

Kidney nontumor / medical renal
Glomerular disease
Fibrillar disease
Renal amyloidosis
Author: Anthony Sisk, D.O.
Editorial Board Members: Nicole K. Andeen, M.D., Jonathan E. Zuckerman, M.D., Ph.D.

Last author update: 18 July 2022
Last staff update: 18 July 2022
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Renal amyloidosis
Table of Contents
Definition / general | Essential features | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Immunofluorescence description | Immunofluorescence images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Genetics | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Sisk A. Renal amyloidosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyamyloidosis.html. Accessed April 24th, 2024.
Definition / general
- A group of insoluble proteins organized into beta pleated sheets, which deposit within extracellular spaces of a variety of tissues leading to organ dysfunction and disease
- Renal involvement largely consists of AL, AA, ALECT2 and rare hereditary types such as ATTR (transthyretin) amyloidosis (BMC Nephrol 2022;23:144)
- Amyloid deposits show similar morphology (irrespective of type) and display orangeophilic staining with Congo red and apple green birefringence under polarization
Essential features
- Deposition of acellular, eosinophilic material within glomeruli, tubulointerstitium and vessel walls; this is negative on Jones silver, pale on PAS / H&E and blue-gray on trichrome
- Immunofluorescence and immunohistochemistry findings can be helpful in determining amyloid type
- Laser dissection of amyloid, followed by tandem mass spectrometry analysis, is the gold standard for definitive amyloid typing
- Treatment options rely on accurate typing of amyloid (Kidney Int 2015;87:516)
ICD coding
Epidemiology
- Dependent on the amyloid type
- AL type:
- Primary amyloidosis is most commonly seen in people over the age of 50
- Most common type in the United States
- Caused by a monoclonal gammopathy of light chains (AH - heavy chains and mixed AH / AL types are much rarer)
- Most frequently associated with lambda light chains
- Related to both monoclonal gammopathy of undetermined significance (MGUS) and multiple myeloma
- AA type:
- Secondary amyloidosis characterized by serum amyloid A protein
- More common in developing countries
- Associated with prolonged inflammatory states including rheumatoid arthritis and other connective tissue disorders, infections, IV drug use, inflammatory bowel disease and paraneoplastic syndromes
- ALECT2 type:
- Leukocyte cell derived chemotaxin 2 associated amyloidosis
- Kidney is the most common organ involved, followed by the liver
- Initial reports indicated that Mexican Americans are preferentially affected but new reports document different ethnicities including Middle Eastern, Indian / Punjabi and Chinese populations (Nephrol Dial Transplant 2018;33:241, J Investig Med 2022;70:348)
- Hereditary types:
- Rare group of diseases involving mutations in a number of genes
- Autosomal dominant with variable penetrance
- Most common type is transthyretin (prealbumin) amyloidosis (ATTR), may be wild type but renal involvement is nearly always hereditary due to Val30Met mutations (Nephron 2022 Mar 18 [Epub ahead of print])
- Fibrinogen alpha chain (AFIB) followed by apolipoprotein A1 (AapoA1) are the next 2 (rarely seen) types (Amyloid 2022 May 3 [Epub ahead of print])
Sites
- Growing number of amyloid protein types, involving various organ systems and tissues, with relatively few involving the kidney specifically
- All components of the kidney may be involved (glomerular, tubular basement membranes and casts, interstitium, vessels)
Pathophysiology
- Specific causes are dependent on amyloid type
- Polypeptides become misfolded, usually into beta sheet secondary structures, where beta strands are oriented perpendicular to the long axis of the fiber
- These misfolded proteins have a propensity for aggregation and deposition within various organs and tissues (including vessels and nerves), causing damage (StatPearls: Amyloidosis [Accessed 8 June 2022])
Etiology
- Dependent on amyloid type
- Monoclonal protein (AL), chronic infection / inflammation (AA), specific populations with unknown cause (ALECT2), hereditary (ATTR, AApol1)
Clinical features
- Varies with type of amyloidosis and component of kidney affected
- Glomerular involvement presents with nephrotic range proteinuria and nephrotic syndrome, which may present suddenly
- Often with AL and AA type amyloidosis
- Tubulointerstitial predominant involvement will present with slow progressive renal failure and less proteinuria
- Often seen with ALECT2 amyloidosis
- Rarely, rapidly progressive renal failure and nephrotic syndrome in the setting of amyloid induced crescentic glomerulonephritis
- Most often with AA type amyloidosis (Indian J Nephrol 2020;30:352)
- Acute renal failure may be seen in the setting of amyloid cast nephropathy
- Glomerular involvement presents with nephrotic range proteinuria and nephrotic syndrome, which may present suddenly
Diagnosis
- May be suspected clinically
- Visualization of the characteristic amyloid deposits, as seen on special stains
- Confirmation with Congo red staining and polarization of amyloid showing typical apple green birefringence
- Thicker tissue sections (8 - 10 microns) are helpful in visualizing the polarized deposits
- Immunohistochemistry stains are available including serum amyloid A, transthyretin and antiserum amyloid P (SAP)
- Immunofluorescence microscopy to determine light or heavy chain restriction
- Electron microscopy findings with characteristic fibrillary deposits
- Amyloid type can be confirmed with mass spectrometry based proteomics
Laboratory
- Serum and urine protein electrophoresis with immunofixation
- Serum free light chain elevation
Prognostic factors
- Unfavorable factors (J Res Med Sci 2014;19:644, Blood 2003;101:827, J Clin Oncol 2016;34:2037):
- Dialysis requirement
- Massive proteinuria
- Renal failure at presentation
- Severe hypoalbuminemia
- Coexisting multiple myeloma
- Heavy organ involvement
- IgM related monoclonal protein
Case reports
- 31 year old woman presents with proteinuria during her second pregnancy (Clin Res Cardiol 2020;109:1438)
- 56 year old man with longstanding Crohn's disease presents with nephrotic syndrome (Ochsner J 2021;21:291)
- 60 year old Caucasian woman with 8 weeks of lower extremity edema, renal failure and a 10 cm kidney mass (Kidney Int Rep 2019;4:882)
Treatment
- Treatment based on amyloid type, underlying cause of disease and symptoms
- Dialysis and kidney transplant in end stage kidney disease
Gross images
Images hosted on other servers:

Pale amyloid deposits in enlarged kidney
Microscopic (histologic) description
- Pale eosinophilic, amorphous and extracellular material on H&E and PAS stains, typically silver negative and gray-blue on trichrome
- Amyloid involving glomerular basement membranes may show long perpendicular spikes
Microscopic (histologic) images
Contributed by Anthony Sisk, D.O.

Glomerulus
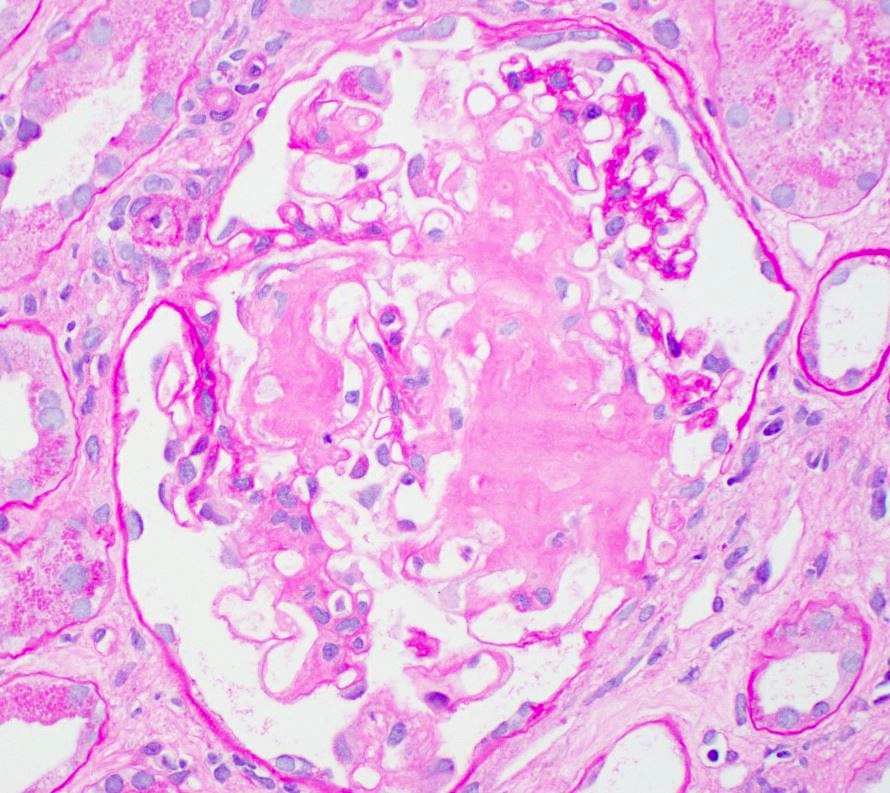
PAS, glomerulus
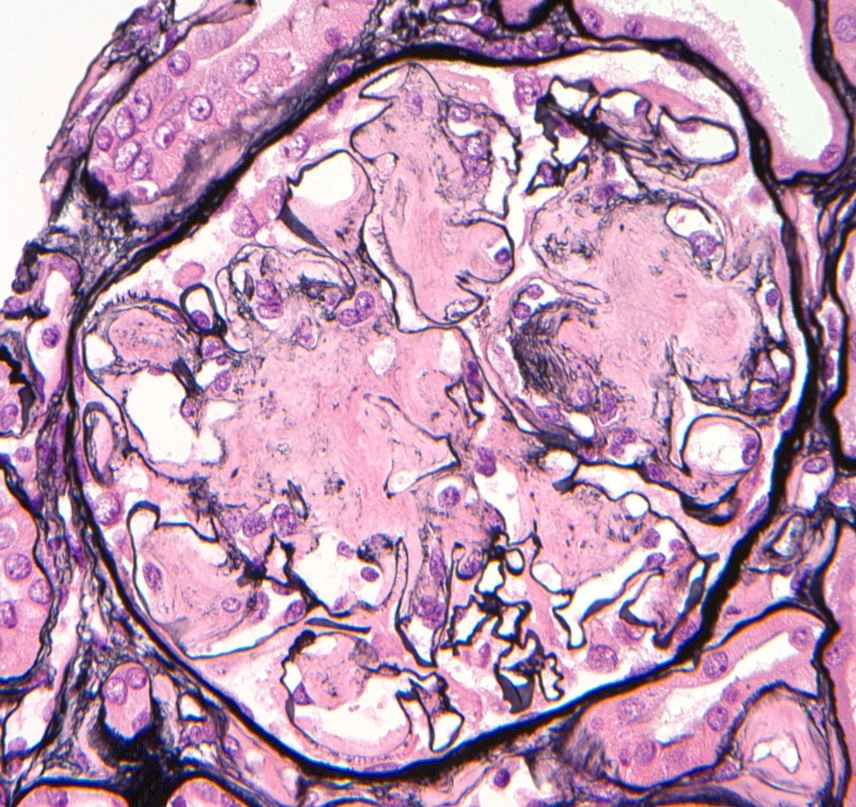
Jones silver, glomerulus
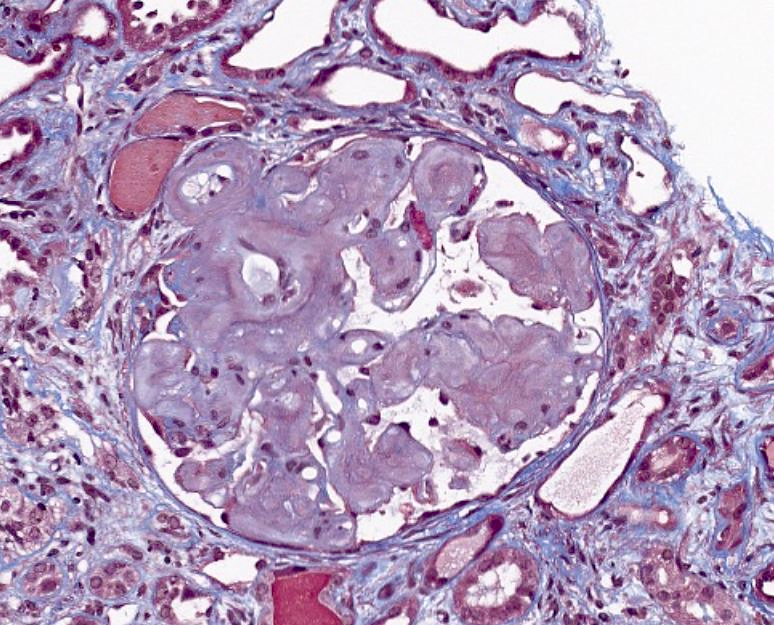
Trichrome, glomerulus
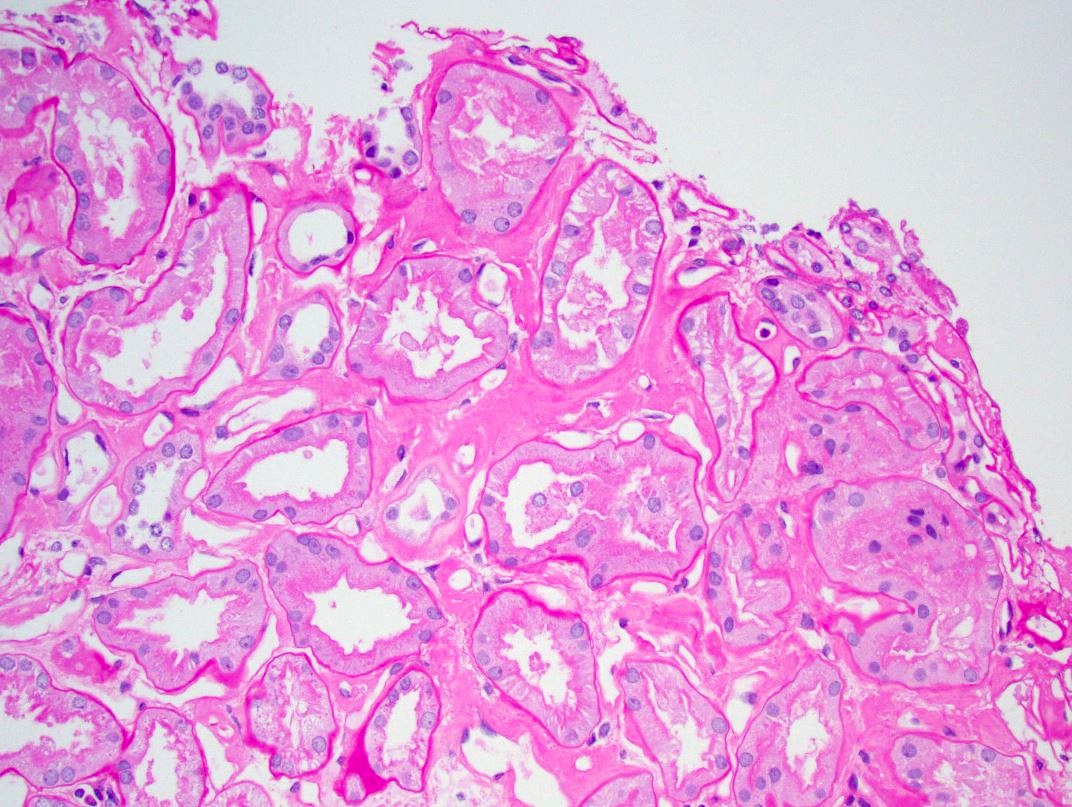
PAS, tubulointerstitium
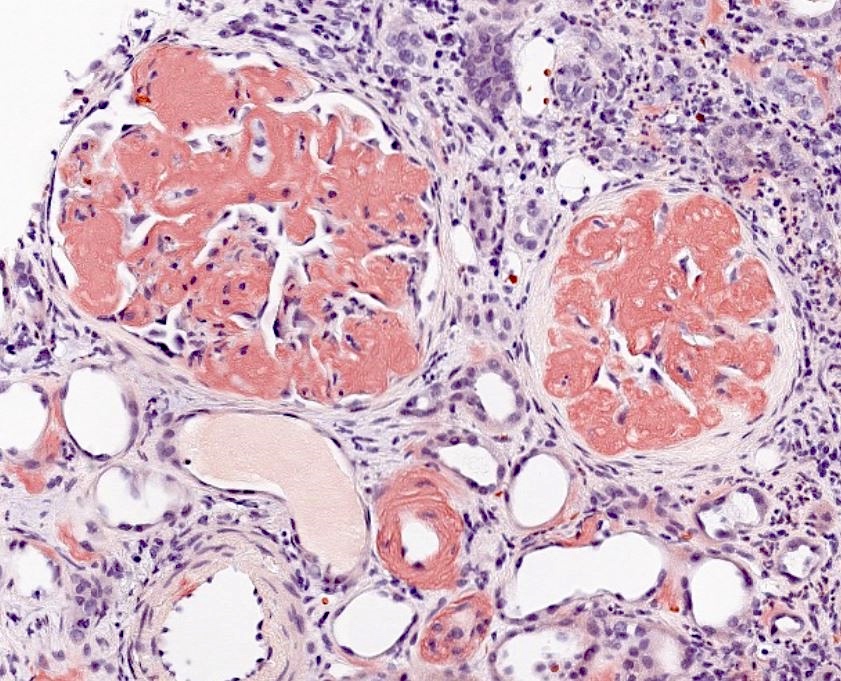
Congo red
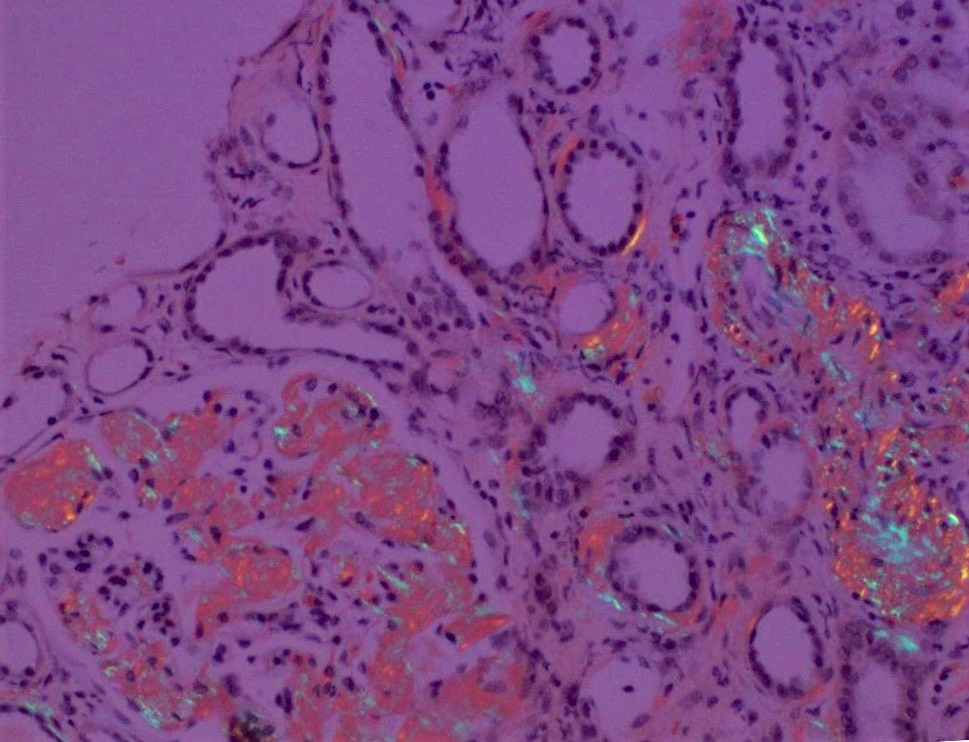
Polarization of Congo red
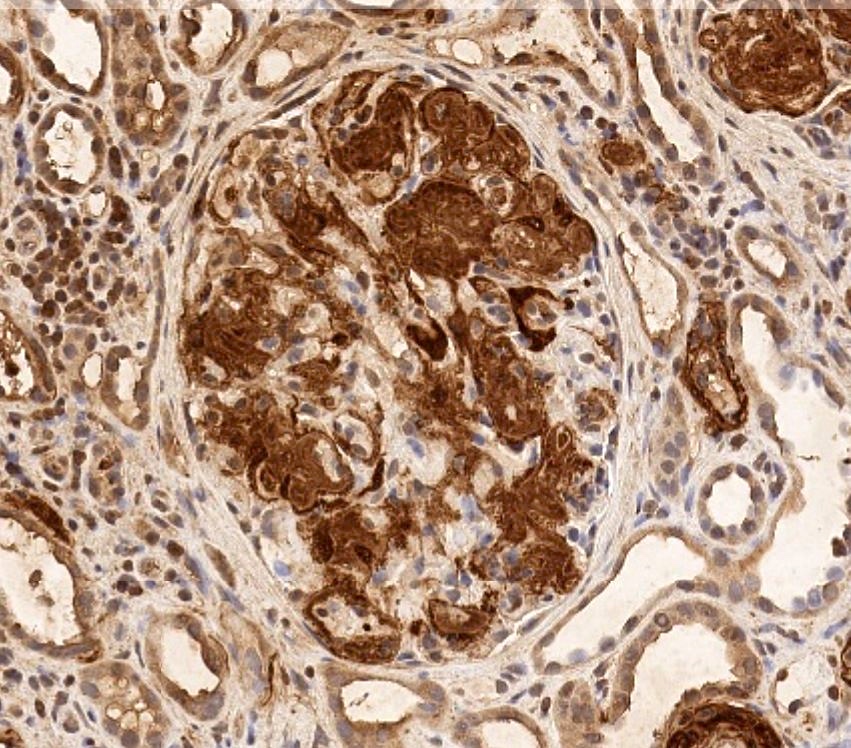
Amyloid A stain
Immunofluorescence description
- Light chain and heavy chain restriction may be seen within amyloid
Immunofluorescence images
Contributed by Anthony Sisk, D.O.
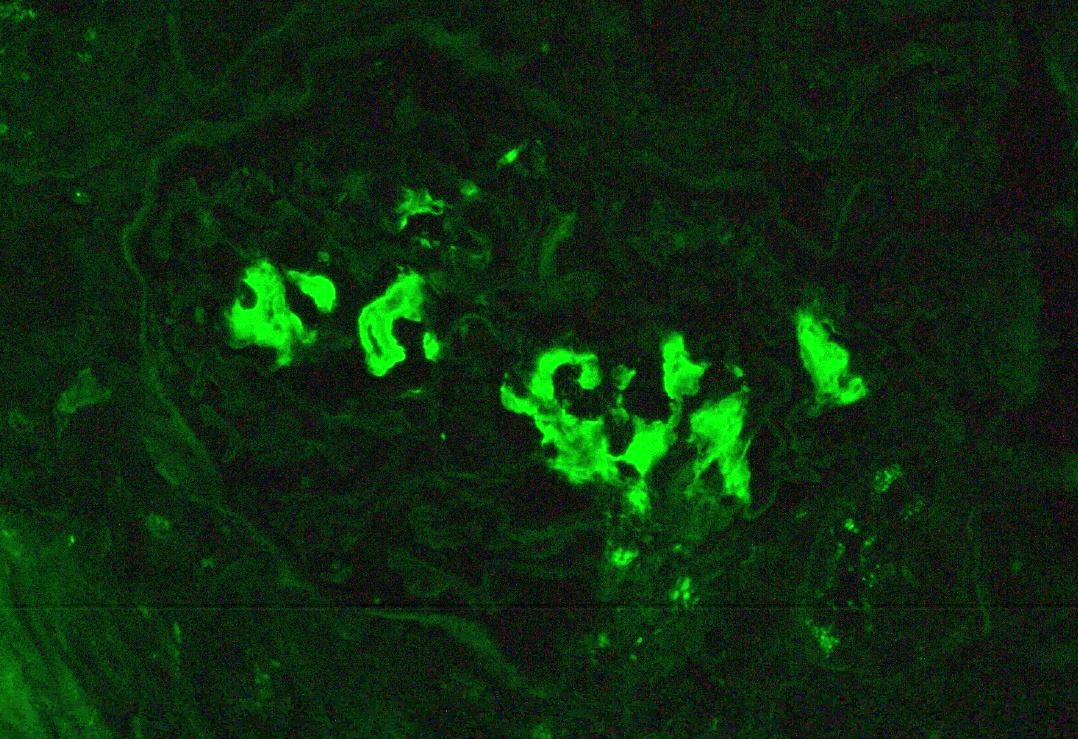
Lambda, glomerulus
Positive stains
Negative stains
- Jones silver: does not typically stain amyloid
Electron microscopy description
- Randomly arranged, nonbranching fibrils with a variable thickness, close to 10 nm
Electron microscopy images
Contributed by Anthony Sisk, D.O.
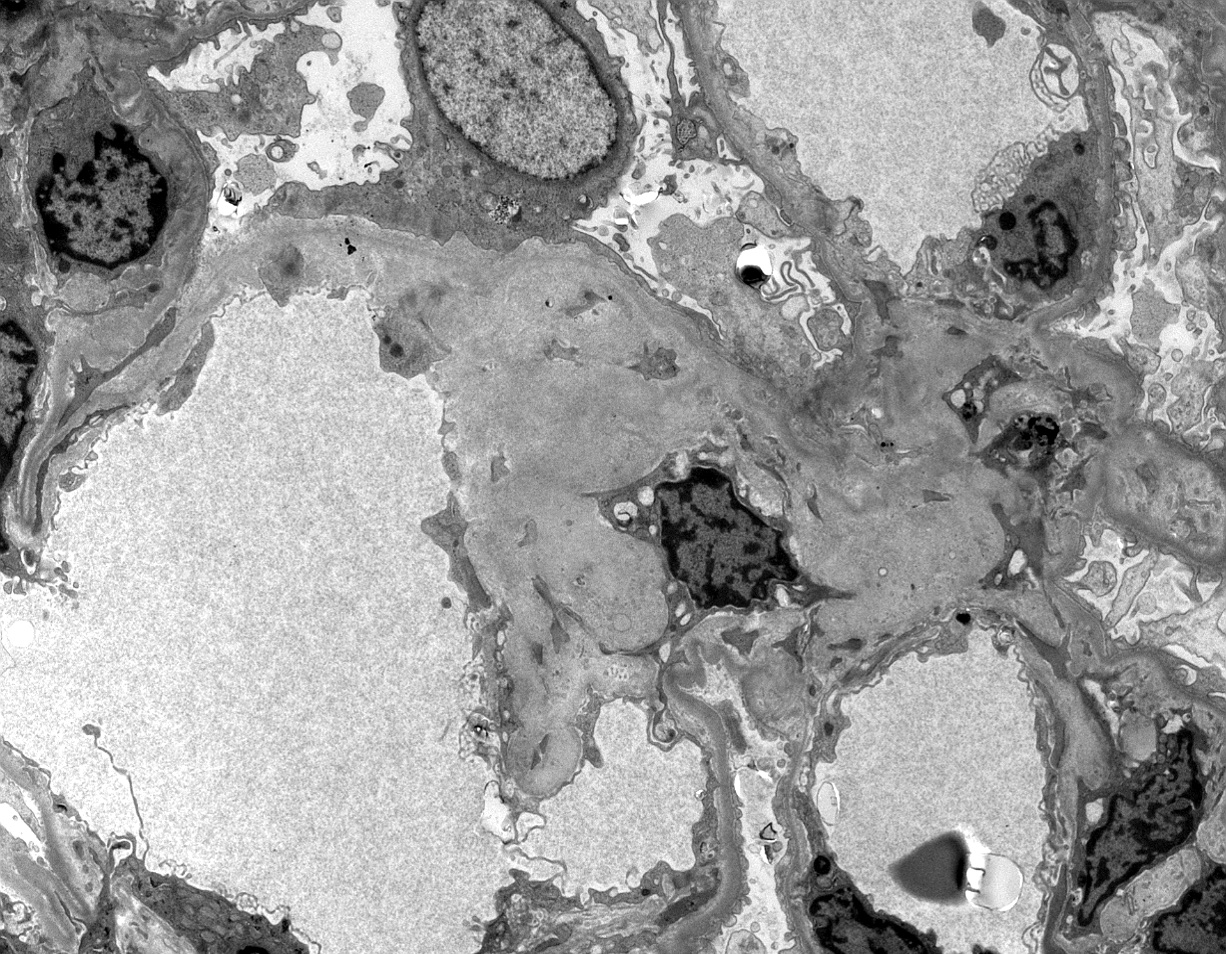
Glomerular involvement
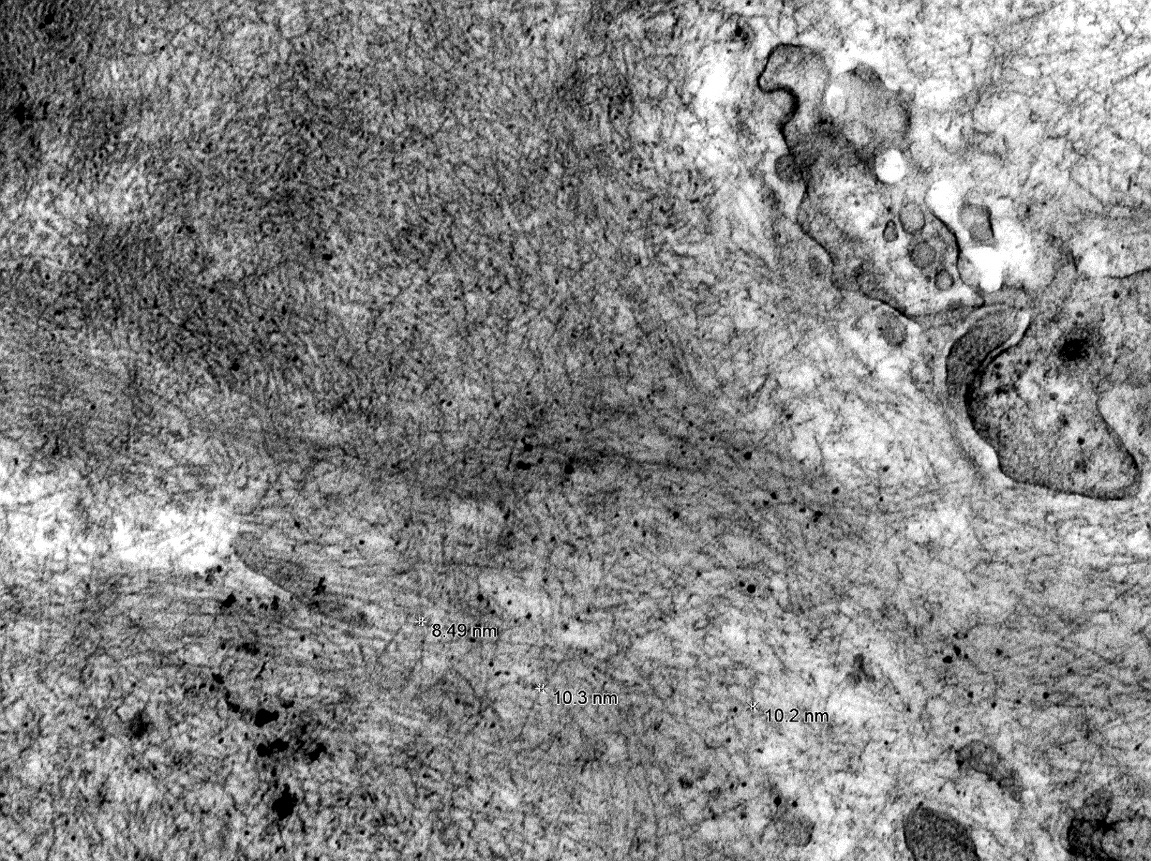
Fibril measurements
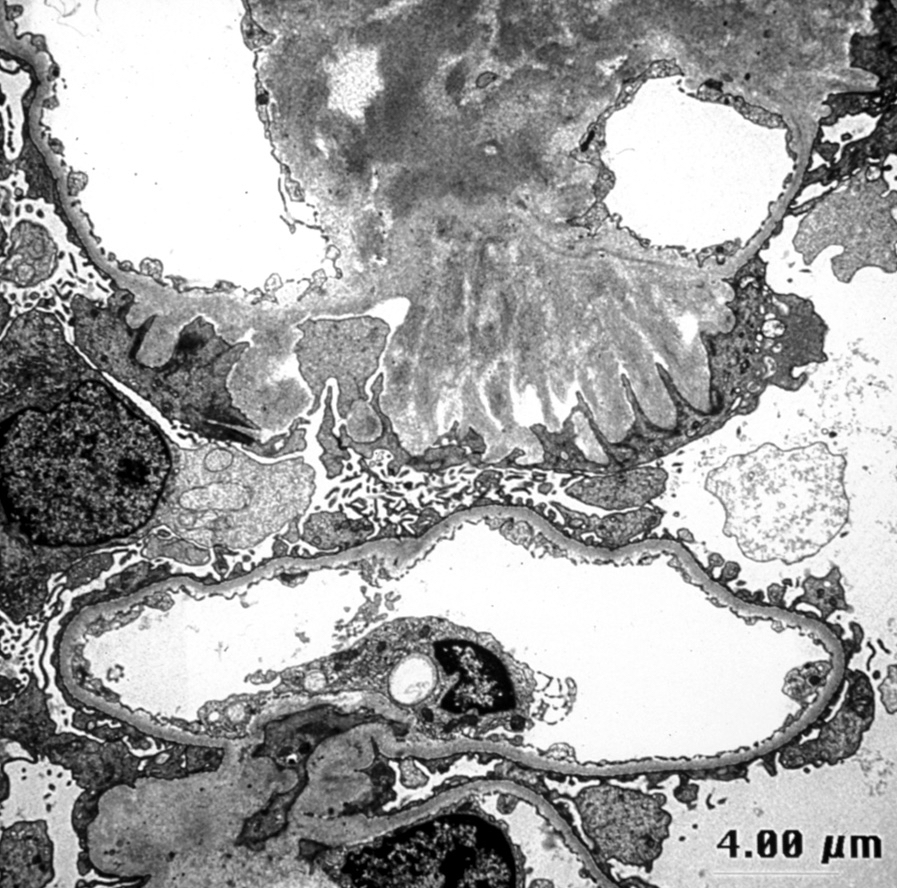
Spicule formation
Genetics
- Mostly acquired with rare inherited forms
- Hereditary forms
- All show an autosomal dominant inheritance
- Transthyretin gene mutation most common - Val30Met seen with renal involvement (Nephron 2022 Mar 18 [Epub ahead of print])
- Other gene mutations include fibrinogen alpha chain, apolipoprotein A1 and A2
Sample pathology report
- Kidney, native, core biopsy:
- Amyloidosis, lambda restricted (AL type), predominantly glomerular
- No significant interstitial fibrosis / tubular atrophy
- Congo red stain confirms amyloid deposits
Differential diagnosis
- Fibrillary glomerulonephritis:
- Immunotactoid glomerulonephritis:
- Extremely rare, microtubules of 20 - 50 nm
- Diabetic nephropathy:
- Strong mesangial silver stain, no well formed fibrils
Board review style question #1
A 73 year old man presents with massive proteinuria. What is the most likely diagnosis based on the image above?
- AL type amyloidosis
- ALECT amyloidosis
- Membranous nephropathy
- Rheumatoid arthritis
Board review style answer #1
Board review style question #2
Which of the following is the typical presentation for ALECT2 amyloidosis?
- Microscopic hematuria
- Nephrotic syndrome and renal failure
- Slow decline in renal function
- Sudden nephrotic range proteinuria
Board review style answer #2
