
Kidney nontumor / medical renal
Glomerular disease
Inherited glomerular disease
Fabry disease
Editorial Board Members: Nicole K. Andeen, M.D., Jonathan E. Zuckerman, M.D., Ph.D.


Last author update: 13 March 2025
Last staff update: 13 March 2025
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Fabry disease

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Clinical images | Microscopic (histologic) description | Microscopic (histologic) images | Immunofluorescence description | Electron microscopy description | Electron microscopy images | Genetics | Videos | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Velagapudi RK, Paueksakon P. Fabry disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyfabry.html. Accessed August 27th, 2025.
Definition / general
- Fabry disease is a lysosomal storage disorder caused by genetic deficiency of lysosomal enzyme alpha galactosidase A (αGal), leading to accumulation of globotriaosylceramide (GL3) in various organs and resulting in dysfunction
- It is inherited in an X linked recessive pattern
Essential features
- Lysosomal storage disorder with an X linked recessive mode of inheritance due to deficiency of enzyme alpha galactosidase A
- Multiorgan involvement including skin, nervous system, heart and kidneys
- Light microscopy shows foamy and vacuolated podocytes and tubular epithelial cells with corresponding lamellated inclusions by electron microscopy
Terminology
- Alpha galactosidase A deficiency
- Anderson-Fabry disease
- Angiokeratoma corporis diffusum
- Ceramide trihexosidase deficiency
- Sweeley-Klionsky disease
- Ruiter-Pompen-Wyers syndrome
ICD coding
- ICD-10: E75.21 - Fabry (-Anderson) disease
Epidemiology
- Rare inherited disease, although it is the second most prevalent inherited lysosomal storage disorder after Gaucher disease (Oxford PharmaGenesis: Fabry Disease - Perspectives From 5 Years of FOS [Accessed 29 November 2024])
- It is inherited in an X linked recessive pattern and primarily affects male patients
- It is a panethnic disease with an estimated birth incidence of 1 per 117,000 in the general population and 1 per 40,000 male live births (Adv Rheumatol 2024;64:22)
- Actual prevalence of this condition may be higher because of challenges in diagnosing this disease, which has a wide range of clinical presentations and ages at presentation (Int Urol Nephrol 2024;56:3161)
Sites
- Multisystem disease
- Kidney (glomeruli)
- Heart
- Sweat glands
- Nerves
Pathophysiology
- Deficiency in lysosomal enzyme exoglycohydrolase, ceramide trihexosidase commonly referred to as alpha galactosidase A (αGal)
- The enzyme catalyzes the cleavage of glycosphingolipids, especially globotriaosylceramide present in cell membranes and deficiency of enzyme leads to accumulation of globotriaosylceramide in lysosomes of endothelial cells of kidney, heart, skin, brain and nerves (Orphanet J Rare Dis 2010;5:30)
Etiology
- X linked inheritance of deficiency of lysosomal enzyme alpha galactosidase A
Diagrams / tables
Images hosted on other servers:
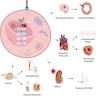
Pathophysiology of Fabry disease

Clinical features of Fabry disease
Clinical features
- Symptoms and signs vary with mutation and functional level of αGal diagnosis (Int Urol Nephrol 2024;56:3161)
- Hemizygous male patients present with multisystem involvement starting in infancy or childhood with acroparesthesias, hypohidrosis and angiokeratomas
- Early renal manifestations result from defects in the ability to concentrate urine, with proteinuria in adulthood and renal failure by 40 years of age
- Younger male patients can have albuminuria and show lesions on kidney biopsy
- Patients with residual enzyme activity and heterozygous female patients vary in symptomatology from asymptomatic to renal failure or cardiomyopathy
- Other systemic manifestations include stroke, autonomic dysfunction, heart attack, deafness and cardiomyopathy
Diagnosis
- Newborn screening using fluorometric enzyme assay for αGal activity
- Genetic testing for disease causing variant in GLA gene that encodes αGal (Adv Rheumatol 2024;64:22)
Laboratory
- For male patients, a very low leukocyte αGal activity (< 1% activity) is highly suggestive of Fabry disease
- Genetic test for mutations in GLA gene is required for female patients and for confirmation in male patients (Ther Clin Risk Manag 2020;16:551)
Radiology description
- Nonspecific findings of medical renal disease, including increased echogenicity, thinned out renal cortex and multiple renal cysts seen on imaging (AJR Am J Roentgenol 2006;186:1184, J Comput Assist Tomogr 2004;28:158)
Prognostic factors
- Prognosis is linked to the extent of organ involvement and timeliness of diagnosis (Int Urol Nephrol 2024;56:3161)
- Urine protein creatinine ratio is the most important indicator of renal disease progression in adult Fabry patients (Clin J Am Soc Nephrol 2010;5:2220)
- Patients may end up with end stage kidney disease requiring transplant
- Although little is known about recurrence of Fabry after kidney transplant, in general it is thought to not recur in patients on enzyme replacement therapy; although biopsies may show inclusions, graft function is not usually compromised (J Am Soc Nephrol 2002;13:S134, Medicina (Kaunas) 2020;56:284)
Case reports
- 21 year old woman with maternal history of Fabry disease (Case Rep Nephrol Dial 2021;11:355)
- 24 year old woman with nocturia and left ventricular hypertrophy (Cureus 2024;16:e63661)
- 37 year old man with recurrent proteinuria and ventricular septal thickening (BMC Nephrol 2024;25:61)
- 45 year old man with hematochezia and proteinuria (Korean J Intern Med 2015;30:925)
- Case series of 5 patients with Fabry disease and end stage renal disease (Ann Med Health Sci Res 2016;6:193)
Treatment
- Enzyme replacement therapy
- Chaperone drugs
- Transplant in patients developing end stage kidney disease
- Reference: Mol Genet Metab 2018;123:416
Clinical images
Images hosted on other servers:
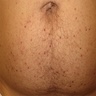
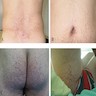
Angiokeratomas
Microscopic (histologic) description
- Standard light microscopy processing may extract the accumulated lipids due to processing in xylene
- Glomeruli show hypertrophic glomerular visceral epithelial cells (podocytes) with vacuolated, foamy or pale and lacy cytoplasm, because of accumulated lipids (J Am Soc Nephrol 2002;13:S134)
- With progressive disease, mesangial expansion, global and segmental glomerulosclerosis and interstitial fibrosis and tubular atrophy develops
- Heterozygotes in female patients may have patchy and irregular distribution of the above findings (Kidney Int 1978;13:223)
- 1 micron (semi-thin) sections of glutaraldehyde or formaldehyde fixed plastic embedded tissue that are stained with toluidine blue are ideal for detecting clusters of lipid inclusions within lysosomes as the processing for electron microscopy studies does not extract the lipids
- Inclusions vary in size and shape and have a lamellated appearance with alternating dark and light layers that are known as myelin bodies or zebra bodies
- These inclusions are most prominent in podocytes but can also be seen in tubular epithelial cells, mesangial cells, endothelial cells and interstitial cells
Microscopic (histologic) images
Contributed by Ramya Krishna Velagapudi, M.D., M.H.A. and @mercury.lin_md on Instagram
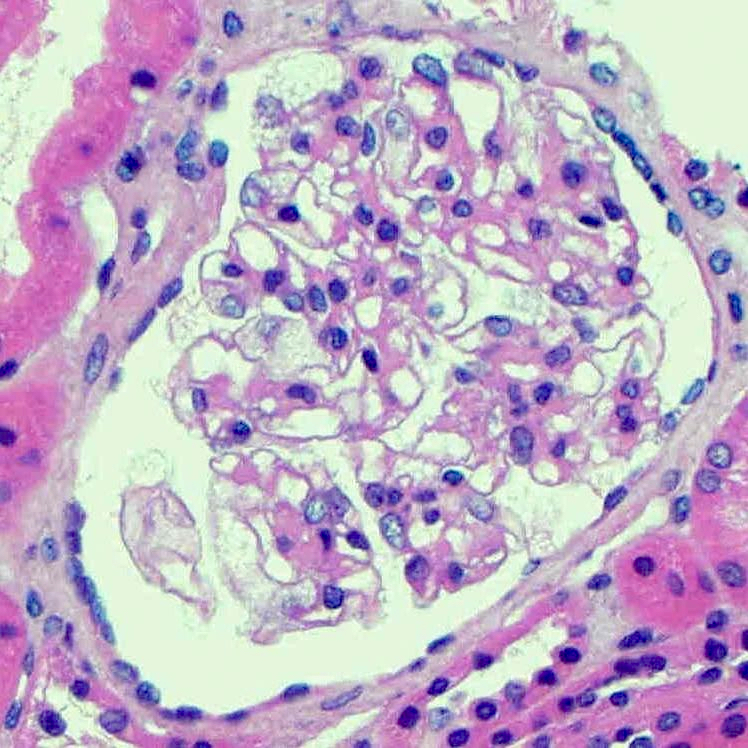
Fabry disease
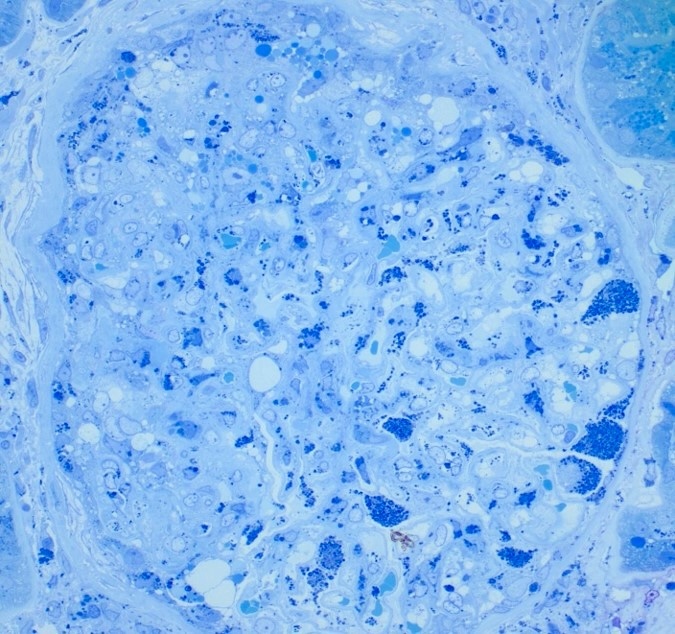
Inclusions (toluidine blue)
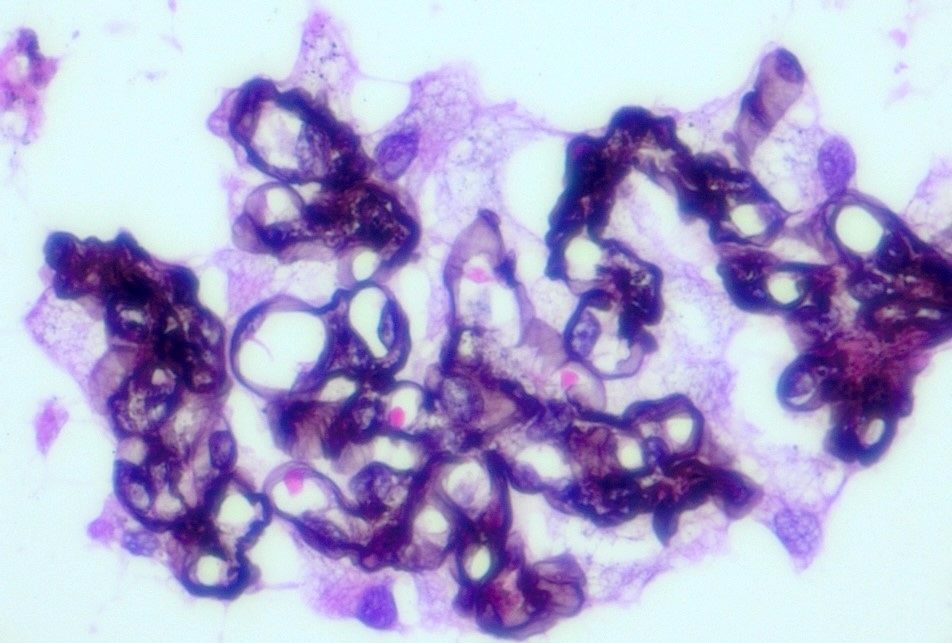
Foamy appearing podocytes (Jones silver)
Immunofluorescence description
- Generally negative
- May have nonspecific trapping of IgM and C3 in mesangial areas
Electron microscopy description
- Lamellated electron dense lipid inclusions within lysosomes, seen predominantly in podocytes and tubular epithelial cells, are diagnostic
- Percentage of podocytes with lipid inclusions decreases with age
Electron microscopy images
Contributed by Paisit Paueksakon, M.D.
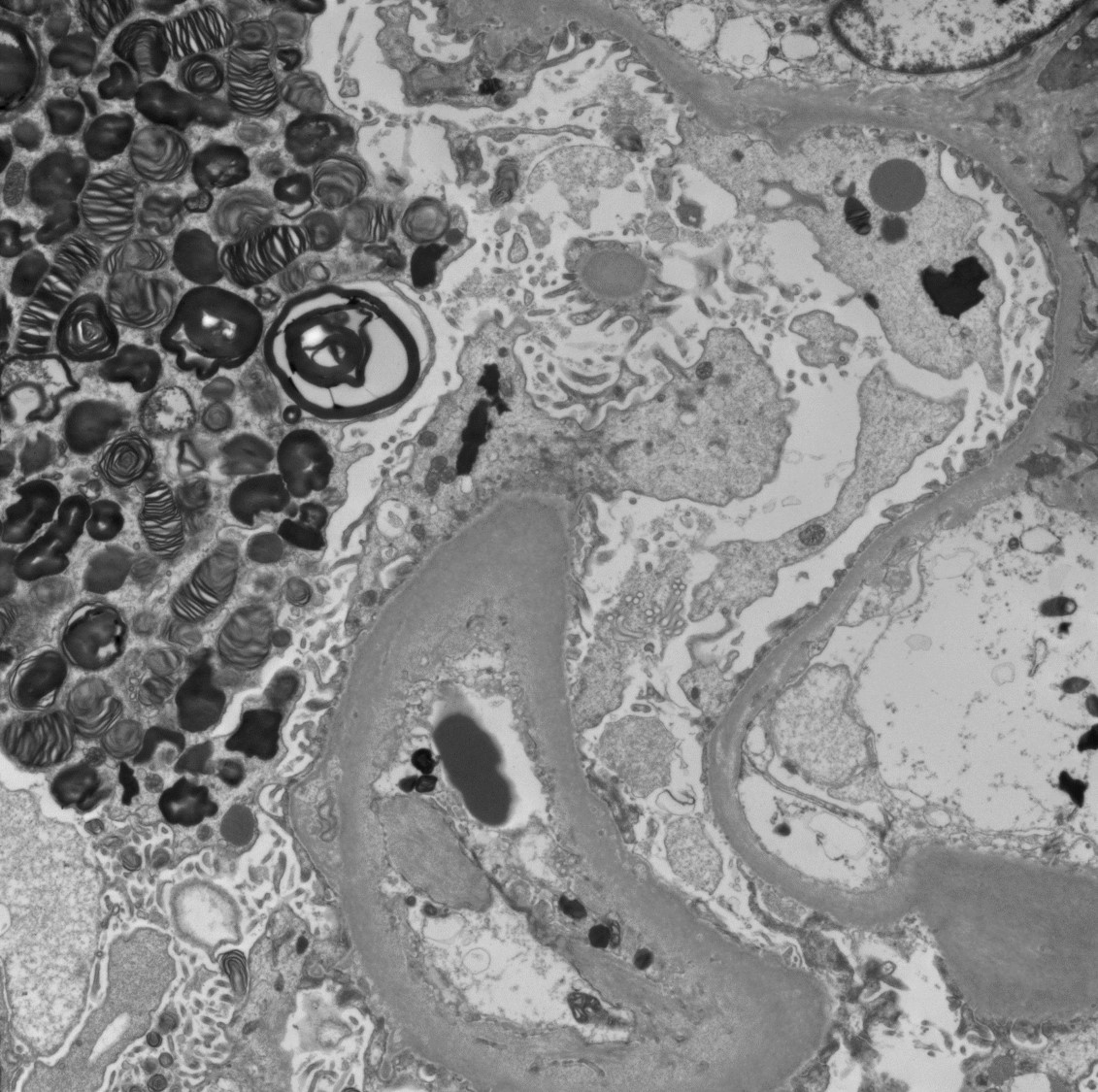
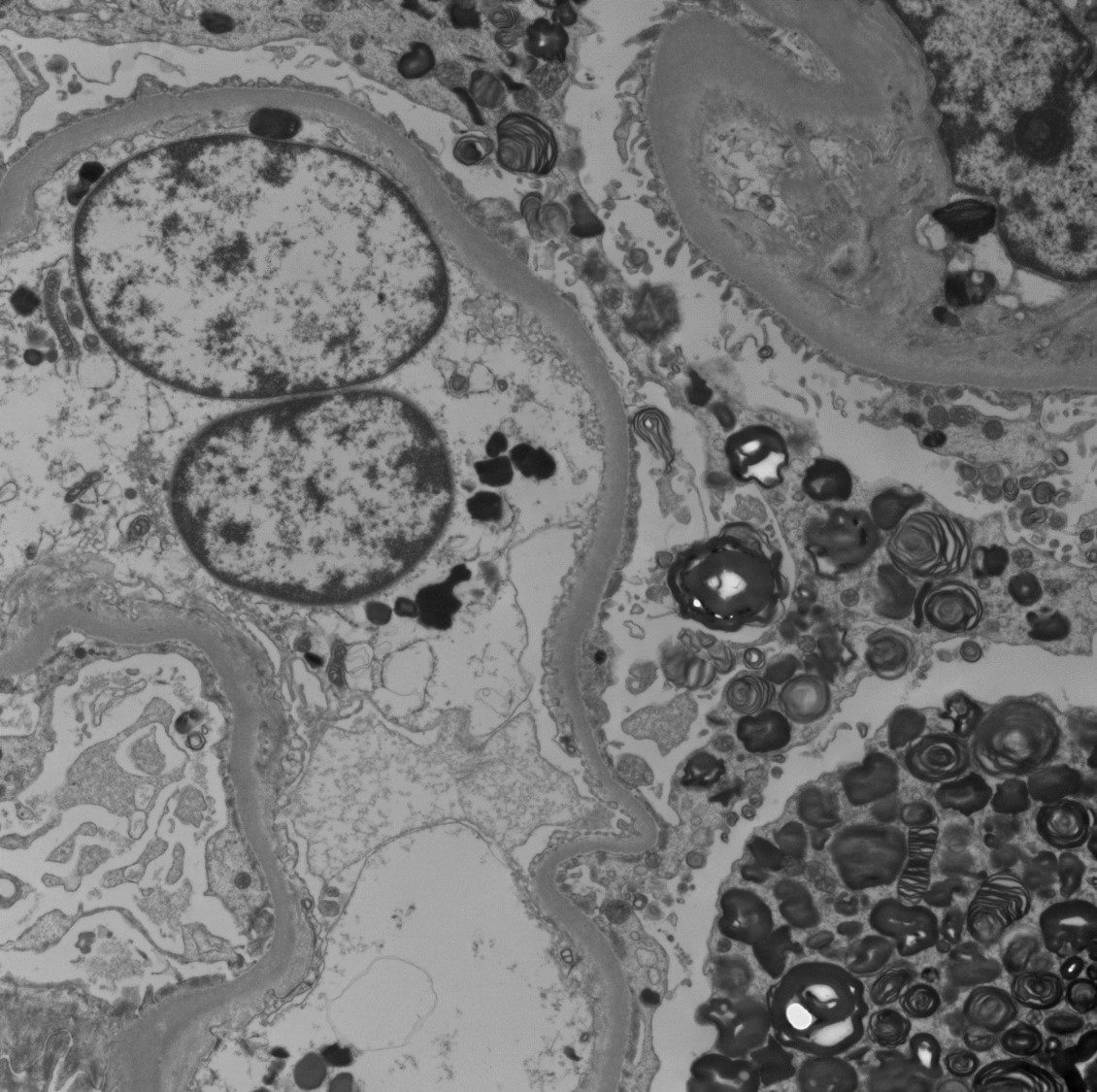
Inclusions
Images hosted on other servers:

Lamellated inclusions in podocytes

Myelin-like inclusions in podocyte cytoplasm
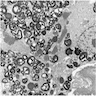
Myelin figures in glomerular epithelial cells

Lamellated inclusions in glomerulus

Inclusions in different cells
Genetics
- X linked disorder with deficient activity of alpha galactosidase A enzyme encoded by GLA gene located on X chromosome (GeneReviews: Fabry Disease [Accessed 29 November 2024])
Videos
Washington University in St. Louis renal pathology teaching series: Fabry disease
Sample pathology report
- Kidney, percutaneous needle biopsy:
- Findings consistent with Fabry disease (see comment)
- Comment: The biopsy shows 22 glomeruli by light microscopy, 7 of which are globally sclerosed, with ~40% interstitial fibrosis. Glomeruli show foamy appearing podocytes but are otherwise unremarkable. Focal tubular epithelial cells show foamy vacuolated cytoplasm. Immunofluorescence performed on frozen tissue does not reveal any positive staining. Electron microscopy shows dense lamellated inclusions within podocytes and proximal tubular epithelial cells. Taken together, these findings are consistent with Fabry disease.
Differential diagnosis
- Iatrogenic phospholipidosis:
- Secondary to drug toxicity of amphophilic drugs, such as chloroquine, hydroxychloroquine and aminoglycosides, that inhibit lysosomal enzyme activity
- Other drugs recently associated with myelin bodies include sertraline, anticholesterol medications (statins and fenofibrate), beta blockers, antidepressants and anti-estrogen therapy (tamoxifen) (Ultrastruct Pathol 2022;46:130)
- Contrast injection medium (BMC Nephrol 2018;19:53)
- Indistinguishable on light microscopy
- Extent and distribution of inclusions by electron microscopy is sparse compared to that of Fabry disease
- Laboratory testing will not reveal any deficiency of lysosomal enzyme alpha galactosidase A (αGal)
- Silicone nephropathy (Am J Nephrol 1983;3:279):
- Negative family history and no other clinical characteristics, including skin angiokeratomas, acroparasthesias, etc.
- Extent and distribution of inclusions by electron microscopy is sparse compared to that of Fabry disease
- Laboratory testing will not reveal any deficiency of lysosomal enzyme alpha galactosidase A (αGal)
- Niemann-Pick disease (NDT Plus 2009;2:448):
- Rarely involves kidney
- Similar light microscopy findings in glomeruli and tubular epithelial cells
- LMX1B associated nephropathy (also called nail patella-like renal disease) (CEN Case Rep 2021;10:588):
- Electron microscopy may show findings of focal and diffuse irregular thickening of glomerular basement membranes with a moth eaten appearance; present in LMX1B associated nephropathy and absent in Fabry disease
- Genetic analysis shows mutations in LMX1B gene
- Longstanding proteinuria:
- Lipid deposition may be an incidental finding in patients with longstanding proteinuria
- Extent and intensity of inclusions by electron microscopy is usually not as extensive as in Fabry disease
Additional references
Practice question #1
A 15 year old boy with a history of paresthesia and a reddish-purple rash on trunk as well as a recent history of shortness of breath was noted to have proteinuria. ECHO showed ventricular septal thickening. There was a family history of unknown kidney disease in his maternal uncles. After multiple serologic tests, a kidney biopsy was performed. Light microscopy revealed vacuolization and foamy changes in podocytes but no segmental sclerotic lesions and no endocapillary or mesangial hypercellularity. Immunofluorescence microscopy did not reveal any deposits or staining. Electron microscopy showed the findings above and no significant foot process effacement. What is the most probable diagnosis?
- Fabry disease / nephropathy
- Focal and segmental glomerulosclerosis
- IgA nephropathy
- Lupus nephritis
Practice answer #1
A. Fabry disease / nephropathy. Light microscopy showing vacuolization and foamy changes in podocytes, negative immunofluorescence microscopy and electron microscopy with extensive lamellated inclusions also called myelin bodies or zebra bodies. Answer D is incorrect because there is no full house pattern of deposit staining by immunofluorescence microscopy and no deposits by electron microscopy. Answer B is incorrect because there are no segmental sclerotic lesions by light microscopy and no foot process effacement by electron microscopy. Answer C is incorrect because there is no IgA dominant staining by immunofluorescence microscopy and no deposits by electron microscopy.
Comment Here
Reference: Fabry disease
Comment Here
Reference: Fabry disease
Practice question #2
An 11 year old boy was recently diagnosed with Fabry disease on genetic testing. What is the most common mode of inheritance for Fabry disease?
- Autosomal dominant
- Autosomal recessive
- Mitochondrial
- X linked recessive
Practice answer #2
D. X linked recessive is the pattern of inheritance for Fabry disease. Answers A, B and C are incorrect because the pattern of inheritance is not autosomal dominant, autosomal recessive or mitochondrial.
Comment Here
Reference: Fabry disease
Comment Here
Reference: Fabry disease
