

Ovary
Disorders of sex development
Disorders of sex development-general
Author: Mohiedean Ghofrani, M.D.

Last author update: 1 November 2011
Last staff update: 15 January 2024
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Gonadal dysgenesis ovary
Table of Contents
Definition / general | Terminology | Etiology | Clinical features | Prognostic factors | Treatment | Molecular / cytogenetics description | Pure (complete) gonadal dysgenesis | Pure (complete) gonadal dysgenesis 46XX | Pure (complete) gonadal dysgenesis 46XY | Testicular feminizationCite this page: Ghofrani M Disorders of sex development-general. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/ovarynontumorgonadaldysgenesis.html. Accessed April 20th, 2024.
Definition / general
- Disorder of sex development (DSD) in which one or both gonad(s) is / are undeveloped (streak gonad)
- Disorders of sex development are either female pseudohermaphroditism (46XX with 2 ovaries), male pseudohermaphroditism (46XY with two testes), true hermaphroditism (ovotestis present) or gonadal dysgenesis (either pure with normal 46XX or 46XY chromosomes and bilateral streak gonads or mixed with streak gonad)
Terminology
- Pure gonadal dysgenesis (PGD): both gonads are streak gonads (i.e. dysfunctional gonads without germ cells)
- Mixed gonadal dysgenesis (MGD): patient has a testis on one side and a streak gonad on the other
Etiology
- Chromosomal abnormality: may be 46XX, 46XY or 45XO
Clinical features
- Patients with pure gonadal dysgenesis have underdeveloped Müllerian organs (sexual infantilism), sometimes with clitoromegaly, while patients with mixed gonadal dysgenesis have ambiguous or female genitalia
- 46XX and 46XY patients present with primary amenorrhea and delayed secondary sexual development; 45XO present with typical Turner syndrome features
Prognostic factors
- Patients with pure gonadal dysgenesis are raised as female since there is no sexual ambiguity at birth; however, gender assignment in patients with mixed gonadal dysgenesis is variable and depends on the degree of virilization
- Approximately 20% of patients with gonadal dysgenesis develop a gonadal neoplasm within the first two decades of life, usually gonadoblastoma; almost all cases of gonadoblastoma have been reported in patients with pure or mixed gonadal dysgenesis or male pseudohermaphroditism
- Other neoplasms include seminomatous germ cell tumors such as seminoma and dysgerminoma, nonseminomatous germ cell tumors such as embryonal carcinoma and choriocarcinoma, as well as nongerm cell tumors such as Wilms tumor
Treatment
- Given the higher risk of malignancy in dysgenetic gonads, early surgical removal is indicated
Molecular / cytogenetics description
- Pure gonadal dysgenesis: 46XX, 46XY or 45XO
- Mixed gonadal dysgenesis: 45XY / 45XO (mosaic)
Pure (complete) gonadal dysgenesis
Definition / general
Terminology
Etiology
Clinical features
Microscopic (histologic) description
- Presence of undeveloped (streak) gonads in a phenotypic female who may have an either 46,XX female or 46,XY male genotype
Terminology
- Term gonadal dysgenesis originally referred to Turner syndrome but it is now applied to other conditions as well
- Under the new nomenclature, pure (complete) gonadal dysgenesis is considered a type of either 46,XY DSD (disorder of sex development) or 46,XX DSD
Etiology
- For a variety of reasons, known and unknown, primordial germ cells do not form or interact with the gonadal ridge or undergo accelerated atresia, leading to extremely hypoplastic (underdeveloped) and dysfunctioning gonads that are mainly composed of fibrous tissue, hence the name streak gonads
- Gonadal dysfunction leads to deficiency of both Müllerian inhibiting factor and testosterone
Clinical features
- During embryonal development, the human reproductive system has an inherent tendency to give rise to female reproductive organs; therefore, in the absence of hormonal influences from the undeveloped and dysfunctional gonads, patients with complete gonadal dysgenesis are phenotypically female, regardless of genotype
- First clinical manifestation is usually absence of expected female secondary sex characteristic development at puberty
Microscopic (histologic) description
- Streak gonads are mainly composed of fibrous tissue
- Absence of testosterone results in regression of Wolffian ducts; i.e. normal male internal reproductive tracts do not develop
- Absence of Müllerian inhibiting factor allows Müllerian ducts to differentiate into oviducts and the uterus
Pure (complete) gonadal dysgenesis 46XX
Definition / general
Terminology
Etiology
Clinical features
Laboratory
Treatment
Microscopic (histologic) description
Additional references
- Form of gonadal dysgenesis (underdeveloped and dysfunctioning ovaries) associated with female 46,XX genotype and female internal and external phenotype
- Phenotypic female 46,XX but no functional ovaries are present to induce puberty (may have streak ovaries)
Terminology
- Term gonadal dysgenesis originally referred to Turner syndrome but it is now applied to other conditions as well
- Under the new nomenclature, this form of gonadal dysgenesis is considered a type of 46,XX DSD (disorder of sex development)
- Perrault syndrome: 46,XX gonadal dysgenesis with sensorineural hearing loss
Etiology
- May be familial (Am J Med Sci 1980;280:157)
- Occasionally due to mutation in FSH receptor (Cell 1995;82:959)
- For reasons that are not clear, primordial germ cells do not form or interact with the gonadal ridge or undergo accelerated atresia
Clinical features
- First clinical manifestation is usually absence of expected female secondary sex characteristic development at puberty
Laboratory
- Low serum estrogen and progesterone (since no functional ovaries), high serum FSH and LH
Treatment
- Estrogen and progesterone therapy
Microscopic (histologic) description
- Progressive loss of primordial germ cells in the developing gonads of the embryo leads to extremely hypoplastic ovaries mainly composed of fibrous tissue, hence the name streak gonads
Additional references
Pure (complete) gonadal dysgenesis 46XY
Definition / general
Terminology
Etiology
Clinical features
Laboratory
Prognostic factors
Case reports
Treatment
Microscopic (histologic) description
Additional references
- Form of gonadal dysgenesis (underdeveloped and dysfunctioning testes) associated with male 46,XY genotype and female internal and external phenotype
- Phenotypic female, hypoplastic (streak) gonads without germ cells
Terminology
- Term gonadal dysgenesis originally referred to Turner syndrome but it is now applied to other conditions as well
- Under the new nomenclature, this form of gonadal dysgenesis is considered a type of 46,XY DSD (disorder of sex development)
- Also called Swyer syndrome
Etiology
- Mutation in SRY, the sex determining region of the Y chromosome, is reported in 10 - 15%
- Defects in the genetic pathways that lead to development of testes from indifferent gonads result in hypoplastic (streak) testes and lack of testosterone or anti-Müllerian hormone (AMH) production
Clinical features
- In the absence of testosterone, external genitalia fail to virilize and the Wolffian ducts fail to develop, leading to normal female genitalia and absent internal male organs
- Presents with primary amenorrhea (delayed puberty) since no functional gonads are present to induce puberty
- May develop pubic hair through androgens produced from adrenal gland
Laboratory
- Low serum estrogen and progesterone, high serum FSH and LH
Prognostic factors
- High risk of gonadoblastoma or germ cell tumor from gonads (Zhonghua Fu Chan Ke Za Zhi 2008;43:442)
- Dysgerminoma may develop by age 10 (BJOG 2008;115:737)
Case reports
- Successful pregnancy using donor oocytes and oocyte transfer (Fertil Steril 2008;90:2015.e1)
Treatment
- Early excision of gonads (J Gynecol Obstet Biol Reprod (Paris) 2009;38:220)
- Estrogen and progesterone therapy
Microscopic (histologic) description
- Hypoplastic (streak) gonads without primordial germ cells
- In the absence of AMH, Müllerian ducts develop into normal internal female organs (uterus, fallopian tubes, cervix, vagina)
Additional references
Testicular feminization
Definition / general
Terminology
Etiology
Clinical features
Prognostic factors
Case reports
Treatment
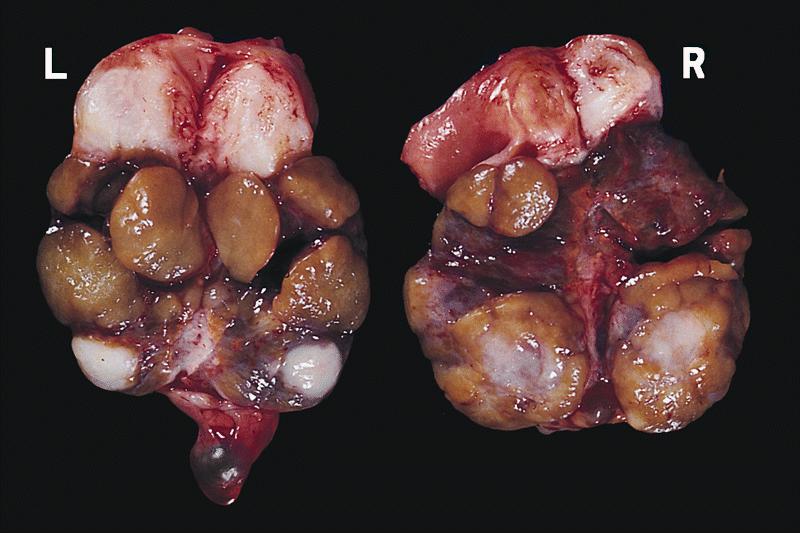
Gross images
AFIP images
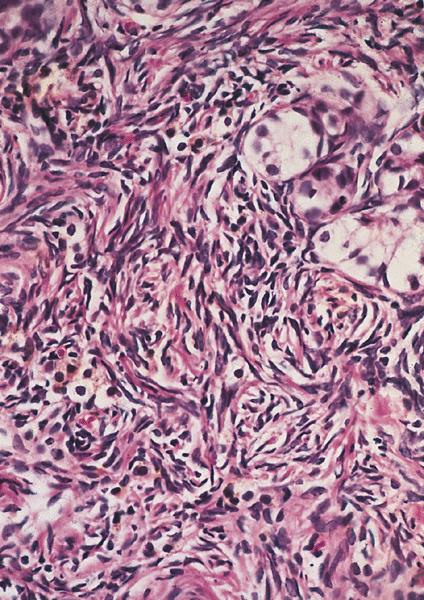
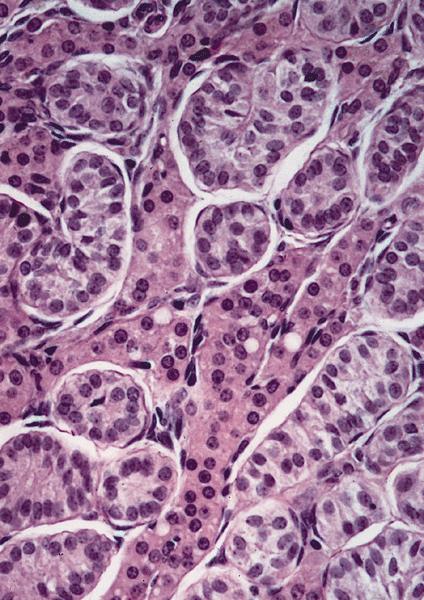
Microscopic (histologic) description
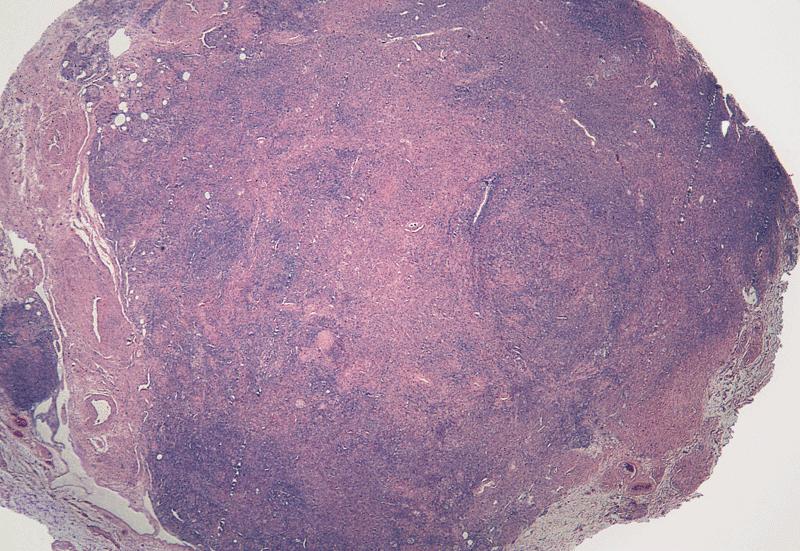
Microscopic (histologic) images
AFIP images
Additional references
- Male genotype (46,XY) but impaired or absent masculinization of male genitalia or development of male secondary sexual characteristics at puberty due to partial or complete inability of cells to respond to androgens (androgen insensitivity)
- Most common type of male pseudohermaphroditism
- Patients present with amenorrhea or sterility
- Vagina but no uterus
- Also, bilateral cryptorchid testes with nodular masses of immature tubules resembling Sertoli-Leydig tumor
Terminology
- Also called androgen insensitivity syndrome (AIS)
- Divided into three categories:
- Complete androgen insensitivity syndrome (CAIS): normal female external genitalia
- Mild androgen insensitivity syndrome (MAIS): normal male external genitalia
- Partial androgen insensitivity syndrome (PAIS): partially but not fully masculinized external genitalia
Etiology
- Most commonly, a mutation in the androgen receptor (AR) gene
- Other etiologies include: chromosomal abnormalities, dysfunctional androgen biosynthesis, developmental syndromes, teratogens
Clinical features
- Depending on the severity of androgen insensitivity, patients may present with male, female or ambiguous phenotype
- Given the presence of a Y chromosome (more specifically, SRY gene), gonads are testes regardless of phenotype
- Physical exam may reveal a vagina but no ovaries or uterus
Prognostic factors
- 9% develop tumors in cryptorchid testis, which should be removed after puberty
Case reports
- 36 year old with abdominal seminoma (J Obstet Gynaecol Res 2004;30:109)
Treatment
- Currently limited to symptomatic management, including sex assignment, genitoplasty, gonadectomy, hormone replacement, counseling
Gross images
AFIP images
Multiple hamartomas
Microscopic (histologic) description
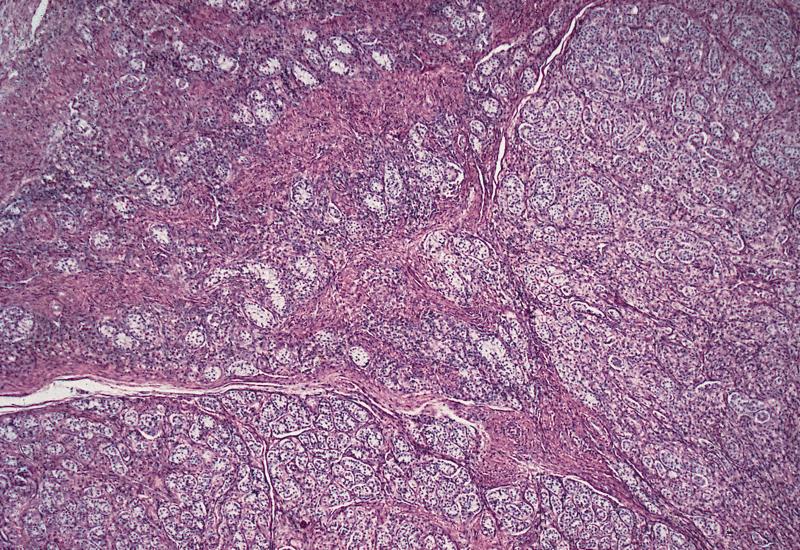
- Bilateral cryptorchid testes with nodular masses of immature tubules resembling Sertoli-Leydig tumor
Microscopic (histologic) images
AFIP images
Ovarian type
stroma with a
storiform pattern
Tubules containing
immature
Sertoli cells
Testis is composed
almost entirely of
stroma resembling
ovarian stroma
Hamartomas are
composed of Sertoli
and Leydig cells
Additional references
