

Lung
Interstitial lung diseases
Lymphoid interstitial pneumonia
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 1 May 2018
Last staff update: 19 August 2022
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed search: lymphoid interstitial pneumonia [title]

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Positive staining - disease | Molecular / cytogenetics description | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Yoshikawa A. Lymphoid interstitial pneumonia. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lungnontumorLIP.html. Accessed April 16th, 2024.
Definition / general
- In 1969, Leibow and Carrinton first described lymphoid interstitial pneumonia (LIP) as interstitial lung disease with diffuse and dense lymphocytic infiltration (Simon: Frontiers of Pulmonary Radiology, 1st Edition, 1969)
- In American Thoracic Society / European Respiratory Society classification of the idiopathic interstitial pneumonia, idiopathic LIP is categorized as a rare idiopathic interstitial pneumonia (Am J Respir Crit Care Med 2002;165:277, Am J Respir Crit Care Med 2013;188:733)
Essential features
- Rare type of interstitial lung disease due to different diseases including Sjögren syndrome, rheumatoid arthritis and human immunodeficiency virus (HIV) infection
- On histology, diffuse infiltration of polyclonal lymphocytes with scant interstitial fibrosis is characteristic
Terminology
- Also called lymphocytic interstitial pneumonia
ICD coding
- J84.2: lymphoid interstitial pneumonia
Epidemiology
- Rare
- Typical onset at ages 40 - 70 years old but can occur at any age (Chest 2002;122:2150)
- More common in women
- No association with smoking history
Sites
- Bilateral lower lobes of the lung
Pathophysiology
- Pathogenic mechanisms of LIP are still unclear
- Has aspects of lymphoproliferative disease and lymphoid hyperplasia of polyclonal T or B cells (Chest 2002;122:2150)
- Although it may transform to lymphoma, especially MALT, the risk is lower than initially reported (Eur Respir J 2006;28:364)
Etiology
- Associated with several systemic diseases and conditions (Chest 2002;122:2150,
Respirology 2016;21:600, Eur Respir J 2006;28:364)
- Autoimmune (most common)
- Sjögren syndrome (SjS); 25% of LIP cases have SjS and 1% of SjS cases present with LIP
- Rheumatoid arthritis
- Systemic lupus erythematosus
- Polymyositis / dermatomyositis
- Hashimoto disease
- Hypothyroidism
- Castleman disease
- Myasthenia gravis
- Autoimmune hemolytic anemia
- Pernicious anemia
- Primary biliary cirrhosis
- Infection
- Human immunodeficiency virus (HIV)
- Epstein-Barr virus
- Human T cell lymphotropic virus type 1
- Legionella pneumonia
- Mycoplasma
- Chlamydia
- Tuberculosis
- Immunodeficiency
- Acquired immunodeficiency syndrome (AIDS); especially in children
- Monoclonal or polyclonal gammopathy
- Common variable immunodeficiency
- Idiopathic LIP accounts for 20% of cases (Eur Respir J 2006;28:364)
- Autoimmune (most common)
Clinical features
- Very slowly progressive respiratory symptoms
- Dyspnea on exertion
- Dry cough
- Systemic symptoms such as malaise, fever and weight loss
- Duration of the symptoms prior to diagnosis can exceed a year
- Bibasilar inspiratory crackles on chest auscultation
Diagnosis
- Based on clinical, radiological and pathological findings (multidisciplinary diagnosis)
- No firm diagnostic criteria currently exist
Laboratory
- Dysproteinemia is often present
- Hypergammaglobulinemia is more common than hypogammaglobulinemia
- Restrictive pattern on pulmonary function tests
- Reduced forced vital capacity (FVC)
- Reduced diffusing capacity of the lung for carbon monoxide (DLCO)
Radiology description
- Chest radiography
- Bibasilar opacities with lower lobe predominance
- High resolution computed tomography (Eur J Radiol 2015;84:542,
Respirology 2016;21:600)
- Ground glass opacity with / without consolidation with lower lobe predominance
- Cyst formation and thickening of bronchovascular bundle and interlobular septa are often present
- Cysts often remain even after resolution of symptoms
Radiology images
Images hosted on other servers:
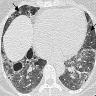
Ground glass opacity with cyst formation and nodules
Prognostic factors
- Prognosis varies from resolution without treatment to progression to end stage (Respirology 2016;21:600)
- Median survival: 11.5 years (Eur Respir J 2006;28:364)
- No characteristic prognostic factor has been found so far
Case reports
- 30 year old woman with LIP related to common variable immunoglobulin deficiency (Intern Med 2008;47:763)
- 35 year old man with HIV related LIP treated with antiretroviral therapy (Int J STD AIDS 2000;11:119)
- 35 year old woman with idiopathic LIP and good response to mycophenolate mofetil (Respir Med Case Rep 2013;9:27)
- 47 year old woman with LIP related to systemic lupus erythematosus and secondary Sjögren syndrome (BMJ Case Rep 2013 Aug 2;2013)
- 52 year old man with HIV related LIP treated with highly active antiretroviral therapy (Sex Transm Infect 2004;80:417)
Treatment
- No treatment data from a controlled study is available so far (Respirology 2016;21:600)
- Corticosteroid therapy is commonly used as a first line treatment and improves the symptoms in most cases
- Immunosuppression (eg, cyclophosphamide, azathioprine, cyclosporine A) may be used as a second line
- Treatment for underlying disease is also essential for secondary LIP
- Antiretroviral therapy can resolve HIV related LIP and its symptoms (Int J STD AIDS 2000;11:119, Sex Transm Infect 2004;80:417)
Gross description
- Ill defined lesion
- Mild increase in lung weight
Microscopic (histologic) description
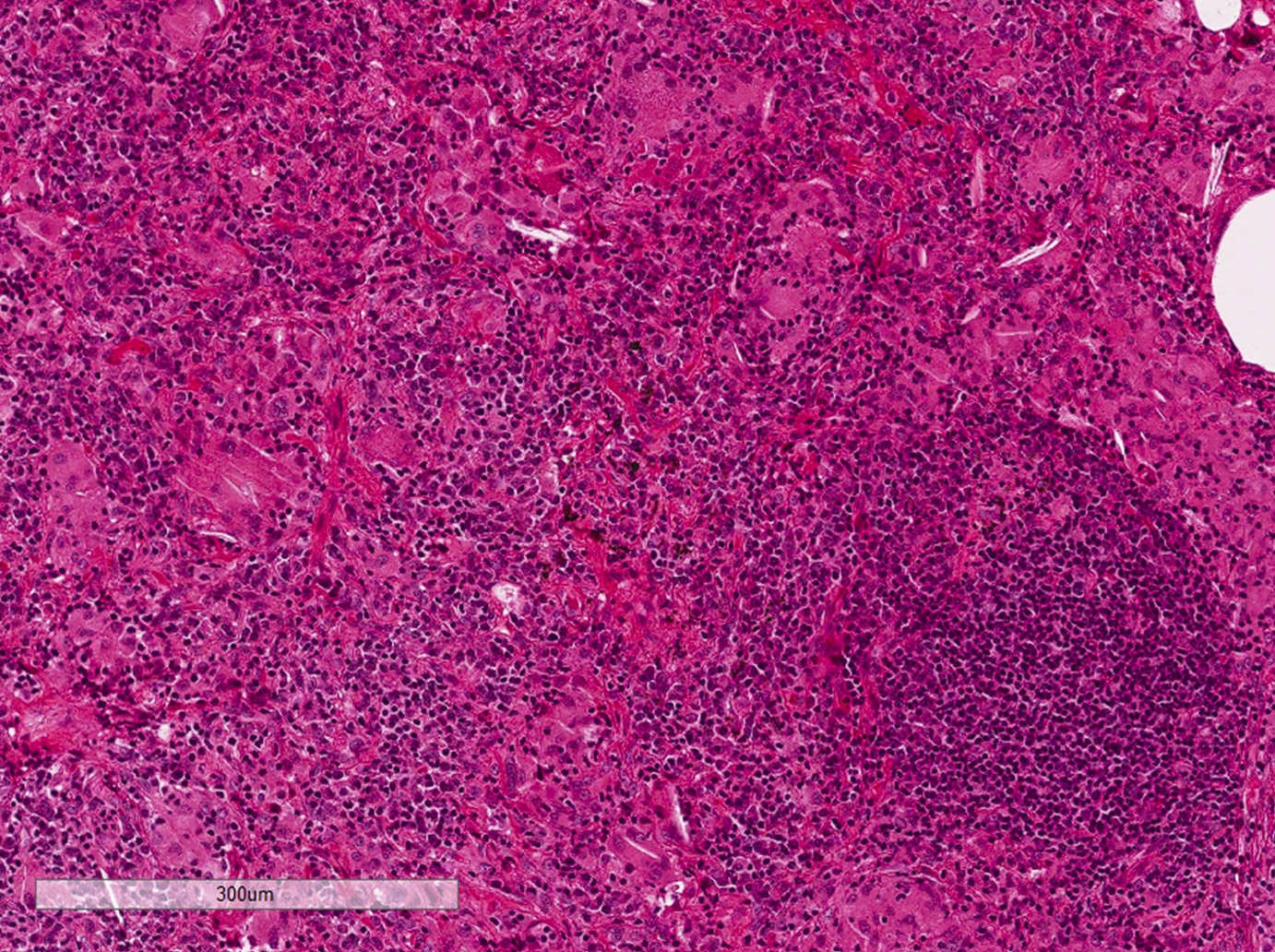
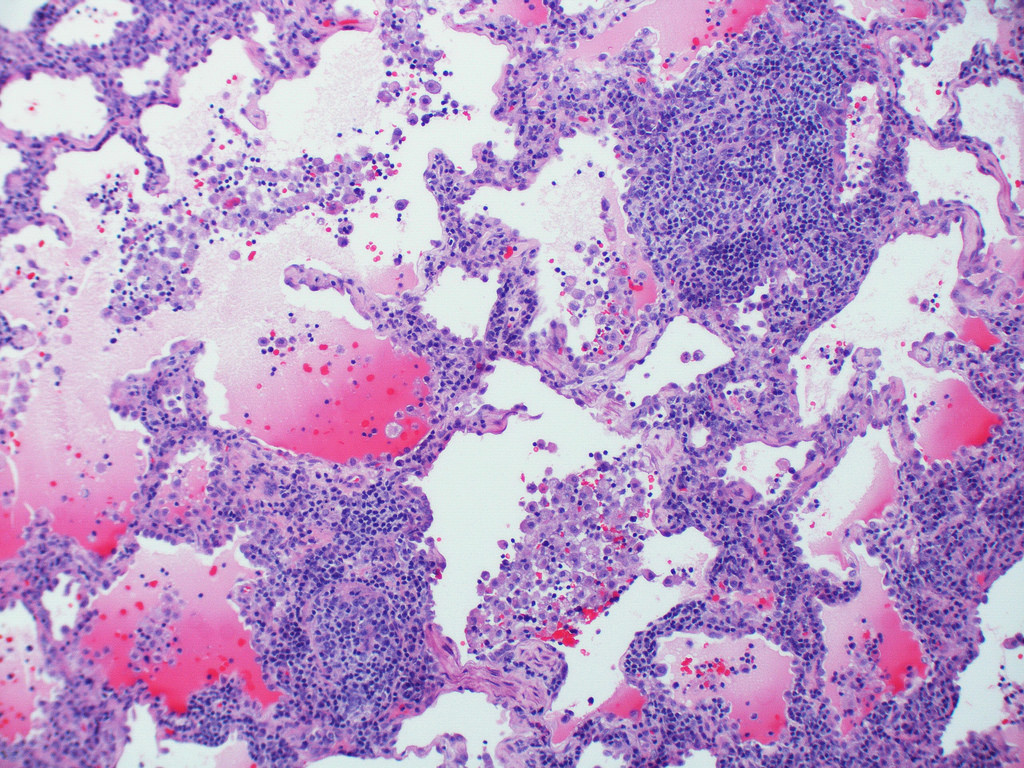
- Diffuse interstitial infiltration of polymorphous lymphocytes and plasma cells
- Lymphoid follicles with germinal centers, histiocytes and macrophages are often present
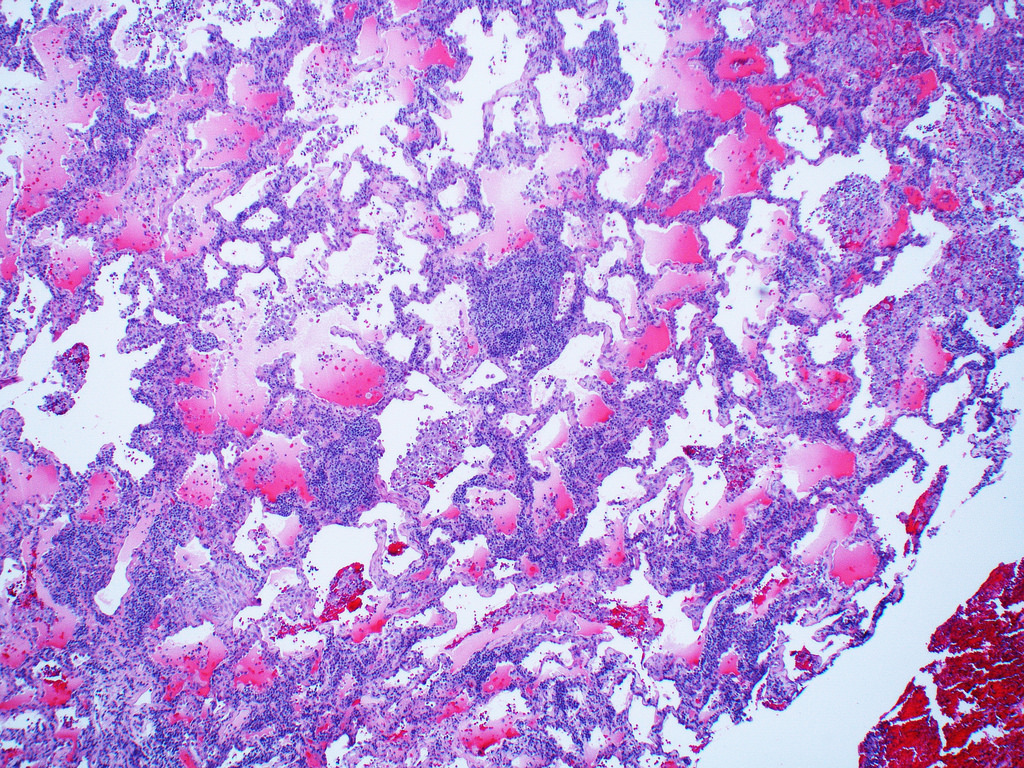
- Bronchovascular bundles and interlobular septa are usually involved
- Alveolar structure is often inflated and disrupted
- Typically CD8+ or CD4+ T cells or B cells predominate, with an admixture of other lymphocytes
- Immunohistochemistry or molecular testing are necessary to confirm polyclonality
- Additional findings
- Loose ill defined epithelioid granulomas
- Interstitial or intra-alveolar giant cells
- Intra-alveolar macrophages
- Type II pneumocyte hyperplasia
- Cyst formation without marked fibrosis
- Pertinent negative findings
- Loose and dense fibrosis (more common in fibrotic / cellular nonspecific interstitial pneumonia (NSIP))
- Fibroblastic focus (more common in usual interstitial pneumonia or fibrotic NSIP)
- Honeycomb change (more common in usual interstitial pneumonia or fibrotic NSIP)
- Organizing pneumonia (more common in hypersensitivity pneumonitis)
- See: Respirology 2016;21:600, Chest 2002;122:2150, Am J Respir Crit Care Med 2002;165:277
Microscopic (histologic) images
Contributed by Akira Yoshikawa M.D. and Yale Rosen, M.D.
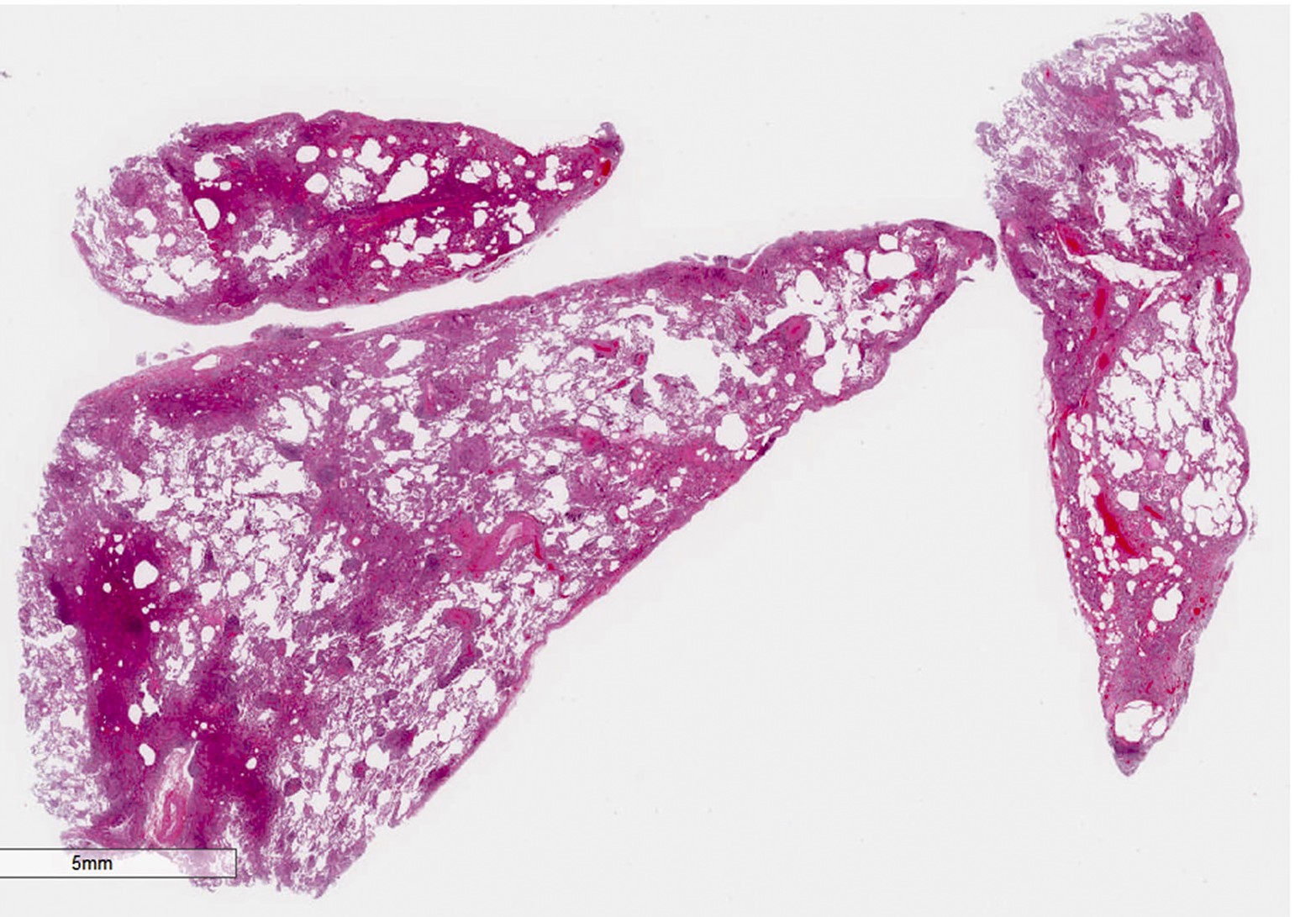
Low power
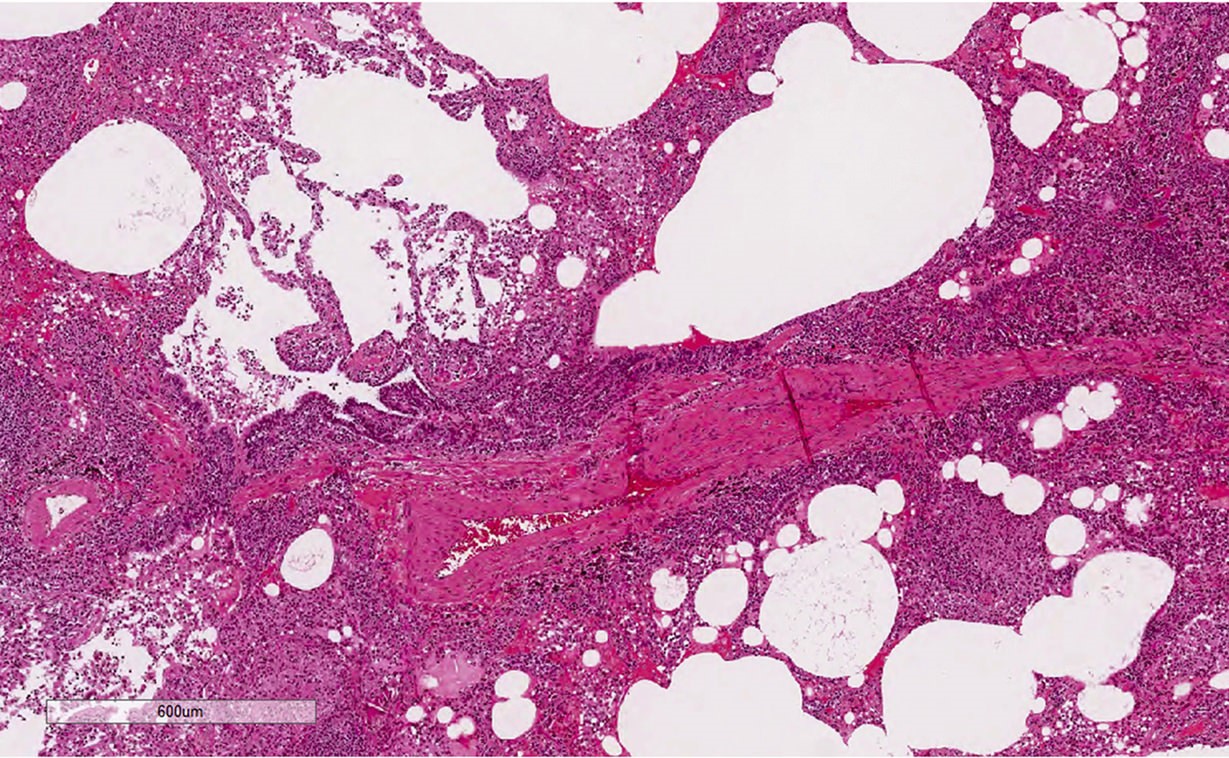
Bronchovascular bundle
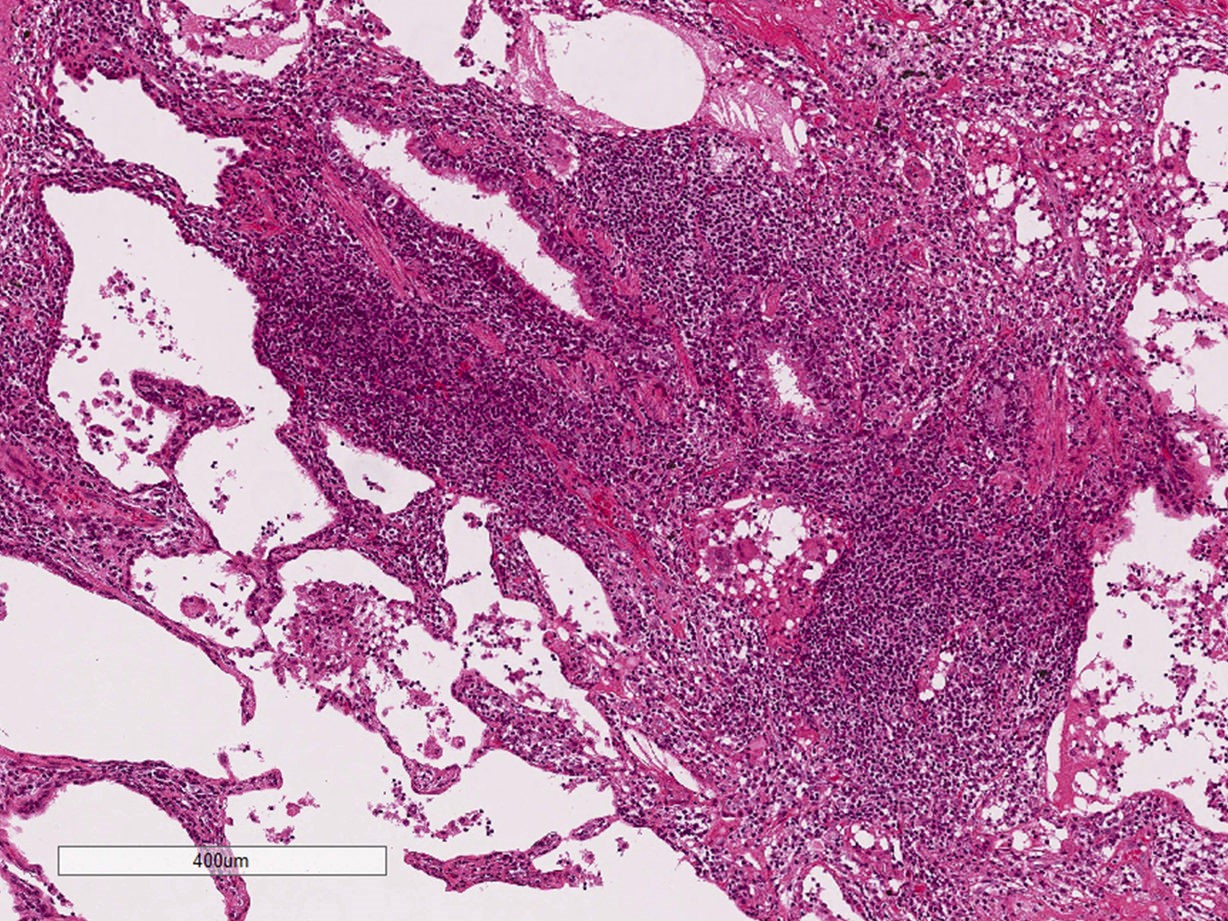
Architectural destruction
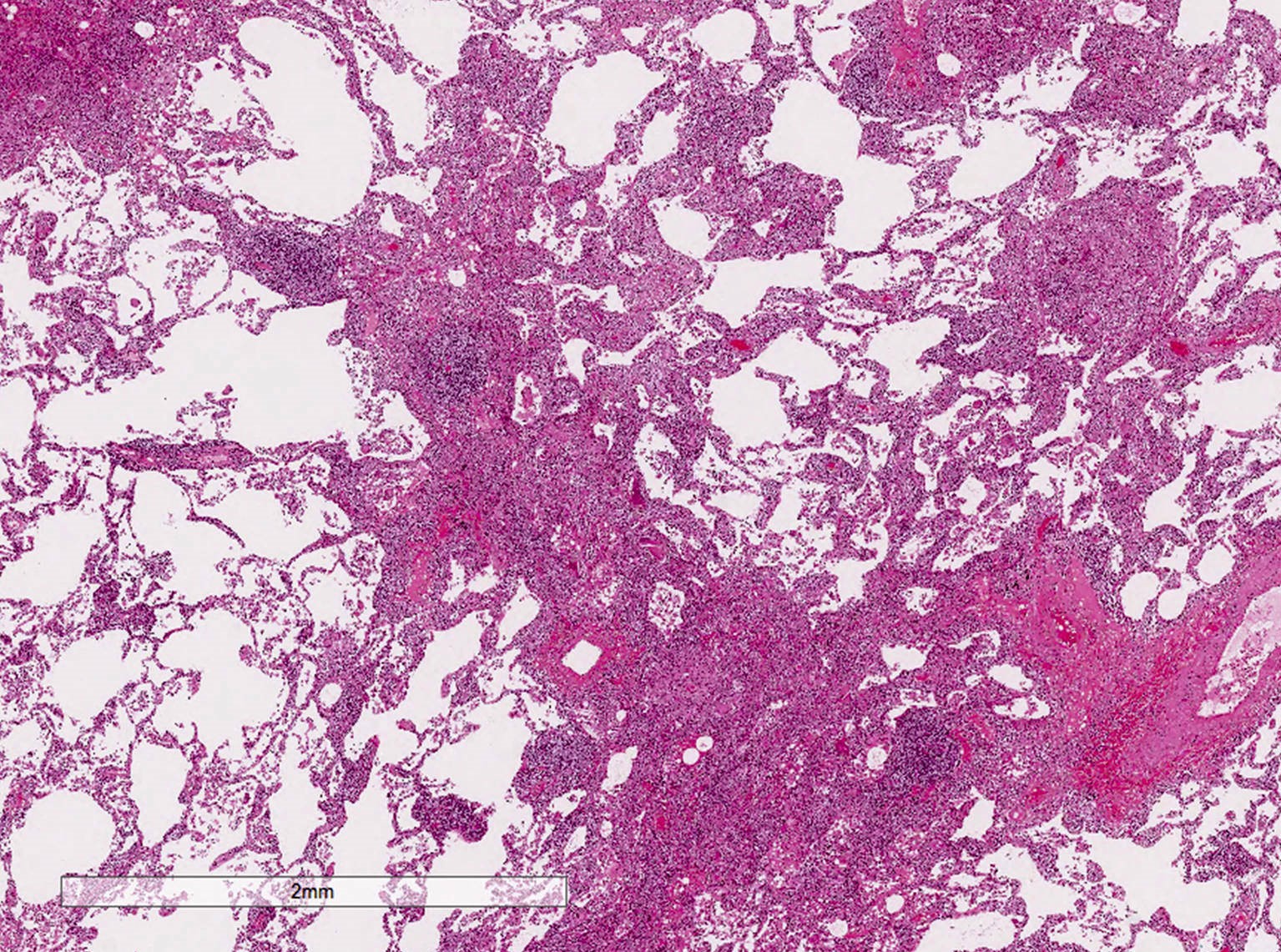
Septal thickening
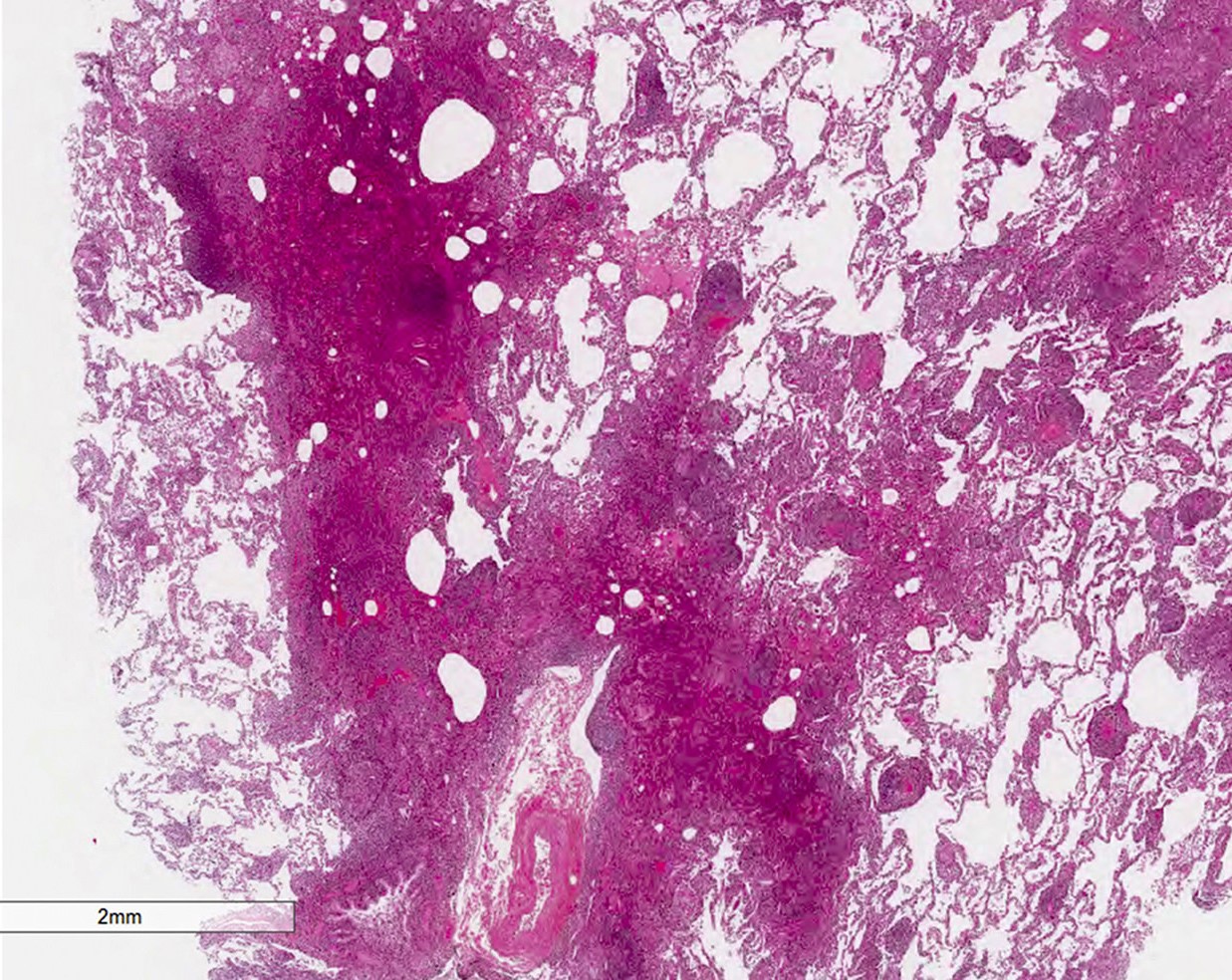
Massive lymphocytic infiltration
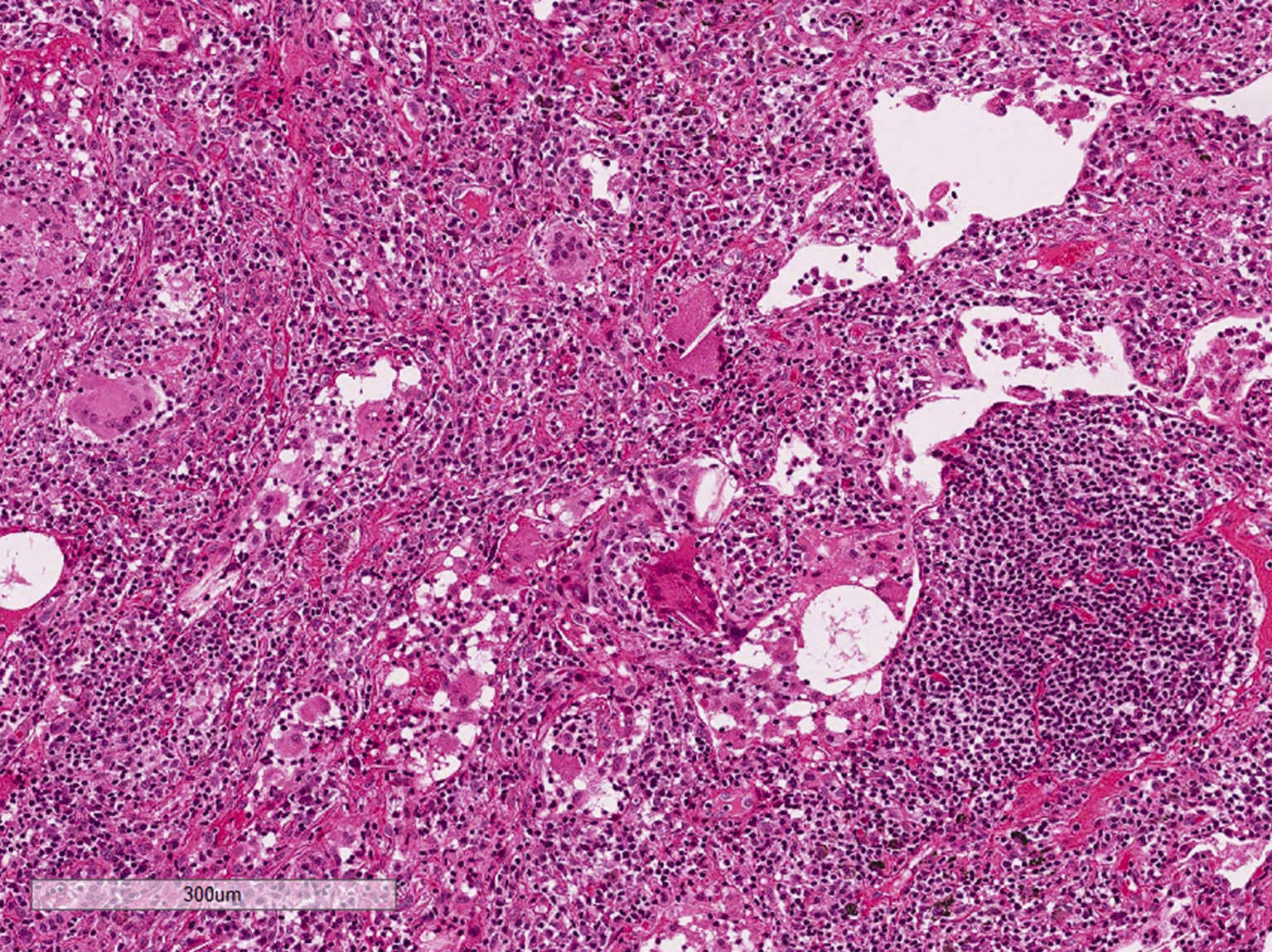
Giant cells and lymphocytes
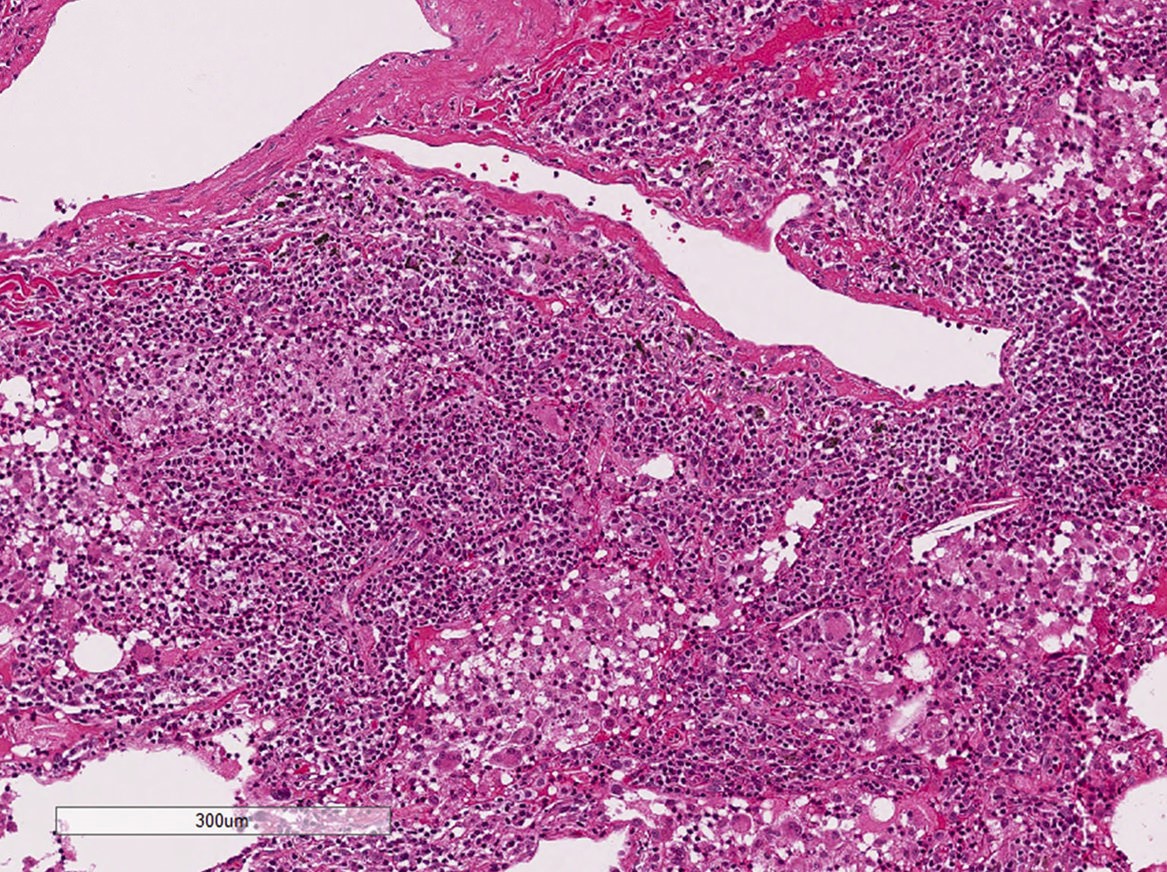
Lymphocytes and histiocytes
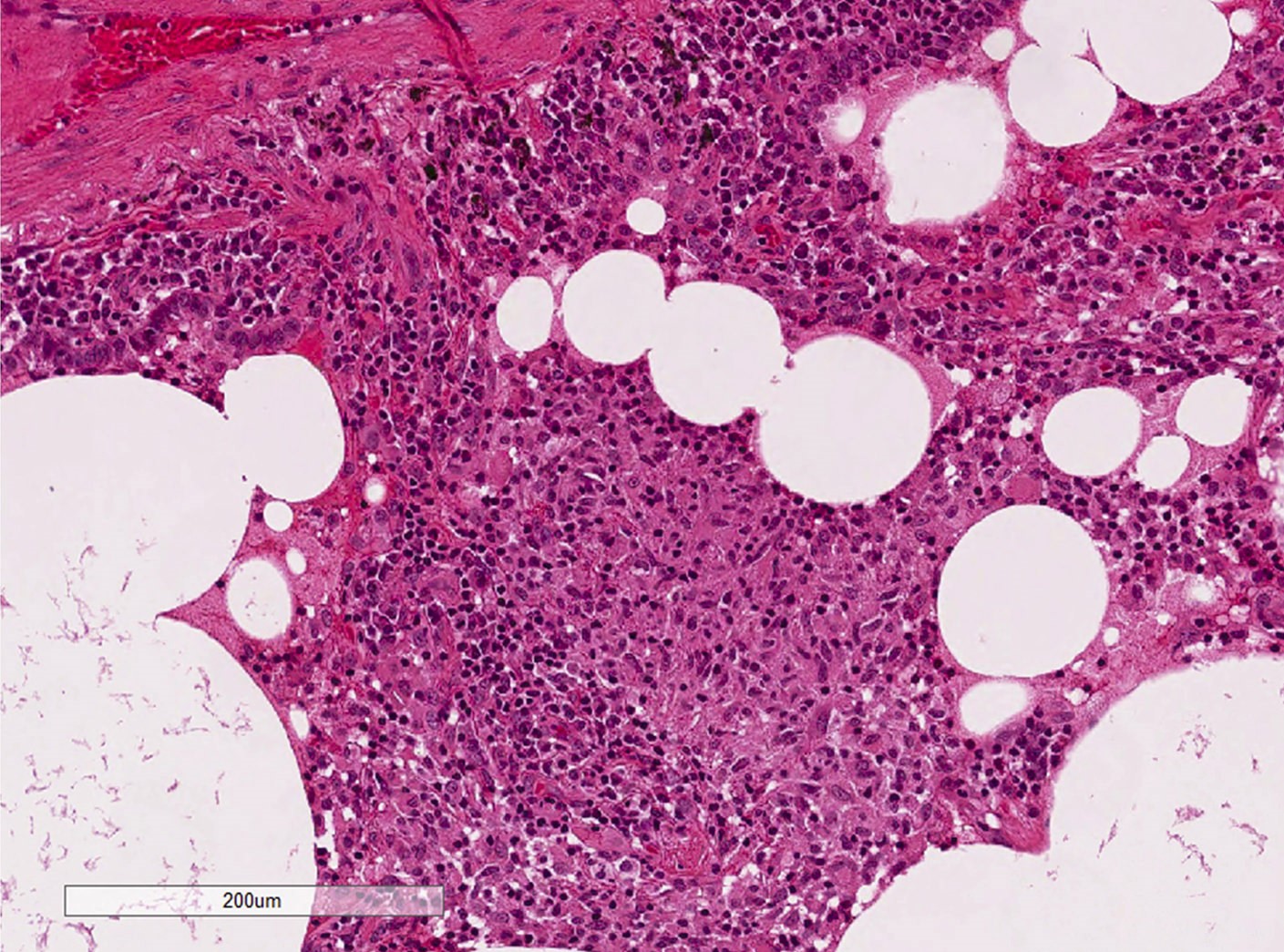
Granuloma and lymphocytes
Lymphoid aggregate and histiocytes
Diffuse cellular pattern
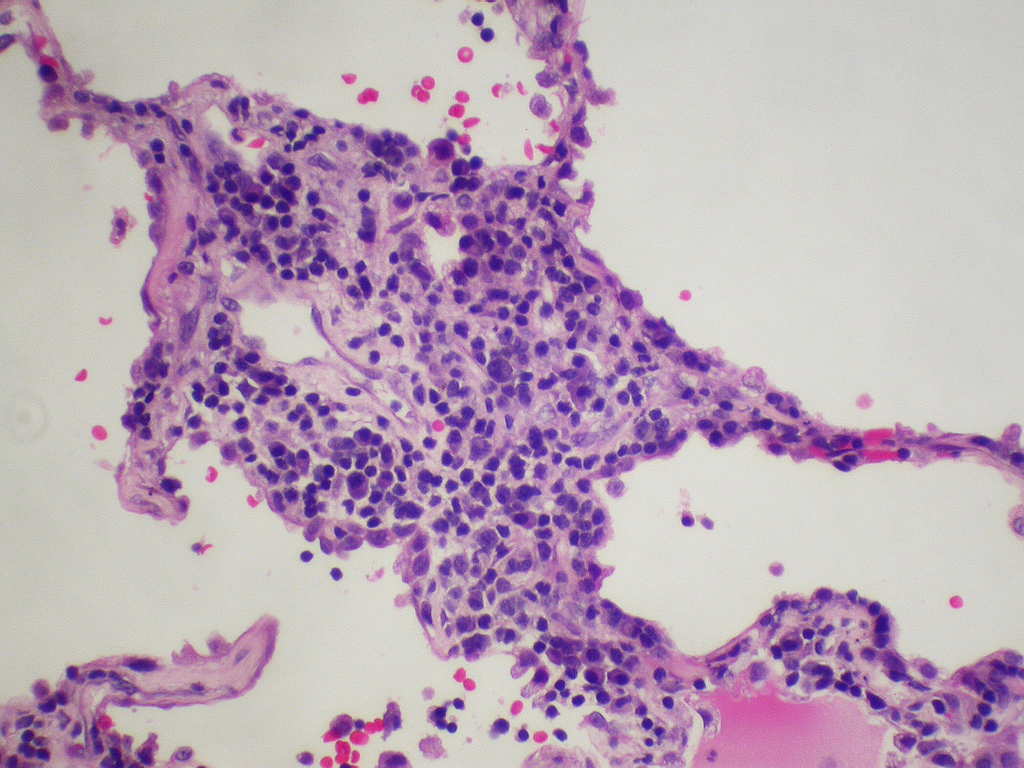
Lymphoplasmacytic infiltration
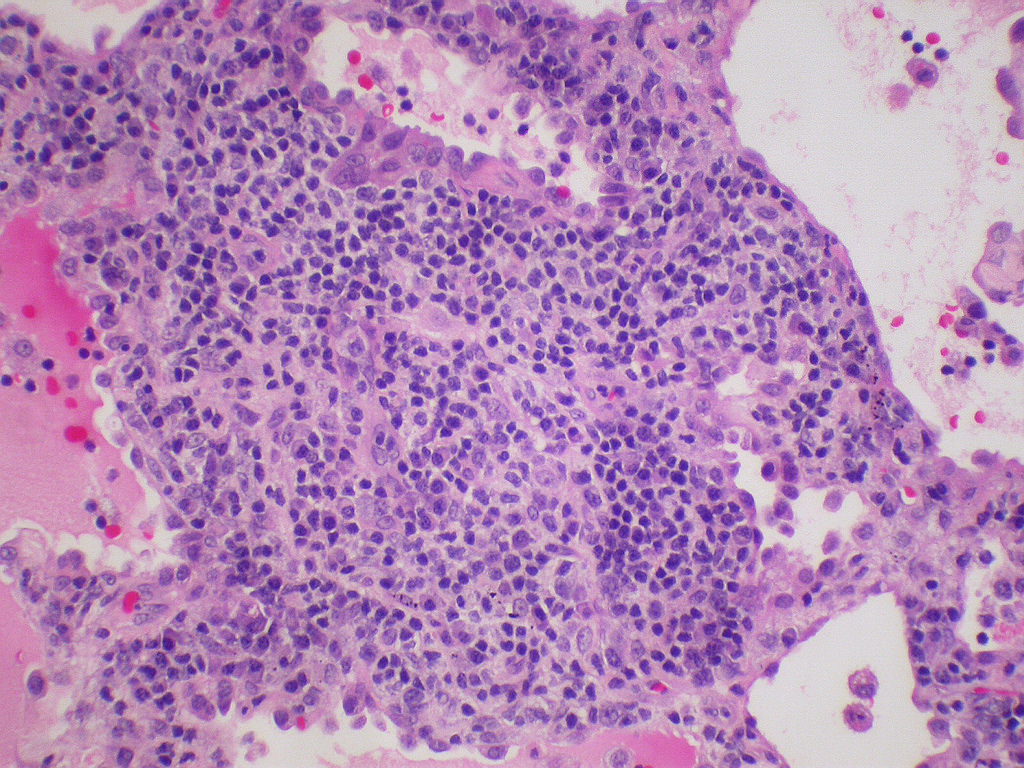
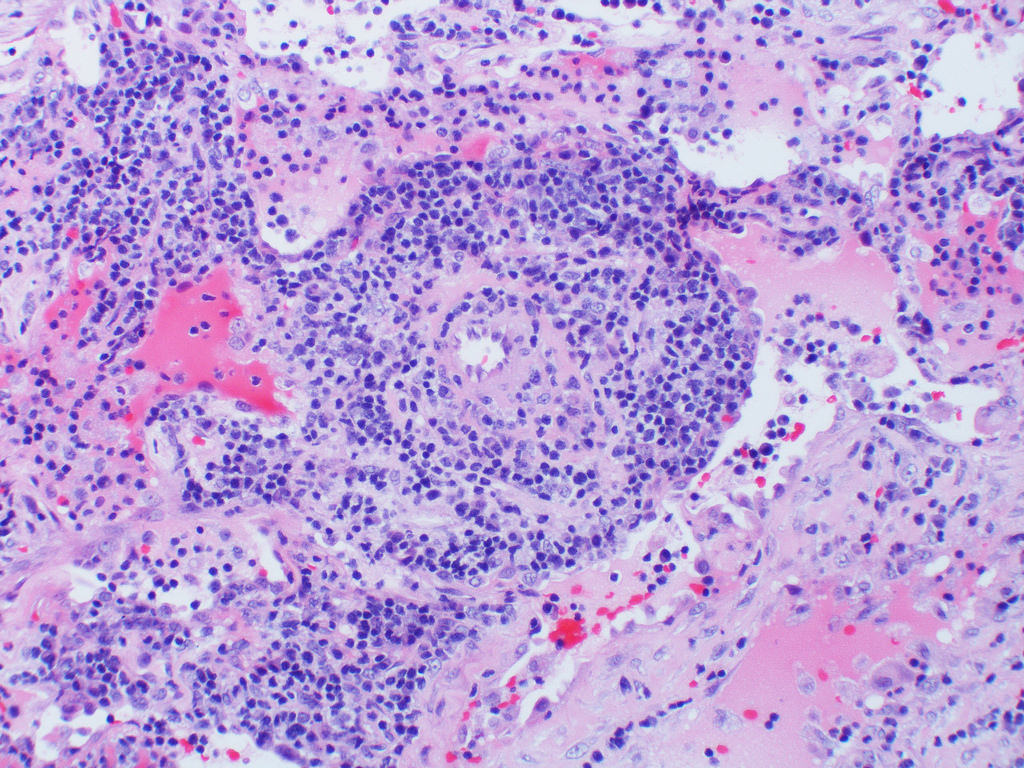
Lymphoplasmacytic infiltration
Images hosted on other servers:

LIP
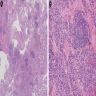
Numerous lymphoid follicles
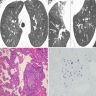
EBV related LIP
Cytology description
- Increase in the number of lymphocytes (especially CD8 positive) of bronchoalveolar lavage, without clonality (Eur Respir J 2006;28:364)
Positive staining - disease
Differential diagnosis
- Follicular bronchitis / bronchiolitis
- Lymphocytic infiltration into bronchial / bronchiolar walls with multiple lymphoid follicles
- No or slight lymphocytic aggregation in intralobular septa
- Hypersensitivity pneumonitis (HP)
- History of an antigen exposure such as animals, birds and chemicals
- Less diffuse but bronchocentric distribution
- Loose ill defined granuloma and giant cells can be seen in both LIP and HP
- Lymphoma, especially mucosa (bronchus) associated lymphoid tissue lymphoma
- Monomorphous infiltration of lymphocytes, distortion of alveolar architecture, Dutcher bodies, pleural infiltration are more common in lymphoma
- Immunohistochemistry is often helpful
- Nonspecific interstitial pneumonia
- Lymphoplasmacytic infiltration is also seen in NSIP but less severe
- Alveolar structure is usually preserved compared with LIP
Additional references
Board review style question #1
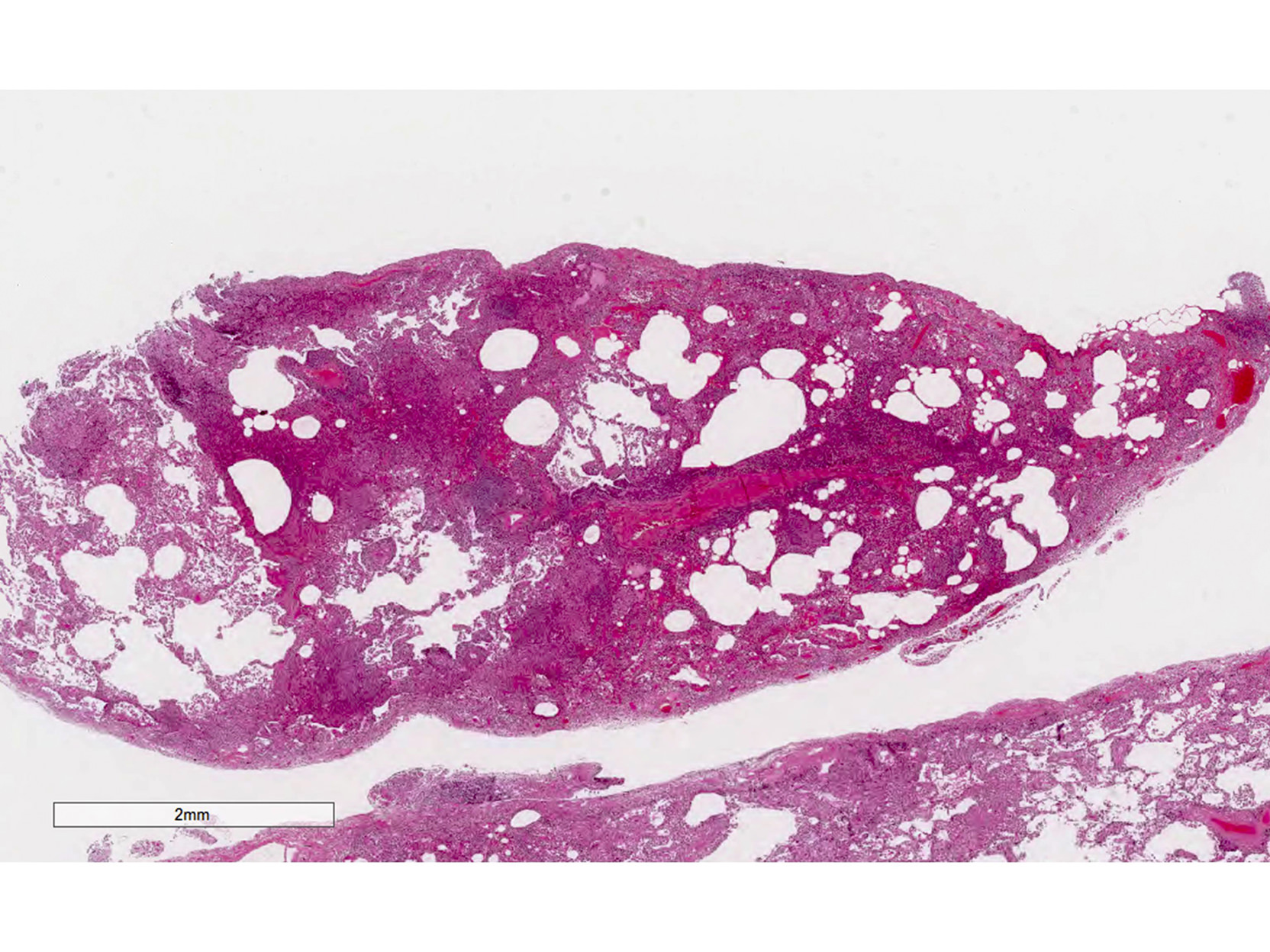

Which of following is not usually seen in this entity?
- Cyst formation
- Giant cells
- Honeycomb change
- Lymphocytic interstitial infiltration
- Nonnecrotizing granuloma
Board review style answer #1
C. Honeycomb change. Lymphoid interstitial pneumonia is predominantly a cellular interstitial pneumonia. Fibrotic processes such as dense fibrosis or honeycomb change usually exclude a diagnosis of LIP.
Comment Here
Reference: Lymphoid interstitial pneumonia
Comment Here
Reference: Lymphoid interstitial pneumonia
