

Muscle & peripheral nerve nontumor
Muscular dystrophies
Facioscapulohumeral muscular dystrophy
Author: Chunyu Cai, M.D., Ph.D.
Editorial Board Members: Meaghan Morris, M.D., Ph.D., Jared T. Ahrendsen, M.D., Ph.D.

Last author update: 10 October 2023
Last staff update: 10 October 2023
Copyright: 2020-2024, PathologyOutlines.com, Inc.
PubMed Search: Facioscapulohumeral muscular dystrophy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Clinical images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Molecular / cytogenetics images | Videos | Sample pathology report | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Cai C. Facioscapulohumeral muscular dystrophy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/muscleFSH.html. Accessed April 23rd, 2024.
Definition / general
- Facioscapulohumeral muscular dystrophy (FSHD) is an inherited muscle disorder, clinically characterized by weakness of the facial and shoulder girdle muscles followed by the leg and trunk muscles and genetically characterized by contraction or hypomethylation of the D4Z4 domain in individuals with 4qA haplotype, leading to toxic aberrant expression of DUX4 on chromosome 4q35 (Science 2010;329:1650)
Essential features
- Clinically characterized by
- Variable age of onset at any time from early childhood until adult life
- Involvement of facial and scapulohumeral muscles and sometimes the pelvic girdle
- Transmission usually by an autosomal dominant inheritance, rarely autosomal recessive
- Variable, nonlinear disease course, usually stable or slowly progressive but may have burst of disease activity with rapid functional decline (Brain 1954;77:169, Nat Rev Neurol 2023;19:91)
- Genetics: 95% of FSHD cases are due to contraction (1 - 10) of the D4Z4 repeats (normally 11 - > 100) on chromosome 4q35 in individuals with the 4qA haplotypes; these are referred to as FSHD1 (Science 2010;329:1650)
- Remaining 5% of patients with FSHD are caused by heterozygous mutations in genes that regulate the methylation of the D4Z4 domain, including SMCHD1, DNMT3B or LRIF1 in individuals with the 4qA haplotypes; these are referred to as FSHD2 (Nat Genet 2012;44:1370, Trends Mol Med 2021;27:123)
- 4qA and 4qB are 2 polymorphic allelic forms directly distal to D4Z4 on chromosome 4q35; although both alleles are equally common in the general population, FSHD is associated solely with the 4qA allele (Nat Genet 2002;32:235)
Terminology
- Landouzy-Dejerine disease
- Landouzy-Déjerine atrophy
- Facioscapulohumeral atrophy
- Facioscapulohumeral myopathy
ICD coding
Epidemiology
- Second most common muscular dystrophy, affecting ~1 in 8,000 individuals, with estimated prevalence of 3.2 - 4.6 per 100,000 people
- Affects all age groups, peak age of presentation is 15 - 30 years
- Affects both males and females
- Does not appear to preferentially affect specific racial groups
- Reference: StatPearls: Facioscapulohumeral Muscular Dystrophy [Accessed 30 August 2023]
Sites
- Skeletal muscle, particularly face, shoulder girdle and upper arms; axial and leg muscles can also be affected (Pract Neurol 2016;16:201)
Pathophysiology
- Muscle wasting found in FSHD is associated with derepression of the DUX4 gene distal to the D4Z4 repeats
- DUX4 codes for a transcription factor that is normally expressed in small amounts in early embryological development and is found in the testis and pluripotent cells but is silenced in adult somatic tissue
- Even small amounts of DUX4 protein in postnatal humans is toxic to skeletal muscle and results in apoptosis through a cascade of events including disruption of RNA metabolism and induction of oxidative stress
- In healthy individuals, the D4Z4 macrosatellite domain consists of 8 to ~100 tandem repeat units of 3.3 kb each
- Each D4Z4 tandem repeat unit contains a retrogene that includes the full open reading frame of DUX4
- D4Z4 domain is epigenetically silenced by methylation that prevents DUX4 expression
- In FSHD, there is gain of function aberrant expression of the toxic DUX4 protein either due to D4Z4 contraction (FSHD1) or mutations in chromatin repressing genes that leads to hypomethylation of the D4Z4 domain (FSHD2)
- FSHD is seen exclusively with the 4qA haplotype, because a DUX4 polyadenylation signal (PAS) that functions to stabilize DUX4 mRNA is only present in the 4qA haplotype
- References: Neurology 2021;96:e1054, Nat Rev Neurol 2023;19:91
Etiology
- Hereditary
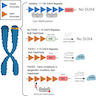

Clinical features
- Hallmark is asymmetric weakness and atrophy of muscles of the face, shoulder girdle and upper arms
- Axial and leg muscles can also be affected
- Degree of muscle involvement is highly variable
- Severe, early onset cases characterized by generalized weakness and extra muscular manifestations include sensorineural hearing loss, retinal vasculopathy, right bundle branch block, restrictive lung disease and the possibility of cognitive impairment and epilepsy
- Severe form is associated with very short D4Z4 domain (1 - 3 repeats)
- Reference: Nat Rev Neurol 2023;19:91
Diagnosis
- Diagnosis is based on clinical phenotype and genetic testing (see Diagrams / tables) (Neuromuscul Disord 2017;27:782)
Laboratory
- Current CLIA laboratory testing for FSHD is by Southern blotting of EcoRI restriction enzyme digests to evaluate the number of D4Z4 repeats for FSHD1 and by genetic sequencing for FSHD2 related genes (Neurology 2021;96:e1054)
- FSHD1: D4Z4 contraction median 6 repeats (interquartile range [IQR] 4 - 7) repeats with 4qA haplotype
- FSHD2: D4Z4 median 15 (IQR 12 - 22) repeats with 4qA haplotype and concurrent SMCHD1, DNMT3B or LRIF1 mutations
- Non-FSHD: D4Z4 median 28 (IQR 19 - 40) repeats
- A new testing method based on methylation of the D4Z4 repeat was published in 2022 which can identify FSHD1 and FSHD2 simultaneously, requires less DNA, less laboratory effort and is more sensitive (Brain 2023;146:1388)
- Creatine kinase (CK) is normal to elevated and usually does not exceed 3 - 5 times normal upper limits (Br Med J 1971;3:464)
Radiology description
- Muscle MRI is used for the diagnosis and monitoring of disease progression
- MRI with short TI inversion recovery (STIR) positive signal significantly correlates with active myopathy and DUX4 target gene expression in FSHD patients (Hum Mol Genet 2019;28:476)
- Rate of muscle fatty replacement has been used as a measure of disease progression and correlates with the severity of STIR positive signals (J Neurol 2019;266:1127)
- Whole body muscle MRI quantitative fat analysis can be used to assess disease progression and potential therapeutic effect (Neurology 2022;99:e877)
Prognostic factors
- There is an inverse correlation between the number of D4Z4 repeats and clinical severity; the shorter the D4Z4 domain, the more severe phenotype and the earlier the age of onset (Ann Neurol 1996;39:744)
Case reports
- 33 year old man with FSHD1 and multiple sclerosis (Acta Myol 2020;39:29)
- 53 year old man with homozygous nonsense variants in LRIF1 (Neurology 2020;94:e2441)
- 59 year old man presented with muscle atrophy, dyspnea and congestive heart failure (eNeurologicalSci 2020;21:100284)
Treatment
- Aerobic exercise and cognitive behavioral therapy may slow down disease progression (Neurology 2016;86:1700)
- A phase 2 clinical trial (NCT04003974) showed preliminary data that losmapimod (Fulcrum Therapeutics) slowed disease progression in FSHD patients (Skelet Muscle 2022;12:1)
Clinical images
Images hosted on other servers:
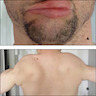
Weakness of lips and shoulder muscles
Microscopic (histologic) description
- Pathology can be quite variable and ranges from mild nonspecific changes to marked dystrophic changes
- Most common feature is chronic myopathy with excessive fiber size variation; small and large fibers are mixed type I and II, mimicking neurogenic atrophy
- May have lobulated myofiber changes (see Cases 1 - 3)
- Often shows inflammation (usually perivascular) that resembles inflammatory myopathy
- May or may not have diffuse MHC1 upregulation in myofibers; necrotic fibers and regenerative fibers are usually scanty
- May have prominent fatty replacement and fibrosis (see Case 2)
- May have rimmed vacuoles (Acta Neuropathol 2004;108:257)
- References: Muscle Nerve Suppl 1995;2:S56, Am J Med Genet A 2018;176:1760
Microscopic (histologic) images
Contributed by Chunyu Cai, M.D., Ph.D.
Case 1: 70 year old man with genetically confirmed FSHD1 (8 D4Z4 repeats on a 4qA haplotype)

Chronic
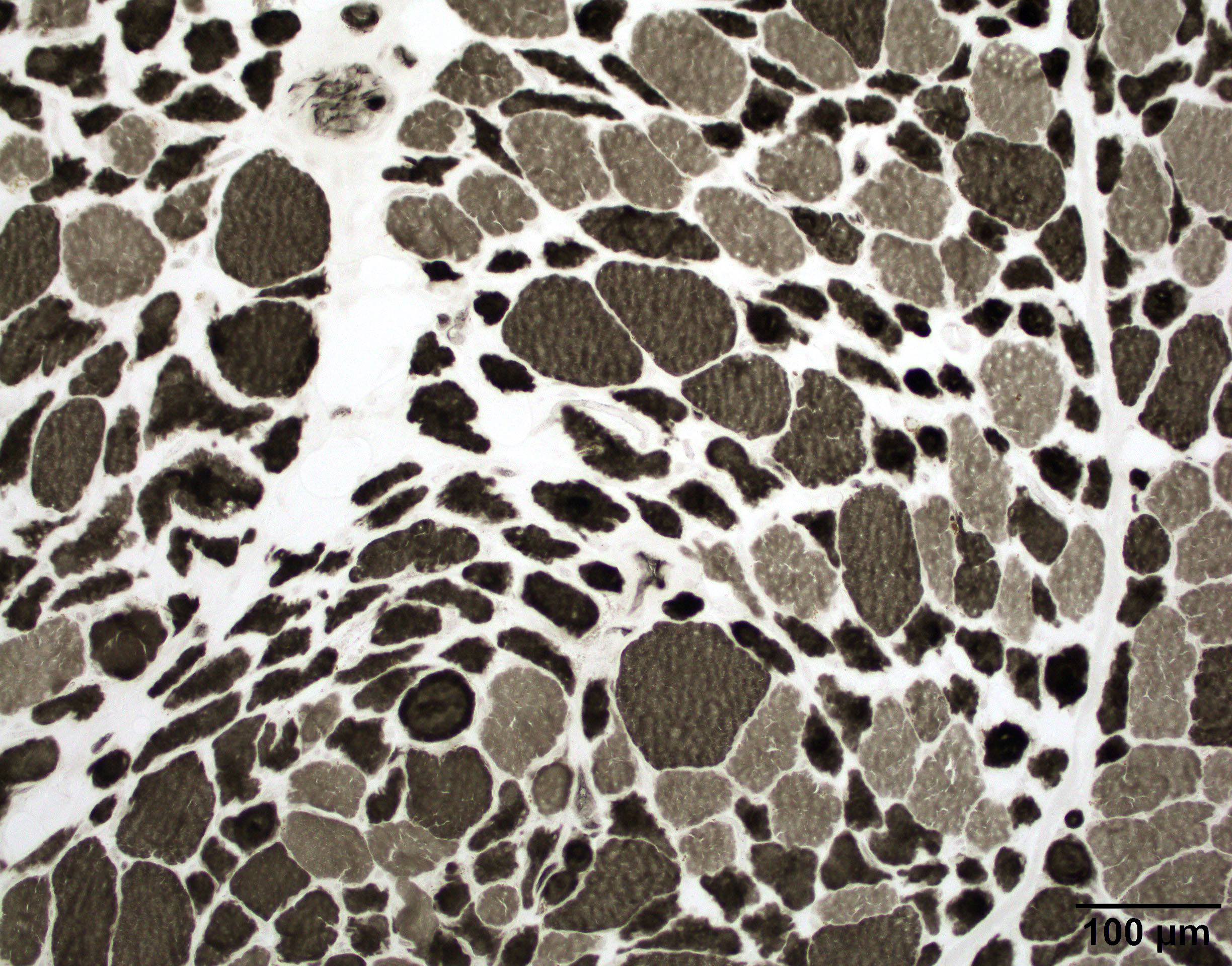
Fiber size variation

Fiber type grouping
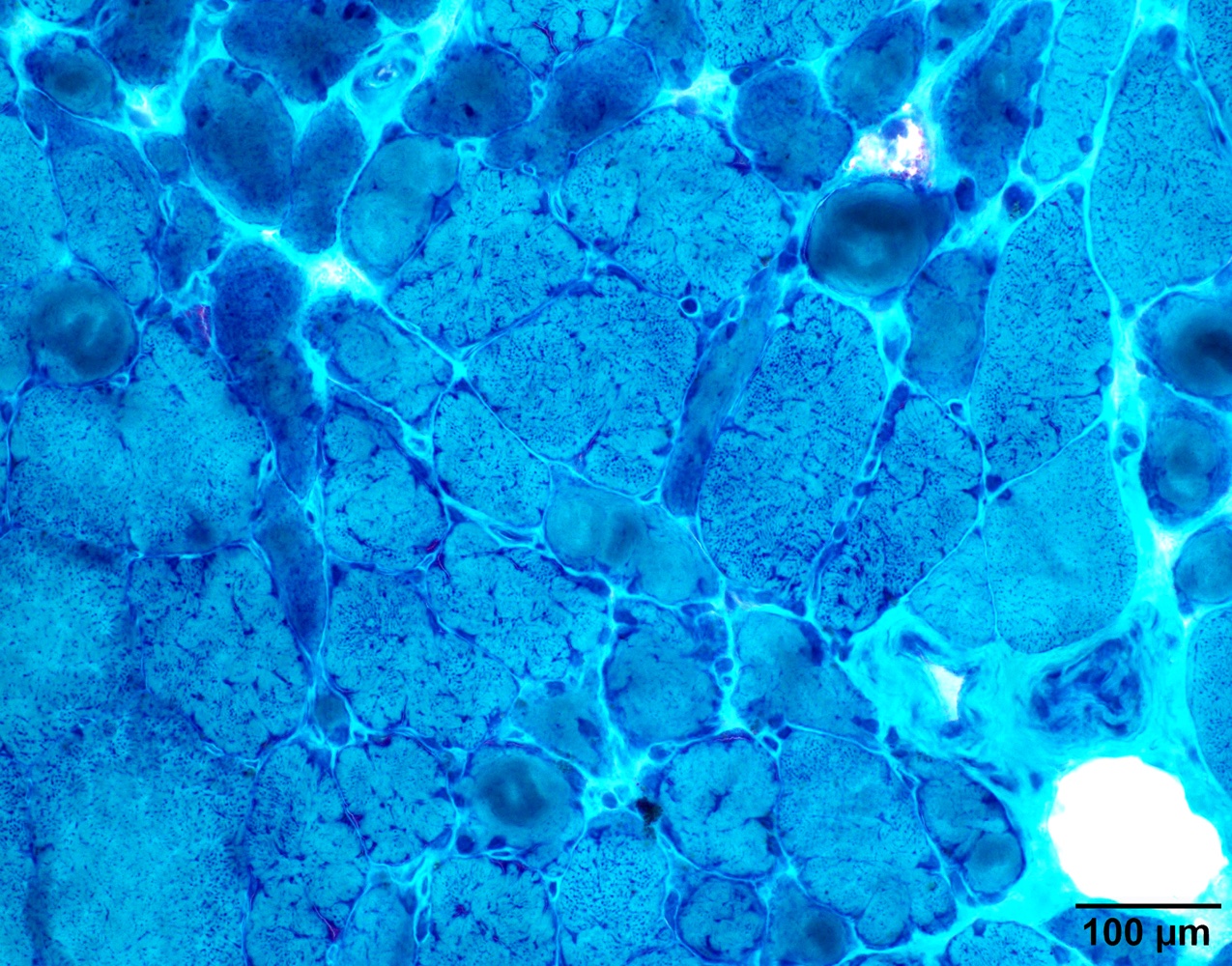
Lobulated internal architecture

Lobulated internal architecture
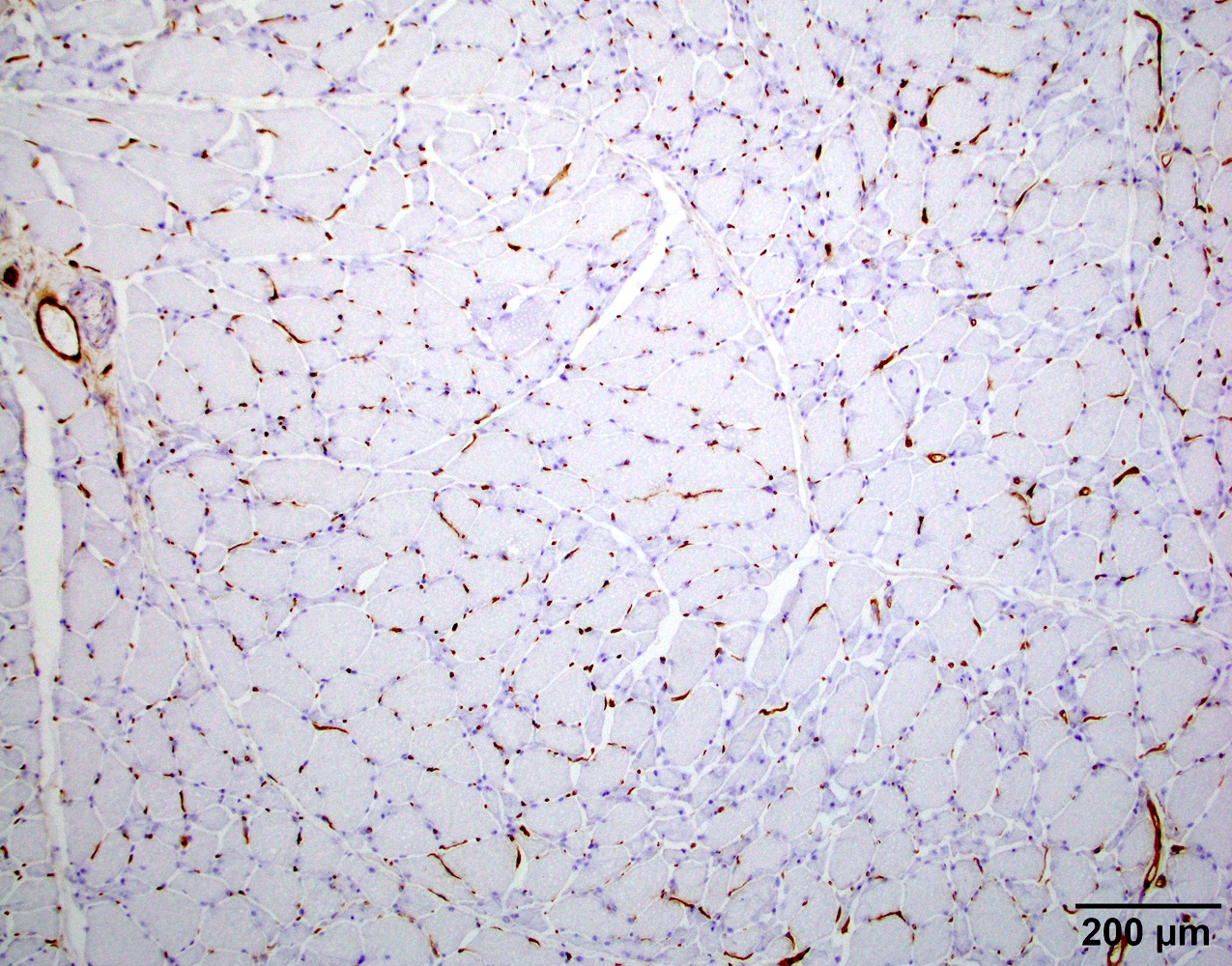
MHC1

Lobulated fiber on EM

Misoriented sarcomere on EM
Case 2: 63 year old woman with genetically confirmed FSHD1 (2 D4Z4 repeats on a 4qA haplotype)

Chronic myopathy with inflammation
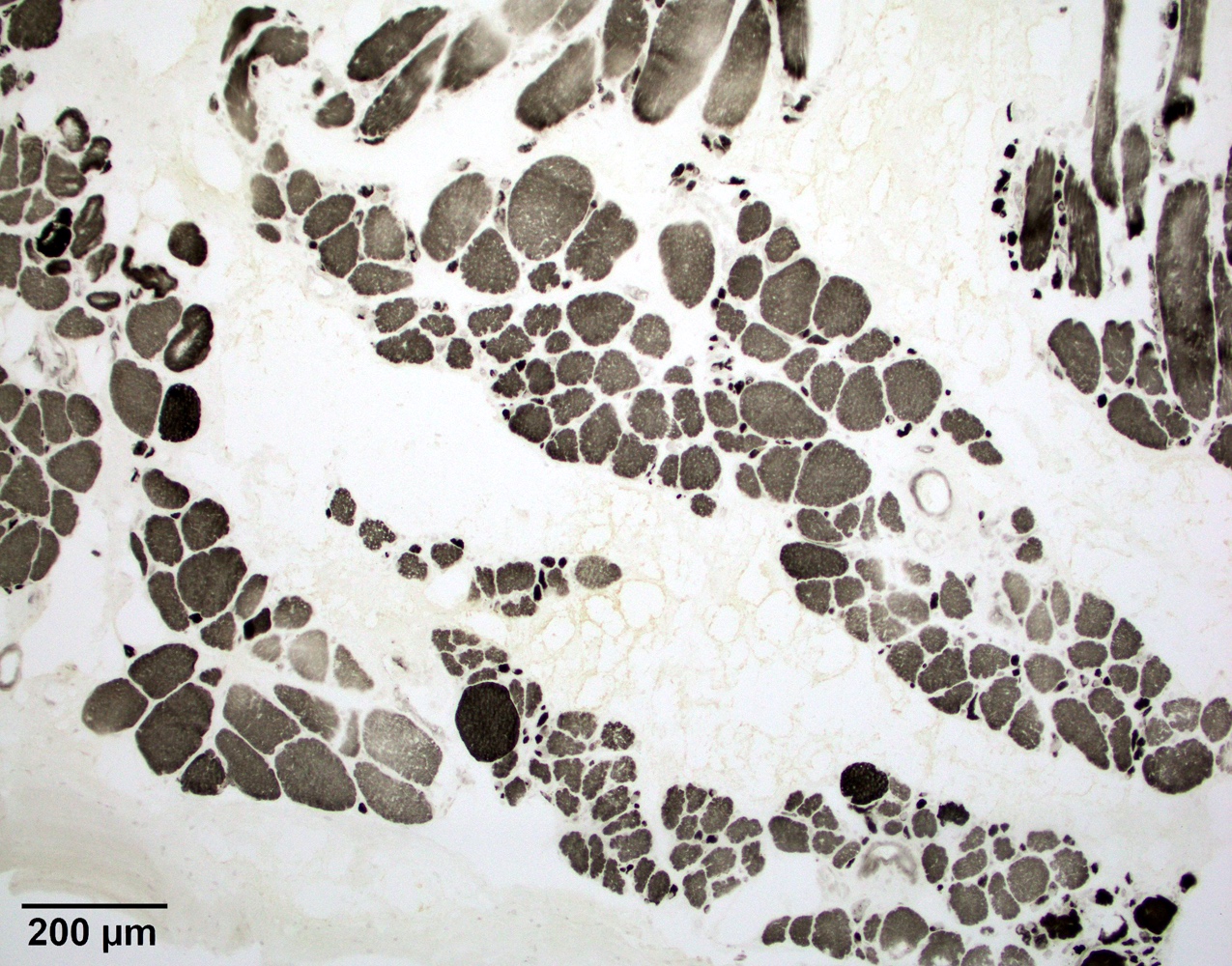
Type 1 predominance
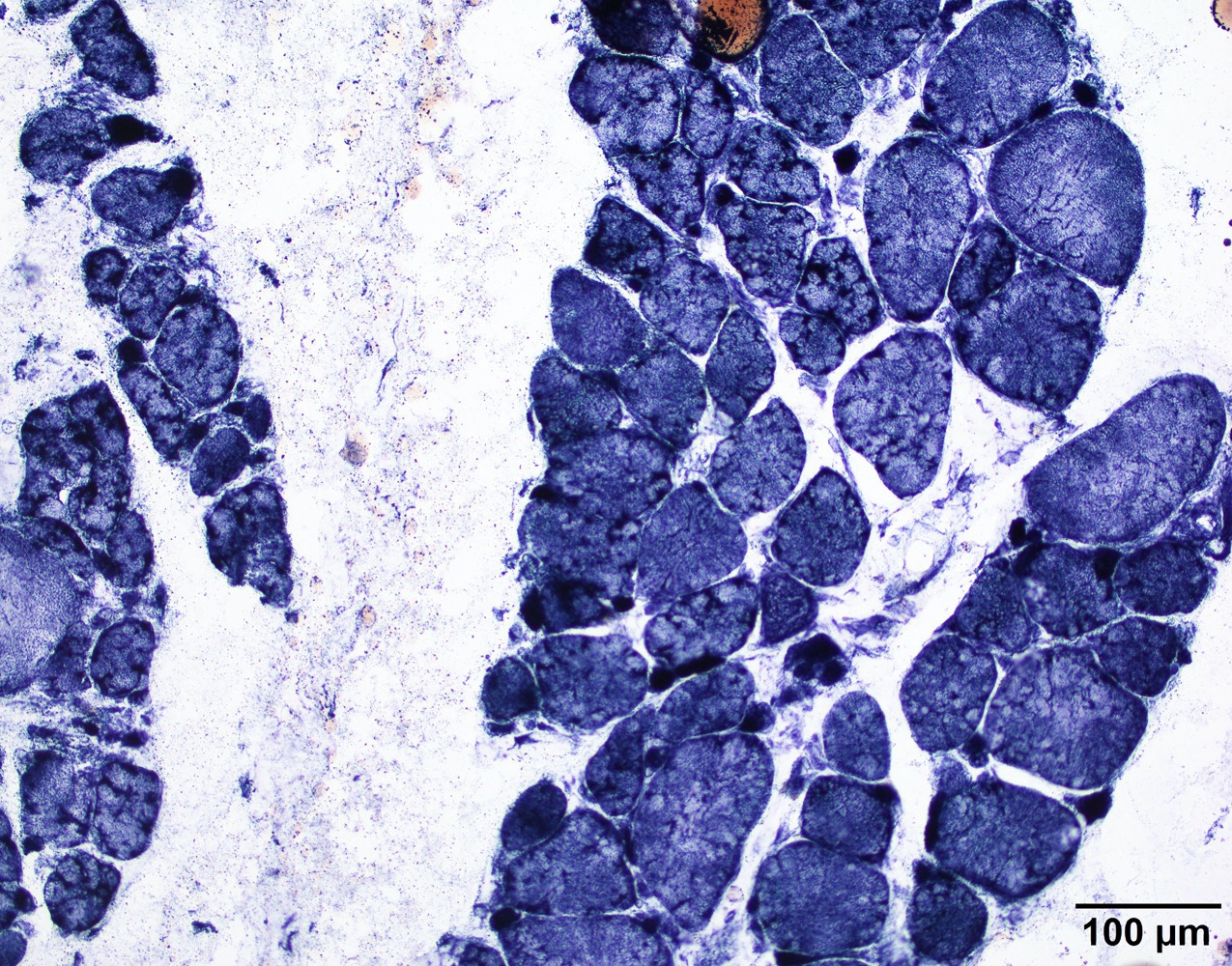
Lobulated internal architecture NADH

MHC1
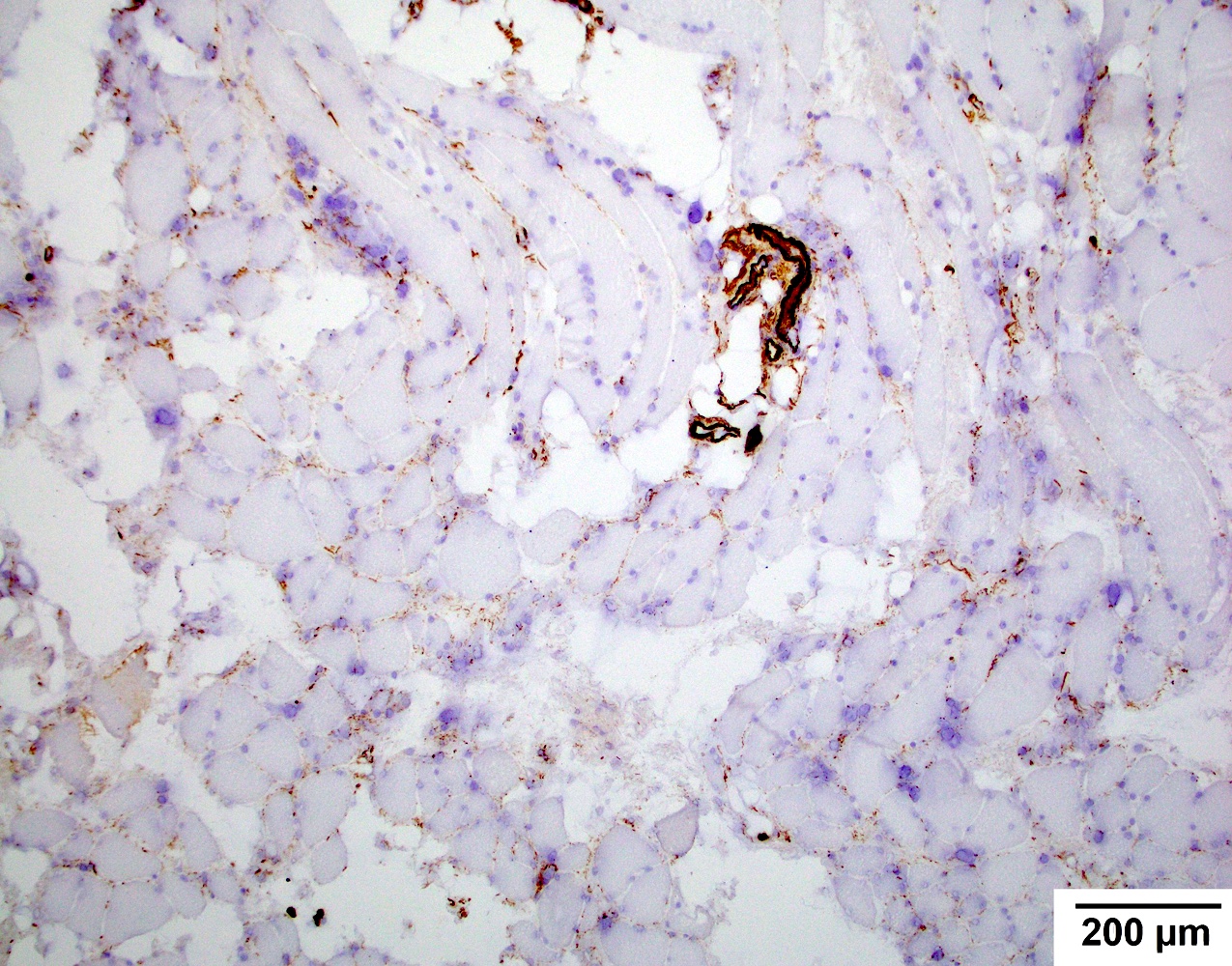
Terminal complement complex
Case 3: 66 year old man with heterozygous pathogenic mutation of SMCHD1 and a FSHD phenotype, consistent with FSHD2

FSHD2 muscle biopsy

FSHD2 ATPase 4.3

Hyaline bodies GT
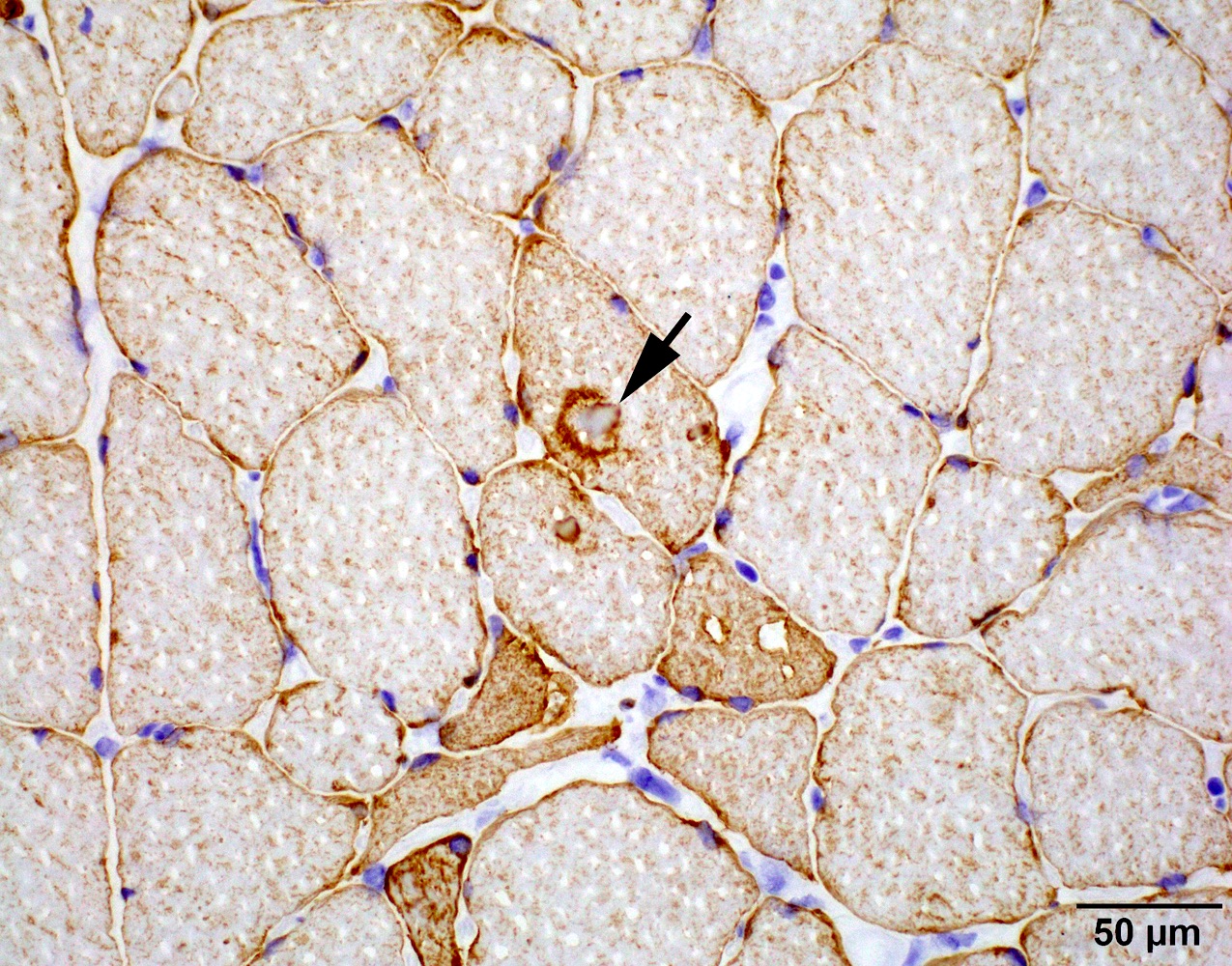
Hyaline bodies, desmin
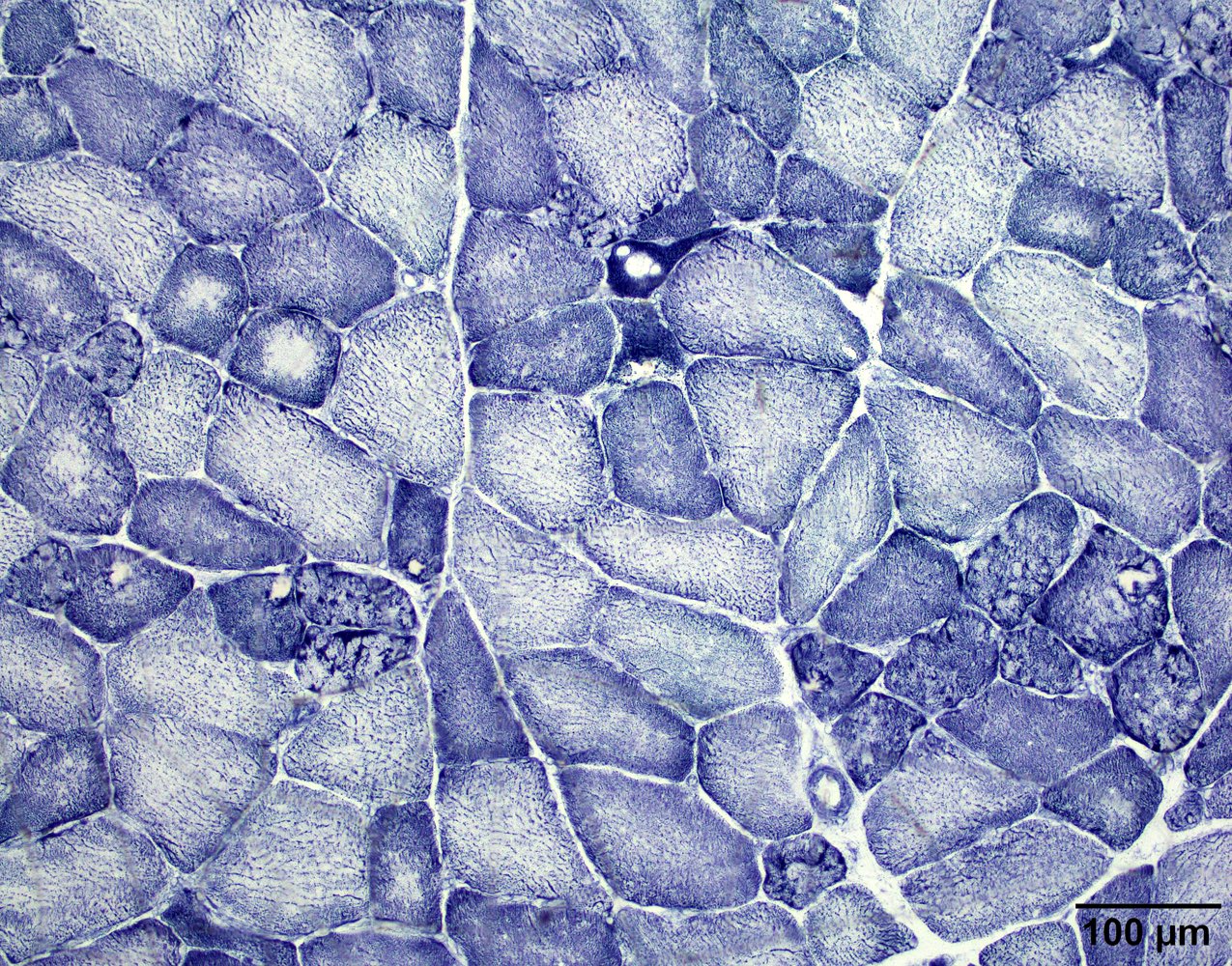
Lobulated internal architecture, NADH
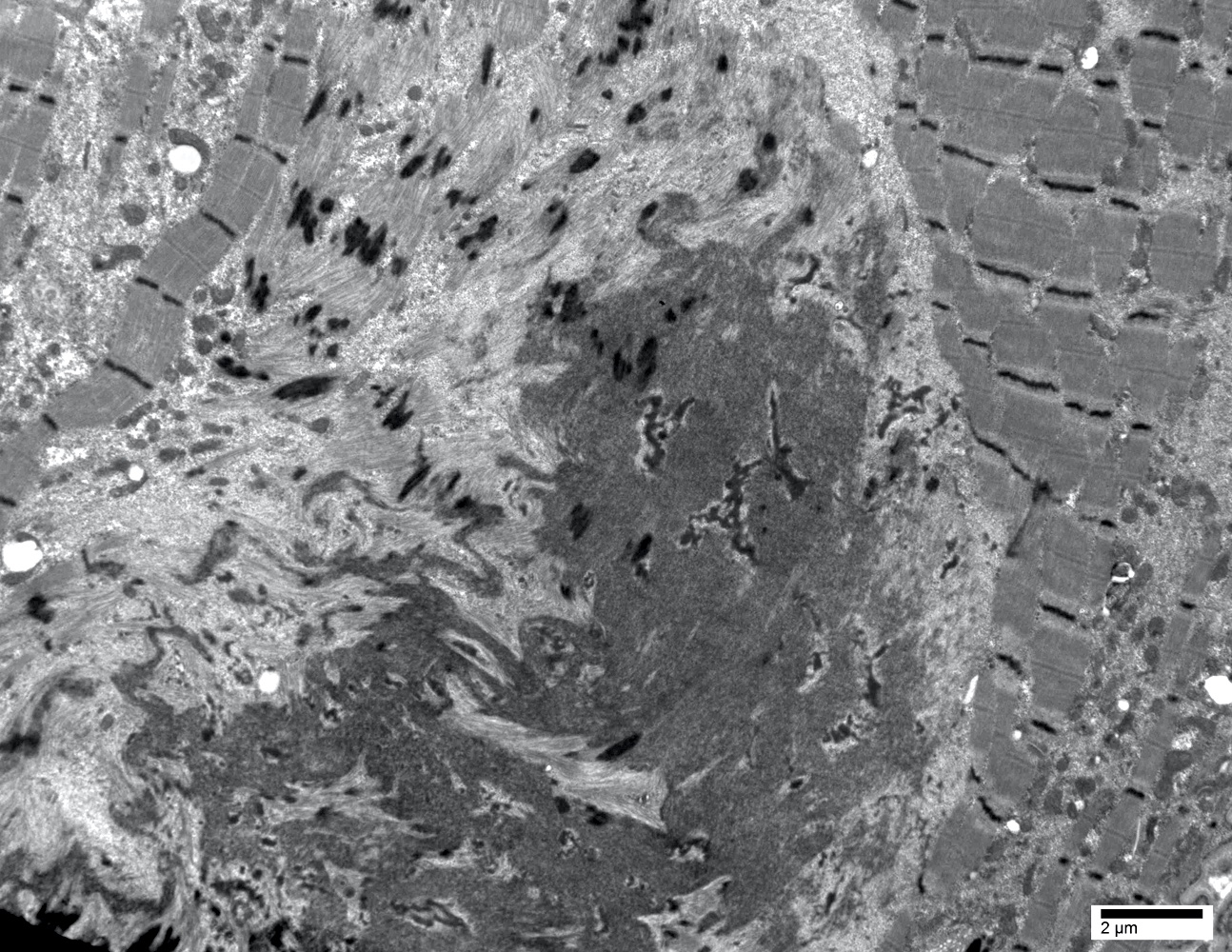
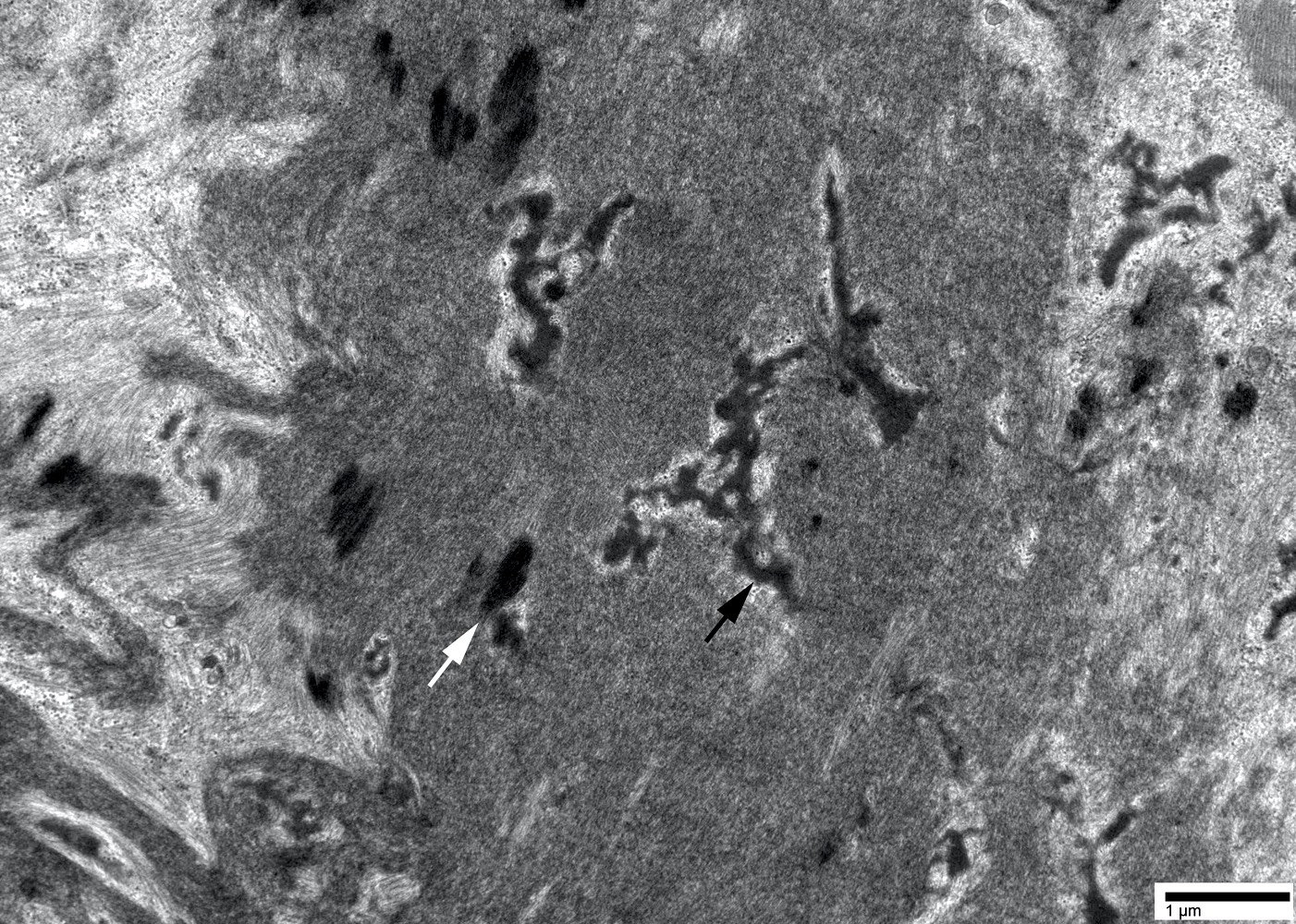
EM hyaline body
Positive stains
- May have capillary or sarcolemma C5b9 (terminal complement complex) deposition (Hum Mol Genet 2019;28:476)
Negative stains
- Most cases did not have MHC1 myofiber expression, although examples of diffuse MHC1 upregulation in myofibers exist (Washington University: Facioscapulohumeral (FSH) Dystrophy [Accessed 30 August 2023])
Electron microscopy description
- Lobulated architecture due to myofibril misorientation and mitochondria maldistribution (see Case 1)
- May have rimmed vacuoles with tubulofilamentous inclusions or cytoplasmic bodies (J Neurol 2010;257:1108)
Electron microscopy images
Molecular / cytogenetics description
- Genetics: 95% of FSHD patients carry 1 allele with a reduced number (1 - 10) of D4Z4 repeat (normally 11 - > 100) units on chromosome 4q35 associated with specific haplotypes (FSHD1) (Science 2010;329:1650)
- Of the remaining 5% of patients with FSHD phenotype (FSHD2), most cases have been explained by heterozygous mutations in the SMCHD1 / SCHMD1 (structural maintenance of chromosomes flexible hinge domain containing 1) gene (Nat Genet 2012;44:1370)
- Each of the repeated segments in the D4Z4 region contains a copy of the DUX4 gene; the copy closest to the end of chromosome 4 is called DUX4, while the other copies are described as DUX4-like or DUX4L
- Entire D4Z4 region is normally hypermethylated; hypermethylation of the D4Z4 region keeps the DUX4-like genes silenced all the time
- Both D4Z4 and SMCHD1 mechanisms result in chromatin relaxation of the D4Z4 repeat in somatic tissue and subsequent expression of the DUX4 transcription factor in skeletal muscle
- DNMT3B is yet another D4Z4 repeat modifier thus a disease modifying gene for FSHD
Molecular / cytogenetics images
Images hosted on other servers:

Schematic of D4Z4
Videos
Facioscapulohumeral muscular dystrophy (Year of the Zebra)
FSHD patient's diagnostic journey
FSHD genetics
Sample pathology report
- Skeletal muscle, right quadriceps muscle, biopsy:
- Myopathy with lobulated myofibers (see comment)
- Comment: The muscle shows prominent lobulated / trabeculated morphology in ~70% of the myofibers. There is no significant active myofiber necrosis, MHC1 upregulation, COX deficient fibers or vacuoles to suggest inclusion body myositis. lmmunostaining shows intact sarcolemmal dystrophin, sarcoglycan, caveolin 3, dysferlin, alpha dystroglycan and nuclear emerin reactivity.
- Myopathy with lobulated myofibers has been reported in FSHD, calpain protein deficiency, a number of other limb girdle myopathies and nonhereditary myopathies (J Neurol Sci 1985;69:345). In addition, lobulated / trabecular change has been described as the predominant abnormality in a subset of elderly patients with limb girdle weakness in the absence of a defined protein abnormality (Neuromuscul Disord 1999;9:208).
Differential diagnosis
- Inclusion body myositis:
- Other inflammatory myopathies:
- FSHD usually does not show diffuse MHC1 expression in myofibers
- Active myofiber damage is usually minimal
- Other muscular dystrophies:
- Lobulated architecture is suggestive of FSHD, although it can also be seen in calpainopathies, dysferlinopathies, myopathy with supervillain mutations and neurogenic changes (J Neurol Sci 1985;69:345, Brain 2021;144:e34)
- Chronic denervation:
- Usually lacks fibrosis, inflammation and active myofiber damage
Board review style question #1
A 63 year old woman presented with progressive weakness over the last 10 years. She initially presented with stooped posture that progressed to head drop followed by proximal arm and leg weakness. No significant distal weakness. She had been treated with steroid therapy for the past few years with no significant improvement. Laboratory study showed mildly elevated creatine kinase (CK, 350 IU). Myositis panel, HMG CoA autoantibody, myasthenia gravis panel were all negative. A muscle biopsy shows a chronic myopathy with inflammation (figure 1) and prominent lobulated myofiber architecture (figure 2). Which of the following is the most likely diagnosis?
- Facioscapulohumeral muscular dystrophy
- Immune mediated necrotizing myopathy
- Mitochondria myopathy
- Neurogenic atrophy
- Sporadic inclusion body myositis
Board review style answer #1
A. Facioscapulohumeral muscular dystrophy (FSHD). This case illustrates a typical presentation and muscle pathology of a FSHD patient. Answer E is incorrect because sporadic inclusion body myositis (sIBM) patients usually have both proximal and distal weakness. The inflammation on muscle biopsy is usually endomysial predominant, not perimysial / perivascular. Answer B is incorrect because these patients usually have high CK and positive HMG CoA or SRP antibodies. Also, muscle usually shows a necrotizing myopathy. These patients usually respond to steroid therapy. Answer D is incorrect because neurogenic atrophy usually does not show dystrophic changes and widespread lobulated changes in myofibers. Answer C is incorrect because patients with mitochondria abnormality usually present with exercise intolerance or rhabdomyolysis. Muscle pathology is characterized by ragged red fibers on Gomori trichrome and COX deficient fibers on COX / SDH stains but usually no dystrophic changes.
Comment Here
Reference: Facioscapulohumeral muscular dystrophy
Comment Here
Reference: Facioscapulohumeral muscular dystrophy
Board review style question #2
A 63 year old woman presented with progressive weakness over the last 10 years. She initially presented with stooped posture that progressed to head drop followed by proximal arm and leg weakness. No significant distal weakness. She had been treated with steroid therapy for the past few years with no significant improvement. Laboratory study showed mildly elevated creatine kinase (CK, 350 IU). Myositis panel, HMG CoA autoantibody, myasthenia gravis panel were all negative. A muscle biopsy shows a chronic myopathy with inflammation (figure 1) and prominent lobulated myofiber architecture (figure 2). What is the next step to confirm the diagnosis?
- Muscle MRI
- Muscular dystrophy immunostaining panel
- Neuromuscular next generation sequencing (NGS) panel
- Specific FSHD gene testing
Board review style answer #2
D. Specific FSHD gene testing. Diagnosis of FSHD requires a specific genetic test that usually involves restriction enzyme digestion and Southern blot analysis that interrogates the size of the D4Z4 domain. Answer B is incorrect because the FSHD protein abnormality (DUX4 aberrant expression) is of very low amount and cannot be detected by immunohistochemistry. Answer C is incorrect because most neuromuscular NGS panels cannot detect D4Z4 domain retraction and thus fail to detect FSHD. Answer A is incorrect because muscle MRI can only be used to monitor fatty replacement and disease progression once the diagnosis has been confirmed. The MRI finding itself is not specific enough to definitively diagnose FSHD.
Comment Here
Reference: Facioscapulohumeral muscular dystrophy
Comment Here
Reference: Facioscapulohumeral muscular dystrophy
