34, CD117, CD123, CD158a, CD158e, TdT, TCRαβ, TCRγδ, myeloperoxidase and HLA-DR.
- Bone marrow biopsy: quality - suboptimal, subcortical; cellularity - 20 - 30% in the most cellular areas; megakaryocytes - significantly decreased, with unremarkable morphology; infiltrate - numerous medium sized cells with dense chromatin and scant cytoplasm in interstitial distribution
- Bone marrow clot section: quality - adequate, particles present; cellularity: 30 - 40%; megakaryocytes - present; infiltrate - numerous medium sized cells with dense chromatin and scant cytoplasm in interstitial distribution
- Stains on clot: Immunohistochemical stains were performed using fixed, paraffin embedded tissue of clot specimen. The neoplastic cells are positive for CD2, CD56 and p53. In situ hybridization studies for Epstein-Barr virus encoded small RNA (EBER) using fixed, paraffin embedded tissue of clot specimen were performed. The neoplastic cells are strongly positive for EBER.
- Bone marrow aspirate: quality / cellularity - adequate, particles present; granulocytes - complete maturation; erythrocytes - complete maturation; megakaryocytes - present; lymphocytes - markedly increased, predominantly medium size, with open nuclear chromatin, inconspicuous nucleoli and moderate amount of deep basophilic cytoplasm with vacuoles
- Extranodal NK / T cell lymphoma (ENKTL):
- Strong association with EBV infection
- Cytokine secretion function
- Overlapping morphologic and genetic features with ANKL
- Less aggressive clinical presentation and outcome
- Predilection to involve upper aerodigestive tract (most commonly nasal cavity), gastrointestinal tract, skin, soft tissue and testes
- Chronic lymphoproliferative disorder of NK cells (CLPD-NK):
- Unknown stimulus, possibly viral
- No EBV association
- Cytotoxic function
- Uniform CD8 positivity
- Loss of CD2 expression
- Less aggressive clinical course
- Patients are mostly asymptomatic or have symptoms related to cytopenias (Eur J Haematol 2018;100:444)
Which of the following statements is true?
- Aggressive NK cell leukemia (ANKL) and extranodal NK / T cell lymphoma (ENKTL) overlap morphologically and genetically
- ANKL harbors specific cytogenetic features
- ANKL is characteristically negative for sCD3 and cCD3 by flow cytometry analysis
- EBER negativity rules out the diagnosis of ANKL
Comment Here
Reference: Aggressive NK cell leukemia
- Aggressive NK cell leukemia (ANKL) tumor burden in bone marrow sample is invariably high
- Cytologic atypia in ANKL is consistently prominent
- Prognosis of ANKL is remarkably ameliorated posttransplant and chemotherapy
- The most commonly involved genetic pathway in ANKL is the JAK / STAT pathway
Comment Here
Reference: Aggressive NK cell leukemia
- Alpha thalassemia is a group of inherited blood disorders characterized by reduced or absent production of α-globin subunits, resulting in low levels of hemoglobin, decreased mean corpuscular volume (MCV) and decreased mean corpuscular hemoglobin (MCH)
- Group of inherited autosomal recessive diseases caused by an α-globin chain synthesis defect
- There are 4 clinical pictures of α-thalassemia, according to the number of genes affected by loss of function with hemoglobin Bart's hydrops fetalis (Hb Bart's) syndrome and HbH disease being clinically significant
- Also classified as α-thalassemia minima (heterozygous α+-thalassemia, -α/αα) and α-thalassemia minor (heterozygous α0-thalassemia, –/αα or homozygous α+-thalassemia, -α/-α) (Dtsch Arztebl Int 2011;108:532)
- ICD-10: D56.0 - alpha thalassemia
- 15% of American blacks are silent carriers for α-thalassemia and about 3% have α-thalassemia trait (Medscape: Alpha Thalassemia [Accessed 23 April 2019])
- In Southeast Asian and Mediterranean populations, HbH disease and Hb Bart’s syndrome (γ4) are common
- The adequacy of the oxygen transport system depends on the affinity of hemoglobin for oxygen
- In adults, HbA is the major hemoglobin (97%), composed of 2 α subunits and 2 β subunits (α₂β₂) with minor amount of HbA2 (approximately 1.5 - 3.5%; α2δ2) and HbF (approximately < 1%; α2γ2)
- 2 α-globin genes are located on each chromosome 16, resulting in 4 α-gene loci (αα/αα)
- Severity of α-thalassemia depends on the number of inactivated or deleted alpha loci
- When both α-globin genes on a chromosome are deleted or inactivated, the allele is referred to as α0 (no output of α-globin from the chromosome)
- An individual with the genotype --/αα is referred to as an α0 carrier (GeneReviews 2005: NBK1435)
- This is common in Southeast Asia and Mediterranean but rare in African Americans
- When 1 α-globin gene on a chromosome is deleted or inactivated by a nondeletion variant, the allele is called α+ (some α-globin is produced)
- An individual with the genotype -α/αα is referred to as an α+ carrier (GeneReviews 2005: NBK1435)
- This is common in African Americans
- α-thalassemia is usually inherited in an autosomal recessive manner



- There are 4 α-thalassemia syndromes, according to the number of genes affected, correlating with different clinical pictures
- Hb Bart's hydrops fetalis syndrome: complete absence of all 4 α chains
- Because of the absence of α chains, no HbA or HbF is present (GeneReviews 2005: NBK1435)
- Large amount of Hb Bart’s, a variable amount of Hb Portland and traces of HbH present
- Red cells with Hb Bart's have an extremely high oxygen affinity and are incapable of effective oxygen delivery
- Incompatible with life, fetuses are still born with severe anemia, marked edema and hepatosplenomegaly
- HbH disease: absence of 3 α chains
- There is chronic hemolytic anemia, mild jaundice and hepatosplenomegaly
- Most individuals are clinically well and survive without treatment (GeneReviews 2005: NBK1435), transfusion is rarely needed
- α-thalassemia trait: absence of 2 α chains either in cis (--/αα, α0 carrier) or in trans (-α/-α) (GeneReviews 2005: NBK1435)
- Benign condition with most patients diagnosed on routine screening
- Does not require treatment
- α-thalassemia silent carrier: absence of 1 α chain
- No clinical abnormalities
- Hb Bart's hydrops fetalis syndrome: complete absence of all 4 α chains
- Electrophoresis or high performance liquid chromatography (HPLC):
- Hb Bart's hydrops fetalis syndrome: Hb Bart’s migrates faster than HbA on alkaline electrophoresis
- Hb Bart’s > 80%, a variable amount of Hb Portland and HbH present (See figure)
- No HbA
- HbH disease: HbH migrates faster than HbA on alkaline electrophoresis (See figure) (Indian J Clin Biochem 2010;25:435)
- HbH > 15% at birth (GeneReviews 2005: NBK1435)
- Normal or decreased HbA2
- α-thalassemia trait:
- Hb Bart’s in newborns (up to 20%) (See figure)
- Normal electrophoresis in adults and the diagnosis is made by excluding iron deficiency, anemia of chronic disease and beta thalassemia
- Normal HbA2 and HbF (GeneReviews 2005: NBK1435)
- α-thalassemia silent carrier:
- Hb Bart’s in newborns (up to 2%)
- Normal electrophoresis in adults and diagnosis is made by molecular or globin chain synthesis studies
- Hb Bart's hydrops fetalis syndrome: Hb Bart’s migrates faster than HbA on alkaline electrophoresis
- Hb Bart's hydrops fetalis syndrome:
- CBC: severe microcytic hypochromic anemia and reticulocytosis
- Hb Bart’s > 80%, HbH and Hb Portland
- HbH disease:
- CBC: decreased MCV and MCH, reticulocytosis (4 - 5%), increased RBCs
- Hb Bart’s:
- 20 - 40% at birth
- 5 - 30% in adults
- α-thalassemia trait:
- CBC: may show mild hypochromic (low MCH), microcytic (low MCV) anemia (GeneReviews 2005: NBK1435)
- Hb Bart’s:
- 2 - 10% in neonate
- None in adults
- α-thalassemia silent carrier:
- CBC: either normal or mild reduction of MCV and MCH (GeneReviews 2005: NBK1435)
- Hb Bart’s:
- 1 - 2% in neonates
- None in adults
- Hb Bart's hydrops fetalis syndrome is suspected in fetuses with increased nuchal thickness, thickened placenta, increased cerebral media artery velocity and increased cardiothoracic ratio on ultrasonography examination at 13 to 14 weeks' gestation (GeneReviews 2005: NBK1435)
Images hosted on other servers:
Hb Bart’s, midpregnancy sonographic features
- Neonate with α-thalassemia major (Pediatr Dev Pathol 2019;22:166)
- 16 year old boy with co-inheritance of heterozygous α+-thalassemia and sickle cell trait (BMC Ophthalmol 2017;17:6)
- 22 year old woman with HbH disease (Biomed Rep 2016;5:23)
- 28 year old Chinese woman with α-thalassemia trait (J Med Case Rep 2015;9:58)
- 36 year old Chinese woman with HbH disease (Case Rep Med 2018;2018:8057045)
- Hb Bart's syndrome is a universally fatal condition and death usually occurs in the neonatal period (GeneReviews 2005: NBK1435)
- Most individuals with HbH disease, thalassemia trait and carriers are clinically well and survive without any treatment (GeneReviews 2005: NBK1435)
- Hb Bart's hydrops fetalis syndrome:
- Large, hypochromic red cells and severe anisopoikilocytosis (GeneReviews 2005: NBK1435) (See figure)
- HbH disease:
- Hypochromia, basophilic stippling, target cells, anispoikilocytosis
- Red blood cell supravital stain show HbH inclusions (β4 tetramers) (GeneReviews 2005: NBK1435)
- Hypochromia, basophilic stippling, target cells, anispoikilocytosis
- α-thalassemia trait:
- Hypochromia and microcytosis
- HbH inclusions found in α0-thalassemia but rarely in α+-thalassemia
- Hypochromia and microcytosis



- Electrophoresis report:
- Features suggestive of α-thalassemia, if other causes of microcytosis are excluded
- Molecular report:
- α-globin (HBA1 and HBA2) deletion / duplication
- Deletion:
- Result: 2 pathogenic deletions detected in the α-globin gene cluster
- DNA variant(s):
- Pathogenic deletion: -α3.7
- Heterozygous pathogenic deletion: -α4.2
- Heterozygous predicted genotype: -α/-α
- Interpretation: 1 copy of the 3.7 Kb α-globin gene deletion and 1 copy of the 4.2 Kb α-globin gene deletion were detected by deletion / duplication analysis of the α-globin gene cluster and its HS-40 regulatory region. This individual is predicted to have a single functional α-globin gene on both chromosomes. This result is consistent with α0 thalassemia (α-thalassemia trait) often associated with mild anemia and microcytosis
-
A 30 year old man presents to his physician for partner screening. Routine laboratory studies are shown:
- α-thalassemia trait
- β-thalassemia trait
- α-thalassemia silent carrier
- δβ-thalassemia
- Hemoglobin S trait
| Test Patient Result | Reference Range | |
| WBC | 6.3 | 2.5 - 7.5 x10 |
| RBC | 5.5 | 4.6 - 5.7 x 10 |
| Hemoglobin | 12 | 13 - 18 g/dL |
| Hematocrit | 36 | 39 - 54% |
| MCV | 65 | 83 - 99 fL |
| MCH | 21.8 | 27 - 30 pg |
| MCHC | 33 | 30.8 - 34.6% |
| Platelet count | 260 | 200 - 400 x10 /mm |
| RDW | 13.8 | < 14 % |
| Ferritin | 112 | 29 - 248 ng/dl |
| Serum ferritin | 104 | 20 - 300 μg/L |
| Hemoglobin A2 | 2.4 | 2.3 - 3.5% |
| Hemoglobin F | 0.6 | 0.5 - 1% |
| Hemoglobin A | 97.0 | 96.8 - 97.8% |
Which one of the following is most likely diagnosis?
Comment Here
Reference: Alpha thalassemia
- Which of the following is associated with deletion of 3 α genes?
- Hemoglobin Bart's hydrops fetalis syndrome
- Hemoglobin H disease
- α-thalassemia trait
- α-thalassemia silent carrier
Comment Here
Reference: Alpha thalassemia
- Which of the following is highly incompatible with life?
- Hemoglobin H disease
- Hemoglobin Bart’s hydrops fetalis syndrome
- α-thalassemia trait
- α-thalassemia silent carrier
Comment Here
Reference: Alpha thalassemia
- Anaplasma phagocytophilia: human granulocytotropic anaplasmosis (HGA, formally termed human granulocytic ehrlichiosis)
- Vector borne disease transmitted through bite of Ixodes ticks
- Bacteria is obligate intracellular pathogen that binds to P selectin glycoprotein ligand 1 (PSGL1 / CD162)
- Susceptibility also associated with expression of CD15s (J Clin Invest 1999;103:407)
- First described in USA in 1994
- Geographic distribution of A. phagocytophilia (HGA) reflects regions of US where their hard tick vectors reside: northeastern states, northwest Wisconsin, eastern Minnesota and Pacific northwest states<
- Presents with fever, leukopenia, thrombocytopenia (70 - 90%) and elevated liver enzymes
- Mortality rate is 0.5 - 1% for HGA
- Particularly severe infections occur in elderly / immunocompromised
- Characteristic intracytoplasmic morulae (morula is Latin for mulberry): cytoplasmic membrane bound vacuoles with irregular edges containing hundreds to thousands of clustered gram negative bacteria
- Infected cells typically contain only 1 or 2 morulae although as many as 15 may be seen in immunosuppressed individuals
- Greatly variable percentage of peripheral blood films with detectable morulae in the literature (3 - 80%) with a higher number seen with HGA infection (50 - 80%) and in immunosuppressed individuals
- 43 year old woman presented with fever, chills and muscle aches after a tick bite (Case of the Month #486)
- 78 year old man with Anaplasma phagocytophilum infection and CML (J Clin Pathol 2004;57:499)
- 3 pancreas transplant recipients with HGA / human granulocytic ehrlichiosis (Transpl Infect Dis 2001;3:34)
- Most patients are seronegative during first few weeks of acute infection (60 - 97%), so therapeutic decisions must be based on clinical suspicion, peripheral blood findings and PCR (sensitivity is 60 - 85%, high degree of false positive results)
- Became a nationally reportable disease to US Centers for Disease Control in 1999
- Organisms are susceptible to tetracyclines and their derivatives, particularly doxycycline
- Peripheral blood: buffy coat examination may reveal intracytoplasmic inclusions (morulae - spherical structures with irregular edges) within neutrophils or monocytes
- Bone marrow: epithelioid granulomas; usually normo or hypercellular with intact trilineage maturation; rare hypoplasia; possible increased megakaryocytes
- Histopathologic bone marrow findings: inconsistent and likely to change during the course of the disease
- HGA organisms preferentially infect more mature rather than immature granulocytic cells in bone marrow
Contributed by Patricia Tsang, M.D.


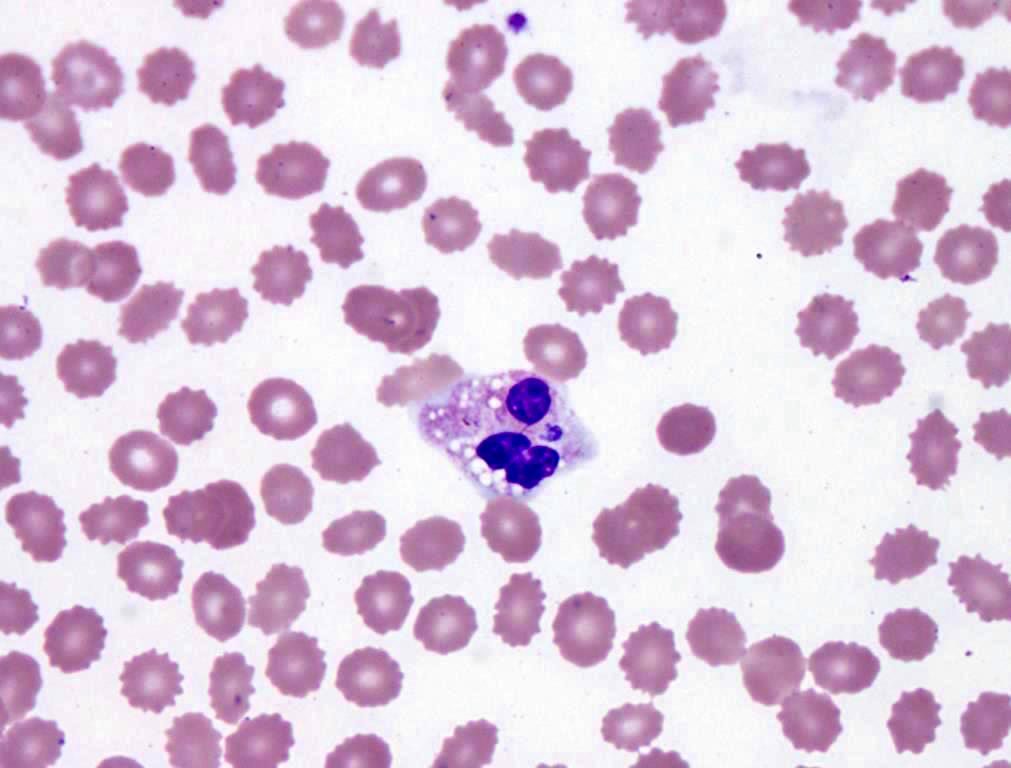
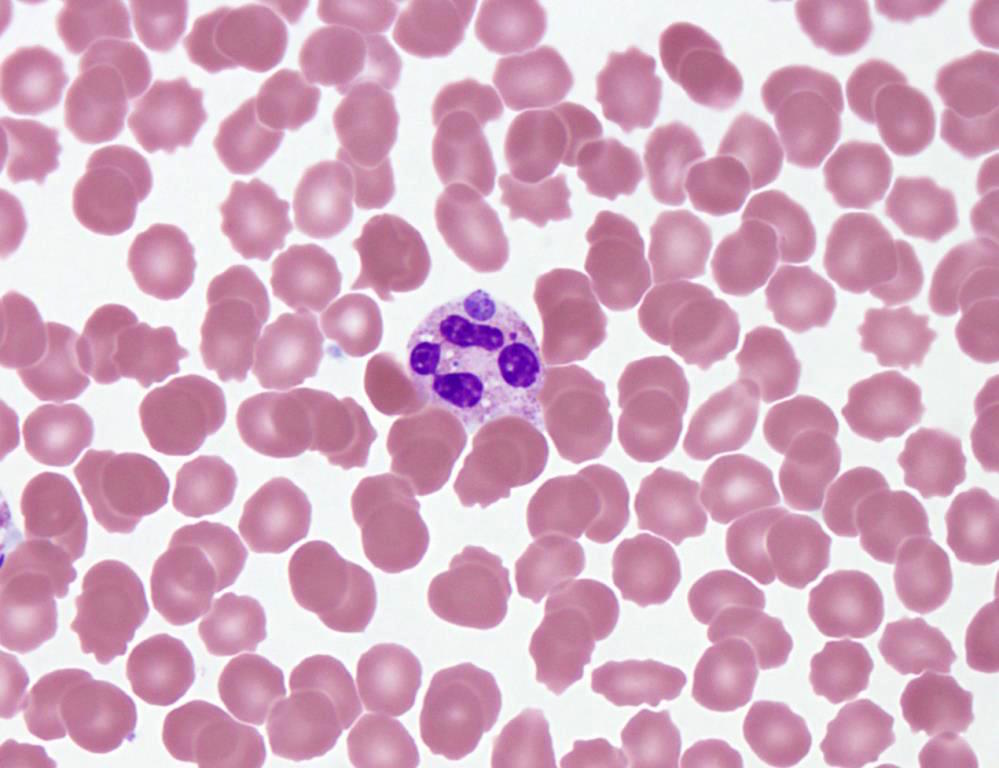
Case of the Month #486
Images hosted on other servers:
HGA: inclusions in granulocytes

Left: HGA (HGE)
A. Anaplasmosis is typically transmitted by exposure to respiratory droplets.
B. Immunocompromised patients traveling to endemic regions should be vaccinated against Anaplasma.
C. A common presentation for patients with anaplasmosis includes relapsing fevers, neutrophilia and reactive thrombocytosis.
D. A diagnostic feature of anaplasmosis is the presence of neutrophilic morulae.
E. Anaplasma and malaria are common coinfections.
Comment Here
Reference: Anaplasmosis
- Also called Canale-Smith syndrome
- First named in 1995 (Cell 1995;81:935)
- Inherited disorder due to defects in Fas/CD95/Apo-1 mediated apoptosis (OMIM 601859: Autoimmune Lymphoproliferative Syndrome [Accessed 28 June 2018])
- Childhood onset of lymphadenopathy, hepatosplenomegaly, hypergammaglobulinemia and autoimmunity; also cytopenias and increased risk of lymphoma (Hematology 2006;11:15)
- 40% have histologic features of sinus histiocytosis with massive lymphadenopathy (Am J Surg Pathol 2005;29:903)
- Type I: functional defects of FAS gene
- Type II: functional FAS deficiency but no FAS gene mutations
- 6 month old girl with cervical lymphadenopathy, hepatosplenomegaly and gamma/delta+ T cell blasts (Am J Surg Pathol 2003;27:546)
- 4 year old girl with type II syndrome and clonal immunoblasts but no evidence of lymphoma (Am J Surg Pathol 1999;23:829)
- 11 year old girl with case due to FasL mutation (Blood 2006;108:1306)
- Paracortical hyperplasia, expanded interfollicular areas; polyclonal plasmacytosis (Am J Pathol 1998;153:1541)
- Frequent mitotic figures
- Often florid follicular hyperplasia with focal progressive transformation of germinal centers
- Occasional follicular involution
- Mutation in death domain of FAS gene
- Babesia spp. are protozoan parasites that infect red blood cells
- Taxonomy:
- Phylum: Apicomplexa
- Order: Piroplasmida
- Family: Babesiidae
- Babesiosis is caused by Babesia spp., transmitted via tick bites (most commonly, Ixodes) (Pathogens 2021;10:1447)
- Infection of erythrocytes leads to hemolytic anemia and cytokine production causing fever, jaundice, hepatosplenomegaly and, in severe cases, multiorgan failure (Pathogens 2022;11:399)
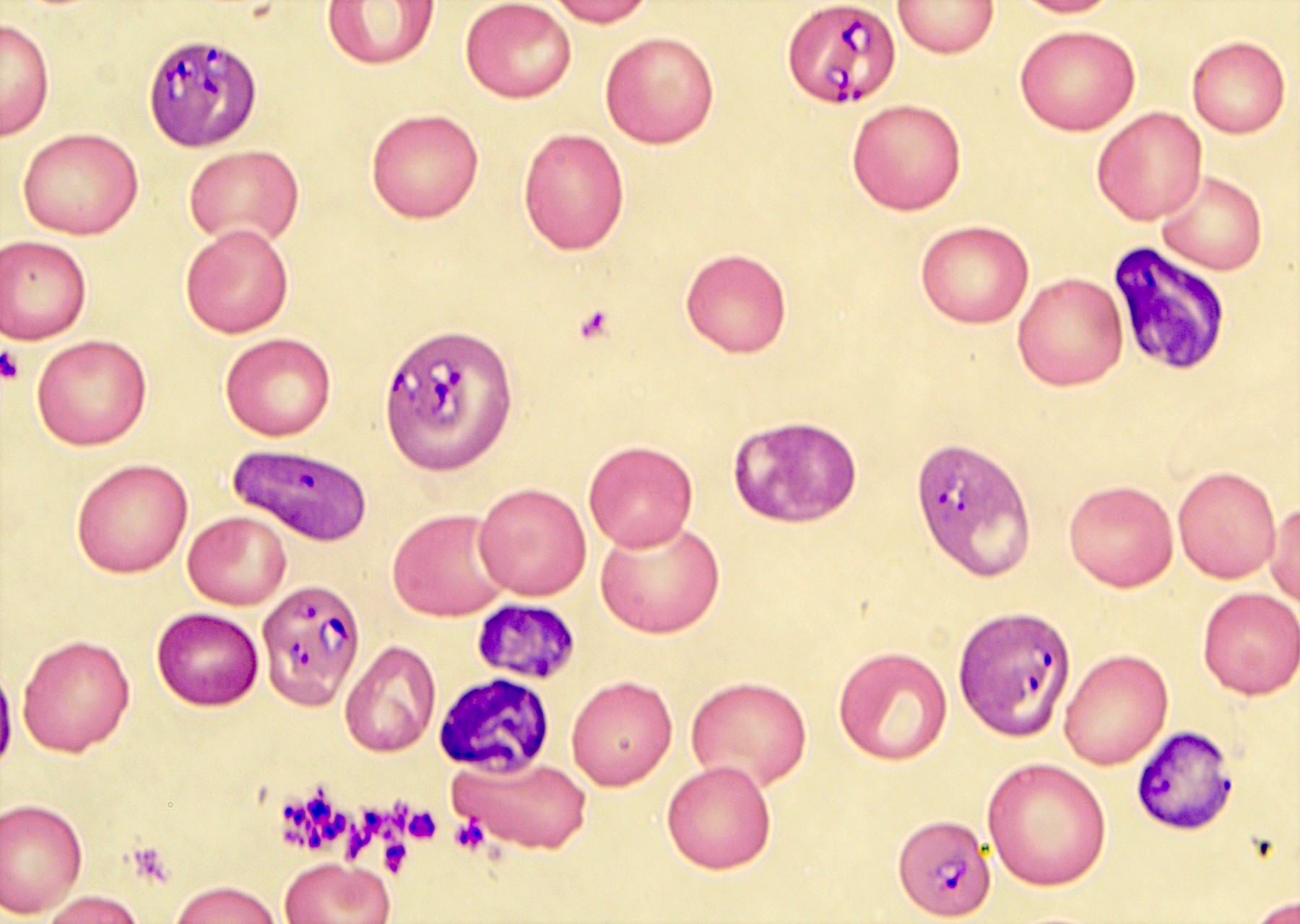
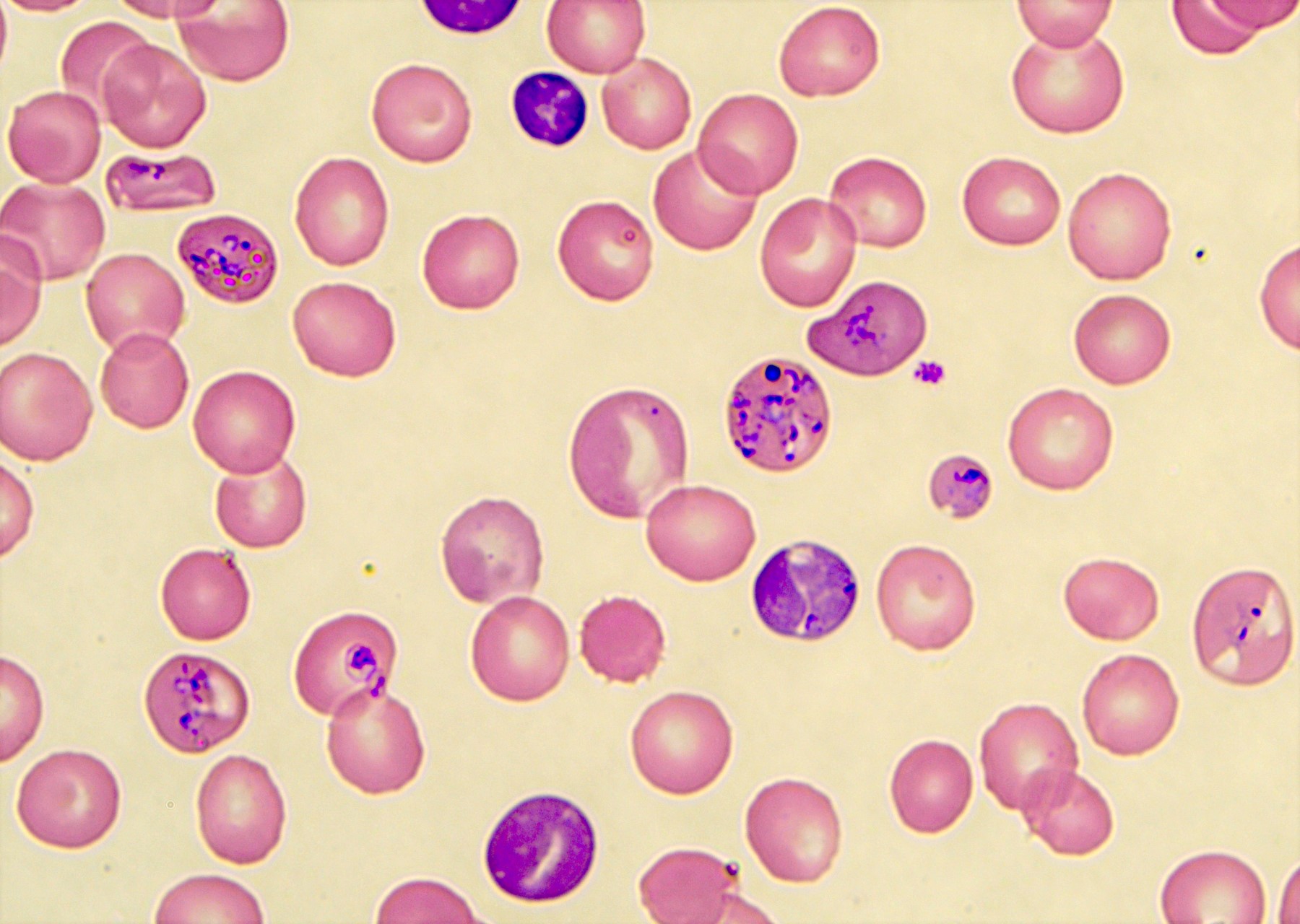
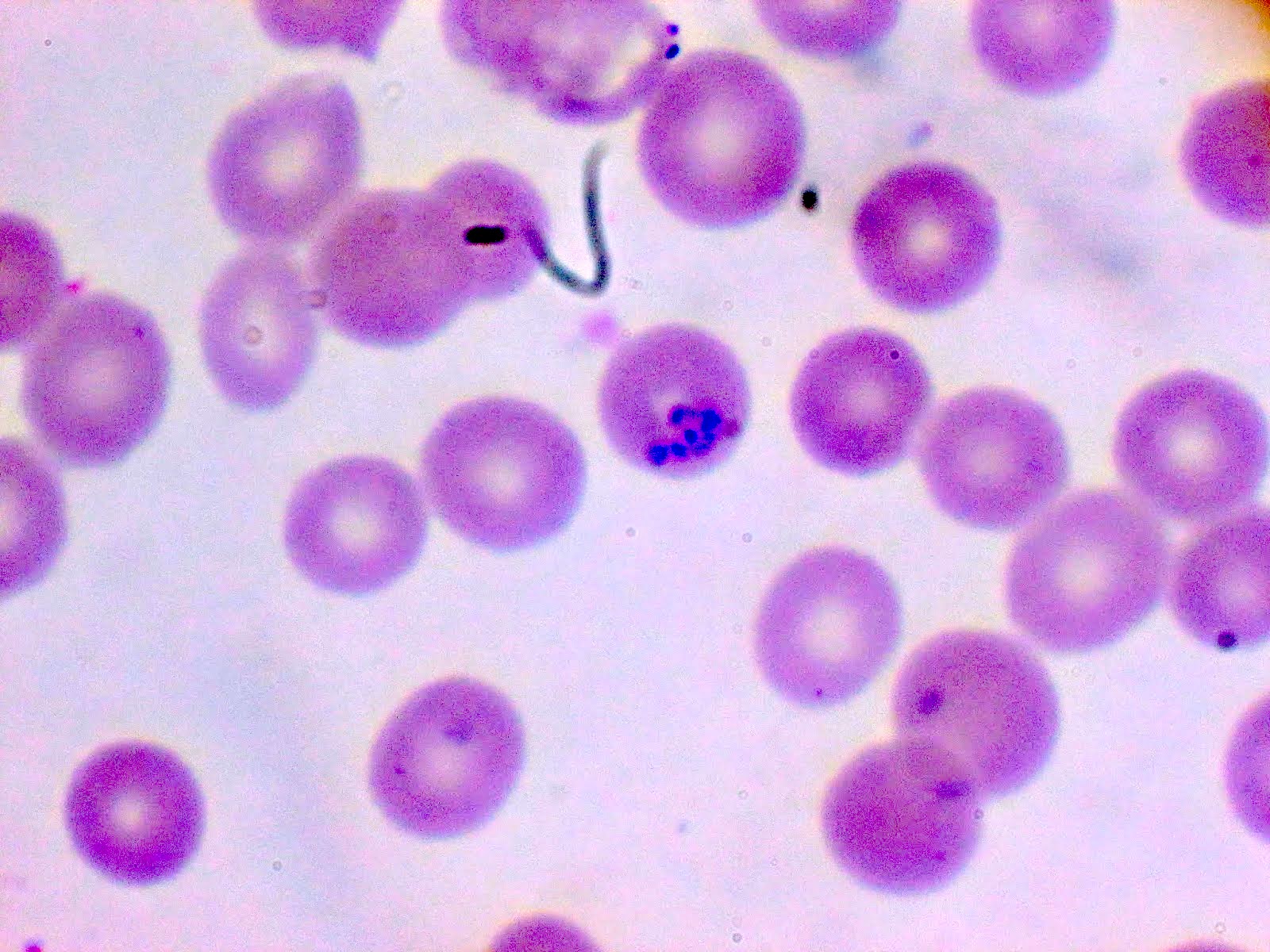
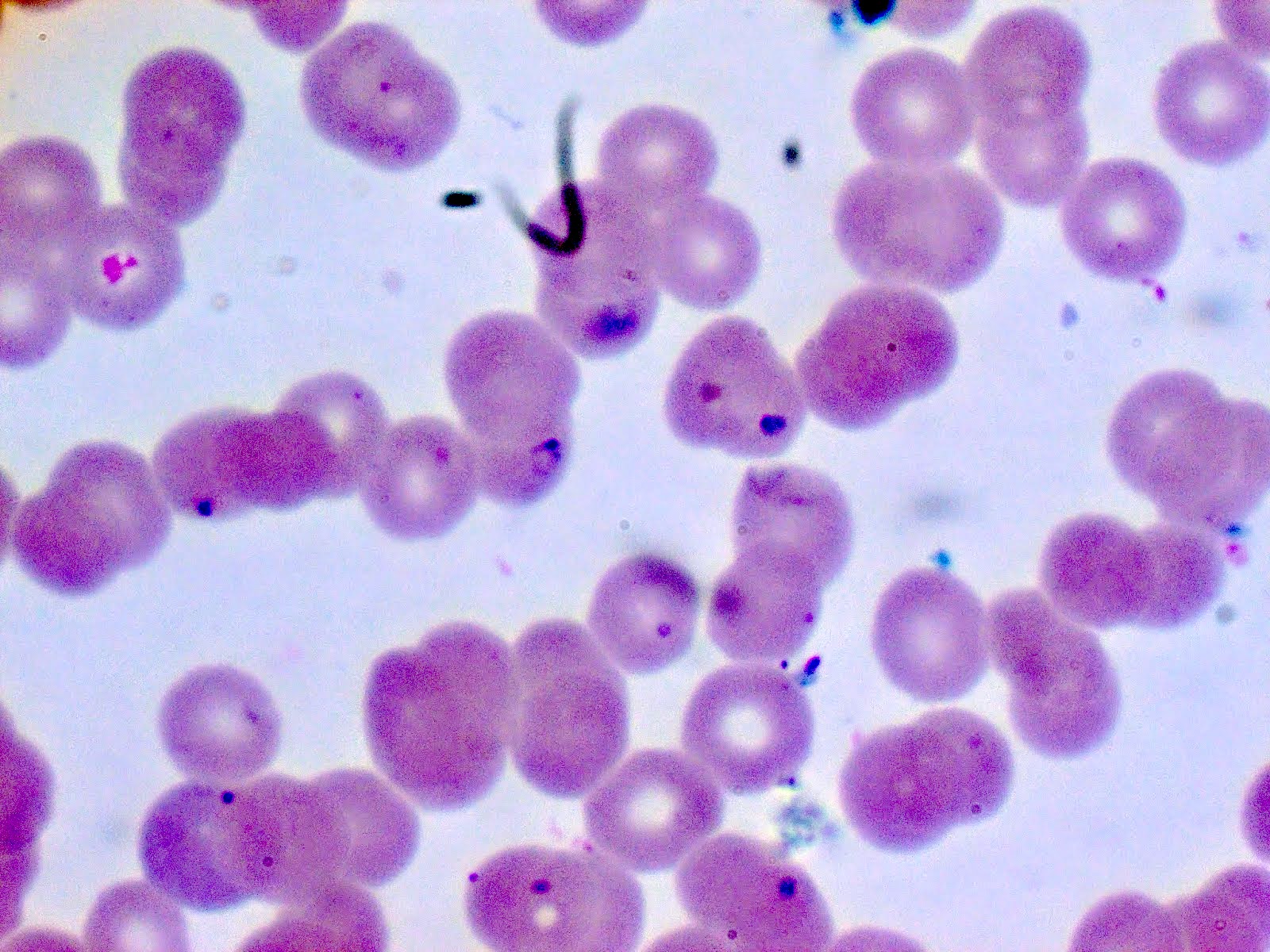
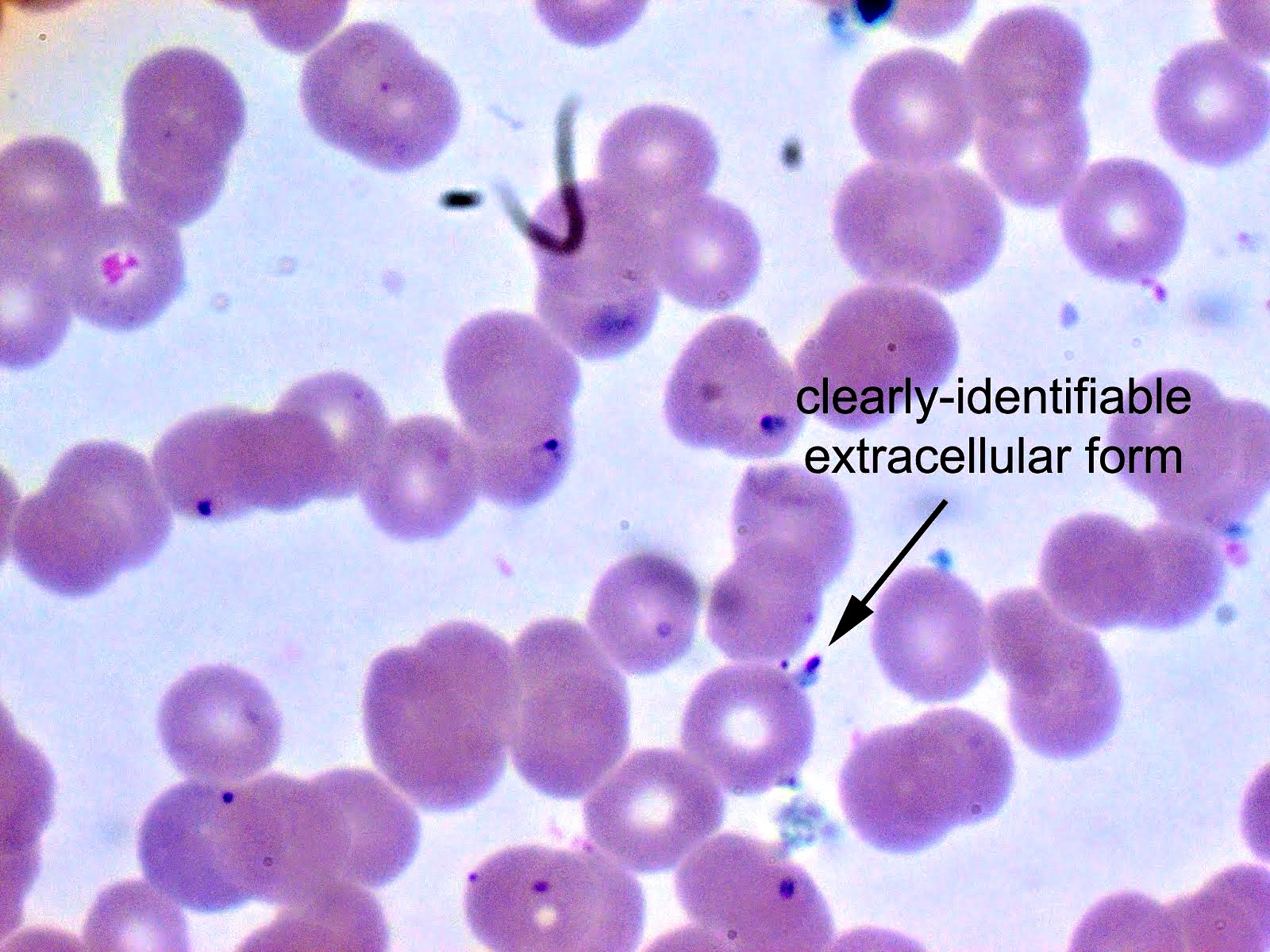
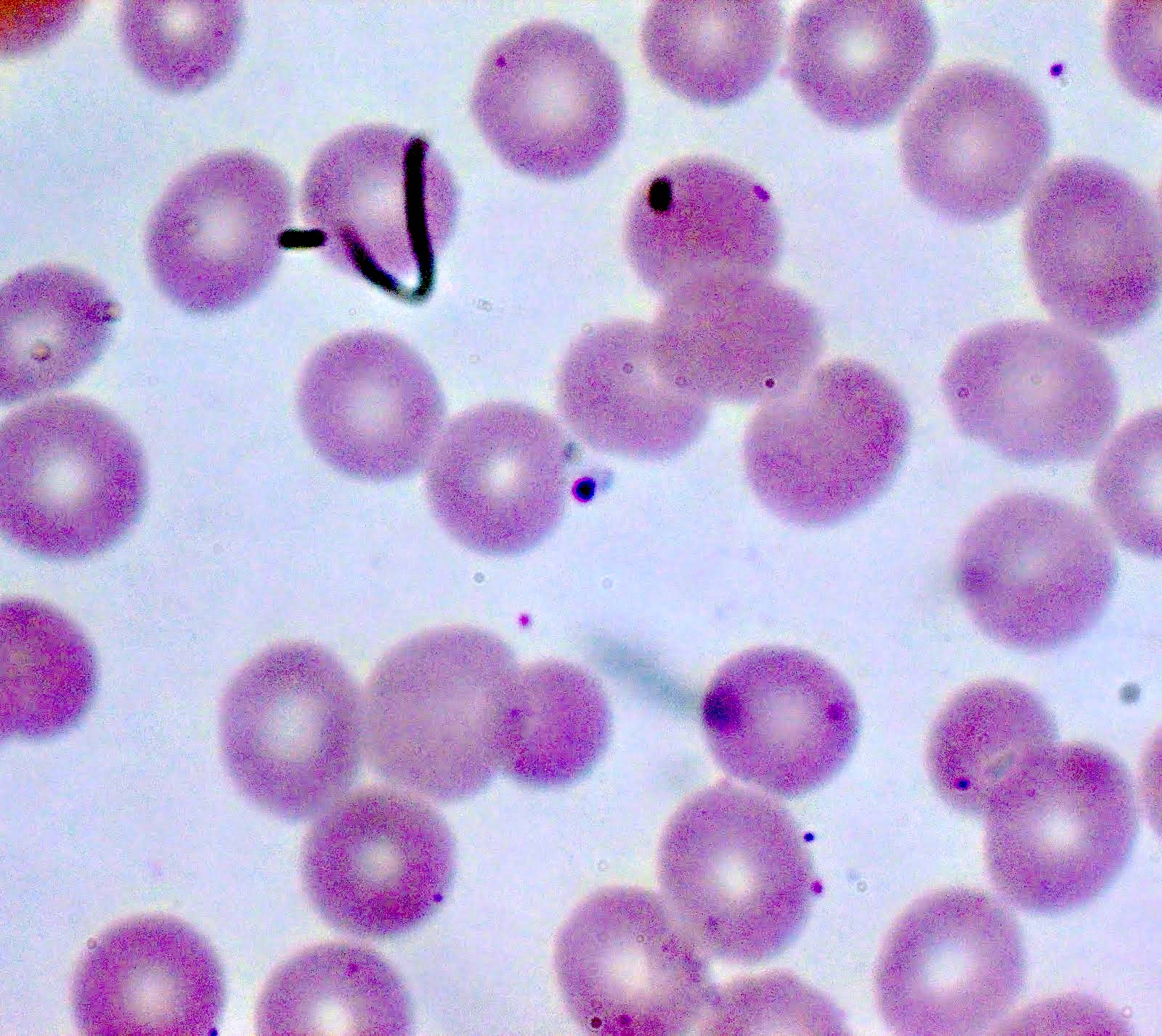
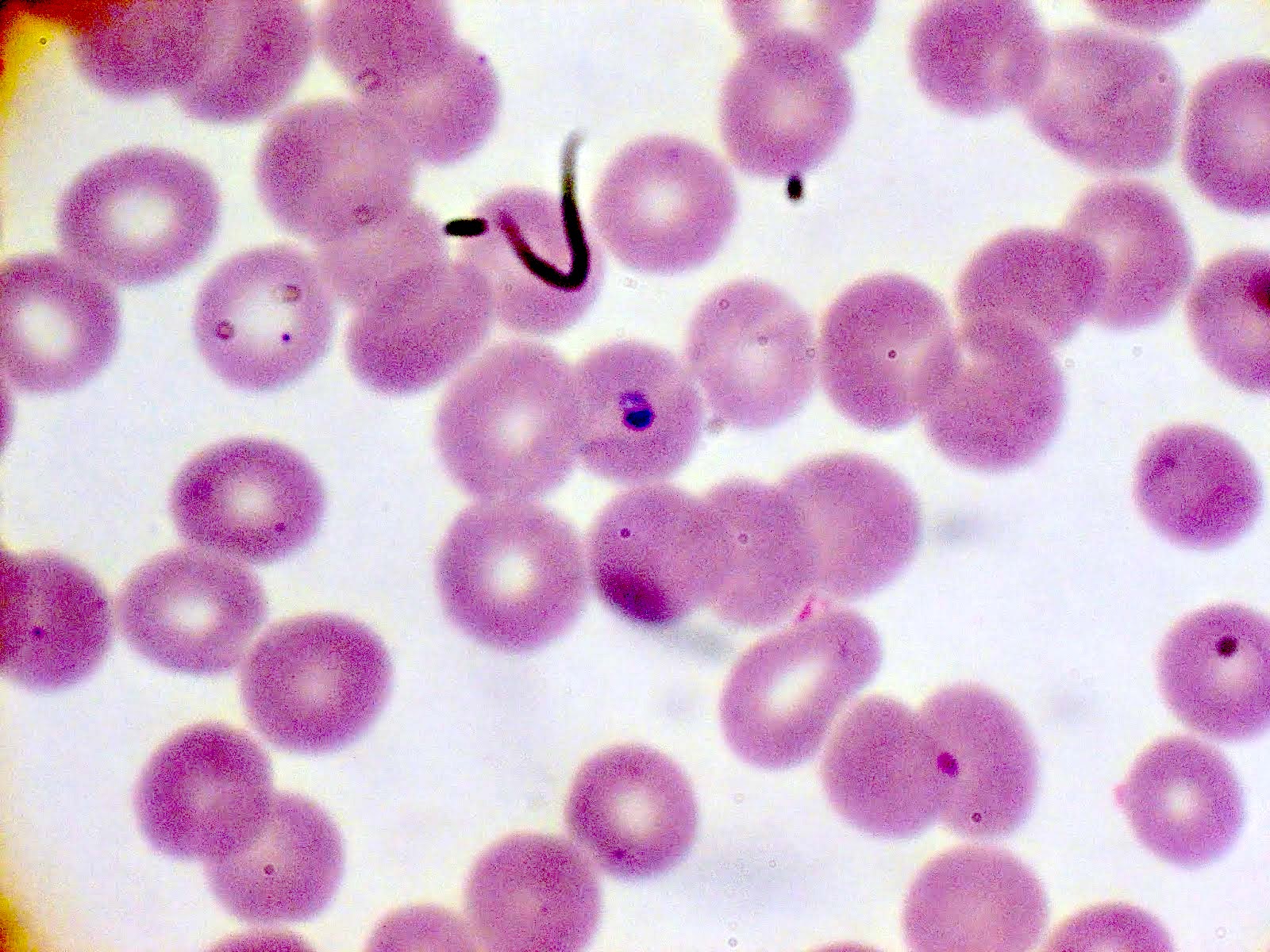
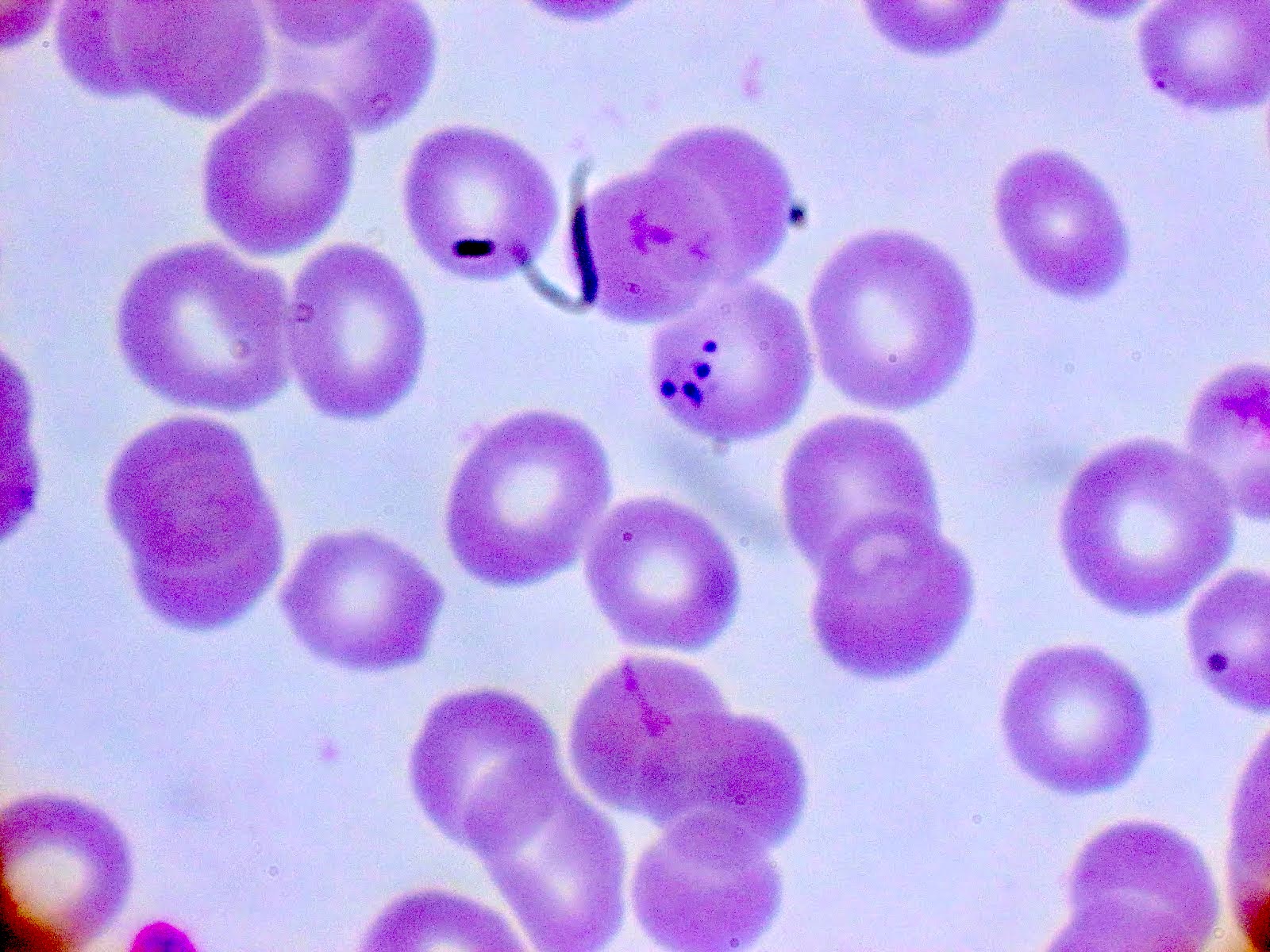
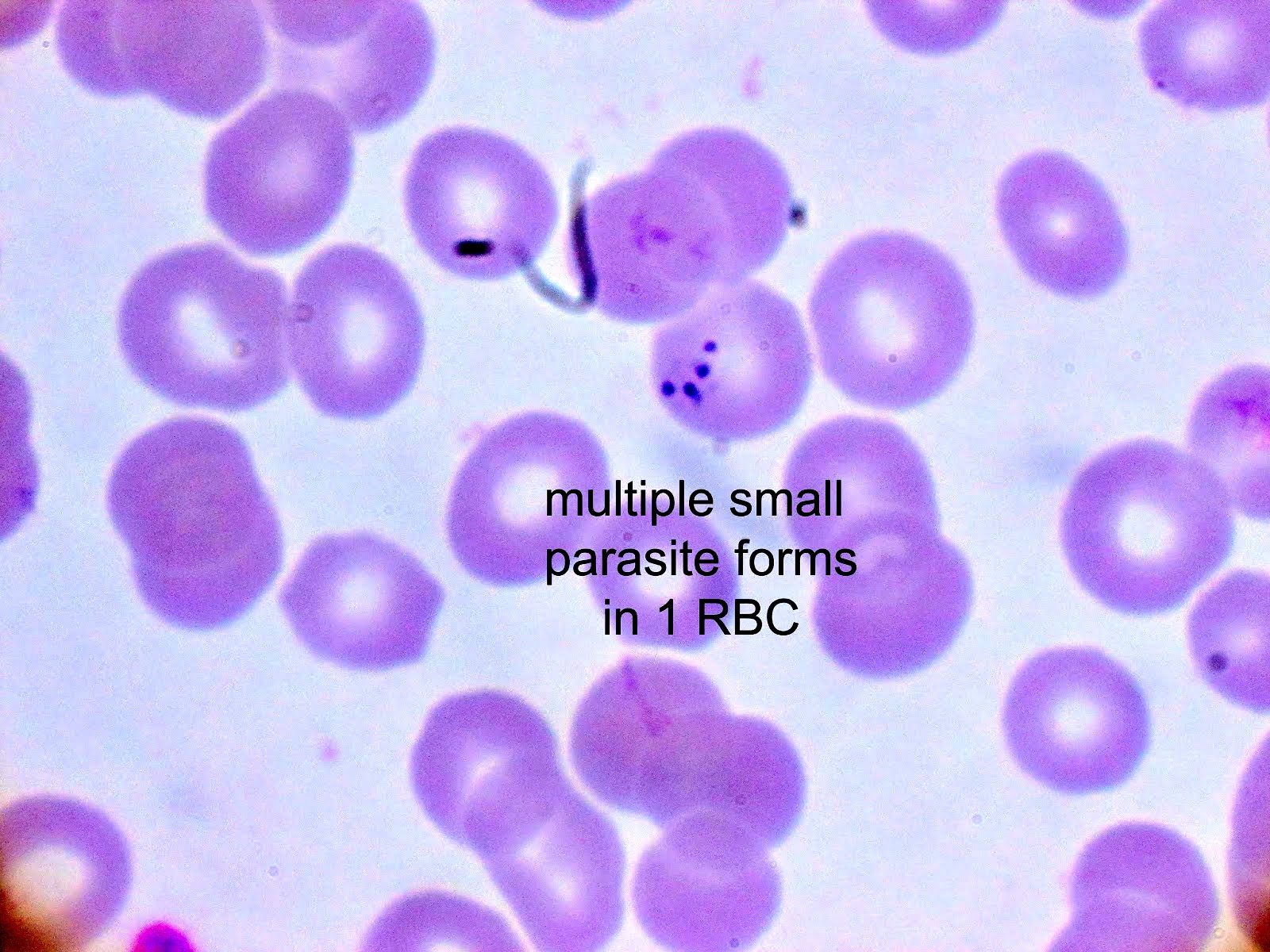
- Diagnosis is made upon seeing multiple infected red cells with extracellular ring forms on blood smear
- Classical blood smear finding is a tetrad of intracellular ring forms (Maltese cross) with extracellular ring forms
- Mild infections resolve spontaneously
- Moderate and severe manifestations may require treatment (moderate: atovaquone and azithromycin; severe: clindamycin, quinine, exchange transfusion)
- More than 70 species exist worldwide but most cases of babesiosis in the United States are due to B. microti
- In Europe, B. divergens is associated with more serious clinical syndrome (Pathogens 2021;10:1165)
- In the Northeastern U.S., Ixodes scapularis, commonly known as deer tick or black legged tick, is the main species of tick that transmits B. microti (Pathogens 2021;10:1447)
- Splenectomy, HIV infection, immunosuppression and advanced age increase the likelihood of severe infection (Pathogens 2022;11:399)
- Humans are incidental hosts; natural hosts include small rodents (voles, field mice, etc.) (Pathogens 2021;10:1447)
- Other modes of transmission include transfusion, organ transplantation and transplacental (Pathogens 2022;11:399)
- Co-infection with Anaplasma and Borrelia can occur, as both diseases are vectored by Ixodes scapularis (Pathogens 2021;10:1447)
- Parasite found in the blood
- Babesia parasites are maintained in animal tick cycles, where ticks have transovarian and stage to stage transmission (Trop Parasitol 2015;5:94)
- Cycle of human infection (Pathogens 2021;10:1447):
- A Babesia infected tick will inject sporozoites via saliva into the bloodstream in the form of pyriform bodies
- Trophozoites infect erythrocytes and asexually reproduce via binary fission
- Erythrocyte lysis releases merozoites that infect other erythrocytes or are taken up by feeding ticks
- Lysis of red cells leads to a cascade of inflammatory responses resulting in fever, malaise with more severe disease (i.e., disseminated intravascular coagulation [DIC], renal failure, shock) in patients with splenectomy, immunosuppression and advanced age (Pathogens 2022;11:399)
- Symptoms include fever without periodicity, malaise, headache, chills, fatigue, weakness
- Signs include hemolytic anemia, hepatosplenomegaly
- Diagnosis is made on thick and thin blood smear with Giemsa stain (gold standard)
- When parasitemia is low and in cases of screening (particularly for blood products), serological assays and antigen capture assays can be performed (Pathogens 2022;11:399)
- During acute disease, polymerase chain reaction (PCR) may be used for diagnosis in the form of organism specific Babesia spp. PCR or as part of larger tick borne disease PCR panels that include anaplasmosis, ehrlichiosis and babesiosis
- 37 year old man in Singapore who acquired Babesia microti infection in the U.S. (Emerg Infect Dis 2020;26:826)
- 66 year old man in the South Bronx presenting with febrile illness (Clin Pract Cases Emerg Med 2018;2:61)
- 70 year old woman presenting with asplenic sepsis (Turk J Haematol 2019;36:284)
- 81 year old man from upstate New York with fevers, malaise, vague abdominal pain and confusion (Proc (Bayl Univ Med Cent) 2020;34:97)
- Mild: resolves spontaneously
- Moderate: combination atovaquone and azithromycin
- Severe: can be treated with clindamycin, quinine, sometimes exchange transfusion
- Thick smear Giemsa stain:
- Red / purple chromatin dot with pale blue cytoplasm forming a ring
- Thin smear Giemsa stain (Pathogens 2022;11:399):
- Intracellular and extracellular ring forms
- Usually multiple forms within each infected red cell
- Maltese cross = classical finding of tetrad of intracellular ring forms
Contributed by Erika Wheeler, M.D. and Bobbi Pritt, M.D.
Intracellular and extracellular Babesia ring forms
Multiple Babesia ring forms

Giemsa stained thick and thin blood films
Giemsa stained thick and thin blood films
Images hosted on other servers:
Tetrad form
- PCR tests for Babesia DNA exist and can be used in cases of low parasitemia and blood product screening
- Plasmodium falciparum (Pathogens 2021;10:1165):
- Up to 2 intracellular ring forms
- No extracellular ring forms
- Can see banana shaped gametocytes
- Clinical history of travel to endemic area
- May exhibit periodicity in fever
- Other Plasmodium spp. (Pathogens 2021;10:1165):
- Red cells with normally 1 ring form, occasionally 2
- Will commonly see other forms: schizonts, gametocytes, etc.
- No extracellular ring forms
- Clinical history of travel to endemic area
- May exhibit periodicity in fever
- If there has been a known Ixodes tick exposure and general symptoms of fever and malaise, differential diagnoses should include the following (Trends Parasitol 2018;34:295):
- Lyme disease:
- No visible parasites on blood smear
- Serology and PCR studies positive for Borrelia burgdorferi, a spirochete bacterium
- Commonly co-infects with Babesia
- Tick borne relapsing fever:
- No visible parasites on blood smear
- Serology and PCR studies positive for Borrelia miyamotoi
- Anaplasmosis:
- No visible intraerythrocytic or extracellular trophozoites
- May have morula observed in granulocytes
- Serology and PCR studies positive for Anaplasma phagocytophilum, a tick borne bacterium
- Ehrlichiosis:
- No visible intraerythrocytic or extracellular trophozoites
- May have morula observed in monocytes or granulocytes
- Serology and PCR studies positive for Ehrlichia spp.
- Powassan virus disease:
- No visible parasites on blood smear
- Laboratory diagnosis by testing serum or cerebrospinal fluid for virus specific antibodies
- Lyme disease:
Which of the following is true about babesiosis?
- It causes more severe disease in asplenic patients
- It commonly co-infects with Plasmodium spp.
- It is endemic to sub-Saharan Africa
- The primary treatment is with doxycycline
Comment Here
Reference: Babesia
- Babesia microti
- Borrelia burgdorferi
- Plasmodium falciparum
- Plasmodium malariae
Comment Here
Reference: Babesia
- Indolent mature B cell lymphoma
- Clonal proliferation of B cells with characteristic phenotype
- Indolent mature B cell lymphoma
- Most common adult leukemia in the Western world
- Characteristic immunophenotype with CD19, CD5, CD20, CD23, CD200 positivity
- 2 types with distinct biological behavior based on mutation status of rearranged IGH gene
- Chronic lymphocytic leukemia (CLL): neoplasm of mature B cells with characteristic immunophenotype showing peripheral lymphocytosis (≥ 5 x 109/L) with or without nodal or extranodal manifestation (Blood 2016;127:2375)
- Monoclonal B cell lymphocytosis: clonal B cells with or without CLL-like immunophenotype in peripheral blood < 5 x 109/L and without nodal manifestation (see B cell monoclonal lymphocytosis)
- Small lymphocytic lymphoma (SLL): < 5 x 109/L CLL-like cells in peripheral blood with nodal or extranodal manifestation, usually with bone marrow involvement
- Most common adult leukemia in the Western world (Future Oncol 2017;13:1873)
- Median age is between sixth and seventh decade, practically nonexistent in children
- Male preponderance (M:F = 1.5 - 2:1)
- Incidence is low in Asian countries (Int J Oncol 2013;43:561)
- Lymph nodes, bone marrow, spleen and peripheral blood
- Usually widespread disease, clinical staging system is used
- Clonal restriction of specific B cell populations with reduced competition of remaining B cell clones due to aging
- B cell receptors show evidence of selection by antigens, B cell receptor signaling is crucial in survival of CLL cells (Front Oncol 2020;10:592205)
- Evidence for autoreactivity of B cell receptors (antigens recognized by studies are related to normal tissue maintenance, apoptosis, atherosclerosis, common infections, etc.) (Front Oncol 2020;10:592205)
- Some evidence indicating self stimulation by B cell receptors (Cell Res 2013;23:182)
- 30% of cases show stereotyped B cell receptors with identical or nearly identical structure (Leukemia 2019;33:287)
- 2 types of CLL that do not show conversion during disease course:
- CLL-UM (unmutated): few mutations in the IGH gene (≥ 98% homology with germline sequence), associated with more proliferation, more aggressive disease course
- CLL-MUT (mutated): many mutations in the IGH gene (< 98% homology), associated with less proliferation, better prognosis
- Gradual accumulation of genetic alterations, however, no clear malignant transformation event recognizable
- Incidence is not increased following radioactive incidents
- Familial predisposition in 5 - 10% cases
- Frequently no symptoms
- Lymphadenopathy, splenomegaly are common
- Anemia, thrombocytopenia and neutropenia related symptoms frequently occur
- Autoimmune hemolytic anemia is common (Best Pract Res Clin Haematol 2010;23:47)
- Extramedullary involvement may occur; liver, skin, GI mucosa, kidneys are most commonly involved
- Laboratory diagnosis of lymphocytosis
- Detection of generalized adenopathy or splenomegaly
- CT scans are helpful to detect lymph node enlargement
- Immunophenotyping is essential: flow cytometry of peripheral blood or bone marrow or immunohistochemistry of biopsy material
- Lymph node biopsy is not generally required, unless to establish diagnosis of Richter transformation (Blood 2018;131:2745)
- Lymphocytosis
- Anemia, thrombocytopenia are common
- Monoclonal gammopathy may occur
- Hypogammaglobulinemia commonly occurs (Blood 2015;126:573)
Images hosted on other servers:
CT scan
- Clinical stage (based on physical manifestations and blood parameters): Rai and Binet systems
- Beta-2 microglobulin: high levels associated with high tumor burden and adverse prognosis
- TP53 mutation / 17p deletion is adverse, is associated with poor response to traditional chemotherapeutic regimens; patients may respond to newer agents (J Oncol Pract 2017;13:371)
- Unmutated IGH gene is adverse
- Complex karyotype (10 - 15%, 3 or more unrelated abnormalities) is adverse (Blood 2019;133:1205)
- 13q deletion is favorable, 11q deletion is unfavorable, trisomy 12 is associated with intermediate prognosis
- CD38 (> 30% positive in CLL cells by flow cytometry) is adverse, ZAP70 expression (> 20%) is adverse
- CD49d (> 30% positive in CLL cells) has been associated with progression (Blood 2020;135:1244)
- Large, confluent proliferation centers (adverse) (Blood 2016;127:2375)
- Methylation profile, various gene mutations may correlate with epigenetic maturation status, response to therapy or prognosis but are not currently used in clinical practice (Nat Genet 2016;48:253)
- Pattern of bone marrow infiltration is no longer considered important
- 47 year old man with clonal evolution, treatment resistance and progression to Richter transformation (Haematologica 2019;104:e38)
- 58 year old man with CLL showing focal cyclin D1 expression (Arch Pathol Lab Med 2005;129:92)
- 66 year old man with Hodgkin transformation (Blood 2017;130:2151)
- 71 year old woman with Richter transformation after 6 years of complete remission of CLL (BMJ Case Rep 2016;2016:bcr2016214361)
- Treatment is tailored based on clinical progression (progressive bone marrow failure, progressive or bulky lymphadenopathy, short lymphocyte doubling time, etc.), biological fitness of patient, presence of TP53 mutation / 17p deletion and IGH mutation status (Curr Oncol Rep 2020;22:36)
- Cytostatic agents: chlorambucil, fludarabine, bendamustine, cyclophosphamide, etc.
- Monoclonal antibodies: rituximab, obinutuzumab (anti-CD20), alemtuzumab (anti-CD52)
- Kinase inhibitors: ibrutinib, acalabrutinib (Btk inhibitors), idelalisib (PI3K inhibitor)
- BCL2 inhibitors: venetoclax
- Lenalidomide
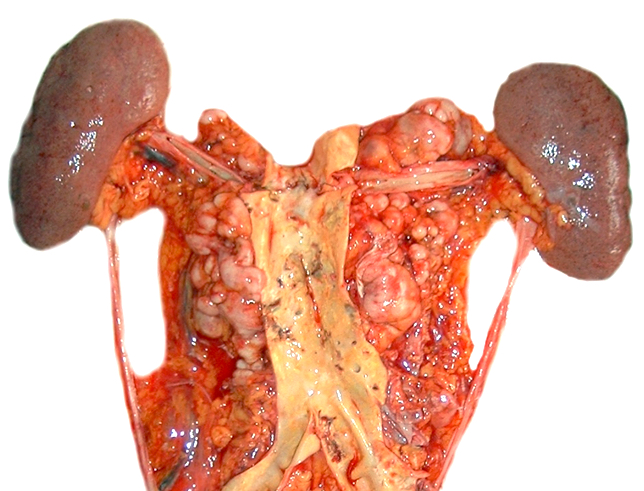
- Lymph node: enlarged lymph node with homogeneous, fleshy cut surface
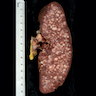
- Spleen: often miliary pattern of white pulp expansion, homogeneous infiltration with massive splenomegaly also occurs (Am J Clin Pathol 2003;120:335)
Contributed by Béla Kajtár, M.D., Ph.D.
Paraaortic lymph nodes
Images hosted on other servers:
Splenic involvement
- Diffuse infiltrate of small, mature lymphocytes with inconspicuous nuclei and scant cytoplasm
- Immunophenotyping is necessary for diagnosis
- Diffuse effacement of parenchyma by small, mature lymphocytes
- Round nucleus, clumped chromatin with only small nucleoli and scant cytoplasm
- Ill defined, pale proliferation centers (pseudofollicles) are common, composed of prolymphocytes and paraimmunoblasts
- Bone marrow: nodular or diffuse infiltration; paratrabecular aggregates are not common
Notes:
- Occasionally extensive plasmacytoid differentiation
- Partial lymph node infiltration is possible with perifollicular or interfollicular infiltration (Haematologica 2011;96:1144)
- Prolymphocyte: small to medium sized mature lymphocyte with clumped chromatin, somewhat larger central nucleolus
- Paraimmunoblast: large cell with round nucleus, dispersed chromatin and central, enlarged nucleolus, often with basophilic cytoplasm
- No grading system is used, prolymphocytic transformation is defined based on peripheral blood
- Histologically aggressive CLL: confluent or very large proliferation center or > 40% Ki67 index; not the equivalent of Richter transformation (Blood 2018;131:2761)
- Richter transformation: usually high grade B cell lymphoma (DLBCL) appearing in the context of CLL; confluent areas of large cells are required for diagnosis
- Hodgkin transformation: rare form of Richter transformation, usually clonally unrelated and EBV positive, requires classical Hodgkin lymphoma pattern; scattered Reed-Sternberg cells may be present in CLL (Blood 2018;131:2761)
- Prolymphocytic transformation: ratio of prolymphocytic cells in peripheral blood increases (< 55%), transformation into B cell prolymphocytic leukemia does not occur, by definition
Contributed by Béla Kajtár, M.D., Ph.D.

Proliferation centers
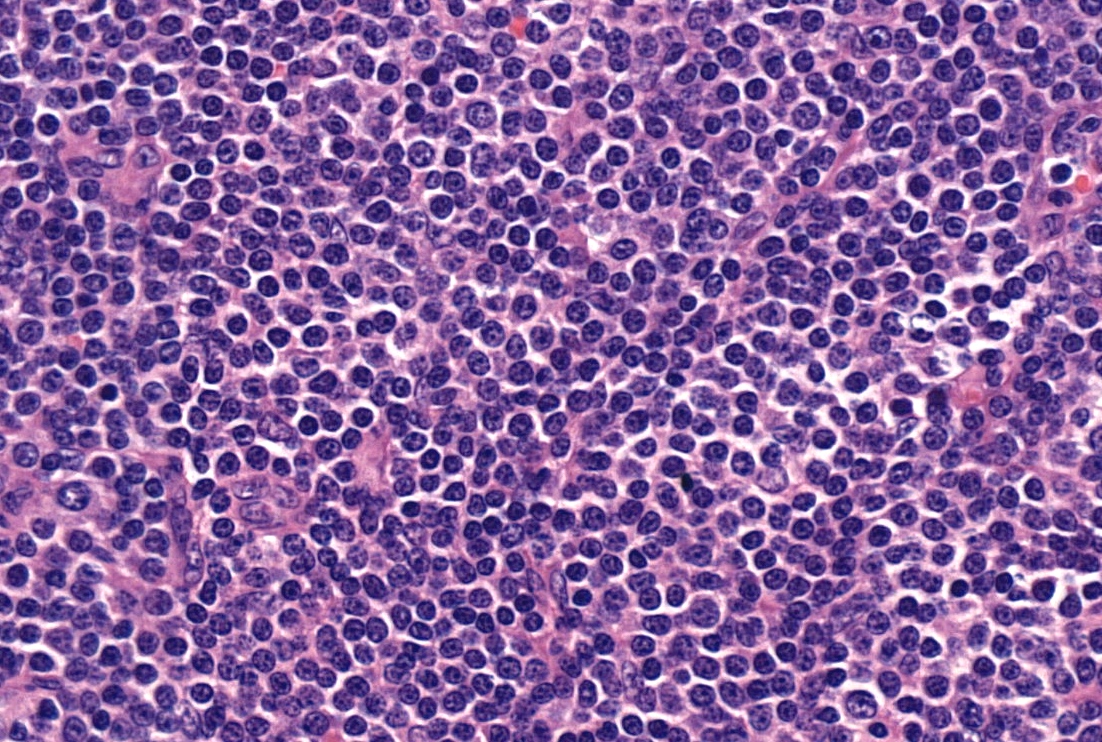
Diffuse lymphocytic infiltrate
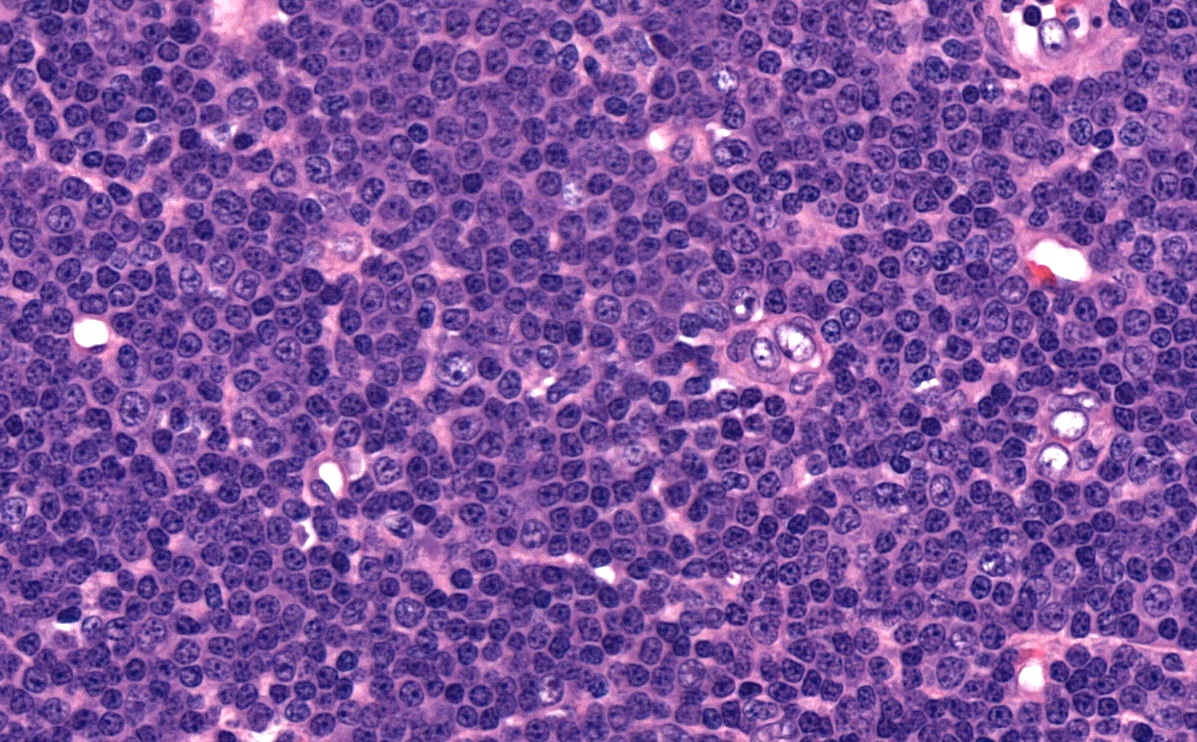
Proliferation center
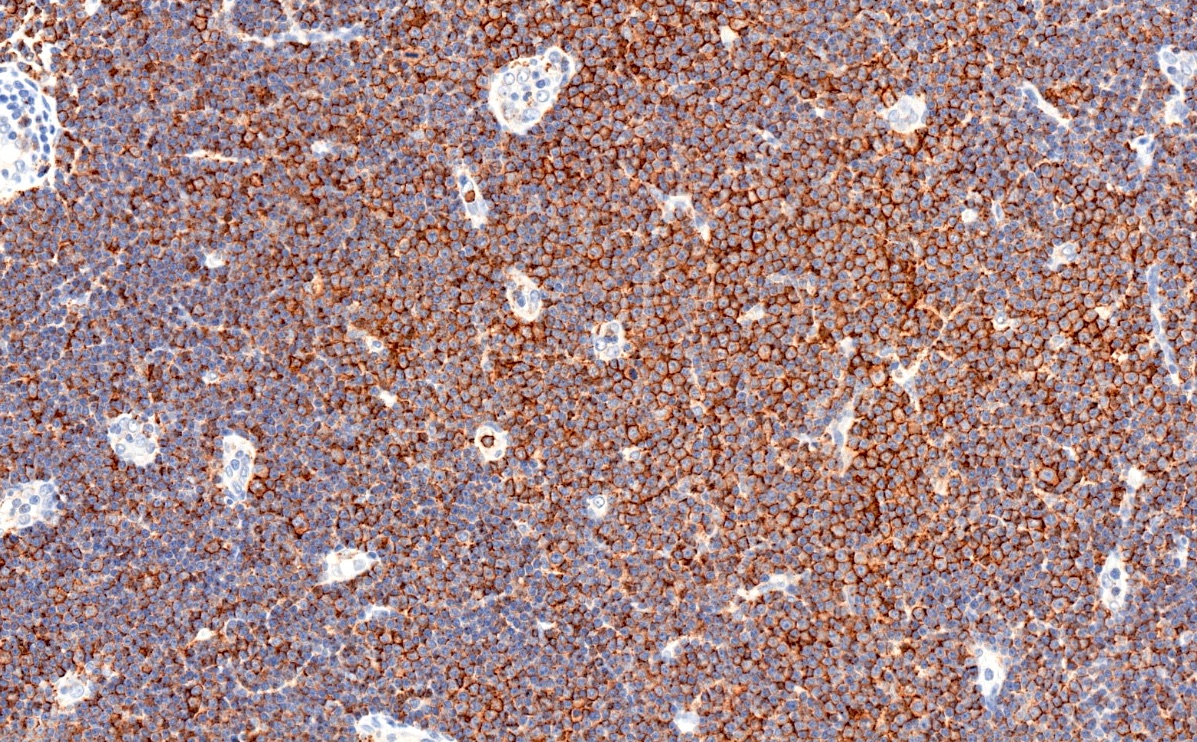
CD20
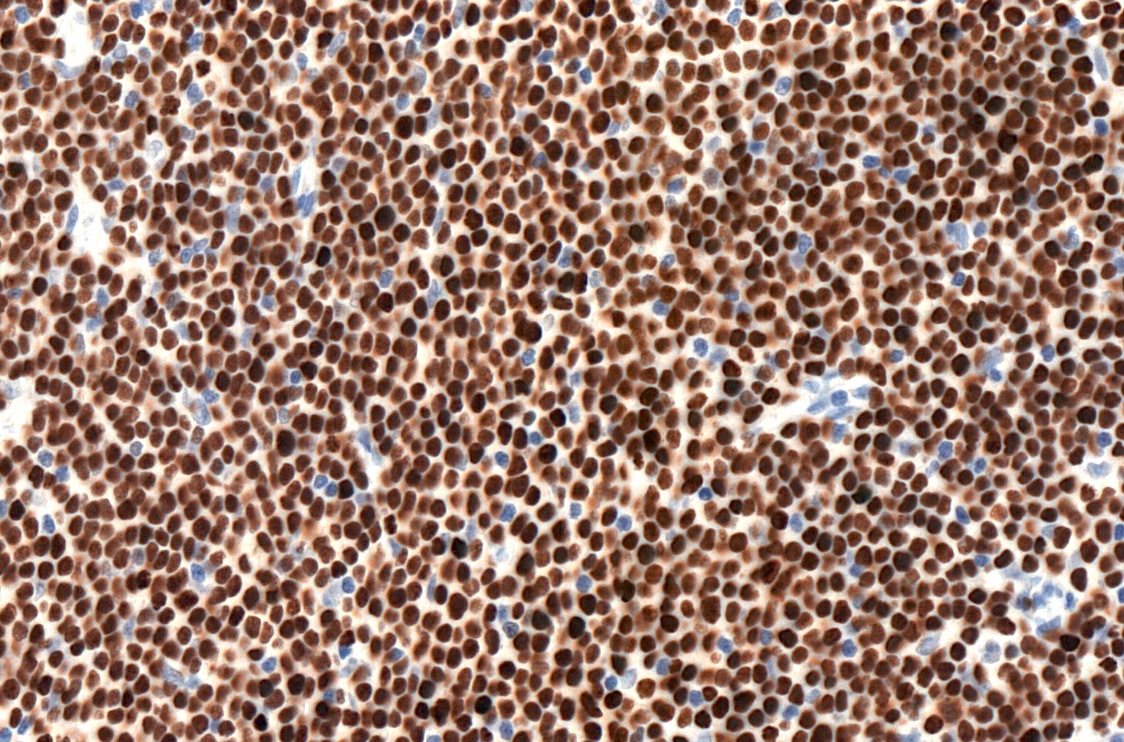
LEF1
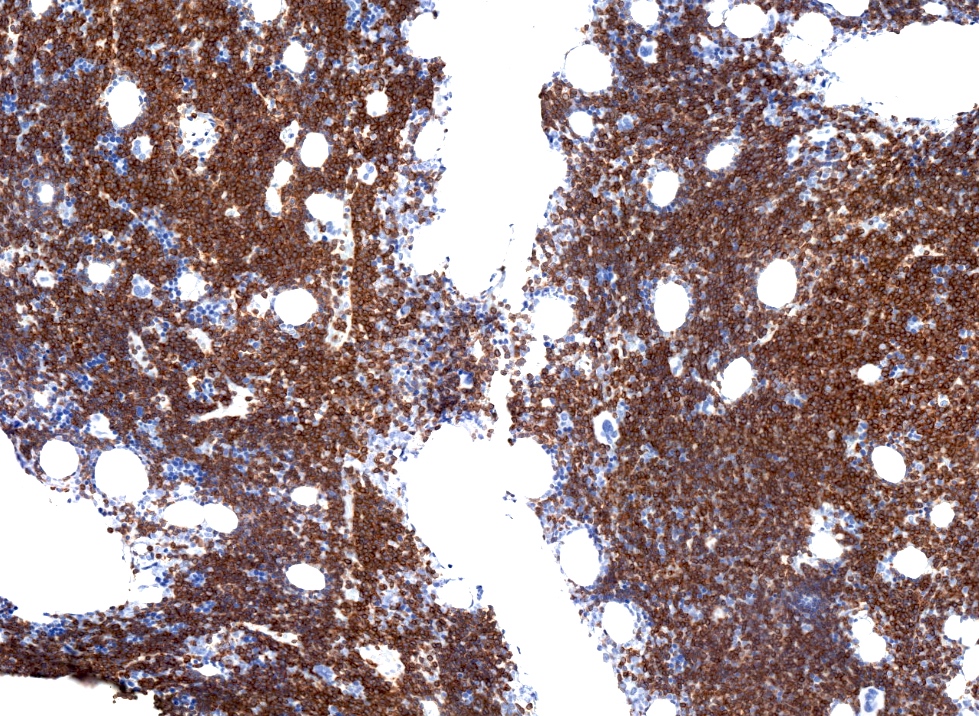
Bone marrow involvement
Images hosted on other servers:


SLL / CLL, H&E
- Small, mature lymphocytes with round nucleus, clumped chromatin and only small nucleoli
Contributed by Béla Kajtár, M.D., Ph.D.
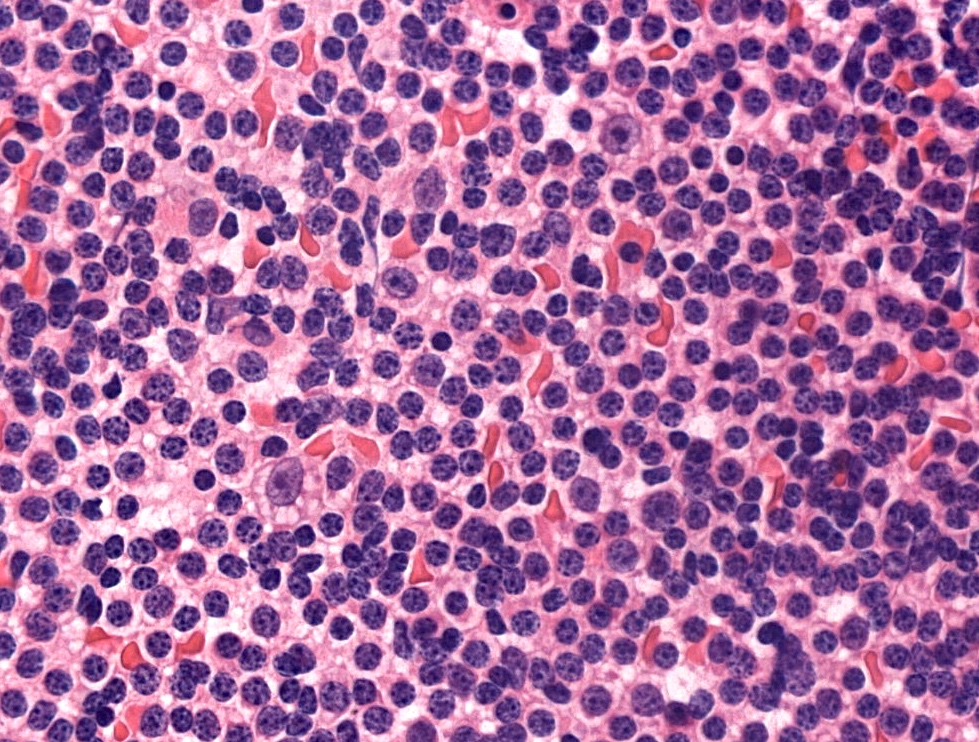
Touch prep SLL
- Lymphocytosis composed of small, mature lymphocytes with round nucleus, clumped chromatin and only small nucleoli
- Smudged cells (Gumprecht cells) commonly seen representing mechanically damaged cells (J Clin Oncol 2009;27:1844)
- Prolymphocytes usually < 15%; 15 - 55% in case of atypical CLL; > 55% defines B cell prolymphocytic leukemia (see Prolymphocytic leukemia)
Contributed by Béla Kajtár, M.D., Ph.D.
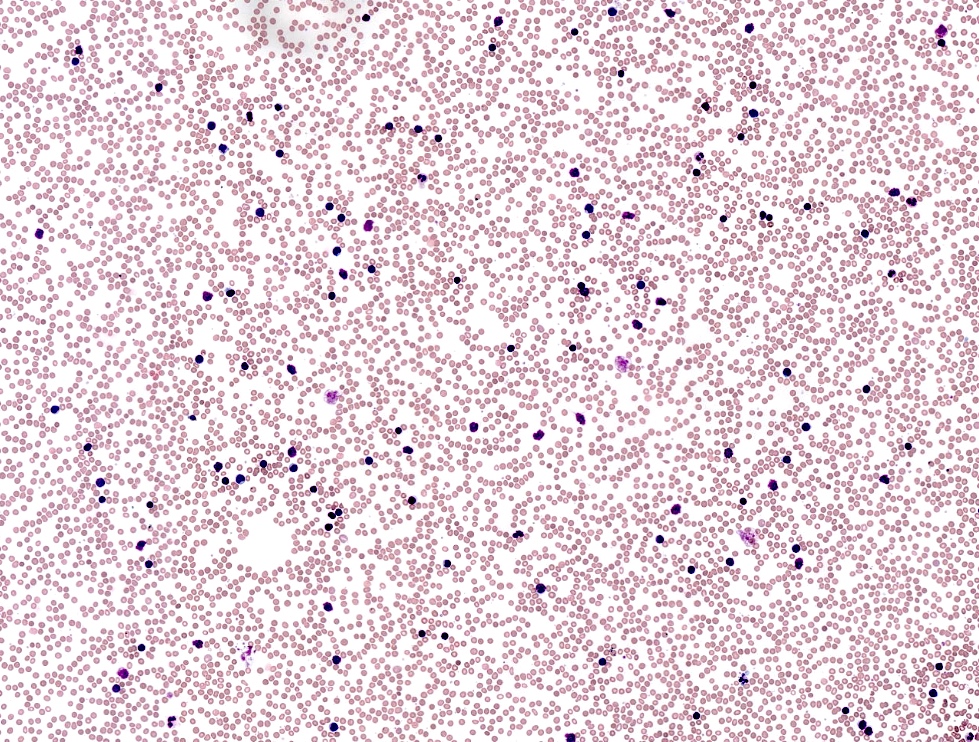
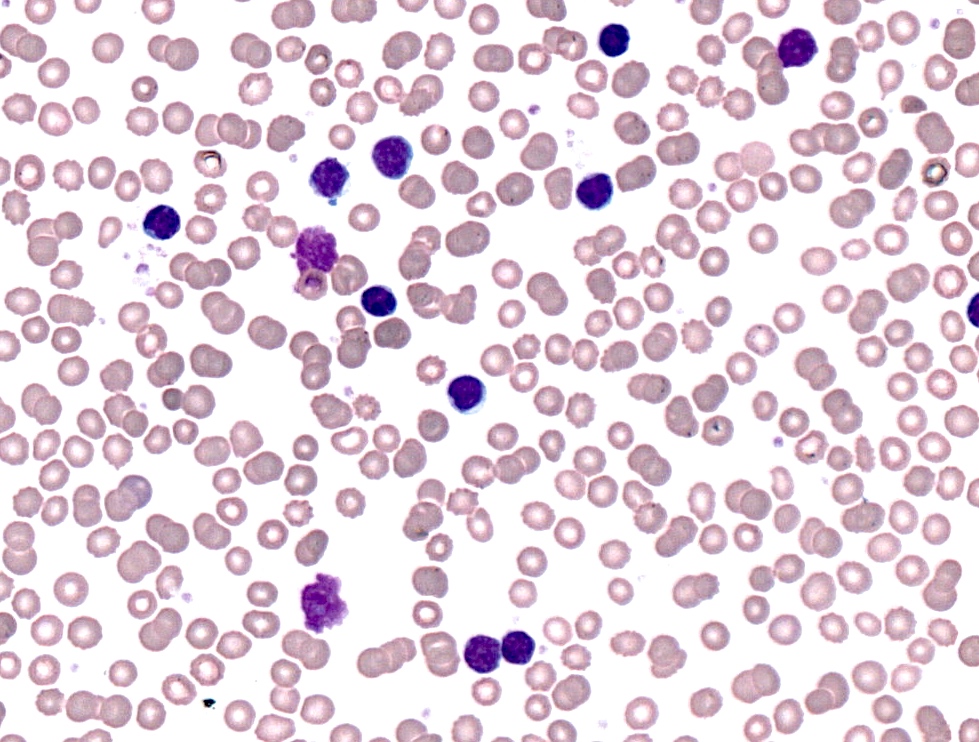
CLL in peripheral blood
- CD5 (rarely negative), CD79a, CD20 (dim), CD23, CD19, PAX5, CD200, BCL2, LEF1, CD43, IgM / IgD (dim) (Arch Pathol Lab Med 2014;138:1666)
- CD38, ZAP70, CD49d may be positive; they are associated with worse prognosis (Blood 2011;118:3470, Leukemia 2003;17:2426, Blood 2020;135:1244)
- MUM1 may be positive, considered as adverse prognostic factor by some
- Richter transformation: only 30% are CD5 positive, CD23 only in 15%, most are nongerminal center type (Blood 2018;131:2761)
- CD10, BCL6, SOX11 (Haematologica 2009;94:1563)
- CD20 may appear negative or very dim
- Cyclin D1 may be positive in scattered prolymphocytes, may show dim staining on proliferation centers, never shows diffuse, strong labeling (Am J Clin Pathol 2012;138:132, Arch Pathol Lab Med 2005;129:92)
Contributed by Béla Kajtár, M.D., Ph.D.
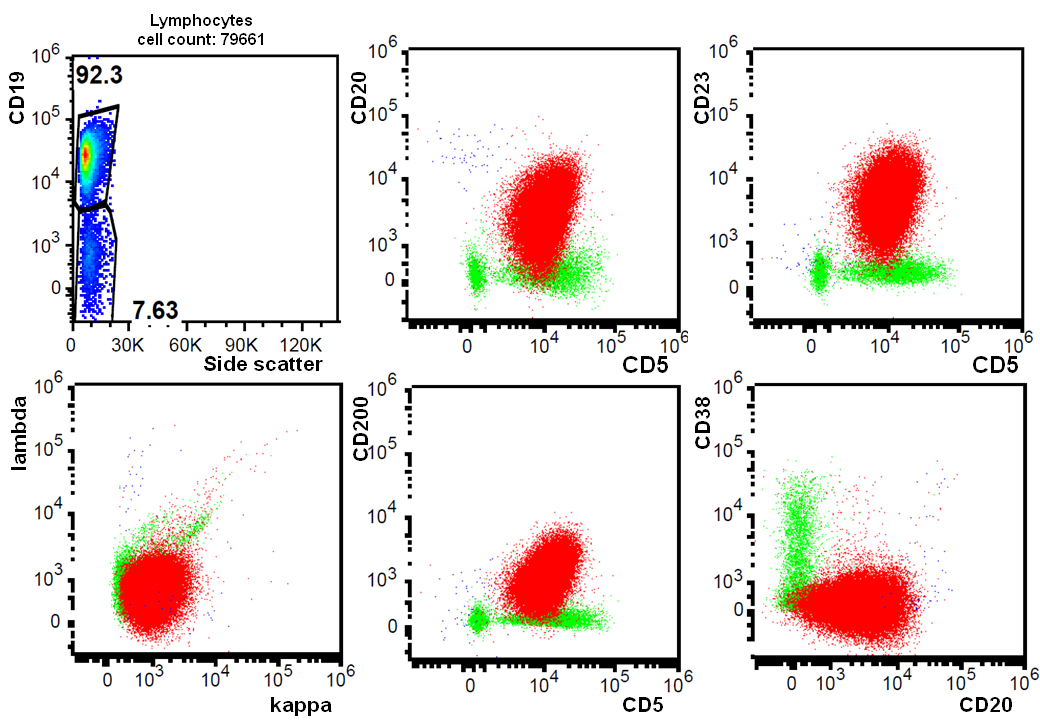
CLL, scatter plot
- Unmutated IGH (sequencing) (40 - 60% of cases), testing is necessary before treatment
- TP53 mutations (sequencing) 10% at diagnosis, 30% at relapse, testing is necessary before treatment (Hematology Am Soc Hematol Educ Program 2016;2016:149)
- 17p deletion (5 - 30%) may be independent from TP53 mutation, testing is necessary before treatment
- 13q deletion (50 - 55%), 11q23 deletion (5 - 20%), 12 trisomy (10 - 20%), 6q deletion by FISH is recommended (Biomed Res Int 2014;2014:435983, Blood 2018;131:2745)
- Karyotyping to show complex karyotype is recommended in clinical studies, not generally indicated in routine clinical practice (Blood 2018;131:2745)
- IGH translocations rarely occur, IGH-BCL2+ CLL does exist (2 - 5%) (Am J Hematol 2019;94:338)
Contributed by Béla Kajtár, M.D., Ph.D.
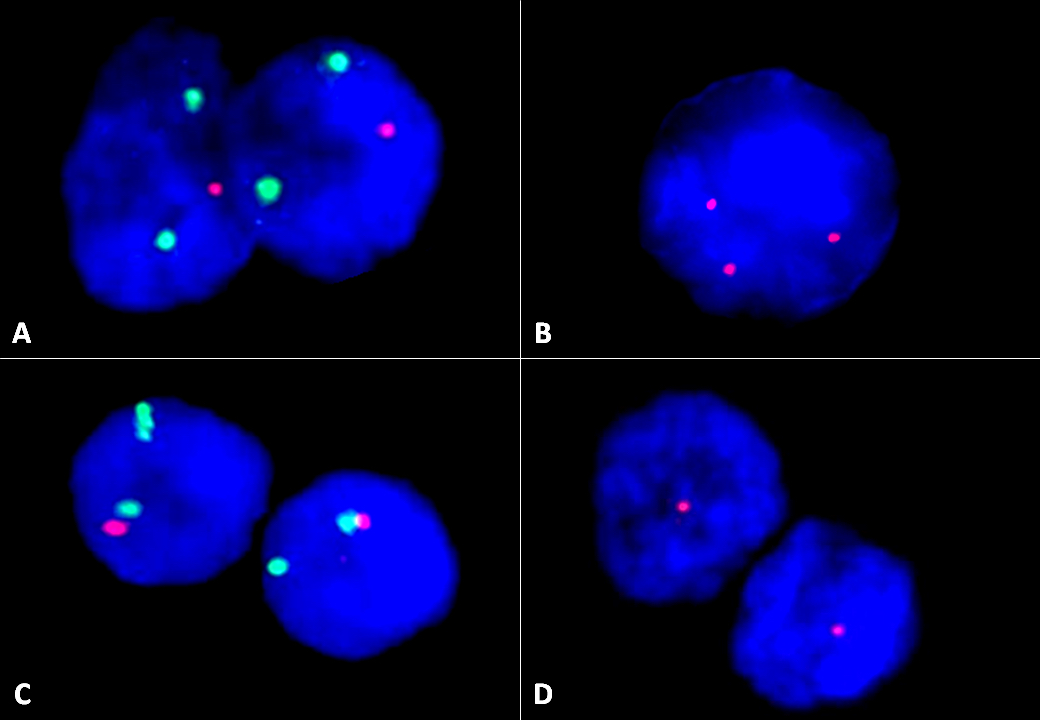
FISH images
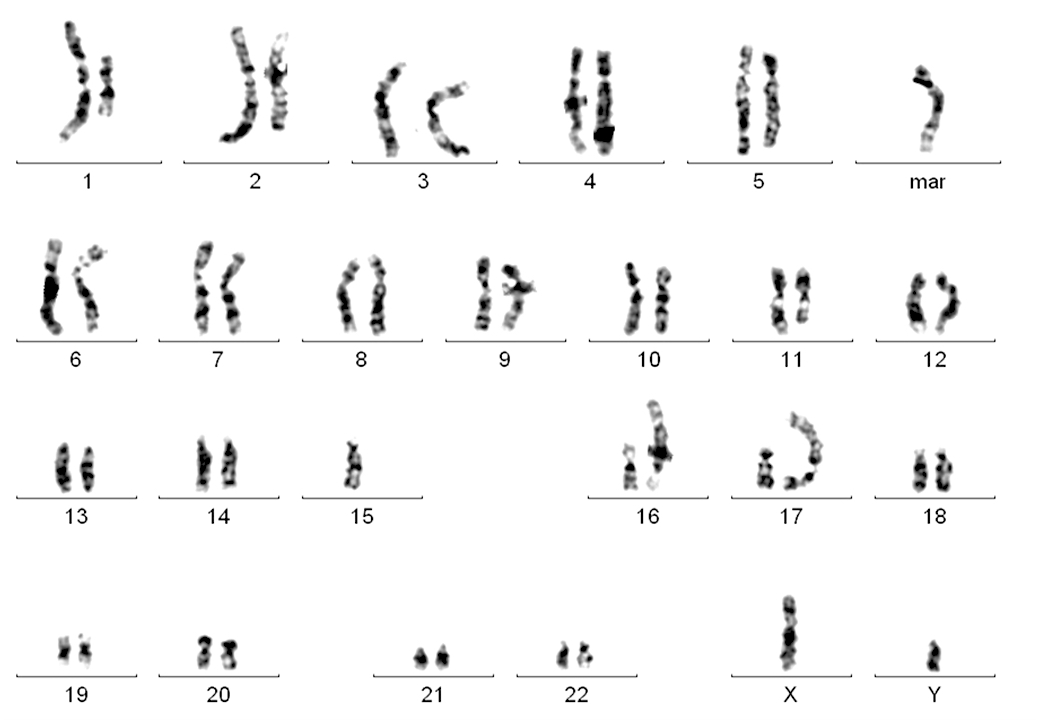
Karyogram
- Bone marrow, left posterior iliac crest, core biopsy, clot section, aspirate smears and peripheral blood smears:
- Hypercellular bone marrow with maturing hematopoietic activity and 85% diffuse lymphocytic infiltrate with immunophenotype consistent with chronic lymphocytic leukemia (CLL) (see comment)
- Comment:
- Flow cytometric analysis of bone marrow demonstrates 62% lymphocytes with CD19+, CD5+, CD10-, CD43+, CD23+, CD200+, CD38-, CD20(dim)+, sIgL-kappa(dim)+ immunophenotype consistent with CLL. Peripheral blood smears indicate > 5 x 109/L CLL cells.
- FISH showed biallelic 13q14 deletion in 58%, 11q23 monoallelic deletion in 32% of cells, no 12 trisomy or 17p deletion was noted (see details in separate cytogenetic report).
- Additional molecular studies (TP53 and IGH sequencing) are in progress and will be reported separately.
- Peripheral smear: Manual review of the peripheral blood shows unremarkable platelets, no morphological alteration of red blood cells and lymphocytosis with few smudged cells. 12% of lymphocytes show enlarged nuclei and nucleoli (prolymphocytes). A manual 500 cell differential count reveals 87% lymphocytes, 2% monocytes, 10% neutrophils and 1% eosinophils.
- Bone marrow biopsy: Quality: adequate. Cellularity: 80%. Hematopoiesis: trilineage maturation is present but diminished without dysplastic features or increased blasts. Megakaryopoiesis: normal, cell number is reduced. Granulopoiesis: normal, no blast increase. Erythropoiesis: normal. Infiltrate: 85% diffuse lymphocytic infiltrate composed of mature lymphocytes, approximately 15% are prolymphocytes. Special stains: Reticulin: loose network of reticulin without significant intersections (minimal reticulin fibrosis). Trichrome: negative for collagen deposition.
- Bone marrow clot section: Quality: adequate. Cellularity: 80% morphologic features are similar to those observed in the core biopsy.
- Bone marrow aspirate: Quality: adequate. Granulocytes: decreased; normal maturation; no dysplastic features. Erythrocytes: decreased, normal maturation, no dysplastic features, no ringed sideroblasts. Megakaryocytes: significantly decreased; no dysplastic features. Blasts: less than 1% of nucleated cells. Infiltrate: diffuse lymphocytic infiltrate (78%) consisting of small, mature lymphocytes as well as 13% prolymphocytes. A manual 500 cell differential count reveals 23% erythroblasts, 1% myelocytes, 7% metamyelocytes, 2% neutrophils, 1% eosinophils and 66% lymphocytes (of which 13% are prolymphocytes).
- Monoclonal B cell lymphocytosis:
- Less than 5 x 109/L B cells in peripheral blood
- Mantle cell lymphoma:
- Marginal zone lymphoma:
- Prolymphocytic B cell leukemia:
- Variable immunophenotype, bright CD20 and sIg
- > 55% of lymphoid cells are prolymphocytes in peripheral blood
- Lymphoplasmacytic lymphoma:
Which of the following is necessary for diagnosis of CLL?
- Immunophenotyping
- Sequencing of IGH genes
- Sequencing of TP53 gene
- Testing for BCL2 rearrangement
- Testing for CCND1 rearrangement
- Appearance of complex karyotype as part of progression of CLL
- Appearance of TP53 mutation as part of progression of CLL
- Diffuse large B cell lymphoma appearing as progression of CLL
- Nodal CD5+ diffuse large B cell lymphoma
- Unmutated CLL turning into mutated CLL
- CR3 deficiency is an autosomal recessive inherited deficiency of the leukocyte beta2 integrin receptor CD11/18 (CR3) causing leukocyte adhesion deficiency syndrome I (LAD I) (Clin Exp Immunol 2000;121:133)
- Migration and adhesion of leukocytes from the bloodstream to site of infection involves multiple steps of the adhesion cascade
- Adhesion molecules are expressed on endothelial cells and leukocytes.
- CD11/CD18 (CR3), also known as the leukocyte beta2 integrin receptor, is one of the main adhesion molecules essential for leukocyte adhesion to endothelial cells and chemotaxis
- Defects in these adhesion molecules as well other adhesion molecules result in recognized clinical syndromes
- Three leukocyte-adhesion deficiency (LAD) syndromes have been delineated (LAD I, LAD II, LAD III) (Blood Cells Mol Dis 2001;27:1000)
- The deficiency / defect leads to impaired phagocytic function
- Four distinct complement receptors, CR1, CR2, CR3 and CR4, have been described for the surface bound complement fraction C3 and its cleavage fragments such as iC3b
- Complement and complement receptors are an integral part of the immune defense
- CR3 (CD11b/18) and CR4 (CD11c/18) both bind to iC3b and facilitate adequate adhesion of leucocytes with the vascular endothelium
- Receptors such as CR3 act as ligands for adhesion molecules, and are present on phagocytic cells
- CR3 and CR4 have an important role in host resistance to infection
- Recurrent cutaneous infections and gingivitis
- Delayed separation of the umbilical cord and secondary omphalitis
- Severe recurrent Staphylococcus aureus and gram negative bacterial infections
- Periodontitis and impaired wound healing
- Leukocytosis with lack of pus formation
- Absence of CD18 and the associated alpha subunit molecules CD11a, CD11b and CD11c on the surface of leukocytes is demonstrated by flow cytometry using CD11 and CD18 monoclonal antibodies
- Sequence analysis can define the exact molecular defect in the beta 2 subunit
- Neutrophilia
- 18 month old girl with leukocyte adhesion deficiency (Pediatr Pathol 1992;12:119)
- 5 year old boy (J Dent Child (Chic) 2012;79:105)
- 31 year old man with pyoderma gangrenosum-like lesions (Pediatr Dermatol 2011;28:156)
- Symptomatic
- Bone marrow and hematopoietic stem cell transplant
Images hosted on other servers:
Skin lesions
Gingivitis, periodontitis
- Severe lymph node hypoplasia with small, poorly delineated germinal centers
- Lymphoid tissue, including the thymus, is depleted of lymphocytes (Pediatr Pathol 1992;12:119)
- Biopsies of infected tissues demonstrate inflammatory infiltrates completely devoid of neutrophils
Images hosted on other servers:

Clinical and histological profile

IL-17 signature
Skin biopsy
- Sequence analysis to define the exact molecular defect in the beta 2 subunit (Prenat Diagn 1991;11:193)
- Common variable immunodeficiency
- Neutrophilia can be caused by
- Infections
- Leukemoid reaction in infants
- Acute leukemia
- Less likely other lymphoproliferative disorders
- Due to congenital enzymatic defect of NADPH oxidase in granulocytes and monocytes
- White blood cells cannot generate superoxide ion which kills microorganisms in lysosomes
- Either Y linked or autosomal recessive
- Diagnose with nitro blue tetrazolium test (almost always positive)
- Clinically, patients have recurrent lymphadenitis, hepatosplenomegaly, skin rash, pulmonary edema
- Anemia, leukocytosis, hypergammaglobulinemia
- Child with Hansenula polymorpha infection (Arch Pathol Lab Med 1980;104:290)
- Granulomas with central necrosis in lymph nodes and other organs
- Pigment laden histiocytes
Contributed by Dr. Mark R. Wick
Childhood disease, AFB stain
Childhood disease
Childhood disease, PAS stain
- Necrotizing granuloma, pigmented histiocytes (Diagn Cytopathol 1991;7:57)
- Pigment is lipofuscin, apparently from lysosomes (Pediatr Pathol 1992;12:839)
- Rare; crystalline material accumulates in cytoplasm of histiocytes; usually kappa light chain origin (Histopathology 2016;68:482)
- Adults are affected with a wide age range; men and women affected nearly equally
- Most commonly affects the head and neck, lung, kidney, bone marrow and lymph nodes, although nearly any site may be involved (Head Neck Pathol 2012;6:111)
- Strongly associated with underlying plasma cell neoplasm or lymphoma with plasma cell differentiation
- A minority of cases are associated with nonneoplastic causes such as infections and autoimmune disease
- Rare disease with crystalline material in cytoplasm of histiocytes
- May affect any organ system
- Strongly associated with underlying lymphoid or plasma cell neoplasm
- 38 year old woman with seizures (Clin Neuropathol 2014;33:23)
- 50 year old woman with lung mass and rheumatoid arthritis (Arch Pathol Lab Med 2005;129:1159)
- 51 year old woman with upper lip and cheek tumor (Head Neck Pathol 2012;6:111)
- 73 year old man with ascites, weight loss and fatigue (Blood 2002;100:1817)
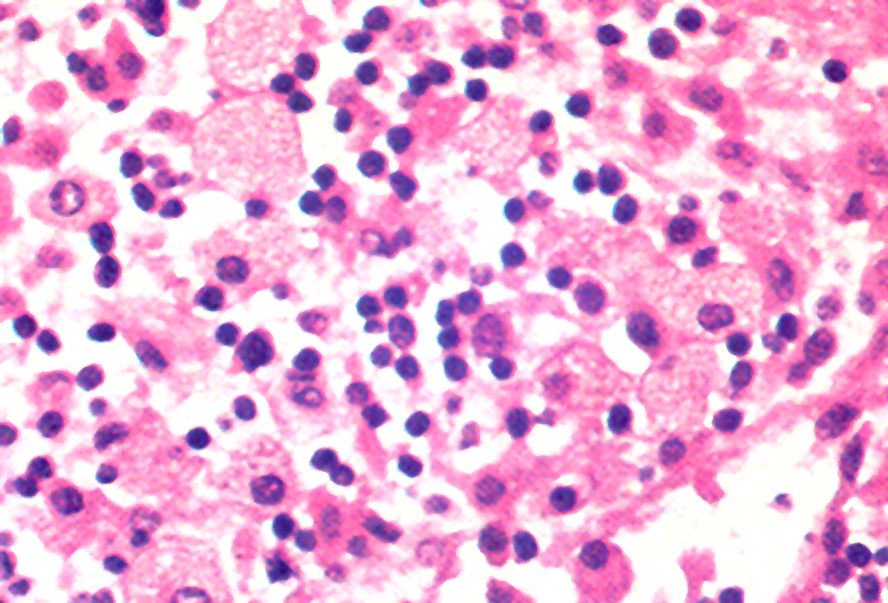
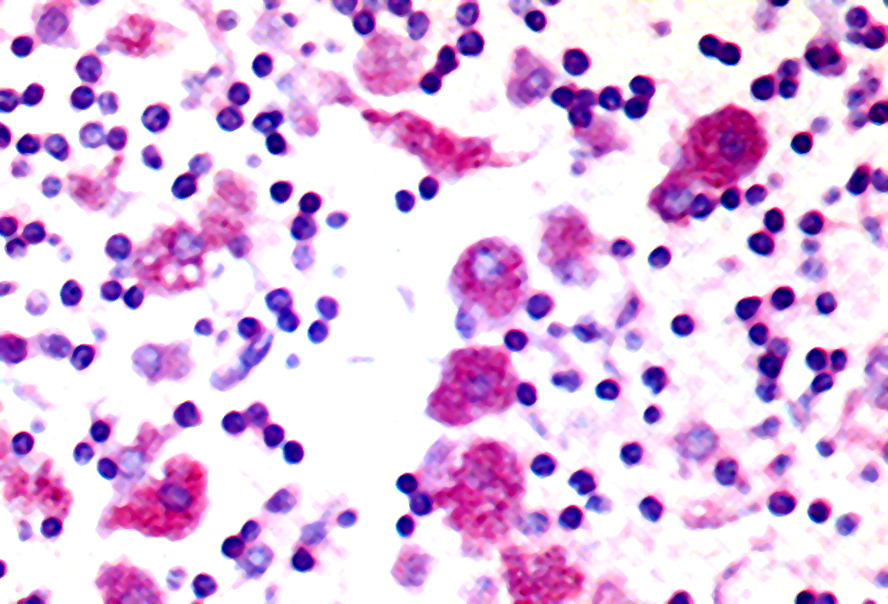
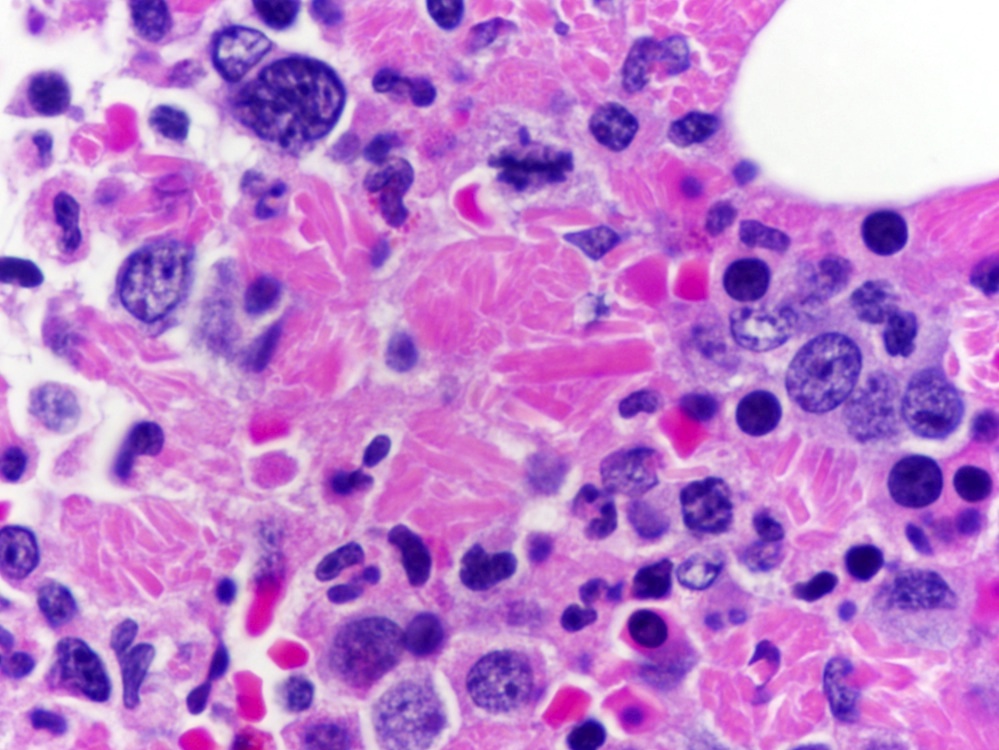
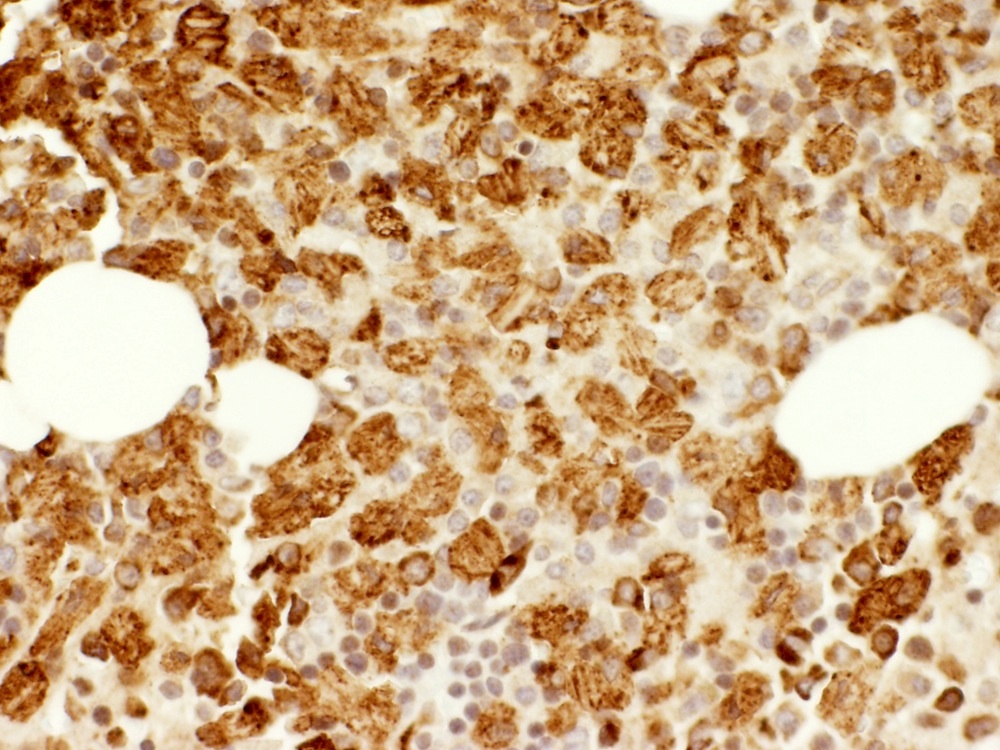
- Cytologically benign histiocytes with abundant cytoplasm filled with many refractile eosinophilic crystals which may be needle-like or rhomboid in shape
- Neoplastic lymphocytes and plasma cells may be present
- Histiocytes often compose the majority of cellular elements, potentially obscuring an underlying neoplasm
Contributed by David Lynch, M.D.
Bone marrow core and CD68
Bone marrow aspirate
- Congenital (hereditary spherocytosis, sickle cell) or acquired
- Acquired cases are usually due to deposition of immune complexes on red blood cell membranes; also bacterial hemolysins, plasma lipid abnormalities, parasites
- Immune related cases often due to brucellosis, Hodgkin lymphoma, leukemia, sarcoidosis, SLE (lupus), tuberculosis
- Coombs test: detects acquired cases via detection of surface immune complexes; first wash patient's red blood cells, then add antihuman globulin rabbit serum, agglutination implies acquired hemolytic anemia
- Direct Coombs test: detects antibody attached to red blood cells (above)
- Indirect Coombs test: detects serum antibodies (i.e. antibodies NOT attached to red blood cells)
- Steroids or immunosuppressives
- Splenectomy if unresponsive
- Firm, deep red tissue, thin capsule, no grossly identifiable Malpighian follicles, 100 - 1000 g
- Congestion in cords and sinuses, hemosiderin deposition, extramedullary hematopoiesis, erythrophagocytosis with neutrophils, reactive follicular hyperplasia
- Folate deficiency is a low level of folic acid (Vitamin B9) in the body
- Characterized by macrocytic anemia
- Also called Vitamin B9 deficiency
- Inadequate ingestion of folate containing foods due to: alcoholism (alcohol dehydrogenase binds folate), psychiatric morbidities, elderly
- Impaired absorption: celiac disease, tropical sprue, achlorhydria, anticonvulsant drugs (Dilantin), zinc deficiency, bacterial overgrowth in blind loops, strictures, jejunal diverticula
- Impaired metabolism, leading to inability to utilize absorbed folate: methotrexate and trimethoprim (folate antagonists)
- Hypothyroidism (decreases hepatic levels of dihydrofolate reductase)
- Congenital deficiency of enzymes of folate metabolism
- Increased requirement: infancy, pregnancy, lactation, malignancy, concurrent infection (immunoproliferative response), chronic hemolytic anemia (increased hematopoiesis)
- Increased excretion/loss: vitamin B12 deficiency (causes "folate trap"), chronic alcoholism (increased excretion of folate into bile), hemodialysis (may have excess folate loss)
- Increased destruction: superoxide can inactivate folate
- Symptoms due to anemia: weakness, fatigue, difficulty concentrating, irritability, headache, palpitations, shortness of breath, cardiac failure
- Gastrointestinal symptoms: anorexia, nausea, vomiting, abdominal pain, diarrhea (especially after meals)
- Neurologic: cognitive impairment, dementia, depression
Complications:
- Coronary artery disease, stroke
- Pregnancy complications include spontaneous abortion, abruption placentae, congenital malformations (neural tube defects), severe language delay
- Serum folate levels < 3 ng/mL and a red blood cell (RBC) folate level < 140 ng/mL indicate folate deficiency
- Normal serum folate level is 2.5-20 ng/mL
- Normal serum cobalamin is 200-900 pg/mL
- The RBC folate level generally indicates folate stored in the body
- Serum folate level tends to reflect acute changes in folate intake
- Mild hyperhomocystinemia is total plasma concentration of 15-25 mmol/L; moderate hyperhomocystinemia is 26-50 mmol/L
- Pyrexia in a patient with megaloblastic anemia (Iran J Med Sci 2013; 38(2 Suppl): 198)
- Folic acid deficiency optic neuropathy (J Med Case Reports 2008;2:299)
- Severe folate deficiency in pregnancy with normal red cell folate level (Clin Lab Haematol 2006 Feb;28:66)
- Important to rule out cobalamin (Vitamin B12) deficiency because folate treatment will not improve neurologic abnormalities due to cobalamin deficiency
Images hosted on other servers:
Megaloblastic erythroid precursors
- Bone marrow biopsy and aspirate may show a hypercellular bone marrow with a megaloblastic maturation of cells, which morphologically resembles changes of vitamin B12 deficiency
Images hosted on other servers:
Hypersegmented neutrophils, macro ovalocytes
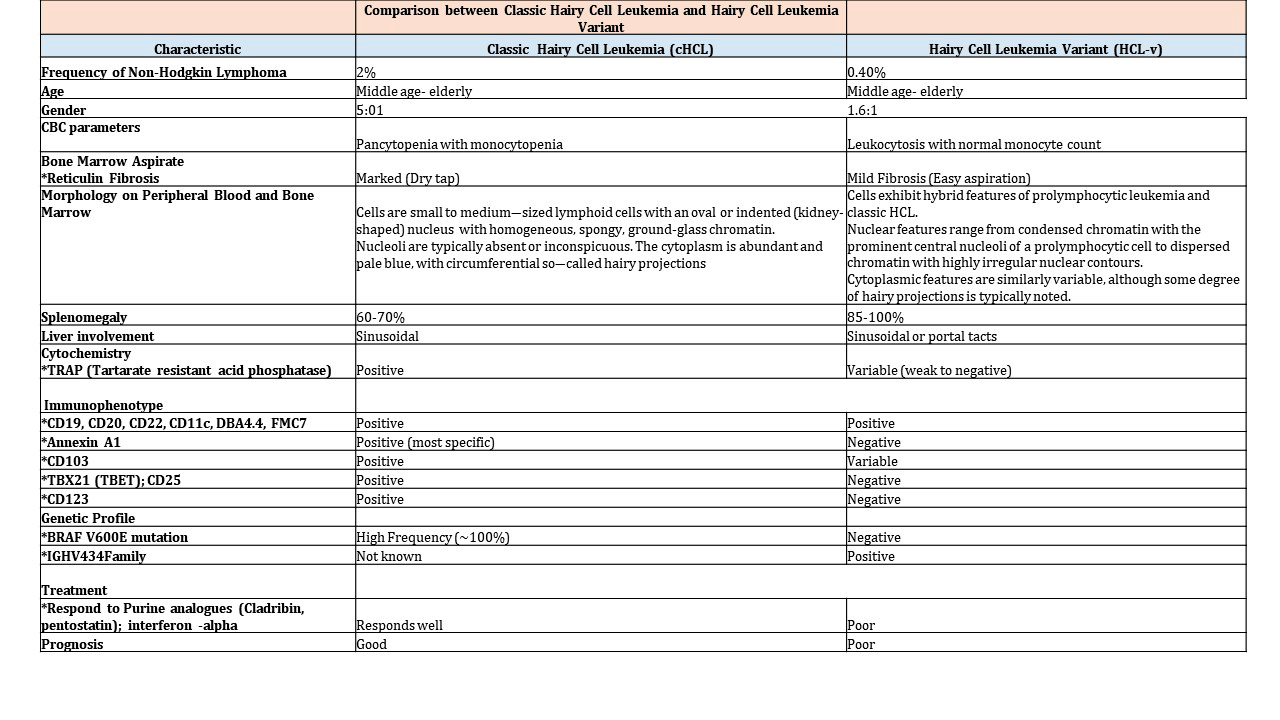
- Rare B cell chronic lymphoproliferative disorder
- Not biologically related to classic hairy cell leukemia (cHCL)
- Exhibits a heterogeneous spectrum of clinical, morphologic, immunophenotypic and genetic features
- Overlap features with classic hairy cell leukemia and other hairy cell-like B cell neoplasms
- Provisional entity within the category of splenic B cell lymphoma / leukemia, unclassifiable, along with splenic diffuse red pulp small B cell lymphoma
- First recognized by Cawley et al. in 1980 (Leuk Res 1980;4:547)
- Clinical laboratory findings
- Leukocytosis and presence of monocytes
- Bone marrow biopsy / aspirate: aspirable (reticulin fibrosis is normal / not increased)
- Morphological findings
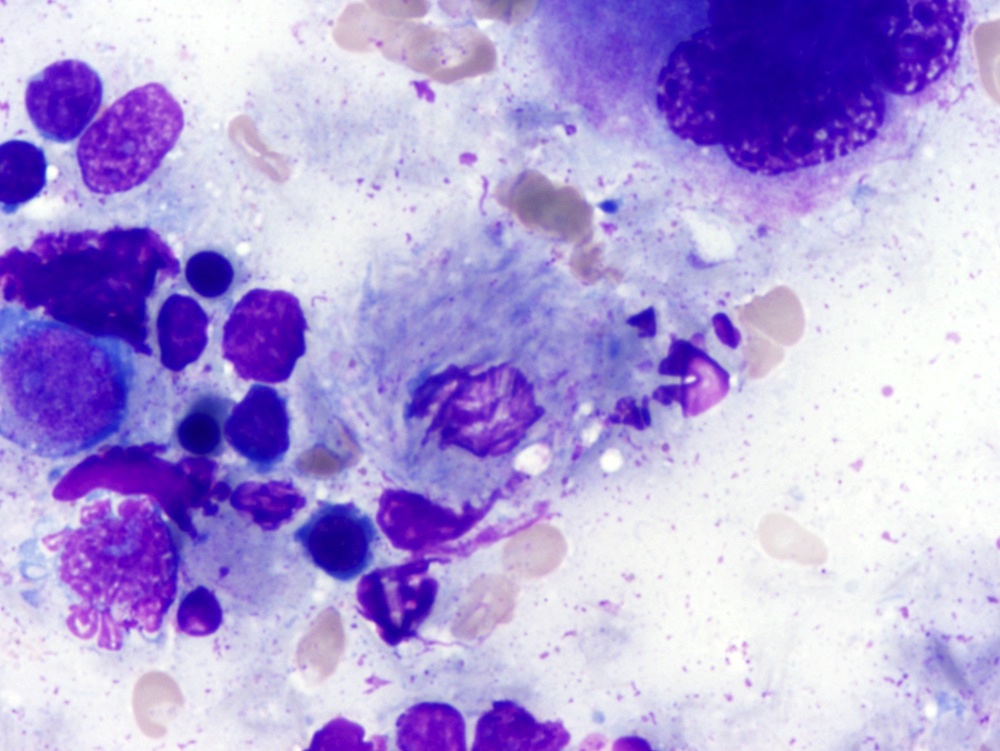
- Intermediate size cells with blastic or convoluted nuclei, prominent nucleoli and circumferential shaggy contours
- Other morphologic subtypes include convoluted and blastoid variant (Best Pract Res Clin Haematol 2015;28:253)
- Immunophenotypic findings
- Absence of CD25, CD123, annexin A1 and tartrate resistant acid phosphatase (TRAP)
- Wild type BRAF
- Resistant to conventional HCL therapy (e.g. cladribine)
- Poorer prognosis (median survival ~ 9 years) (Leukemia 2001;15:184)
- Prolymphocytic variant of hairy cell leukemia
- Atypical hairy cell leukemia
- Japanese variant (HCL JV): predominant in females and may respond better to treatment (Leukemia 1993;7:181)
- Synonyms
- HCL variant
- Incidence
- Constitutes 0.4% of chronic lymphoid malignancies; ~ 10% of all HCL cases
- Age (Cancer Treat Rev 2011;37:3)
- Middle aged to elderly patients
- Reported median age 71 years
- Sex
- Slight male predominance (Leuk Res 2013;37:401)
- Mainly spleen, bone marrow and peripheral blood
- Liver: less than 33% of patients
- Lymph node, other solid organs: uncommon
- Activated B cell at a late stage of maturation
- Most common symptom: abdominal discomfort / pain (Blood 2011;117:5019)
- Attributed to splenomegaly, hypersplenism or bone marrow infiltration (Best Pract Res Clin Haematol 2015;28:253)
- Requires constellation of clinical features, peripheral blood smear, bone marrow, immunophenotyping and molecular studies
- Clinical laboratory values
- Leukocytosis (average WBC ~ 30 x 109/L) and lymphocytosis (versus splenic diffuse red pulp small B cell lymphoma (SDRPL) with low lymphocytosis and cHCL with pancytopenia and monocytopenia)
- Anemia (~ half of the patients)
- Thrombocytopenia (~ half of the patients)
- Absolute monocyte count is typically within the normal range (versus classic hairy cell leukemia (cHCL) with monocytopenia) (Mod Pathol 2018;31:1717)
- Abdominal CT scan
- Splenomegaly
- Hepatomegaly
- Chronic clinical course with a tendency for more aggressive behavior than cHCL
- Median survival 9 years with only 15% survival over 15 years (Leukemia 2001;15:184, Cancer Treat Rev 2011;37:3)
- Reported complete remission (combined purine analog and rituximab treatment or immunotherapy alone)
- Unfavorable prognostic factors: (Br J Haematol 2012;158:347)
- Significant anemia
- Older age
- Mutations in TP53
- 63 year old woman with splenomegaly and lymphocytosis (Blood 2016;128:1018)
- 64 year old man with massive splenomegaly (Clin Case Rep 2019;7:1161)
- 65 year old man with leukocytosis (WBC 39.6 x 109/L), splenomegaly and periportal lymphadenopathy (Case #437)
- 67 year old Taiwanese man with HCL variant with leukocytosis and splenomegaly (Int J Clin Exp Pathol 2011;4:183)
- 72 year old man with leukocytosis with KDM6A mutation (Clin Case Rep 2019;7:1161)
- Therapeutic algorithm for hairy cell leukemia variant (HCL variant; see Diagram 1) (Ann Oncol 2015;26:v100)
- Observation and close monitoring: asymptomatic patients with moderate splenomegaly and no cytopenia
- Symptomatic disease (progressive splenomegaly or elevated leukocyte counts with cytopenia)
- A combination of purine analog (cladribine) and a monoclonal anti-CD20 antibody (rituximab) (Br J Haematol 2016;174:760)
- Rituximab alone or splenectomy followed by rituximab (Ann Hematol 2013;92:711)
- Splenectomy in some patients has been reported, resulting in improvement in anemia and thrombocytopenia (Haematologica 1990;75:54)
- Relapsed / refractory cases (Am J Hematol 2017;92:1382)
- Rituximab (anti-CD20) and moxetumomab pasudotox (anti-CD22 recombinant immunotoxins)
- BCR inhibitors: ibrutinib (Leuk Lymphoma 2017;58:1224)
- Bendamustine plus rituximab (anti-CD20) (Oncotarget 2017;8:110727)
- Alemtuzumab (anti-CD52 antibody)
- Allogenic or autologous stem cell transplantation (Bone Marrow Transplant 2010;45:1117)
- Clinical trials (Am J Hematol 2017;92:1382)
- Cell cycle inhibitors (CDK4 / 6 inhibitors)
- Antiapoptotic histone methyl transferase (HMT) inhibitor (HMT inhibitors)
- Interferon alpha and purine analogs alone are unsatisfactory and fail to achieve a high complete remission rate
- Peripheral smear
- Leukemic cells
- Variable morphology
- Exhibit hybrid features of prolymphocytic leukemia and cHCL
- Morphological subtypes e.g. blastic and convoluted
- Cells are intermediate size
- Nuclear features range from condensed chromatin with the prominent central nucleoli of a prolymphocytic cell to dispersed chromatin with highly irregular nuclear contours (Blood 2016;128:1018)
- Cytoplasm abundant basophilic cytoplasm, circumferential stellate / hairy projections
- Bone marrow
- Aspirable: no significant reticulin fibrosis (MF = 0) (versus hairy cell leukemia showing marked fibrosis and dry tap)
- Infiltration may be subtle; usually interstitial and lesser intrasinusoidal distribution (Mod Pathol 2018;31:1717)
- Immunohistochemical staining is helpful to highlight the pattern and extent of infiltration
- Spleen
- Red pulp: diffusely involved and expanded
- White pulp: follicles atretic / absent
- Leukemic cells are present in the dilated sinusoids
- Blood lakes may be present
- Liver
- Leukemic cell infiltration involves both the portal tract and sinusoids
- Leukemic cells
Contributed by Pallavi Khattar, M.D. and Case #437
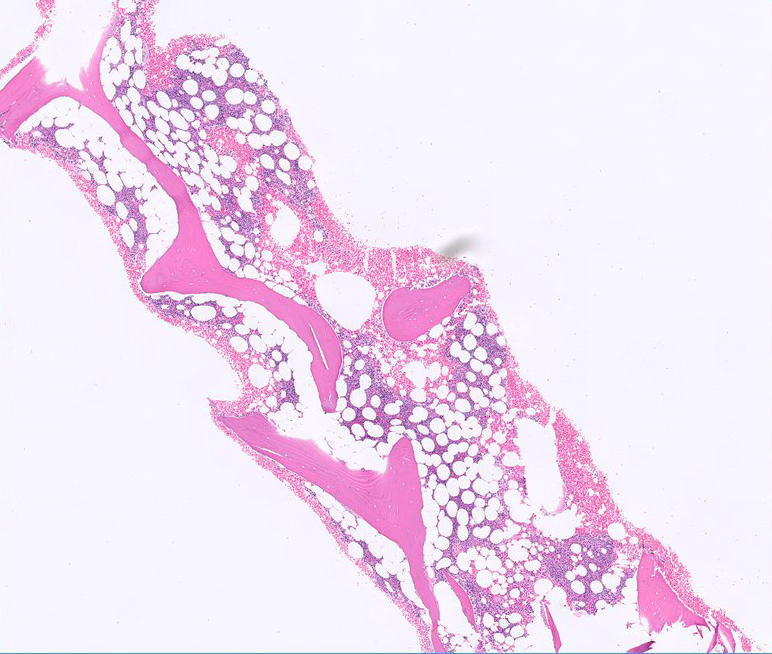
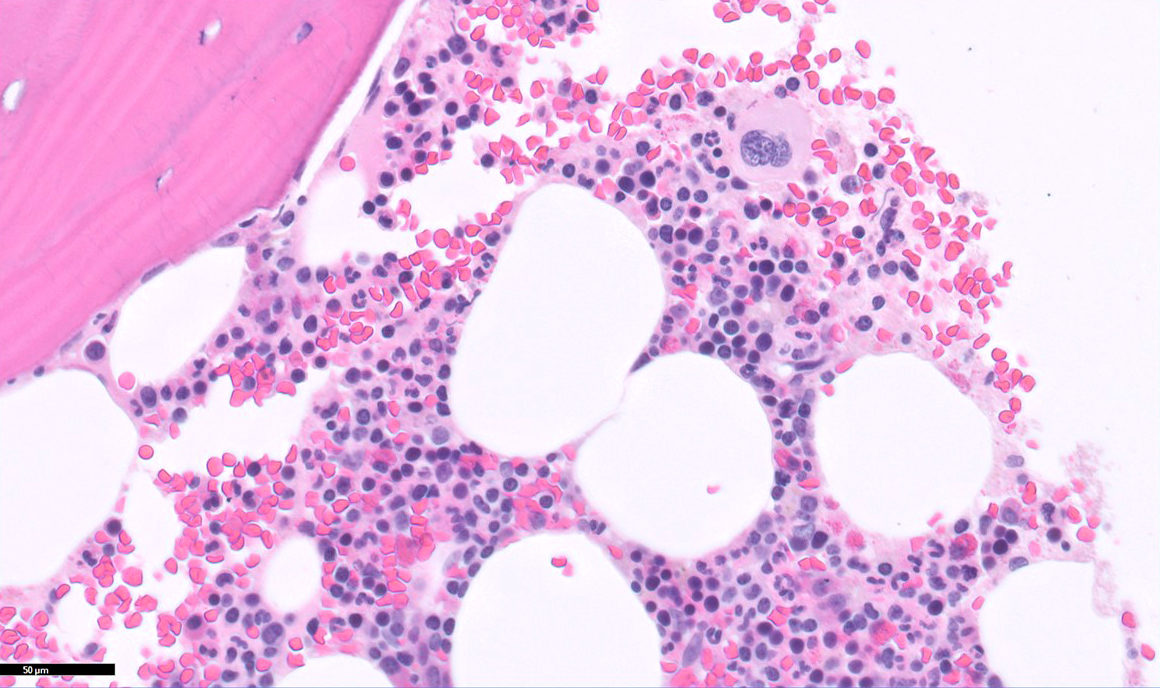
Bone marrow biopsy
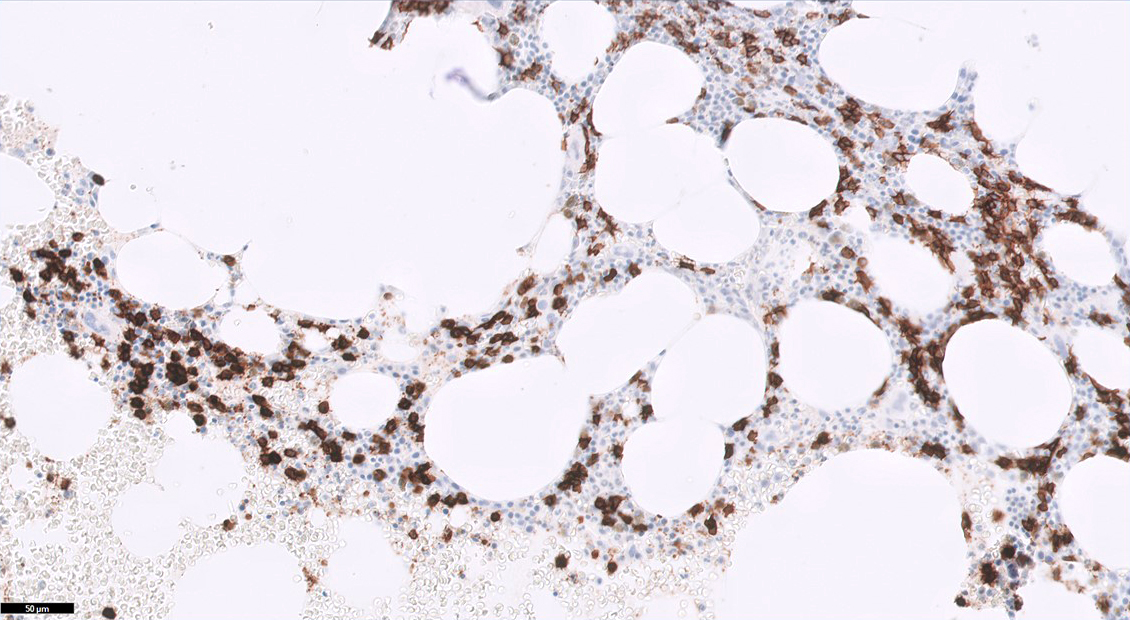
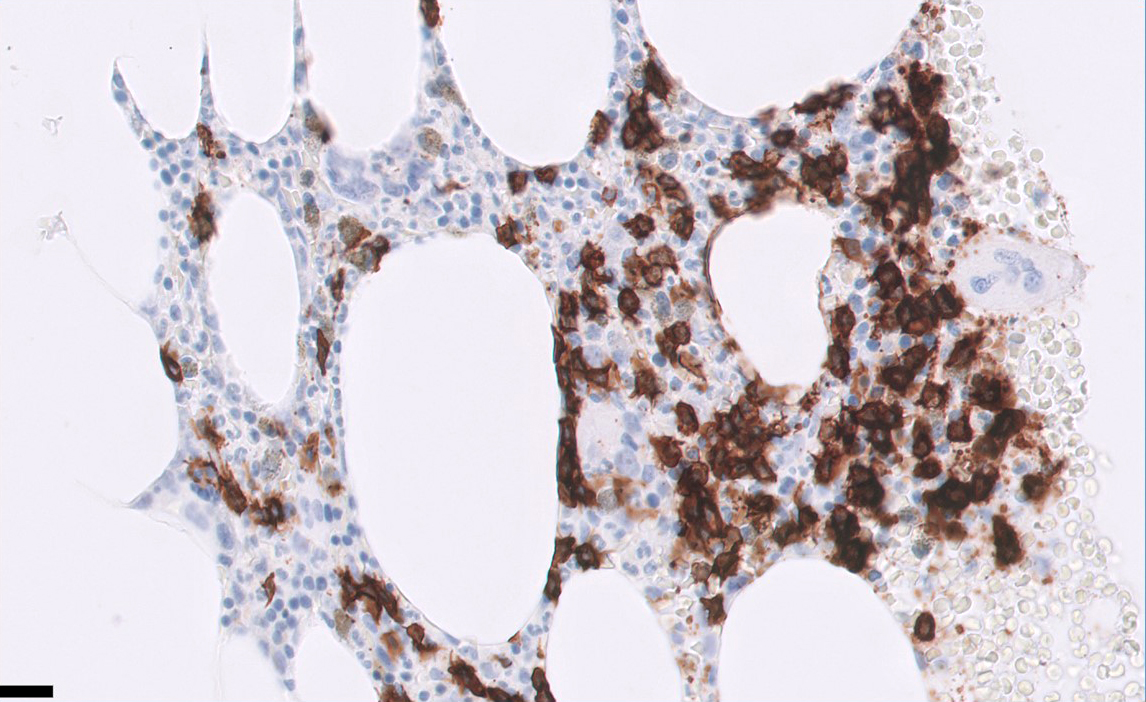
CD20 immunostain
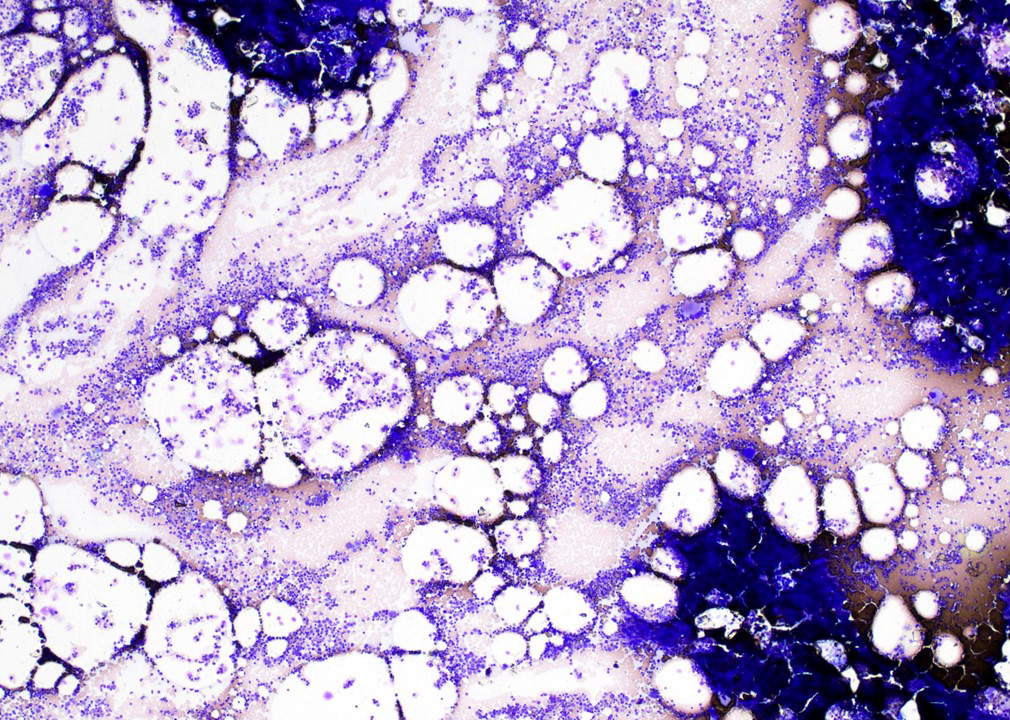
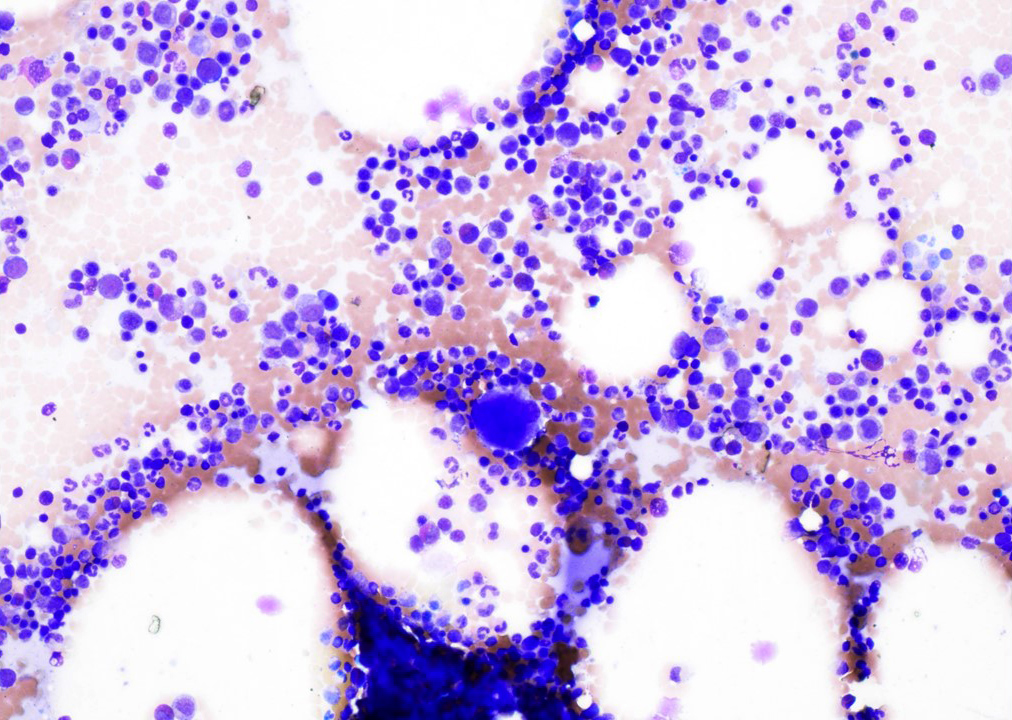
Bone marrow aspirate
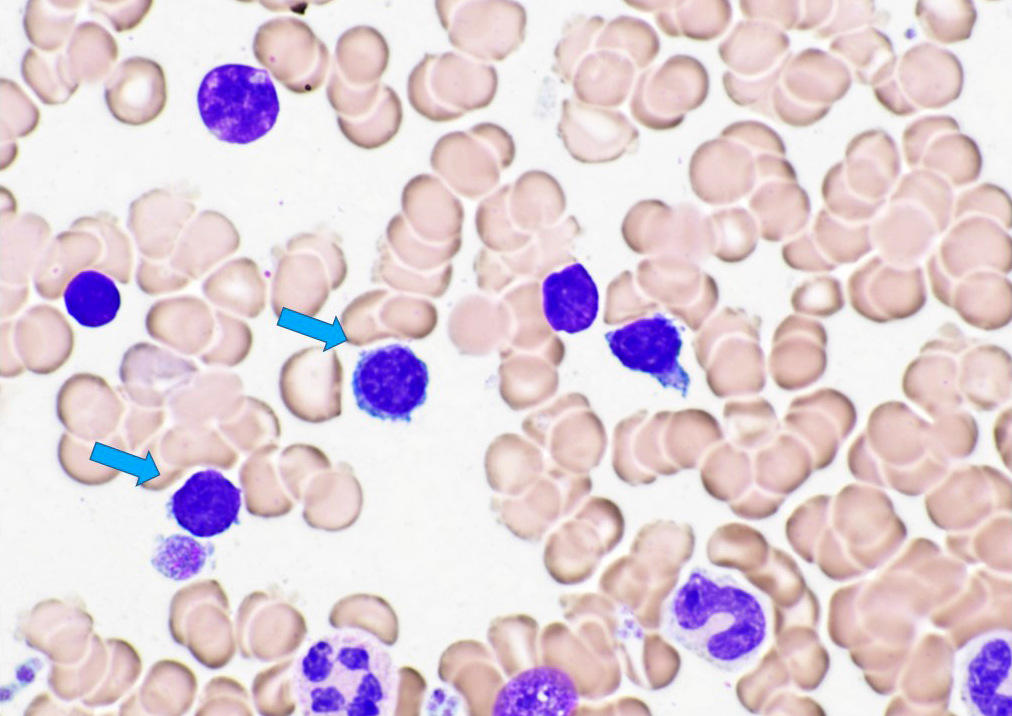
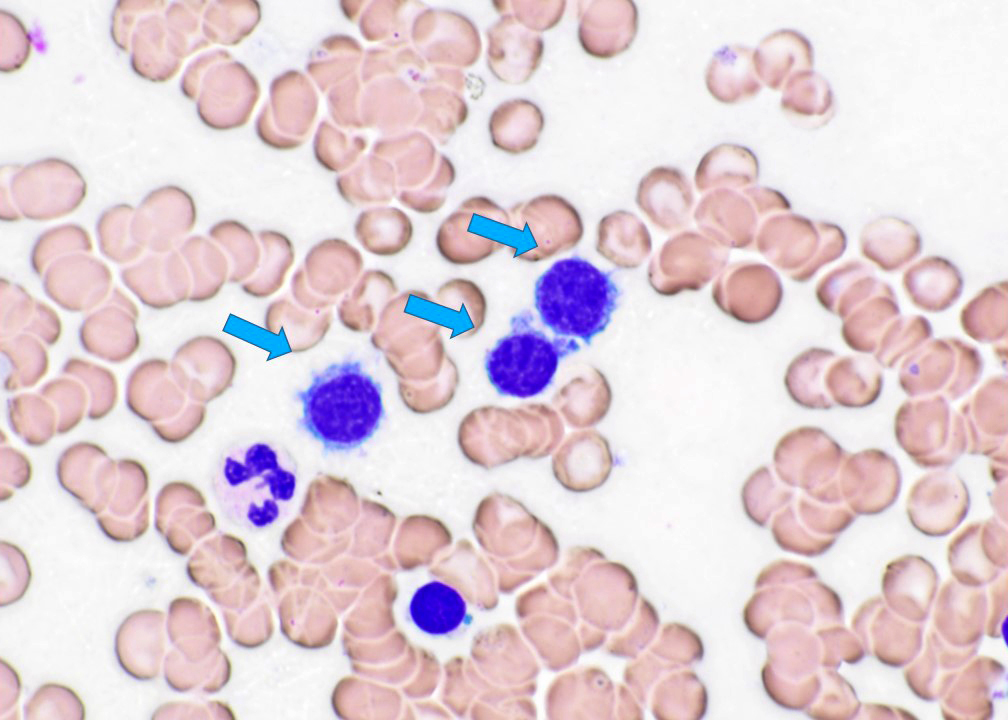
Bone marrow aspirate
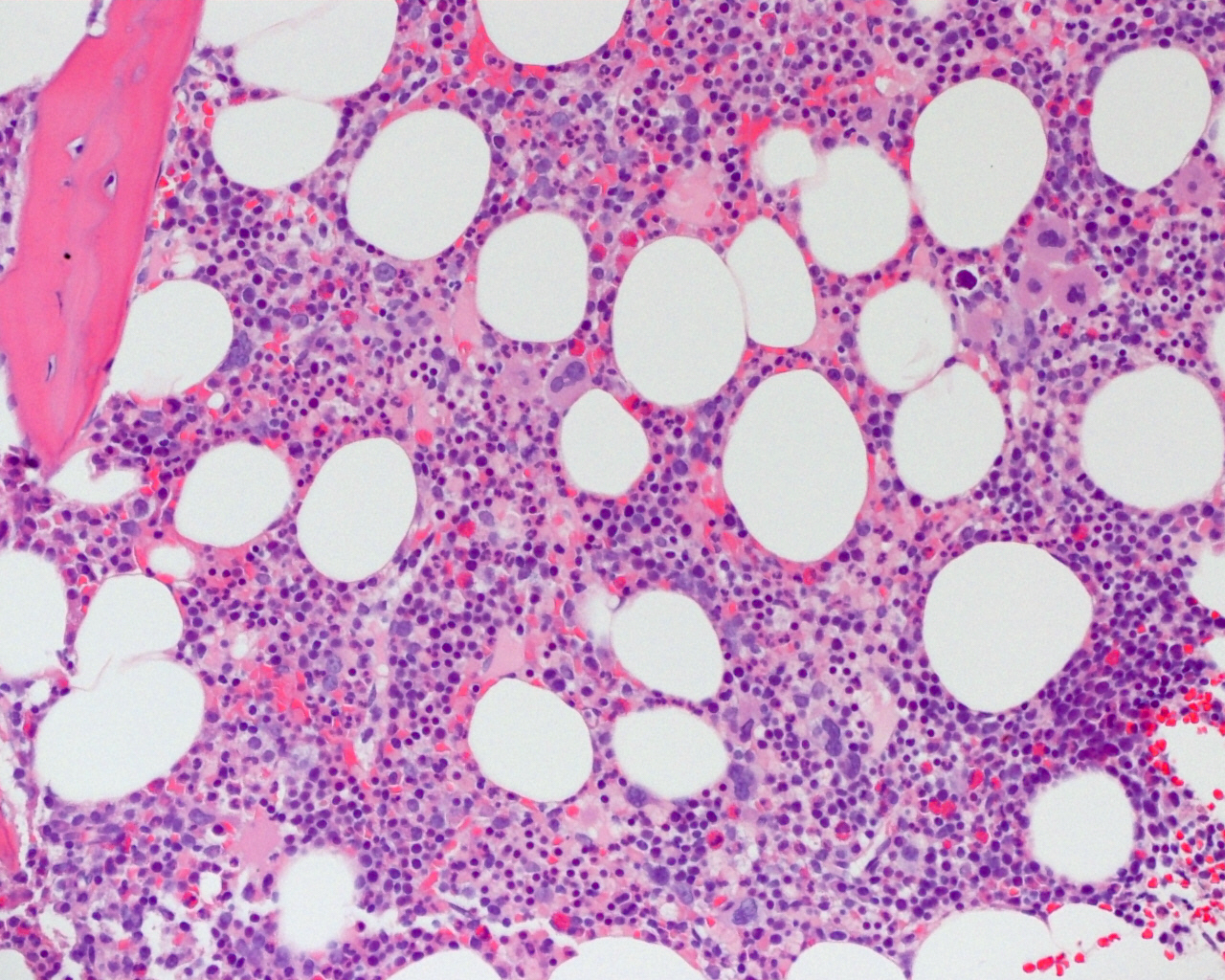
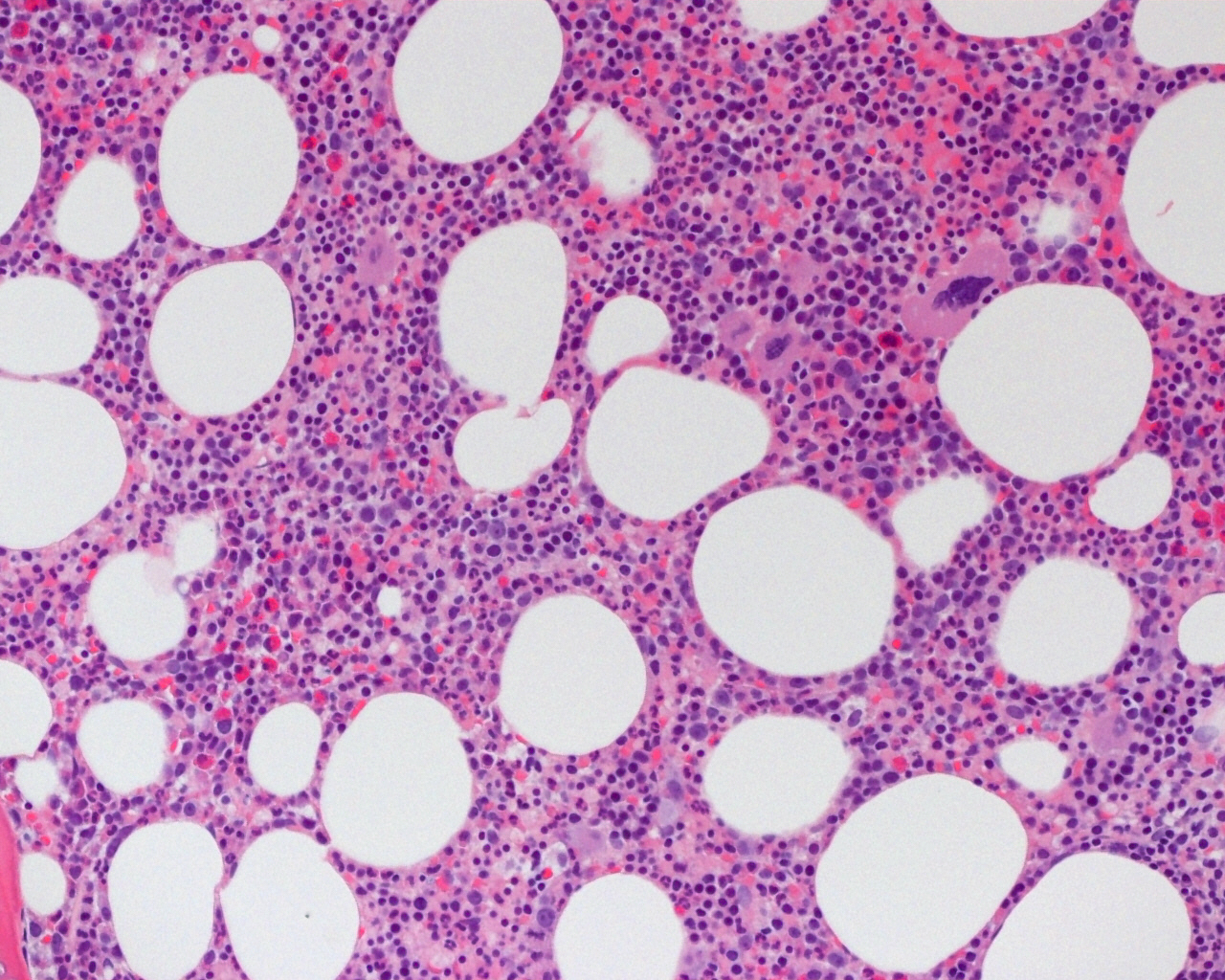
Bone marrow biopsy
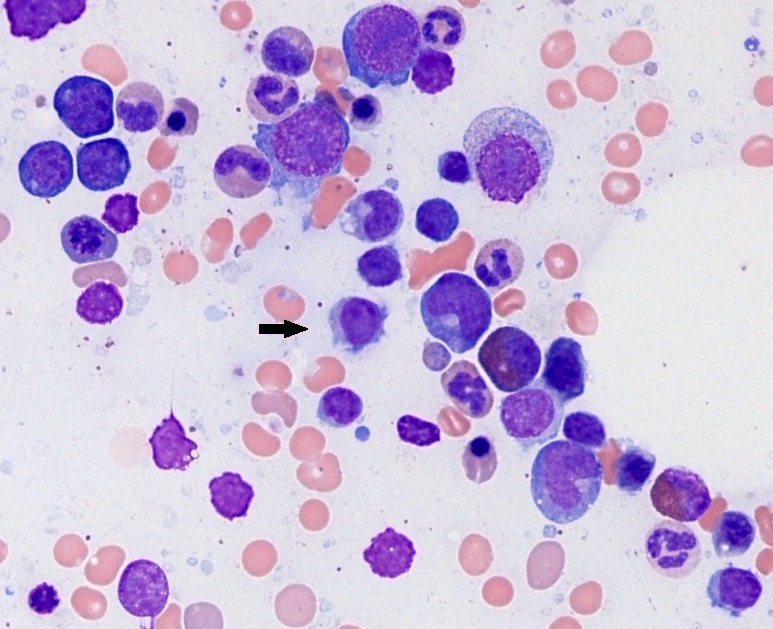
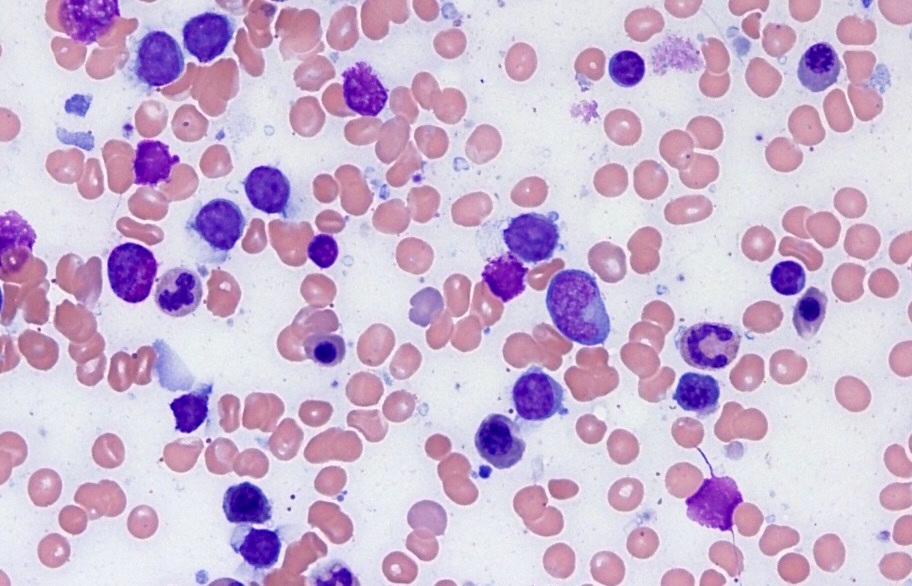
Bone marrow aspirate
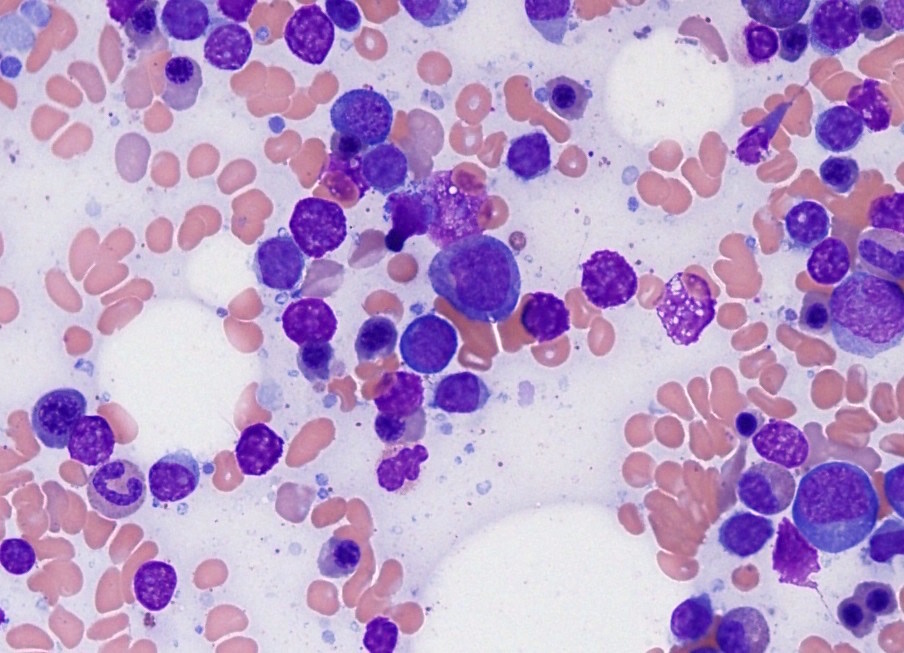
Bone marrow aspirate
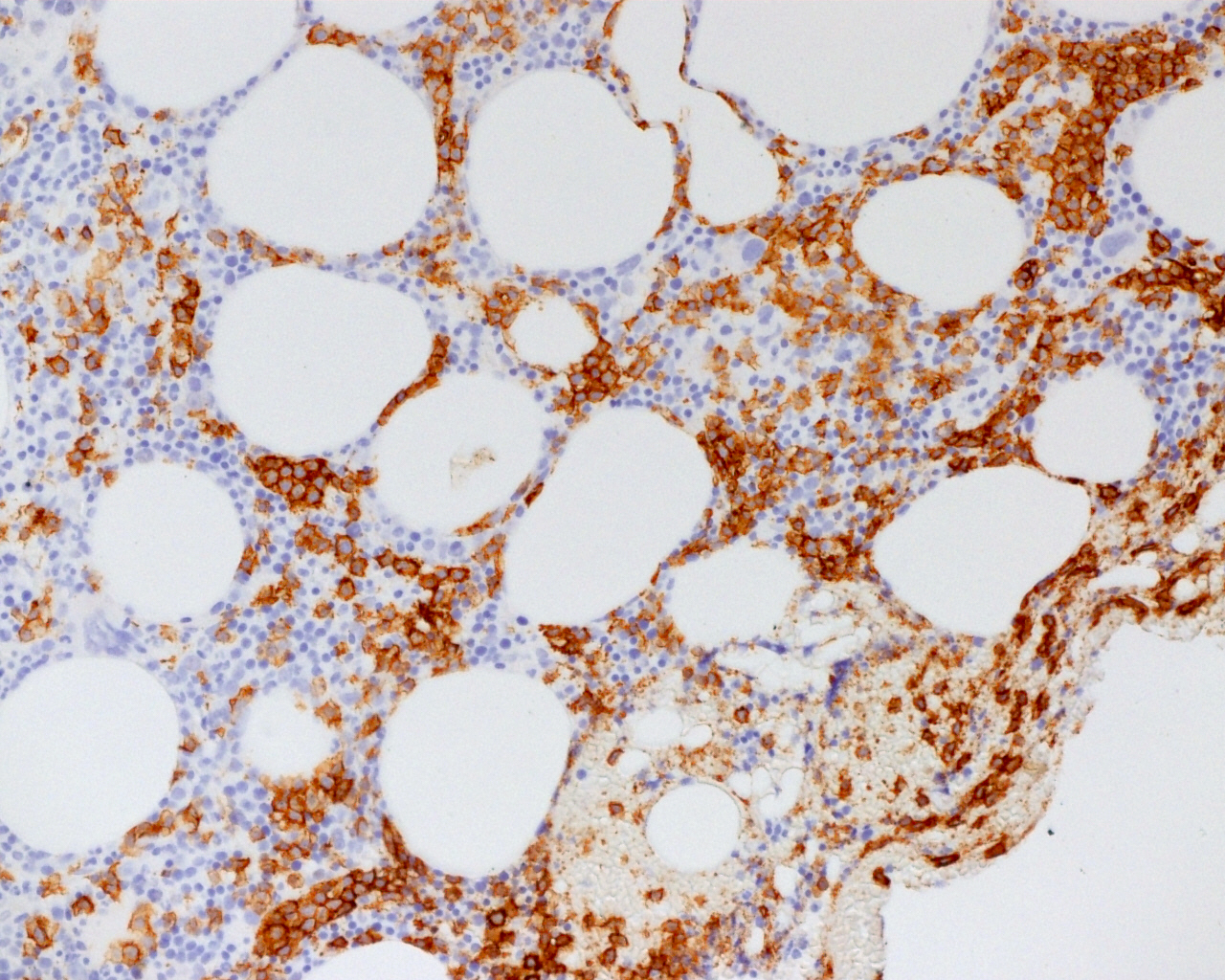
CD20 immunostain
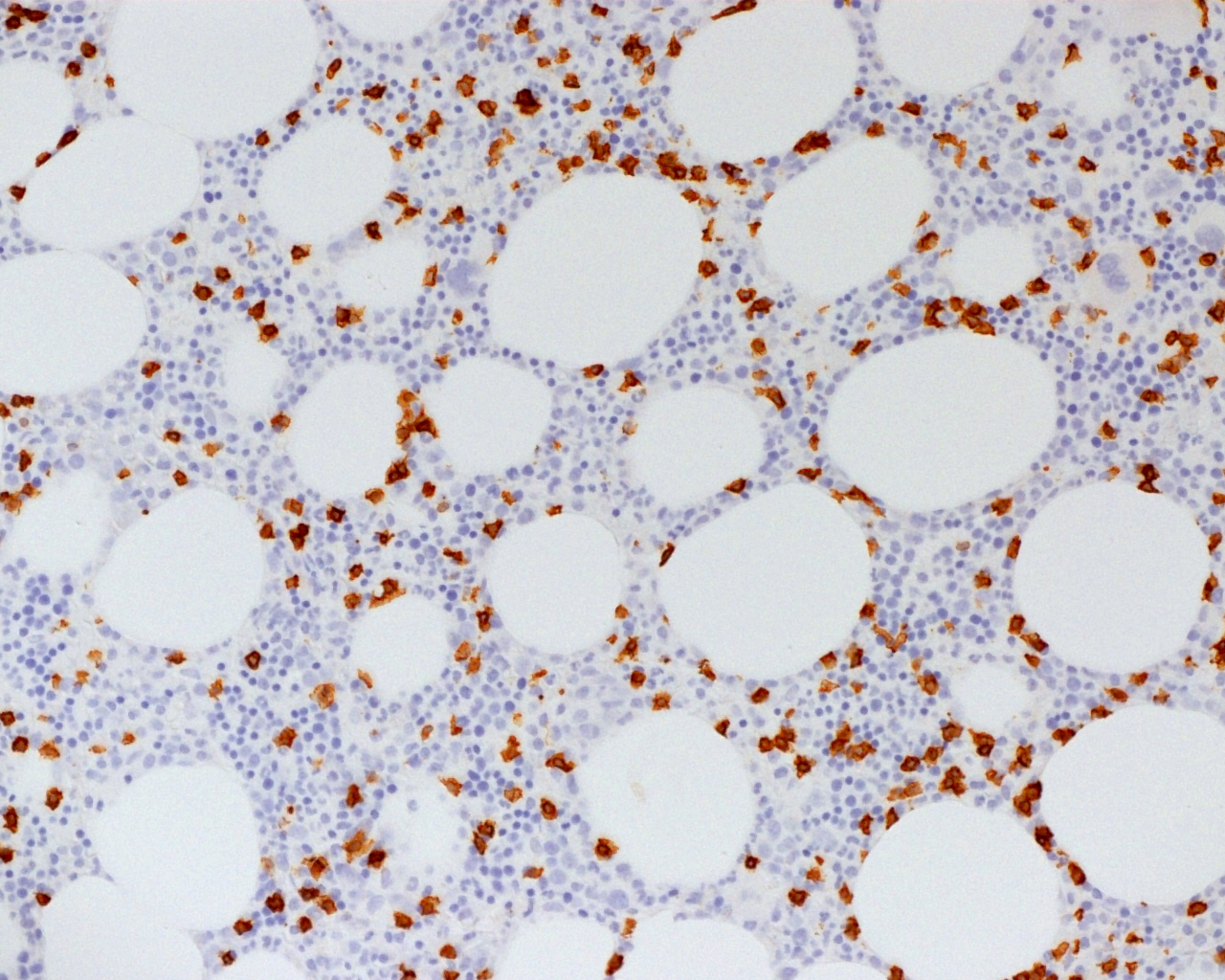
CD3 immunostain

DBA-44 immunostain
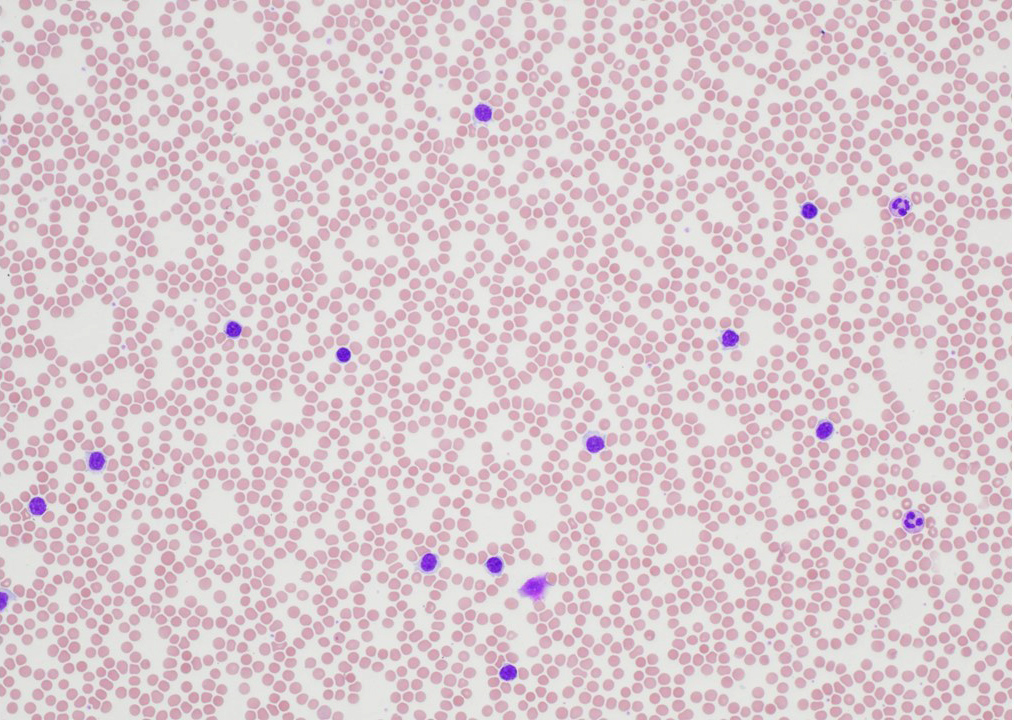
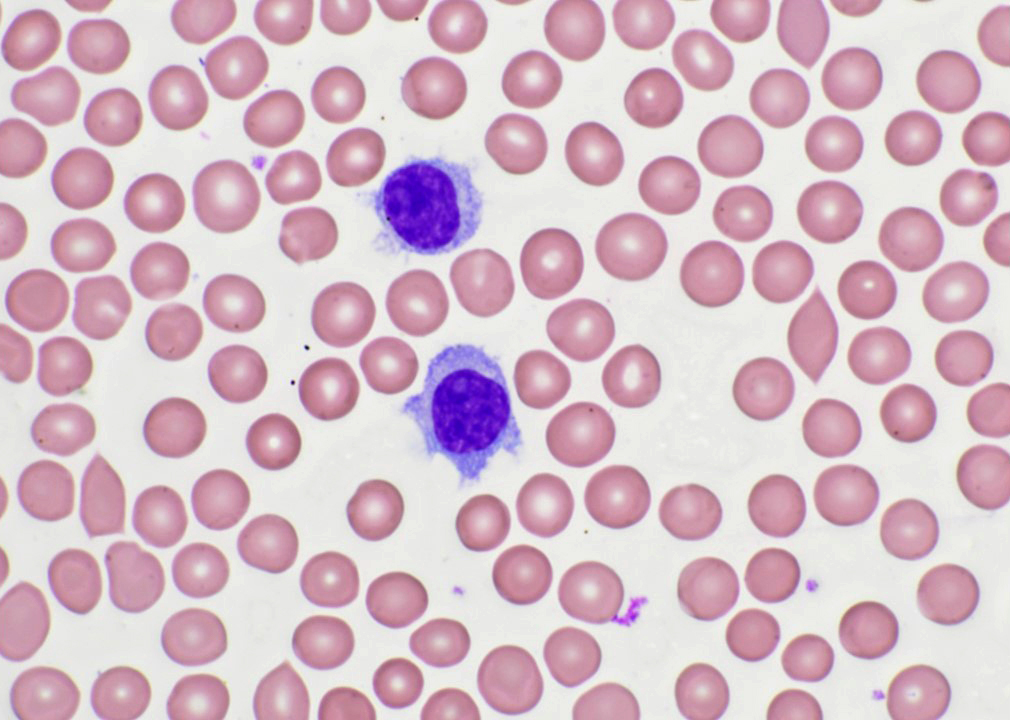
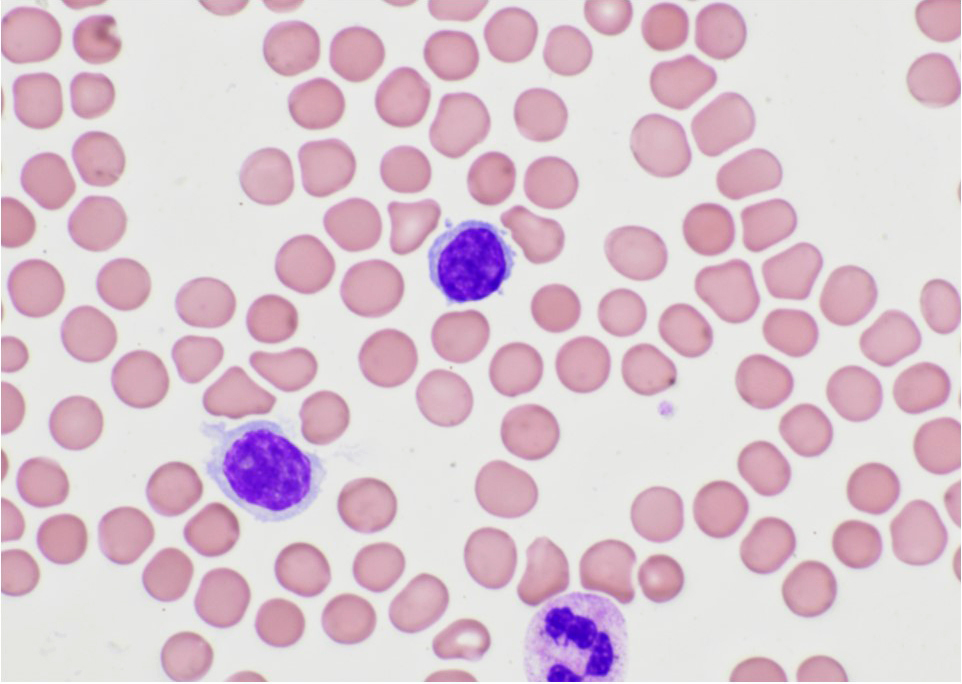
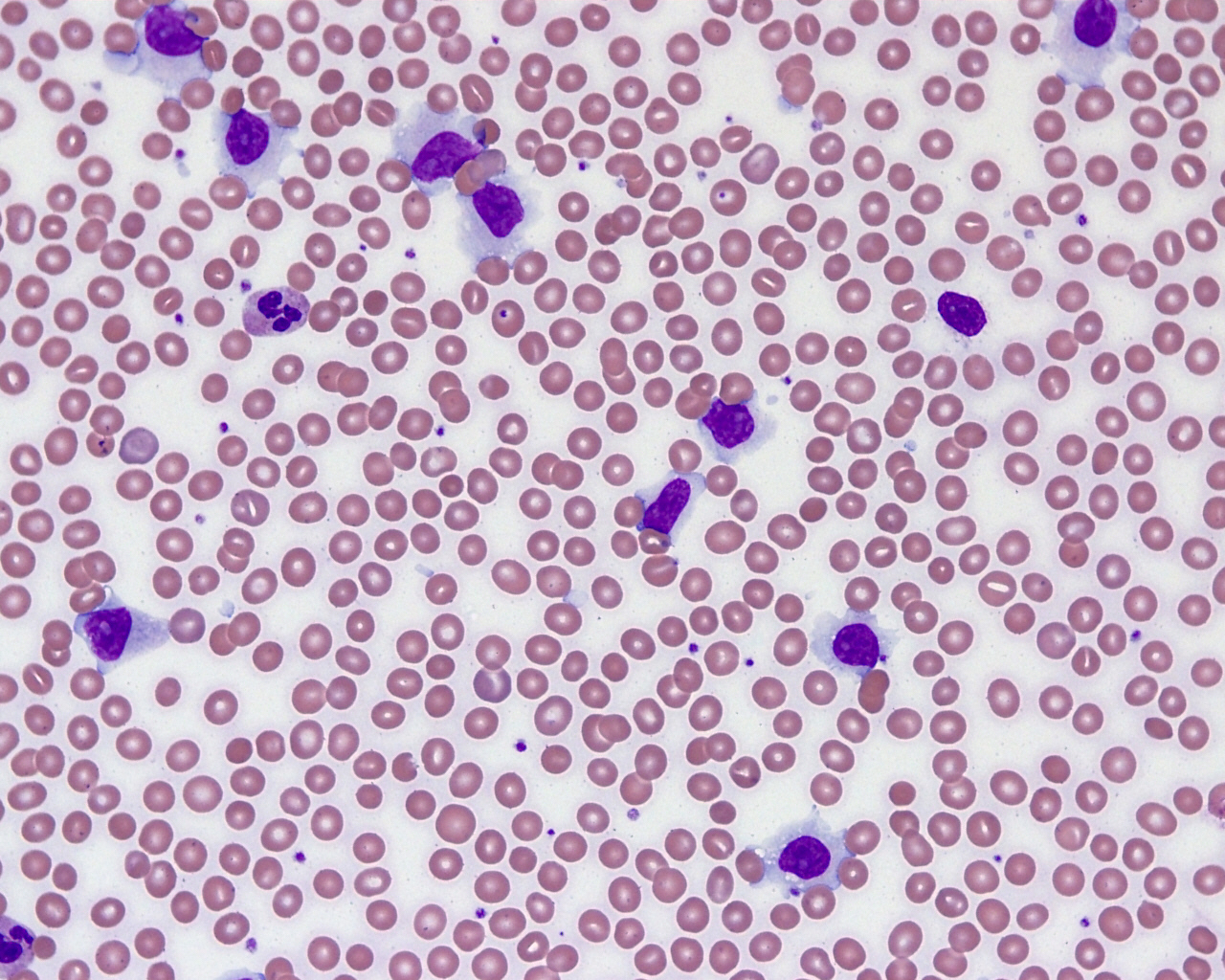
- Immunohistochemical stains
- References: Mod Pathol 2018;31:1717, Best Pract Res Clin Haematol 2015;28:253
- Positive
- CD19, CD20, CD22 (variable)
- CD11c (bright)
- CD103 (variable)
- FMC7 (variable)
- Monotypic surface immunoglobulin (most frequently IgG; bright)
- Negative
- CD5, CD10, CD25 and CD123
- CD200 differential expression diminished or negative (Cytometry B Clin Cytom 2019;96:275, Am J Clin Pathol 2013;140:536)
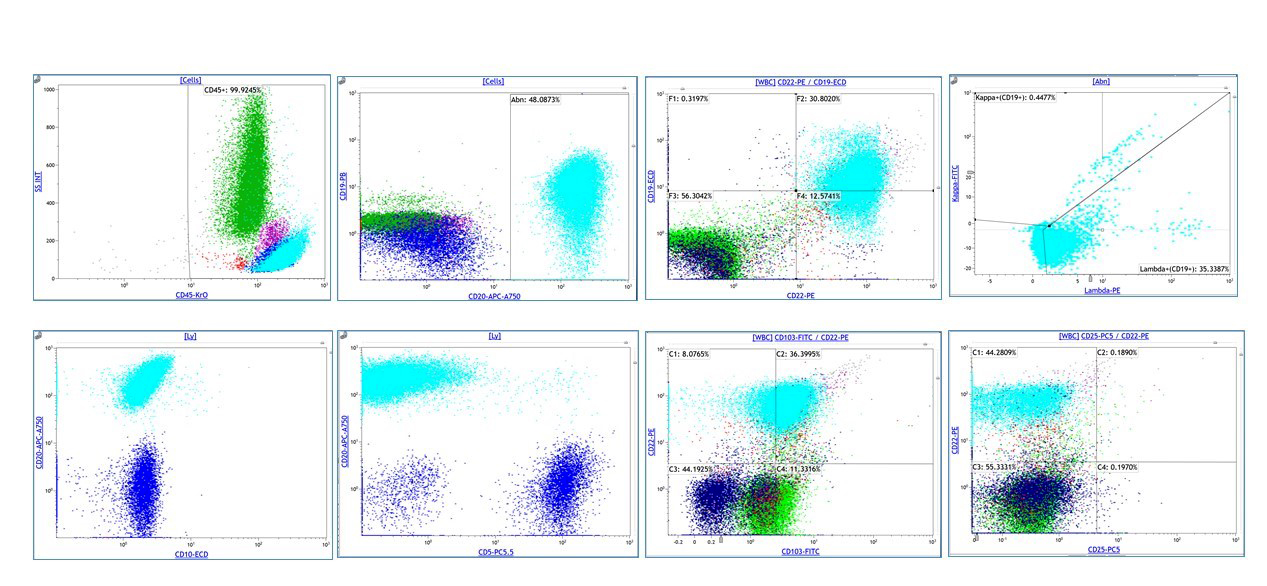
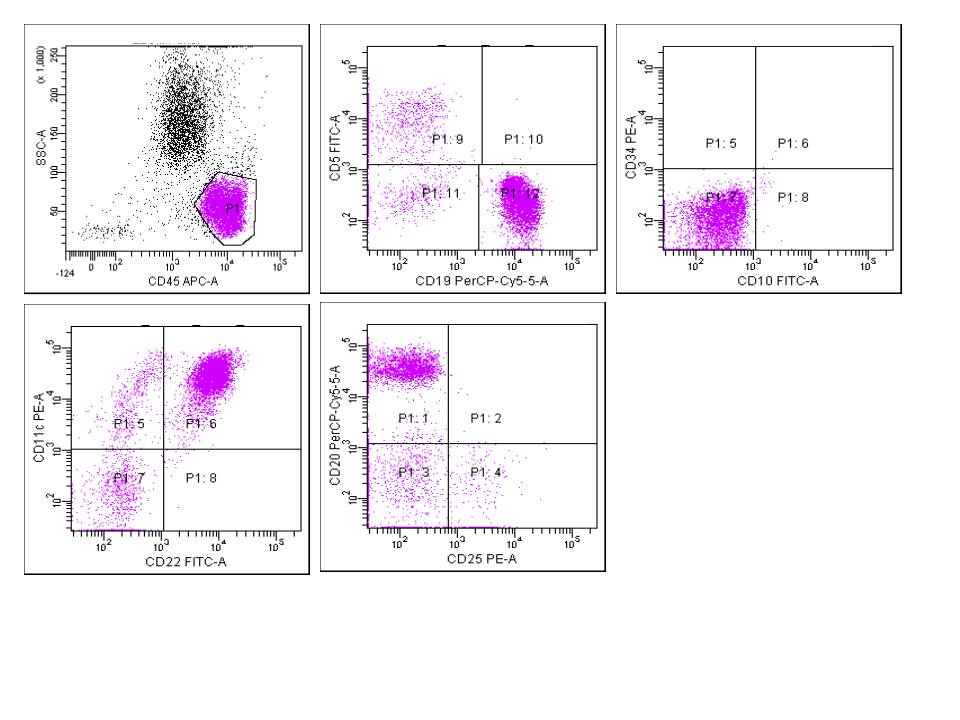
- Negative for BRAF V600E mutation
- No somatic mutations of IGHV: 33% of cases
- Unmutated cases have a high frequency of TP53 dysfunction
- VH gene family, IGHV4-34 (immunoglobulin heavy variable) overexpression (Blood 2012;119:3330)
- Recurrent mutations in the gene MAP2K1 (15q22.31) (Nat Genet 2014;46:8)
- Mutations in KMT2C, CCND3 and U2AF1 seen in a subset (Blood 2017;130:1644)
- Large number of DNA copy number alterations
- Most frequent gains on chromosome 5 and losses on 7q and 17p (Leukemia 2011;25:1189)
- A - D: bone marrow, left posterior iliac crest, trephine biopsy, aspirate smears, peripheral blood and flow cytometry analysis:
- CD103 positive / CD5 negative / CD10 negative B cell lymphoma (see comment)
- Comment: Bone marrow biopsy demonstrates increased CD20+ atypical lymphoid cells in the interstitial distribution. Flow cytometry analysis exhibits monotypic lambda restricted CD5 negative / CD10 negative B cell population with bright CD20, CD11c, variable expression of CD103 and lack of CD25. Overall findings, in conjunction with morphologic, immunophenotypic features (lack of CD25 (on flow cytometry) and annexin A1 (by IHC)) are suggestive of splenic B cell lymphoma / leukemia, unclassifiable, favor hairy cell leukemia variant (HCL variant). However, the differential diagnosis includes splenic diffuse red pulp small B cell lymphoma (SDRPL). Additional cytogenetic and molecular studies are in progress and will be reported separately.
- Peripheral smear: Manual review of the peripheral blood shows normochromic, normocytic anemia, thrombocytopenia and increased atypical mononuclear cells.
- RBCs: Mild normochromic, normocytic anemia with anisopoikilocytosis.
- WBCs: Increased atypical lymphocytes (~ 15%) on smear that are intermediate in size with cytoplasmic projections and occasional prominent nucleoli.
- Platelets: Thrombocytopenia with unremarkable morphology.
- Bone marrow biopsy: Quality: adequate. Cellularity: 30 - 40% (normocellular marrow for age). Hematopoiesis: trilineage maturation with increased interstitial lymphocytes (better highlighted on CD20 immunostain ~ 10 - 20% of biopsy cellularity).
- Immunohistochemical stains: Show interstitial small lymphocytes that are positive for CD20 and PAX5, while negative for CD3, CD5, CD10, BCL6, BCL1, BRAF / VE1, CD25 and annexin A1.
- Bone marrow clot section: Quality: adequate. Morphologic features are similar to those observed in the core biopsy.
- Special stains: Reticulin stain: no increase in reticulin fibrosis (MF = 0).
- Bone marrow aspirate: Quality: adequate. Increased atypical lymphocytes that are intermediate in size with cytoplasmic projections and occasional prominent nucleoli. Myeloid and erythroid lineage shows progressive maturation with no significant dysplastic features. Megakaryocytes are adequate in number with unremarkable morphology.
- Iron stores (PERL stain): Yes positive stores, no ring sideroblasts.
- Flow cytometry studies: Flow cytometry immunophenotypic studies: abnormal B cell population detected. Monotypic lambda restricted B cell population (40% of total, 72% of cells in lymphocyte gate) exhibiting the following immunophenotype: CD5-, CD10-, CD19+, CD20+ (bright), CD22+, CD25- and CD103+. Remaining antigens are negative or noncontributory.
- See Diagram 1
- Hairy cell leukemia:
- Pancytopenia and monocytopenia with characteristically few circulating cells
- Reniform or oval nuclei, circumferential long villi, inconspicuous nucleoli
- Spleen: diffuse infiltrate in red pulp cords, effaced white pulp, formation of blood lakes
- Marked reticulin fibrosis (dry tap)
- Immunophenotype: CD25+, CD103+, CD123+
- Genetics: BRAF V600E (~ 100%)
- Treatment: purine analogs, rituximab
- Splenic diffuse red pulp small B cell lymphoma:
- Splenic marginal zone lymphoma:
- No monocytopenia
- Smaller cells may have short cytoplasmic projections
- Cells have clumpy chromatin, indistinct nucleoli, different staining pattern
- Spleen: white pulp is infiltrated by the neoplastic cells, marginal zone differentiation
- Immunophenotype: CD25+, CD103 variable, CD123-
- Cytogenetic: deletion of 7q
- Genetic: NOTCH2, VH1 / 2
- Treatment: splenectomy, rituximab
- Asymptomatic patient with normal counts
- IGHV mutation status
- Mutation in TP53
- Young age
Comment Here
Reference: Hairy cell leukemia variant

Which of the following stains are most useful for differentiating classic hairy cell leukemia and hairy cell leukemia variant?
- CD21, CD23, CD35
- CD103, CD11c, CD25, annexin A1
- CD34, CD117, myeloperoxidase
- CD3, CD10, CD20, BCL2
Comment Here
Reference: Hairy cell leukemia variant
- Affects adult patients
- BRAF V600E mutation
- CD103 positivity
- Splenomegaly
Comment Here
Reference: Hairy cell leukemia variant
- Classic hairy cell leukemia
- Hairy cell leukemia variant
- Splenic diffuse red pulp small B cell lymphoma
- Splenic marginal zone lymphoma
Comment Here
Reference: Hairy cell leukemia variant
- Homozygosity for C (βC) allele of hemoglobin beta chain (HBB) gene
- Mild chronic hemolysis
- Characteristic blood smear morphology
- Relative protection against severe Plasmodium falciparum malaria
- Diagnosed by electrophoresis or high performance liquid chromatography (HPLC)
- HbC disease, HbCC
- ICD-10: D58.2 - other hemoglobinopathies
- Found worldwide
- Most prevalent in West African countries such as Ghana and Ivory Coast (BMC Res Notes 2018;11:215)
- Prevalence of hemoglobin C gene in Black Americans approximately 2% (Hemoglobin 2013;37:16)
- Bone marrow, spleen
- HbC has lysine (AAG) substituted for glutamate (GAG) at position 6 of the beta chain
- Has poor solubility, leading to precipitation in red blood cells
- Activation of potassium / chloride channels leads to cellular dehydration
- Slightly shortened red blood cell survival (hemolytic anemia)
- Inherited genetic mutation
- Mild chronic hemolysis, usually well compensated and asymptomatic
- Some patients have mild anemia, splenomegaly or gallstones (Hemoglobin 2013;37:16)
- Protects against severe Plasmodium falciparum malaria (Sci Rep 2017;7:14267)
- Note that heterozygous C train (HbAC) is clinically asymptomatic, manifesting only as target cells in the peripheral blood smear but does confer some protection against severe P. falciparum malaria
- Hemoglobin electrophoresis or HPLC shows about 95% HbC with no HbA and less than 5% HbA2 and HbF
- Molecular genetic testing important in some cases; beta gene sequencing currently best method
- Normal to mildly decreased hemoglobin (Hemoglobin 2013;37:16)
- Normal to slightly increased reticulocytes (Hemoglobin 2013;37:16)
- Normal to slightly increased bilirubin and lactate dehydrogenase (LDH)
- Decreased mean corpuscular volume (microcytosis), on average about 55 fL
- Normal to increased mean corpuscular hemoglobin concentration (MCHC)
- Normal to slightly increased red cell distribution width (RDW)
- Relative reduction in glycated HbA1C (Clin Chim Acta 2012;413:819)
- Overall prognosis good; no known reduction in overall life expectancy
- 21 month old girl with anemia (Am J Hematol 2019;94:144)
- 23 year old woman with sickle cell disease (Transfusion 2012;52:466)
- 56 year old man with gallstones (Am J Hematol 2015;90:174)
- No treatment required
- Genetic counseling may be indicated to address risk of compound heterozygosity in offspring
- Bone marrow is hypercellular with relative erythroid hyperplasia
- Irregular nuclear contours and other dyspoietic changes may be seen
- Microcytosis
- Numerous target cells
- Irregularly contracted cells
- Occasional rhomboid crystals, more numerous postsplenectomy
Contributed by Daniel D. Mais, M.D.

Peripheral blood smear (Wright stain)

Hemoglobin electrophoresis
- Beta globin (HBB) gene located at 11p15.5
- Point mutation in sixth codon, GAG > AAG
- Lysine encoded instead of glutamate at sixth amino acid (β6 glu→lys)
- Same amino acid position affected in HbS (β6 glu→val)
- Hemoglobin electrophoresis:
- Consistent with hemoglobin C disease (see comment)
- Comment: Blood smear demonstrates microcytic red cells, frequent target cells, boat cells and occasional blunt ended hemoglobin crystals. Electrophoresis shows 96.7% HbC.
- On cellulose acetate electrophoresis at alkaline pH, migrates with:
- HbE
- HbO Arab
- HbC Harlem
- HbA2
- Distinguished on citrate agar
- On HPLC, migrates very close to:
- HbA2
- Hb Lepore
- Distinguished by electrophoresis
- Rarely, transient acquired HbC after exchange transfusion (Ann Clin Lab Sci 2015;45:627)
- Affected patients are more prone to severe malaria
- Affected patients are usually transfusion dependent
- Affected patients may have falsely low glycated HbA1c
- HbC comigrates with HbE and HbO Arab on citrate agar
- It is clinically more severe than sickle cell disease
Comment Here
Reference: Hemoglobin C disease
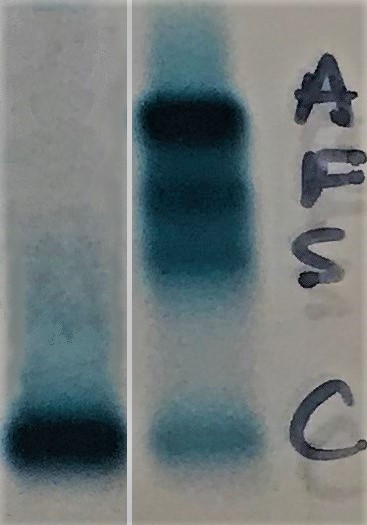
In the image shown above of an alkaline electrophoretic gel, what other hemoglobin variant comigrates with the abnormal band in the left panel?
- HbCS
- HbD
- HbF
- HbO
- HbS
- Rare, aggressive extranodal neoplasm that originates from nonactivated cytotoxic T cells; it usually expresses the T cell receptor (TCR) γδ (gamma delta) (Ann Diagn Pathol 2017;26:16, Am J Surg Pathol 2016;40:676, Am J Surg Pathol 2017;41:82, Cancer Treat Res 2019;176:185)
- First described in 1981 by Marshall Kadin as erythrophagocytic Tγ lymphoma; recognized as a distinct entity in 1994 (N Engl J Med 1981;304:648)
- Rare, aggressive extranodal neoplasm that originates from cytotoxic T cells; it usually expresses the T cell receptor (TCR) γδ (gamma delta) (Am J Surg Pathol 2017;41:82)
- Hepatomegaly and splenomegaly are the most common clinical manifestations (Ann Diagn Pathol 2017;26:16)
- Liver and bone marrow: sinusoidal infiltration (Histopathology 2014;64:171)
- Median bone marrow tumor burden ~30% (5 - 80%) (Am J Surg Pathol 2016;40:676)
- Spleen
- Diffuse involvement; expansion of the red pulp cords and sinusoids, with atrophy or absence of white pulp (Hum Pathol 2018;74:5)
- Cytology: variable
- Usually monotonous, small to intermediate size cells with pale agranular cytoplasm, round / oval nuclei, moderately condensed chromatin and inconspicuous nucleoli (Blood 2003;102:4261)
- Obsolete term: erythrophagocytic Tγ lymphoma
- ICD-O: 9716/3 - hepatosplenic T cell lymphoma
- < 1% of all non-Hodgkin lymphomas and 1 - 2% of all T / NK cell lymphomas (Hum Pathol 2018;74:5, Semin Diagn Pathol 2020;37:47, Clin Lymphoma Myeloma Leuk 2021;21:106)
- Most common T cell lymphoma that affects the spleen (Semin Diagn Pathol 2020;37:47)
- M > F (Am J Surg Pathol 2017;41:82, Blood 2020;136:2018, Int J Gen Med 2021;14:8399)
- More common in young adult men, median age ~35 years (range: 4 - 82 years) (Ann Diagn Pathol 2017;26:16, Hum Pathol 2016;50:109, Am J Surg Pathol 2016;40:676, Am J Surg Pathol 2017;41:82, Blood 2003;102:4261)
- Increased incidence in the United States in the last 2 decades (Am J Hematol 2017;92:E99)
- Neoplastic clonal proliferation of γδ T cells induced by the upregulation of the JAK / STAT (STAT3 and STAT5B) and PI3K signaling pathways or mutations of chromatin modifiers (SETD2, INO80 and ARID1B) (Hum Pathol 2018;74:5, Haematologica 2019;104:e104, Blood 2020;136:2018)
- Unknown in 80% of cases
- Up to 20% of cases arise in a context of chronic immunosuppression (Hum Pathol 2018;74:5, Blood 2020;136:2018, Curr Opin Oncol 2021;33:406)
- Posttransplantation (kidney or liver) (Am J Clin Pathol 2000;113:487, Cancer Treat Res 2019;176:185)
- Patients with immunodysregulatory disorders (inflammatory bowel disease, rheumatoid arthritis) that are using immunosuppressive agents (mercaptopurine, azathioprine, infliximab) (Ann Diagn Pathol 2017;26:16, Am J Surg Pathol 2016;40:676, Cancer Treat Res 2019;176:185, Blood 2020;136:2018)
- TNFα inhibitors in Crohn's disease: fourfold increased risk of lymphomas (B cell lymphomas and hepatosplenic T cell lymphoma) (Am J Gastroenterol 2013;108:99)
- Risk factors for hepatosplenic T cell lymphoma (HSTCL) in patients with inflammatory bowel disease: male sex and long term thiopurine therapy (≥ 2 years)
- Genetic predisposition and chronic antigenic stimulation
- Hepatomegaly and splenomegaly are the most common clinical manifestations (Ann Diagn Pathol 2017;26:16, Am J Surg Pathol 2017;41:82, Clin Lymphoma Myeloma Leuk 2021;21:106)
- 70 - 80% of patients
- Massive splenomegaly is found in most patients, defined as ≥ 6 cm below costal margin, or ≥ 20 cm in greatest dimension, or splenectomy > 1,000 g
- Abdominal discomfort
- B symptoms (Am J Surg Pathol 2016;40:676)
- Lymphadenopathy is uncommon (< 25%); when present, splenic peripheral lymph nodes are most commonly involved (Histopathology 2014;64:171, Blood 2020;136:2018, Semin Diagn Pathol 2020;37:47)
- Peripheral blood involvement at initial presentation is uncommon
- Cytopenia or pancytopenia (Blood 2003;102:4261, Cancer Treat Res 2019;176:185, Blood 2020;136:2018):
- Moderate anemia, thrombocytopenia and neutropenia
- Hypersplenism, bone marrow infiltration or immune related
- Moderate anemia, thrombocytopenia and neutropenia
- Lymphocytosis in ~10% of the cases
- Cytopenia or pancytopenia (Blood 2003;102:4261, Cancer Treat Res 2019;176:185, Blood 2020;136:2018):
- Elevated levels of lactate dehydrogenase (LDH), β2 microglobulin and bilirubin (Hum Pathol 2018;74:5)
- Bone marrow (BM) is usually involved at initial presentation (Hum Pathol 2016;50:109)
- Cutaneous involvement is rare
- CT or MRI (Blood 2020;136:2018):
- Homogeneous hepatosplenomegaly
- Fluorodeoxyglucose (FDG) PET / CT (Blood 2020;136:2018):
- Diffuse FDG uptake in affected areas
- Usually liver, spleen and bone marrow
- Diffuse FDG uptake in affected areas
- Poor prognosis, with relapse in most cases (Am J Surg Pathol 2016;40:676)
- Median overall survival (OS) is 12 months (range: 3 - 34 months) (Hum Pathol 2018;74:5, Hum Pathol 2016;50:109, Int J Gen Med 2021;14:8399)
- 3 year overall survival: 37.6%
- 5 year overall survival: 31.6%
- Median progression free survival (PFS): 9.5 months 95% (CI, 1.8 - 16.3) (Clin Lymphoma Myeloma Leuk 2021;21:106)
- Prognostic factors correlated with worse outcomes:
- Serum bilirubin level > 1.5 mg/dl
- Expression of TCRαβ
- Trisomy 8
- 20 year old man hepatosplenic T cell lymphoma αβ subtype presenting with Coombs negative hemolytic anemia (Clin Med Insights Oncol 2015;9:123)
- 47 year old man with Crohn's disease on thiopurine monotherapy (World J Gastroenterol 2016;22:10465)
- 53 year old woman with coexisting hairy cell leukemia and hepatosplenic T cell lymphoma (Diagn Pathol 2014;9:58)
- 56 year old woman with hemophagocytic lymphohistiocytosis associated with hepatosplenic T cell lymphoma (Autops Case Rep 2014;4:19)
- 59 year old man with hepatosplenic αβ T cell lymphoma masquerading as cirrhosis (J Gastrointest Oncol 2013;4:131)
- 59 year old man with hepatosplenic T cell lymphoma successfully treated with PD-1 inhibitor, chidamide and chemotherapy (J Dig Dis 2022;23:50)
- No standard therapeutic guideline
- Cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP), or CHOP-like, hyper CVAD (cyclophosphamide, vincristine sulfate, adriamycin and dexamethasone) or hyper CVAD-like; or non CHOP induction regimen (2' deoxycoformycin) (Ann Diagn Pathol 2017;26:16, Am J Surg Pathol 2016;40:676)
- Consolidative stem cell transplant (SCT) for eligible patients (Haematologica 2005;90:ECR14, Clin Lymphoma Myeloma Leuk 2013;13:8, Cancer Treat Res 2019;176:185, Curr Opin Oncol 2021;33:406)
- Potential target therapy: CD52, JAK1 / 2, STAT3, STAT5B and PI3KCD (Hum Pathol 2018;74:5, Cancer Treat Res 2019;176:185)
- Diffuse and homogeneous, enlarged liver and spleen; without distinct macroscopic lesions; lymph nodes are usually not enlarged (Blood 2003;102:4261)
- Spleen
- Diffuse involvement; expansion of the red pulp cords and sinusoids, with atrophy or absence of white pulp (Hum Pathol 2018;74:5, Blood 2020;136:2018)
- Hemophagocytosis may be present (Blood 2020;136:2018)
- Liver and bone marrow: sinusoidal infiltration (Histopathology 2014;64:171, Blood 2003;102:4261, Blood 2020;136:2018)
- Median bone marrow tumor burden ~30% (5 - 80%) (Am J Surg Pathol 2016;40:676, Blood 2020;136:2018)
- Bone marrow tumor burden directly related to neutropenia (Hum Pathol 2016;50:109)
- Median bone marrow tumor burden ~30% (5 - 80%) (Am J Surg Pathol 2016;40:676, Blood 2020;136:2018)
Contributed by Roberto N. Miranda, M.D.
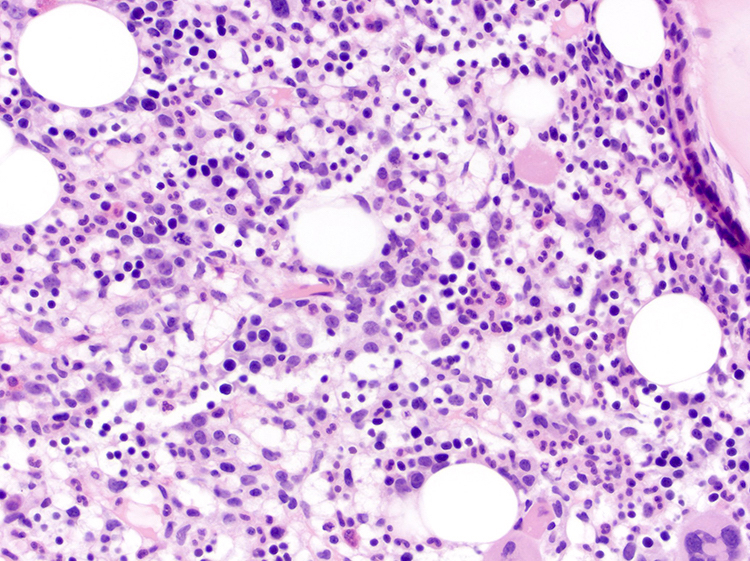

Bone marrow infiltration
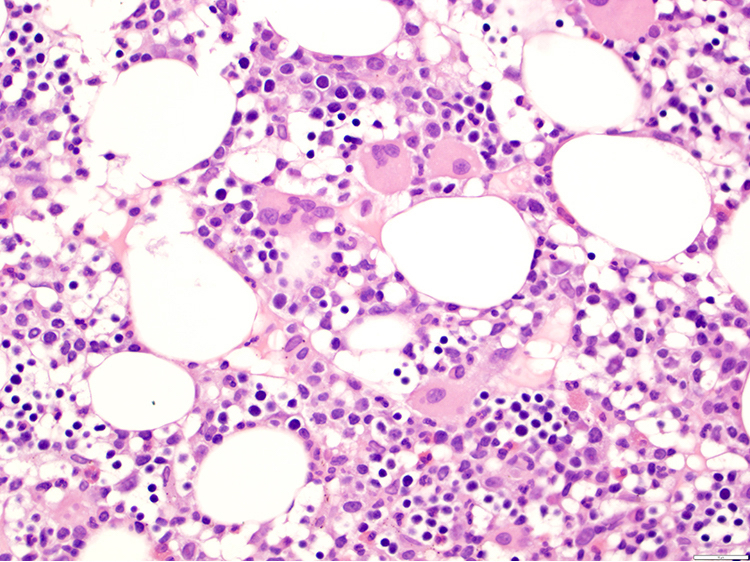
Bone marrow with dyspoietic changes
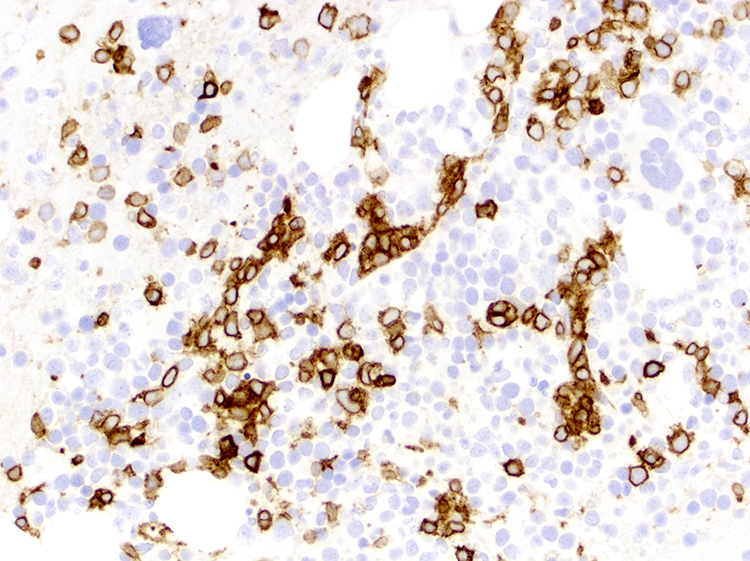
CD3 positive
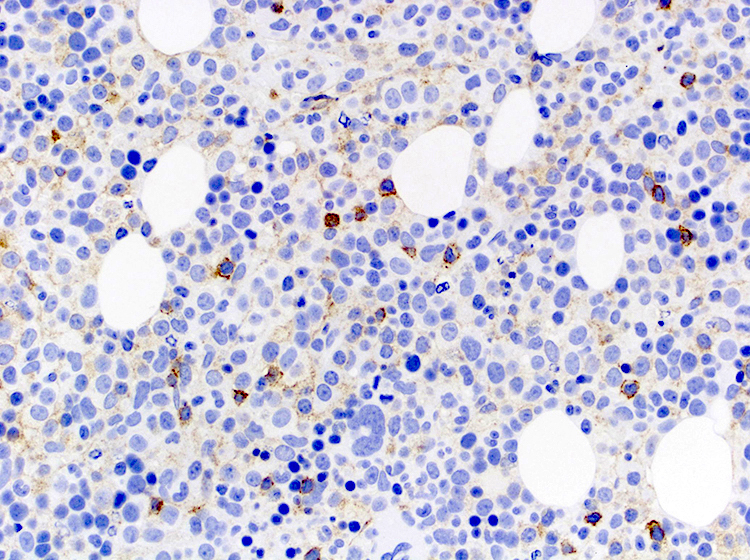
CD4 negative

CD8 negative
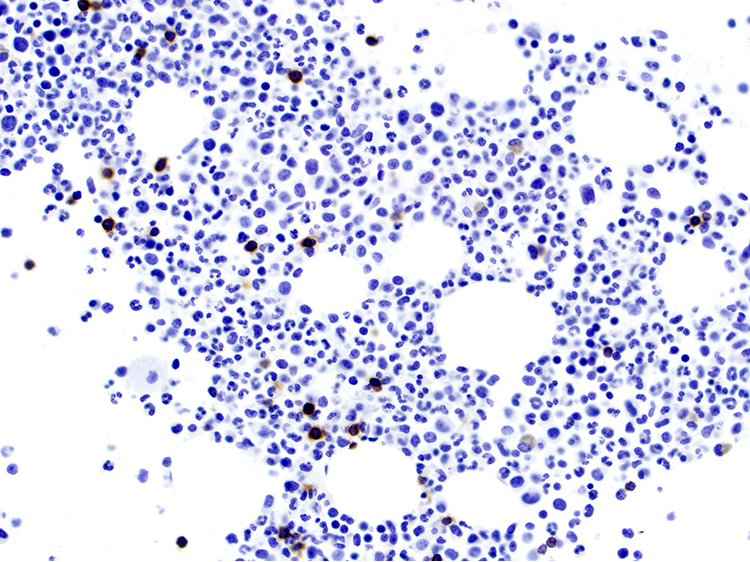
TCR βF1 negative
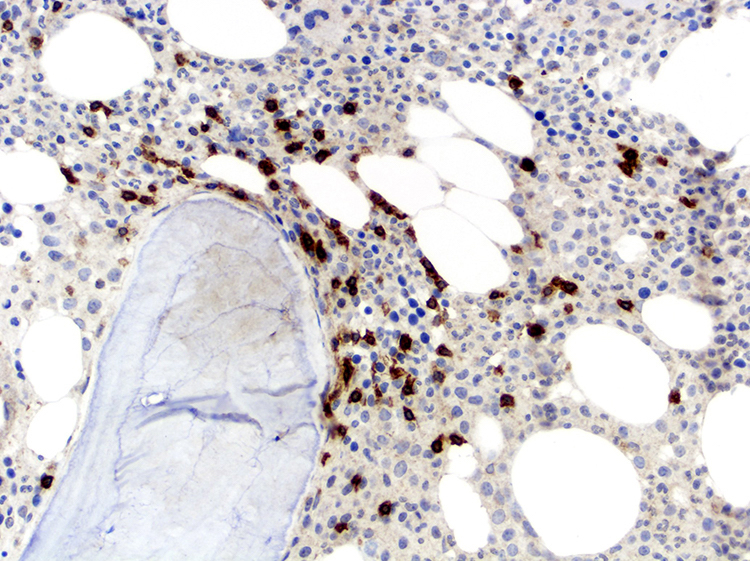
TCRγδ positive
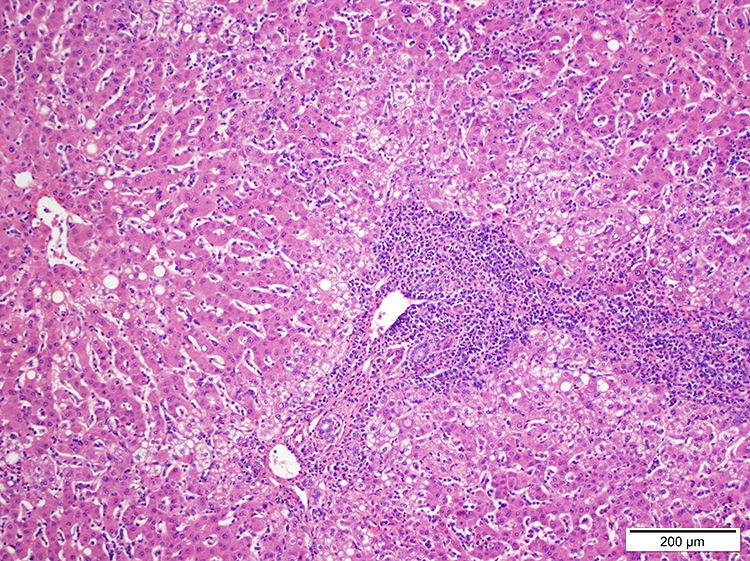
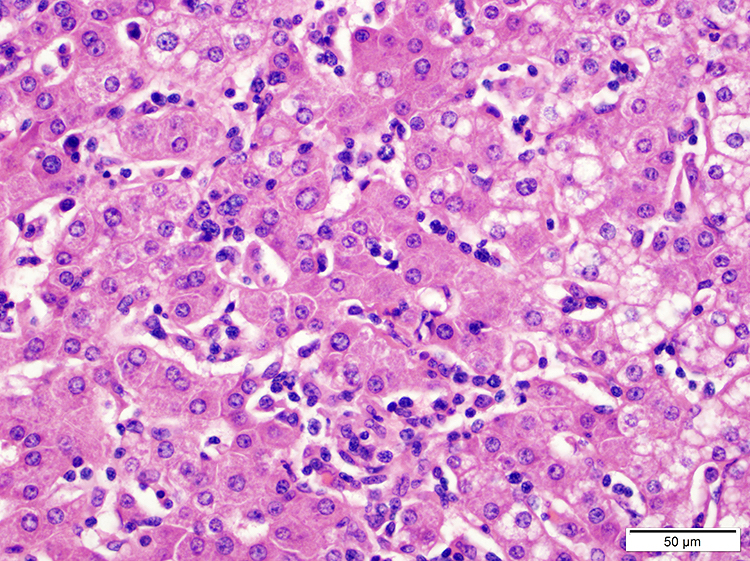
Liver infiltration
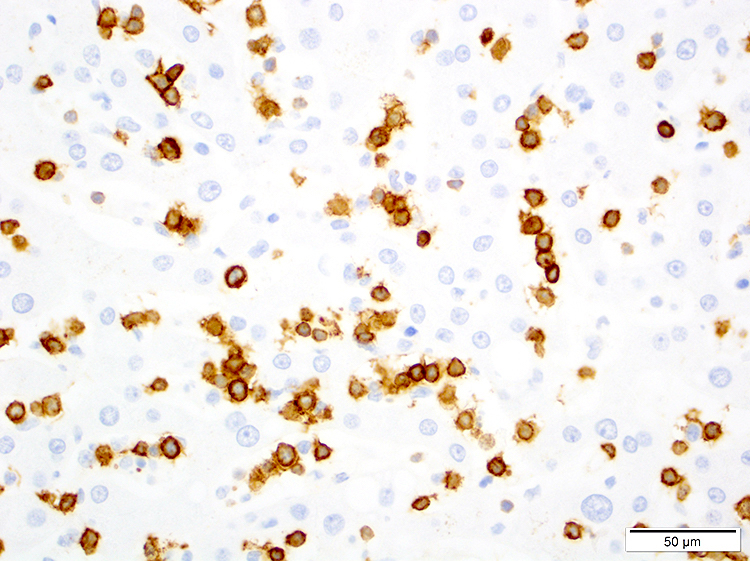
HSTCL: CD3 positive
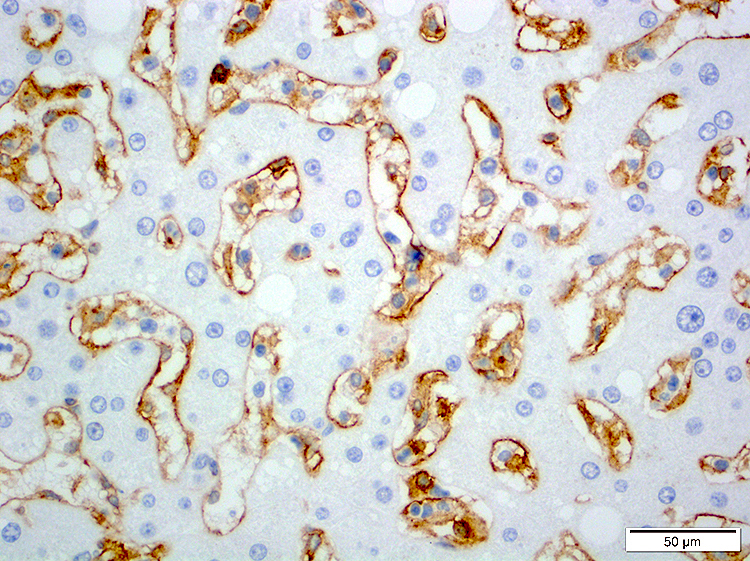
HSTCL: CD4 negative

CD8 positive
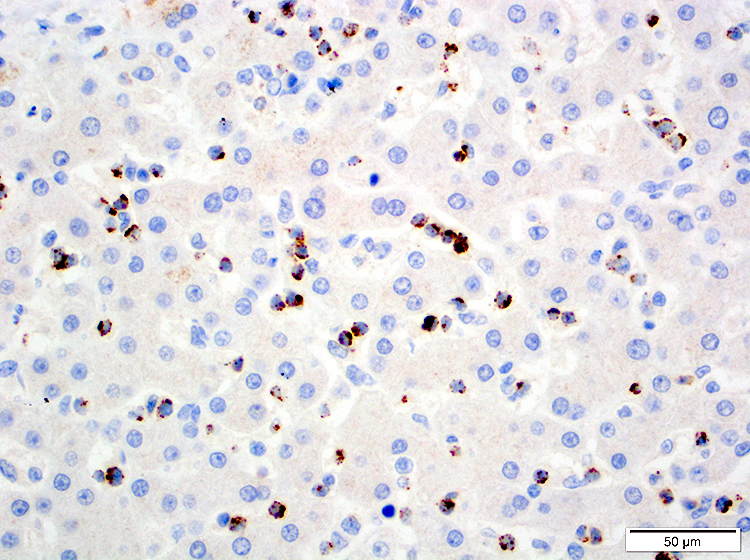
TIA1 positive
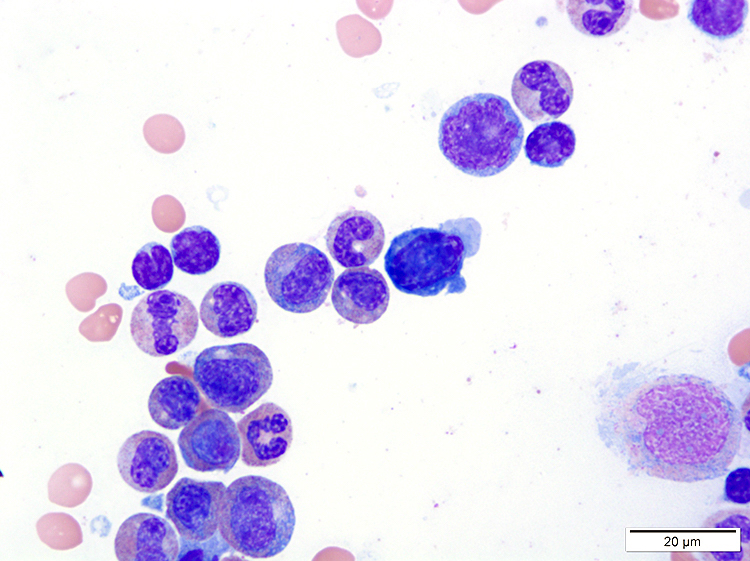
Small size morphology
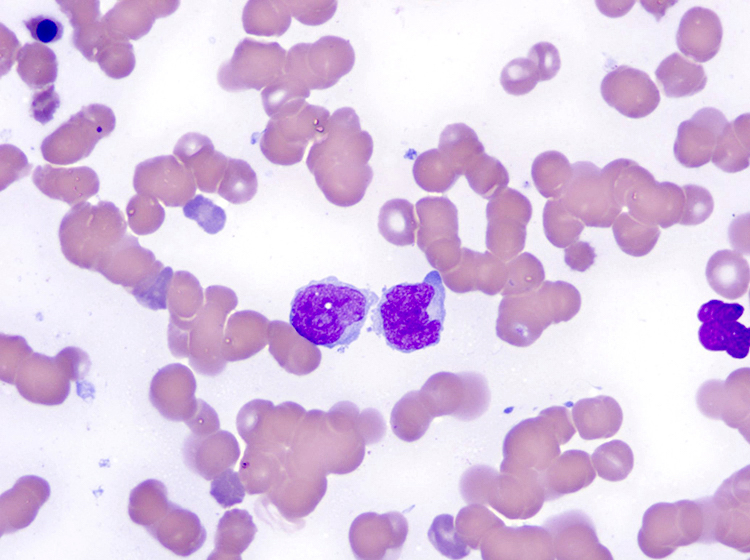
Intermediate size morphology
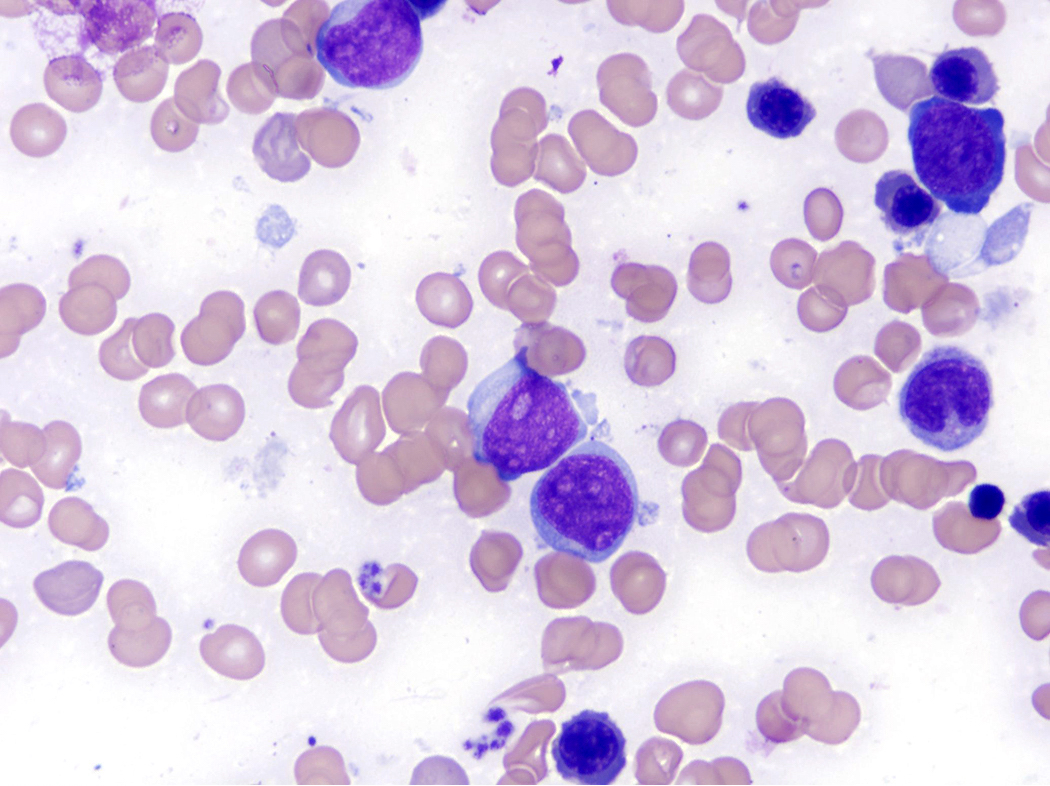
Blastic morphology
- Variable
- Usually monotonous, small to intermediate size cells with pale agranular cytoplasm, round / oval nuclei, moderately condensed chromatin and inconspicuous nucleoli (Am J Surg Pathol 2016;40:676, Am J Surg Pathol 2017;41:82, Blood 2003;102:4261, Blood 2020;136:2018, Semin Diagn Pathol 2020;37:47)
- Blastic cells (medium to large cells with high nucleus to cytoplasm ratio, fine chromatin, without nucleoli) (Am J Clin Pathol 2001;116:410)
- Usually monotonous, small to intermediate size cells with pale agranular cytoplasm, round / oval nuclei, moderately condensed chromatin and inconspicuous nucleoli (Am J Surg Pathol 2016;40:676, Am J Surg Pathol 2017;41:82, Blood 2003;102:4261, Blood 2020;136:2018, Semin Diagn Pathol 2020;37:47)
- Background trilineage dyspoietic changes in bone marrow (Hum Pathol 2016;50:109)
- Dysmegakaryopoiesis is most common: hypolobated or small size nuclei
- Hemophagocytosis (< 5%) (Hum Pathol 2018;74:5, Semin Diagn Pathol 2020;37:47)
- T cell antigens: CD2, CD3, CD7, TIA1, granzyme M, Fas ligand (Ann Diagn Pathol 2017;26:16, Hum Pathol 2016;50:109, Am J Surg Pathol 2016;40:676, Am J Surg Pathol 2017;41:82, Histopathology 2014;64:171, Blood 2003;102:4261, Blood 2020;136:2018)
- CD56: 67% of cases; related to immune disorders
- Ki67 variable (Hum Pathol 2018;74:5)
- TCRγδ
- CD1a, CD4, CD5, CD8, CD10, CD57, CD94, TdT, Epstein-Barr virus encoded small RNAs (EBER) (Ann Diagn Pathol 2017;26:16, Hum Pathol 2016;50:109, Am J Surg Pathol 2016;40:676, Am J Surg Pathol 2017;41:82, Histopathology 2014;64:171, Blood 2003;102:4261, Blood 2020;136:2018)
- B cell antigens: CD19, CD20 and CD25
- Mature and nonactivated cytotoxic T cell immunophenotype: granzyme B (albeit positive in 38% of cases) and perforin
- TCRαβ (20%): when present appears to carry a poor prognosis
- Follicular T cell markers (Semin Diagn Pathol 2020;37:47):
- Rare cases negative for TCRγδ and αβ (TCR silent) (Hum Pathol 2018;74:5)
- Helpful for diagnosis (Semin Diagn Pathol 2020;37:47, Leuk Res 2021;108:106614):
Contributed by Roberto N. Miranda, M.D.
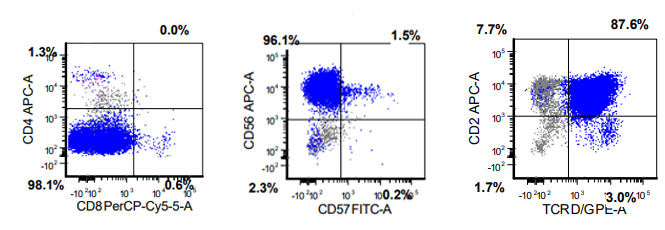
Positivity for CD2, TCRγδ and CD56
- Gamma delta origin: biallelic rearrangement of TRG (Am J Surg Pathol 2017;41:82, Blood 2003;102:4261)
- Chromatin modifying genes (~60%): SETD2, INO80, TET3 and SMARCA2 (Hum Pathol 2018;74:5, Blood 2020;136:2018)
- Mutations in JAK / STAT pathway
- STAT3 (10%) and STAT5B (30%) mutations
- RHOA, CD28 and CCR4 are usually absent (more common in other T cell lymphomas) (PLoS One 2014;9:e102977)
- DNA methylation profiling (Haematologica 2019;104:e104)
- Hypermethylated genes: CD5, BCL11B, CXCR6, GIMAP7
- Hypomethylated genes: ADARB1, NFIC, NR1H3, ST3GAL3
- Mutation of PIK3CD in up to 10% of cases (Cancer Discov 2017;7:369)
- Aberrant karyotype (67%)
- Isochromosome (7q) or ring chromosome 7 (Ann Diagn Pathol 2017;26:16, Genes Chromosomes Cancer 2002;33:243, Cancer Treat Res 2019;176:185, Blood 2020;136:2018)
- ~50% of cases
- Primary chromosomal aberration
- Loss of 7p22.1p14.1
- Gain of 7q22.11q31.1
- Trisomy 8 is usually present (Hum Pathol 2016;50:109, Am J Surg Pathol 2016;40:676, Blood 2020;136:2018)
- ~ 70% of cases
- Secondary event
- If present at time of diagnosis, there is a significantly shorter overall survival and progression free survival
- Bone marrow (right iliac crest) core and clot specimen; aspirate smears and peripheral blood:
- Hepatosplenic T cell lymphoma
- Flow cytometry immunophenotype demonstrated aberrant T cells with loss of CD4 and CD8, positive for T cell receptor gamma delta chain (see comment)
- Comment: According to clinical records, patient is a 28 year old man who has a history of Crohn's disease, for which he was receiving azathioprine and tumor necrosis alpha receptor antagonist. Patient reports malaise and fever for 3 months. On physical exam, patient had massive splenomegaly (8 cm below costal margin) and hepatomegaly. Laboratory findings showed leukopenia, severe anemia and thrombocytopenia. Additional testing proved negative for antiglobulin testing, negative for HIV, hepatitis B, hepatitis C and HTLV1.
- Bone marrow core biopsy and clot sections show similar features. There is 70% cellularity with trilineage hematopoiesis. Megakaryocytes are adequate in number and range from small, hypolobated forms to megakaryocytes of normal size and shape. There are scattered small clusters of small to intermediate size lymphocytes with hyperchromatic nuclei with irregular nuclear contours.
- Bone marrow aspirate smears show trilineage hematopoiesis with mild dysmegakaryopoiesis. There are approximately 15% lymphocytes, of small to intermediate size, with clear and agranular cytoplasm and central hyperchromatic nuclei with irregular nuclear contours.
- Immunohistochemical studies were performed in bone marrow core biopsy and the T cell marker CD3 highlights lymphocytes in an interstitial and sinusoidal pattern, representing approximately 10% of marrow cells. Atypical lymphocytes are also highlighted with CD2, CD7, CD56, TIA1 and granzyme M. The T cell receptor gamma delta is positive in lymphocytes, while the T cell receptor alpha beta (βF1 clone) is negative. CD4 and CD8 are negative in atypical lymphocytes. The following markers are also negative in lymphocytes: CD1a, CD5, CD10, CD57, EBER (EBV in situ hybridization), TCL1 and TdT.
- Concurrent flow cytometry immunophenotype demonstrates that approximately 15% of marrow cells are T lymphocytes, positive for CD2, CD7, CD52, CD56 and T cell receptor gamma delta. The atypical lymphocytes are negative for CD4, CD5, CD8, CD57 and T cell receptor alpha beta. The B lymphocytes are polytypic.
- Molecular testing revealed monoclonal T cell receptor gene rearrangement of gamma (V gamma 4 family) and beta chains. Next generation sequencing showed mutation of STAT5B. The specimen was negative for IGH gene rearrangement.
- Cytogenetic analysis revealed isochromosome 7 and trisomy 8
- Clinical correlation and followup is recommended.
- T cell large granular lymphocytic (LGL) leukemia (Am J Clin Pathol 2015;144:607, Am J Surg Pathol 2017;41:82):
- Elderly patients
- Absent or mild splenomegaly
- Liver may be involved but not enlarged
- Neoplastic lymphocytes with azurophilic granules and hyperchromatic nuclei
- Expression of CD5, CD8, CD57, granzyme B and TCRαβ
- Diagnostic challenge in cases of γδ T LGL:
- When compared with HSTCL, has a higher presence of azurophilic granules and unlikely to have massive splenomegaly
- Marrow infiltration is interstitial and may be focally sinusoidal but without expanding sinuses
- Aggressive NK cell leukemia / lymphoma:
- T cell prolymphocytic leukemia (Blood 2019;134:1132):
- T cell lymphoblastic leukemia / lymphoma (Haematologica 2020;105:2524):
- Splenic marginal zone lymphoma:
- Infiltration of white pulp and secondarily red pulp
- Small sized lymphocytes with abundant pale cytoplasm in spleen and villous lymphocytes in peripheral blood
- Chronic active EBV infection (Blood 2020;136:2018):
- Bone marrow infiltration of EBV+ small to intermediate sized lymphocytes (B, T or NK cells)
- Hemophagocytosis may be present
- No significant lymphocytic atypia or phenotypic abnormalities
- Nonsplenic cytotoxic T cell lymphoma with γδ+ phenotype (Histopathology 2014;64:171):
- Skin involvement
- Absence of hepatosplenomegaly
Which of the following immunophenotypic patterns corresponds to hepatosplenic T cell lymphoma?
- CD2+, CD3+, CD4+, CD5-, TIA1+, granzyme B+, TCRγδ+
- CD2+, CD3+, CD4-, CD5-, TIA1+, granzyme M+, TCRγδ+
- CD2+, CD3-, CD4+, CD5+, CD7+, CD8+, TCRγδ+
- CD2+, CD3-, CD4+, CD5+, CD7-, CD8-, TCRγδ+
- CD2-, CD3-, CD4-, CD5+, CD7-, CD8-, TCRγδ+
Comment here
Reference: Hepatosplenic T cell lymphoma
- Del(6q) and RHOA1 mutation
- Inversion 14 and TCL1 rearrangement
- Isochromosome 7 and STAT5B mutation
- t(2;5) and JAK / STAT activation
- 6p25 and DUSP22 / IRF4
- Most common type of anemia worldwide; occurs due to decreased iron levels in the body, which leads to decreased erythropoiesis
- Characterized by hypochromic microcytic red blood cells (RBCs)
- Iron deficiency anemia (IDA) is the most frequent presentation of iron deficiency
- Common causes are blood loss, reduced absorption, inadequate dietary intake, pregnancy, intestinal worm colonization and chronic inflammation
- Low ferritin levels; ferritin is an indicator of iron stores and is the most sensitive and specific biomarker
- Hypochromia, microcytosis and anisopoikilocytosis of the RBCs are the characteristic morphologic abnormalities
- Iron deficiency anemia (IDA)
- Microcytic, hypochromic anemia with anisopoikilocytosis
- Anemia of chronic disease or anemia of inflammation or acute blood loss anemia or nutritional anemia (depending on the cause)
- Iron deficiency is defined as ferritin < 15 ng/mL and transferrin saturation < 10% (PLoS One 2020;15:e0232125)
- Iron deficiency is more prevalent than IDA
- F > M; common in women of childbearing age, children and elderly
- Approximately 25% of people worldwide have anemia and 50% of the anemias are due to iron deficiency (StatPearls: Iron Deficiency Anemia [Accessed 16 February 2022])
- Affects > 1.2 billion population worldwide; iron deficiency in the absence of anemia is even more frequent (Blood 2019;133:30)
- Iron deficiency is more common in developing countries than in developed countries like the United States (StatPearls: Iron Deficiency Anemia [Accessed 16 February 2022])
- Iron is essential for hemoglobin synthesis during erythropoiesis
- Impaired delivery of iron to erythroid precursors results in decreased erythropoiesis
- Iron deficiency leading to IDA is a chronic process
- Initially normal RBCs are produced
- Later, decreased iron transport to bone marrow results in microcytic hypochromic RBCs
- Daily iron requirement is about 20 - 25 mg
- About 1 - 2 mg is absorbed daily and the remaining comes from recycled iron
- Iron absorption pathway
- Dietary iron is absorbed into the bloodstream via the small intestine
- Following absorption, iron may be stored in enterocytes or transported to other tissues via transferrin (Medicines (Basel) 2019;6:85)
- Iron homeostasis
- Role of hepcidin
- Peptide hormone produced and secreted by the liver; responsible for regulation of iron levels
- Binds to iron transporter ferroportin, causes its internalization and degradation, thus preventing iron transport and decreasing the circulating levels
- Levels directly correlate with serum ferritin levels, increases in systemic inflammation or infection
- Increased hepcidin causes iron sequestration and reduces supply of erythropoietic iron
- Its production is inhibited with increased erythropoiesis, iron deficiency and tissue hypoxia to facilitate iron absorption and release of iron from body stores (See Diagrams)
- When the iron stores are low, activation of hypoxia inducible factor 2α increases intestinal iron uptake through divalent metal transporter 1 (DMT1) (N Engl J Med 2015;372:1832)
- Iron recycling:
- Majority of the iron required by the body is recycled from senescent RBCs undergoing phagocytosis by the reticuloendothelial macrophages; this iron is either used for hematopoiesis or stored as ferritin and hemosiderin (N Engl J Med 2015;372:1832, Merck Manual: Iron Deficiency Anemia [Accessed 7 March 2022])
- Role of hepcidin
- Major causes:
- Blood loss: menstruation, gastrointestinal (GI) bleeding, intestinal worm colonization (developing countries)
- Women of childbearing age: menorrhagia; fibroid
- Adults > 50 years: GI bleeding is the most common cause and should be evaluated for malignancy
- Reduced absorption: celiac disease, Helicobacter pylori, gastritis, bariatric surgery
- Infants: iron is the most common single nutrient deficiency
- Children consuming cow’s milk are at greater risk of developing iron deficiency
- Higher concentration of calcium in cow’s milk competes with iron for absorption
- Inadequate dietary intake
- Increased demand: pregnancy, lactation and rapid growth phase in children (ASH: Iron Deficiency Anemia [Accessed 16 February 2022])
- Blood loss: menstruation, gastrointestinal (GI) bleeding, intestinal worm colonization (developing countries)
- Medications
- Proton pump inhibitors directly affect iron metabolism by suppressing iron absorption through the inhibition of duodenal ferroportin via hepcidin upregulation (Toxicol Lett 2020;318:86)
- Recombinant erythropoietin used in treatment of chronic kidney disease may increase need and utilization of iron by the hematopoietic tissues (Kidney Int Suppl 1999;69:S107)
- Procedures such as hemodialysis and frequent phlebotomies
- Blood remaining in the dialysis tubing and an increased rate of blood loss during dialysis can cause iron deficiency anemia (Acta Haematol 2019;142:44)
- Rare causes: iron refractory iron deficiency anemia
- Totally or partially resistant (refractory) to treatment with iron
- Occurs due to mutations in TMPRSS6, a hepcidin regulatory gene
- Mutations in other genes such as divalent metal transporter 1 (DMT1), ceruloplasmin (CP), transferrin (TF) (N Engl J Med 2015;372:1832)

- Can frequently be asymptomatic (N Engl J Med 2015;372:1832)
- In addition to anemia, may present with features specific to iron deficiency (Clin Med (Lond) 2021;21:107)
- Anemia symptoms:
- Fatigue
- Altered behavior, irritability
- Exertional dyspnea
- Can lead to decreased cognitive performance and delay mental and motor development in children (N Engl J Med 2015;372:1832)
- Headache
- Sleep disturbance
- Decrease cognitive function and impaired memory
- Palpitations
- Muscle and joint pain
- Features of iron deficiency:
- Glossitis, angular cheilitis
- Craving for ice or clay (pica or pagophagia), especially in pregnant women (American Pregnancy Association: Pica Cravings During Pregnancy [Accessed 16 February 2022])
- Dysphagia with solid foods
- Koilonychia and brittle nails (see Clinical images) (N Engl J Med 2018;379:e13)
- Restless legs syndrome (Willis-Ekbom disease)
- Severe IDA in pregnancy is associated with an increased risk of preterm labor, low neonatal weight and increased newborn and maternal mortality (N Engl J Med 2015;372:1832)
- See Laboratory
- Complete blood count
- Low hemoglobin (Hb) and hematocrit (Hct)
- Low mean corpuscular volume (MCV) and mean corpuscular hemoglobin concentration (MCHC)
- Increased red cell distribution width (RDW)
- Decreased reticulocyte index
- Low serum iron and ferritin levels (best initial test)
- Low iron saturation
- Elevated total iron binding capacity (TIBC) or transferrin
- Reticulocyte hemoglobin content (CHr) value is a strong predictor of the early measurement of functional iron deficiency (ASH: Clinical utility of the reticulocyte hemoglobin content in the diagnosis of iron deficiency [Accessed 16 February 2022])
- In adult men, postmenopausal women or younger women with severe anemia, tests to determine the reason for iron deficiency should be performed:
- Fecal occult blood test
- Upper and lower endoscopy
- Test for hematuria or hemoglobinuria
- Gynecologic evaluation in women
- In adult men, postmenopausal women or younger women with severe anemia, tests to determine the reason for iron deficiency should be performed:
- 42 year old Hispanic woman with a chief complaint of increasing fatigue and dizziness for 2 weeks (J Med Case Rep 2021;15:472)
- 52 year old man on omeprazole (a proton pump inhibitor) for 25 years developed iron deficiency anemia (Intern Med 2018;57:899)
- 54 year old woman with a 7 year history of hemorrhoid bleeding and developing iron deficiency anemia; although her anemia was treated, her koilonychia persisted (N Engl J Med 2018;379:e13)
- Dietary supplementation
- Iron therapy:
- Oral iron for mild to moderate symptoms
- Parenteral iron: unable to absorb oral iron or anemia worsening despite oral iron therapy
- RBC transfusions in patients with acute bleeding or in severe symptoms of anemia or coronary insufficiency (ASH: Iron Deficiency Anemia [Accessed 3 January 2022])


- Peripheral blood smear:
- Microcytic and hypochromic RBCs with marked anisopoikilocytosis in chronic cases of iron deficiency anemia
- Platelets may be increased (reactive thrombocytosis) (Am J Hematol 2014;89:524)
- Target cells are absent, unlike in thalassemia
- In more acute cases, dimorphic population of RBCs with increased RDW is the earliest evidence of iron deficient erythropoiesis
- Normocytic cells produced before bleeding and microcytic cells produced after bleeding
- Blood smear in iron deficiency anemia shows many hypochromic (pale, relatively colorless) and small red blood cells and may also show poikilocytosis (variation in shape) and anisocytosis (variation in size)
- With more severe iron deficiency anemia, the peripheral blood smear may show hypochromic, pencil shaped cells and, occasionally, small numbers of nucleated RBCs
- Platelet count may be slightly above the high limit of normal in IDA (mild thrombocytosis) but severe cases can present with thrombocytopenia (low platelet count)
- Reference: Wikipedia: Iron deficiency anemia [Accessed 17 Januuary 2022]
Contributed by Fnu Raja, M.B.B.S.

Microcytic and hypochromic RBCs
- Prussian blue stain of the bone marrow is considered the gold standard for evaluating marrow iron stores (see Cytology images)
Iron deficiency anemia
- Anemia of chronic disease / anemia of inflammation:
- Normal to increased ferritin levels
- Normal MCV and serum transferrin receptors
- C reactive protein (CRP) increases unlike in iron deficiency anemia, where it is normal
- Thalassemia:
- Serum iron levels normal to high
- Serum ferritin levels normal to slightly decreased
- Serum TIBC is normal unlike in iron deficiency anemia, where it is high
- Hb electrophoresis is diagnostic
- Lead poisoning:
- Also causes hypochromic, microcytic anemia with characteristics basophilic stippling
- Serum iron levels are variable; however, serum ferritin and TIBC are at normal levels
- Copper deficiency:
- Deficiency can cause sideroblastic anemia (iron containing red blood cells); morphology varies from microcytic to macrocytic
- Increased serum iron concentration (decrease utilization) and ferritin levels
- Normal to mildly decreased TIBC
A 27 year old Asian woman, gravida 1, para 0, comes to the office at 26 weeks gestation for prenatal care. The patient denies abdominal pain, vaginal bleeding and leakage of fluid. Fetal movement is normal. For the past few weeks, she has become increasingly fatigued and has been having exertional dyspnea. She is taking proper vitamins as prescribed. On examination, she has general pallor and scooping of nails. Her pregnancy has been uncomplicated. Hemoglobin (Hb) electrophoresis does not reveal abnormal Hb and her initial complete blood count (CBC) results are shown below:
RBCs: 4.2 (4.3 - 5.6 x 1012/L)
Hb: 8.0 (11.6 - 15 grams/dL)
MCV: 60 (80 - 100 fL)
Platelets: 452 x 103/uL (150 - 450 x 103/uL)
WBC count: 5.2 k/ul (4.5 - 11.5/uL)
What is most likely diagnosis and treatment for the patient?
- Anemia due to hemoglobinopathy and requires immediate blood transfusion
- Anemia due to increased nutrient requirement during pregnancy and requires iron supplementation
- Anemia due to physiologic changes of pregnancy and requires no further treatment
- Anemia due to the lack of prenatal vitamins; folic acid supplementation is indicated
Comment Here
Reference: Iron deficiency anemia
- Decreased serum ferritin
- Decreased serum iron
- Decreased soluble serum transferrin receptor
- Decreased total iron binding capacity
Comment Here
Reference: Iron deficiency anemia
- Mantle cell lymphoma involving the peripheral blood, bone marrow, occasionally spleen and gastrointestinal tract but not lymph nodes
- Must lack significant lymphadenopathy (< 1 cm on physical exam and not detected via imaging / CT scans)
- t(11;14)(q13;q32) = fusion of CCND1 and IGH genes
- Positive for Cyclin D1 (also known as BCL1) and negative for SOX11
- Leukemic mantle cell lymphoma
- Nonnodal leukemic mantle cell lymphoma (NN-L-MCL)
- Monoclonal asymptomatic lymphocytosis, cyclin D1 positive (MALD1)
- ICD-10: C83.10 - mantle cell lymphoma, unspecified site
- M > F = ~2:1
- Mean age = ~60 years old
- Peripheral blood, bone marrow and occasionally spleen or gastrointestinal tract
- Circulating cells may reversibly infiltrate extranodal sites and typically remain in the mantle zones of follicles, overlapping with in situ mantle cell lymphoma
- t(11;14)(q13;q32), fusion of CCND1 gene and IGH promoter increases production of the Cyclin D1 protein, regulator of cyclin dependent kinase (CDK4) and CDK6 which controls the G1 / S cell cycle transition (J Clin Invest 2012;122:3416)
- Cell of etiology thought to be peripheral B cell of inner mantle zone
- Typically asymptomatic at presentation
- Mild leukocytosis and mild absolute lymphocytosis
- 10 - 20% patients will have a monoclonal protein
- Better prognosis than classic mantle cell lymphoma (mean survival of leukemic nonnodal = 79 months) (Blood 2003;101:4975)
- TP53 gene mutations = more aggressive course (Clin Lymphoma Myeloma Leuk 2019;19:e93, Leukemia 2012;26:1895)
- Deletions of TP53, ATM or 13q14 = worse prognosis (Ann Diagn Pathol 2014;18:214)
- 48 year old woman with disease progression 20 years after splenectomy (Clin Case Rep 2017;5:477)
- 53 year old man with TP53 loss and response to rituximab / ibrutinib therapy (Clin Lymphoma Myeloma Leuk 2019;19:e93)
- 84 year old woman with associated pure red blood cell aplasia (Int J Hematol 2014;99:777)
- May be observed until progression, then treated as typical mantle cell lymphoma with combination chemotherapy, rituximab and subsequent stem cell transplant depending on age and performance status (Curr Oncol Rep 2018;20:79)
- Diffuse interstitial infiltrate or small scattered nonparatrabecular lymphoid aggregates
- Small to intermediate size lymphoid cells with scant cytoplasm, round to slightly irregular nuclear contours and mature chromatin
- Some cases will show dispersed chromatin and indistinct nucleoli
Contributed by Nicholas Nowacki, M.D.
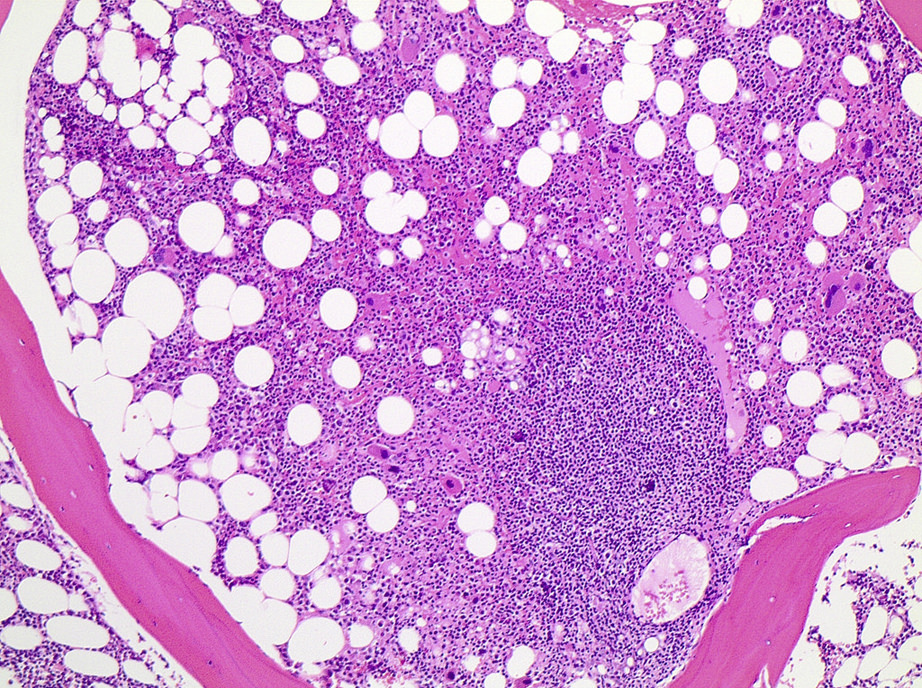
Lymphoid infiltrate

Small lymphoma cells
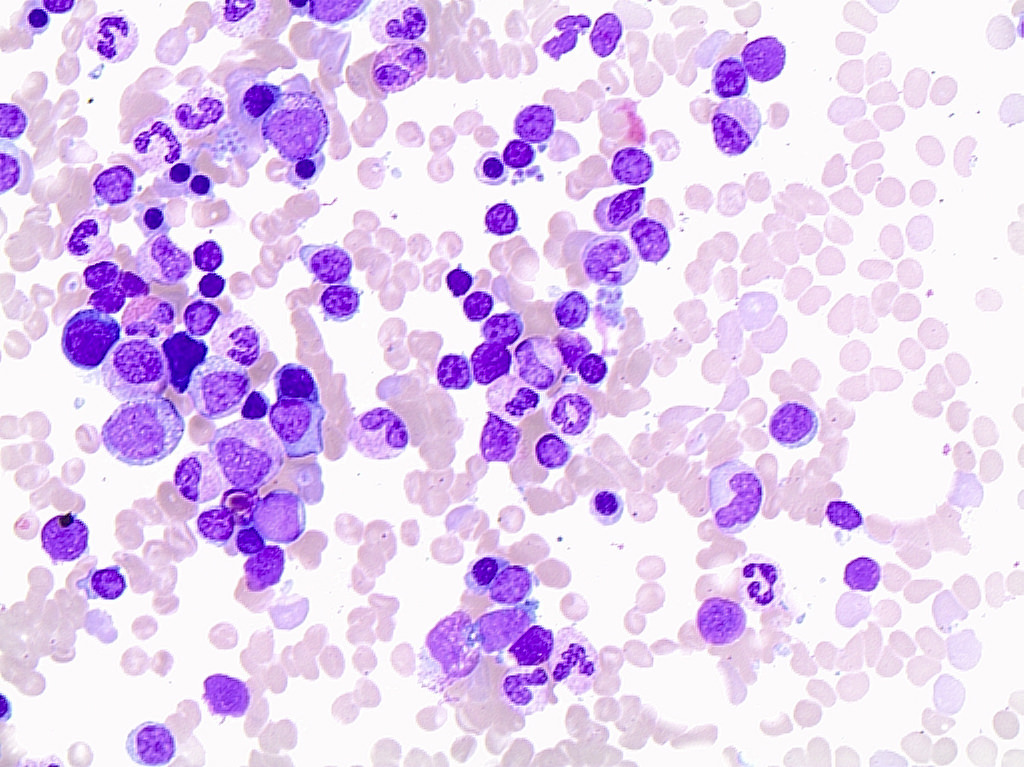
Marrow aspirate

CD3 highlights T cells
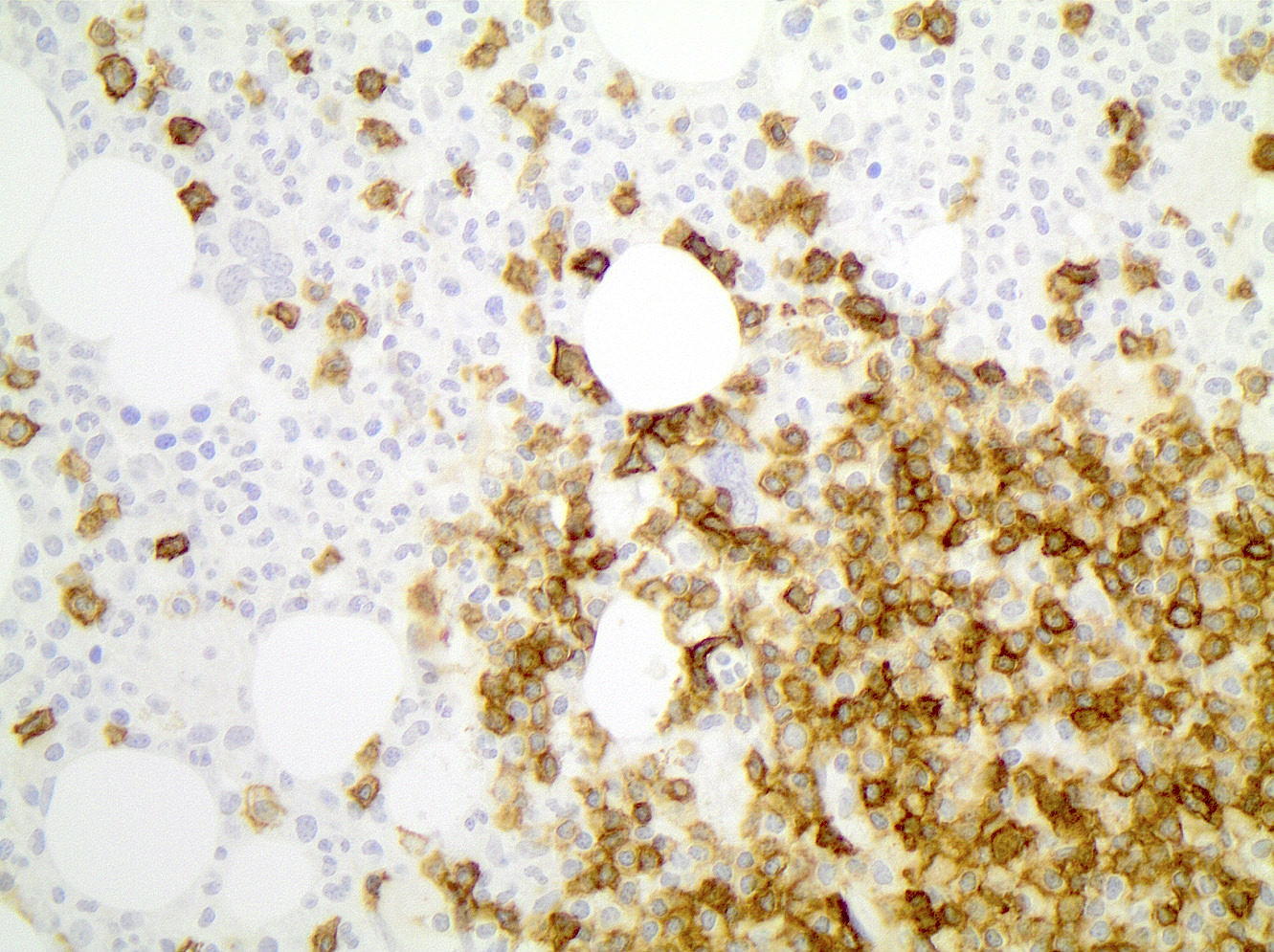
CD5 positive
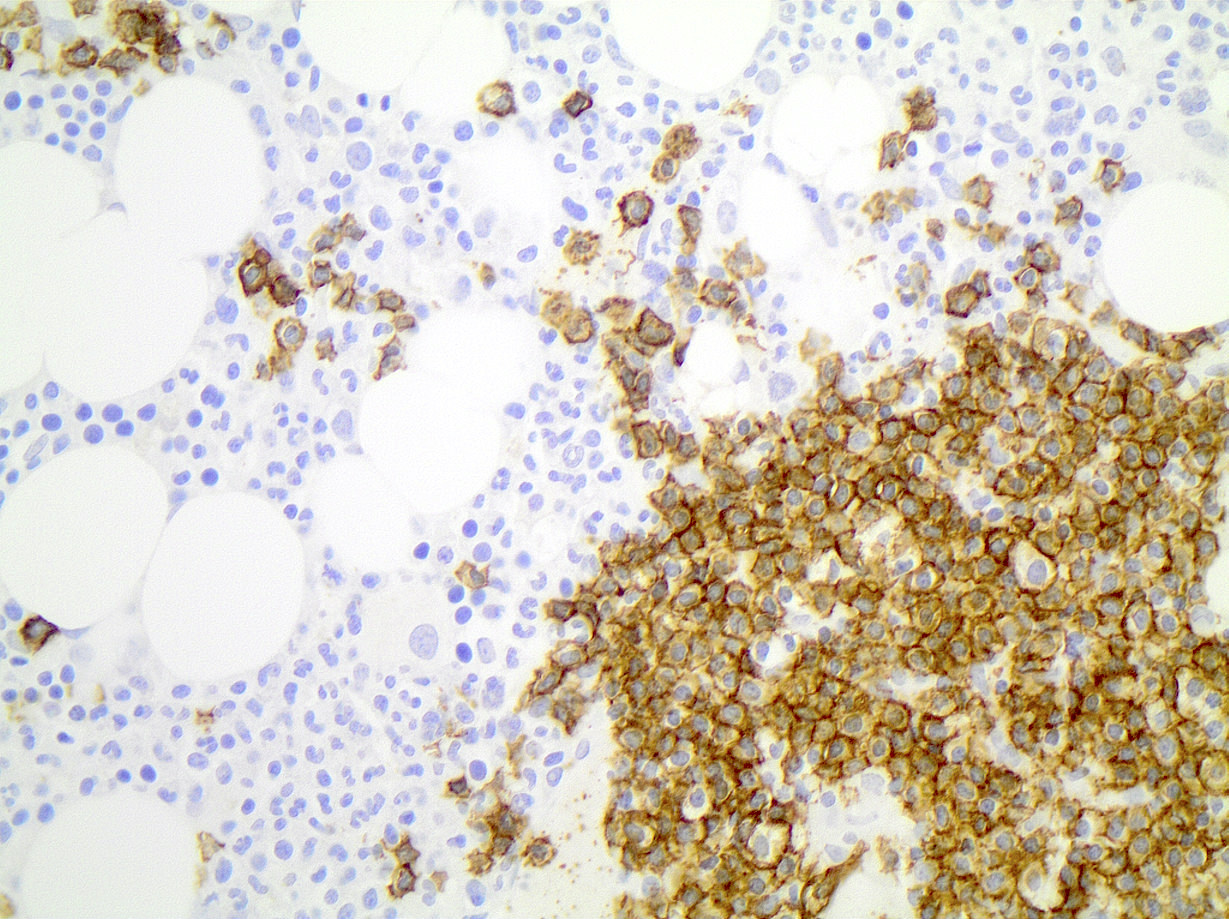
CD20 positive

Cyclin D1 positive
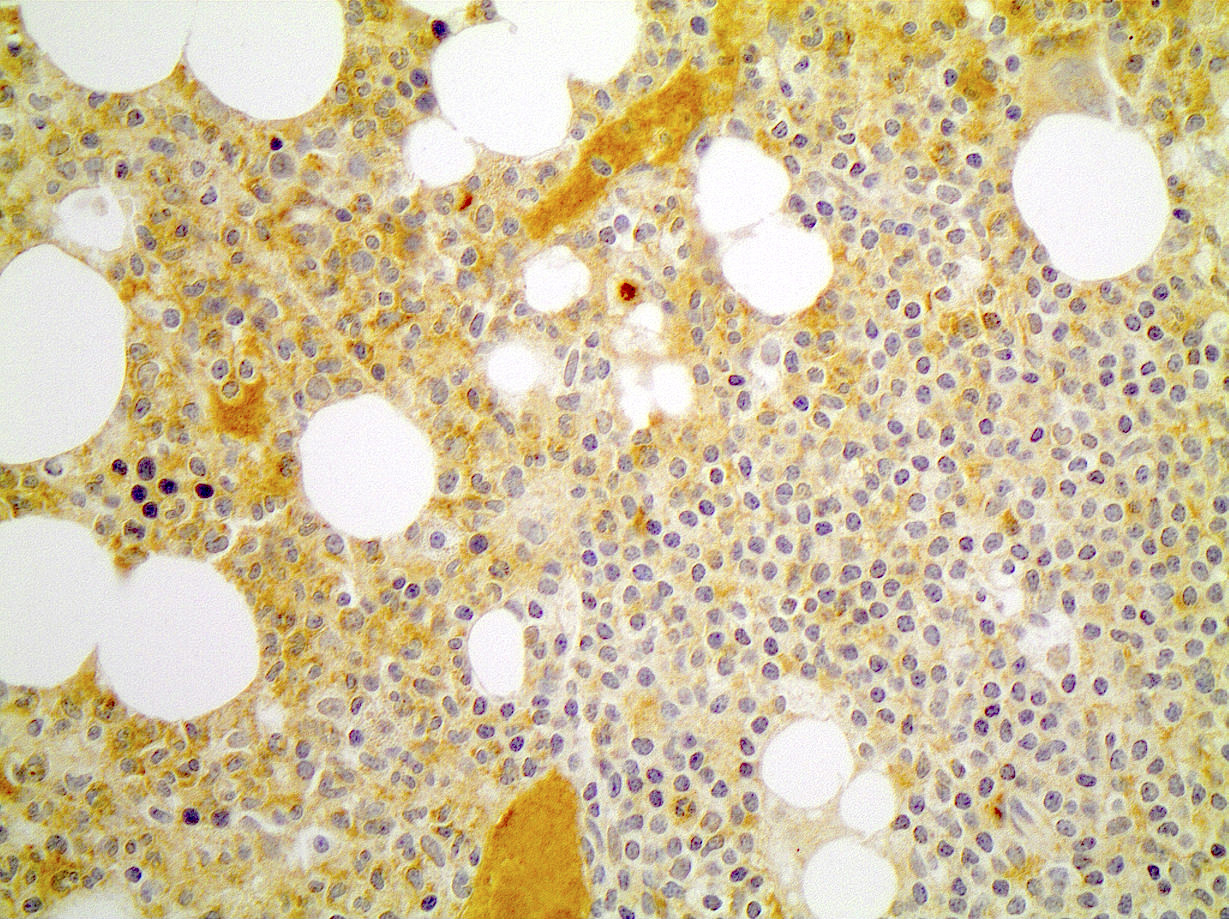
SOX11 negative
- Variable morphologic appearance
- Majority (~70%) show chronic lymphocytic leukemia-like morphology = small with scant cytoplasm, round nuclear contours and condensed chromatin (Cancer Res 2010;70:1408)
- Fewer (~30%) with typical mantle cell lymphoma, centrocyte-like morphology = small to medium sized lymphoid cells with scant cytoplasm, irregular nuclear contours, dispersed chromatin and indistinct nucleoli
- Same as cytology description
Contributed by Nicholas Nowacki, M.D.
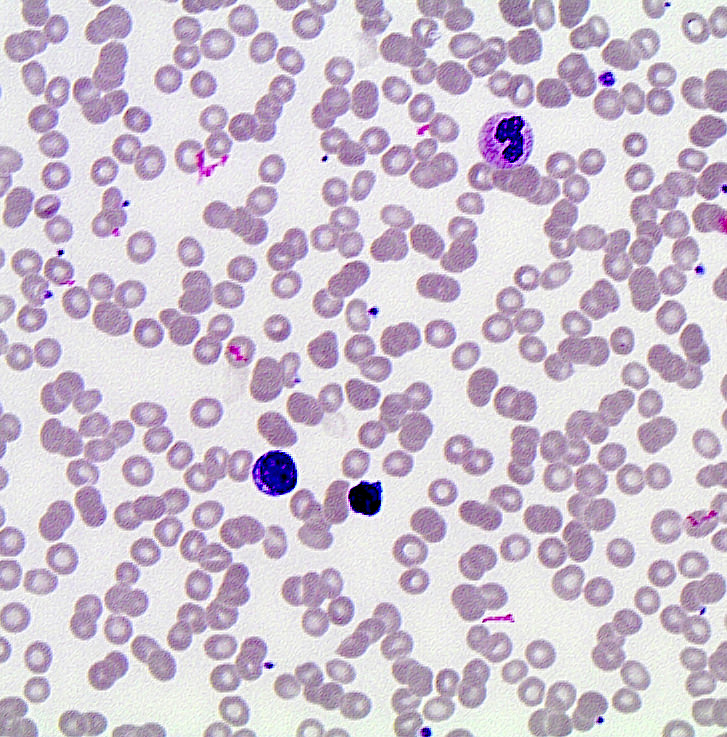
Peripheral blood
- Typical immunophenotype:
- Positive: CD20 (moderate to bright), surface light chain restriction (moderate to bright), CD5 (subset), CD79b, FMC7
- Negative: CD10, CD103, CD123, CD11c
Contributed by Nicholas Nowacki, M.D.
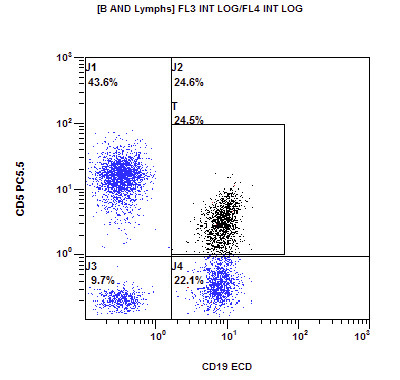
Aberrant CD5 coexpression

CD20 moderate expression
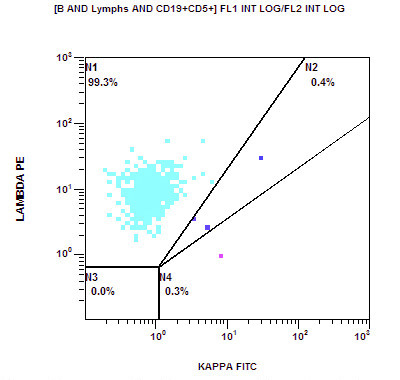
Surface light chain restriction
- t(11;14)(q13;q32), fusion of CCND1 and IGH genes
- Most cases have somatic hypermutation / IgVH gene hypermutated (Clin Cancer Res 2014;20:1007)
- Typically few additional cytogenetic abnormalities and much less likely to have a complex karyotype than classic mantle cell (Leukemia 2012;26:1895)
- Bone marrow, biopsy, aspirate and peripheral blood smear:
- Mantle cell lymphoma involving a normocellular marrow with preserved trilineage hematopoiesis (see comment)
- Comment: The overall findings are consistent with marrow involvement by mantle cell lymphoma. Given the lack of SOX11 staining, the possibility of the leukemic nonnodal mantle cell lymphoma variant should be considered. By definition, patients with this variant must lack significant lymphadenopathy (< 1 cm). The presence of lymphadenopathy would argue for the classic variant of mantle cell lymphoma. Clinical correlation and correlation with cytogenetic / FISH studies, including t(11;14)(q13;q32) IGH-CCND1, is recommended.
- Classical mantle cell lymphoma: often lymphadenopathy (> 1 cm) is present and typically SOX11+
- Chronic lymphocytic leukemia with variant immunophenotype: negative for cyclin D1 and t(11;14)(q13;q32) IGH-CCND1
- Hairy cell leukemia (HCL): frequent cyclin D1 positivity with t(11;14) translocation in hairy cell leukemia can mimic mantle cell lymphoma but hairy cell leukemia typically shows unique morphology and immunophenotype CD103+, CD123+, CD11c+ and CD5-
What is the most likely immunophenotype for leukemic nonnodal mantle cell lymphoma?
- CD20+, CD5+, CD10+, CD103-, CD123-, Cyclin D1+, SOX11+
- CD20+, CD5+, CD10-, CD103+, CD123+, Cyclin D1+, SOX11-
- CD20+, CD5+, CD10-, CD103-, CD123-, Cyclin D1+, SOX11+
- CD20+, CD5+, CD10-, CD103-, CD123-, Cyclin D1+, SOX11-
- CD20+, CD5-, CD10-, CD103-, CD123-, Cyclin D1-, SOX11+
Comment here
Reference: Nonnodal mantle cell lymphoma
- deletion 17p and t(8;14)(q24;q32) IGH-MYC
- t(8;14)(q24;q32) IGH-MYC
- t(11;14)(q13;q32) IGH-CCND1
- t(14;18)(q32;q231) IGH-BCL2
- trisomy 12
- Chronic lymphoproliferative disorders of NK cells (CLPD-NK, provisional entity) are characterized by a persistent (> 6 months) increase in peripheral blood NK cells (usually ≥ 2×109/L) without a clearly identified cause (WHO 2008)
- Chronic NK cell lymphocytosis, chronic NK-LGL LPD, NK cell lineage granular lymphocyte proliferative disorder, NK cell LGL lymphocytosis, indolent large granular NK cell LPD
- Rare and heterogeneous; difficult to distinguish between reactive and neoplastic without highly specialized techniques
- Predominantly adults (median age 60 years)
- No gender predominance, no racial or genetic predisposition
- Autoimmune disorders or viral infections may be accompanied by a transient increase in circulating NK cells
- Viral stimulus postulated as an activation factor leading to selection of NK cell clones (no evidence of direct NK cell infection observed so far)
- May have genetic susceptibility related to haplotypes containing higher numbers of KIR
- Predominantly peripheral blood and bone marrow
- Most patients asymptomatic
- May have systemic symptoms; variable cytopenias (mainly neutropenia and anemia)
- Rarely lymphadenopathy, hepatosplenomegaly and skin lesions
- Sometimes associated with solid tumors, hematologic neoplasms, vasculitis, splenectomy, neuropathy and autoimmune disorders
- 65 year old man with a preexisting, untreated indolent NK cell lymphoproliferative disorder evolving into an aggressive extranodal NK cell lymphoma (Am J Surg Pathol 2005;29:1540)
- Usually indolent clinical course over a prolonged period; occasional disease progression with increasing lymphocytosis and worsening cytopenias (Br J Haematol 1999;106:960)
- Cytopenias, recurrent infections, comorbidity and cytogenetic abnormalities may indicate worse prognosis
- Rare cases of spontaneous regression or transformation to an aggressive NK cell disorder
- Mature NK cell
- Peripheral blood: NK cells of intermediate size with moderate amounts of slightly basophilic cytoplasm and coarse azurophilic granules, round nuclei, condensed chromatin
- Bone marrow: intrasinusoidal and interstitial infiltration by cells with modest amounts of pale cytoplasm and small, minimally irregular nuclei; difficult to identify without immunostains
- CD16, CD56 (usually weak), often cytoplasmic CD3ε, TIA1, granzyme B & M (Am J Pathol 2004;165:1117)
- May have diminished or lost expression of CD2, CD7 and CD57
- May see aberrant coexpression of CD5 and abnormal uniform expression of CD8
- Abnormal KIR expression (restricted KIR isoforms or complete lack of KIR), uniform bright CD94 / NKG2A, diminished CD161)
Images hosted on other servers:

Characteristic immunophenotype


Immunophenotypic comparison
- Usually normal karyotype with no Ig or TCR gene rearrangements
- May be able to use X chromosome inactivation to prove clonality in female patients
- Neoplastic hematolymphoid cells circulating in the peripheral blood
- Morphologic identification of neoplastic hematolymphoid cells on peripheral blood smears
- Chronic lymphocytic leukemia (CLL) / small lymphocytic lymphoma
- Mature B cell neoplasm
- Monomorphic small lymphocytes with a minor proportion of prolymphocytes
- Round to slightly irregular nuclear contours
- Clumped chromatin
- Scant amount of pale blue cytoplasm
- Smudge cells are often seen
- CD5+, CD19+, CD20+ (dim), CD23+, CD79a+, LEF1+, CD200+, FMC7-, cyclin D1-, SOX11-, kappa or lambda surface immunoglobulin light chain (dim)
- IGHV genes are mutated in the majority of cases, while del13q is the most common cytogenetic finding
- See figure 1
- Mantle cell lymphoma
- Mature B cell neoplasm
- Monomorphic small - intermediate sized lymphocytes
- Irregular nuclear contours (indented, folded or cleaved)
- Moderately clumped chromatin
- Scant amount of pale blue cytoplasm
- Morphologically indistinguishable from CLL cells
- Prolymphocytic-like morphology is common during leukemic phase
- CD5+, CD19+, CD20+, cyclin D1+, SOX11+, CD200-, CD23-, FMC7+
- Associated with t(11;14)(q13;q32) / CCND1::IGH translocation (95%)
- See figure 2
- Follicular lymphoma
- Mature B cell neoplasm
- Irregular nuclear contour with frequent indentations, folding, angulations and cleaved nuclei
- Moderately clumped chromatin
- Occasional small nucleoli
- Scant amount of basophilic cytoplasm
- CD19+, CD20+, CD79a+, CD10+, FMC7+, CD5-, CD23-
- Genetically characterized by t(14;18)(q32;q21), a translocation resulting in fusion of the IGH and BCL2 genes
- See figure 3
- Hairy cell leukemia
- Mature B cell neoplasm
- Monomorphic small to intermediate sized lymphocytes
- Smooth chromatin with inconspicuous nucleoli
- Moderate amount of pale blue cytoplasm classically seen with fine, hair-like projections
- CD19+, CD20+ (bright), TRAP+, CD11c+, CD25+, CD103+, CD200+, annexin A1+ (most specific), CD5-, CD10- (80 - 90%), CD23-
- Splenic B cell lymphoma / leukemia with prominent nucleoli (WHO 5th edition) and hairy cell leukemia variant (ICC 2022), a chronic lymphoproliferative disorder whose morphologic characters resemble hairy cell leukemia; neoplastic cells have prominent nucleoli and are negative for HCL markers (CD25, annexin A1 and TRAP)
- BRAF V600E mutation is found in most (> 90%) classic hairy cell leukemia cases
- Associated with pancytopenia and splenomegaly
- See figure 4
- Reference: Leuk Lymphoma 2015;56:2000, Leukemia 2022;36:1720, Blood 2022;140:1229
- T cell prolymphocytic leukemia
- Mature T cell neoplasm
- Small to intermediate sized lymphocytes
- Round to irregular nuclei
- Distinct nucleoli
- Basophilic cytoplasm with cytoplasmic blebs
- CD2+, CD3+, CD5+, CD7+, TdT-, CD4+/-, CD8+/-, TCL1+
- Most cases are found with inv(14)(q11q32) and TCL1 gene rearrangement detectable by FISH, while deletion or mutations in ATM have also been observed
- Aggressive disease
- See figure 5
- Sézary syndrome
- Mature T cell neoplasm
- Medium sized to larger lymphocytes
- Irregular nuclear contour, often described as cerebriform pattern
- Condensed chromatin
- Inconspicuous nucleoli
- Abundant basophilic cytoplasm
- CD2+, CD3+, CD5+, TCRαβ+, CLA+, usually CD4+, CD8-, CD7- and CD26-
- T cell receptor gene rearrangements are present; recurrent balanced chromosomal translocations have not been identified
- Aggressive form of mycosis fungoides
- See figure 6
- Adult T cell leukemia / lymphoma
- Mature T cell neoplasm, caused by human retrovirus HTLV1
- Several morphological variants have been identified: small, medium, large cell and anaplastic variants
- Convoluted and polylobated nuclei, often described as flower cells
- Condensed chromatin
- Small or inconspicuous nucleoli
- Moderate amount of basophilic cytoplasm
- CD2+, CD3+, CD5+, CD4+/- and CD8-/+, CD25+, CD7-
- Clonal T cell receptor gene rearrangement
- Associated with hypercalcemia
- See figure 7
- Reference: Am J Hematol 2019;94:1027
- Multiple myeloma - plasma cell leukemia (primary or secondary)
- Plasma cell neoplasm
- Medium sized neoplastic cells
- Eccentric round nucleus
- Coarse chromatin, often seen in a clockface pattern
- Abundant amount of pale to basophilic cytoplasm with perinuclear clearing
- According to the 2021 International Myeloma Working Group recommendation, primary plasma cell leukemia is defined as "5% or more circulating plasma cells in peripheral blood smears in patients otherwise diagnosed with symptomatic multiple myeloma" (Blood Cancer J 2021;11:192)
- CD38+, CD138+, CD56+ (80%), CD45-, CD19- (95%)
- Immunoglobulin heavy and light chain gene rearrangement
- Associated with Rouleaux formation of RBCs and monoclonal gammopathy
- See figure 8
- Reference: Blood Rev 2011;25:107
- Diffuse large B cell lymphoma (example: CLL status post-Richter transformation)
- T cell large granular lymphocytic leukemia
- Mature cytotoxic T cell neoplasm
- Large sized lymphocytes
- Irregular nuclear contour
- Coarse chromatin
- Indistinct nucleoli
- Abundant clear cytoplasm with azurophilic granules; cytologically indistinguishable from natural killer cells (CD3-)
- CD2+, CD3+, CD4-, CD8+, CD57+, CD56 variable, CD16+ (variable), loss of CD5 or CD7 is common
- Clonal T cell receptor gene rearrangement
- Indolent disease; associated with cytopenia, particularly neutropenia, and rheumatoid arthritis
- See figure 10
- Reference: Autops Case Rep 2016;6:41
- Myeloid: acute myeloid leukemia (example: AML with inv(16))
- Immature myeloid neoplasm
- Medium to large sized myeloblasts
- Round to irregular or folded nuclei
- Dispersed chromatin
- Distinct nucleoli
- Moderately abundant cytoplasm
- Auer rods present in some cases, abundant in acute promyelocytic leukemia
- Typically, CD34+ and CD117+, may exhibit granulocytic lineage (positive for CD13, CD33, CD15, CD65 and MPO) or monocytic lineage (positive for CD14, CD4, CD11c, CD64, CD36 and lysozyme)
- See figure 11 showing an AML with inv(16)(p13.1;q22)
- Lymphoid: B lymphoblastic leukemia / lymphoma (example: B ALL with KMT2A rearrangement)
- Immature lymphoid neoplasm
- Intermediate sized lymphoblasts
- Round to irregular nuclei
- Dispersed chromatin
- Distinct nucleoli
- Scant amount of basophilic cytoplasm
- Typically, CD19+, CD22+, CD79a+, CD10+, CD45 (moderate intensity) and variable expression of TdT and CD34
- Recurrent genetic abnormalities include t(9;22)(q34.1;q11.2) BCR::ABL1, t(v;11q23.3) KMT2A rearranged, t(12;21)(p13.2;q22.1) ETV::RUNX1, t(5;14)(q31.1;q32.1) IGH::IL3 or t(1;19)(q23;p13.3) TCF3::PBX1
- See figure 12
- Blast equivalents (promonocytes) (example: acute myelomonocytic leukemia)
- Immature monocytic precursors that are counted towards the total blast count in monocytic and myelomonocytic neoplasms
- Irregular and convoluted nuclear contours with fine folds
- Fine chromatin
- Distinct nucleoli
- Moderate amount of gray to pale blue cytoplasm with occasional small vacuoles
- Immature monocytic precursor cells are usually CD14+/-, CD64+, CD15+, CD11b+, CD11c+, CD4+, CD36+, CD68+, CD163+ and lysozyme+
- Nonspecific cytogenetic abnormalities
- See figure 13
- Reference: StatPearls: Leukemia [Accessed 11 January 2023]
Table 1: B cell lymphoproliferative disorders and markers
| CD45RA | CD19 | CD20 | CD79a | CD5 | CD10 | CD23 | Cyclin D1 | SOX11 | LEF1 | CD200 | CD103 | CD11c | CD25 | |
| Follicular lymphoma | + | + | + | + | - | + | - | - | -/+ | Unknown | - | - | -/+ | -/+ |
| Chronic lymphocytic leukemia / small lymphocytic lymphoma | + | + | + | + | + | - | + | - | - | +/- | + | - | -/+ | -/+ |
| Diffuse large B cell lymphoma | + | + | + | + | - | +/- | - | - | -/+ | -/+ | +/- | -/+ | -/+ | -/+ |
| Mantle cell lymphoma | + | + | + | + | + | - | - | + | + | - | - | - | - | -/+ |
| Marginal zone lymphoma | + | + | + | + | - | - | - | - | -/+ | - | - | - | -/+ | - |
| Lymphoplasmacytic lymphoma | + | + | + | + | - | - | - | - | Unknown | - | +/- | - | +/- | +/- |
| Hairy cell leukemia | + | + | + | + | - | - | - | + | +/- | - | + | +/- | + | + |
| +: positive; -: negative; +/-: majority positive; -/+: majority negative | ||||||||||||||
Table 2: T cell lymphoproliferative disorders and markers
| CD4 / CD8 | Pan-T cell markers (CD2, CD3, CD5, CD7) | CD30 | T cell receptor | |
| T cell prolymphocytic leukemia | CD4+ / CD8- > CD4+ / CD8+ | All expressed | - | Alpha / beta |
| Sézary syndrome | CD4 | CD7 loss | +/- | Alpha / beta |
| Adult T cell leukemia / lymphoma | CD4 | CD7 loss | -/+ | Alpha / beta |
| T cell large granular lymphocytic / leukemia | CD8 | CD2, CD5 and CD7 variably loss | - | Alpha / beta |
| +: positive; -: negative; +/-: majority positive; -/+: majority negative | ||||
Contributed by Jingwei Li, M.D., Ph.D., Olga Pozdnyakova, M.D., Ph.D. and Karry Charest, MT (ASCP)
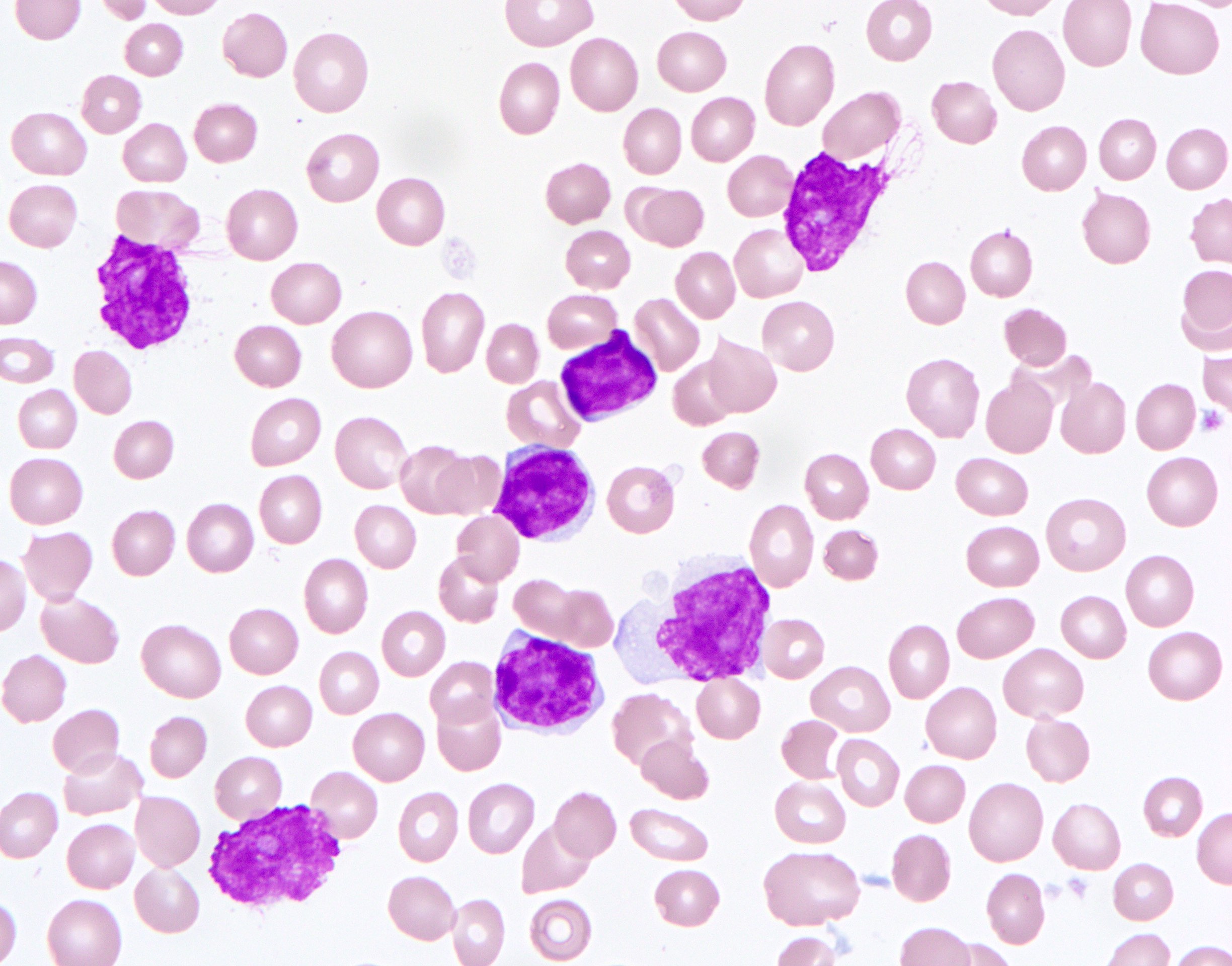
Clumped chromatin, smudge cells
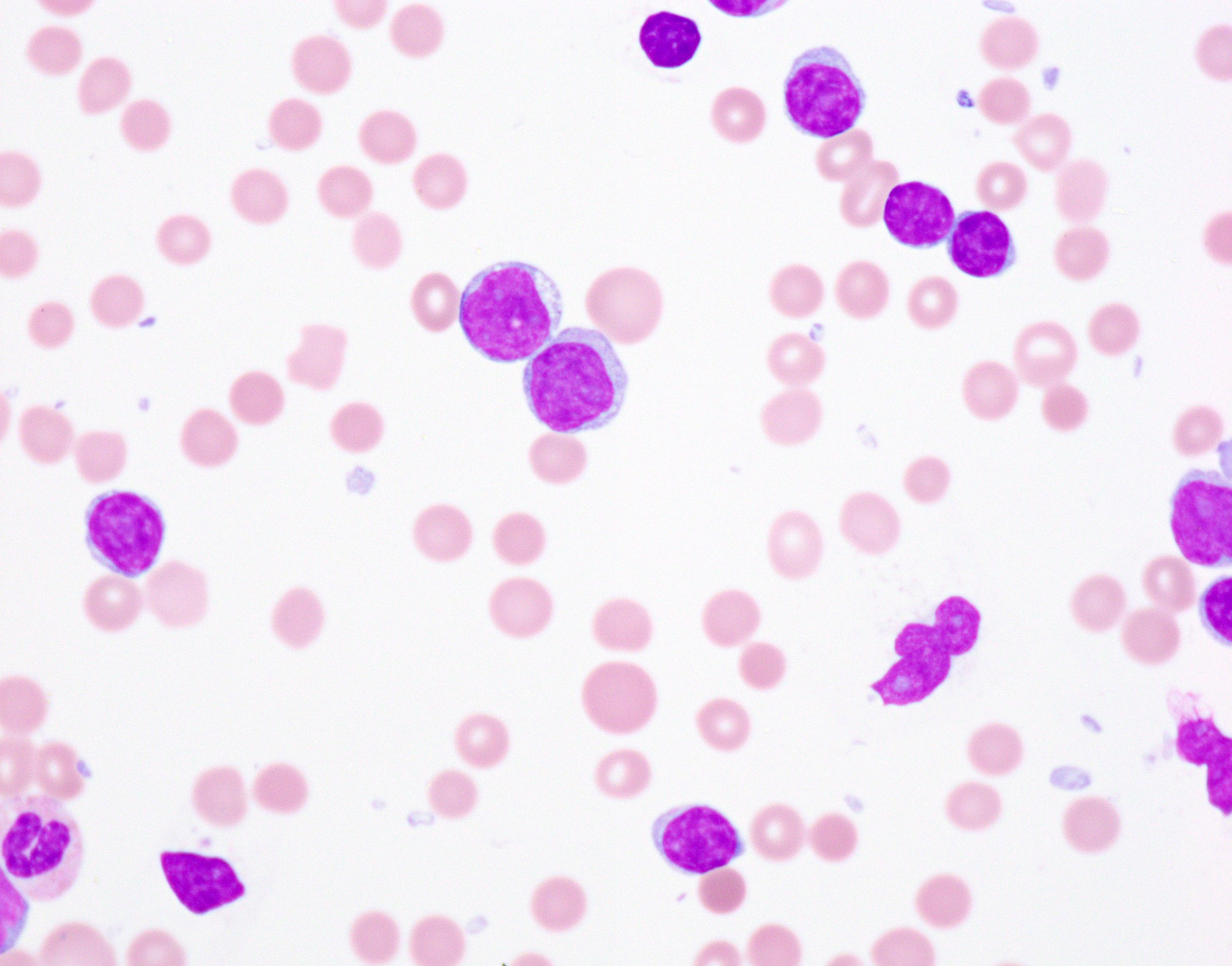
Moderately clumped chromatin
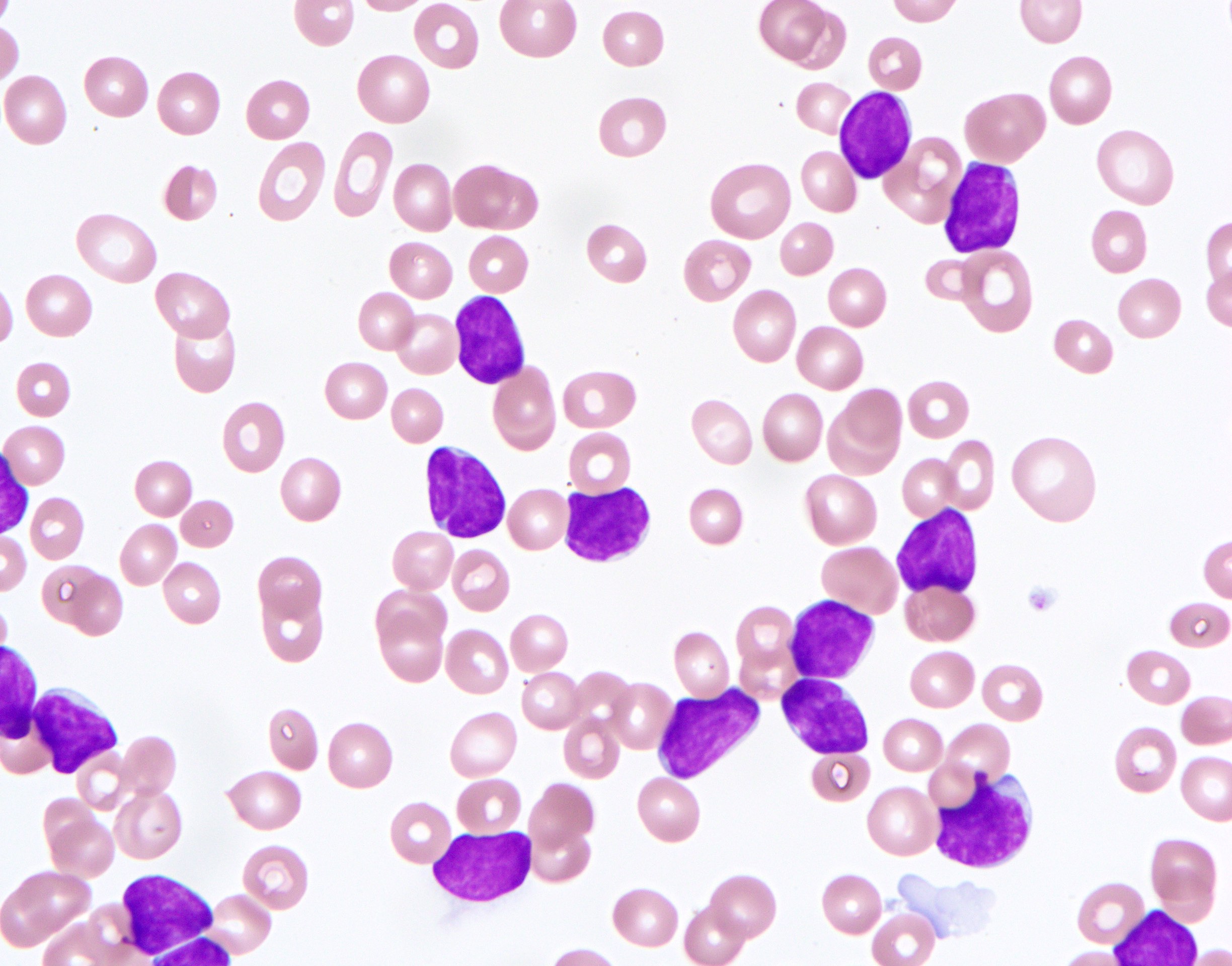
Cleaved nuclei
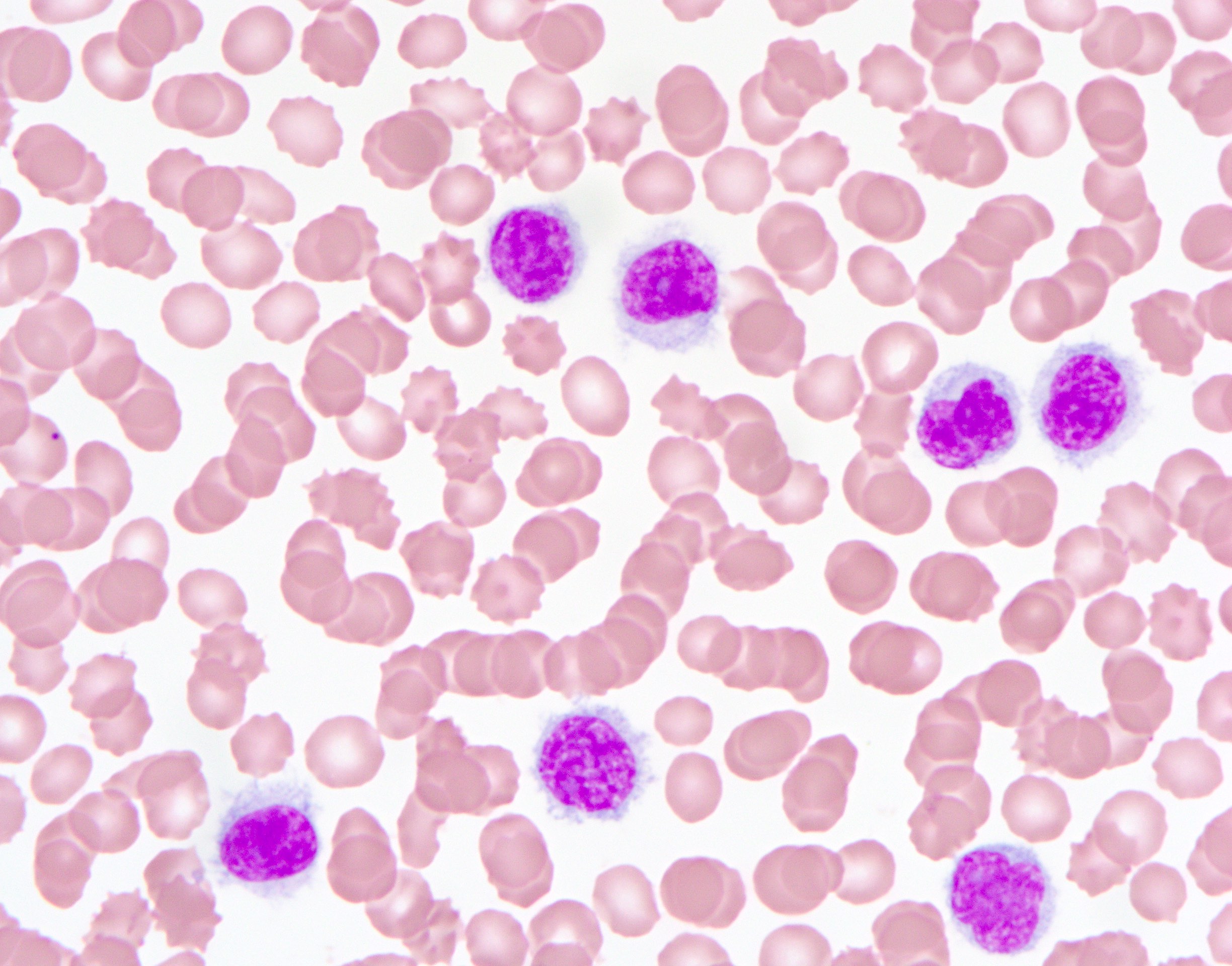
Fine, hair-like cytoplasmic projections
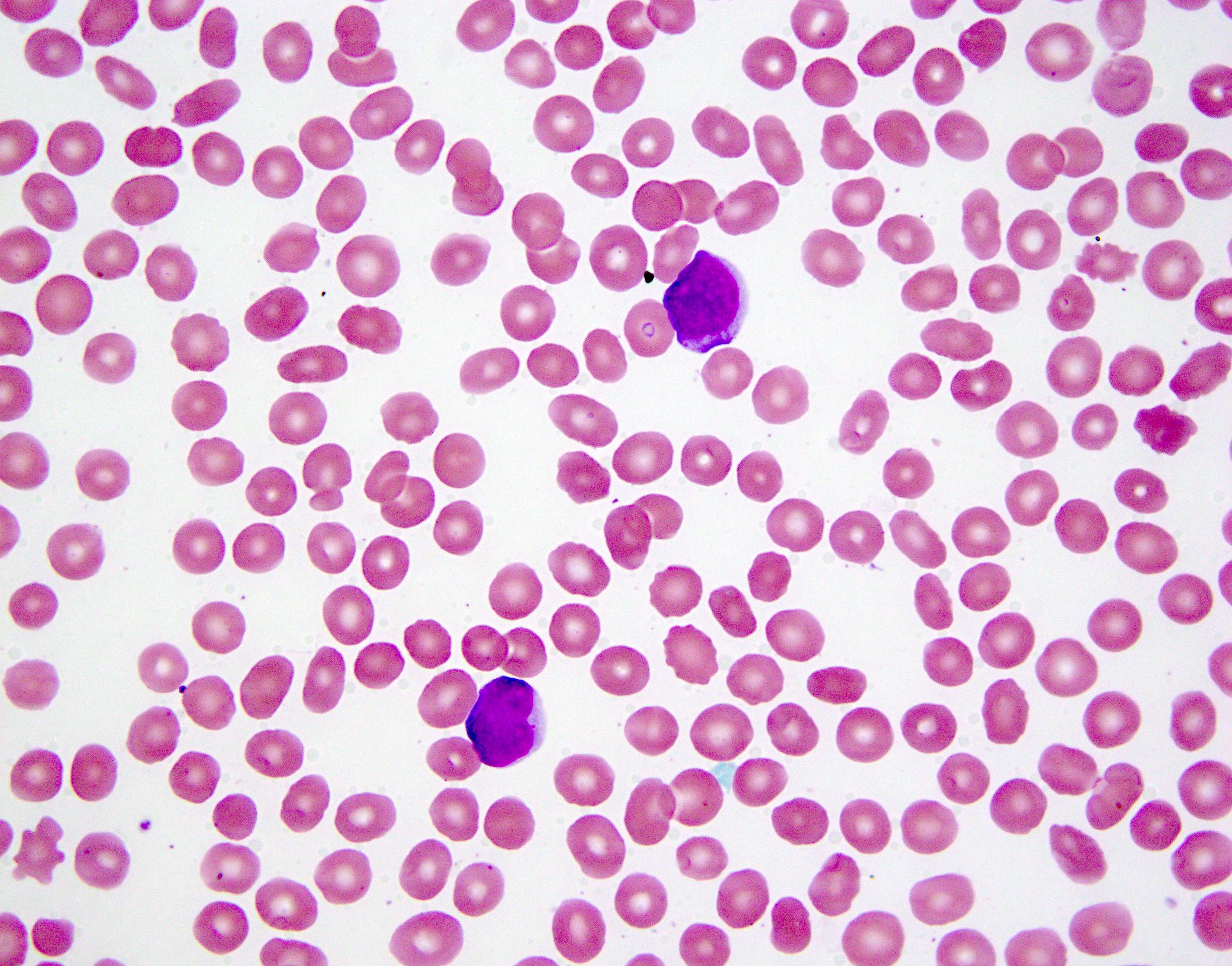
Round to irregular nuclei
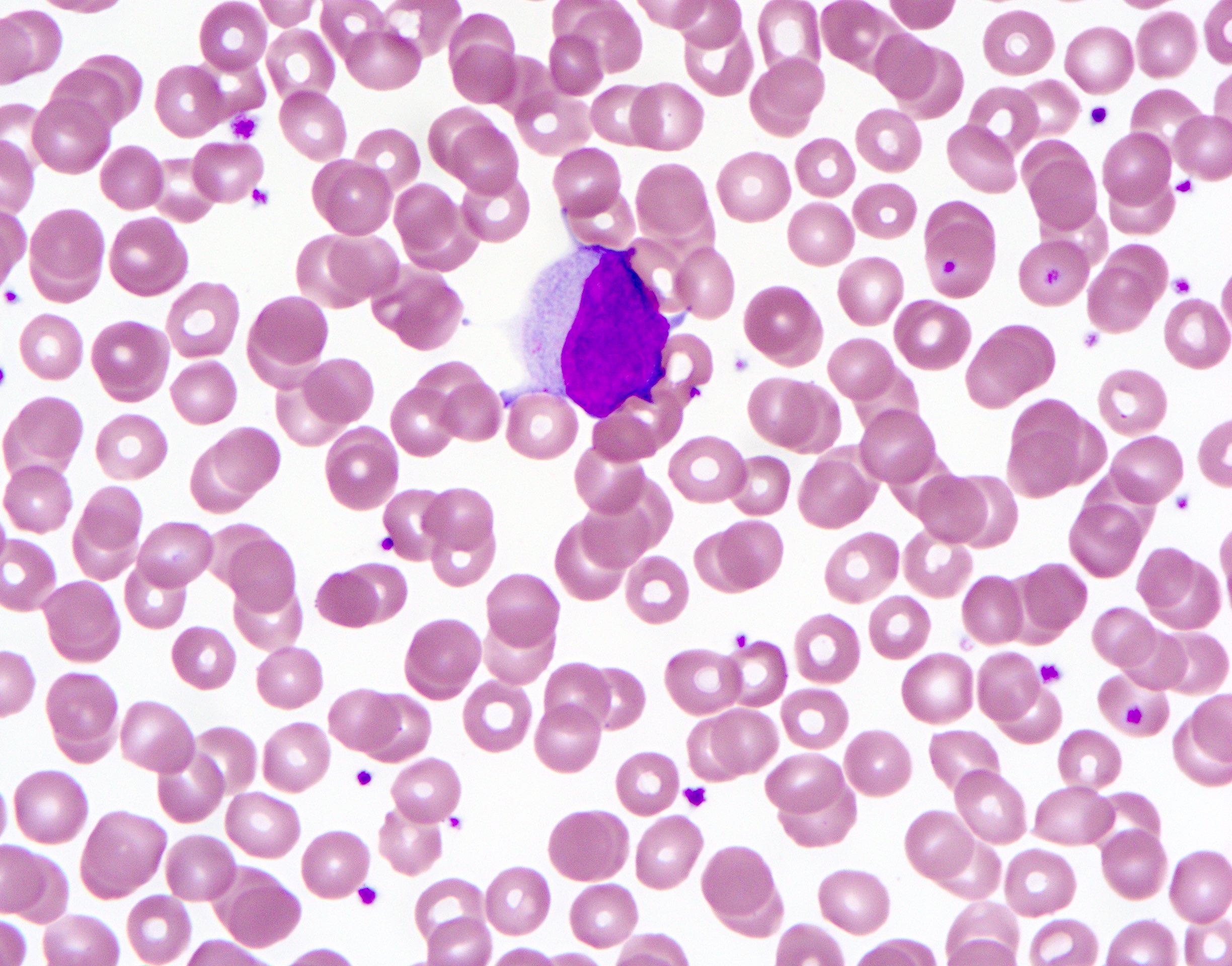
Irregular, cerebriform nuclear contour
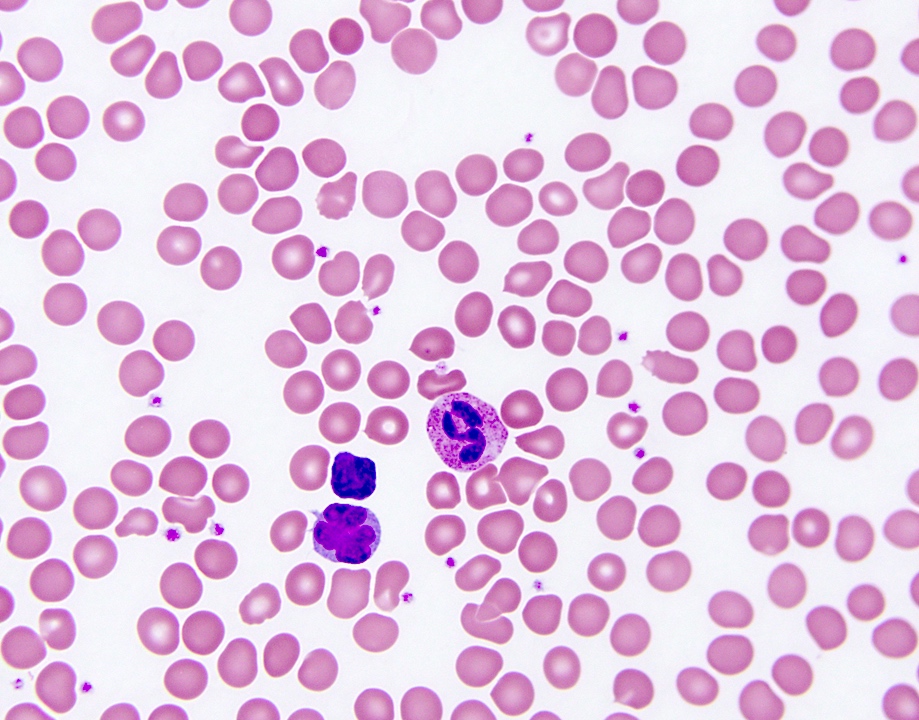
Polylobed nuclei, flower cells
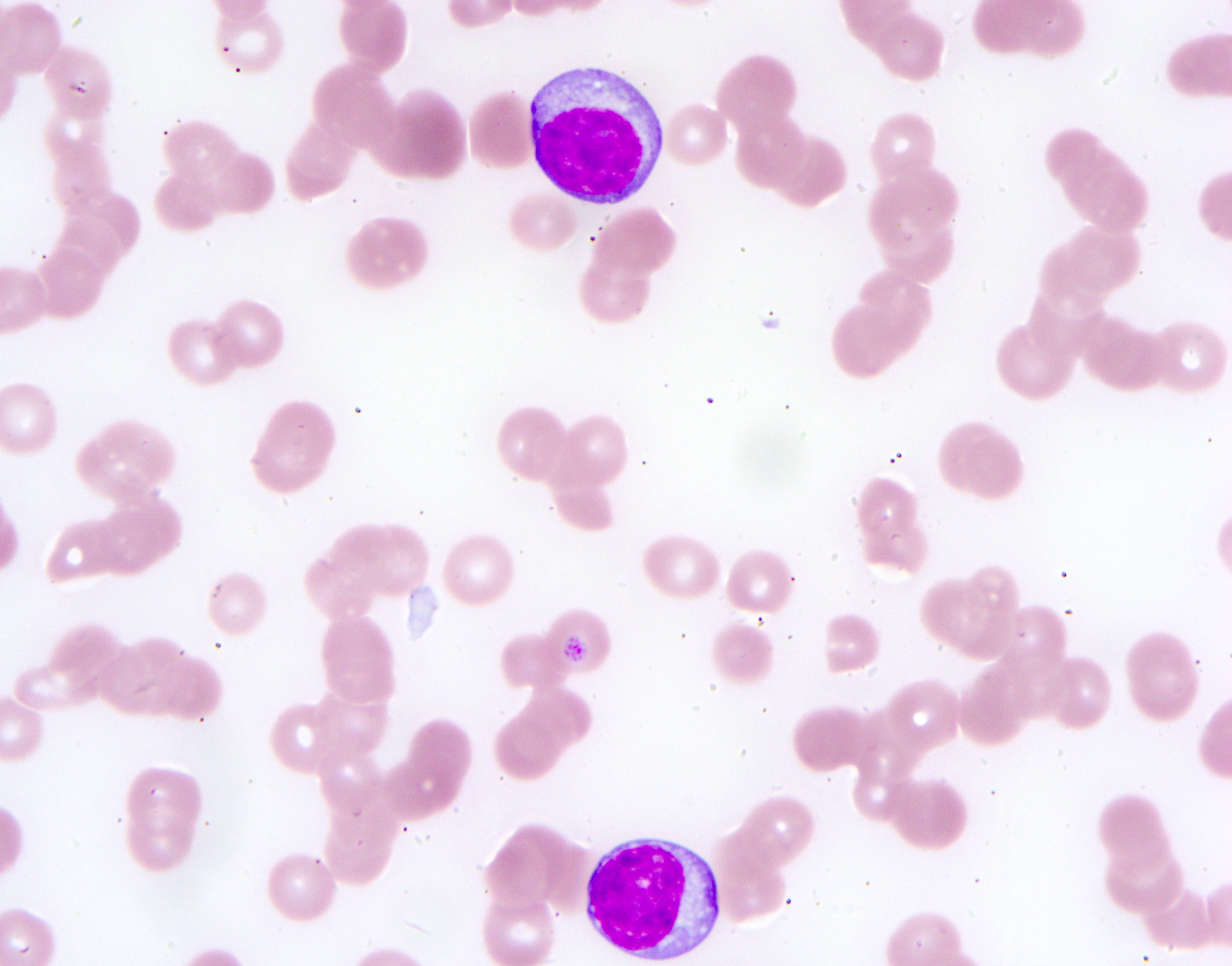
Eccentric, round nuclei
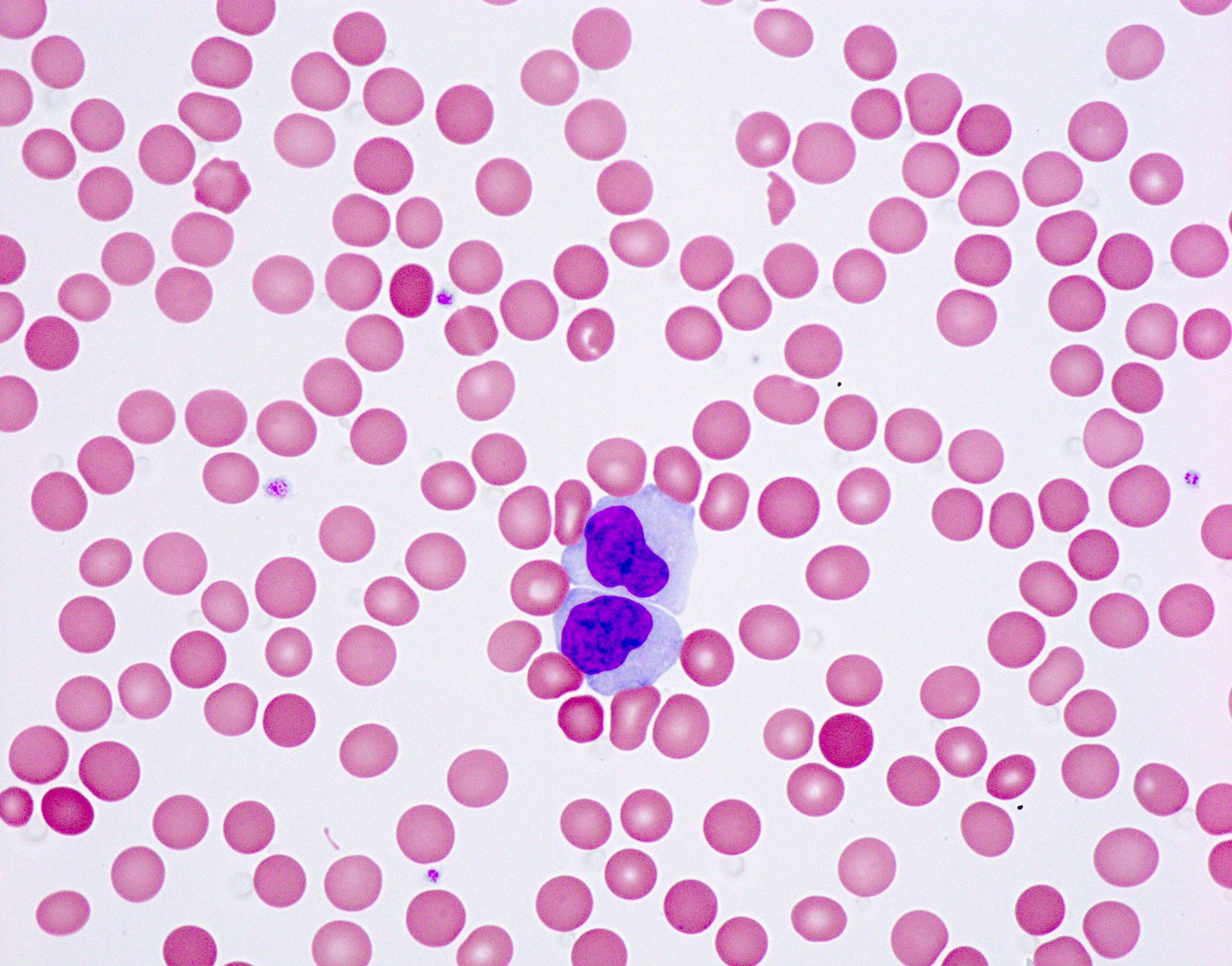
Large sized neoplastic cells
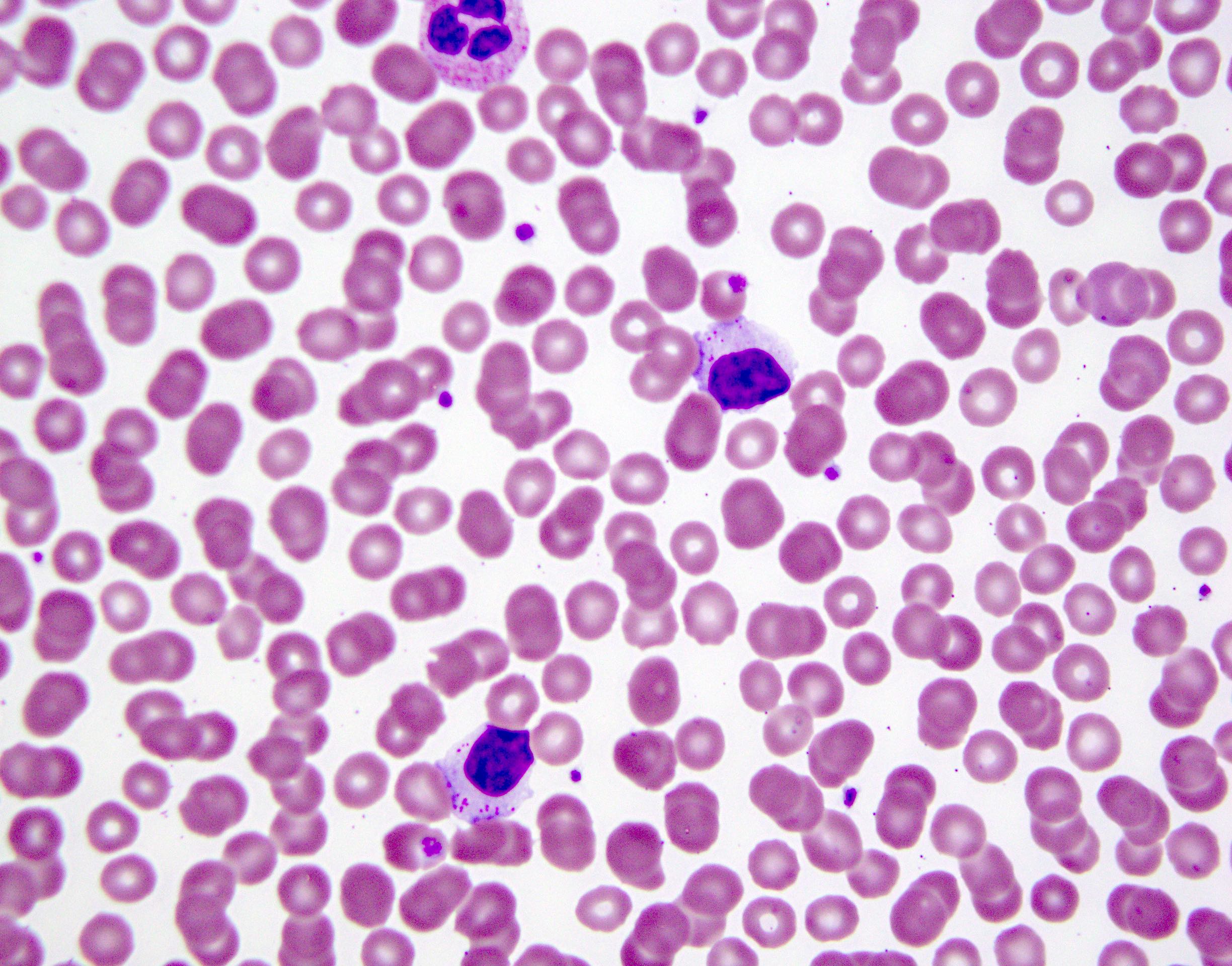
Abundant cytoplasm, azurophilic granules
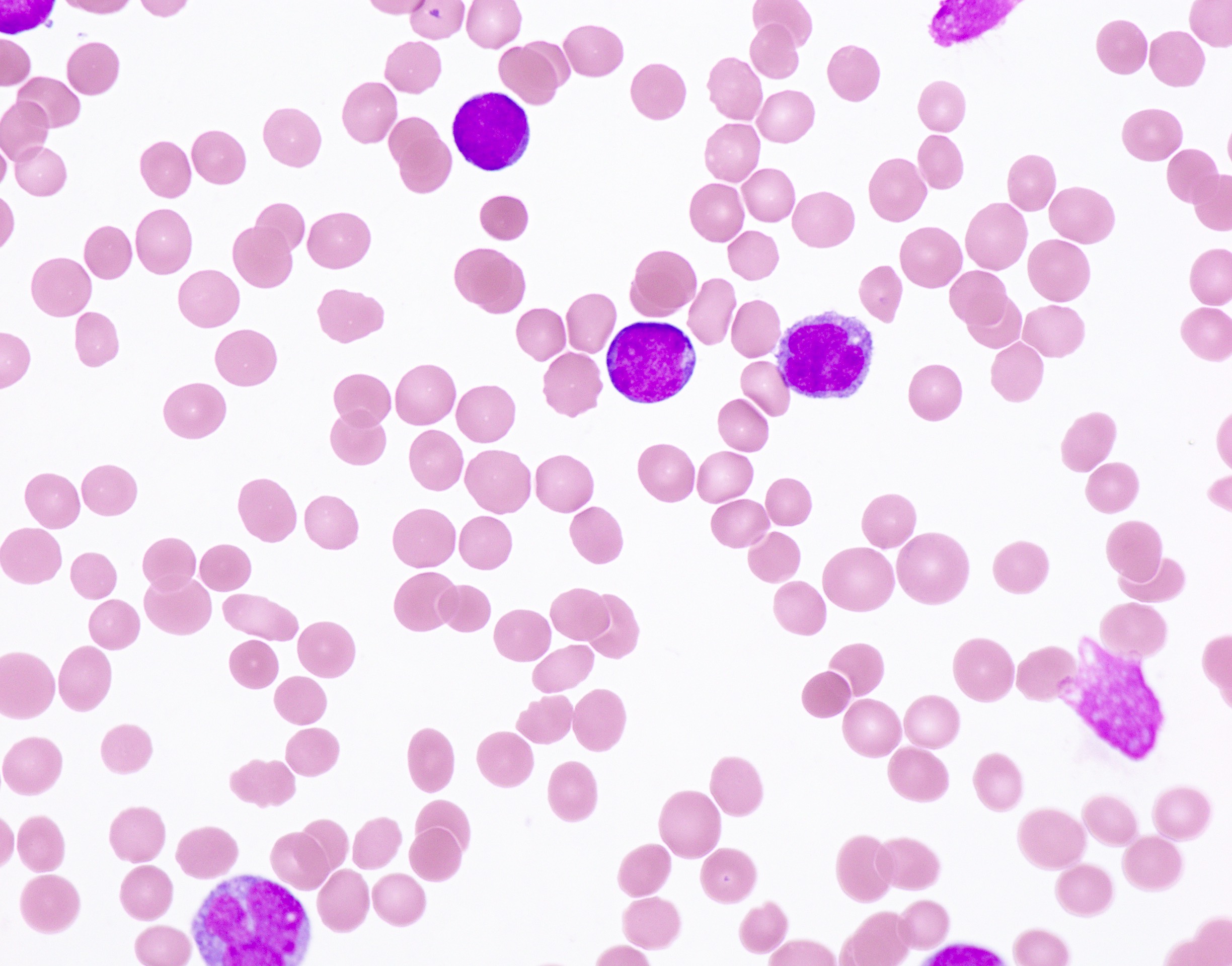
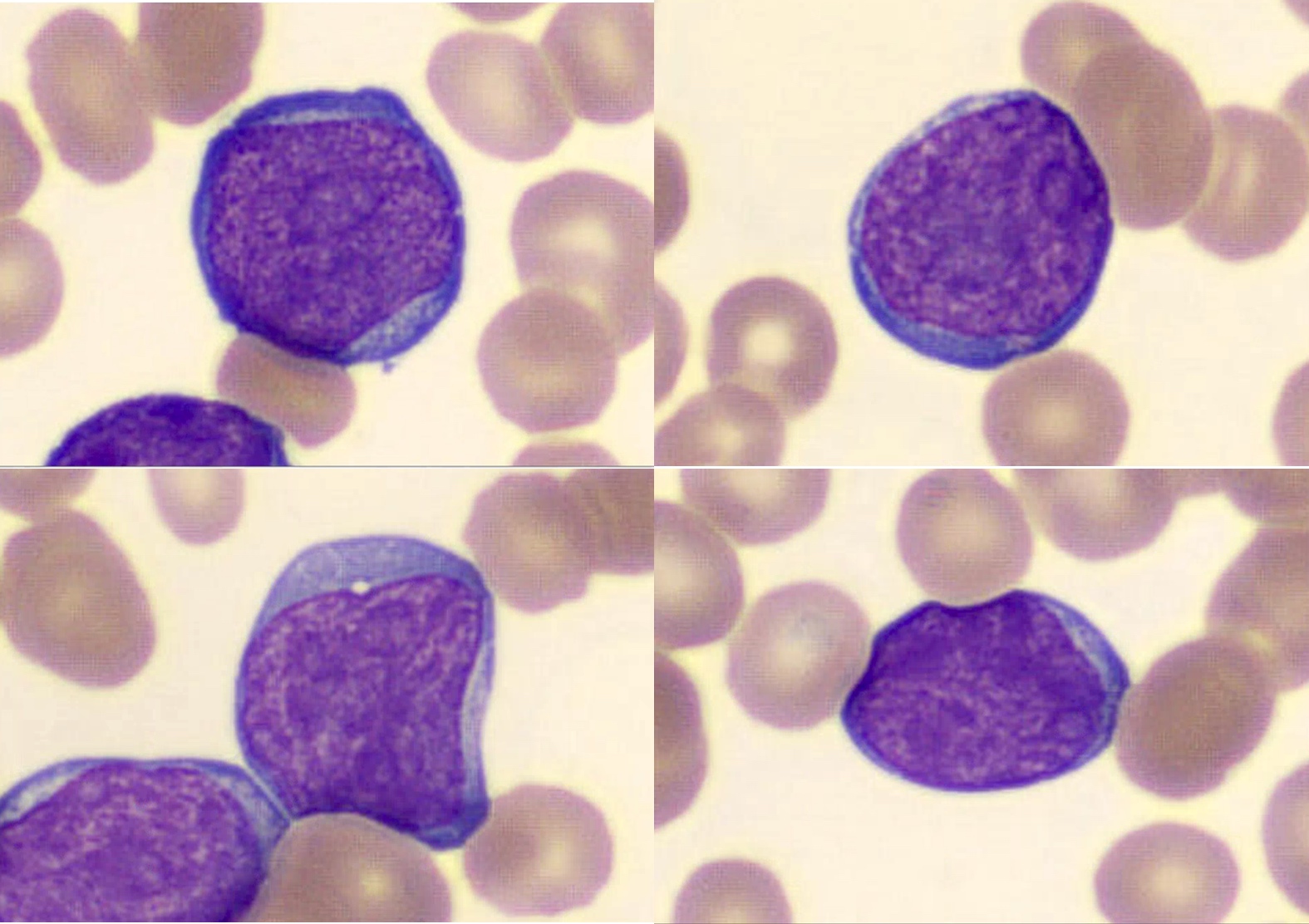
Dispersed chromatin, distinct nucleoli
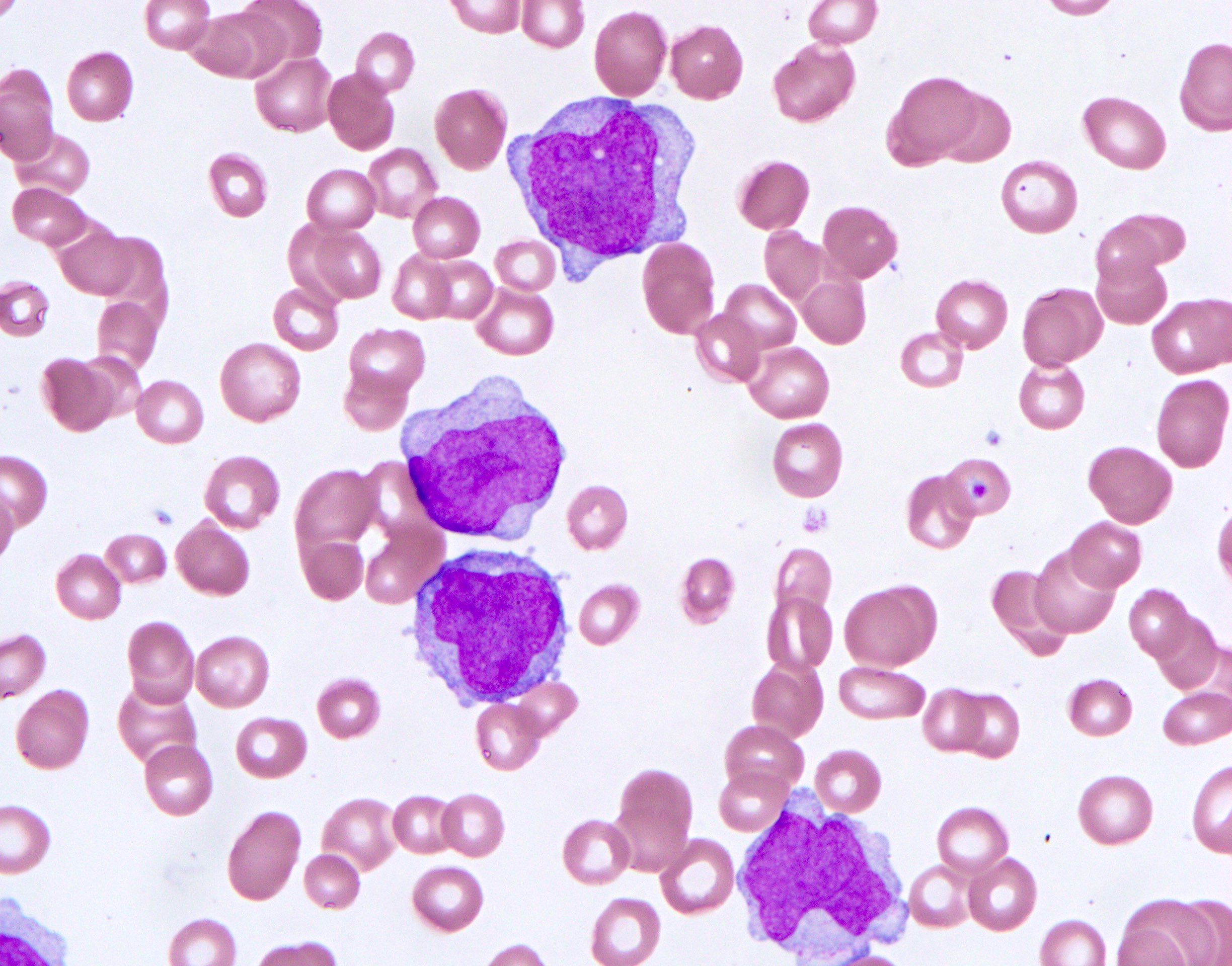
Irregular, convoluted nuclear contours
- Adult T cell leukemia / lymphoma
- Hairy cell leukemia
- Sézary syndrome
- T cell large granular lymphocytic leukemia
- T cell prolymphocytic leukemia
Which of the following lymphoproliferative disorders is associated with BRAF V600E mutation?
- Chronic lymphocytic leukemia / small lymphocytic lymphoma
- Classic hairy cell leukemia
- Mantle cell lymphoma
- Multiple myeloma
- Sézary syndrome
- Rare, autosomal recessive form of severe combined immunodeficiency (SCID) of infancy
- Recurrent infections, early diffuse erythrodermia, failure to thrive, protracted diarrhea, hepatosplenomegaly, elevated serum IgE and eosinophilia, lymphadenopathy, oligoclonal T cell expansions
- Often recombinase activating gene mutations (Clin Immunol 2005;116:246)
- Most common genetic alteration is the partial loss of function for recombinase activating genes RAG (RAG1 and RAG2)
- The RAG genes are essential for gene recombination in the T cell receptor and B cell receptor
- This mutation leads to the Inability to productively rearrange VDJ regions in T cell and B cell receptors, which leads to abnormal T cell proliferation and absent B cells
- As a result, the immune system has difficulty recognizing specific pathogens
- An impaired V(D)J recombination process leads to the generation of a few T cells expanding in the periphery, infiltrating target organs such as skin and gut, resulting in the erythroderma and colitis typical of this syndrome
- The disease is characteristically associated with an oligoclonal expansion of Th2 population
- Recurrent infections
- Early diffuse erythrodermia, failure to thrive, protracted diarrhea, hepatosplenomegaly
- Elevated serum IgE and eosinophilia, lymphadenopathy, oligoclonal T cell expansions
- Suppurative lymphadenitis and disseminated sometimes fatal tuberculosis as a reaction to the immunization for Bacille Calmette-Guerin (BCG)
- Lymphoproliferative disorders associated with Primary Immune Disorders (PID) such as SCID
- WBC count may be normal or elevated with a predominance of lymphocytes
- Eosinophilia is invariably present
- Molecular studies show presence of an oligoclonal set of activated antigen stimulated Th2 cells
- B cells are absent, and NK cells are present
- Immunoglobulin levels show absent immunoglobulin A (IgA) and immunoglobulin M (IgM), elevated IgE levels, and immunoglobulin G (IgG) that is maternal in origin
- IgG antibodies against T dependent antigens, such as tetanus, are nonprotective
- Specific IgM antibodies, such as isohemagglutinins, are absent
- Infant with Omenn syndrome and reticular dysgenesis (J Allergy Clin Immunol 2013;131:1227)
- Isolation to prevent infection
- Prophylactic treatment with nystatin, antiviral agents (e.g. acyclovir) or antibiotics may be considered
- Immunosuppression with cyclosporine (Arch Dis Child 2002;87:231)
- Bone marrow or other stem cell reconstitution is first line conventional therapy
- Diagnosis at birth may lead to protection from infection and improve transplantation outcome (Blood 2011;117:3243)
Images hosted on other servers:
Pre- and post-treatment
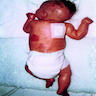
Alopecia and erythroderma
- Total effacement of nodal architecture with no distinct cortex and no follicles
- Also B cell depletion and accumulation of interdigitating reticulum cells (Am J Surg Pathol 1995;19:1082)
- Hassall corpuscles are poorly formed, and lymphocytes are deficient in the thymus
- Paracortical lymphocytes are absent in the spleen
- The spleen, skin and mucosa associated lymphoid tissues contain only a few scattered CD3+ T lymphocytes, and B cells are totally absent (Blood 1999;94:3468)
- Non caseating granulomatous inflammation with BCG / Bacille Calmette-Guerin lymphadenitis (Am J Clin Pathol 2000;113:703)
- Lymphoproliferative disease associated with primary immune disorders:
- EBV is involved in most cases
- Morphology includes reactive lymphoid hyperplasia, polymorphous infiltrate with immunoblast proliferation and overt lymphoma
- Diffuse Large B-cell Lymphoma (DLBCL) is the most common type of lymphoma
- May present as Fatal Infectious Mononucleosis (FIM)
Contributed by Shamayel Mohammed, M.D.
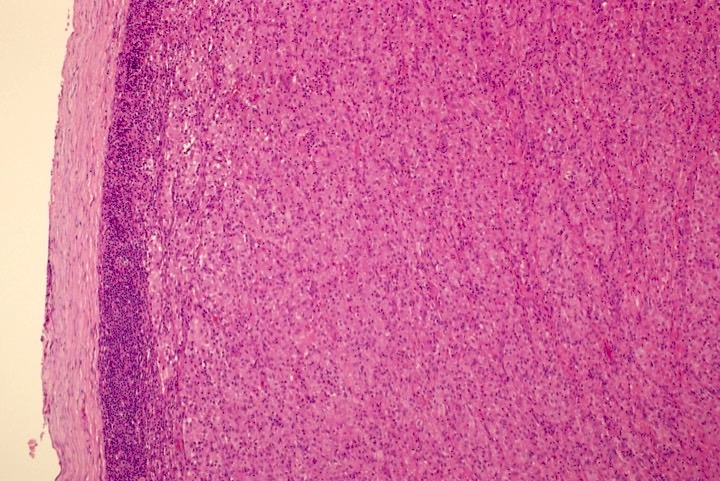
Distinct cortex

Diffuse histiocytic infiltrate

Patient with SCID
Ziehl Neelsen+
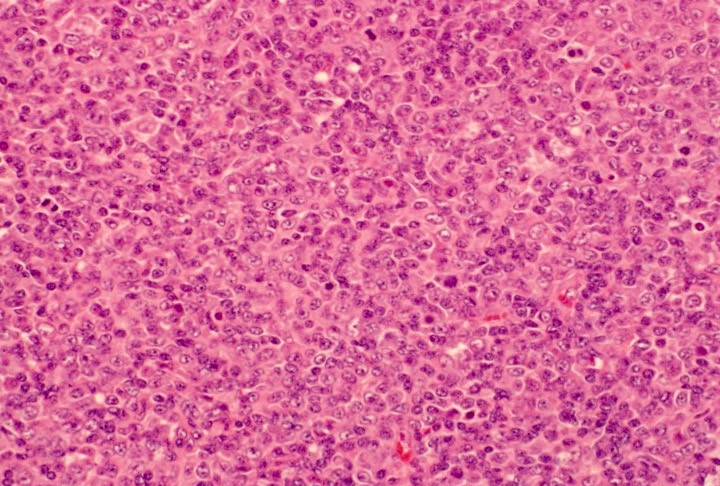
EBV+
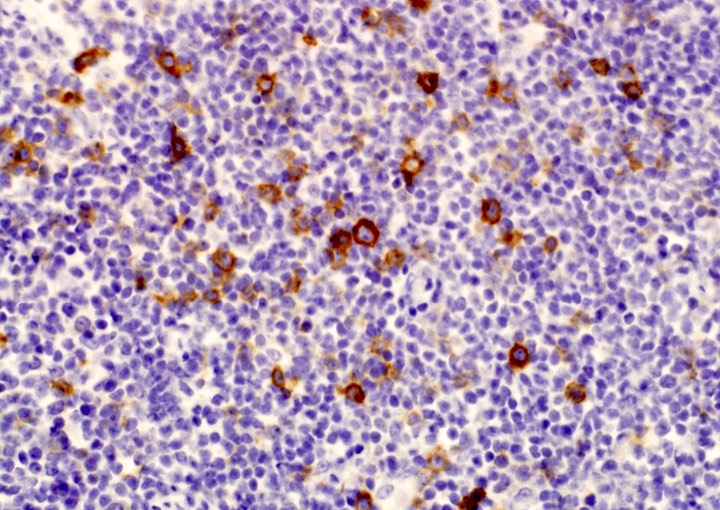
CD30+ immunoblasts
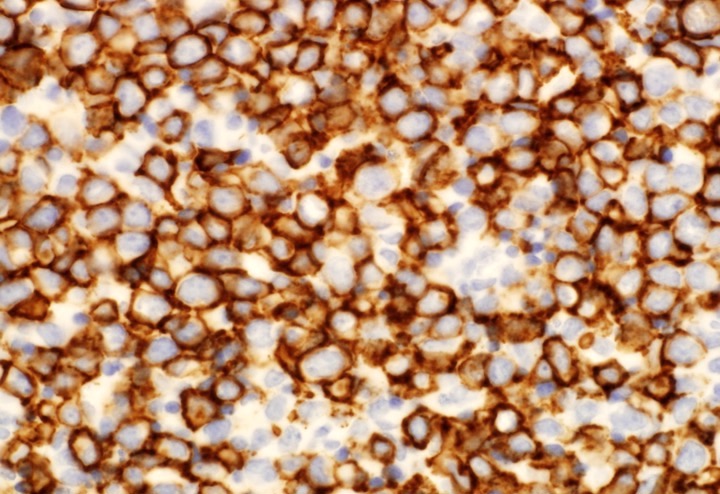
CD20+
Images hosted on other servers:
Parakeratosis and lymphohistocytic infiltrate
H&E, S100
CD30
- Large number of histiocytes with abundant streaked cytoplasm
- Ziehl-Neelsen (ZN) stain for acid-fast bacilli positive (Diagn Cytopathol 2001;24:333)
Contributed by Shamayel Mohammed, M.D.
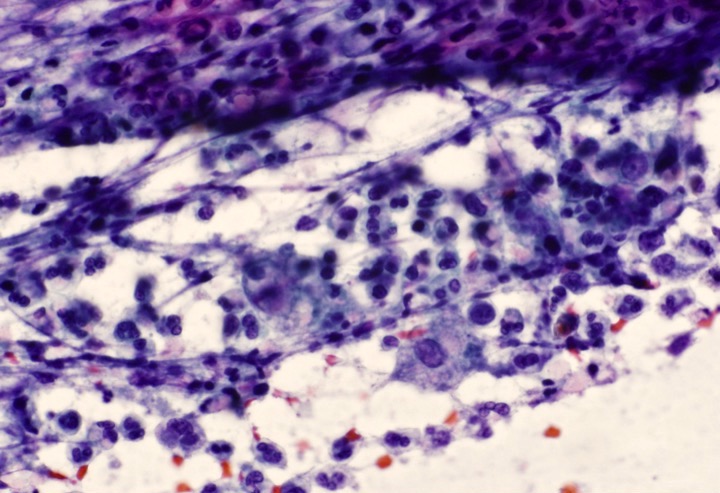
Ziehl-Neelsen+
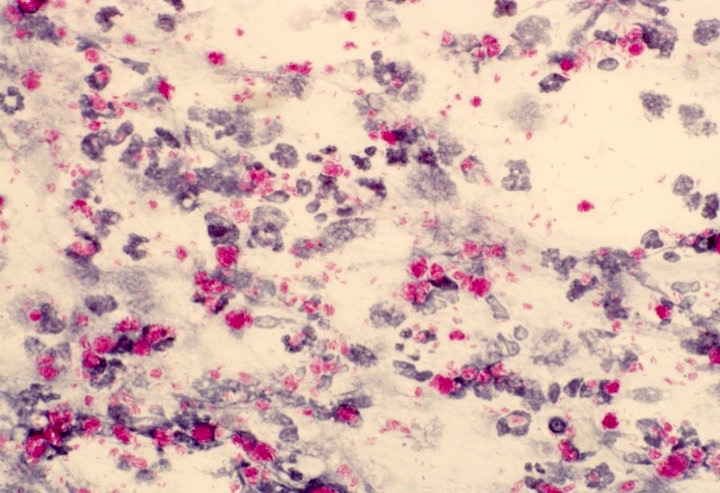
Histiocytes
- Immunohistochemistry staining show T lymphocyte (CD3+) proliferation
- Majority of the cells express CD30 (Sultan Qaboos Univ Med J 2007;7:133)
- EBV positive in associated lymphoproliferative disorders
- Eczema associated with diarrhea raises the possibility of a food allergy
- Hyperimmunoglobulin E (HIE) syndrome
- T-cell disorders
- Topical dermatitis
- See Thyroid gland topic
- Acquired complement mediated intravascular hemolytic anemia due to biphasic autoantibody, usually against the P red blood cell (RBC) antigen
- Paroxysmal cold hemoglobinuria (PCH) is an acquired complement mediated hemolytic process associated with polyclonal biphasic IgG autoantibodies against the P antigen, which bind at cold temperatures, resulting in activation of the complement cascade and intravascular hemolysis on warming to 37 °C (Hematol Oncol Clin North Am 2015;29:473)
- Most commonly seen in children < 5 years old with acute hemolysis 1 - 3 weeks after a viral or bacterial infection (Transfusion 2017;57:1332)
- Rarely seen in adults (because of the common use of antibiotics) but historically seen in association with congenital or tertiary syphilis, which causes chronic relapsing PCH (Hematol Oncol Clin North Am 2015;29:473)
- Generally transient and self limiting
- Paroxysmal cold hemoglobinuria (PCH)
- Donath-Lansteiner hemolytic anemia
- Donath-Landsteiner test
- Limited population data due to disease rarity
- Annual incidence estimated at 0.04 cases per 100,000 persons (Blood Adv 2023;7:2520)
- M:F ≈ 1 - 2:1 (J Clin Med 2021;10:216, Blood Adv 2023;7:2520)
- No ethnicity predilection (Lab Med 2014;45:253)
- Median age at diagnosis is 5 years old (Transfusion 2017;57:1332)
- Estimated to account for 5% in adults and associated with tertiary syphilis
- Estimated to account for up to 30% of cases of autoimmune hemolytic anemia (AIHA) in the pediatric population
- Vascular system: intravascular hemolysis (Hematol Oncol Clin North Am 2015;29:473)
- Kidneys: renal insufficiency or acute renal failure (Hematol Oncol Clin North Am 2015;29:473)
- Caused by a weak affinity biphasic autoantibody, usually IgG
- Also referred to as the Donath-Landsteiner antibody
- Reports of biphasic IgA and IgM autoantibodies (Pediatr Blood Cancer 2015;62:2044, Pediatr Int 2013;55:664)
- Autoantibodies are most commonly directed against the P red cell antigen but there have been reports of other carbohydrate antigen targets (i, I, HI, Pr) (Transfusion 2017;57:1332)
- Autoantibody most commonly forms ~1 - 3 weeks post-viral or bacterial infection (Transfusion 2017;57:1332)
- Other causes include vaccination, autoimmune disorders, hematopoietic malignancy, tertiary or congenital syphilis (historically)
- Rarely seen in adults in developed countries due to the introduction of antibiotic therapy (Hematol Oncol Clin North Am 2015;29:473)
- Mechanism of autoantibody sensitization is unknown; hypotheses include viral modification of RBC membrane, production of crossreactive antibodies or altered immune regulation (Transfusion 2017;57:1332)
- Other causes include vaccination, autoimmune disorders, hematopoietic malignancy, tertiary or congenital syphilis (historically)
- 2 phase process
- Autoantibody binds to the RBC membrane at temperatures < 37 °C
- Components of the complement cascade also bind at colder temperatures (Hematol Oncol Clin North Am 2015;29:473)
- On recirculation and rewarming to 37 °C, the autoantibody dissociates but the complement components remain bound, thus activating the terminal complement pathway with formation of the membrane attack complex on the RBC surface and resulting in intravascular RBC lysis (Hematol Oncol Clin North Am 2015;29:473)
- Free hemoglobin released during hemolytic episodes can result in acute kidney injury (Blood 2018;131:2506)
- Autoantibody binds to the RBC membrane at temperatures < 37 °C
- More prevalent in the pediatric population
- Pediatrics: > 70% of cases post-upper respiratory tract infection (URI), viral or bacterial
- Most commonly enveloped viral infections (Transfusion 2017;57:1332)
- Also associated with gastrointestinal illness, vaccination (most common association: pneumococcal vaccination), autoimmune disorder, hematopoietic malignancy, very rarely parvovirus B19 infection (Transfusion 2017;57:1332)
- Adults: rare
- Reports of cases with non-Hodgkin lymphoma, myeloproliferative disorder and post-varicella infection (Hematol Oncol Clin North Am 2015;29:473)
- Historically, 90% of adult cases associated with tertiary syphilis, chronic relapsing form with worsening in winter (Hematol Oncol Clin North Am 2015;29:473)
- Pediatrics: > 70% of cases post-upper respiratory tract infection (URI), viral or bacterial
- Presentation is variable but includes some degree of intravascular hemolysis (J Clin Med 2021;10:216)
- Intravascular hemolysis at presentation can be severe, necessitating immediate intervention
- Possible symptoms include anemia, jaundice, fatigue, pallor, fever, chills, back or abdominal pain
- Most common symptom: dark urine (Blood Adv 2023;7:2520)
- Symptoms appear 1 - 3 weeks postinfection; strong relationship to viral infections in children (Transfusion 2017;57:1332)
- Symptom onset can be associated with cold exposure (e.g., outside in cold weather, air conditioning, drinking cold beverage) (J Clin Med 2021;10:216)
- Prognosis is favorable (Lab Med 2014;45:253)
- Transient, self limiting and generally does not recur
- Classic presentation warrants immediate Donath-Landsteiner testing
- Hemolytic anemia in anyone under 18 years old with recent infection (viral or bacterial) (Immun Ageing 2020;17:38)
- Hemolysis may be sudden and severe
- New onset Coca-Cola colored urine reported
- Direct antiglobulin test (DAT) negative for IgG, positive for complement (most commonly)
- Hemolytic anemia in anyone under 18 years old with recent infection (viral or bacterial) (Immun Ageing 2020;17:38)
- Presence of Donath-Landsteiner antibody is pathognomonic
- Labs indicating hemolysis (Immun Ageing 2020;17:38)
- Increased indirect (unconjugated) bilirubin
- Low / absent haptoglobin
- Elevated lactate dehydrogenase
- Increased urinary urobilogen
- Hemosiderin in urine ~1 week after start of hemolysis
- Organ damage resulting from acute hemolysis (Hematol Oncol Clin North Am 2015;29:473)
- Relative reticulocytopenia is possible despite significant anemia; more common in children (Transfusion 2017;57:1332)
- May reflect sudden severity of hemolysis and cause depletion of reticulocytes in marrow pool
- Reticulocytes may be a target of the autoantibody
- Reticulocytopenia is not considered a poor prognostic sign in children with PCH as in adult populations with AIHA; children normally recover in 1 - 4 weeks
- May reflect sudden severity of hemolysis and cause depletion of reticulocytes in marrow pool
- Direct Coombs / DAT test is negative for IgG and positive for C3 (Clin Exp Rheumatol 2015;33:588)
- 5 - 10% of AIHA cases do not have a positive DAT (Immun Ageing 2020;17:38)
- Absence of warm autoantibodies (Transfusion 2017;57:1332)
- Positive Donath-Landsteiner test: in vitro test to measure biphasic hemolysins with anti-P specificity (Hematol Oncol Clin North Am 2015;29:473)
- Sample is required to be collected and maintained at 37 °C during transport
- Sample must be collected in tubes with no anticoagulant (red top Vacutainer)
- Can result in false negative results due to incorrect specimen handling or transient nature of the autoantibody
- Antibody titers are highest during periods of clinical hemolysis
- 15 month old boy presented with an upper respiratory infection and sudden onset of pallor with dark urine (J Pediatr Hematol Oncol 2022;44:60)
- 23 month old girl and 4 year old girl both admitted to the hospital with acute hemolytic anemia following upper respiratory infections and otitis media in the weeks prior (Lab Med 2014;45:253)
- 4 year old boy presented with presumed viral infection and sudden onset severe hemolysis successfully treated with eculizumab therapy (Blood Adv 2019;3:3575)
- 18 year old woman in early pregnancy with acute hemolytic anemia was found to be D - L antibody positive (BMC Hematol 2015:15:3)
- 27 year old woman presented with cough and acute hemolytic anemia leading to acute kidney injury and oliguric renal failure requiring hemodialysis (Blood 2018;131:2506)
- Supportive care: transient, self limiting disease (Transfusion 2017;57:1332)
- Warm ambient temperature
- Blood transfusions as necessary
- Hemoglobin frequently falls below 6 g/dL during active hemolysis (Blood Adv 2019;3:3575)
- P antigen negative blood is not necessary for support; only considered in refractory hemolysis despite supportive measures (Clin Exp Rheumatol 2015;33:588)
- P antigen negative RBC units are rare and difficult to find due to the high prevalence of P antigen in the population
- P antigen negative blood is not necessary for support; only considered in refractory hemolysis despite supportive measures (Clin Exp Rheumatol 2015;33:588)
- Iron, folate and B12 supplementation to support hematopoiesis if deemed necessary (Immun Ageing 2020;17:38)
- Hemoglobin frequently falls below 6 g/dL during active hemolysis (Blood Adv 2019;3:3575)
- Corticosteroid treatment is common but efficacy is unclear (J Clin Med 2021;10:216, Blood Adv 2023;7:2520)
- Chronic, refractory or recurrent cases: corticosteroids, intravenous immunoglobulin, rituximab and eculizumab have all been used with limited efficacy (Indian J Pathol Microbiol 2021;64:613)
- Plasmapheresis can help clear antibody in severe hemolytic or refractory cases (Clin Exp Rheumatol 2015;33:588)
- Treat the underlying condition: syphilis related PCH resolves with curative antibiotic therapy for syphilis (Hematol Oncol Clin North Am 2015;29:473)
Contributed by Samantha Mack, M.D.

Donath-Landsteiner
assay interpretation
worksheet

Donath-Landsteiner tube assay example
- Peripheral smear may show (Transfusion 2017;57:1332)
- Evidence of hemolysis: spherocytosis (50% of cases), autoagglutination / rouleaux formation (17 - 25% of cases)
- Bone marrow compensation: polychromasia (if reticulocytosis present)
- Neutrophil erythrophagocytosis and neutrophil rossettes, while rare, are strongly associated with PCH (Hematol Oncol Clin North Am 2015;29:473, Blood 2020;135:393)
- Assessment: Johnny is a 5 year old boy who presented with 1 week of symptoms consistent with an upper respiratory infection (cough, fever, congestion), anemia and new onset hemoglobinuria. Send out testing identified the presence of a biphasic antibody (Donath-Landsteiner antibody).
- Plan: The patient presentation and laboratory workup are consistent with a diagnosis of paroxysmal cold hemoglobinuria.
- Transfuse pRBCs as clinically indicated. A blood warmer is recommended.
- Recommend patient avoid cold climates while antibody is present.
- No special transfusion requirements or precautions.
- Autoimmune hemolytic anemia (AIHA) (Immun Ageing 2020;17:38):
- Mixed autoimmune hemolytic anemia (J Clin Med 2021;10:216):
- Cold agglutinin syndrome (CAS) (Immun Ageing 2020;17:38):
- Presents similarly to PCH with intravascular hemolysis precipitated by recent infection (Mycoplasma pneumoniae or Epstein-Barr virus most frequently)
- DAT positive for C3 but high titer IgM autoantibody with anti-Ia specificity
- Cold agglutinin titer positive
- Symptoms of both intravascular and extravascular hemolysis
Which of the following is considered the definitive test for diagnosis of paroxysmal cold hemoglobinuria (PCH)?
- Bone marrow biopsy
- Coombs test
- Donath-Landsteiner test
- Flow cytometry
Comment Here
Reference: Paroxysmal cold hemoglobinuria (PCH)
- A antigen
- I antigen
- O antigen
- P antigen
Comment Here
Reference: Paroxysmal cold hemoglobinuria (PCH)
- Acquired autosomal clonal mutation of hematopoietic stem cells (HSCs) characterized by altered surface protein expression of hematopoietic elements and increased hemolysis, risk of thrombosis and impaired bone marrow function
- Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired somatic mutation in the X linked phosphatidylinositol glycan class A (PIGA) gene, which leaves hematopoietic cells unable to produce the glycosylphosphatidylinositol (GPI) anchor that links cell surface proteins to the plasma membrane (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Blood 2014;124:2804)
- PNH leads to episodic hemolytic anemia, recurrent thrombosis and bone marrow failure and cytopenias (EJIFCC 2019;30:355)
- Despite the name, PNH patients may experience hemoglobinuria at night, during the day or not at all (EJIFCC 2019;30:355)
- PNH clones frequently appear in patients with aplastic anemia and myelodysplastic syndromes (Int J Lab Hematol 2017;39:329)
- Paroxysmal nocturnal hemoglobinuria (PNH)
- Marchiafava-Micheli (Q J Med 1949;18:105)
- Global incidence: 1 - 2 per million (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Global prevalence: 10 - 20 per million (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Median age at diagnosis: early to mid-30s (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Globally, M = F (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- In a comparative analysis of 1,665 patients between Asia and Europe / America:
- Proportion of European / American women was significantly higher than in Asia
- Incidence of hemoglobinuria and thromboembolism: Europe / U.S. (F) > Asia (F) (Int J Hematol 2016;103:649)
- Vascular system: intravascular hemolysis, thrombosis (especially venous thrombosis) in atypical locations such as hepatic vein (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Bone marrow (J Hematopathol 2013;6:71):
- Classic PNH: 65% of patients have normal or hypercellular bone marrow; erythroid hyperplasia is common; myelopoiesis is decreased in 86% and megakaryopoiesis is decreased in 46%
- PNH patients with aplastic anemia: 95% have hypocellularity in bone marrow; myelopoiesis decreased in 85% and megakaryopoiesis decreased in 100%
- Kidneys: renal insufficiency or renal failure (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Esophagus: esophageal spasm, free hemoglobin in plasma depletes nitric oxide (NO), causing smooth muscle dystonia (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Blood 2014;124:2804)
- Penis: priapism or erectile dysfunction, also due to effect of nitric oxide scavenging by free hemoglobin (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Anemia 2011;2011:297364)
- PNH is caused by an acquired mutation in the phosphatidylinositol glycan class A (PIGA) gene; this gene produces the glycosylphosphatidylinositol (GPI) anchor proteins (GPI-APs), a posttranslational modification that links cell surface proteins to cell membranes (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Mechanism of mutation is unknown: proposed theory of immune attack on normal HSCs and sparing of PNH stem cell clones due to close association between PNH and acquired aplastic anemic (AA) (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Other factors leading to proliferation and expansion of PNH clones include:
- Mutation or aberrant expression of recombinant signal binding protein of immunoglobulin kJ region (RBPJ) (Exp Ther Med 2019;17:4536)
- A small subset of patients may alternately have a mutation in the phosphatidylinositol glycan anchor biosynthesis class T (PIGT) gene, responsible for transferring GPI to precursor proteins; patients with this mutation tend to have additional autoinflammatory symptoms (J Clin Invest 2019;129:5123)
- HSCs containing PIGA mutations lack GPI anchored cell surface markers, including complement inhibitors CD59, which interferes with C9 binding to the cell membrane and formation of C3 convertase; decay accelerating factor (DAF / CD55), homologous restriction factor (HRF), C8 binding protein, membrane inhibitor of reactive lysis (MIRL) (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2016;32:383)
- Mutants HSCs undergo clonal expansion under bone marrow (BM) failure (J Clin Invest 2019;129:5123)
- Mature erythrocytes lacking GPI-APs are unprotected from the membrane attack complex (MAC or C5b9), leading to paroxysmal hemolysis (Indian J Hematol Blood Transfus 2016;32:383, Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- During hemolytic episodes, free hemoglobin (Hb) is released into the blood, which causes renal damage; free Hb can also contribute to hypercoagulability, thrombosis (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Free hemoglobin scavenges nitric oxide in the bloodstream, which impairs its ability to regulate vascular tone and inhibit platelet activation, leading to smooth muscle dystonia and thrombosis (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2017;33:453, Blood 2014;124:2804)
- Extravascular hemolysis: C3 fragments accumulate on GPI-AP negative red blood cells (RBCs) and act as an opsonin, which causes reticuloendothelial destruction in the liver and spleen (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- PNH clone found in:
- 40 - 70% of patients with aplastic anemia (AA); T cell mediated autoimmune attack may give PNH stem cells a survival advantage due to fewer surface markers (Indian J Hematol Blood Transfus 2017;33:453, Blood 2014;124:2804)
- 48% of patients with hemoglobinuria (Cytometry B Clin Cytom 2017;92:361)
- 33% of bone marrow failure patients have a PNH clone (Cytometry B Clin Cytom 2017;92:361)
- 10 - 19% of direct antiglobulin test (DAT) negative hemolytic anemia patients (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
- 5 - 10% of patients with myelodysplastic syndrome (MDS) (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
- 3 - 9% of patients with unexplained cytopenias (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
- 0.4 - 2% of patients with thrombosis (Int J Lab Hematol 2017;39:329, Cytometry B Clin Cytom 2017;92:361)
Images hosted on other servers:

Suggested pathophysiology of PNH

Suggested pathophysiology of hemolysis in PNH
Complement action in healthy subjects and PNH patients
- Presentation varies, including intravascular hemolysis, thrombosis, AA and cytopenia, which precede or follow PNH (EJIFCC 2019;30:355)
- Possible symptoms include pulmonary hypertension, dyspnea, impaired renal function, erectile dysfunction, pain in the abdomen, fatigue and difficulty swallowing, although PNH may also be asymptomatic (EJIFCC 2019;30:355)
- Hemolytic episodes may be precipitated by infection, surgery or transfusions (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Hemolytic episodes may cause fever, headache, fatigue and back or abdominal pain (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Hemoglobinuria occurs in 90% of patients; it may occur at night, during the day or not at all (EJIFCC 2019;30:355)
- Thrombosis occurs in 50% of PNH patients, often in unusual locations, such as intraabdominal visceral veins or hepatic veins (Blood Cells Mol Dis 2020;80:102372, Pract Lab Med 2020;20:e00158)
- These complications are among the primary causes of morbidity and mortality for PNH patients (Sci Rep 2018;8:13458)
- Chronic kidney disease occurs in up to 65% of PNH patients, including stage III - V disease in 21% of PNH patients (Hematology 2018;23:558)
- Indications for testing for PNH: cytopenia, diagnosis or suspicion of aplastic anemia, thrombosis, Coombs negative hemolysis or hemoglobinuria (Eur J Haematol 2019;102:36)
- Annual flow cytometry screening for PNH may be useful in patients with AA or MDS (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Other causes of anemia should be excluded before testing for PNH (Cytometry B Clin Cytom 2018;94:16)
- PNH is diagnosed by flow cytometry analysis of at least 2 GPI anchored markers on at least 2 cell types (Int J Lab Hematol 2016;38:5, Indian J Hematol Blood Transfus 2017;33:453)
- 3 classifications of PNH clones (Cytometry B Clin Cytom 2018;94:16, Pract Lab Med 2020;20:e00158):
- Classic or hemolytic PNH: patients have a PNH clone and symptoms related to intravascular hemolysis without evidence of a bone marrow failure disorder
- PNH in the context of other primary bone marrow disorders: patients have aplastic anemia or myelodysplastic syndrome with a PNH clone and symptoms primarily related to bone marrow failure
- Subclinical or asymptomatic PNH: patients have a PNH clone but lack clinical or laboratory signs of hemolysis or thrombosis
- Whole body magnetic resonance imaging can be used to detect and evaluate vascular complications (Sci Rep 2018;8:13458)
- Hemolysis with negative direct Coombs (DAT) test (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Ham test to RBC fragility when placed in a mild acid (replaced by flow cytometry) (Pract Lab Med 2020;20:e00158, Ann Clin Lab Sci 2003;33:401)
- Sucrose hemolysis test to RBC fragility when placed in hypotonic sucrose solution triggers complement activation in PNH patients (replaced by flow cytometry) (Pract Lab Med 2020;20:e00158)
- Normocytic anemia with inadequate reticulocytosis and variable neutropenia (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Thrombocytopenia is rare (Hematol Rep 2017;9:6862)
- BM appearance from erythroid hyperplasia to marked hypoplasia (J Hematopathol 2013;6:71)
- Increased levels of lactate dehydrogenase (LDH) and bilirubin and decreased levels of haptoglobin (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Flow cytometry (gold standard): absence of GPI anchored proteins (GPI-APs): CD14, CD16, CD24, CD48, CD52, CD55, CD58, CD59, CD66b, CD66c, CD73, CD87, CD157, CD177, with loss of at least 2 GPI-APs on RBCs and neutrophils (see below) (Pract Lab Med 2020;20:e00158, Cytometry B Clin Cytom 2007;72:167)
- Fluorescein labeled proaerolysin (FLAER) often used in conjunction, as it binds selectively to the GPI moiety on GPI-APs, resulting in increased sensitivity (Int J Lab Hematol 2016;38:5, Cytometry B Clin Cytom 2007;72:167)
- Organ damage resulting from hemolysis or thrombosis (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- 12 month remission rate: 79% (J Clin Lab Anal 2020;34:e23008)
- 5 year mortality rate: 35% with median survival time at 10 - 15 years (J Clin Lab Anal 2020;34:e23008)
- 10 year overall survival at 70 - 75% (J Clin Lab Anal 2020;34:e23008, Nihon Ketsueki Gakkai Zasshi 1989;52:1386, Blood 2008;112:3099)
- Other factors related to poor survival (J Korean Med Sci 2016;31:214):
- Initial clinical features at the time of diagnosis (Blood 2008;112:3099)
- Hemoglobin (Hb) < 10g/dL and neutropenia (Blood 2008;112:3099)
- Absence of specific treatment during the first year (hazard ratio [HR], 2.2) (Blood 2008;112:3099)
- Independent prognostic factors:
- Thromboembolism: odds ratio (OR) 7.11; 14 times higher risk of mortality (J Korean Med Sci 2016;31:214, J Clin Lab Anal 2020;34:e23008)
- Bone marrow failure (J Clin Lab Anal 2020;34:e23008)
- Elevated LDH > 1.5 times the upper limit of normal (ULN): OR 3.204; 4.8 times higher risk of mortality compared with general (non-PNH) population (J Korean Med Sci 2016;31:214, J Clin Lab Anal 2020;34:e23008)
- Renal impairment: OR 2.953; 8 times higher risk of mortality (J Korean Med Sci 2016;31:214)
- PNH cytopenia: OR 2.547; 6.2 times higher risk of mortality (J Korean Med Sci 2016;31:214)
- Patients with both hemolysis and 1 clinical symptom have a greater risk of mortality compared with patients with just hemolysis or just a clinical symptom (J Korean Med Sci 2016;31:214)
- Leading cause of death for PNH patients in Europe / North America is thromboembolism and for patients in Asia, serious infections (Int J Hematol 2016;103:649)
- Renal failure leads to death in 8 - 18% of PNH patients (Hematology 2018;23:558)
- 17 year old Caucasian boy presented with several months of abdominal pain, fever and dark colored urine (Case Rep Pediatr 2019;2019:4930494)
- 23 year old woman was admitted to the emergency department due to bleedings of the jawbone after inflammation (Clin Case Rep 2018;7:175)
- 24 year old woman presented with fevers, epigastric pain, nausea and vomiting (Blood 2013;122:2795)
- 32 year old woman, G2P2A0, with no past medical history of any systemic illnesses (Bol Asoc Med P R 2015;107:9)
- 36 year old Hispanic man presented to the emergency department with 4 days of diffuse abdominal pain (Cureus 2020;12:e8941)
- Pegcetacoplan is a pegylated peptide targeting complement protein C3
- New Food and Drug Administration (FDA) approved PNH therapy that showed its superiority to eculizumab in improving hemoglobin (Hb) and clinical and hematological outcomes (N Engl J Med 2021;384:1028)
- In a phase 3 open label controlled trial in adults with PNH and Hb levels lower than 10.5 g/dL despite eculizumab therapy, 85% of patients received pegcetacoplan as compared to 15% receiving eculizumab who no longer required transfusions (N Engl J Med 2021;384:1028)
- Eculizumab: anti-C5 antibody; interferes with activation of the complement system (Int J Lab Hematol 2016;38:5)
- Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved PNH therapy (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Not curative but symptomatic management (Indian J Hematol Blood Transfus 2016;32:383)
- In the initial randomized controlled trial of eculizumab, 49% of PNH patients had stabilized Hb levels following treatment; eculizumab also led to improved quality of life (N Engl J Med 2006;355:1233)
- A subsequent study found that eculizumab improved overall survival (92% survival at 6 years compared with 80% in nontreated historical controls), led to fewer thrombotic events (4% compared with 27%) and caused fewer transitions to MDS or leukemia (Am J Hematol 2016;91:366)
- Eculizumab improved renal function in some patients, especially in those with chronic kidney disease (CKD) stage I - II, while showing significant decrease in major clinical kidney event rates from 4.22 to 2.10 events per 100 patient years (Hematology 2018;23:558)
- Meningococcal vaccine is required before treatment with eculizumab due to increased risk of Neisseria meningitidis infections (Indian J Hematol Blood Transfus 2016;32:383)
- Ravulizumab is a new C5 inhibitor given every 8 weeks; 2 phase 3 trials showed it was noninferior to eculizumab and had a similar safety profile (Blood 2019;133:530, Blood 2019;133:540)
- Allogenic hematopoietic stem cell transplantation
- Only curative treatment (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Indicated for patients with very severe or life threatening disease or who have had eculizumab treatment failure (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2016;32:383)
- Only curative treatment (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print])
- Supportive care (Hematol Transfus Cell Ther 2020 Jul 6 [Epub ahead of print], Indian J Hematol Blood Transfus 2016;32:383):
- Iron, folate and B12 supplementation
- Transfusion of RBCs
- Steroid therapy with glucocorticoids during hemolytic crises
- Immunosuppressive therapies in patients with bone marrow deficiency
- Anticoagulants to manage thrombosis
- Flow cytometry should be repeated every 3 - 6 months to monitor the size of the PNH clone (Indian J Hematol Blood Transfus 2017;33:453)
- Bone marrow:
- Bone marrow analysis is primarily done to determine if PNH arose in association with aplastic anemia (AA) or myelodysplastic syndrome (MDS) (Pract Lab Med 2020;20:e00158, Br J Haematol 2018;182:758)
- There are no specific morphologic features for PNH (Pract Lab Med 2020;20:e00158, Br J Haematol 2018;182:758, J Hematopathol 2013;6:71)
- Bone marrow cellularity may be increased, decreased or within a normal range for age (J Hematopathol 2013;6:71)
- Erythroid hyperplasia, decreased myelopoeisis and decreased megakaryocytes are features that can be seen in PNH bone marrow (J Hematopathol 2013;6:71, Pract Lab Med 2020;20:e00158, Br J Haematol 2018;182:758, Semin Hematol 2019;56:65)
- Peripheral smear may show spherocytosis or presence of sphere shaped erythrocytes; however, these findings are nonspecific (Clin Adv Hematol Oncol 2018;16:1)
- At least 2 different hematopoietic cell types should be evaluated using antibodies against GPI anchored cell surface marker or using FLAER, which binds to all GPI anchored molecules (Int J Lab Hematol 2016;38:5)
- Because the majority of potential PNH cases will be negative, an initial screen with fewer antibodies can be performed before using a diagnostic confirmation assay using a full antibody panel (EJIFCC 2019;30:355)
- Screens may be accomplished with a simpler 2 color panel consisting of FLAER and CD15 labeling of neutrophils, although this is not a substitute for a full diagnostic assay (Int J Lab Hematol 2020;42:589)
- PNH clones will appear with these immunophenotypes:
- Granulocytes: absence of CD16, CD24, FLAER (Int J Lab Hematol 2016;38:5)
- Neutrophils or monocytes: absence of CD16, CD24, CD14, CD157, FLAER (Int J Lab Hematol 2016;38:5)
- Erythrocytes: absence of CD55, CD59, FLAER (Int J Lab Hematol 2016;38:5)
- Platelets: absence of CD55, CD59, FLAER (Blood Cells Mol Dis 2020;80:102372)
- Different types of RBC clones (Ann Hematol 2019;98:1083):
- Type I: presence of GPI-APs (CD55 and CD59; i.e. normal, non-PNH RBCs)
- Type II: partial absence of GPI-APs
- Type III: complete absence of GPI-APs
- 10% of patients present with both type II and type III clones (Ann Hematol 2019;98:1083)
- Analyze the size of PNH clone; if 1 - 5% of a given cell type is GPI deficient, treatment should be considered (Int J Lab Hematol 2016;38:5)
- Quantitation of the PNH clone is not as accurate when measuring RBCs if the patient has received blood transfusions containing normal RBCs or has had recent hemolytic crises (EJIFCC 2019;30:355)
- Quantification of RBCs is more accurate when adding anti-CD71 antibodies to the flow cytometry assay and using immature RBC populations, which are resistant to destruction by the complement system (Cytometry B Clin Cytom 2020;98:179)
- Because diagnosis relies on absence rather than the presence of specific markers, care should be taken to exclude apoptotic cells using cell viability dye (EJIFCC 2019;30:355)
- Using an 8 color panel can more accurately detect PNH clones compared with a 4 color panel; the sensitivity of the optimized 8 color panel is 0.01% when measuring granulocytes and 0.05% when measuring monocytes, after acquiring 100,000 events (Biomed Rep 2018;8:224)
Contributed by Saja Asakrah, M.D.

High sensitivity PNH flow cytometry
- Peripheral blood sample
- Paroxysmal nocturnal hemoglobinuria (PNH) (see comment)
- Comment:
- Flow cytometric analysis of the submitted blood sample demonstrates the presence of population(s) of white blood cells (neutrophils or monocytes) or red blood cells (RBCs) with deficiency of tested glycosylphosphatidyl inositol (GPI) linked proteins.
- Percentages of the GPI deficient cell populations are:
- Total GPI deficient RBCs (type III plus type II): 13.8%
- Type III (GPI deficient) RBCs (CD235a+ CD59-): 10.2%
- Type II (partial GPIdeficient) RBCs (CD235a+ CD59- intermediate): 3.6%
- GPI deficient neutrophils (CD15+ CD24- FLAER-): 28.7%
- GPI deficient monocytes (CD64+ CD14- FLAER-): 59.4%
- The flow cytometric findings are consistent with a diagnosis of paroxysmal nocturnal hemoglobinuria (PNH). However, PNH cells may be seen in PNH, aplastic anemia, myelodysplastic syndromes and other conditions. Correlation with clinical and laboratory findings is suggested.
- Note: Discordance between the size of GPI deficient RBC and WBC populations may be due to hemolysis or transfusion.
- The lower limit of quantification (lower limit of enumeration) for the RBC assay is 0.01%, for the neutrophil assay is 0.05% and for the monocyte assay is 0.5%. Below these levels, PNH cells may be reported as detected but below the level of quantification. For leukopenic patients, the neutrophil and monocyte assay sensitivity may be decreased.
- References: Cytometry B Clin Cytom 2018;94:16, Cytometry B Clin Cytom 2018;94:23, Cytometry B Clin Cytom 2018;94:49, Cytometry B Clin Cytom 2018;94:67, CLSI: H52-A2 - Red Blood Cell Diagnostic Testing Using Flow Cytometry, 2nd Edition, 2014, EJIFCC 2019;30:355, Int J Lab Hematol 2019;41:73
- Broad differential diagnosis includes other causes of hemolytic anemia and bone marrow failure
- Aplastic anemia (AA):
- Patients tend to have smaller clone sizes compared with patients with classic PNH (35% compared with 83%, although these clone sizes are variable) (Cytometry B Clin Cytom 2018;94:16)
- Have PNH clones that primarily affect granulocytes, rather than both granulocytes and erythrocytes (Exp Hematol 2019;77:41)
- Patients generally have leukopenia or thrombocytopenia (Pract Lab Med 2020;20:e00158)
- Patients tend to have smaller clone sizes compared with patients with classic PNH (35% compared with 83%, although these clone sizes are variable) (Cytometry B Clin Cytom 2018;94:16)
- Myelodysplastic syndrome (MDS):
- Patients also have GPI deficient cells but these arise in colony forming cells rather than hematopoietic stem cells and T cells are not affected (Cytometry B Clin Cytom 2018;94:16)
- Patients generally have leukopenia or thrombocytopenia (Pract Lab Med 2020;20:e00158)
- Autoimmune hemolytic anemia:
- Positive Coombs test, evidence of extravascular rather than intravascular hemolytic anemia (Oxf Med Case Reports 2020;2020:omz125)
- Various presentations of microangiopathic and macroangiopathic anemia
- Sickle cell disease:
- Vaso-occlusion and vasculopathy with early onset and chronic progression (Intern Emerg Med 2019;14:1051)
- Glucose-6-phosphate dehydrogenase (G6PD) deficiency:
- Acute hemolytic anemia occurs in response to infection, fava beans or drugs like primaquine (Hematol Oncol Clin North Am 2016;30:373)
- Pyruvate kinase deficiency:
- Congenital
- Family history
- Low enzyme activity detected during pyruvate kinase enzyme tests (Am J Hematol 2015;90:825)
- Bone marrow biopsy
- Coombs test
- Flow cytometry
- Serum haptoglobin level
- CD11b
- CD11c
- CD14
- CD64
- Malaria (from the Italian "mal' aria," meaning "bad air") is an acute and sometimes chronic bloodstream infection characterized by fever, anemia and splenomegaly, caused by apicomplexan parasites of the genus Plasmodium
- Four species of plasmodia causing human malaria are Plasmodium vivax, Plasmodium falciparum, Plasmodium malariae and Plasmodium ovale
- Plasmodium falciparum causes 85% of malaria cases
- Because its infection is potentially life threatening, its presence must be considered in the differential diagnosis of unexplained fever and history of travel to endemic geographic areas should always be sought
- Falciparum infection occurs principally in tropical areas worldwide
- U.S. civilian cases of malaria increased from 151 in 1970 to 1,838 in 1980 (McPherson: Henry's Clinical Diagnosis and Management by Laboratory Methods, 22nd Edition, 2011 [Chapter 62]) and reached a 40 year high of 1,925 cases in 2011 (CDC: Malaria Facts [Accessed 9 January 2018])
- Worldwide, disease generally occurs between 45° N and 40° S and rates have been declining (WHO: Malaria [Accessed 9 January 2018])
- Spreads exclusively by female anopheline mosquitoes
- Fever paroxysm (see below) occurs over 6 - 10 hours; is initiated by synchronous rupture of erythrocytes with release of new infectious blood stage forms known as merozoites
- Transfusion induced malaria occurs when blood donors have subclinical malaria and may prove fatal for recipient
- Similarly, congenital malaria may occur in infants of mothers from endemic areas; infant acquires infection at birth due to rupture of placental blood vessels with maternal fetal transfusion
- Typically, no relapse with transfusion or congenital malaria because exoerythrocytic schizogony does not occur
Images hosted on other servers:

Fever paroxysm
Life cycle
- Most patients become symptomatic within 1 month of exposure
- Defining clinical features of a malarial attack or paroxysm consist of initial shaking chills, fever (up to 40° C or higher) and generalized diaphoresis, followed by resolution of fever
- Brain involvement, known as cerebral malaria, causes disorientation, progressing to delirium, coma and often death
- Exchange transfusion may be lifesaving in severe cases (McPherson: Henry's Clinical Diagnosis and Management by Laboratory Methods, 22nd Edition, 2011 [Chapter 62])
- Identification of malarial parasites on thin blood films requires a systematic approach
- Three major factors should be considered: appearance of infected erythrocytes, appearance of parasites and stages found
- Appearance of RBCs and size: normal size; multiple infected red blood cells are common
- Schüffner stippling: Maurer dots (comma shaped spots, dark blue by Giemsa staining, on RBC surface) are occasionally seen
- Parasite cytoplasm: young rings are small, delicate, often with double chromatin dots; gametocytes are crescent or elongate
- Appearance of parasite pigment: black; coarse and conspicuous in gametocytes
- Number of merozoites: 6 - 32; average is 20 - 24
- Stages found in circulating blood: rings or gametocytes; other stages develop in blood vessels of internal organs but are not seen in peripheral blood except in severe infection
- Laboratory evaluation relies on timely examination of thick and thin blood films to demonstrate the intraerythrocytic parasites
- More advanced laboratory methods include acridine orange staining and detection of parasite specific DNA (Exp Parasitol 2007;116:427)
- Morphologic stages seen in erythrocytes include trophozoites (growing forms), schizonts (dividing forms) and gametocytes (sexual forms)
- Malarial parasites undergo a sexual phase (sporogony) in Anopheles mosquitoes that produces infectious sporozoites and an asexual stage (schizogony) in humans that produces schizonts and merozoites
- In the bloodstream, some merozoites eventually differentiate into gametocytes (gametogony), which when ingested by female anopheline mosquitoes, mature into male microgametes and female macrogametes
- Fusion of a microgamete and a macrogamete results in the formation of the motile ookinete, which migrates to the outside of the mosquito stomach wall and forms an oocyst
- Within the oocyst, numerous spindle shaped sporozoites are formed
- Mature oocyst ruptures into the body cavity, releasing sporozoites, which then migrate through the tissues to the salivary glands, from which they are injected into the vertebrate host as the mosquito feeds
- Time required for development in the mosquito ranges from 8 - 21 days
- Sporozoites injected into the vertebrate host reach the hepatic parenchymal cells within minutes and initiate the proliferative phase known as exoerythrocytic schizogony
- Release of merozoites from ruptured hepatic schizonts initiates the bloodstream infection or erythrocytic schizogony and eventually the clinical symptoms of malaria
- P. vivax and P. ovale differ from P. falciparum and P. malariae in that true disease relapses of the former species may occur weeks to months following subsidence of previous attacks
- This occurs due to renewed exoerythrocytic and eventually, erythrocytic schizogony from latent hepatic sporozoites, which are known as hypnozoites
- Recurrence of disease due to P. falciparum or P. malariae, called recrudescences, arise from increased numbers of persisting blood stage forms to clinically detectable levels, not from persisting liver stage forms
- Liver cells are infected only by sporozoites from the mosquito; thus, transfusion acquired P. vivax or P. ovale infection does not relapse
- Merozoites released from infected hepatocytes subsequently infect erythrocytes
- Following amplification of parasites in the bloodstream for a period of time and the development of synchrony in their appearance, clinical attacks of malaria occur
- P. falciparum infects erythrocytes of all ages; P. vivax and P. ovale parasites primarily infect young erythrocytes; P. malariae infects older erythrocytes
- Individuals with sickle cell trait are less susceptible to P. falciparum malaria and persons who lack certain Duffy blood group determinants are protected against P. vivax infection
- Glucose-6-phosphate dehydrogenase (G6PD) deficiency has been associated with protection from malaria but evidence is less striking than with these other genetic abnormalities
- 45 year old man with disseminated intravascular coagulation in malaria (Niger Med J 2014;55:171)
- 45 year old man with high grade fever, rigors, chills and shortness of breath (Case of the Week #277)
- 50 year old woman with recent travel to Kenya presents with acute onset of fever and chills (Pritt: Creepy Dreadful Wonderful Parasites Blog - Case of the Week 558 [Accessed 13 September 2019])
- 51 year old man with severe Plasmodium falciparum infection mimicking acute myocardial infarction (Mala J 2014;13:341)
- A patient with recent travel to southern Africa presented with fever and myalgias (Pritt: Creepy Dreadful Wonderful Parasites Blog - Case of the Week 494 [Accessed 29 November 2018])
- If parasitemia > 2%, contact the clinical team as if a critical value since patient is at high risk of death (Pritt: Creepy Dreadful Wonderful Parasites Blog - Answer to Case 494 [Accessed 29 November 2018])
- Therapy and prophylaxis of malaria have become highly complex due to widespread resistance by P. falciparum to chloroquine and other antimalarials and to a lesser extent, resistance by P. vivax to chloroquine
- These drugs are usually recommended by national malaria control programs but may not be effective due to local drug resistant strains (CDC: Guidelines for Treatment of Malaria in the United States [Accessed 9 January 2018]):
- Artemesinin containing combination treatments (for example, artemether-lumefantrine, artesunate-amodiaquine)
- Atovaquone-proguanil
- Chloroquine
- Doxycycline
- Mefloquine
- Quinine
- Sulfadoxine-pyrimethamine
- In addition to antimalarial treatment, red blood cell exchange may also be performed for patients with > 10% parasitemia (CDC paper arguing against red cell exchange for severe malaria: Clin Infect Dis 2013;57:923; ASA paper supporting red cell exchange: Clin Infect Dis 2014;58:302)
Images hosted on other servers:

Adult of Anopheles freeborni
- Rings of P. falciparum can be found in blood smears
- Some may exhibit Maurer clefts
- Gametocyte may be present in blood smears
- Schizont of P. falciparum can be seen
- Rules for determining parasitemia (% RBC infected) (Pritt: Creepy Dreadful Wonderful Parasites Blog - Case of the Week 494 [Accessed 29 November 2018]):
- Count multiple infected RBCs as 1 only
- Don't count gametocytes, since they are a dead end stage in the human host
Contributed by Bobbi Pritt, M.D.
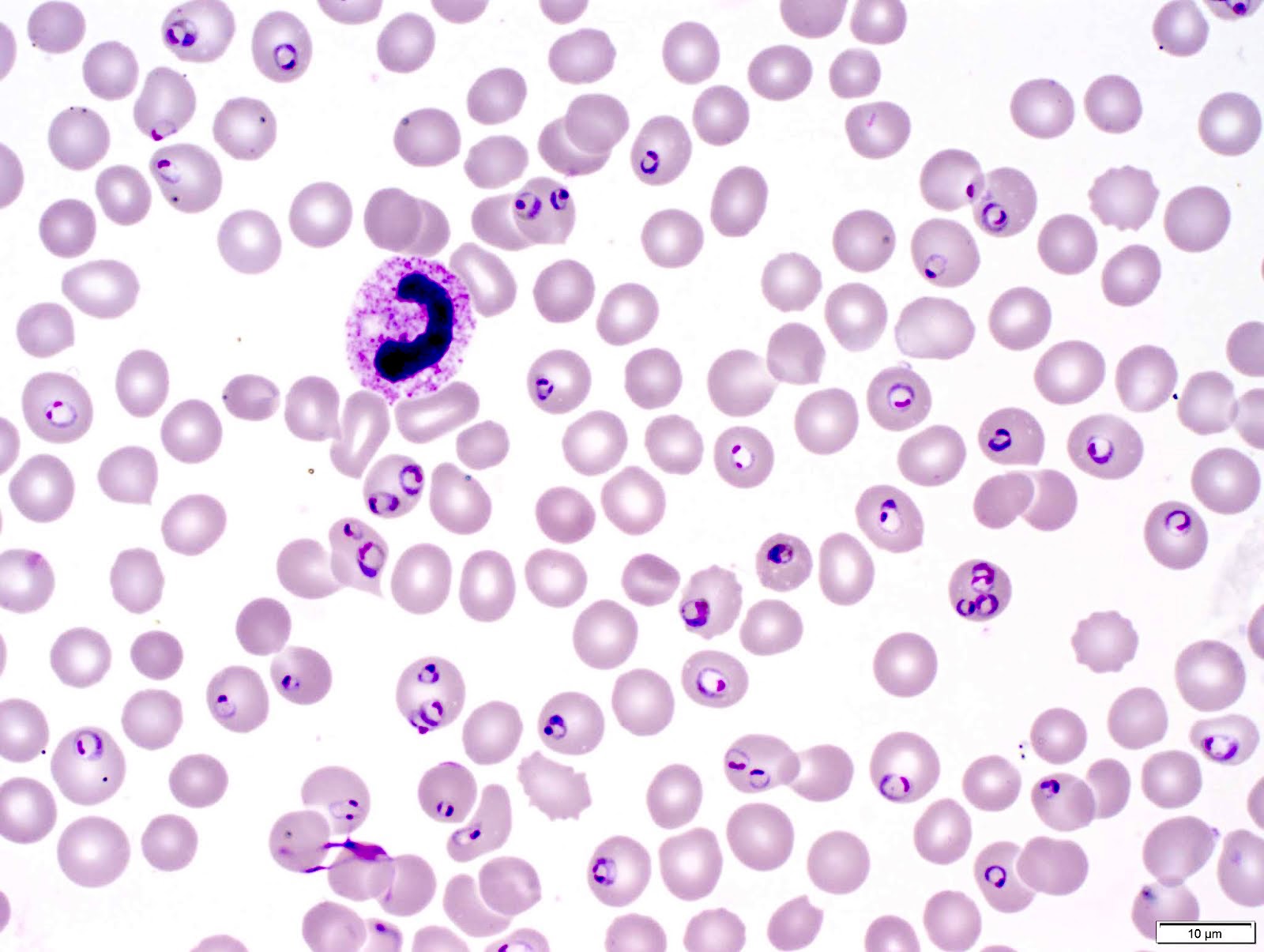
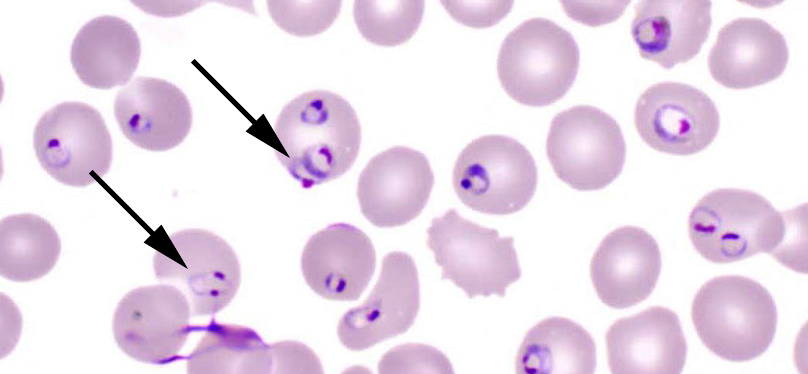
P. falciparum malaria, > 10% parasitemia, negative rapid antigen, Giemsa stained thin blood smear

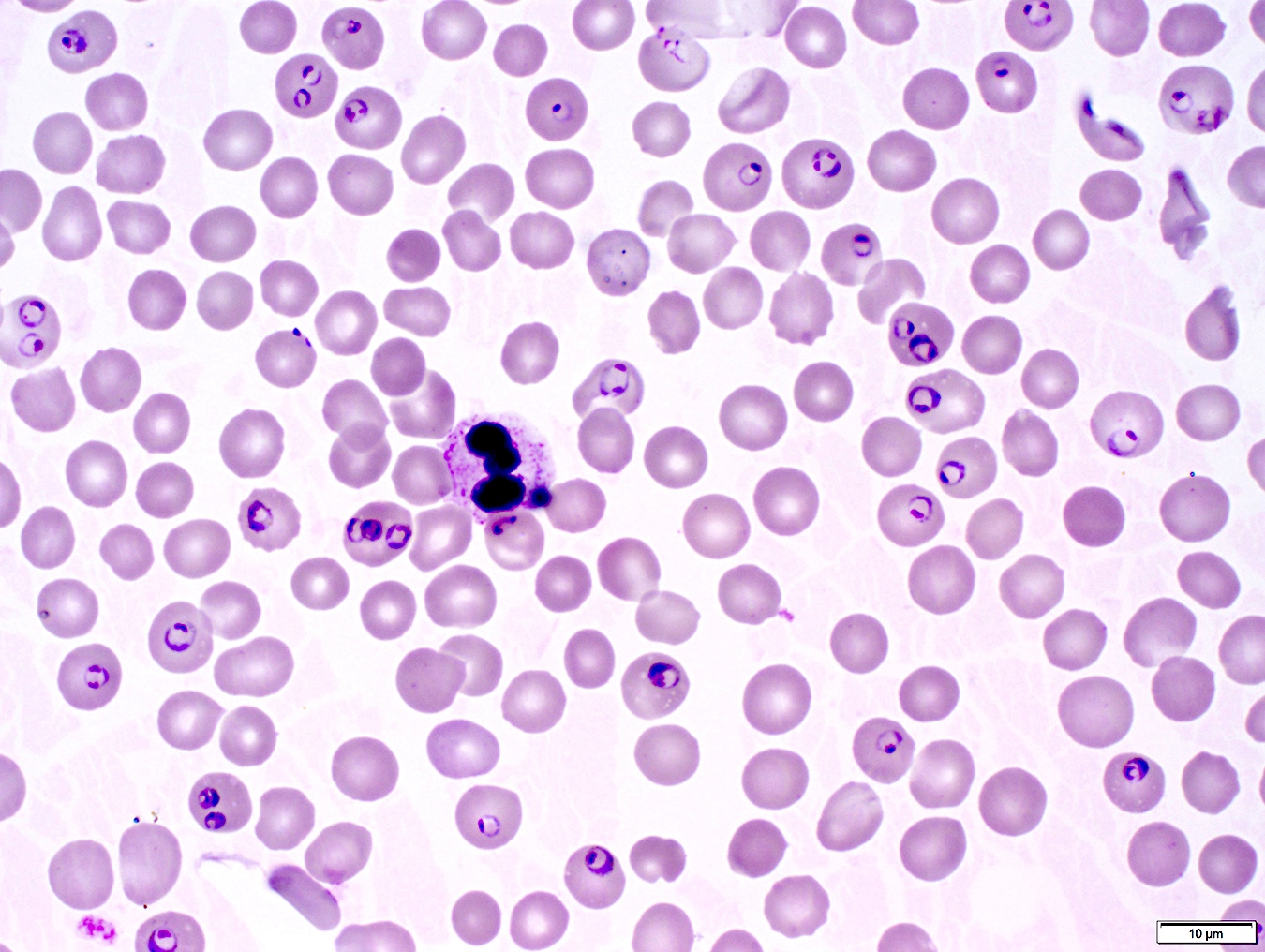
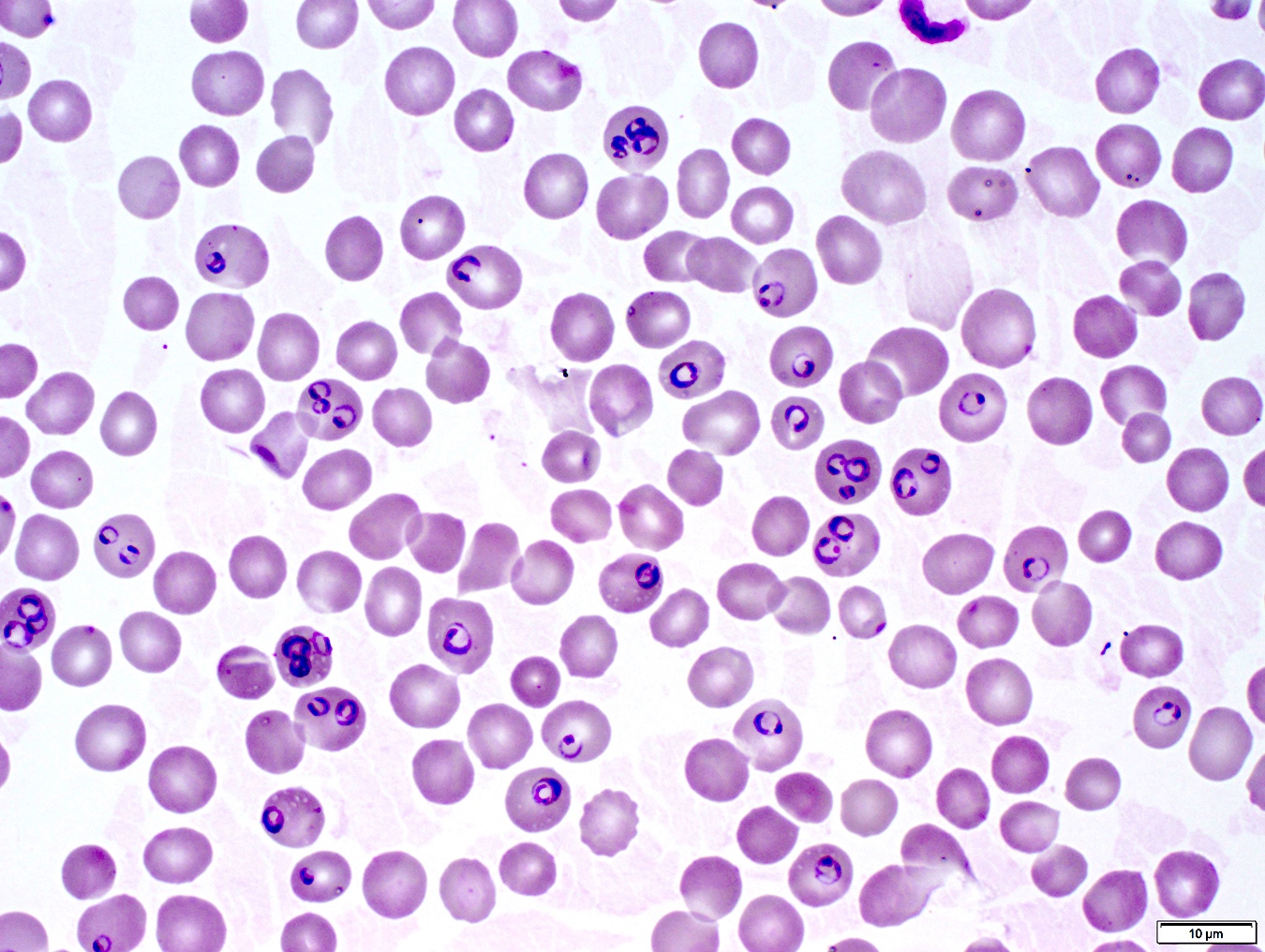
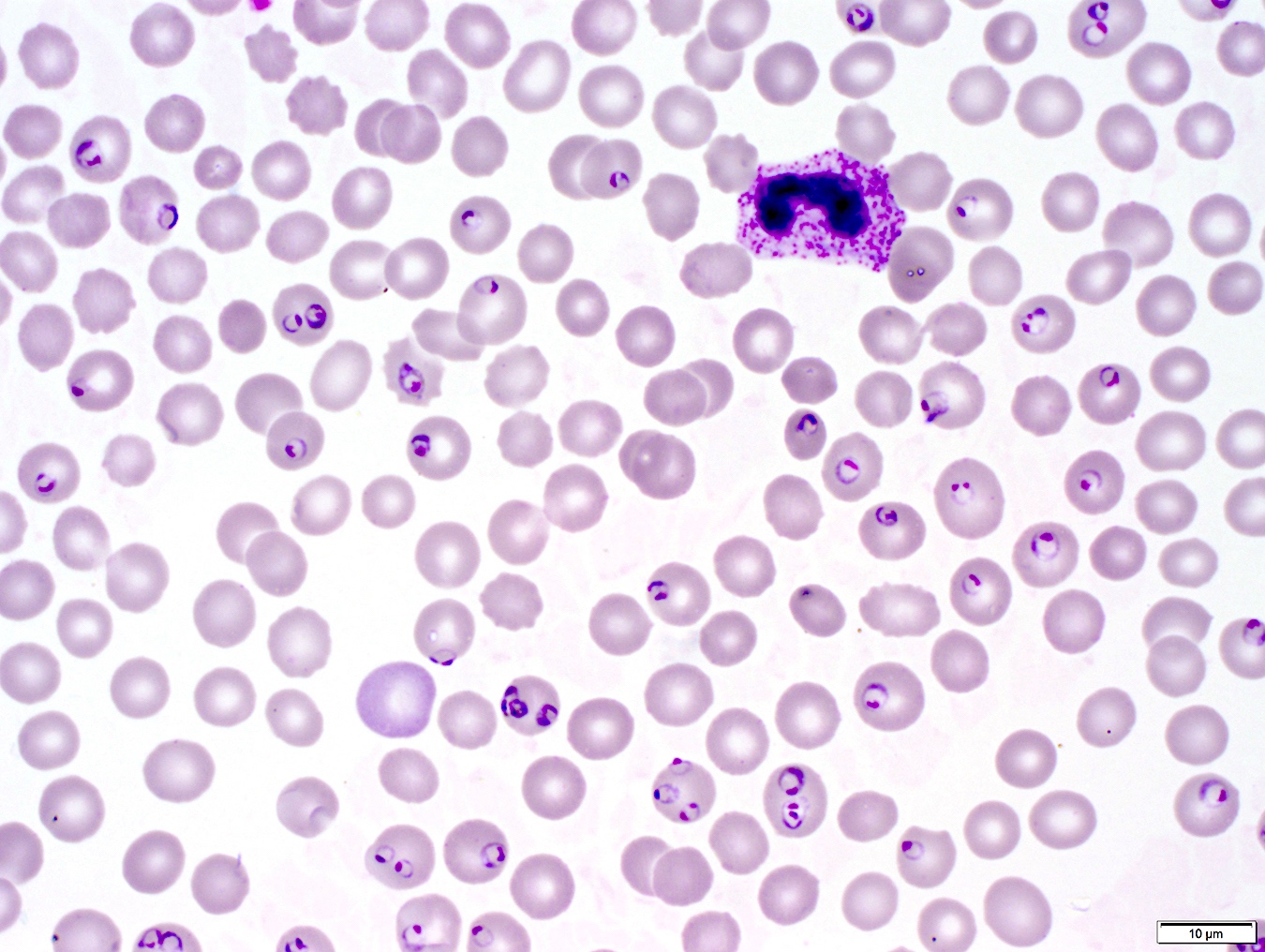
Giemsa stained thick and thin peripheral blood films from a patient with malaria due to P. falciparum; > 20% parasitemia
Contributed by Patricia Tsang, M.D., M.B.A.
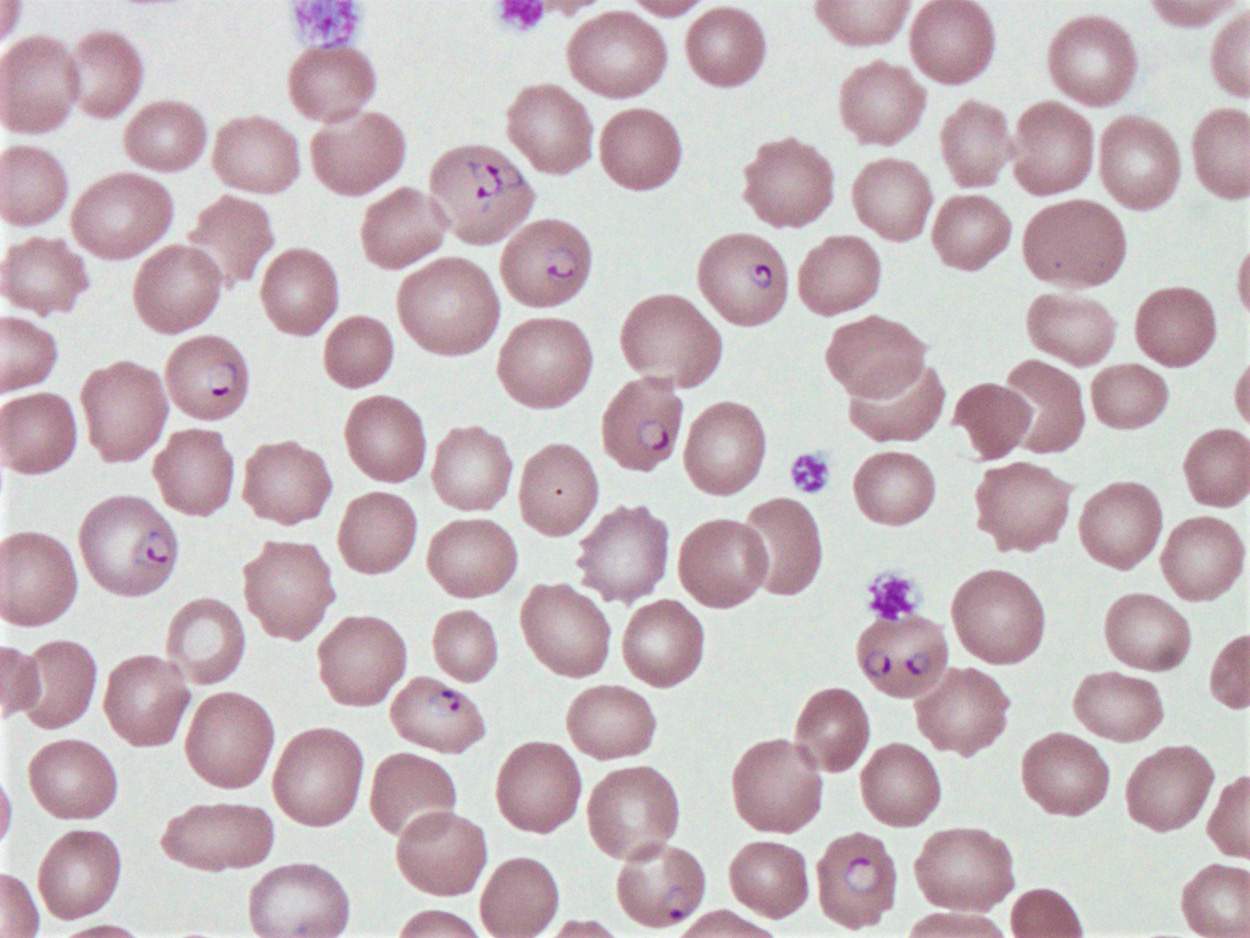
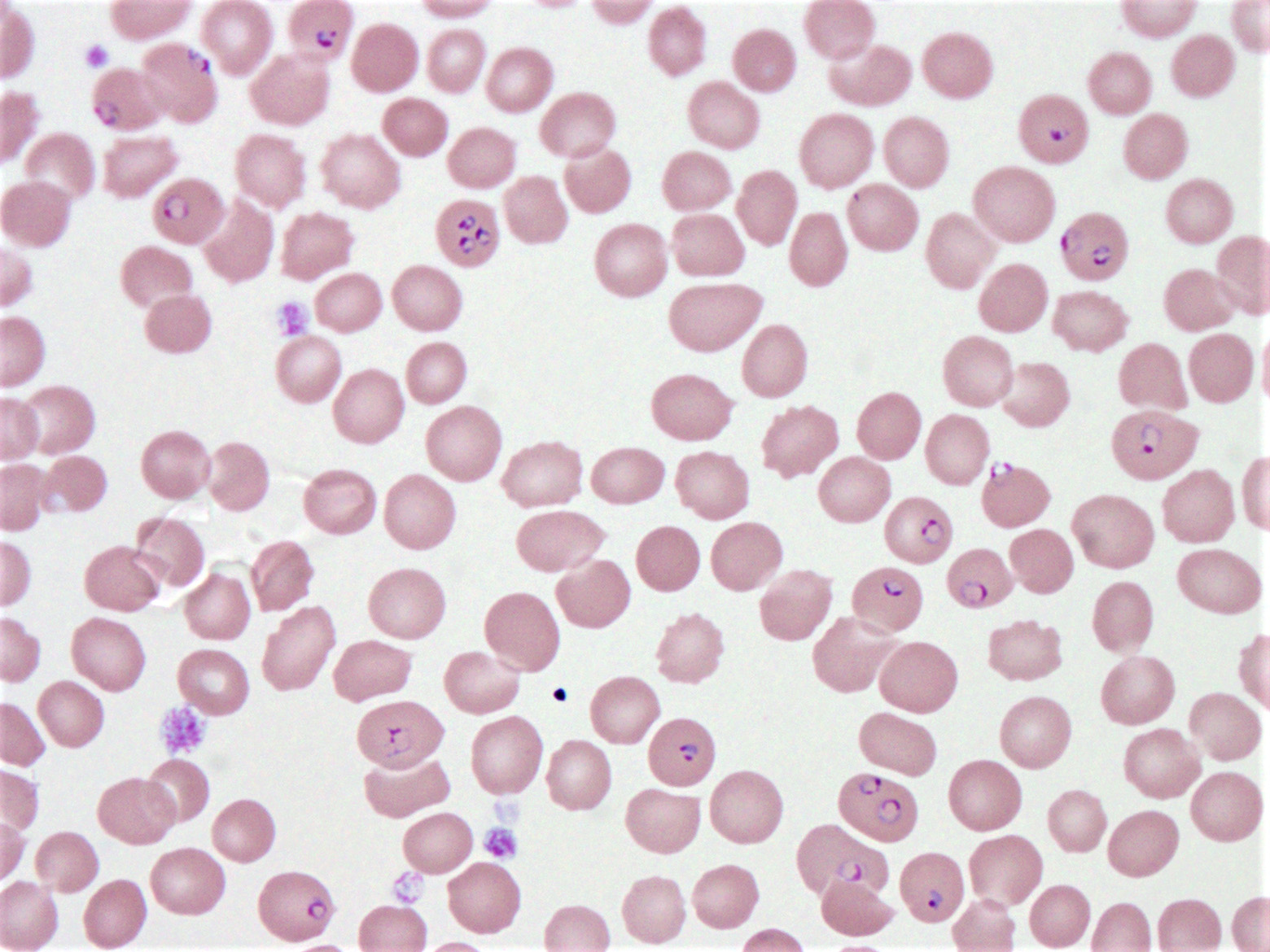
Plasmodium falciparum (blood smear)
Case #277
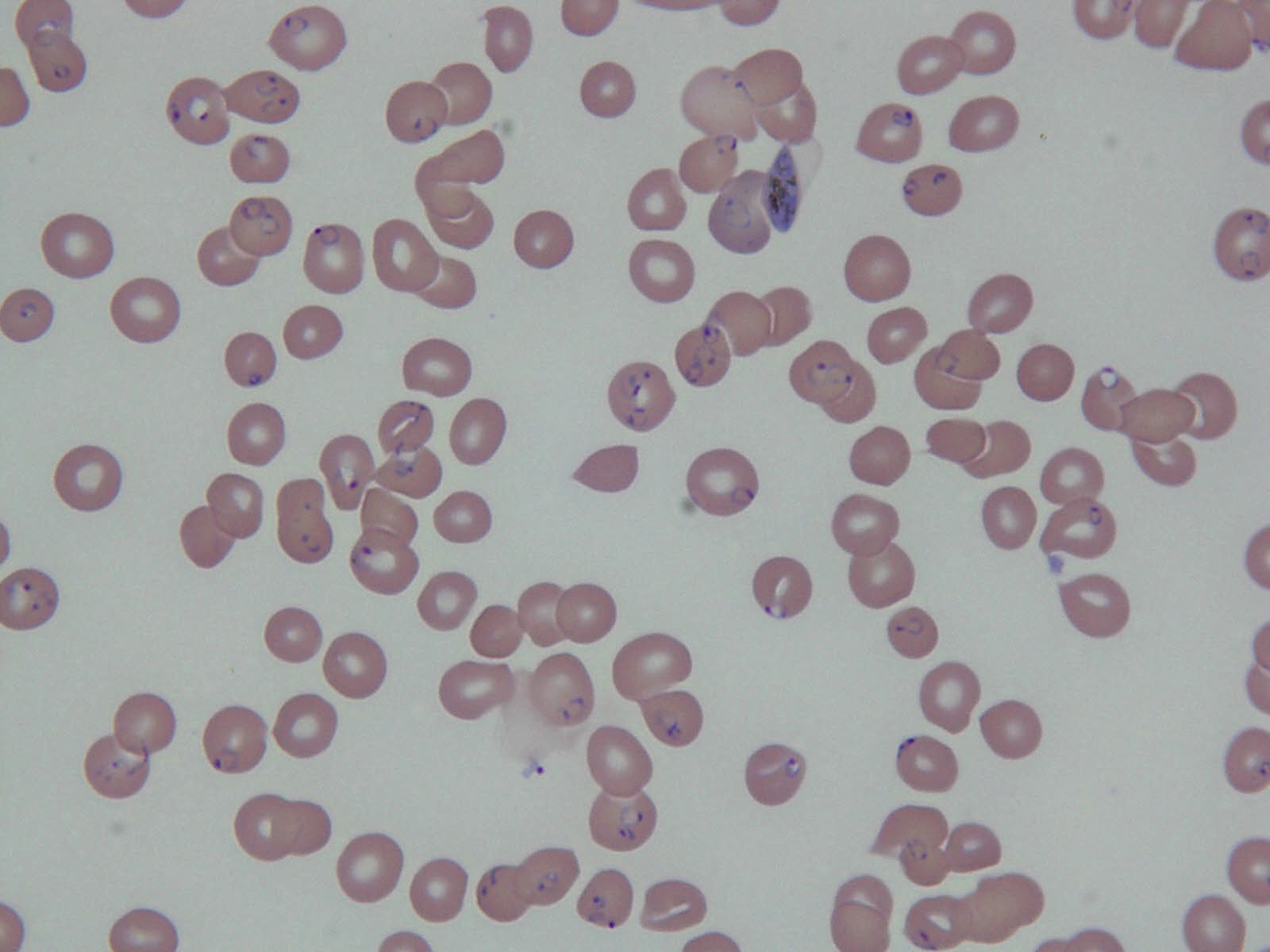
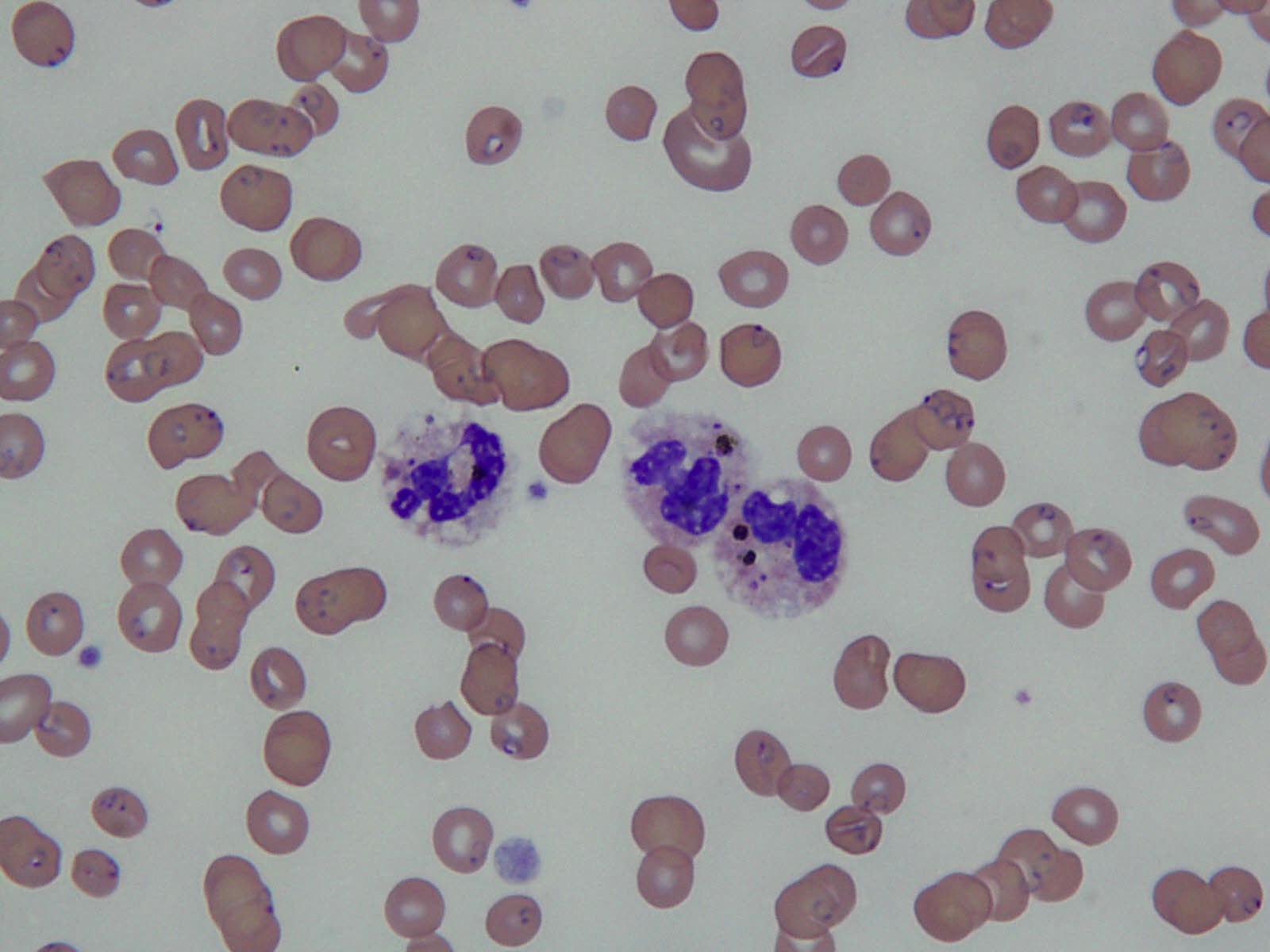
Blood smear
Images hosted on other servers:
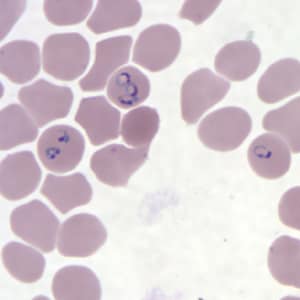
Ring form trophozoites of P. falciparum

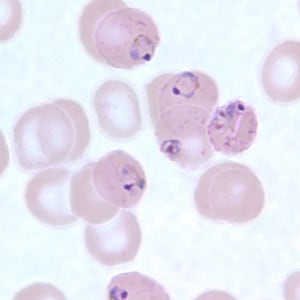
Maurer clefts
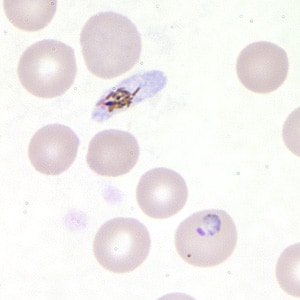
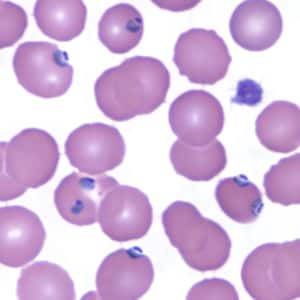
Older trophozoites of P. falciparum
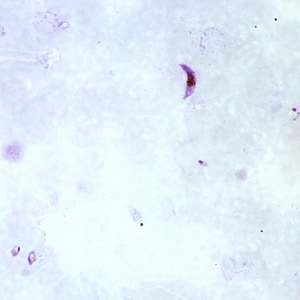

Gametocytes of P. falciparum
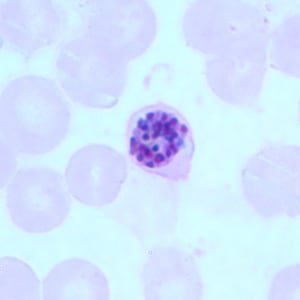
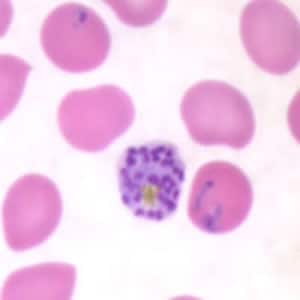
Schizonts of P. falciparum
- Endocarditis, bacterial
- Gastroenteritis
- Pharyngitis
- Plague
- Pneumococcal bacteremia
- Rare aggressive mature B cell neoplasm that represents less than ~1% of all leukemias
- Characterized by the presence of large lymphoid cells (prolymphocytes) accounting for at least 55% of total circulating cells in the peripheral blood
- Prolymphocytes are defined as large cells (> 2 erythrocytes) with clumped chromatin (but more open than in chronic lymphocytic leukemia [CLL]), a large single prominent vesicular nucleolus and abundant cytoplasm (Br J Haematol 2016;174:767)
- Aggressive B cell neoplasm defined by large prolymphocytes accounting for 55% of total circulating cells in the peripheral blood
- Main sites of involvement are peripheral blood, spleen and bone marrow
- Marked absolute lymphocytosis (usually > 100 x 109/L)
- Poor prognosis in general; the degree of treatment response is dictated by anemia, degree of lymphocytosis and molecular prognostic markers
- Prolymphocytic leukemia, B cell type (B-PLL)
- ICD-O: 9833/3 - Prolymphocytic leukemia, B cell type
- Rare, accounting for less than 1% of all leukemias
- Patients are typically > 60 years old (median age of 65 - 69 years)
- M = F (Br J Haematol 1986;63:377)
- Additional reference: Cancer Causes Control 2008;19:379
- Sites of involvement are peripheral blood, bone marrow and typically the spleen
- Limited data on this apart from molecular characteristics
- Very rare malignancy; no specific etiology or causal lesion associated with it
- Peripheral blood lymphocytosis usually > 100 x 109/L
- Patients often present with constitutional symptoms and massive splenomegaly
- Lymphadenopathy is typically absent or mild (Semin Oncol 2006;33:257)
- Peripheral blood: CBC count, morphology review and flow cytometry
- Bone marrow biopsy: morphology, flow cytometry and immunohistochemistry studies
- Molecular and cytogenetic analysis have prognostic significance but are not necessary for a diagnosis
- Additional laboratory findings to the presence of marked absolute lymphocytosis in the CBC include anemia and thrombocytopenia; these are present ~50% of the time
- No specific radiology findings
- Aberrations in MYC and TP53 portend worst prognosis (Blood 2019;134:1821)
- Lymphocytosis > 100 x 109/L and anemia associated with shorter survival (Leuk Lymphoma 1999;33:169)
- 48 year old man with 17p deletion shows response to sequential kinase inhibition (Case Rep Hematol 2017;2017:8563218)
- 53 year old man with transformation from CLL and unexpected response to R-CHOP (Perm J 2016;20:74)
- 84 year old man with 17p and 13q14 deletions with ibrutinib resistance (Cureus 2019;11:e5629)
- 84 year old man with B-PLL and concurrent warm and cold autoimmune hemolytic anemia (Acta Haematol 2019;141:222)
- Rare cases that present as indolent disease and are asymptomatic can be followed with a watch and wait approach without immediate treatment; however, cases often progress rapidly and will require close monitoring
- Chemotherapy is the treatment indicated; in the absence of 17p deletion or TP53 mutation, rituximab in association to FC (fludarabine, cyclophosphamide) or bendamustine is the recommended first line therapy (Hematology Am Soc Hematol Educ Program 2015;2015:361)
- Chemotherapy for poor prognosis cases is different; in del(17p) or TP53 mutated tumors, treatment with alemtuzumab or more recently ibrutinib are used (Hematology Am Soc Hematol Educ Program 2015;2015:361, Br J Haematol 2017;179:501)
- Bone marrow involvement shows neoplastic infiltrates with a mixed nodular, interstitial or diffuse patterns composed of medium to large lymphoid cells
- Spleen shows expanded white pulp with red pulp infiltration by medium to large neoplastic cells with ample cytoplasm, round or irregular nuclei and a central large, eosinophilic nucleolus
- Lymph node involvement when present shows diffuse or nodular infiltration by neoplastic lymphoid cells without proliferation centers
Contributed by Richard K. Wood, M.D. and Dietrich Werner, M.D.
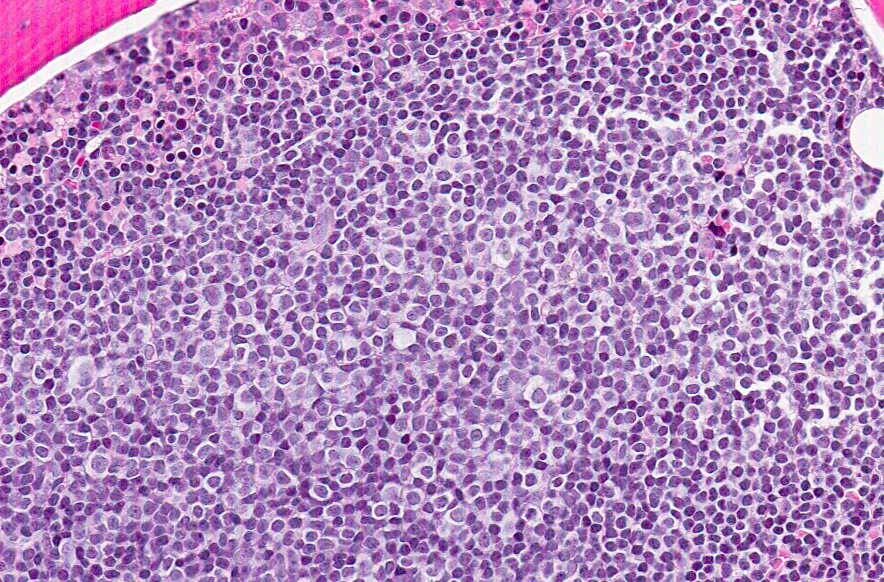
Bone marrow involvement
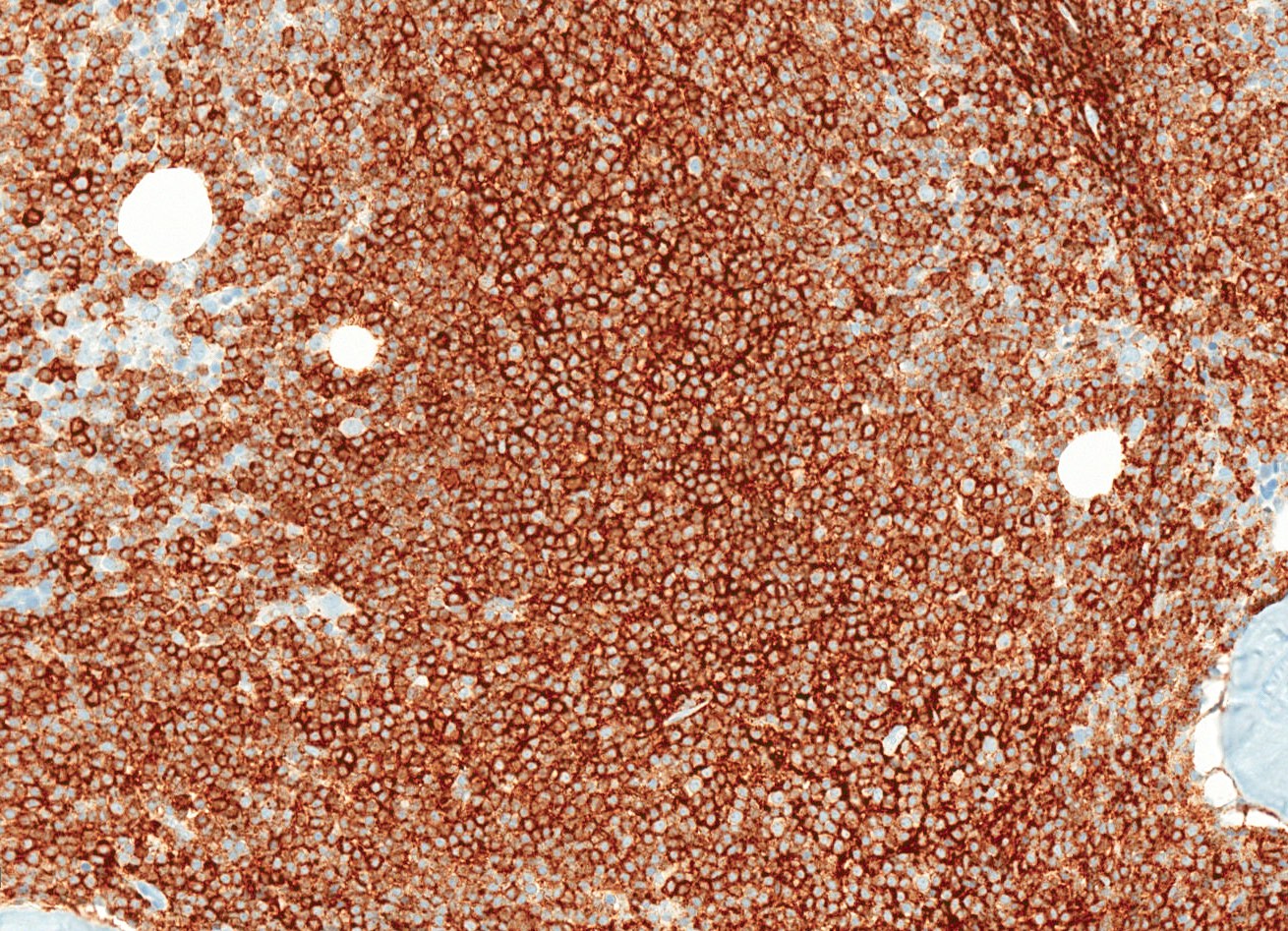
CD20 positivity in bone marrow
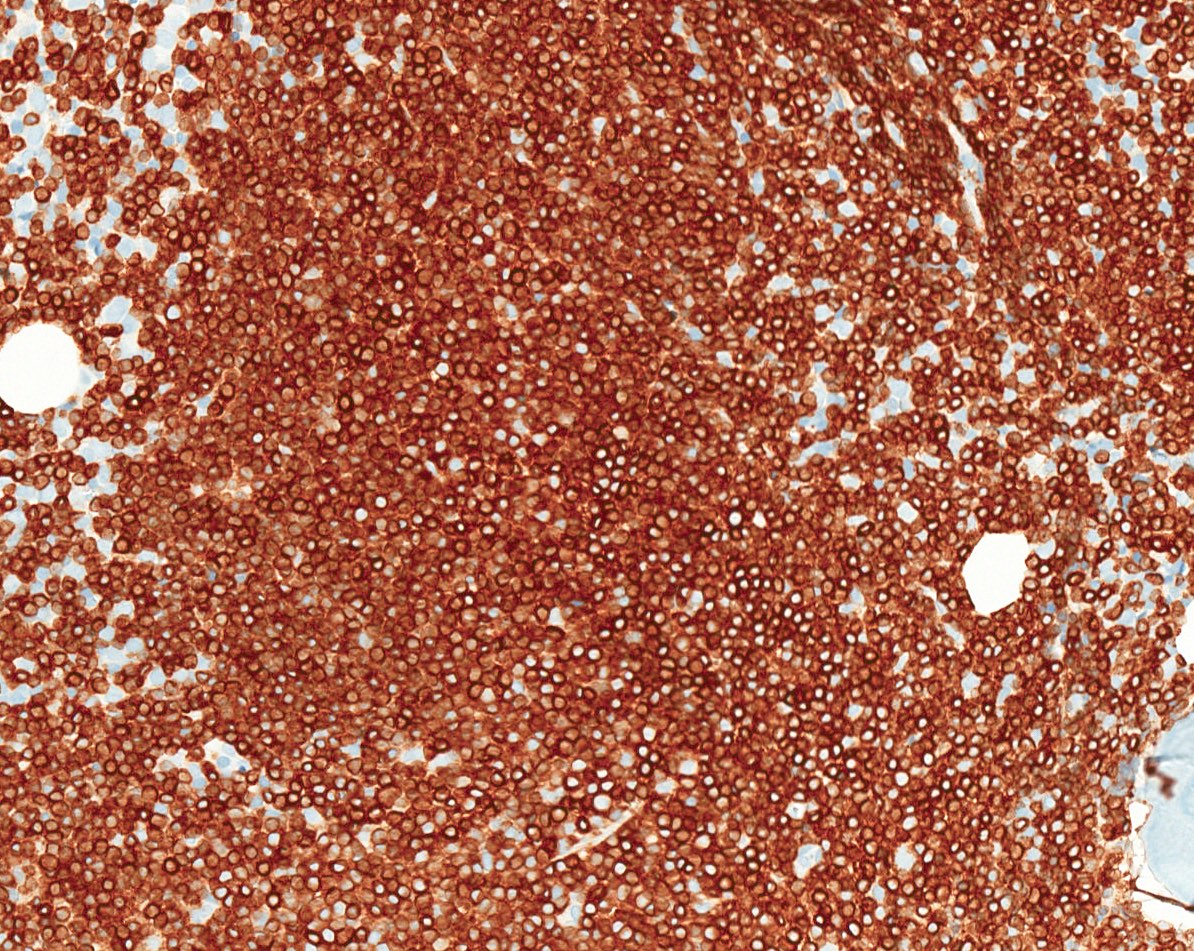
CD79a positivity in bone marrow

Cyclin D1 negativity in bone marrow
- Marked absolute lymphocytosis with at least a 55% prolymphocyte count out of total circulating cells
- Prolymphocytes:
- Medium to large sized lymphoid cells with round or occasionally indented nuclei
- Moderately condensed to open chromatin
- Single large prominent central nucleolus
- Moderate amount of basophilic cytoplasm
- Reference: Br J Haematol 1974;27:7
Contributed by Allam Shawwa, M.D.
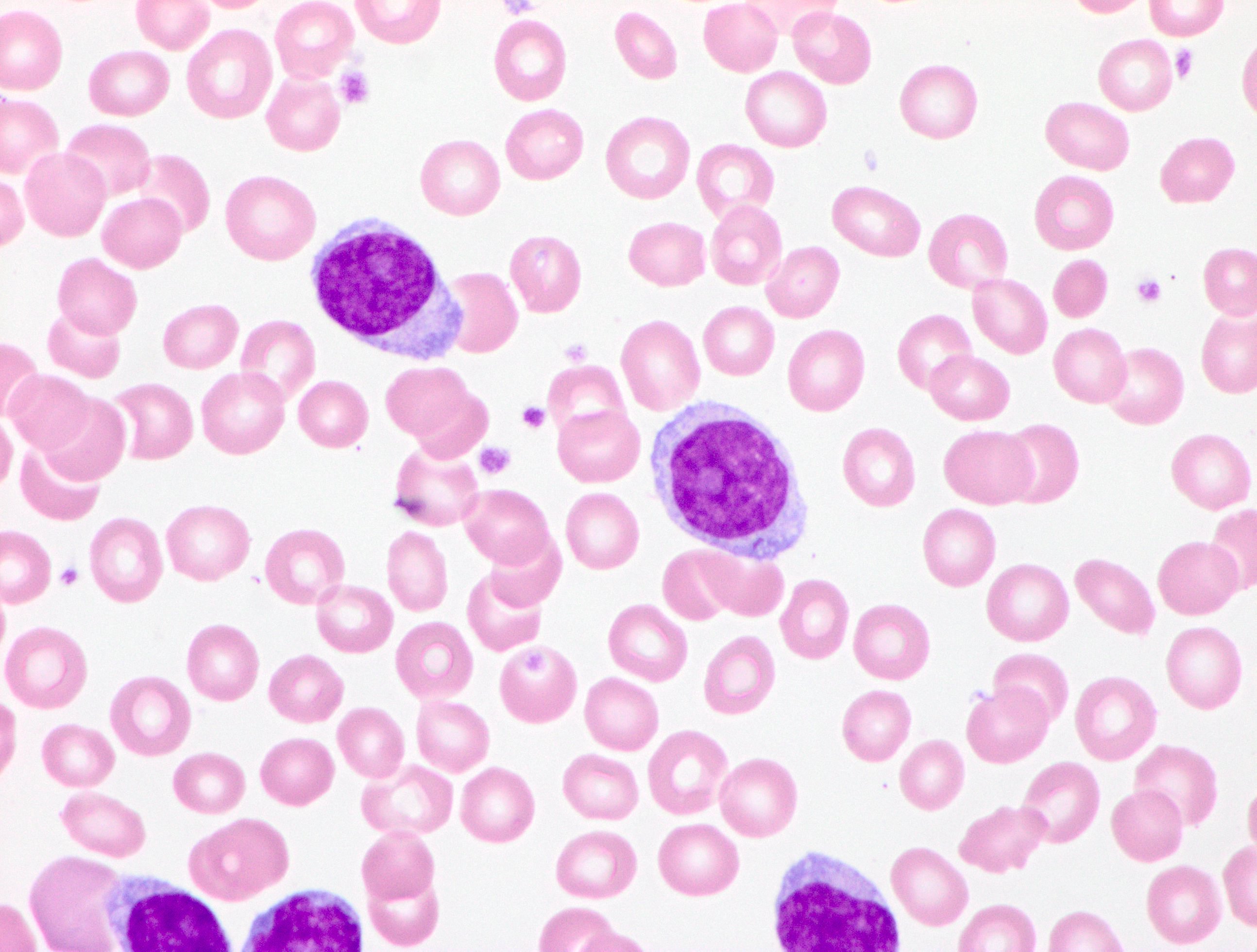
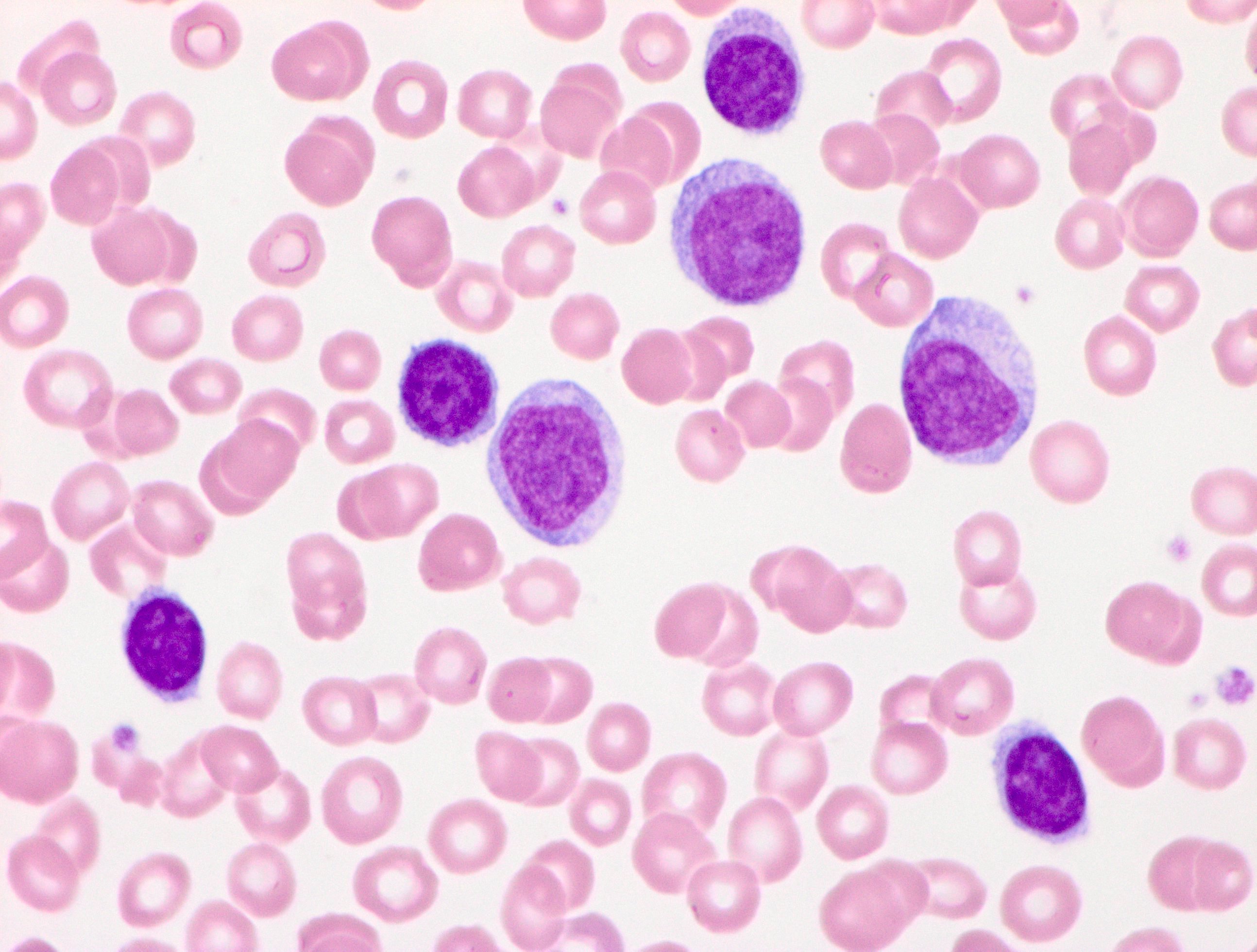
Prolymphocytes in peripheral blood

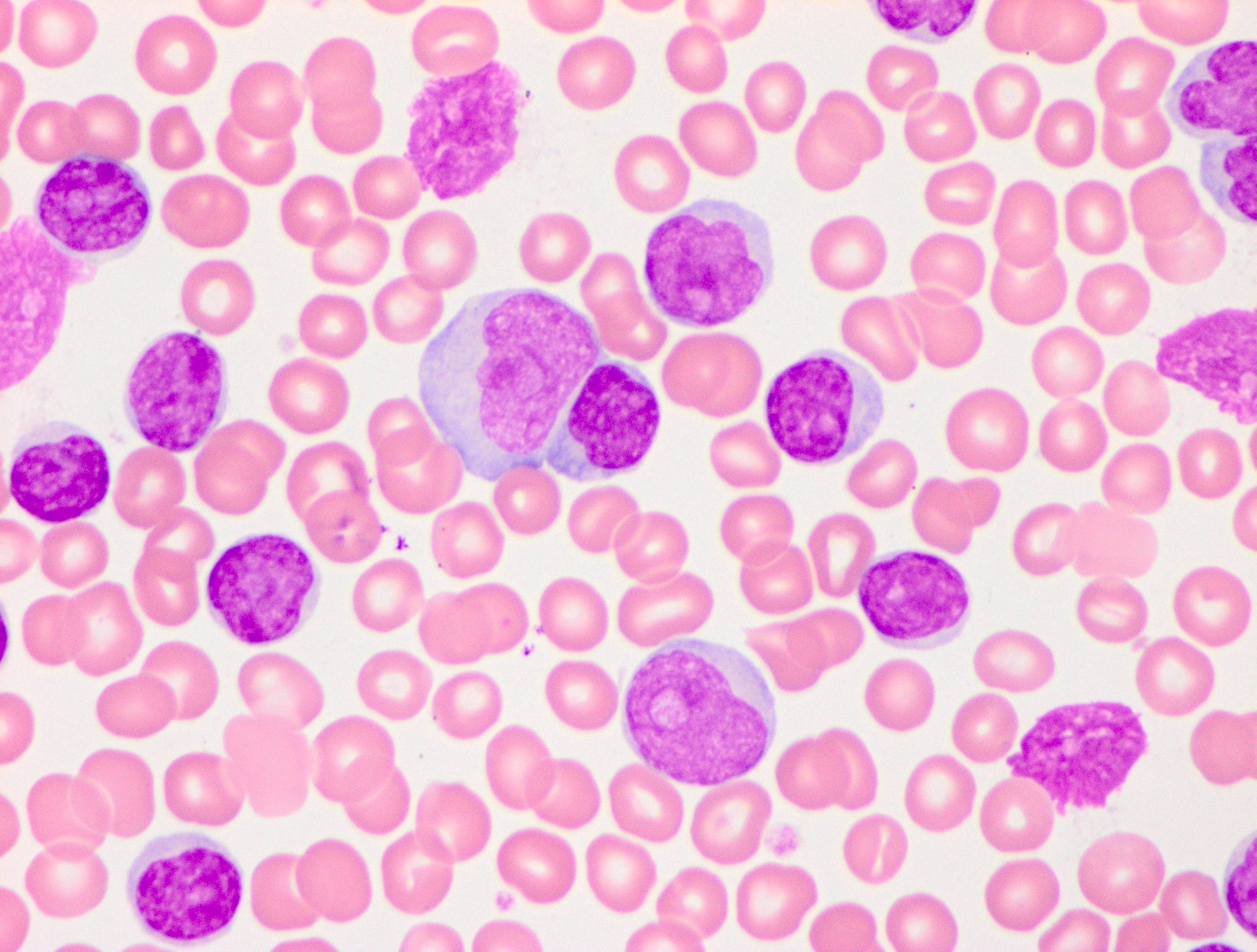
Prolymphocytes in peripheral blood
- In contrast to chronic lymphocytic leukemia, CD5 and CD23 are usually negative (expressed in 20 - 30% and 10 - 20%, respectively) (Best Pract Res Clin Haematol 2019;32:217)
- Negative for cyclin D1, CD10 and BCL6 (Semin Oncol 2006;33:257)
- Positive
- Negative
- Reference: Best Pract Res Clin Haematol 2019;32:217

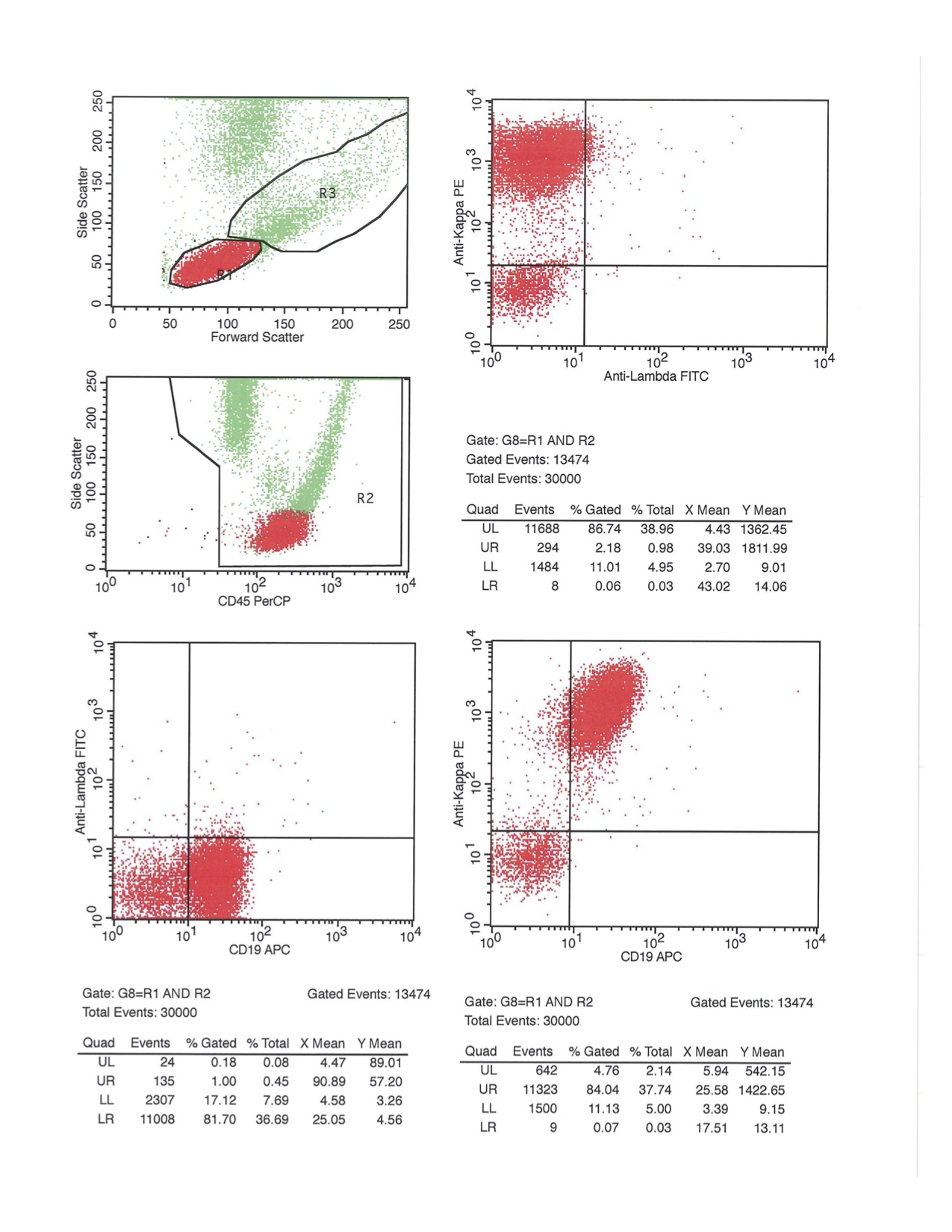
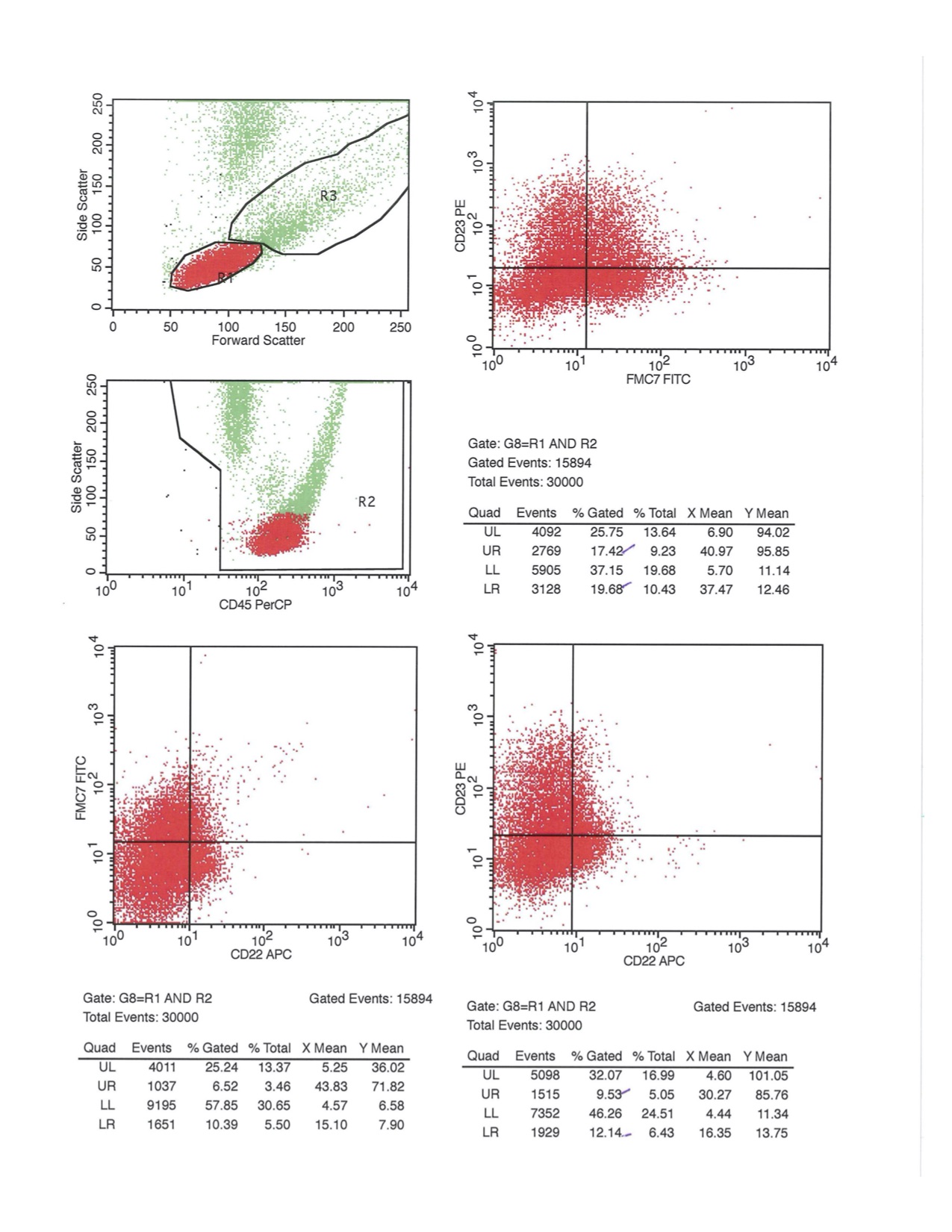
- No molecular or cytogenetic specific signature exists for B-PLL and data is limited given its rarity
- Clonal rearrangement of IGHV is commonly found, with preferential use of IGHV3 and IGHV4 (Leukemia 2006;20:1231)
- Deletion of 17p13 was found in 53% of B-PLL and is associated with TP53 mutations (Leukemia 2006;20:1231)
- Deletions of 11q23 and 13q14 are frequently found (Leukemia 2006;20:1231, Leukemia 2000;14:427)
- Mutations in MYC have been reported, including translocations involving IGH, IGK or IGL (Am J Clin Pathol 2014;142:347)
- Bone marrow, right posterior iliac crest, biopsy, aspirate and clot section:
- B cell prolymphocytic leukemia (B-PLL) (see comment)
- Comment: This is a 79 year old man presenting with an elevated CBC count of 153 x 109/L and an absolute lymphocyte count of 124 x 109/L. A manual peripheral blood smear count shows a prolymphocyte count of 74%.
- Bone marrow aspirate:
- Quality of specimen: particulate
- Cellularity: hypercellular
- Approx. M:E ratio: 4:1
- Erythropoiesis: normoblastic and decreased
- Granulopoiesis: all stages of maturation present and decreased
- Megakaryocytopoiesis: normal
- Lymphocytes: increased with two monomorphic populations noted. The first is characterized by medium sized cells with prominent central nucleoli and abundant cytoplasm. The second consists of small lymphocytes with densely packed chromatin and minimal cytoplasm.
- Plasma cells: normal; not increased
- Bone marrow aspirate differential:
- Metamyelocytes: 3%
- Bands: 7%
- Seg. neutrophils: 10%
- Eosinophils: 1%
- Lymphocytes: 72%
- Monocytes: 2%
- Erythroblasts: 8%
- Total cells counted: 500
- Bone marrow biopsy:
- A trephine biopsy with a length of 1.5 cm is submitted. The sample is adequate for examination. The specimen is hypercellular at approximately 70% cellularity. Trilineage hematopoiesis is decreased. Megakaryopoiesis is nondysplastic. Granulopoiesis is decreased but still shows a normal range of maturation and geographic distribution. There is a diffuse, interstitial and nodular infiltrate of lymphocytes consisting of two discrete populations. The first shows medium to large sized cells with prominent nucleoli and vesicular chromatin while the second consists of small, monomorphic lymphocytes with densely packed chromatin.
- Ancillary testing:
- Immunohistochemical evaluation of the larger cell population shows expression of CD20, CD19 and CD79a while CD5 and CD23 are not expressed. The population of smaller cells expresses CD20, CD19, CD5 and CD23. Both populations are negative for expression of CD3, SOX11 and cyclin D1.
- The flow cytometry report shows two neoplastic cell populations surface light chain kappa restricted. The first larger population represents 69% of the sample and is positive for CD19, CD20 bright and kappa bright and is negative for CD5, CD10 and CD23. A second smaller population in 18% of the sample is positive for CD19, CD20 dim, CD5, CD23 and kappa dim.
- Molecular and cytogenetic studies demonstrated deletion of 17p13. Translocation (11;14)(q13;q32) was not detected.
- Mantle cell lymphoma:
- T cell prolymphocytic leukemia:
- Chronic lymphocytic leukemia with increased prolymphocytes:
- Hairy cell leukemia variant:
- Splenic marginal zone lymphoma:
- Neoplastic cells show characteristic short polar villi and do not have prominent nucleoli
- Splenic involvement by MZL shows prominent marginal zone
- Marrow involvement may show follicular arrangement with surrounding marginal zone B cells
- Molecular analysis shows in some cases NOTCH2 and NFκB mutations (not reported in B-PLL)
- Follicular lymphoma:
- Typically follicular pattern and often involving lymph nodes
- Expresses CD10
- Lymphoplasmacytic lymphoma:
- Plasmacytoid appearance of neoplastic cells often accompanied by rouleaux
- MYD88 mutation in > 90% of cases
You review the peripheral blood smear of a 74 year old man with a high WBC count of 117 x 109/L, anemia and thrombocytopenia. The majority of the cells (75%) have the appearance shown in the above image. Flow cytometry shows that these cells have the following immunophenotype: CD20+, CD19+, FMC7+, CD200+ and CD10-, CD5-, CD23-. What is your diagnosis?
- Acute myeloid leukemia
- B cell prolymphocytic leukemia
- Chronic lymphocytic leukemia
- Mantle zone lymphoma
- Splenic marginal zone lymphoma
- Anemia
- Bone marrow involvement
- Deletion of 17p13
- Lymphocytosis over 100 x 109/L
- Medium to large sized lymphocytes with central, large nucleoli that are greater than 55% of the total WBC
Comment Here
Reference: Prolymphocytic leukemia
- Basophilic nuclear remnants, i.e. clusters of DNA in circulating erythrocytes as well as erythroid precursors
- Named after William Henry Howell and Justin Marie Jolly (Am J Med Sci 2012;343:407)
- Also called "micronucleated reticulocytes"
- Circulating erythrocytes
- Erythroid precursors in bone marrow
- During maturation in bone marrow, erythrocytes normally expel their nuclei but sometimes a small portion of DNA remains
- In healthy people, Howell-Jolly bodies are pitted out by spleen during erythrocyte circulation
- Howell-Jolly bodies persist in those with functional hyposplenia or asplenia:
- Autosplenectomy due to sickle cell anemia
- Celiac disease (~10% have splenic atrophy)
- Hereditary spherocytosis
- Megaloblastic anemia
- Myelodysplastic syndrome (MDS)
- Radiation therapy
- Severe hemolytic anemia
- Splenectomy following trauma
- 16 month old boy with congenital asplenia (Paediatr Child Health 2011;16:391)
- 60 year old man with Babesia (N Engl J Med 2003;349:2467)
- Patient with malignant histiocytosis (Acta Cytol 1988;32:680)
- By itself, does not need to be treated
- Peripheral blood smear preparation with standard Wright-Giemsa stain shows smooth, round basophilic (purple) particles in eosinophilic erythrocytes
- Single Howell-Jolly bodies may be seen in megaloblastic anemia, hemolytic anemia, postsplenectomy
- Multiple Howell-Jolly bodies in a single cell usually indicates megaloblastic anemia or other abnormal erythropoiesis
Images hosted on other servers:

Inclusions of nuclear chromatin remnants
- EM confirms Howell-Jolly bodies are intracellular, located beneath red cell membrane, which have small circular membrane defect in their concavity beneath the membrane bulge (Arch Intern Med 1973;131:236)
- Basophilic stippling (punctate basophilia):
- Irregular basophilic granules within erythrocytes, fine to coarse, deep blue with Wright stain, due to abnormal instability of RNA in young red cell
- Fine stippling is associated with increased polychromatophilia, increased production of red cells
- Coarse stippling is associated with lead poisoning, other diseases of impaired hemoglobin synthesis, megaloblastic anemia, other severe anemia
- Howell-Jolly-like bodies:
- Detached nuclear fragments in cytoplasm of neutrophils that resemble Howell-Jolly bodies
- Malarial stippling:
- Fine minute granules in enlarged erythrocytes that harbor Plasmodium vivax
- With Wright stain, these "Schüffner granules" stain purplish red; may be so numerous that they hide the parasites
- Pappenheimer bodies:
- Iron containing granules of siderocytes which may stain with Wright stain
- Usually few in number in a given red cell
- Less commonly seen in peripheral blood except after splenectomy
- Siderocytes:
- Red cells with inorganic iron containing granules, as demonstrated by Prussian stain for iron
- Defined by an absolute neutrophil count (ANC) that exceeds the age related normal range
- Normal reference interval (individually established for each laboratory) is approximately 1.8 - 7.0 x 109/L in adults and 1.0 - 8.5 x 109/L in young children
- Absolute neutrophil count is higher in infants, with newborns having the highest absolute neutrophil count of any age (7.8 - 14.4 x 109/L)
- Neutrophil count also differs by race, with individuals of African descent having lower absolute neutrophil counts
- References: McPherson: Henry's Clinical Diagnosis and Management by Laboratory Methods, 22nd Edition, 2011, Curr Opin Hematol 2014;21:43
- Commonly encountered laboratory finding and can be seen in benign and neoplastic disorders
- Clinical findings are extremely helpful in identifying the underlying etiology
- Leukemoid reaction: prominent granulocytosis with a variable degree of left shift
- Granulocytosis
- Blood
- Demargination:
- Neutrophils attached to endothelium are released back into circulation
- Seen in stressful conditions that are accompanied by the release of endogenous adrenaline
- Absolute neutrophil counts are doubled within a few minutes but not associated with a left shift
- Decreased transit to solid tissues:
- Neutrophils remain in the blood for about 12 hours, before leaving the circulation and entering solid tissues
- Administration of corticosteroids can delay the transit from blood to solid tissues, resulting in a mild to moderate neutrophilia but not associated with left shift
- Mobilization from the maturation storage pool of the bone marrow:
- A large reserve compartment of neutrophils, bands and metamyelocytes in the bone marrow is released into the circulation, which may cause a mild left shift
- Infections and other inflammatory processes are the most common causes
- Increased production:
- Granulocyte colony stimulating factor (G-CSF) mediated increases of granulopoiesis within the bone marrow
- Seen with administration of G-CSF or in chemokine producing neoplasms
- Reference: Weksler: Wintrobe's Atlas of Clinical Hematology, 2nd Edition, 2017
- Infection: bacterial infection is the most common cause; occasional viral infections such as severe acute respiratory distress syndrome (SARS) or Hantavirus pulmonary syndrome (HPS)
- Medication effect: most commonly granulocyte colony stimulating factor (G-CSF) and glucocorticoids
- Others: acute stressful events such as burns or trauma; smoking; paraneoplastic syndrome
- Reference: Kjeldsberg: Practical Diagnosis of Hematologic Disorders, 5th Edition, 2010
- Abnormal CBC with neutrophilia
- Clinical features are variable, based on the etiology of the neutrophilia (e.g., infection, medication use, stressful events)
- Reactive neutrophilia typically does not cause splenomegaly
- Reference: StatPearls: Neutrophilia [Accessed 9 February 2023]
- Based on automated CBC with differential testing or peripheral blood smear examination
- Normal neutrophil contains 3 to 5 nuclear lobes
- Provide first line of defense against microorganisms
- Mediates acute inflammatory response
- Most abundant leukocyte in the circulation
- Antimicrobial:
- Degranulates and releases myeloperoxidase, an enzyme stored in azurophilic granules of the neutrophils, into the extracellular space
- Phagocytosis of pathogens
- Absolute neutrophilia
- Reactive neutrophilia, as seen in infections, usually demonstrates toxic granulation, Döhle bodies and cytoplasmic vacuolization
- Döhle bodies are pale cytoplasmic inclusions that are parallel stacks of rough endoplasmic reticulum with bound ribosomes
- Toxic granulation represents either retained primary granules or altered uptake of stain by secondary granules
- Prominent cytoplasmic vacuoles in neutrophils are usually associated with sepsis
- Variable degree of left shift in the granulocytic lineage can be seen
- In adults with infections, the left shift is usually mild (bands and metamyelocytes) with absent or rare blasts
- In pediatric patients and in patients receiving G-CSF, the left shift can be prominent
- In these adult and pediatric patients, binucleated neutrophils or very large neutrophils can sometimes be present and should not be interpreted as dysplasia
- Hypergranulation induced by G-CSF therapy shows a high density of granules, which stain redder than toxic granulation and often obscure the nucleus; left shifted granulocytic maturation can be seen in the marrow with G-CSF administration
- In reactive neutrophilia, white blood cell (WBC) count is usually < 30 x 109/L, except in pediatric patients with infection or patients receiving G-CSF; basophils are usually not increased
- Blasts are very rare and are usually seen only in patients receiving G-CSF or in pediatric patients
- With peripheral reactive neutrophilia, the bone marrow demonstrates granulocytic hyperplasia with a full spectrum of maturation
- References: Weksler: Wintrobe's Atlas of Clinical Hematology, 2nd Edition, 2017, Int J Lab Hematol 2014;36:279, Hematol Pathol 1994;8:55
Contributed by Linlin Wang, M.D., Ph.D. and Sonam Prakash, M.B.B.S.
Peripheral neutrophilia
Toxic granulation and Döhle bodies
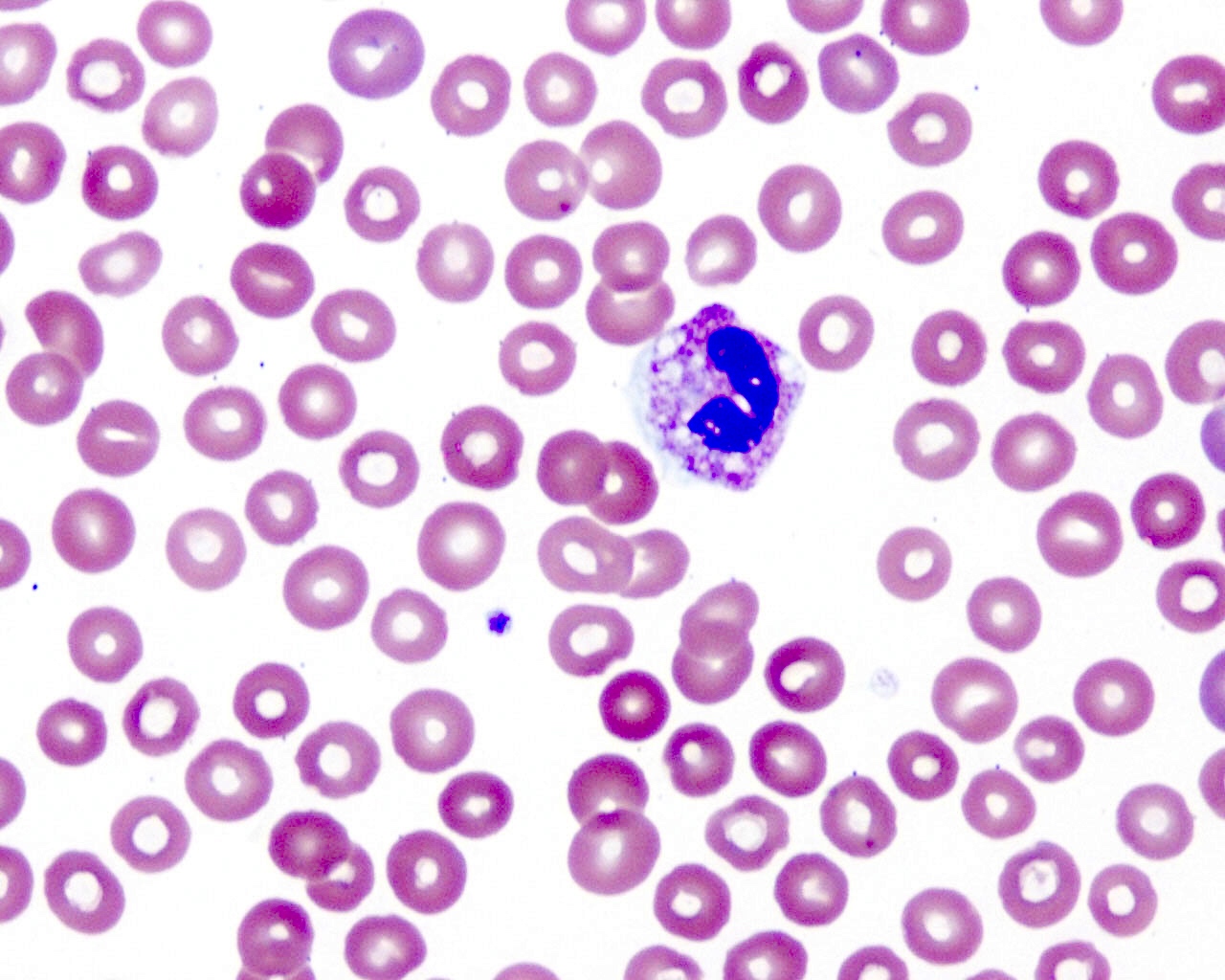
Cytoplasmic vacuoles in neutrophil
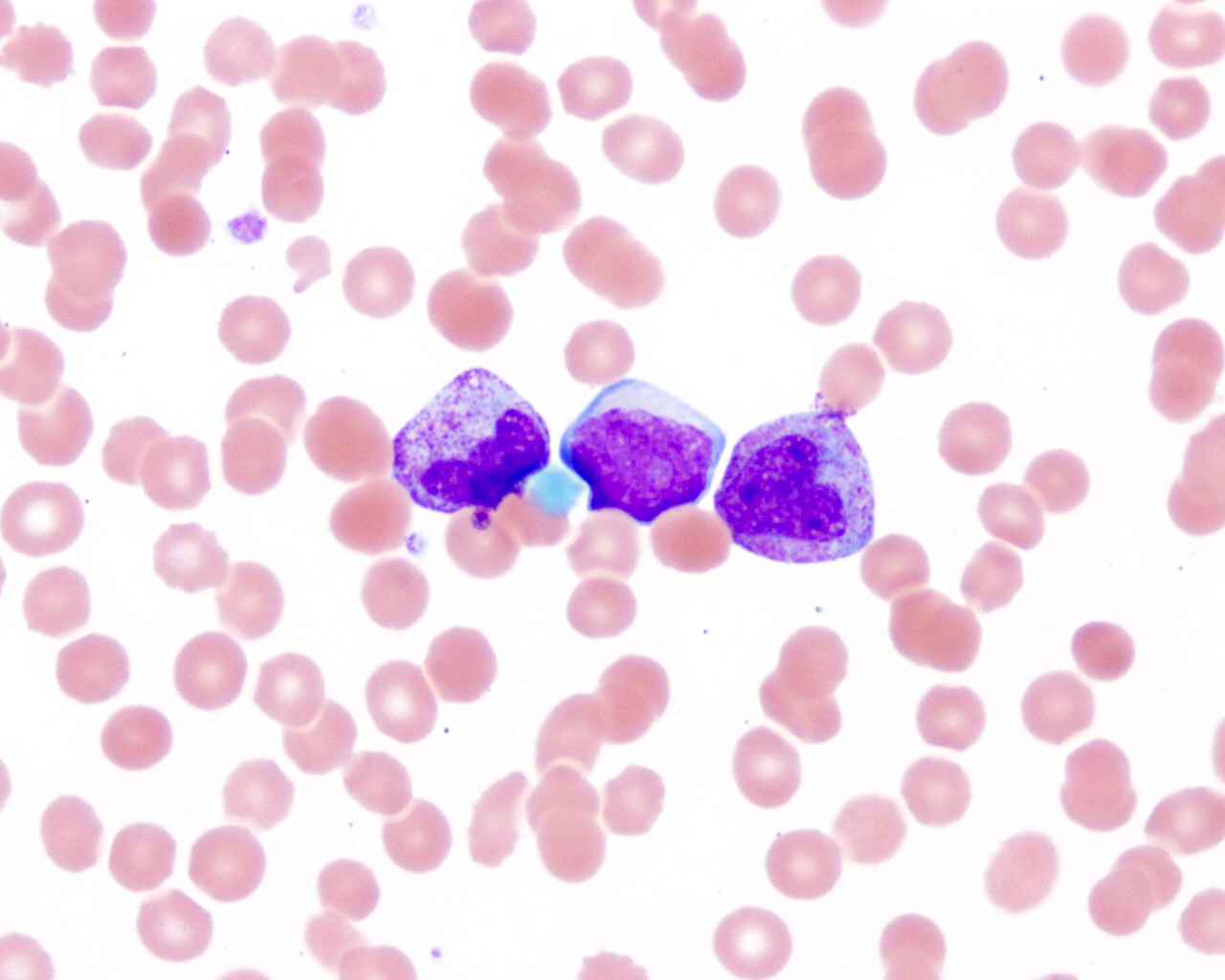
Immature granulocytes and rare blast
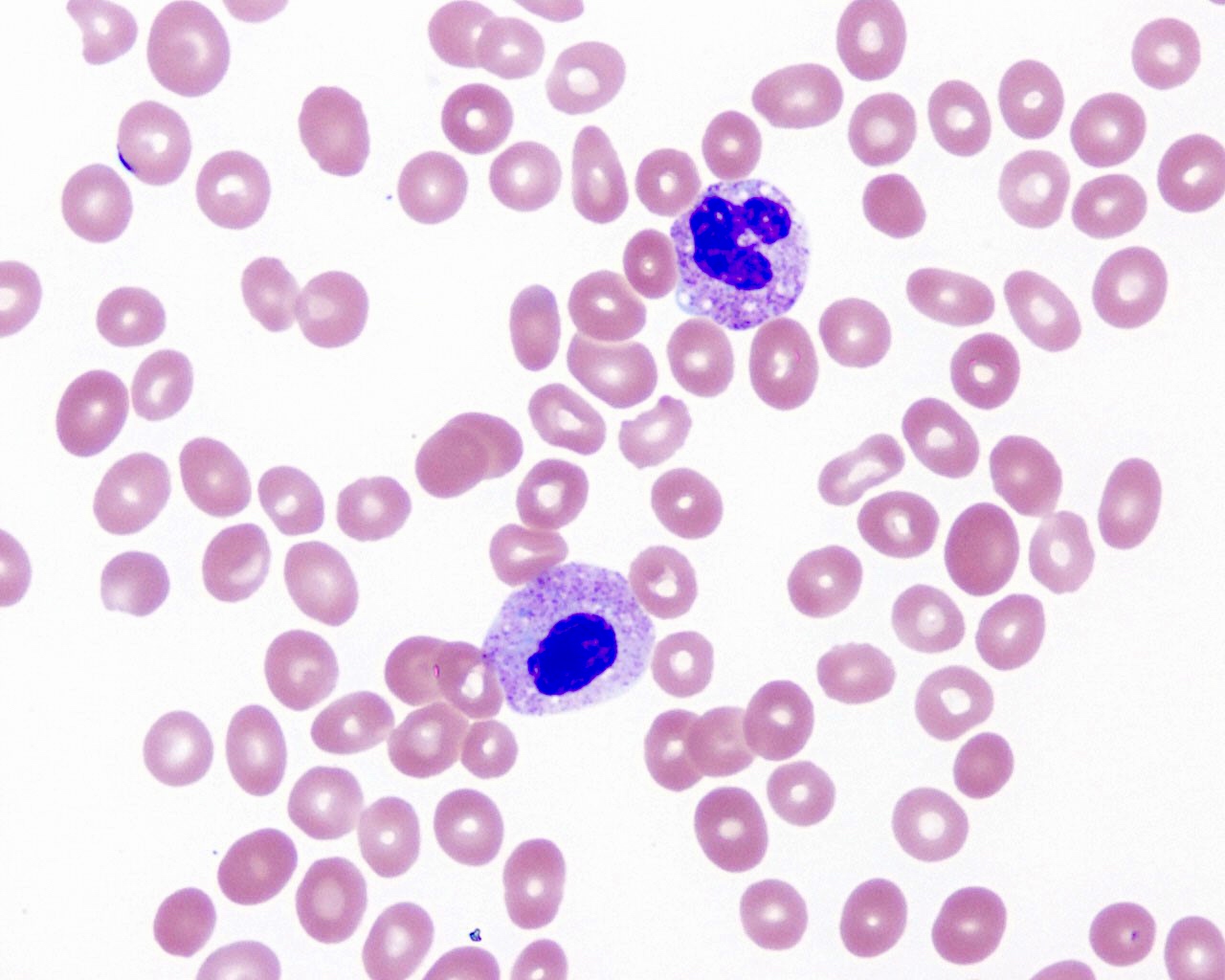
Pseudo-Pelger-Huët neutrophils

Large neutrophils
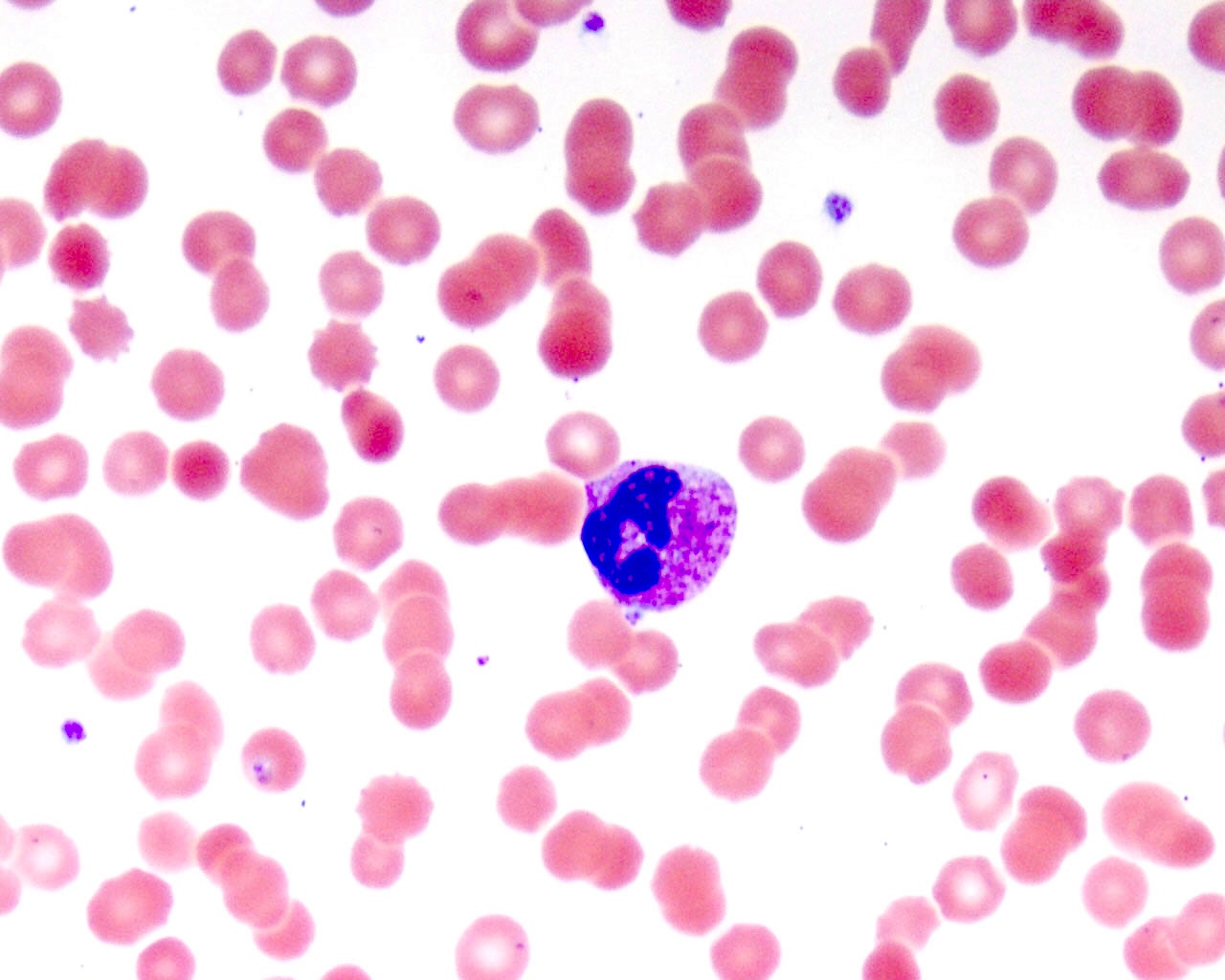
Toxic granulation (red granules)
- Blood smear:
- Reactive neutrophilia (see comment)
- Comment: Neutrophilia with toxic changes, left shift and rare blasts, consistent with cytokine effect or inflammatory changes. Clinical correlation is recommended.
- Chronic myeloid leukemia (CML):
- Usually has very high WBC (> 50 x 109/L) with prominent left shift with a myelocyte bulge and associated basophilia
- Toxic granulation is usually absent
- Blasts can be increased
- Splenomegaly is common
- Positive for BCR::ABL1 gene rearrangement or t(9;22)(q34;q11)
- Chronic neutrophilic leukemia (CNL):
- Marked leukocytosis (> 30 x 109/L) with absolute neutrophilia
- No significant left shift in peripheral blood (immature granulocytes are < 10% of WBC)
- Splenomegaly
- Mutational analysis is positive for CSF3R mutations in > 80% of cases
- In a patient with absolute neutrophilia, the distinction between reactive and neoplastic etiology (especially CML) requires integration of clinical, hematologic, morphologic and other laboratory parameters; it is extremely important to carefully review the clinical history for possible reactive etiologies of neutrophilia
A 45 year old man presented with leukocytosis of 28 x 109/L with prominent left shifted granulocytes, rare blasts and no basophilia in the peripheral smear review. What is the next step?
- Check clinical history and physical examination record
- Order BCR::ABL PCR
- Send peripheral blood for flow cytometry
- Suggest a marrow biopsy
Comment here
Reference: Reactive neutrophilia
- Need flow cytometry study for diagnosis
- Need marrow biopsy for diagnosis
- Neoplasm, since the white cell count is too high
- Reactive neutrophilia
- Leukemic variant of cutaneous T cell lymphoma (CTCL) defined by the presence of erythroderma, generalized lymphadenopathy and clonal T cells with cerebriform nuclei (Sézary cells); diagnosis is stronger if the same clone is demonstrated in the skin, lymph nodes and peripheral blood (J Am Acad Dermatol 2012;67:1189, Lancet 2008;371:945, Blood Adv 2018;2:2115, Am J Hematol 2019;94:1027, Eur J Cancer 2018;93:47, Curr Hematol Malig Rep 2016;11:468)
- Closely related to mycosis fungoides
- Sézary syndrome (SS) may be indistinguishable from mycosis fungoides when mycosis fungoides becomes erythrodermic and clinically aggressive, although mycosis fungoides usually lacks leukemic phase (J Am Acad Dermatol 2014;70:205.e1)
- Despite the overlapping features, Sézary syndrome (SS) and mycosis fungoides are considered distinct entities; SS is a disease arising de novo, while erythroderma and lymphadenopathy in a patient known to have mycosis fungoides is better referred to as SS preceded by mycosis fungoides (Eur J Cancer 2017;77:57)
- Leukemic variant of cutaneous T cell lymphoma (CTCL) defined by the presence of erythroderma, generalized lymphadenopathy and clonal T cells with cerebriform nuclei (Sézary cells); diagnosis is stronger if the same clone is demonstrated in the skin, lymph nodes and peripheral blood (J Am Acad Dermatol 2012;67:1189, Lancet 2008;371:945, Blood Adv 2018;2:2115, Am J Hematol 2019;94:1027, Eur J Cancer 2018;93:47, Curr Hematol Malig Rep 2016;11:468)
- Histologically similar to mycosis fungoides but variable and sometimes the findings of mycosis fungoides, such as epidermotropism, are not present (Lancet 2008;371:945, Curr Hematol Malig Rep 2016;11:468, J Am Acad Dermatol 2011;64:352)
- Chronic condition and incurable: the main aim of treatment is controlling symptoms and improvement of the quality of life (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Blood 2016;127:3142, J Am Acad Dermatol 2011;64:352)
- Erythrodermic CTCL includes all primary cutaneous lymphoma that evolve to erythroderma, such as Sézary syndrome (SS) and erythrodermic mycosis fungoides
- SS was first described by Albert Sézary in 1938 (Lancet 2008;371:945, Eur J Cancer 2017;77:57)
- Rare: corresponds to 2.5 - 5.0% of all CTCLs (J Am Acad Dermatol 2012;67:1189, Lancet 2008;371:945, Eur J Cancer 2017;77:57)
- Age adjusted incidence of 6.4 cases per million (Lancet 2008;371:945)
- Sixth and seventh decade (J Am Acad Dermatol 2012;67:1189, Blood Adv 2019;3:1145, Am J Clin Pathol 2011;136:944, Clin Cancer Res 2012;18:5051)
- M:F = 2:1 (J Am Acad Dermatol 2012;67:1189, Lancet 2008;371:945, Blood Adv 2019;3:1145)
- More common in Caucasians (J Am Acad Dermatol 2012;67:1189, Clin Cancer Res 2012;18:5051)
- Mycosis fungoides and SS are thought to arise from chronic antigenic stimulation; early mycosis fungoides lesions show increased numbers of dendritic cells and upregulation of their antigen presenting cell (APC) ligands B7 and CD40 and their respective T cell costimulatory ligands CD28 and CD40L (J Am Acad Dermatol 2014;70:205.e1)
- Both mycosis fungoides and SS have as cell of origin the skin resident CD45RO+ effector memory T cell (Lancet 2008;371:945)
- Antigen presenting dendritic cells could maintain the survival and proliferation of clonal T cells: higher specific human leukocyte antigen (HLA) class II alleles than the general population (Lancet 2008;371:945)
- Skin microbiomes (e.g. as Chlamydia spp) could play a role in the pathogenesis by the stimulation of clonality in T cells but it is still controversial (Lancet 2008;371:945)
- Abnormalities in pathways: NFκB / JAK STAT activation, cell cycle dysregulation / apoptosis and DNA structural dysregulation affecting gene expression (Nat Genet 2015;47:1465)
- Micro RNA expression profile detected differences between erythrodermic mycosis fungoides and SS (Acta Derm Venereol 2019;99:1148)
- Chemokines receptors (Lancet 2008;371:945, Curr Hematol Malig Rep 2016;11:468, J Am Acad Dermatol 2011;64:352)
- CCR4 and CCR10 play a role in the homing of malignant T cells to skin where they bind to ligands on endothelial cells, keratinocytes or Langerhans cells
- CCL17 is a major CCR4 ligand and it is increased in the serum of patients with mycosis fungoides and SS
- May explain the lower epidermotropism observed in SS compared with mycosis fungoides
- Loss of CD26 promotes the inactivation of CXCL12, which is the CXCR4 ligand
- CCR4 and CCR10 play a role in the homing of malignant T cells to skin where they bind to ligands on endothelial cells, keratinocytes or Langerhans cells
- T helper 2 (Th2) response is characteristic of SS as well as that of tumor stage mycosis fungoides; overexpression of CD47 in Sézary cells is under the influence of Th2 cytokines, such as IL-4, IL-7 and IL-13 (Blood Adv 2019;3:1145, Curr Hematol Malig Rep 2016;11:468, J Am Acad Dermatol 2011;64:352)
- Erythroderma is the key feature for diagnosis and is defined by involvement of > 80% of body surface and may range from mild to intense (J Am Acad Dermatol 2012;67:1189, Lancet 2008;371:945, Eur J Cancer 2018;93:47, Curr Hematol Malig Rep 2016;11:468)
- Alopecia and plaques at initial clinical presentation are uncommon (systemic symptoms usually absent)
- Nail dystrophy, blepharoconjunctivitis and ectropium can be present in advanced disease
- Palms and sole usually show thickening, scales and fissures
- Median duration of all dermatologic symptoms before diagnosis: 3.5 years
- Erythroderma is present usually 1.7 years before diagnosis
- Clinical presentation with erythroderma with < 1 x 109/L is defined as pre-Sézary and some patients progress to SS, while others follow a more indolent clinical course
- Pruritus is often present (Lancet 2008;371:945)
- Systemic symptoms usually absent (J Am Acad Dermatol 2012;67:1189)
- Adenopathy is usually present (J Am Acad Dermatol 2012;67:1189)
- Bone marrow involvement in approximately 20% of the cases (J Am Acad Dermatol 2012;67:1189)
- Cases may be preceded by mycosis fungoides and should be designated as SS preceded by mycosis fungoides; these cases are different from de novo SS (Am J Hematol 2019;94:1027)
- Characterized by the presence of erythroderma, generalized lymphadenopathy and clonal T cells (same clone in skin, lymph node and peripheral blood) in addition to 1 or more of the following criteria (Am J Hematol 2019;94:1027, Eur J Cancer 2018;93:47, Am J Clin Pathol 2011;136:944, Curr Hematol Malig Rep 2016;11:468, J Am Acad Dermatol 2011;64:352):
- Absolute Sézary cell count ≥ 1 x 109/L (manual count or by flow cytometry)
- Alternative diagnostic criteria: ≥ 40% of CD4+ / CD7- and ≥ 30% of CD4+ / CD26- T cells (flow cytometry)
- TRBC1 is more sensitive and specific for detection of clonal T cells by flow cytometry
- Expanded CD4+ T cell population with CD4:CD8 ≥ 10
- It is reliant on the absolute CD8 count and the ratio may decrease after treatment
- Loss of one or more T cell antigens
- Demonstration of T cell clonality by southern blot or PCR based methods
- Use of V beta antibodies allows quantification of the clone (obsolete)
- Clonality by flow cytometry: constant chain of T cell receptor: TRBC1 (Int J Mol Sci 2021;22:1817)
- Use of high throughput sequencing is more sensitive but not widely used
- Cytogenetic demonstration of an abnormal clone
- Bone marrow involvement is not a defined criteria for SS
- Criteria for blood response (see staging table below):
- Complete response (CR): B2 should change to B0
- Partial response (PR): at least 50% reduction of tumor burden
- Progressive disease (PD): B0 or B1 to B2 with an increase in absolute counts of ≥ 50% or B2 with an increase in absolute counts of ≥ 50% or loss of response with an increase in absolute counts ≥ 1x109/L and ≥ 50% from nadir
- Relapse: increase in absolute counts of ≥ 1x109/L in the context of CR
- Staging of mycosis fungoides and SS is performed according to the International Society for Cutaneous Lymphomas (ISCL) and the European Organization for Research and Treatment of Cancer (EORTC) (Eur J Cancer 2017;77:57, Blood 2016;127:3142)
| Skin | |
| T1 | Limited patches, papules or plaques covering < 10% of the skin surface: T1a: patch only T1b: plaque with or without patch |
| T2 | Patches, papules or plaques covering ≥ 10% of the skin surface: T2a: patch only T2b: plaque with or without patch |
| T3 | One or more tumors (≥ 1cm diameter) |
| T4 | Confluence of erythema covering ≥ 80% body surface area |
| Node | |
| N0 | No clinically abnormal peripheral lymph node |
| N1 | Clinically abnormal peripheral lymph nodes; histopathology Dutch grade 1 or NCI LN0-2 N1a: clone negative N1b: clone positive |
| N2 | Clinically abnormal peripheral lymph nodes; histopathology Dutch grade 2 or NCI LN3 N2a: clone negative N2b: clone positive |
| N3 | Clinically abnormal peripheral lymph nodes; histopathology Dutch grade 3 - 4 or NCI LN4; clone positive or negative |
| Nx | Clinically abnormal peripheral lymph nodes with no histological confirmation |
| Visceral | |
| M0 | No visceral organ involvement |
| M1 | Visceral involvement (must have pathological confirmation) |
| Peripheral Blood (PB) | |
| B0 | Absence of significant blood involvement: ≤ 5% Sézary cells in PB B0a: clone negative B0b: clone positive |
| B1 | Low blood tumor burden: > 5% Sézary cells in PB and does not meet criteria of B2 B1a: clone negative B1b: clone positive |
| B2 | High blood tumor burden: ≥ 1000/µL Sézary cells in PB with clone positive |
- SS is defined as stage IVA1 (T1-T4, N0-N2, M0, B2) and above
- Poor prognosis (J Am Acad Dermatol 2012;67:1189, Blood Adv 2019;3:1145, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1, Clin Cancer Res 2012;18:5051)
- Median overall survival after diagnosis is 3.1 - 4 years
- Cases with visceral involvement have a median overall survival of 2.5 years
- Median time to death after diagnosis is 3.5 years
- 5 year overall survival range is 18 - 37% and 10 year overall survival range is 0 - 18%
- Median overall survival after diagnosis is 3.1 - 4 years
- Overexpression of CD47 by Sézary cells correlates with worse overall survival (Blood Adv 2019;3:1145)
- Poor prognostic factors (J Am Acad Dermatol 2012;67:1189, Blood 2016;127:3142, Leuk Lymphoma 2012;53:868, Clin Cancer Res 2012;18:5051)
- Advanced age at diagnosis (> 65 years)
- Diagnosis of mycosis fungoides before diagnosis of SS
- Presence of TCR gene rearrangement in skin or blood
- Usually present
- Higher lactate dehydrogenase levels at presentation
- Number of Sézary cells in the peripheral blood does not appear to be a prognostic factor (J Am Acad Dermatol 2014;70:223.e1)
- Increased risk of a secondary lymphoma in patients with SS / mycosis fungoides (Arch Dermatol 2007;143:45)
- 49 year old man with clinical presentation of erythematous papules (J Dermatol 2019;46:61)
- 64 year old man with pulmonary involvement by Sézary syndrome (Int J Dermatol 2016;55:81)
- 66 year old man with renal involvement by Sézary syndrome (Am J Kidney Dis 2018;72:890)
- 70 year old woman with bullous Sézary syndrome (Dermatol Online J 2020;26:13030)
- Chronic condition and incurable: the main aim of treatment is control of symptoms and improvement of quality of life (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Blood 2016;127:3142,J Am Acad Dermatol 2011;64:352)
- Systemic therapies
- Retinoids (derivates of vitamin A) and bexarotene (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Curr Hematol Malig Rep 2016;11:468, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1)
- Well tolerated
- Efficacy in cases of extracutaneous involvement
- Moderate rates of response can be achieved if used as monotherapy
- Chlorambucil associated with prednisone
- Methotrexate (MTX) low dose
- Recombinant interferon α (Lancet 2008;371:945, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1)
- Used as monotherapy or associated with retinoids
- Conventional chemotherapy (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Blood 2016;127:3142)
- Cyclophosphamide, hydroxydaunorubicin, oncovin and prednisone (CHOP) or CHOP-like regimen
- Rarely achieves complete durable remission
- Target therapy (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Blood Adv 2019;3:1145, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1)
- Alemtuzumab
- Monoclonal antibody against CD52 cell surface glycoprotein
- May induce long term remission
- Second line therapy
- Anti-PD1
- CD47 / SIRPα
- Alemtuzumab
- Extracorporeal photopheresis (ECP) (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1)
- Considered first line of treatment
- May induce long term remission
- Excellent safety profile and response rates up to 60%
- Alone or in combination with systemic therapy
- Histone deacetylase inhibitors (HDACi) (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Curr Hematol Malig Rep 2016;11:468, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1)
- Vorinostat and romidepsin
- Overall response rate 24 - 38%
- Vorinostat and romidepsin
- Allogenic stem cell transplantation (Blood 2016;127:3142, J Clin Oncol 2010;28:2365)
- There is no consensus regarding the indication
- May be preceeded by skin electron beam radiation
- Acute and chronic graft versus host disease occur in more than 50% of patients
- Retinoids (derivates of vitamin A) and bexarotene (Lancet 2008;371:945, Am J Hematol 2019;94:1027, Curr Hematol Malig Rep 2016;11:468, Blood 2016;127:3142, J Am Acad Dermatol 2014;70:223.e1)
Contributed by Roberto N. Miranda, M.D.
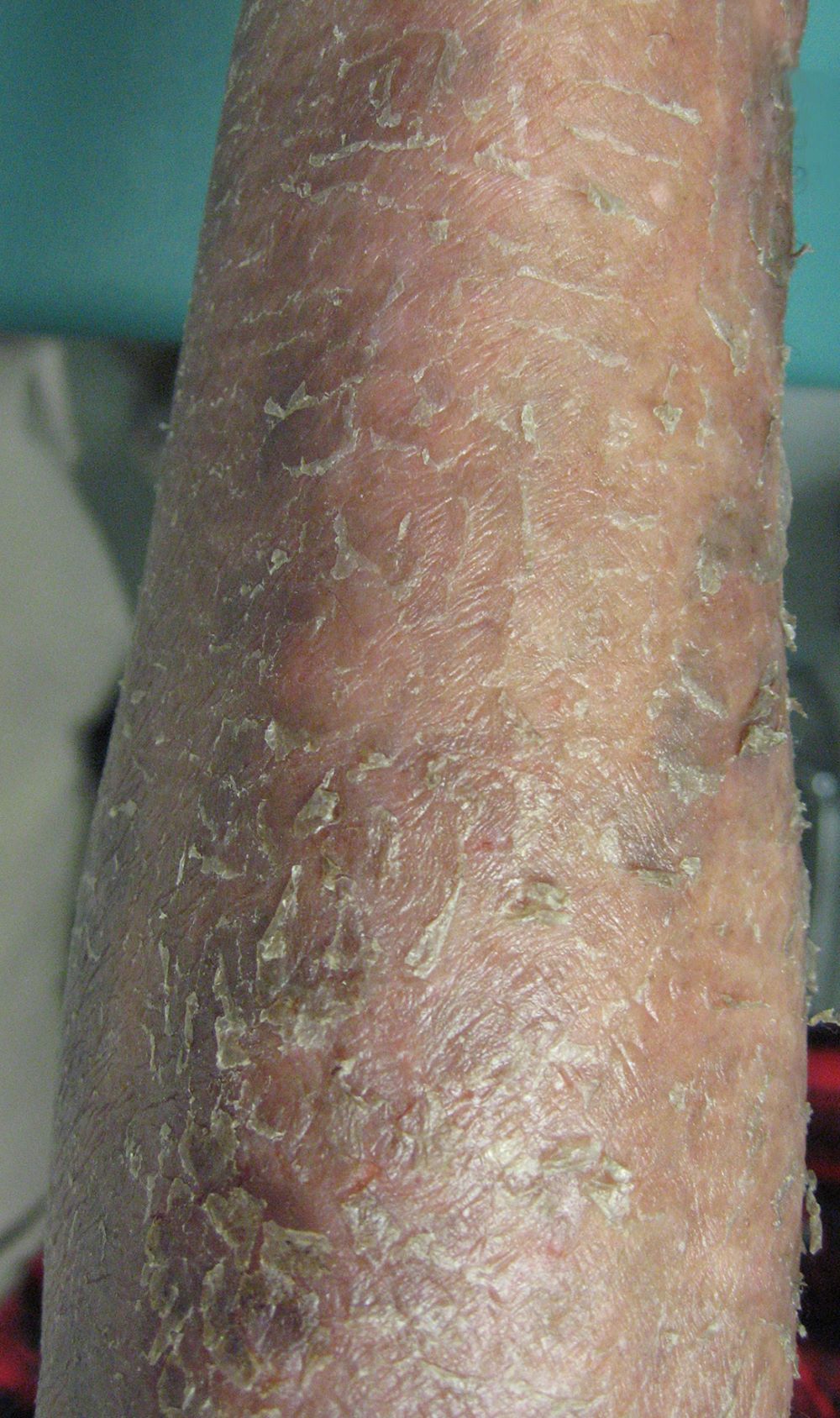
Prominent exfoliative lesion
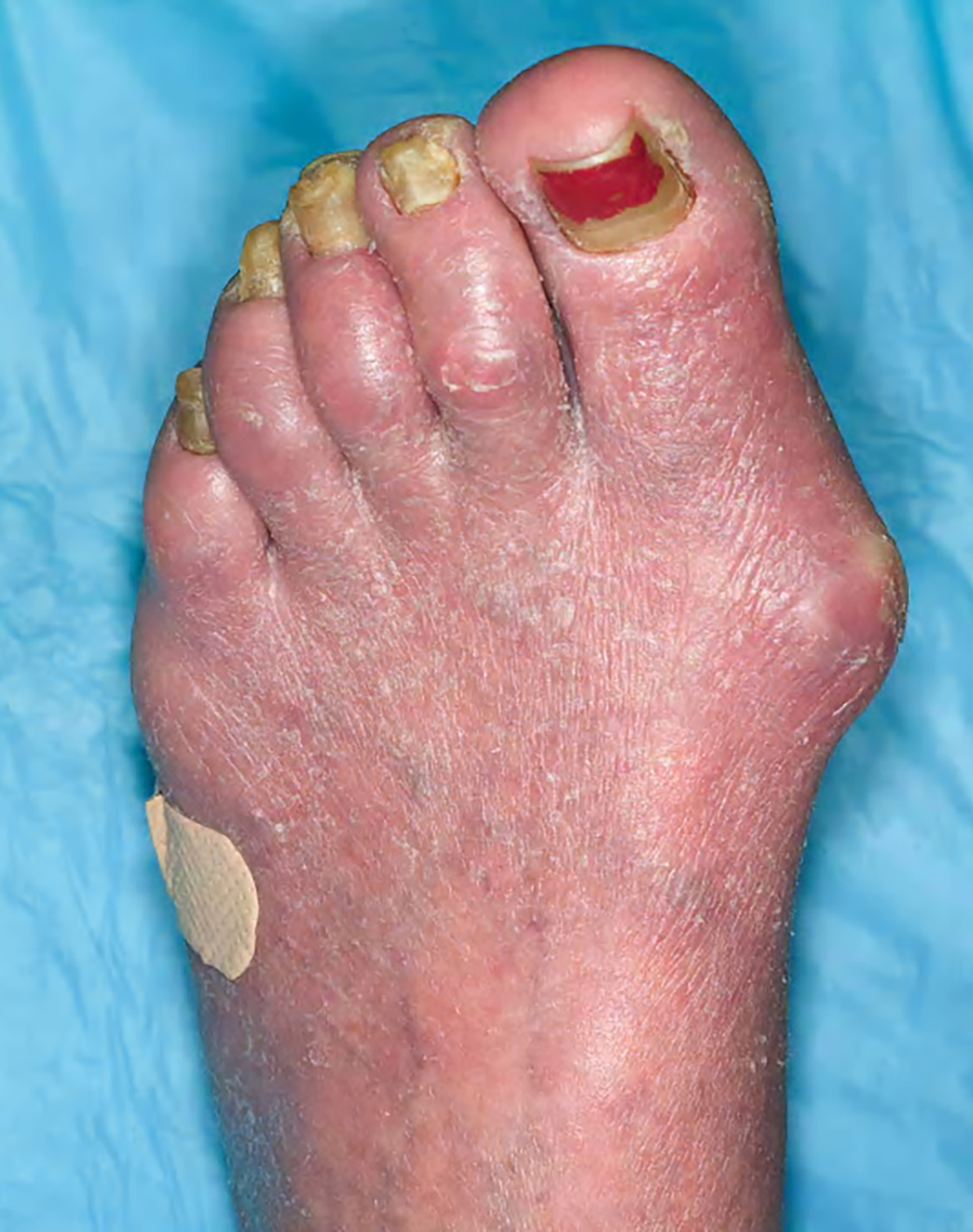
Prominent erythroderma
Contributed by Roberto N. Miranda, M.D.
Central nervous system (CNS) involvement
- SS is histologically similar to mycosis fungoides but sometimes epidermotropism is not present (Lancet 2008;371:945, Curr Hematol Malig Rep 2016;11:468, J Am Acad Dermatol 2011;64:352)
- May range from limited to superficial perivascular lymphocytic and eosinophilic dermatitis with or without spongiosis (atopic-like lesions) to lichenoid lymphocytic infiltrate
- Pautrier microabscesses are not common
- Peripheral blood (Lancet 2008;371:945)
- Sézary cells: atypical lymphocytes of intermediate to large size and cerebriform nuclei
- Similar cells, albeit few or rare may be found in healthy individuals or in patients with other inflammatory skin diseases
- Sézary cells: atypical lymphocytes of intermediate to large size and cerebriform nuclei
- Lymph node (Am J Hematol 2019;94:1027, Cancer 1986;57:237)
- Complete effacement of the nodal architecture by monotonous infiltrating population of Sézary cells
- Large cell or blastoid morphology may occur in advanced stages.
Contributed by Roberto N. Miranda, M.D.
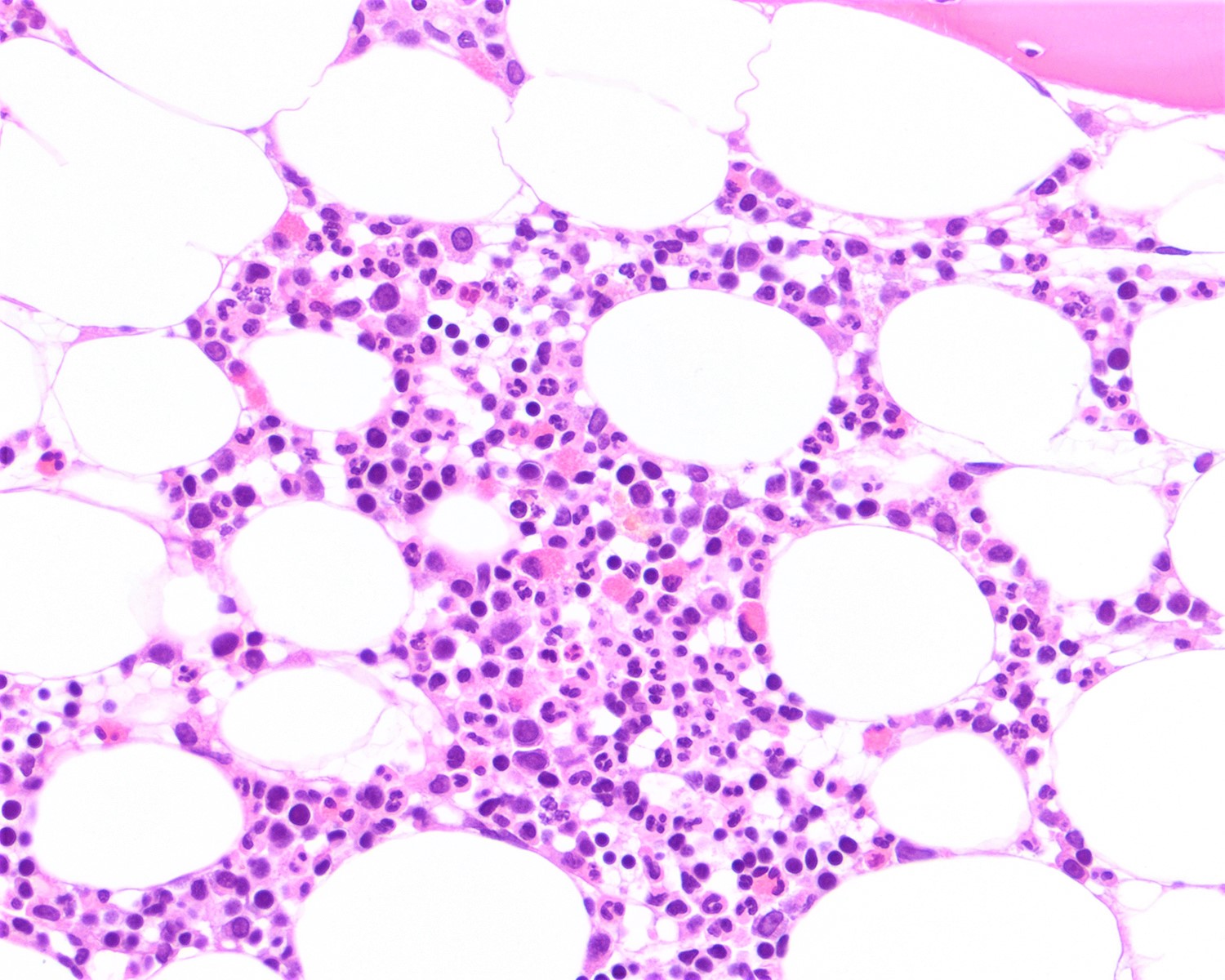
Bone marrow involvement
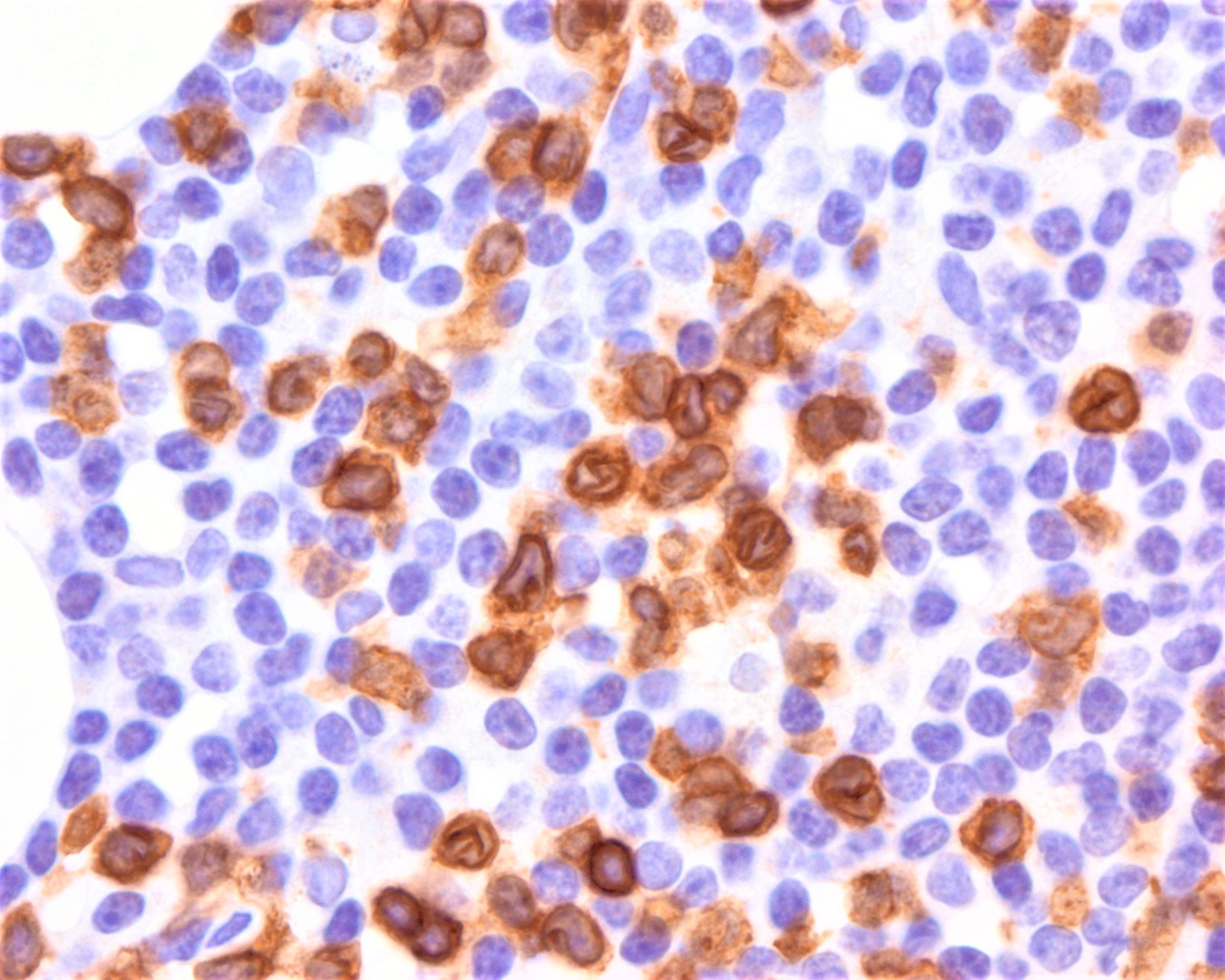
CD3 positivity
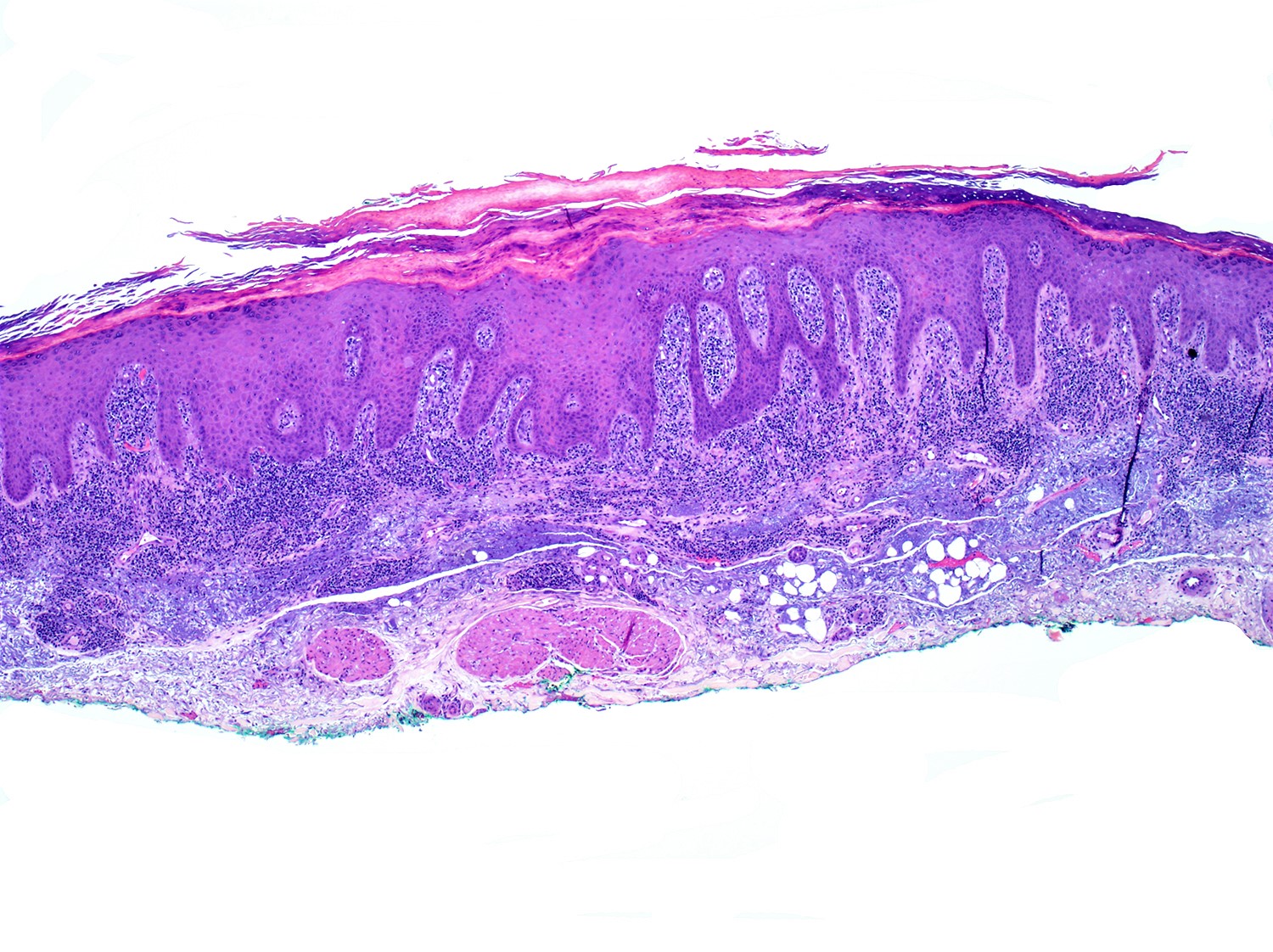
Epidermotropic and lichenoid lymphoid infiltrate
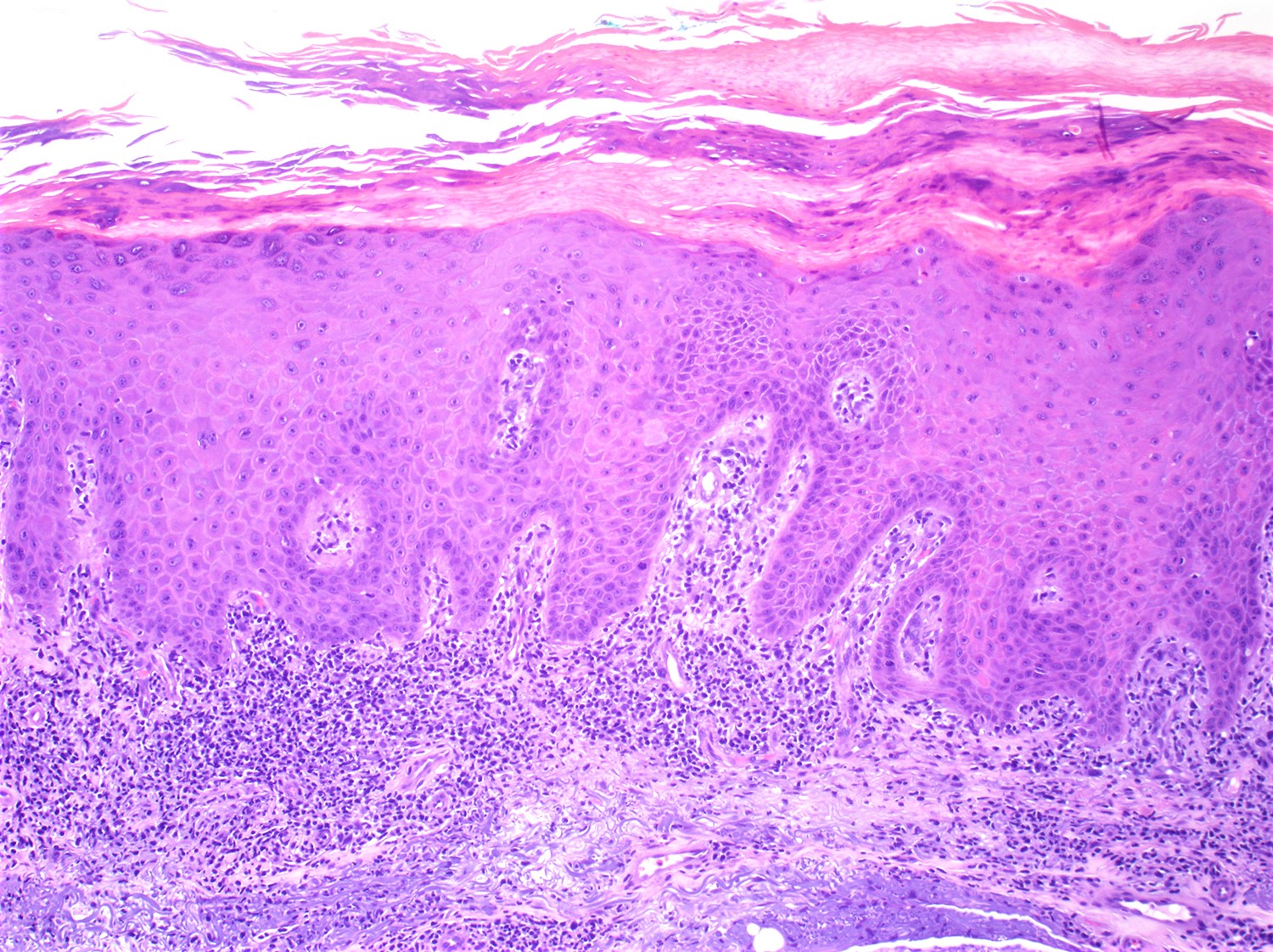
Epidermotropic infiltrate
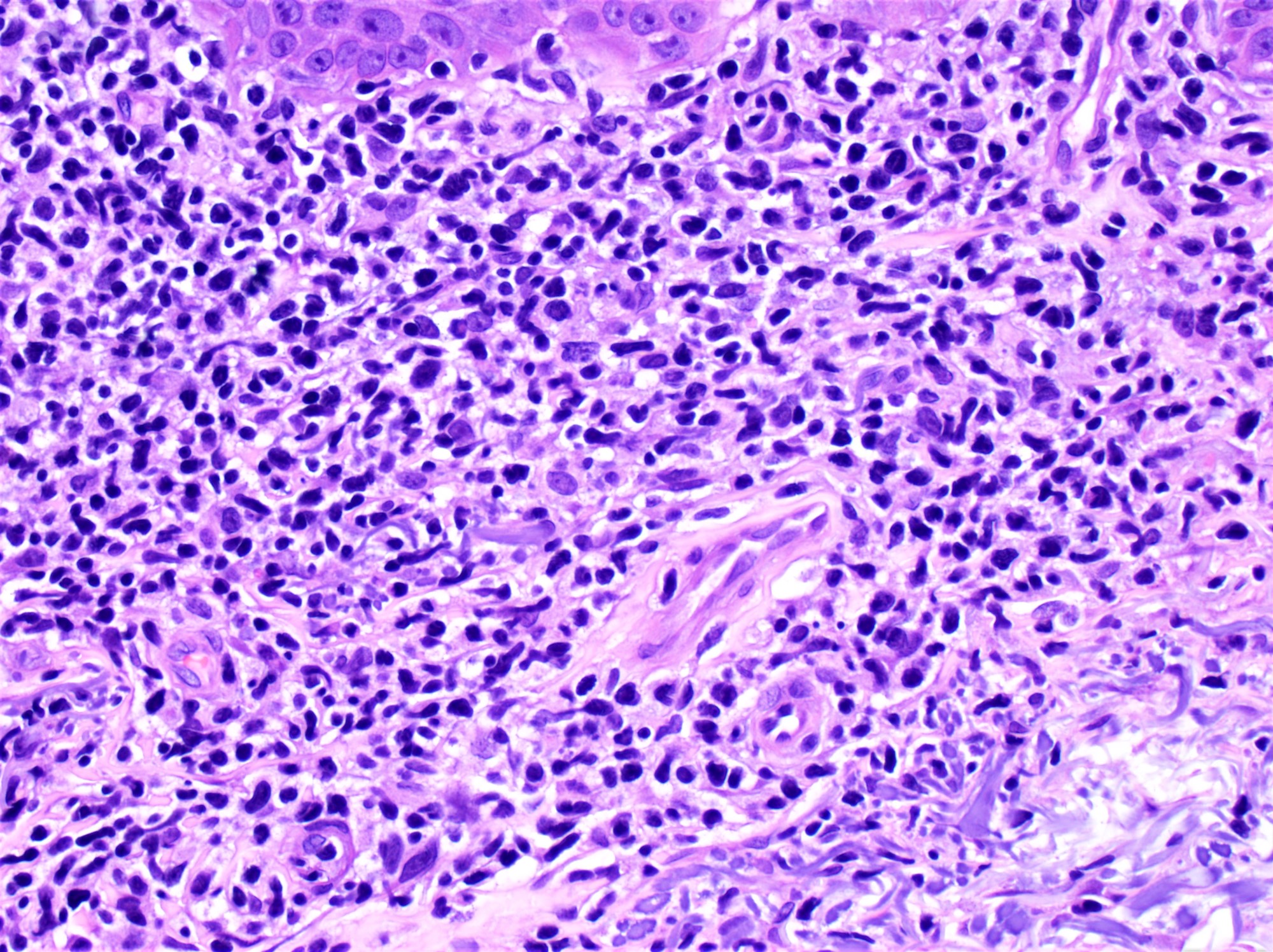
Cerebriform cytology
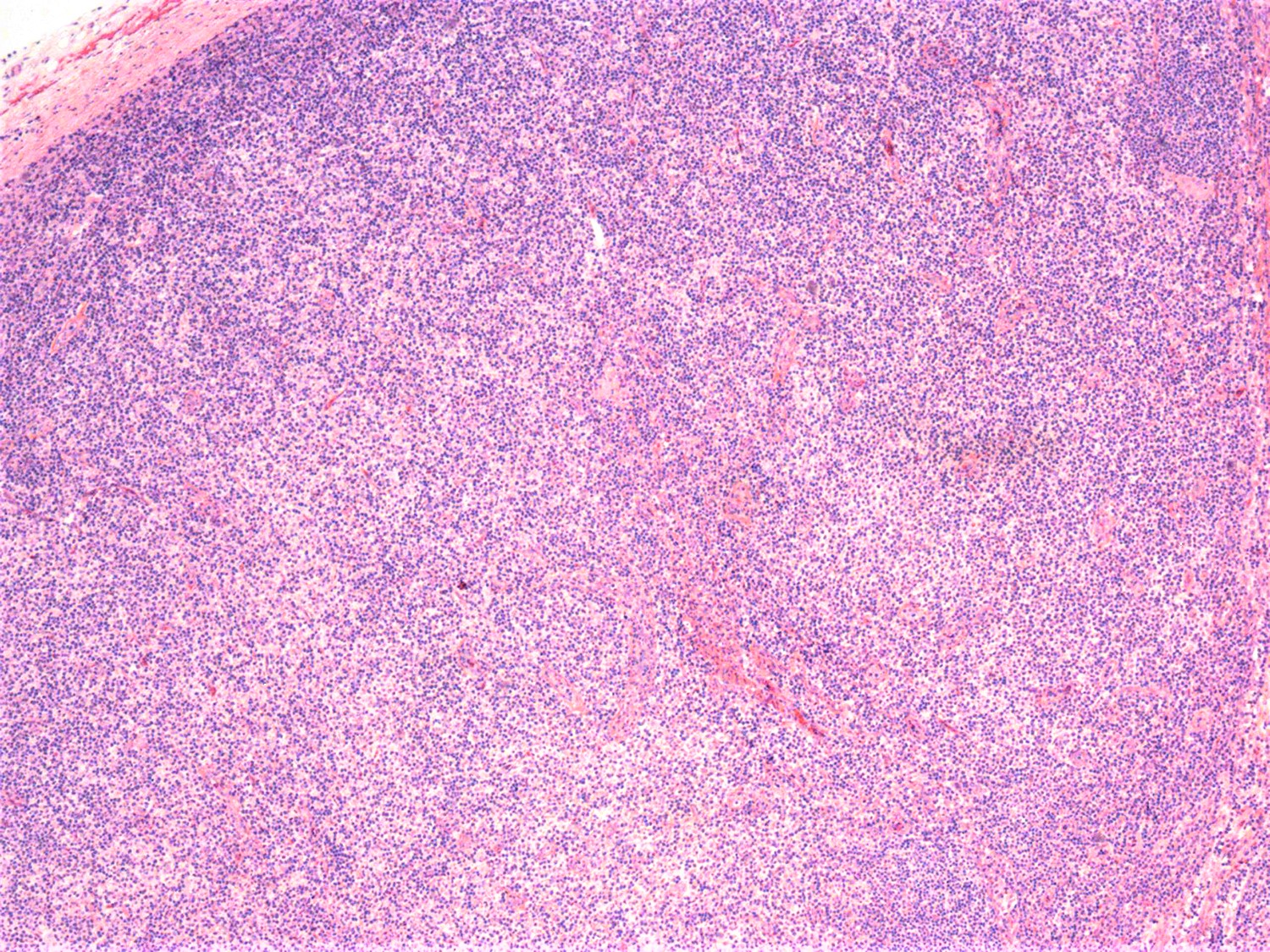
Lymph node involvement
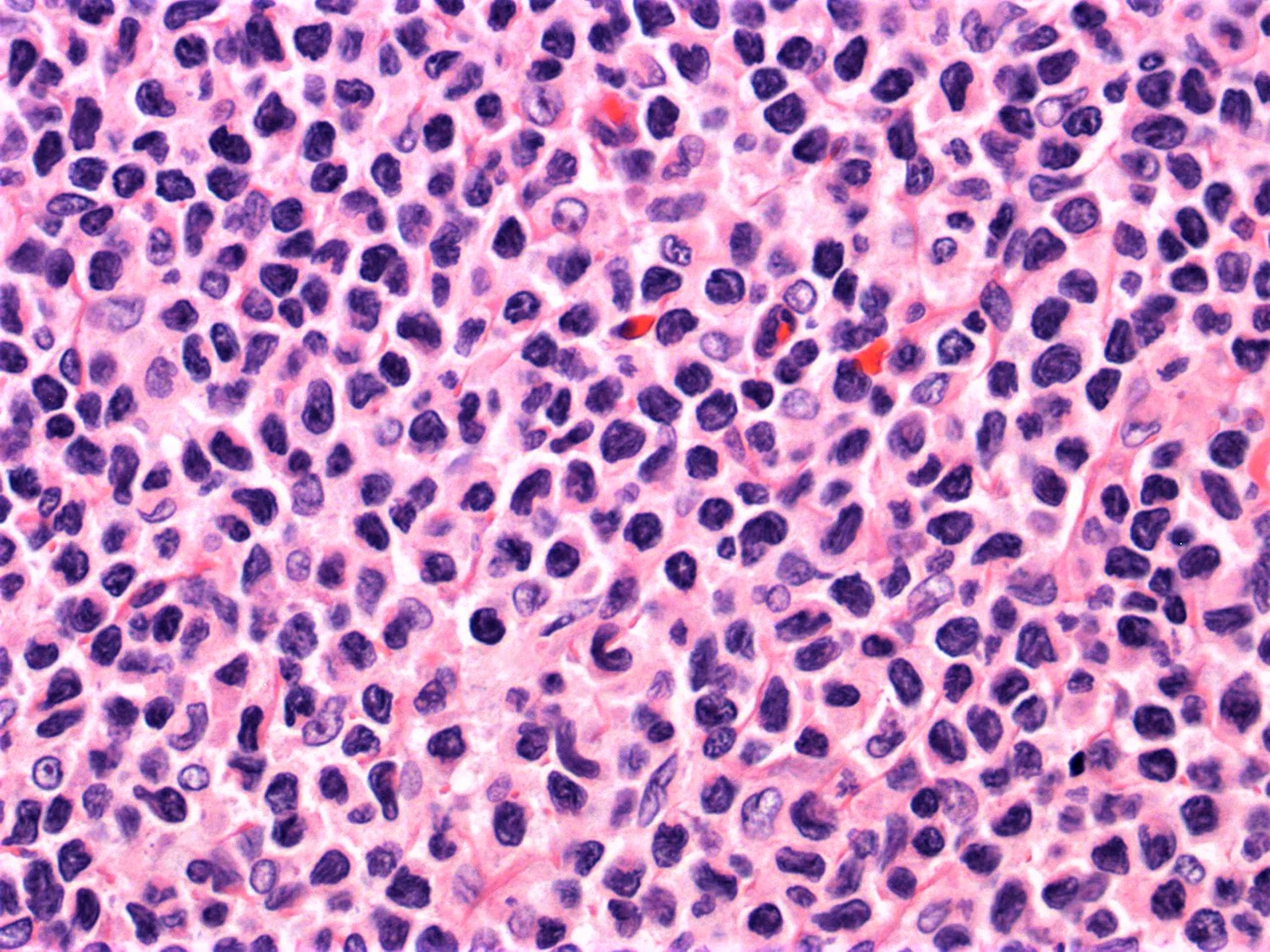
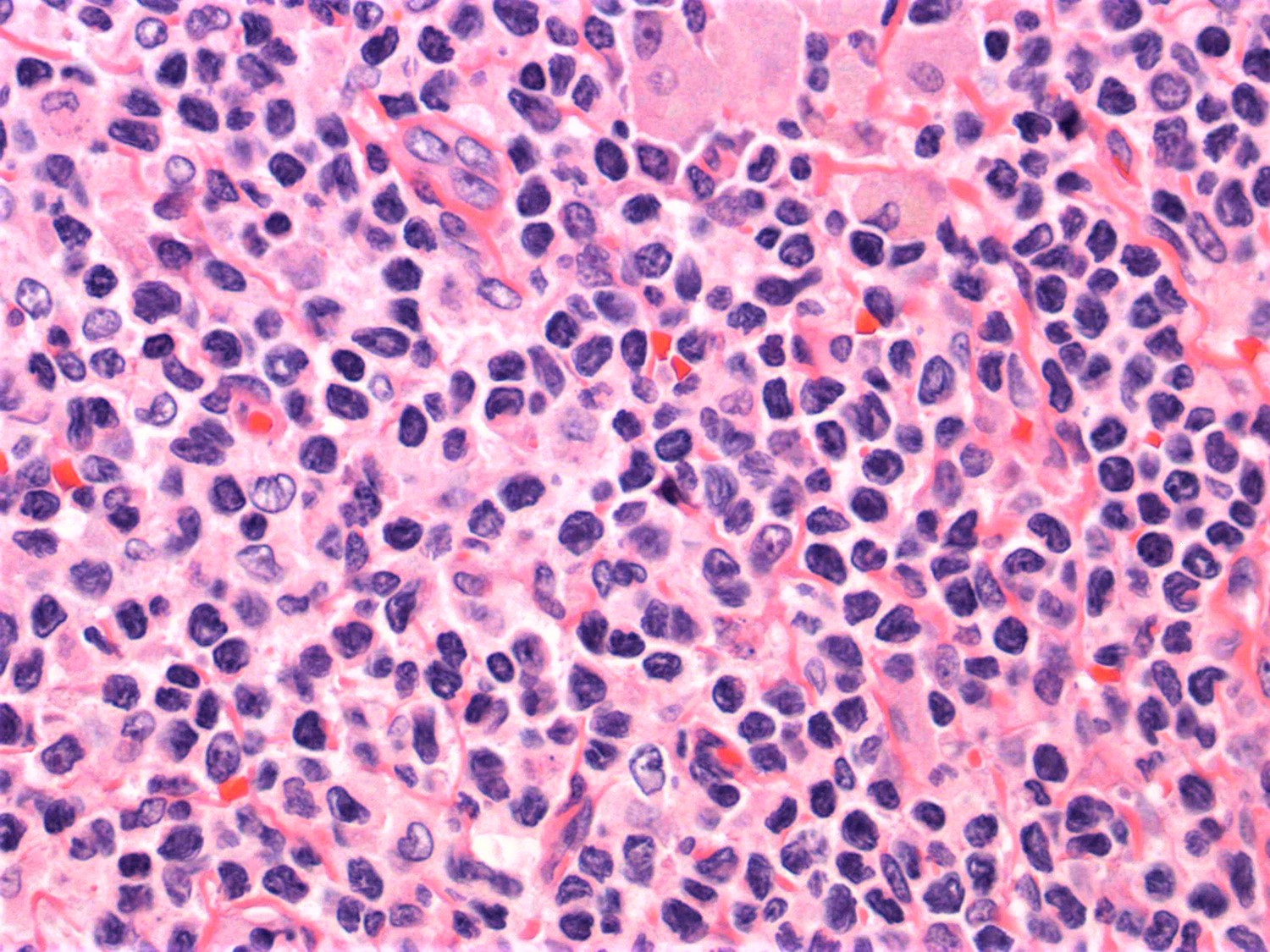
Cytological atypia
Contributed by Roberto N. Miranda, M.D.
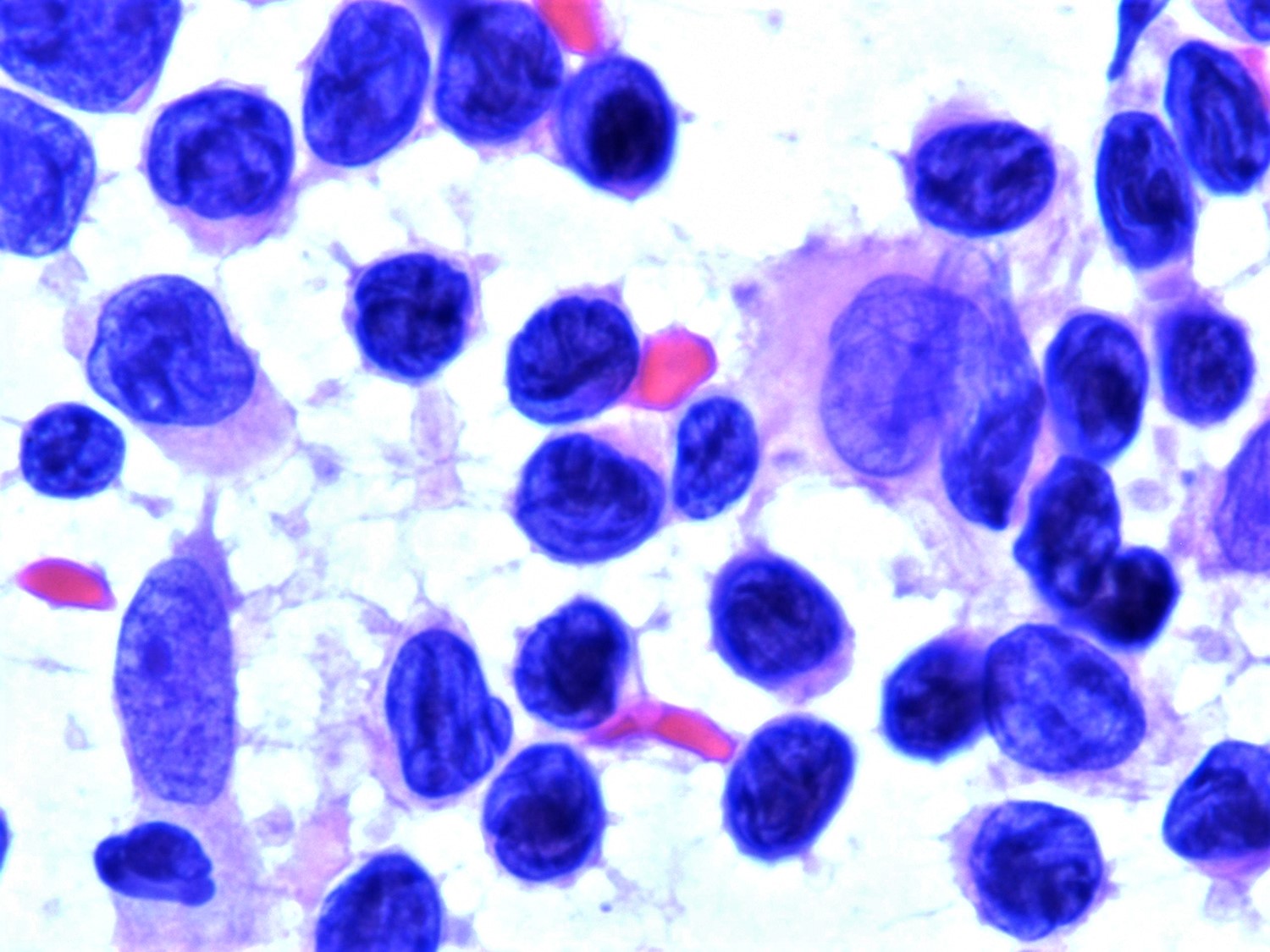
Lymph node touch print
Contributed by Roberto N. Miranda, M.D.
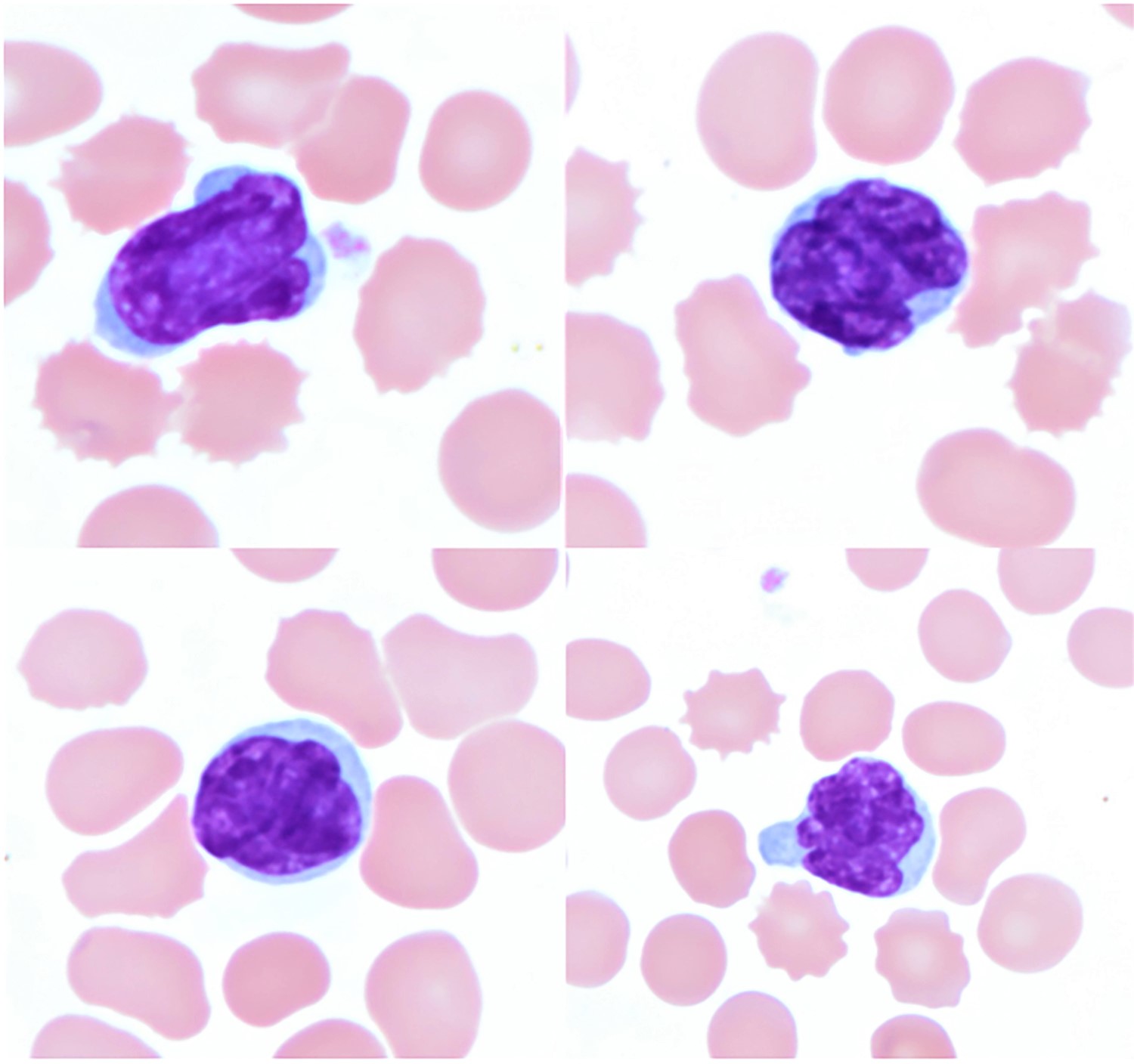
Cerebriform lymphocytes in peripheral blood smear
- Heterogeneous expression of T cell markers (Blood Adv 2018;2:2115, Am J Hematol 2019;94:1027, Eur J Cancer 2018;93:47, Am J Clin Pathol 2011;136:944, Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468, Blood 2013;122:3511, Leuk Lymphoma 2012;53:868)
- T central memory phenotype: CD45RO, CD62L and CD197 (Blood Adv 2018;2:2115, Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468)
- Some cases may have a T naïve or stem cell memory T and express CD45RA
- CCR4 and CCR7 (Lancet 2008;371:945, Curr Hematol Malig Rep 2016;11:468)
- CD47 (Blood Adv 2019;3:1145)
- Possible target therapy
- CTLA4 (Exp Dermatol 2017;26:668)
- PD-1 (Arch Dermatol 2012;148:1379)
- When positive, it is usually expressed in > 50% of cells
- NOTCH1 (N terminal sequence of icNOTCH1 antibody) (J Invest Dermatol 2012;132:2810)
- Syndecan-4 (Exp Dermatol 2017;26:668)
- Aberrant expression of MHC class I killer immunoglobulin-like receptor KIR / CD158κ (Am J Hematol 2019;94:1027, Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468)
- TCRVβ (Blood Adv 2018;2:2115, Exp Dermatol 2017;26:668, Leuk Lymphoma 2012;53:868, Mod Pathol 2010;23:284)
- Loss of one or more conventional T cell markers: CD2, CD5, CD7, CD8, CD25, CD26, CD30, FOXP3 (Blood Adv 2018;2:2115, Am J Hematol 2019;94:1027, Eur J Cancer 2018;93:47, Am J Clin Pathol 2011;136:944, Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468,Leuk Lymphoma 2012;53:868, J Am Acad Dermatol 2011;64:352)
- B cell markers (Exp Dermatol 2017;26:668)
Contributed by Roberto N. Miranda, M.D.
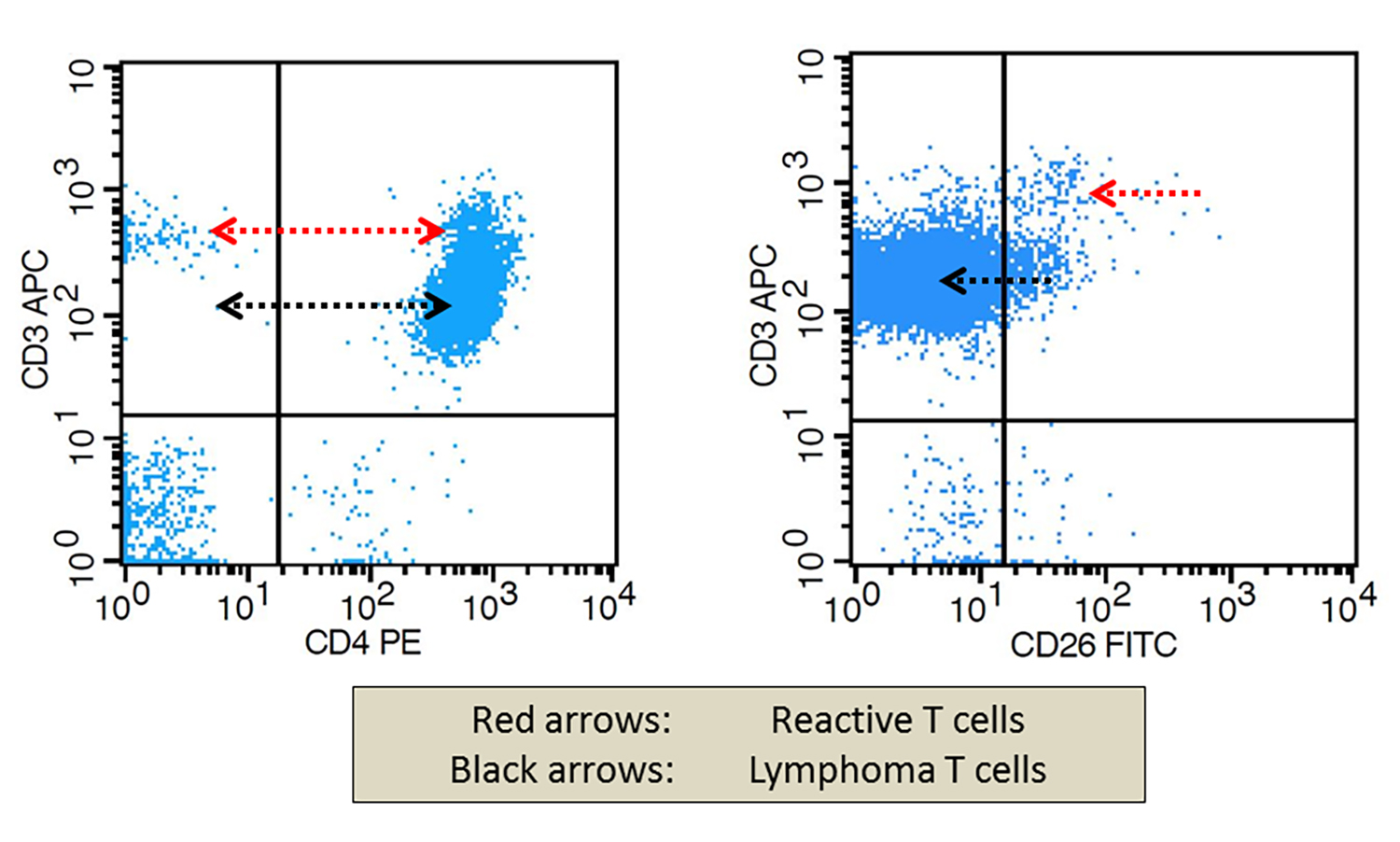
Flow cytometry in SS
- No driver mutations identified (Blood Adv 2018;2:2115)
- Somatic mutations in TCR/NFκB signaling, Th2 differentiation (ZEB1), cell survival (e.g. JAK / STAT), epigenetic regulation (e.g. DNMT3A, TET2, SMARCA4), homologous recombination (e.g. BRCA2) and cell cycle control (e.g. TP53) (Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468, Nat Commun 2015;6:8470, Nat Genet 2015;47:1465)
- Activation of NOTCH1 signaling (J Invest Dermatol 2012;132:2810)
- Heterogeneous expression of transcripts (Blood Adv 2018;2:2115, Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468)
- S100A4, S100A10, GATA3, Twist1, TOX, IL7R, CCR7 and CXCR4 may be upregulated
- ARID1A, SATB1, STAT4 and Fas may be downregulated
- Genomic complexity (Exp Dermatol 2017;26:668, Curr Hematol Malig Rep 2016;11:468, Nat Commun 2015;6:8470, Blood 2013;122:3511)
- No specific recurrent translocations or gene fusions
- Cytogenetic abnormalities
- Gain of 17p11.2-q25.3 and 8q24.1-8q24.3
- Loss of 17p13.2-p11.2 and 10p12.1-q26.3
- Deletion of PTEN on locus 10q23 and it is usually monoallelic
- Skin, shave biopsy of the arm:
- Primary cutaneous T cell lymphoma, most consistent with Sézary syndrome (see comment)
- Comment: Section shows an adequate skin shave biopsy with mild spongiosis and perivascular and lichenoid infiltrate in superficial dermis with mild epidermotropism. The infiltrate is composed of small lymphocytes, with irregular nuclear contours (cerebriform-like), hyperchromatic nuclei and scant cytoplasm. Immunohistochemical studies show that the abnormal lymphocytes are diffusely positive for CD3, CD4, CD5 and CD45RO and negative for CD2, CD7 and CD30. The concurrent complete blood count (CBC) reveals lymphocytosis (7 x 109/L) with abnormal Sézary cell count and the flow cytometry immunophenotype of the peripheral blood revealed CD4+ lymphocytosis (5 x 109/L) with low CD8+ count (0.8 x 109/L) and expanded CD4+ CD26- (63%) and CD4+ CD7- (58%). According to clinical notes, the patient is a 67 year old man who presented to the department of dermatology with a 1 month history of generalized erythroderma (90% of skin involvement) and inguinal and axillary bilateral lymphadenopathy. In summary, patient presented with erythroderma and skin is involved by cerebriform T lymphocytes with minimal epidermotropism. Peripheral blood is involved morphologically by cerebriform lymphocytes that by flow cytometry express CD3, CD4 and lack CD7 and CD26. These features support the diagnosis of Sézary syndrome.
- Nonneoplastic erythroderma (pseudo E CTCL) (J Cutan Pathol 2006;33:27)
- Adult T cell leukemia / lymphoma (Semin Diagn Pathol 2020;37:81)
- Leukemia cutis (Int J Dermatol 2012;51:383)
- Secondary cutaneous involvement of peripheral T cell lymphoma (Am J Clin Pathol 2013;139:491)
- Exclusion diagnosis: always rule out other defined entities that involve skin secondarily
- Small size morphology and CD4+
- Usually do not present cerebriform cells
- Secondary skin involvement; unusual confined to cutaneous sites
Which of the following is true about Sézary syndrome (SS) shown above?
- Erythroderma is essential for the diagnosis
- Frequently presents as an indolent disease with favorable prognosis
- Lymph node involvement is uncommon
- Most common dermatologic lesion is plaque
- Absolute Sézary cell count ≥ 1 x 109/L
- Different T cell clones in skin, blood or lymph node
- Expanded CD8+ T cells with CD8:CD4 ≥ 10
- Loss of CD3 (T cell marker) expression
- A chronic T cell lymphoproliferative disorder characterized by a clonal proliferation of mature cytotoxic T cells without an identified cause
- Patients commonly present with cytopenia(s) and eventually require treatment
- Indolent clonal proliferation of mature cytotoxic T cells
- Neutropenia or anemia
- Associated with autoimmune disorders
- Commonly CD8+ T cells coexpressing NK cell associated markers
- Characteristic intrasinusoidal involvement in bone marrow, spleen and liver
- Approximately half of the patients carry a STAT3 / STAT5b mutation
- T cell large granular lymphocytosis; T cell lymphoproliferative disease of granular lymphocytes
- 2 - 5% of mature lymphocytic leukemias
- M = F
- Commonly occurs in elderly individuals with a median age of 60 years (Blood 2017;129:1082)
- Peripheral blood, bone marrow, spleen, liver
- Oligoclonal expansion of cytotoxic large granular T cells in response to antigen stimulation
- Subsequent clonal proliferation of large granular T cells due to constitutive upregulation of cellular survival signals or downregulation of apoptotic pathways via a secondary event
- STAT3 / STAT5b mutation as a secondary event (N Engl J Med 2012;366:1905, Blood 2012;120:3048)
- Other secondary events include resistance to Fas / FasL mediated cell death, increased cell survival by interleukin 15 and platelet derived growth factor and activation of NFkB pathway (Blood 2018;131:2803)
- Neutropenia and anemia are caused by direct cytotoxicity from the clonal T cells
- Chronic antigen exposure from autoimmune disorders such as rheumatoid arthritis and Sjögren syndrome
- Chronic antigen exposure due to viral infection such as human T lymphotropic virus
- Approximately 70% of patients have neutropenia or anemia
- Approximately 30% of patients are asymptomatic but regular complete blood count reveals lymphocytosis and cytopenia(s) (Blood 2017;129:1082)
- Splenomegaly in approximately 30% of patients
- Approximately 30% of patients carry autoimmune disorders with rheumatoid arthritis being by far the most common (Blood 2017;129:1082)
- Could be associated with other hematologic malignancies such as B cell lymphoma, myelodysplastic syndrome and aplastic anemia
- Persistent (more than 6 months) increase in the number of peripheral blood large granular lymphocytes, usually 2-20 x 10⁹
- Distinct T cell population with coexpression of one or more NK cell associated antigens (CD16, CD56 or CD57) and decreased CD2, CD5 or CD7 expression
- Proof of T cell clonality by a molecular or flow cytometry study
- Intrasinusoidal cytotoxic T cell infiltrates in bone marrow, spleen or liver (Blood 2002;99:268)
- STAT3 or STAT5b mutation (N Engl J Med 2012;366:1905, Blood 2012;120:3048)
- Variable lymphocytosis (Blood 2017;129:1082)
- Neutropenia occurs in approximately 85% of patients, anemia in 50% with pure red cell aplasia in 10% and thrombocytopenia in < 25%
- Rheumatoid factor and antinuclear antibodies could be positive
- Splenomegaly in 30% of patients (Blood. 2017;129:1082)
- Hepatomegaly < 30% of patients
- Rarely lymphadenopathy
- Overall an indolent disease
- Deaths infrequently occur because of infections related to severe neutropenia
- STAT3 / STAT5b mutations may adversely impact overall survival (Leukemia 2019 Nov 18 [Epub ahead of print], Blood 2013;121:4541)
- 16 year old boy developed donor derived T cell large granular lymphocytic leukemia after hematopoietic stem cell transplant (J Natl Compr Canc Netw 2016;14:939)
- 54 year old man with giant intracytoplasmic inclusion in T cell large granular lymphocytic leukemia cells (Blood 2019;134:492)
- 55 year old woman suffered from panuveitis caused by T cell large granular lymphocytic leukemia (Ocul Immunol Inflamm 2019:1)
- 57 year old woman with immunoglobulin heavy chain amyloidosis (Am J Case Rep 2019;20:43)
- 65 year old man developed late graft rejection by recipient origin T cell large granular lymphocytic leukemia (Transplant Proc 2016;48:3222)
- The majority of patients eventually require treatment because of severe or symptomatic neutropenia or anemia
- No standard therapy established
- Immunosuppressive therapy (methotrexate, cyclophosphamide or cyclosporine) as initial agents
- Bone marrow biopsy does not show apparent abnormal lymphocytic infiltrates by hematoxylin and eosin stain; immunohistochemical studies reveal the characteristic linear / intrasinusoidal distribution of cytotoxic T cells (Blood 2002;99:268)
- Spleen biopsy demonstrates linear arrays of cytotoxic T cells in the sinusoids and red pulp cords
- Liver biopsy reveals linear arrays of cytotoxic T cells in the sinusoids
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
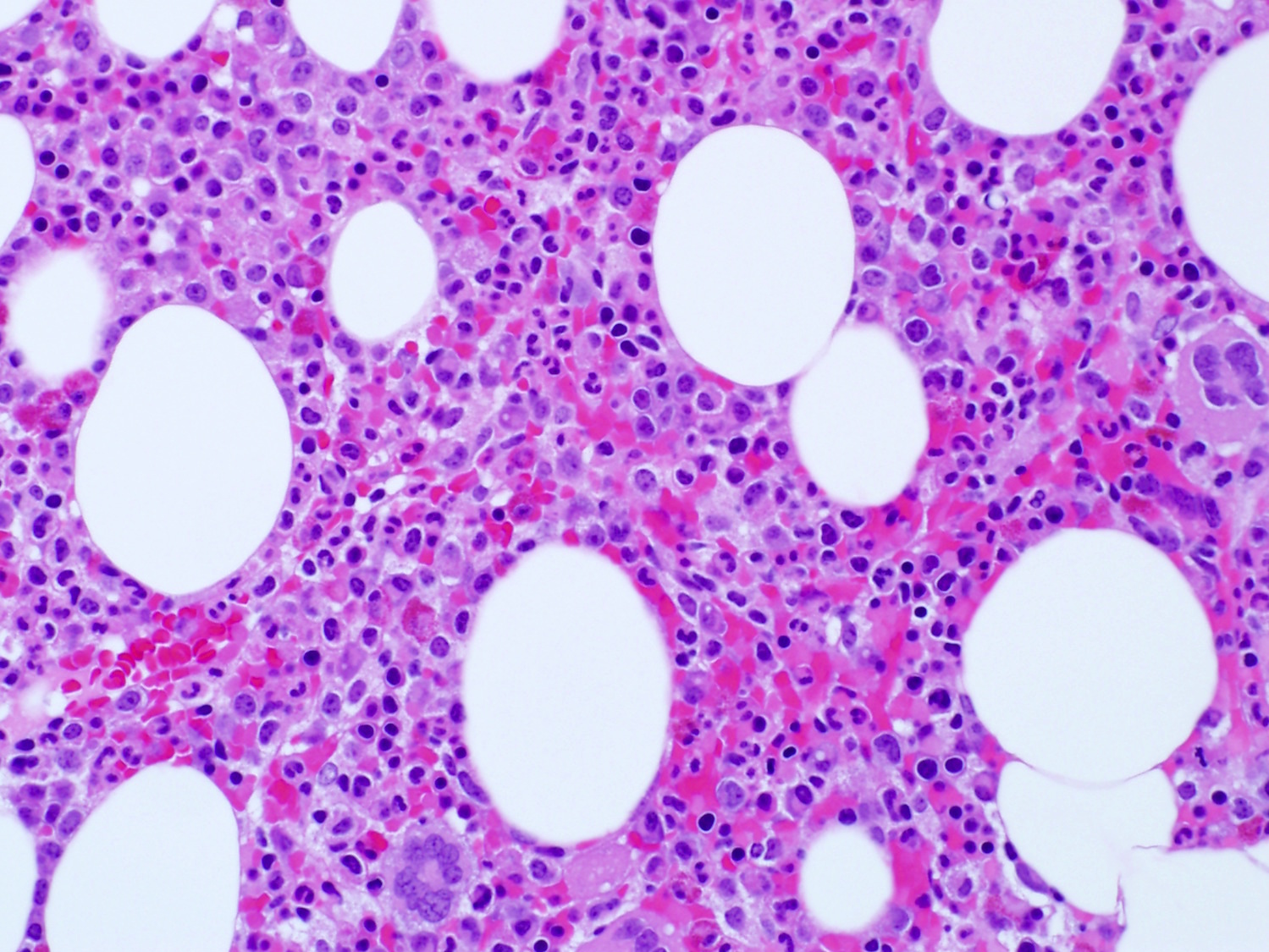
T LGLL bone marrow involvement

Intrasinusoidal CD3+ T LGLL
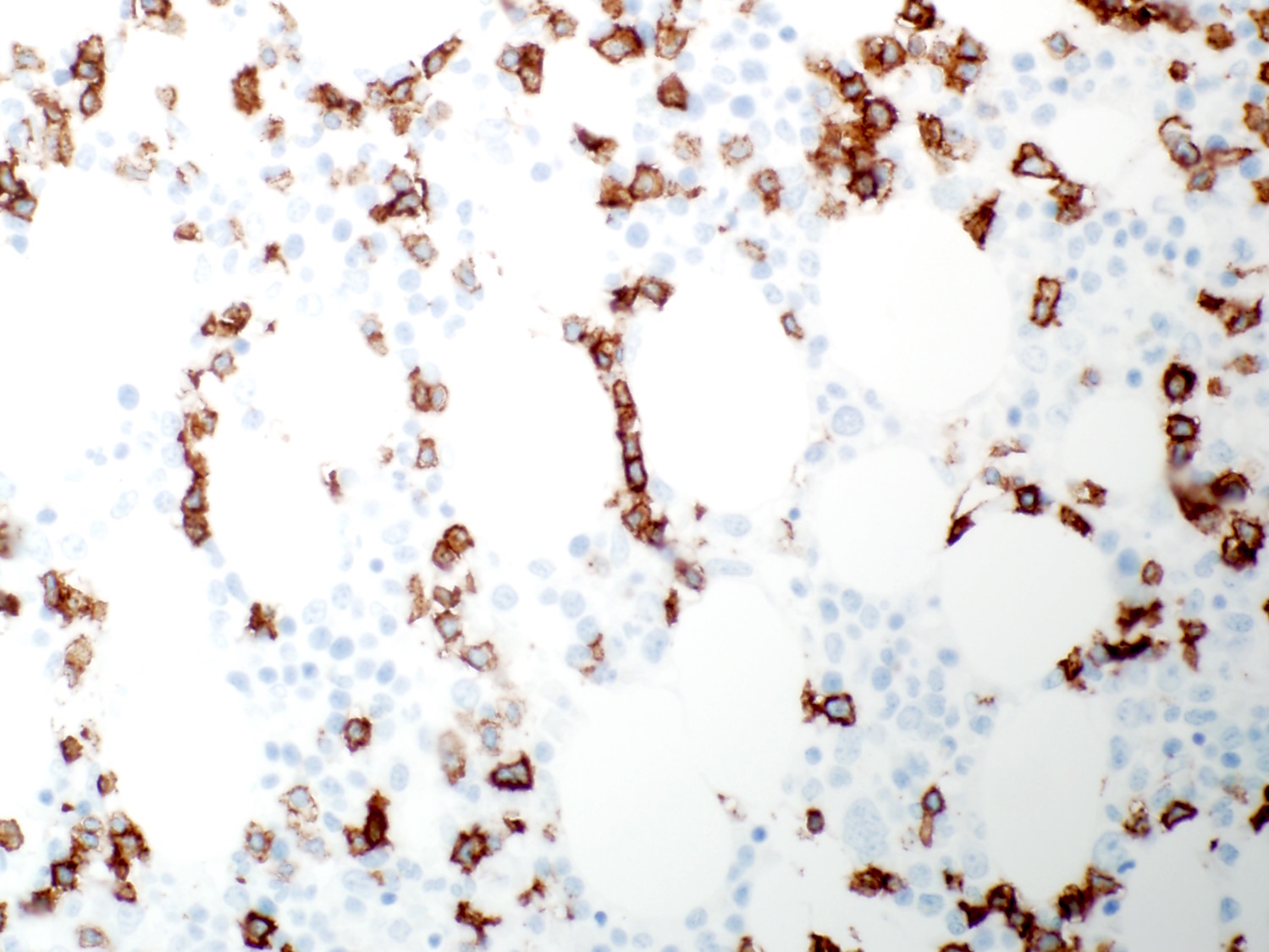
Intrasinusoidal CD8+ T LGLL
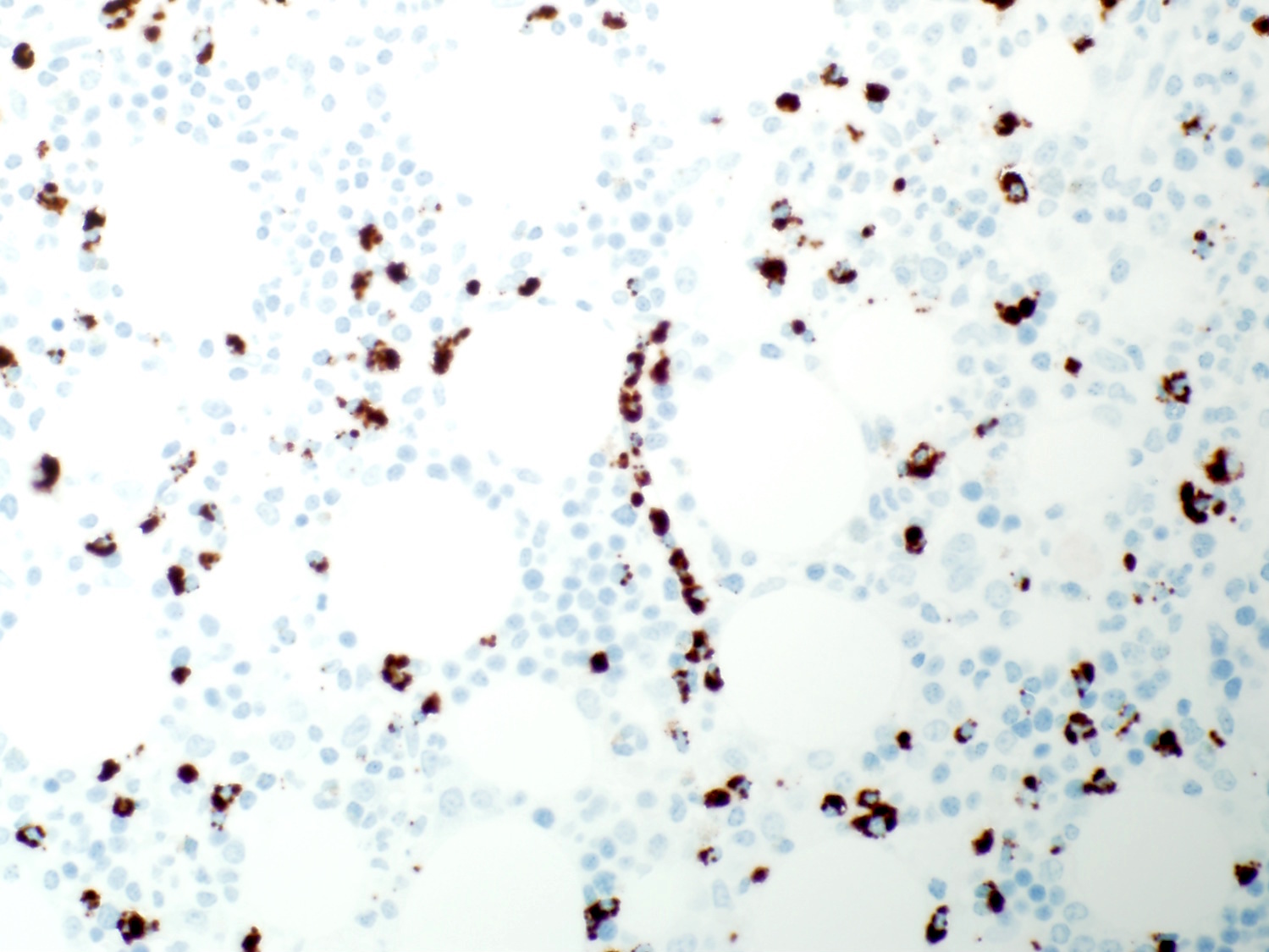
Intrasinusoidal granzyme B+ T LGLL

Intrasinusoidal TIA1+ T LGLL
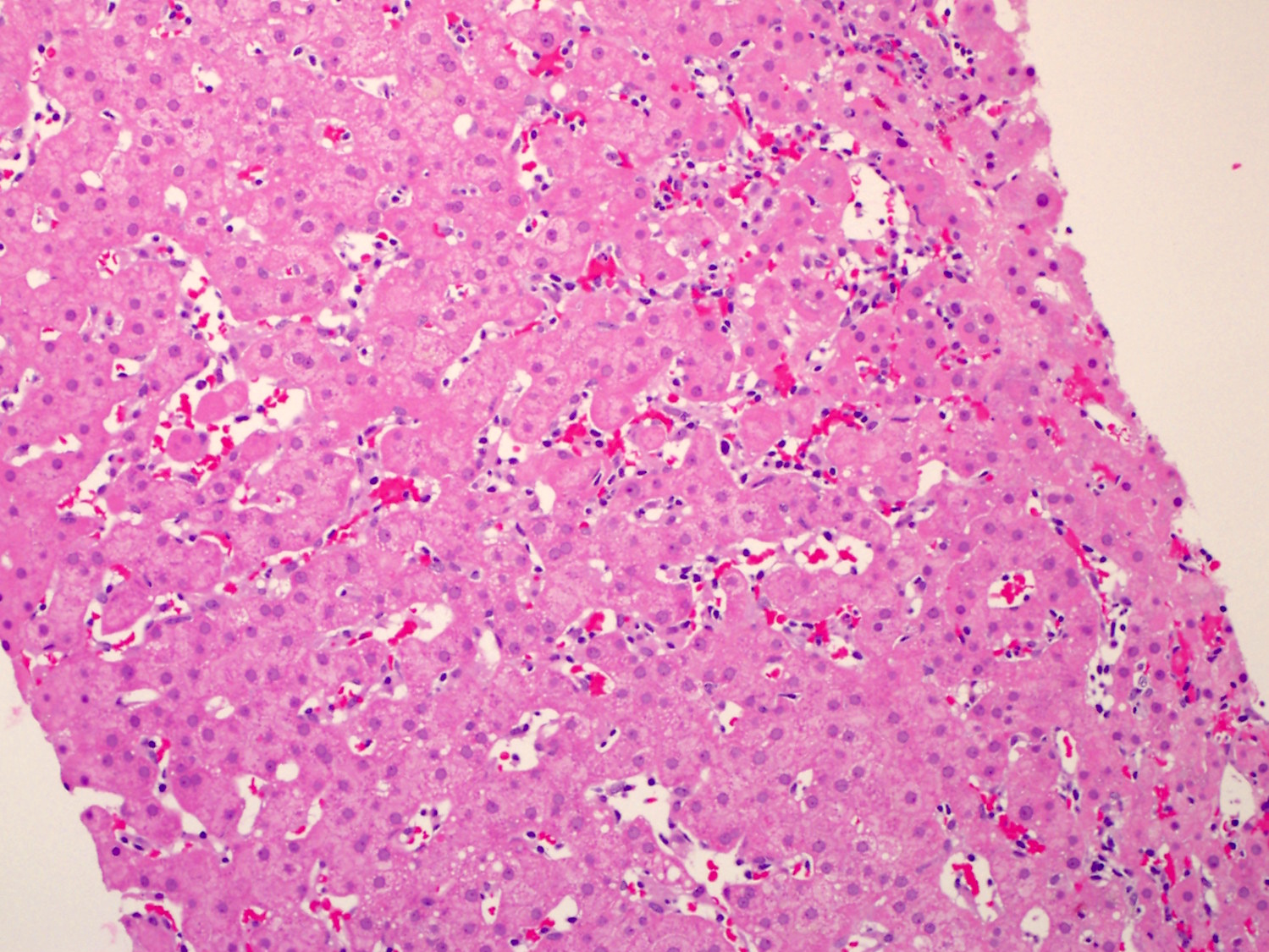
T LGLL liver involvement
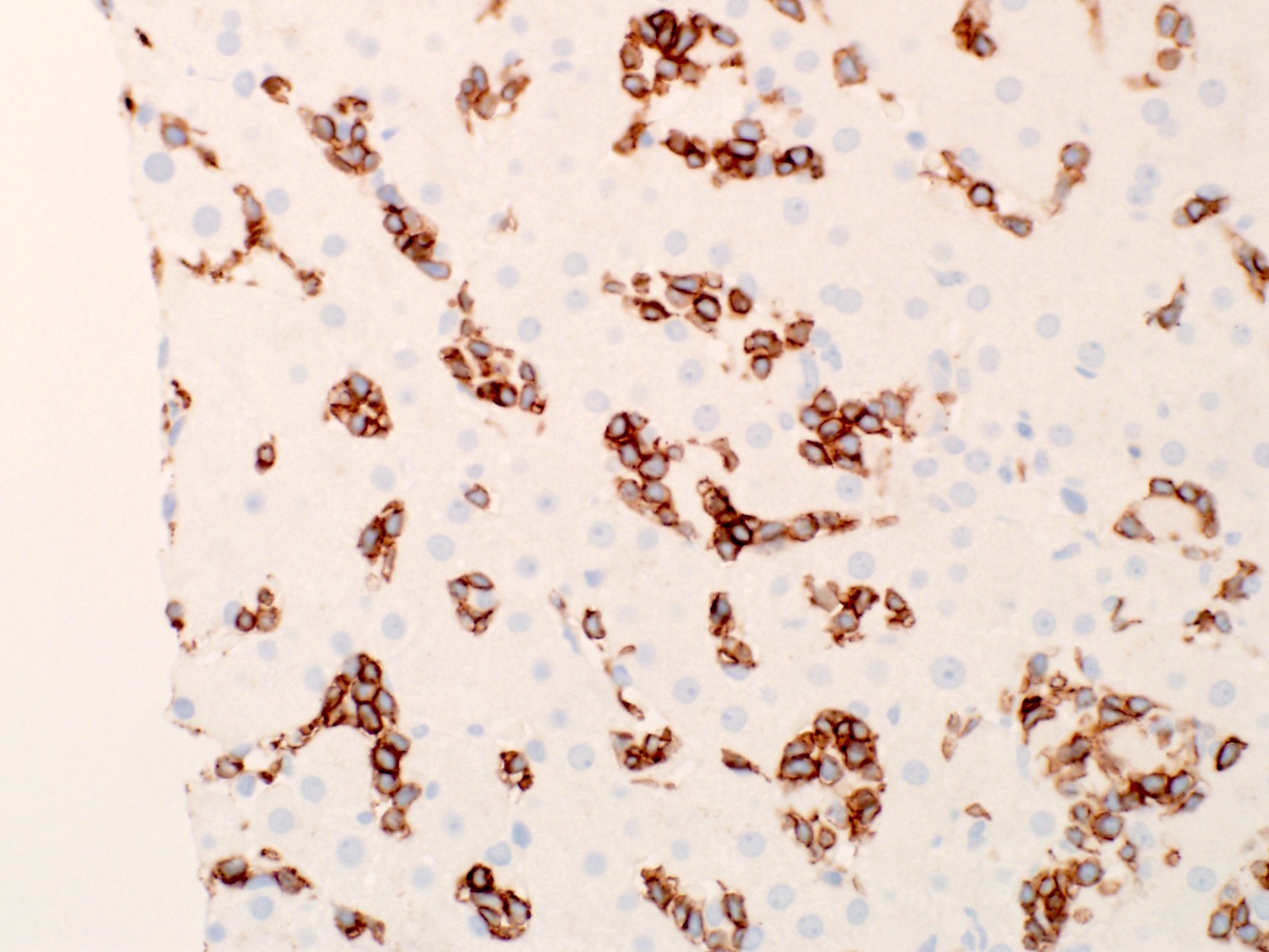
CD8+ T LGLL in liver

CD5- T LGLL in liver
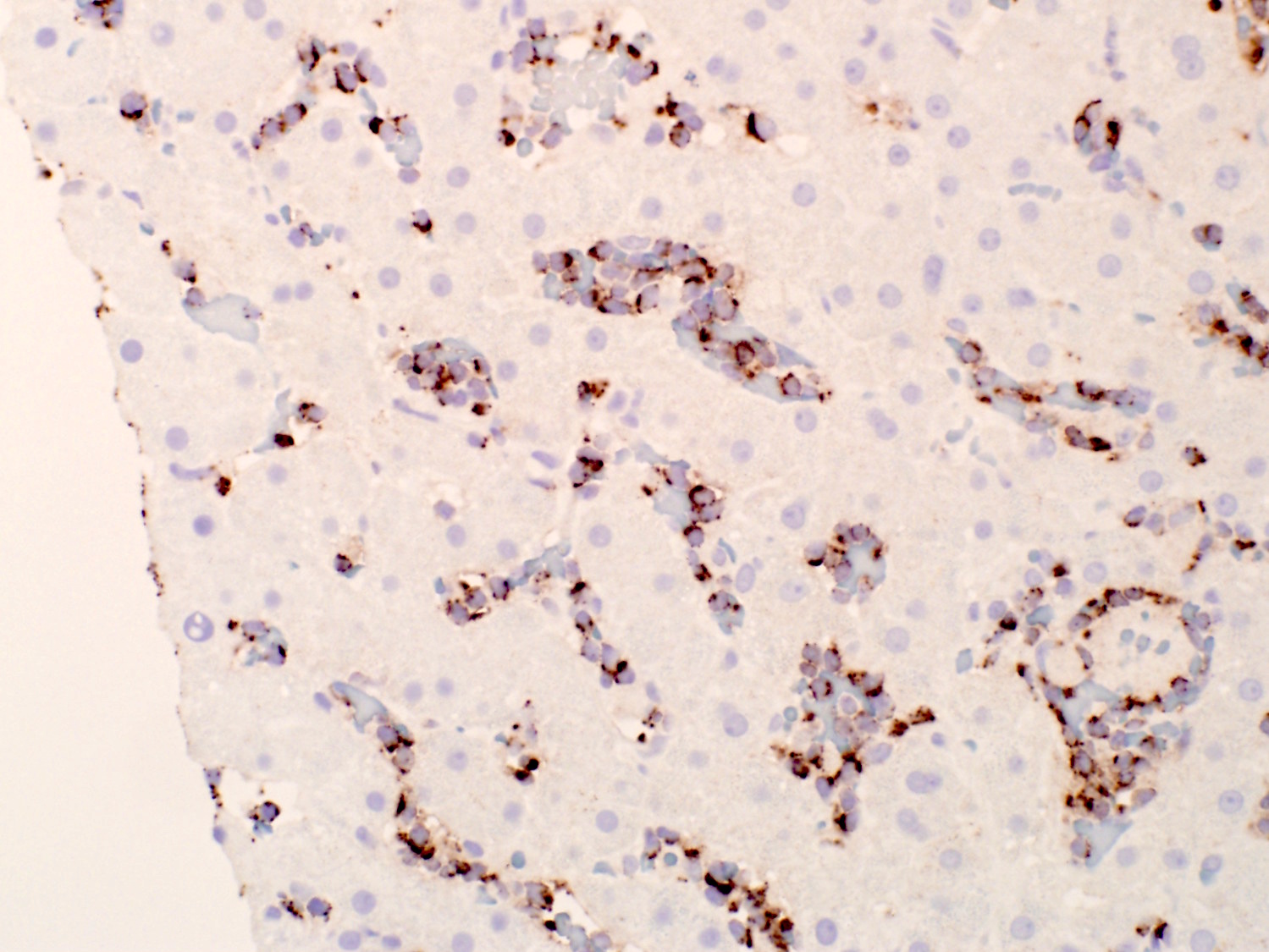
TIA1+ T LGLL in liver
- Increased lymphocytes containing small to intermediate sized reticulated nuclei and fine to coarse azurophilic cytoplasmic granules
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
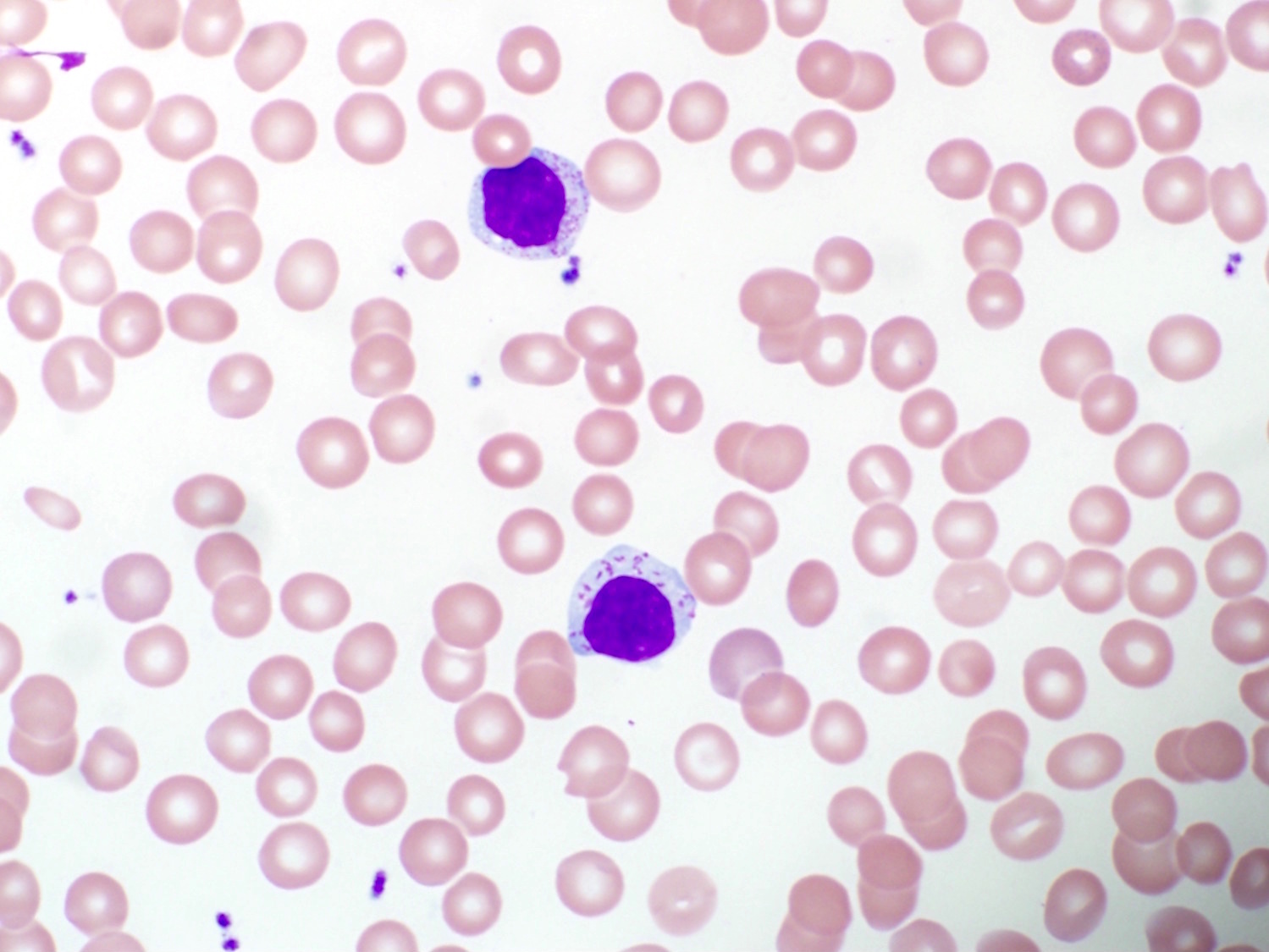
T LGLL in peripheral blood
- Mature CD3 positive T cells, usually coexpress NK cell associated markers (CD16 and CD57), with variable expression of other pan T cell markers such as CD2, CD5, CD7; 25% of cases are KIR restricted
- CD4- CD8+ T cell type being the most common, followed by TCRγδ+ T cell type, CD4+ CD8- T cell type, TCRαβ+ CD4- CD8- T cell type and rarely mixed phenotype (Hum Pathol 2018;81:96)
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
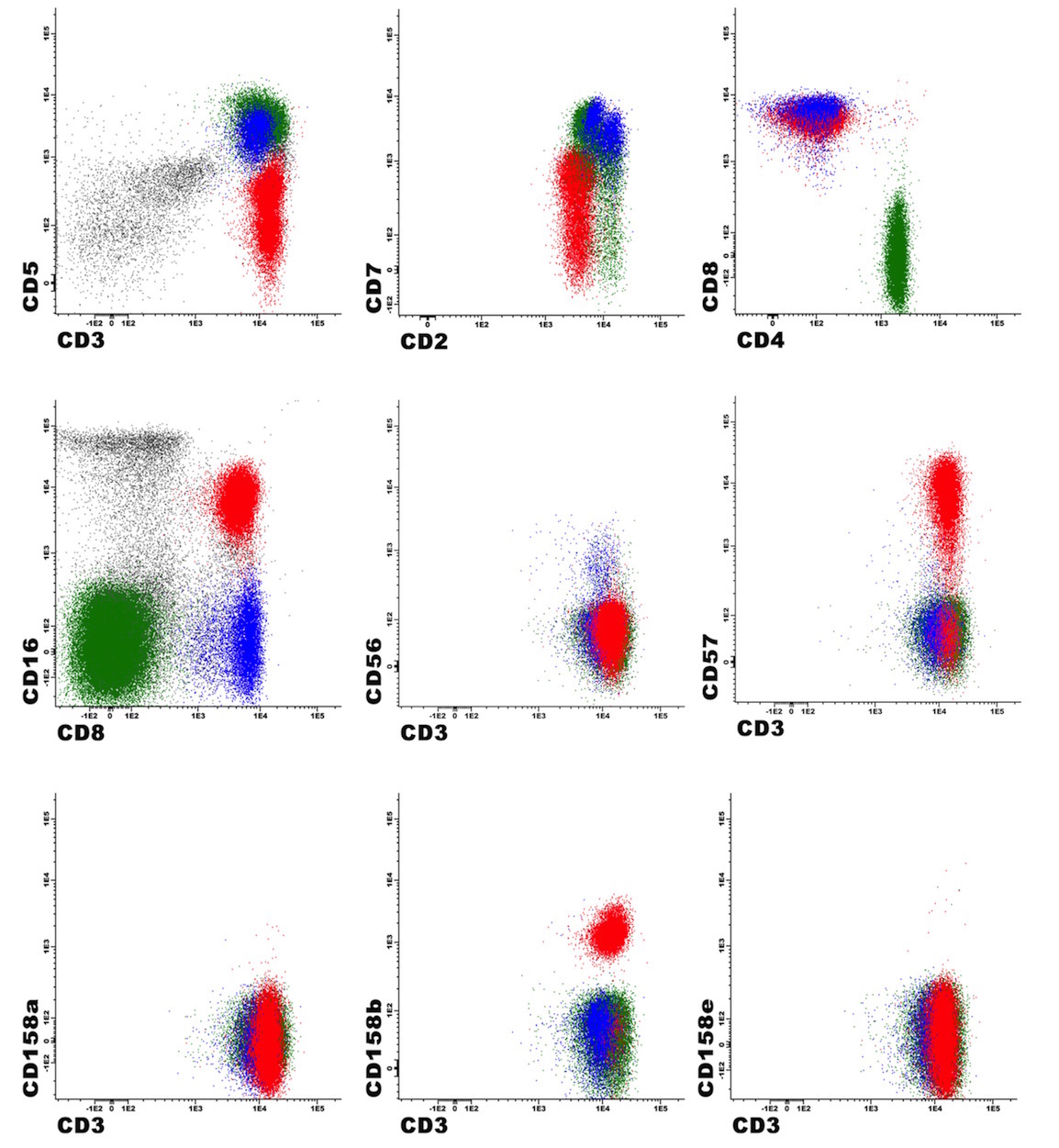
T LGLL flow cytometry
- Clonal T cell receptor (TCR) gene rearrangements
- STAT3 mutation in 40 - 50% of T cell large granular lymphocytic leukemia (N Engl J Med 2012;366:1905, Blood 2012;120:3048)
- STAT5b mutation in 2% of T cell large granular lymphocytic leukemia (Blood 2013;121:4541)
- No specific cytogenetic abnormalities
- Peripheral blood, bone marrow aspirate and biopsy, iliac crest:
- T cell large granular lymphocytic leukemia involving 15% marrow cellularity with normal trilineage hematopoiesis
- Peripheral blood smear: neutropenia; absolute lymphocytosis with increased large granular cells
- Bone marrow aspirate and biopsy: adequate quality; normal M:E ratio; slightly hypercellular marrow with left shift granulopoiesis, otherwise normal hematopoiesis; slight interstitial lymphocytic infiltrates of small lymphocytes
- Flow cytometric immunophenotyping: distinct T cell population (70% of gated lymphoid events, 30% of total analyzed events) with expression of CD2, CD3, CD5 (dim), CD7 (dim), CD8, CD16, CD57, CD94 (partial), CD158b (restricted); they do not express CD4, CD56, CD158a, CD158e, NKG2A and TCR gamma / delta
- Molecular study for TCR gene rearrangement: positive for clonal TCR gene rearrangement
- Reactive T cell expansion:
- Clinical presentation (viral infection or an autoimmune disorder)
- Polyclonal
- No linear arrays of cytotoxic T cells in the bone marrow
- Chronic lymphoproliferative disorder of NK cells:
- Peripheral T cell lymphoma:
- Aggressive clinical presentation (B symptoms, lymphadenopathy)
- Neoplastic cells have very irregular nuclear contours
- Loss of pan T cell markers
- Hepatosplenic T cell lymphoma:
- Aggressive clinical presentation
- TCRγδ+, TIA1+, perforin-, granzyme B-
- Isochromosome 7q
- CD2+ CD3- CD4- CD5- CD7+ CD8+ CD16+ CD56- CD57+
- CD2+ CD3+ CD4- CD5- CD7+ CD8+ CD16+ CD56- CD57+
- CD2+ CD3+ CD4+ CD5- CD7+ CD8+ CD16+ CD56- CD57+
- CD2- CD3+ CD4- CD5- CD7+ CD8+ CD16- CD56- CD57-
- CD2+ CD3- CD4- CD5- CD7+ CD8- CD16- CD56+ CD57-
Comment Here
Reference: T cell large granular lymphocytic leukemia
- Which genetic abnormality is most commonly identified in T cell large granular lymphocytic leukemia?
- STAT5b mutation
- NOTCH mutation
- TP53 mutation
- STAT3 mutation
- TCL1A rearrangement
Comment Here
Reference: T cell large granular lymphocytic leukemia
- T prolymphocytic leukemia (T PLL) is an aggressive, mature T cell leukemia, composed of small to medium sized mature T cells, usually with high white blood cell (WBC) count and widespread organ involvement
- No significant changes in WHO 5 and ICC classifications (Leukemia 2022;36:1720, Blood 2022;140:1229)
- Aggressive leukemia of mature T cells
- High white blood cell count (lymphocytosis)
- Usually CD4+
- Usually TCL1 positive by immunophenotyping and TCL1 rearranged by FISH
- Difficult to treat, poor prognosis
- T cell chronic lymphocytic leukemia (small cell variant of T cell prolymphocytic leukemia)
- Rare (2% of mature lymphocytic leukemias)
- Adults and elderly (> 30 years, median age: 65 years)
- Widespread: peripheral blood, bone marrow, lymph nodes, spleen, liver, skin
- Combination of overexpression of TCL1 family of proteins (which stimulate AKT / protein kinase B driven proliferation) and functional deficit of ATM protein (Nat Commun 2018;9:697)
- Unknown at this time
- Higher risk in patients with ataxia-telangiectasia (germline ATM mutations)
- High tumor burden with high white blood cell count (median 50 - 60 x 109/L), bone marrow involvement (in 100% of patients) with cytopenias, hepatosplenomegaly, lymphadenopathy, skin and mucosal lesions, other organ involvement / dysfunction (Am J Hematol 2017;92:441)
- Prominent constitutional symptoms
- 20 - 30% present with inactive disease and progress to active disease within 1 - 2 years (criteria for progression: lymphocyte doubling time (LDT) of less than 6 months or lymphocyte count increase by > 50% in 2 months) (Blood 2019;134:1132)
- Peripheral blood: morphology + flow cytometry with or without fluorescent in situ hybridization (FISH)
- Bone marrow or solid tissue biopsy (lymph node, spleen, liver, skin, other): morphology + phenotyping (immunohistochemistry or flow cytometry) with or without FISH
- Note: FISH not necessary if TCL1 overexpression in neoplastic T cells can be shown by IHC or flow (J Clin Pathol 2018;71:309)
- Consensus criteria for diagnosis by the T PLL International Study Group (Blood 2019;134:1132)
- All 3 major criteria or the first 2 major and 1 minor criteria are required for diagnosis
- Major criteria:
- 5 x 109/L cells of T PLL phenotype in peripheral blood or bone marrow
- T cell clonality by molecular or flow cytometry methods
- Abnormalities of 14q32 or Xq28 or expression of TCL1A/B or MTCP1
- Minor criteria:
- Abnormalities involving chromosome 11 (11q22.3; ATM)
- Abnormalities in chromosome 8: idic(8)(p11), t(8;8), trisomy 8q
- Abnormalities in chromosome 5, 12, 13, 22 or complex karyotype
- Involvement of specific sites (spleen, effusions)
- Increased peripheral blood lymphocytes, often > 100 x 109/L, increased lactate dehydrogenase (LDH) and beta2 microglobulin (Ann Oncol 2017;28:1554)
- Negative serology for HTLV1
- Prominent hepatosplenomegaly and widespread lymphadenopathy; moderate to high FEV on PET scan
- Overall poor prognosis; median survival with active disease is 1 - 2 years
- Worse prognosis: pleural effusion, high LDH (> 1668 IU/l), low hemoglobin (< 9.3 g/dl), complex karyotype (Ann Oncol 2017;28:1554, Am J Hematol 2017;92:441)
- 59 year old man with the history of renal transplant (J Med Case Rep 2019;13:223)
- 60 year old woman with a history of chronic myeloid leukemia (CML) (J Clin Pathol 2019;72:511)
- 64 year old man with TCL1 positive hematogones after T cell prolymphocytic leukemia therapy (Hum Pathol 2017;65:175)
- 68 year old man with heart failure (Blood 2017;130:691)
- 75 year old man with unusual phenotype of T cell prolymphocytic leukemia (Blood 2018;132:111)
- Only for active disease
- Standard treatment: alemtuzumab (anti-CD52) variable allogeneic bone marrow transplant
- Experimental therapies with BCL2, JAK3 or HDAC inhibitors (Blood 2019;134:1132)
- Perivascular and diffuse tissue infiltrates of uniform small to medium sized lymphocytes
- Red pulp involvement in the spleen
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
Diffuse bone marrow involvement
Splenic red pulp infiltrate
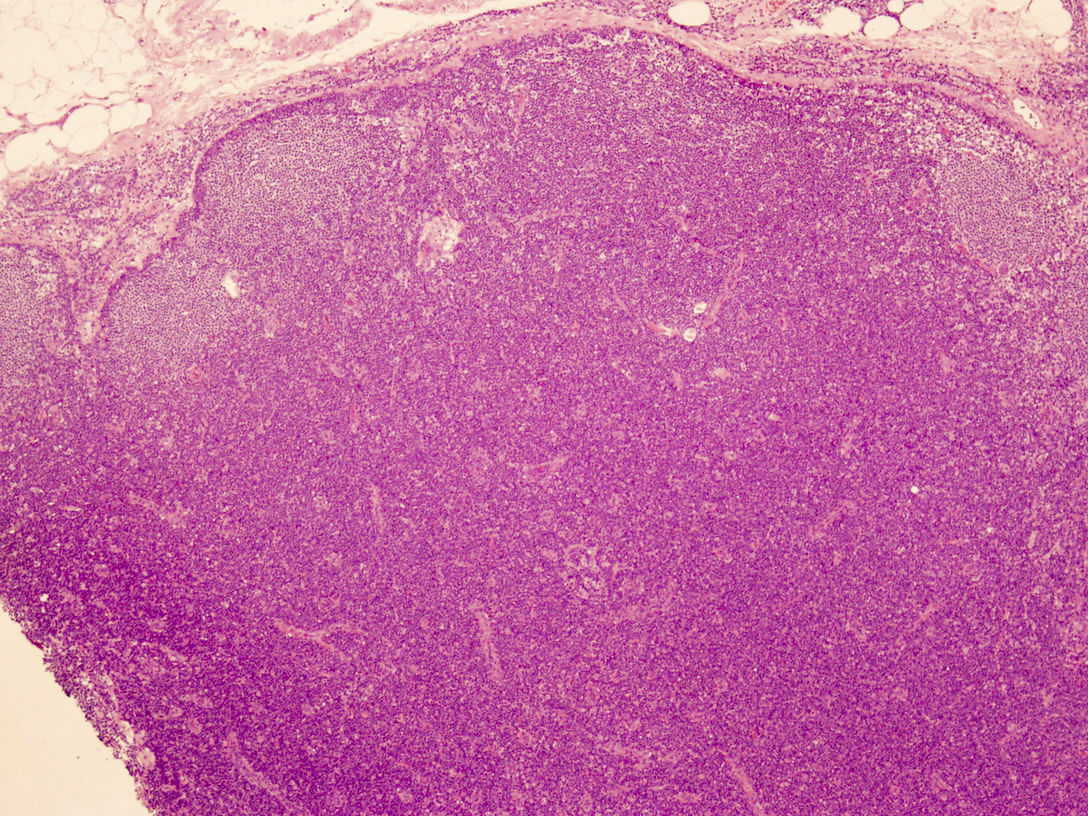
Monotonous, paracortical lymphoid infiltrate
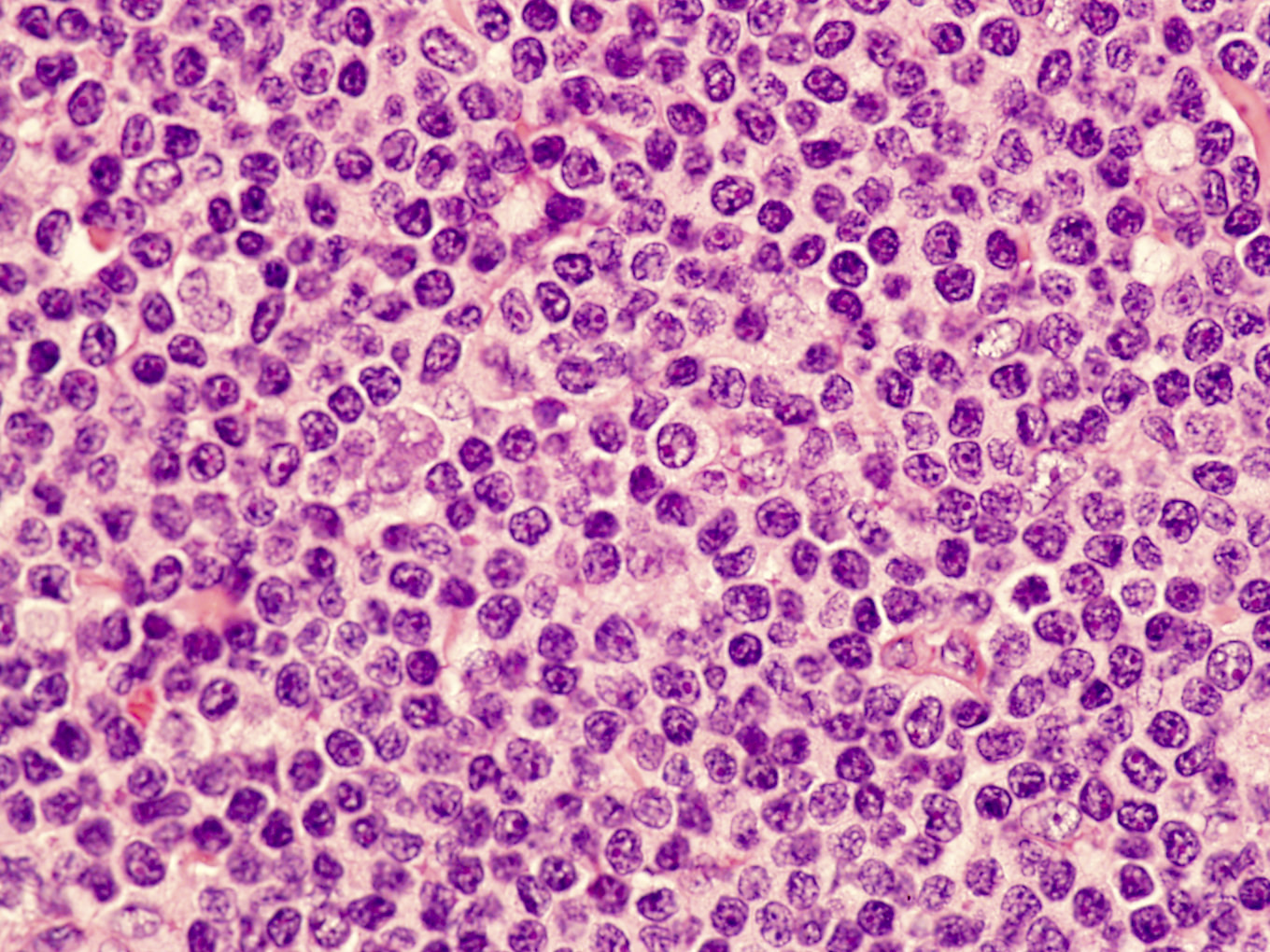
Intermediate size, atypical lymphocyte
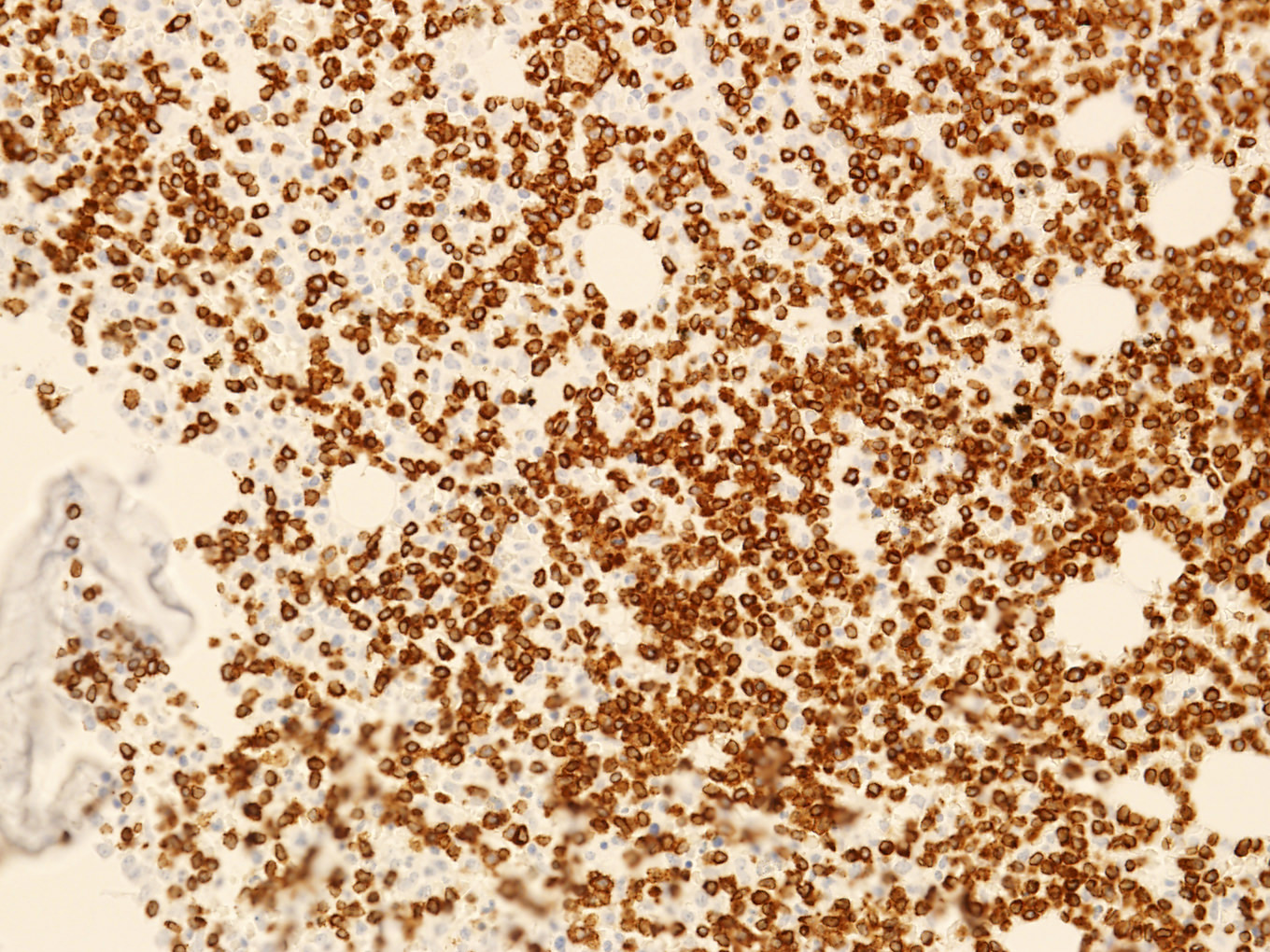
CD3 positivity in bone marrow
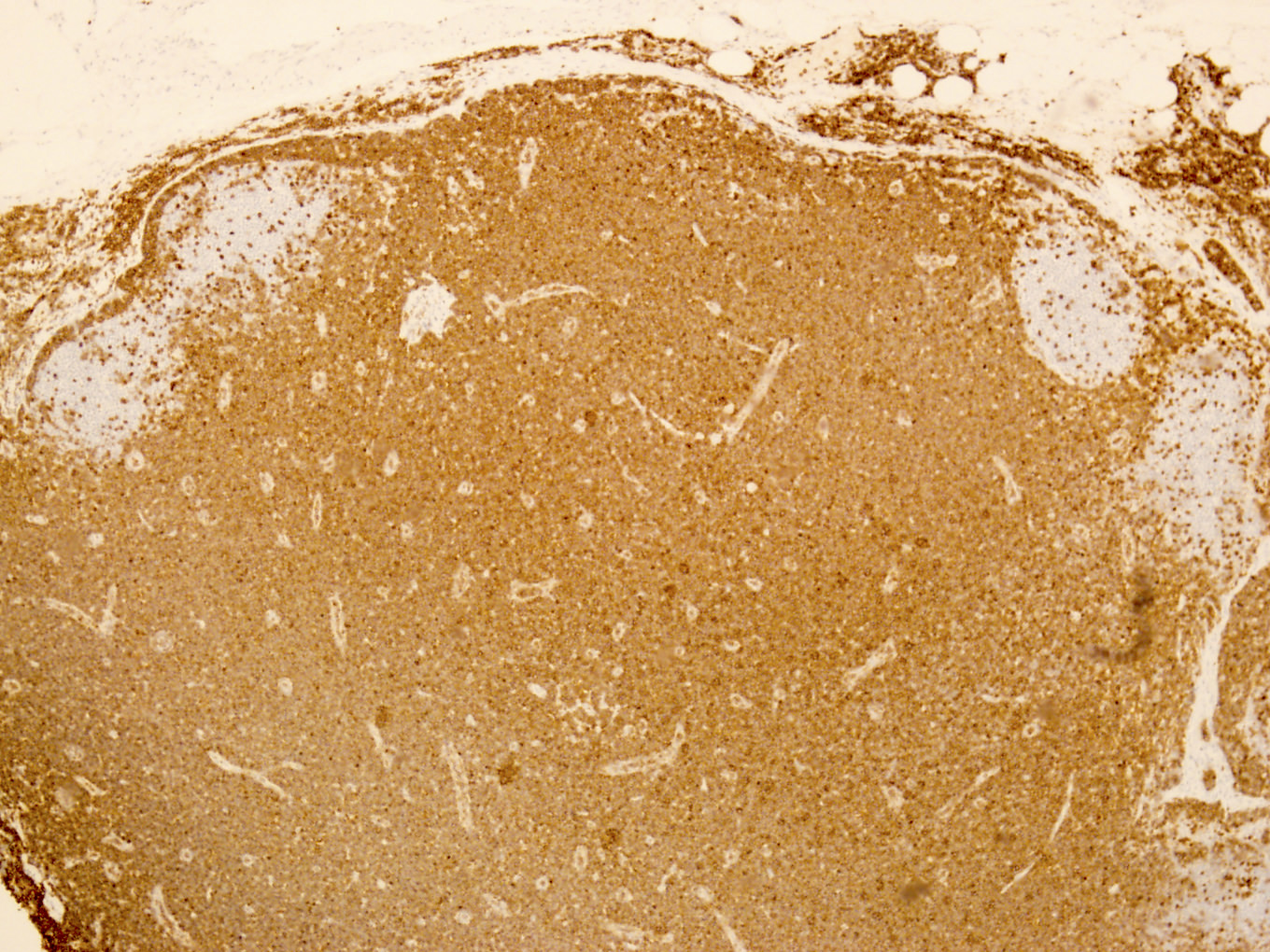
Monotonous T cell infiltrate
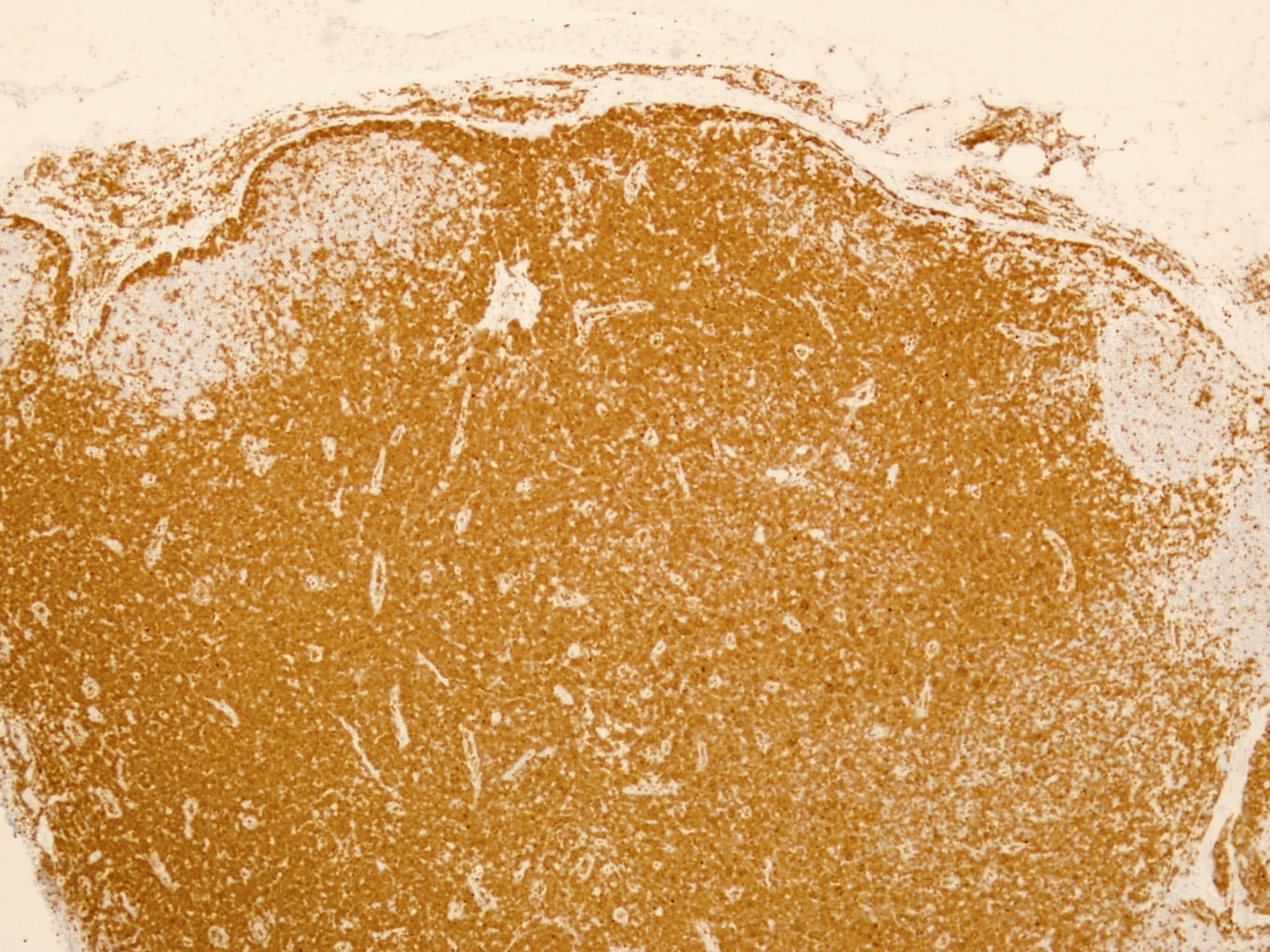
TCL1A positivity
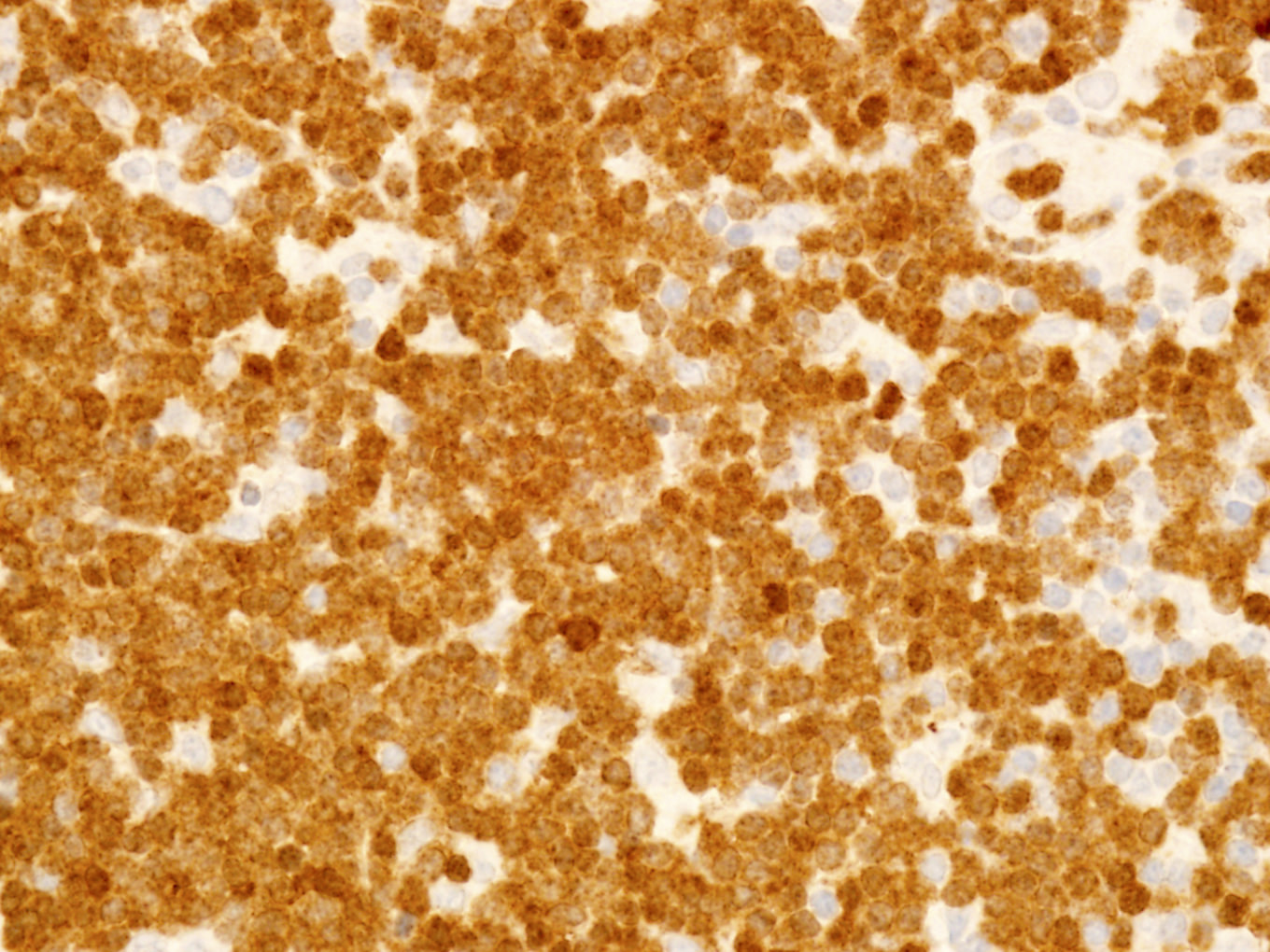
Uniform strong TCL1A staining
- Lymphocytosis with small to medium sized lymphocytes with cytoplasmic blebs, clumped chromatin and variably prominent central nucleolus
- Small cell variant in 25% of cases: smaller cells without obvious nucleolus
- Cerebriform variant in 5% of cases
- Reference: Blood 2019;134:1132
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
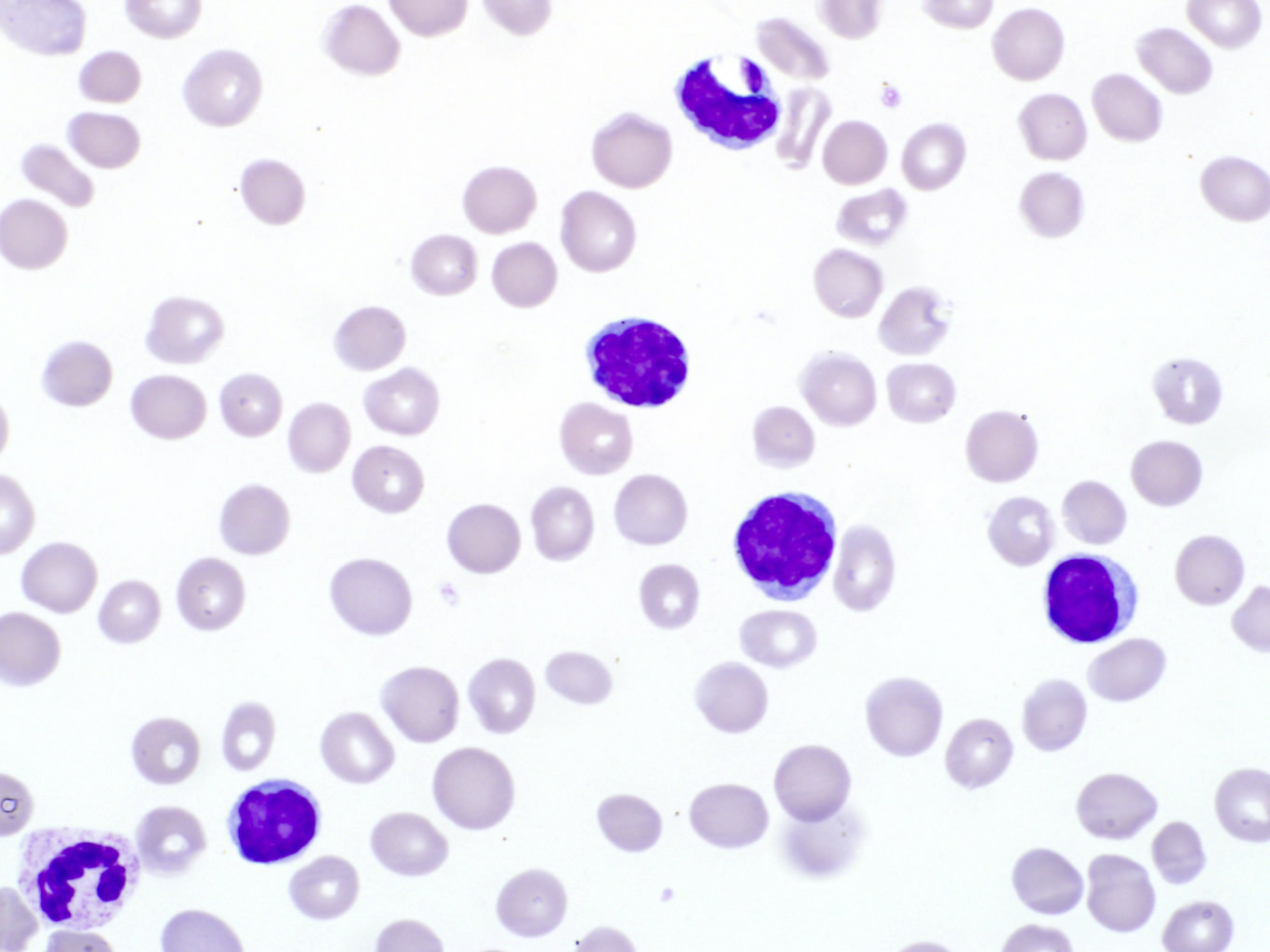
Cerebriform variant
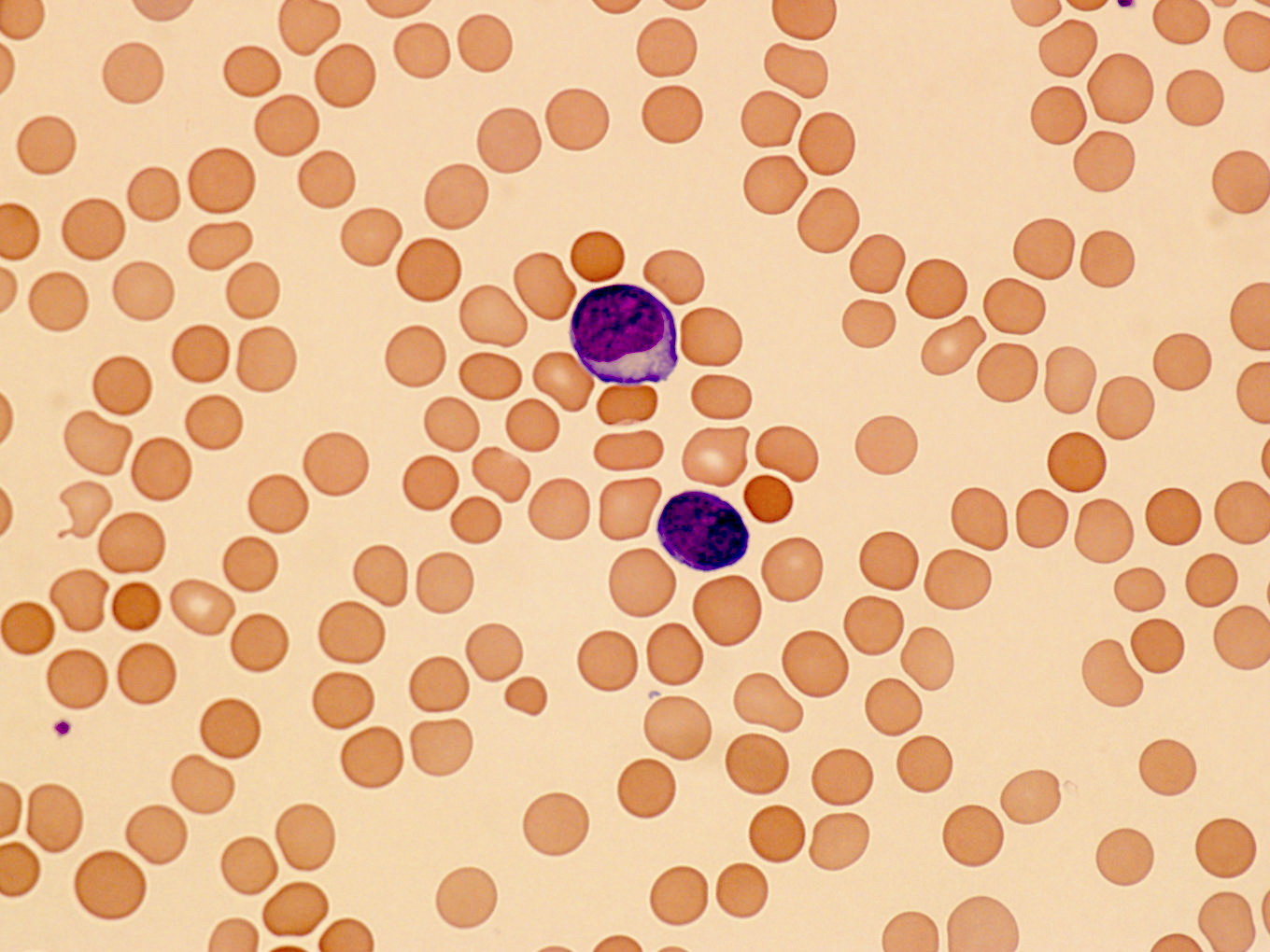
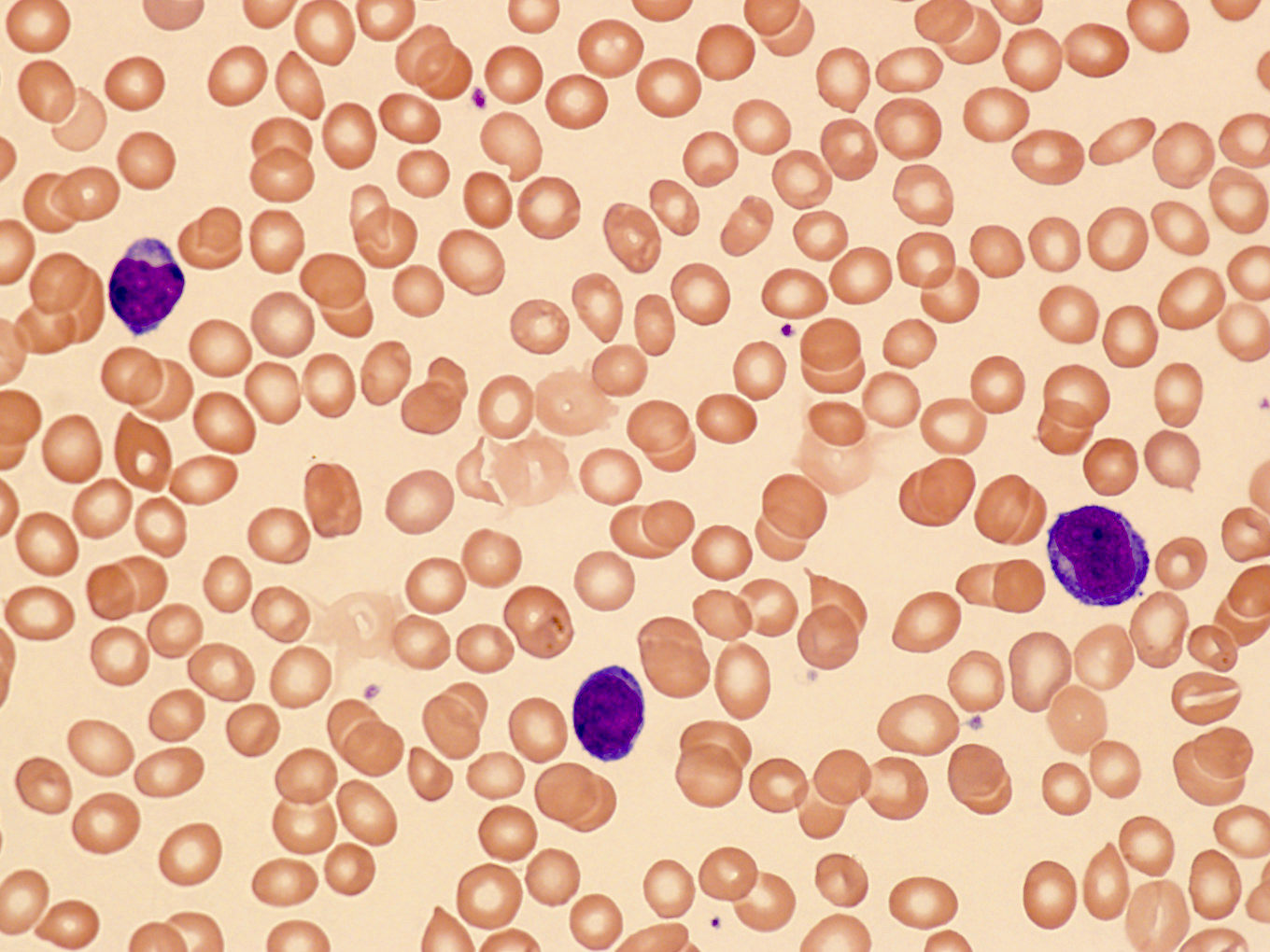
Prolymphocytes in peripheral blood
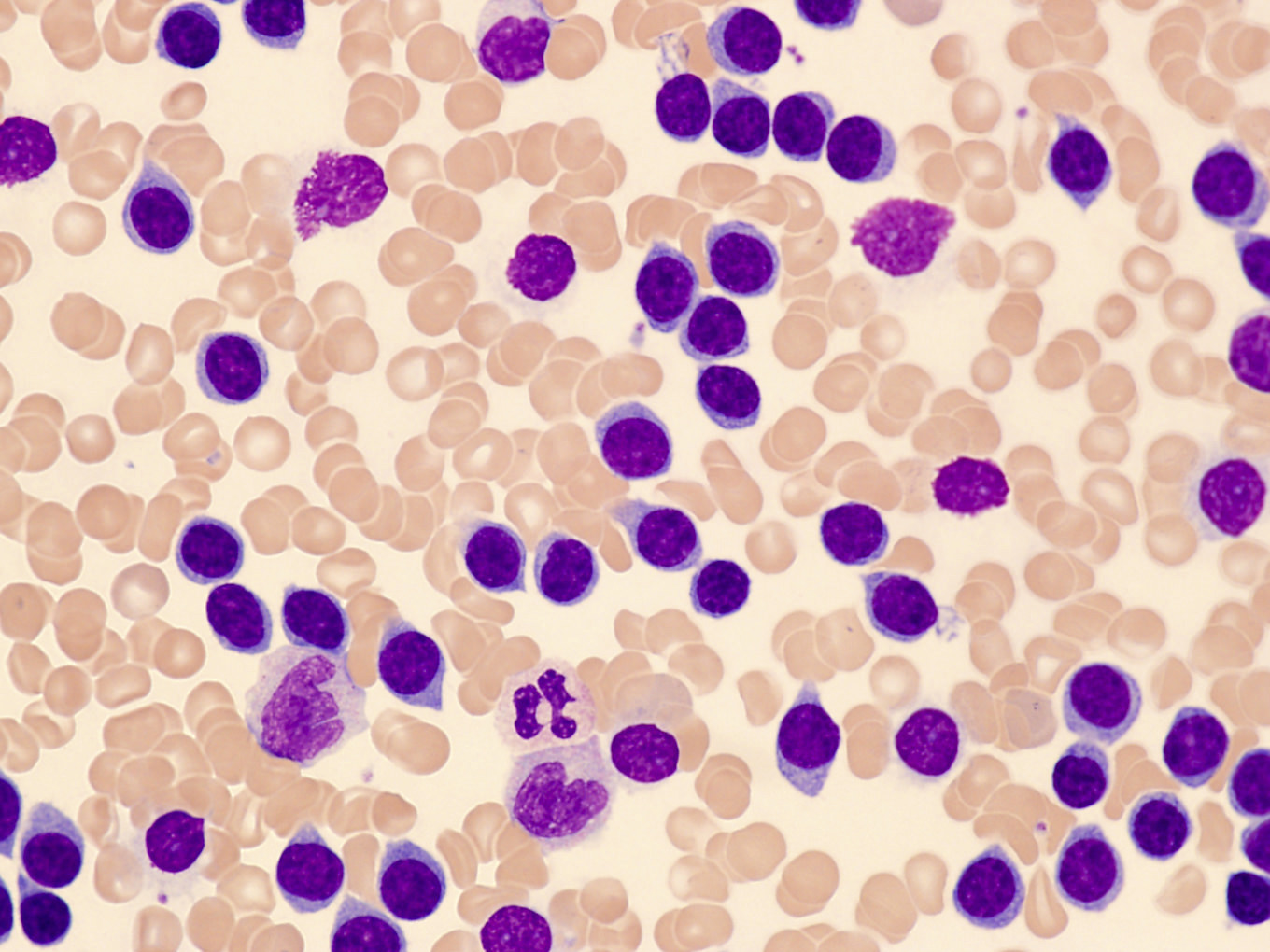
Small cell variant
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.

T PLL flow cytometry
- Clonal T cell receptor gene rearrangements (TRB@ and TRG@)
- FISH is commonly used for diagnosis; T cell receptor locus rearranged with the TCL1 family of genes:
- TCL1A / TCL1B rearrangement: inv(14)(q11;q32) in 80%, t(14;14)(q11;q32) in 10%
- Rarely MTCP1 rearrangement t(X;14)(q28;q11)
- Rarely negative for TCL1A / TCL1B or MTCP1 rearrangements (Blood 2019;134:1132)
- Complex karyotype in 70 - 80%; abnormalities of chromosomes 6, 8, 12p, 17p
- Mutations in ATM gene on 11q23 in 80 - 90%
- Other mutations / alterations: TP53, JAK / STAT pathway genes IL2RG, JAK1, JAK3, STAT5B (Blood 2014;124:1460)
Contributed by Min Shi, M.D., Ph.D. and Dragan Jevremovic, M.D., Ph.D.
FISH for TCL1A separation
- Peripheral blood, bone marrow aspirate and biopsy, iliac crest:
- T cell prolymphocytic leukemia, extensively involving peripheral blood and bone marrow, with decreased trilineage hematopoiesis.
- Peripheral blood:
- Complete blood count: hemoglobin 11.2 g/dL; red blood cell 3.55 x 1012/L; mean corpuscular volume 97.2 fL; red blood cell distribution width 15.2%; white blood cell 470.0 x 109/L; PLT 111 x 109/L
- Cell percentage of total cells: neutrophils (4%), lymphocytes (95%), monocytes (1%)
- Peripheral smear: lymphocytosis; small to intermediate lymphocytes with mature chromatin, prominent nucleoli, eccentric nuclei and moderate amounts of basophilic cytoplasm
- Bone marrow aspirate and biopsy:
- Aspirate quality: cellular
- Biopsy quality: adequate
- Myeloid to erythroid (M:E) ratio: normal, 3:1
- Cellularity: hypercellular, 90%
- Erythroid precursors: markedly decreased quantity, normal morphology
- Myeloid precursors: markedly decreased quantity, normal morphology, blasts not increased
- Megakaryocytes: markedly decreased quantity, normal morphology and distribution
- Lymphocytes: abnormal (diffuse) infiltrates of small to intermediate sized cells present (90% of the total marrow cellularity)
- Plasma cells: not increased
- Ancillary studies:
- Flow cytometry, bone marrow:
- Blasts: not increased by CD45 / side scatter and CD34
- B cells: no monotypic; normal expression pattern of CD19, CD10, surface kappa and lambda
- T cells / NK cells: distinct T cell population
- Express: CD4, CD2, CD3, CD5, CD7
- Do not express: CD8, CD16, TCR gamma / delta, CD1a, nTdT, cMPO, cCD79a, cCD22
- Estimated size: 95% gated lymphoid events; 87% total analyzed events
- T cell lymphoma FISH, bone marrow: the result is abnormal and indicates 88.5% of nuclei had a rearrangement involving TCL1A; this observation may indicate a clone with inv(14) or t(14;14), which are common rearrangements in T cell prolymphocytic leukemia
- Flow cytometry, bone marrow:
- CD3-, CD19+, CD20+, CD5+, CD23+
- CD3+, CD4+, CD5+, CD7-, CD8-, CD26-
- CD3-, CD4+, CD8+, TdT+
- CD3+, CD4+, CD5+, CD7+, CD8-
- CD3+, CD4-, CD7-, CD8+, CD16+
Comment Here
Reference: T cell prolymphocytic leukemia
Which genetic abnormality is used to define T cell prolymphocytic leukemia (T PLL)?
- 17p (TP53) deletion
- CCND1 rearrangement
- MYC rearrangement
- TCL1 rearrangement
- TP53 mutations
Comment Here
Reference: T cell prolymphocytic leukemia
- Vitamin B12 deficiency, also known as hypocobalaminemia, refers to low blood levels of vitamin B12
- 6% of those under age 60 and 20% over age 60
- Rates may be as high as 80% in parts of Africa and Asia
- Total amount of vitamin B12 stored in the body is between 2 - 5 mg in adults
- Of this, 50% is stored in the liver, 0.1% is lost each day as secretions into the gut
- Most of the vitamin B12 excreted into the bile is recycled via enterohepatic circulation
- Because of this mechanism, the liver can store three to five years’ worth of vitamin B12 under normal conditions and functioning
- Deficiency of Vitamin B12 affects the metabolic pathway in 2 critical points:
- Homocysteine to methionine, catalyzed by methionine synthase needs cyanocobalamin as a cofactor; resulting hyperhomocysteinemia may lead to a prothrombotic state
- The end product methionine aids in purine and thymidine synthesis, myelin production, protein / neurotransmitters / fatty acid / phospholipid production and DNA methylation, which becomes deficient in these individuals
- Methylmalonyl-CoA to succinyl-CoA also requires Vitamin B12 as a cofactor
- Homocysteine to methionine, catalyzed by methionine synthase needs cyanocobalamin as a cofactor; resulting hyperhomocysteinemia may lead to a prothrombotic state
- Poor absorption from the stomach or intestines
- Pernicious anemia
- Surgical removal of the stomach
- Chronic inflammation of the pancreas
- Intestinal parasites, certain medications
- Some genetic disorders
- Decreased intake
- Vegan diet
- Malnutrition
- Increased requirements
- HIV / AIDS
- Rapid red blood cell breakdown
- Extreme tiredness
- Lack of energy
- Pins and needles (paraesthesia)
- Sore and red tongue
- Mouth ulcers
- Muscle weakness
- Disturbed vision
- Psychological problems, which may include depression and confusion
- Problems with memory, understanding and judgement
- Rarely hyperpigmentation (Indian J Endocrinol Metab 2013;17:S254)
- Vitamin B12 levels in blood below 120 – 180 picomol/L in adults
- Elevated methylmalonic acid levels (values > 0.4 micromol/L)
- Hyperhomocysteinuria is a non-specific marker of deficiency
- Hyperhomocysteinuria is a non-specific marker of deficiency
- Methylmalonic acid is a more specific test of B12 deficiency
- 14 year old girl with neuropathy and ileal tuberculosis (J Med Case Rep 2008;2:90)
- 64 year old woman with confusion (Encephale 2003;29:560)
- 66 year old woman with major depressive disorder (Prim Care Companion J Clin Psychiatry 2009;11:269)
- Oral or parenteral supplements (J Hum Nutr Food Sci 2013;1: 1008)
- When large doses (1 - 2 mg) are given by mouth, its absorption does not rely on the presence of intrinsic factor or an intact ileum - the free crystalline B12 is absorbed along the entire intestine by passive diffusion
Images hosted on other servers:

Hyperpigmentation before treatment
- Individuals with autoimmune gastritis may develop pernicious anemia because of extensive loss of parietal cell mass and anti-intrinsic factor antibodies
- Autoimmune gastritis is usually restricted to the gastric corpus and fundus; lymphocytes infiltrate the gastric mucosa, destroy epithelial cells and cause gastric atrophy
Images hosted on other servers:
Atrophic gastritis with intestinal metaplasia
- Decreased red blood cell (RBC) count and hemoglobin levels
- Increased mean corpuscular volume (MCV > 100 fL) and mean corpuscular hemoglobin (MCH)
- Reticulocyte count is decreased due to destruction of fragile and abnormal megaloblastic erythroid precursor
- Platelet count may be reduced
- Neutrophil granulocytes may have hypersegmented nuclei
- Anisocytosis (increased variation in RBC size) and poikilocytosis (abnormally shaped RBCs)
- Macrocytes (larger than normal RBCs) are present
- Ovalocytes (oval-shaped RBCs) are present
- Howell-Jolly bodies (chromosomal remnant) also present
Images hosted on other servers:
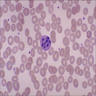
Hypersegmented neutrophil

Macrocytes, macro-ovalocytes,
nucleated RBCs and hypersegmented neutrophils
- X linked primary immunodeficiency disorder with the following classic presentation: thrombocytopenia with abnormal bleeding, eczema and recurrent infections
- Rare genetic disorder of the immune system, with variable phenotype
- Absent protein in typical Wiskott-Aldrich syndrome (WAS) (Blood 2004;104:4010)
- Due to mutations in WASP gene, a key gene involving T cell antigen receptor (TCR) signaling, cytoskeletal remodeling, synapse formation (Blood 2009;113:6288, Hematology Am Soc Hematol Educ Program 2019;2019:443)
- Distinguished from other WAS related disorders: partial protein in X linked thrombocytopenia or activating mutation in X linked neutropenia (Immunol Rev 2019;287:121)
- Wiskott-Aldrich syndrome, X linked thrombocytopenia (XLT), X linked neutropenia (XLN), all known as WAS related disorders
- ICD-10: D82.0 - Wiskott-Aldrich syndrome
- 1 - 4 per 1,000,000 live male births
- WASP protein structure:
- WASP gene encodes a 502 amino acid protein expressed exclusively in the cytoplasm of hematopoietic cells
- C terminal tripartite domain of WASP contains a common actin monomer binding motif, the verprolin homology domain (V) and a central acidic region (A) that is capable of activating the actin related protein (Arp)2/3 complex, a potent nucleator of actin polymerization (Annu Rev Cell Dev Biol 2002;18:247)
- A proline rich region in exon 10 required for optimal actin polymerization activity has been shown to be indispensable for effective recruitment of WASP to the immunologic synapse in T cells (Immunity 2003;18:141)
- N terminal region of WASP allows recruitment of the WASP interacting protein (WIP); in resting lymphocytes, WASP constitutively associates with WIP, stabilizing the inactive conformation of WASP (Mol Cell 2002;10:1269)
- Following interaction with the T cell antigen receptor, WIP is phosphorylated, resulting in disengagement of WASP from the WIP / WASP complex, thus allowing WASP activation by Cdc42, which leads to actin polymerization and stabilization of actin filaments (Cell 2002;111:565)
- WASP mutation spectrum:
- Total of 141 unique WASP mutations were detected in 262 patients; the most common WAS mutations were missense mutations (35.3%), followed by splice site mutations (19.4%), deletions (17.6%) and nonsense mutations (15.4%); insertions and complex mutations made up less than 13% of the mutations identified (Blood 2004;104:4010)
- Missense mutants that cause the immune disorder Wiskott-Aldrich syndrome (WAS) map primarily to the enabled / VASP homology 1 (EVH1) domain of the actin regulatory protein WASP; more than 300 such mutations have been described (Ann Diagn Pathol 2019;43:151413)
- Functional mechanisms:
- Affects functions of both T and B cells
- WAS mutants can cause a downregulation of T cell receptor signaling through reduced stability of the immune synapse following its formation, which may result in failure of immune activation (Immunology 2010;131:466, Haematologica 2011;96:1415)
- Besides the WASP regulation of the cytoskeleton dependent immune synapse formation, the interaction between the translocation of the transcription factor of nuclear factor of activated T cells (NFAT) and the Ena-VASP homology domain (EVH1) of WASP, prevents Th1 cytokines secretion from the CD4+ cells, while increasing the relative amount of Th2 cytokines (J Exp Med 2018;215:1009, Ann Diagn Pathol 2019;43:151413)
- Thrombocytopenia and very small platelets usually present at birth (Blood 2004;103:456)
- Life threatening bleeding (occurs in 30% of males prior to diagnosis; sites including brain, gastrointestinal tract)
- Skin eczema in 80% of the cases; other skin lesions include impetigo, cellulitis and abscesses
- Increased risk of infections, mostly recurrent ear infections and some viruses such as cytomegalovirus (CMV), herpes simplex virus (HSV), Epstein-Barr virus (EBV)
- Increased risk of autoimmune disorders that may include hemolytic anemia, immune thrombocytopenic purpura, rheumatoid arthritis, vasculitis of small and large vessels and immune mediated damage to the kidneys and liver
- Increased risk of developing cancer, such as lymphoma, especially in setting of EBV infection or older patients with autoimmune disease; high risk for EBV+ non-Hodgkin lymphoma and other malignancies (leukemia, myelodysplastic syndrome, solid tumor) at mean age of 9.5 years (Blood 2009;113:6288)
- Isolated EBV lymphoproliferative disease resembling cutaneous lymphomatoid granulomatosis was reported in a child with Wiskott-Aldrich syndrome (J Clin Pathol 2003;56:555, Clin Immunol Immunopathol 1986;41:479)
- Vaccination often ineffective due to body’s inability to mount normal protective antibody response
- Wiskott-Aldrich syndrome is usually suspected by clinical presentations, including thrombocytopenia with small platelets, eosinophilia, skin eczema, infections, immunodeficiency and autoimmunity; diagnosis is confirmed by mutations of the WASP gene (J Allergy Clin Immunol 2006;117:725, Biol Blood Marrow Transplant 2009;15:84)
- Complete blood count shows anemia, eosinophilia, microthrombocytopenia; determining platelet size and shape in a boy with thrombocytopenia is a rapid diagnostic clue (Curr Opin Hematol 2008;15:30)
- Patients typically have elevated levels of IgE and IgA, sometimes with low levels of IgM; the T cell function is often abnormal, such as T cell lymphopenia, abnormal response to mitogen or impaired antibody production (J Clin Immunol 2018;38:13)
- WAS is well known to have associated peripheral eosinophilia, induced by cytokines such as IL4 and IL5 (Ann Diagn Pathol 2019;43:151413)
- 12 year old boy with WAS who developed a pulmonary vasculitis associated with lymphoreticular proliferation (Clin Immunol Immunopathol 1986;41:479)
- 16 year old boy with WAS who presented with an isolated nonhealing ulcerating skin lesion (J Clin Pathol 2003;56:555)
- 24 year old man diagnosed at age 7 with Wiskott-Aldrich syndrome (Ital J Pediatr 2011;37:42)
- A case series of Wiskott-Aldrich syndrome, CARMIL2 and DOCK8 deficiency (Ann Diagn Pathol 2019;43:151413)
- Treatment for Wiskott-Aldrich syndrome is based on a person’s clinical condition; possible treatment options include immunoglobulin (antibody) infusions, platelet transfusions, topical creams for eczema, steroids or similar medications to control autoimmunity
- Stem cell transplant: allogeneic HCT for WAS has been a standard curative therapy to be considered immediately at the time of diagnosis; patients undergoing sibling matched transplant can achieve great survival rate (Lancet 2003;361:553, Immunol Allergy Clin North Am 2010;30:179)
- Splenectomy: improves platelet counts, platelet size normalizes; potential need for prophylactic antibiotics given further increase in risk of infection / sepsis and death; splenectomy may be reserved only for patients who are unlikely to ever be transplanted (Immunol Allergy Clin North Am 2010;30:179, N Engl J Med 1980;302:892)
- Gene therapy clinical trial ongoing (Boston Children's Hospital: Wiskott-Aldrich Syndrome [Accessed 6 July 2021])
- Based on transplanting genetically modified stem cells from the patient's own bone marrow or blood
- Spleen in Wiskott-Aldrich syndrome (WAS) patients shows:
- Significant depletion of the white pulp, affecting both T and B cell areas
- Splenic marginal zone in patients with WAS is severely depleted
- Follicles are heterogeneous in morphologic appearance
- Red pulp plasma cells in WAS patients are similar to those in control subjects (Am J Surg Pathol 1999;23:182)
- Lymph node biopsy shows preserved architecture with prominent reactive follicles and eosinophils in paracortex; however, the reduction in germinal center has been confirmed in WAS protein deficient murine model (Cell Immunol 2019;341:103919)
- Peripheral blood smear shows thrombocytopenia with small platelets and eosinophilia; lymphopenia is characteristic for classic WAS during childhood (Blood 2004;104:4010)
- Screen for WASP mutations with flow cytometry with anti-WAS protein antibody (Blood 1999;93:756, J Allergy Clin Immunol 2018;142:333)
- See Pathophysiology
- Deficiency of WAS protein interacting protein (WIP deficiency) causes an autosomal recessive, WAS-like syndrome with early onset combined immunodeficiency that has been described in few pedigrees (Front Immunol 2018;9:2554, J Leukoc Biol 2014;96:713, J Exp Med 2012;209:29)
- Mutation analysis alone is of limited value in predicting the clinical phenotype; the vast majority of XLT patients carry missense mutations in exons 1 and 2 of the WAS gene (Blood 2004;104:4010)
- Missense mutations in the GTP-ase binding domain (GBD) or Cdc42 binding domain of WAS result in constitutive activation of the protein, causing X linked neutropenia (XLN), with neither thrombocytopenia nor signs of T cell immunodeficiency (Blood 2006;108:2182, Br J Haematol 2009;144:120, Nat Genet 2001;27:313)
- CARMIL2 (RLTPR) is a protein involved in cytoskeletal organization and cell migration, which also plays a role in CD28 cosignaling of T cells; homozygous mutation of the CARMIL2 gene, like decreased WASP, dysregulates cell to cell signaling by actin dysregulation; patients present with dermatitis, esophagitis, recurrent skin and chest infections, as well as an increased risk for EBV associated smooth muscle tumors (Front Immunol 2018;9:203)
- DOCK8 is linked to WASP through the WASP interacting protein (WIP), which also stabilizes WASP from degradation and itself can result in a primary immune deficiency syndrome identical to WAS when deficient (Front Immunol 2021;11:604206, Clin Immunol 2017;181:75)
- CXCR4
- FAS
- IL7R
- RAG1/2
- WASP
Glassy: 2018
Hoyer: 2003
Kaushansky: 2021
Keohane: 2015
Medeiros: 2017
Sallman: 2016
Turgeon: 2017
Find related Pathology books: hematopathology
