

Muscle & peripheral nerve nontumor
Congenital myopathies
Nemaline myopathy
Authors: Wesley Hiser, M.D., Jesse L. Kresak, M.D.


Last author update: 1 June 2017
Last staff update: 12 August 2022
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Muscle Nemaline Myopathy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Videos | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Hiser W, Kresak JL. Nemaline myopathy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/musclenemalinemyo.html. Accessed April 25th, 2024.
Definition / general
- One of the most common non dystrophic congenital myopathies, with a heterogeneous clinical presentation and characteristic rod-like inclusions (nemaline bodies) within myofibers
Essential features
- Skeletal muscle with red to purple rod-like inclusions on Gomori trichrome stain and electron dense deposits on electron microscopy
- Congenital myopathy primarily involving proximal musculature and with variable age of onset and severity
- Most common mutations are NEB (recessive) and ACTA1 (dominant)
Terminology
- Nemaline myopathy (NM), nemaline rod myopathy
- "Nemaline" means thread-like, describes the appearance of rods (Semin Pediatr Neurol 2011;18:230)
- Greek nema = thread
ICD coding
- ICD-10: G71.2 - congenital myopathies
Epidemiology
- Most cases present in infancy and childhood but also seen in adults (Mol Med Rep 2014;10:175)
- Incidence reported as 1:50,000 i(J Neuromuscul Dis 2017;4:99) to 1:500,000 births (Clin Genet 2016;90:199)
Sites
- Preferentially involves proximal muscles, namely the face, neck flexors and proximal extremities (Neuropathol Appl Neurobiol 2017;43:5)
- Bulbar and respiratory muscles are affected (Semin Pediatr Neurol 2011;18:230)
- Cardiac and extraocular muscles are typically spared
Pathophysiology
- Congenital forms caused by mutations involving the thin filament of the sarcomere and cause contractile dysfunction, leading to muscle weakness (J Neuromuscul Dis 2017;4:99)
Etiology
- To date, mutations in eleven genes have been identified which result in NM (J Neuromuscul Dis 2017;4:99)
- Ten genes encode proteins involved with the thin filament of the sarcomere, either as components or having roles in stability and turnover of the filament proteins
- The most recently identified gene encodes myosin 18B (J Neuromuscul Dis 2017;4:99, Circulation 1989 Jun;79:1282)
- Inherited in autosomal recessive or autosomal dominant manner, with 50% of cases occurring de novo (GeneReviews® [Internet];1993-2017)
- The most frequently affected genes are recessive mutations in NEB and de novo dominant mutations in ACTA1 (Curr Opin Neurol 2016;29:642)
Clinical features
- Progressive myopathy which most commonly has a congenital onset
- Large variation in clinical presentation, ranging from normal lifespan with mild symptoms to neonatal death (Mol Med Rep 2014;10:175)
- Subdivided into six clinical categories (Semin Pediatr Neurol 2011;18:230)
- Typical congenital form
- Intermediate congenital form
- Severe congenital form
- Mild nemaline myopathy with childhood onset
- Adult onset nemaline myopathy
- Other forms with unusual associated features (Amish NM)
- Usually symmetric, generalized weakness with preference for neck flexors, facial muscles, axial muscles and proximal extremities (Neuropathol Appl Neurobiol 2017;43:5)
- Can have late involvement of distal musculature
- Bulbar and respiratory muscles are frequently affected and may be a
presenting feature (Semin Pediatr Neurol 2011;18:230)
- Patients are at risk for nocturnal hypoxia and recurrent lower respiratory tract infections
- Feeding and nutritional difficulties are common
- Cardiac and extraocular muscles typically spared, but involvement can be seen with specific mutations (Curr Opin Neurol 2016;29:642, Semin Cell Dev Biol 2017;64:191, Circulation 1989;79:1282)
- Frequently show weak deep tendon reflexes and joint hypermobility
- May develop joint and spine abnormalities (J Med Genet 1997;34:705)
- Congenital forms often show facial elongation, tent shaped mouth and high arched palate (J Med Genet 1997;34:705)
Diagnosis
- Clinicopathologic diagnosis
- Muscle biopsy
- Laboratory findings are typically nonspecific
Radiology description
- MRI frequently shows patchy fatty degeneration of muscle (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- CT often shows decreased muscle density with normal volume (J Med Genet 1997;34:705)
- Ultrasound can reveal increased echogenicity of affected muscle (J Med Genet 1997;34:705)
Prognostic factors
- Neonatal hypotonia was historically identified as the single most important prognostic sign, however later studies have not shown this to be true (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Severe neonatal respiratory disease and arthrogryposis multiplex congenita associated with death in first year of life (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Independent ambulation prior to 18 months is predictive of survival (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- KLHL40 mutations are associated with poor prognosis (death within 5 months on average) and prenatal presentation in 80% (Clin Genet 2016;90:199)
- ACTA1 mutations are associated with up to 50% of lethal congenital cases, but exhibit significant clinical variability (Clin Genet 2016;90:199)
- Presence of intranuclear rods is often associated with more severe disease (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
Case reports
- 6 year old with typical congenital nemaline myopathy and sudden cardiac arrest (Ital J Pediatr 2015;41:20)
- 42 year old with congenital nemaline myopathy and associated mitochondrial abnormalities (Rinsho Shinkeigaku 2000;40:452)
Treatment
- No proven effective therapy other than symptomatic treatment
- Symptomatic treatment has shown to be helpful in many patients (Semin Pediatr Neurol 2011;18:230)
- Mechanical nighttime ventilation
- Orthoses
- Nasogastric feeding and nutritional support
- Aggressive treatment of respiratory infections
- Physical and speech therapy
- While lacking in objective evidence, low impact exercise and L-tyrosine may provide some benefit
- Medications targeting thin filaments or muscle atrophy have been developed and show promise
Microscopic (histologic) description
- Variation in myofiber size with areas of atrophy, hypertrophy and grouping
- Fatty replacement or fibrosis may be seen but necrosis, fiber regeneration, or inflammation is uncommon (J Med Genet 1997;34:705)
- Gomori trichome stain shows red to purple colored inclusions,
frequently rod shaped, in sarcoplasm and subsarcolemmal region of
myofibers (Semin Pediatr Neurol 2011;18:230, J Med Genet 1997;34:705)
- Often more prominent in type I myofibers
- No correlation between number of rods and clinical severity
- Not specific for nemaline myopathies
- Nemaline rods can also be visualized on plastic sections with toluidine blue (Semin Pediatr Neurol 2011;18:230)
- Fiber typing will typically show predominantly type I fibers with atrophy and grouping
Microscopic (histologic) images
Contributed by Wesley M. Hiser, M.D.
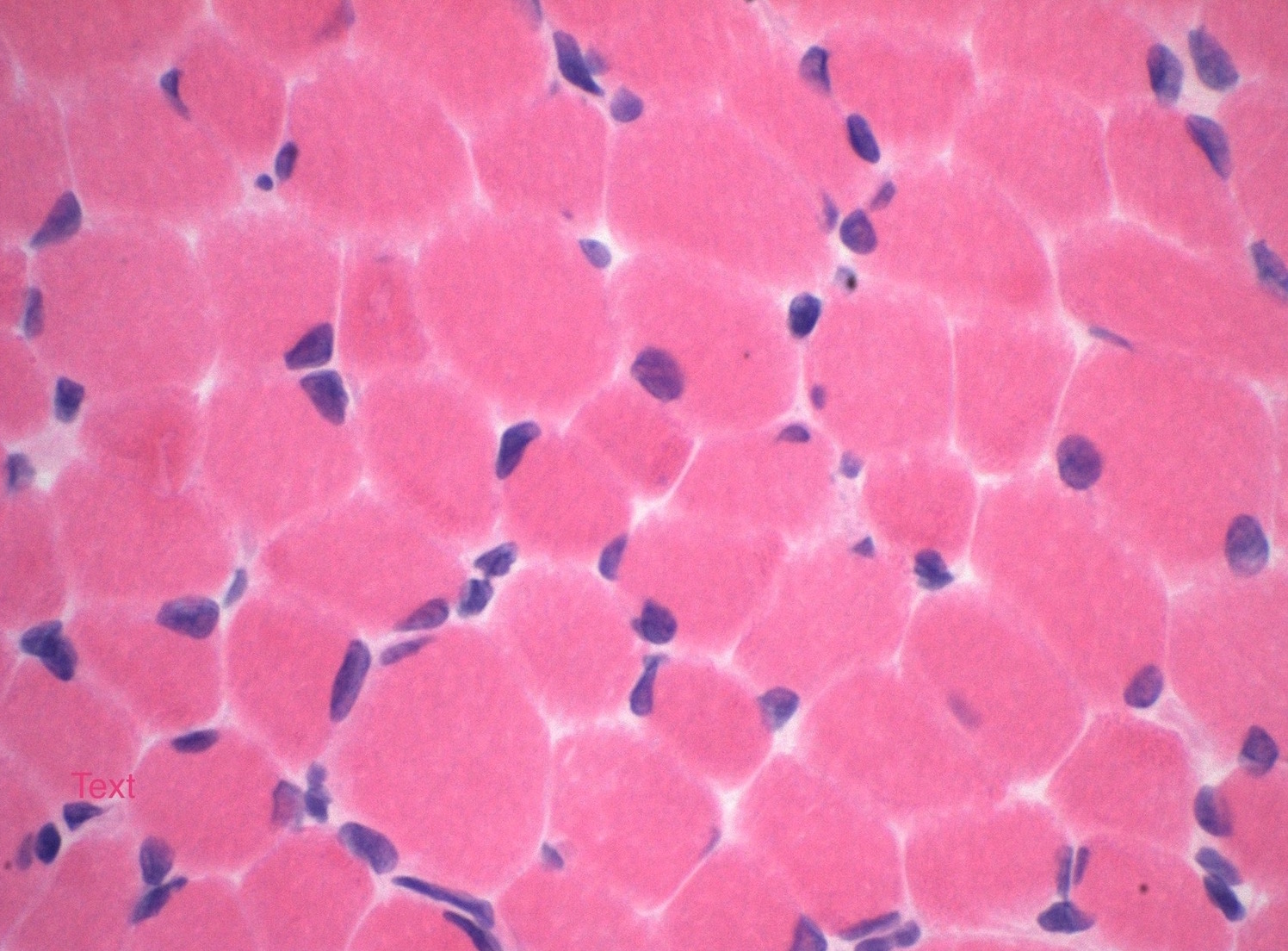
H&E
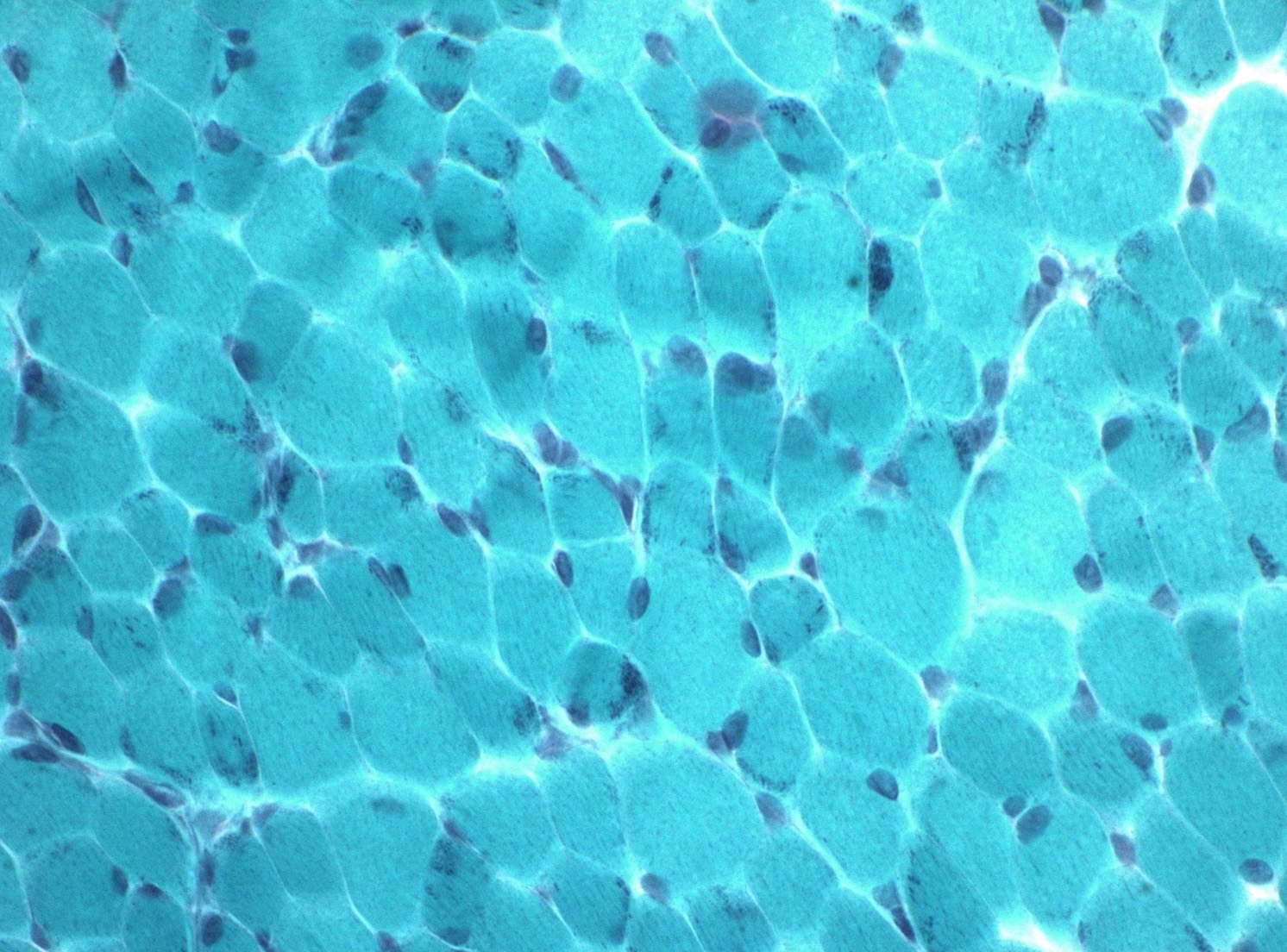

Gomori trichrome
Positive stains
- Gomori trichrome stain highlights red to purple colored rods within sarcoplasm of myofibers
- Nemaline bodies positive for alpha-actinin
- Myofiber typing shows type I predominance
Electron microscopy description
- Rods appear as electron dense rod to ovoid shaped deposits measuring 1 - 7 um in length (J Med Genet 1997;34:705, GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Present in sarcoplasm or nucleus, often parallel to the long axis of the sarcomere (Semin Pediatr Neurol 2011;18:230)
- Deposits can show continuity with Z-disks and exhibit similar lattice structure
Electron microscopy images
Images hosted on other servers:

Nemaline rods
Molecular / cytogenetics description
- 11 genes have been implicated in NM (J Neuromuscul Dis 2017;4:99)
- 10 genes involved with thin filament of sarcomere:
- ACTA1
- NEB
- TPM3
- TPM2
- CFL2
- TNNT1
- LMOD3
- KBTBD13
- KLHL40
- KLHL41
- ACTA1
- More recently described myosin 18B associated with severe NM with cardiomyopathy
- 10 genes involved with thin filament of sarcomere:
- Multigene sequence analysis is typically performed first, followed by deletion/duplication analysis when necessary (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
Videos
Differential diagnosis
- Other congenital myopathies
- Core myopathies (Neuropathol Appl Neurobiol 2017;43:5)
- Centronuclear myopathies (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Congenital fiber type disproportion
- Muscular dystrophies
- Metabolic myopathies such as Pompe disease
- Spinal muscular atrophy
- Prader-Willi syndrome
- Can present with marked hypotonia in neonates (GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993-2017)
- Sporadic late onset nemaline myopathy (Semin Pediatr Neurol 2011;18:230, Rinsho Shinkeigaku 2016;56:605)
- Associated with HIV, autoimmune disease and monoclonal gammopathy
Additional references
Board review style question #1
In which gene are de novo mutations most commonly associated with nemaline myopathy?
A. NEB
B. KLHL40
C. TPM3
D. ACTA1
E. TNNT1
A. NEB
B. KLHL40
C. TPM3
D. ACTA1
E. TNNT1
Board review style answer #1
D. ACTA1
De novo mutations in ACTA1 are the most common cause of nemaline myopathy. Mutations in NEB are the most common cause of autosomal recessive nemaline myopathy.
Comment Here
Reference: Nemaline myopathy
De novo mutations in ACTA1 are the most common cause of nemaline myopathy. Mutations in NEB are the most common cause of autosomal recessive nemaline myopathy.
Comment Here
Reference: Nemaline myopathy
