

Thyroid & parathyroid
Congenital / metabolic anomalies
DiGeorge syndrome
Author: Andrey Bychkov, M.D., Ph.D.

Last author update: 1 May 2016
Last staff update: 16 August 2023
Copyright: 2016-2024, PathologyOutlines.com, Inc.
PubMed Search: DiGeorge syndrome [title] thyroid

Table of Contents
Definition / general | Terminology | Epidemiology | Sites | Pathophysiology / etiology | Diagrams / tables | Clinical features | Diagnosis | Radiology description | Radiology images | Case reports | Treatment | Clinical images | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Molecular / cytogenetics images | Videos | Differential diagnosis | Additional referencesCite this page: Bychkov A. DiGeorge syndrome. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/thyroiddigeorge.html. Accessed April 24th, 2024.
Definition / general
- DiGeorge syndrome is a chromosomal disorder due to 22q11.2 deletion, characterized by failure of development of the third to fourth pharyngeal pouches and fourth branchial arch, which leads to a combination of congenital heart disease, parathyroid abnormalities (hypocalcemia) and thymic abnormalities (immunodeficiency)
- In 1965 DiGeorge described a group of infants with congenital absence of the thymus and parathyroid glands (J Pediatr 1965;67:907); however a constellation of clinical signs was known at least since 1829 (London Medical Gazette 1829;3:314)
- See also Thymic dysplasia
Terminology
- There are numerous synonyms, which were used historically to describe various manifestations of chromosome 22q11.2 deletion (OMIM #188400)
- Most common synonyms: DiGeorge syndrome / anomaly, velocardiofacial syndrome (Shprintzen syndrome), conotruncal anomaly face syndrome (Takao syndrome)
- Other synonyms: hypoplasia of thymus and parathyroids, III - IV pharyngeal pouch syndrome, Opitz G / BBB syndrome, CATCH 22 syndrome, Sedlackova syndrome, Cayler cardiofacial syndrome, Strong syndrome, congenital thymic aplasia
- It is now recommended that patients with the classic chromosome 22q11.2 deletion be described according to the genetic nomenclature, i.e. "22q11.2 deletion syndrome", and patients with a clinical phenotype but a distinct cause or no known cause be described using syndromic nomenclature, i.e. "DiGeorge syndrome" (Medicine (Baltimore) 2011;90:1)
Epidemiology
- Estimated prevalence is 1:4,000 births, range 1:3,000 - 1:6,000 (Lancet 2007;370:1443)
- M = F
- Spontaneous : familial = 10:1 (Genet Med 2001;3:23)
- 22q11.2 deletion syndrome is one of the most common genetic abnormalities (slightly less frequent than Down syndrome), and the most common chromosomal deletion syndrome in humans
- 5% of congenital cardiac defects and 2% - 3% of childhood onset schizophrenia are due to 22q11.2 deletion (J Med Genet 1993;30:852)
- Infant mortality in 22q11 deletion syndrome is now relatively low (4%) due to adequate cardiac care, however the overall mortality rate compared to general population is elevated, especially in adults (J Pediatr 2011;159:332)
Sites
- The syndrome crosses all organ systems, and is classically defined as polytopic developmental field defect (Am J Med Genet Suppl 1986;2:113)
- Major targets are the organs derived from branchial apparatus; see details in Clinical Features
- Thyroid is one of the organs involved, however clinical impact in the thyroid is low compared to other manifestations
Pathophysiology / etiology
- Due to the hemizygous (only one of the chromosome pair) 22q11.2 deletion
- The occurrence of 22q11.2 deletions is related to the genomic architecture of the chromosome 22q11.2 region with several common rearrangement breakpoints (J Pediatr 2011;159:332)
- The characteristic deletion of chromosome 22q11.2 is at least 10 times more common than the next most frequent human deletion syndrome, suggesting that this region is inherently unstable (Dev Disabil Res Rev 2008;14:11)
- The deletion typically arises via unequal meiotic crossover / exchange, facilitated by asynchronous replication at the site of the deletion (J Med Genet 2004;41:413)
- Alternative mechanism is 22q11.2 microduplication (Eur J Med Genet 2009;52:88)
- Approximately 85% - 90% of individuals with the syndrome have 3 Mb deletion, 8% - 10% have 1.5 Mb deletion with mild phenotype, and individuals with atypical deletions (so called central and distal) have also been reported (Cytogenet Genome Res 2015;146:89)
- There are approximately 40 genes within the commonly deleted region of chromosome 22q11.2
- TBX1 is the main gene responsible for DiGeorge phenotype
- TBX1 is a transcription factor normally expressed in the pharyngeal pouches, which give rise to the face, neck (parathyroids and thymus) and upper thorax (Medicine (Baltimore) 2011;90:1)
- TBX1 is involved in retinoic acid signaling (Circ Res 2010;106:630)
- TBX1 is not expressed in the thyroid primordium, but in surrounding mesoderm, and determines thyroid size and positioning (Hum Mol Genet 2007;16:276)
- Other candidate genes are CRKL, COMT, DCGR8 (Cytogenet Genome Res 2015;146:89)
- Over 90% of the deletions occur de novo, while 6% - 10% of new cases are inherited from an affected parent (Hum Mol Genet 2004;13:417)
- Risk of recurrence in the offspring is up to 50% in each pregnancy
Diagrams / tables
Images hosted on other servers:

Pathophysiology

Mode of inheritance

Pedigree

Chromosome 22 abnormalities



Genetic mapping
Clinical features
- Classic phenotype is a triad of heart defects (usually involving the aorta and the part of the heart from which the aorta develops), thymus gland abnormalities with related immunodeficiency / autoimmunity, and parathyroid gland abnormalities with hypocalcemia
- However, it is widely acclaimed today that DiGeorge / 22q11.2 deletion syndrome has many more manifestations beyond the classical triad
- Phenotypic features grouped by frequency and clinical significance (Medicine (Baltimore) 2011;90:1)
- Major:
- Cardiac anomalies mainly involving outflow tract, i.e. conotruncal (80%): tetralogy of Fallot, pulmonary atresia, truncus arteriosus, interrupted aortic arch, ventricular septal defect
- Immune deficiency (> 75%): thymic hypoplasia, rarely aplasia, with T cell lymphopenia
- Palatal anomalies (70%): velopharyngeal insufficiency and cleft palate, both contributing to poor feeding and speech
- Developmental delay with low IQ (50%)
- Intermediate: hypocalcemia, various structural anomalies (renal, GI, CNS, etc.), and psychiatric illnesses
- Minor: facial dysmorphism (bulbous nasal tip, hypertelorism, hooded eyelid, micrognathia, low set ears)
- Major:
- Depending on whether thymic hypoplasia or aplasia is present (in addition to heart defects and hypoparathyroidism), DiGeorge syndrome can be classified as partial or complete (Eur J Pediatr 1989;149:96):
- Complete phenotype (< 1%) is used to describe patients who are athymic and have no circulating T cells
- Patients with partial or incomplete phenotype have thymic hypoplasia with circulating T cells
- Thyroid issues:
- Up to 10% of patients have overt thyroid disease, with hypo- or hyperthyroidism (4:1, Am J Med Genet A 2015;167:1560)
- Subclinical thyroid dysfunction often converts to apparent disease
- Recommended to monitor TSH and thyroxine levels in all 22q11.2 deletion syndrome patients (J Pediatr 2011;159:332)
- Rarely presents with Graves disease (16× risk compared to general population, Hormones (Athens) 2005;4:200)
- Structural defects are detailed in Gross Description
Diagnosis
- Most patients are identified shortly after birth due to the presence of cardiac anomaly, hypocalcemia or recurrent infections
- Genetic testing (see Molecular / Cytogenetics Descriptions / Images):
- May be warranted if affected parent/sibling or suspicious prenatal US findings;
- Advised in children with certain combinations: cardiac anomaly + any major / intermediate phenotypic feature; speech delay + any major / intermediate phenotypic feature; hypocalcemia alone or with any major / intermediate phenotypic feature (J Allergy Clin Immunol Pract 2013;1:589)
- DiGeorge syndrome needs to be considered for some pediatric autopsies; for example, thymus and parathyroid should be located in cases of congenital heart disease, and in turn, absent parathyroids / thymus should prompt a detailed heart examination (J Pediatr 1979;94:883)
Radiology description
- US and CT are useful to evaluate precise thyroid anatomy (Clin Endocrinol (Oxf) 2010;72:839, Laryngoscope 2009;119:1495)
Radiology images
Images hosted on other servers:

Absent thymus on CT

Subglottic stenosis on CT
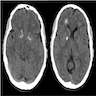
Basal ganglia and periventricular calcification
Case reports
- Male newborn with thyroid hemiagenesis (Horm Res Paediatr 2013;79:243)
- 1 month old girl with congenital hypothyroidism (J Pediatr Endocrinol Metab 1998;11:273)
- 3 month old girl with hypoplastic thyroid (J Postgrad Med 1989;35:114)
- 3 year old boy and 18 year old woman with Graves disease in DiGeorge syndrome (J Pediatr Endocrinol Metab 2004;17:1575, Endocr J 2000;47:91)
- 34 year old man with follicular adenomata and a nodular goiter (Case Rep Med 2013;2013:923129)
Treatment
- Varies by age and phenotype
- May include cardiac and palatal surgery, thymus transplantation, calcium supplementation, etc.
Clinical images
Images hosted on other servers:
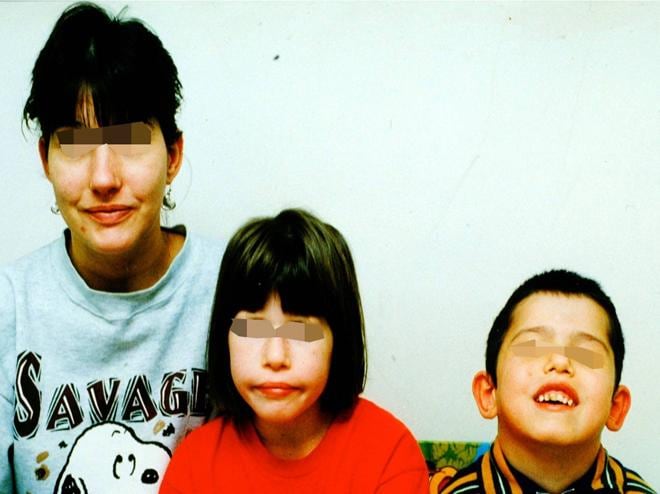
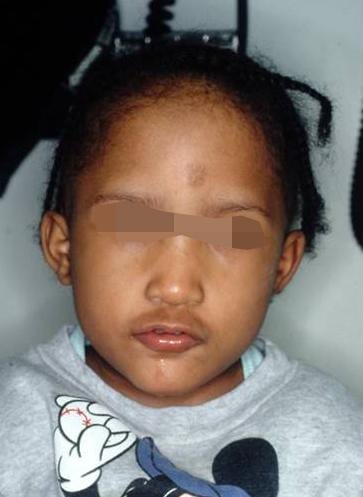
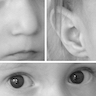



Facial dysmorphism
Gross description
- Unremarkable thyroid in half of the cases
- Reported anomalies include (J Pediatr 1979;94:883, Pediatr Pathol 1993;13:463):
- Thyroid hypoplasia, with a left lobe predilection (Clin Endocrinol (Oxf) 2010;72:839)
- Absence of the isthmus of thyroid
- Agenesis of lobe of thyroid
- Retrocarotid or retroesophageal extension (Laryngoscope 2009;119:1495)
- Hypoplasia and persistent fetal shape of thyroid cartilage (Pediatr Pathol 1986;6:209)
Microscopic (histologic) description
- Consistent with normal thyroid histology (most specimens come from pediatric autopsies)
- Rarely, pathological changes has been described:
- Predominant fetal follicles
- Hyperplasia
- Follicular adenoma and nodular goiter (Case Rep Med 2013;2013:923129)
- Thyroid carcinoma (Am J Med Genet A 2006;140:906)
- C cells can be absent or significantly reduced in number, consistently with their pharyngeal pouch origin (Hum Pathol 1987;18:355, Am J Med Genet 1993;46:641, Pediatr Pathol 1993;13:463)
Microscopic (histologic) images
Images hosted on other servers:

Hypoplastic thyroid in mice


Various malformations in mice
Molecular / cytogenetics description
- FISH is usually performed with a chromosome 22 specific probe that identifies the chromosome and a second probe that hybridizes to the commonly deleted region; if the second probe is absent on a "tagged" chromosome, then the diagnosis is established (Medicine (Baltimore) 2011;90:1)
- Turnaround time is 3 - 14 days
- Cost of the commercial test is $750
- Alternative genetic tests
- Multiplex PCR (MLPA): rapid and cost effective
- CGH array: identifies atypical deletions and duplications not recognized by FISH
Molecular / cytogenetics images
Images hosted on other servers:

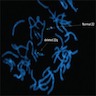
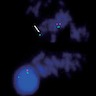
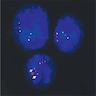
Deletion of 22q11.2 on FISH
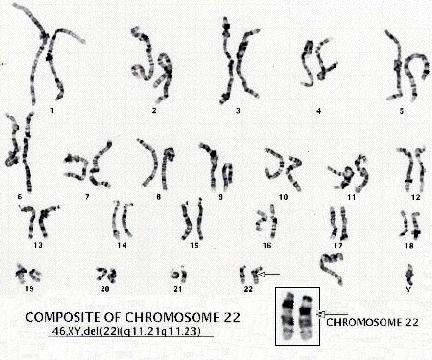

Karyotype

Various techniques
Videos
DiGeorge syndrome
Pharyngeal apparatus
Differential diagnosis
- Some genetic syndromes have overlapping features with 22q11 deletion syndrome (resolved by genetic testing):
- CHARGE syndrome: coloboma, heart, atresia, retarded growth / development (or both), genital, ear (Wikipedia)
- Smith-Lemli-Opitz syndrome (Genetics Home Reference)
- Kabuki syndrome (Genetics Home Reference)
- Goldenhar syndrome (Wikipedia)
Additional references

