![]()
5 April 2007 Case of the Week #79
These cases can also be accessed by clicking on the Case of the Week button on the left hand side of our Home Page at www.PathologyOutlines.com. This email is sent only to subscribers. To view the images or references, you must click on the links in blue.
To subscribe or unsubscribe, email info@PathologyOutlines.com, indicating subscribe or unsubscribe to Case of the Week. We do not sell, share or use your email address for any other purpose. We also have emails for new Pathologist jobs (biweekly), new Laboratory (not pathologist) jobs (biweekly), website news (monthly), new books (monthly), and a newsletter (twice a year). You must subscribe or unsubscribe separately to these email lists.
We recently updated the section on lipomatous tumors in the Soft Tissue chapter. We added more content from textbooks, journals, the AFIP fascicle and the WHO tumor book, as well as images from the AFIP fascicle and elsewhere. We emphasize references to journals with free full text, which are highlighted in green. Let us know what you think.
Share your tips on using PathologyOutlines.com by entering our Contest (click on the Contests button on the Home Page) to win a $50 Amazon.com gift card.
We thank Dr. Julia Braza, Beth Israel Deaconess Medical Center, Boston, Massachusetts (USA), for contributing this case and the discussion. We invite you to contribute a Case of the Week by emailing info@PathologyOutlines.com with microscopic images (any size, we will shrink if necessary) in JPG or TIFF format, a clinical history, your diagnosis and any other images (gross, immunostains, EM, etc.) that may be helpful or interesting. We will write the discussion (unless you want to), list you as the contributor, and send you a check for $35 (US dollars) for your time after we send out the case. Please only send cases with a definitive diagnosis.
Case of the Week #79
Clinical History
Three years ago, a 69 year old man was found to have leukocytosis and lymphocytosis on a routine visit. He had no constitutional symptoms or weight loss. Physical examination was normal, with no lymphadenopathy or hepatosplenomegaly. His white blood count was 18.2 K/L (normal 4-11) with 46% neutrophils (normal 50-70%); 44% lymphocytes (18-42%); 3% monocytes (2-11%); 1% eosinophils (0-4%) and 0.5% basophils (0-2%). The platelet count was 401K/L (150-440).
Flow cytometry of peripheral blood showed a monoclonal kappa (dim) restricted B cell population coexpressing CD19, CD20, CD5, CD38 and FMC7. The lymphocytes did not express CD23 or CD10. The peripheral blood smear showed medium sized lymphoid cells with moderate cytoplasm and occasional nucleoli, but no prolymphocytes.
Cytogenetics revealed a complex karyotype of 46, XY, add (14)(q32), inv(19)(p13.1,q13.3). FISH showed rearrangement in 78% of cells of the immunoglobulin heavy chain at 14q32, trisomy 12 (using the CEP1 probe for 12p11.1-q11.1) in 12% of nuclei and a p53 mutation (a single hybridization signal was seen with the p53 probe at 17p13.1). No abnormalities were seen with the probe for ATM (11q22.3) or 13q14.3. FISH showed no fusion signals for IgH-CCND1 / bcl1 associated with t(11;14), although three signals were observed with the IgH probe in keeping with the IgH rearrangement noted previously.
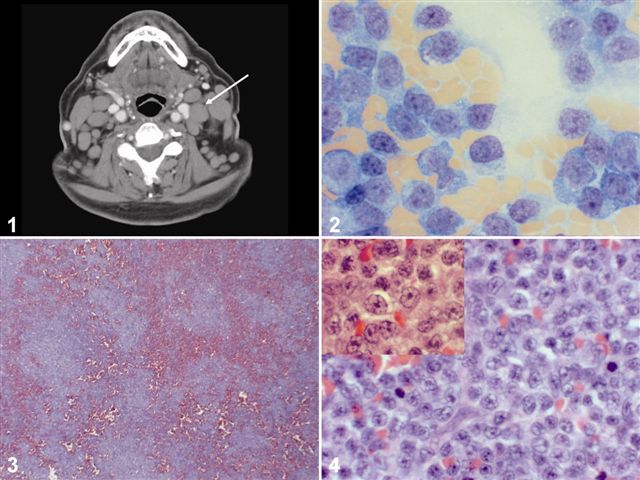
The patient presented recently with fatigue, nausea, and painful cervical (Figure 1) and pre-auricular adenopathy. His peripheral smear showed 30% prolymphocytes. His left supraclavicular lymph node was biopsied, showing large atypical lymphocytes with central, prominent nucleoli on touch prep (Figure 2). On H&E, the nodal architecture was effaced, with infiltration of the capsule and perinodal adipose tissue. On low power examination, a vaguely nodular pattern was noted (Figure 3), that revealed medium sized lymphocytes with vesicular chromatin and small, central nucleoli at high power (Figure 4).
{kind=link}
The lymphocytes were immunoreactive for CD20 and CD79a. The Ki-67 fraction was overall 10-20%. Flow cytometry of the nodal tissue showed coexpression of CD19, CD20, CD5, CD23 (bright), and HLA-DR. There was no expression of FMC-7, CD10 or bcl-1. No cytogenetic studies were available.
What is your diagnosis?
Diagnosis:
Small Lymphocytic Lymphoma / Chronic Lymphocytic Leukemia (SLL/CLL), Paraimmunoblastic Variant
Discussion
The initial flow cytometry showed CD20 and CD5 co-expression without CD23 expression, generating a differential diagnosis of atypical CLL (atypical since CD23 was not expressed), prolymphocytic leukemia (either de novo or secondary to CLL) and mantle cell lymphoma. A diagnosis of atypical CLL was favored based on the lack of organomegaly / lymphadenopathy, only moderately increased and stable WBC counts, prolymphocytes < 55% and lack of t(11;14). The subsequent lymph node biopsy had the characteristic CLL/SLL immunophenotype of CD5+, CD23+ B cells.
The paraimmunoblastic variant of SLL/CLL is a rare morphologic variant characterized by a diffuse to nodular proliferation of paraimmunoblasts, the cells usually seen in pseudoproliferation centers of SLL. In classic SLL, the predominant population is small lymphocytes with scant cytoplasm, coarsely clumped chromatin and inconspicuous nucleoli. In the paraimmunoblastic variant, the predominant cells are slightly larger, with moderately abundant cytoplasm, more open/vesicular chromatin and a single prominent, central nucleolus. The paraimmunoblasts have the same staining pattern as classic CLL/SLL (i.e. expression of CD19, CD20, CD5 and CD23, negative for CD10 and FMC7). It is considered the tissue counterpart of a prolymphocytic transformation of CLL.
The paraimmunoblastic variant was first described by Pugh as an aggressive variant that presents with generalized lymphadenopathy, and occasionally splenomegaly (Am J Surg Pathol 1988;12:907). The term paraimmunoblast was first used by Lennert (Malignant Lymphomas other than Hodgkins disease. Berlin/Heidelberg: Springer-Verlag, 1978:111-36) to describe a mitotically active, medium-sized cell with weakly staining eosinophilic cytoplasm, irregular nuclear borders, vesicular chromatin and a single, prominent, central nucleolus. In contrast, immunoblasts are larger cells with moderate basophilic cytoplasm, large round nuclei and a similar prominent, central nucleolus. Centroblasts are larger cells with multiple basophilic, peripherally-placed nucleoli.
The differential diagnosis in this case includes a Richters transformation and blastoid variant of mantle cell lymphoma. Richters transformation of CLL/SLL usually presents with a single area of marked nodal growth, unlike the diffuse, symmetrical lymphadenopathy in this and other paraimmunoblast variant cases. The Richters node may show complete or partial involvement by sheets of centroblasts or immunoblasts with a proliferation fraction in transformed areas greater than 40% (Cancer J 2005;11:161).
Blastoid variant of mantle cell lymphoma also has a vaguely nodular architecture and slightly larger cells with less clumped chromatin. However, cyclin D1 is overexpressed and t(11;14) is detected by FISH analysis.
The original flow cytometric examination demonstrated several features associated with a poor prognosis, including CD38 expression, a complex karyotype and p53 mutation (Jaffe: WHO Classification of Tumors, Pathology and Genetics of Tumours of the Haematopoietic and Lymphoid System, Blood 1998;91:4342, Blood 2001;98:181). In addition, patients with the paraimmunoblastic variant of CLL/SLL have an aggressive clinical course (Hum Path 2002;33:1145).
Nat Pernick, M.D., President
PathologyOutlines.com, Inc.
30100 Telegraph Road, Suite 404
Bingham Farms, Michigan (USA) 48025
Telephone: 248/646-0325
Fax: 248/646-1736
Email: NPernick@PathologyOutlines.com