
Liver & intrahepatic bile ducts
Metabolic diseases
Hemochromatosis
Editorial Board Member: Aaron R. Huber, D.O.
Deputy Editor-in-Chief: Catherine E. Hagen, M.D.


Last author update: 2 September 2021
Last staff update: 8 October 2021
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Hemochromatosis[title] liver pathology "last 5 years"[DP]
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Frozen section description | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Leonard N, Evason KJ. Hemochromatosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverhemochromatosis.html. Accessed April 20th, 2024.
Definition / general
- Most common autosomal recessive disorder in Caucasians, caused by decreased hepcidin activity due to mutations in iron metabolism genes such as HFE (Nat Rev Dis Primers 2018;4:18016)
- Characterized by increased iron absorption, toxic iron accumulation and end organ damage
Essential features
- Caused by mutations in iron metabolism genes, such as HFE, resulting in hepcidin deficiency and increased iron absorption, toxic iron accumulation and end organ damage (Nat Rev Dis Primers 2018;4:18016)
- Diagnosis can be confirmed with HFE genetic testing but liver biopsy may still be necessary to assess fibrosis
- Excess iron accumulates predominantly in hepatocytes, in a periportal (early) to panlobular (advanced) distribution
- Hemochromatosis patients with cirrhosis have up to 100 times the chance of developing hepatocellular carcinoma than the general population (J Hepatol 2010;53:3)
- Distinct from secondary hemochromatosis (secondary iron overload), which occurs in the setting of inherited or acquired disorders of erythropoiesis or frequent blood transfusions (Dtsch Arztebl Int 2009;106:499)
Terminology
- Type 1 hemochromatosis (specifically refers to HFE associated hemochromatosis)
- Primary or inherited iron overload
- Bronze diabetes
- Hereditary hemochromatosis
ICD coding
Epidemiology
- M:F = 2:1
- Men typically develop symptoms at 40 - 60 years old
- Women typically develop symptoms after menopause
- Predominantly seen in people of northern European descent
- Estimated prevalence: 1 in 300 - 500 (StatPearls: Hemochromatosis [Accessed 16 July 2021])
- Due to relatively high prevalence of HFE mutations (Semin Liver Dis 2005;25:411)
- Non HFE hemochromatosis is seen worldwide (StatPearls: Hemochromatosis [Accessed 16 July 2021])
Sites
- Liver, heart, pancreas, skin, gonads, joints
Pathophysiology
- Iron is absorbed by duodenal enterocytes via divalent metal transporter 1 (DMT1) (Semin Liver Dis 2005;25:411)
- Can be transported into plasma by ferroportin (FPN1) or can be stored as ferritin and excreted in feces when enterocyte is sloughed
- Iron in plasma is bound to transferrin (Semin Liver Dis 2005;25:411)
- Cannot be excreted
- Hepatocytes take up transferrin bound iron via transferrin receptor 1 (TFR1) or TFR2 (Semin Liver Dis 2005;25:411)
- Can also take up nontransferrin bound iron if transferrin highly saturated
- Kupffer cells take up iron mainly by phagocytosing senescent erythrocytes (Semin Liver Dis 2005;25:411)
- Can be transported into plasma via ferroportin
- Hepcidin (HAMP) is master regulator of iron homeostasis (Semin Liver Dis 2005;25:411, StatPearls: Hemochromatosis [Accessed 16 July 2021], Nat Rev Dis Primers 2018;4:18016)
- Peptide hormone produced in the liver in the presence of sufficient transferrin bound iron
- Inhibits dietary iron absorption in gut and release of iron from macrophages by inhibiting ferroportin
- Hemojuvelin (HJV) binds bone morphogenetic protein receptor (BMPR) (Nat Rev Dis Primers 2018;4:18016)
- BMP6 production increases with increased cellular iron stores
- Signaling through BMP6-BMPR-HJV promotes hepcidin transcription
- Hereditary hemochromatosis protein (HFE) normally promotes hepcidin transcription (Nat Rev Dis Primers 2018;4:18016)
- Interacts with TFR1
- Loss of function mutations in HFE → decreased hepcidin → increased ferroportin activity → increased iron absorption (hemochromatosis type 1) (Semin Liver Dis 2005;25:411, StatPearls: Hemochromatosis [Accessed 16 July 2021], Nat Rev Dis Primers 2018;4:18016)
- Loss of function mutations in HAMP (gene encoding hepcidin)→ increased ferroportin activity → increased iron absorption (hemochromatosis type 2b) (Nat Rev Dis Primers 2018;4:18016)
- Loss of function mutations in HJV → decreased hepcidin → increased ferroportin activity → increased iron absorption (hemochromatosis type 2a) (Nat Rev Dis Primers 2018;4:18016)
- Loss of function mutations in TFR2→ decreased hepcidin → increased ferroportin activity → increased iron absorption (hemochromatosis type 3) (Nat Rev Dis Primers 2018;4:18016)
- Gain of function mutations in FPN1 (gene encoding ferroportin) → increased iron absorption (hemochromatosis type 4) (Nat Rev Dis Primers 2018;4:18016)
- Excess iron accumulation in organs and tissues leads to increased oxidative stress, cell death and organ damage (StatPearls: Hemochromatosis [Accessed 16 July 2021])
- Liver
- Fibrosis and cirrhosis
- Increased risk of hepatocellular carcinoma by up to 100 times (J Hepatol 2010;53:3)
- Pancreas
- Diabetes
- Other affected organs include:
- Heart
- Arrhythmias, dilated cardiomyopathy, congestive heart failure
- Joints
- Arthropathy (pain)
- Pituitary gland
- Hypogonadotropic hypogonadism
- Skin
- Hyperpigmentation, bronzing of skin
- Heart
- End organ damage is exacerbated by alcohol use and viral hepatitis
- Liver
Etiology
- Usually caused by loss of function point mutations in HFE gene (Nat Rev Dis Primers 2018;4:18016, Semin Liver Dis 2005;25:411)
- Autosomal recessive
- Most common mutation is C282Y
- Iron overload develops clinically in 25 - 60% of homozygotes
- Other mutations include H63D and S65C
- More rare and generally less severe disease
- Can also be caused by mutations in other iron related genes, including HAMP, HJV and TFR2 (Nat Rev Dis Primers 2018;4:18016)
Diagrams / tables
Images hosted on other servers:

Hemochromatosis diagnostic algorithm

Iron metabolism
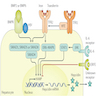
Hepcidin regulation

Scheuer iron scoring system
Clinical features
- Classic triad: liver cirrhosis, diabetes mellitus, bronzing of skin (Am J Hematol 2006;81:202)
- May also have joint pain, fatigue, abdominal pain
Diagnosis
- Laboratory testing (Nat Rev Dis Primers 2018;4:18016)
- If iron overload is suspected, screen with transferrin saturation and serum ferritin
- If transferrin saturation is ≥ 45% and ferritin is elevated, perform genetic testing
- PCR testing targeted for C282Y variable H63D and S65C
- Screening asymptomatic patients without family history of hemochromatosis with genetic testing is not recommended, due to high frequency of HFE mutations and variable penetrance (J Hepatol 2010;53:3)
- First degree relatives of patients with hemochromatosis should undergo genetic testing
- Liver biopsy:
- Was the gold standard but is no longer required for diagnosis in the presence of genetic testing
- Tissue can be used to determine hepatic iron concentration (HIC) via mass spectrometry
- Useful to confirm diagnosis of hepatic iron overload when genetic testing for common HFE mutations is negative (Clin Liver Dis (Hoboken) 2021;17:232)
- Hepatic iron index (HII) = hepatic iron concentration / patient’s age in years (Clin Liver Dis (Hoboken) 2021;17:232)
- HII values < 1.0 are normal
- HII values 1.0 to 1.9 indicate mild iron accumulation
- Heterozygous for HFE or other hemochromatosis associated mutation
- Alcoholic liver disease
- HII values > 1.9 indicate iron overload
- Homozygous HFE or other hemochromatosis associated mutation
- Cirrhosis
- Recommended to assess fibrosis when serum ferritin > 1,000 ug/L (J Hepatol 2010;53:3)
- Imaging:
- MRI can be used to quantify iron and determine distribution of iron in the liver
- Transient elastography may help assess fibrosis and cirrhosis
Laboratory
- Transferrin saturation ≥ 45% (J Hepatol 2010;53:3)
- Elevated ferritin
- May have elevated liver enzymes (AST, ALT) with disease progression
Radiology description
- MRI can be used to determine distribution of iron deposition and quantify the iron (Insights Imaging 2012;3:173)
- Increased iron leads to accelerated T2 relaxation and reduced signal intensity
- Dual energy CT can also be used to quantitate iron (Radiology 2015;277:95)
Radiology images
Images hosted on other servers:
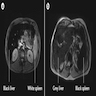
MRI findings in hemochromatosis

Iron quantification by MRI
Prognostic factors
- Life expectancy is normal if diagnosed and managed before end organ damage develops
- Prognosis is poor after development of cirrhosis, hepatocellular carcinoma, diabetes or cardiomyopathy
- Patients with cirrhosis have up to a 100 times greater chance of developing hepatocellular carcinoma than the general population (J Hepatol 2010;53:3)
Case reports
- 28 year old Chinese man with abnormal liver function tests (Front Genet 2020;11:77)
- 45 year old man with recurrent intermittent skin changes (ACG Case Rep J 2019;6:e00247)
- 48 year old man with atrial fibrillation (Eur Heart J Case Rep 2020;4:1)
- 65 year old man with disseminated Cryptococcus (Autops Case Rep 2020;10:e2020180)
Treatment
- Therapeutic phlebotomy / venesection
- Frequency based on hemoglobin, serum ferritin and transferrin saturation (Br J Haematol 2018;181:293)
- Chelation therapy may be considered if therapeutic phlebotomy is not available or contraindicated or as an adjunct to phlebotomy in severe cases
Clinical images
Images hosted on other servers:
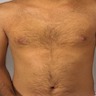
Skin
hyperpigmentation
(bronze diabetes)
Gross description
- Dark brown discoloration of liver and other involved organs
- Hepatomegaly (StatPearls: Hemochromatosis [Accessed 16 July 2021])
Gross images
Images hosted on other servers:

Dark brown discoloration of the liver
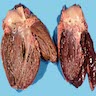
Dark brown discoloration of the myocardial muscle
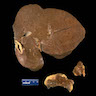
Liver, pancreas, lymph nodes
Frozen section description
- Granular brown-black pigment within periportal (milder) or panlobular (more severe) hepatocytes (Arch Pathol Lab Med 2013;137:270)
- Potential donor livers with severe hepatocellular iron overload are generally not accepted for transplantation (Arch Pathol Lab Med 2013;137:270)
Microscopic (histologic) description
- Liver:
- Iron appears as dark brown granular pigment, predominantly within hepatocytes
- Pericanalicular pattern (Clin Liver Dis (Hoboken) 2021;17:232)
- Periportal deposition initially, extending towards zones 2 and 3 with disease progression
- Iron pigment may also be seen in cholangiocytes and Kupffer cells, especially at later stages
- Fibrosis develops over time
- Amount of iron can be graded semi quantitatively (Mod Pathol 2007;20:S31)
- Scheuer methodology (scale 0 - 4) based on ease of observation of iron granules at various magnifications (World J Gastroenterol 2007;13:4755, J Pathol Bacteriol 1962;84:53)
- Iron appears as dark brown granular pigment, predominantly within hepatocytes
- Heart:
- Iron within cardiomyocytes (Int Heart J 2018;59:427)
- Pancreas:
- Iron within acinar cells (Diabetes Care 2014;37:e137)
- Interstitial fibrosis
- Joints:
- Acute synovitis
- Calcium pyrophosphate deposition (pseudogout) (N Engl J Med 2016;374:2575)
- Skin:
- Iron in dermal macrophages and fibroblasts (Transl Sci Rare Dis 2017;2:101)
- Increased melanin production
- Testes:
- Atrophy (hypogonadism), most likely due to iron deposition in anterior pituitary gonadotropin producing cells (Basic Clin Androl 2017;27:13)
Microscopic (histologic) images
Contributed by Kimberley J. Evason, M.D., Ph.D.
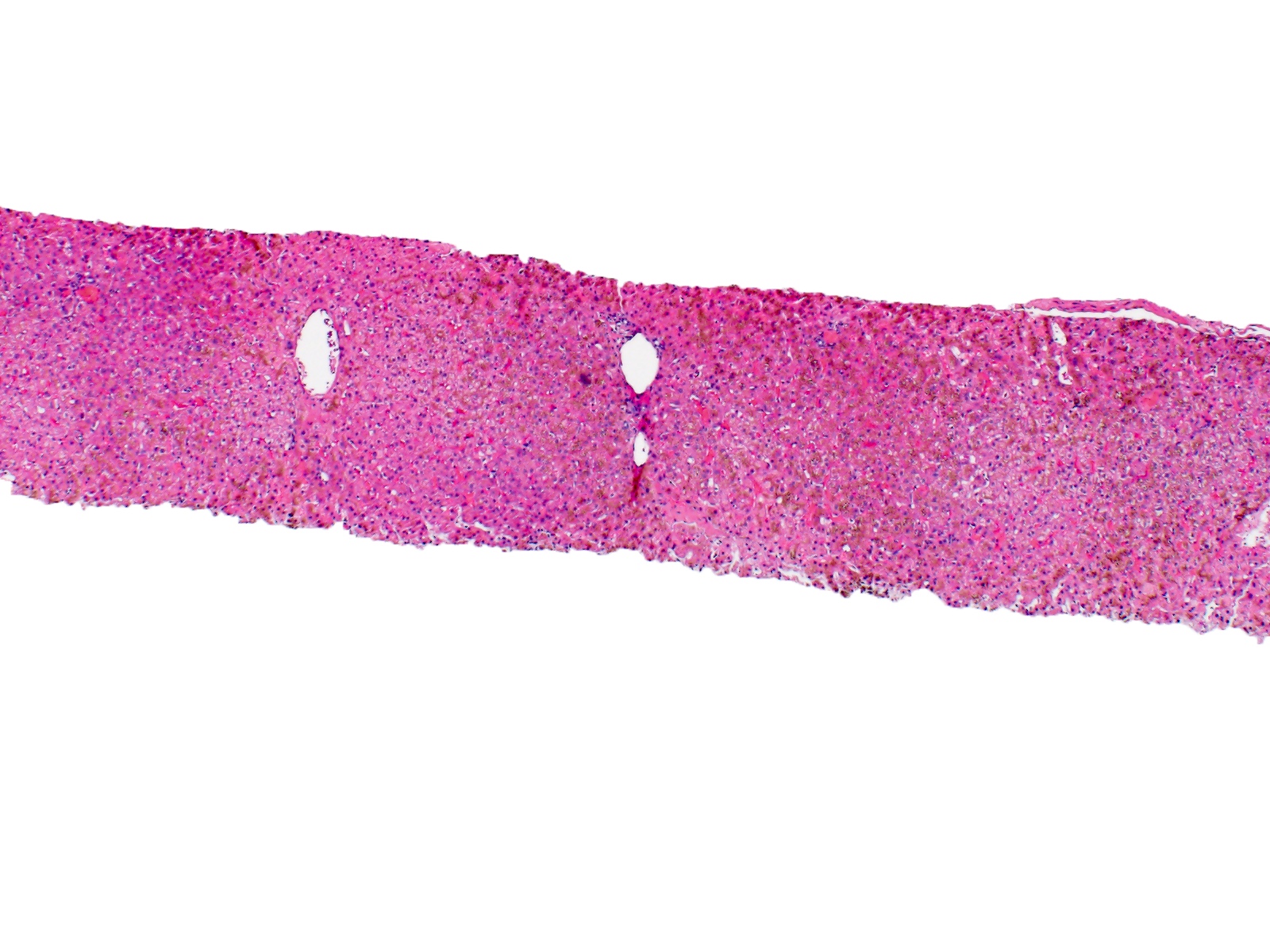
Liver with hepatocellular iron
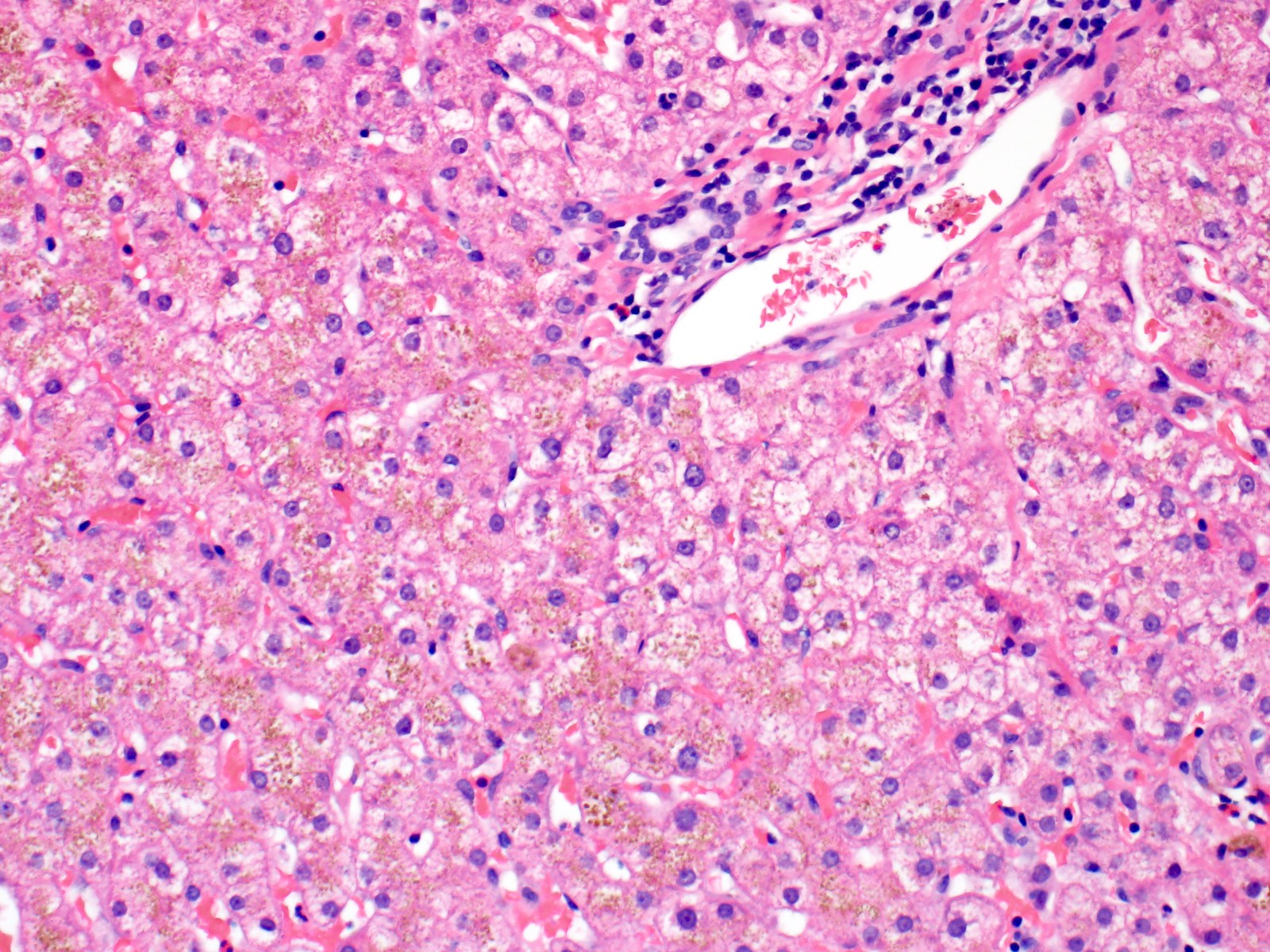
Liver, iron stain
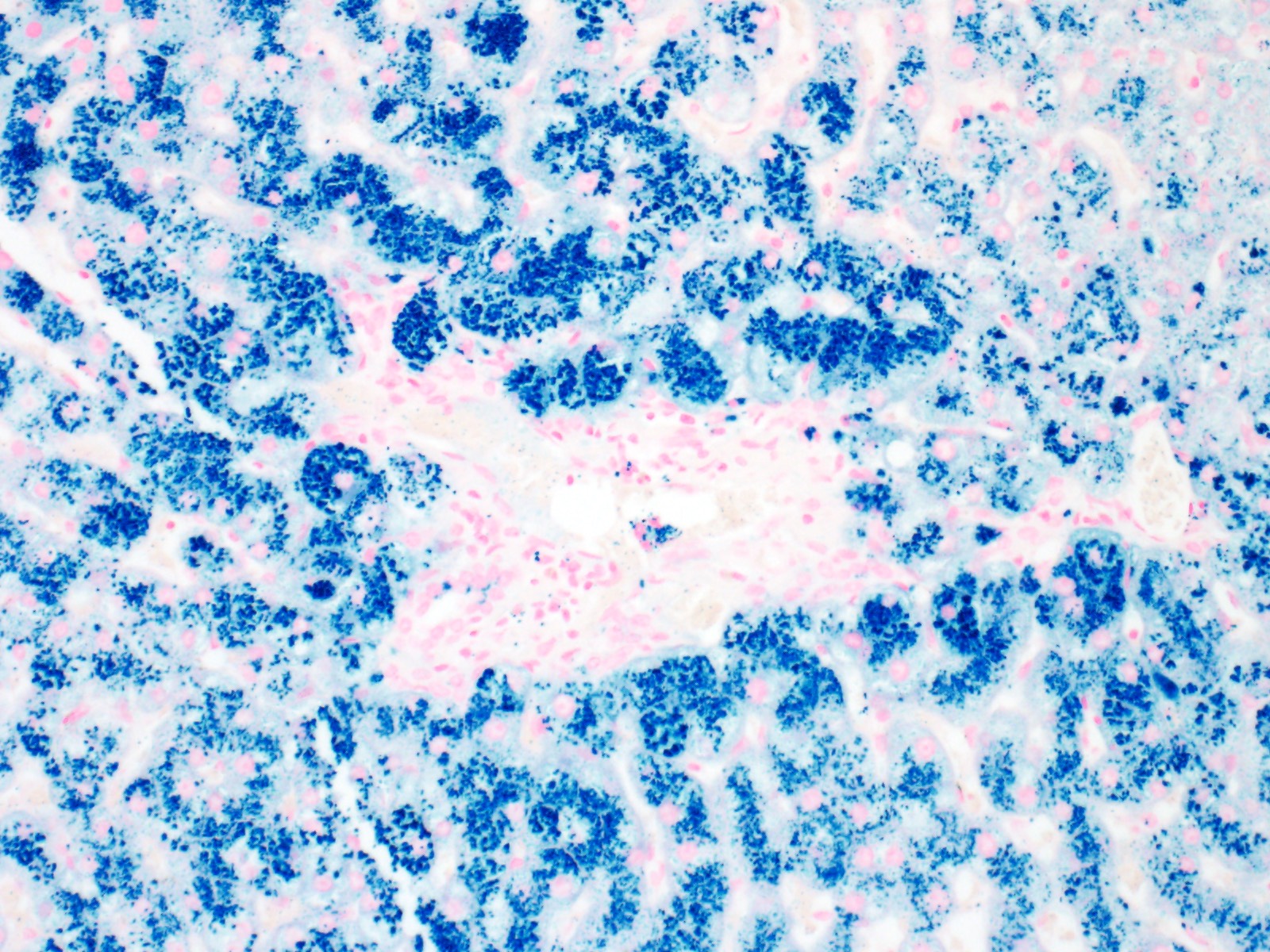
Periportal hepatocytes, iron stain
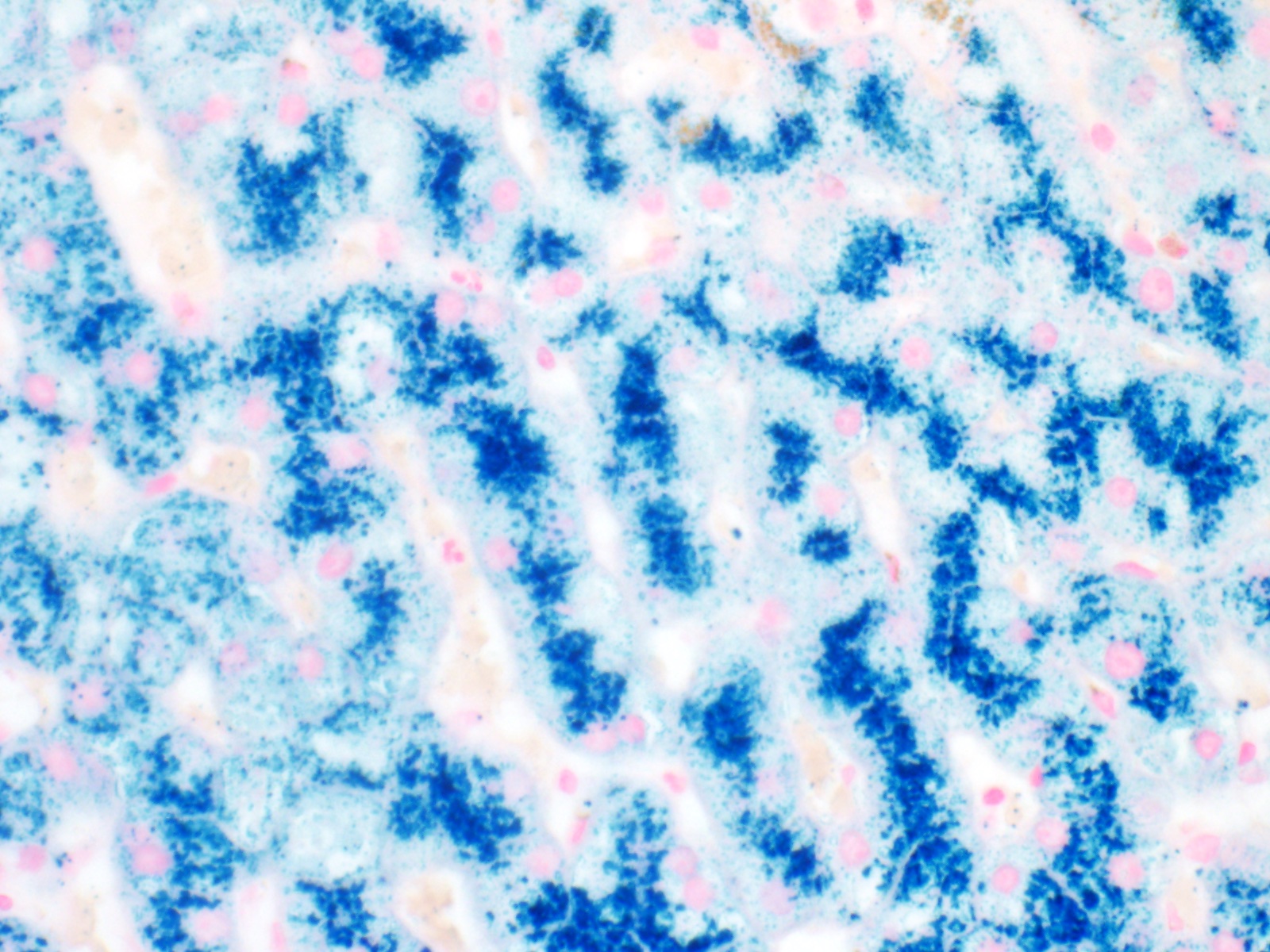
Canalicular pattern, iron stain
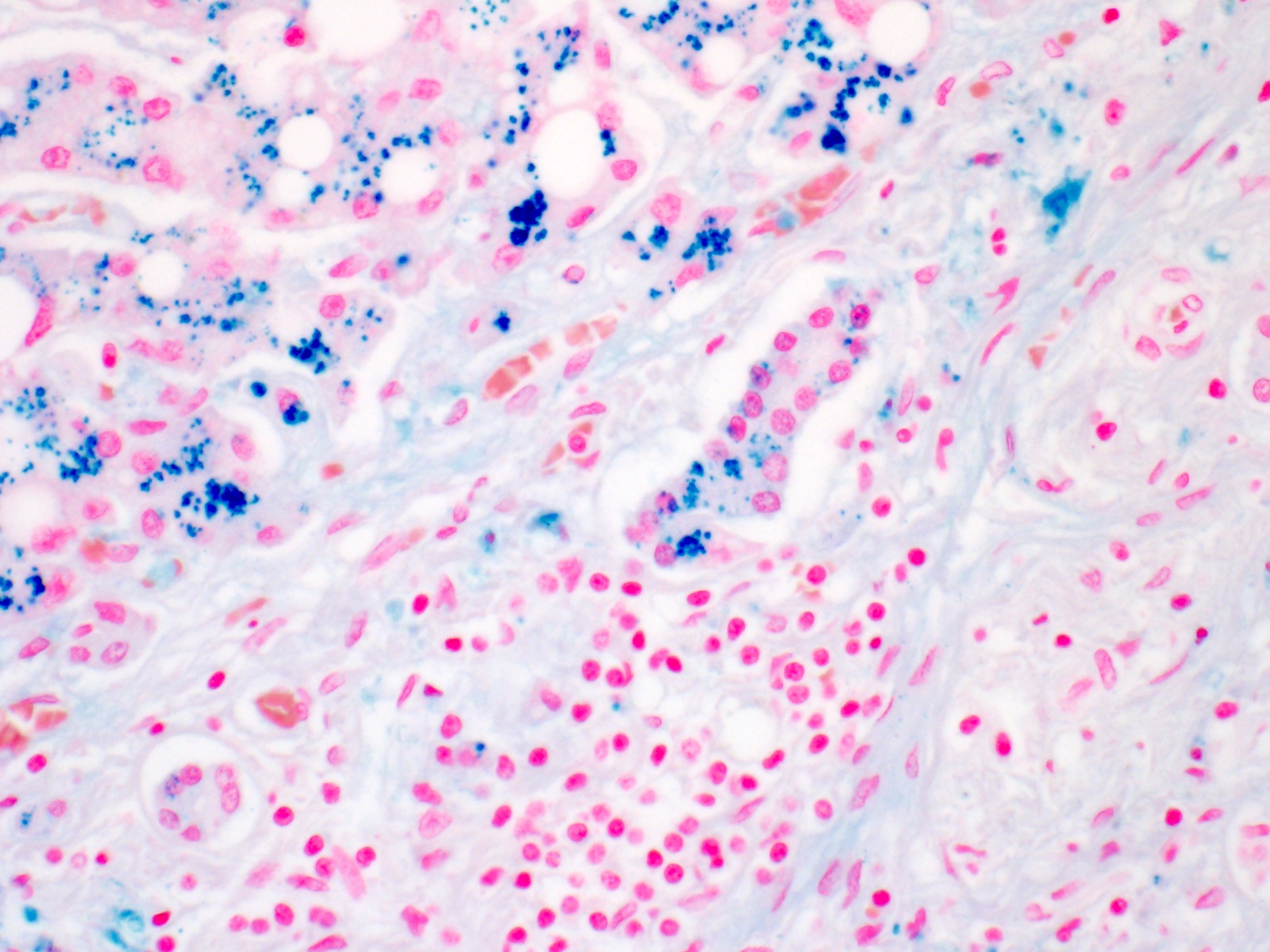
Bile duct epithelium iron
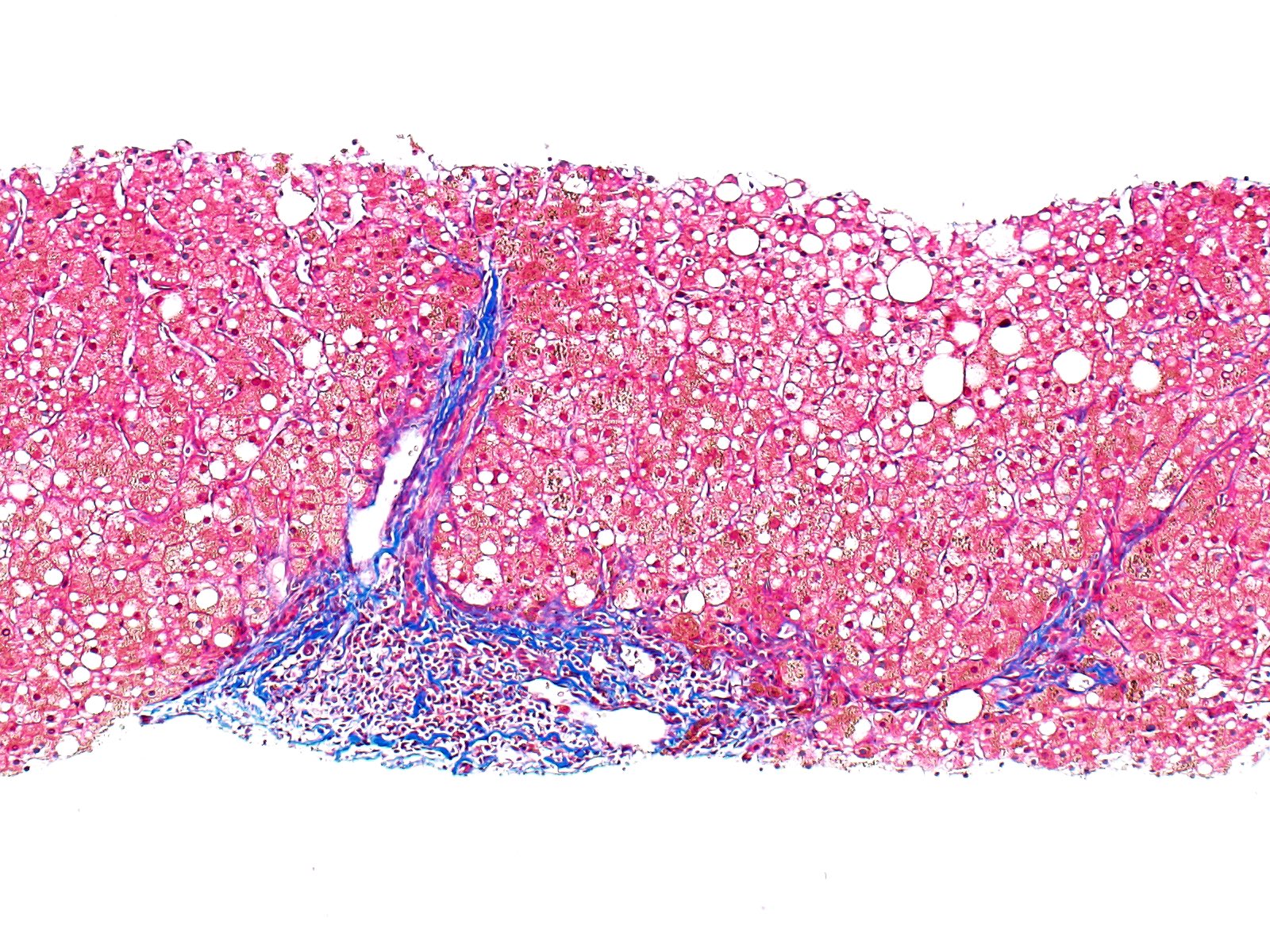
Trichrome
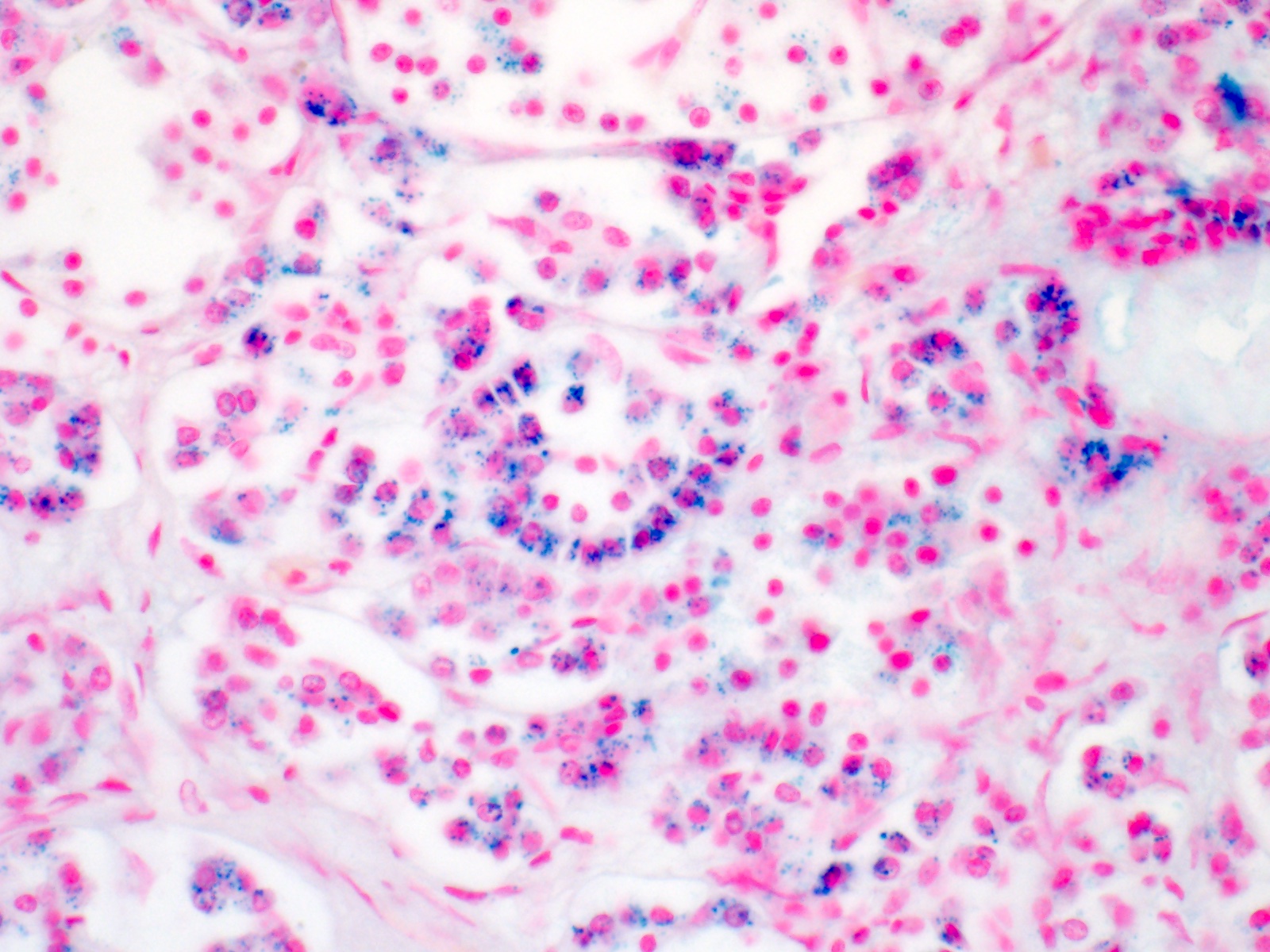
Pancreas, iron stain
Positive stains
Negative stains
- Rhodanine (copper) stain (Semin Diagn Pathol 2018;35:381)
- Hall’s (bilirubin) stain (Am J Clin Pathol 1960;34:313)
Molecular / cytogenetics description
- Type 1 (HFE hemochromatosis)
- Hemochromatosis (HFE) mutation
- Most common mutation is C282Y
- Compound heterozygosity with H63D, S65C and mutations at other loci have been reported (J Hepatol 2010;53:3)
- Non HFE hemochromatosis (StatPearls: Hemochromatosis [Accessed 16 July 2021])
- Type 2 (juvenile hemochromatosis)
- Hepcidin (HAMP) or hemojuvelin (HJV) mutation
- Often have hypogonadism at presentation
- Cardiac iron deposition typically results in heart failure, usually before the age of 30 (Am J Hematol 2006;81:202)
- Type 3
- Transferrin receptor 2 (TFR2) mutation
- Type 4
- Ferroportin 1 (FPN1, SLC40A1) mutation
- Autosomal dominant
- Type 2 (juvenile hemochromatosis)
Molecular / cytogenetics images
Contributed by Pinar Bayrak-Toydemir, Ph.D.
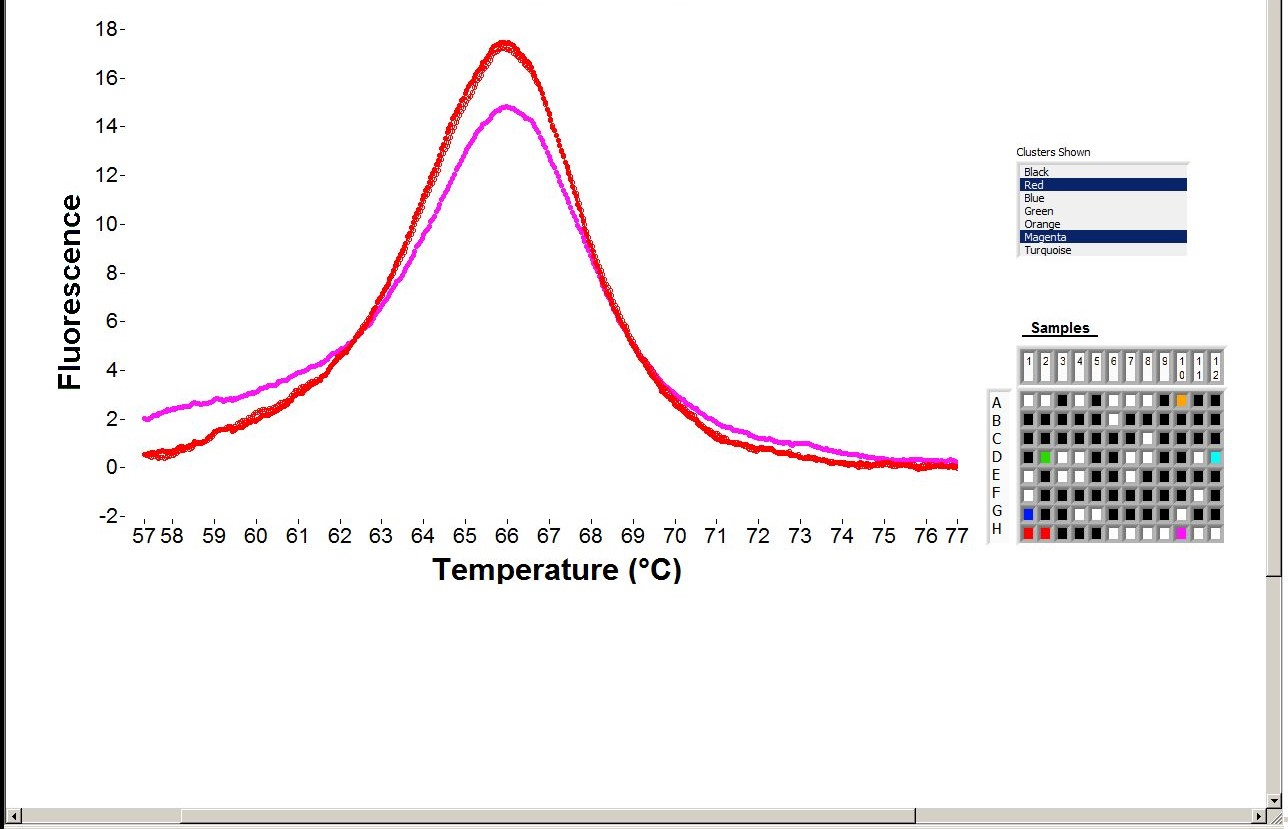


Melting curve: wild type
Images hosted on other servers:

Compound heterozygote sequence analysis
Sample pathology report
- Liver, biopsy:
- Extensive hepatocellular siderosis consistent with hereditary hemochromatosis (see comment)
- Comment: An iron stain shows extensive iron accumulation in hepatocytes with periportal accentuation. There is no evidence of fatty liver disease or chronic hepatitis, the other entities that can have siderosis. The pattern of intracellular iron deposition in hepatocytes in the clinical setting of homozygous HFE C282Y mutation is consistent with hereditary hemochromatosis.
Differential diagnosis
- Secondary hemochromatosis (StatPearls: Hemochromatosis [Accessed 16 July 2021]):
- Excess hepatic iron is usually predominantly sinusoidal, within Kupffer cells
- Iron overload due to transfusions, chronic liver disease, congenital atransferrinemia, oral intake of iron, ineffective erythropoiesis
- Hepatitis C:
- Positive HCV antibody screen
- Presence of portal inflammation with lymphoid aggregates
- Fatty liver disease:
- Steatosis, ballooned hepatocytes, centrizonal fibrosis
- Cholestasis:
- Bile is canalicular (extracellular) as well as intracellular
- Lipofuscin pigment:
- Lipofuscin pigment has a centrizonal distribution and is usually light brown
- Neonatal hemochromatosis (Hepatology 2006;43:654):
- Hepatic iron overload presenting soon after birth
- Thought to be a consequence of intrauterine liver failure (J Hepatol 2012;56:1226)
Board review style question #1
Which of the following genes is most commonly mutated in the heritable disease shown here in this liver biopsy stained with Prussian blue?
- FPN1 (encoding ferroportin)
- HAMP (encoding hepcidin)
- HFE
- HJV
- PTEN
Board review style answer #1
Board review style question #2
Which of the following patterns of iron deposition is most consistent with hemochromatosis?
- Centrilobular, intracellular deposition within hepatocytes
- No specific pattern of deposition
- Panlobular, intracellular deposition within Kupffer cells
- Periportal, intracellular deposition within hepatocytes
- Periportal, intracellular deposition within Kupffer cells
Board review style answer #2
