

Heart & vascular pathology
Cardiomyopathy
Hypertrophic cardiomyopathy
Last author update: 1 May 2016
Last staff update: 9 May 2023
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed Search: Hypertrophic cardiomyopathy[title]


Table of Contents
Definition / general | Terminology | Epidemiology | Etiology | Pathophysiology | Clinical features | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy description | Molecular / cytogenetics description | Differential diagnosis | Additional referencesCite this page: Singaravel S, Vaideeswar P. Hypertrophic cardiomyopathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/heartHCM.html. Accessed April 16th, 2024.
Definition / general
- Hypertrophic cardiomyopathy (HCM) is defined by the presence of increased left ventricular (LV) wall thickness (in a non dilated chamber) that is not solely explained by abnormal loading conditions (Eur Heart J 2014;35:2733)
- This term is preferred for hypertrophy associated with mutations in sarcomeric protein genes
Terminology
- Idiopathic hypertrophic subaortic stenosis (IHSS) or hypertrophic obstructive cardiomyopathy (HOCM) are not preferred terms, as obstruction to left ventricular outflow is not invariably present in HCM
- One third of patients have no obstruction either at rest or with physiologic provocation
Epidemiology
- HCM is the most common inherited cardiac disease, with a prevalence of 1 in 500 in the general adult population
- Prevalence of HCM in children is unknown, but population based studies report an annual incidence of 0.3 to 0.5 per 100,000
- Occurs in all ethnic groups and equally in both sexes
- Most affected individuals remain unidentified
Etiology
- Heterogenous genetic disease, may be familial or non familial
- 60% of familial cases are autosomal dominant with variable expressivity and incomplete, age related penetrance
- Mutations have been identified in numerous sarcomeric protein genes; most common are the beta myosin heavy chain and myosin binding protein C
- Private mutations are unique to individual families
- Identification of these mutations is important for family screening
- Phenotypic diversity suggests the role of modifier genes and environmental factors in disease expression
- Sporadic HCM can reflect an inaccurate family history, incomplete penetrance or a de novo mutation which may be heritable
- Patients who are genotype positive may be phenotypically negative without overt hypertrophy (subclinical HCM)
Pathophysiology
- HCM is characterized by multiple interrelated abnormalities, including left ventricular outflow tract (LVOT) obstruction, diastolic dysfunction, mitral regurgitation, myocardial ischemia and arrhythmias
- A third of patients with HCM have obstruction under basal (resting) conditions
- In these patients, mechanical obstruction occurs due to systolic anterior motion (SAM) of the anterior mitral leaflet and mitral septal contact
- Muscular obstruction occurs in the mid cavitary region due to hypertrophy of the papillary muscles or anomalous papillary muscle insertion into the anterior mitral leaflet
- In another third, obstruction is labile, attributable to increased myocardial contractility, decreased ventricular volume or decreased afterload
- One third of patients have the non obstructive form of HCM
- Diastolic dysfunction in HCM occurs due to abnormal dissociation of active and myosin filaments in diastole and increase in chamber stiffness due to hypertrophy
- Myocardial ischemia occurs due to supply demand mismatch
- Compromised coronary blood blood flow may be present as a result of medial hypertrophy and thickening of arteriolar walls, associated with luminal narrowing
- Mitral regurgitation may be present due to distortion of the mitral valve apparatus from the SAM; intrinsic valvular abnormalities may also be present
- Autonomic dysfunction in HCM presents as systemic vasodilatation with fall in blood pressure associated with bradycardia during exercise
Clinical features
- Many individuals with HCM are asymptomatic, and diagnosis may be incidental or identified during screening
- Some individuals are symptomatic, presenting with angina, dyspnea, palpitations and syncope
- In a small number of affected individuals, the condition is fatal, producing sudden cardiac death due to ventricular arrhythmia
- Other causes of death include progressive heart failure and atrial fibrillation with risk of stroke
Laboratory
- Recommended laboratory tests include routine hematology, glucose, cardiac enzymes, renal, thyroid and liver function tests, pH, electrolytes and uric acid
- These may help to detect extra cardiac conditions causing or exacerbating left ventricular dysfunction
Radiology description
- Standard 12 lead ECG shows variable combination of LVH, ST and T wave abnormalities
- Pathological Q waves can suggest an underlying diagnosis or provide clues to the distribution of hypertrophy and myocardial scar
- 48 hour ambulatory ECG is used to identify ventricular and atrial tachyarrhythmias and assess the risk of sudden cardiac death and stroke
- Transthoracic echocardiography (TTE) is recommended in the initial evaluation of patients suspected of having HCM
- TTE is also useful at follow up to assess the degree of myocardial hypertrophy, dynamic obstruction, systolic anterior motion of the mitral valve leaflet and myocardial diastolic function
- Short axis view is used to obtain septal measurements at all levels
- M Mode is used to determine the outflow gradient
- A transesophageal echocardiogram (TEE) is recommended to establish apical hypertrophy and for intraoperative guidance of surgical myectomy
- Cardiac Magnetic Resonance imaging (CMR) is recommended when echocardiography is inconclusive
- CMR can establish magnitude and distribution of hypertrophy, anatomy of the mitral valve apparatus or papillary muscles and quantity of scarring
- It can also identify localized apical disease
Prognostic factors
- Disease progression is difficult to predict
- Genetic testing cannot identify high risk patients due to differences in penetrance and expressivity
Case reports
- 61 year old man with hypertrophic obstructive cardiomyopathy with mitral valve leaflet elongation (Circulation 2015;131:1605)
- 68 year old woman with apical hypertrophic cardiomyopathy (N Engl J Med 2015;373:e22)
Treatment
- Surgical myectomy or alcohol septal ablation are performed in cases with outflow obstruction to alleviate symptoms
- Implantable cardioverter defibrillators (ICDs) can prevent sudden death
- Catheter based procedures can control atrial fibrillation
- Heart transplantation is performed for end stage failure
Gross description
- HCM in adults is characterised by unexplained LV wall thickness ≥15 mm, septal/posterior wall thickness ratio >1.3 in normotensive patients or septal/posterior wall thickness ratio >1.5 in hypertensive patients
- In children, hypertrophy is defined as wall thickness ≥2 standard deviations above the mean (z score ≥2) for age, sex or body size
- Due to age related penetrance, a normal sized heart in childhood does not rule out HCM
- Hypertrophy is characteristically asymmetric with disproportionate septal thickening, particularly at the confluence of the anterior septum and anterior free wall
- Symmetric, apical and other atypical distributions of hypertrophy are also described
- A four chamber view (longitudinal section of the heart) may show sigmoid deformation of the hypertrophied septum
- Only a small portion of the left ventricle may be involved (segmental hypertrophy) and calculated left ventricular mass may be normal
- Any degree of wall thickness may be seen with the HCM genetic substrate
- Myocardium has a glistening, leiomyoma-like appearance
- Papillary muscle abnormalities including hypertrophy and displacement may be observed
- Mitral leaflet abnormalities include enlargement or elongation and presence of accessory tissues
- Coronary arteries show medial hypertrophy
- Myocardial bridging may be seen with tunneled coronary arteries
- Myocardial fibrosis may be seen due to demand and coronary supply mismatch or as an intrinsic part of the disease
- Presence of fibrosis (end stage disease) may be associated with a dilated phenotype
Gross images
Contributed by Saranya Singaravel, M.B.B.S.
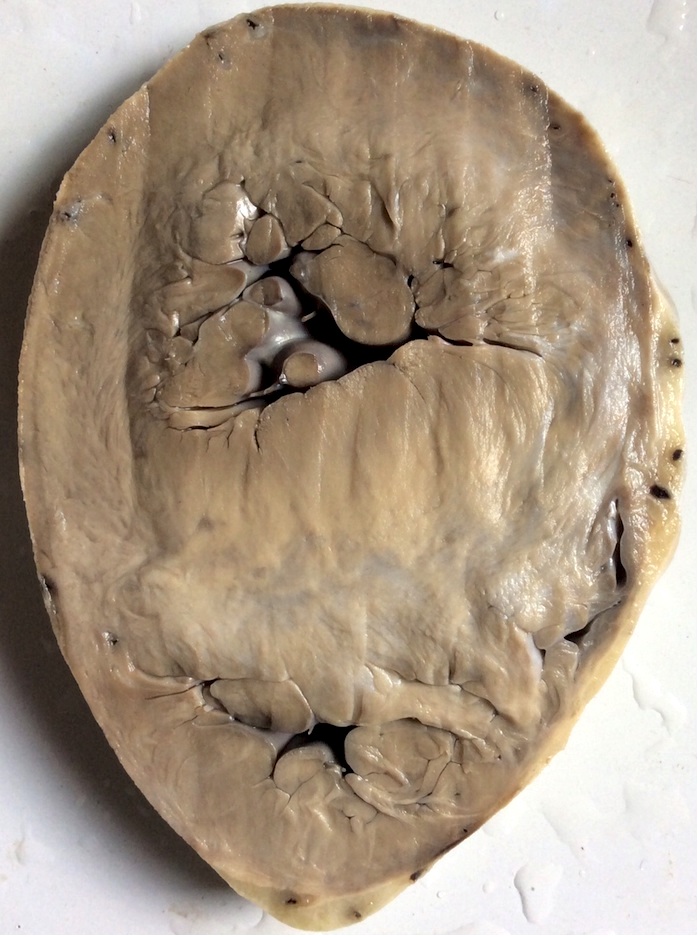
Transverse section
Microscopic (histologic) description
- Cardiomyocytes are hypertrophied with abundant eosinophilic cytoplasm and box shaped nuclei
- Myocytes display bizarre forms with Y shaped branching, frequent side to side junctions or a characteristic whorled appearance, usually around a central fibrous core
- Myocardial architecture is disorganized, with bundles of cardiomyocytes arranged at perpendicular and oblique angles to each other (myocardial disarray)
- Myocardial disarray is specific for HCM when it is diffuse or when it involves at least 20% of one or more tissue blocks (2x2 cm)
- Replacement myocardial fibrosis resulting from microvascular ischemia and resultant cell death may be seen
- Interstitial fibrosis may be seen forming arrythmogenic foci
Microscopic (histologic) images
Contributed by Saranya Singaravel, M.B.B.S.
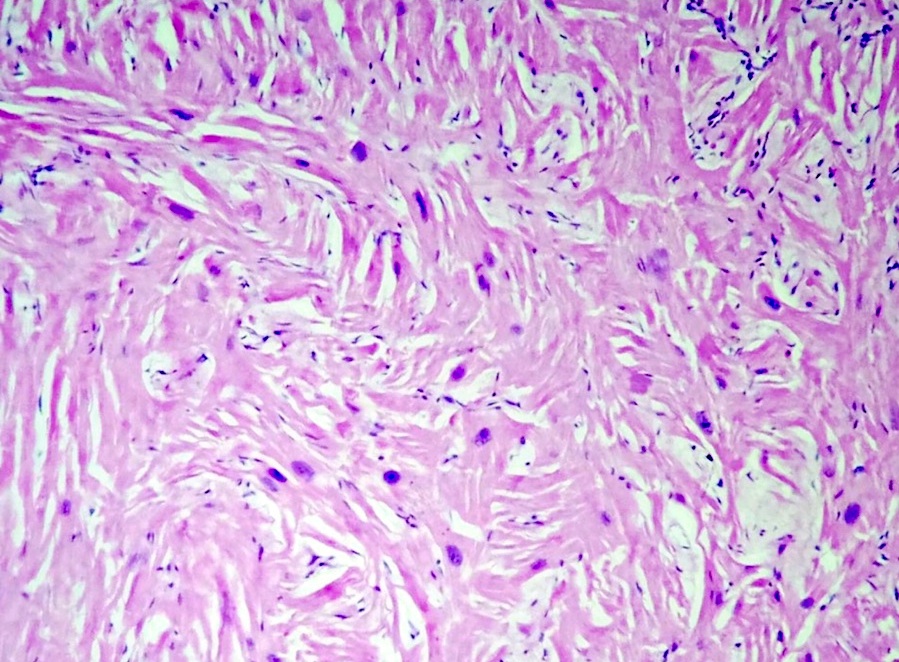
Myocardial hypertrophy and disarray
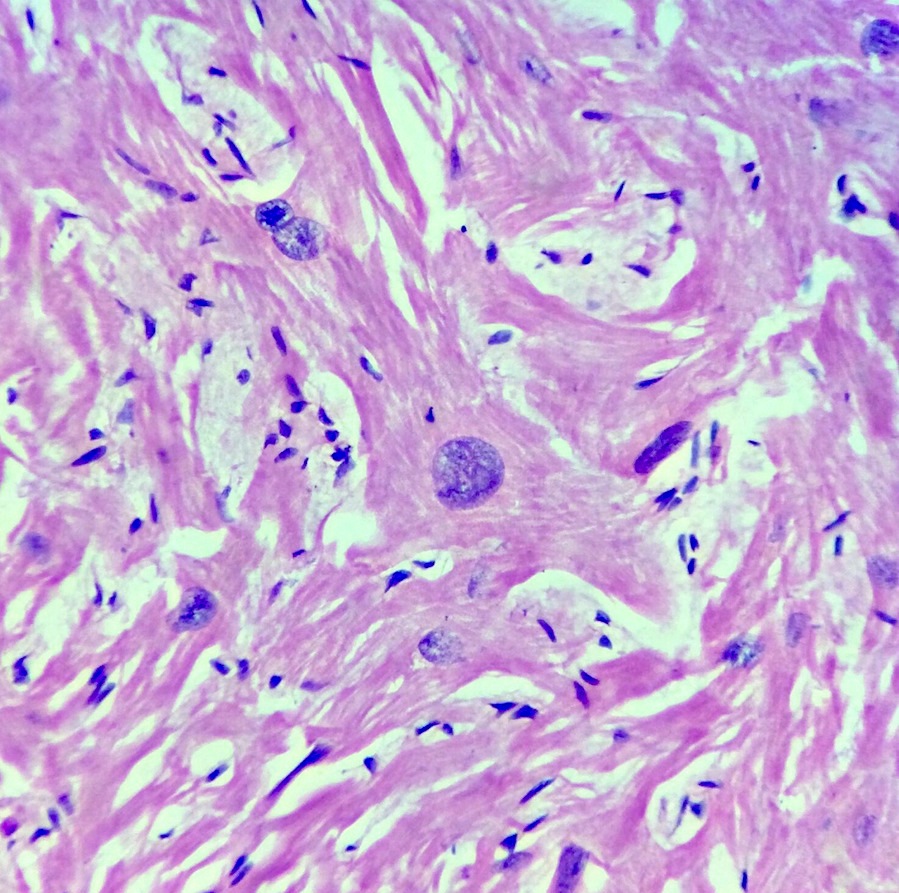
Hypertrophied cardiomyocyte with a whorled apperance
Positive stains
- Masson's trichrome, picrosirius red - fibrosis
Electron microscopy description
- Structural derangement is evident at the subcellular level, with loss of the normal alignment of myofibrils and Z disks
Molecular / cytogenetics description
- Genetic testing by rapid automated DNA sequencing is used to identify family members who carry the mutation but have no left ventricular hypertrophy (i.e. genotype positive, phenotype negative)
Differential diagnosis
- Physiologic hypertrophy (athlete's heart): absence of sarcomeric mutations or family history of HCM, enlarged LV cavity, normal diastolic function, pattern of LV hypertrophy is contiguous with decrease in wall thickness during short deconditioning periods
- Metabolic disorders:
- Inherited as autosomal recessive traits; most common metabolic disorders with hypertrophy are Anderson-Fabry disease, (mutations in the γ2 subunit of the adenosine monophosphate activated protein kinase, PRKAG2) and Danon disease (mutations in lysosome associated membrane protein 2, LAMP2)
- These are disproportionately better represented in childhood and adolescence
- Mitochondrial cardiomyopathies: mutations in mitochondrial DNA coding for the respiratory chain protein complexes, transmitted as maternally inherited traits
- Neuromuscular disease: cardiac hypertrophy is a rare manifestation of neuromuscular disease, including Friedreich's ataxia, muscular dystrophies and congenital skeletal myopathies
- Mutations in desmin may cause dilated, restrictive or hypertrophic phenotypes
- Malformation syndromes such as Noonan syndrome, LEOPARD syndrome or Costello syndrome may show cardiac hypertrophy as part of the constellation of manifestations
- Infiltrative diseases: cardiac amyloidosis may be part of a generalized disease
- Light chain (AL) and hereditary transthyretin (TTR) related amyloidosis or localized senile amyloidosis predominantly affects the heart and the carpal tunnel ligament
- Endocrine disorders: infants of diabetic mothers
- Drug use: anabolic steroids
Additional references
- 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy (Circulation 2011;124:2761)
- Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases (Eur Heart J 2008;29:270)
