
Bone marrow neoplastic
Bone marrow - plasma cell and lymphoid neoplasms
B lymphoblastic leukemia / lymphoma with recurrent genetic abnormalities
BCR::ABL1-like
Editorial Board Member: Genevieve M. Crane, M.D., Ph.D.
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 16 May 2022
Last staff update: 29 September 2023
Copyright: 2018-2024, PathologyOutlines.com, Inc.
PubMed Search: B lymphoblastic leukemia BCR-ABL1-like
Table of Contents
Definition / general | Essential features | ICD coding | Epidemiology | Pathophysiology | Clinical features | Laboratory | Prognosis | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Peripheral smear images | Positive stains | Negative stains | Molecular / cytogenetics description | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Kaseb H, Hudnall SD. BCR::ABL1-like. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/leukemiaprebbcrabl1like.html. Accessed April 19th, 2024.
Definition / general
- Introduced as a provisional entity in the WHO 2017 (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Does not have BCR-ABL1 translocation but shows a pattern of gene expression similar to that seen in ALL with BCR-ABL1
Essential features
- Exhibits National Cancer Institute (NCI) high risk features: (a) WBC ≥ 50,000/microL or age ≥ 10 years; up to 13 years if treated on a COG protocol; (b) persistent, postinduction, minimal residual disease; and (c) genetic alterations in kinase signaling including cytokine receptors and tyrosine kinases (Pediatr Blood Cancer 2017;64:10.1002)
- Important to recognize because:
- Associated with a poor prognosis
- Some are sensitive to tyrosine kinase inhibitors similar to BCR-ABL1 (Eur J Cancer 2017;82:203, Cancer Cell 2016;29:186)
- Use of tyrosine kinase inhibitors with treatment protocols may improve outcome (Eur J Cancer 2017;82:203)
- Identified in 15 - 20% of patients with B ALL (Lancet Oncol 2009;10:125, N Engl J Med 2009;360:470)
ICD coding
- ICD-10: C91.00 - acute lymphoblastic leukemia not having achieved remission
Epidemiology
- Relatively common subtype, representing 7 - 25% of B ALL patients (Swerdlow: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th Edition, 2017)
- Reported in both pediatric and adult patients
- More common in Down syndrome patients; uniquely shows CRLF2 translocation
Pathophysiology
- Harbors a large number of kinase activating gene rearrangements primarily involving the ABL class, JAK / STAT or RAS pathway associated signaling pathways
- Common genes involved include ABL1, ABL2, CRLF2, CSF1R, EPOR, NTRK3, PDGFRB, JAK1, JAK2, JAK3, FLT3, IL7R, SH2B3 and IKZF1
- These genes encode proteins involved in B cell development, proliferation and differentiation, cell cycle regulation and cell signaling
- Overexpression of cytokine receptors such as cytokine receptor-like factor 2 (CRLF2) occurs in both ALL with BCR-ABL1-like / Philadelphia-like and other B ALL categories (Eur J Cancer 2017;82:203)
Clinical features
- B ALL in general usually presents with symptoms related to bone marrow suppression by lymphoblasts
- Patients can present with anemia, leucopenia or thrombocytopenia or a combination of these
- Symptoms include bruising or bleeding due to thrombocytopenia, pallor or fatigue due to anemia and recurrent infections caused by neutropenia / leucopenia or bone pains
- May also present with lymphadenopathy (> 10 mm in single dimension of the lymph node), hepatomegaly or splenomegaly
- B ALL with BCR-ABL1-like and other B ALL with recurrent genetic abnormalities show no unique clinical presentation or microscopic findings
- Further, similar to some other B ALL with recurrent genetic abnormalities, B ALL with BCR-ABL1-like shows no unique immunophenotypic profile (Pediatr Blood Cancer 2017;64:10.1002)
- Essentially a diagnosis of exclusion
- Identifying patients with B ALL with BCR-ABL1-like can influence the management of the patient
- Those with PDGFRB translocation can benefit from tyrosine kinase inhibitors, while patients with JAK translocations may benefit from JAK inhibitors (Pediatr Blood Cancer 2017;64:10.1002)
- Adults tend to have poor outcome even with high intensity chemotherapy regimens
Laboratory
- B ALL with BCR-ABL1-like patients do not show a BCR-ABL1 fusion protein expressed from t(9;22)(q34;q11.2); however, they have a gene expression profile similar to ALL with BCR-ABL1
- Almost none of the genetic alterations are detected by standard genetic diagnostic methods such as conventional karyotyping and FISH because these genetic alterations are diverse and often cryptic
- To date, there are over 60 different identified rearrangements and approximately 16 different targetable genes
- Patients can only be diagnosed by gene expression panels (genome wide Affymetrix gene expression arrays):
- Currently 2 gene expression signatures have been verified for diagnosis of B ALL with BCR-ABL1-like / Ph-like
- BCR-ABL1-like gene expression array consists of 110 expression probe sets and was originally developed to classify B ALL in a population based Dutch / German cohort
- Gene expression of a group of cases were similar to ALL with BCR-ABL1 and therefore named the category ALL with BCR-ABL1-like
- These had an overall unfavorable outcome (Lancet Oncol 2009;10:125)
- Philadelphia-like signature was first demonstrated in cases of B ALL with IKZF1 deletions in a high risk United States cohort (N Engl J Med 2009;360:470)
- Differences between these 2 gene expression signatures can be explained by their different origins and discovery cohorts
- Few reference laboratories are currently offering low density microarray classifiers (8 or 15 gene) that may help screen patients
- One of these reference laboratories is Tricore reference laboratories (BCR-ABL1 like ALL (Ph-like ALL): TriCore Reference Laboratories [Accessed 8 May 2019])
- Some institutions and reference laboratories have developed multiplex FISH testing that may help screen cases
- Some institutions such as St. Jude utilize transcriptome and whole exome sequencing
- Flowcytometry:
- There is evidence that B-ALL with BCR-ABL1-like present as common-ALL on IPT, however this maturation stage is not distinct of the entity and is common in other B-ALL subt
- ypes B-ALL with BCR-ABL1-like patients with CRLF translocations show high levels of surface expression of the protein on flow cytometry, which can be used as a screening tool
Prognosis
- Prognostic factors include age, white blood cell count, immunophenotype, genetics and detection of measurable residual disease
- Numerous stratification schemes to assess risk in B ALL
- NCI risk group:
- NCI standard risk group: WBC < 50,000/microL and age 1 to < 10 years
- NCI high risk group: WBC ≥ 50,000/microL or age ≥ 10 years (up to 13 years if treated on a COG protocol)
- One scheme stratifies patients into standard risk and high risk based on age (< 10 years = standard, > 10 years = high) and WBC count (< 50,000/mm3 = standard, > 50,000/mm3 = high)
- Recurrent genetic abnormalities are identified in the majority of B ALL cases
- Prognosis of B ALL with BCR-ABL1-like is generally unfavorable
Case reports
- 10 year old boy treated with tyrosine kinase inhibitor for EBF1-PDGFRB fusion (J Clin Oncol 2013;31:e413)
- 16 year old boy treated with tyrosine kinase inhibitor for EBF1-PDGFRB fusion (Haematologica 2013;98:e146)
- 16 year old girl and 29 year old man with B ALL and novel ABL1 fusion genes (Br J Haematol 2011;153:43)
- 17 year old girl with novel JAK2 F694L mutation treated with JAK2 inhibitor and stem cell transplant (Pediatr Blood Cancer 2017;64:10.1002)
- 20 year old patient with a relapse of Philadelphia chromosome negative ALL who responded to tyrosine kinase inhibitor therapy (Oncol Res Treat 2018;41:550)
Treatment
- B ALL with BCR-ABL1-like is managed by standard combination chemotherapy
- Effective treatment modality for B ALL since the 1950s
- Usually administered in 3 distinct phases (induction, consolidation and maintenance) and should include intrathecal treatment, which is directed to the central nervous system
- Addition of tyrosine kinase inhibitors in B ALL with BCR-ABL1-like depends on the specific genetic alteration and has been shown to improve patient's outcome
- Both imatinib and dasatinib specifically inhibit ABL1, ABL2, PDGFRB and CSF1R kinases; ruxolitinib inhibits JAK / CRLF2 / EPOR alterations (Eur J Cancer 2017;82:203)
- Sensitivity to ruxolitinib in patients with JAK class aberrations is promising and has been successful in 2 JAK class fusion positive patients and 1 JAK2 mutated ALL (Eur J Cancer 2017;82:203)
- Adults tend to have poor outcome even with high intensity chemotherapy regimens; therefore, bone marrow transplantation becomes an important treatment option
Microscopic (histologic) description
- Blasts have scant agranular cytoplasm, coarse to fine chromatin, often with indistinct nucleoli
- No Auer rods and no dysplastic myeloid cells
Microscopic (histologic) images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
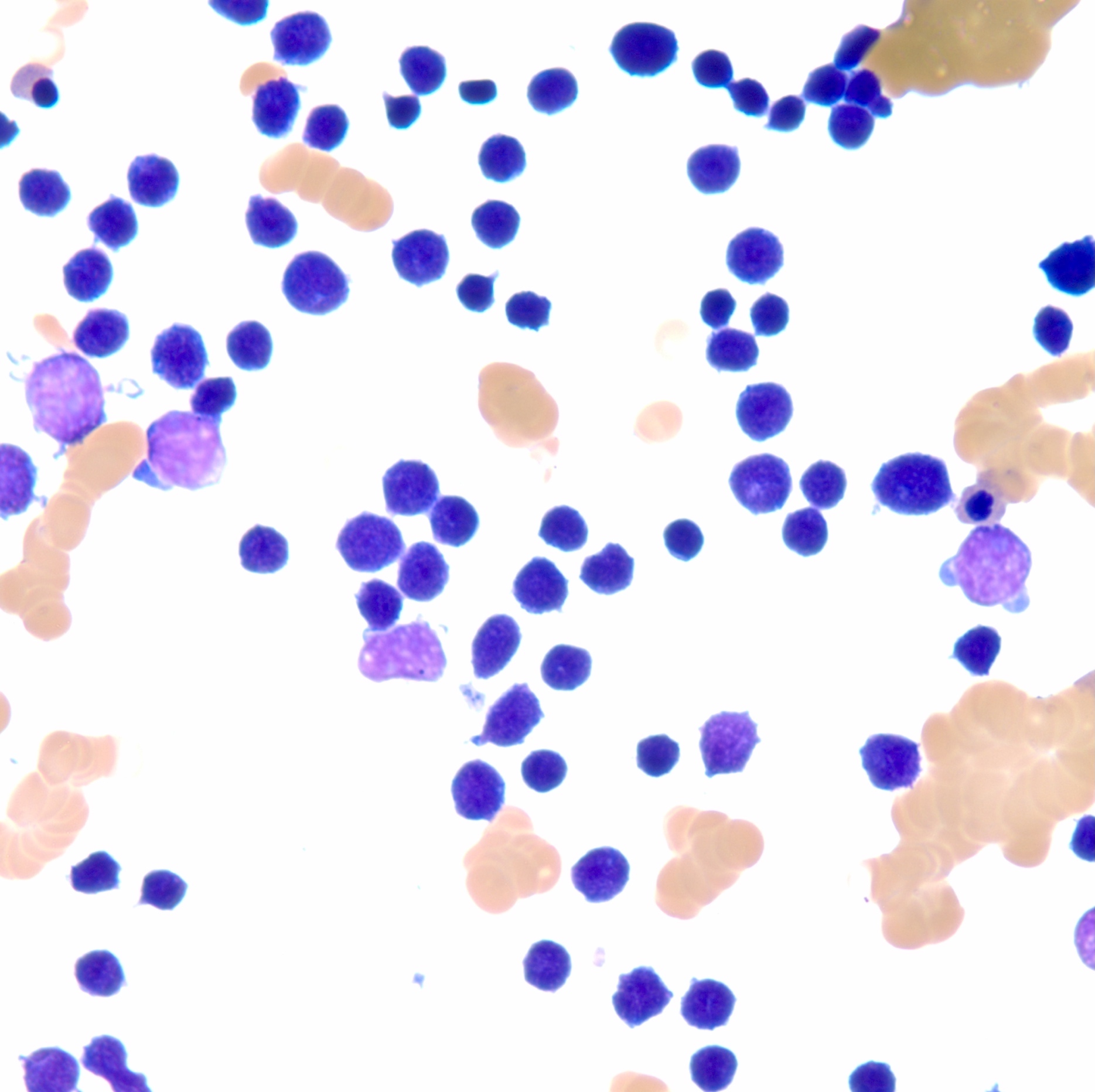
Lymphoblasts
Peripheral smear images
Contributed by Hatem Kaseb, M.D., Ph.D., M.P.H.
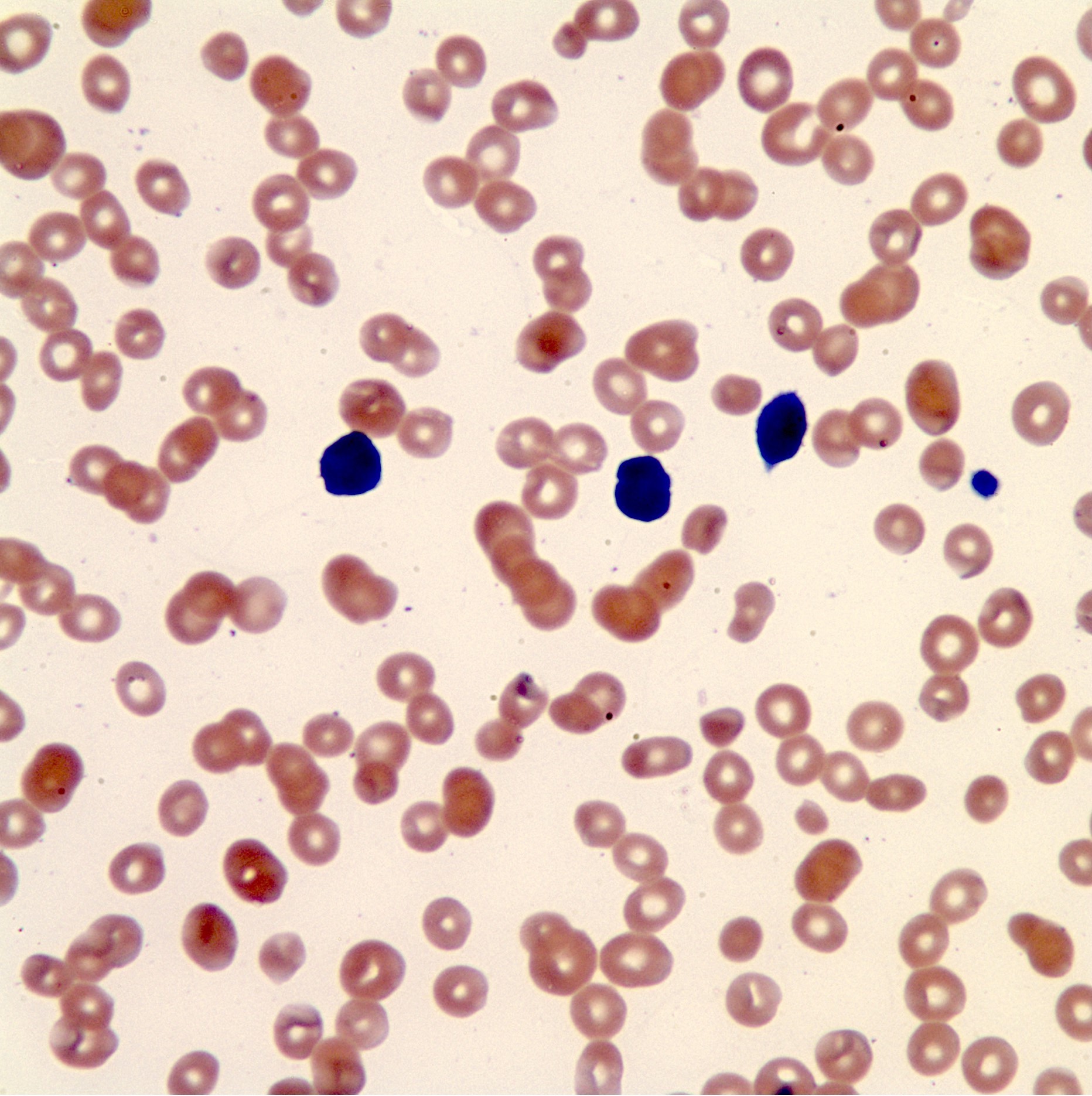
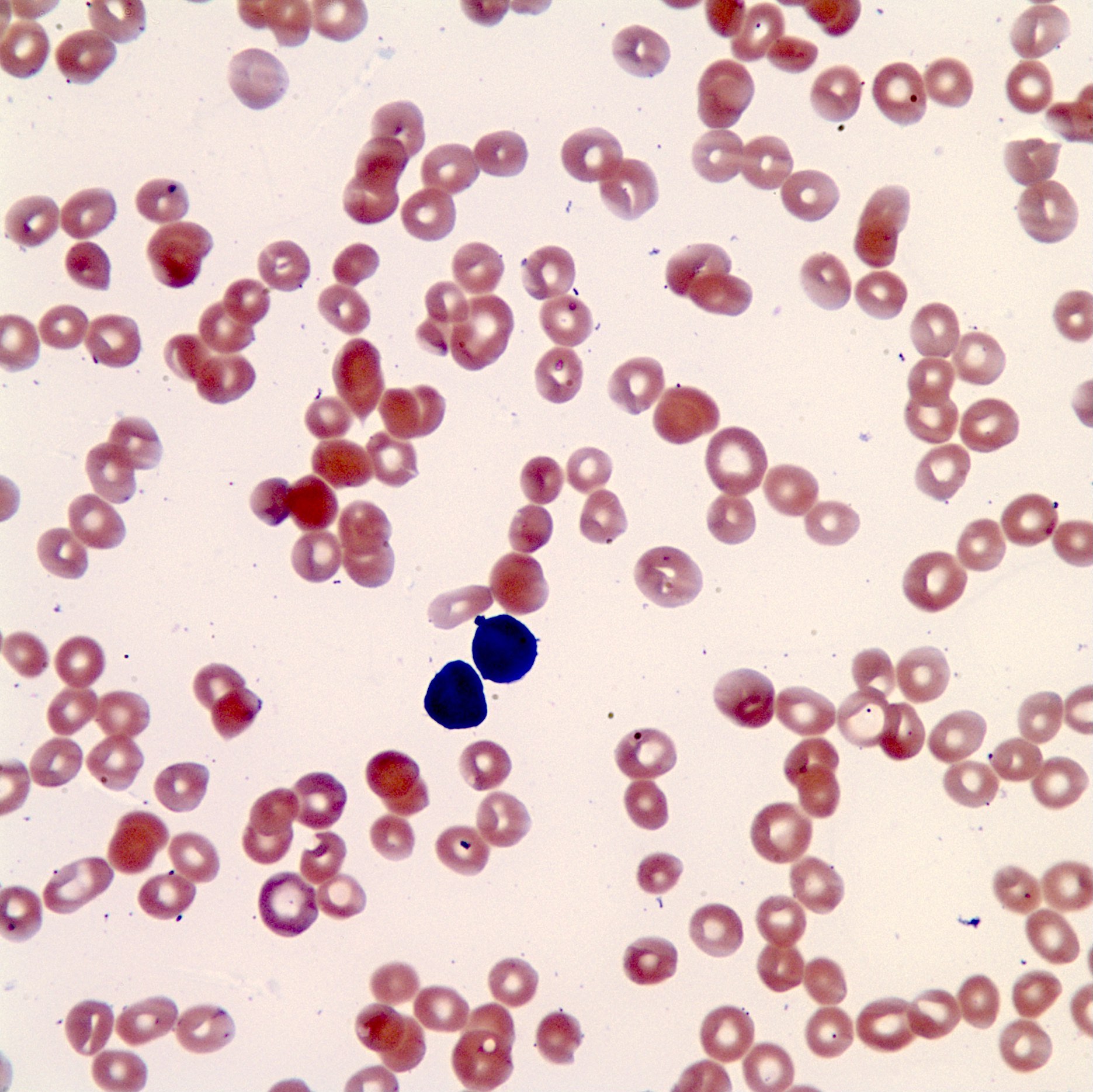
Lymphoblasts
Positive stains
Molecular / cytogenetics description
- Gene expression profiling is the gold standard for diagnosis of B ALL with BCR-ABL1-like / Ph-like (Lancet Oncol 2009;10:125, N Engl J Med 2009;360:470)
- Genetic alterations in B ALL with BCR-ABL1-like are harder to detect by standard genetic diagnostic approaches (Eur J Cancer 2017;82:203)
- Common gene rearrangements include CRLF2, EPOR and IGH
- Down syndrome patients uniquely show CRLF2 translocation
- ABL1 oncogene translocation with ABL2, PDGFRB, NTRK3, TYK2, CSF1R and JAK2 have all been reported
- Many cases of B ALL with BCR-ABL1-like may additionally show other deletions or mutations that have a clear role in leukemogenesis such as IKZFA and CDKN2A / B
- FISH and karyotyping are helpful in ruling out other B ALL entities, especially ALL with recurrent genetic abnormalities
- Multiplex FISH testing can be useful in ruling out B ALL with recurrent genetic abnormalities
- Identifying patients with B ALL with BCR-ABL1-like can influence patients' prognosis and management of the patient
- PDGFRB translocation: may benefit from tyrosine kinase inhibitors
- JAK translocations: may benefit from JAK inhibitors
Differential diagnosis
- B lymphoblastic leukemia / lymphoma, NOS: diagnosis should only be rendered after the exclusion of all other entities
- Burkitt lymphoma / leukemia
- B lymphoblastic leukemia / lymphoma with recurrent genetic abnormalities: must perform genetic testing including karyotyping, FISH or gene expression array; the entities in this group include:
- B ALL / LBL with t(9;22)(q34;q11.2); BCR-ABL1
- B ALL / LBL with t(v;11q23.3); KMT2A rearrangement
- B lymphoblastic leukemia / lymphoma with t(12;21)(p13.2;q22.1); ETV6-RUNX1
- B lymphoblastic leukemia / lymphoma with hyperdiploidy
- B lymphoblastic leukemia / lymphoma with hypodiploidy
- B lymphoblastic leukemia / lymphoma with t(5;14)(q31.1;q32.3); IL3-IGH
- B lymphoblastic leukemia / lymphoma with t(1;19)(q23;p13.3); TCF3-PBX1
Board review style question #1
What is the most sensitive test in diagnosing B lymphoblastic leukemia / lymphoma with BCR-ABL1-like / Ph-like?
- Flow cytometry
- Gene expression profiling
- Karyotyping
- Multiplex FISH
Board review style answer #1
B. Only gene expression profiling can diagnose the entity B ALL with BCR-ABL1-like / Ph-like
Comment Here
Reference: BCR-ABL1-like
Comment Here
Reference: BCR-ABL1-like
Board review style question #2
Which of the following is a characteristic of B lymphoblastic leukemia / lymphoma with BCR-ABL1-like / Ph-like?
- NCI high risk features
- NCI intermediate risk features
- NCI low risk features
- NCI risk features is not applicable
Board review style answer #2
A. ALL with BCR-ABL1-like shows the following characteristics: NCI high risk features, persistent postinduction minimal residual disease (MRD), genetic alterations that activate kinase signaling (Pediatr Blood Cancer 2017;64:10.1002). NCI risk stratifications include NCI standard risk group: WBC < 50,000/microL and age 1 to < 10 years; NCI high risk group: WBC ≥ 50,000/microL or age ≥ 10 years (up to 13 years if treated on a COG protocol).
Comment Here
Reference: BCR-ABL1-like
Comment Here
Reference: BCR-ABL1-like
