
Liver & intrahepatic bile ducts
Metabolic diseases
Gaucher disease
Editorial Board Member: Wei Chen, M.D., Ph.D.
Deputy Editor-in-Chief: Aaron R. Huber, D.O.


Last author update: 3 July 2023
Last staff update: 2 October 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Gaucher disease
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Virtual slides | Cytology description | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Videos | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Samdani R, Tsang P. Gaucher disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/livergauchers.html. Accessed April 20th, 2024.
Definition / general
- Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by mutation of the GBA1 gene that codes for glucocerebrosidase (GCase)
- Impaired activity of GCase causes its substrate, glucocerebroside, to accumulate in lysosomes of reticuloendothelial system (liver, spleen, bone marrow)
- Macrophages (Gaucher cells) become laden with lipid and dysfunctional
Essential features
- One of the most common lysosomal storage disorders with a predisposition among those of Ashkenazi Jewish descent
- Caused by mutation in GBA1 gene on chromosome 1q that codes for GCase enzyme
- Impaired enzymatic activity leads to accumulation of Gaucher cells in reticuloendothelial system (liver, spleen, bone marrow), which leads to hepatosplenomegaly and bone marrow infiltration with cytopenia and bone pain
- Diagnosis is made by assessing GCase activity
- Treatment includes enzyme replacement therapy (GCase infusions) and substrate reduction therapy
Terminology
- Gaucher disease (pronounced as GO-SHEY)
ICD coding
- ICD-10: E75.22 - Gaucher disease
Epidemiology
- First described in 1882 in a patient with massive splenomegaly (Int J Mol Sci 2017;18:441)
- Prevalence is highest among the Ashkenazi Jewish population (Eastern European) with a frequency of 6% compared to 0.7 - 0.8% of all other ethnic groups (Hematology 2017;22:65)
Sites
- Liver
- Hepatomegaly with or without elevated liver enzymes (alkaline phosphatase, aminotransferase) due to intracellular accumulation of Gaucher cells and secondary inflammatory response
- Concurrent chronic liver diseases such as chronic viral hepatitis, steatohepatitis and vascular diseases are often seen
- Liver fibrosis (20 - 50% of cases) and cirrhosis with portal hypertension due to accumulation of Gaucher cells inciting an inflammatory response with fibrosis causing ischemia, infarction and progression of liver fibrosis (Mol Genet Metab Rep 2020;22:100564)
- Focal liver lesions also known as Gaucheroma (benign cluster of Gaucher cells associated with inflammation, fibrosis and iron accumulation)
- Increased risk of chronic liver disease and hepatocellular carcinoma due to cirrhosis
- Hypergammaglobulinemia and hyperferritinemia with probable underlying factor include chronic inflammation, impaired macrophage function and dysregulation in hepcidin - ferroportin axis
- Spleen
- Splenomegaly with focal splenic lesions characterized by clusters of Gaucher cells involving mostly the splenic cords in the red pulp
- Extramedullary hematopoiesis
- Bone marrow
- Infiltration of bone marrow by Gaucher cells causes reduced hematopoiesis and cytopenia
- Reduced proportion of marrow fat
- Bone
- Skeletal remodeling is also affected by replacement of marrow adipocytes with Gaucher cells; this causes fractures, remodeling defects, loss of bone mineral density and bone infarction
- Renal involvement is rare but proteinuria with or without renal insufficiency can occur
- Reference: Am J Hematol 2015;90:S6
Pathophysiology
- Mutation in GBA1 gene causes decrease in GCase activity with accumulation of glucocerebroside within activated macrophages (Gaucher cells) that infiltrate liver, spleen, bone marrow and other organs, leading to organomegaly
- Gaucher cells release cytokines and chemokines (IL1 beta, IL6, TNF alpha, IL10, IL18 and CCL18)
- Accumulated glucocerebroside is metabolized into sphingosine that can cause direct damage to hepatocytes, hematopoietic cells and the CNS
- GGase deficiency causes reduced osteoblast differentiation and function, reduced bone mineral deposition, defective bone remodeling caused by decreased bone formation leading to bone weakening, cortical thinning and trabecular resorption
- Infiltration of marrow by Gaucher cells leads to change in medullary microenvironment and compression of intraosseous blood vessels with potential bone infarction and necrosis
- References: Int J Mol Sci 2017;18:441, Am J Hematol 2015;90:S6
Etiology
- Autosomal recessive disease caused by mutation in gene GBA1 that codes for GCase causing impaired function and excessive accumulation of glucocerebroside in the reticuloendothelial system (liver, spleen, bone marrow)
- GD patients inherit 2 copies of mutated GBA1 (homozygous) from parents who are carriers
- Reference: Am J Hematol 2015;90:S6
Diagrams / tables
Images hosted on other servers:

Glucosylceramide metabolic pathway
Clinical features
- Splenomegaly is often the initial presentation
- Primary hypersplenism with anemia and thrombocytopenia, leukopenia also occurs frequently
- Hepatomegaly occurs late in the disease course with an increase in liver enzymes
- Bone marrow involvement / infiltration leading to anemia, thrombocytopenia, plasma cell dyscrasia
- Skeletal involvement in the form of osteopenia, osteoporosis, defective bone remodeling, osteonecrosis
- Pulmonary involvement in the form of interstitial lung disease
- Central nervous system (CNS) involvement
- Impaired cognition (type I GD)
- Convulsions, intellectual disability, apnea (type II GD)
- Myoclonus, seizures, dementia (type III GD)
- Categorized into 3 main types
- Type I: nonneuronopathic; most common type (90 - 95% of GD); variable presentation from asymptomatic patients discovered incidentally to early childhood presentation (OMIM: Gaucher Disease, Type I; GD1 [Accessed 1 May 2023])
- Type II: acute and severe neuronopathic type; affects CNS in infants, usually death before third year of life (OMIM: Gaucher Disease, Type II; GD2 [Accessed 1 May 2023])
- Can cause hydrops fetalis
- Perinatal lethal / collodion type: most severe form of type II with clinical manifestations presenting before birth and causing neonatal death
- Type III: subacute or juvenile neuronopathic, with systemic and CNS involvement, including oculomotor symptoms (OMIM: Gaucher Disease, Type III; GD3 [Accessed 1 May 2023])
- Intermediate in severity
- Associated with increased risk of multiple myeloma (relative risk: 5.9) (Blood 2005;105:4569)
- References: Expert Rev Endocrinol Metab 2018;13:107, Hematology 2017;22:65, J Bone Miner Res 2019;34:996
Diagnosis
- Diagnosis is made by assessing GCase activity in circulating lymphocytes or fibroblast culture; < 10 - 15% of mean normal activity is diagnostic
- Disease activity or response to therapy is evaluated by measuring biochemical markers in activated macrophages (e.g., chitotriosidase, angiotensin converting enzyme, ferritin, chemokine CCL18 / PARC)
- Genotype testing in Ashkenazi Jewish patients for the most common alleles (L444P, N370S, IVS2+1g>a, V394L and R496H)
- References: Int J Mol Sci 2017;18:441, Am J Hematol 2015;90:S6
Laboratory
- Anemia
- Thrombocytopenia due to hypersplenism
- Hyperferritinemia
- Hypergammaglobulinemia
- GCase enzyme activity assay
- Serum protein electrophoresis
- Bone marrow
- Large (30 - 100 micron) macrophages with fibrillary pale blue to grey cytoplasm
- Liver biopsy
- Performed for hepatomegaly and shows clusters of Gaucher cells (macrophages with amphophilic cytoplasm resembling wrinkled tissue paper)
- Reference: Mol Genet Metab Rep 2020;22:100564
Radiology description
- Radiographic / Xray findings
- Erlenmeyer flask deformity (enlargement of metaphysis and absence of the typical concave diametaphyseal curve) in distal femur
- Osteonecrosis, proximal femur
- Osteopenia
- Lytic lesions
- MRI performed to assess skeletal involvement
- Osteonecrosis
- Gaucher cell infiltration produce low signal on T1 - T2 weighted images
- Degree of bone marrow infiltration by Gaucher cells
- CT / ultrasound findings
- Hepatosplenomegaly
- Bone scan
- Decreased uptake
- EEG and swallowing studies for neuronopathic GD
- Reference: J Bone Miner Res 2019;34:996
Radiology images
Images hosted on other servers:
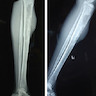
Gaucher lytic lesions

Erlenmeyer flask deformity
Prognostic factors
- Variable prognosis depending on clinical type of disease, ranging from asymptomatic to fatal outcome
- Type I: normal life span
- Type II: patients die within first 3 years of life
- Type III: intermediate between type I and type II
- Early diagnosis and treatment can have a good outcome
- Reference: Mol Genet Metab 2021;132:49
Case reports
- 4 year old Albanian boy diagnosed with GD type 1 through newborn screening presented with multiple osteonecrosis in femur on follow up (JIMD Rep 2022;63:414)
- 15 year old boy who presented with fever, pulmonary interstitial fibrosis and hepatomegaly and was ultimately diagnosed with GD (J Med Case Rep 2018;12:306)
- 18 year old man diagnosed with neuroblastoma, also found to have GD (Blood Cells Mol Dis 2018;68:106)
- 24 year old woman diagnosed as carrier of GD when she presented with gestational thrombocytopenia and anemia that did not respond to any medications (J Med Case Rep 2022;16:203)
- 38 year old man who presented with solitary swelling of proximal tibia, mimicking a musculoskeletal tumor (J Orthop Case Rep 2022;12:64)
Treatment
- Enzyme replacement therapy, which includes glucocerebrosidase IV infusions
- FDA approved Cerezyme (imiglucerase), VPRIV (velaglucerase alfa) and Elelyso (taliglucerase alfa) effective against hematologic, visceral and bone symptoms
- Not known to be effective against neurologic symptoms
- Substrate reduction therapy (oral medication) to reduce the accumulation of toxic substrate
- Cerdelga (eliglustat), a glucosylceramide synthase inhibitor, is indicated only for type 1 GD
- Zavesca (miglustat) for type 1 GD; not known to be effective against neurological symptoms (Ann Neurol 2008;64:514)
- Symptomatic treatment
- Bone remodeling: targeted treatments and vitamin D
- Splenectomy for hypersplenism
- Analgesics for bone pain and joint pain
- Bone marrow transplant only in cases of neuronopathic GD
- Somatic cell gene therapy under investigation
- References: Expert Rev Endocrinol Metab 2018;13:107, Mol Genet Metab 2021;132:49, Eur J Pharmacol 2022;926:175023
Clinical images
Images hosted on other servers:
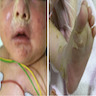
Perinatal collodion Gaucher (type 2)
Gross description
- Bone
- Mass-like lesions that are soft, hemorrhagic and irregular, resulting in extensive localized bone loss
- Osteonecrosis of bone appears as pale yellow zones
- Spleen
- Splenomegaly with pale homogenous cut surface with reddish yellow nodules of infarction
- References: Am J Hematol 2016;91:736, J Orthop Case Rep 2022;12:64
Gross images
Images hosted on other servers:

Splenomegaly in Gaucher disease
Microscopic (histologic) description
- Bone marrow, liver, spleen
- Infiltration by Gaucher cells containing abundant cytoplasm with fine, fibrillary, amphophilic characteristics resembling wrinkled tissue paper
- May have increased reticulin fibers and reduced fat in bone marrow biopsy
- Liver often demonstrates infiltration in the zone 3 region by Gaucher cells
- Small focal accumulations (Gaucheroma) or diffuse replacement by large, ovoid histiocytes (30 - 100 microns) with abundant, fibrillary eosinophilic granular cytoplasm resembling wrinkled tissue paper
- Small, bland nucleus that may be centrally or eccentrically located
- Cytoplasm has periodic acid Schiff diastase (PASD) resistant granules
- Reference: Mol Genet Metab Rep 2020;22:100564
Microscopic (histologic) images
Contributed by Patricia Tsang, M.D. and Mowafak Hamodat, M.B.Ch.B., M.Sc. (Case #164)

Gaucher in marrow biopsy
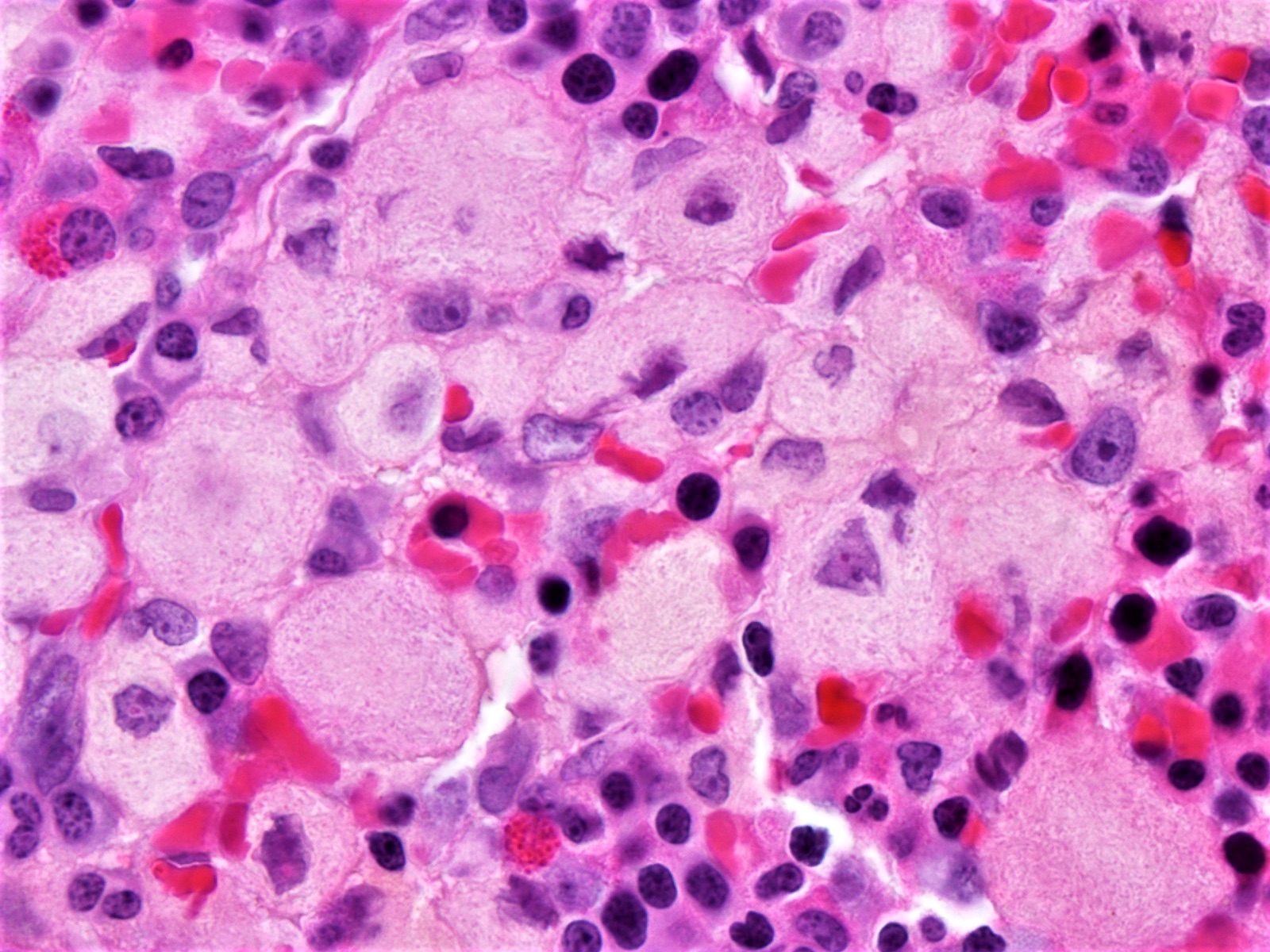
Fibrillary Gaucher cell cytoplasm

Gaucher cells marrow clot
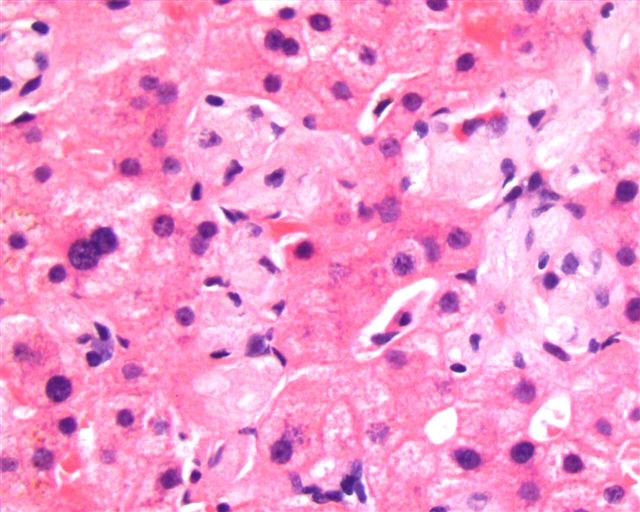
Liver biopsy

PASD positive Gaucher cells
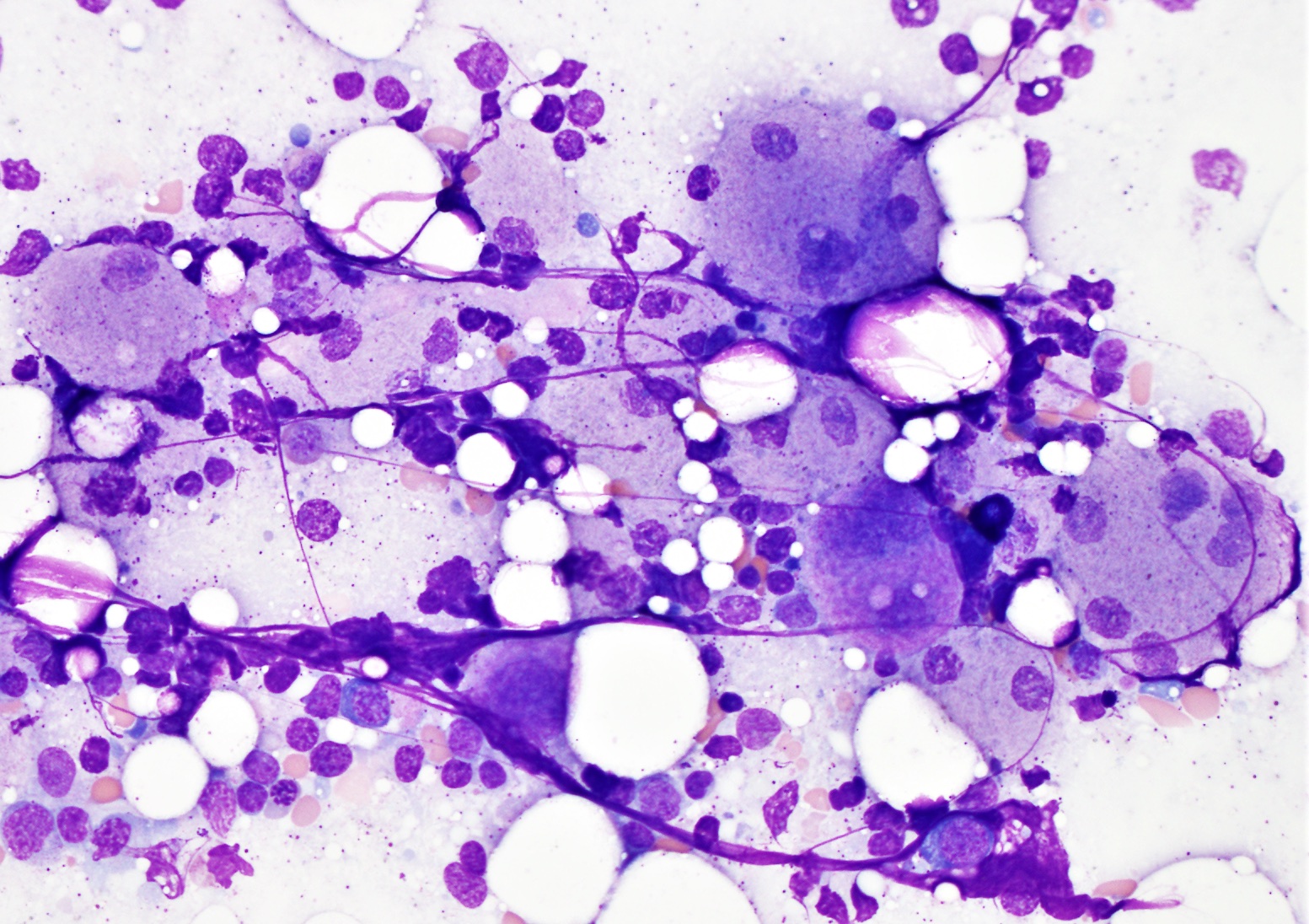
Marrow aspirate
Virtual slides
Images hosted on other servers:
Granular, fibrillary blue gray cytoplasm
Cytology description
- Wright-Giemsa stained bone marrow aspirate shows Gaucher cell containing abundant granular or fibrillary, blue-gray cytoplasm with wrinkled tissue paper-like appearance
Positive stains
- PAS and PASD are positive in Gaucher cells (diastase resistant)
- Iron
- CD68
- Reference: Arch Pathol Lab Med 2008;132:851
Negative stains
- Phospholipid stains, acid fast stains, CD11b, CD40
Electron microscopy description
- Elongated / rod shaped lysosomes filled with tubular structures
Electron microscopy images
Images hosted on other servers:

Gaucher cell cytoplasmic fibrils
Molecular / cytogenetics description
- Gene encoding glucocerebrosidase (Gcase) GBA1 is located on the long arm of chromosome 1 (1q21) and genetic testing is performed using saliva or blood samples
- DNA result is obtained by whole gene sequencing or selective sequencing within families
- More than 400 different mutations have been described in GBA1 gene; most prevalent are N370s, L444P
- c.1226A>G (N370S) mutation is common in the Ashkenazi Jewish population and most of these patients are asymptomatic
- L444P mutation seen in GD is at high risk for developing CNS complications
- Homozygous (D409H) mutation presents with cardiac complications
- References: Int J Mol Sci 2017;18:441, Mol Genet Metab 2021;132:49
Videos
Gaucher disease involving bone
Sample pathology report
- Liver, right lobe, needle core biopsy:
- Gaucher disease involving liver (see comment)
- Comment: Sections of the liver biopsy show clusters of Gaucher cells (pale staining histiocytes with striated cytoplasm, PASD positive) involving mainly the perivenular zone with atrophy of hepatocyte plates.
Differential diagnosis
- Pseudo-Gaucher cells:
- Mimic Gaucher cells on light microscopy but iron stain negative and lack the typical electron microscopy findings seen in GD (Turk J Haematol 2014;31:428)
- Seen in conditions that cause rapid cell turnover (e.g., myeloproliferative and myelodysplastic neoplasms, myeloma)
- Mycobacterial infection (AFB stain+)
- Sea blue histiocytosis (Arch Pathol Lab Med 2008;132:851):
- Seen in various conditions with increased marrow cell turnover (e.g., myeloproliferative or myelodysplastic neoplasms)
- Macrophage contains ceroid (insoluble lipid pigment); inclusions are globular (not fibrillary)
- Stains more intensely blue with Wright-Giemsa than Gaucher cells
- Niemann-Pick disease (Humpath.com: Niemann-Pick Diseases [Accessed 6 June 2023]):
- Lipid storage disorder caused by sphingomyelinase deficiency, which leads to accumulation of lipids in histiocytes that appear foamy
- Electron microscopy shows lamellar inclusions in lysosomes
Board review style question #1
A 5 year old boy presents to the clinic for evaluation of peripheral pancytopenia. His parents are of Ashkenazi Jewish descent. On physical examination, he has hepatosplenomegaly. Bone marrow aspirate smears show large cells with fibrillary cytoplasm that are positive for periodic acid Schiff (PASD) stain. Which of the following lysosomal enzymes are most likely deficient?
- Alpha galactosidase
- Glucocerebrosidase
- Hexosaminidase A
- Sphingomyelinase
Board review style answer #1
B. Glucocerebrosidase. Gaucher disease is caused by deficiency of glucocerebrosidase due to mutation of the GBA1 gene. Glucocerebrosidase deficiency causes abnormal accumulation of its substrate, glucocerebroside, in the macrophages of liver, spleen, bone marrow, etc. Patients can present with pancytopenia, hepatosplenomegaly and bone pain with fractures. Diagnosis can be made on finding a low glucocerebrosidase enzyme level in peripheral blood leukocytes. Answer D is incorrect because sphingomyelinase A deficiency is seen in Niemann-Pick Disease. The clinical presentation may overlap with Gaucher disease; however, there is an accumulation of foamy vacuolated macrophages in Niemann-Pick disease. Answer A is incorrect because alpha galactosidase deficiency is seen in Fabry disease, which typically presents with neurological manifestations. Answer C is incorrect because hexosaminidase deficiency is seen in Tay-Sachs disease.
Comment Here
Reference: Gaucher disease
Comment Here
Reference: Gaucher disease
Board review style question #2
Which of the following clinical characteristics is associated with type I Gaucher disease?
- Gastric ulcers
- Hydrops fetalis
- Normal expected life span
- Oculomotor symptoms
Board review style answer #2
C. Normal expected life span. Type 1 Gaucher disease is nonneuronopathic and associated with a normal life span. Answer A is incorrect because Gaucher disease is not typically associated with gastric symptoms. Answers B and D are incorrect because hydrops fetalis is associated with type 2 Gaucher disease and oculomotor symptoms are associated with type 3.
Comment Here
Reference: Gaucher disease
Comment Here
Reference: Gaucher disease
