


- Peripheral neuropathy due to extracellular deposition of amyloid fibrils within the endoneurium, epineurium or the wall of endo or epineurial vessels
- Most common cause of amyloid neuropathy is primary systemic immunoglobulin light chain (AL) amyloidosis in patients with plasma cell dyscrasia
- Most common hereditary amyloidosis is caused by mutations in transthyretin (hATTR)
- Other hereditary forms include mutations in apolipoprotein A1, gelsolin, lysozyme, fibrinogen, amyloid β and cystatin C
- Senile amyloidosis occurs in the elderly population (> 70 years old) and the amyloid deposits are composed of wild type transthyretin
- AA amyloidosis and β2 microglobulin dialysis associated amyloidosis rarely involve peripheral nerves
- Involved peripheral nerve pathology characterized by axonal degeneration preferentially affecting small myelinated and unmyelinated axons in early stages and extending to involve large myelinated axons in later stages
- Familial amyloid polyneuropathy (FAP)
- Neuromuscular amyloidosis
- AL amyloidosis is the most common form of systemic amyloidosis with a prevalence of 2.5 per 100,000 in the U.S.
- 17 - 35% of patients with AL amyloidosis have peripheral neuropathy (Semin Neurol 2019;39:578)
- Hereditary transthyretin amyloidosis polyneuropathy overall incidence rate is 0.87/100,000 but is endemic in some regions such as Portugal, Japan and Sweden (Neuroepidemiology 2018;51:177, Orphanet J Rare Dis 2019;14:34)
- Systemic disease with multiorgan involvement
- Peripheral nerves, cardiovascular system, kidneys and musculoskeletal system are commonly involved
- In the peripheral nerve system, small myelinated and unmyelinated fiber damage predominates in amyloidosis
- References: J Neurol Sci 1983;59:237, Arch Neurol 1969;20:490
- Amyloidosis is caused by extracellular deposition of insoluble aggregates of amyloid fibrils in tissue
- Different types of amyloidosis are classified by the amyloid precursor proteins
- Amyloid deposition leads to direct damage in blood vessels, disruption of the blood nerve barrier, Schwann cell damage, local compression and potentially toxic effects on peripheral nerve axons
- Reference: Neurology 2016;87:2220
- Acquired forms arise from excessive or misfolded monoclonal κ or λ light chains in primary systemic amyloid (AL), serum amyloid A protein in secondary amyloidosis (AA) and β2 microglobulin (β2M) in dialysis associated amyloidosis
- Hereditary forms include transthyretin (TTR), apolipoprotein A1, gelsolin, lysozyme, fibrinogen, amyloid β and cystatin C; TTR is by far the most common hereditary form of amyloidosis
- References: Semin Neurol 2019;39:578, J Neurol Sci 1983;59:237
Images hosted on other servers:
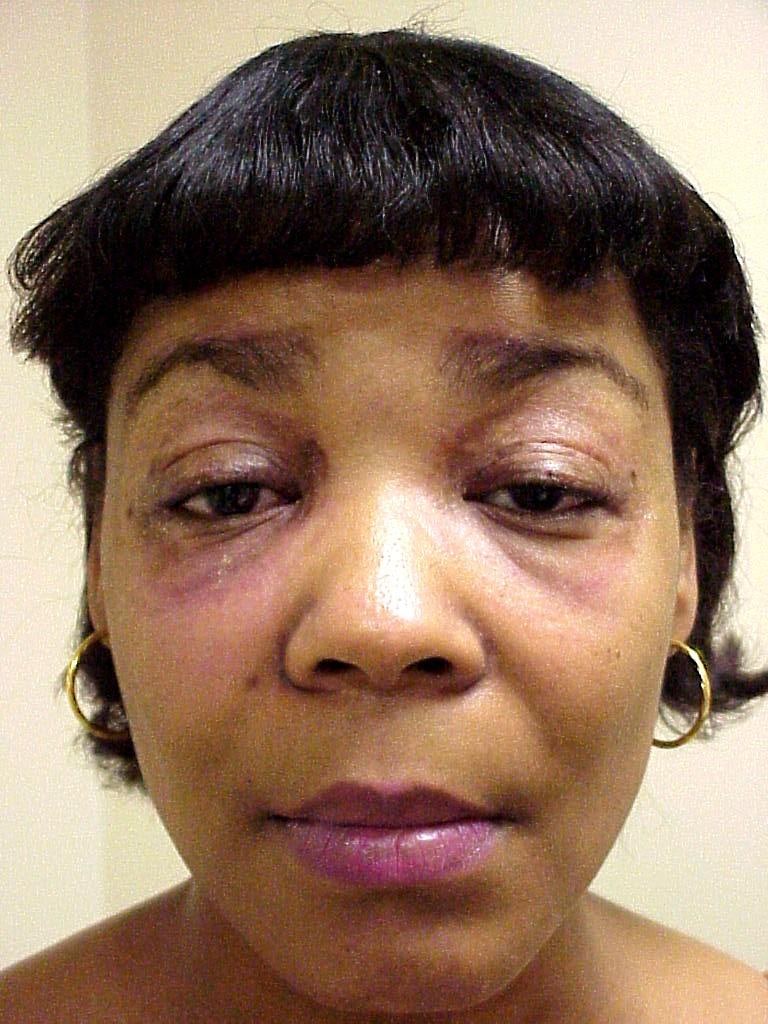
hATTR clinical epidemiologic characteristic
- Length dependent sensory neuropathy is the most common clinical presentation of amyloid neuropathy; other presentations include predominant upper limb neuropathy, pure small fiber neuropathy or carpal tunnel syndrome
- Early in the disease course there is selective involvement of distal thermal and pain sensation and later involvement of touch, vibration, joint sensations and motor function; this is due to selective small fiber involvement and later larger fiber involvement
- Autonomic dysfunction is common due to small fiber involvement, including orthostatic hypotension, alternating postprandial diarrhea and constipation, erectile dysfunction and neurogenic bladder dysfunctions
- Besides neurologic symptoms; patients may also have cardiac conduction defects or cardiomyopathy and other organ involvement, such as kidneys, gastrointestinal tract and brain
- References: Semin Neurol 2019;39:578, Arch Neurol 1969;20:490
- Hereditary forms of amyloid neuropathy may be diagnosed on the grounds of family history, clinical examination / genetic analysis or demonstration of amyloid in tissue and genetic analysis
- Serum free light chain assay (SFLC) is more sensitive than serum and urine protein electrophoresis (SPEP or UPEP) in detection of abnormal immuno light chains and plasma cell dyscrasia
- Liquid chromatography with tandem mass spectrometry (LC MS / MS) is the current gold standard for amyloidosis diagnosis and subtyping on pathology tissue
- Reference: Lancet 2016;387:2641
- AL amyloidosis: SPEP identifies monoclonal gammopathies
- SFLC is more sensitive than SPEP in detecting abnormal immuno light chains and plasma cell dyscrasia
- Reference: Semin Neurol 2019;39:578
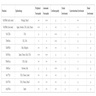
- 99mTc-3,3-diphosphono-1,2-pyrophosphate (99mTc-DPD) whole body scintigraphy with single photon emission computed tomography (SPECT) / CT may help detect AL and ATTR type amyloid in vivo (Medicine (Baltimore) 2020;99:e18905, Amyloid 2023 Aug 2 [Epub ahead of print])
Images hosted on other servers:
99mTc-DPD scan of cardiac ATTR
- Prognosis is influenced by the extent of organ damage, especially by cardiac involvement
- Median survival of AL amyloidosis patients presenting with neuropathy is 25 - 35 months
- Usual cause of death is from congestive heart failure or kidney failure
- Reference: Semin Neurol 2019;39:578
- 45 year old woman with painful neuropathy, deafness and cardiac pacemaker (Ann Afr Med 2022;21:296)
- 64 year old man with diarrhea, anemia and peripheral neuropathy (J Med Case Rep 2022;16:248)
- 70 year old woman being treated for refractory painful neuropathy (J Palliat Med 2021;24:1579)
- Principle of treatment is to reduce the supply of amyloid precursor protein
- Mainstay treatment for AL type amyloidosis is to eliminate abnormal plasma cells or lymphocytes by chemotherapy and autologous peripheral blood stem cell transplantation (Ann Intern Med 2004;140:85)
- Treatment for hATTR disease
- Liver transplant: eliminate the source of mutant TTR
- Tafamidis, diflunisal: TTR tetramer stabilizers
- TTR gene silencers: patisiran (siRNA) and inotersen (antisense oligonucleotide) decrease abnormal TTR production in liver
- Reference: Curr Neuropharmacol 2023;21:471
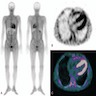
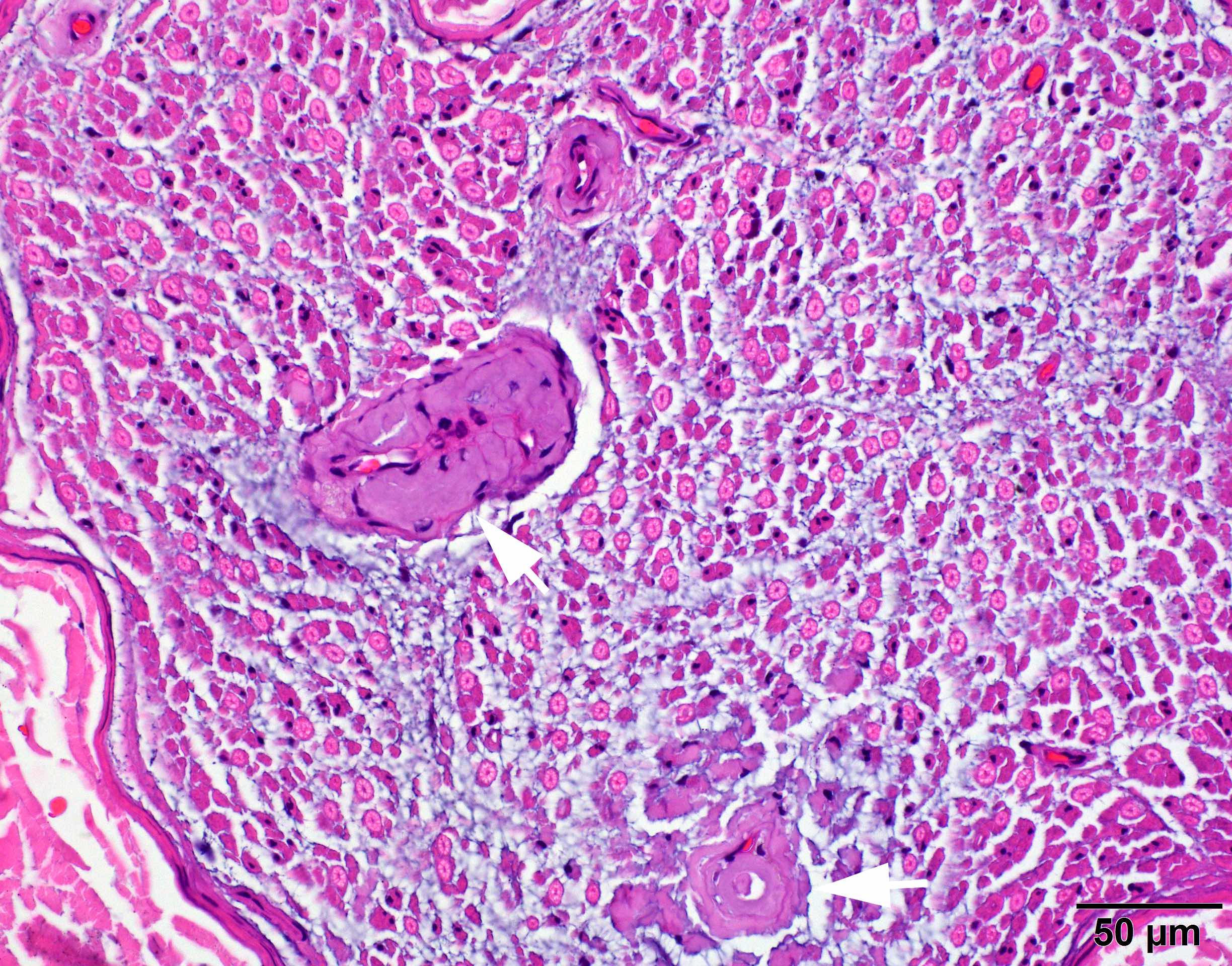
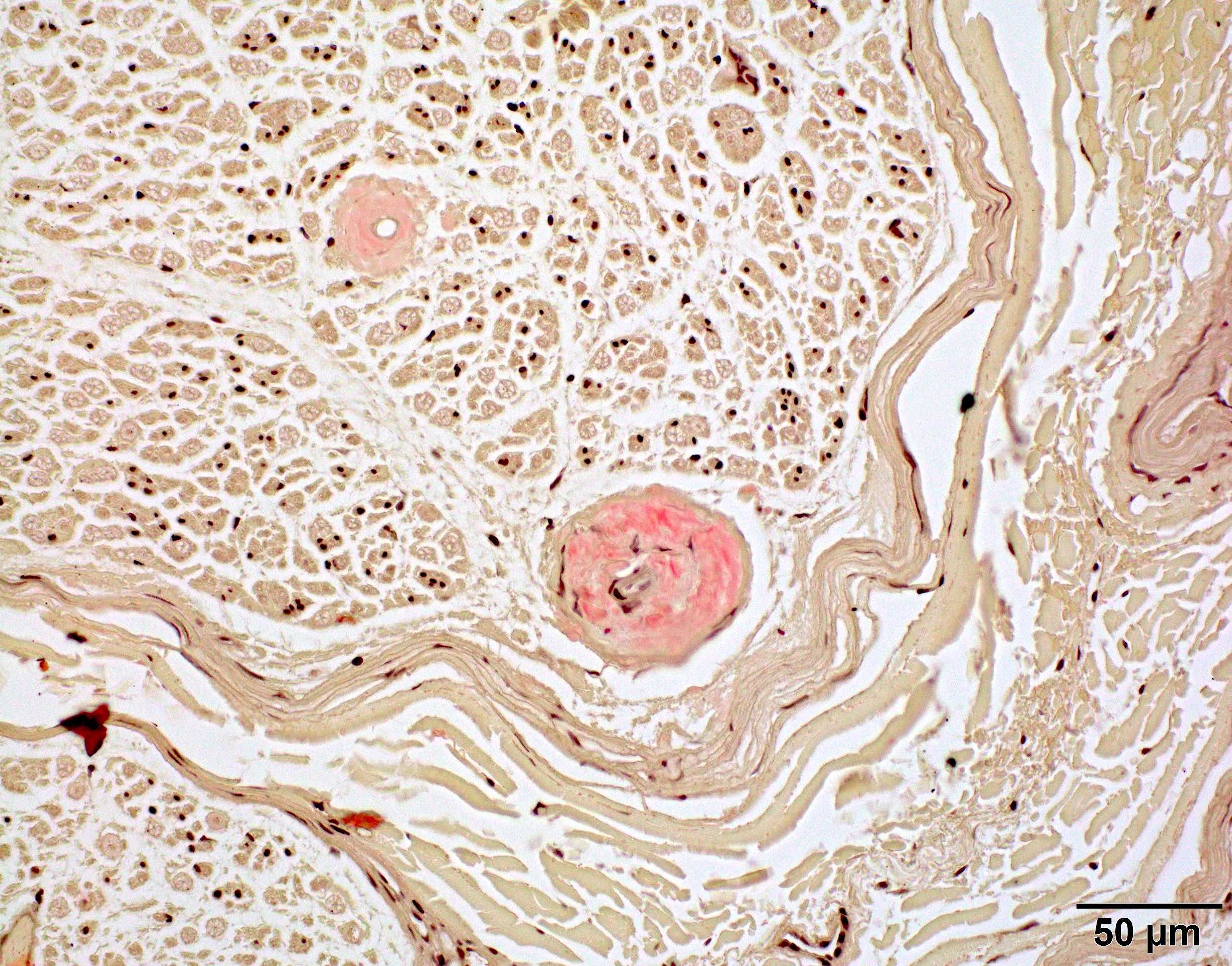
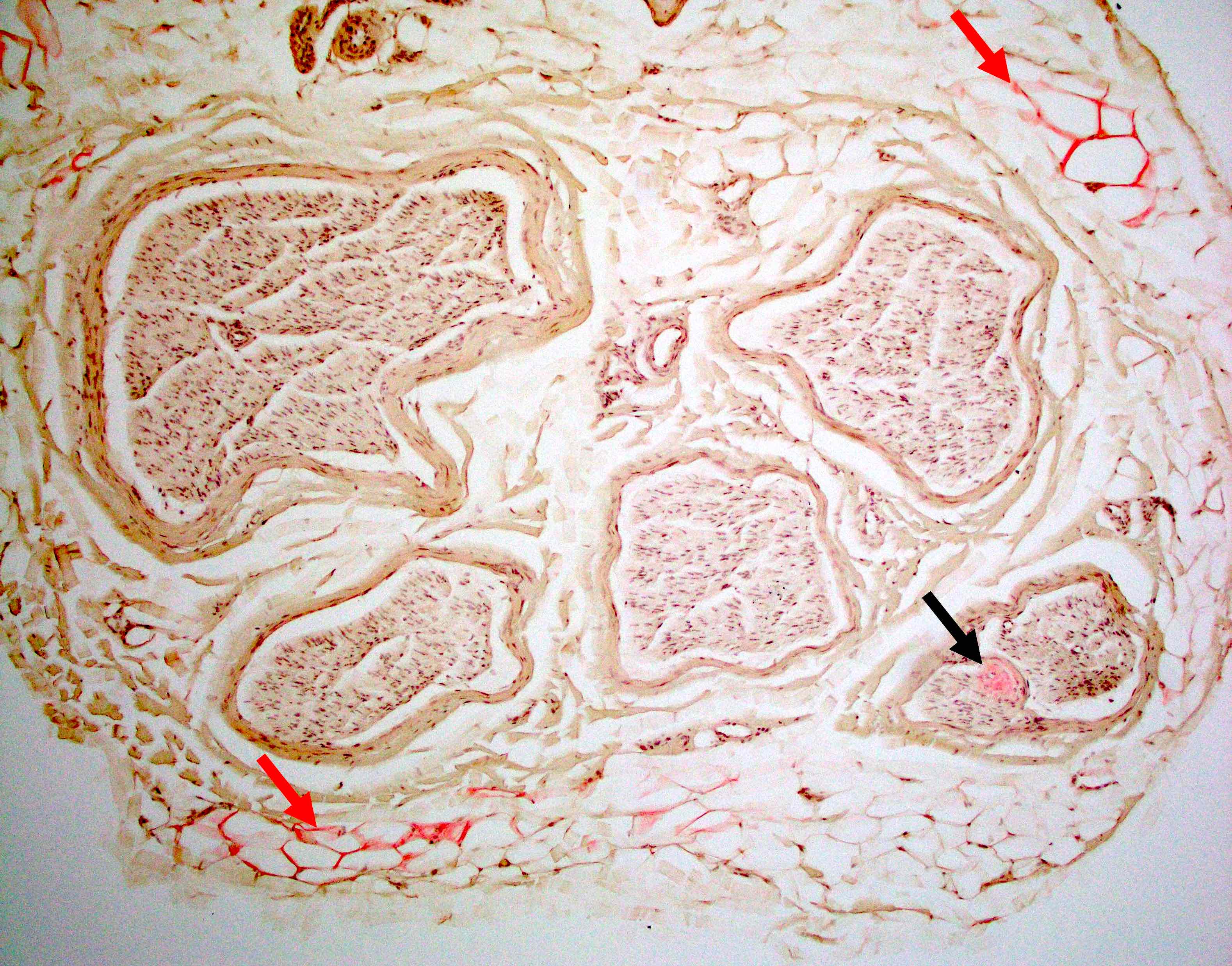
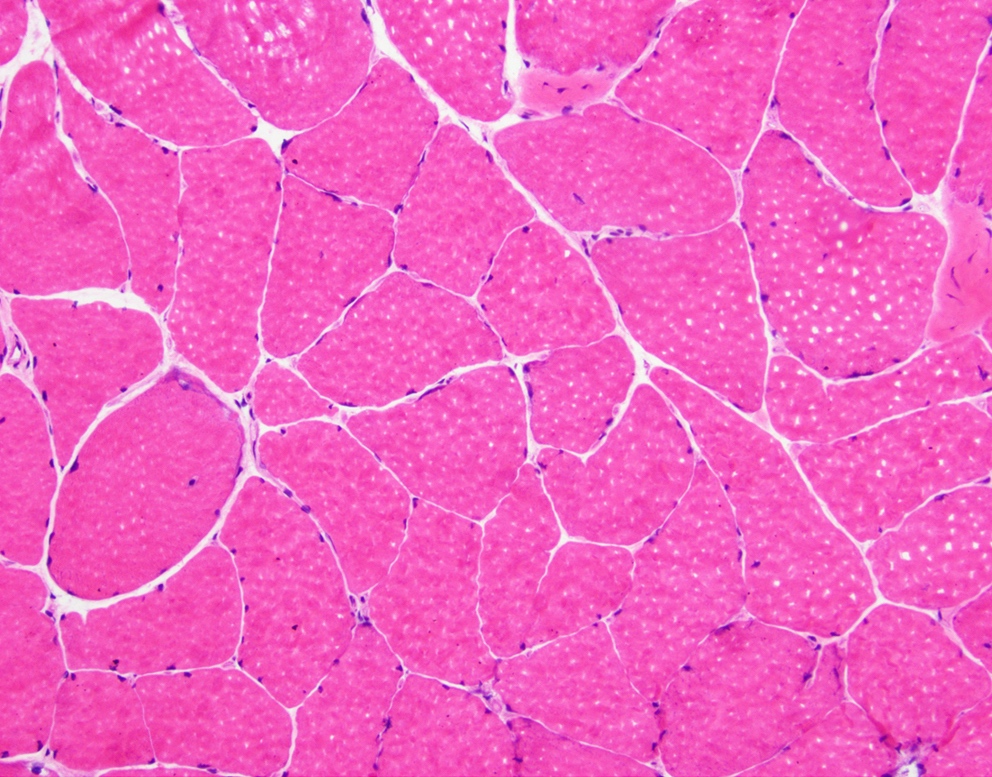
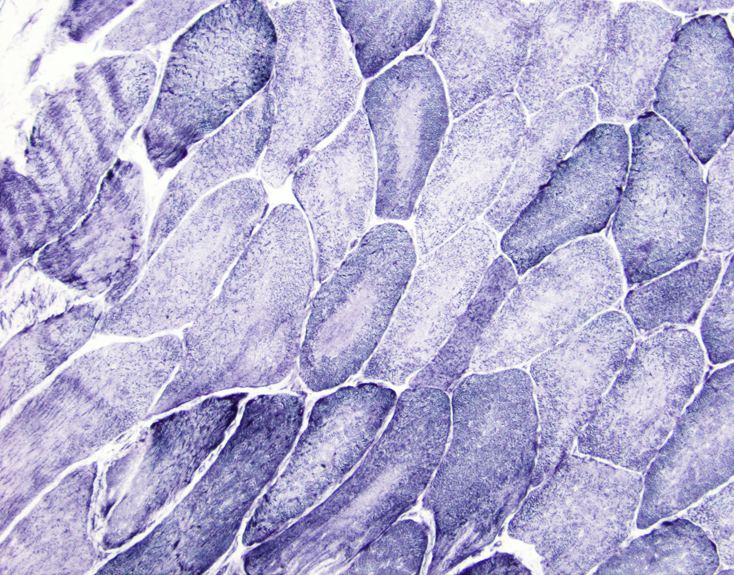
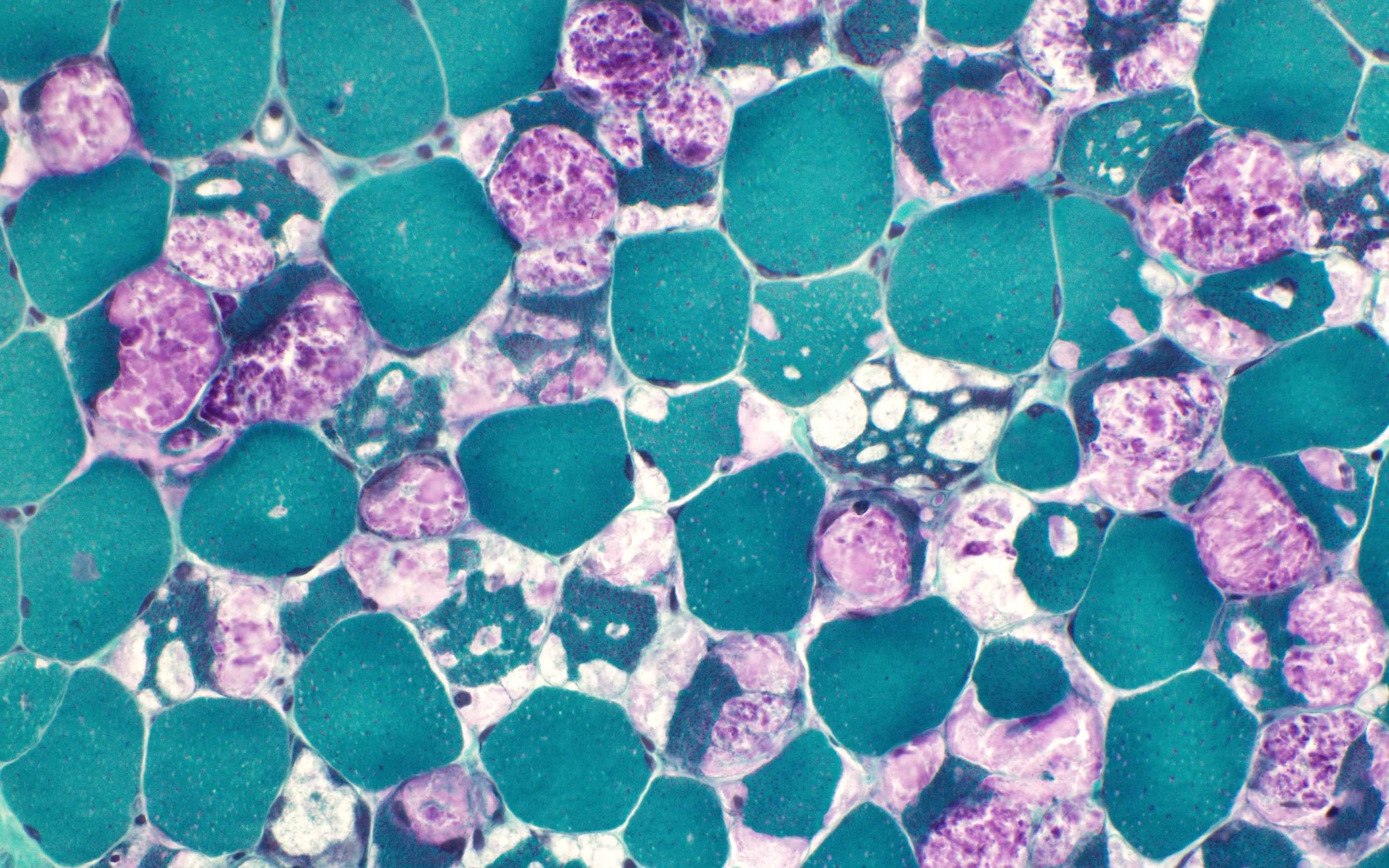
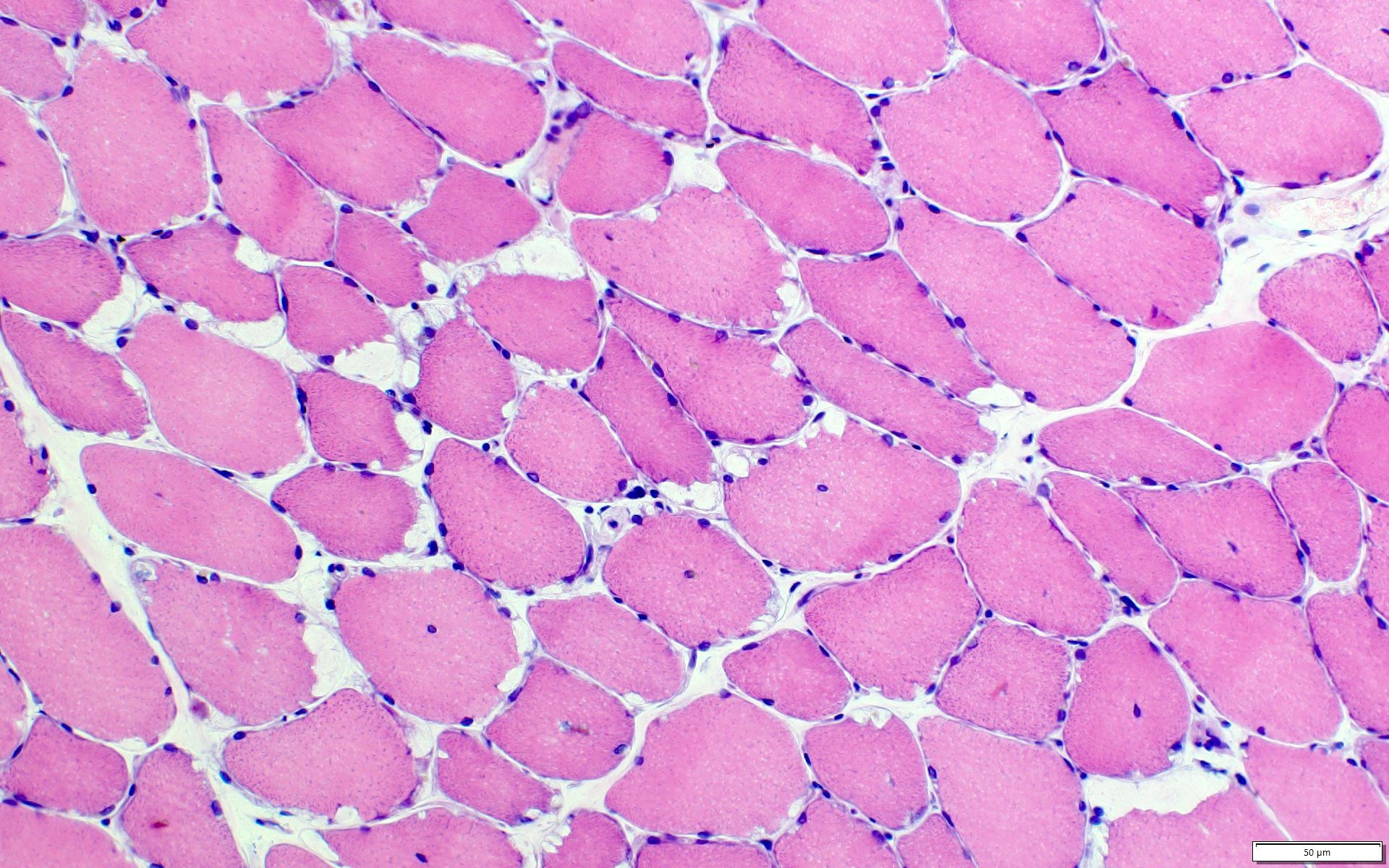
- Amorphous pink material, usually in association with irregular expansion of blood vessel wall
Contributed by Chunyu Cai, M.D., Ph.D.
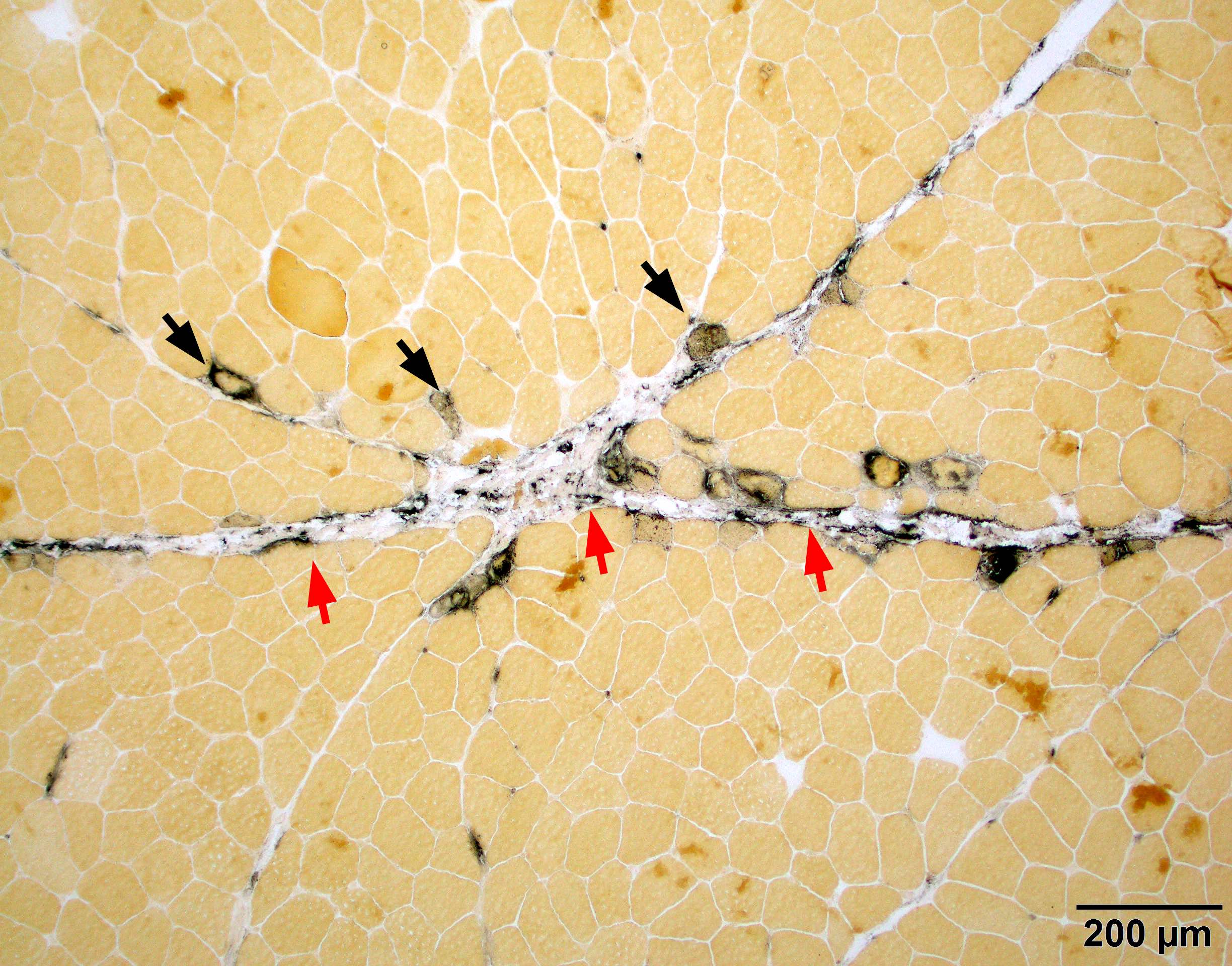
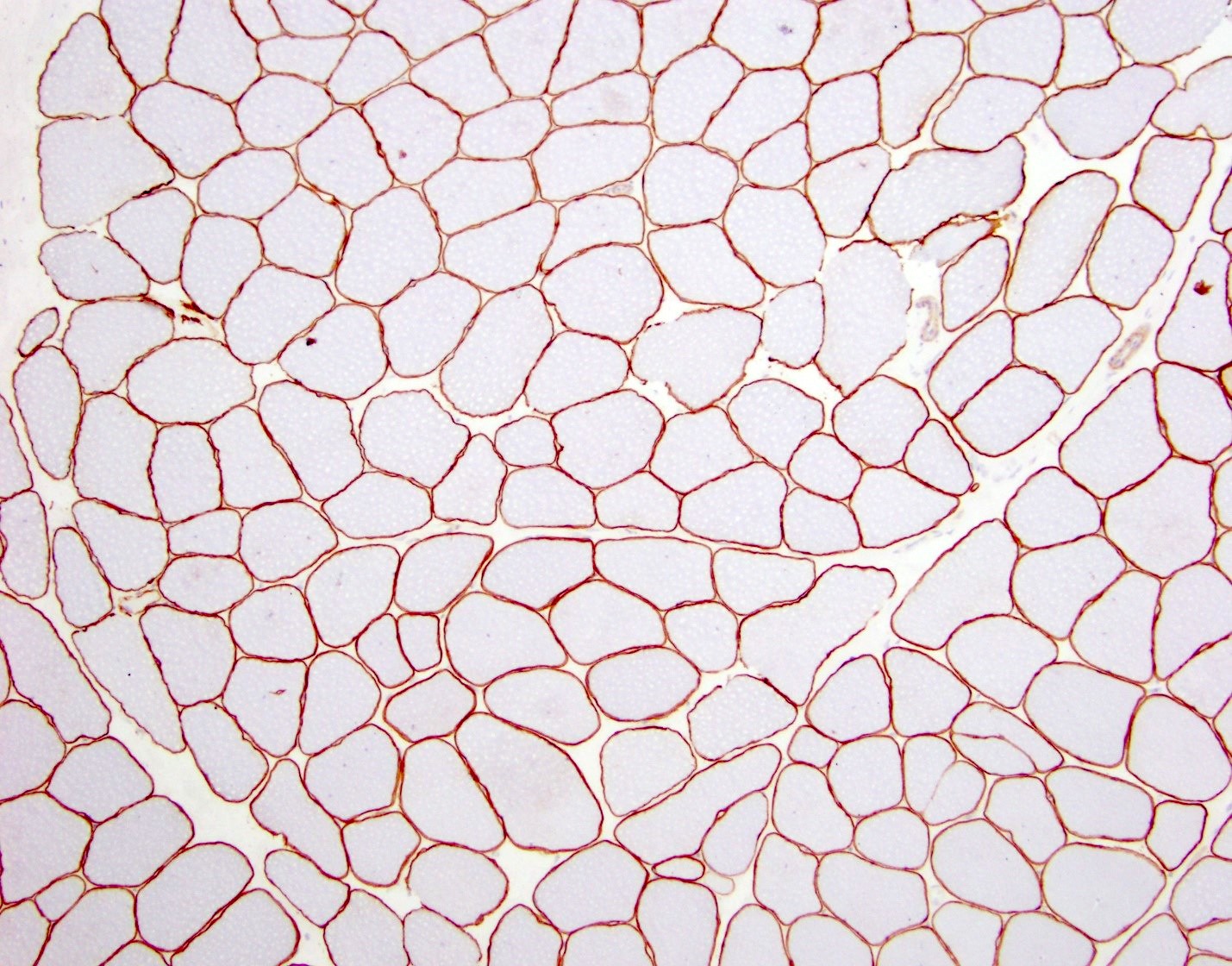
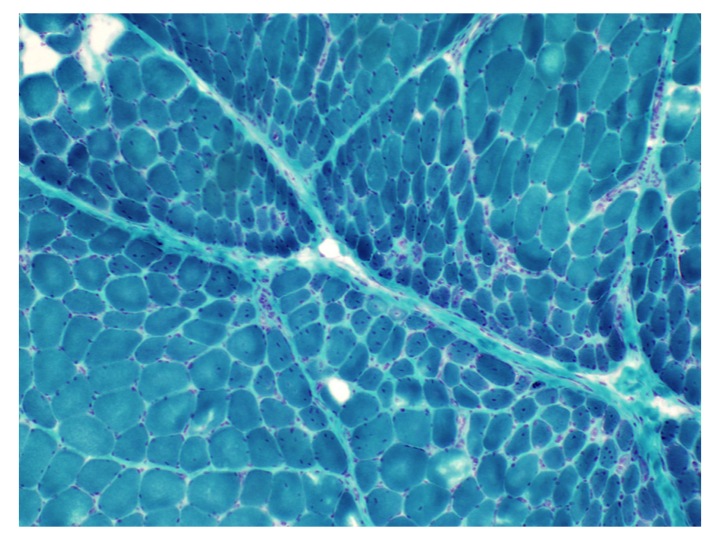
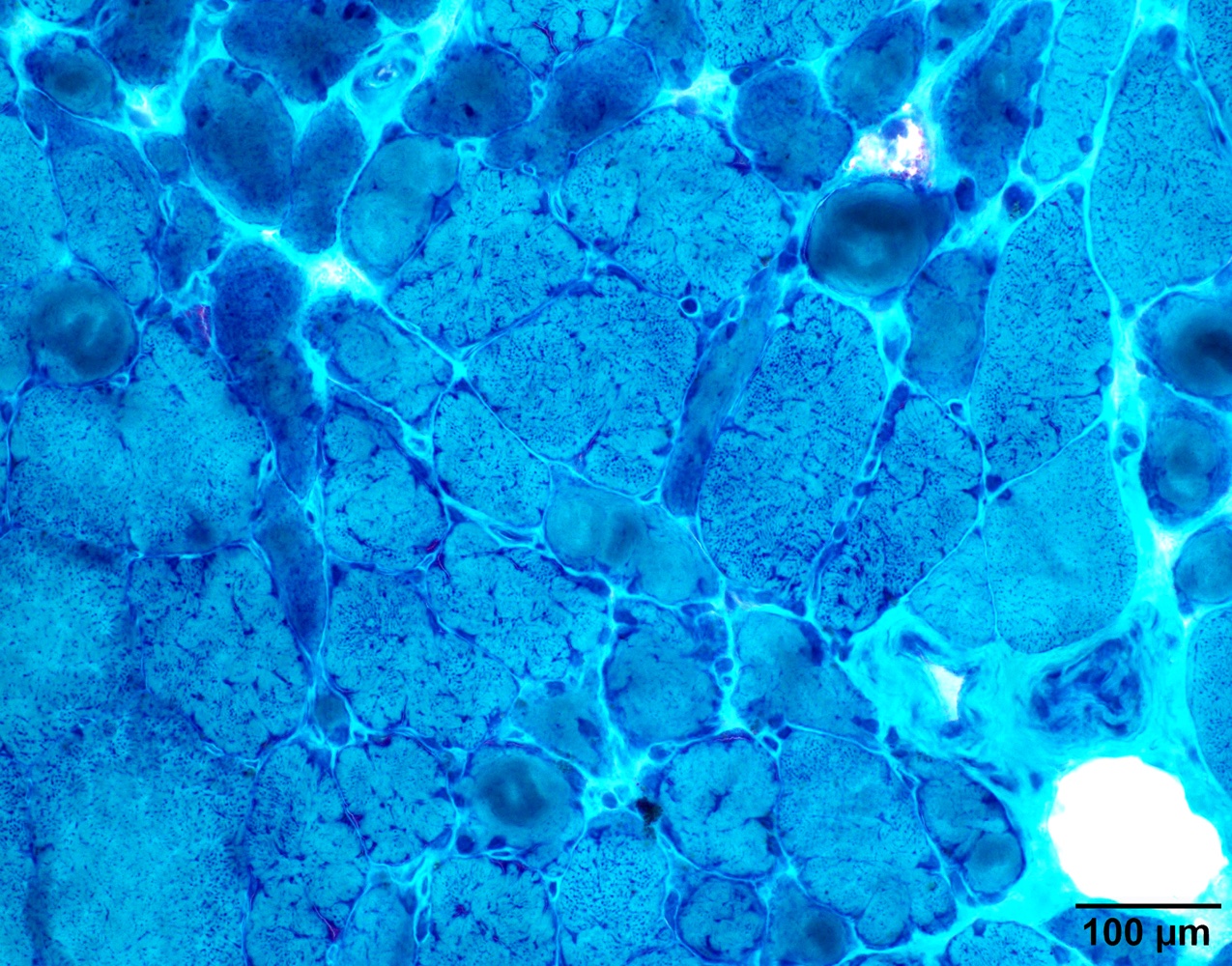
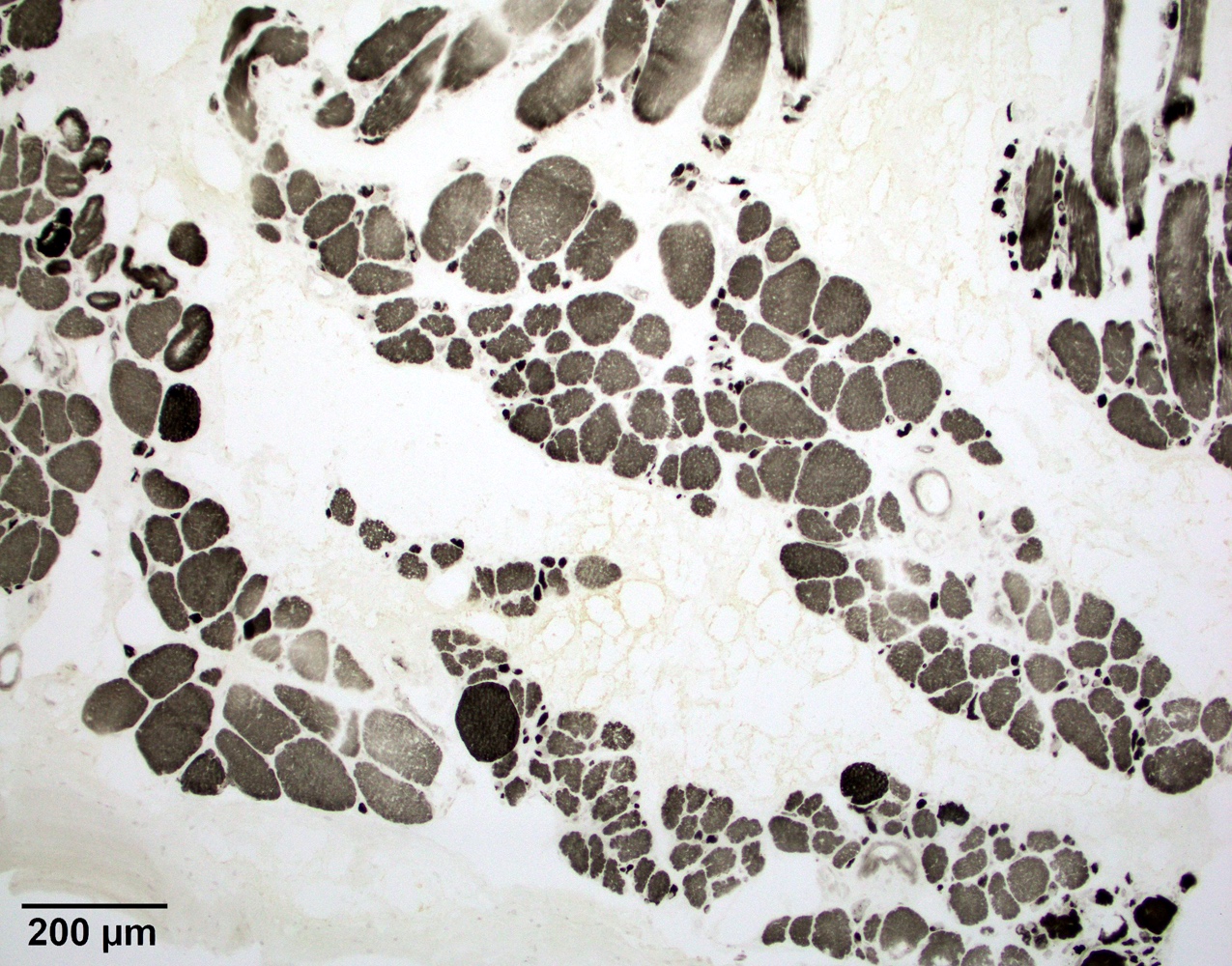
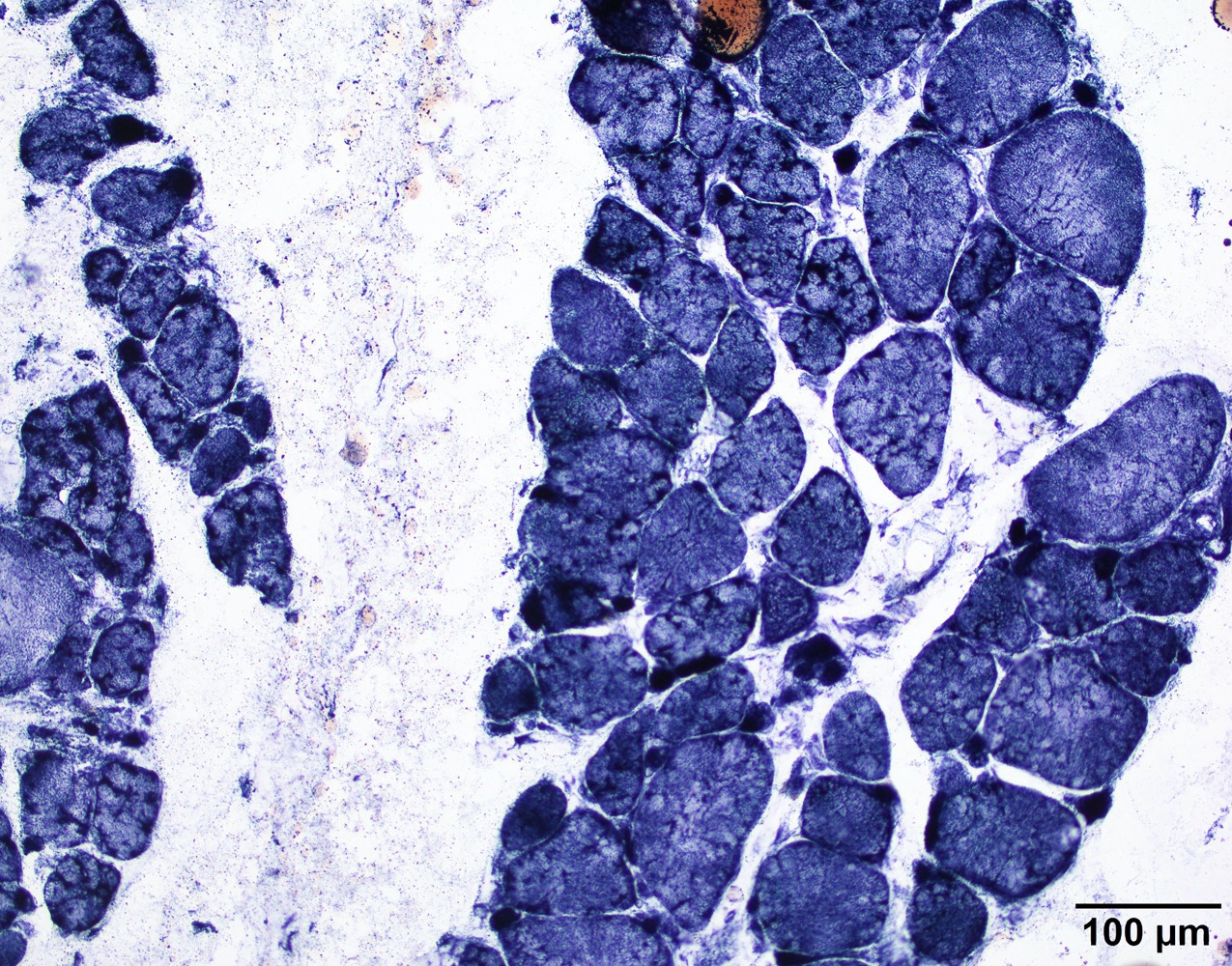
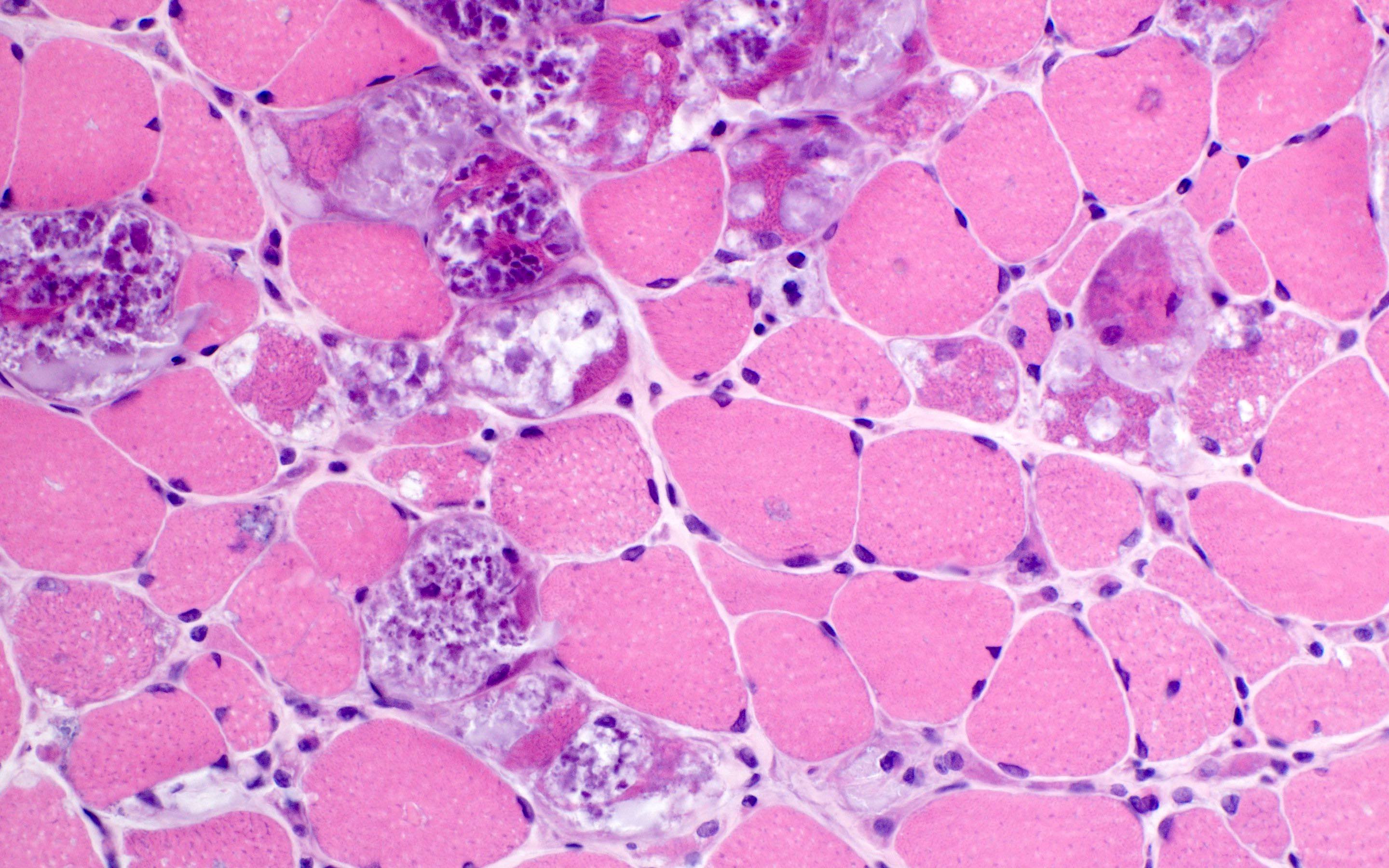
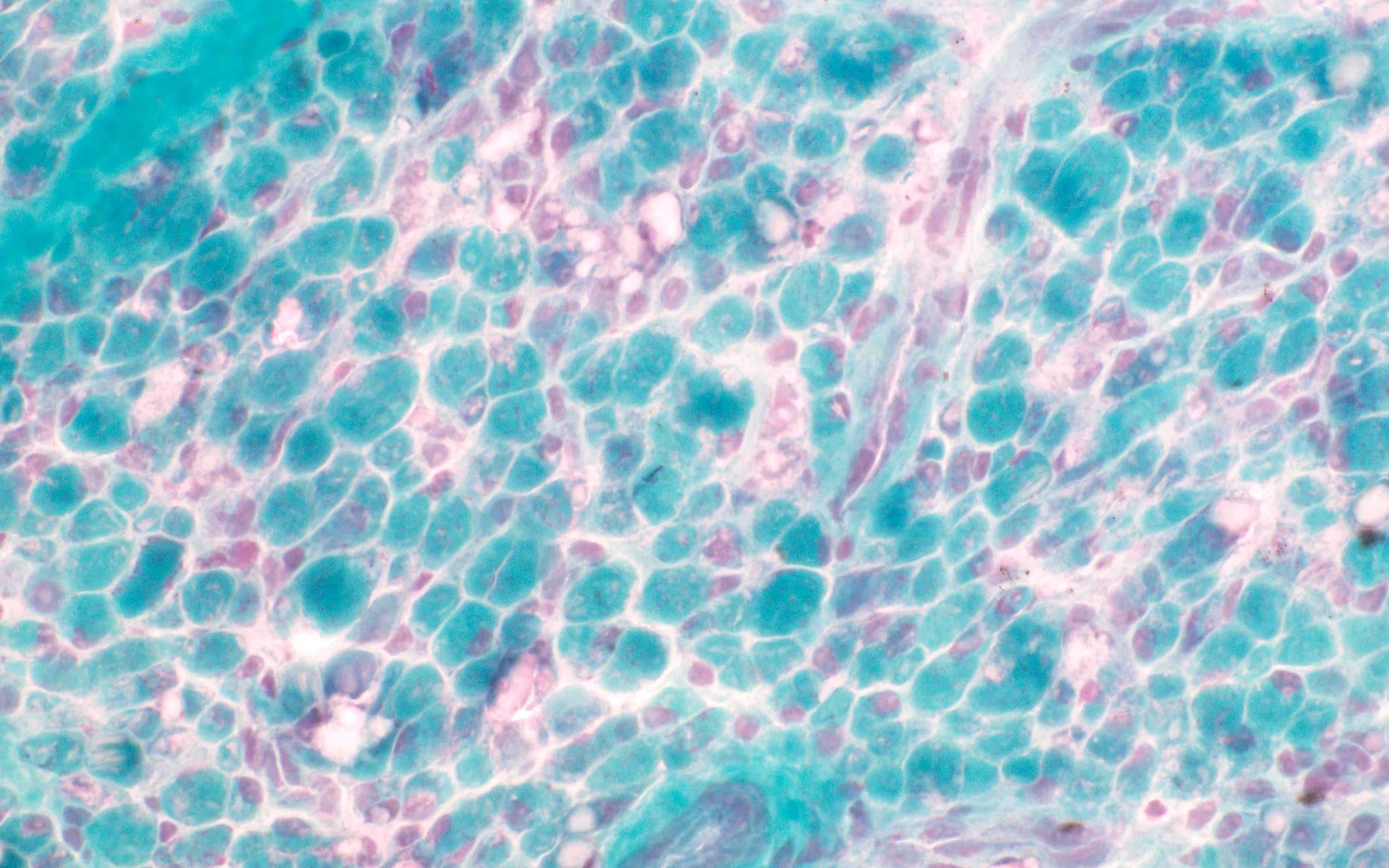
Amyloid in peripheral nerve
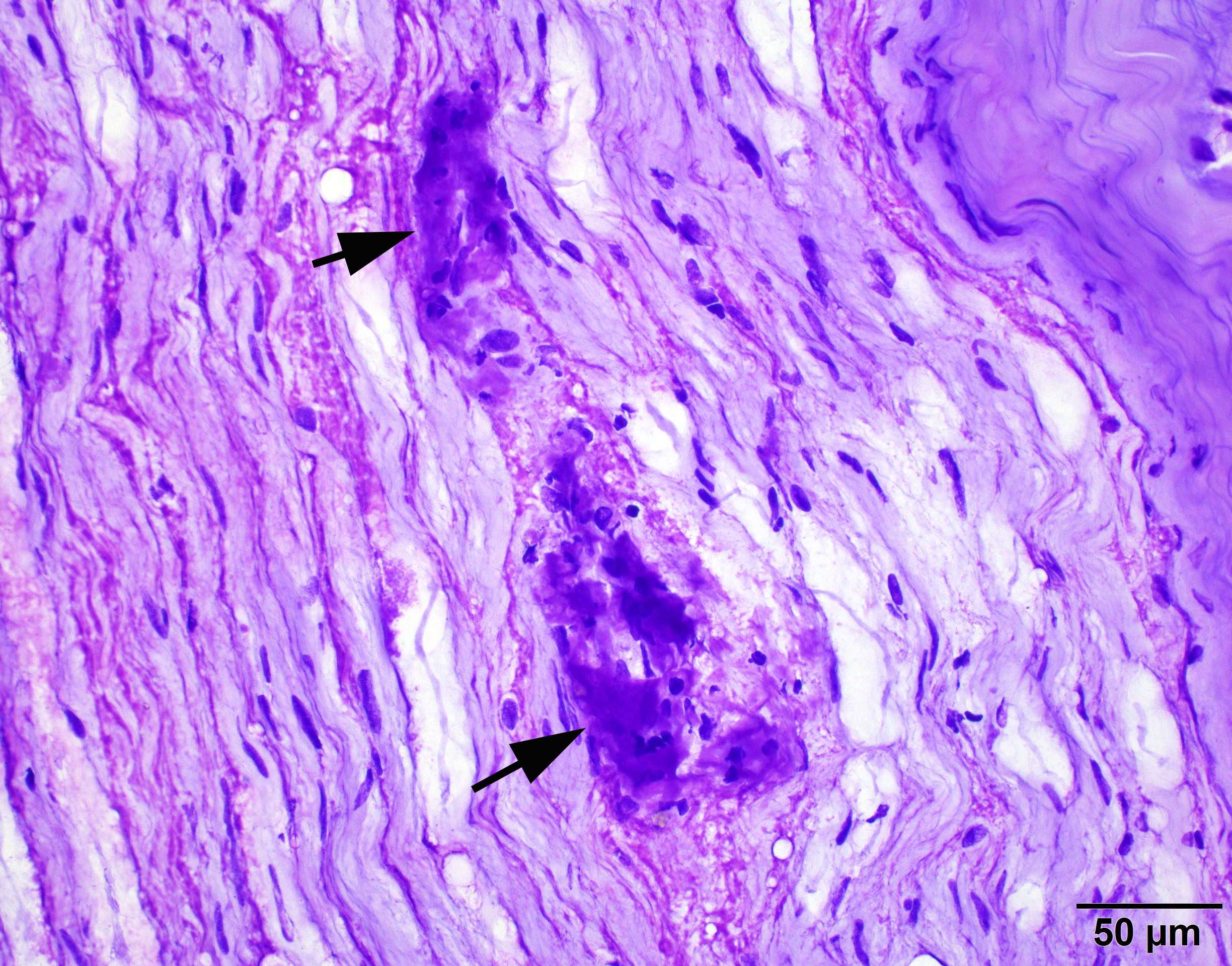
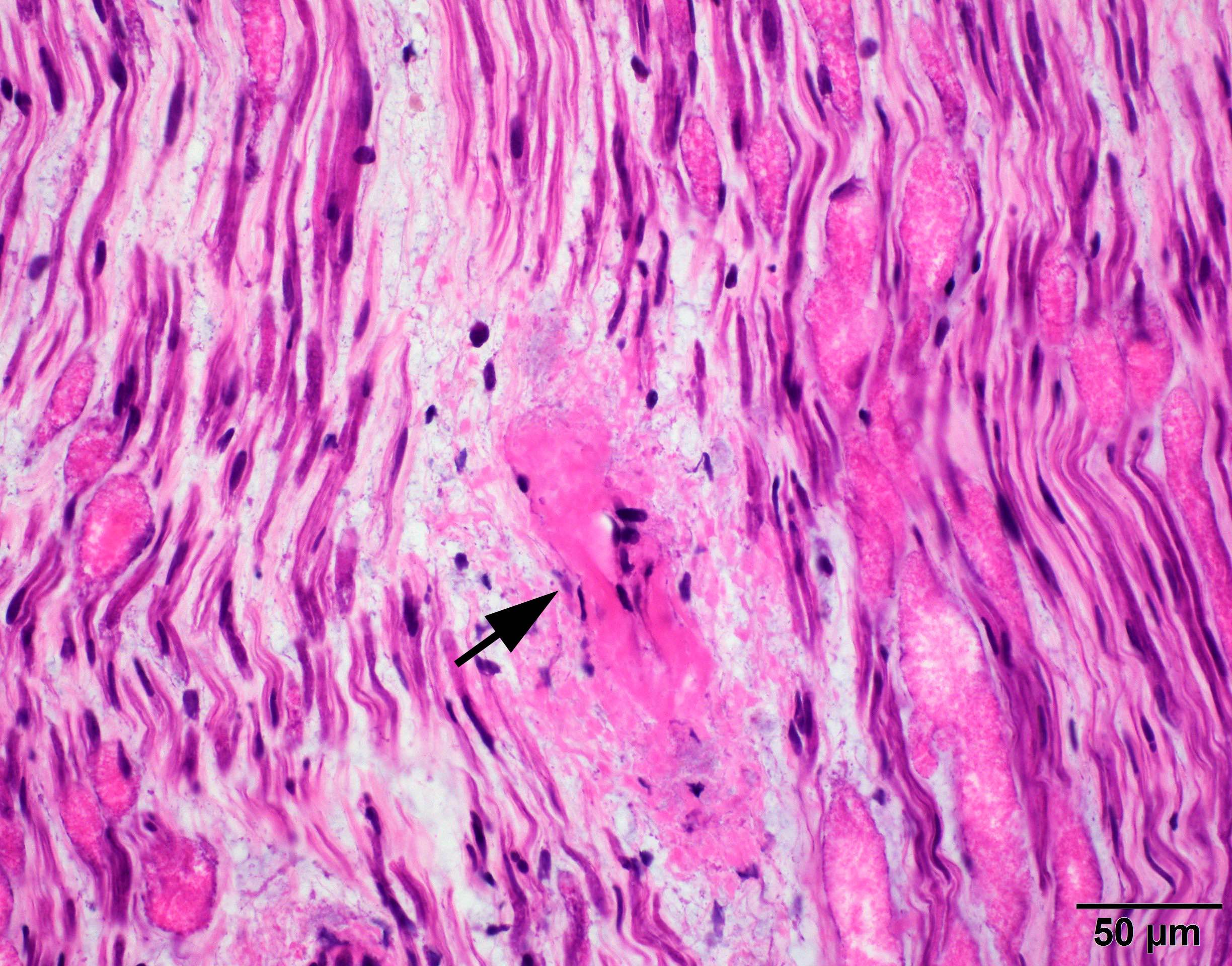
Crystal violet stain amyloid
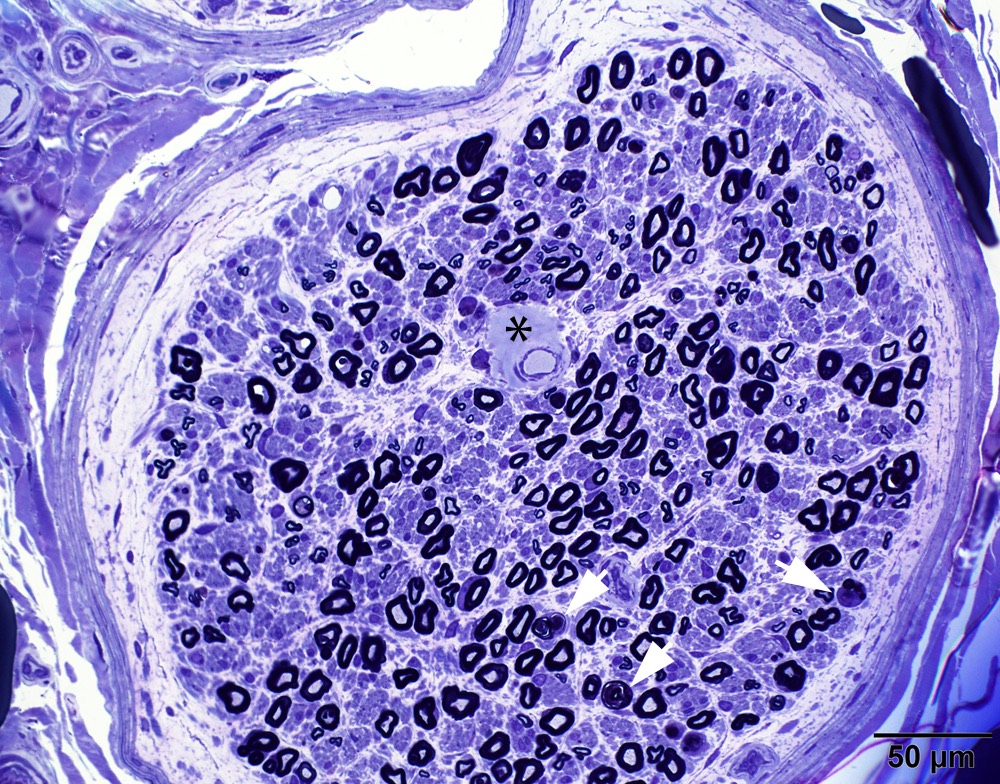
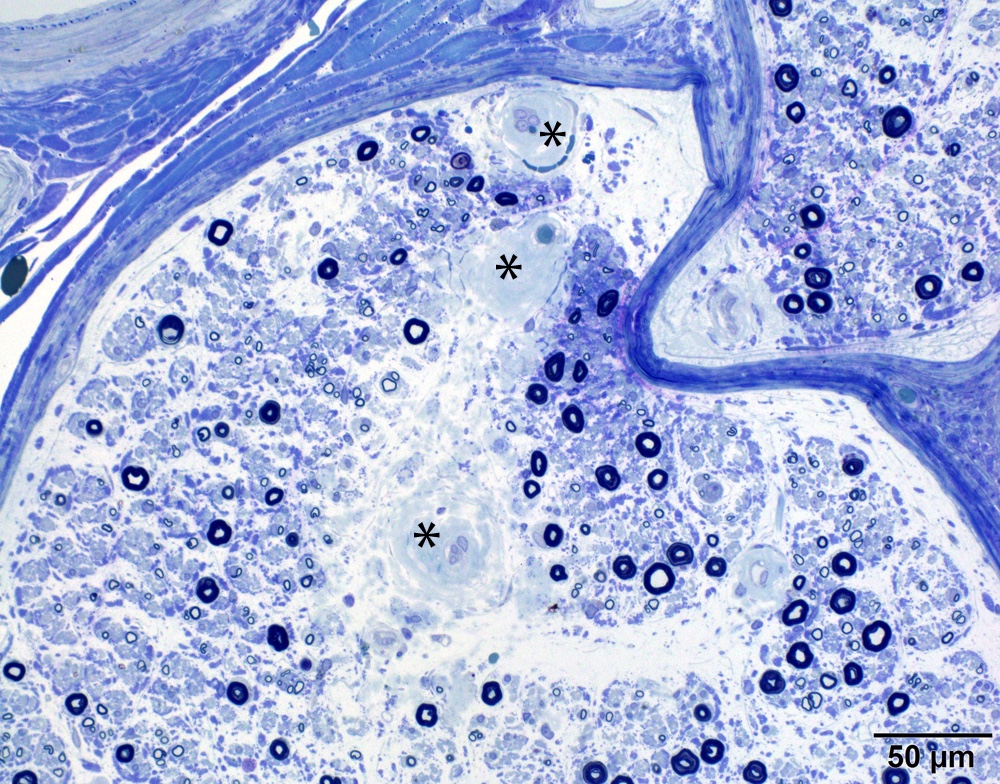
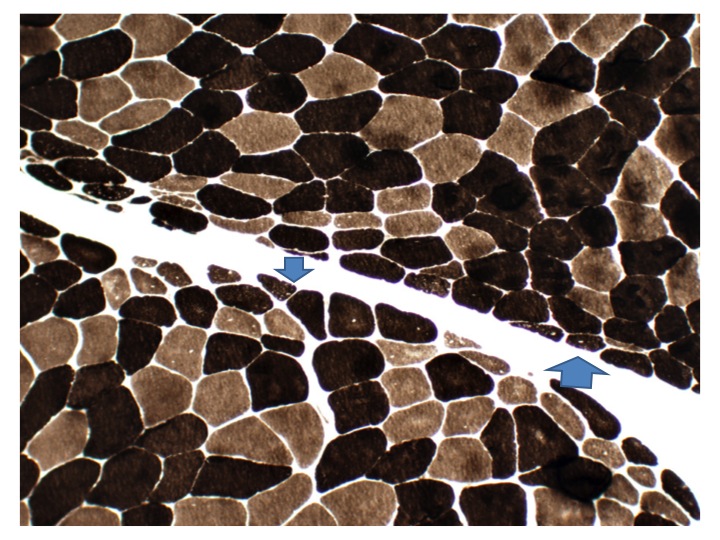
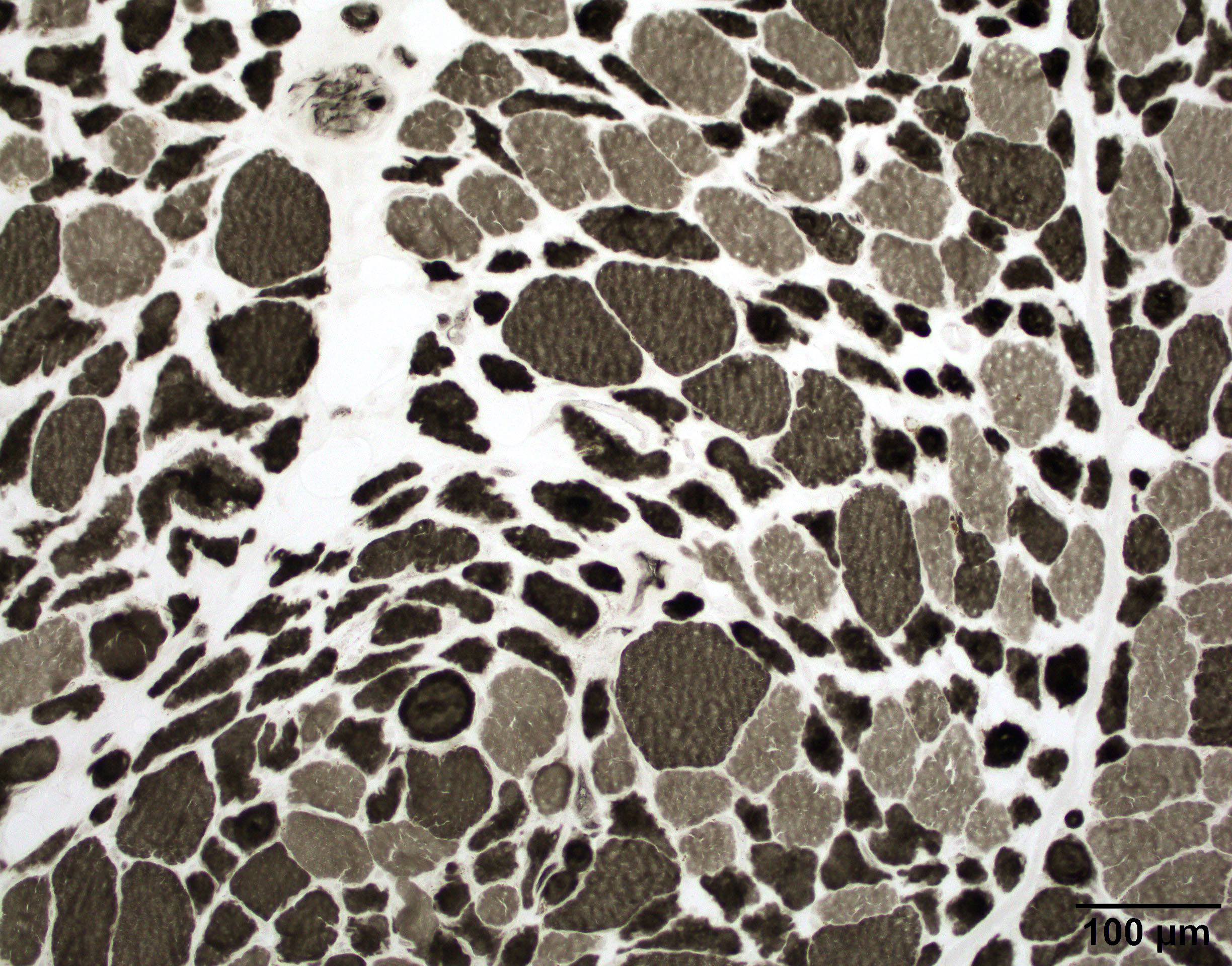
- Axonal degeneration preferentially involving small myelinated and unmyelinated axons
- Amyloid deposits can be found in vessel walls or connective tissue in the epineurium, perineurium and endoneurium
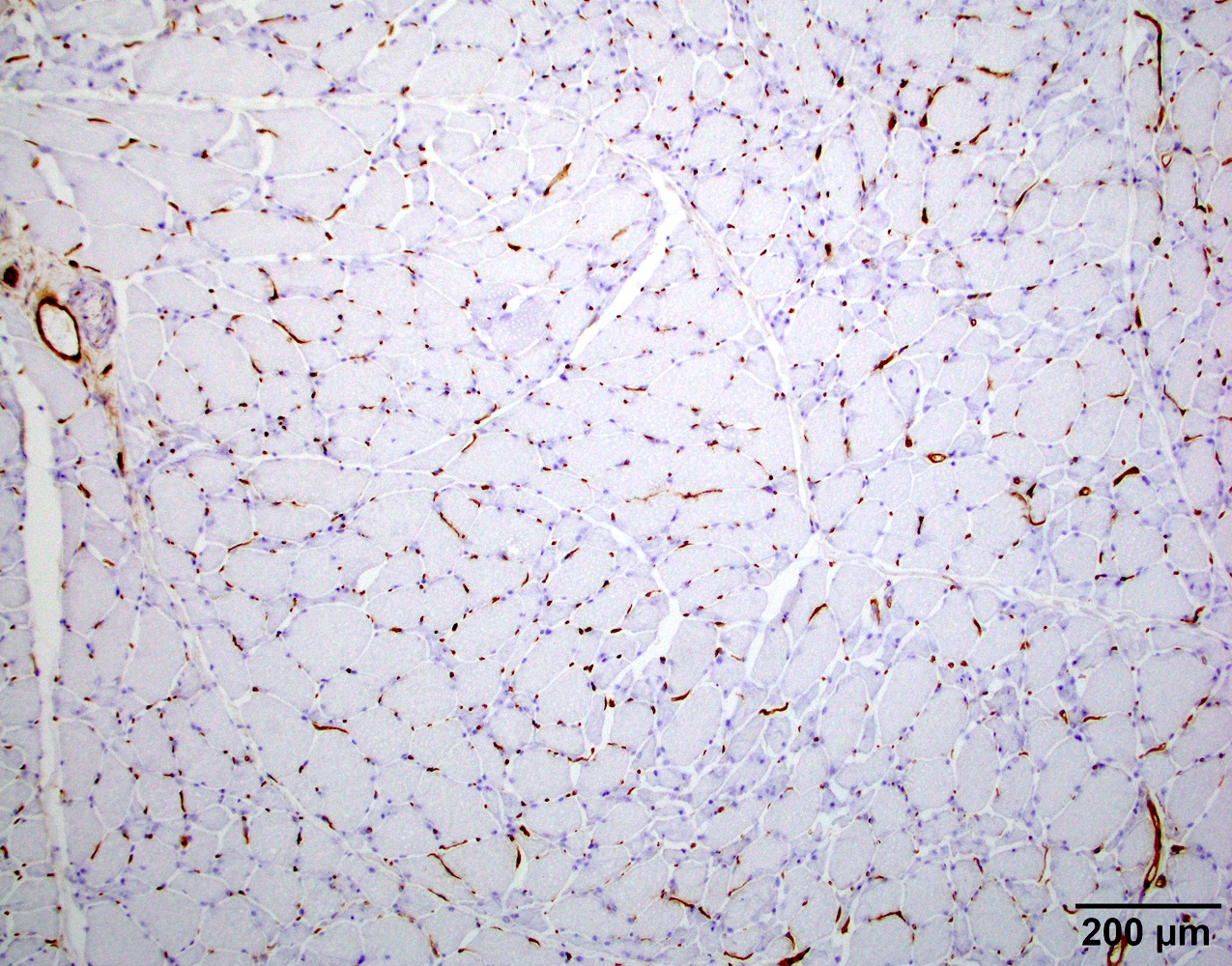
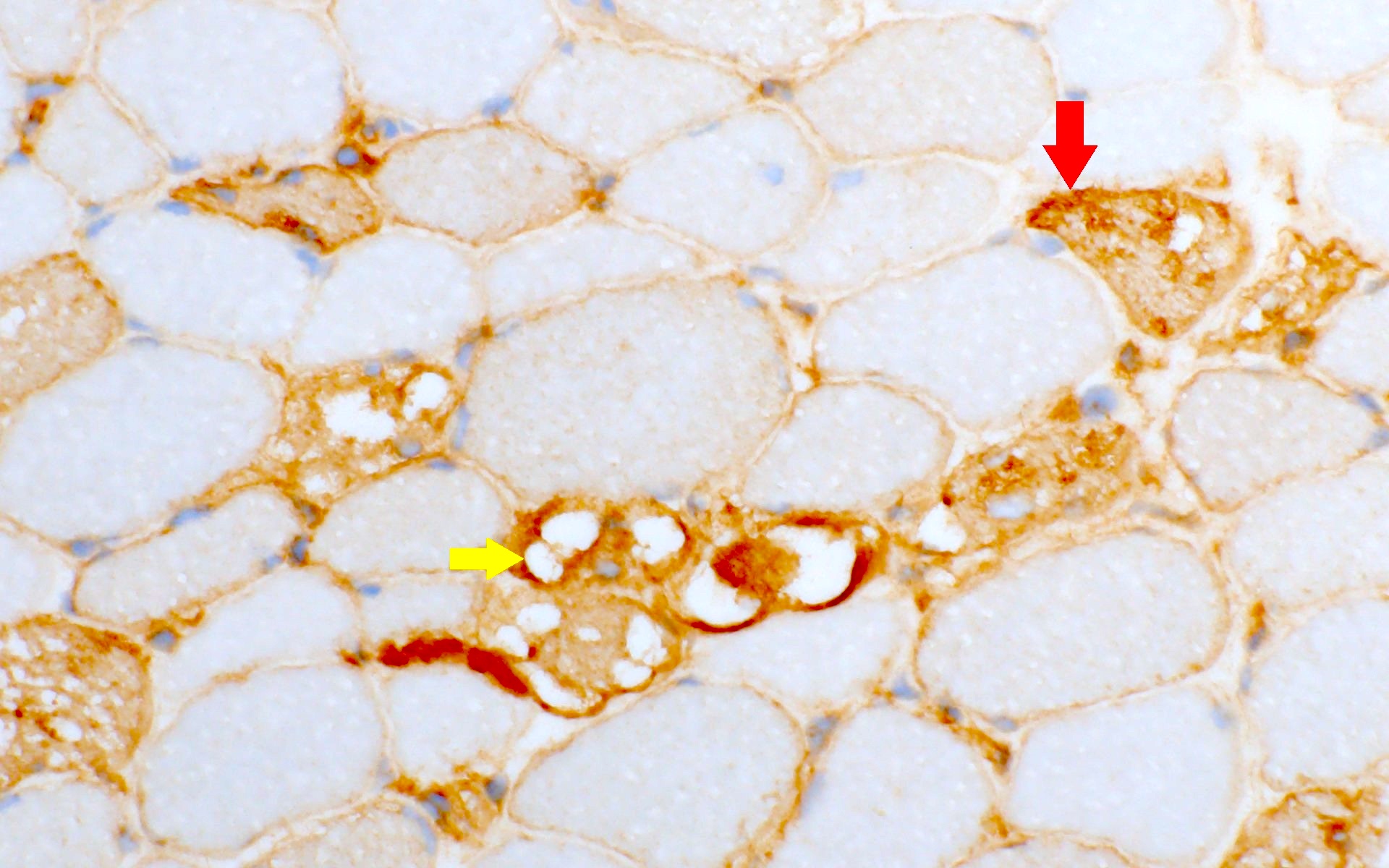
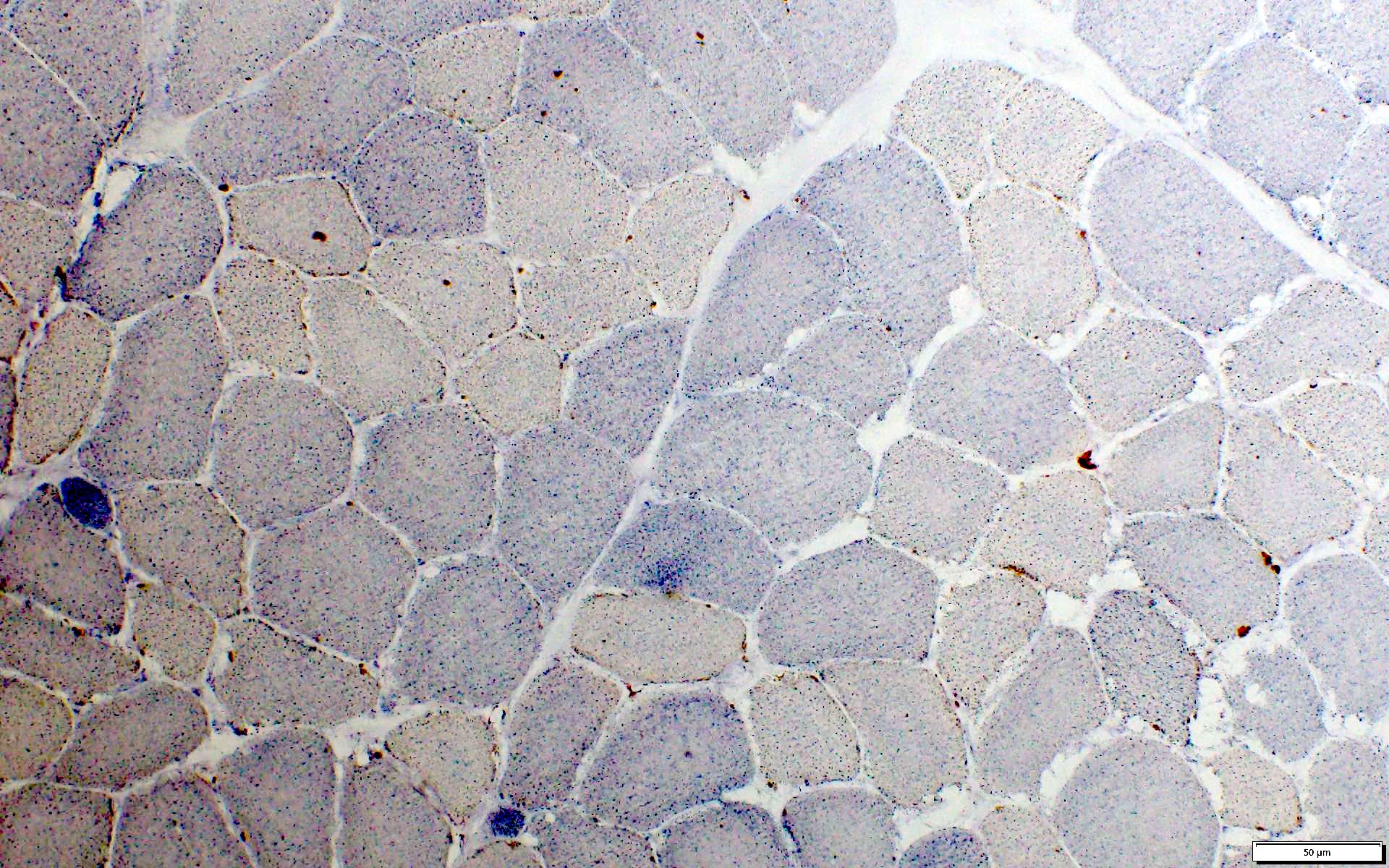
- Transthyretin IHC stain highlights both mutant and wild type forms of TTR amyloid deposits
- References: Blood 2012;119:488, Arch Neurol 1969;20:490
Contributed by Chunyu Cai, M.D., Ph.D.
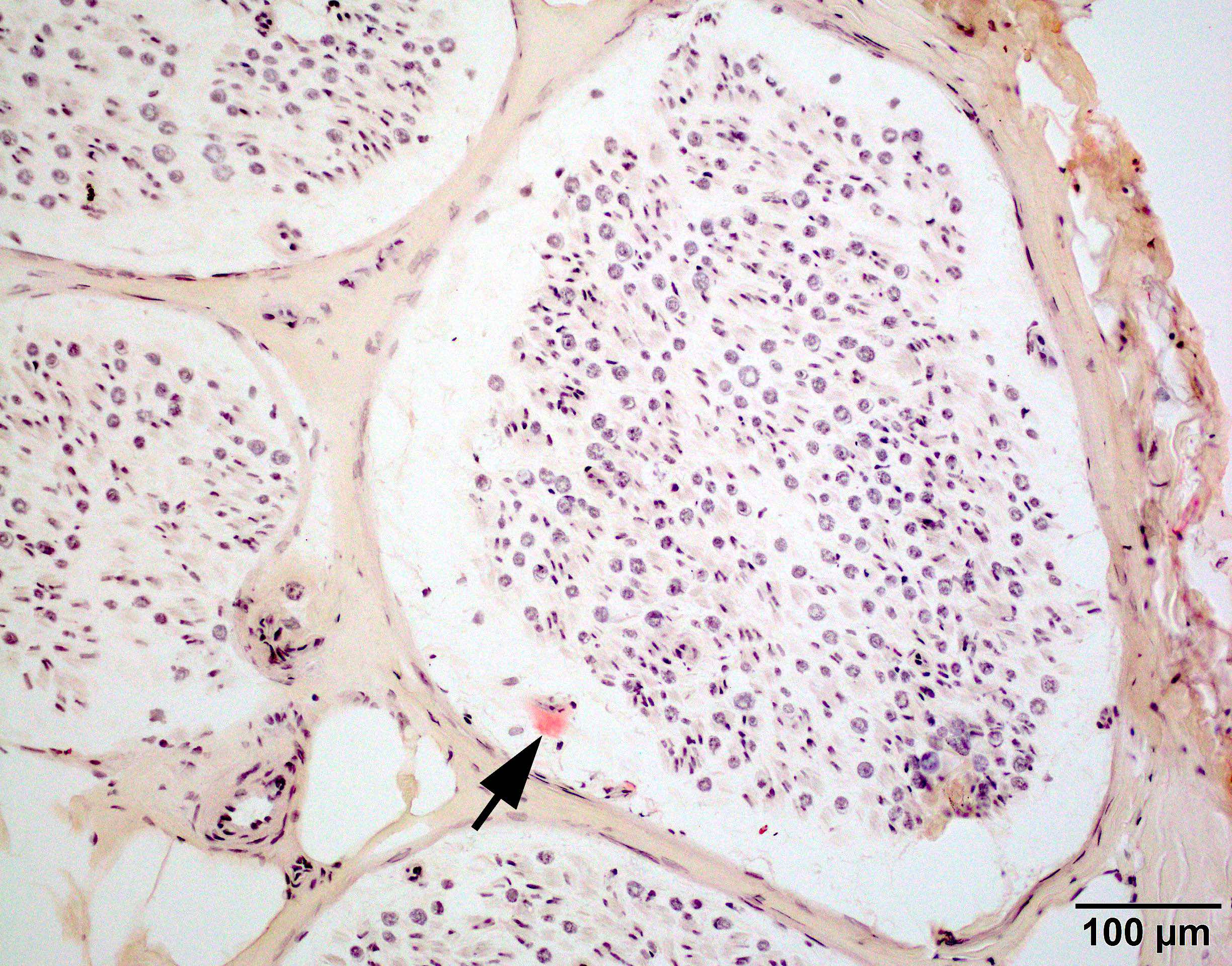
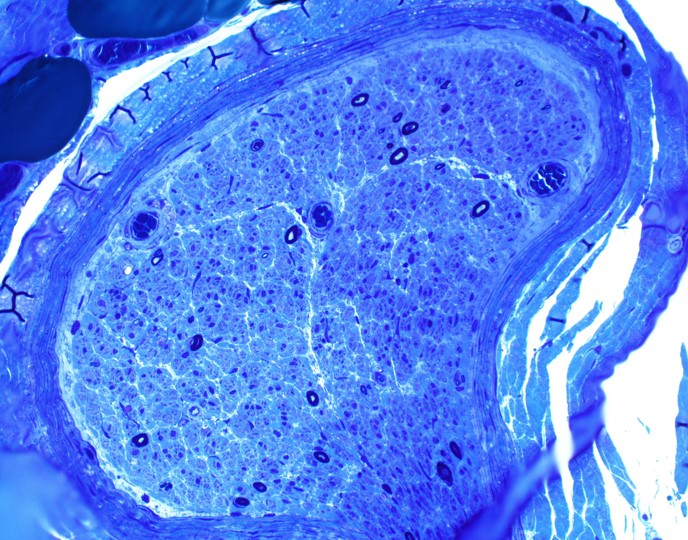
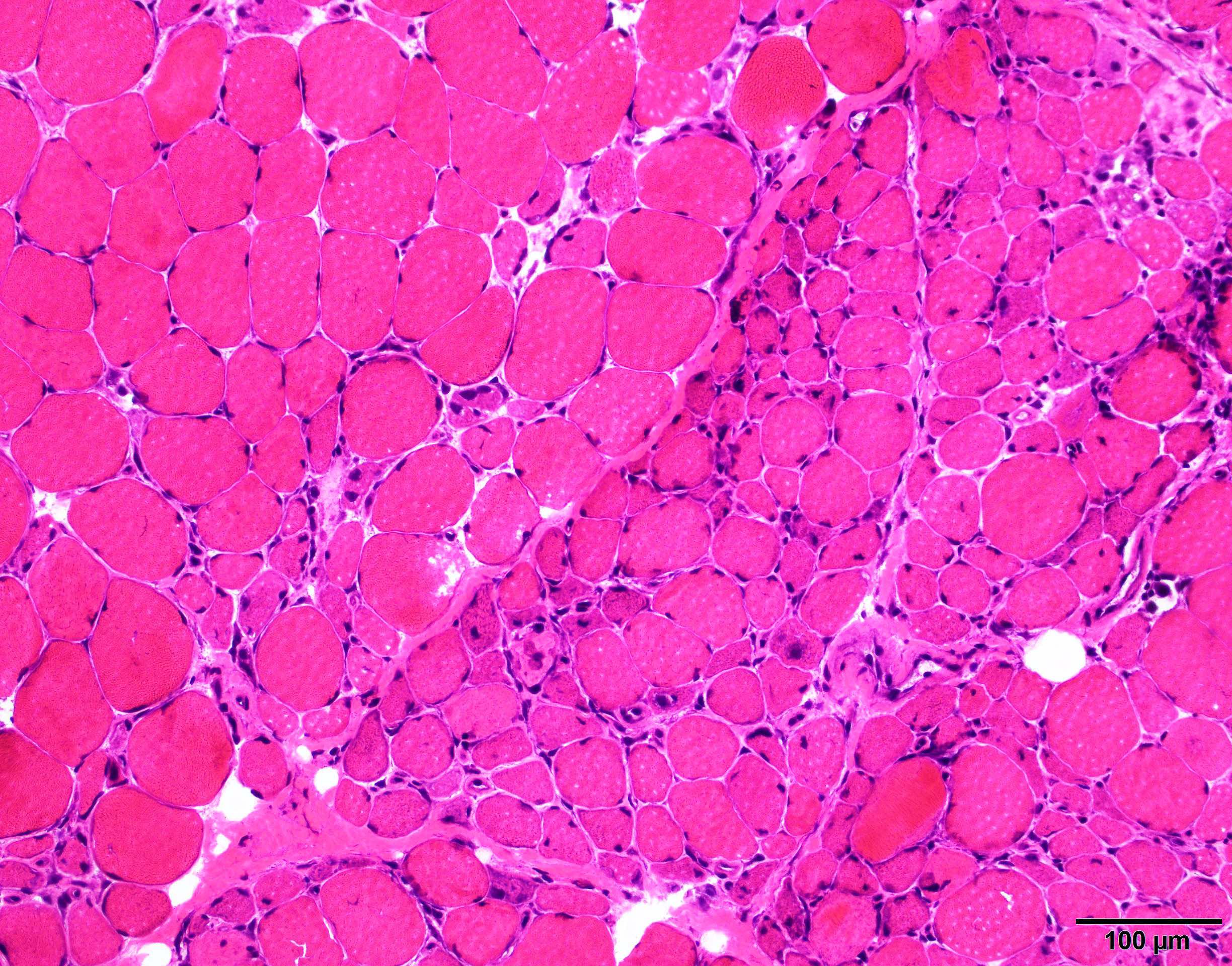
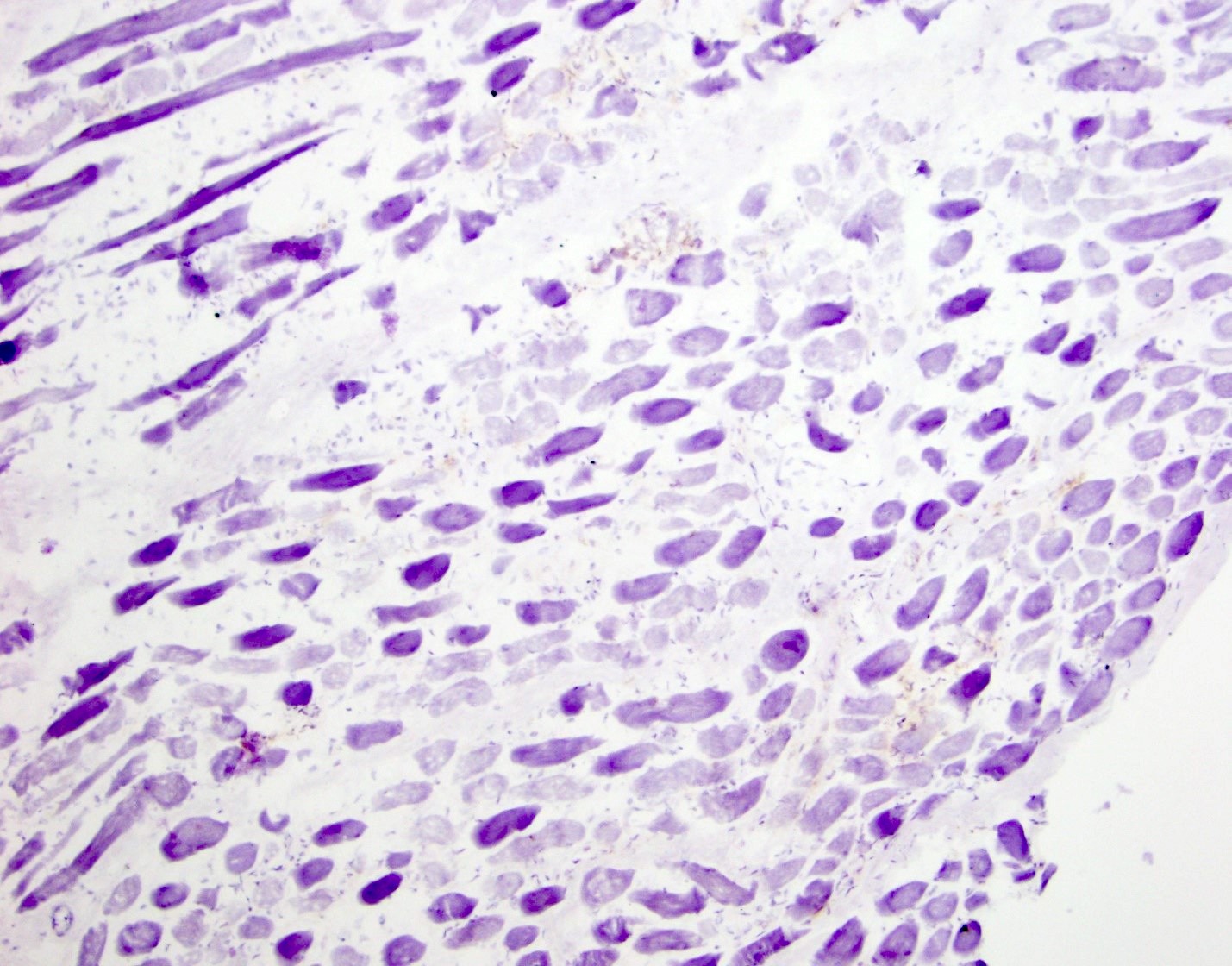
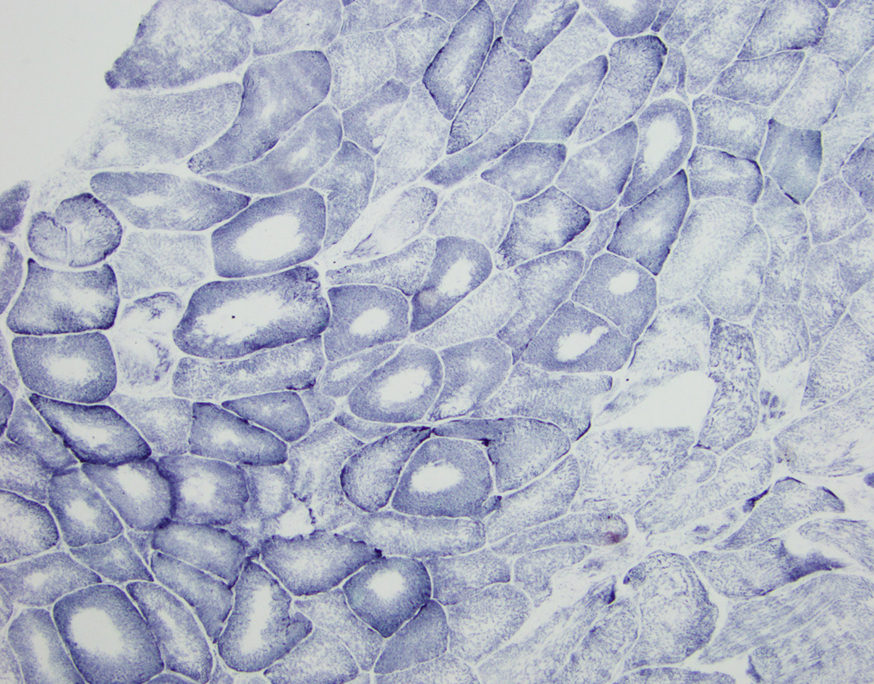
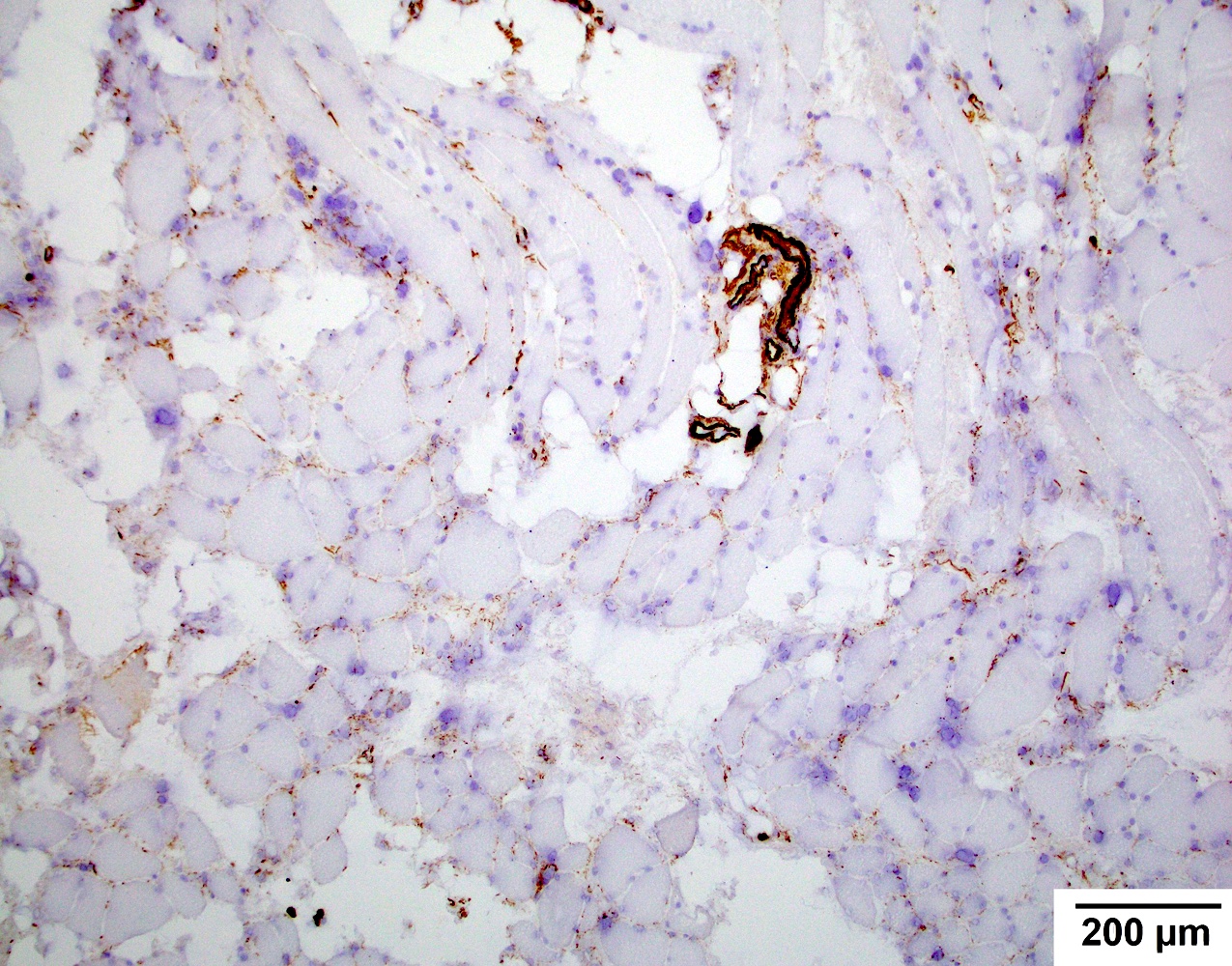
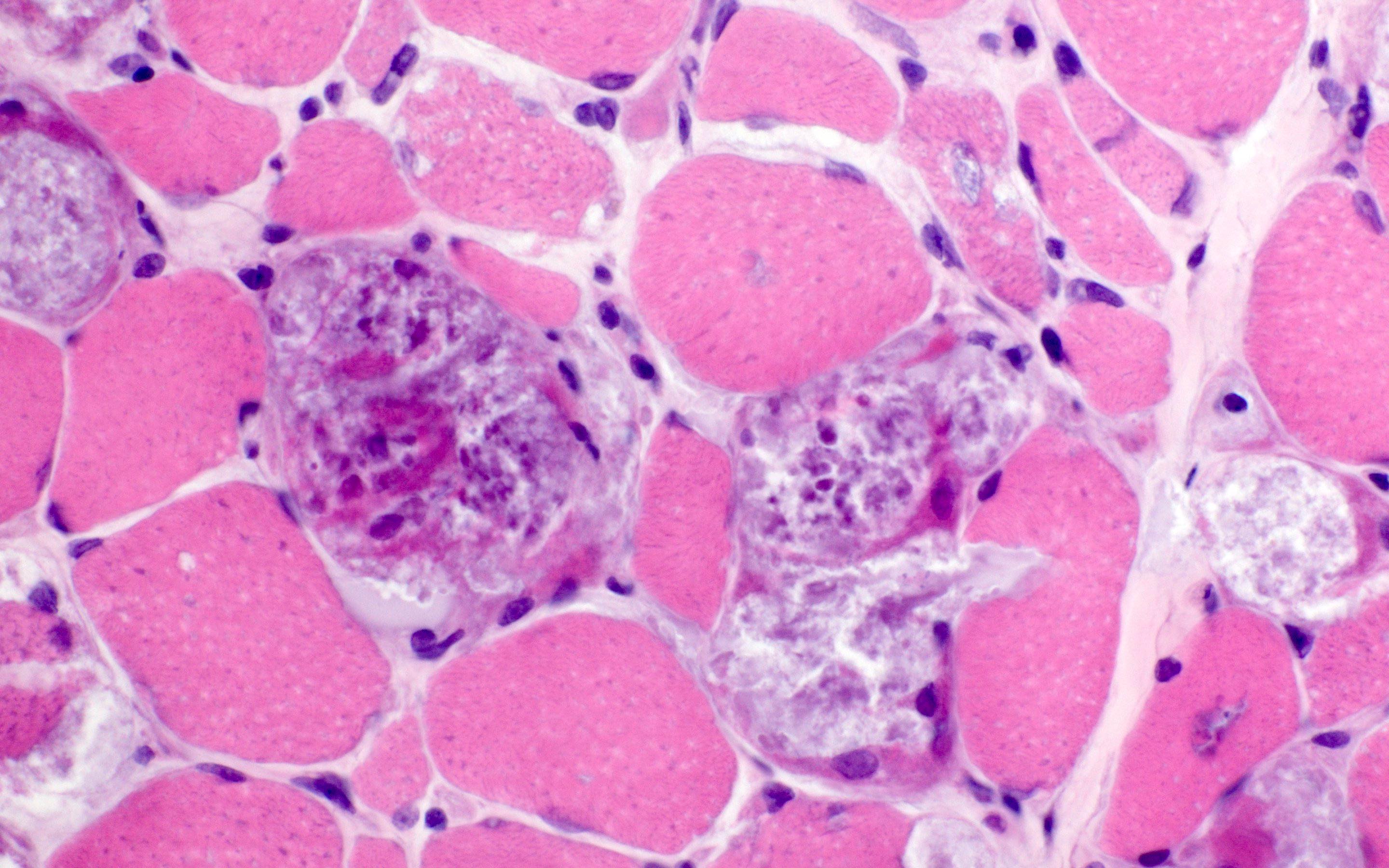
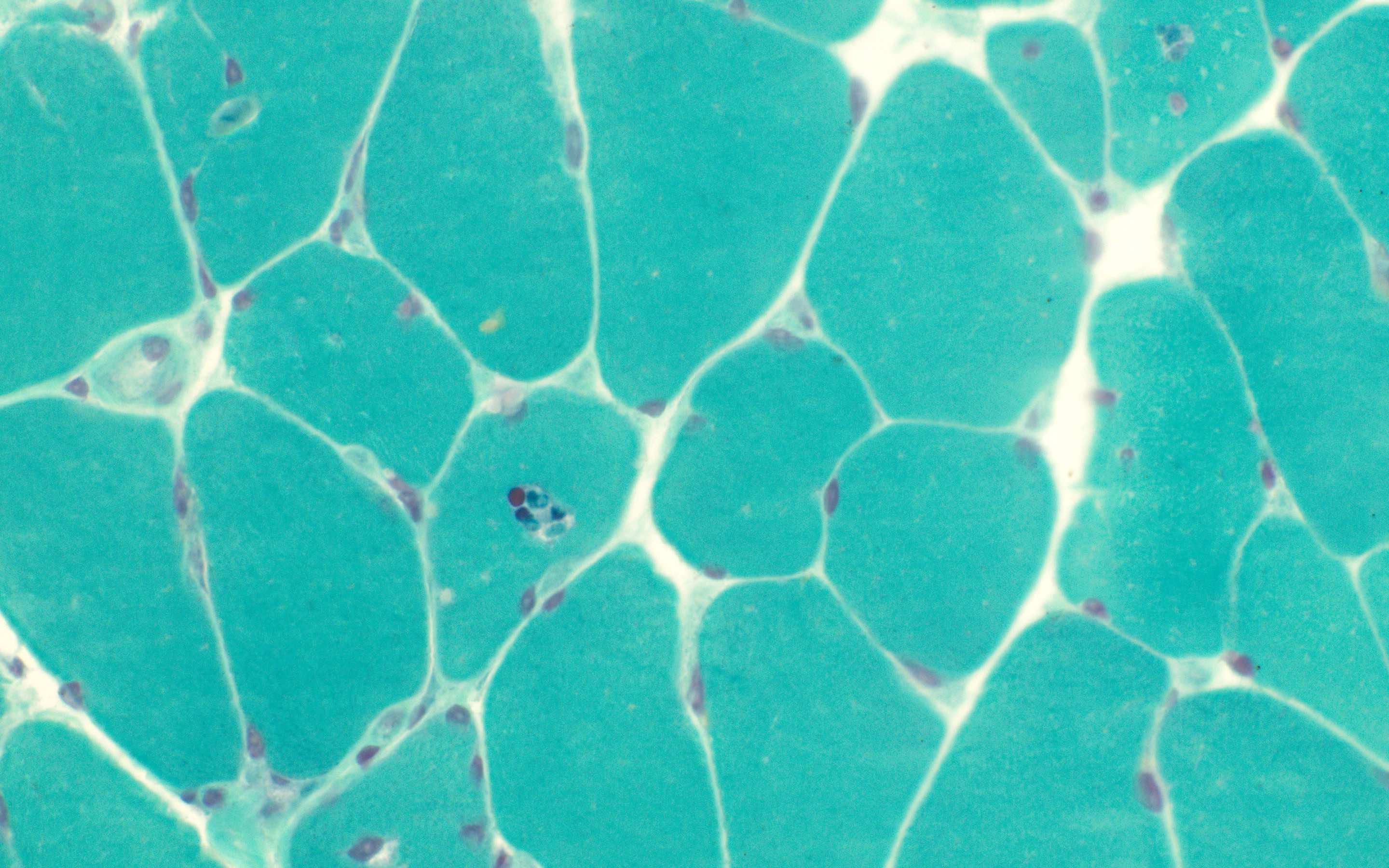
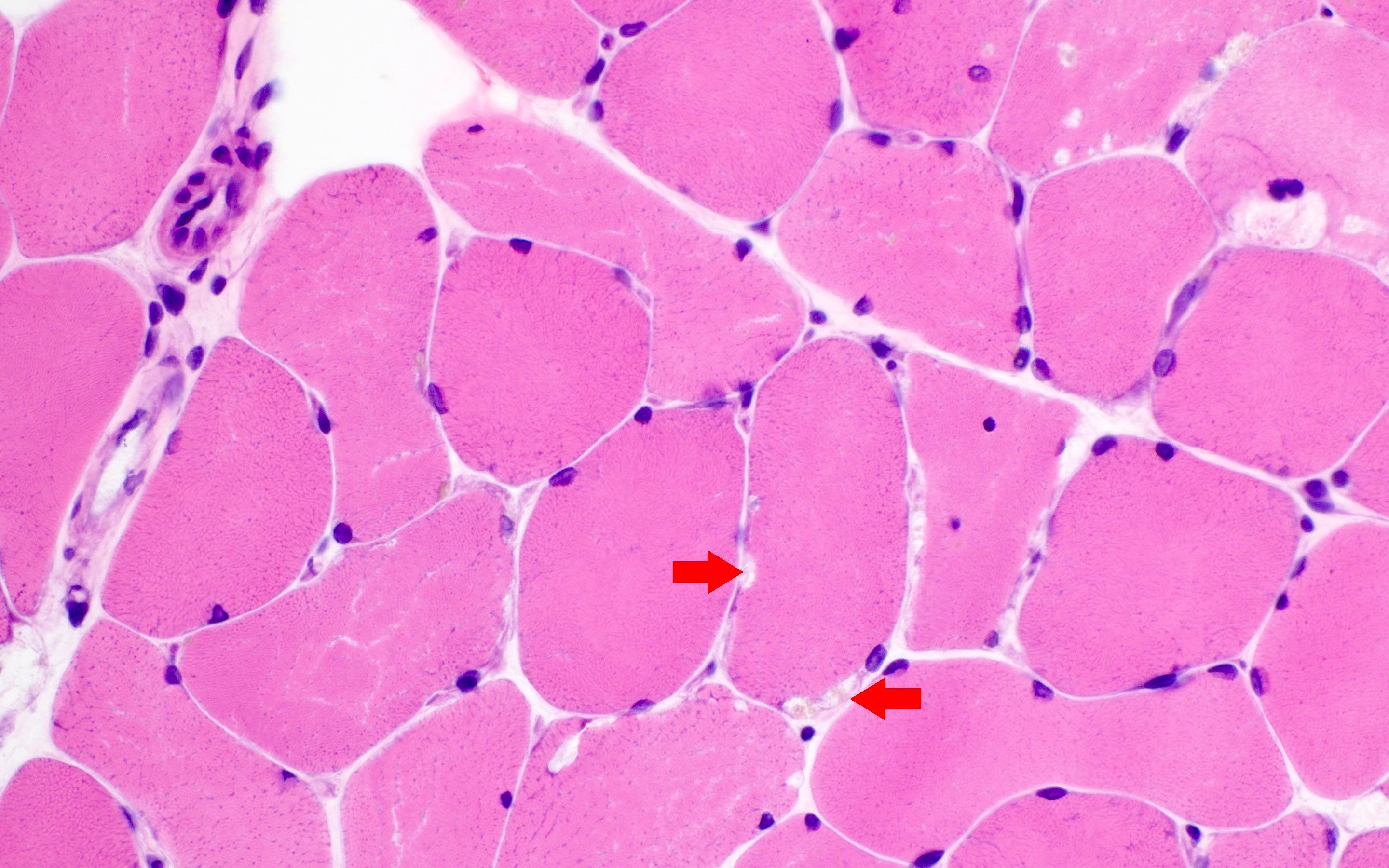
Early amyloid neuropathy

Nerve edema
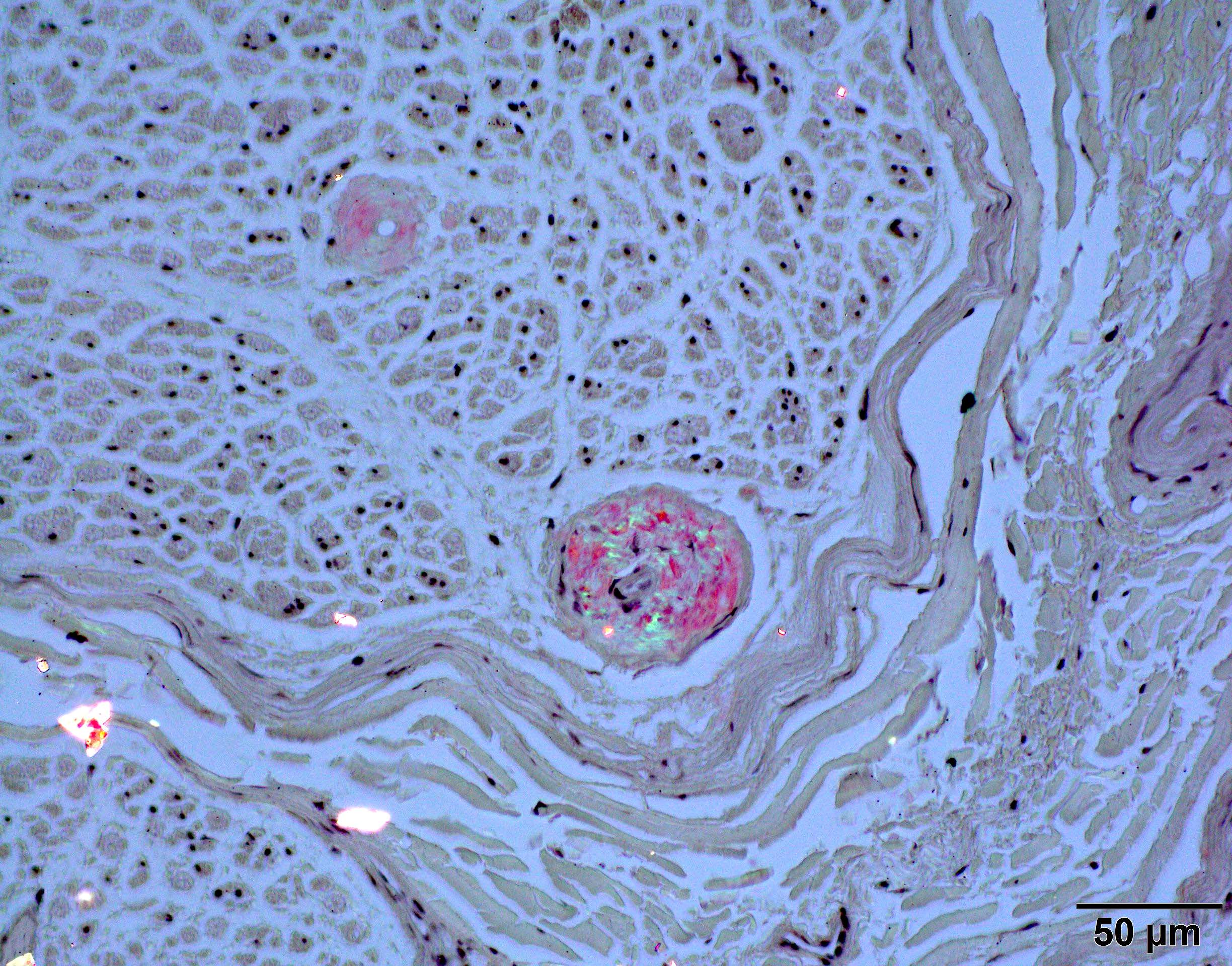
Congo red amyloid deposit
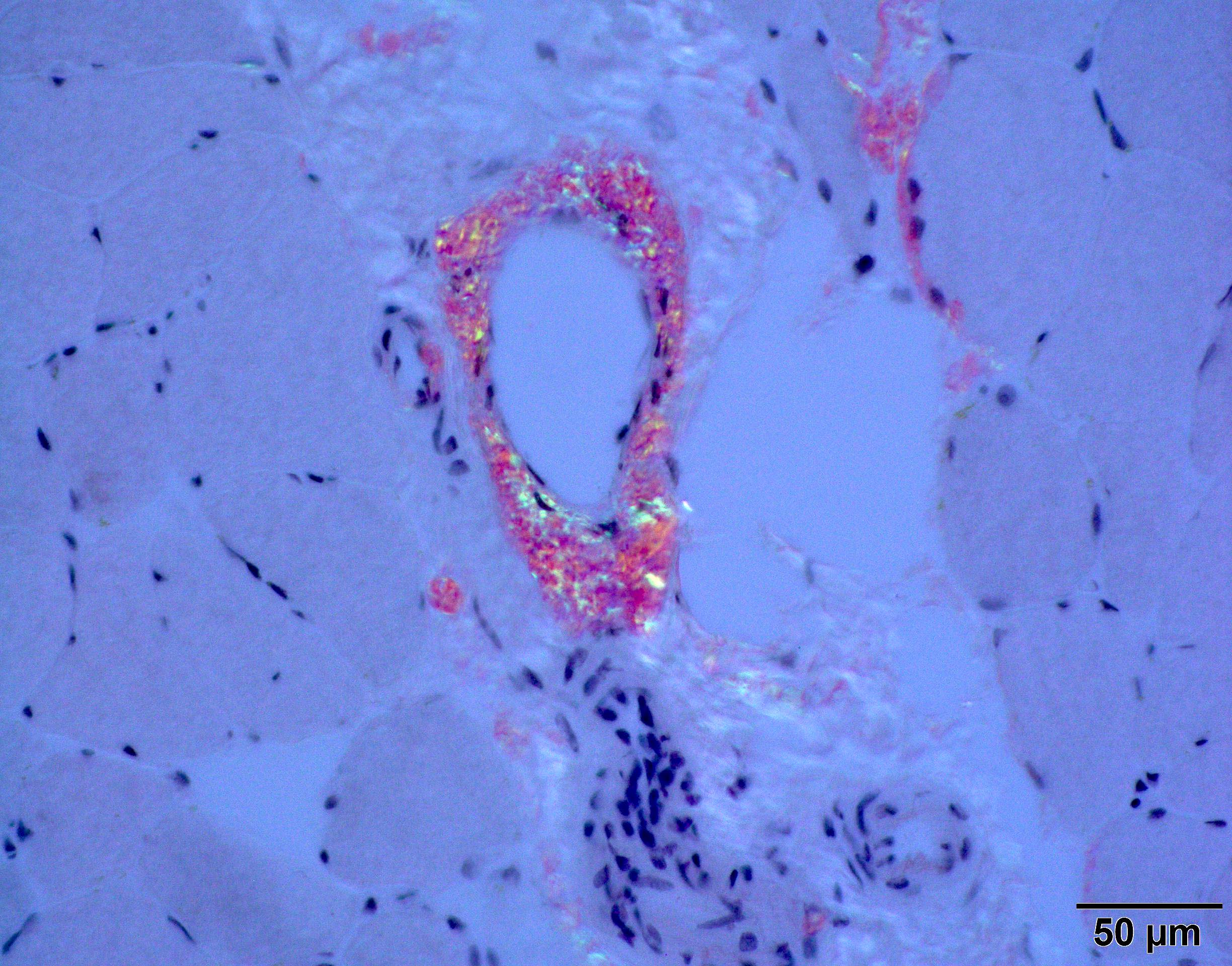
Congo red polarized
Loss of small myelinated axons
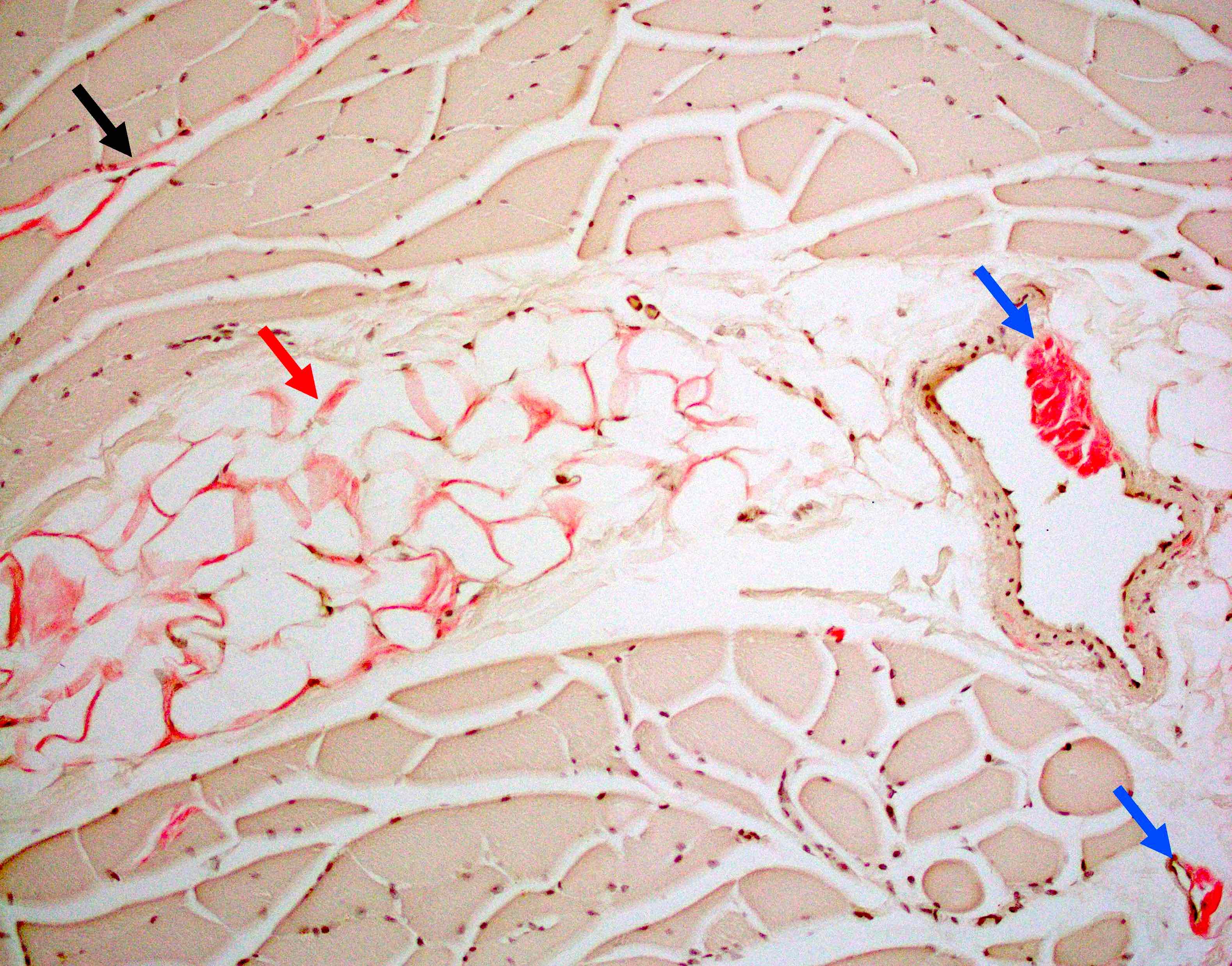
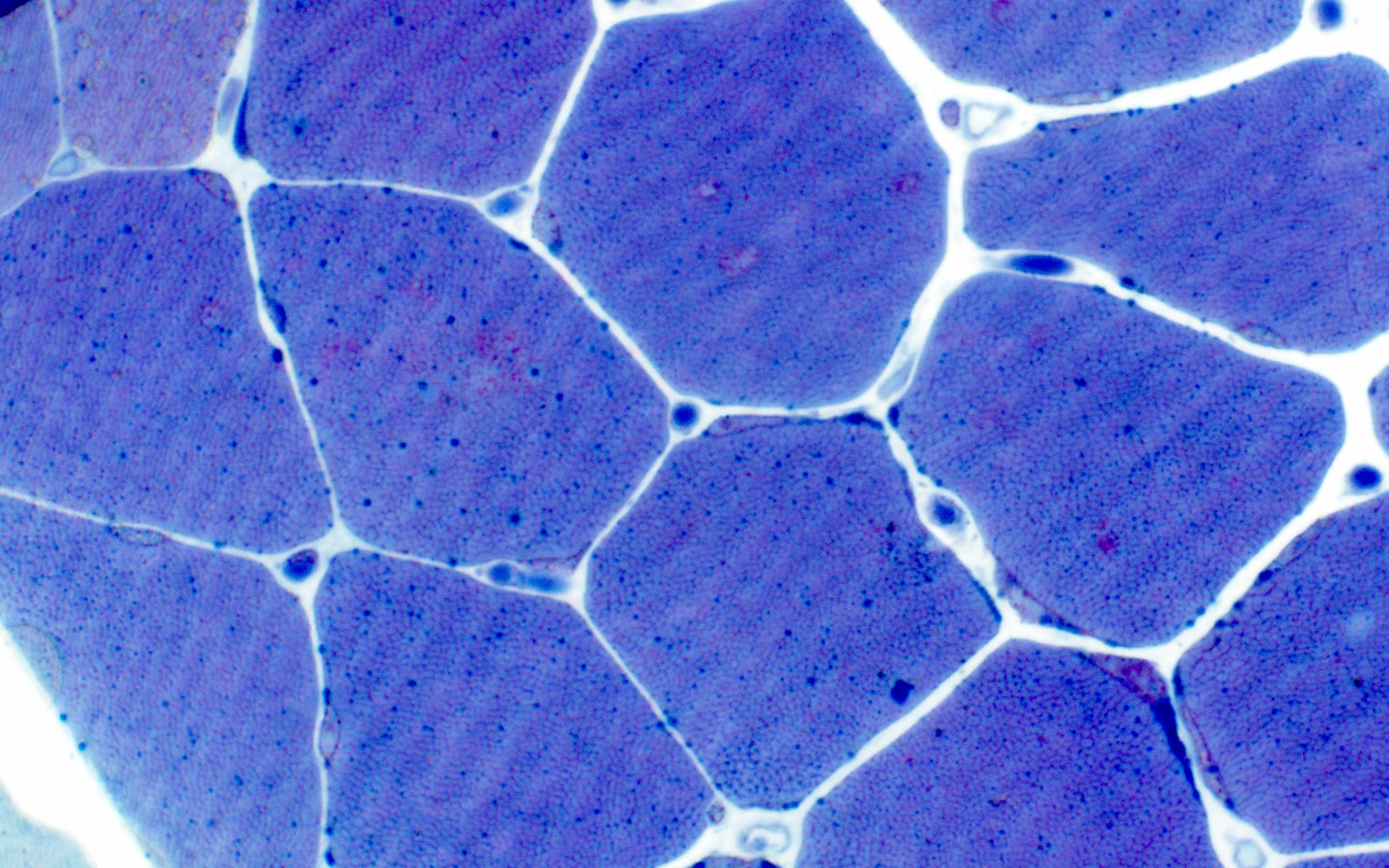
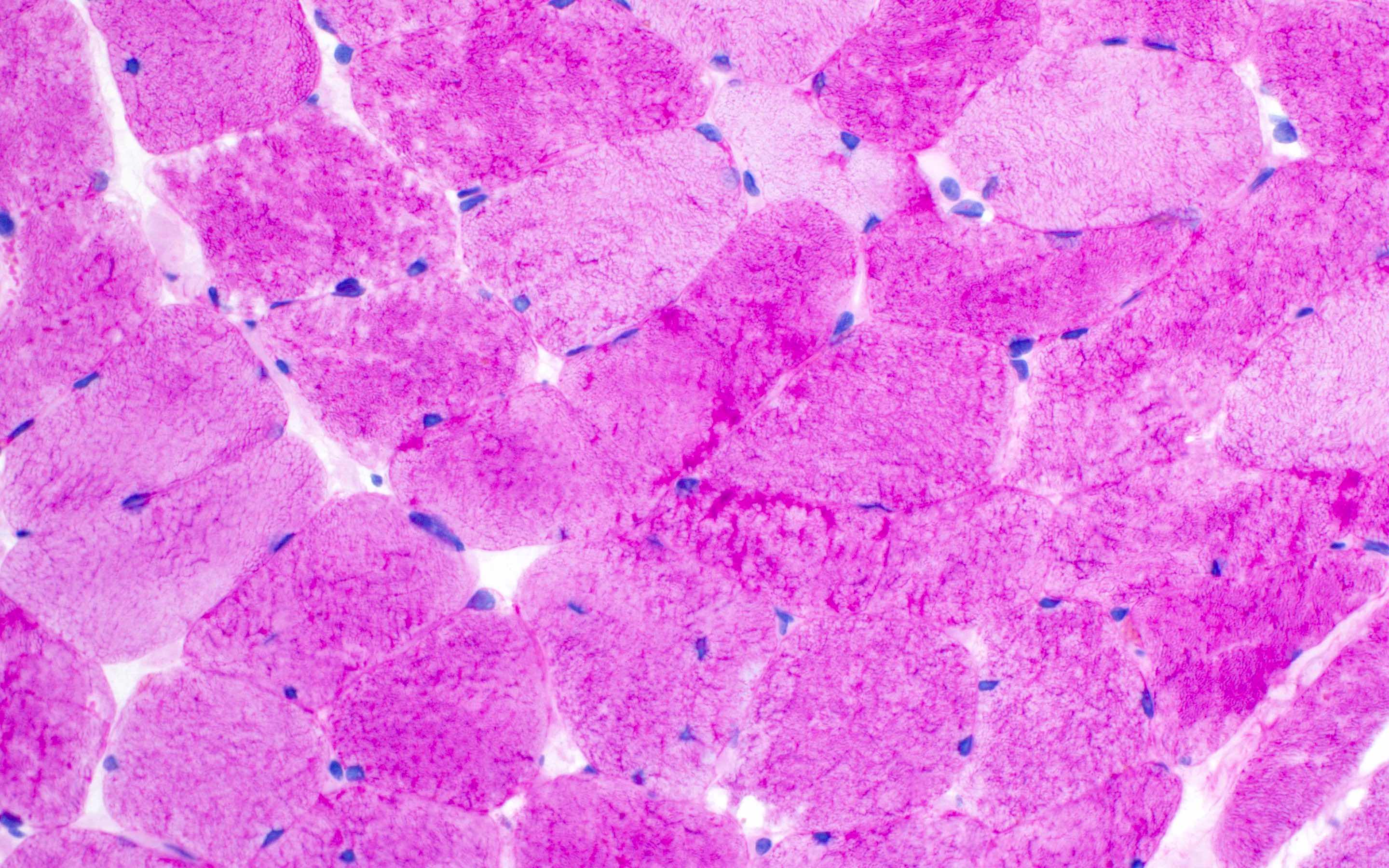
Late amyloid neuropathy
Nerve edema
Congo red amyloid deposit
Congo red polarized
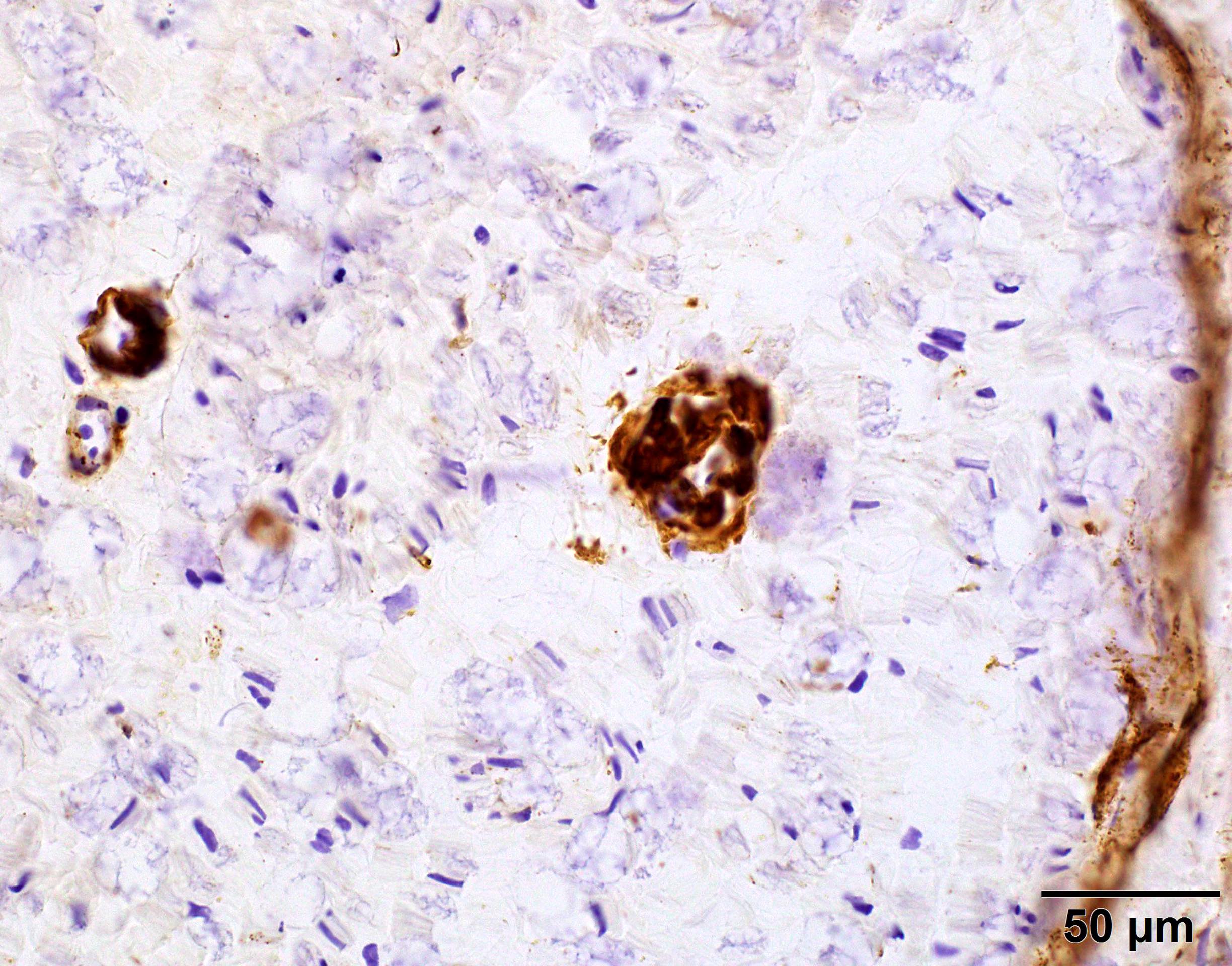
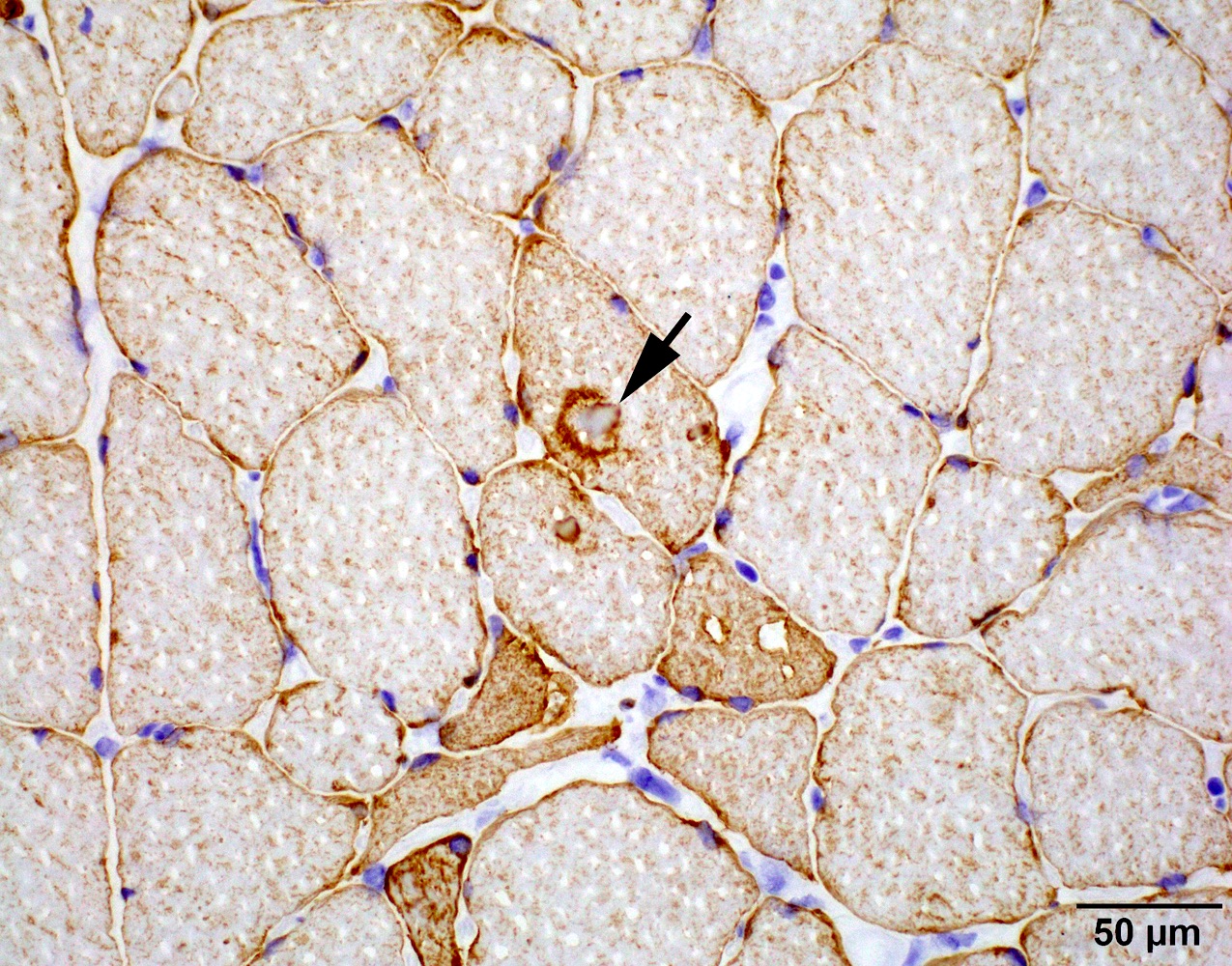
C5b9 amyloid deposit
Small and large axon loss
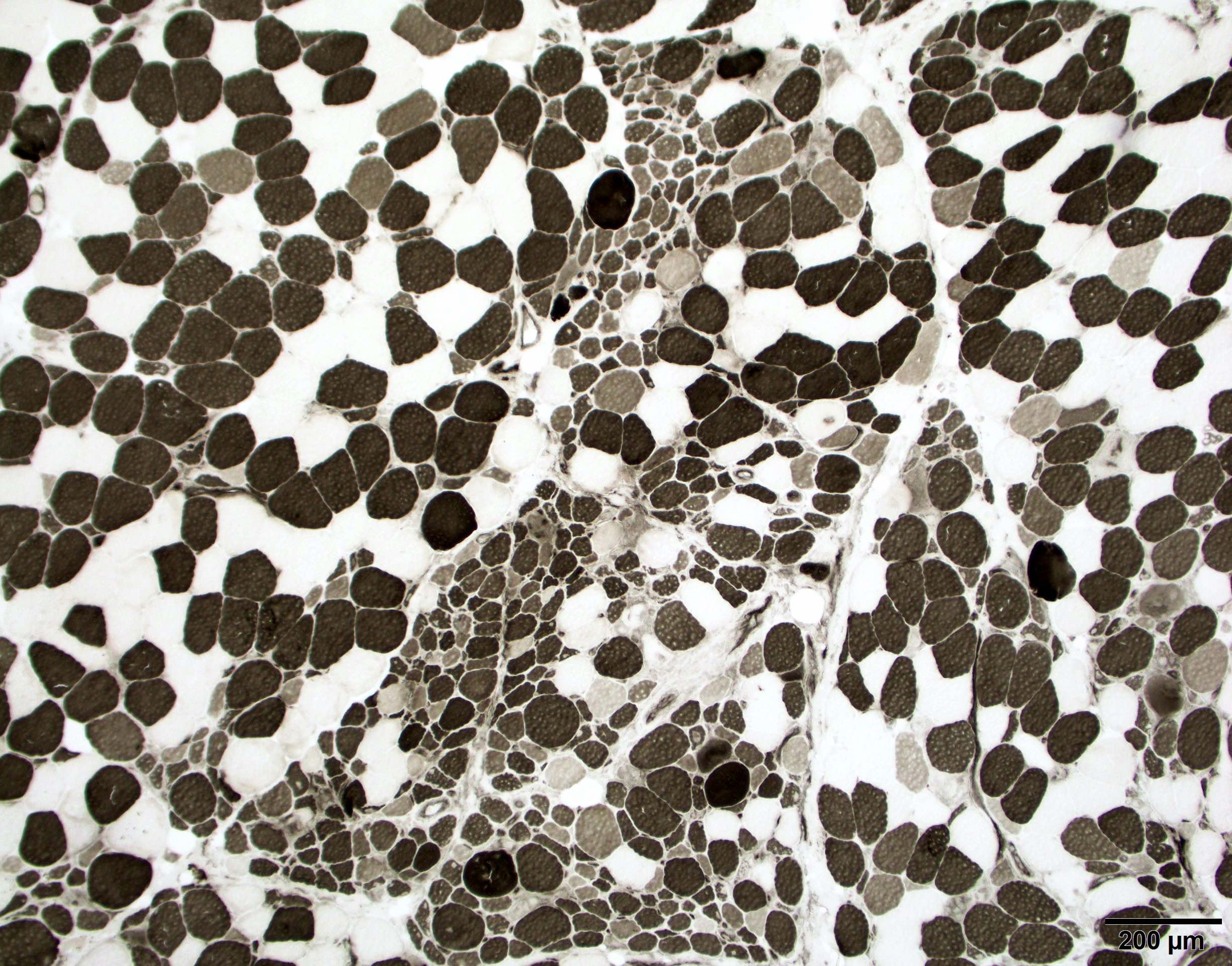
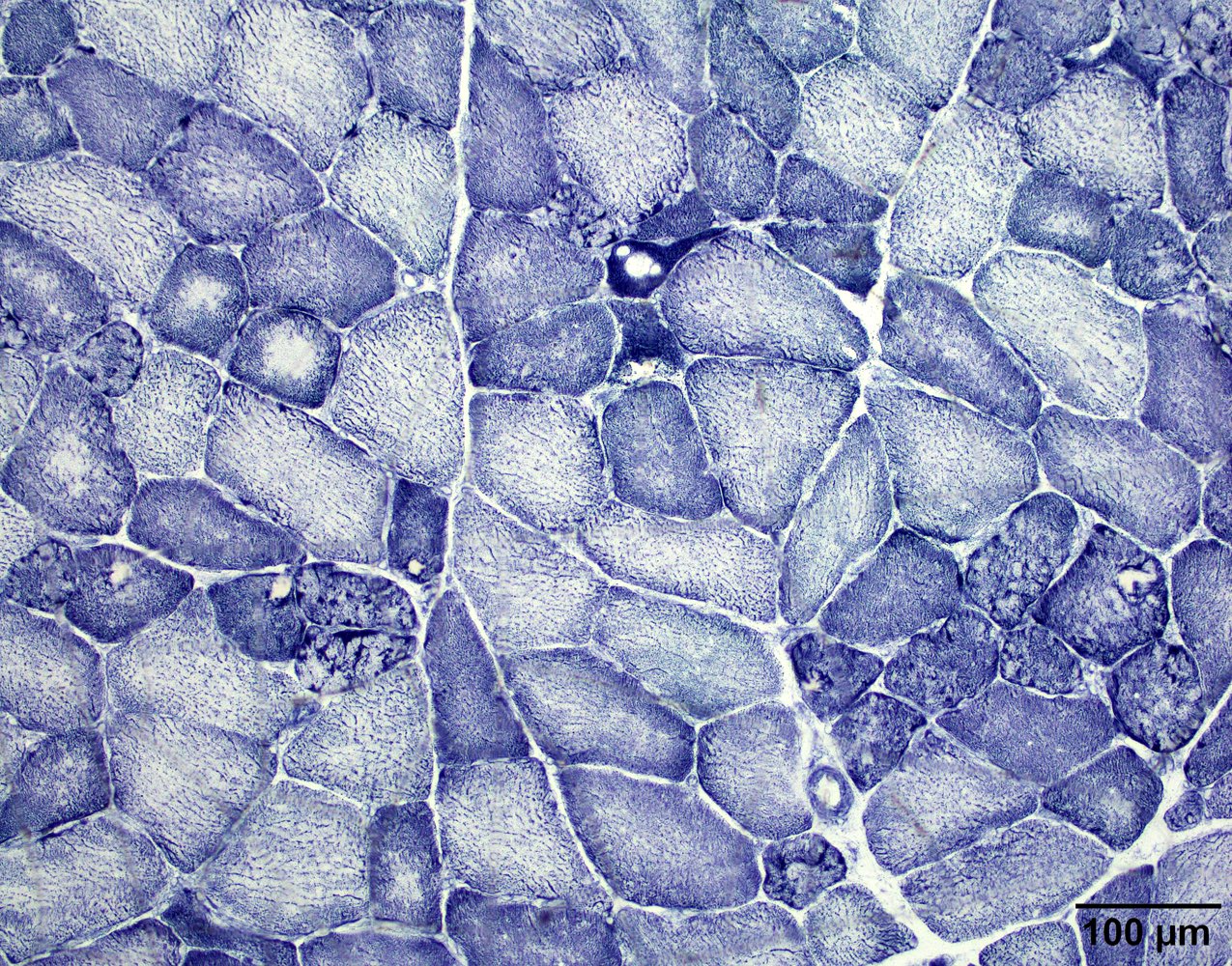
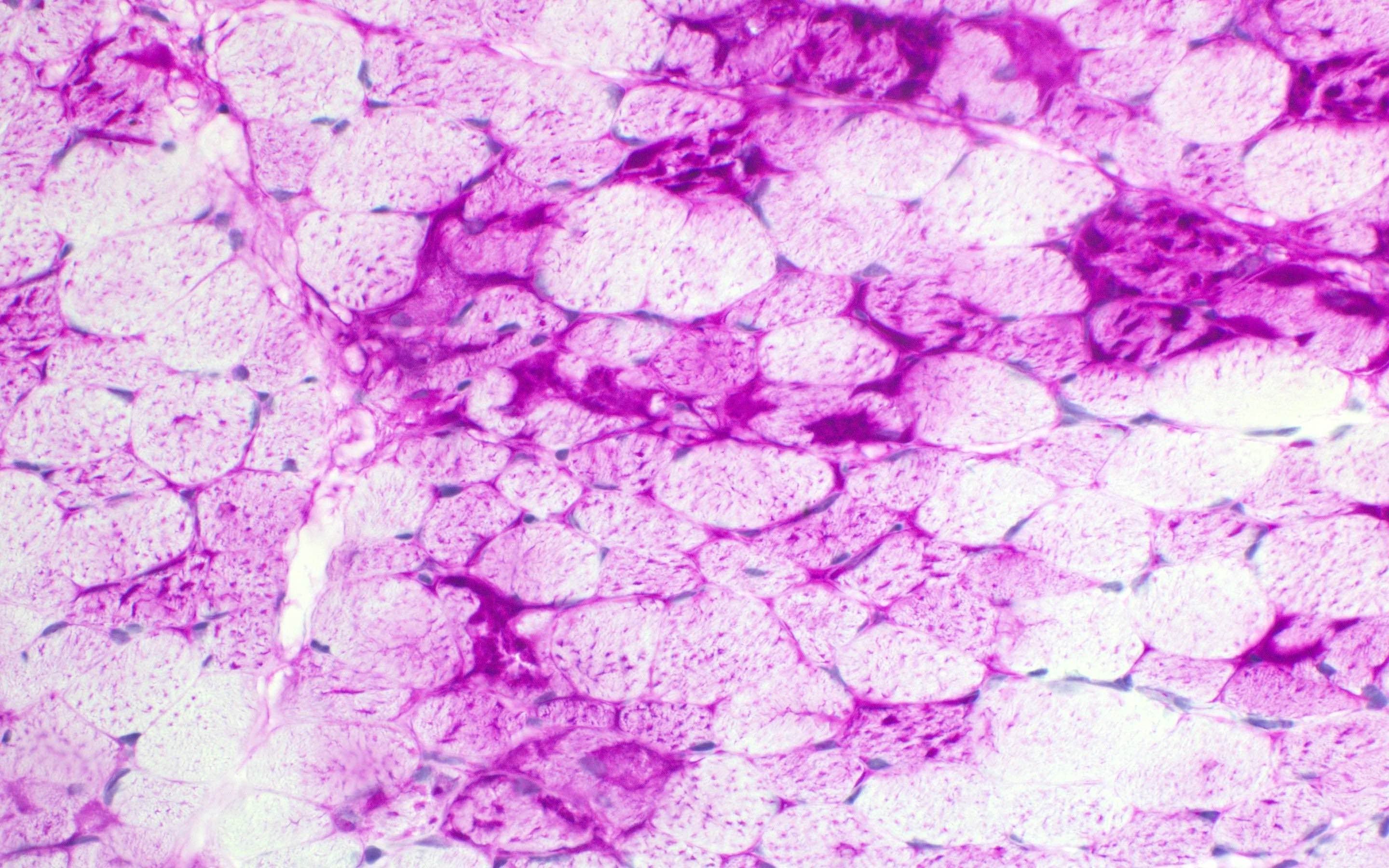
Hereditary transthyretin amyloid neuropathy
Nerve Congo red
Nerve plastic section
Muscle Congo red
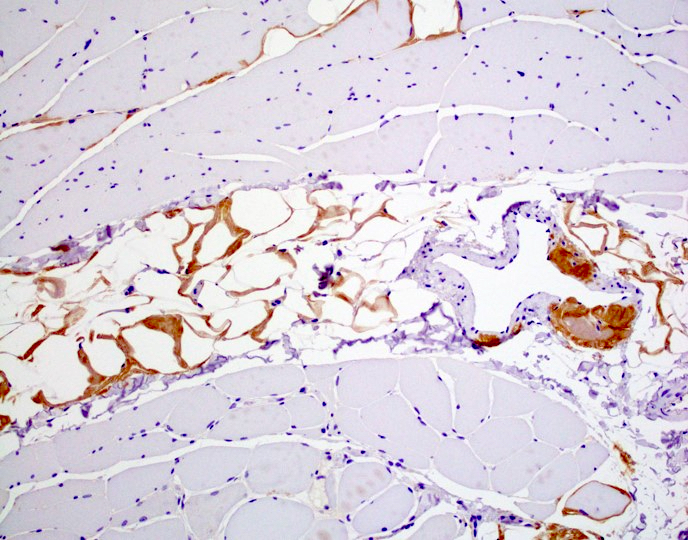
Muscle TTR
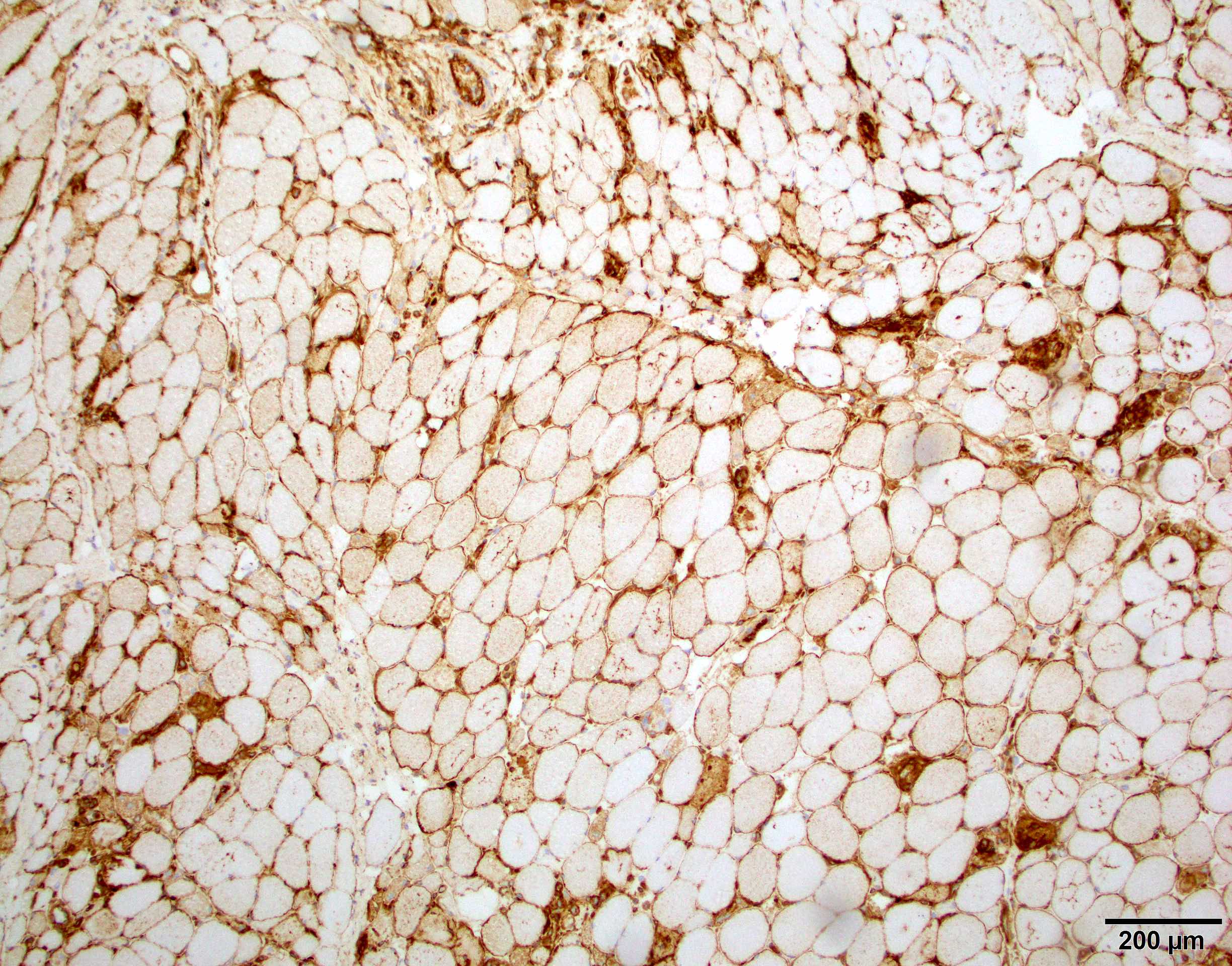
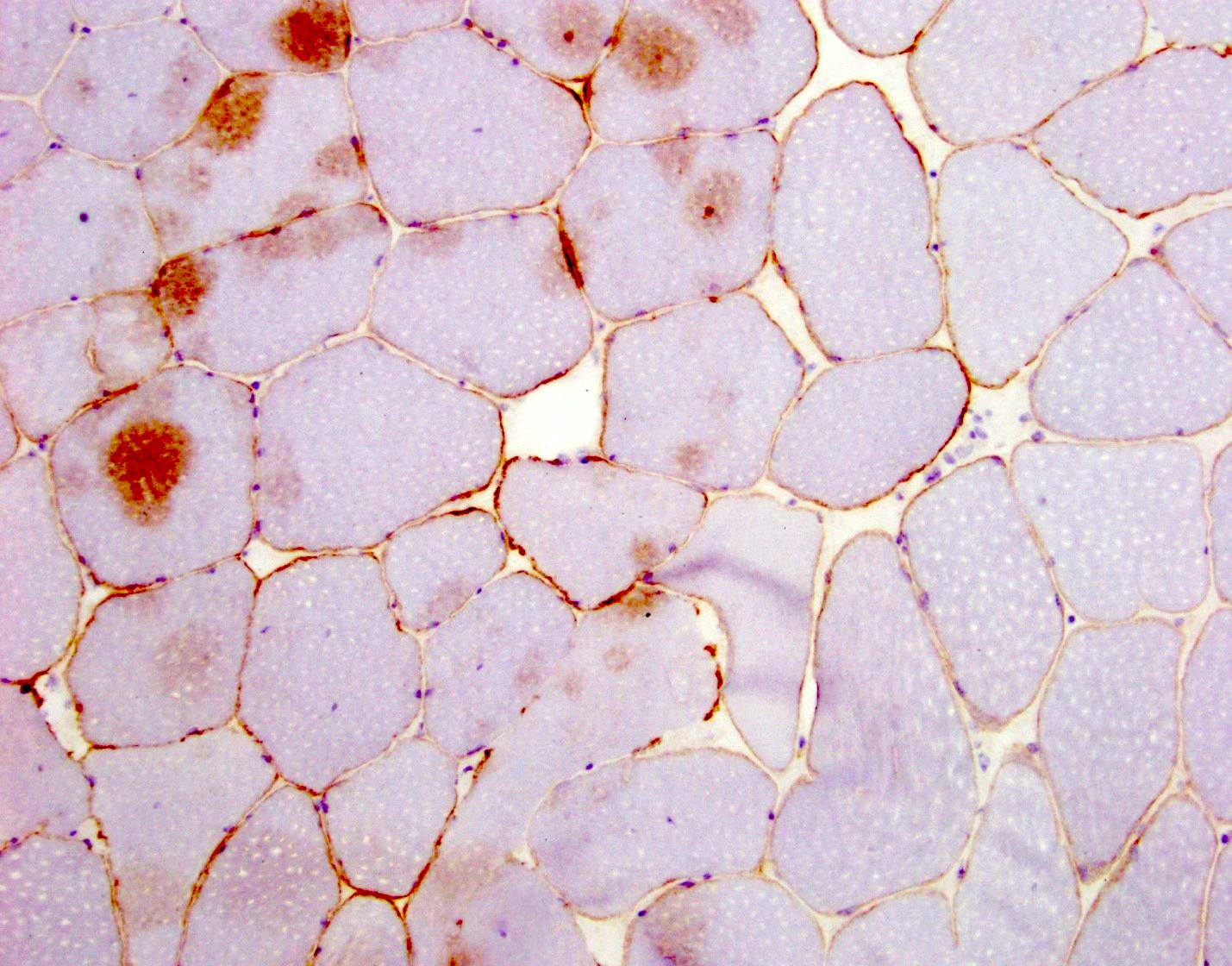
AL amyloid neuropathy
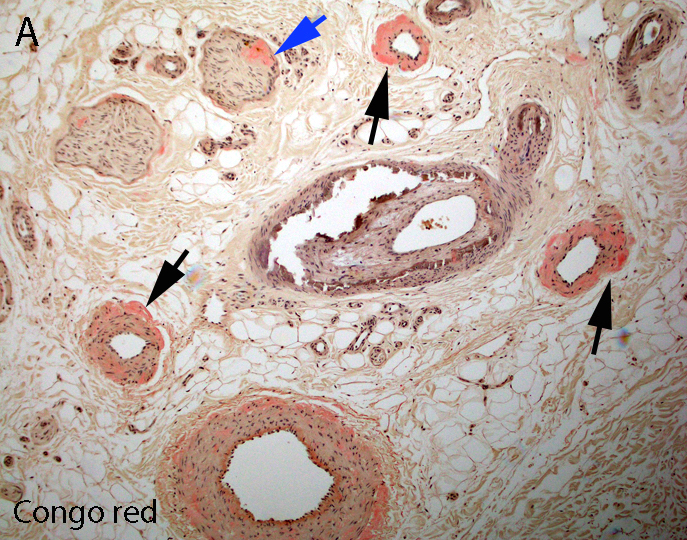
Congo red
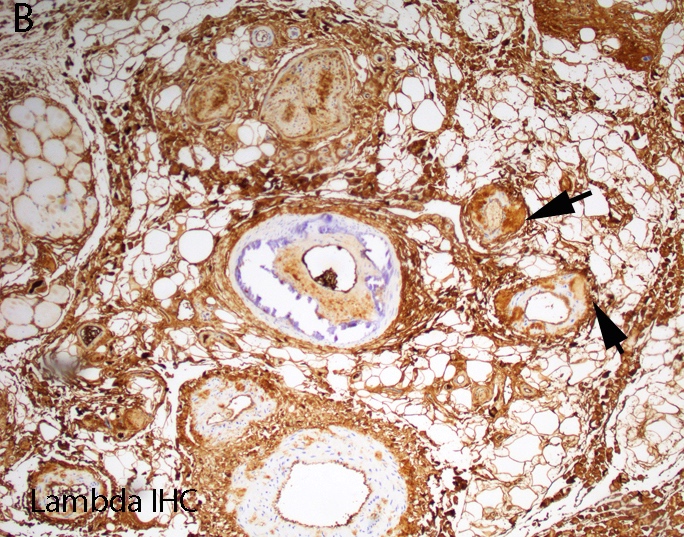
Lambda IHC
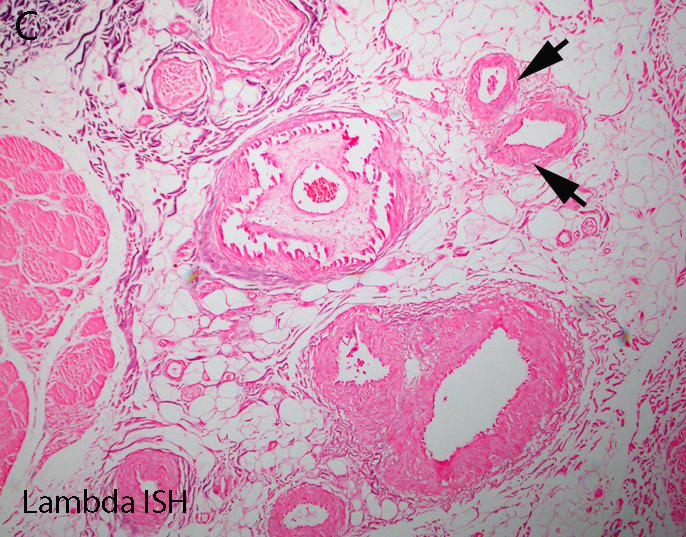
Lambda in situ hybridization
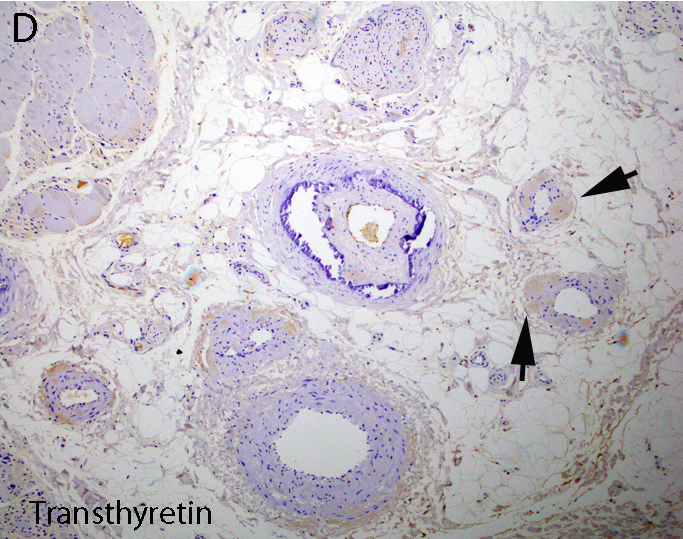
Transthyretin IHC
- General amyloid markers
- Congo red
- Thioflavin T or S
- Terminal complement complex (C5b9)
- Crystal violet
- Subtype specific amyloid markers
- Transthyretin (Blood 2012;119:488)
- Nerve damage (J Neurol Sci 2021:421:117305, Neurology 2016;87:2220)
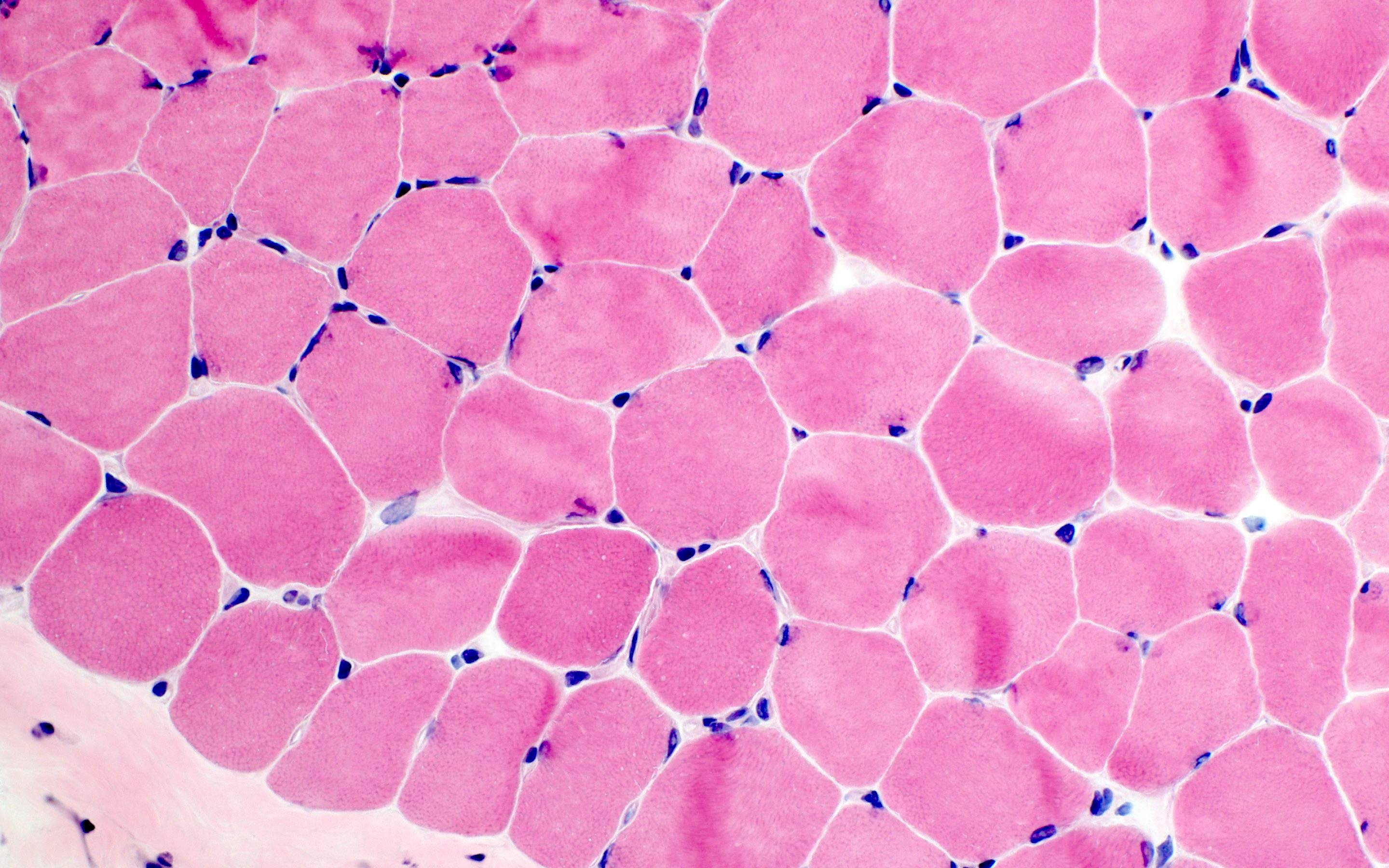
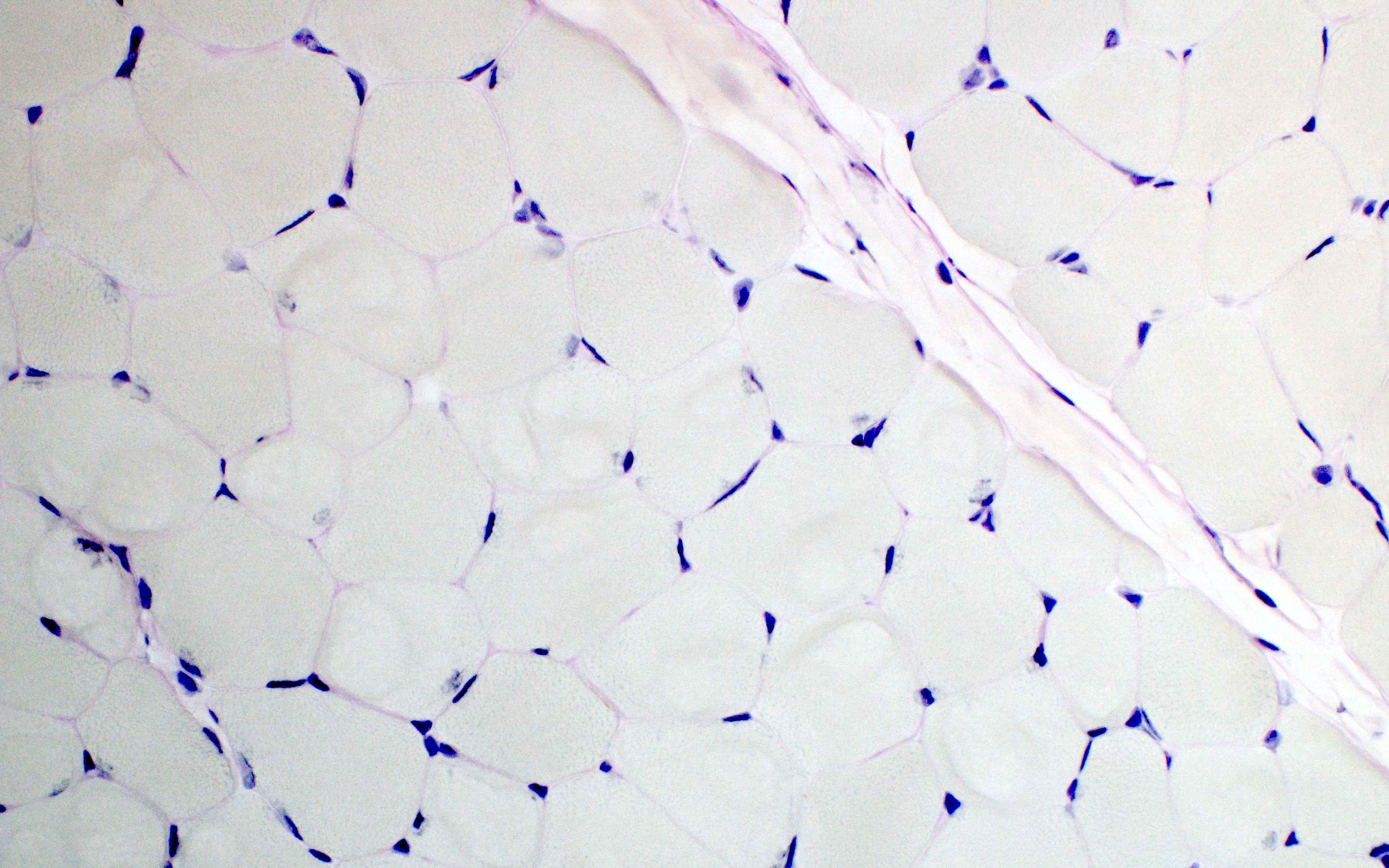
- Prominent loss of small myelinated and unmyelinated axons; relatively preserved large myelinated axons
- Atrophy of Schwann cells
- Damage of endothelial cells and disrupted blood nerve barrier
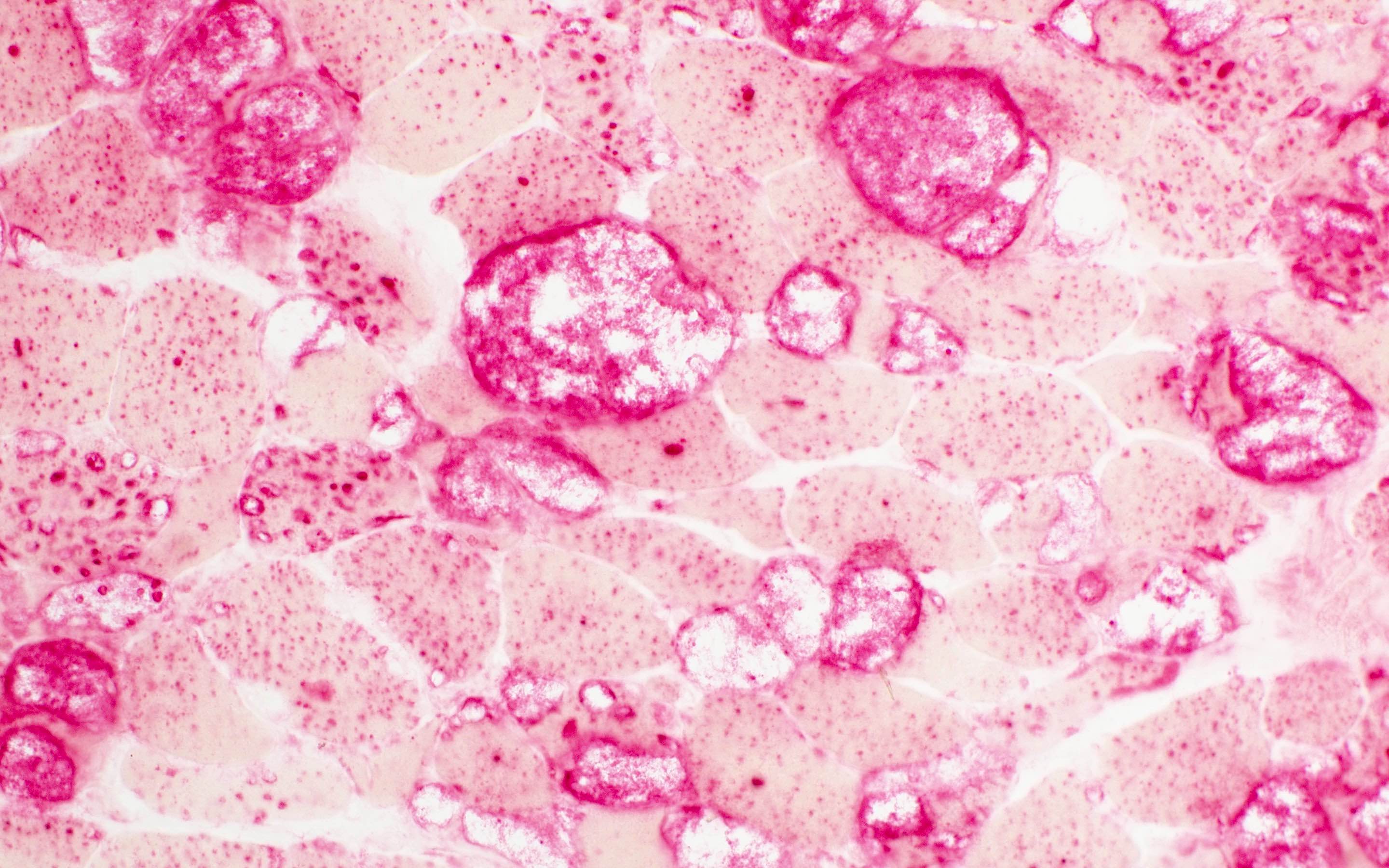
- Amyloid fibrils
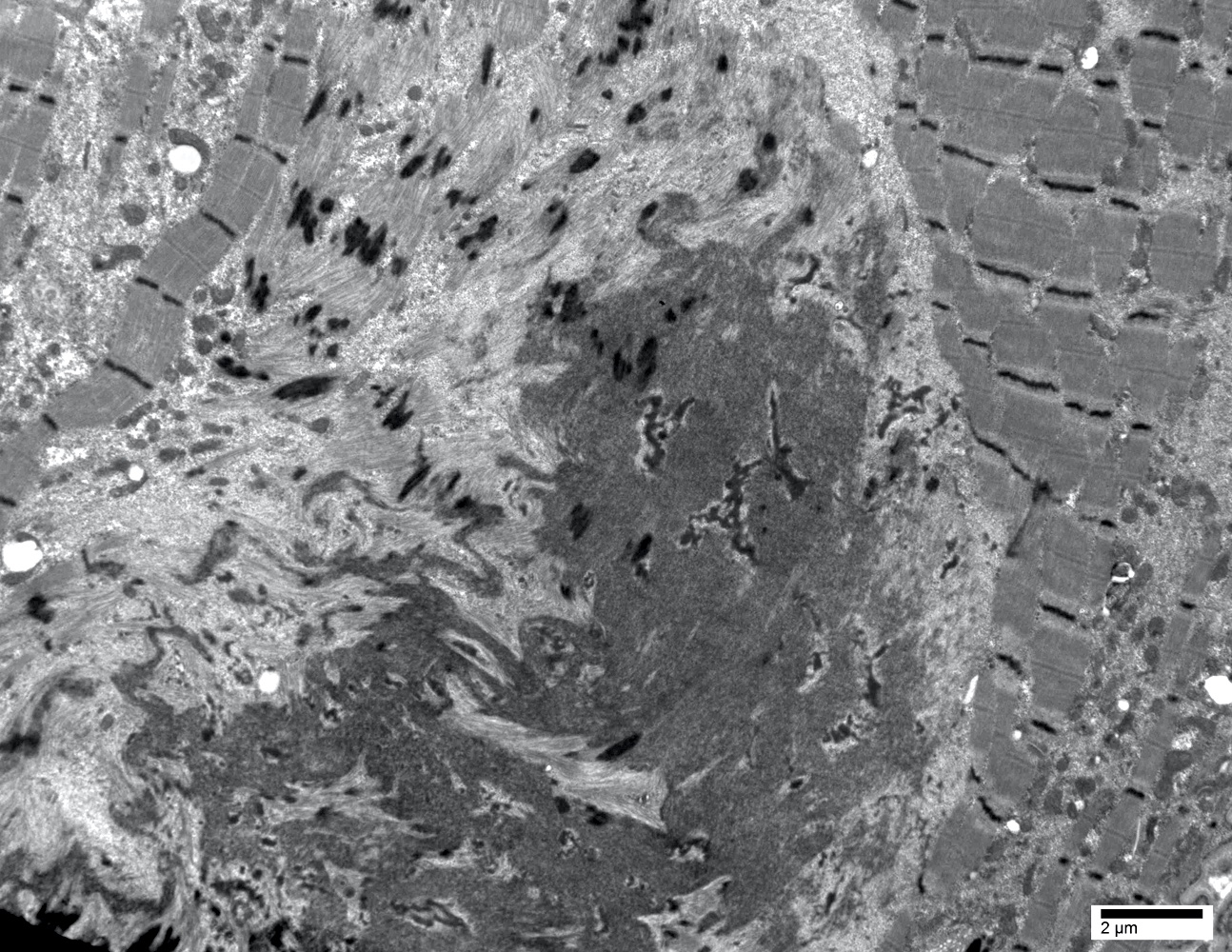
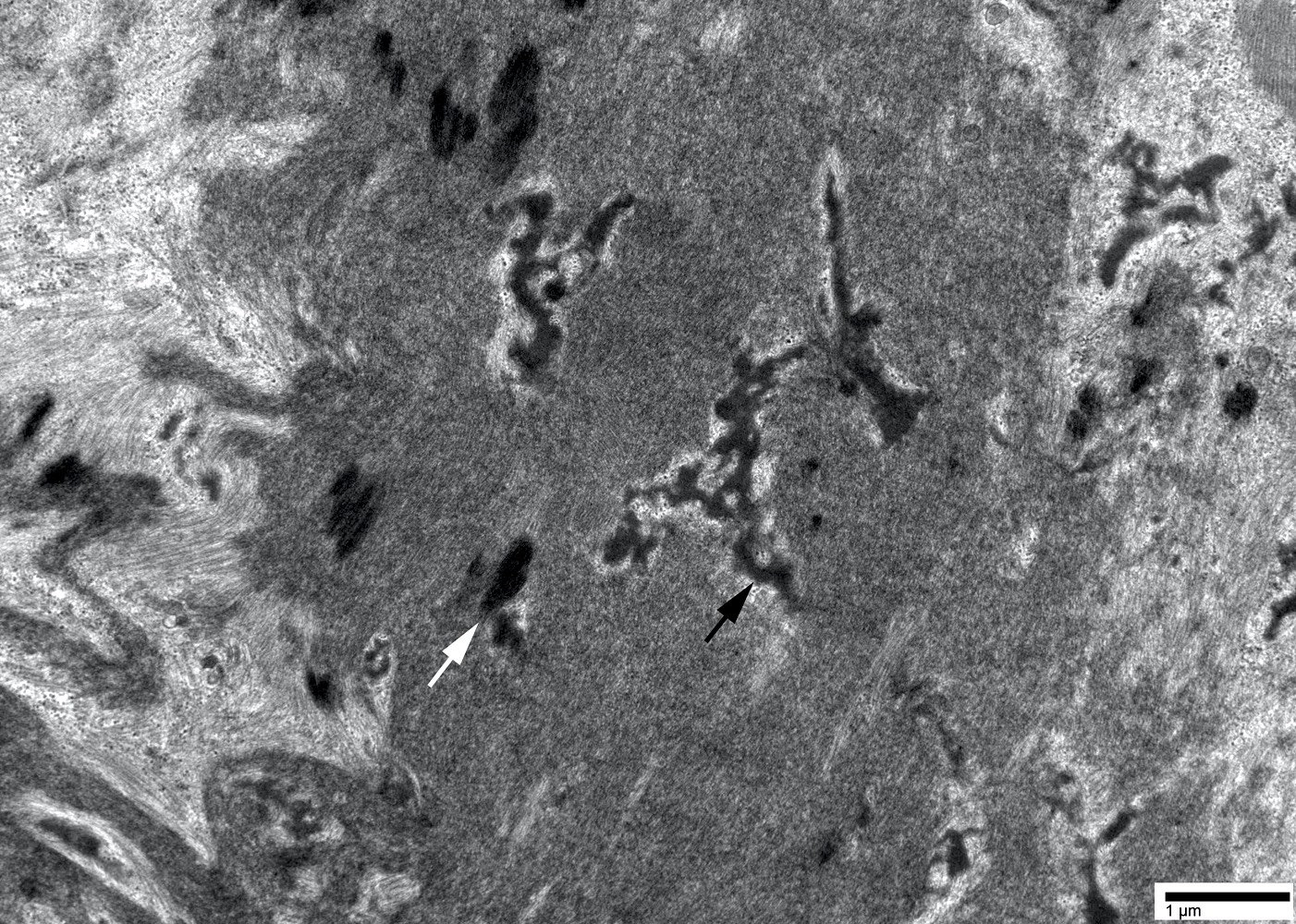
- Amyloid deposits are composed of haphazardly arranged, nonbranching, nonperiodic fibrils with a diameter of 7 - 15 nm (Ultrastruct Pathol 2020;44:325)
Contributed by Chunyu Cai, M.D., Ph.D.
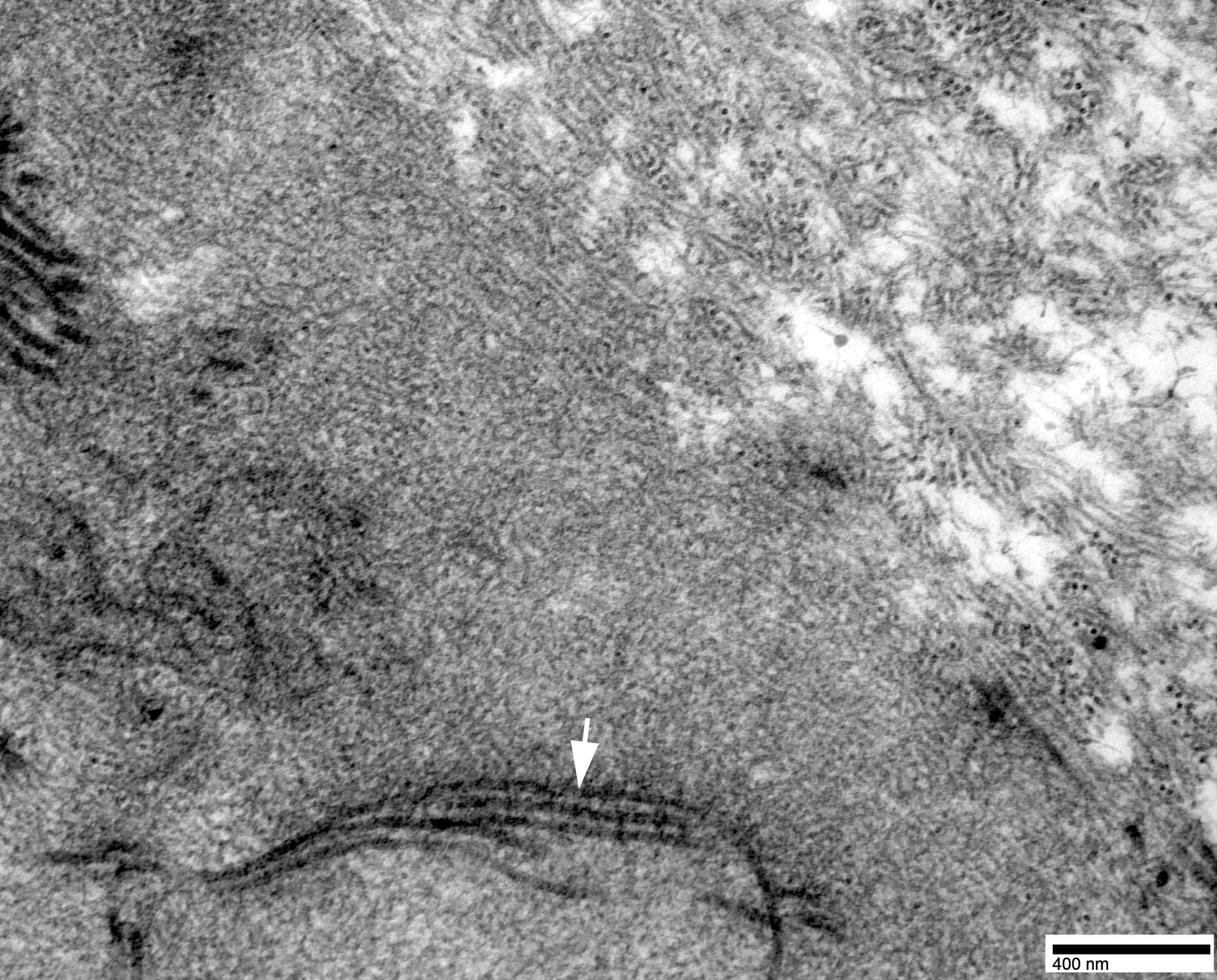
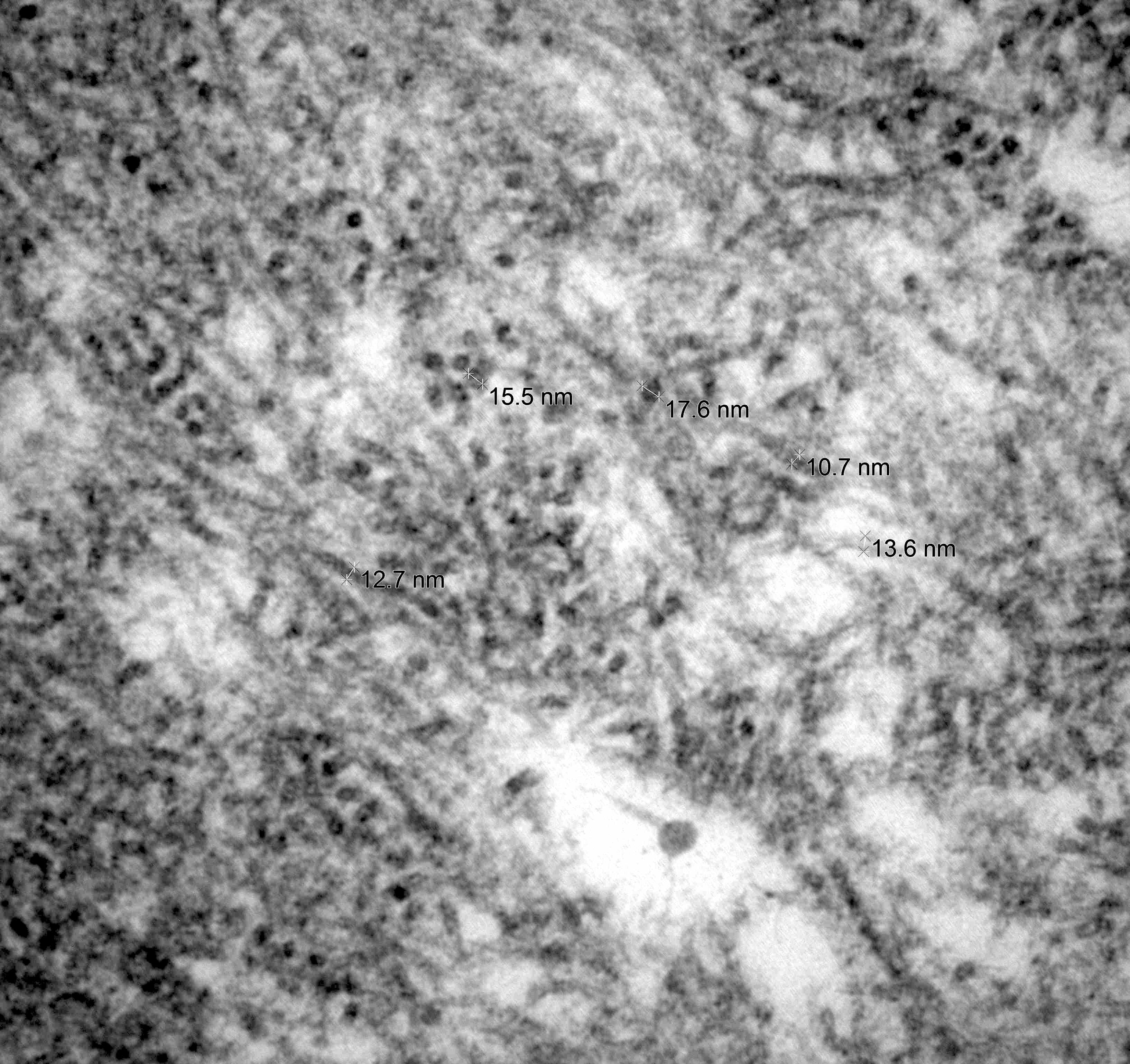
Amyloid deposit
- TTR amyloidosis is by far the most common cause for hereditary amyloidosis
- Of the > 140 mutations reported in the TTR gene, the p.Val50Met mutation is by far the most common (Curr Neuropharmacol 2023;21:471)
The principles for pathology confirmation of amyloidosis and significance of subtyping
- Peripheral nerve, right sural, biopsy:
- Amyloid neuropathy (see comment)
- Comment: The nerve shows amyloid deposition in multiple endoneurial blood vessels. There is severe loss of small myelinated and unmyelinated axons but relatively preserved large myelinated axons, compatible with amyloid neuropathy. Transthyretin immunostain is negative in the amyloid deposits. Given the patient’s history of Waldenström lymphoma, immune light chain associated amyloidosis (AL) is suspected. Liquid chromatography mass spectrometry (LC MS) is recommended for definitive subtyping of amyloid.
- Positive Congo red stain with green-yellow birefringence under polarized light is considered pathognomonic for amyloidosis
- Main amyloid subtypes in peripheral nerve include AL, hereditary and senile types
- Precise amyloid subtyping relies on LC MS / MS
- Absence of amyloid on nerve biopsy:
- Since amyloid deposition is patchy, absence of amyloid on nerve biopsy does not completely exclude the possibility of amyloidosis
- Paraproteinemic neuropathy:
- Diseases with circulating paraprotein such as Waldenström macroglobulinemia and POEMS syndrome may cause peripheral neuropathy without amyloid deposition
- Other small fiber neuropathies:
- Other peripheral neuropathies with predominant involvement of small myelinated or unmyelinated fibers and relatively preserved large myelinated axons include diabetic neuropathy, alcoholic neuropathy, Fabry disease, Tangier disease, hereditary sensory and autonomic neuropathies (Bilbao: Biopsy Diagnosis of Peripheral Neuropathy, 2nd Edition, 2015)
Congo red
Transthyretin IHC
A 70 year old man with history of progressive weakness and sensory loss for 4 years has reported several falls. No bulbar or respiratory symptoms are reported. The symptoms progressed despite receiving intravenous immunoglobulin treatment. Electrophysiologic studies demonstrated severe sensory motor polyneuropathy with demyelinating features and evidence of active denervation in proximal and distal limb muscles. A right sural nerve and right quadriceps muscle biopsy demonstrated the findings above. Which of the following is the best choice to confirm the diagnosis?
- Cardiac ultrasound
- Electron microscopy of the nerve
- Liquid chromatography with tandem mass spectrometry (LC MS / MS)
- Serum protein electrophoresis (SPEP)
- Whole body scintigraphy with single photon emission computed tomography (SPECT) / CT
Answer D is incorrect because SPEP will be useful in identifying serum paraproteins for AL type amyloidosis but not in cases of hATTR or senile amyloidosis. Answer E is incorrect because whole body scintigraphy with SPECT / CT is useful in assessing the extent of amyloid deposition in the whole body but has a limited role in differentiating amyloid subtypes. Answer B is incorrect because electron microscopy of the nerve can help assess the extent of nerve damage and visualize the amyloid fibrils but cannot determine the type of amyloid by ultrastructural morphology. Answer A is necessary since the heart is often involved in amyloidosis and the extent of cardiac involvement is an important prognostic factor; however, it is not the correct answer since it cannot determine the amyloid subtype.
Comment Here
Reference: Amyloid neuropathy
- AA
- Aβ
- Aβ2 microglobuli
- AL
- ATTR
Comment Here
Reference: Amyloid neuropathy
- Defined by the presence of 1 of the antisynthetase syndrome autoantibodies and at least 1 of the following 3 clinical features: interstitial lung disease, inflammatory myopathy or inflammatory polyarthritis
- Other common symptoms include Raynaud phenomenon, mechanic hands, skin rashes, sicca syndrome and constitutional symptoms, such as fever
- To date, 8 antisynthetase autoantibodies have been identified (Jo1, PL12, PL7, EJ, OJ, KS, Zo and Ha), all of which are directed against aminoacyl tRNA synthetases (ARS: enzymes that attach amino acid onto its corresponding tRNA)
- Reference: Autoimmun Rev 2014;13:367
- Jo1 is the first identified and the most common anti-aminoacyl tRNA synthetase (ARS) autoantibody, affecting 18% of European patients with idiopathic immune myopathies, other anti-aminoacyl tRNA synthetase autoantibodies collectively accounted for 3% (Ann Rheum Dis 2001;60:116)
- Majority of patients with anti-aminoacyl tRNA synthetase autoantibodies also have the anti-Ro52 / SSA autoantibody that is commonly associated with Sjögren disease (Autoimmun Rev 2009;8:632)
- Histopathologically, Jo1 antibody positive patients show a characteristic necrotizing perifascicular myositis (Brain 2015;138:2485)
- This injury pattern has also been referred to as immune myopathies with perimysial pathology (Curr Opin Rheumatol 2011;23:595)
- Myositis associated with PL7, EJ, OJ or KS autoantibodies demonstrates similar perifascicular necrotizing myopathy as Jo1 myositis (Brain 2016;139:e50)
- Patients with PL12 autoantibody may demonstrate severe interstitial lung disease but much milder muscle disease (Brain 2016;139:e50)
- When myositis is present, it demonstrates pathologically as immune mediated necrotizing myopathy: pauci-inflammation and randomly distributed necrotic fibers (Muscle Nerve 2012;46:282)
- Patients with anti-Jo1 are more likely to have cancer than patients with anti-PL7 / PL12 (13% versus 5%), while anti-PL7 / PL12 positive patients are far more likely to exhibit interstitial lung disease than Jo1 (90% versus 68%) (Autoimmun Rev 2012;11:739)
- Coexistence of Jo1 and anti-Ro52 seems to be associated with increased risk of malignancy (19%) (Semin Arthritis Rheum 2012;41:890)
- Small series of patients with OJ, KS, Zo and Ha antibodies indicate they have a similar clinical profile as other antisynthetase syndrome (Best Pract Res Clin Rheumatol 2020;34:101503)
- Antisynthetase syndrome
- Aminoacyl tRNA synthetases (ARS)
- Necrotizing perifascicular myositis
- Immune myopathies with perimysial pathology
- ICD-10: M35.8 - other specified systemic involvement of connective tissue
- ~25% of all immune and inflammatory myopathies patients may have antisynthetase syndrome, providing a prevalence estimate of 1/25,000 - 33,000 worldwide
- F:M = 2:1
- Mean age at presentation is 40 - 59
- Reference: Autoimmun Rev 2014;13:883
- Muscle, joints and lung (Autoimmun Rev 2014;13:367)
- Unknown
- Unknown; genetic predisposition, viral infections and medication use may play a role (Curr Med Chem 2015;22:1963)
- Hallmark clinical features of antisynthetase syndrome are myositis, polyarthritis (62%) and interstitial lung disease (70%)
- Other common symptoms include Raynaud phenomenon (47%), fever (43%), skin rashes (32%), mechanic hands (28%), sclerodactyly (12%) and cancer (9%)
- Patients with non anti-Jo1 anti-aminoacyl tRNA synthetase autoantibodies are more likely to present with interstitial lung disease, while those with anti-Jo1 autoantibodies are more likely to present with myositis and arthralgia
- Reference: Autoimmun Rev 2014;13:883
- Based on the clinical features and confirmed in the presence of positive serologic testing for anti-aminoacyl tRNA synthetase antibodies (anti-Jo1, anti-PL12, anti-PL7, anti-OJ, anti-KS, anti-Ha, anti-Zo) (Autoimmun Rev 2014;13:367)
- Interstitial lung disease is diagnosed by high resolution computed tomography of the lungs (see radiology description)
- Absence of myositis or interstitial lung disease does not exclude the diagnosis of antisynthetase syndrome
- Features on the muscle biopsy include necrotizing perifascicular myositis and perimysial connective tissue damage or necrotizing myopathy (Brain 2015;138:2485, J Neurol Neurosurg Psychiatry 2000;68:472, Muscle Nerve 2012;46:282)
- Presence of positive anti-aminoacyl tRNA synthetase antibodies (anti-Jo1, anti-PL12, anti-PL7, anti-OJ, anti-KS, anti- Ha, anti-Zo)
- Creatine kinase levels are often significantly elevated
- Majority of patients with anti-aminoacyl tRNA synthetase autoantibodies also have the anti-Ro52 / SSA autoantibody that is commonly associated with Sjögren disease (Autoimmun Rev 2009;8:632)
- Most frequent patterns are nonspecific interstitial pneumonia (70% of the patients) and organizing pneumonia (20% of the patients)
- Nonspecific interstitial pneumonia pattern characterized by patchy or diffuse ground glass opacities with associated reticular opacities
- Organizing pneumonia pattern characterized by peribronchial or subpleural consolidation or ground glass opacities without fibrosis
- Muscle MRI finding is not specific
- Reference: Eur Radiol 2019;29:5349
- Chronic, requiring long term treatment
- Most common causes of death were pulmonary fibrosis (49%) and pulmonary hypertension (11%)
- Patients with non-Jo1 anti-aminoacyl tRNA synthetase autoantibodies have worse survival than Jo1 positive patients
- 5 and 10 year unadjusted cumulative survivals were 90% and 70% for Jo1 patients and 75% and 47% for non-Jo1 patients (p < 0.005)
- References: Ann Rheum Dis 2014;73:227, Autoimmun Rev 2012;12:210
- 21 year old man with fever, arthralgia and pulmonary infiltrates (N Engl J Med 2012;367:2134)
- 36 year old woman with fatigue, weight loss, progressive weakness in a scapuloperoneal distribution and dysphagia (J Clin Neuromuscul Dis 2017;18:223)
- 52 year old man with asymmetric polyarthralgia, myalgia, weight loss, dyspnea and progressive muscle weakness (Clin Rheumatol 2013;32:715)
- 68 year old woman with eosinophilic pleural effusion (Intern Med 2018;57:2227)
- No FDA approved medication for antisynthetase syndrome
- Treatment of antisynthetase syndrome should target the most severe or life threatening disease manifestation, often interstitial lung disease
- Treatment of antisynthetase syndrome associated myositis is not significantly different from other idiopathic inflammatory myopathies
- Reference: Best Pract Res Clin Rheumatol 2020;34:101503
- See diagnosis
Contributed by Chunyu "Hunter" Cai, M.D., Ph.D.
Jo1 myositis

Jo1 myositis MCH1
Jo1 myositis C5b9
Jo1 myositis myopathy alkaline phosphatase
PL7 myositis
PL7 myositis ATPase pH 4.3

PL7 myositis alkaline phosphatase
PL7 myositis MCH1

PL7 myositis C5b9
- Strong sarcoplasmic C5b9 expression is seen in acutely necrotic fibers
- Sarcolemmal C5b9 expression in a subset of viable myofibers, mostly perifascicular fibers
- Capillary C5b9 expression can be seen but usually less frequent than dermatomyositis
- MHC1 is diffusely positive or patchy and accentuated in perifascular fibers
- Alkaline phosphatase shows prominent perimysial connective tissue damage
- CD8+ T cells are found in both perimysium and endomysium
- CD4+ T cells are mainly found in perimysium and around vessels
- CD20+ B cells are either absent or restricted to perimysium
- References: Brain 2015;138:2485, J Neurol Neurosurg Psychiatry 2000;68:472
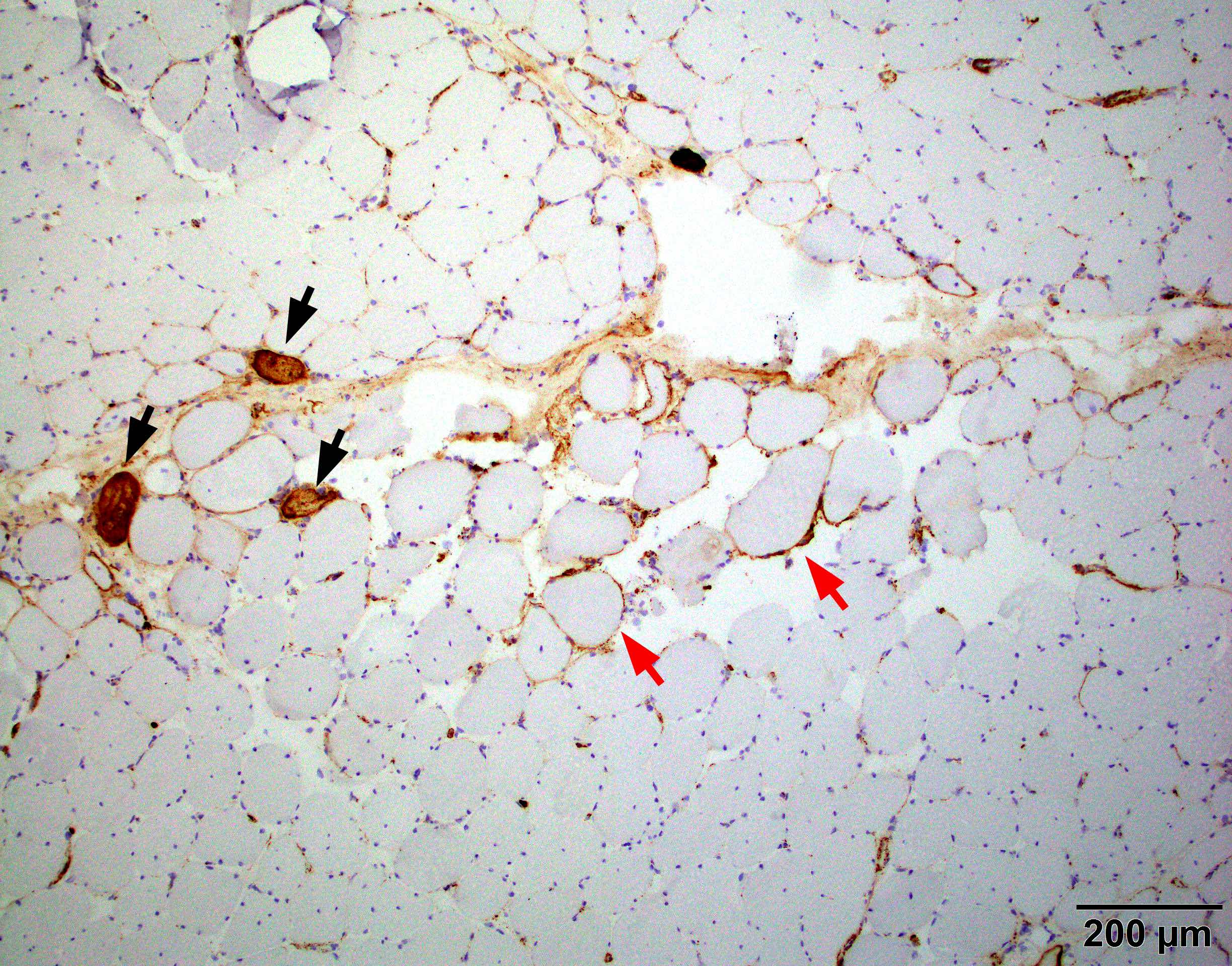
- Endothelial tubuloreticular inclusions can be present but much less frequent than dermatomyositis (Brain 2015;138:2485)
- Intranuclear actin aggregation is an electron microscopy hallmark for antisynthetase syndrome (Neurology 2015;84:1346)
Contributed by Dennis Burns, M.D. and Chunyu "Hunter" Cai, M.D., Ph.D.


Intranuclear actin filament aggregates
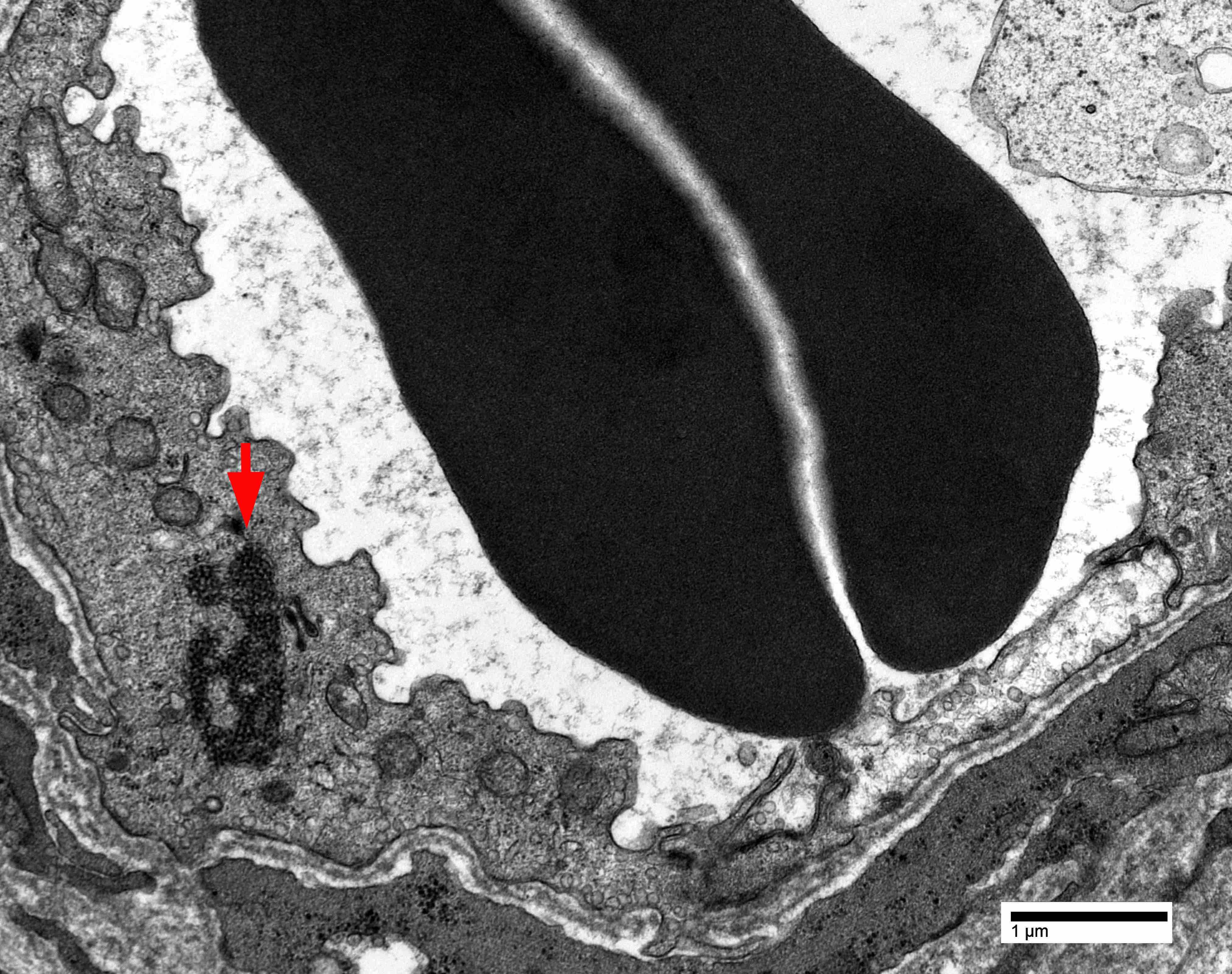
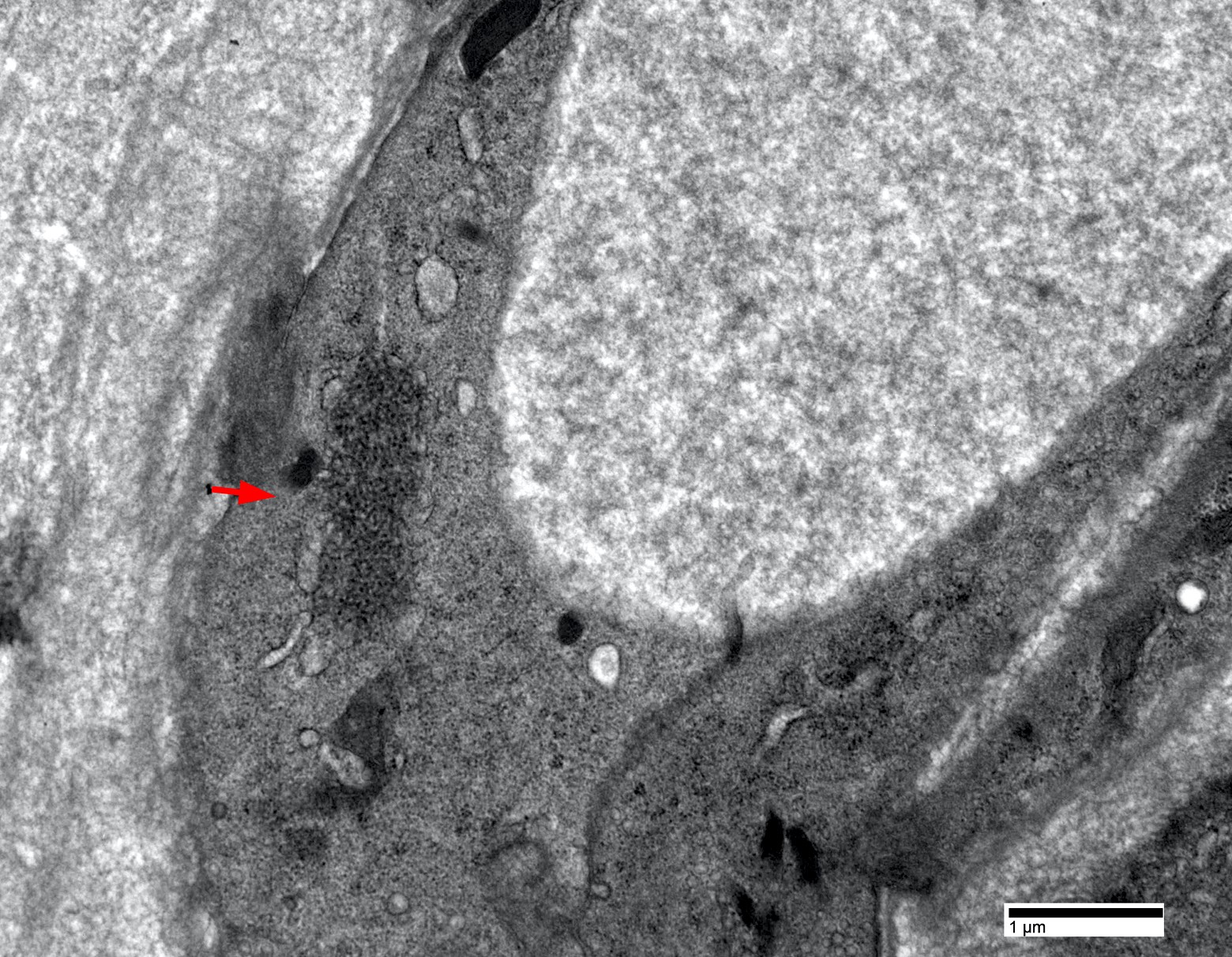
Endothelial tubuloreticular inclusion
- Skeletal muscle, left quadriceps (biopsy):
- Inflammatory myopathy with abundant necrotic fibers, perifascicular atrophy and perimysial connective tissue damage (see comment)
- Comment: The quadriceps muscle biopsy shows multifocal perimysial lymphohistiocytic inflammation, abundant necrotic fibers, perifascicular atrophy and perimysial connective tissue damage. Immunostaining shows patchy myofiber MHC1 upregulation with perifascicular accentuation. Ultrastructural examination demonstrates rare but unequivocal endothelial tubuloreticular inclusions in capillary. The main diagnostic considerations include dermatomyositis and necrotizing perifascicular myositis, which may show overlapping features exemplified by this case (Brain 2015;138:2485). The latter is highly associated with serum anti-Jo1 antibody and interstitial lung disease. Correlation with serology and chest imaging is recommended.
- Dermatomyositis:
- Significant overlap between the histology of dermatomyositis and antisynthetase syndrome associated myositis
- Differentiation is not always possible solely based on histology, correlation with serology is highly recommended
- Generally shows more perifascicular atrophy and less perifascicular necrosis than Jo1 myositis (Brain 2015;138:2485)
- However, dermatomyositis with Mi2 autoantibody may show a perifascicular necrotizing myopathy pattern identical to Jo1 myositis (Acta Neuropathol Commun 2020;8:125)
- Immune mediated necrotizing myopathy:
- Typically lacks visible lymphocytic inflammation or significant MHC1 upregulation
- Necrotic fibers are randomly scattered (Curr Rheumatol Rep 2018;20:21, J Neurol Neurosurg Psychiatry 2016;87:1038)
- Other overlap myositis, such as systemic sclerosis / myositis with anti-PM / Scl antibodies, Sjögren syndrome, lupus:
- Differentiation relies on correlations of muscle pathology, clinical symptoms and serum autoantibodies (Autoimmun Rev 2014;13:883)
- Polymyositis:
- Due to the rapid discovery of serum autoantibodies, diagnosis has been drastically reduced or entirely abandoned (Neuromuscul Disord 2015;25:268, JAMA Neurol 2018;75:1528)
- Most patients with the traditional polymyositis diagnosis now fall in the immune mediated necrotizing myopathy or antisynthetase syndrome categories (JAMA Neurol 2018;75:1528)
A 57 year old man presents with shortness of breath, weakness and elevated creatine kinase levels. Chest CT shows bilateral lung ground glass opacities. Muscle biopsy shows necrotic fibers and myophagocytosis predominantly involving perifascicular fibers. The perimyisial connective tissue appears edematous and fragmented. Which of the following is the most likely diagnosis?
- Antisynthetase syndrome
- Dermatomyositis
- Immune mediated necrotizing myopathy
- Inclusion body myositis
- Polymyositis
- Endothelial tubuloreticular inclusions
- Intranuclear actin aggregate
- Myofiber sarcolemma C5b9 deposition
- Perifascicular atrophy
- Perifascicular myofiber necrosis
Comment Here
Reference: Antisynthetase syndrome associated myositis
- Becker muscular dystrophy (BMD) is caused by dystrophin (DMD) gene mutations on chromosome Xp21, which decreases / alters dystrophin production and causes variable progressive proximal weakness in childhood, progressing to paralysis by adulthood
- Duchenne muscular dystrophy (DMD) is also caused by DMD gene mutations, which causes severe progressive muscle weakness, progressive cardiorespiratory compromise in adulthood and death
- X linked muscular dystrophies include DMD and BMD
- Both are caused by mutations in the dystrophin gene
- DMD is caused by frameshift mutations which disrupt the normal reading frame, therefore little to no dystrophin protein is produced
- BMD is caused by mutations which do not alter the reading frame and some protein is produced
- Clinically, DMD is a more severe, lethal disorder with earlier onset and more rapid deterioration
- Histologically, both show myopathic changes and decreased staining with dystrophin
- DMD shows more severe changes
- X linked dystrophinopathy includes both BMD and DMD
- ICD-10: G71.0: muscular dystrophy
- Both BMD and DMD are X linked and therefore are seen almost exclusively in males
- DMD is the most common muscular dystrophy, with an incidence of 1:5,000 live male births (Curr Opin Neurol 2019;32:722)
- X linked typically affects proximal muscle groups, especially the lower extremities, with variable cardiomyopathy
- Mutations in the DMD gene lead to reduced production of a truncated dystrophin protein, which normally functions to stabilize the sarcolemmal membrane (Continuum (Minneap Minn) 2019;25:1619)
- Because dystrophin is located on the X chromosome, dystrophinopathies are X linked
- Men are affected; women can be carriers and exhibit cardiomyopathy (Pediatr Clin North Am 2015;62:723)
- Heterogenous presentation, typically proximal muscle weakness affecting lower extremities more than upper extremities
- Course is more severe for DMD
- Symptoms begin at earlier age (mean age of diagnosis is 4 years)
- Delayed motor milestones
- Waddling gait
- Difficulty rising from floor (Gower maneuver)
- Difficulty climbing stairs, running and jumping
- Hypertrophy of calf muscles
- Wheelchair needed by age 10
- Contractures
- Most patients expire by third decade of life (Semin Neurol 2015;35:369)
- BMD is less severe
- Symptoms begin between 5 - 15 years of age
- Wheelchair needed after age 16
- More severe cardiomyopathy (Continuum (Minneap Minn) 2019;25:1619)
- Relies on clinical features in combination with genetic testing and creatinine kinase levels
- Timed function tests (patient asked to walk for 6 minutes)
- Muscle biopsy is less frequently performed but is useful for assessment of dystrophin expression (Pediatr Clin North Am 2015;62:723)
- DMD has extremely elevated creatinine kinase levels, usually 50 - 100 times normal
- Elevation may decrease over course of disease
- Up to 70% of carriers may have elevated creatinine kinase levels
- BMD has elevated creatinine kinase levels
- Highest levels seen in 10 - 15 years of age
- Alanine transaminase (ALT) and aspartate transaminase (AST) may be elevated
- Can see occasional myoglobinuria following strenuous activity (Pediatr Clin North Am 2015;62:723)
- Skeletal muscle ultrasound and magnetic resonance imaging can show amount of fibrosis and atrophy in muscle groups (Continuum (Minneap Minn) 2019;25:1619)
- DMD is progressive and invariably fatal, with death typically occurring in the third or fourth decade of life
- BMD has an extremely variable prognosis with some patients following a course like DMD and some having only mild muscle weakness (Semin Neurol 2015;35:369)
- Symptomatic Duchenne muscular dystrophy carrier (J Neurol Sci 2014;336:36)
- 13 year old boy with a mutation in DMD causing Becker muscular dystrophy associated with intellectual disability (J Dev Behav Pediatr 2016;37:239)
- 15 year old boy with Duchenne muscular dystrophy (BMJ Case Rep 2014;2014:bcr2014205296)
- 18 year old man with a case of Becker muscular dystrophy with early manifestation of cardiomyopathy (Korean J Pediatr 2012;55:350)
- No curative treatment for the X linked dystrophinopathies
- Corticosteroids are the primary therapy in DMD (N Engl J Med 1989;320:1592)
- Intensive physical therapy is the primary treatment in BMD
- Corticosteroids are used in severe cases
- Numerous clinical trials for gene therapy are currently in progress (Pediatr Clin North Am 2015;62:723)
- One of the most promising is adeno-associated virus (AAV) based gene replacement therapy (Curr Opin Neurol 2019;32:722)
- X linked dystrophinopathies include similar histologic changes with difference in severity
- DMD has more pronounced changes than BMD
- Variation in myofiber size with small atrophic fibers admixed with large, rounded, hypertrophic fibers
- Increased internal nuclei
- Myofiber splitting, necrosis, phagocytosis and regeneration
- Increased endomysial fibrosis and fatty replacement of muscle (more prominent later in disease course)
- May have inflammation (macrophages, T cells) in association with necrosis
- Architectural changes (whorled fibers, moth eaten fibers) may be seen (Neurology 2011;76:346)
- Carriers of DMD may demonstrate histologic abnormalities as well as a mosaic pattern of dystrophin expression
Contributed by Jesse L. Kresak, M.D.
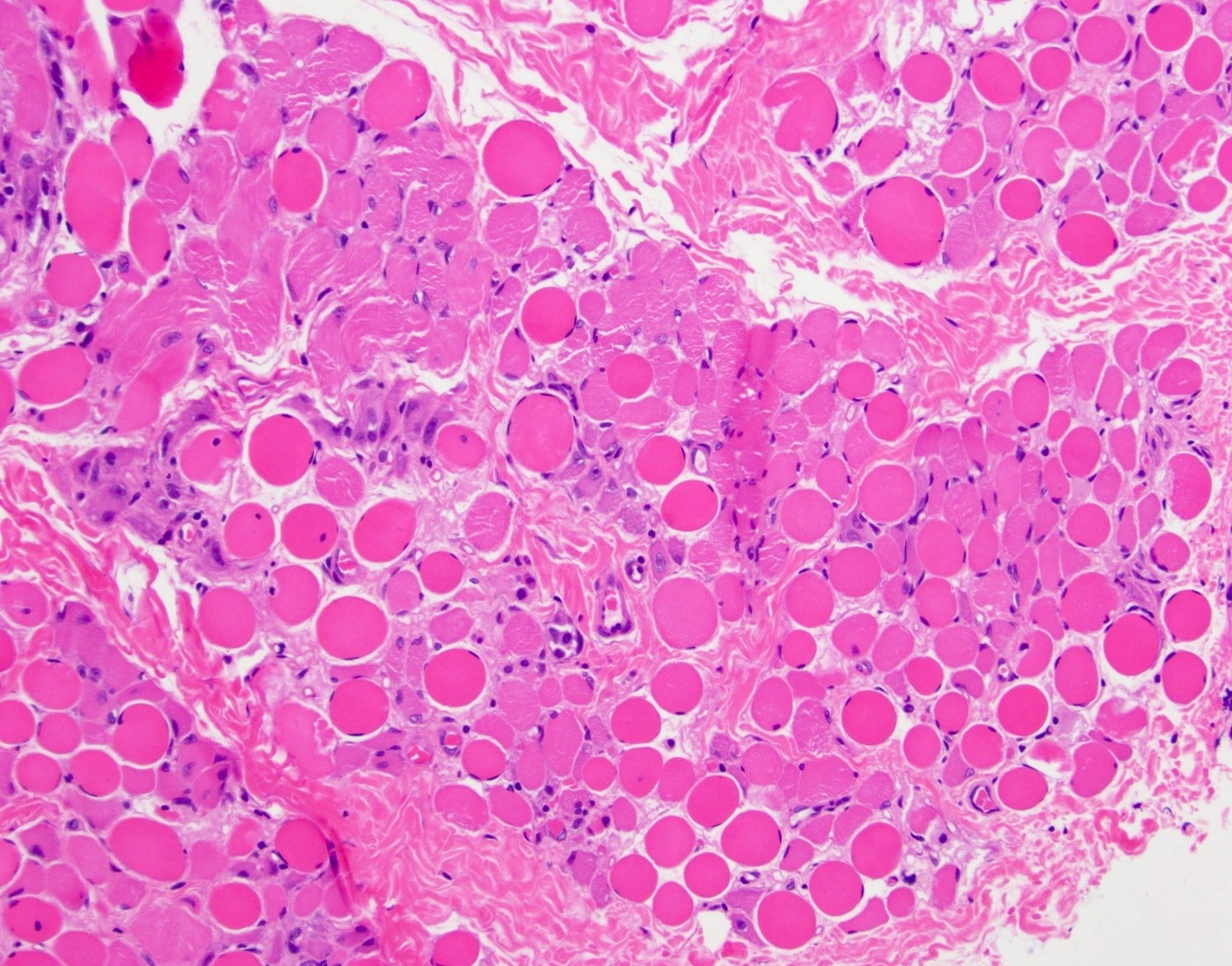
Myopathic changes
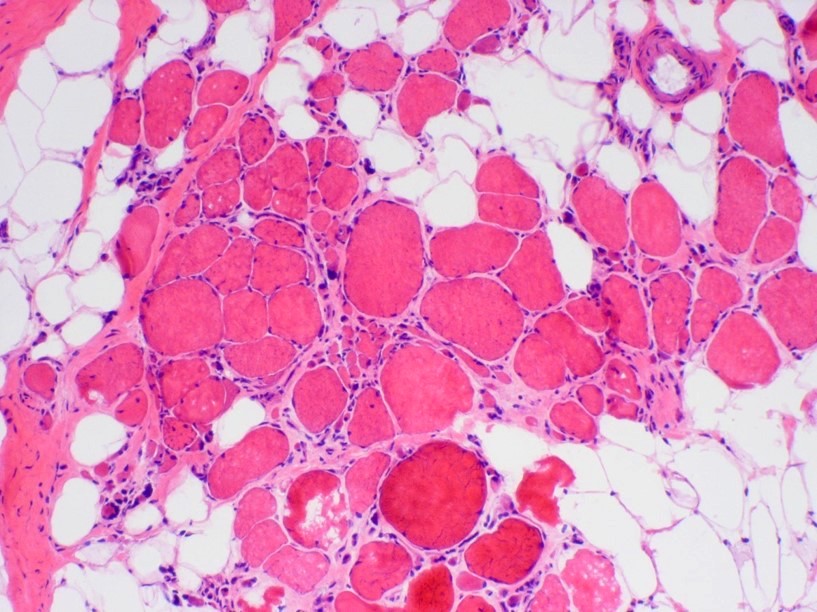
Fatty replacement

End stage muscle
Dystrophin IHC control
Dystrophin IHC in Duchenne muscular dystrophy
Dystrophin IHC in Becker muscular dystrophy
- NADH and SDH oxidative stains may show nonspecific myofibrillar changes, such as moth eaten, lobulated or whorled fibers
- Expression of other proteins is sometimes increased including utrophin and fetal myosins (Neuromuscul Disord 1992;2:177)
- 3 antibodies which recognize epitopes in different domains are used to assess dystrophin expression (DYS1 - rod domain, DYS2 - C terminal, DYS3 - N terminal)
- In general, most cases of DMD show an absence of the C terminus, while in most BMD cases it is preserved (Dubowitz: Muscle Biopsy - A Practical Approach, 5th Edition, 2020)
- In DMD, staining for dystrophin will show absent to markedly reduced expression in sarcolemma of myofibers
- 95 - 100% of fibers will be negative
- May have secondary reduced expression of proteins in the dystrophin associated complex, such as dystroglycan and sarcoglycans to variable degrees
- BMD will show reduced dystrophin expression
- 50 - 80% of fibers will be negative
- Female carriers can have greater than 60% of fibers that stain for dystrophin (Semin Neurol 2015;35:369)
- Both X linked dystrophinopathies are caused by mutations in the dystrophin gene on Xp21
- In DMD, mutations commonly disrupt the reading frame, which severely reduces or eliminates normal protein production
- In BMD, mutations commonly do not alter the reading frame and some protein is still produced (Pediatr Clin North Am 2015;62:723)
- Quadriceps, muscle biopsy:
- Skeletal muscle with late stage myopathy, consistent with dystrophinopathy (see comment)
- Comment: H&E stained sections demonstrate skeletal muscle with features of myopathy, including marked myofiber size variation ranging from minute, atrophic myofibers to hypertrophic forms exhibiting splitting, increased myofibers with internal nuclei and scattered degenerating / regenerating myofibers. The endomysium is expanded by collagen deposition and is replaced by adipose tissue in areas, indicative of a chronic process evolving into end stage muscle changes. A targeted immunohistochemical panel for muscular dystrophy reveals loss of expression of dystrophin 1, dystrophin 2 and dystrophin 3 proteins, with reduced / attenuated expression of sarcoglycan B and sarcoglycan D proteins. Considering the results of this panel together with the advanced stage changes within this patient's skeletal muscle, these findings are consistent with muscular dystrophy, specifically a dystrophinopathy. The differential diagnosis includes Duchenne and Becker muscular dystrophies. Definitive determination of muscular dystrophy type is based on genetic study results. Clinicopathologic correlation is highly suggested.
- Congenital muscular dystrophies:
- Heterogenous group of disorders that present in neonates and infants with hypotonia, muscle weakness, atrophy and joint deformities
- Limb girdle muscular dystrophy:
- Heterogeneous group of inherited muscular dystrophies, which can clinically present like BMD / DMD
- May require molecular testing to differentiate
- Spinal muscular atrophy
- Group of autosomal recessive disorders due to degeneration of spinal anterior horn motor neurons
- Clinically will have tongue fasciculations
- Congenital myopathies such as central core disease, centronuclear myopathy, nemaline myopathy:
- Frequently have earlier onset
- Characteristic histologic findings and staining pattern based on disease type
- Metabolic myopathy:
- Heterogenous group of disorders in cellular energy metabolism that range for severe infantile disease to adult onset mild disease
Mutations in what gene, whose product is shown in this immunohistochemical stain, are responsible for the Duchenne and Becker muscular dystrophies?
- BKR
- DMD
- MTM1
- NEB
- RYR1
- Autosomal dominant
- Autosomal recessive
- De novo
- Mitochondrial
- X linked recessive
- First described in 1956 by Magee and Shy (Brain 1956;79:610)
- One of the more common forms of congenital myopathy
- Characterized by the presence of central cores in skeletal muscle histologically in addition to the clinical features of congenital myopathy
- Skeletal muscle with areas of reduced to absent staining of enzymes such as NADH, SDH and COX
- Most commonly associated with mutation in RYR1 gene
- Presence of cores in a muscle biopsy without associated clinical symptoms and weakness is insufficient for diagnosis of central core disease
- "Central core" refers to areas of reduced oxidative and glycolytic enzymatic activity along the longitudinal axis of skeletal muscle fibers, as seen on enzymatic stains such as NADH
- ICD-10: G71.2 - congenital myopathies
- One of the more common congenital myopathies but true incidence is unknown
- Predominantly involves proximal musculature
- Most frequently hip girdle and axial muscles
- Two models of mutation induced receptor malfunction:
- Leaky channel hypothesis: depletion of sarcoplasmic reticulum calcium stores and increase in cytoplasmic calcium levels
- EC uncoupling hypothesis: disturbance of excitation contraction coupling (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Typically caused by mutations in the skeletal muscle ryanodine receptor (RYR1)
- Less commonly caused by selenoprotein N (SEPN1) mutations (Semin Pediatr Neurol 2011;18:239)
- Variable presentation
- Static to slowly progressive disease course
- May worsen or progress during or after pregnancy
- Usually presents in infancy or early childhood
- Most common symptoms: myalgias, muscle stiffness, exertional weakness
- Common orthopedic symptoms include congenital hip dislocation, scoliosis and foot deformities (Neurology 2013;80:1584), but most patients can walk independently
- Extraocular, respiratory, cardiac muscle involvement is uncommon
- Precautions with general anesthesia due to risk of malignant hyperthermia (associated with RYR1 mutation)
- Histologic finding of central cores in skeletal muscle combined with clinical features of congenital myopathy
- Presence of cores in a muscle biopsy without associated clinical symptoms and weakness is insufficient for diagnosis of central core disease
- CK levels are typically within normal range but may be elevated
- Ultrasound shows localized increased echogenicity within quadriceps
- MRI shows pattern of selective muscle involvement with predilection for vasti muscles, sartorius and adductor magnus of thigh, as well as soleus and peroneal group of lower leg
- Relative sparing of gracilis, adductor longus and rectus femoris (Neurology 2013;80:1584)
- Autosomal dominant mutations are typically associated with a favorable prognosis
- Autosomal recessive mutations may be associated with more severe complications
- Infant boy presenting with motor delay and muscle weakness (Eur J Paediatr Neurol 2011;15:70)
- Infant girl with bilateral congenital lumbar hernias, multiple joint contractures, decreased muscle bulk and symptoms of malignant hyperthermia (Neuromuscul Disord 2016;26:56)
- No current treatment
- Supportive care
- Large areas of reduced oxidative and glycolytic enzymatic activity along longitudinal axis of muscle fiber
- Fibers may have multiple cores; cores may be central or eccentrically placed
- Usually involves type 1 fibers, with some degree of hypertrophy or predominance
- Internal nuclei may be seen
- Myofiber necrosis and regeneration are not seen (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
Contributed by Jesse L. Kresak, M.D.
H&E

NADH and NADH1
SDH
- Desmin positivity in region of core
- May display nemaline rods with Gömöri trichrome (core rod myopathy)
- NADH stain shows absence of enzyme activity within the cores
- SDH and COX stains may also show absence of enzyme activity within cores
- Reduced or absent mitochondria within cores
- Two types of cores: structured and unstructured
- Structured: preserved myofibrillar architecture and retained ATPase activity
- Unstructured: severe myofibrillar disruption with accumulation of smeared Z line material and absent ATPase activity (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Most commonly caused by autosomal dominant mutations in RYR1 gene on chromosome 19q13.1
- Less commonly caused by selenoprotein N (SEPN1) gene mutations (Semin Pediatr Neurol 2011;18:239)
- RYR1 encodes ryanodine receptor, which is ligand gated release channel for calcium stored in terminal cisterna
- RYR1 gene is also implicated in malignant hyperthermia sensitivity (MHS) phenotype
- Dominant mutations affecting N terminal or central domains of RYR protein give rise to MHS phenotype, while those affecting C terminal give rise to central core disease phenotype
- Recessive mutations are uncommon and typically present as multiminicore disease on histology (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Cores are nonspecific and can be seen in a number of other conditions:
- Denervation – often with "targetoid" fibers
- Following exercise in healthy individuals
- Associated with other gene defects (ACTA1, MYH7) and congenital myopathy (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Metabolic conditions
- Tenotomy: surgical act which involves the division of a tendon
- Areas of increased oxidative and glycolytic enzymatic activity along the longitudinal axis of skeletal muscle fibers, as seen on enzymatic stains such as NADH
- Areas of reduced oxidative and glycolytic enzymatic activity along the longitudinal axis of skeletal muscle fibers, as seen on enzymatic stains such as NADH
- Centrally located accumulation of red to purple, rod-like inclusions within skeletal muscle fibers, visible with Gömöri trichrome stain
- Centrally placed, rimmed vacuole within skeletal muscle fibers, visible with Gömöri trichrome stain
Comment Here
Reference: Central core disease
- Genetically heterogeneous group of myopathies defined by the presence of multiple centrally placed nuclei on histologic sections
- Diagnosis requires the clinical features of a congenital myopathy in combination with histologic finding of multiple centrally placed nuclei on muscle biopsy
- Most frequent mutations include MTM1, DNM2 and BIN1
- X linked form is often referred to as myotubular myopathy
- ICD-10: G71.2 - congenital myopathies
- Uncertain incidence and prevalence but less frequent than central core and nemaline myopathies
- X linked myotubular myopathy estimated at 1 per 100,000 male births per year (Brain Behav 2013;3:476)
- Predominantly involves proximal musculature but may extend distally
- Ocular muscles are typically involved
- Autosomal recessive form more frequently involves facial muscles of mastication (Orphanet J Rare Dis 2008;3:26)
- Disease occurs as a result of varying mutations affecting different proteins involved with multiple cellular pathways
- Most proteins affected are involved with various pathways of membrane trafficking and remodeling, including endocytosis and autophagy
- Multiple mutations with differing inheritance patterns have been implicated in CNM:
- X linked: MTM1 (90% of affected men)
- Autosomal dominant: DNM2 and CCDC78
- Autosomal recessive: BIN1 and TTN
- Less frequently mutations of RYR1, MTMR14, SPEG (Neuropathol Appl Neurobiol 2017;43:5)
- Clinical presentation is variable and partly based on mutation
- Marked proximal muscle weakness but may also involve distal musculature, particularly on lower extremities
- X linked form is severe and presents at birth with significant weakness, hypotonia, external ophthalmoplegia and respiratory distress
- Fetal signs include polyhydramnios, reduced fetal movement and thinning of the ribs
- Large head circumference and length > 90th percentile
- Cryptorchidism, pyloric stenosis and hepatic cavernous hemangiomas may also be seen
- Most carriers are asymptomatic but some may have mild muscle weakness
- Autosomal dominant form tends to be the mildest and occur later than the X linked
- Severity varies based on what part of the protein is affected
- Presentation frequently in adolescence / early adulthood but some mutations present in neonatal period
- Progressive and typically begins in adolescence but rarely results in loss of ambulation before the sixth decade
- May present with exercise induced myalgias
- Neonatal presentation is often more severe but symptoms typically improve over time
- Ocular involvement with ptosis is almost always seen
- Autosomal recessive form is characterized by facial muscle weakness, particularly those involved with mastication, in addition to ocular involvement with ptosis and external ophthalmoplegia
- Intermediate severity between X linked and autosomal dominant
- Skeletal abnormalities (scoliosis, high arched palate) often seen
- Variable degrees of respiratory distress but may be severe
- Associated cardiomyopathy has been reported (Orphanet J Rare Dis 2008;3:26)
- Must have characteristic histologic findings of centrally placed nuclei in addition to compatible clinical presentation
- DNA sequencing is used for molecular confirmation of the diagnosis
- Screening for MTM1 mutations should be performed in females with appropriate clinical or histologic findings
- Creatinine kinase (CK) normal to slightly elevated
- Muscle MRI of patients with DNM2 mutations shows a characteristic progressive pattern of early ankle plantarflexor involvement with later changes in hamstring muscles and ending with anterior thigh
- Also shows adductor longus and rectus femoris involvement (Orphanet J Rare Dis 2008;3:26)
Images hosted on other servers:
Selective muscle involvement in a 59 year-old man
- X linked: typically fatal within first few months of life, though a small portion may live into teenage years or beyond with significant medical intervention
- Autosomal recessive: More favorable prognosis with the absence of cardiorespiratory involvement
- Infant boy at birth presenting with severe hypotonia (J Child Neurol 2007;22:447
- Two infant boys with X linked MTM (J Clin Neurol 2013;9:57)
- 2 month old boy with hypotonia (Brain Pathol 2015;25:651)
- 8 year old boy with genetically confirmed X linked myotubular myopathy (Pediatr Neurol 2009;40:483)
- 17 year old girl with proximal muscle weakness (Autops Case Rep 2017;7:43)
- Two first degree cousins with a novel BIN1 stop mutation (Orphanet J Rare Dis 2010;5:35)
- Patient with dynamin centronuclear myopathy (J Clin Neuromuscul Dis 2016;18:84)
- No curative treatment
- Supportive therapy
Images hosted on other servers:
Elongated face and inverted V shaped mouth
- Small muscle fibers with centrally located nuclei, often with a peripheral halo
- Halos lack mitochondria and are highlighted by oxidative stains (Neuropathol Appl Neurobiol 2017;43:5)
- Type 1 myofiber predominance, myofiber size variation and fatty infiltration
- Necklace fibers: basophilic ring along the periphery of the cell membrane visible with H&E, PAS, Gömöri trichrome and oxidative stains in patients with MTM1 mutation (Acta Neuropathol 2009;117:283)
- Radial arrangement of sarcoplasmic strands, visible with NADH stain, seen with DNM2 mutation (Orphanet J Rare Dis 2008;3:26)
Contributed by Marie Rivera-Zengotita, M.D.
H&E
NADH
- Desmin
- Variable vimentin
- RYR1 and DHPR (Brain Behav 2013;3:476)
- X linked: prominent nucleoli and central region with mitochondrial aggregates, glycogen granules and reduced myofilaments; increased mitochondria, glycogen granules and sarcoplasmic reticulum within necklace fibers
- Autosomal dominant: central region with radial sarcoplasmic strands, tapering toward the center
- Autosomal recessive: central region with filamentous, amorphous material comprised of mitochondria, tubules and glycogen (Brain Pathol 2015;25:651)
- Congenital myotonic dystrophy
- Motor neuropathies
- Myasthenia
- Other congenital myopathies
- Spinal muscular atrophy
- BIN1
- DNM2
- MTM1
- MTMR14
- RYR1
- Idiopathic process that leads to an inflammatory myopathy with skin manifestations
- Myositis with perifascicular muscle fiber atrophy and generally inflammatory infiltrates around intramuscular vessels
- Clinical history can be supportive, with the classic skin finding being a heliotrope rash of the eyelids, face, neck and MCP joints
- Dermatomyositis, DM
- Dermatomyositis sine myositis or amyopathic myositis: without muscle involvement
- Dermatomyositis sine dermatitis: either no skin findings or skin findings not noted in darker skin individuals:
- There are two forms, adult and juvenile
- Adult dermatomyositis peaks ~ age 50; twice as common in women than men
- Juvenile dermatomyositis tends to occur between 5-10 years
- Dermatomyositis is the most common form of inflammatory myopathy in children (as opposed to polymyositis and inclusion body myositis)
- Symmetric weakness that affects the proximal limb muscles
- This weakness is progressive and occurs over weeks to months
- There are rare acute cases of weakness
- Patients describe difficulty rising from a seated position or chair, lifting objects or climbing stairs
- Distal weakness, in general, is not a presenting symptom
- The primary process is attack on the endothelium of the capillaries of myofibers, with deposition of complement on the vessel walls and eventual formation of membrane attack complex (N Engl J Med 1986;314:329)
- This causes perivascular inflammation and can eventually reduce the number of intramuscular small vessels
- This causes hypoxic change in the muscle, characterized by perifascicular atrophy, since these fibers are more distal to the vessels (N Engl J Med 1991;325:1487)
- In chronic disease, the number of capillaries can be significantly reduced in a biopsy
- There is up regulation of MHC-1 in myofibers and also increased expression of ICAM1 (N Engl J Med 1993;329:1993)
- No viral etiology has been associated with dermatomyositis
- The exact etiology is idiopathic; however, juvenile dermatomyositis is associated with HLA DQA1 0501
- In adult forms, there is a 15% chance of an underlying malignancy (N Engl J Med 1992;326:363)
- There is also an association with other connective tissue diseases such as SLE, systemic sclerosis and mixed connective tissue disease (Yachnis: Neuropathology: A Volume in the High Yield Pathology Series, 2012)
- The classic symptoms are a rash followed by mild to severe myopathy
- Some cases have no rash or an unrecognized rash in darker skinned individuals (dermatomyositis sine dermatitis)
- Some cases lack muscle involvement (dermatomyositis sine myositis or amyopathic myositis)
- The skin rash is helicotrophic (violaceous, purple-blue) with edema over the upper eyelids; it can also involve the face, neck, anterior chest, back, shoulders, elbow and knees
- The rash is called the "V sign" when it occurs on the chest, and the "shawl sign" when it occurs on the back / shoulders (N Engl J Med 1991;325:1487)
- Classic findings also include Gottron's papules (elevated, purple rash on MCP joints), dilated capillaries at the base of the nails, skin calcinosis in chronic cases and a tiptoe gait from contractures in children when chronic (N Engl J Med 1991;325:1487)
- Occasionally patients have idiopathic interstitial lung disease (Clin Rheumatol 2007;26:1647)
Images hosted on other servers:
Heliotrope rash: the "classic" violaceous rash over the eyes and the malar region of the face
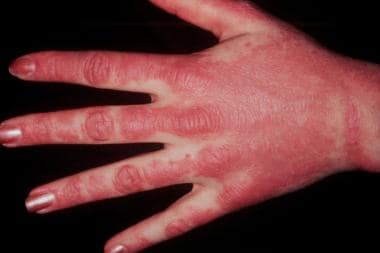
Gottron's papules: erythematous papules on the dorsum of MCP or interphalangeal joints; biopsy shows acanthosis and hyperkeratosis with vacuolar change and a scattered perivascular inflammatory infiltrate

Calcinosis: subcutaneous cases occur in long term, intractable cases, usually of juvenile type
- A clinical-pathologic diagnosis
- Skin and muscle biopsies can be performed at the same time, although a clinical history of skin rash may override the need for a skin biopsy
- EMG/NCS [nerve conduction studies] findings may show increased membrane irritability (Up To Date)
- Elevated sedimentation rate, CK level (generally 5-50x normal, Yachnis: Neuropathology: A Volume in the High Yield Pathology Series, 2012), aldolase
- Antibodies may be found, but also associated with other connective tissue diseases
- ANA antibodies are more common in juvenile forms and anti-Jo antibodies are more common in adult forms
- MRI of musculature using T2 and STIR sequences can be helpful to establish a muscle group for biopsy (Rheumatology (Oxford) 2004;43:603, Arch Dermatol 1999;135:721)
- Response to therapy and presence of an underlying malignancy are useful factors
- 24 year old woman with severe abdominal symptoms 3 months after diagnosis of dermatomyositis (Surgery 1998;123:356)
- 25 year old woman with spontaneous abortion after prednisone treatment for dermatomyositis (Scand J Rheumatol 1999;28:192)
- 32 year old man with osteosarcoma arising in heterotopic bone from dermatomyositis treated at age 3 (Cancer 1981;48:1256)
- 42 year old man with pneumomediastinum from interstitial lung disease with dermatomyositis (Clin Rheumatol 2001;20:359)
- 52 year old man with dermatomyositis and Lyme disease (Clin Infect Dis 1994;18:166)
- Patient with Lafora disease with phenytoin-induced dermatomyositis (J Child Neurol 1998;13:577)
- Corticosteroids and other immunomodulators
- Rituximab for refractory cases (Reumatol Clin 2013;9:117)
- Newer therapies include high dose immunoglobulins (Int J Inflam 2016;2016:3523057)
- The skeletal muscle gross findings are non-specific
- Perifascicular atrophy is the hallmark of dermatomyositis
- Muscle may have altered muscle fiber sizes, but there is less of a tendency to hypertrophy muscle fibers (more common in dystrophy)
- May be increased internal nuclei and basophilic myofibers
Contributed by Meggen Walsh, D.O., M.S., P.A. and Jesse L. Kresak, M.D.

Perifascicular atrophy
Perifascicular atrophy stain
GMS


Myosin I/II immunostain
- Cytology is of no benefit since the main feature is atrophy in the perifascicular region
- Biopsy shows increased CD4+ T cells
- H&E cross sections are best to examine perifascicular atrophy
- Dystrophy panel is normal
- No loss of enzyme histochemical stains
- Intramuscular vessels will occasionally show tubuloreticular inclusions
- Myasthenia gravis: also causes muscle weakness, but has ophthalmologic muscular fatigue (DM does not)
- Polymyositis: Similar inflammatory myopathy, but no prominent perifascicular atrophy
- Facioscapulohumeral muscular dystrophy (FSHD) is an inherited muscle disorder, clinically characterized by weakness of the facial and shoulder girdle muscles followed by the leg and trunk muscles and genetically characterized by contraction or hypomethylation of the D4Z4 domain in individuals with 4qA haplotype, leading to toxic aberrant expression of DUX4 on chromosome 4q35 (Science 2010;329:1650)
- Clinically characterized by
- Variable age of onset at any time from early childhood until adult life
- Involvement of facial and scapulohumeral muscles and sometimes the pelvic girdle
- Transmission usually by an autosomal dominant inheritance, rarely autosomal recessive
- Variable, nonlinear disease course, usually stable or slowly progressive but may have burst of disease activity with rapid functional decline (Brain 1954;77:169, Nat Rev Neurol 2023;19:91)
- Genetics: 95% of FSHD cases are due to contraction (1 - 10) of the D4Z4 repeats (normally 11 - > 100) on chromosome 4q35 in individuals with the 4qA haplotypes; these are referred to as FSHD1 (Science 2010;329:1650)
- Remaining 5% of patients with FSHD are caused by heterozygous mutations in genes that regulate the methylation of the D4Z4 domain, including SMCHD1, DNMT3B or LRIF1 in individuals with the 4qA haplotypes; these are referred to as FSHD2 (Nat Genet 2012;44:1370, Trends Mol Med 2021;27:123)
- 4qA and 4qB are 2 polymorphic allelic forms directly distal to D4Z4 on chromosome 4q35; although both alleles are equally common in the general population, FSHD is associated solely with the 4qA allele (Nat Genet 2002;32:235)
- Landouzy-Dejerine disease
- Landouzy-Déjerine atrophy
- Facioscapulohumeral atrophy
- Facioscapulohumeral myopathy
- Second most common muscular dystrophy, affecting ~1 in 8,000 individuals, with estimated prevalence of 3.2 - 4.6 per 100,000 people
- Affects all age groups, peak age of presentation is 15 - 30 years
- Affects both males and females
- Does not appear to preferentially affect specific racial groups
- Reference: StatPearls: Facioscapulohumeral Muscular Dystrophy [Accessed 30 August 2023]
- Skeletal muscle, particularly face, shoulder girdle and upper arms; axial and leg muscles can also be affected (Pract Neurol 2016;16:201)
- Muscle wasting found in FSHD is associated with derepression of the DUX4 gene distal to the D4Z4 repeats
- DUX4 codes for a transcription factor that is normally expressed in small amounts in early embryological development and is found in the testis and pluripotent cells but is silenced in adult somatic tissue
- Even small amounts of DUX4 protein in postnatal humans is toxic to skeletal muscle and results in apoptosis through a cascade of events including disruption of RNA metabolism and induction of oxidative stress
- In healthy individuals, the D4Z4 macrosatellite domain consists of 8 to ~100 tandem repeat units of 3.3 kb each
- Each D4Z4 tandem repeat unit contains a retrogene that includes the full open reading frame of DUX4
- D4Z4 domain is epigenetically silenced by methylation that prevents DUX4 expression
- In FSHD, there is gain of function aberrant expression of the toxic DUX4 protein either due to D4Z4 contraction (FSHD1) or mutations in chromatin repressing genes that leads to hypomethylation of the D4Z4 domain (FSHD2)
- FSHD is seen exclusively with the 4qA haplotype, because a DUX4 polyadenylation signal (PAS) that functions to stabilize DUX4 mRNA is only present in the 4qA haplotype
- References: Neurology 2021;96:e1054, Nat Rev Neurol 2023;19:91
- Hereditary

- Hallmark is asymmetric weakness and atrophy of muscles of the face, shoulder girdle and upper arms
- Axial and leg muscles can also be affected
- Degree of muscle involvement is highly variable
- Severe, early onset cases characterized by generalized weakness and extra muscular manifestations include sensorineural hearing loss, retinal vasculopathy, right bundle branch block, restrictive lung disease and the possibility of cognitive impairment and epilepsy
- Severe form is associated with very short D4Z4 domain (1 - 3 repeats)
- Reference: Nat Rev Neurol 2023;19:91
- Diagnosis is based on clinical phenotype and genetic testing (see Diagrams / tables) (Neuromuscul Disord 2017;27:782)
- Current CLIA laboratory testing for FSHD is by Southern blotting of EcoRI restriction enzyme digests to evaluate the number of D4Z4 repeats for FSHD1 and by genetic sequencing for FSHD2 related genes (Neurology 2021;96:e1054)
- FSHD1: D4Z4 contraction median 6 repeats (interquartile range [IQR] 4 - 7) repeats with 4qA haplotype
- FSHD2: D4Z4 median 15 (IQR 12 - 22) repeats with 4qA haplotype and concurrent SMCHD1, DNMT3B or LRIF1 mutations
- Non-FSHD: D4Z4 median 28 (IQR 19 - 40) repeats
- A new testing method based on methylation of the D4Z4 repeat was published in 2022 which can identify FSHD1 and FSHD2 simultaneously, requires less DNA, less laboratory effort and is more sensitive (Brain 2023;146:1388)
- Creatine kinase (CK) is normal to elevated and usually does not exceed 3 - 5 times normal upper limits (Br Med J 1971;3:464)
- Muscle MRI is used for the diagnosis and monitoring of disease progression
- MRI with short TI inversion recovery (STIR) positive signal significantly correlates with active myopathy and DUX4 target gene expression in FSHD patients (Hum Mol Genet 2019;28:476)
- Rate of muscle fatty replacement has been used as a measure of disease progression and correlates with the severity of STIR positive signals (J Neurol 2019;266:1127)
- Whole body muscle MRI quantitative fat analysis can be used to assess disease progression and potential therapeutic effect (Neurology 2022;99:e877)
- There is an inverse correlation between the number of D4Z4 repeats and clinical severity; the shorter the D4Z4 domain, the more severe phenotype and the earlier the age of onset (Ann Neurol 1996;39:744)
- 33 year old man with FSHD1 and multiple sclerosis (Acta Myol 2020;39:29)
- 53 year old man with homozygous nonsense variants in LRIF1 (Neurology 2020;94:e2441)
- 59 year old man presented with muscle atrophy, dyspnea and congestive heart failure (eNeurologicalSci 2020;21:100284)
- Aerobic exercise and cognitive behavioral therapy may slow down disease progression (Neurology 2016;86:1700)
- A phase 2 clinical trial (NCT04003974) showed preliminary data that losmapimod (Fulcrum Therapeutics) slowed disease progression in FSHD patients (Skelet Muscle 2022;12:1)
Images hosted on other servers:
Weakness of lips and shoulder muscles
- Pathology can be quite variable and ranges from mild nonspecific changes to marked dystrophic changes
- Most common feature is chronic myopathy with excessive fiber size variation; small and large fibers are mixed type I and II, mimicking neurogenic atrophy
- May have lobulated myofiber changes (see Cases 1 - 3)
- Often shows inflammation (usually perivascular) that resembles inflammatory myopathy
- May or may not have diffuse MHC1 upregulation in myofibers; necrotic fibers and regenerative fibers are usually scanty
- May have prominent fatty replacement and fibrosis (see Case 2)
- May have rimmed vacuoles (Acta Neuropathol 2004;108:257)
- References: Muscle Nerve Suppl 1995;2:S56, Am J Med Genet A 2018;176:1760
Contributed by Chunyu Cai, M.D., Ph.D.
Case 1: 70 year old man with genetically confirmed FSHD1 (8 D4Z4 repeats on a 4qA haplotype)

Chronic
Fiber size variation

Fiber type grouping
Lobulated internal architecture

Lobulated internal architecture
MHC1

Lobulated fiber on EM

Misoriented sarcomere on EM
Case 2: 63 year old woman with genetically confirmed FSHD1 (2 D4Z4 repeats on a 4qA haplotype)

Chronic myopathy with inflammation
Type 1 predominance
Lobulated internal architecture NADH

MHC1
Terminal complement complex
Case 3: 66 year old man with heterozygous pathogenic mutation of SMCHD1 and a FSHD phenotype, consistent with FSHD2

FSHD2 muscle biopsy

FSHD2 ATPase 4.3

Hyaline bodies GT
Hyaline bodies, desmin
Lobulated internal architecture, NADH
EM hyaline body
- May have capillary or sarcolemma C5b9 (terminal complement complex) deposition (Hum Mol Genet 2019;28:476)
- Most cases did not have MHC1 myofiber expression, although examples of diffuse MHC1 upregulation in myofibers exist (Washington University: Facioscapulohumeral (FSH) Dystrophy [Accessed 30 August 2023])
- Lobulated architecture due to myofibril misorientation and mitochondria maldistribution (see Case 1)
- May have rimmed vacuoles with tubulofilamentous inclusions or cytoplasmic bodies (J Neurol 2010;257:1108)
- Genetics: 95% of FSHD patients carry 1 allele with a reduced number (1 - 10) of D4Z4 repeat (normally 11 - > 100) units on chromosome 4q35 associated with specific haplotypes (FSHD1) (Science 2010;329:1650)
- Of the remaining 5% of patients with FSHD phenotype (FSHD2), most cases have been explained by heterozygous mutations in the SMCHD1 / SCHMD1 (structural maintenance of chromosomes flexible hinge domain containing 1) gene (Nat Genet 2012;44:1370)
- Each of the repeated segments in the D4Z4 region contains a copy of the DUX4 gene; the copy closest to the end of chromosome 4 is called DUX4, while the other copies are described as DUX4-like or DUX4L
- Entire D4Z4 region is normally hypermethylated; hypermethylation of the D4Z4 region keeps the DUX4-like genes silenced all the time
- Both D4Z4 and SMCHD1 mechanisms result in chromatin relaxation of the D4Z4 repeat in somatic tissue and subsequent expression of the DUX4 transcription factor in skeletal muscle
- DNMT3B is yet another D4Z4 repeat modifier thus a disease modifying gene for FSHD
Images hosted on other servers:

Schematic of D4Z4
Facioscapulohumeral muscular dystrophy (Year of the Zebra)
FSHD patient's diagnostic journey
FSHD genetics
- Skeletal muscle, right quadriceps muscle, biopsy:
- Myopathy with lobulated myofibers (see comment)
- Comment: The muscle shows prominent lobulated / trabeculated morphology in ~70% of the myofibers. There is no significant active myofiber necrosis, MHC1 upregulation, COX deficient fibers or vacuoles to suggest inclusion body myositis. lmmunostaining shows intact sarcolemmal dystrophin, sarcoglycan, caveolin 3, dysferlin, alpha dystroglycan and nuclear emerin reactivity.
- Myopathy with lobulated myofibers has been reported in FSHD, calpain protein deficiency, a number of other limb girdle myopathies and nonhereditary myopathies (J Neurol Sci 1985;69:345). In addition, lobulated / trabecular change has been described as the predominant abnormality in a subset of elderly patients with limb girdle weakness in the absence of a defined protein abnormality (Neuromuscul Disord 1999;9:208).
- Inclusion body myositis:
- Other inflammatory myopathies:
- FSHD usually does not show diffuse MHC1 expression in myofibers
- Active myofiber damage is usually minimal
- Other muscular dystrophies:
- Lobulated architecture is suggestive of FSHD, although it can also be seen in calpainopathies, dysferlinopathies, myopathy with supervillain mutations and neurogenic changes (J Neurol Sci 1985;69:345, Brain 2021;144:e34)
- Chronic denervation:
- Usually lacks fibrosis, inflammation and active myofiber damage
A 63 year old woman presented with progressive weakness over the last 10 years. She initially presented with stooped posture that progressed to head drop followed by proximal arm and leg weakness. No significant distal weakness. She had been treated with steroid therapy for the past few years with no significant improvement. Laboratory study showed mildly elevated creatine kinase (CK, 350 IU). Myositis panel, HMG CoA autoantibody, myasthenia gravis panel were all negative. A muscle biopsy shows a chronic myopathy with inflammation (figure 1) and prominent lobulated myofiber architecture (figure 2). Which of the following is the most likely diagnosis?
- Facioscapulohumeral muscular dystrophy
- Immune mediated necrotizing myopathy
- Mitochondria myopathy
- Neurogenic atrophy
- Sporadic inclusion body myositis
Comment Here
Reference: Facioscapulohumeral muscular dystrophy
A 63 year old woman presented with progressive weakness over the last 10 years. She initially presented with stooped posture that progressed to head drop followed by proximal arm and leg weakness. No significant distal weakness. She had been treated with steroid therapy for the past few years with no significant improvement. Laboratory study showed mildly elevated creatine kinase (CK, 350 IU). Myositis panel, HMG CoA autoantibody, myasthenia gravis panel were all negative. A muscle biopsy shows a chronic myopathy with inflammation (figure 1) and prominent lobulated myofiber architecture (figure 2). What is the next step to confirm the diagnosis?
- Muscle MRI
- Muscular dystrophy immunostaining panel
- Neuromuscular next generation sequencing (NGS) panel
- Specific FSHD gene testing
Comment Here
Reference: Facioscapulohumeral muscular dystrophy
- Glycogen storage diseases (GSD) are inherited metabolic disorders of glycogen metabolism
- Occur due to a lack of specific enzymes that control the synthesis, regulation and degradation of glycogen
- After the discovery of deficient glucose 6 phosphatase in von Gierke disease (GSD I) by Carl F. Cori and Gerty T. Cori, a number of inborn errors of glycogen metabolism have been recognized (J Biol Chem 1929;81:389, J Med Biogr 2021;29:143)
- Classified numerically in the order of recognition and identification of the enzyme defect causing the disorder
- There are 15 types, most of them are autosomal recessive, except for X linked GSD IXd, phosphoglycerate kinase deficiency (J Endocrinol 2018;238:R131)
- The most common presenting symptoms are hypoglycemia and exercise intolerance
- Danon disease (lysosomal glycogen storage disease without acid maltase deficiency, pseudoglycogenosis II) caused by deficiency of lysosome associated membrane protein 2 (LAMP2), led to failure of cellular autophagy with accumulation of glycogen granules and intracytoplasmic vacuoles containing autophagic material; recently not recognized as GSD (Cell Death Dis 2017;8:e2565)
- GSD affecting the skeletal muscles are mainly involved
- GSD 0 (glycogen synthase 1 deficiency)
- GSD II (acid maltase deficiency, Pompe disease)
- GSD III (debrancher enzyme deficiency, Cori-Forbes disease)
- GSD IV (branching enzyme deficiency, Andersen disease)
- GSD V (glycogen phosphorylase deficiency, McArdle disease)
- GSD VII (phosphofructokinase deficiency, Tarui disease)
- GSD IXd (phosphorylase kinase deficiency)
- Phosphoglycerate kinase deficiency
- GSD X (phosphoglycerate mutase deficiency)
- GSD XI (lactate dehydrogenase deficiency)
- GSD XII (aldolase A deficiency)
- GSD XIII (β enolase deficiency)
- GSD XIV (phosphoglucomutase deficiency)
- GSD XV (glygogenin 1 deficiency)
- Rare
- 1 per 20,000 - 43,000 live births (Pediatrics 2000;105:e10)
- GSD I / GSD VI only cause liver disease, while GSD II / GSD V / GSD VII / GSD X / GSD XI cause predominantly muscle disease; the rest cause mixed muscle / systemic disease (Amato: Neuromuscular Disorders, 2nd Edition, 2015)
- GSD 0 / GSD II / GSD III / GSD IV / GSD XIV / GSD XV can cause cardiomyopathy; GSD IV can involve the nervous system (adult polyglucosan body disease) (Dubowitz: Muscle Biopsy - A Practical Approach, 5th Edition, 2020)
- Primary physiologic function of glycogen is to store and provide glucose
- Liver glycogen stores are utilized to maintain glucose homeostasis in the serum; the muscle glycogen stores are utilized as a source of energy for muscular activity (Semin Hematol 2002;39:103)
- Aerobically, glucose is metabolized into pyruvate and acetyl CoA, which enters the citric acid cycle to produce water, carbon dioxide and adenosine triphosphate (ATP) or is used for the synthesis of fatty acids
- Under anaerobic conditions, glucose is metabolized to lactate (Curr Mol Med 2002;2:101)
- Failure to maintain glycolytic pathway (glycogenesis, glycogenolysis or glycolysis) leads to glycogen storage diseases
- Defects in the enzymes caused by inherited mutations that are required for glycolytic pathway, such as glycogen synthase (encoded by the GYS1 and GYS2 genes), acid maltase (GAA gene), branching enzyme (GBE1 gene), debrancher enzyme (AGL gene), myophosphorylase (PYGM gene), phosphorylase (PYGM), phosphofructokinase (PFKM), phosphorylase kinase (PHKA1), phosphoglycerate kinase (PGK1), phosphoglycerate mutase (PGAM2), lactate dehydrogenase (LDHA), aldolase A (ALDOA), β enolase (ENO3), phosphoglucomutase (PGM1), glygogenin 1 (GYG1) (World J Gastroenterol 2007;13:2541)
Contributed by Truong Phan Xuan Nguyen, M.D.

Metabolic glycolytic pathway
| GSD 0 | GYS1 (skeletal muscle) or GYS2 (liver) | Glycogen synthase 1 | Autosomal recessive | Glycogen synthase 1 deficiency |
| GSD II | GAA | Acid maltase | Autosomal recessive | Acid maltase deficiency; Pompe disease |
| GSD III | AGL | Debrancher enzyme | Autosomal recessive | Debrancher enzyme deficiency; Cori-Forbes disease |
| GSD IV | GBE1 | Branching enzyme | Autosomal recessive | Branching enzyme deficiency; Andersen disease |
| GSD V | PYGM | Glycogen phosphorylase | Autosomal recessive | Glycogen phosphorylase deficiency; McArdle disease |
| GSD VII | PFKM | Phosphofructokinase | Autosomal recessive | Phosphofructokinase deficiency; Tarui disease |
| GSD IXd | PHKA1 | Phosphorylase kinase | X linked inheritance | Phosphorylase kinase deficiency |
| Phosphoglycerate kinase deficiency | PGK1 | Phosphoglycerate kinase | X linked inheritance | N/A |
| GSD X | PGAM2 | Phosphoglycerate mutase | Autosomal recessive | Phosphoglycerate mutase deficiency |
| GSD XI | LDHA | Lactate dehydrogenase | Autosomal recessive | Lactate dehydrogenase deficiency |
| GSD XII | ALDOA | Aldolase A | Autosomal recessive | Aldolase A deficiency |
| GSD XIII | ENO3 | β enolase | Autosomal recessive | β enolase deficiency |
| GSD XIV | PGM1 | Phosphoglucomutase | Autosomal recessive | Phosphoglucomutase deficiency |
| GSD XV | GYG1 | Glygogenin 1 | Autosomal recessive | Glygogenin 1 deficiency |
- Muscle cramps, exercise intolerance, muscle weakness (hypotonia), hypoglycemia and fatigue are typical symptoms of GSDs that relate to the energy deficiency in skeletal muscle
- Rhabdomyolysis and myoglobinuria may lead to renal failure in severe cases (Goebel: Muscle Disease, 2nd Edition, 2013)
- Clinical spectrum is highly variable, ranging from severe exercise intolerance or multisystem involvement in infancy to isolated progressive muscle weakness in adulthood (Gaspar: Myopathology A Practical Clinico-pathological Approach to Skeletal Muscle Biopsies, 1st Edition, 2019)
- Late onset GSD II is characterized by proximal muscle weakness and respiratory insufficiency (Aging 2020;12:15856)
- In GSD V, the second wind phenomenon is the hallmark, which is the alleviation of myalgia and exhaustion after a swift increase in physical activity (Ann Neurol 2003;54:539)
- GSD II is part of the newborn screening panel by measurement of acid α glucosidase (GAA) enzyme activity in dried blood spots for early diagnosis (J Neurol Neurosurg Psychiatry 2022 Apr 25 [Epub ahead of print], Int J Neonatal Screen 2020;6:31)
- Genetics and muscle pathology can confirm diagnosis
- Nonischemic forearm exercise shows a flat lactate response and marked hyperammonemia in GSD V, GSD VII (Ann Neurol 2002;52:153)
- Exercise to exhaustion on a cycle ergometer shows severely impaired maximal oxidative capacity in GSD V, GSD VII (Acta Neurol Scand 2018;138:301)
- Serum creatine kinase (CK) levels are high in GSDs but may be normal in late onset cases of GSD II (Gaspar: Myopathology A Practical Clinico-pathological Approach to Skeletal Muscle Biopsies, 1st Edition, 2019, Genet Med 2006;8:267)
- Cutoff in the GAA activity assay using dried blood spots is set between 0.1 and 0.5 percentile values for the population or 20 - 30% of the mean value of the population (6.5 pmol/h/disk)
- A second GAA activity assay and GAA gene analysis are performed if newborns have values that are less than the cutoff (Int J Neonatal Screen 2020;6:31)
- Chest Xray: massive cardiomegaly in GSD II infant onset (Genet Med 2006;8:267)
- MRI in GSD II:
- In late onset, T1 weighted shows fatty replacement commonly in the tongue, subscapularis, latissimus dorsi, paraspinal and abdominal muscles, psoas, glutei, adductor magnus, posterior muscles of the thigh (especially the semimembranosus) and vastus intermedius
- Vastus lateralis and medialis can be affected in the progression of the disease
- Noticeable loss of muscle volume in the psoas and adductor magnus muscles
- In infant onset, T1 weighted shows normal or identifies only mild fatty replacement, although patients may have significant muscle weakness; muscles commonly affected include the tongue, glutei and adductor magnus muscles (Muscle Nerve 2021;63:640)
- In late onset, T1 weighted shows fatty replacement commonly in the tongue, subscapularis, latissimus dorsi, paraspinal and abdominal muscles, psoas, glutei, adductor magnus, posterior muscles of the thigh (especially the semimembranosus) and vastus intermedius
- Infant onset is aggressive and can be fatal in some GSDs (Ultrastruct Pathol 2011;35:183)
- Early diagnosis and proper treatment are important for good prognosis (Ann Transl Med 2018;6:474)
- Infant boy with hypertrophic cardiomyopathy in infant onset Pompe disease (GSD II) (Medicine (Baltimore) 2017;96:e9186)
- 14 year old boy with seizures and rhabdomyolysis in GSD XII (Ital J Pediatr 2022;48:39)
- 23 year old man with several episodes of rhabdomyolysis in phosphoglycerate kinase deficiency (Intern Med 2022 May 7 [Epub ahead of print])
- 35 year old woman with cardiomyopathy after a pregnancy in late onset Pompe disease (GSD II) (JIMD Rep 2017;31:79)
- 40 year old woman with severe left forearm pain after moving furniture was diagnosed with McArdle disease (GSD V) (J Hand Surg Am 2015;40:2377)
- Most treatments involve symptomatic management and surveillance (Hum Mol Genet 2019;28:R31)
- GSD II can be treated with enzyme replacement therapy (ERT) and gene therapy (Neurotherapeutics 2018;15:928)
- GSD 0, muscle:
- Glycogen depletion in all fibers
- Mitochondrial proliferation
- Type I fiber predominance
- Phosphorylase is deficient in all muscle fibers
- GSD II:
- Histopathological features vary with the phenotypic forms: infant form, childhood form and adult form
- Marked sarcoplasmic membrane bound vacuoles containing basophilic amorphous material (glycogen) in most fibers
- Type 1 muscle fibers are more involved than type 2
- Little muscle fiber degeneration or increased connective tissue
- Abundant acid phosphatase activity (increased lysosomes)
- GSD III:
- Subsarcolemmal nonmembrane bound vacuoles with glycogen accumulation
- At a later stage, the muscle may appear dystrophic with atrophy and connective tissue infiltration and no apparent glycogen storage
- GSD IV:
- Adult form shows PAS positivity and sarcoplasmic rounded opalescent inclusions
- Perinatal forms show polyglucosan bodies which have variable size, shape and reaction to PAS
- GSD V, VII, IXd:
- Muscle pathology is variable from minor nonspecific changes to subsarcolemmal nonrimmed vacuoles and occasional fibers with accumulation
- Excess glycogen on PAS staining may be visible but may only be apparent at the periphery of the fiber
- GSD X, XIV, XV, phosphoglycerate kinase deficiency:
- May reveal variation in myofiber size with normal to mild glycogen distribution
- Reference: Goebel: Muscle Disease, 2nd Edition, 2013
Contributed by Ichizo Nishino, M.D., Ph.D.
GSD 0: no specific changes
GSD 0: glycogen depletion
GSD II childhood: marked sarcoplasmic vacuoles
GSD II childhood: PAS

GSD II childhood: epon PAS

GSD II childhood: NADH TR

GSD II childhood:
predominant
involved
type 1 fibers
GSD II childhood:
high acid
phosphatase
activity
GSD II childhood: modified Gomori trichrome
GSD II childhood:
MHC I

GSD II adult: small vacuoles
GSD II adult: modified Gomori trichrome
GSD II adult: epon PAS
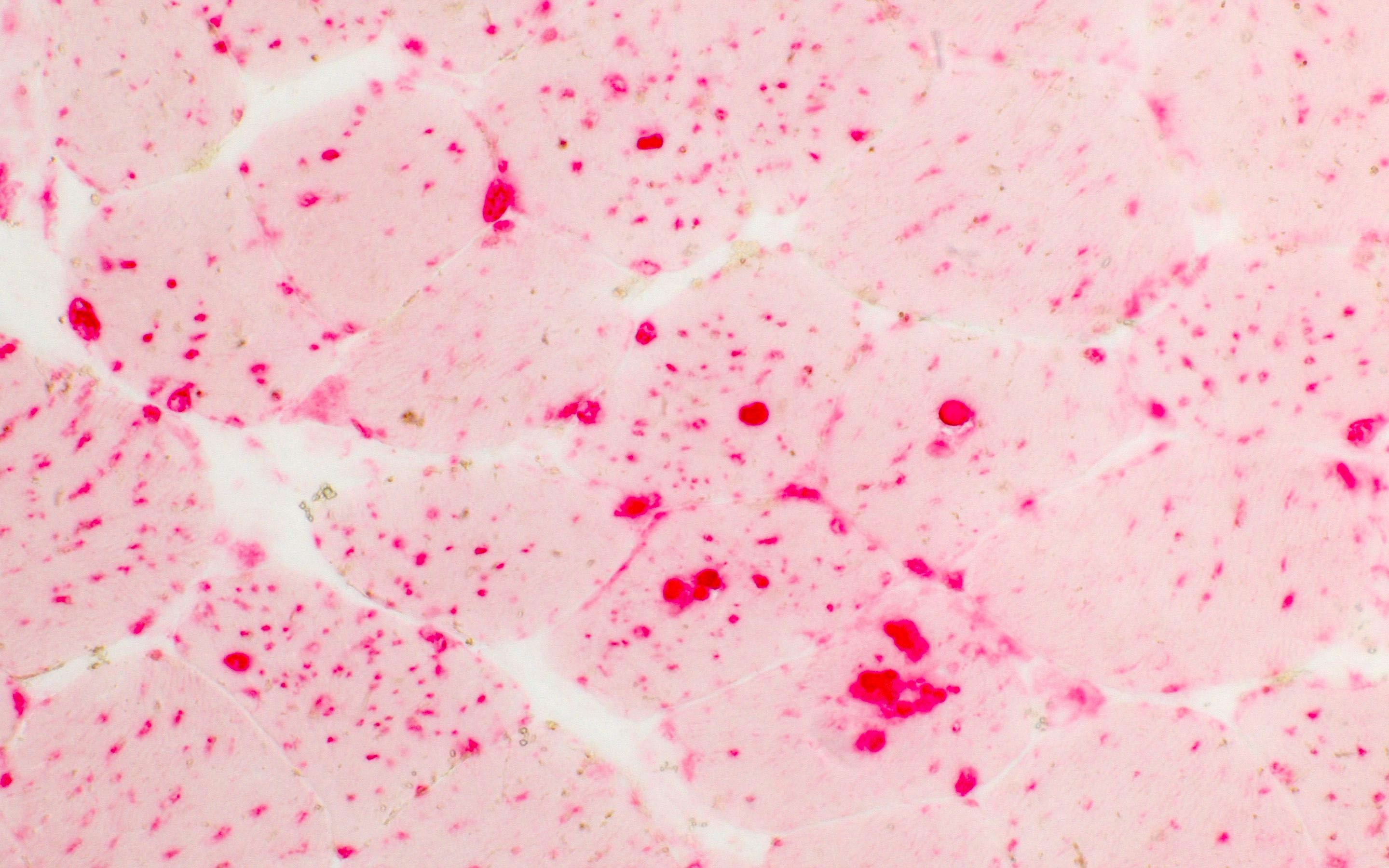
GSD II adult: increased acid phosphatase activity
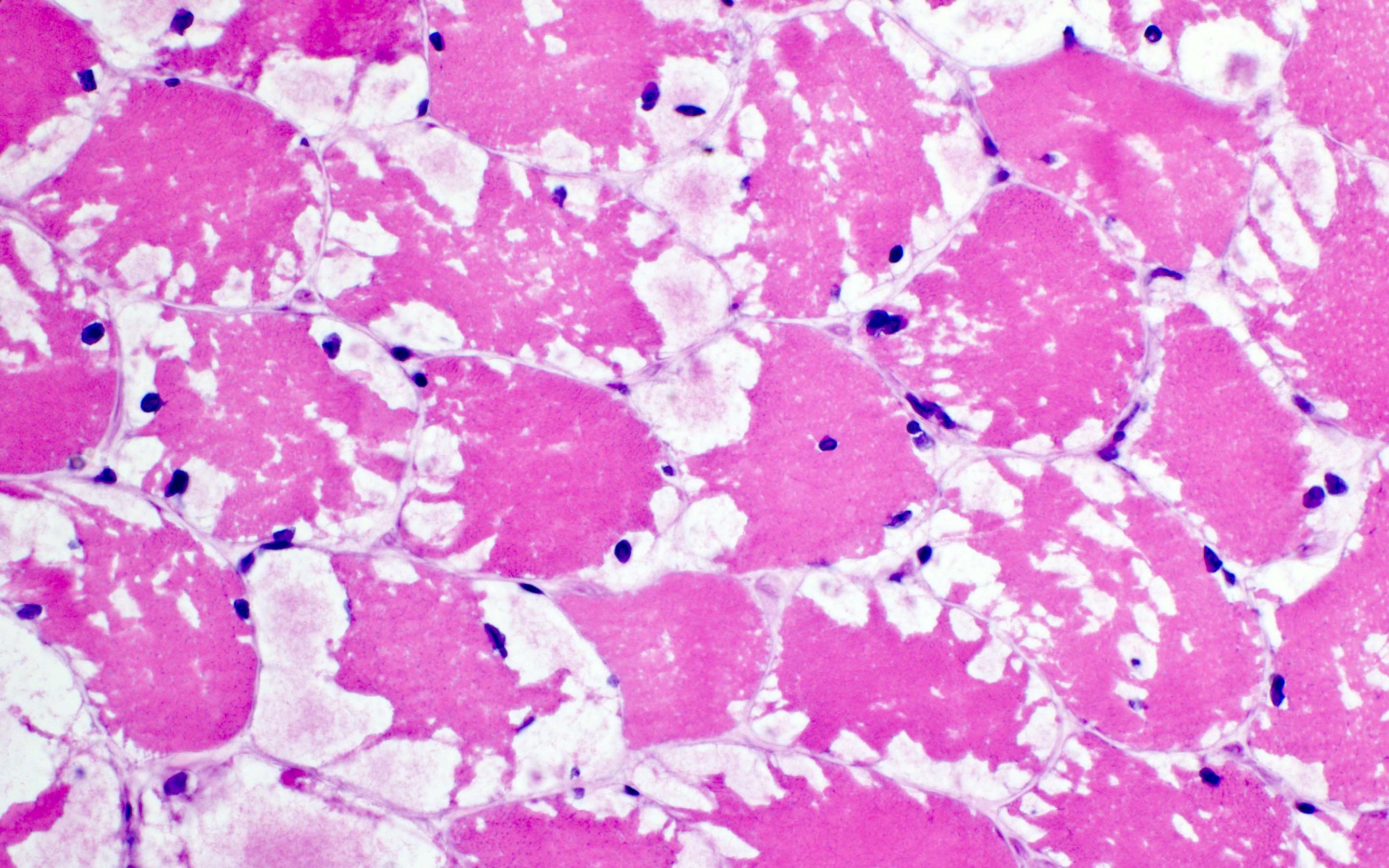
GSD III: nonmembrane bound vacuoles
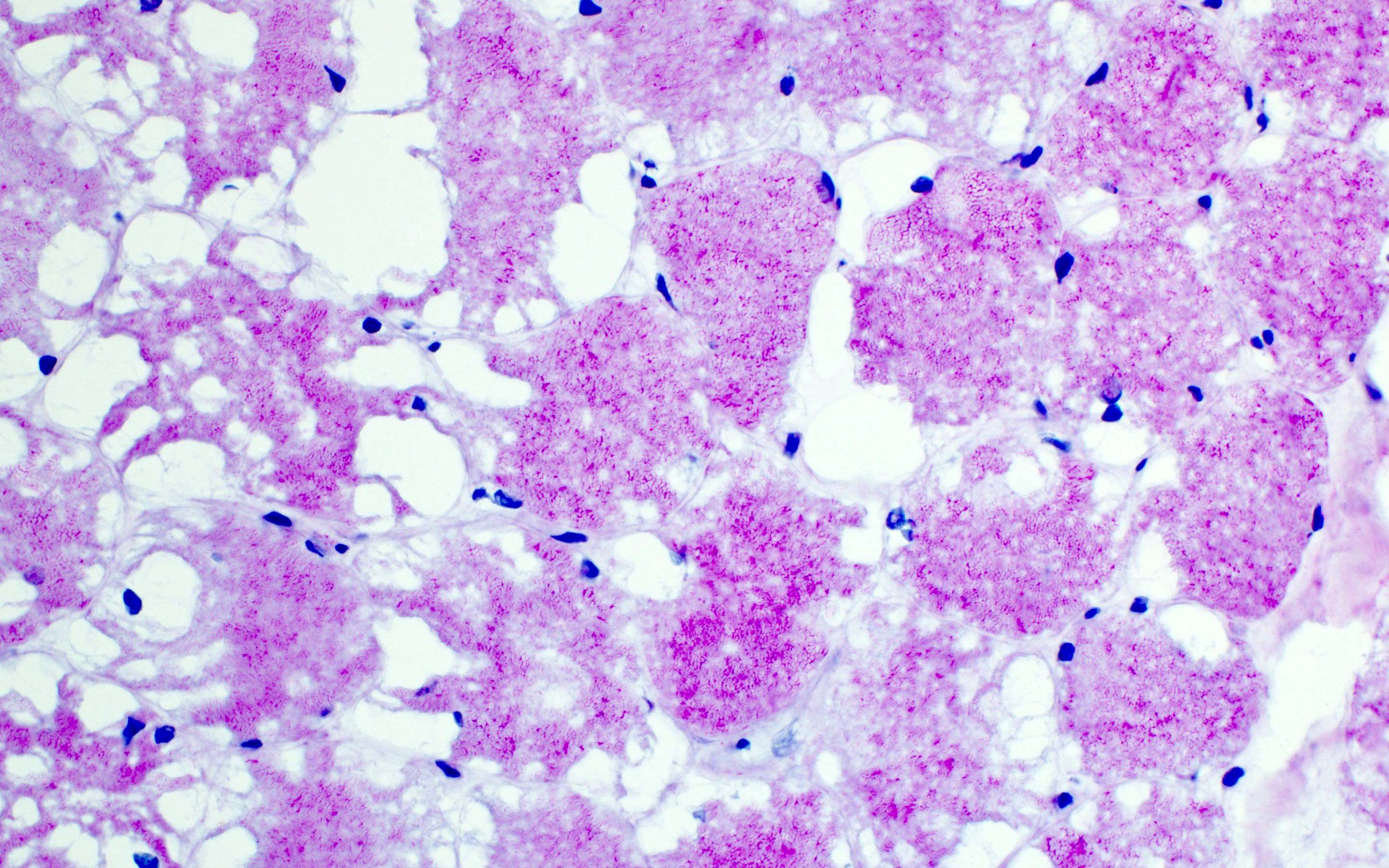
GSD III: PAS positive
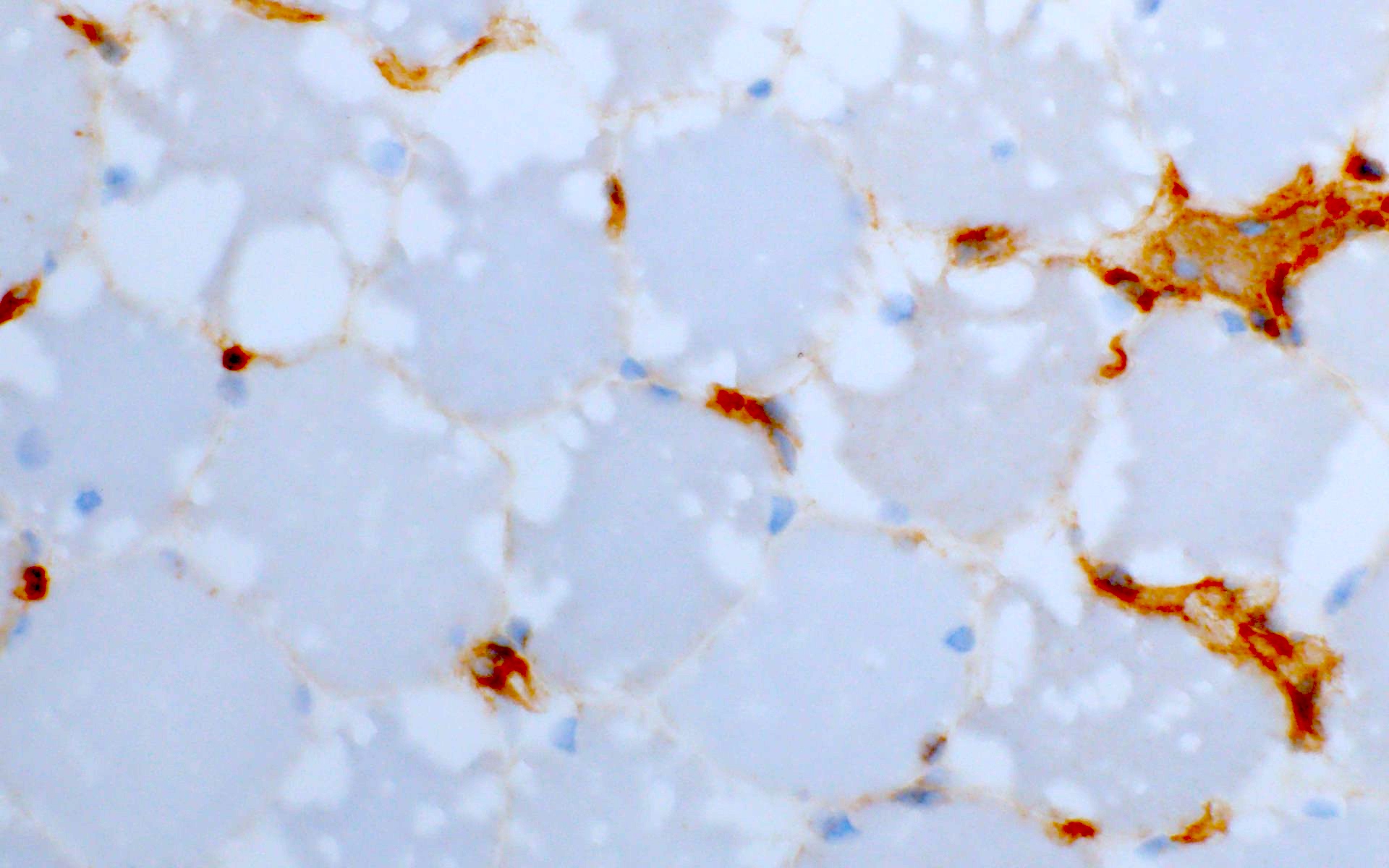
GSD III: MHC I

GSD IV: fibrosis
fibers and rounded
opalescent
inclusions

GSD IV: PAS
GSD IV: modified Gomori trichrome
GSD V: small sarcolemmal vacuoles
GSD V: PAS
GSD V: negative PHS
GSD VII: small sarcolemmal vacuoles

GSD VII: PAS
GSD VII:
phosphofructokinase
is negative
Images hosted on other servers:

McArdle disease
- PAS stain is intense positive in cytoplasm and vacuoles (except GSD 0)
- PAS stain is normal or mild positive in GSD X, XIV, XV, phosphoglycerate kinase deficiency
- ACP stain: acid phosphatase activity increases (increased lysosomes in GSD II)
- In GSD II, vacuoles are surrounded by MHC I (Goebel: Muscle Disease, 2nd Edition, 2013)
- PAS stain is reduced (in GSD 0)
- PHS stain: phosphorylase activity is deficient (in GSD V and when the GYS1 gene is mutated) (Dubowitz: Muscle Biopsy: A Practical Approach, 5th Edition, 2020, Biomedicines 2020;8:33)
- PFK stain: phosphofructokinase activity is reduced (< 10% of normal) or mildly reduced (in GSD VII) (Neuromuscul Disord 2021;31:1296)
- Inflammatory myopathy panel is negative
- GSD II: glycogen is seen within membrane bound vacuoles in the sarcoplasm of muscle fibers and disrupted myofibrils in the fiber
- Other GSDs: extensive areas of myofibrillar loss, subsarcolemmal and intermyofibrillar accumulation of accumulated nonmembrane bound glycogen
- Occasional fibers with accumulation of polyglucosan that look filamentous or amorphous (GSD IV, GSD VII) (Gaspar: Myopathology A Practical Clinico-pathological Approach to Skeletal Muscle Biopsies, 1st Edition, 2019)
Contributed by Ichizo Nishino, M.D., Ph.D.

GSD II childhood: membrane bound vacuoles

GSD II adult: membrane bound vacuoles

GSD III: nonmembrane bound glycogen
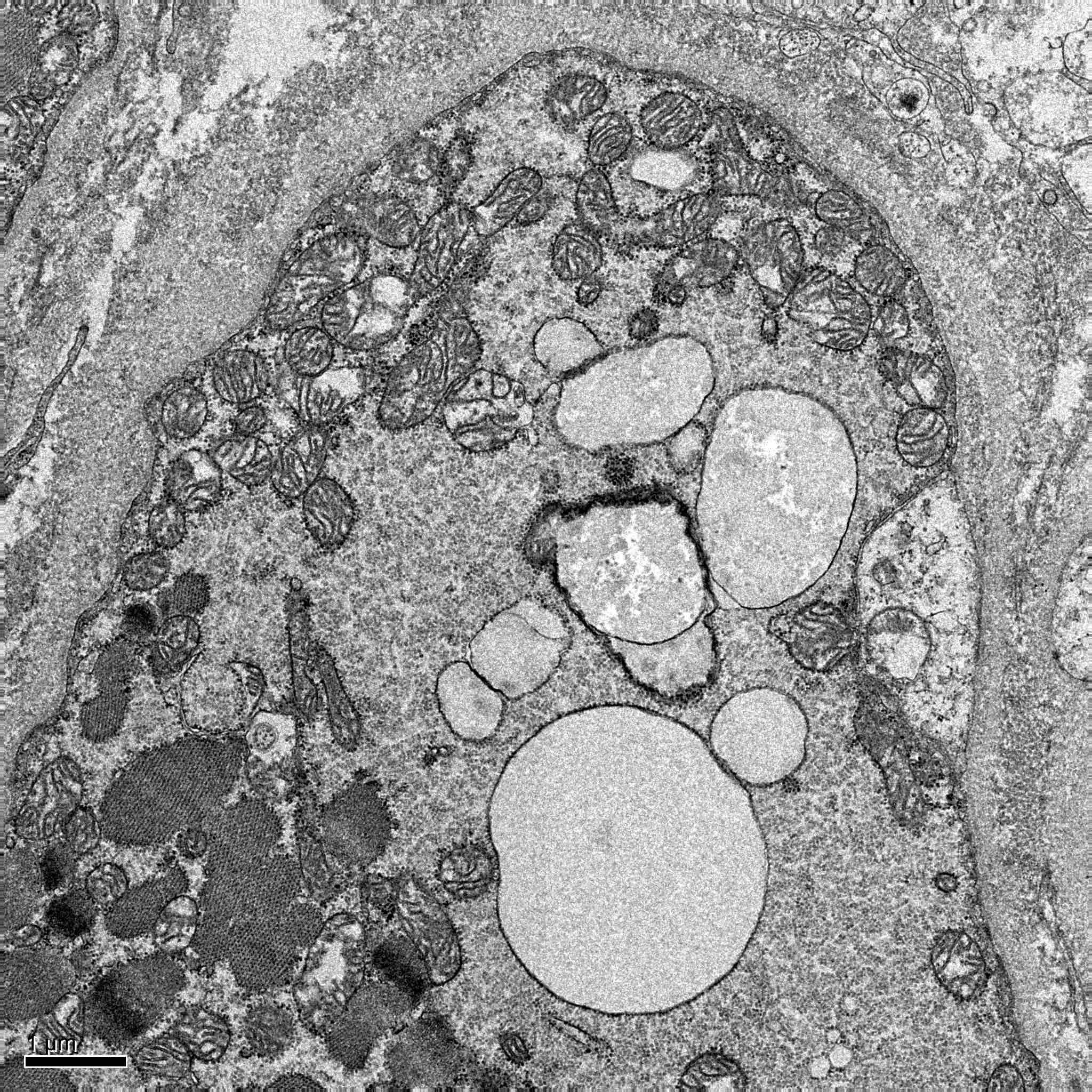
GSD IXd: sarcoplasmic glycogen deposits
- GSDs are inherited by an autosomal recessive trait, except for X linked GSD IXd and phosphoglycerate kinase deficiency (Ann Transl Med 2018;6:474)
- GSD 0: gene GYS1 (skeletal muscle) or GYS2 (liver) mutation
- GSD II: gene GAA mutation
- GSD III: gene AGL mutation
- GSD IV: gene GBE1 mutation
- GSD V: gene PYGM mutation
- GSD VII: gene PFKM mutation
- GSD IXd: gene PHKA1 (X linked) mutation
- Phosphoglycerate kinase deficiency, gene PGK1 (X linked) mutation
- GSD X: gene PGAM2 mutation
- GSD XI: gene LDHA mutation
- GSD XII: gene ALDOA mutation
- GSD XIII: gene ENO3 mutation
- GSD XIV: gene PGM1 mutation
- GSD XV: gene GYG1 mutation
- Left vastus lateralis, muscle biopsy, serial frozen sections were stained with H&E, modified Gomori trichrome (mGT) and a battery of histochemical methods:
- Compatible with glycogen storage disease (see comment)
- Comment:
- On H&E, there is mild to moderate variation in fiber size ranging from 20 to 105 microns in diameter. There are some fibers with multiple cytoplasmic vacuoles mainly in the subsarcolemmal region. No necrotic and regenerating fibers are seen. Scattered fibers with internal nuclei are seen. Mononuclear cell infiltration is not seen in endomysium. Perifascicular atrophy is not seen. Endomysial fibrosis is not seen.
- On mGT, no ragged red fibers, fibers with rimmed vacuoles or nemaline rods are seen.
- On NADH TR, intermyofibrillar networks are mildly disorganized in many fibers.
- On SDH, strongly SDH reactive blood vessels (SSVs) are not highlighted.
- On ALP, enzymatic activity is not seen in perimysium.
- On acid phosphatase, enzymatic activity is increased.
- On ATPase, moderate type 2 fiber atrophy is seen. Some type 2C fibers are seen.
- On PAS, strongly positive in vacuoles.
- On PHS, enzymatic activity is decreased.
- Other stains, including COX, AChE, NSE, AMP and MAG, show no additional abnormalities.
- The above findings are compatible with glycogen storage disease. Genetic analysis should be tested.
- Limb girdle muscular dystrophy:
- Can resemble GDS II in late onset
- Has a similar pattern of muscle involvement and disease course
- Muscle MRI: usually shows (in contrast to GSD II) fatty replacement of leg muscles
- Immunohistochemistry and genetic analysis help to differentiate
- Duchenne-Becker muscular dystrophy:
- Can resemble GDS II in late onset
- Progressive proximal muscle weakness, respiratory insufficiency and difficulty ambulating are seen
- Primarily affects males; inheritance is X linked
- Immunohistochemistry and genetic analysis help to differentiate
- Acid maltase
- Debrancher enzyme
- Glucose 6 phosphatase
- Phosphoglucomutase
- Phosphorylase
Comment Here
Reference: Glycogen storage diseases

Comment Here
Reference: Glycogen storage diseases
- Pathological definition of immune mediated necrotizing myopathy (IMNM) requires prominent myofiber necrosis, the absence of significant inflammatory infiltrates, negative major histocompatibility complex (MHC) class I expression and variable complement deposition on capillaries (Neuromuscul Disord 2018;28:87)
- Histological hallmark of IMNM is pauci-inflammatory necrotizing myopathy
- Necrotic fibers are randomly distributed and at different temporal stages
- Necrotizing autoimmune myopathy (NAM)
- HMGCR: 3-Hydroxy-3-Methylglutaryl-CoA Reductase
- ICD-10: G72.4 - inflammatory and immune myopathies, not elsewhere classified
- Prevalence of IMNM is approximately 1 per 100,000 people; accounts for ~10% of autoimmune myopathies
- More common in women
- Mean age of anti-SRP and statin exposed anti-HMGCR is 40 years
- Mean age of nonstatin exposed anti-HMGCR is 55 years
- Reference: Curr Rheumatol Rep 2018;20:21
- Major target organ of anti-SRP and anti-HMGCR are muscles, leading to myofiber necrosis and weakness (J Neurol Neurosurg Psychiatry 2016;87:1038)
- Statin exposure is highly associated with anti-HMGCR myopathy (Arthritis Rheum 2010;62:2757)
- In Asians, majority of anti-HMGCR myopathy is not associated with statin exposure (Medicine (Baltimore) 2015;94:e416)
- In adults, anti-HMGCR myopathy is strongly associated with the human leukocyte antigen (HLA) class DRB1*11:01 (Arthritis Care Res (Hoboken) 2012;64:1233)
- In children, anti-HMGCR myopathy is associated with HLA class DRB1*07:01 (Arthritis Care Res (Hoboken) 2017;69:1088)
- Hallmark clinical features of immune mediated necrotizing myopathy patients are proximal muscle weakness and elevated creatine kinase
- Severe limb muscle weakness, neck weakness, dysphagia, respiratory insufficiency and muscle atrophy are more frequent in patients with anti-SRP antibodies than in those with anti-HMGCR antibodies
- In anti-SRP positive patients, extramuscular organ involvement includes the heart, joints and lungs, with interstitial lung disease more common than in anti-HMGCR positive patients
- Reference: J Neurol Neurosurg Psychiatry 2016;87:1038
- IMNM is further classified by serum autoantibodies into anti-SRP, anti-HMGCR and seronegative groups (Neuromuscul Disord 2018;28:87)
- SRP group:
- Titer of anti-SRP antibody directly correlates with myofiber necrosis and serum creatine kinase levels (Arthritis Rheum 2011;63:1961)
- This group usually has more severe, rapidly progressive muscle weakness, frequent lung involvement, occasional cardiac muscle involvement and poor response to treatment but is not associated with risk of malignancy (Curr Opin Rheumatol 2016;28:619)
- HMGCR group:
- Majority of this group is developed in the setting of statin usage and generally responds very well to immune suppressive treatment (Arthritis Rheum 2010;62:2757)
- Serum creatine kinase level correlated with anti-HMGCR level only in the patients with statin exposure but not in the statin unexposed patients (Arthritis Rheum 2012;64:4087)
- There is a moderate risk of malignancy (Neurol Neuroimmunol Neuroinflamm 2016;3:e290)
- Seronegative group:
- This group has negative serum autoantibodies but a similar muscle pathology to IMNM
- Associated with a high risk of malignancy (Curr Opin Rheumatol 2018;30:655)
- Anti-SRP and anti-HMGCR autoantibodies are considered specific for IMNM
- Patients with dermatomyositis-like rash but with positive anti-SRP or anti-HMGCR autoantibodies are classified as IMNM with dermatomyositis-like rashes (Neuromuscul Disord 2020;30:70)
- Patients without anti-SRP or anti-HMGCR but with elevated creatine kinase, proximal weakness and muscle biopsy with features of IMNM (outlined below) are diagnosed as seronegative IMNM
- Pathological diagnosis of IMNM requires (Neuromuscul Disord 2018;28:87):
- Presence of necrotic fibers with scattered distribution
- Different stages of necrosis, myophagocytosis and regeneration
- Macrophage predominant, pauci-lymphocytic inflammation
- Following features can be seen in IMNM (presence of these features does not exclude IMNM) (Neuromuscul Disord 2018;28:87):
- Sarcolemmal MHC class I expression in nonnecrotic / nonregenerating myofibers (typically small clusters)
- Patchy sarcolemmal C5b9 (terminal complement complex) deposition
- Endomysial fibrosis
- Enlarged capillaries
- Markedly elevated creatine kinase or persistently elevated creatine kinase after statin cessation
- Positive anti-HMGCR or anti-SRP
- Negative for autoantibodies specific to dermatomyositis (e.g. Mi2, NXP2, TIF1γ, MDA5, p155 / 140, SAE) or antisynthetase syndrome (e.g. Jo1, PL7, PL12)
- Reference: Neuromuscul Disord 2018;28:87
- Hyperintensities on short tau inversion recovery sequence (STIR) reflect active muscle edema associated with inflammation or myofiber necrosis
- T1 MRI can be used to assess fat replacement
- MRI does not distinguish IMNM from other types of myositis; however, it can be used to guide / target muscle biopsy and monitor the evolution of muscle disease
- Reference: Ann Rheum Dis 2017;76:681
- Prognosis of IMNM is in general worse than other types of myositis (Curr Rheumatol Rep 2018;20:21)
- Anti-SRP myopathy has more severe, rapidly progressive muscle weakness and poor response to treatment (Curr Opin Rheumatol 2016;28:619)
- Statin induced anti-HMGCR myopathy generally responds well to immune suppressive therapy (Arthritis Rheum 2010;62:2757)
- 40 year old woman with anti-SRP antibody interstitial lung disease requiring double lung transplant (Cureus 2020;12:e7962)
- 63 year old woman with previous statin exposure presented with muscle weakness and raised creatinine kinase (BMC Rheumatol 2020;4:29)
- 87 year old woman with heart failure symptoms as the first clinical presentation (BMC Neurol 2020;20:29)
- Initial treatment includes intravenous or oral steroids, along with the addition of another agent (e.g. methotrexate, rituximab, intravenous immunoglobulin) at the same time or within 1 month, depending on severity and treatment response
- Intravenous immunoglobulin (IVIG) should always be added within 6 months if other strategies are not effective; IVIG monotherapy can be considered in patients with steroid contraindications
- In patients requiring intensive care as well as in refractory patients, plasma exchange, cyclophosphamide or cyclosporine may be considered
- Reference: Neuromuscul Disord 2018;28:87
- See Diagnosis section
Contributed by Chunyu "Hunter" Cai, M.D., Ph.D.
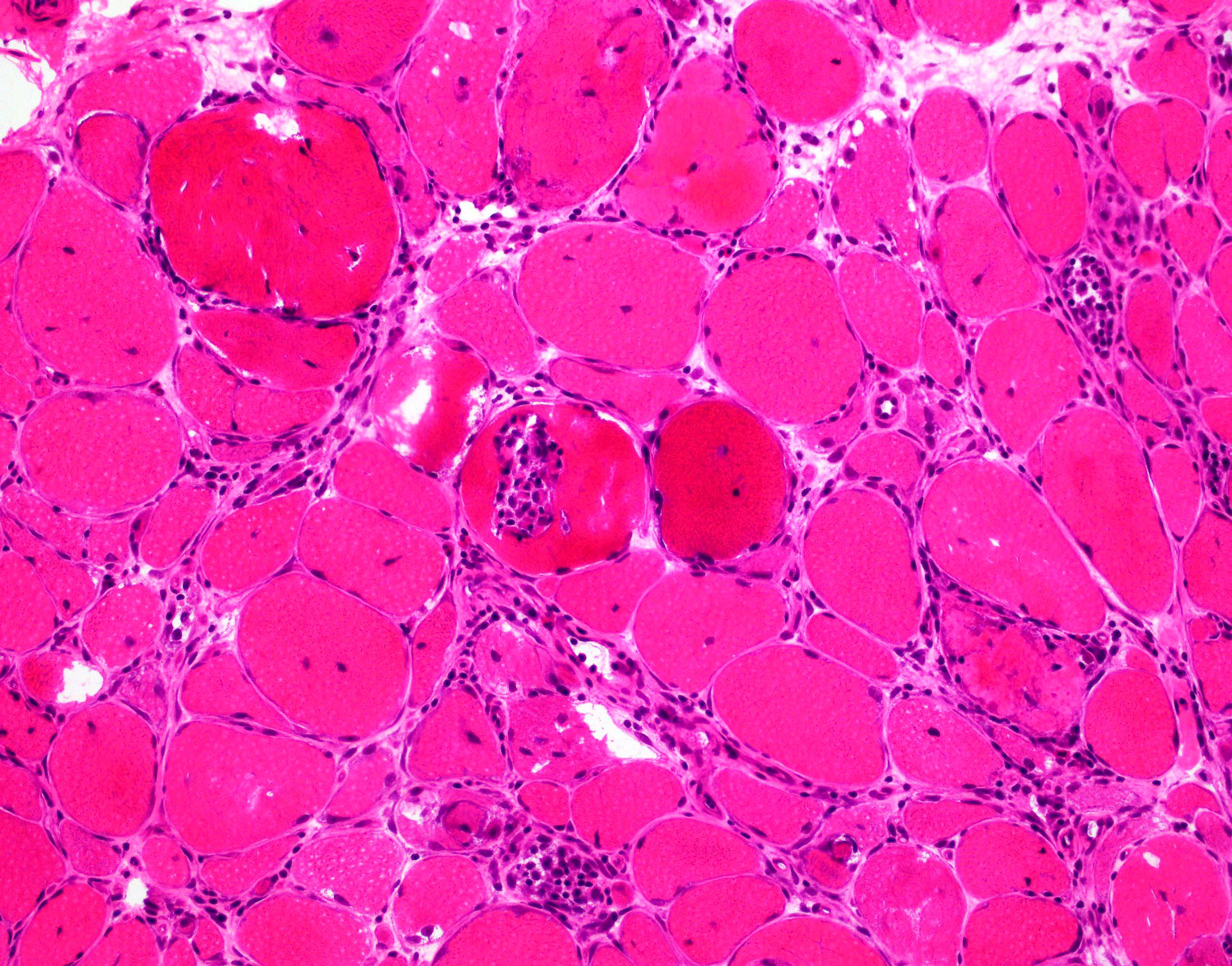
HMGCR myopathy
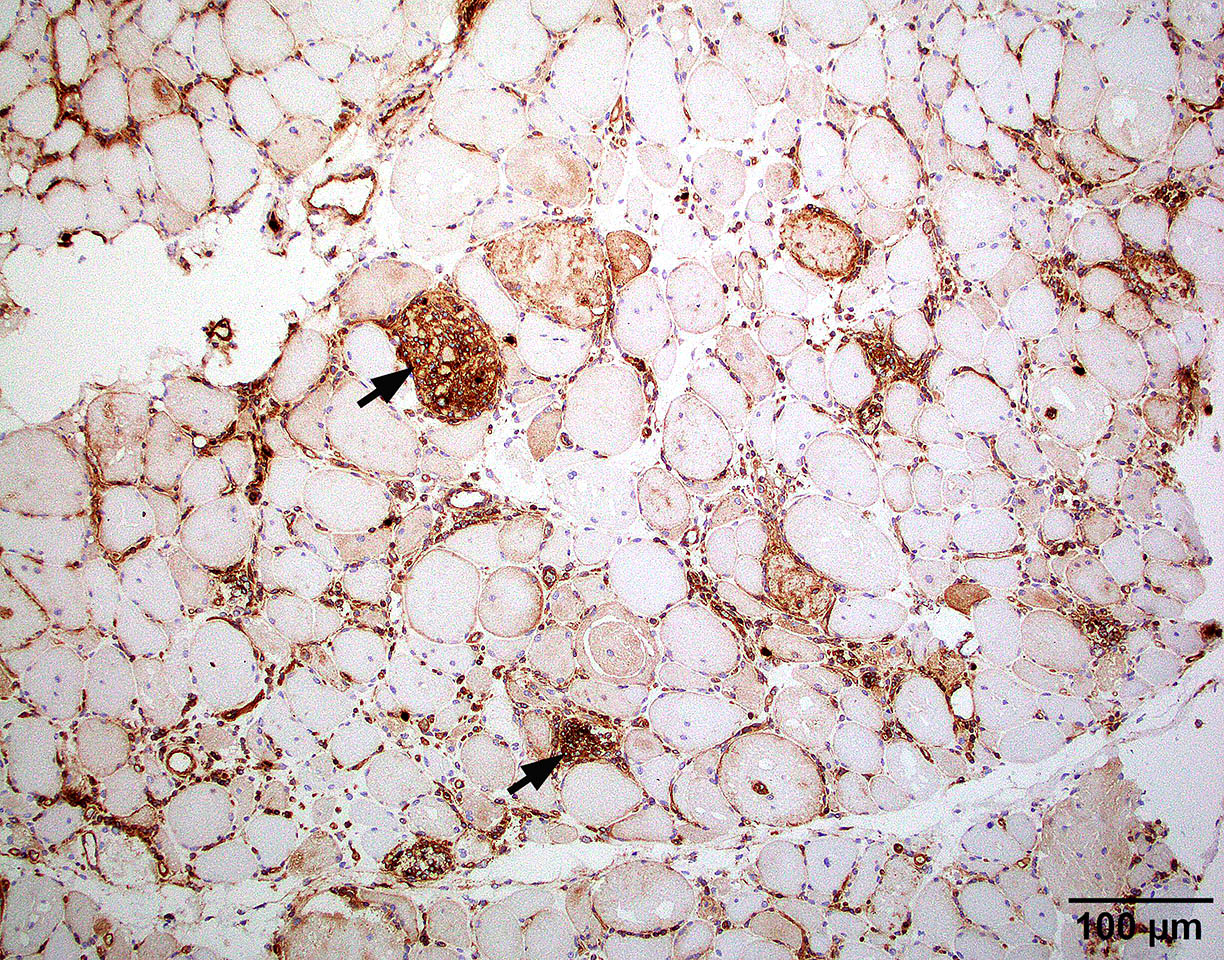
HMGCR myopathy - MHC1
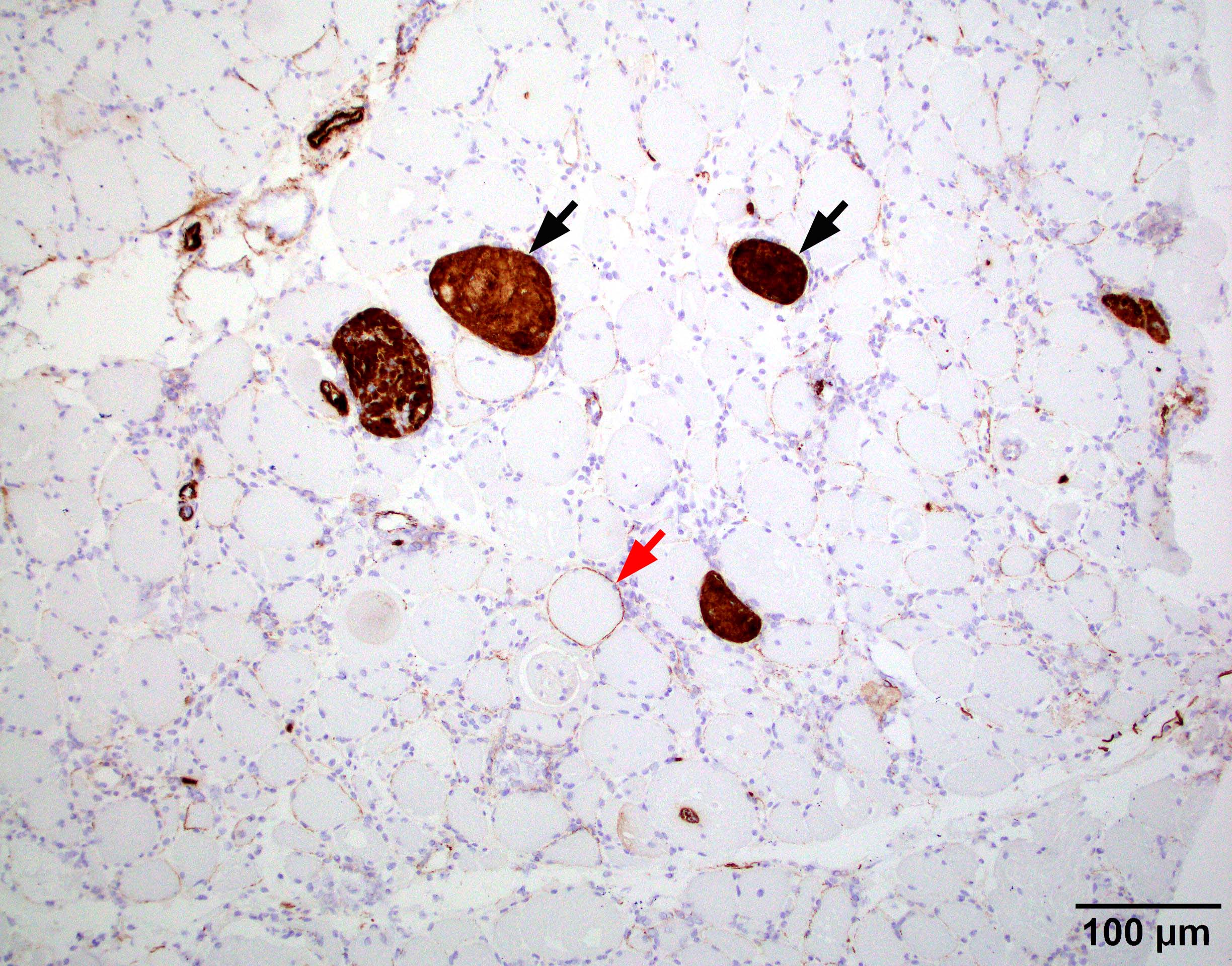
HMGCR myopathy - C5b9
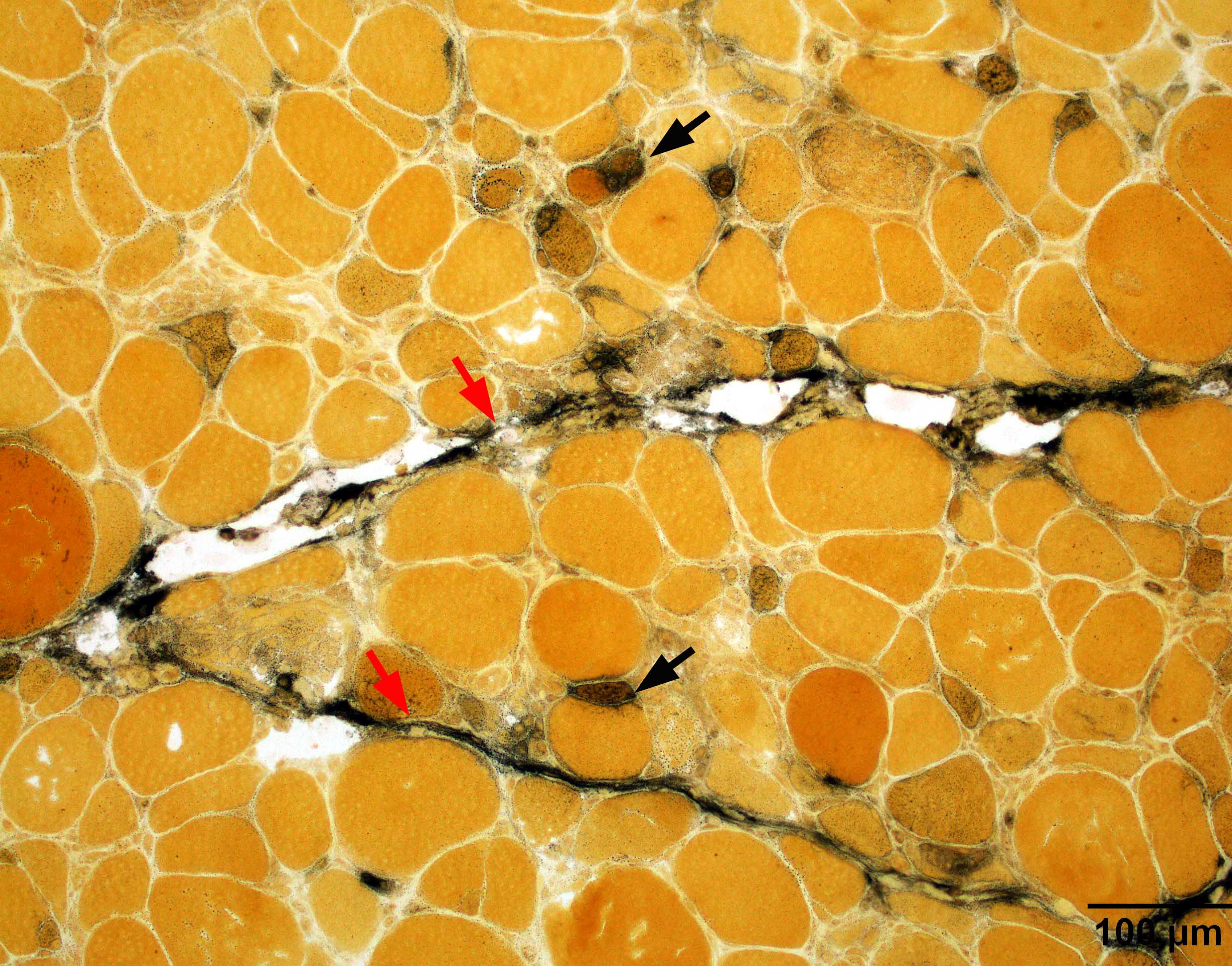
HMGCR myopathy - alkaline phosphatase
- Strong sarcoplasmic C5b9 expression is seen in acutely necrotic fibers of any causes (usually abundant but not limited to IMNM)
- Sarcolemmal C5b9 expression in a subset of viable myofibers can be seen in IMNM (Neuromuscul Disord 2018;28:87)
- IMNM is usually negative for MHC1; a subset of IMNM with anti-HMGCR may have some MHC1 expression in scattered myofibers but no diffuse expression (Arthritis Rheum 2010;62:2757)
- There are no endothelial tubuloreticular inclusions in IMNM
- Skeletal muscle, left quadriceps, biopsy:
- Necrotizing myopathy (see comment)
- Comment: The muscle demonstrates a pauci-inflammation necrotizing myopathy. Necrotic fibers are randomly distributed and temporally heterogeneous, including all stages of acute necrosis, myophagocytosis and regeneration. There are no specific features, such as perifascicular atrophy, lymphocytic invasion in myofibers or rimmed vacuoles, to suggest dermatomyositis, polymyositis or inclusion body myositis, respectively.
- The primary differential consideration is an immune mediated necrotizing myopathy (IMNM). Clinical correlation with serological myositis specific markers including anti-SRP and anti-HMGCR antibodies may be of additional diagnostic value. Alternatively, exogenous toxins or medication such as statins may cause acute myofiber damage. Clinical correlation is needed. Metabolic disease can present as acute myofiber damage or rhabdomyolysis but this is less likely given the age of the patient, presentation and lack of storage material in muscle fibers.
- Rhabdomyolysis:
- Necrotic fibers in rhabdomyolysis are more monophasic (temporally homogeneous), while necrotic fibers in IMNM are more temporally heterogeneous (include all stages of acute necrosis, myophagocytosis, regeneration)
- Polymyositis:
- Polymyositis requires the presence of lymphocytic inflammation surrounding or invading myofibers; IMNM typically lacks visible lymphocytic inflammation
- MHC1 is usually diffuse upregulated in polymyositis but negative in IMNM
- Muscular dystrophy:
- Besides abundant necrotic fibers, muscular dystrophy typically has prominent chronic dystrophic changes (i.e. fatty replacement, interstitial fibrosis, prominent fiber size variation), which are usually lacking in IMNM
- Antisynthetase syndrome associated myositis:
- Antisynthetase syndrome associated myositis typically shows prominent perifascicular necrotizing myopathy and perimysial connective tissue damage that is distinctive from the randomly distributed pattern from IMNM (Brain 2015;138:2485, Brain 2016;139:e50)
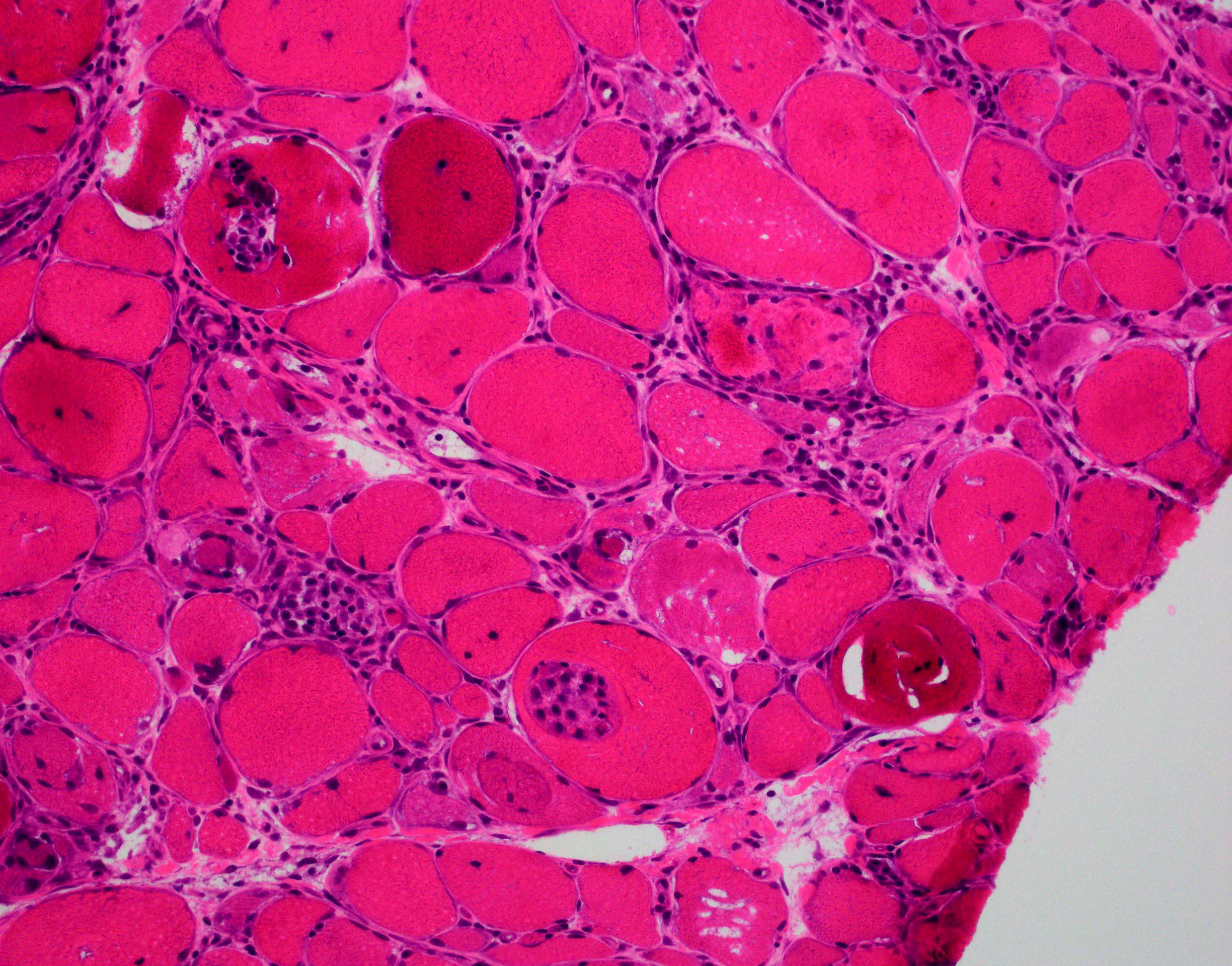
A 55 year old man presented with 2 weeks of proximal muscle weakness and myalgia. Muscle biopsy shows finding in the above image. Which of the following serum autoantibodies is most supportive of the diagnosis of an immune mediated necrotizing myopathy?
- Anti-ANA
- Anti-Jo1
- Anti-Mi2
- Anti-NT5c1a
- Anti-SRP
Comment Here
Reference: Immune mediated necrotizing myopathy
A 55 year old man presented with 2 weeks of proximal muscle weakness, myalgia and elevated creatine kinase. Muscle biopsy shows necrotizing myopathy. Serology workup is positive for anti-SRP autoantibody. Which of the following additional tests is highly recommended?
- Abdominal CT
- Cancer screening tests
- Chest CT
- EKG
Comment Here
Reference: Immune mediated necrotizing myopathy
- An inflammatory myopathy of predominantly skeletal muscle usually seen in ages 50+
- The main histologic finding is rimmed vacuoles with accumulation of specific proteins of autophagy
- Myopathic muscle with increased internal nuclei and myofiber size variation
- Inflammatory response is primarily centered around myofibers and is not perivascular
- "Rimmed vacuoles" are seen with Gomori trichrome stain and protein inclusions of autophagy with special stains
- Inclusion body myositis (Lab Invest 1971;25:240), IBM, inflammatory myopathy with rimmed vacuoles
- In North America, constitutes 16 - 28% of inflammatory myopathies
- Male to female ratio is 3:1
- Most occur in older individuals, although congenital and childhood forms have been described (reference)
- The average age is between 40s - 70s in men and slightly older in females
- Generally affects the proximal leg and distal arm musculature; however, any skeletal muscle can be involved
- Dysphagia can also occur
- This is a progressive muscle weakness with an insidious onset
- Patients may have other autoimmune disorders
- Generally, it affects proximal leg and distal arm musculature and patients can have asymmetry of their weakness
- The non-dominant side may be affected to a greater extent than the dominant side
- The most common complaint is quadriceps weakness
- Finger flexor and ankle dorsiflexion weakness can be noted clinically
- Dysphagia is a common symptom (J Neurol 2005;252:1448)
- Generally, patients do not report myalgias (muscle pain or cramps)
- Muscle biopsy
- Serum creatine kinase (CK) can be normal or elevated
- Typically, patients do not have autoantibodies
- Physical therapy and occupational therapy (exercise program) may be of some assistance (J Rehabil Med 2003;34:31)
- Recalcitrant to steroid therapy and will progress despite high dose steroids, in contrast to polymyositis, which generally responds to steroid therapy
- CK, aldolase and AST levels can be elevated (all are non-specific markers of muscle damage)
- Has a limited role for diagnosis
- Refractory to steroids or immunotherapy
- Generally, weakness slowly progresses, although complete wheelchair dependence occurs in only 3% (J Neurol 2005;252:1448)
- 53 year old man with associated collagen vascular disease (J Neurol Neurosurg Psychiatry 1985;48:270)
- 62 year old man with dermatomyositis (Br J Dermatol 1999;141:926)
- Inclusion body myositis in connective tissue disorders (Clin Rheumatol 2003;22:324)
- Inclusion body myositis with cricopharyngeus muscle involvement and severe dysphagia (Muscle Nerve 1991;14:470)
- Macrophagic myofasciitis associated with inclusion body myositis (Neuromuscul Disord 2001;11:452)
- Creutzfeldt-Jakob disease and inclusion body myositis (Ann Neurol 2004;55:121)
- Sporadic inclusion body myositis in a patient with HTLV1 associated myelopathy (Clin Infect Dis 2001;32:510)
- Variation in myofiber sizes with small angulated myofibers, either individually or in groups
- Hypertrophied myofibers can also be seen
- An increase in internal nuclei (normal muscle can have up to 3% of myofibers having internal nuclei) can be seen
- There can be a brisk inflammatory response including CD8+ lymphocytes, which may invade non-necrotic myofibers
- Occasional myofibers undergoing phagocytosis by CD68+ macrophages can be identified
- Regenerating, basophilic myofibers can be seen
- The Gomori Trichrome stain shows "rimmed vacuoles", although the extent of myofibers having classic rimmed vacuoles varies (Dubowitz: Muscle Biopsy: A Practical Approach, 2013, 4th Edition)
- The vacuoles disrupt the myofiber architecture and can lack NADH-TR staining (Dubowitz: Muscle Biopsy: A Practical Approach, 2013, 4th Edition)
Contributed by Meggen Walsh, D.O., M.S., P.A.
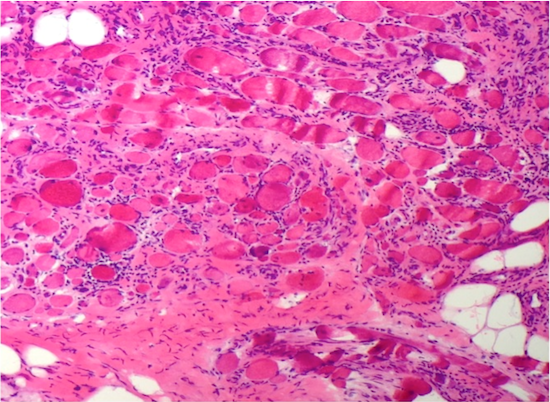
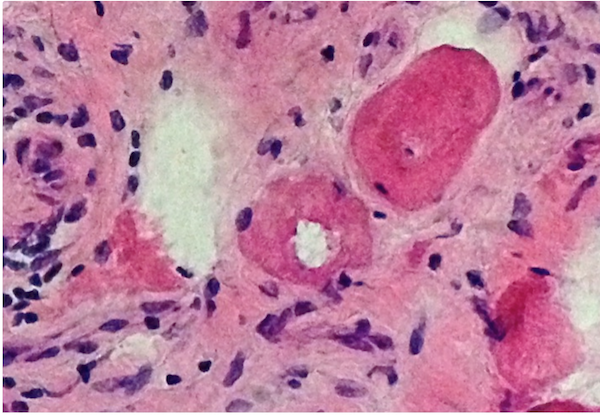
Myopathic features
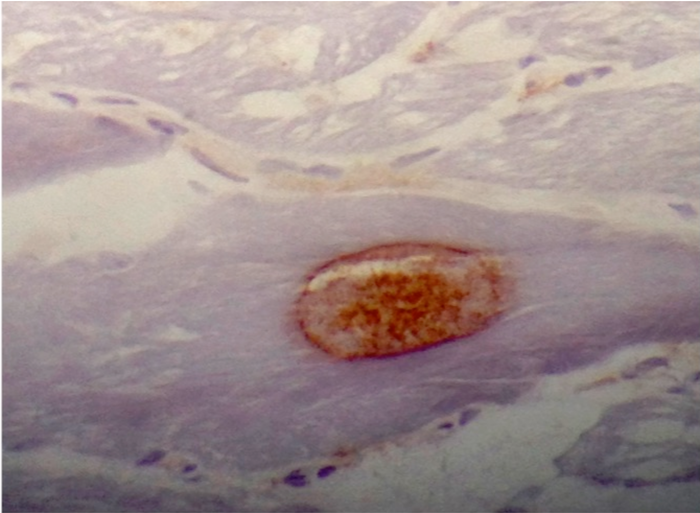
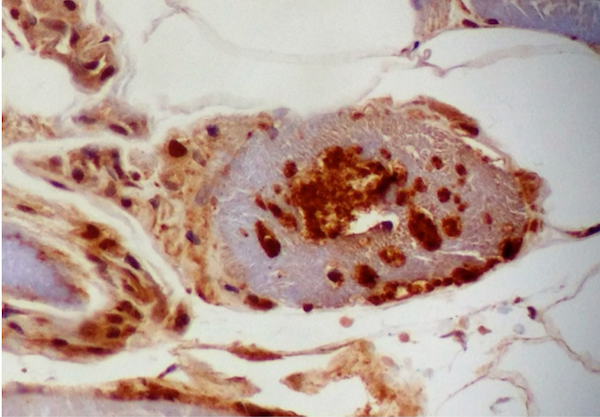
Ubiquitin
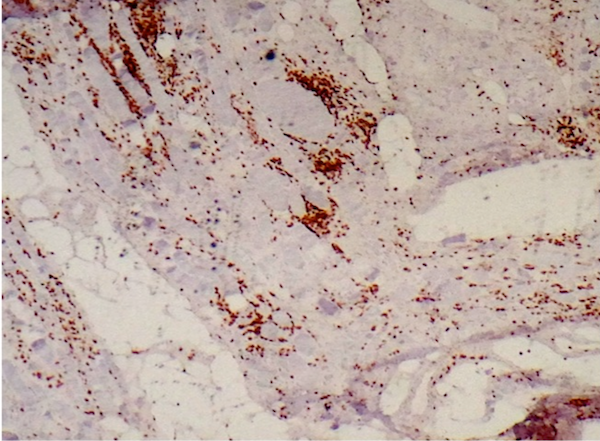
CD3
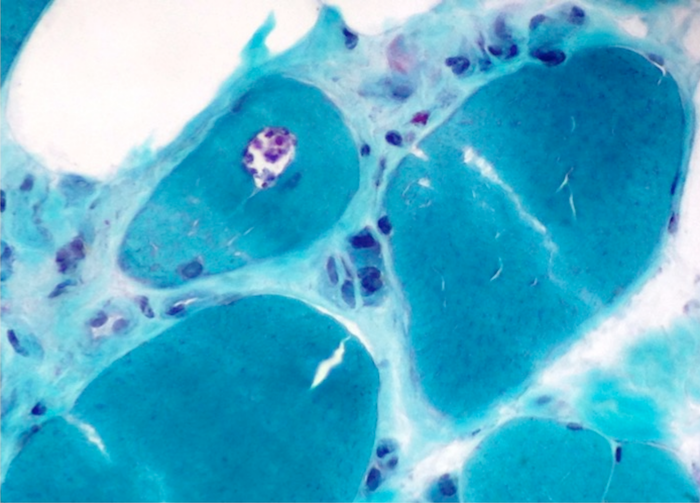
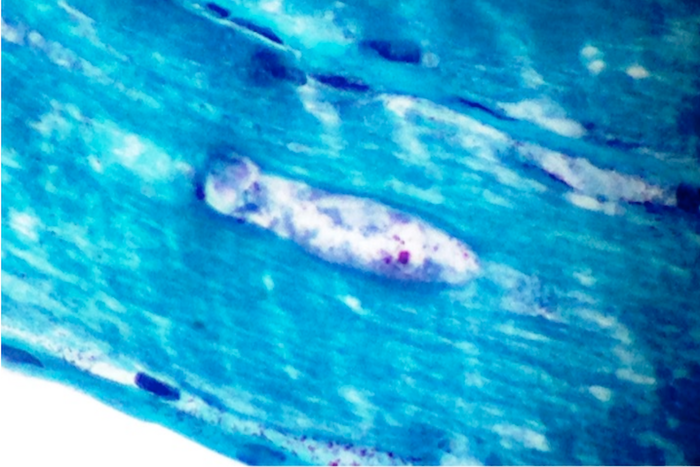
Gomori trichrome
- Cytology is of limited to no benefit - muscle needle biopsy may not provide a large enough sample to obtain myofibers with rimmed vacuoles
- Gomori trichrome shows rimmed vacuoles and can show ragged red fibers in areas
- Congo Red (amyloid) positive "apple-green" birefringence positive inclusions (Arch Neurol 1991;48:1229)
- Ubiquitin and LC3B highlight the inclusions
- Ubiquitin, B-amyloid, B-amyloid precursor protein (APP), α-synuclein, tau, TDP43, and LC3B can be seen in the inclusion bodies
- CD8+ T lymphocytes highlight the inflammatory response and infiltrate non-necrotic myofibers
- Rarely, cytochrome oxidase / COX negative fibers are seen
- 15-21 nm tubulofilaments in sarcoplasm
- Mitochondria may be structurally abnormal (Yachnis: Neuropathology: A Volume in the High Yield Pathology Series, 2013, 1st Edition)
- Associated with HLA-DR3, DR52 and B8 (Yachnis: Neuropathology: A Volume in the High Yield Pathology Series, 2013, 1st Edition)
- Upregulation of MHCI in myofibers (Dubowitz: Muscle Biopsy: A Practical Approach, 2013, 4th Edition)
- Amyotrophic lateral sclerosis: progressive muscle weakness despite therapy, with predominantly, neurogenic changes on muscle biopsy and minimal to no inflammation
- Dermatomyositis: an inflammatory myositis, but inflammation is generally perivascular with CD4+ T cells and perifascicular myofiber atrophy
- Polymyositis: in the differential if rimmed vacuoles are not present in inflammatory myositis, because immunostudies for ubiquitin or LC3 show coarse granular staining which suggests inflammatory myositis
- Inherited muscular dystrophy characterized by muscle weakness, myotonia and additional systemic manifestations including cardiac and neurologic
- Myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2) are caused by differing nucleotide repeat expansions but have similar pathophysiologic mechanisms
- DM1 is the most common type of adult onset muscular dystrophy
- Autosomal dominant (AD) muscular dystrophy caused by expansions of different nucleotide repeats which affect RNA splicing and processing, leading to muscle weakness, myotonia and systemic effects
- Variable clinical course, from late onset of mild symptoms to death in infancy
- Frequently involves cardiac conduction system and CNS
- Significantly increased internal nuclei on histologic examination
- Dystrophia myotonica (DM)
- Classic type DM1 was first clinically described in 1909 by German physician Hans Steinert and termed, "Steinert's disease" (Biochim Biophys Acta 2015;1852:594)
- ICD-10: G71.11 - myotonic muscular dystrophy
- Prevalence varies by region and is most common in individuals of European descent
- DM1 is more common than DM2 in the U.S. but some studies suggest similar prevalence of DM1 and DM2 in Europe (Neurol Clin 2014;32:705)
- Combined prevalence reported as 1 in 8,000 (12.5 per 100,000) but this is likely an underestimate due to clinical heterogeneity (Curr Opin Genet Dev 2017;44:30)
- DM1:
- Involvement of distal limb muscles with preferential involvement of finger, wrist and ankle flexors
- Diaphragm involvement can occur early in disease course
- Neck flexors involved
- Facial muscles involved with wasting of temporalis muscles (hatchet appearance)
- DM2:
- Proximal musculature affected including limb girdle, neck flexors and elbow extensor muscles (Neurol Clin 2014;32:705)
- Less involvement of facial or respiratory musculature
- Both forms are a result of toxicity from abnormal mRNA caused by expanded repeats
- DM1 has been more extensively studied
- Mutant RNA with expanded repeats is not exported to cytoplasm but is retained in the nucleus where it forms multiple clumps or foci
- RNA binding proteins such as MBNL1 and DDX6 exhibit high affinity for the mutant RNA and become sequestered in the nucleus
- These proteins are normally involved in splicing, as well as mRNA transport, stability and decay
- Protein function is lost once sequestered, leading to incorrect splicing and defects of proteins including insulin receptor, dystrophin, BIN1 and ClC-1 and L type calcium channels
- Mutated RNA may also produce peptides which are directly cytotoxic (Neurol Clin 2014;32:705)
- Autosomal dominant (AD) inheritance in both DM1 and DM2
- DM1 is caused by expansion of a CTG repeat in the 3' noncoding region of the DMPK gene on chromosome 19q13.3, which codes for myotonic dystrophy protein kinase
- Normal individuals have between 5 and 37 repeats but symptomatic patients typically have > 50 repeats
- Anticipation is frequently seen
- Symptoms appear earlier and with greater severity in successive generations
- Individuals with borderline elevated CTG repeats (> 50) may be asymptomatic but offspring are at risk
- Clinical presentation correlates with CTG repeat size (Neurol Clin 2014;32:705)
- DM2 is caused by expansion of a CCTG repeat in the first intron of the CNTB gene (previously ZNF9) on chromosome 3q21, which codes for CCHC type zinc finger nucleic acid binding protein
- Normal individuals typically have between 10 and 33 repeats but symptomatic patients usually have greater than 1,000 repeats (range, 75 to greater than 11,000)
- Anticipation is less prominent
- No correlation between repeat size and clinical presentation (Neurol Clin 2014;32:705)
- DM1 is typically broken down into four subtypes: congenital, childhood, classic and minimal / late onset
- Spectrum of clinical severity: from death in infancy to onset in late adulthood with extremely mild symptoms
- Congenital DM1: fetal onset, involving musculature and CNS; severe
- Prenatal features: decreased fetal movement, polyhydramnios
- Neonatal features: hypotonia with feeding or respiratory distress
- 79% require nasogastric feeding, 53% require ventilator support (Neurol Clin 2014;32:705)
- Childhood: delayed motor milestones, intellectual impairment, prominent oropharyngeal weakness with tenting of upper lip
- Degenerative features develop by second or third decade, resembling classic DM1
- More than half of mothers do not carry DM1 diagnosis so diagnosis can be delayed (Neurol Clin 2014;32:705)
- Childhood DM1: between 1 and 10 years of age
- Predominantly cognitive and behavioral issues
- Facial weakness and conduction abnormalities
- Approximately half with intellectual impairment
- Range of psychiatric disorders
- Classic DM1: onset usually between second and fourth decades
- Myotonia is the most common presenting symptom
- More pronounced after rest, improves with activity
- Involves forearms and hands (grip), tongue and jaw
- Muscle weakness of distal limbs and craniofacial muscles
- Wasting of fascial muscles with characteristic ptosis and hatchet appearance
- Respiratory distress secondary to diaphragmatic involvement
- Cardiac conduction abnormalities common
- Risk of sudden cardiac death as high as 1.1% per year
- Cataracts located on the posterior lens capsule with a multicolored, iridescent appearance on slit lamp examination (Neurol Clin 2014;32:705)
- Sleep disturbances (80% with daytime hypersomnolence)
- Gastrointestinal involvement: cholelithiasis, intestinal dysmotility
- Insulin resistance, metabolic syndrome, frontal balding and hypogonadism in men
- Evidence for increased risk of malignancy (thyroid, ovarian, colorectal, endometrial, Mayo Clin Proc 2012;87:130, JAMA 2011;306:2480)
- Myotonia is the most common presenting symptom
- Minimal / late onset DM1: small expansions (70 - 100 repeat) with mild weakness, myotonia and development of cataracts, usually after age 40 (range 20 to 70 years, Neurol Clin 2014;32:705)
- DM2: overall milder disease; most often presents in the third decade of life (range second to sixth decades)
- Presents with proximal muscle weakness as well as myotonia
- May resemble limb girdle dystrophy
- Less muscle wasting and respiratory involvement than DM1
- Frontal balding, hypogonadism, cataracts and insulin resistance
- Less cardiac and CNS involvement than DM1 (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Some patients exhibit calf and thigh hypertrophy (true hypertrophy)
- Patients often have a history of unexplained pain and may have a diagnosis of fibromyalgia (Neurol Clin 2014;32:705)
- Presents with proximal muscle weakness as well as myotonia
- Molecular testing is definitive and may be the only test performed in the appropriate clinical setting
- PCR is most commonly used for detection of repeat expansion
- Southern blot is sometimes utilized in addition to PCR testing (Eur J Hum Genet 2012;20:1203)
- Muscle biopsy is infrequently performed if clinical suspicion is high
- Not specific
- Liver function tests (LFTs) commonly abnormal
- Creatine kinase (CK) can be normal to slightly elevated
- Electromyography (EMG) shows combination of myotonic features and myopathic changes (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- No specific imaging features of the involved musculature have been identified
- Imaging of CNS may show alterations in the white matter signal intensity, most notable in the frontotemporal region
- Prenatal ultrasound in congenital DM1 patients may show borderline ventriculomegaly or talipes equinovarus (club foot) (Neurol Clin 2014;32:705)
- Classic DM1 has a slowly progressive course
- Respiratory failure is the leading cause of death in DM1, followed by sudden cardiac death (Neurol Clin 2014;32:705)
- Congenital DM1 patients may live to adulthood and typically die of cardiorespiratory complications (similar to classic DM1)
- DM2 patients typically have a milder clinical course
- 38 year old man with myotonic dystrophy mimicking postpolio syndrome in a polio survivor (Am J Phys Med Rehabil 2009;88:161)
- 61 year old man with normal pressure hydrocephalus (Neurosurgery 2006;58:E796)
- Woman with gastric bypass surgery for obesity in DM1 (Neuromuscul Disord 2015;25:414)
- Patient with hydrocephalus and cognitive decline in myotonic dystrophy (Arch Phys Med Rehabil 1998;79:1022)
- No curative treatment - supportive therapy only
- Many patients require nighttime respiratory support
- May require pacemaker or defibrillator placement
- Ongoing research into curative genetic therapies
- Variation in myofiber size, ranging from 10 um to 100 um
- Ring fibers and sarcoplasmic masses (dark staining regions) are frequently seen in DM1
- DM1: type 1 myofiber atrophy with type 2 hypertrophy
- DM2: greater variation in both type 1 and 2 fibers with predominantly type 2 myofiber atrophy (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Pyknotic nuclear clumps in atrophic fibers
- May see moth eaten or whorled fibers
Contributed by Jesse L. Kresak, M.D
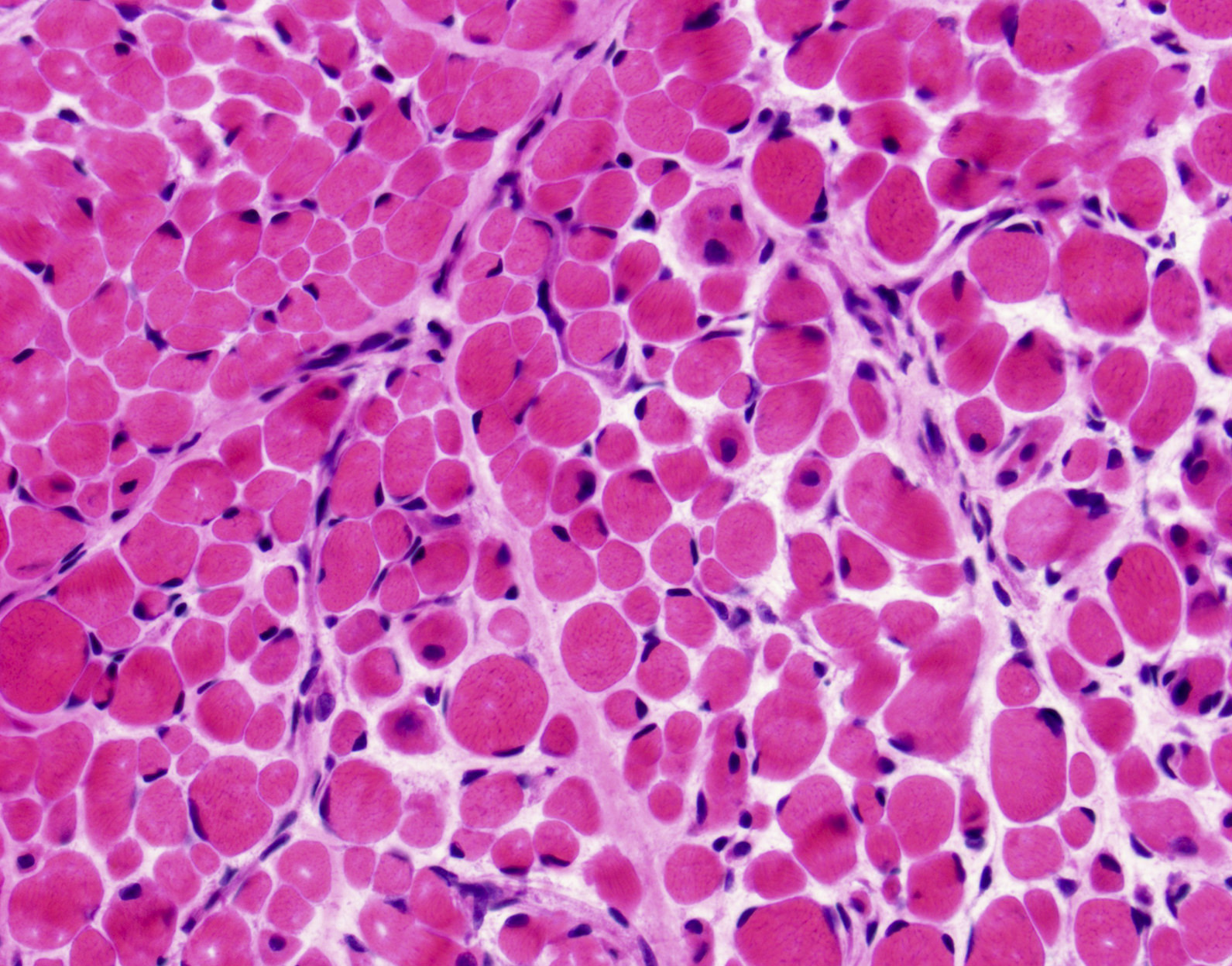
Scattered internal nuclei
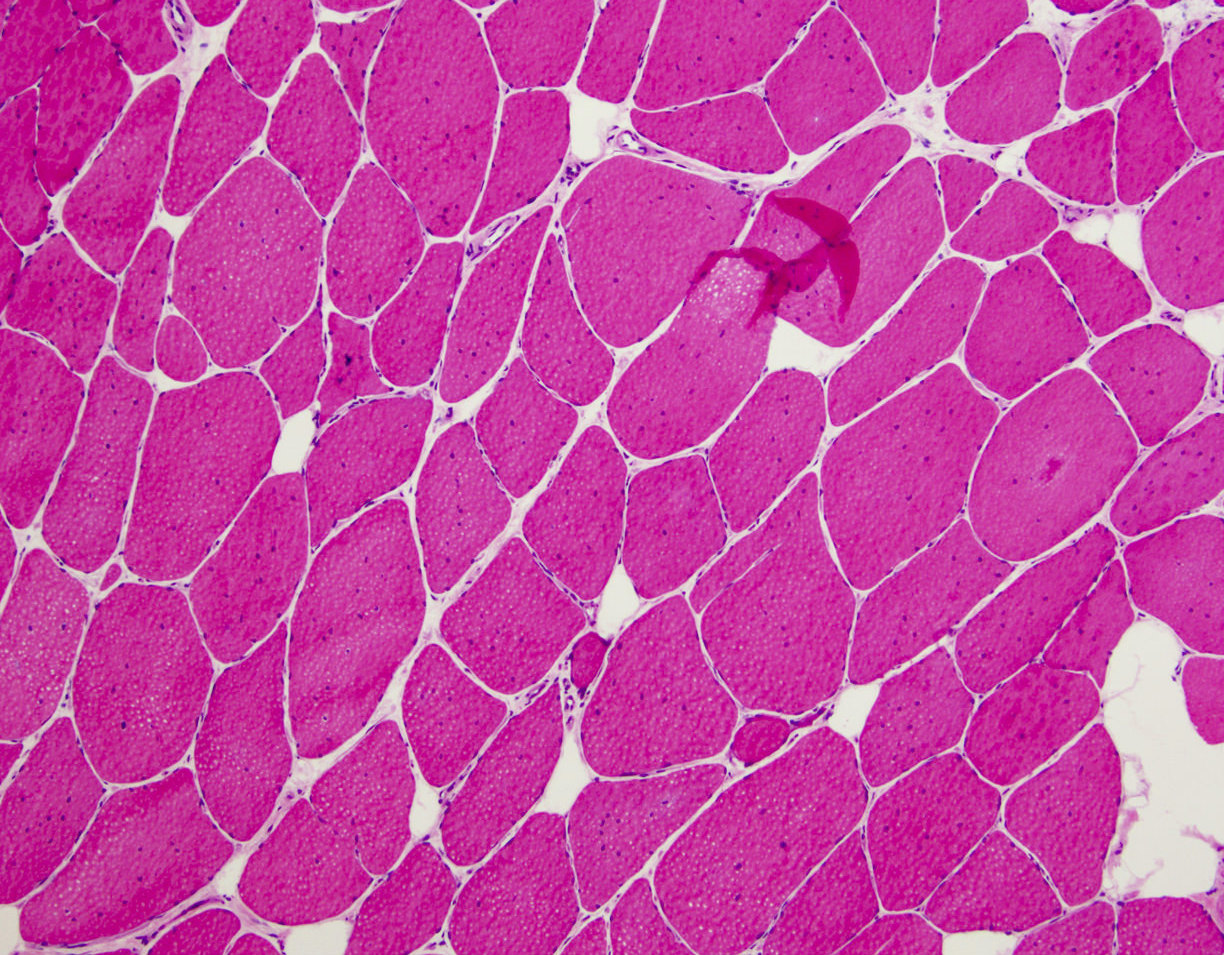
Increased internal nuclei
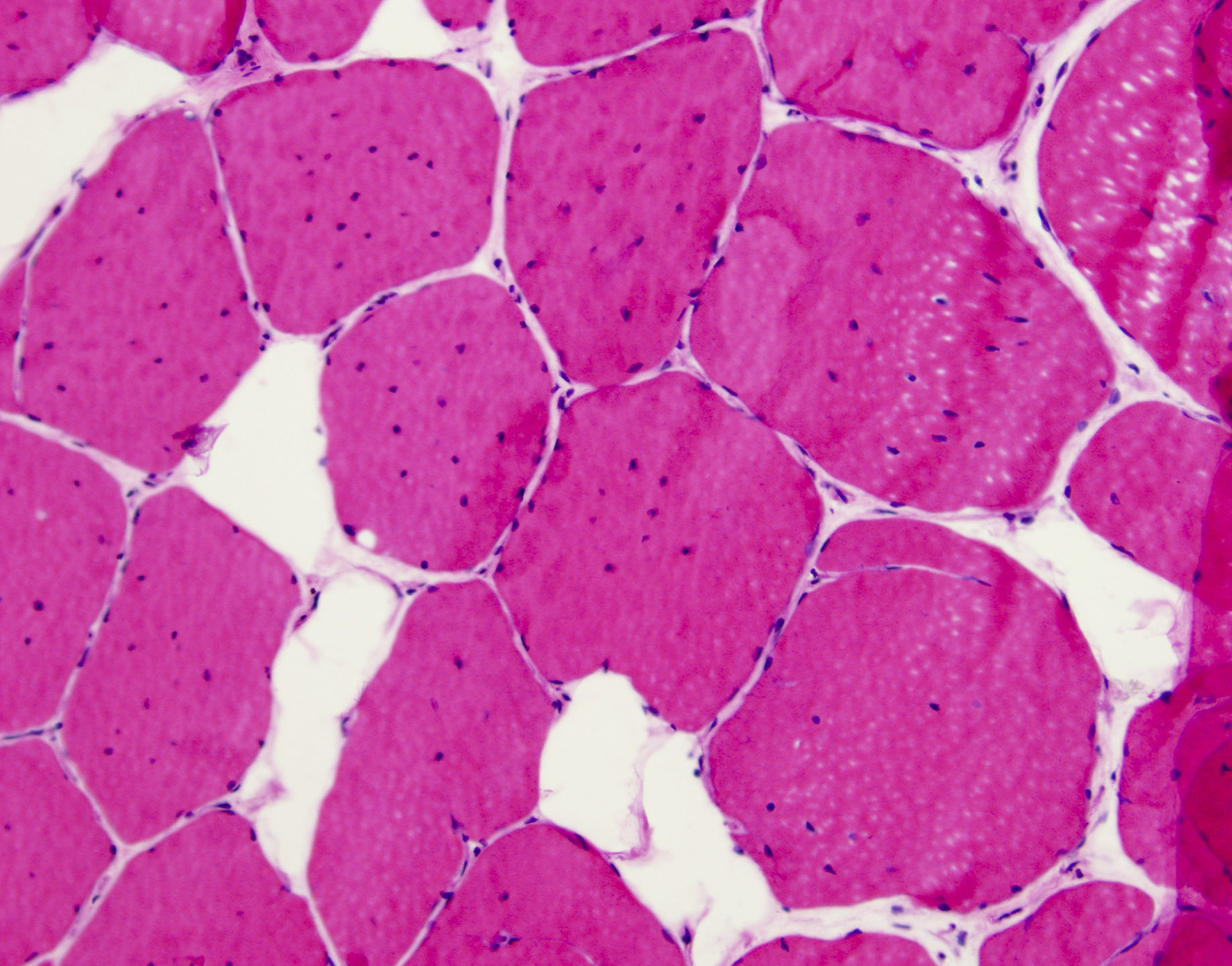
Markedly increased internal nuclei
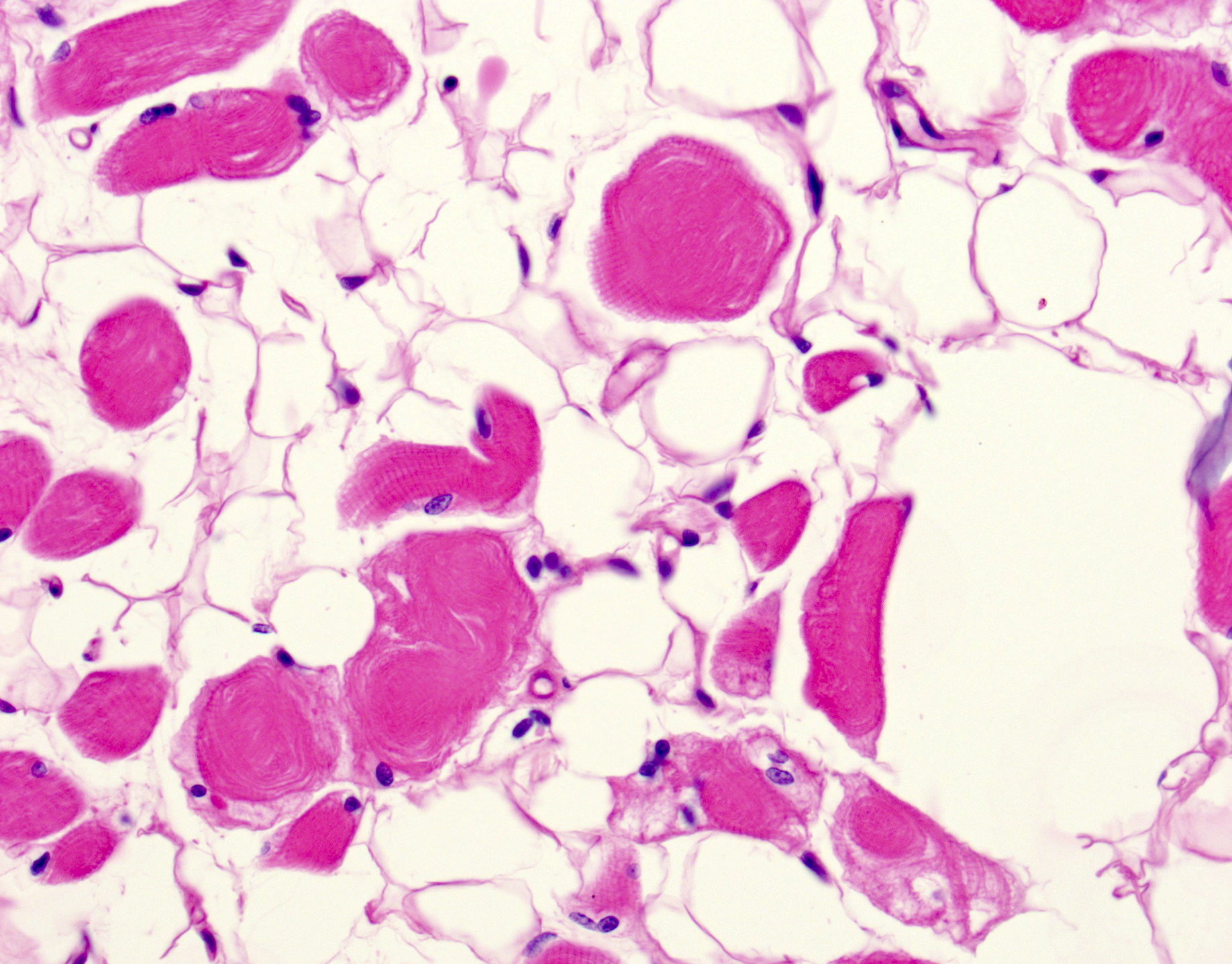
Fatty replacement and ring fibers
- No specific special or immunohistochemical stains are typically utilized for the diagnosis
- In situ hybridization (ISH) specific for CTG or CCTG repeats may be utilized in some laboratories to allow for direct visualization of aberrant mRNA in muscle biopsies (Acta Myol 2013;32:154)
- Staining for MBNL1 is also available in some laboratories (Neuromuscul Disord 2012;22:225)
- Involved neurons of the limbic system and brain stem contain tau protein positive neurofibrillary tangles (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Sarcoplasmic masses composed of disorganized myofibrils, dilated sarcoplasmic reticulum and free ribosomes (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
Images hosted on other servers:
DM2 muscle biopsy: FISH and MBNL1 immunofluorescence
- Limb girdle dystrophy (DM2): often has significantly increased CK; molecular testing and immunohistochemistry on muscle biopsy can also be utilized to make the diagnosis
- Other muscular dystrophies: molecular studies will aid in differentiation; myotonic dystrophy shows a greater number of and more consistent internal nuclei
- Myotubular myopathy (in congenital DM1): greater number of internal nuclei in DM1 without peripheral halos seen in myotubular myopathy; can also look for MTM1 gene mutation
- Myopathic conditions (i.e. inflammatory myopathies): degenerating and regenerating fibers, as well as inflammatory cell infiltrates, are not commonly seen in myotonic dystrophy
- Autosomal dominant
- Autosomal recessive
- Mitochondrial
- X linked
Comment Here
Reference: Myotonic dystrophy
- One of the most common non dystrophic congenital myopathies, with a heterogeneous clinical presentation and characteristic rod-like inclusions (nemaline bodies) within myofibers
- Skeletal muscle with red to purple rod-like inclusions on Gomori trichrome stain and electron dense deposits on electron microscopy
- Congenital myopathy primarily involving proximal musculature and with variable age of onset and severity
- Most common mutations are NEB (recessive) and ACTA1 (dominant)
- Nemaline myopathy (NM), nemaline rod myopathy
- "Nemaline" means thread-like, describes the appearance of rods (Semin Pediatr Neurol 2011;18:230)
- Greek nema = thread
- ICD-10: G71.2 - congenital myopathies
- Most cases present in infancy and childhood but also seen in adults (Mol Med Rep 2014;10:175)
- Incidence reported as 1:50,000 i(J Neuromuscul Dis 2017;4:99) to 1:500,000 births (Clin Genet 2016;90:199)
- Preferentially involves proximal muscles, namely the face, neck flexors and proximal extremities (Neuropathol Appl Neurobiol 2017;43:5)
- Bulbar and respiratory muscles are affected (Semin Pediatr Neurol 2011;18:230)
- Cardiac and extraocular muscles are typically spared
- Congenital forms caused by mutations involving the thin filament of the sarcomere and cause contractile dysfunction, leading to muscle weakness (J Neuromuscul Dis 2017;4:99)
- To date, mutations in eleven genes have been identified which result in NM (J Neuromuscul Dis 2017;4:99)
- Ten genes encode proteins involved with the thin filament of the sarcomere, either as components or having roles in stability and turnover of the filament proteins
- The most recently identified gene encodes myosin 18B (J Neuromuscul Dis 2017;4:99, Circulation 1989 Jun;79:1282)
- Inherited in autosomal recessive or autosomal dominant manner, with 50% of cases occurring de novo (GeneReviews® [Internet];1993-2017)
- The most frequently affected genes are recessive mutations in NEB and de novo dominant mutations in ACTA1 (Curr Opin Neurol 2016;29:642)
- Progressive myopathy which most commonly has a congenital onset
- Large variation in clinical presentation, ranging from normal lifespan with mild symptoms to neonatal death (Mol Med Rep 2014;10:175)
- Subdivided into six clinical categories (Semin Pediatr Neurol 2011;18:230)
- Typical congenital form
- Intermediate congenital form
- Severe congenital form
- Mild nemaline myopathy with childhood onset
- Adult onset nemaline myopathy
- Other forms with unusual associated features (Amish NM)
- Usually symmetric, generalized weakness with preference for neck flexors, facial muscles, axial muscles and proximal extremities (Neuropathol Appl Neurobiol 2017;43:5)
- Can have late involvement of distal musculature
- Bulbar and respiratory muscles are frequently affected and may be a
presenting feature (Semin Pediatr Neurol 2011;18:230)
- Patients are at risk for nocturnal hypoxia and recurrent lower respiratory tract infections
- Feeding and nutritional difficulties are common
- Cardiac and extraocular muscles typically spared, but involvement can be seen with specific mutations (Curr Opin Neurol 2016;29:642, Semin Cell Dev Biol 2017;64:191, Circulation 1989;79:1282)
- Frequently show weak deep tendon reflexes and joint hypermobility
- May develop joint and spine abnormalities (J Med Genet 1997;34:705)
- Congenital forms often show facial elongation, tent shaped mouth and high arched palate (J Med Genet 1997;34:705)
- Clinicopathologic diagnosis
- Muscle biopsy
- Laboratory findings are typically nonspecific
- MRI frequently shows patchy fatty degeneration of muscle (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- CT often shows decreased muscle density with normal volume (J Med Genet 1997;34:705)
- Ultrasound can reveal increased echogenicity of affected muscle (J Med Genet 1997;34:705)
- Neonatal hypotonia was historically identified as the single most important prognostic sign, however later studies have not shown this to be true (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Severe neonatal respiratory disease and arthrogryposis multiplex congenita associated with death in first year of life (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Independent ambulation prior to 18 months is predictive of survival (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- KLHL40 mutations are associated with poor prognosis (death within 5 months on average) and prenatal presentation in 80% (Clin Genet 2016;90:199)
- ACTA1 mutations are associated with up to 50% of lethal congenital cases, but exhibit significant clinical variability (Clin Genet 2016;90:199)
- Presence of intranuclear rods is often associated with more severe disease (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- 6 year old with typical congenital nemaline myopathy and sudden cardiac arrest (Ital J Pediatr 2015;41:20)
- 42 year old with congenital nemaline myopathy and associated mitochondrial abnormalities (Rinsho Shinkeigaku 2000;40:452)
- No proven effective therapy other than symptomatic treatment
- Symptomatic treatment has shown to be helpful in many patients (Semin Pediatr Neurol 2011;18:230)
- Mechanical nighttime ventilation
- Orthoses
- Nasogastric feeding and nutritional support
- Aggressive treatment of respiratory infections
- Physical and speech therapy
- While lacking in objective evidence, low impact exercise and L-tyrosine may provide some benefit
- Medications targeting thin filaments or muscle atrophy have been developed and show promise
- Variation in myofiber size with areas of atrophy, hypertrophy and grouping
- Fatty replacement or fibrosis may be seen but necrosis, fiber regeneration, or inflammation is uncommon (J Med Genet 1997;34:705)
- Gomori trichome stain shows red to purple colored inclusions,
frequently rod shaped, in sarcoplasm and subsarcolemmal region of
myofibers (Semin Pediatr Neurol 2011;18:230, J Med Genet 1997;34:705)
- Often more prominent in type I myofibers
- No correlation between number of rods and clinical severity
- Not specific for nemaline myopathies
- Nemaline rods can also be visualized on plastic sections with toluidine blue (Semin Pediatr Neurol 2011;18:230)
- Fiber typing will typically show predominantly type I fibers with atrophy and grouping
Contributed by Wesley M. Hiser, M.D.
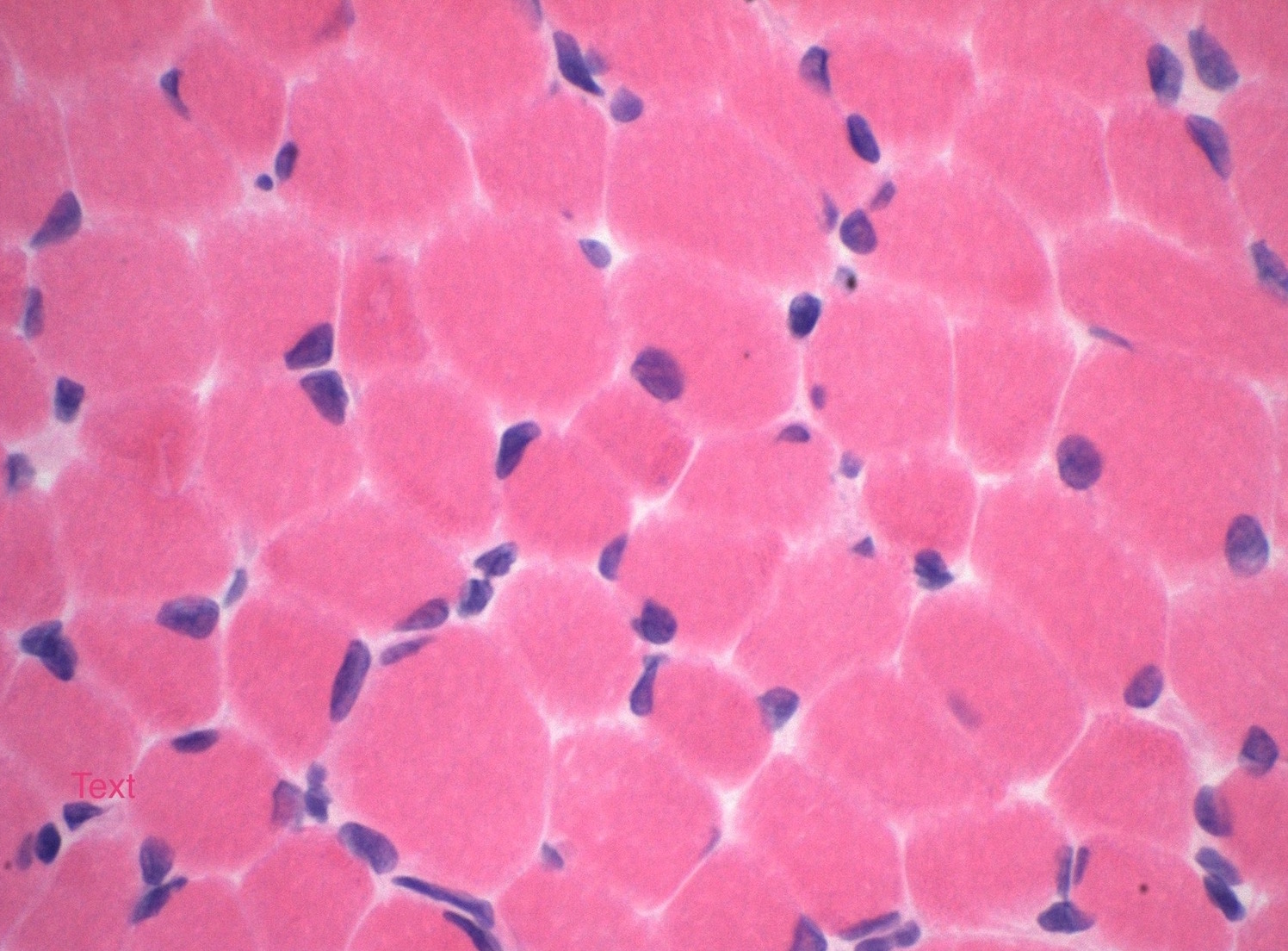
H&E
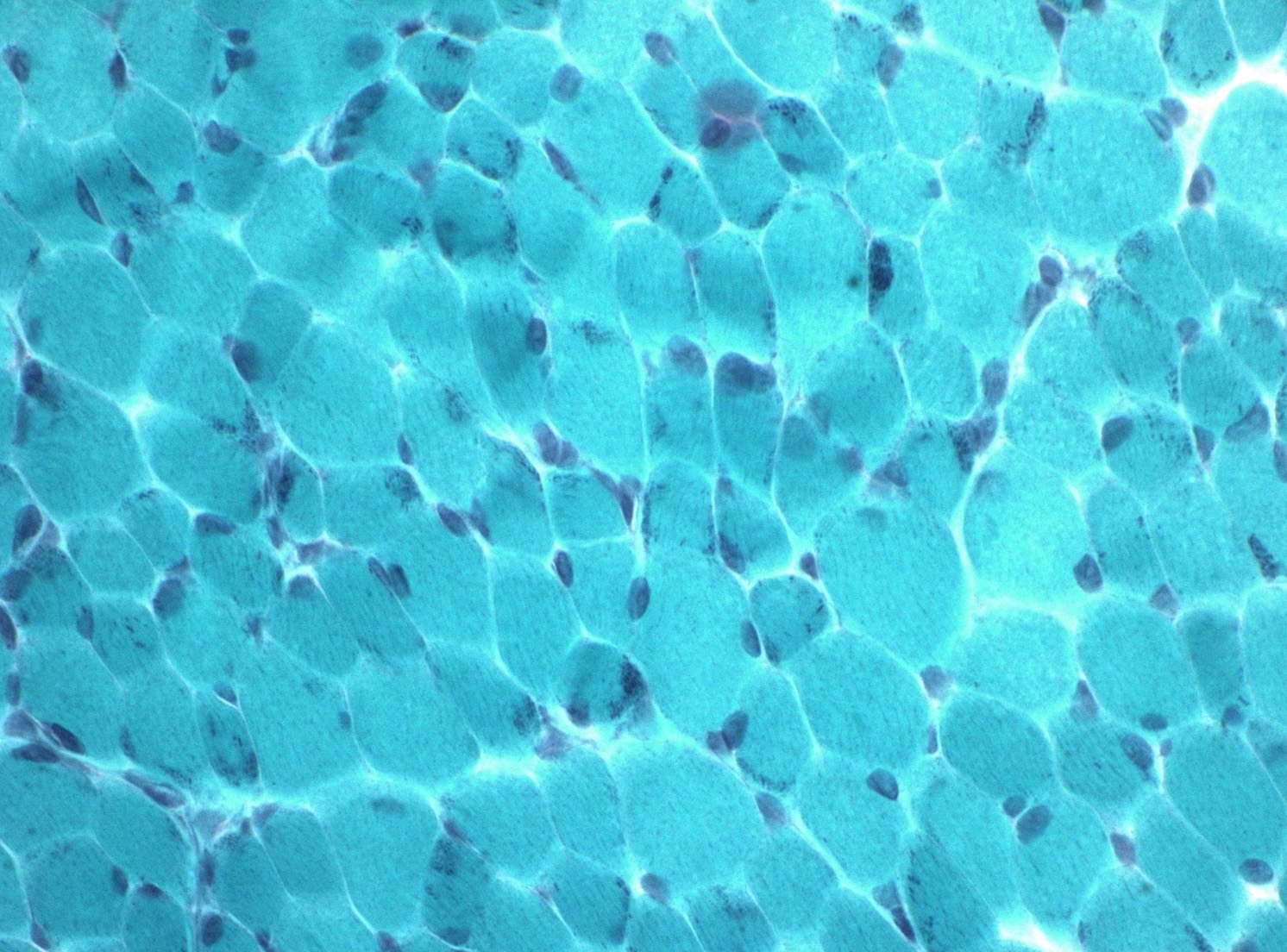

Gomori trichrome
- Gomori trichrome stain highlights red to purple colored rods within sarcoplasm of myofibers
- Nemaline bodies positive for alpha-actinin
- Myofiber typing shows type I predominance
- Rods appear as electron dense rod to ovoid shaped deposits measuring 1 - 7 um in length (J Med Genet 1997;34:705, GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Present in sarcoplasm or nucleus, often parallel to the long axis of the sarcomere (Semin Pediatr Neurol 2011;18:230)
- Deposits can show continuity with Z-disks and exhibit similar lattice structure
Images hosted on other servers:

Nemaline rods
- 11 genes have been implicated in NM (J Neuromuscul Dis 2017;4:99)
- 10 genes involved with thin filament of sarcomere:
- ACTA1
- NEB
- TPM3
- TPM2
- CFL2
- TNNT1
- LMOD3
- KBTBD13
- KLHL40
- KLHL41
- ACTA1
- More recently described myosin 18B associated with severe NM with cardiomyopathy
- 10 genes involved with thin filament of sarcomere:
- Multigene sequence analysis is typically performed first, followed by deletion/duplication analysis when necessary (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Other congenital myopathies
- Core myopathies (Neuropathol Appl Neurobiol 2017;43:5)
- Centronuclear myopathies (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Congenital fiber type disproportion
- Muscular dystrophies
- Metabolic myopathies such as Pompe disease
- Spinal muscular atrophy
- Prader-Willi syndrome
- Can present with marked hypotonia in neonates (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Sporadic late onset nemaline myopathy (Semin Pediatr Neurol 2011;18:230, Rinsho Shinkeigaku 2016;56:605)
- Associated with HIV, autoimmune disease and monoclonal gammopathy
A. NEB
B. KLHL40
C. TPM3
D. ACTA1
E. TNNT1
De novo mutations in ACTA1 are the most common cause of nemaline myopathy. Mutations in NEB are the most common cause of autosomal recessive nemaline myopathy.
Comment Here
Reference: Nemaline myopathy
- Neurogenic type atrophy is a descriptive diagnosis that has multiple different etiologies; underlying etiology generally cannot be further elucidated by the muscle biopsy itself and needs clinicopathologic or radiologic correlation
- Amyotrophic lateral sclerosis (ALS): sporadic and familial forms, progressive painless motor weakness with upper and lower motor symptoms; symptoms vary from fatigue, twitching, fasciculations, dropping items, tripping, difficulty with speech and swallowing; eye muscles and bowel and bladder are generally spared
- Primary lateral sclerosis (PLS): only upper motor neurons are affected
- Spinal muscular atrophy (SMA): lower limbs are affected more than upper, proximal muscles are affected more than distal
- 3 different types:
- I - infantile onset, has hypotonia with sparing of the diaphragm; patients usually succumb to respiratory infections around ages 1-2
- II - onset 6-12 months of age, able to sit but not stand / walk unaided, symmetrical weakness, hand tremor, fasciculation of tongue and joint laxity
- III - onset 2 years of age to adulthood, able to walk, difficulty with other activities (running, jumping), weakness can be progressive, static or regress
- Hereditary motor and sensory neuropathy (HMSN): CMT (Charcot Marie Tooth) is a broad category with mild deficits such as clumsiness, distal motor weakness, classic "stork leg deformity" and possible scoliosis; in more severe forms (CMT3) such as Dejerine-Sottas and congenital hypomyelination neuropathy (with hypotonia at birth) and congenital hypomyelination neuropathy, life expectancy may be only a few months
- Atrophic myofibers with myofiber type grouping (groups of myofibers of the same histochemical type)
- No / minimal inflammation
- No specific terminology
- Skeletal muscle with neurogenic atrophy
- Skeletal muscle with features of neurogenic atrophy
- Specific to the underlying cause
- ALS incidence is 2 per 100,000
- Incidence for SMA type I is 1:10,000 and types II and III are 1:24,000
- HMSN incidence is approximately 1 in 2500 to 1 in 1214 in Scandinavian countries
- Muscle biopsy can be obtained from the more distal muscles depending on the disease process and the muscle that is affected
- Generally, distal is more affected than proximal in many of these disorders
- Has a variety of different causes
- Central theme is loss of innervation to the muscle
- Can be due to radiculopathy from tumor, cyst or herniated nucleus pulposus
- Other causes are loss of the anterior horn cell in SMA / ALS
- In CMT1, CMT3 and CMT4, there is a problem with myelination; in CMT2, there is axonal disease
- Varies as to the cause of the neurogenic atrophy
- Main causes and significant chromosomal mutations are listed under Molecular Description
- Dependent on underlying etiology
- Varies by etiology
- Most of the neurogenic muscle biopsies are descriptive
- Radiography, such as MRI, is ultimately required for diagnosis of radiculopathy
- Clinical correlation (upper motor neuron findings and lower motor neuron findings) and EMG / NCS findings are required for diagnosis of ALS
- Mutation studies will identify exact causes of HMSN, clinical and EMG findings may point to the type of mutation
- UpToDate has an excellent chart for clinicians to follow based on EMG / NCS and clinical findings on which mutation to test for first and then what to test if negative for mutation
- SMA is a clinicopathologic diagnosis, type is based on clinical presentation and genetic mutations can be assessed
- Occasional cases of SMA can have mild elevations in CK levels
- May reveal cause of nerve impingement
- Specifically, MRI can show herniated nucleus pulposus or spinal stenosis
- Prognosis rests on etiology
- ALS runs a fatal course; some mutations cause a fairly rapid progression (FUS), but other mutations (SOD-1 D90A) have survival of 10 years or greater
- Spinal muscular atrophy type I generally has a poor outcome and most succumb to respiratory compromise at age 1-2; other types have prognosis based on the type of mutations and underlying factors
- HMSN varies; form with Dejerine Sottas has a poor prognosis; CMT1 has minimal impact on survival
- Therapy is aimed at the underlying etiology
- SMA III may require physical therapy
- ALS is generally progressive and requires additional support if bulbar symptoms; ie. PEG tube and ventilator dependency
- No gross features of neurogenic atrophy are identifiable on the biopsy
- General / ALS / HMSN:
- Myofibers are smaller and angulated
- No increase in central nucleation
- In areas, myofiber may become atrophic to the point that there is only a "nuclear clump" or "nuclear bag" left
- In cases with deinnervation / reinnervation, there will be myofiber type grouping on ATPase staining or myofiber immunohistochemical stain
- Surrounding unaffected myofibers that are innervated differently may compensate and become hypertrophic
- Care should be also taken to ensure that both myofiber type groups are affected and not just a select type II atrophy for instance
- SMA:
- Large groups of atrophic myofibers
- Myofibers will be smaller and rounded
- There is associated hypertrophy of Type I myofibers (type I and type II)
- Type III may have no changes or show similar findings to type I and II
Contributed by Meggen Walsh, D.O., M.S., P.A.
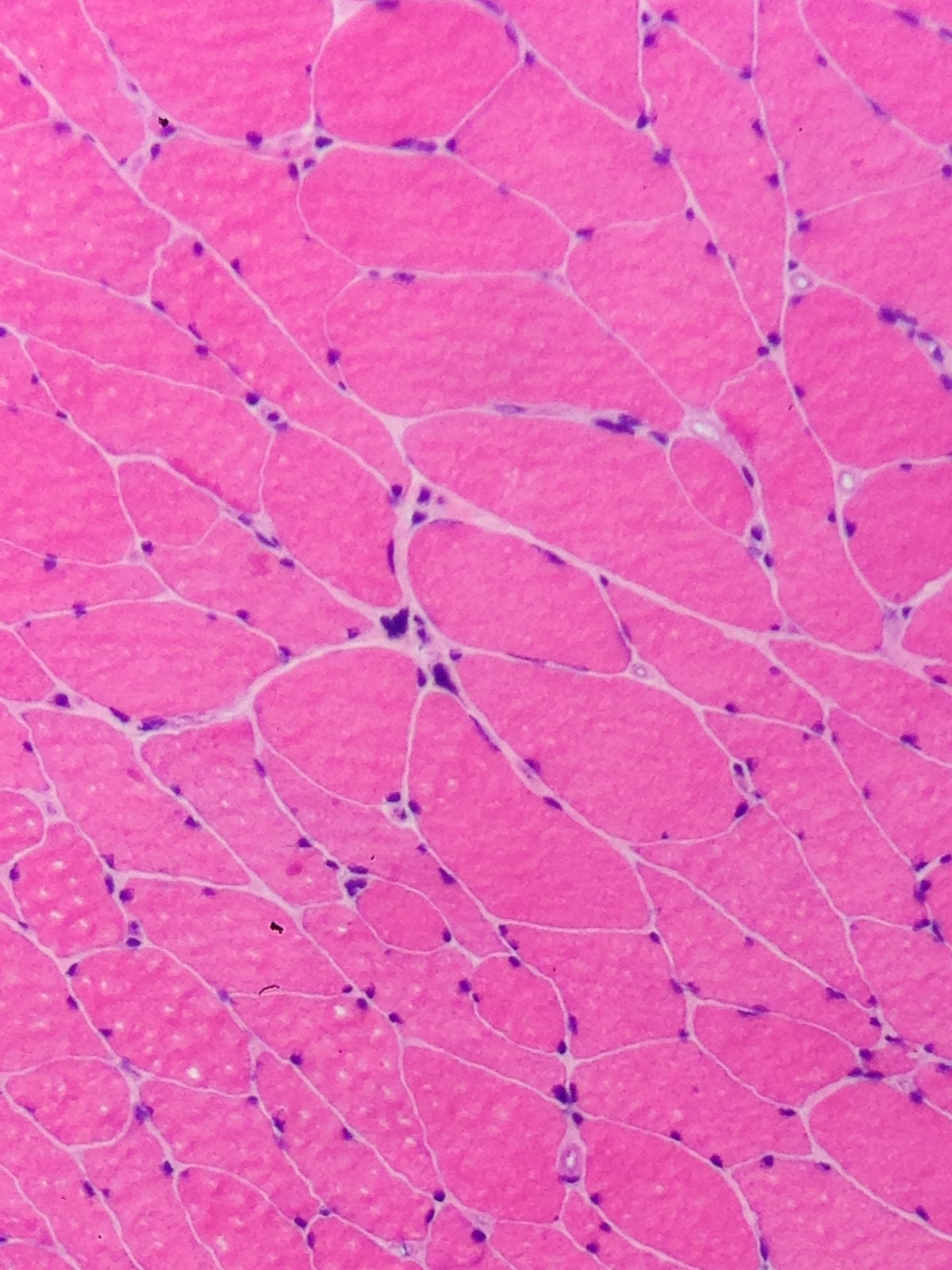
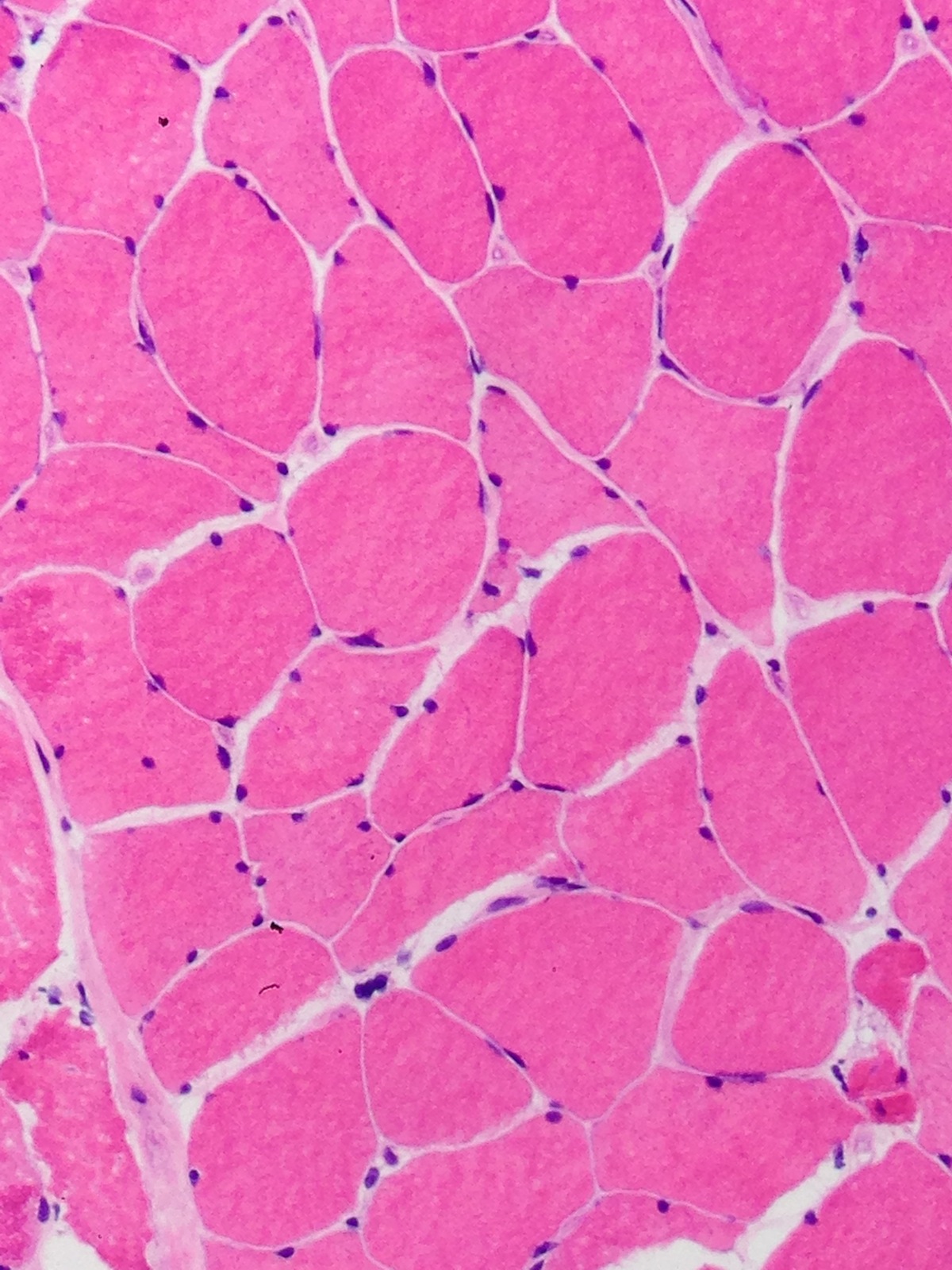
Skeletal muscle: multiple nuclear "clumps" or nuclear "bags"; also few atrophic angulated myofibers (H&E)
- In some angulated myofibers there can be expression of fetal myosin (Dubowitz: Muscle Biopsy: A Practical Approach, 4th ed, 2013)
- "Grouping of myofibers by type" is not a hard and fast criteria; grouping occurs if a myofiber of one type is completely surrounded by myofibers of the same type; normally, there are groups of both type I and type II myofibers
- Must rule out an "abundance" of one myofiber type, which resembles myofiber type grouping
- NADH-TR can show "target fibers", which have a dark staining rim around a pale central zone, similar to a central core
- Deinnervated myofibers may loose glycogen and not stain as strongly with PAS (Dubowitz: Muscle Biopsy: A Practical Approach, 4th ed, 2013)
- ALS: molecular and neurodegenerative features
- C9ORF72: (familial 40%, sporadic 5-7%) 9q21-22; hexanucleotide repeat in the noncoding region of chromosome 9p (normal is 3 repeats and ALS is more than 30); ubiquitylated and TDP-43 positive neuronal cytoplasmic inclusions; bunina bodies can be seen; can see cytoplasmic inclusions in the granular cell layer of the cerebellum, dentate nucleus of the hippocampus and CA3/4
- SOD-1: (familial 15-20%,) 21q22; multiple forms, ubiquitylated only inclusions in some forms or hyalin inclusions that stain with non-phosphorylated neurofilament or phosphorylated neurofilament
- FUS: (familial 5%) 16q12.1-q12.2; glial and neuronal cytoplasmic inclusions that are positive with FUS-antisera and are negative for TDP-43 and few if any ubiquitylated inclusions
- Alsin2: (juvenile ALS form) 2q33; mutation can cause PLS, ALS and hereditary spastic paraplegia; therefore can have upper motor neuron only findings, upper and lower motor neuron findings
- Spinal muscular atrophy:
- SMA: most common is deletion of exon 7 or 8 on 5q
- Hereditary motor and sensory neuropathy:
- CMT1: pathology shows "onion bulb" formation on sural nerve biopsies; divided by mutations but all are clinically similar and have distal weakness, nerve hypertrophy but ambulation is maintained; CMT1A commonly is a mutation of PMP22 (17p11.2-p1); CMT1B is commonly MPZ, myelin protein zero (1q22); CMT1C generally LITAF, lipopolysaccharide-induced tumor necrosis factor alpha factor (16p13.1-p12.3); CMT1D EGR2, early growth response 2 gene (10q21.1-q22.1); CMT1E PMP22; CMT1F NEFL (8p21)
- CMT2: axonal loss is present but very little to no "onion bulb formation" as this is more an axonopathy than myelinating problem; can be wallerian degeneration of the nerves; sensation is lost more than motor skills and hypertrophic nerves are not clinically apparent; in early clinically presenting forms of the disease, ambulation can be severely affected; CMT2A is MFN2, mitochondrial fusion protein mitofusin 2 (1p35-36); CMT2B is RAB7 (6q21.3); CMT2C TRPV4 (12q24.1); CMT2D is GARS, glycyl tRNA synthetase (7p15); CMT2E is NEFL, neurofilament light polypeptide (8p); CMT2F is HSPB1, heat shock protein (7q11.23); CMT2G is 12q12-q13; CMT2I is MPZ; CMT2L HSPB8, heat shock protein (12q24.23); CMT2S IGHMBP2 (missense mutation)
- CMT3: these are the more severe forms; Dejerine-Sottas has "onion bulb formation" on biopsy and shows motor delays with distal weakness and contracture formation may occur; in congenital hypomyelinating neuropathy there is absent myelin, hence no onion bulb; congenital hypomyelination neuropathy presents with hypotonia and respiratory distress; death can occur in infancy; Dejerine Sottas has recessive and dominant forms mutations; mutations can be seen in PMP22, MPZ and EGR2 gene; congenital hypomyelinating neuropathy has mutations in the same genes and also MTMR2, myotubularin related 2 gene
- CMT4: CMT4 is more severe than 1 and 2; they are also rare; CMT4A shows mutations in GDAP1 (8q13-q21.1) and may be a possible mitochondrial disorder; CMT4B1 is MTMR2 (11q22-23); CMT4B2 is SBF2 (11p15); CMT4C SH3TC2 (5q32); CMT4D NDRG1 (8q24.3); CMT4E is EGR2 (10q21-22); CMT4F is PRX, periaxin (19q13.1-13.3); CMT4H is FGD4 (12p11.2); and CMT4J FIG4 gene (6q21)
- CMTX: CMT X linked is the most common form after CMT1A; weakness with occasional deafness is noted; cases with transient "stroke like episodes" have been noted; CMX1 is caused by GJB1, gap junction protein connexin (Xq13.1)
- Myotonic dystrophies and limb girdle dystrophies can resemble SMA type III
- Prototypical oculopharyngeal muscular dystrophy (OPMD) is a late adult onset myopathy clinically characterized by progressive ptosis, dysphasia and proximal weakness; also genetically characterized by GCN trinucleotide repeat expansion in exon 1 of the PABPN1 gene on chromosome 14q11
- Broader definition of OPMD includes a group of diseases that share the common features of ptosis, dysphagia and multinucleotide repeat expansion involving several different genes (see Table 1)
- This topic will focus on OPMD PABPN1
- Prototypical OPMD is caused by GCN repeat expansion in exon 1 of the PABPN1 gene on chromosome 14q11 and is usually late adult onset; this is the most common type of OPMD and the only OPMD that can currently be confirmed by commercial genetic testing
- First described by E. W. Taylor in 1915 (J Nerv Ment Dis 1915;42:129)
- Oculopharyngeal muscular dystrophy was first termed in 1962 (N Engl J Med 1962;267:1267)
- Cause of OPMD was identified in 1998 as GCN trinucleotide repeat expansions in the PABPN1 gene on chromosome 14q11 (Nat Genet 1998;18:164)
- Muscle pathology was myopathy with rimmed vacuoles (Neuromuscul Disord 1997;7:S63)
- Diagnostic electron microscopy feature was intranuclear filamentous inclusions composed of short (0.1 μm), unbranched, 8.5 nm wide filaments, present in 3 - 6.5% of myofiber nuclei (Can J Neurol Sci 1989;16:446, Neurology 1996;46:1324)
- ICD-10: G71.0 - muscular dystrophy
- Largest population of patients with OPMD are those with French Canadian ancestry who reside in Quebec (Surg Clin North Am 1983;63:825)
- Other clusters of this disease are found in Bukhara Jewish immigrants in Israel and individuals of Hispanic descent in the state of New Mexico (Neuromuscul Disord 1997;7:S38, JAMA 2001;286:2437)
- Mean age of presentation is 48; younger age at onset (< 30 years) can be observed in longer GCN expansion or in individuals who are compound heterozygous or homozygous for the GCN expansion (GeneReviews: Oculopharyngeal Muscular Dystrophy [Accessed 28 June 2023])
- Levator palpebrae superioris and pharyngeal muscles are most affected
- Proximal muscle groups can also be affected in late stage of disease
- Reference: N Engl J Med 1962;267:1267
- PABPN1 is an RNA binding nuclear protein that regulates the length of the mRNA poly(A) tail, the mRNA export from the nucleus and the processing of long noncoding RNA (lncRNA)
- GCN repeat expansion in the exon 1 of the PABPN1 gene leads to a short alanine expansion in the N terminal of PABPN1 protein, which results in nuclear accumulation of PABPN1 and other proteins and RNAs that are resistant to degradation
- Misregulation of PABPN1 dependent lncRNA affects downstream gene expression
- Reference: FEBS J 2013;280:4230
Table 1: Genetic diseases with shared features of progressive ptosis and dysphagia
| Gene | Site | Heritance | Genetic defect | Clinical | Reference |
| PABPN1 | 14q11.2 | AD | GCN repeats | Late onset OPMD | Acta Neuropathol 2022;144:1157 |
| HNRNPA2B1 | 7p15.2 | AD | Frameshift | Early onset OPMD | Nat Commun 2022;13:2306 |
| LRP12 | 8q22.3 | AD | CGG repeats | Oculopharyngeal distal myopathy (OPDM) type 1 | JAMA Neurol 2021;78:853 |
| GIPC1 | 19p13.12 | AD | CGG repeats | OPDM type 2 | Am J Hum Genet 2020;106:793 |
| NOTCH2NLC | 1q21.2 | AD | CGG repeats | OPDM type 3 | Nat Genet 2019;51:1222 |
| RILPL1 | 12q27.31 | AD | CGG repeats | OPDM type 4 | Am J Hum Genet 2022;109:533 |
| NUTM2B::AS1 | 10q22.3 | AD | CGG repeats | Oculopharyngeal myopathy with leukodystrophy (OPML) | Nat Genet 2019;51:1222 |
- OPMD (PABPN1) usually presents insidiously in the 50s - 60s (Neurology 2017;88:359)
- Progressive, bilateral, myogenic ptosis
- Dysphagia, initially solid foods, progressing to difficulty swallowing liquids
- Aspiration pneumonia and malnutrition are the leading cause of death but usually do not shorten life expectancy because these tend to occur late in the disease
- Proximal limb weakness of varying severity, with some patients suffering from profound limb girdle weakness
- OPMD patients are often misdiagnosed as having myasthenia gravis
- Definitive diagnosis is by genetic testing on blood showing expansion of the GCN trinucleotide repeat in the first exon of PABPN1 gene, usually by PCR (Neurology 2017;88:359)
- Normal individual has ≤ 10 GCN repeats
- 90% of OPMD patients had GCN trinucleotide repeat expansion of 11 to 18 repeats in the first exon of PABPN1
- 10% of OPMD patients had compound heterozygous or homozygous GCN trinucleotide repeat expansions
- Serum creatine kinase (CK) can be normal, mildly increased or > 1,000 IU/L, depending on number of GCN repeats (Neurology 2017;88:359)
- Muscle MRI: fatty involvement of the combination of tongue, soleus and adductor magnus muscles are highly suggestive of OPMD (J Neurol Neurosurg Psychiatry 2019;90:576)
- OPMD (PABPN1) patients with longer repeats present earlier and have higher CK (Neurology 2017;88:359)
- 11 - 12 GCN repeats: mean age of diagnosis is in the 70s, CK normal
- 13 - 15 GCN repeats: mean age is in the 60s, CK of 100 - 500
- 16 - 17 GCN repeats: mean age is in the 50s, CK of 500 - 1,000
- Patients with both alleles affected may present at even earlier ages (30s - 40s)
- 53 year old woman with fatal choking (Medicine (Baltimore) 2018;97:e12935)
- 75 year old woman with OPMD and dementia (BMJ Case Rep 2019;12:e230521)
- 80 year old man with progressive dysphagia, frequent aspiration and change of voice (Ann Gastroenterol 2015;28:291)
- Currently no cure; treatment is mostly supportive
- Ptosis repair / improve levator muscle function (Ann Plast Surg 2002;49:419, Ophthalmic Plast Reconstr Surg 2009;25:103)
- Cricopharyngeal muscle botulinum injection / dilatation / myotomy for dysphagia (J Clin Med 2021;10:1375)
- PABPN1 gene therapy is at early clinical trial stage (Nat Commun 2017;8:14848)
- First patient enrolled in a phase 1b / 2a clinical trial (BB-301, Benitec Biopharma) in January 2023 (Benitec Biopharma: Benitec Biopharma Receives FDA Clearance of the IND for BB-301 for the Treatment of Oculopharyngeal Muscular Dystrophy [Accessed 3 July 2023])
- All OPMD and OPDM subtypes share muscle pathology of myopathy with rare rimmed vacuoles (see references in Table 1)
- Significant fiber size variation
- May have fatty replacement and interstitial fibrosis
- Infrequent red rimmed vacuoles on Gomori trichrome and acid phosphatase
- Usually no inflammation or MHC1 myofiber upregulation
- Usually no significant increase in COX deficient fibers
- One of the most useful distinguishing feature is intranuclear filamentous inclusions identifiable by electron microscopy (see Electron microscopy description) or p62 stain (see Positive stains)
- References: Acta Neuropathol Commun 2022;10:176, Neurology 1996;46:1324
Contributed by Chunyu Cai, M.D., Ph.D.

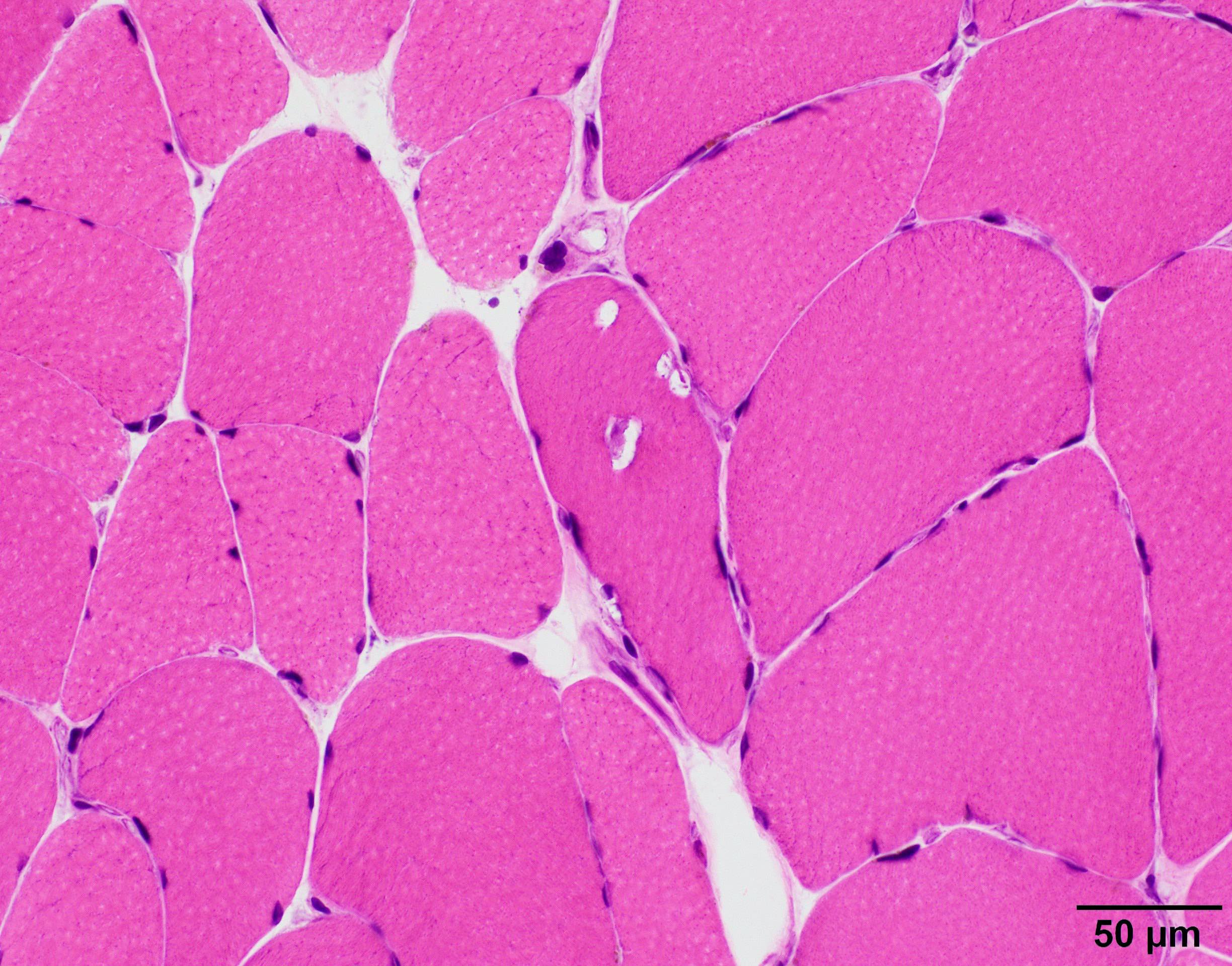
Myopathy with rimmed vacuoles

Gomori trichrome
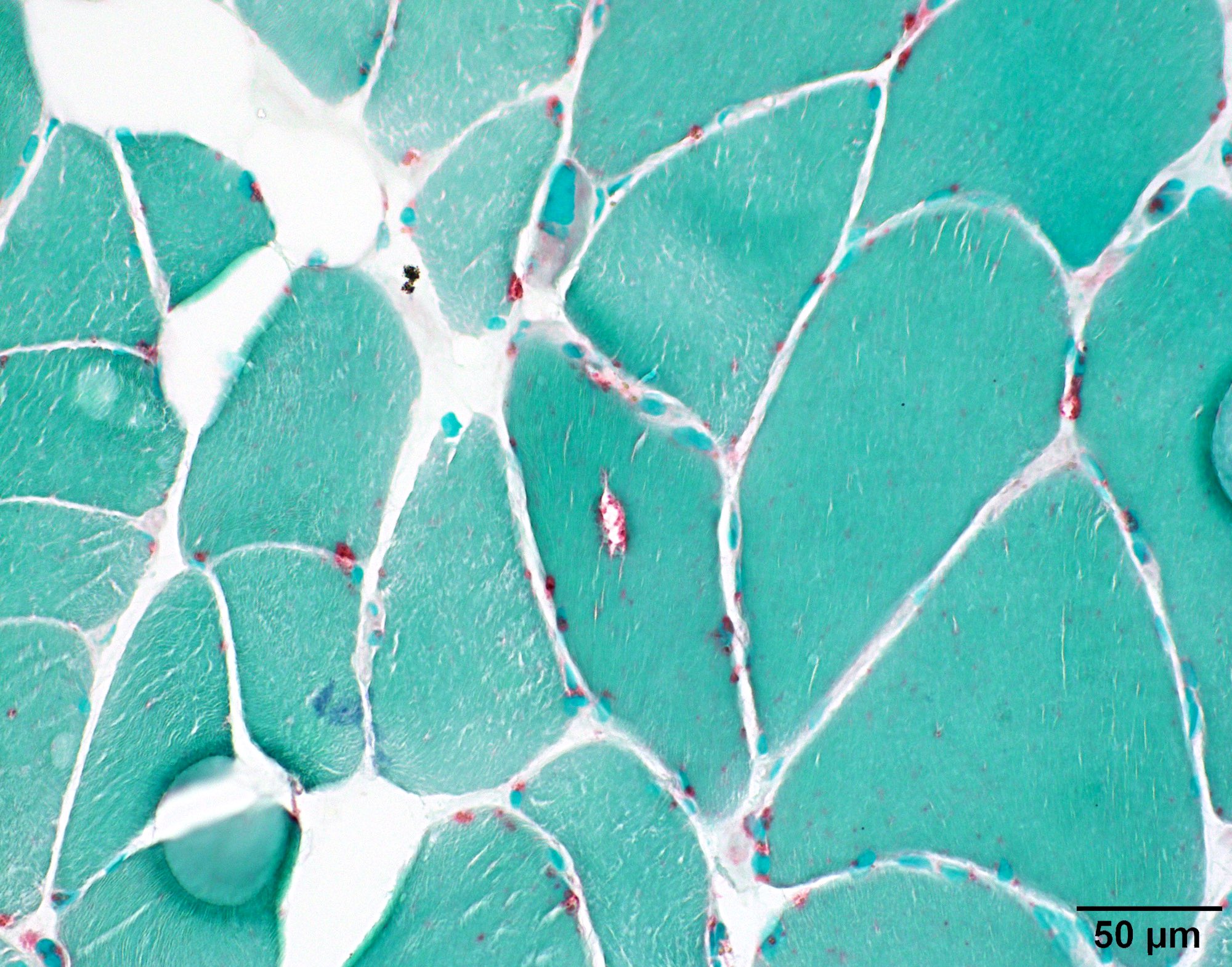
Acid phosphatase
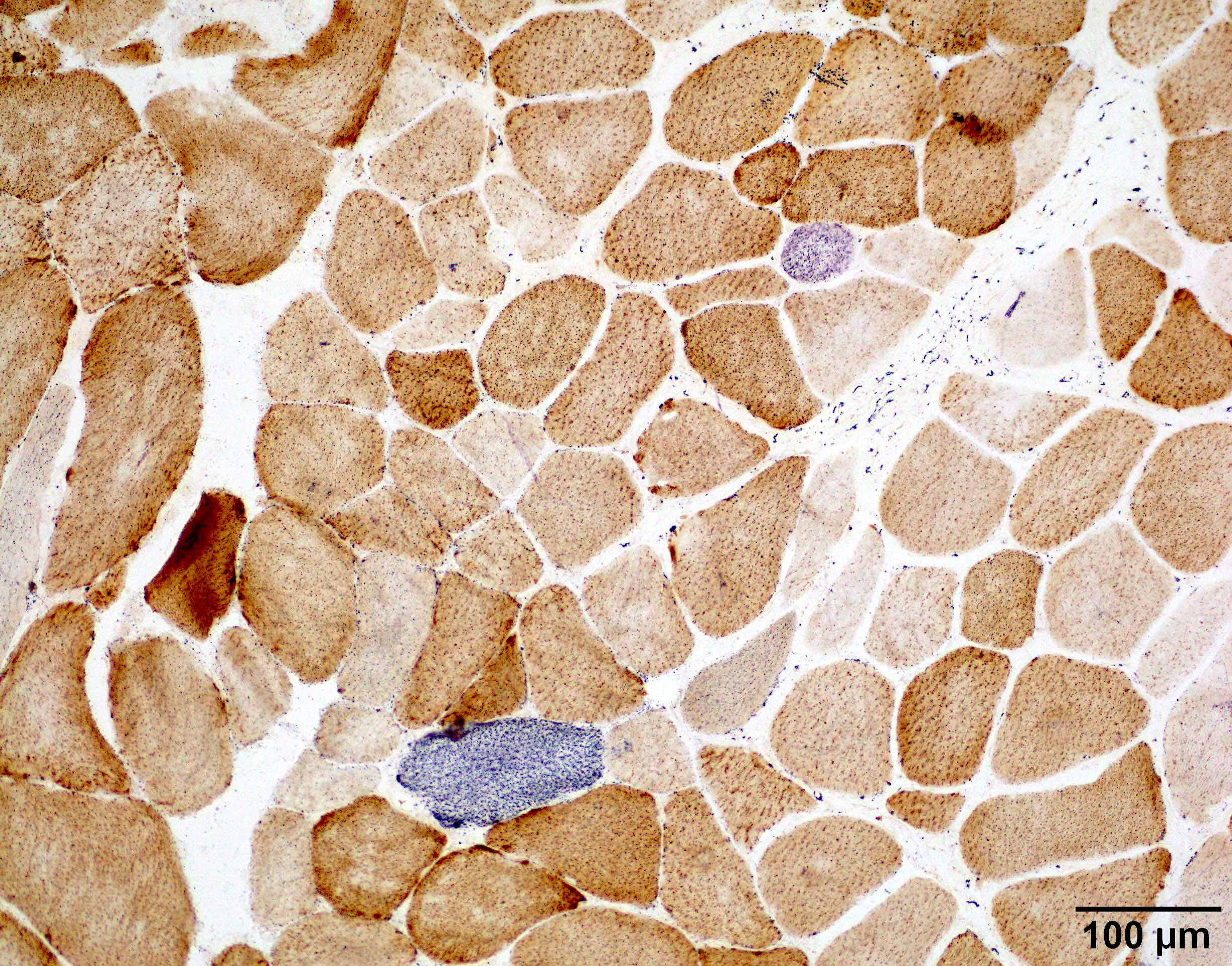
COX SDH double stain

MHC1
- Intranuclear inclusions in OPMD are positive for p62 and PABPN1 immunostains (Acta Neuropathol Commun 2022;10:176)
- Autophagic markers LC3, LAMP2 and p62 are negative in the sarcoplasmic vacuoles or intranuclear inclusions of OPMD (Acta Myol 2017;36:191)
- Ubiquitin and beta amyloid are negative in the sarcoplasmic vacuoles or intranuclear inclusions of OPMD (Neuromuscul Disord 1993;3:283)
- Diagnostic electron microscopy feature was intranuclear filamentous inclusions composed of short (0.1 μm), unbranched, 8.5 nm wide filaments, present in 3 - 6.5% of myofiber nuclei (Can J Neurol Sci 1989;16:446, Neurology 1996;46:1324)
Contributed by Chunyu Cai, M.D., Ph.D.
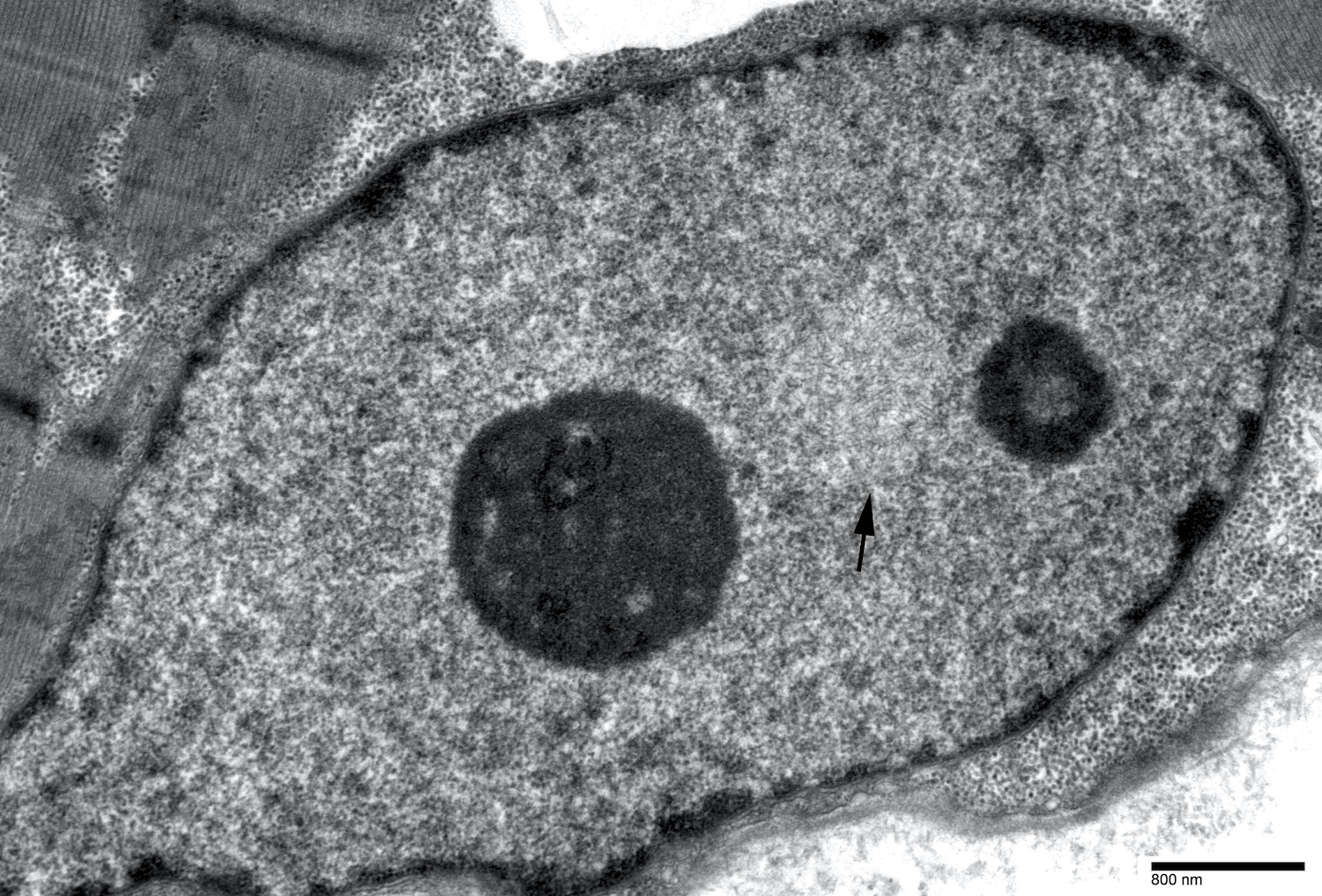
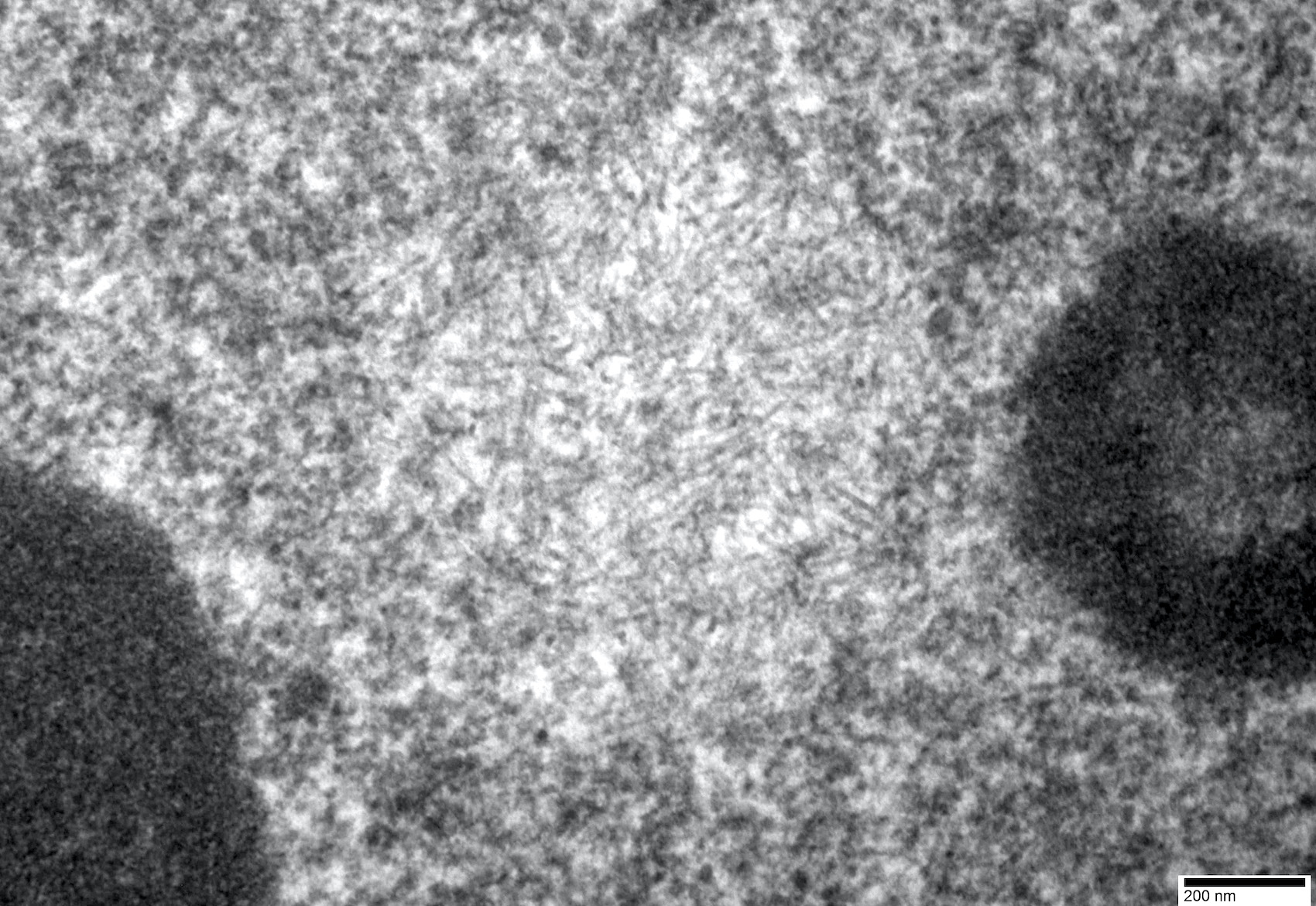
Intranuclear tubulofilamentous inclusion

Filaments reveal tubular nature
- Definitive diagnosis is by genetic testing on blood showing expansion of the GCN trinucleotide repeat in the first exon of PABPN1 gene, usually by PCR (Neurology 2017;88:359)
- Normal individual: ≤ 10 GCN repeats
- OPMD patients: 11 - 18 GCN repeats
- Skeletal muscle, right vastus lateralis, biopsy:
- Chronic myopathy with rimmed vacuoles and intranuclear filamentous inclusions (see comment)
- Comment: The muscle biopsy shows mild myopathic changes with abnormal fiber size variation and rare red rimmed vacuoles but no significant inflammation or COX deficient fibers. Electron microscopy identified rare cytoplasmic bodies and rare intranuclear filamentous inclusions with diameter of 8.5 nm. These findings, in the correct clinical settings, are suggestive of oculopharyngeal muscular dystrophy (OPMD). Genetic analysis for GCN expansions in the PABPN1 gene may be of additional diagnostic value.
- Sporadic inclusion body myositis (sIBM):
- Besides rimmed vacuoles, sIBM muscle usually contains lymphocytic inflammation and substantially increased COX deficient fibers; both are usually absent in OPMD
- Intranuclear inclusion of 8.5 nm filaments are nearly always present in OPMD, while sIBM only rarely has intranuclear tubulofilamentous inclusions, which are thicker (15 - 18 nm) in diameter (Semin Neurol 2012;32:237)
- Rimmed vacuoles and ubiquitin positive sarcoplasmic inclusions are less frequent in OPMD than sIBM (Neuromuscul Disord 1993;3:283)
- Oculopharyngeal distal myopathy (OPDM) and other rimmed vacuolar myopathies:
- p62 positive intranuclear inclusions are substantially more frequent in OPMD (~12% myofiber nuclei) than OPDM (~1% myofiber nuclei) and other rimmed vacuolar myopathies / hereditary inclusion body myositis (Acta Neuropathol Commun 2022;10:176)
- Otherwise, OPDM muscle pathology might be indistinguishable from OPMD; definitive diagnosis might require correlation with clinical weakness pattern and genetic test confirmation
A 63 year old man presented with bilateral ptosis and difficulty in swallowing. The patient had 5 years of progressive difficulty in swallowing, so now he can barely swallow solid food. The ptosis has been gradually progressive for 3 years. Tests for myasthenia gravis were negative. Aside from ptosis and dysphagia, a neurophysiological examination is also significant for proximal lower extremity weakness. A muscle biopsy shows pathology demonstrated in the image above. Electron microscopy of the muscle revealed rare intranuclear inclusions composed of aggregations of short filaments with diameter of 8.5 nm. Which of the following genetic defects are most likely to be present in this patient?
- CTG trinucleotide repeat of the DMPK gene
- GCN trinucleotide repeat of PABPN1 gene
- In frame deletion of the dystrophin (DYS) gene
- Missense mutation of the VCP gene
- Reduced D4Z4 microsatellite repeats upstream of the DUX4 gene
Comment Here
Reference: Oculopharyngeal muscular dystrophy
- COX and SDH combination stains
- Dystrophin epitopes (DYS1 / DYS2 / DYS3)
- MHC class 1
- p62
- PAS with and without diastase
Comment Here
Reference: Oculopharyngeal muscular dystrophy
- Very rare idiopathic inflammatory myopathy; often overdiagnosed (Neurology 2003;61:316)
- Many cases previously diagnosed as polymyositis may be classified as dermatomyositis, inclusion body myositis, immune mediated necrotizing myopathy or antisynthetase syndrome based on serologic testing for myositis specific autoantibodies (J Neuromuscul Dis 2015;2:13, JAMA Neurol 2018;75:1528, Curr Opin Neurol 2019;32:704)
- Existence of polymyositis as a distinct entity has been questioned and may not be included in some idiopathic inflammatory myopathy classification systems (JAMA Neurol 2018;75:1528, Rheumatol Int 2015;35:915)
- Myopathic features with endomysial inflammatory infiltrate of predominantly CD8+ T cells surrounding and invading nonnecrotic muscle fibers expressing MHC I antigen (J Neuromuscul Dis 2018;5:109)
- Diagnosis of exclusion made after all other inflammatory disorders have been ruled out, including other idiopathic inflammatory myopathies (dermatomyositis, inclusion body myositis, antisynthetase syndrome and immune mediated necrotizing myopathy), toxin / drug induced myopathies and muscular dystrophies
- Polymyositis, idiopathic inflammatory polymyositis, idiopathic polymyositis, pure polymyositis
- Incidence of 3.8 per 100,000 in U.S. (J Neuromuscul Dis 2018;5:109, Muscle Nerve 2012;45:676)
- < 5% of all inflammatory myopathies (Arthritis Rheumatol 2017;69:878)
- Predominantly affects adults (N Engl J Med 2015;372:1734)
- More common in women
- Predominantly affects proximal muscles of the extremities (N Engl J Med 2015;372:1734)
- Clonal expansion of CD8+ T cells that expand and destroy myocytes via perforin and granzyme B (Rheum Dis Clin North Am 2011;37:159)
- Exact etiology unknown
- Known association with human leukocyte antigen (HLA) DRB1*03:01 and HLA-B*08:01 (Ann Rheum Dis 2016;75:1558)
- PTPN22 gene single nucleotide polymorphisms (SNPs) associated with increased susceptibility to development of polymyositis (Muscle Nerve 2017;55:270, Ann Rheum Dis 2016;75:1558)
- Subacute onset of proximal muscle weakness
- Extramuscular manifestations include systemic symptoms (e.g. fever, weight loss) and Raynaud phenomenon
- May be associated with other connective tissue disorders such as rheumatoid arthritis and Sjögren syndrome
- May also be associated with cardiac and pulmonary dysfunction including arrhythmias and interstitial lung disease, respectively
- References: Lancet 2003;362:971, Clin Exp Rheumatol 2014;32:188, RMD Open 2017;3:e000507
- Diagnosis of exclusion after other inflammatory myopathies have been ruled out
- Diagnosis of definite polymyositis requires the demonstration of CD8+ lymphocytes surrounding and invading nonnecrotic muscle fibers that express MHC class I antigen in the absence of rimmed vacuoles (Neuromuscul Disord 2015;25:268, Neuromuscul Disord 2004;14:337)
- In the absence of endomysial inflammation, a diagnosis of probable polymyositis can be made if there is ubiquitous expression of MHC class I antigen, in the absence of rimmed vacuoles and dystrophic changes (Neuromuscul Disord 2015;25:268, Neuromuscul Disord 2004;14:337)
- The European League Against Rheumatism / American College of Rheumatology (EULAR / ACR) Classification criteria may clinically help distinguish major subgroups of idiopathic inflammatory myopathies and distinguish them from mimicking conditions (Arthritis Rheumatol 2017;69:2271)
- Score based on age of onset, muscle weakness, skin manifestations, dysphagia or esophageal dysmotility, laboratory measurements (anti-Jo1 autoantibodies; elevated serum creatine kinase [CK], lactate dehydrogenase [LDH], aspartate aminotransferase [AST] or alanine aminotransferase [ALT]), with or without muscle biopsy findings
- Elevated creatine kinase (CK) and aldolase
- Elevated aspartate aminotransferase (AST) and alanine aminotransferase (ALT)
- Elevated lactate dehydrogenase (LDH)
- Positive human leukocyte antigen (HLA) DRB1*03:01 and HLA-B*08:01 haplotypes (Ann Rheum Dis 2016;75:1558)
- References: Lancet 2003;362:971, Clin Exp Rheumatol 2014;32:188, RMD Open 2017;3:e000507
- T2 weighted MRI may show muscle edema and fatty infiltration (Mod Rheumatol 2018;28:913)
- 10 year survival rate of 90% (Clin Exp Rheumatol 2014;32:188)
- Age > 60 and malignancy are associated with worse survival (Mod Rheumatol 2016;26:115)
- 1 study found that intravenous corticosteroid use was associated with worse prognosis when compared to oral corticosteroids, likely secondary to disease severity (Arthritis Res Ther 2012;14:R22)
- 51 year old man with polymyositis presenting with acute heart failure and rhabdomyolysis (Reumatismo 2017;69:78)
- 54 year old man with concomitant polymyositis, myasthenia gravis and aplastic anemia treated with bone marrow transplantation (Case Rep Neurol 2018;10:108)
- 73 year old woman with polymyositis and elevated serum IgG4 (Medicine (Baltimore) 2017;96:e8710)
- Oral glucocorticosteroids are first line therapy
- Intravenous (IV) steroids, immunosuppressants, intravenous immunoglobulin (IVIG) and immunomodulators may be considered in cases which do not respond to initial therapy (Arthritis Rheumatol 2018;70:1532)
- Skeletal muscle usually submitted as 1 large piece but may also be submitted in small, multiple pieces
- Gross findings are nonspecific
- Myopathic changes including myofiber size variation with small rounded myofibers and increased internalized nuclei
- Endomysial lymphocytic inflammation surrounding and invading nonnecrotic myofibers
- Lymphocytic infiltrate may be accompanied by macrophages
- Degenerating and basophilic regenerating myofibers with large nuclei with prominent nucleoli
- May see rare necrotic myofibers with phagocytosis
- Lacks perifascicular atrophy
- Lacks rimmed vacuoles
- References: Lancet 2003;362:971, Clin Exp Rheumatol 2014;32:188, RMD Open 2017;3:e000507
Contributed by Carrie A. Mohila, M.D., Ph.D., Matthew Cykowski, M.D. and Chunyu Cai, M.D., Ph.D.
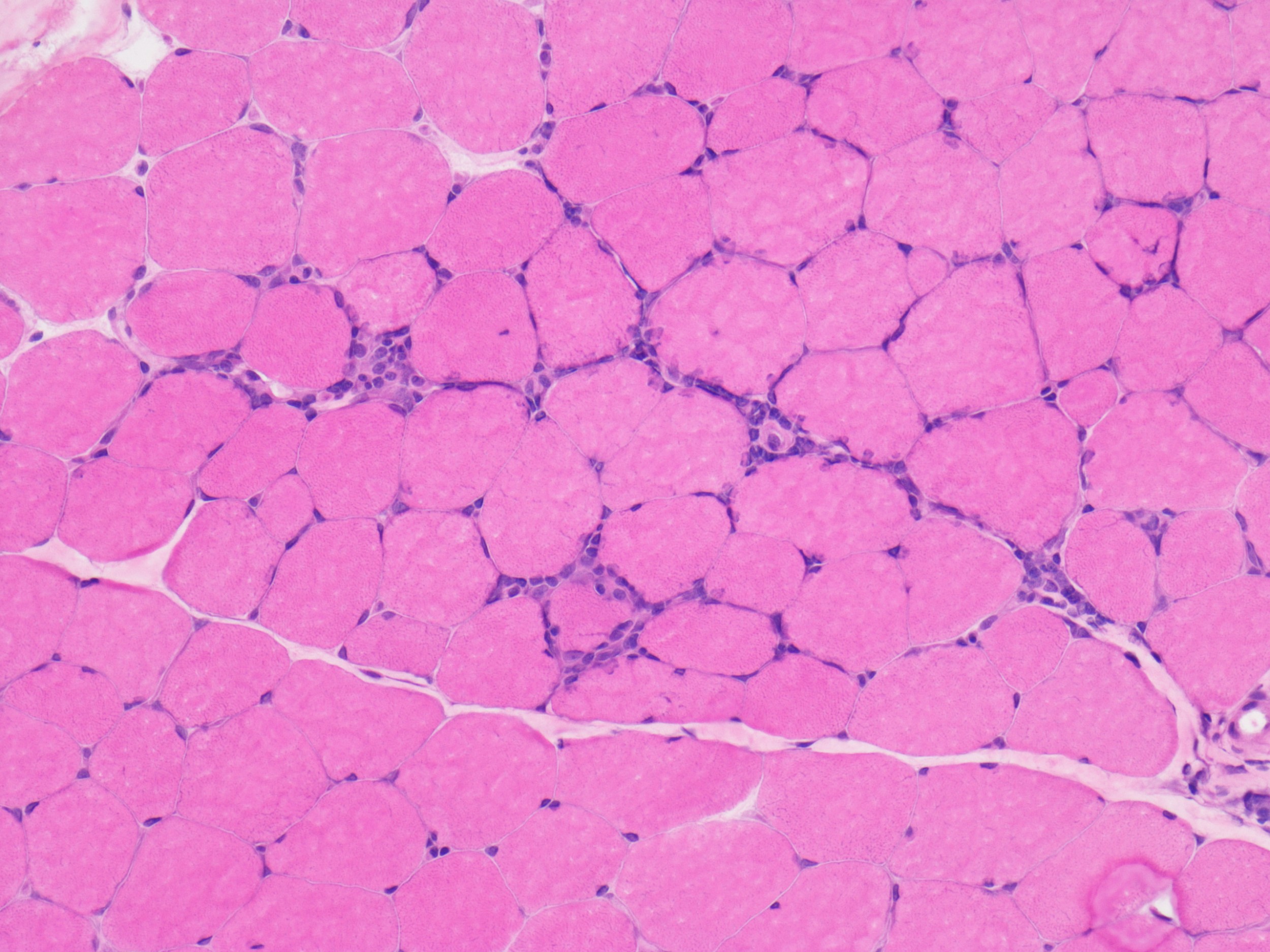
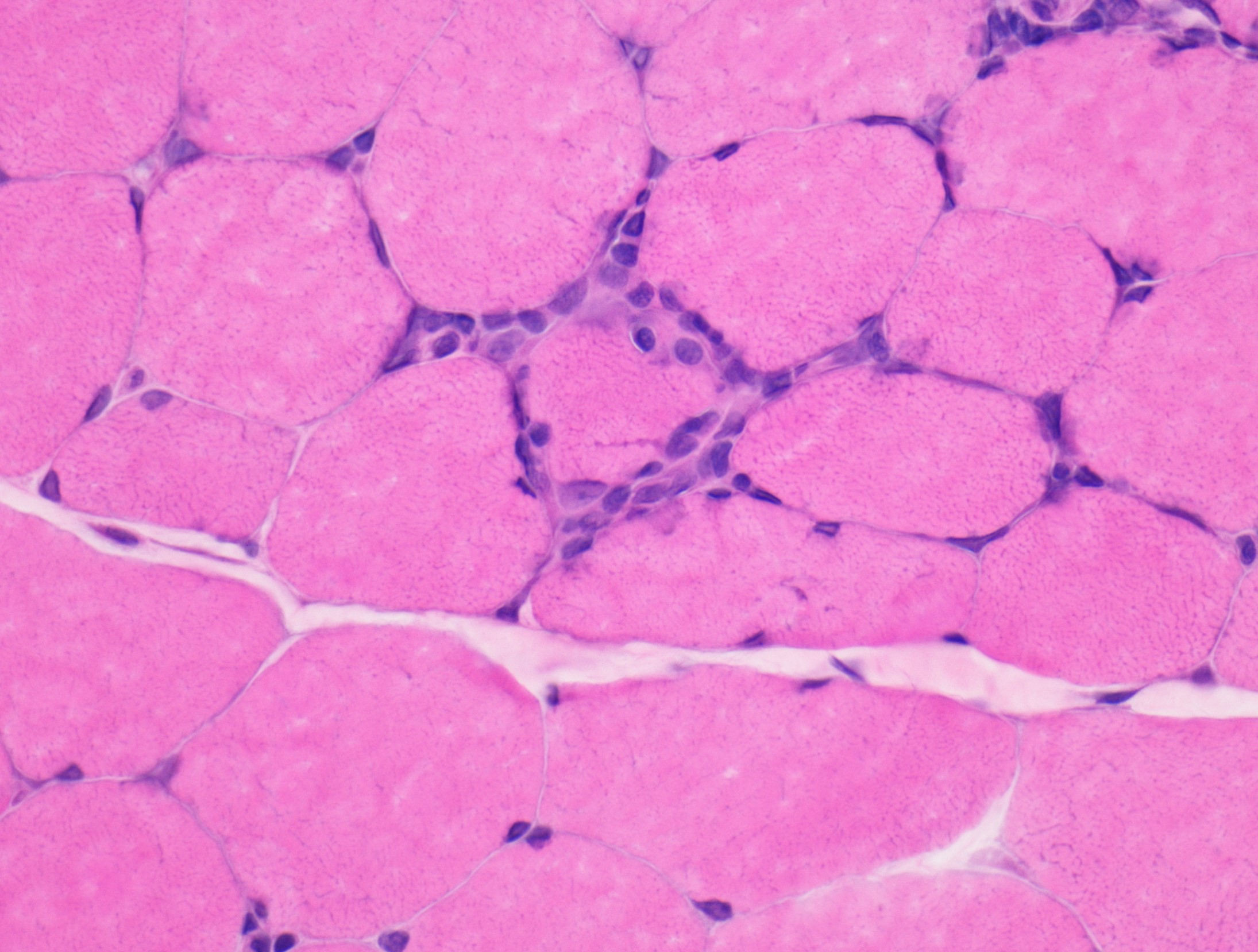
Endomysial lymphocytic inflammation
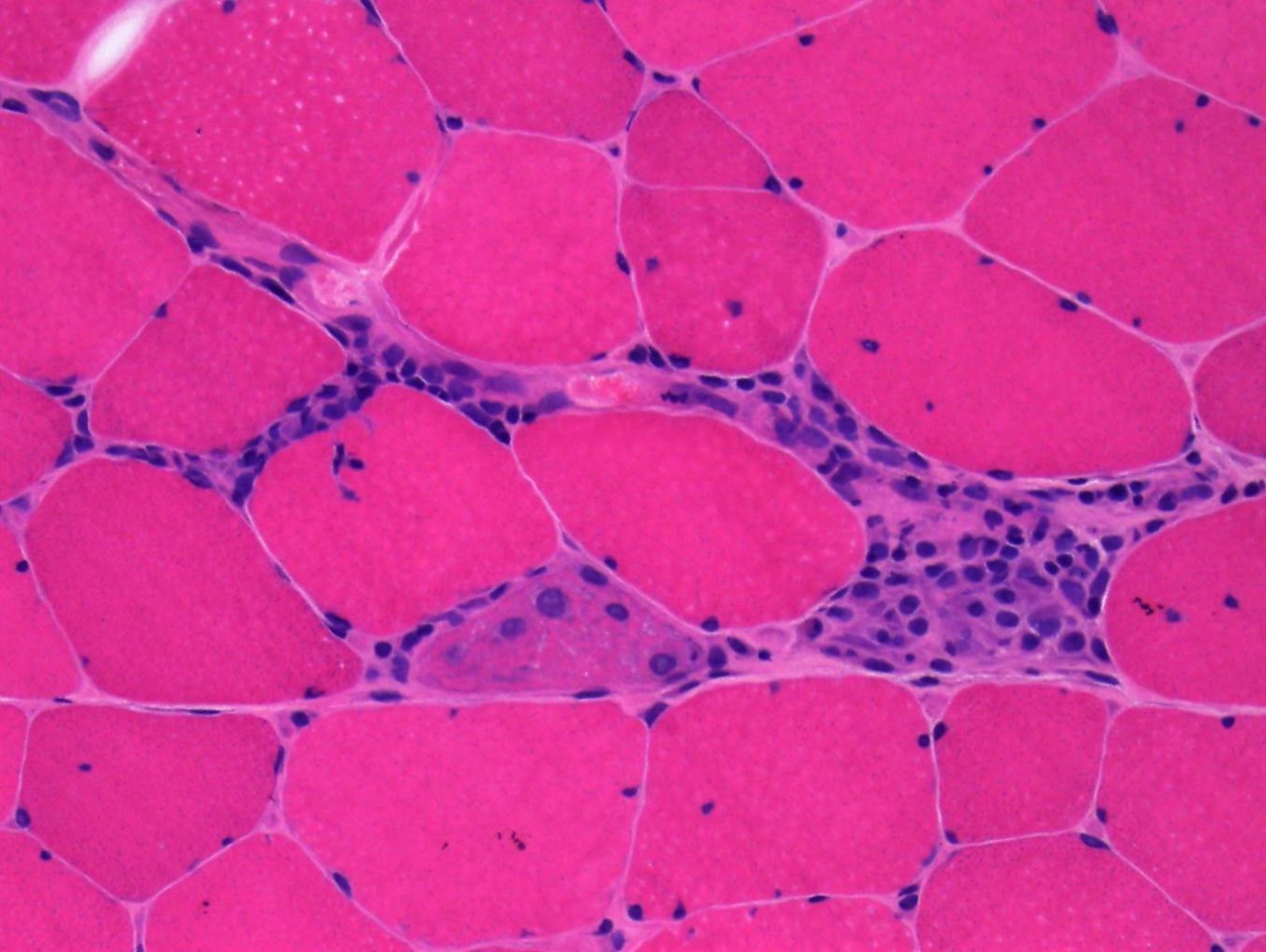
Endomysial lymphocytic infiltrate
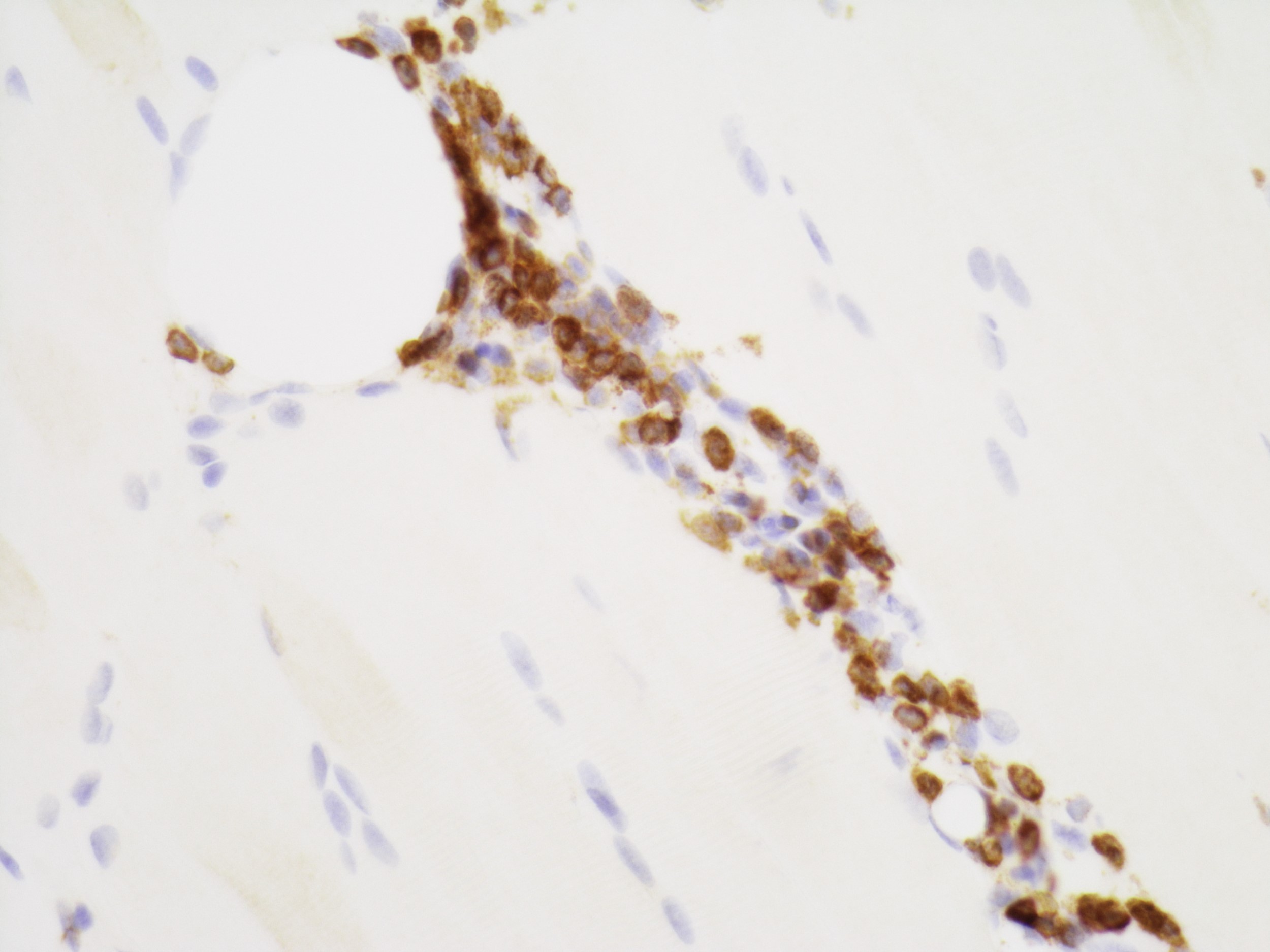
CD3+ lymphocytic infiltrate
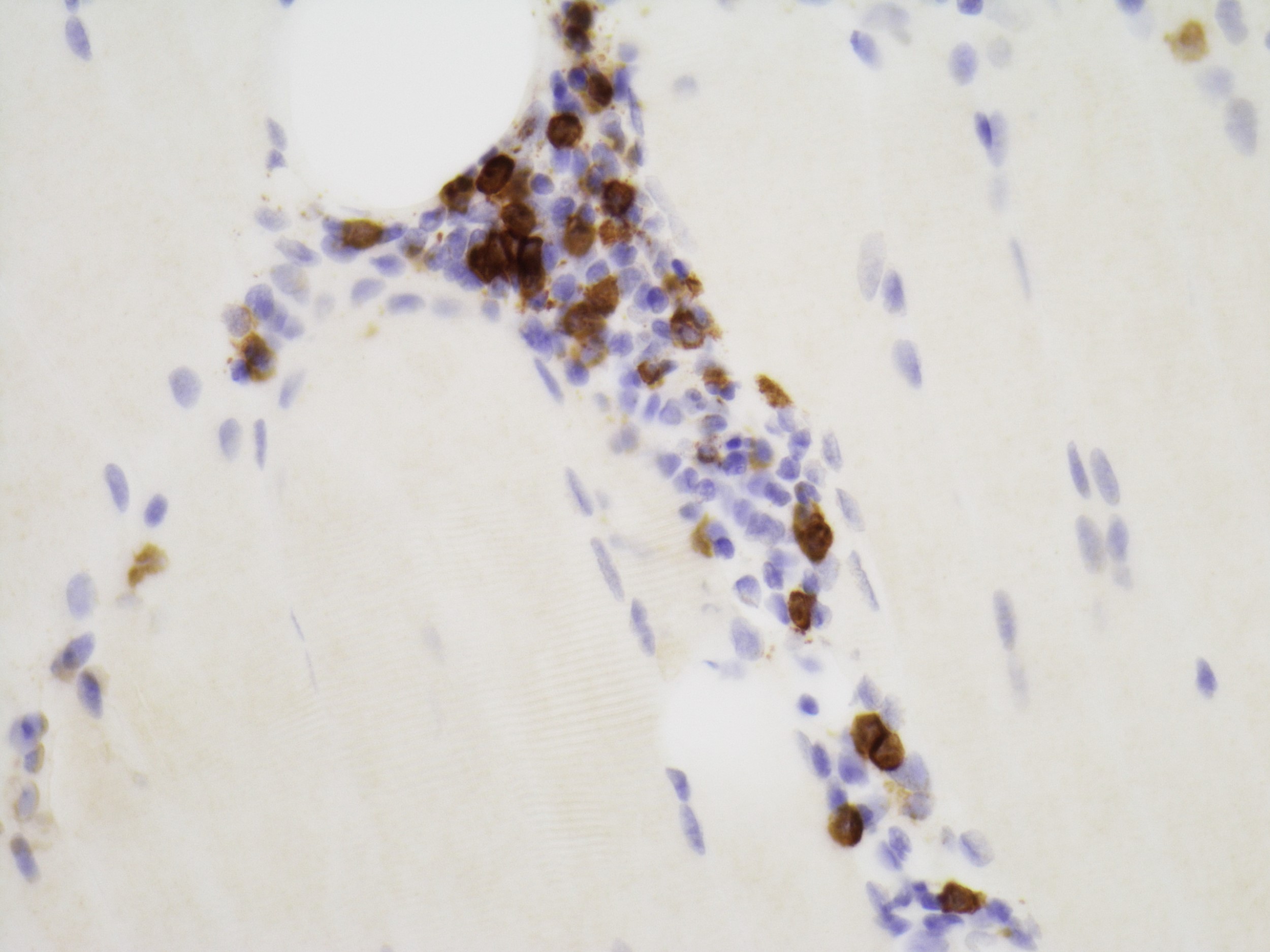
Predominantly CD8+ lymphocytic infiltrate
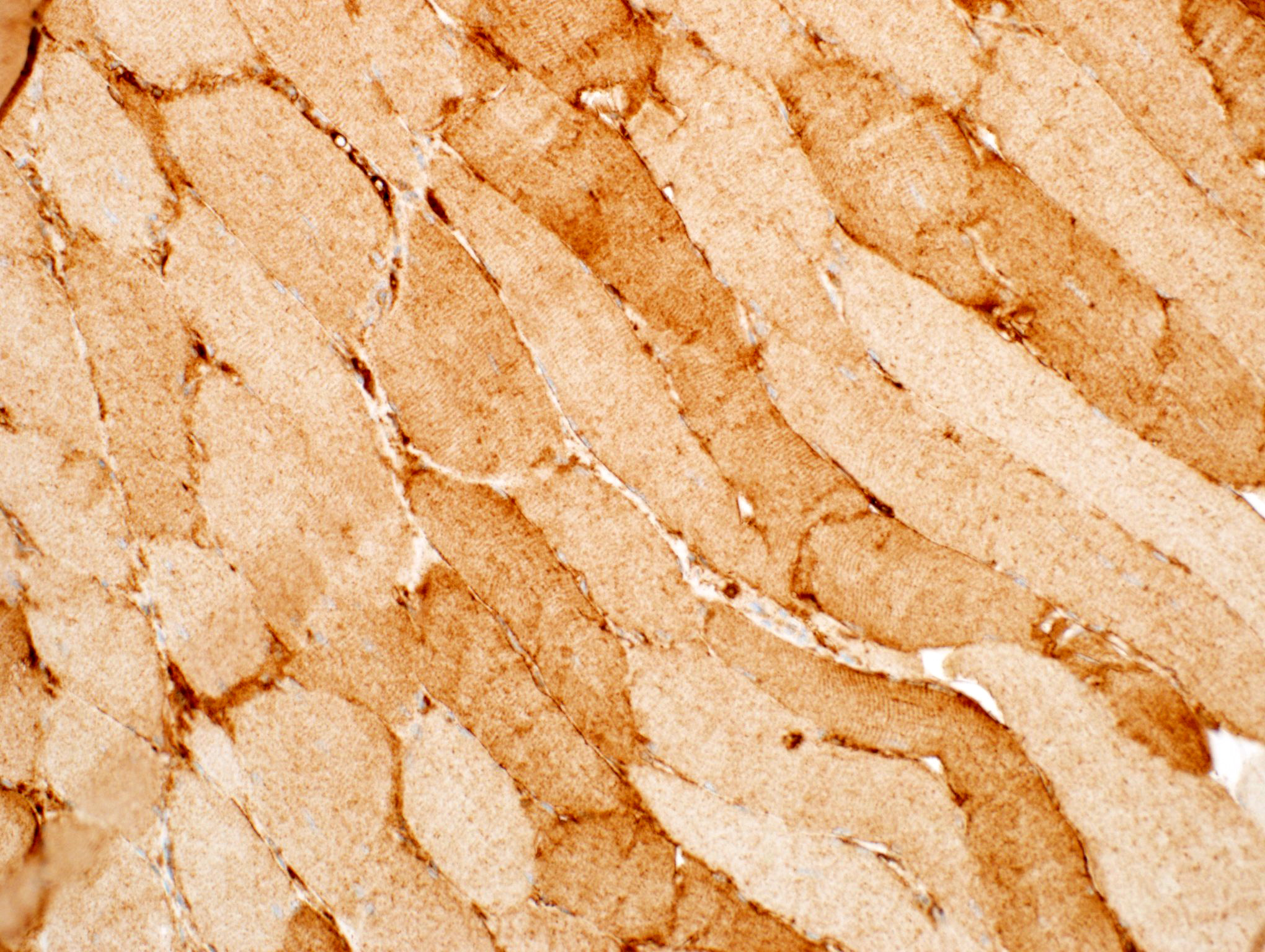
MHC1
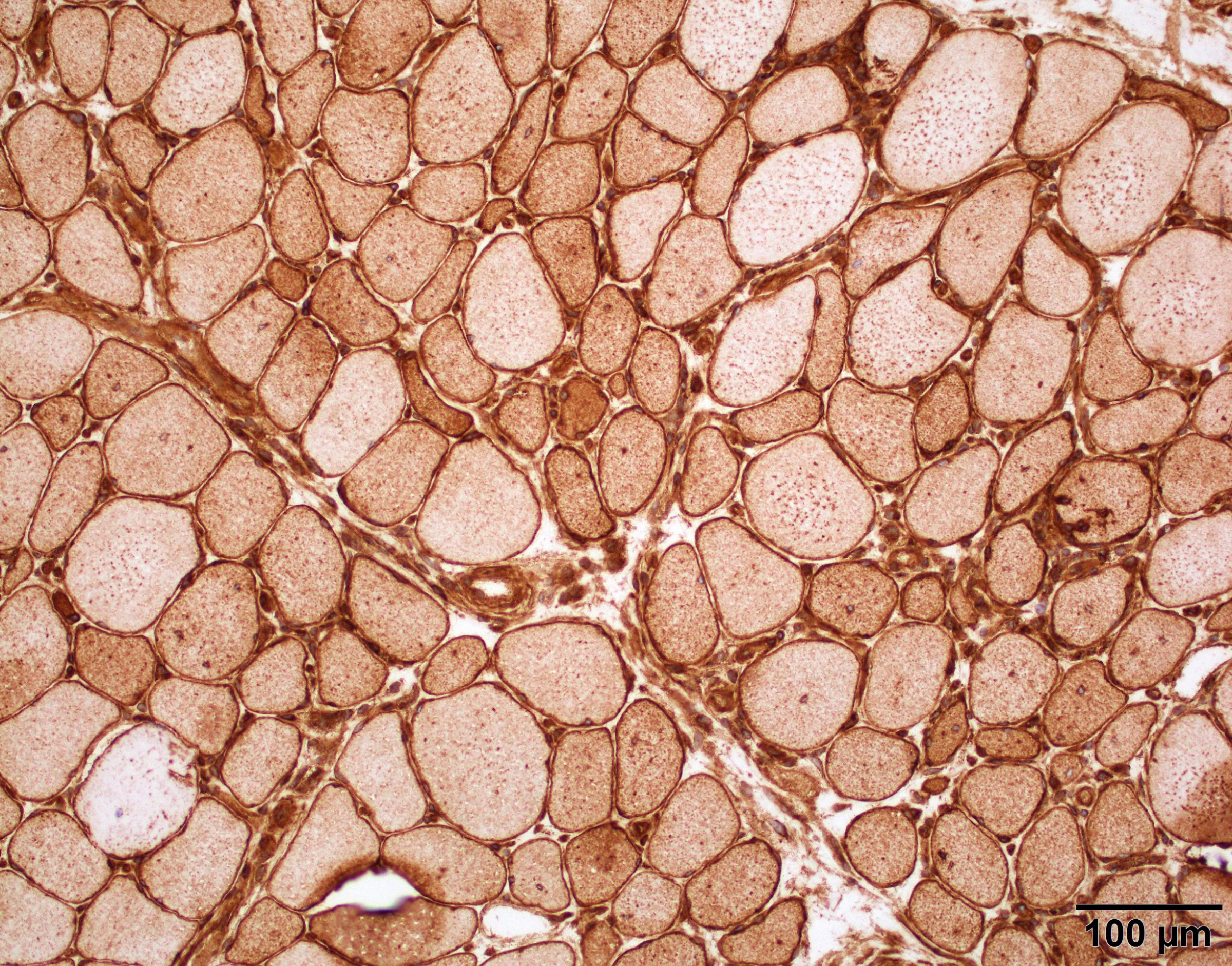
Polymyositis MHC1
- CD3+, CD8+ T lymphocytes
- Myofibers with sarcolemmal and sarcoplasmic immunostaining for MHC class I antigen (nonspecific)
- References: Lancet 2003;362:971, Clin Exp Rheumatol 2014;32:188, RMD Open 2017;3:e000507
- No rimmed vacuoles identified on Gomori trichrome
- No loss of dystrophy associated proteins (e.g. dystrophin) on immunohistochemistry
- Absence of C5b9 (terminal complement complex / membrane attacking complex) on capillaries or sarcolemma of nonnecrotic fibers (Neuromuscul Disord 2004;14:337)
- References: Lancet 2003;362:971, Clin Exp Rheumatol 2014;32:188, RMD Open 2017;3:e000507
- Lack of rimmed vacuoles and tubuloreticular inclusions
- Skeletal muscle, left vastus lateralis, biopsy:
- Inflammatory myopathy compatible with polymyositis (see comment)
- Comment: This biopsy shows myopathic changes associated with a lymphocytic inflammatory infiltrate of CD8 positive T cells invading nonnecrotic muscle fibers that express MHC class I antigens. There is no evidence of perifascicular atrophy, capillary loss or tubuloreticular inclusions on electron microscopy to suggest dermatomyositis. Rimmed vacuoles are not present. There are no dystrophic features. In the correct clinical setting, this biopsy is compatible with polymyositis. Correlation of biopsy findings with clinical features and myositis specific autoantibodies is required to exclude other idiopathic inflammatory myopathies.
- Inclusion body myositis:
- Endomysial CD8+ lymphocytic inflammatory infiltrate
- Rimmed vacuoles present on H&E and Gomori trichrome
- Does not respond to typical steroid or immunosuppressive treatments as seen in other myopathies
- May show tau, p62, alpha synuclein and amyloid beta positive immunohistochemistry staining (Acta Neuropathol 2009;118:407, Presse Med 2011;40:e219, J Neuropathol Exp Neurol 2012;71:680)
- Immune mediated necrotizing myopathy:
- Necrotic and regenerating myofibers
- Minimal lymphocytic infiltration
- Associated with autoantibodies including anti-SRP, anti-HMGCR; may also be negative for autoantibodies
- p62+ myofibers may be present (Muscle Nerve 2019;60:315)
- Drug induced myopathy:
- Statin induced myopathy may show similar histologic findings to polymyositis
- Review of medications is required
- Dermatomyositis (Lancet Neurol 2018;17:816):
- Often associated with skin rash (heliotrope rash) or Gottron papules (CMAJ 2013;185:148)
- Perivascular and perimysial B cell lymphocytic inflammatory infiltrate
- Perifascicular atrophy is characteristic
- Tubuloreticular inclusions in vascular endothelial cells on electron microscopy
- Associated with dermatomyositis specific autoantibodies including Mi2, TIF1y, NXP2, MDA5 or SAE
Which of the following statements is true regarding the skeletal muscle biopsy image shown above?
- CD20+ B cells are the predominant inflammatory component
- CD8+ T cells are the predominant inflammatory component
- Histologic findings are classic for dermatomyositis
- IVIG is the first line therapy
- Presence of rimmed vacuoles favors polymyositis
- Histologic findings alone are characteristic and diagnostic of disease
- Patients often present with skin rash
- Polymyositis is a diagnosis of exclusion
- Polymyositis is associated with a specific autoantibody
- Sarcolemmal staining for MHC class I antigen is specific for polymyositis

Amato: 2015

Bilbao: 2014
Cooper: 2015

Dubowitz: 2020
Gray: 2018
Husain: 2021

Zhou: 2019
Find related Pathology books: muscle and peripheral nerve nontumor, neuropathology, pediatric
