
15 April 2021 - Case of the Month #502
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. Chunyu "Hunter" Cai, University of Texas Southwestern Medical Center, Dallas, Texas (USA), for contributing this case and discussion and to Dr. Maria Martinez-Lage Alvarez, Harvard Medical School, Boston, Massachusetts (USA), for reviewing the discussion.

Advertisement
Case of the Month #502
Clinical history:
A 3 year old girl presented with a large heterogeneously enhancing pineal mass and obstructive hydrocephalus.
Histopathology images and MRI:
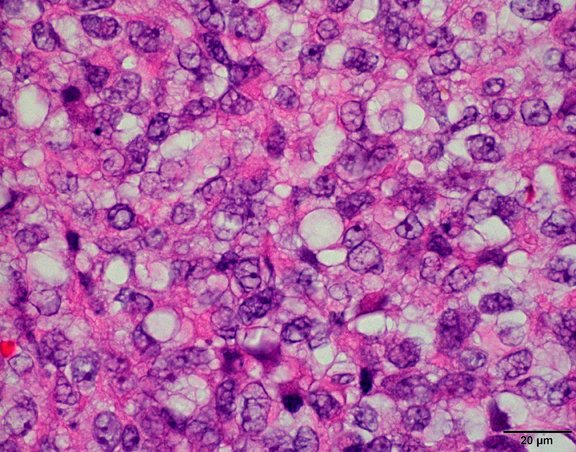
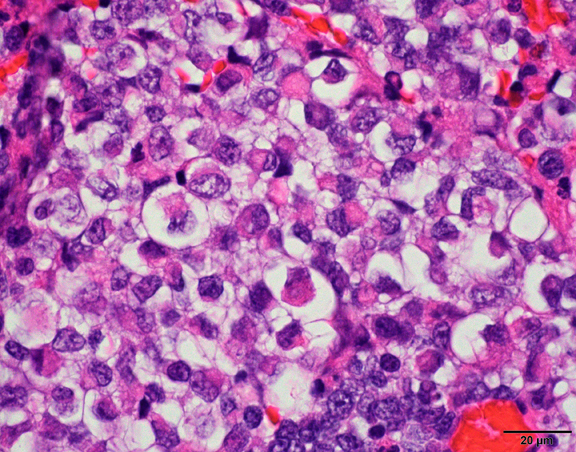
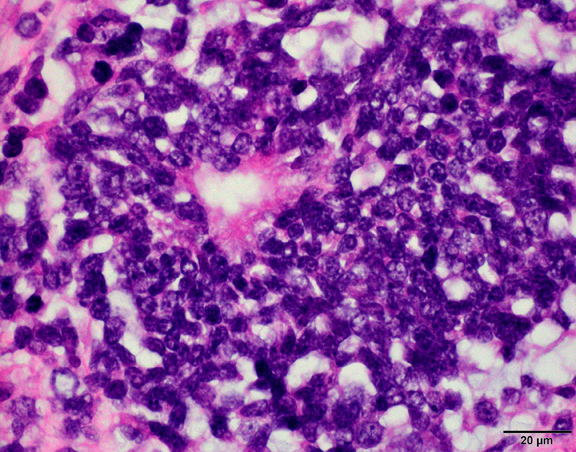

What is your diagnosis?
Diagnosis: Atypical teratoid / rhabdoid tumor (AT/RT)
Test question (answer at the end):
Which of the following is the most common age range when AT/RT presents?
A. 0 - 3 years
B. 3 - 20 years
C. 20 - 50 years
D. 50 - 80 years
Stains:
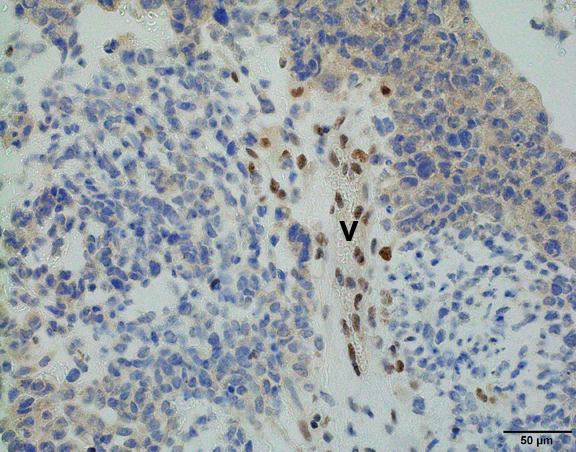
Discussion:
Atypical teratoid / rhabdoid tumor (AT/RT) is a malignant rhabdoid tumor of the CNS that also contains primitive neuroectodermal components, as well as areas with heterologous differentiation. AT/RT predominantly occurs in infants or children, with more than 80% of AT/RT diagnosed before age 3. There is a male to female predominance of 1.6 - 2:1. 52% of pediatric AT/RT occurs in the posterior fossa, 39% in the cerebral hemispheres or suprasellar region, 5% in the pineal region and 2% in the spinal cord. Approximately 33% have evidence of leptomeningeal dissemination at the time of diagnosis (Neurooncol Pract 2019;6:163). Adult cases are very rare, with approximately 50 cases reported in the English literature (Front Oncol 2018;8:567). Interestingly, nearly half of the adult cases (46%) occurred in the sellar region, with a 10:1 female predominance, followed by cerebral hemispheres (32%).
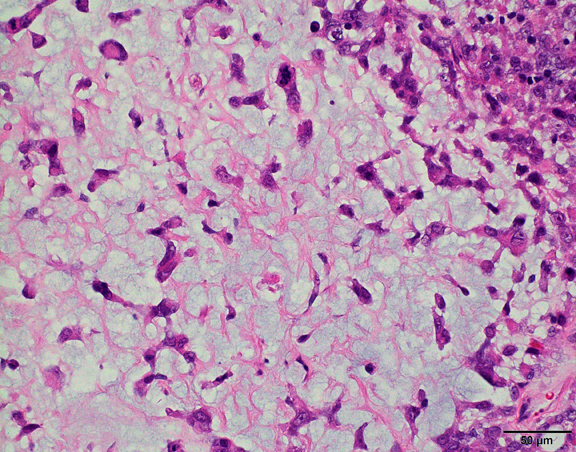
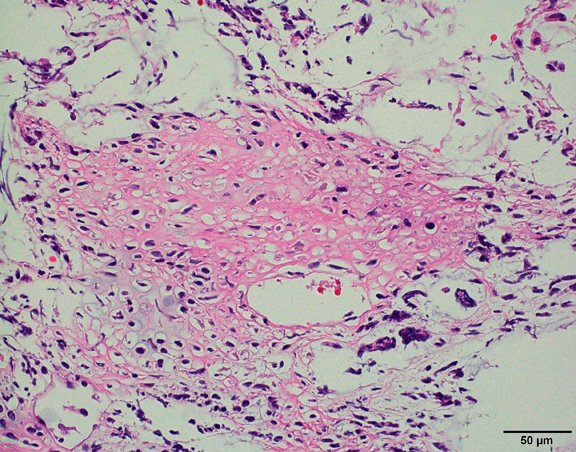
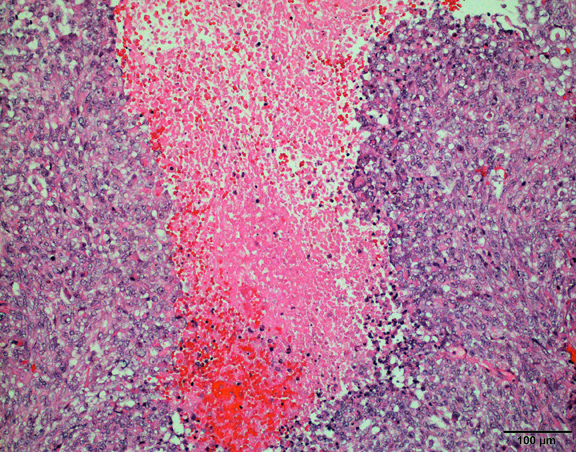
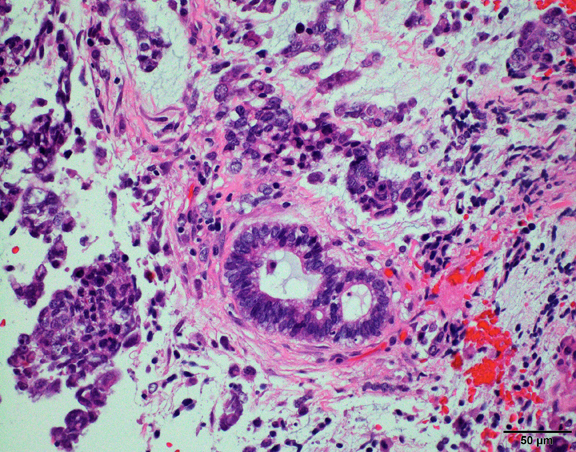
The histological hallmark of AT/RT are rhabdoid cells with eosinophilic, paranuclear inclusions, ultrastructurally composed of intermediate filamen. However, presence of rhabdoid cells is prominent only in a minority of cases and can be focal or inconspicuous in a substantial subset. The primitive blue round cell component can be large pale cells with vesicular chromatin, prominent nucleolus and abundant vacuolated cytoplasm or small blue round cells that resemble primitive neuroectodermal tumor (PNET). Focal fascicular areas of spindle cells are often present. Geographic necrosis is usually widespread, with abrupt transition from viable tumor to necrosis. Common heterologous components include myxoid matrix, glandular formation and chondroid differentiation.
Immunohistochemically, the hallmark for AT/RT is absence of nuclear INI1 protein expression in tumor cells but retained by vascular endothelial cells or inflammatory cells (Am J Surg Pathol 2004;28:644), although this is not 100% sensitive or specific. Approximately 2% of AT/RT cases have intact nuclear INI1 protein expression but loss of another SWI/SNF complex protein, SMARCA4/BRG1 (Am J Surg Pathol 2011;35:933). Genetically AT/RT is characterized by biallelic loss of the SMARCB1/INI1 gene located on chromosome 22q11.2 (Neurosurg Focus 2006;20:E11).
Survival is generally poor, with median survival around 17 months in children (Front Oncol 2012;2:114) and 20 months in adults (Front Oncol 2018;8:567). However, individual prognosis is highly variable and long term survival without evidence of disease is seen in both pediatric and adult patients.
Test question answer:
A. More than 80% of AT/RTs are diagnosed before age 3.
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. Chunyu "Hunter" Cai, University of Texas Southwestern Medical Center, Dallas, Texas (USA), for contributing this case and discussion and to Dr. Maria Martinez-Lage Alvarez, Harvard Medical School, Boston, Massachusetts (USA), for reviewing the discussion.
Advertisement
Case of the Month #502
Clinical history:
A 3 year old girl presented with a large heterogeneously enhancing pineal mass and obstructive hydrocephalus.
Histopathology images and MRI:
What is your diagnosis?
Click here for diagnosis, test question and discussion:
Diagnosis: Atypical teratoid / rhabdoid tumor (AT/RT)
Test question (answer at the end):
Which of the following is the most common age range when AT/RT presents?
A. 0 - 3 years
B. 3 - 20 years
C. 20 - 50 years
D. 50 - 80 years
Stains:
INI1
Discussion:
Atypical teratoid / rhabdoid tumor (AT/RT) is a malignant rhabdoid tumor of the CNS that also contains primitive neuroectodermal components, as well as areas with heterologous differentiation. AT/RT predominantly occurs in infants or children, with more than 80% of AT/RT diagnosed before age 3. There is a male to female predominance of 1.6 - 2:1. 52% of pediatric AT/RT occurs in the posterior fossa, 39% in the cerebral hemispheres or suprasellar region, 5% in the pineal region and 2% in the spinal cord. Approximately 33% have evidence of leptomeningeal dissemination at the time of diagnosis (Neurooncol Pract 2019;6:163). Adult cases are very rare, with approximately 50 cases reported in the English literature (Front Oncol 2018;8:567). Interestingly, nearly half of the adult cases (46%) occurred in the sellar region, with a 10:1 female predominance, followed by cerebral hemispheres (32%).
The histological hallmark of AT/RT are rhabdoid cells with eosinophilic, paranuclear inclusions, ultrastructurally composed of intermediate filamen. However, presence of rhabdoid cells is prominent only in a minority of cases and can be focal or inconspicuous in a substantial subset. The primitive blue round cell component can be large pale cells with vesicular chromatin, prominent nucleolus and abundant vacuolated cytoplasm or small blue round cells that resemble primitive neuroectodermal tumor (PNET). Focal fascicular areas of spindle cells are often present. Geographic necrosis is usually widespread, with abrupt transition from viable tumor to necrosis. Common heterologous components include myxoid matrix, glandular formation and chondroid differentiation.
Immunohistochemically, the hallmark for AT/RT is absence of nuclear INI1 protein expression in tumor cells but retained by vascular endothelial cells or inflammatory cells (Am J Surg Pathol 2004;28:644), although this is not 100% sensitive or specific. Approximately 2% of AT/RT cases have intact nuclear INI1 protein expression but loss of another SWI/SNF complex protein, SMARCA4/BRG1 (Am J Surg Pathol 2011;35:933). Genetically AT/RT is characterized by biallelic loss of the SMARCB1/INI1 gene located on chromosome 22q11.2 (Neurosurg Focus 2006;20:E11).
Survival is generally poor, with median survival around 17 months in children (Front Oncol 2012;2:114) and 20 months in adults (Front Oncol 2018;8:567). However, individual prognosis is highly variable and long term survival without evidence of disease is seen in both pediatric and adult patients.
Test question answer:
A. More than 80% of AT/RTs are diagnosed before age 3.
