

CNS nontumor
Movement disorders
X linked bulbospinal neuronopathy (Kennedy disease)
Authors: Pouya Jamshidi, M.D., Rachel A. Multz, M.D., Matthew McCord, M.D., Margaret E. Flanagan, M.D.
Editorial Board Member: Meaghan Morris, M.D., Ph.D.
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.


Last author update: 7 June 2023
Last staff update: 7 June 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: X linked bulbospinal neuronopathy (Kennedy disease)

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy description | Videos | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Jamshidi P, Multz RA, McCord M, Flanagan ME. X linked bulbospinal neuronopathy (Kennedy disease). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/cnsbulbospinal.html. Accessed April 19th, 2024.
Definition / general
- X linked recessive disorder that predominantly affects men, usually between the ages of 20 and 50, causing slowly progressive weakness of the proximal limbs, lower motor neuron and facial (bulbar) muscles as well as facial (perioral) fasciculation, loss / reduction of reflexes, diabetes mellitus and gynecomastia
- Often associated with gynecomastia due to an expansion of a repeat of the trinucleotide CAG encoding glutamine in the androgen receptor gene (J Mol Neurosci 2016;58:313, Neurol Clin 2015;33:847)
- Commonly associated with infertility and primary sensory neuronopathy
- Neuronopathy: polyneuropathy involving destruction of the cell bodies of neurons
- Patients with X linked bulbospinal neuronopathy generally live a normal life span; milder form of the disease has been reported in elderly men (Neurology 2008;70:1967)
Essential features
- Rare neurodegenerative disorder characterized by slowly progressive weakness of the proximal limbs, lower motor neuron and facial (bulbar) muscles as well as facial (perioral) fasciculation, loss / reduction of reflexes, diabetes mellitus and gynecomastia (Neurol Clin 2015;33:847)
- X linked recessive disorder that predominantly affects men
- Expansion of tandem CAG repeat region in first exon of androgen receptor gene on proximal part of long arm of X chromosome (Xq11-12) (Nature 1991;352:77)
- Characteristic clinical features and genetic testing can help distinguish X linked bulbospinal neuronopathy from amyotrophic lateral sclerosis (ALS)
- Patients with X linked bulbospinal neuronopathy generally live a normal life span, despite not having treatment options that halt the progression of the disease
Terminology
- X linked bulbospinal neuronopathy
- Spinobulbar muscular atrophy
- Kennedy disease
- Spinal and bulbar muscular atrophy
- Spinal bulbar muscular atrophy
- X linked spinal and bulbar muscular atrophy
- X linked spinal bulbar muscular atrophy
ICD coding
Epidemiology
- Rare disorder with distinct populations and varying prevalence
- In the U.S., reported prevalence of ~2:100,000 men (J Mol Neurosci 2016;58:313)
- In Ireland, prevalence is estimated be 0.83:100,000 (Neurology 2017;88:304)
- In western Finland, a surprisingly high prevalence of 13:85,000 was reported (Acta Neurol Scand 1998;98:128)
- Affected men typically develop weakness in third and fourth decades of life, plus androgen insensitivity with reduced fertility and gynecomastia (Neurol Clin 2015;33:847)
- Age range: 18 - 64 years
- General impression is that X linked bulbospinal neuronopathy, due to mild symptoms exhibited by some patients, may be underdiagnosed (Arch Neurol 2002;59:1921)
Sites
- Proximal limb muscles
- Bulbar muscles
- Sensory nerve
Pathophysiology
- X linked recessive disorder
- Expansion of tandem CAG repeat region in first exon of androgen receptor (AR) gene on proximal part of long arm of X chromosome (Xq11-12)
- There is variable phenotypic expression between and within families, which is not clearly related to the size of the CAG expansion
- Repeat lengths of 38 - 62 CAGs have been reported in patients, versus 11 - 32 CAGs in normal individuals (Muscle Nerve 1995;18:1378)
- CAG repeat is expressed as an expanded polyglutamine tract in the androgen receptor; studies indicate that androgen dependent gain of function by the receptor results in toxicity of the mutant protein
- In addition to the toxic effects of the AR, loss of normal receptor function also contributes to the androgen insensitivity aspects of the disease phenotype (Neuron 2014;82:251)
- Translocation of the mutant AR into the nucleus also seems necessary for toxicity
- Deletion of the nuclear localization signal prevents toxicity in a mouse model
- Presence of nuclear inclusion bodies is a pathological hallmark of the polyglutamine disease
- In X linked bulbospinal neuronopathy, nuclear inclusions contain mutated truncated androgen receptor and are most commonly seen within motor neurons in the brainstem and spinal cord (Ann Neurol 1998;44:249)
- There is evidence for mitochondrial dysfunction in X linked bulbospinal neuronopathy, which is likely a result of complex interactions of elongated polyglutamine (polyQ) AR with mitochondrial protein and nuclear / DNA, resulting in oxidative stress and ultimately activation of the mitochondrial caspase pathway or decreased mitochondrial membrane potential (Hum Mol Genet 2009;18:27)
Etiology
- Trinucleotide CAG repeat expansion in the gene encoding the androgen receptor located on the X chromosome (Xq11-12)
Diagrams / tables
Images hosted on other servers:

Impacts of spinal and bulbar muscular atrophy
Clinical features
- Facial, hypoglossal and spinal cord motor neuron degeneration with neurogenic wasting of corresponding skeletal muscles (particularly tongue)
- Third, fourth and sixth (oculomotor, trochlear and abducens) cranial nerves are spared
- Slowly progressive lower motor neuron weakness of facial, bulbar and proximal limb muscles with fasciculations (Neurol Clin 2015;33:847)
- Progression of weakness is slow, with ~2% decrease in muscle strength by quantitative muscle testing per year
- Tremor and cramping are also common
- Often associated with gynecomastia, infertility and primary sensory neuronopathy
- Family history may be absent in as many as 26 - 60% of patients (Arch Neurol 2002;59:1921)
Diagnosis
- Electrophysiology
- Laboratory (serum levels)
- Imaging
- Magnetic resonance imaging (MRI)
- Proton magnetic resonance spectroscopy
- Molecular diagnosis (Rev Neurol (Paris) 2017;173:326)
Laboratory
- Laboratory (serum)
- Elevated serum creatine kinase
- Normal or elevated serum testosterone levels
- Elevated serum myoglobin may help differentiate from amyotrophic lateral sclerosis (Int J Neurosci 2021;131:1209)
- Electrophysiology
- Generalized chronic damage of motor neurons with fasciculations and pseudomyotonic discharges (Clin Neuropathol 2015;34:199)
- Genetic test
- DNA analysis of peripheral blood detection of CAG triplet repeat expansion in the AR gene (Neurology 1997;49:568)
Radiology description
- Magnetic resonance imaging (MRI)
- Atrophy of cervical and thoracic spinal cord (Eur Neurol 2005;53:74)
- Proton magnetic resonance spectroscopy
- Significant reduction in the ratio of N-acetyl-aspartate to phosphocreatine in the motor cortex (J Neurol Sci 2009;277:71)
Prognostic factors
- Slowly progressive disease but life expectancy does not seem to be significantly affected (Neurology 2008;70:1967)
- Study of 223 cases of X linked bulbospinal neuronopathy shows that those patients with longer CAG repeat size displayed an earlier onset of disability in activities of daily living but the rate of subsequent declines was independent of the triplet repeat size (Brain 2006;129:1446)
- Urinary levels of the oxidative stress marker 8 hydroxydeoxyguanosine (8-OHdG) correlates with severity of motor dysfunction (Muscle Nerve 2012;46:692)
- Development of insulin resistance is a clinical feature of X linked bulbospinal neuronopathy and the degree of this resistance has been reported to correlate with disease severity (J Neurol 2017;264:839)
Case reports
- 31 and 56 year old monozygotic male twins with discordant presentation of Kennedy disease (J Clin Neuromuscul Dis 2019;21:112)
- 43 year old man with progressive limb weakness, gynecomastia, erectile dysfunction and tongue atrophy with no family history of Kennedy disease (Medicine (Baltimore) 2017;96:e6792)
- 50 year old man with lumbar disc herniation secondary to progressive proximal limb weakness (Am J Transl Res 2021;13:7412)
- 57 year old Brazilian man with progressive brachiocrural paresis over 3 years with fasciculations and tremors of extremities (Einstein (Sao Paulo) 2018;16:eRC4011)
Treatment
- There is currently no effective therapy to prevent progression of the disease
- Management is focused on preventing complications and improving mobility and function
- Clinical trials have focused on reducing AR ligand
- Androgen reduction with leuprorelin and castration mitigates disease manifestations in transgenic animal models (J Neurosci 2004;24:4778, Nat Med 2003;9:768)
Clinical images
Images hosted on other servers:
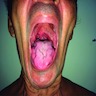
Tongue atrophy
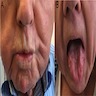
Asymmetric facial muscle atrophy
Gross description
- Degeneration of facial, hypoglossal and spinal cord motor neurons with neurogenic wasting of corresponding skeletal muscles (particularly the tongue) (Ellison: Neuropathology - A Reference Text of CNS Pathology, 3rd Edition, 2013)
- Third, fourth and sixth cranial nerve nuclei are spared
Microscopic (histologic) description
- Depletion of lower motor neurons through spinal cord segments and brainstem motor nuclei
- Muscle histopathology is atypical for a motor neuron disease
- Cytoplasmic inclusions are lacking and both neurogenic and myogenic alterations are seen, with neurogenic alterations being more common
- Commonly described morphological changes include groups of small atrophic myofibers with pyknotic nuclei and hypertrophic fibers, sometimes with central nuclei (Clin Neuropathol 2015;34:199)
- Nuclear inclusions may be found in spinal neurons and brainstem motor neurons
- Nuclear inclusions can also be found in nonneural tissues, including skin, dermis, kidney, heart and testis (Ellison: Neuropathology - A Reference Text of CNS Pathology, 3rd Edition, 2013)
- Ultrastructurally, most nuclei contain clusters of dense heterochromatin (Clin Neuropathol 2015;34:199)
Microscopic (histologic) images
Images hosted on other servers:
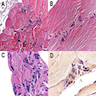
Muscle biopsy

Pathology of Kennedy disease
Positive stains
- Nuclear inclusions: N terminus of AR protein, ubiquitin (Ellison: Neuropathology - A Reference Text of CNS Pathology, 3rd Edition, 2013)
Electron microscopy description
- Ultrastructurally, most nuclei contain clusters of dense heterochromatin (Clin Neuropathol 2015;34:199)
Videos
Hand tremor, tongue fasciculation
Sample pathology report
- Peripheral blood:
- Positive for CAG repeat expansion in AR (46 repeats) (see comment)
- Comment: Genetic testing on peripheral blood identified 46 CAG repeats in the androgen receptor gene, which would support a clinical impression of X linked bulbospinal neuronopathy (Kennedy disease).
Differential diagnosis
- Amyotrophic lateral sclerosis (ALS):
- Clinically, characterized by upper and lower motor neuron signs / symptoms: gross atrophy of anterior spinal nerve roots, neuronal loss and gliosis affecting anterior horn of spinal cord and primary motor cortex, pallor of corticospinal tracts, Bunina bodies (cystatin C positive) and skein-like inclusions (TDP-43 and ubiquitin positive) in motor neurons
- No CAG repeat expansion in the androgen receptor gene
- Familial cases show C9orf72 expansion and SOD1 mutation, which are the 2 most common inherited genetic alterations
- Hereditary spastic paraplegia:
- Different clinical findings
- Primarily affects upper motor neurons with slow degeneration
- Slow degeneration causes the muscles to not receive the correct messages, causing progressive spasticity
- No CAG repeat expansion in the androgen receptor gene
- 80+ genetic mutations cause various types of hereditary spastic paraplegia but some patients have no known mutation
- Different clinical findings
- Myasthenia gravis:
- Different clinical findings
- Symptoms typically occur or are exacerbated after exertion, better in morning and worse at night
- Positive nerve electrical stimulation
- Often thymic hyperplasia and hypertrophy by CT
- Normal tendon reflexes
- No CAG repeat expansion in the androgen receptor gene
- Treatment with cholinesterase inhibitors is effective
- Different clinical findings
- Secondary causes of motor neuron disease (Ellison: Neuropathology - A Reference Text of CNS Pathology, 3rd Edition, 2013):
- All have distinctive clinical findings and lack CAG repeat expansion in the androgen receptor gene
- Infectious:
- Acute poliomyelitis
- HIV infection
- Metabolic:
- Immune:
- Paraproteinemia
- Paraneoplastic:
- Hodgkin lymphoma
- Non-Hodgkin lymphoma
- Infectious:
- Spinal muscular atrophy
- All have distinctive clinical findings and lack CAG repeat expansion in the androgen receptor gene
Additional references
Board review style question #1
A middle aged adopted man with progressive proximal weakness and perioral fasciculation is suspected to suffer from X linked bulbospinal neuronopathy. Peripheral blood is drawn. What is the expected genetic finding?
- C9orf72 mutation
- Expansion of tandem CAG repeat
- Homozygous deletion of exon 7 in the SMN1 gene
- LGMDR1 mutation
- SOD1 mutation
Board review style answer #1
B. Expansion of tandem CAG repeat. X linked bulbospinal neuronopathy (Kennedy disease) is a rare, neurodegenerative disorder resulting from the expansion of tandem CAG repeat region in first exon of androgen receptor gene on proximal part of long arm of X chromosome. Answers A, C, D and E are incorrect because these mutations are associated with other disorders. C9orf72 expansion and SOD1 are associated with ALS. Homozygous deletion of exon 7 in the SMN1 gene and SOD1 mutations are associated with spinal muscular atrophy. SMN1 gene is associated with spinal muscular atrophy (Neuro Asia 2022;27;955). LGMDR1 mutation is associated with limb girdle muscular dystrophy (J Neuromuscul Dis 2021;8:125).
Comment Here
Reference: X linked bulbospinal neuronopathy
Comment Here
Reference: X linked bulbospinal neuronopathy
Board review style question #2
Which of the following features is characteristic of Kennedy disease (spinal and bulbar muscular atrophy)?
- It has similar clinical features as amyotrophic lateral sclerosis (ALS)
- It is a neurodegenerative disease of upper motor neurons
- It is due to expansion of tandem CAG repeat region in the androgen receptor gene
- It primarily affects women
Board review style answer #2
C. It is due to expansion of tandem CAG repeat region in the androgen receptor gene. Answer A is incorrect because the clinical features of Kennedy disease differ from amyotrophic lateral sclerosis. Answer B is incorrect because Kennedy disease is a lower neuron degenerative disease. Answer D is incorrect because Kennedy disease primarily affects men.
Comment Here
Reference: X linked bulbospinal neuronopathy
Comment Here
Reference: X linked bulbospinal neuronopathy
