

Kidney nontumor / medical renal
Vascular disease
Thrombotic microangiopathy and acute endothelial injury
Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura
Editorial Board Members: Jonathan E. Zuckerman, M.D., Ph.D., Nicole K. Andeen, M.D.


Last author update: 15 March 2024
Last staff update: 15 March 2024
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear images | Immunofluorescence description | Immunofluorescence images | Positive stains | Electron microscopy description | Electron microscopy images | Genetics | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Caballero C, Bruner ET. Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyhusttp.html. Accessed April 17th, 2024.
Definition / general
- Thrombotic microangiopathy (TMA) is a pathologic term used to describe thrombi within microvasculature; TMA is clinically characterized by ischemic organ injury, particularly acute kidney injury, thrombocytopenia and microangiopathic hemolytic anemia (MAHA)
- TMA is a heterogeneous disease that is classically seen in hemolytic uremic syndrome and thrombotic thrombocytopenic purpura but can also be seen in a variety of clinical settings including severe hypertension, scleroderma renal crisis and preeclampsia / eclampsia, among others (Semin Diagn Pathol 2020;37:121)
- Classic / typical hemolytic uremic syndrome (HUS) is an infection mediated process caused by Shiga toxin producing bacteria most commonly Escherichia coli O157:H7 or Shigella dysenteriae; numerous other infections have also been associated with TMA
- Atypical HUS is due to an inherited or acquired abnormality in complement proteins causing dysregulation of the alternative pathway
- Thrombotic thrombocytopenic purpura (TTP) is an inherited or acquired deficiency of ADAMTS13 enzyme which normally cleaves large multimers of von Willebrand factor (vWF)
Essential features
- Endothelial injury with microvascular thrombi
- Correlation with clinical and laboratory data necessary to determine etiology
- Often due to infection, complement disorders or ADAMTS13 deficiency
- Many secondary causes including severe hypertension, sclerodermal renal crisis, preeclampsia / eclampsia
Terminology
- Infection associated TMA, Shiga toxin producing Escherichia coli (STEC HUS), postdiarrheal HUS
- Complement mediated atypical hemolytic uremic syndrome (aHUS), complement mediated TMA (CM TMA), diarrhea negative HUS
- ADAMTS13 mediated TMA
ICD coding
- ICD-10
- D50 - D89 - diseases of the blood and blood forming organs and certain disorders involving the immune mechanism
- D59.3 - hemolytic uremic syndrome
- D59.30 - hemolytic uremic syndrome, unspecified
- D59.31 - infection associated hemolytic uremic syndrome
- D59.32 - hereditary hemolytic uremic syndrome
- D59.39 - other hemolytic uremic syndrome
Epidemiology
- HUS is most common in infants and young children, peak age 3 - 5 years
- Streptococcus pneumoniae HUS (SP HUS) usually affects younger children than STEC HUS (Pediatr Clin North Am 2019;66:235)
- aHUS and TTP are more common in adults
- ~10% of TTP occurs in children
- Most patients with aHUS are < 60 years of age (Am J Kidney Dis 2023;81:591)
- HUS affects men and women equally
- TTP: F > M
Sites
- HUS / aHUS: kidney, followed by central nervous system
- TTP: strongly associated with central nervous system and cardiac involvement, less commonly kidney involvement; kidney involvement does not exclude TTP (Expert Rev Hematol 2019;12:383)
- Other organ systems can be involved including cardiovascular, respiratory, gastrointestinal
Pathophysiology
- Endothelial injury is fundamental feature of all causes of TMA
- Endothelial cell injury activates platelet aggregation within the microvasculature and forms thrombi
- Results in microangiopathic hemolytic anemia and tissue ischemia / end organ damage
- TTP: von Willebrand factor ultralarge multimers accumulate due to deficiency of ADAMTS13 and promotes platelet adhesion and aggregation (Expert Rev Hematol 2019;12:383)
- aHUS: dysregulation of the alternative pathway with C3b deposition leading to endothelial injury
- STEC: Shiga toxin (Stx), from the intestines, enters circulation via binding with globotriaosylceramide (Gb3) receptors on endothelial cells and inhibits protein synthesis leading to endothelial cell injury
- Kidney has high expression of Gb3 receptors
- Reference: Am J Kidney Dis 2023;81:591
Etiology
- Highly variable, can be primary or secondary
- Primary TMA
- Hereditary: ADAMTS13 mutation, complement gene mutation, cobalamin C (cblC) deficiency, diacylglycerol kinase epsilon (DGKE)
- Acquired: factor H (FH) autoantibody, ADAMTS13 autoantibody
- Secondary TMA
- Pregnancy associated
- Hemolysis, elevated liver enzymes, low platelet count (HELLP)
- Solid organ / bone marrow transplantation
- Severe hypertension
- Drug induced
- Autoimmune (systemic lupus erythematosus [SLE], scleroderma renal crisis [SRC], cryopyrin associated periodic syndromes [CAPS])
- Malignancy associated
- Glomerular diseases (antineutrophilic cytoplasmic antibody [ANCA] associated, immunoglobulin A nephropathy [IgAN], membranous nephropathy [MN], focal segmental glomerulosclerosis [FSGS], complement 3 glomerulopathy [C3G] / membranoproliferative glomerulonephritis [MPGN])
- Infection associated TMA
- STEC HUS
- Undercooked, contaminated beef, unpasteurized milk or contaminated fruits and vegetables
- E. coli OH157:H7 in United States and Europe
- Shigella dysenteriae in other countries
- Pneumococcal
- Human immunodeficiency virus (HIV) associated
- STEC HUS
- Other infections
- Reference: Clin J Am Soc Nephrol 2018;13:300
Diagrams / tables
Images hosted on other servers:

Etiologies of TMA

Diagnostic algorithm for TMA
Clinical features
- HUS: classic clinical triad includes acute kidney injury, anemia and thrombocytopenia
- STEC HUS typically presents with gastrointestinal symptoms followed by thrombocytopenia and acute kidney injury
- aHUS may present in a fulminant manner following a triggering event, such as upper respiratory or gastrointestinal infection; in a smaller number of patients, aHUS presents in an insidious, indolent manner
- TTP: classic pentad includes fever, thrombocytopenia, MAHA, neurological disorders, acute kidney injury (occurs in < 10% of patients) (Expert Rev Hematol 2019;12:383)
- Extrarenal manifestations dependent upon organ(s) involved but can include petechiae, purpura, neurologic symptoms including seizures and coma, pancreatitis, myocardial infarction, hepatitis (Clin J Am Soc Nephrol 2018;13:300)
Diagnosis
- Recognize laboratory findings and investigate underlying disease cause (Am J Kidney Dis 2023;81:591)
- Includes ADAMTS13 activity study
- Screening for Shiga toxin or other infections
- Complement studies
- Evaluation for secondary causes: autoimmune screen, metabolic (vitamin B12, methylmalonic acid [MMA] and homocysteine) pregnancy test, transplant rejection markers (Am J Kidney Dis 2023;81:591)
- Because decreased platelet counts and elevated blood pressures are common at presentation, kidney biopsy is not often performed at onset; it may be performed in more severe cases of acute kidney injury (AKI) but is not required for diagnosis (Clin J Am Soc Nephrol 2018;13:300)
Laboratory
- Anemia
- Thrombocytopenia
- Impaired renal function: oligouria, hematuria or low grade proteinuria; elevated creatinine in more severe cases
- Elevated bilirubin, lactate dehydrogenase
- Reticulocytosis
- Schistocytes
- Low haptoglobin
- Negative Coombs test
- Reference: Clin J Am Soc Nephrol 2018;13:300
Prognostic factors
- STEC HUS has 3 - 5% acute mortality with neurologic, cardiovascular and intrabdominal issues as primary cause of death
- SP HUS is a generally more severe disease than STEC HUS and has worse outcomes
- Long term renal sequelae reported in 25% of patients (Pediatr Clin North Am 2019;66:235)
- Poorer prognosis
- Prolonged anuria
- Increased length of dialysis requirement
- TTP: older age, high lactate dehydrogenase (LDH) and cardiac troponin levels (Expert Rev Hematol 2019;12:383)
- Progression to end stage renal disease (ESRD) in patients with aHUS has declined; 79% patients with aHUS progressed to ESRD or death prior to use of escluzumab (Pediatr Clin North Am 2019;66:235)
Case reports
- 21 year old woman developing aHUS after vaccination with mRNA vaccine against SARS-CoV-2 (Front Immunol 2022;13:1001366)
- 27 year old post partum woman with TTP (Ann Med Surg (Lond) 2022;84:104828)
- 42 year old woman developing aHUS after myomectomy (Case Rep Womens Health 2022;35:e00424)
- 62 year old woman developing TTP after receiving the first dose of the Ad26.COV.S vaccine (Am J Emerg Med 2021;49:441)
- 62 year old man presenting with immune mediated TTP (iTTP) 1 week after diagnosis of COVID-19 (IDCases 2021;26:e01256)
- 65 year old man with gemcitabine induced HUS (Cureus 2022;14:e20926)
Treatment
- Highly dependent upon the underlying etiology of the TMA
- Supportive care, primarily
- Eculizumab (anti-C5 monoclonal antibody) for complement mediated and pregnancy associated aHUS
- Recommended to start within first 24 hours in patients with aHUS, with time to treatment being a significant factor in renal function recovery (Pediatr Clin North Am 2019;66:235)
- Ravulizumab (longer acting anti-C5 antibody)
- Immunosuppression (steroids, rituximab, cyclophosphamide)
- Systemic antibiotic administration for infections
- Therapeutic plasma exchange (TPE) is first line in the setting of TTP (Expert Rev Hematol 2019;12:383)
- Caplacizumab prevents interaction of platelets and vWF and has been approved for acute TTP treatment (Am J Kidney Dis 2023;81:591)
Microscopic (histologic) description
- Acute
- Glomerular and arteriolar fibrin thrombi, red blood cell fragmentation
- Other glomerular changes include endothelial cell swelling, giving the glomerulus a bloodless appearance and mesangiolysis
- Arterial endothelial swelling and intimal mucoid / myxoid change of the intima
- Chronic
- Glomerular basement membrane double contouring (duplication)
- Multilayering of the peritubular capillary basement membrane
- Arterial myointimal hyperplasia (onion skinning)
- Reference: Kidney Res Clin Pract 2022;41:524
Microscopic (histologic) images
Contributed by Evelyn T. Bruner, M.D.
Prominent fibrin thrombi
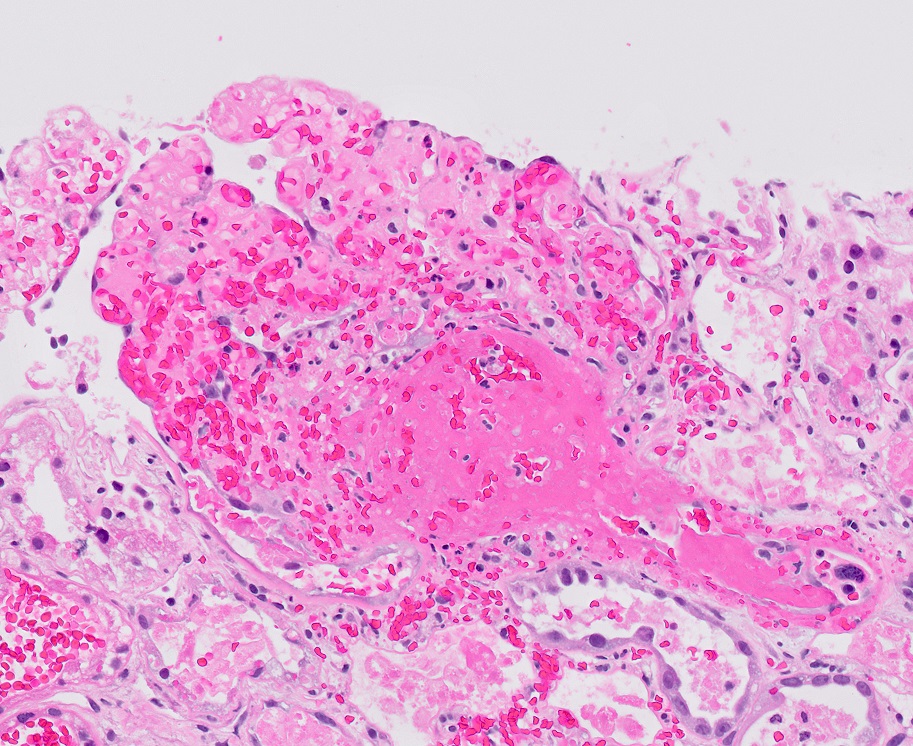
Glomerulus with prominent fibrin thrombi
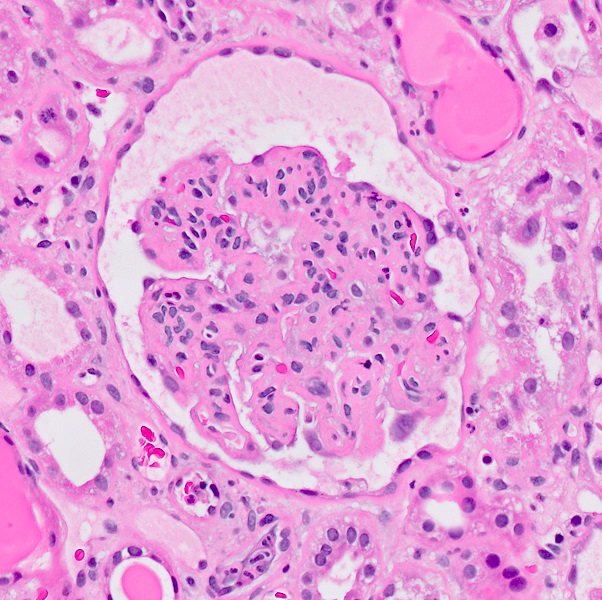
Glomerulus with endothelial swelling
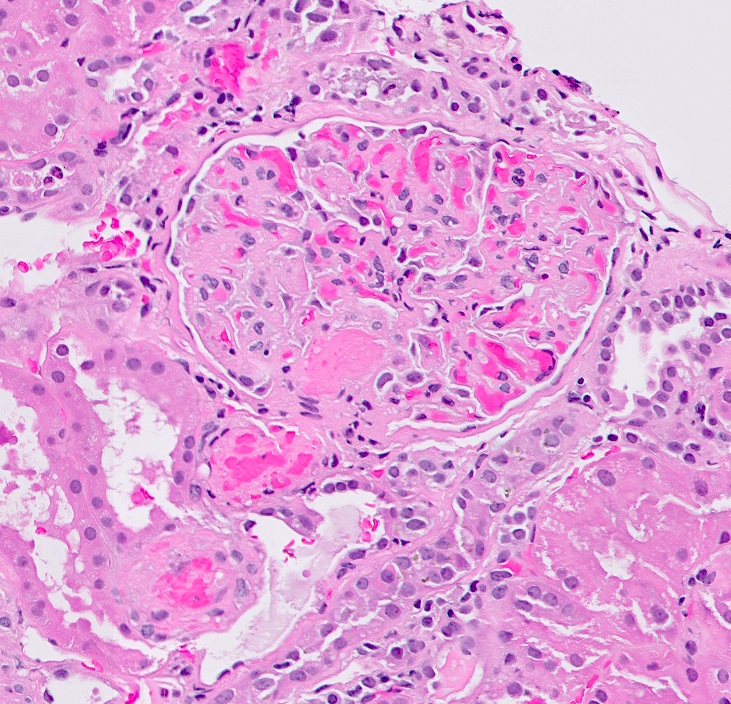
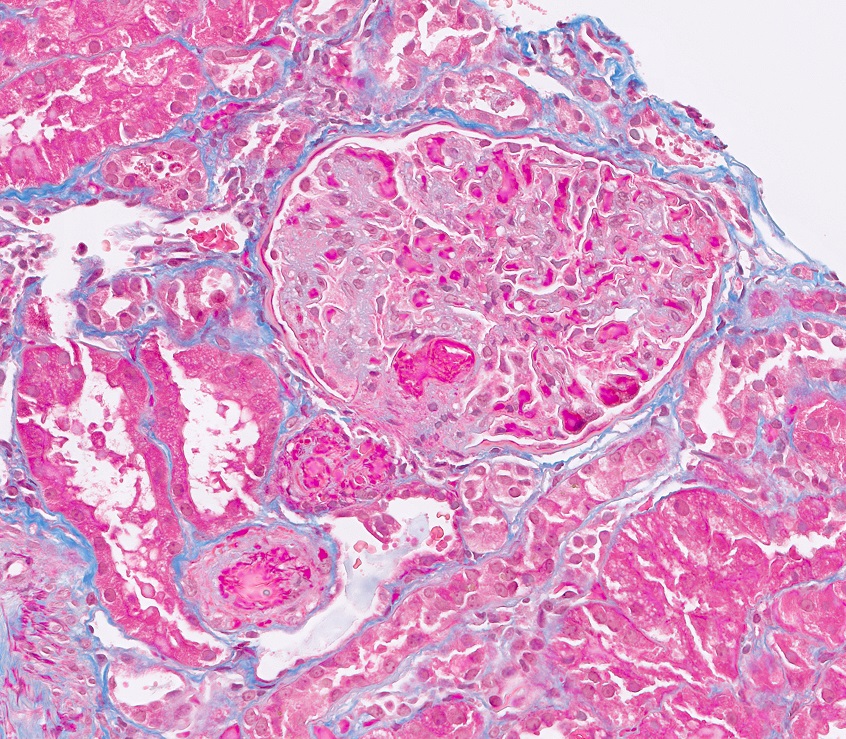
Glomerulus with prominent fibrin thrombi
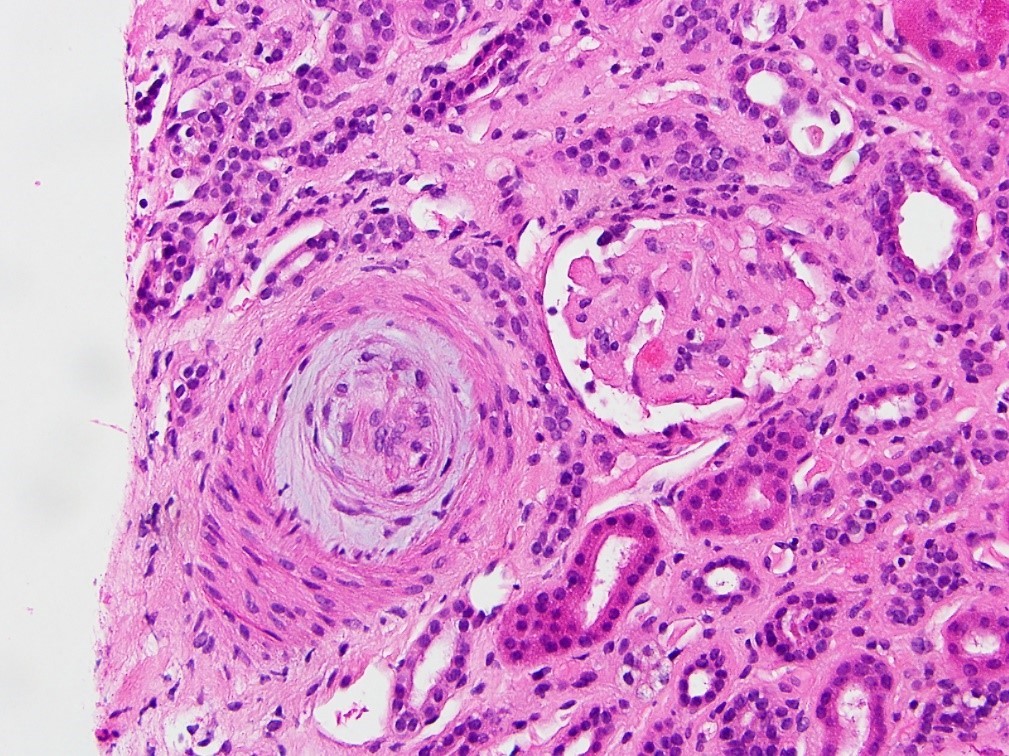
Artery with mucoid intimal thickening
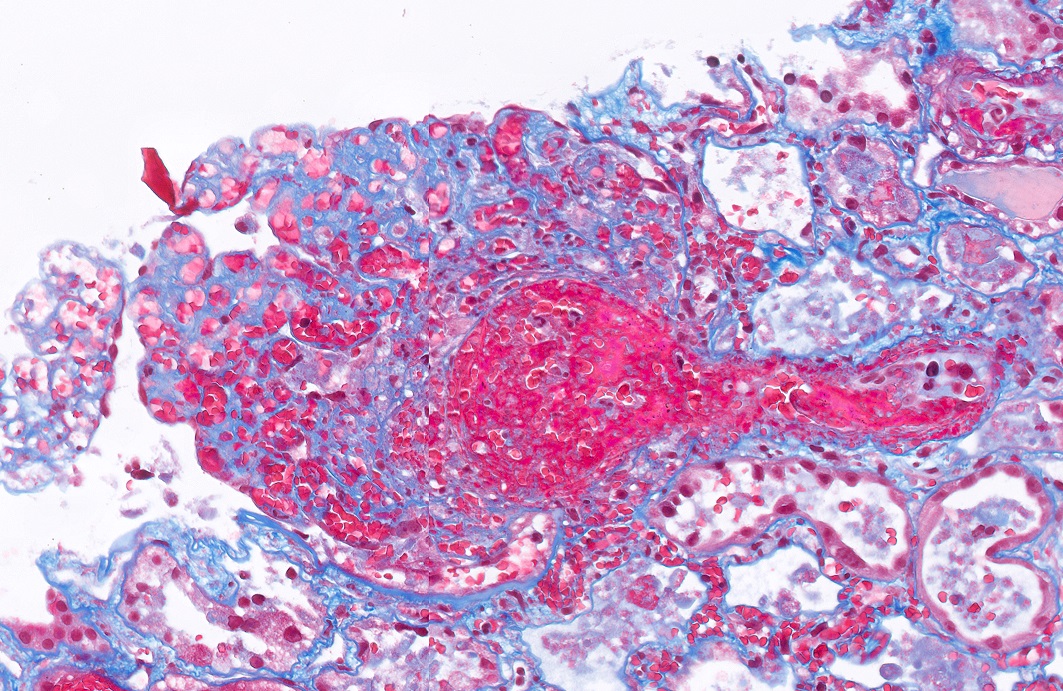
Glomeruli with prominent fibrin thrombi
Peripheral smear images
Contributed by Dr. Avinash Mane (Case #403)
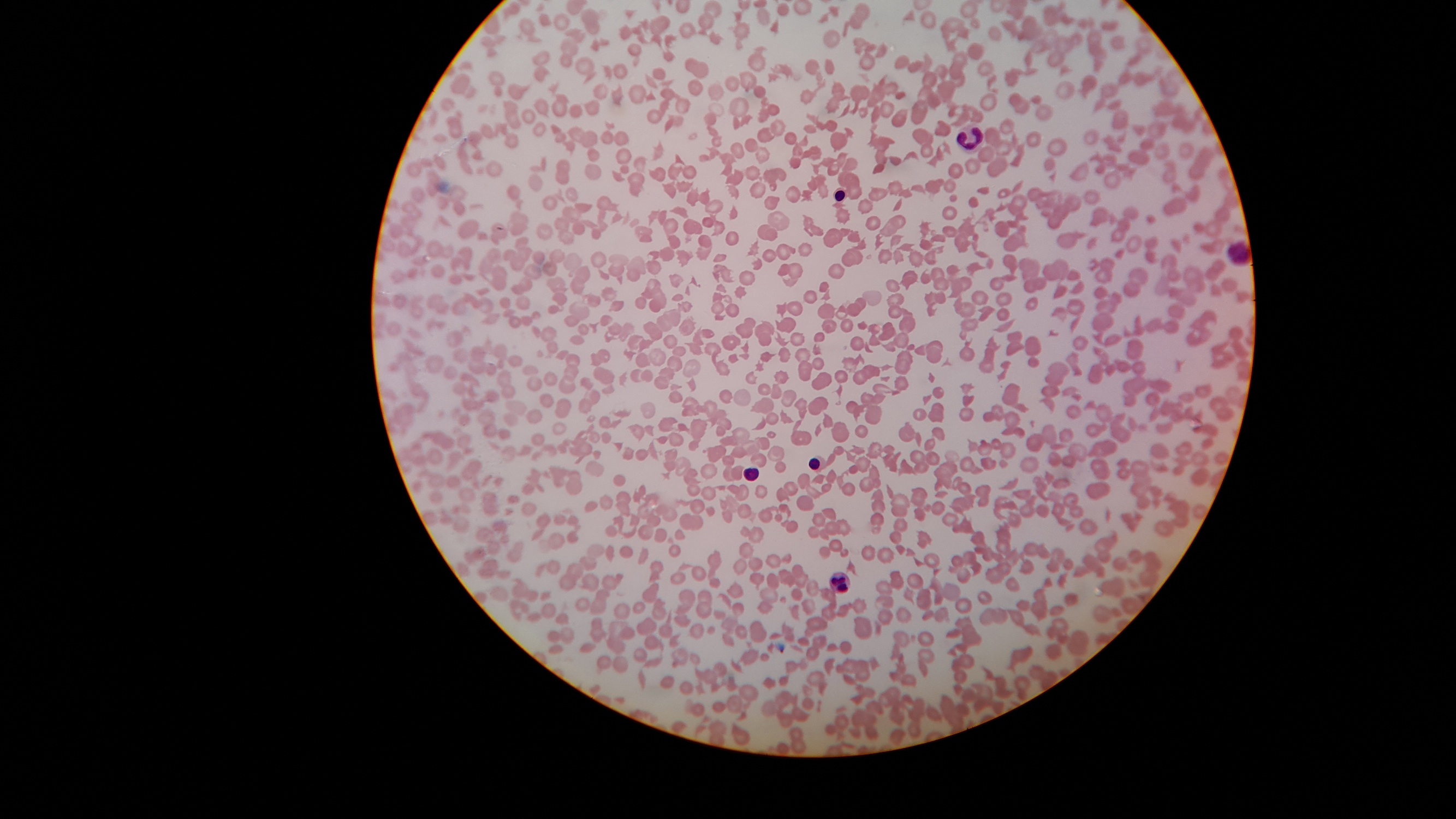
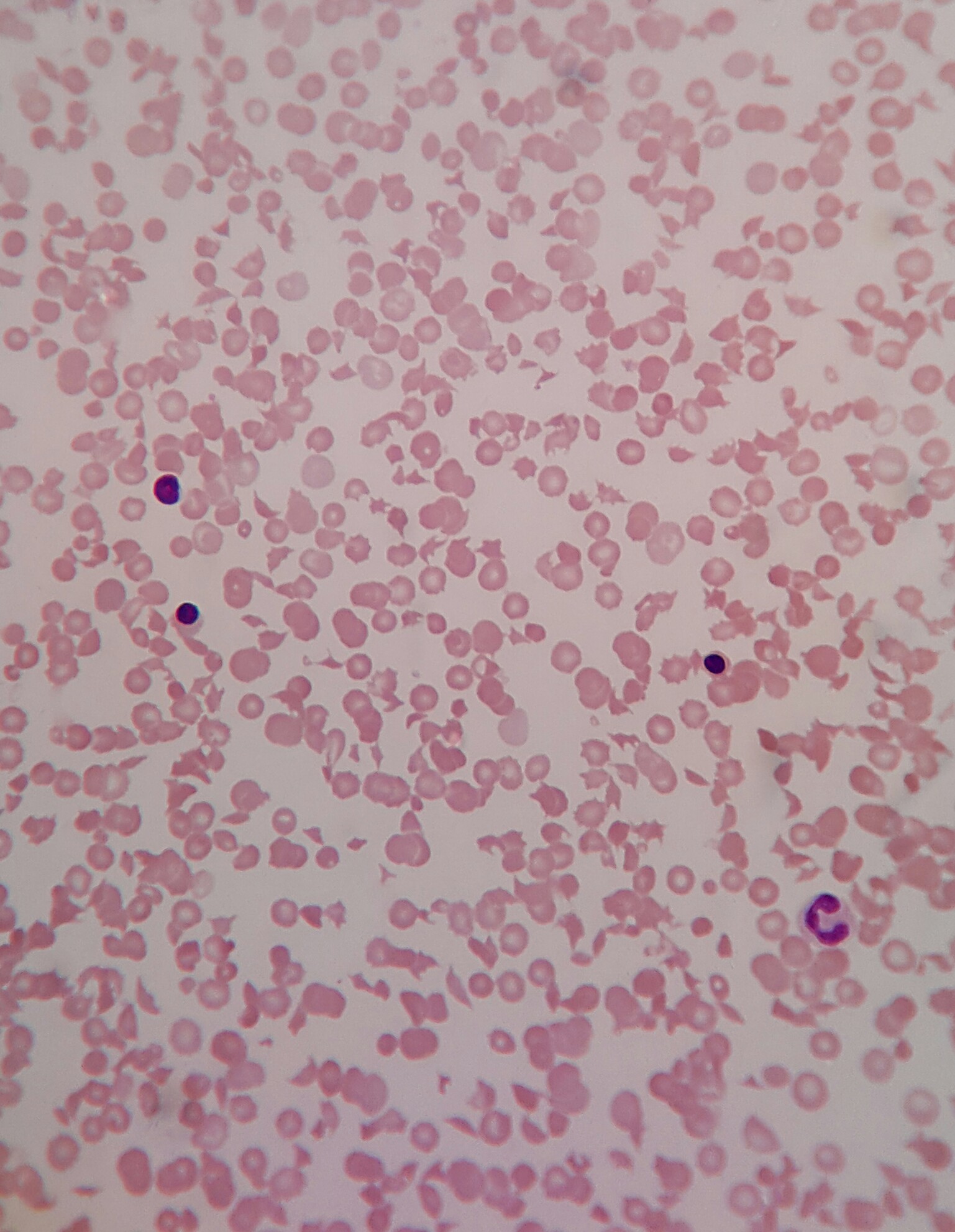
Numerous schistocytes and nucleated RBCs with extremely low platelets on peripheral smear
Immunofluorescence description
- Fibrin / fibrinogen within glomeruli capillaries and arterioles; otherwise, negative aside from nonspecific trapping of immunoglobulins and complement
Immunofluorescence images
Contributed by Evelyn T. Bruner, M.D.
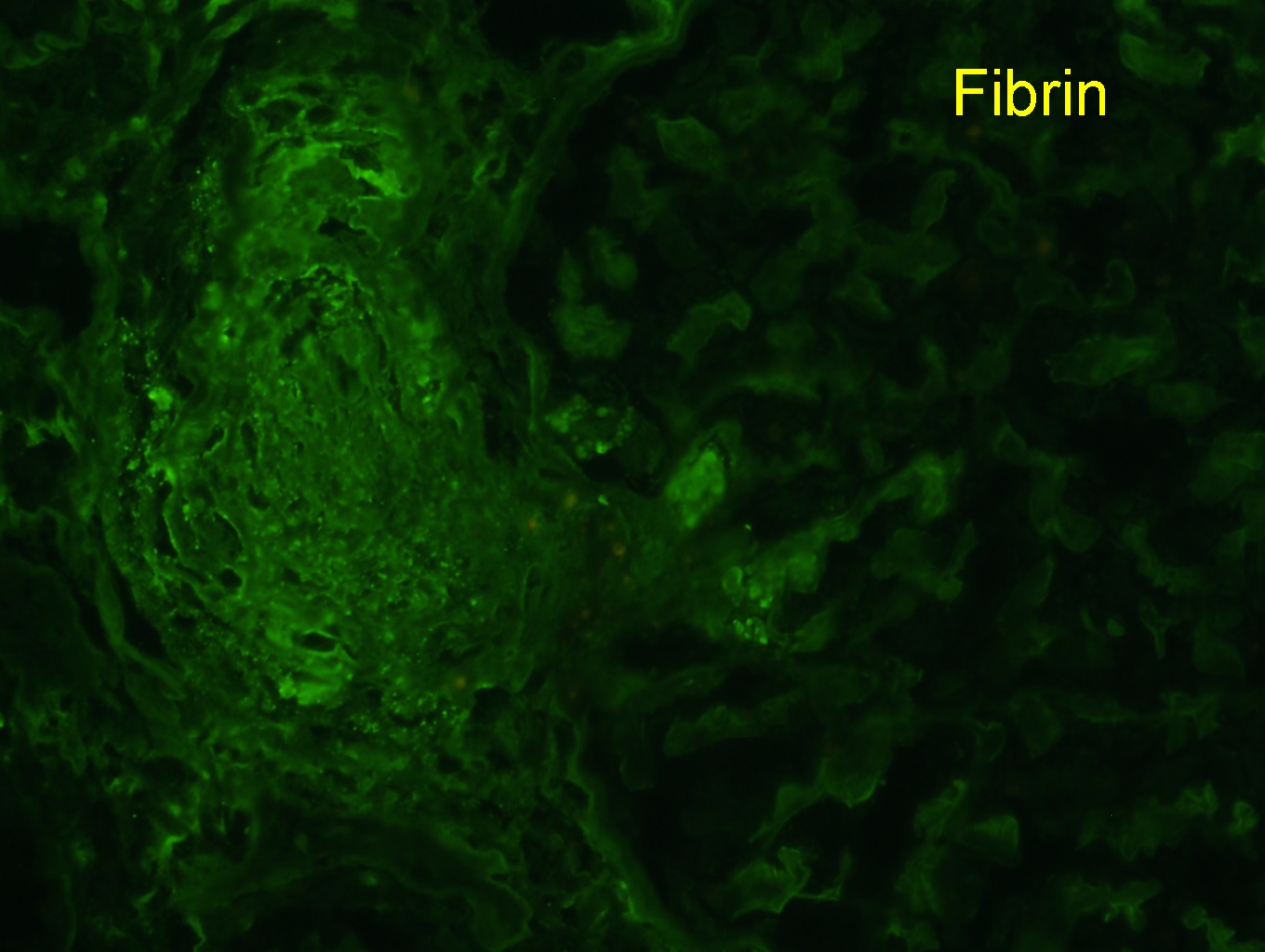
Fibrin
immunofluorescence
Positive stains
- Fibrin thrombi are best identified with H&E, trichrome and Jones silver stains
Electron microscopy description
- Subendothelial widening may be seen in early stage with acellular subendothelial fibrillary material (J Clin Med 2022;11:3124)
- Glomerular capillary microthrombi composed of fibrin tactoids, platelets
- Red blood cell (RBC) fragmentation
- Reduplication of the glomerular basement membrane in chronic TMA
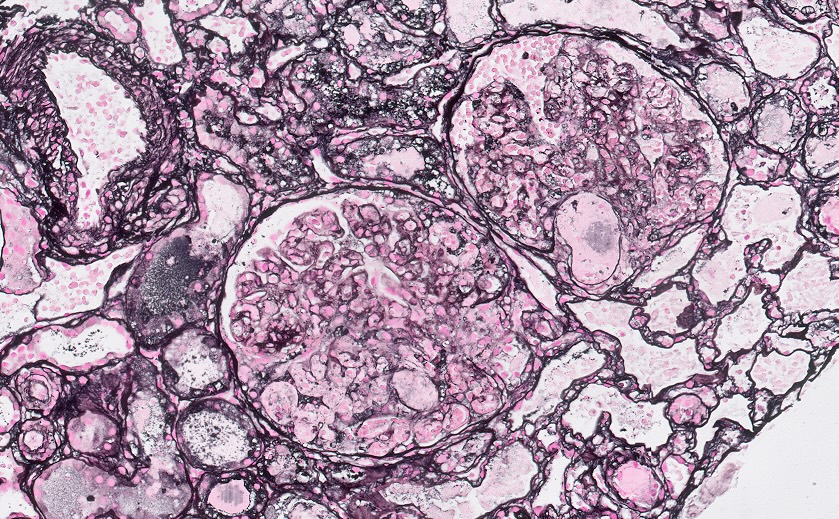
Electron microscopy images
Contributed by Evelyn T. Bruner, M.D.
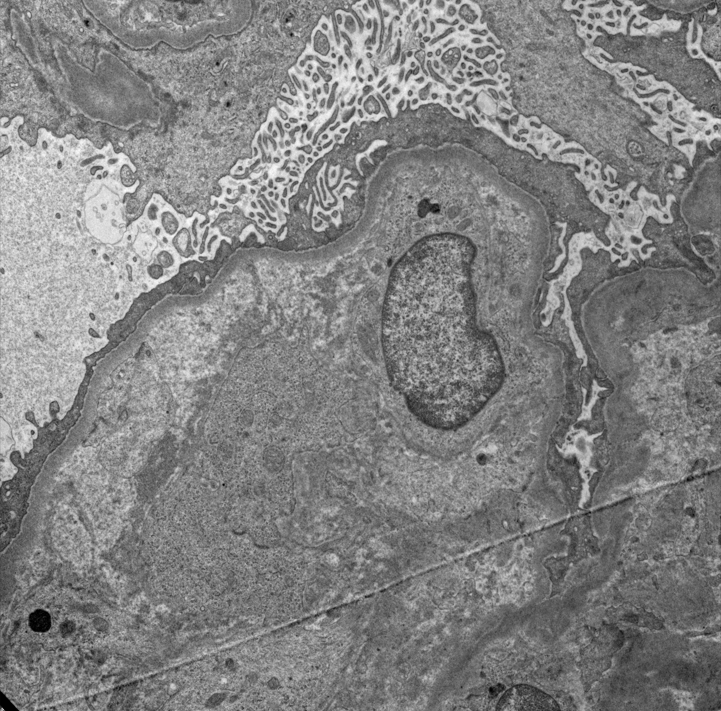
Endothelial swelling and lucency
Genetics
- > 50 - 60% of aHUS cases are due to genetic variants in genes such as CFH, CFI, MCP / CD46, C3, CFB, CFH::CFHR, THBD, DGKE; incomplete penetrance (Am J Kidney Dis 2023;81:591)
- Variants in the ADAMTS13 gene can cause congenital TTP (Am J Kidney Dis 2023;81:591)
Sample pathology report
- Kidney, core biopsy:
- Thrombotic microangiopathy (see comment)
- Comment: The biopsy findings show fibrin thrombi within the microvasculature compatible with thrombotic microangiopathy. Determination of the underlying etiology requires clinical and laboratory correlation.
Differential diagnosis
- Malignant hypertension:
- Clinically severe hypertension, usually a pre-existing condition
- Mucoid intimal thickening / onion skinning of arteries, fibrinoid necrosis
- Systemic sclerosis:
- Systemic disease
- Mucoid intimal thickening / onion skinning of arteries
- Antinuclear antibody (ANA)
- Antiphospholipid antibody syndrome:
- Positive lupus anticoagulant or anticardiolipin antibody
- Drug induced TMA:
- Immune and nonimmune mediated
- Careful medication history
- Pregnancy associated TMA:
- Typically in the setting of preeclampsia, HELLP syndrome
Board review style question #1
A 50 year old patient presents with acute kidney injury and a biopsy is performed. A representative glomerulus is shown in the H&E image above. What is the most important laboratory test in the initial workup of this patient?
- ADAMTS13 activity
- Antinuclear antibody (ANA)
- Bacterial culture
- Full complement evaluation
- Imaging workup for malignancy
Board review style answer #1
A. ADAMTS13 activity. While all the above may aid in determining the underlying etiology of the thrombotic microangiopathy (TMA), the ADAMTS13 activity is initially critical in ruling out thrombotic thrombocytopenic purpura and to help guide treatment. An activity level of < 10% confirms the diagnosis of thrombotic thrombocytopenic purpura (TTP). Answer B is incorrect because ANA would be seen in a lupus associated TMA. Answer C is incorrect because positive bacterial culture is infection associated. Answer D is incorrect because a complement abnormality would be seen in atypical hemolytic uremic syndrome (aHUS). Answer E is incorrect because imaging could assess for malignancy associated TMA but would not be ideal for initial workup.
Comment Here
Reference: Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura
Comment Here
Reference: Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura
Board review style question #2
A biopsy is performed on a patient with suspected chronic thrombotic microangiopathy (TMA). What is the most supportive finding by electron microscopy?
- Basketweave appearance of the glomerular basement membrane
- Diffuse foot process effacement
- Mesangial electron dense deposits
- Reduplication of the glomerular basement membrane
- Tubuloreticular inclusions
Board review style answer #2
D. Reduplication of the glomerular basement membrane (GBM). Reduplication of the GBM is supportive of chronic thrombotic microangiopathy (TMA). Answer A is incorrect because a basketweave appearance of the GBM would be seen in Alport syndrome. Answer B is incorrect because diffuse foot process effacement is seen in minimal change disease. Answer C is incorrect because mesangial electron dense deposits are seen in IgA nephropathy. Answer E is incorrect because tubuloreticular inclusions are seen in lupus nephritis and viral infections (human immunodeficiency virus [HIV], cytomegalovirus [CMV]).
Comment Here
Reference: Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura
Comment Here
Reference: Hemolytic uremic syndrome / thrombotic thrombocytopenic purpura
