
Hematology & immune disorders
Hemoglobinopathies
Alpha thalassemia
Authors: Momal Tara Chand, M.D., Daniel Snower, M.D.
Editorial Board Member: Genevieve M. Crane, M.D., Ph.D.
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 22 May 2019
Last staff update: 4 October 2023
Copyright: 2019-2024, PathologyOutlines.com, Inc.
PubMed Search: Alpha thalassemia
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Case reports | Treatment | Peripheral smear description | Peripheral smear images | Sample pathology report | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2 | Board review style question #3 | Board review style answer #3Cite this page: Chand MT, Snower D. Alpha thalassemia. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/hematoalphathalassemia.html. Accessed April 24th, 2024.
Definition / general
- Alpha thalassemia is a group of inherited blood disorders characterized by reduced or absent production of α-globin subunits, resulting in low levels of hemoglobin, decreased mean corpuscular volume (MCV) and decreased mean corpuscular hemoglobin (MCH)
Essential features
- Group of inherited autosomal recessive diseases caused by an α-globin chain synthesis defect
- There are 4 clinical pictures of α-thalassemia, according to the number of genes affected by loss of function with hemoglobin Bart's hydrops fetalis (Hb Bart's) syndrome and HbH disease being clinically significant
Terminology
- Also classified as α-thalassemia minima (heterozygous α+-thalassemia, -α/αα) and α-thalassemia minor (heterozygous α0-thalassemia, –/αα or homozygous α+-thalassemia, -α/-α) (Dtsch Arztebl Int 2011;108:532)
ICD coding
- ICD-10: D56.0 - alpha thalassemia
Epidemiology
- 15% of American blacks are silent carriers for α-thalassemia and about 3% have α-thalassemia trait (Medscape: Alpha Thalassemia [Accessed 23 April 2019])
- In Southeast Asian and Mediterranean populations, HbH disease and Hb Bart’s syndrome (γ4) are common
Pathophysiology
- The adequacy of the oxygen transport system depends on the affinity of hemoglobin for oxygen
- In adults, HbA is the major hemoglobin (97%), composed of 2 α subunits and 2 β subunits (α₂β₂) with minor amount of HbA2 (approximately 1.5 - 3.5%; α2δ2) and HbF (approximately < 1%; α2γ2)
- 2 α-globin genes are located on each chromosome 16, resulting in 4 α-gene loci (αα/αα)
- Severity of α-thalassemia depends on the number of inactivated or deleted alpha loci
- When both α-globin genes on a chromosome are deleted or inactivated, the allele is referred to as α0 (no output of α-globin from the chromosome)
- An individual with the genotype --/αα is referred to as an α0 carrier (GeneReviews 2005: NBK1435)
- This is common in Southeast Asia and Mediterranean but rare in African Americans
- When 1 α-globin gene on a chromosome is deleted or inactivated by a nondeletion variant, the allele is called α+ (some α-globin is produced)
- An individual with the genotype -α/αα is referred to as an α+ carrier (GeneReviews 2005: NBK1435)
- This is common in African Americans
Etiology
- α-thalassemia is usually inherited in an autosomal recessive manner
Diagrams / tables



Clinical features
- There are 4 α-thalassemia syndromes, according to the number of genes affected, correlating with different clinical pictures
- Hb Bart's hydrops fetalis syndrome: complete absence of all 4 α chains
- Because of the absence of α chains, no HbA or HbF is present (GeneReviews 2005: NBK1435)
- Large amount of Hb Bart’s, a variable amount of Hb Portland and traces of HbH present
- Red cells with Hb Bart's have an extremely high oxygen affinity and are incapable of effective oxygen delivery
- Incompatible with life, fetuses are still born with severe anemia, marked edema and hepatosplenomegaly
- HbH disease: absence of 3 α chains
- There is chronic hemolytic anemia, mild jaundice and hepatosplenomegaly
- Most individuals are clinically well and survive without treatment (GeneReviews 2005: NBK1435), transfusion is rarely needed
- α-thalassemia trait: absence of 2 α chains either in cis (--/αα, α0 carrier) or in trans (-α/-α) (GeneReviews 2005: NBK1435)
- Benign condition with most patients diagnosed on routine screening
- Does not require treatment
- α-thalassemia silent carrier: absence of 1 α chain
- No clinical abnormalities
- Hb Bart's hydrops fetalis syndrome: complete absence of all 4 α chains
Diagnosis
- Electrophoresis or high performance liquid chromatography (HPLC):
- Hb Bart's hydrops fetalis syndrome: Hb Bart’s migrates faster than HbA on alkaline electrophoresis
- Hb Bart’s > 80%, a variable amount of Hb Portland and HbH present (See figure)
- No HbA
- HbH disease: HbH migrates faster than HbA on alkaline electrophoresis (See figure) (Indian J Clin Biochem 2010;25:435)
- HbH > 15% at birth (GeneReviews 2005: NBK1435)
- Normal or decreased HbA2
- α-thalassemia trait:
- Hb Bart’s in newborns (up to 20%) (See figure)
- Normal electrophoresis in adults and the diagnosis is made by excluding iron deficiency, anemia of chronic disease and beta thalassemia
- Normal HbA2 and HbF (GeneReviews 2005: NBK1435)
- α-thalassemia silent carrier:
- Hb Bart’s in newborns (up to 2%)
- Normal electrophoresis in adults and diagnosis is made by molecular or globin chain synthesis studies
- Hb Bart's hydrops fetalis syndrome: Hb Bart’s migrates faster than HbA on alkaline electrophoresis
Laboratory
- Hb Bart's hydrops fetalis syndrome:
- CBC: severe microcytic hypochromic anemia and reticulocytosis
- Hb Bart’s > 80%, HbH and Hb Portland
- HbH disease:
- CBC: decreased MCV and MCH, reticulocytosis (4 - 5%), increased RBCs
- Hb Bart’s:
- 20 - 40% at birth
- 5 - 30% in adults
- α-thalassemia trait:
- CBC: may show mild hypochromic (low MCH), microcytic (low MCV) anemia (GeneReviews 2005: NBK1435)
- Hb Bart’s:
- 2 - 10% in neonate
- None in adults
- α-thalassemia silent carrier:
- CBC: either normal or mild reduction of MCV and MCH (GeneReviews 2005: NBK1435)
- Hb Bart’s:
- 1 - 2% in neonates
- None in adults
Radiology description
- Hb Bart's hydrops fetalis syndrome is suspected in fetuses with increased nuchal thickness, thickened placenta, increased cerebral media artery velocity and increased cardiothoracic ratio on ultrasonography examination at 13 to 14 weeks' gestation (GeneReviews 2005: NBK1435)
Radiology images
Images hosted on other servers:
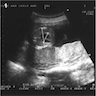
Hb Bart’s, midpregnancy sonographic features
Case reports
- Neonate with α-thalassemia major (Pediatr Dev Pathol 2019;22:166)
- 16 year old boy with co-inheritance of heterozygous α+-thalassemia and sickle cell trait (BMC Ophthalmol 2017;17:6)
- 22 year old woman with HbH disease (Biomed Rep 2016;5:23)
- 28 year old Chinese woman with α-thalassemia trait (J Med Case Rep 2015;9:58)
- 36 year old Chinese woman with HbH disease (Case Rep Med 2018;2018:8057045)
Treatment
- Hb Bart's syndrome is a universally fatal condition and death usually occurs in the neonatal period (GeneReviews 2005: NBK1435)
- Most individuals with HbH disease, thalassemia trait and carriers are clinically well and survive without any treatment (GeneReviews 2005: NBK1435)
Peripheral smear description
- Hb Bart's hydrops fetalis syndrome:
- Large, hypochromic red cells and severe anisopoikilocytosis (GeneReviews 2005: NBK1435) (See figure)
- HbH disease:
- Hypochromia, basophilic stippling, target cells, anispoikilocytosis
- Red blood cell supravital stain show HbH inclusions (β4 tetramers) (GeneReviews 2005: NBK1435)
- Hypochromia, basophilic stippling, target cells, anispoikilocytosis
- α-thalassemia trait:
- Hypochromia and microcytosis
- HbH inclusions found in α0-thalassemia but rarely in α+-thalassemia
- Hypochromia and microcytosis
Peripheral smear images



Sample pathology report
- Electrophoresis report:
- Features suggestive of α-thalassemia, if other causes of microcytosis are excluded
- Molecular report:
- α-globin (HBA1 and HBA2) deletion / duplication
- Deletion:
- Result: 2 pathogenic deletions detected in the α-globin gene cluster
- DNA variant(s):
- Pathogenic deletion: -α3.7
- Heterozygous pathogenic deletion: -α4.2
- Heterozygous predicted genotype: -α/-α
- Interpretation: 1 copy of the 3.7 Kb α-globin gene deletion and 1 copy of the 4.2 Kb α-globin gene deletion were detected by deletion / duplication analysis of the α-globin gene cluster and its HS-40 regulatory region. This individual is predicted to have a single functional α-globin gene on both chromosomes. This result is consistent with α0 thalassemia (α-thalassemia trait) often associated with mild anemia and microcytosis
Additional references
Board review style question #1
-
A 30 year old man presents to his physician for partner screening. Routine laboratory studies are shown:
- α-thalassemia trait
- β-thalassemia trait
- α-thalassemia silent carrier
- δβ-thalassemia
- Hemoglobin S trait
| Test Patient Result | Reference Range | |
| WBC | 6.3 | 2.5 - 7.5 x10 |
| RBC | 5.5 | 4.6 - 5.7 x 10 |
| Hemoglobin | 12 | 13 - 18 g/dL |
| Hematocrit | 36 | 39 - 54% |
| MCV | 65 | 83 - 99 fL |
| MCH | 21.8 | 27 - 30 pg |
| MCHC | 33 | 30.8 - 34.6% |
| Platelet count | 260 | 200 - 400 x10 /mm |
| RDW | 13.8 | < 14 % |
| Ferritin | 112 | 29 - 248 ng/dl |
| Serum ferritin | 104 | 20 - 300 μg/L |
| Hemoglobin A2 | 2.4 | 2.3 - 3.5% |
| Hemoglobin F | 0.6 | 0.5 - 1% |
| Hemoglobin A | 97.0 | 96.8 - 97.8% |
Which one of the following is most likely diagnosis?
Board review style answer #1
A. α-thalassemia trait. A presumptive diagnosis of α-thalassemia trait can be made when there is microcytosis, normal hemoglobin A2, hemoglobin F and iron studies. β-thalassemia trait patients have increased hemoglobin A2. Silent carriers have normal hemoglobin (no anemia). δβ-thalassemia presents with microcytosis, but elevated hemoglobin F. Hemoglobin S trait presents with increased hemoglobin S.
Comment Here
Reference: Alpha thalassemia
Comment Here
Reference: Alpha thalassemia
Board review style question #2
- Which of the following is associated with deletion of 3 α genes?
- Hemoglobin Bart's hydrops fetalis syndrome
- Hemoglobin H disease
- α-thalassemia trait
- α-thalassemia silent carrier
Board review style answer #2
B. Hemoglobin H disease. Hemoglobin Bart's hydrops fetalis syndrome: absence of all 4 α chains. Hemoglobin H disease: absence of 3 α chains. α-thalassemia trait: absence of 2 α chains. α-thalassemia silent carrier: absence of 1 α chain.
Comment Here
Reference: Alpha thalassemia
Comment Here
Reference: Alpha thalassemia
Board review style question #3
- Which of the following is highly incompatible with life?
- Hemoglobin H disease
- Hemoglobin Bart’s hydrops fetalis syndrome
- α-thalassemia trait
- α-thalassemia silent carrier
Board review style answer #3
B. Hemoglobin Bart’s hydrops fetalis syndrome. Hemoglobin Bart's hydrops fetalis syndrome is characterized by complete absence of α chains. Because of the absence of α chains, no HbA or HbF is present. The affected fetuses die in utero or shortly after birth. Hemoglobin H disease is caused by the absence of 3 α chains. It presents with microcytosis and hypochromia, but most diseased individuals are clinically well and survive without treatment. α-thalassemia trait: absence of 2 α chains, benign condition, doesn’t require treatment. α-thalassemia silent carrier is characterized by absence of 1 α chain, patients usually do not have any clinical abnormalities.
Comment Here
Reference: Alpha thalassemia
Comment Here
Reference: Alpha thalassemia
