

Kidney nontumor / medical renal
Tubulointerstitial disease
Genetic
Oxalosis
Editorial Board Member: Nicole K. Andeen, M.D.
Editor-in-Chief: Debra L. Zynger, M.D.

Last author update: 9 February 2022
Last staff update: 10 February 2022
Copyright: 2019-2024, PathologyOutlines.com, Inc.
PubMed Search: Oxalosis[TI] pathology full text[sb]
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Immunofluorescence description | Immunohistochemistry & special stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Zuckerman JE. Oxalosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyoxalosis.html. Accessed April 17th, 2024.
Definition / general
- Excess oxalate accumulation in the kidney resulting in acute and chronic tubulointerstitial injury
- May be primary genetic (primary hyperoxaluria) resulting in oxalate overproduction or secondary due to excess intake or decreased clearance of oxalate
Essential features
- Detection of excess calcium oxalate crystals in the kidney
- Crystals are rhomboid / fan shaped and polarizable
Terminology
- Primary: hyperoxaluria, hyperoxalosis, primary hyperoxaluria
- Secondary: oxalate nephropathy, oxalosis
ICD coding
Epidemiology
- Primary:
- Autosomal recessive
- Prevalence is < 3:1,000,000 individuals worldwide (J Am Soc Nephrol 2015;26:2559)
- 1 - 2% of all children with end stage renal disease (N Engl J Med 2013;369:649, Clin J Am Soc Nephrol 2012;7:458)
- 3 types (Nat Rev Nephrol 2012;8:467)
- Type 1 is the most common (~80%) and most severe form
- Type 2 (~10%)
- Type 3 (~10%)
- Secondary:
- 5 - 24% of all patients with gastrointestinal diseases associated with malabsorption including those with a history of (Nephrol Dial Transplant 2016;31:375)
- Bariatric surgery
- Celiac
- Crohn's
- Pancreatic insufficiency
- Cystic fibrosis
- 5 - 24% of all patients with gastrointestinal diseases associated with malabsorption including those with a history of (Nephrol Dial Transplant 2016;31:375)
Sites
- Primary:
- Insoluble calcium oxalate salts deposit primarily in the kidneys
- Other sites include retina, myocardium, vascular walls, skin, bone and central nervous system (Kidney Int 2009;75:1264)
- Secondary:
- Kidney, both native and allograft
Pathophysiology
- Primary:
- Defective glyoxylate metabolism in hepatocytes resulting in excessive oxalate production
- Oxalate is renally cleared as calcium salts
- Excess oxalate results in oversaturation leading to crystallization in the renal tubules
- Crystals cause direct renal tubular toxicity and obstruction resulting in acute tubular injury and chronic renal failure
- Urolithiasis may also occur
- Secondary:
- Mechanism of renal injury is similar to primary
- Excess intake of oxalate results in increased deposition within the kidney, usually mild
- Enteric hyperoxaluria (Nephrol Dial Transplant 2016;31:375):
- Fat malabsorption results in increased calcium sequestration in the gut resulting in increased oxalate absorption from the small intestine and, to some degree, colon
- Free fat and bile acids may also contribute to colonic permeability of oxalate
- Oxalate degrading bacteria such as Oxalobacter formigenes may be a factor (Kidney Int 2013;83:1144)
Etiology
- Primary:
- 3 most common subtypes are caused by mutations in the AGXT (type 1), GRHPR (type 2) and HOGA1 (type 3) genes
- Secondary:
- Increased oxalate intake of foods or supplements rich in oxalate (Curr Rheumatol Rep 2013;15:340)
- Rhubarb
- Parsley
- Spinach
- Black tea
- Vitamin C (an oxalate precursor)
- Ethylene glycol toxicity
- Increased gut absorption of oxalate in malabsorptive states (enteric hyperoxaluria)
- Bariatric surgery
- Inflammatory bowel disease (Crohn's disease)
- Celiac disease
- Pancreatic insufficiency
- Short gut syndrome
- Increased oxalate intake of foods or supplements rich in oxalate (Curr Rheumatol Rep 2013;15:340)
Clinical features
- Acute renal failure
- Chronic renal failure
- Recurrent nephrolithiasis
- In primary disease, clinical presentation and disease course is quite variable (Am J Nephrol 2005;25:290)
- Median age at symptom onset is ~6 years
- Onset reported to range from birth to > 50 years
Diagnosis
- Renal biopsy demonstrating excess oxalate accumulation with compatible clinical history
- Presence of characteristic molecular findings in hereditary forms
Laboratory
- Urinalysis / stone analysis with oxalate stones
- Molecular testing
Prognostic factors
- Primary:
- Type 1 is most severe form
- Patients with the Gly170Arg or Phe152Ile mutation in AGXT gene have better overall outcome due to sensitivity to pyridoxine (Kidney Int 2005;67:1704)
- Type 2 has less severe course
- Type 3 is least severe; nephrocalcinosis and chronic kidney failure are uncommon (N Engl J Med 2013;369:649)
- Type 1 is most severe form
- Secondary (Kidney Int Rep 2018;3:1363):
- Prognosis is guarded with up to 58% progressing to end stage renal disease
- Complete recovery of renal function is rare
Case reports
- 3 month old boy with hyperoxaluria type 1 and autosomal dominant polycystic kidney diesease (Pediatr Radiol 2011;41:107)
- 4 year old girl with end stage renal disease and primary hyperoxaluria type 1 (Pediatr Dev Pathol 2009;12:229)
- 51 year old man with hyperoxaluria nephropathy after Roux-en-Y bypass induced by vitamin C (Oxf Med Case Reports 2016;2016:omw054)
- 57 year old woman with oxalate nephropathy following vitamin C intake (Clin Nephrol 2017;88:354)
- 65 year old woman with oxalate rich green smoothie juice cleanse causing acute oxalate nephropathy (Am J Kidney Dis 2018;71:281)
- 67 year old man with calcium oxalate crystal related kidney injury after Roux-en-Y hepaticojejunostomy (BMC Nephrol 2017;18:106)
- 70 year old woman with calcium oxalate nephropathy due to short bowel syndrome and cholecystectomy (Case Rep Nephrol Dial 2018;8:147)
- Acute kidney injury due to oxalate nephropathy after chelating therapy with oral ethylenediamine tetra acetic acid (Am J Kidney Dis 2017;70:722)
- Acute oxalate nephropathy due to pancreatic atrophy in newly diagnosed pancreatic carcinoma (Hum Pathol 2016;48:163)
Treatment
- Primary:
- Hydration
- Pyridoxine supplementation for type 1 (N Engl J Med 2013;369:649)
- Combined liver / kidney transplant (inevitable recurrence without liver transplantation in type 1 disease)
- Secondary:
- Elimination of sources of excess oxalate
- Enteric oxalosis (Nephrol Dial Transplant 2016;31:375):
- Oxalate binding agents, calcium
- Calcium supplementation
- Citrate supplements
- Hemodialysis
- Otherwise supportive care
Microscopic (histologic) description
- Specific findings:
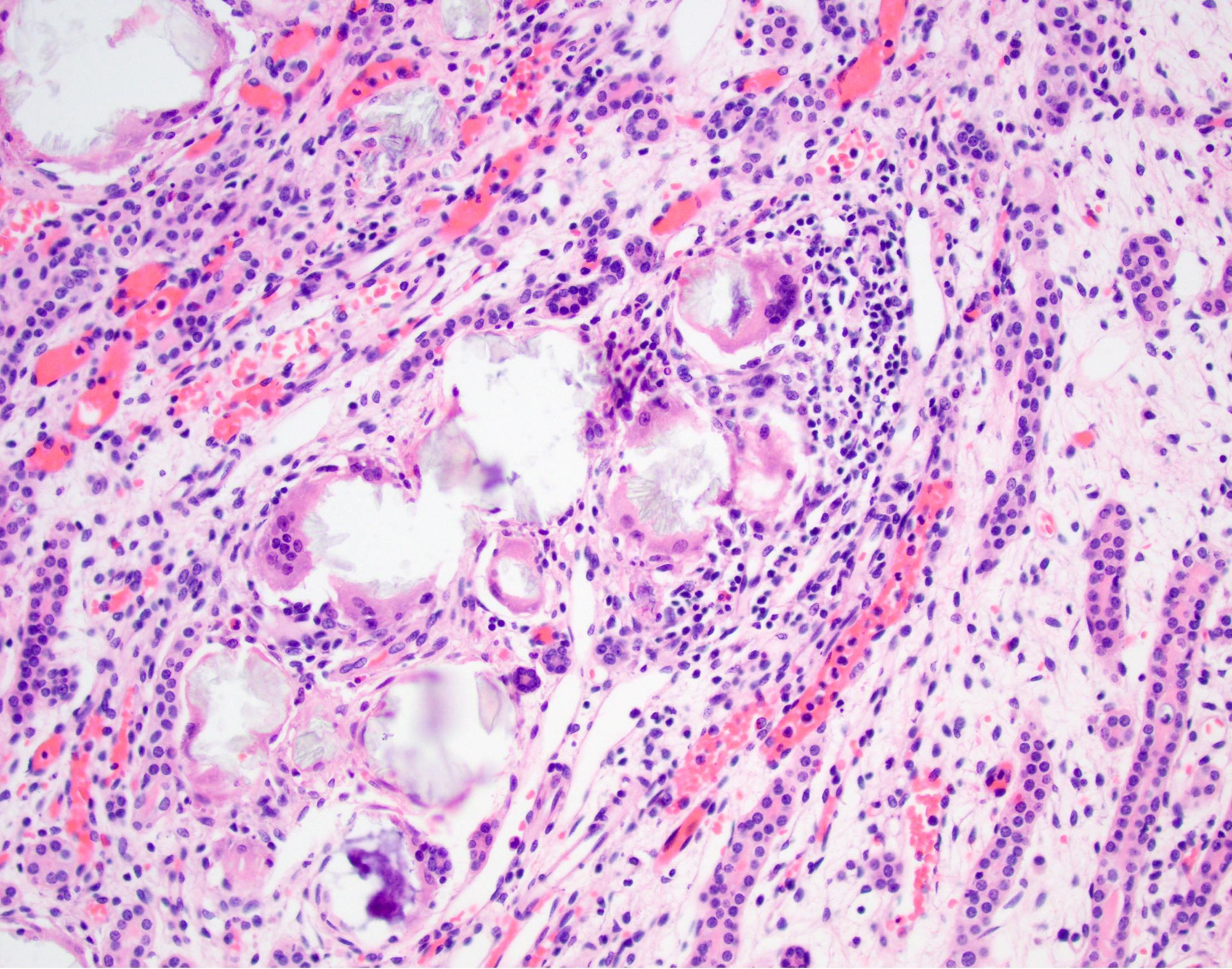
- Translucent polyhedral, rhomboid and fan-like calcium oxalate crystals found within cortical and medullary tubular lumens and interstitial spaces
- Crystals are birefringent under polarized light
- Crystal deposition can be associated with giant cell reaction
- Occasionally arterial / arteriolar deposits (usually in cases of primary hyperoxaluria)
- Advanced primary hyperoxaluria generally shows massive oxalate deposition with extensive interstitial fibrosis / tubular atrophy
- Nonspecific findings:
- Acute tubular injury
- Interstitial fibrosis and tubular atrophy
- Generally mild tubulointerstitial inflammation
- Early primary hyperoxaluria difficult to distinguish from secondary causes
Microscopic (histologic) images
Contributed by Jonathan E. Zuckerman, M.D., Ph.D.
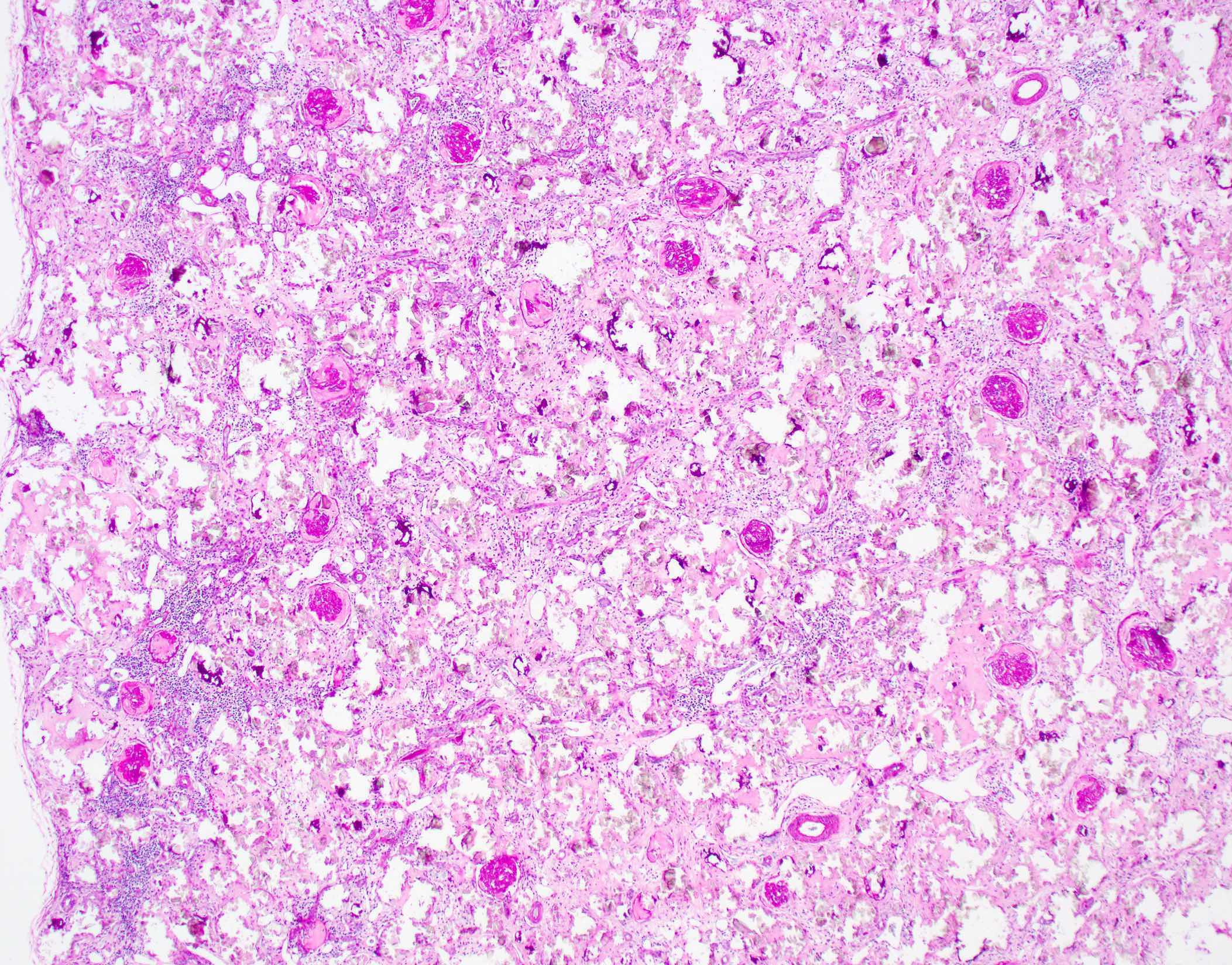

Type 1 primary hyperoxaluria
Secondary oxalosis: calcium phosphate deposition
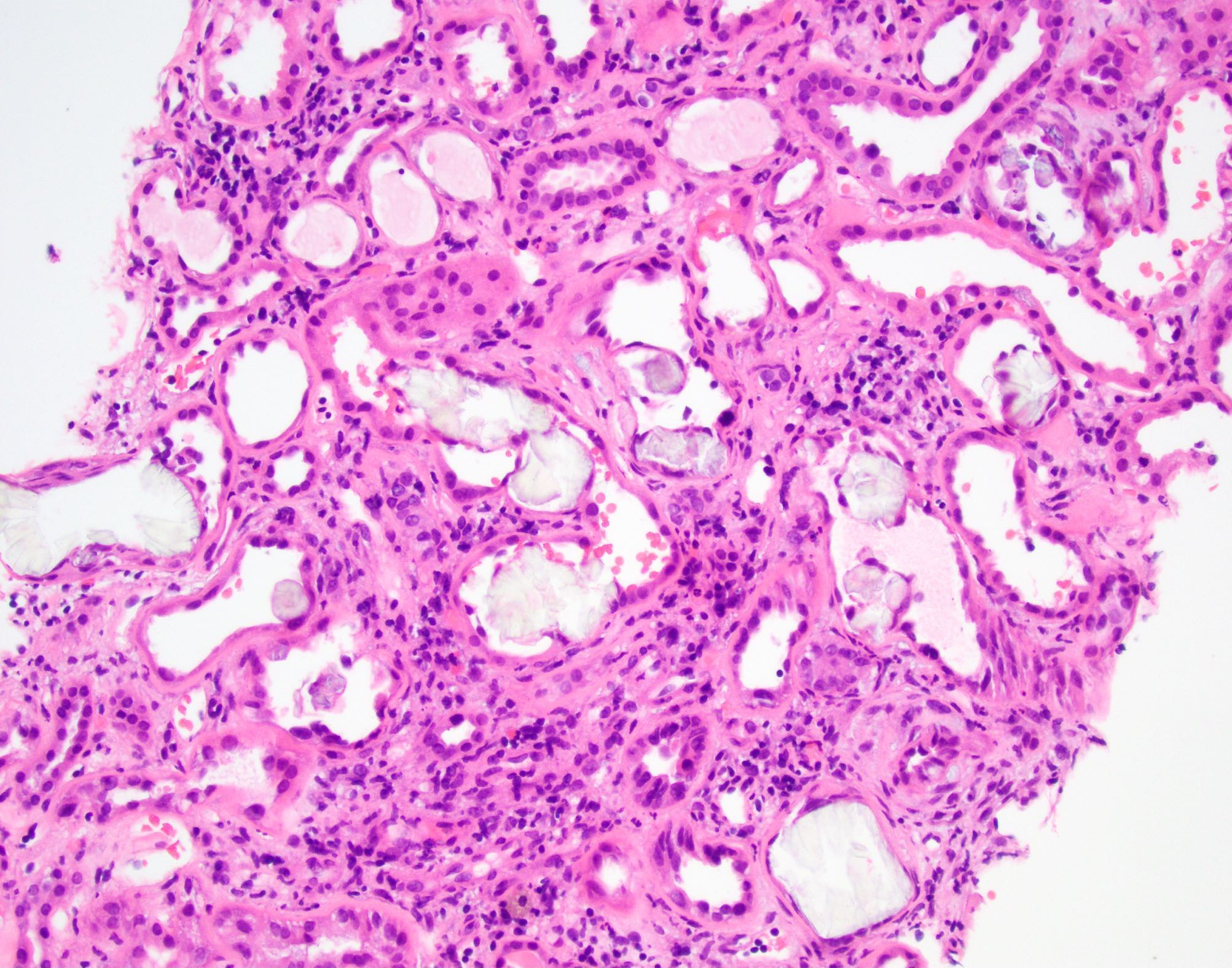

Secondary oxalosis: intraluminal calcium phosphate deposition

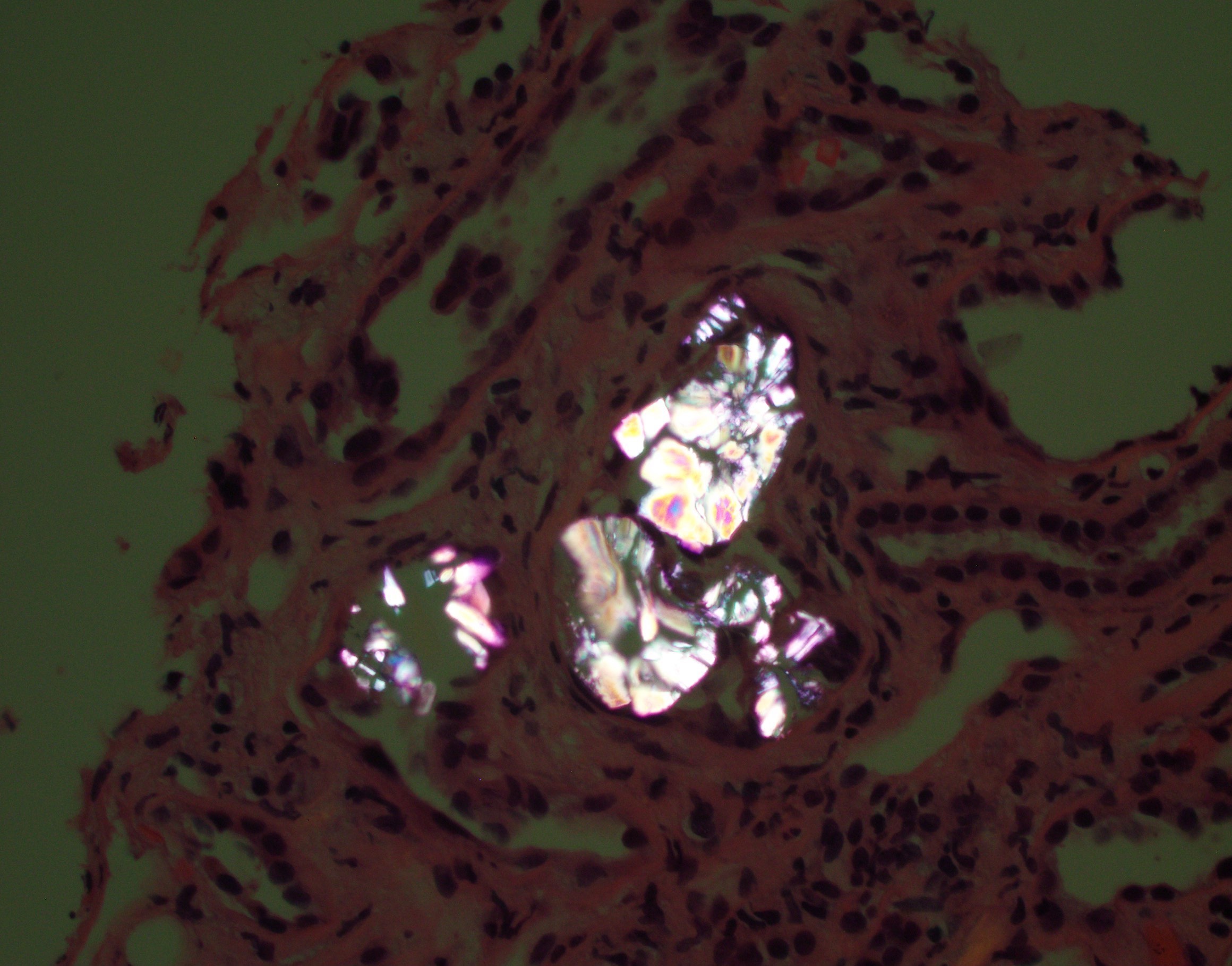
Fan shaped calcium oxalate deposits
Immunofluorescence description
- No immunofluorescence staining of oxalate depositions
- Usual nonspecific background IgA and light chain staining of adjacent tubular casts
Immunohistochemistry & special stains
- von Kossa stain: oxalate crystals will be negative (calcium phosphate crystals will be positive)
Electron microscopy description
- Oxalate crystals or outlines of crystals (may not survive processing) may be seen in tubule lumens or interstitial spaces
Electron microscopy images
Contributed by Jonathan E. Zuckerman, M.D., Ph.D.

Oxalate crystals
Molecular / cytogenetics description
- Primary:
- All forms autosomal recessive
- AGXT gene mutations (type 1), which encodes the hepatic peroxisomal enzyme alanine:glyoxylate aminotransferase (AGT)
- GRHPR gene (type 2), which encodes the GRHPR enzyme
- HOGA1 gene (type 3), which encodes the mitochondrial 4-hydroxy-2-oxoglutarate (HOG) aldolase enzyme
- ~5% do not have demonstrable mutations in any of these 3 genes (N Engl J Med 2013;369:649, J Am Soc Nephrol 2015;26:2559)
Sample pathology report
- Left kidney, biopsy:
- Acute tubular injury (acute tubular necrosis) with marked calcium oxalate deposition suggestive of oxalate nephropathy
- Comment: The differential diagnosis for these findings includes both secondary and primary causes. Secondary hyperoxaluria / oxalate nephropathy can be seen in the setting of gastrointestinal malabsorption (enteric hyperoxaluria) or exogenous / dietary oxalate ingestion (e.g. excess vitamin C, rhubarb, parsley, spinach, beet greens, star fruit, nuts, black tea) usually in a background of underlying chronic kidney disease. Of note, oxalate deposition can also be seen in the setting of ethylene glycol ingestion. Primary oxaluria can present in adulthood; however, this possibility should be investigated after all secondary causes are excluded.
Differential diagnosis
- Calcium phosphate:
- Purple crystals, not polarizable
- 2,8-Dihydroxyadenine (DHA) crystals / adenine phosphoribosyltransferase deficiency:
- Crystals polarizable, brown in color (Clin J Am Soc Nephrol 2012;7:1521)
- Urate crystals:
- Needle shaped birefringent crystal
- Positive for uric acid stain (Arch Pathol Lab Med 2000;124:774)
- Drug crystals:
- E.g. triamterene (Am J Kidney Dis 2014;63:148)
- Acute tubular injury:
- May show nonspecific calcium oxalate crystals in tubular lumens
- Usually less extensive deposition than in cases of true oxalosis
- May be difficult to distinguish from secondary oxalosis; requires clinical correlation
- Nonspecific accumulation can be seen in end stage kidney disease due to any cause:
- Usually more focal than in true oxalosis; clinical correlation
Board review style question #1
A 60 year old man presents with acute kidney injury. His past medical history is significant for morbid obesity treated by gastric bypass therapy. He also notes that he recently went on a green smoothie juice cleanse. What is the most likely cause of this patient's acute kidney injury?
- Acute interstitial nephritis
- Obesity related glomerulopathy
- Oxalate nephropathy
- Phosphate nephropathy
- Urate nephropathy
Board review style answer #1
Board review style question #2

You receive a liver and bilateral kidneys from an 8 year old boy undergoing combined liver / kidney transplantation. The liver shows no significant gross or histologic findings. The kidneys appear atrophic with a gritty cut surface. Microscopic examination reveals extensive rhomboid and fan shaped translucent crystals that are polarizable and shows extensive cortical scarring. This boy most likely suffers from which of the following conditions?
- Adenine phosphoribosyltransferase deficiency
- Congenital nephrocalcinosis
- Primary hyperoxaluria type 1
- Primary hyperoxaluria type 2
- Uromodulin kidney disease (ADTKD-UMOD)
Board review style answer #2
