

CNS & pituitary tumors
General
DNA methylation classification of CNS tumors
Authors: Chris Dampier, M.D., P.J. Cimino, M.D., Ph.D.
Editorial Board Member: Jared T. Ahrendsen, M.D., Ph.D.
Deputy Editor-in-Chief: Chunyu Cai, M.D., Ph.D.


Last author update: 24 July 2023
Last staff update: 7 August 2023
Copyright: 2023, PathologyOutlines.com, Inc.
PubMed Search: DNA methylation classification of CNS tumors

Table of Contents
Definition / general | Essential features | Terminology | Technologies for measuring DNA methylation | Utility of whole genome DNA methylation for CNS tumors | Limitations and potential pitfalls of methylation profiling | Molecular / cytogenetics images | Sample pathology report | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Dampier C, Cimino PJ. DNA methylation classification of CNS tumors. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/CNStumorDNAmethylationclass.html. Accessed April 25th, 2024.
Definition / general
- Genome wide DNA methylation based tumor classification is a valuable complementary method of CNS tumor classification that relies on computational analyses of tumor specific epigenetic modifications (Nature 2018;555:469, Acta Neuropathol 2018;136:181, Neuro Oncol 2022;24:571, Neuro Oncol 2021;23:S16)
Essential features
- DNA methylation is a type of epigenetic modification in which a covalent bond in a nucleotide is enzymatically altered to produce a subtly different compound
- A common example and one of the best studied is cytosine converted to 5 methylcytosine (5mC); this is when a DNA methyl transferase enzyme replaces the hydrogen attached to the carbon atom in position 5 of the cytosine molecule with a methyl group
- In mammalian genomes, methylation typically occurs at the 5 prime cytosine of a cytosine guanine dinucleotide, which is connected by a phosphodiester bond (CpG) (Trends Genet 2022;38:676)
Terminology
- DNA methylation classification
- Genome wide DNA methylation
- Whole genome DNA methylation
- DNA methylation
- Methylation classifier
- DNA methylation profiling
Technologies for measuring DNA methylation
- Most contemporary technologies for DNA methylation profiling rely on bisulfite conversion of DNA extracted from formalin fixed paraffin embedded tissue
- Bisulfite treatment of DNA selectively deaminates cytosine and converts it to uracil but leaves 5 methylcytosine unaffected (Nucleic Acids Res 1980;8:4777, Proc Natl Acad Sci U S A 1992;89:1827)
- Resultant bisulfite treated DNA can then be amplified by polymerase chain reaction, which amplifies uracil and thymine as thymine and 5 methylcytosine as cytosine (Proc Natl Acad Sci U S A 1992;89:1827)
- Cytosine residues in the PCR products are therefore markers of 5 methylcytosine in the original input DNA (Proc Natl Acad Sci U S A 1992;89:1827)
- As an alternative to the amplification and sequencing approach, the similar hybridization behavior of uracil and thymine can be exploited to detect cytosine to uracil / thymine polymorphisms in bisulfite treated DNA using DNA hybridization technology (Genome Res 2006;16:383)
- Bisulfite sequencing
- Bisulfite treated and PCR amplified DNA can be sequenced by, for example, massively parallel short read sequencing
- All cytosines in the output sequences are interpreted as 5mC, so genome wide DNA methylation can be identified at nucleotide resolution (Proc Natl Acad Sci U S A 1992;89:1827, Nucleic Acids Res 2001;29:E65)
- Genome wide bisulfite sequencing is still prohibitively expensive for large scale implementation
- Early whole genome DNA methylation arrays
- Illumina GoldenGate DNA Methylation BeadArray; first generation genome wide DNA methylation array, introduced in 2006 (Genome Res 2006;16:383)
- Illumina Infinium HumanMethylation27K BeadChip; second generation genome wide DNA methylation array, introduced in 2009 (Epigenomics 2009;1:177)
- Illumina Infinium HumanMethylation450K BeadChip; third generation genome wide DNA methylation array, introduced in 2011 (Genomics 2011;98:288, Epigenetics 2011;6:692)
- Current whole genome DNA methylation array
- Illumina Infinium MethylationEPIC BeadChip; fourth generation genome wide DNA methylation array, introduced in 2015
- Version 1.0 includes 853,307 CpG sites, including 439,562 of the 482,421 CpGs (91%) included in the 450K microarray (Epigenomics 2016;8:389, Nucleic Acids Res 2017;45:e22)
- Adds 333,265 CpGs located in enhancer regions identified by ENCODE and FANTOM5 (Epigenomics 2016;8:389)
- Version 2.0 includes more than 935,000 CpG sites
- Over 186,000 new probes to target known enhancers, super enhancers, CTCF binding domains and open regions of chromatin associated with primary tumors identified by ATAC seq and ChIP seq experiments
- DNA input requirement: 250 ng
Utility of whole genome DNA methylation for CNS tumors
- Definitive classification of CNS tumor types, especially useful for diagnostically challenging cases or to confirm suspected diagnoses, as discussed above (Nature 2018;555:469, Acta Neuropathol 2018;136:181, Neuro Oncol 2022;24:571, Neuro Oncol 2021;23:S16, Clin Epigenetics 2019;11:185)
- DKFZ CNS tumor classifier: first comprehensive CNS tumor DNA methylation based classifier, published in 2018, based on random forest classifier trained on a reference cohort of 2801 samples from 91 methylation classes (Nature 2018;555:469)
- Emerging bioinformatic approaches have been introduced for clinical utility and validation (Neuro Oncol 2022;24:571, J Mol Diagn 2022;24:924)
- Tumor purity greatly influences methylation class prediction and interpretability (Acta Neuropathol 2023;145:365, Neuro Oncol 2022;24:571, PLoS One 2022;17:e0265557)
- Determining well established, clinically relevant medulloblastoma subtypes (Neuro Oncol 2019;21:214, Nature 2017;547:311)
- Determining proposed surrogate grading subtypes of meningioma (Lancet Oncol 2017;18:682, Nat Genet 2022;54:649, J Clin Oncol 2021;39:3839, Nature 2021;597:119)
- Discovery of new CNS tumor types
- Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (DGONC) (Neuropathol Appl Neurobiol 2020;46:422)
- High grade astrocytoma with piloid features (Acta Neuropathol 2018;136:273)
- Infant type hemispheric glioma (Cancer Discov 2020;10:942)
- Cribriform neuroepithelial tumor (Brain Pathol 2017;27:411)
- High grade glioma with pleomorphic and pseudopapillary features (HPAP) (Acta Neuropathol 2022;143:403)
- Glioneuronal tumor with ATRX alteration, kinase fusion and anaplastic features (GTAKA) (Acta Neuropathol 2023;145:667)
- PATZ1 fusion positive neuroepithelial tumor (Acta Neuropathol 2021;142:841)
- PLAGL1 fusion positive neuroepithelial tumor (Acta Neuropathol 2021;142:827)
- Oligosarcoma, IDH mutant (Acta Neuropathol 2022;143:263)
- Resolving historical tumor types into molecularly coherent tumor classes
- Historical diffuse astrocytoma, IDH wild type; reclassified as glioblastoma, diffuse midline glioma and others (J Neuropathol Exp Neurol 2018;77:422, Acta Neuropathol 2015;130:407)
- Historical primitive neuroectodermal tumors of the central nervous system (PNET CNS); reclassified as several well defined entities (e.g., embryonal tumor with multilayered rosettes) and 4 newer tumor entities: CNS neuroblastoma, FOXR2 activated; CIC rearranged sarcoma; astroblastoma, MN1 altered; CNS tumor with BCOR internal tandem duplication (Cell 2016;164:1060)
- Historical embryonal tumor with abundant neuropil and true rosettes; reclassified as embryonal tumor with multilayered rosettes (ETMR) (Acta Neuropathol 2014;128:279)
- Historical ependymoblastoma; reclassified as ETMR (Acta Neuropathol 2014;128:279)
- Historical medulloepithelioma; reclassified as ETMR (Acta Neuropathol 2014;128:279)
- Defining prognostic tumor subtypes
- Diffuse pediatric type, high grade glioma (Acta Neuropathol 2017;134:507)
- Pineal parenchymal tumors (Acta Neuropathol 2020;139:243)
- Choroid plexus tumors (Neuro Oncol 2016;18:790)
- Intracranial mesenchymal tumor, FET::CREB fusion positive (Brain Pathol 2022;32:e13037)
- Glioblastoma (Acta Neuropathol 2022;144:155, Acta Neuropathol 2021;142:179, Brain Pathol 2018;28:656)
- Myxopapillary ependymoma (Neuro Oncol 2022;24:1689)
- Chordoma (Neuro Oncol 2022;24:442)
- Posterior fossa ependymoma (Acta Neuropathol 2022;144:373)
- IDH mutant astrocytoma (Acta Neuropathol 2021;141:85, Acta Neuropathol 2020;140:569)
- Diffuse leptomeningeal glioneuronal tumor (Acta Neuropathol 2022;144:1185)
- High grade astrocytoma with piloid features (Acta Neuropathol 2023;145:71)
- In addition to CNS tumor classification, the DNA methylation array allows for determination of alterations routinely assayed in surgical neuropathology on a single platform, including 1p / 19q codeletion status, somatic copy number alterations (chromosomal and gene level), MGMT promoter methylation status and chromosomal breakpoints inferring gene fusions (Neuro Oncol 2014;16:1630, Neuropathol Appl Neurobiol 2021;47:406)
- Some of these features may not be clinically validated in certain settings but their presence may suggest further orthogonal testing
Limitations and potential pitfalls of methylation profiling
- Like other studies, methylation profiling should be considered in conjunction with other histopathologic, genetic and clinical data in order to reach a diagnosis
- Misleading methylation profiles can occur (i.e., methylation matches that apparently conflict with histologic, genetic, radiographic or clinical features)
- Requires a relatively large amount of optimal input DNA (~250 ng)
- Some methylation classes do not necessarily correspond to a CNS WHO tumor type
- High confidence methylation classifier score may not be seen in up to 1/3 of cases (Neuro Oncol 2022;24:571, Clin Epigenetics 2019;11:185)
- Factors that may contribute to low classifier score (no methylation match)
- Low tumor cell content (low purity of sample)
- Failed DNA bisulfite conversion
- Poor quality DNA (e.g., old tissue blocks)
- Single bioinformatic pipelines are reliant on a single reference dataset
- Rare tumor types may not be present in reference dataset, so no class exists for them
Molecular / cytogenetics images
Contributed by P.J. Cimino, M.D., Ph.D. and Chris Dampier, M.D.
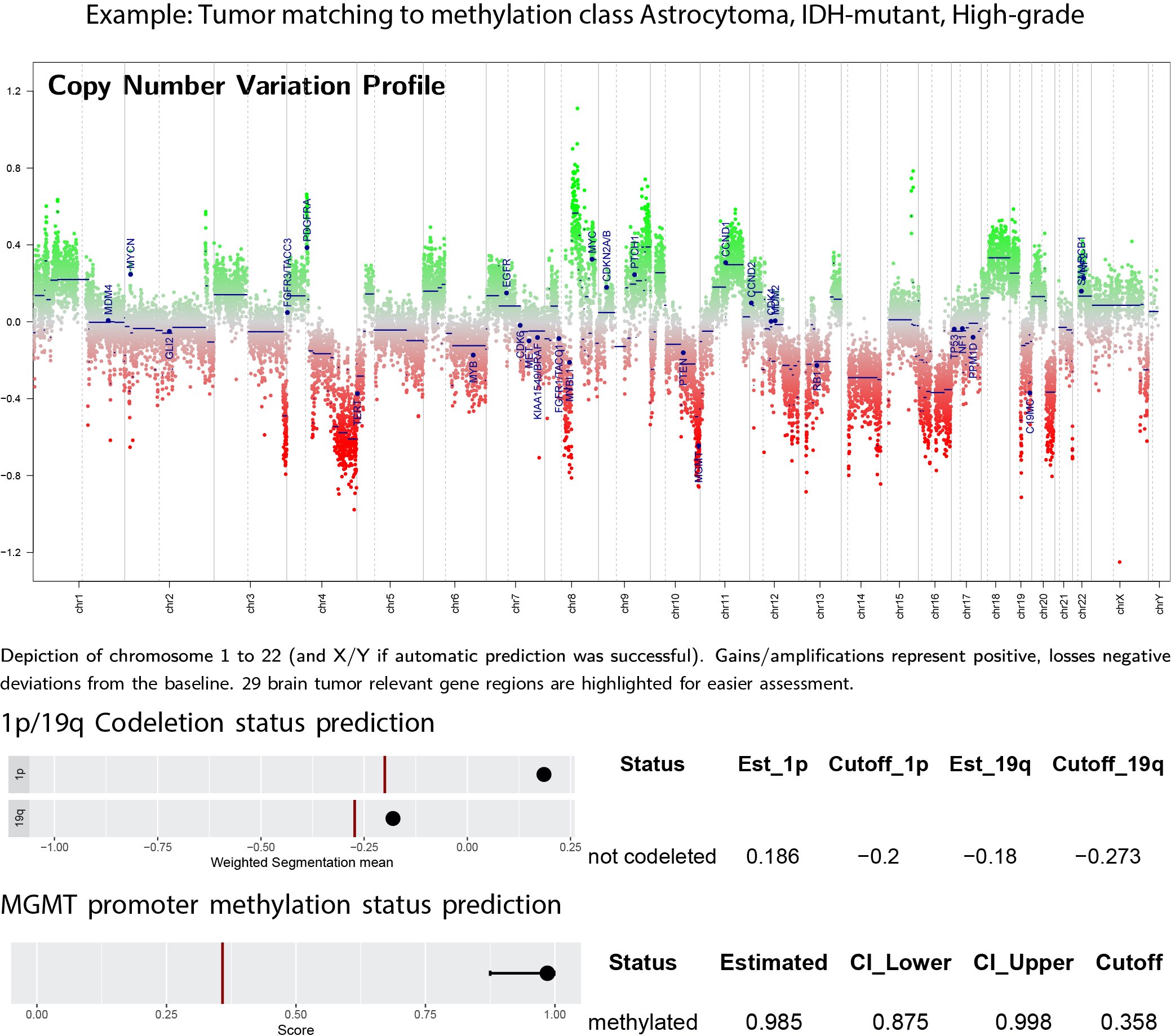
Methylation copy number plot
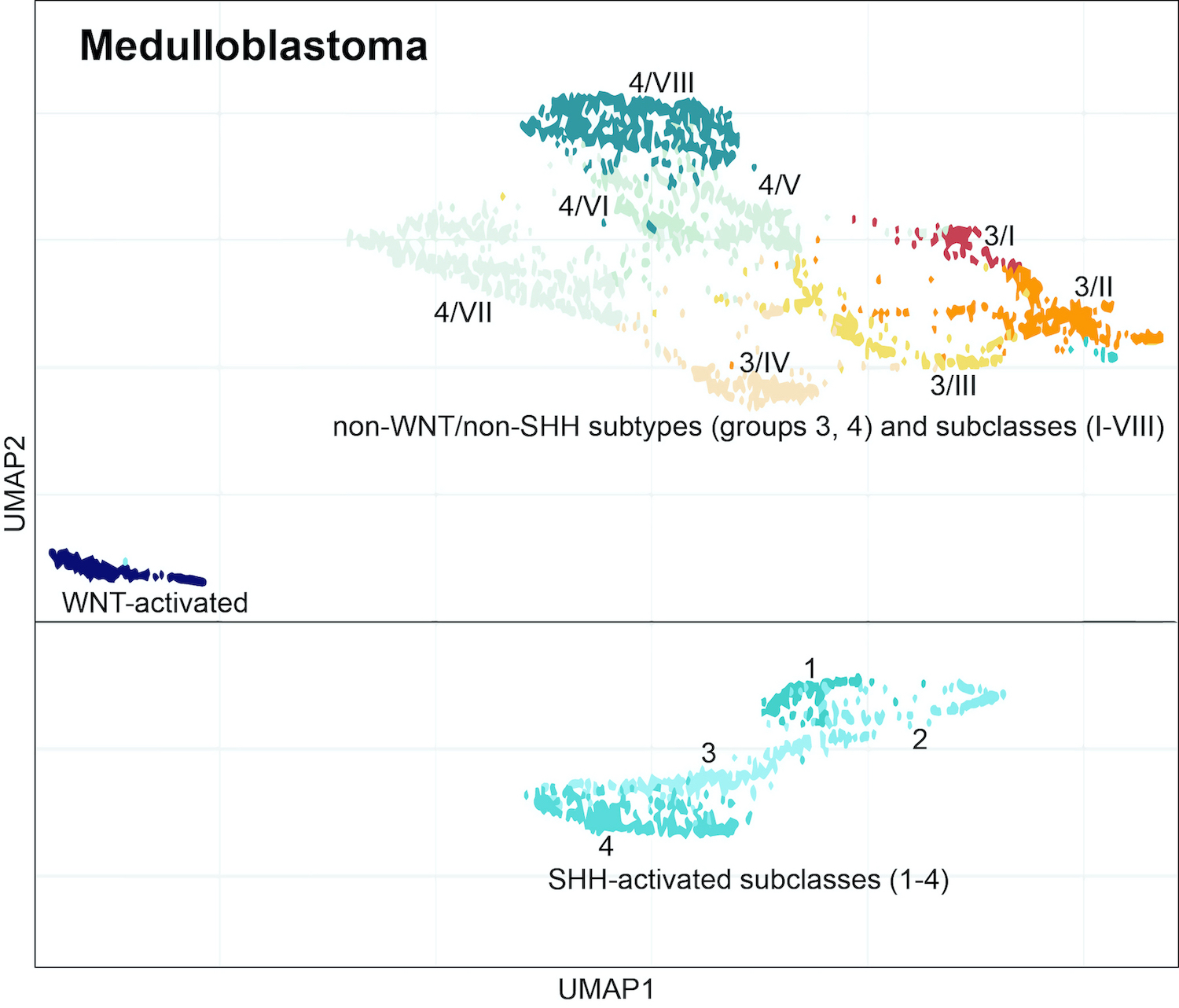
Dimensionality reduction of medulloblastoma
Images hosted on other servers:

Methylation based CNS tumor clustering

Impact of tumor purity

Workflow in surgical neuropathology

Heterogeneity of histologic diagnoses
Sample pathology report
- Brain, cervical spinal cord, biopsy:
- Histological classification: glial neoplasm with focal ependymal features
- CNS WHO grade 2
- Consensus methylation profiling class: spinal ependymoma
- Integrated diagnosis: spinal ependymoma, CNS WHO grade 2
Additional references
Board review style question #1
A 7 year old girl undergoes stereotactic needle biopsy for diagnosis of an intracranial intrinsic right parietal lobe mass. There is scant tissue in the biopsy and nucleic acid extraction is expected to yield material for only a single genome wide assay (e.g., cytogenomic array, next generation sequencing, DNA methylation array). Which of these features are specifically detected by DNA methylation array profiling?
- Chromosomal copy number alterations
- Genomic breakpoints to suggest gene fusions
- Methylation class
- MGMT promoter methylation status
- Single gene mutations (e.g., specific H3 G34 mutation)
Board review style answer #1
C. Methylation class is specifically detected by DNA methylation array profiling. Answer D is incorrect because MGMT promoter methylation status can also be detected by methylation specific PCR. Answers A and B are incorrect because chromosomal copy number alterations and genomic breakpoints to suggest gene fusions can also be detected by other modalities such as cytogenetic microarrays, next generation sequencing and fluorescence in situ hybridization. Answer E is incorrect because single gene mutations may be inferred by DNA methylation array but the DNA sequence is not directly detectable.
Comment Here
Reference: DNA methylation classification of CNS tumors
Comment Here
Reference: DNA methylation classification of CNS tumors
Board review style question #2
A 5 year old boy undergoes resection of a medulloblastoma from the posterior fossa. DNA methylation profiling of the tumor can determine which of the following?
- Medulloblastoma histologic subtype
- Medulloblastoma molecular subtype
- Mitotic count
- Patient age
- TP53 mutational status in SHH activated medulloblastoma
Board review style answer #2
B. Medulloblastoma molecular subtype. DNA methylation profiling is useful for determining medulloblastoma molecular subtype. Answer D is incorrect because patient age is not discernable by methylation profiling. Answer E is incorrect because TP53 status requires DNA sequencing, not methylation. Answers A and C are incorrect because medulloblastoma histologic subtype and mitotic counts are determined by microscopic examination, not methylation.
Comment Here
Reference: DNA methylation classification of CNS tumors
Comment Here
Reference: DNA methylation classification of CNS tumors
