Kidney tumor
Childhood tumors
Congenital mesoblastic nephroma
Editorial Board Member: Maria Tretiakova, M.D., Ph.D.
Editor-in-Chief: Debra L. Zynger, M.D.


Last author update: 10 December 2020
Last staff update: 1 May 2025 (update in progress)
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed Search: Mesoblastic nephroma[TI] kidney[TIAB]
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Positive stains | Negative stains | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: D'Hooghe E, Vujanic GM. Congenital mesoblastic nephroma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneytumormesoblastic.html. Accessed September 2nd, 2025.
Definition / general
- Mesoblastic nephroma is a mesenchymal / myofibroblastic renal tumor of low grade malignancy
- It typically occurs in infancy, often congenital but not always
- First described by Bolande et al. in 1967 (Pediatrics 1967;40:272)
Essential features
- Most common renal tumor in the first month of life
- 90% of cases diagnosed in the first 9 months of life
- Virtually never after 3 years of age
- Adult mesoblastic nephroma does not exist
- 3 histologic subtypes: classic (~25% of cases), cellular (~65%) and mixed (~10%)
- Cellular type: ETV6-NTRK3 gene fusion in ~70% of cases
- Classic type: EGFR internal tandem duplication mutation
- Most important prognostic factor is completeness of surgical resection
Terminology
- Historic synonyms: renal leiomyomatous hamartoma, congenital mesoblastic nephroma
- Although "congenital" is a part of the WHO stated name for this tumor, it is sometimes a misnomer as the tumor is not always present from birth
ICD coding
Epidemiology
- Represents 3 - 4% of all renal tumors of childhood (Pediatr Blood Cancer 2011;56:744)
- Most common renal tumor in the first month of life (Pediatr Blood Cancer 2008;50:1130)
- 90% of cases occur in the first 9 months of life (Pediatr Blood Cancer 2008;50:1130)
- Median age < 1 month in 63% of cases (Pediatr Blood Cancer 2017;64:e26437)
- Median age differs between classic (7 days), mixed (1.9 months) and cellular (4.6 months) types (Pediatr Blood Cancer 2011;56:744)
- Virtually never occurs after the age of 3 years (Pediatr Blood Cancer 2017;64:e26437)
- Adult mesoblastic nephroma does not exist
- Not associated with syndromes or congenital anomalies
Sites
- Kidney; never bilateral, never multifocal
Pathophysiology
- Unknown
Etiology
- No recognized risk factors
- No familial cases have been reported
- Cellular type associated with t(12;15)(p13;q25) in ~70% of cases (Pediatr Blood Cancer 2018;65:e26925)
- Classic type associated with EGFR internal tandem duplication mutation (Histopathology 2020;77:611)
- Not associated with nephrogenic rests
Clinical features
- Abdominal mass (~75% of patients) (Pediatr Blood Cancer 2017;64:e26437)
- ~15% detected prenatally, usually associated with polyhydramnios
- Hypertension (~20%)
- Hematuria (~10%)
- Never presents as metastatic or bilateral disease
- Virtually never associated with syndromes or anomalies typical for Wilms tumor
Diagnosis
- Imaging techniques cannot distinguish it from other renal tumors (J Pediatr Surg 2008;43:1301)
- Biopsy not recommended
- Diagnosis made on nephrectomy
Laboratory
- No specific laboratory findings
- Hypercalcemia (4% of cases)
- Hyperreninemia (1% of cases) (Pediatr Blood Cancer 2017;64:e26437)
Radiology description
- Unilateral, well demarcated, hypoechogenic mass
Radiology images
Images hosted on other servers:
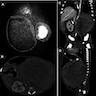
Large renal mass
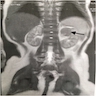
Upper pole mass
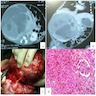
CT large mass
Prognostic factors
- Most important prognostic factor is completeness of surgical resection
- Histologic type is of no prognostic significance
- Overall survival reported as ~96% but is almost certainly higher since cases with unfavorable outcome are more likely to be reported (Pediatr Blood Cancer 2008;50:1130)
- Of 12/276 patients with mesoblastic nephroma who died, 5 died of tumor related and 7 of treatment related causes (Pediatr Blood Cancer 2017;64:e26437)
- Very rare local relapses and metastases; 38 cases reported in the literature (Pediatr Surg Int 2017;33:1183)
- Relapses and metastases occur within 12 months after the diagnosis (range 1 - 12 months, median 6 months)
- Local relapses in 27/38 (71%) of these cases
- 21/27 (76%) had incomplete resection (stage 3)
- All but 1 patient had cellular type but even then, cellular type had no significant difference in prognosis
- Metastases developed in 18 children (7 also had local recurrence)
- Lung metastases in 7/18 (39%), liver in 5/18 (28%), brain in 4/18 (22%) and heart, bone and peritoneum in 5.5% each
- 38% of patients with metastases died (all who had brain, bone, heart and peritoneum metastases)
Case reports
- Prematurely born boy presented with neonatal hypertension (Turk J Pediatr 2018;60:198)
- Prematurely born boy with a prenatal diagnosis of polyhydramnios (Urol Case Rep 2015;3:157)
- 7 day old boy presented with abdominal distension and vomiting (J Neonatal Surg 2017;6:45)
- 11 month old boy presented with a renal mass (Urol Case Rep 2019;26:100979)
Treatment
- Complete surgical resection (total nephrectomy) with wide resection margins (Pediatr Blood Cancer 2017;64:e26437, Pediatr Surg Int 2017;33:1183)
- Even stages II and III treated with surgery only
- Relapses also treated with surgery
- Chemotherapy treatment of relapses only in unresectable cases
Gross description
- Always unilateral and solitary (Adv Anat Pathol 2003;10:243)
- Typically found in the medial renal sinus
- Classic type:
- Firm, whorled, leiomyomatous consistency
- Poorly demarcated from the normal renal parenchyma
- Cellular type:
- Usually soft
- Tumor-kidney junction relatively clearly demarcated
- Cystic areas
- Can show hemorrhage and necrosis
Gross images
Contributed by Ellen D’Hooghe, M.D., Gordan M. Vujanic, M.D., Ph.D. and Case #57

Classic type
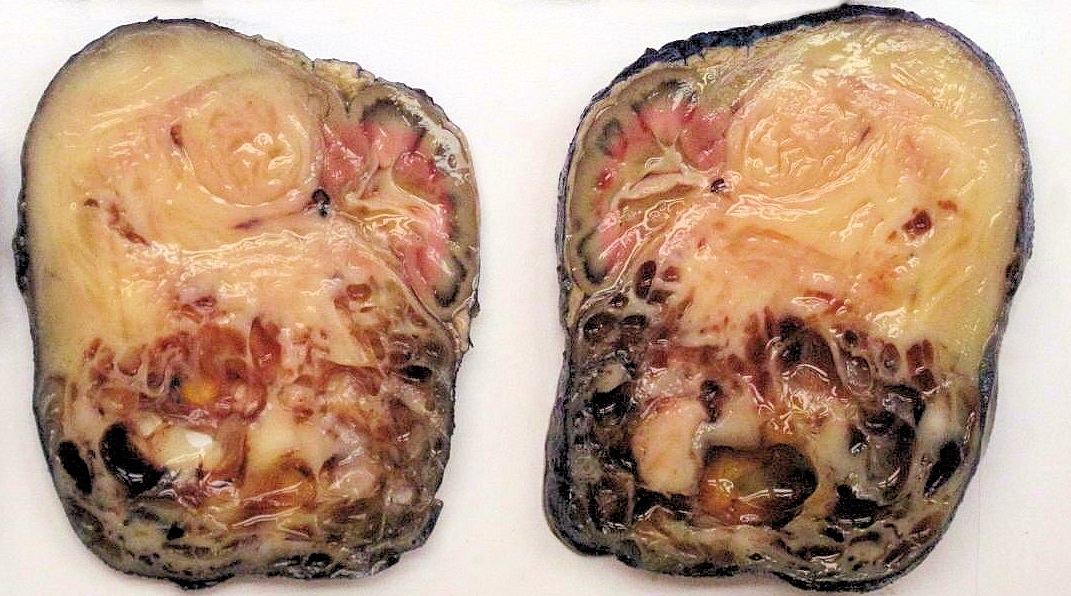
Mixed type
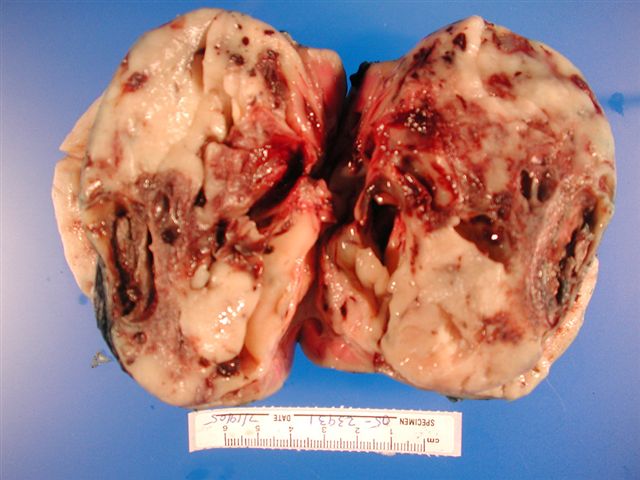
Cellular type
Microscopic (histologic) description
- Classic type (~25% of cases) (Adv Anat Pathol 2003;10:243, Histopathology 1985;9:741):
- Intermingling fascicles of spindle cells with low mitotic activity
- Collagen deposition
- Often prominent dilated, thin walled vascular spaces
- No capsule
- Tumor-kidney border is irregular with finger-like protrusions of the tumor into the renal parenchyma
- Islands of hyaline cartilage can be seen at the tumor-kidney interface
- Entrapped islands of renal parenchyma
- Foci of extramedullary hematopoiesis common
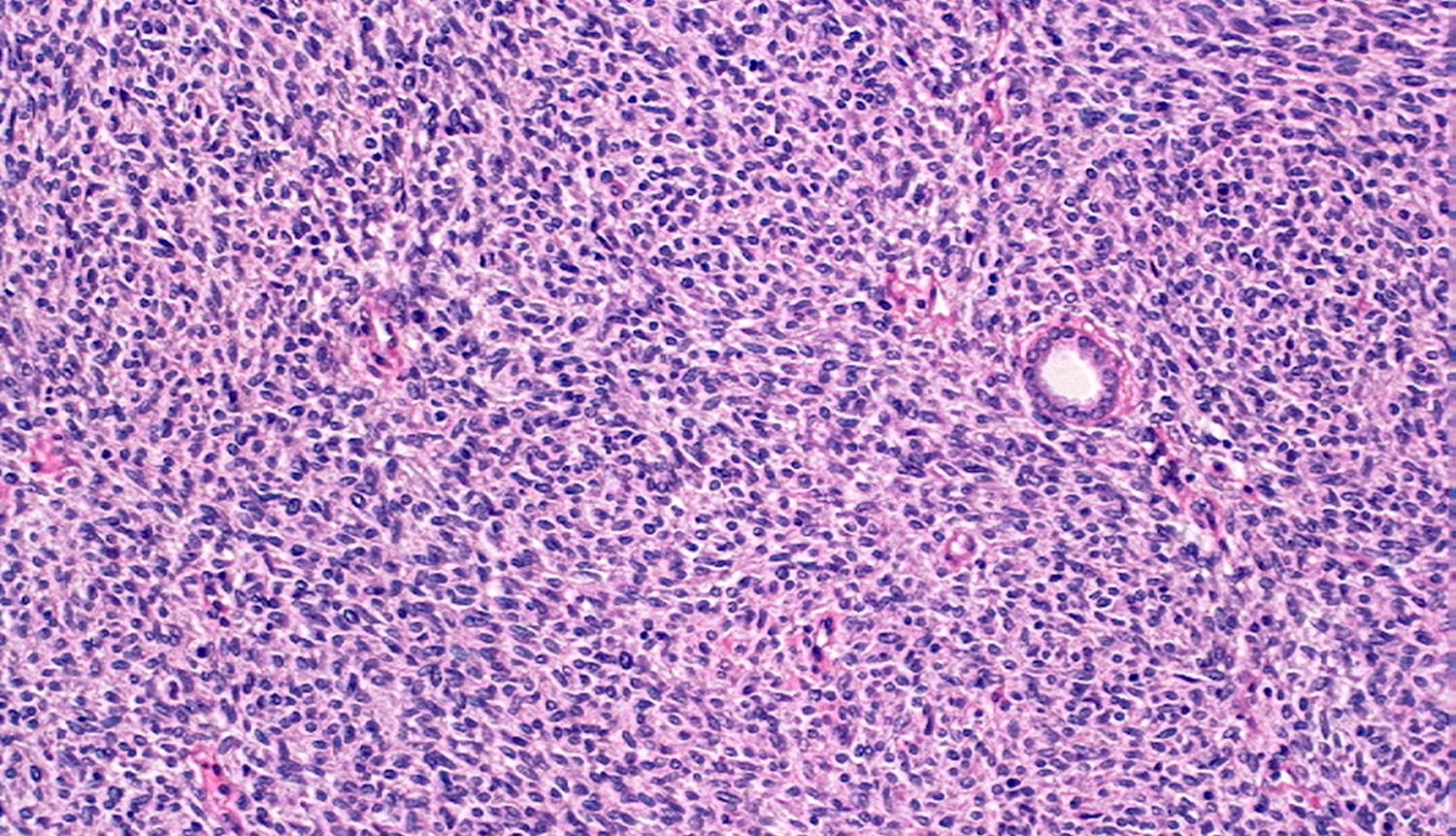
- Cellular type (~65% of cases):
- High cellularity of plump cells with vesicular nuclei, moderate amount of cytoplasm
- Sheet-like growth pattern
- High mitotic activity (which is of no prognostic significance)
- No capsule
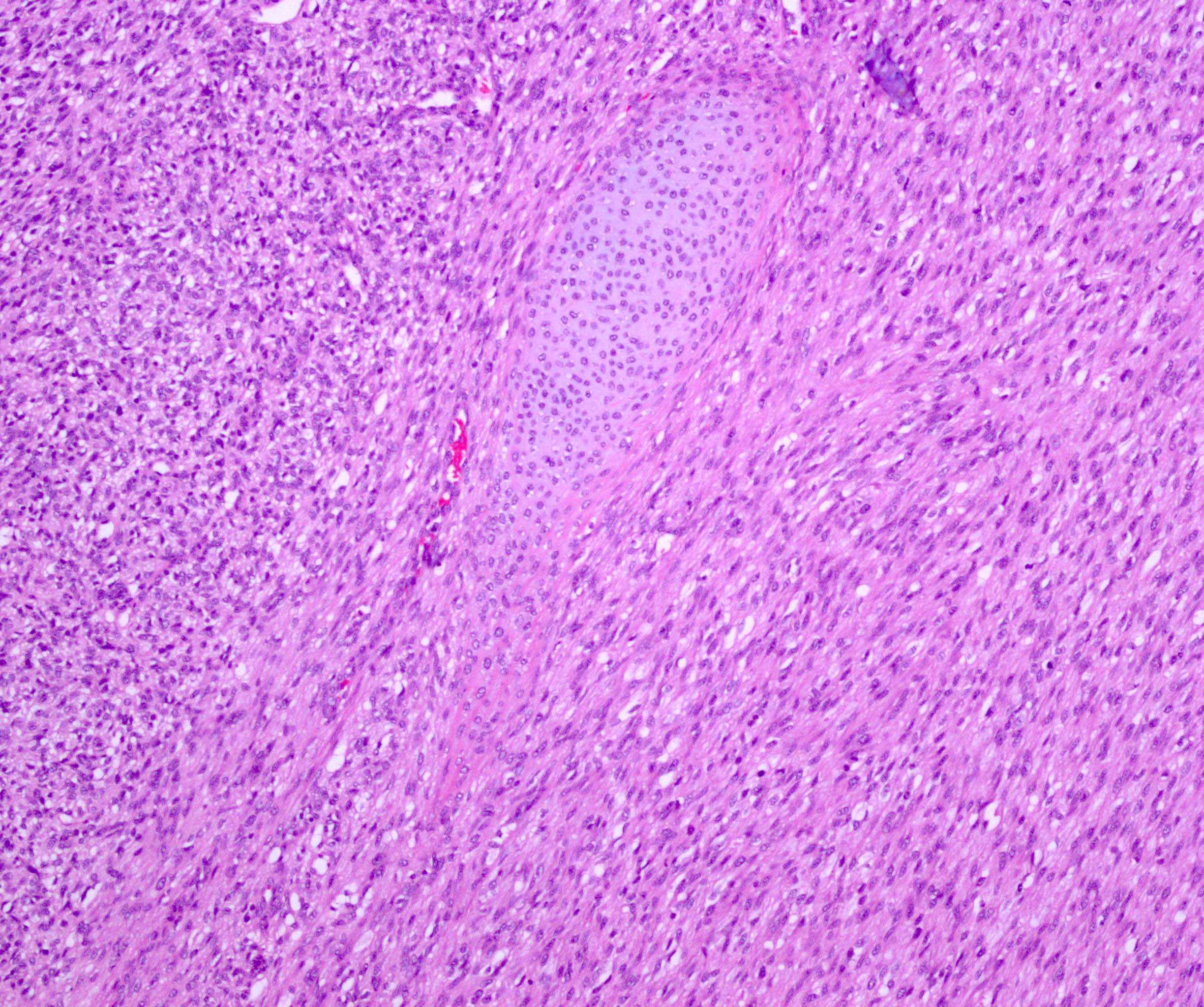
- Clear tumor-kidney border but subtle infiltration into the renal parenchyma
- Isolated entrapped tubules, which may be mistaken for neoplastic tubules
- Mixed type (~10% of cases):
- Features of both types in a variable proportion
- Staging criteria are as for nephroblastoma
- Vast majority are stage 2 due to tumor's infiltrative growth into the renal sinus or perirenal fat
Microscopic (histologic) images
Contributed by Ellen D’Hooghe, M.D. and Gordan M. Vujanic, M.D., Ph.D.
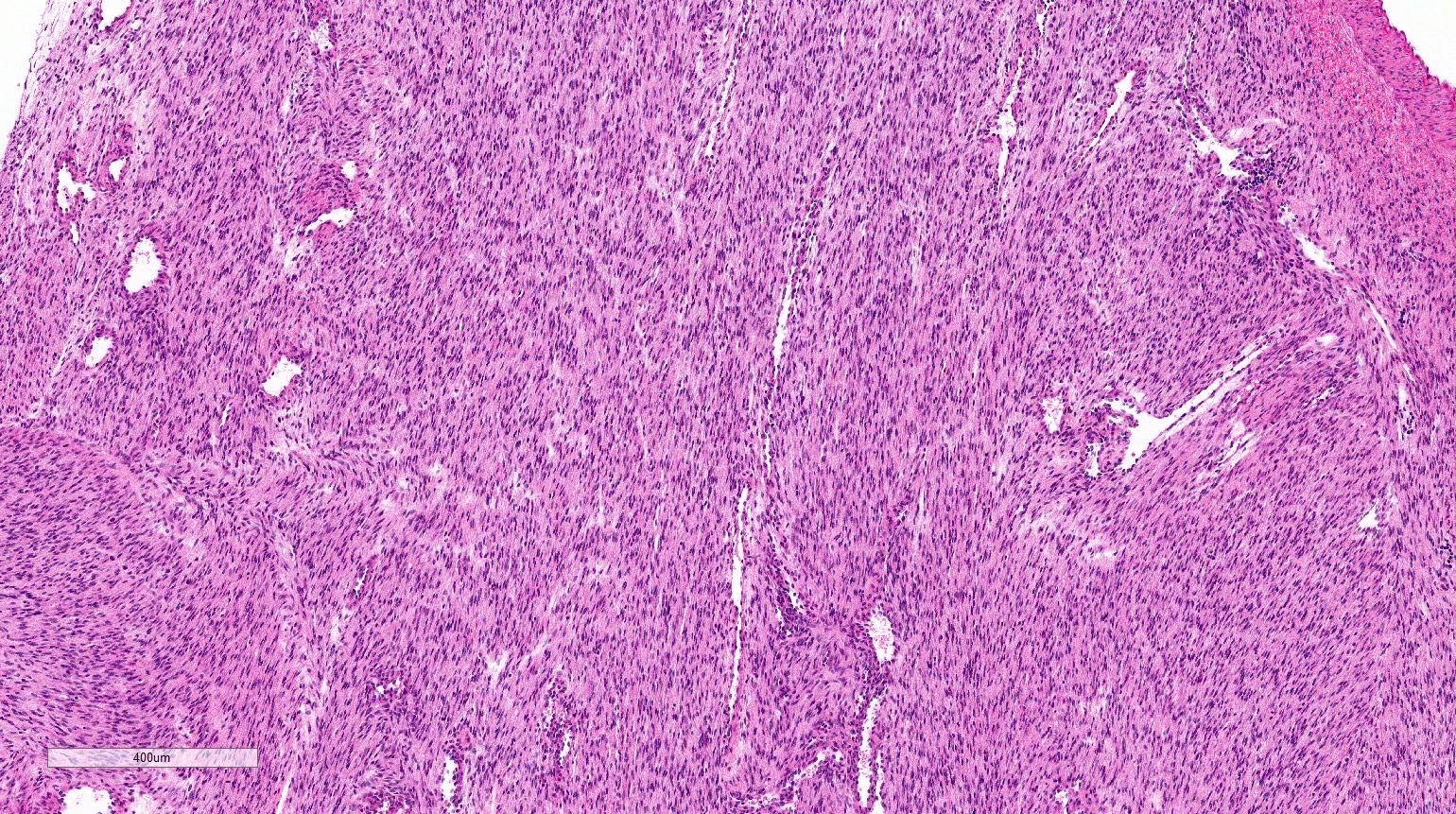
Classic type
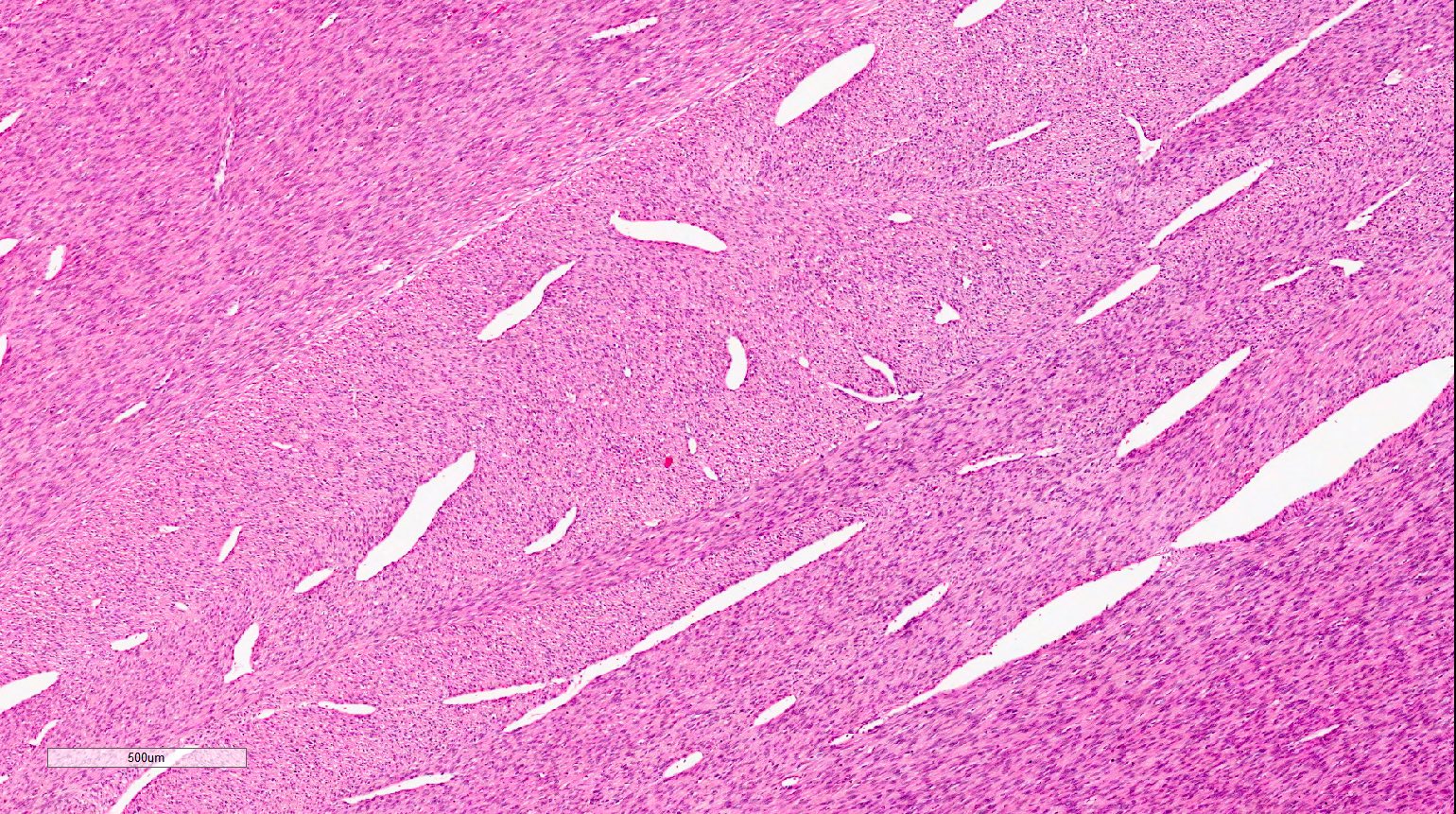
Classic type vessels
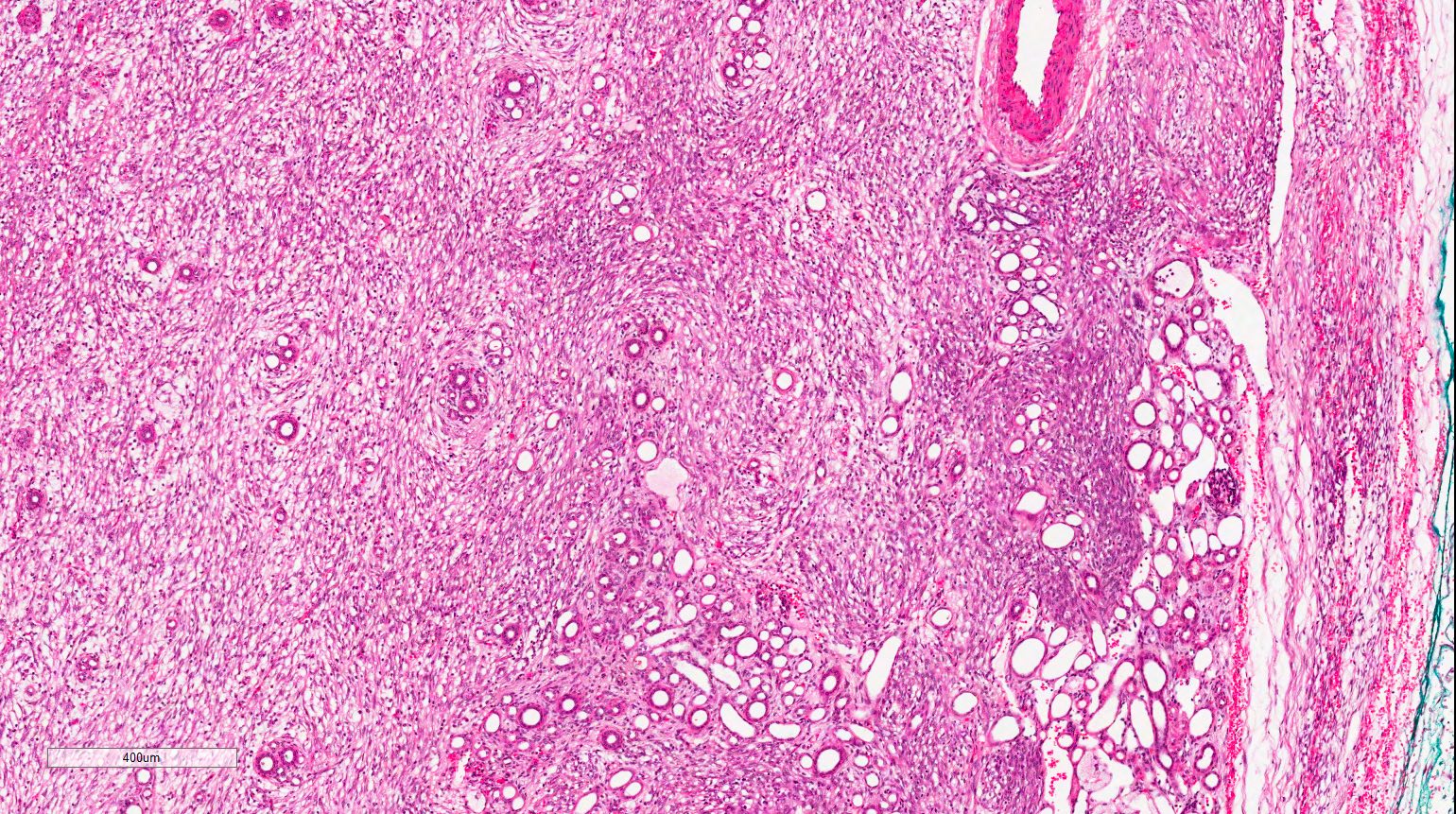
Entrapped normal renal parenchyma
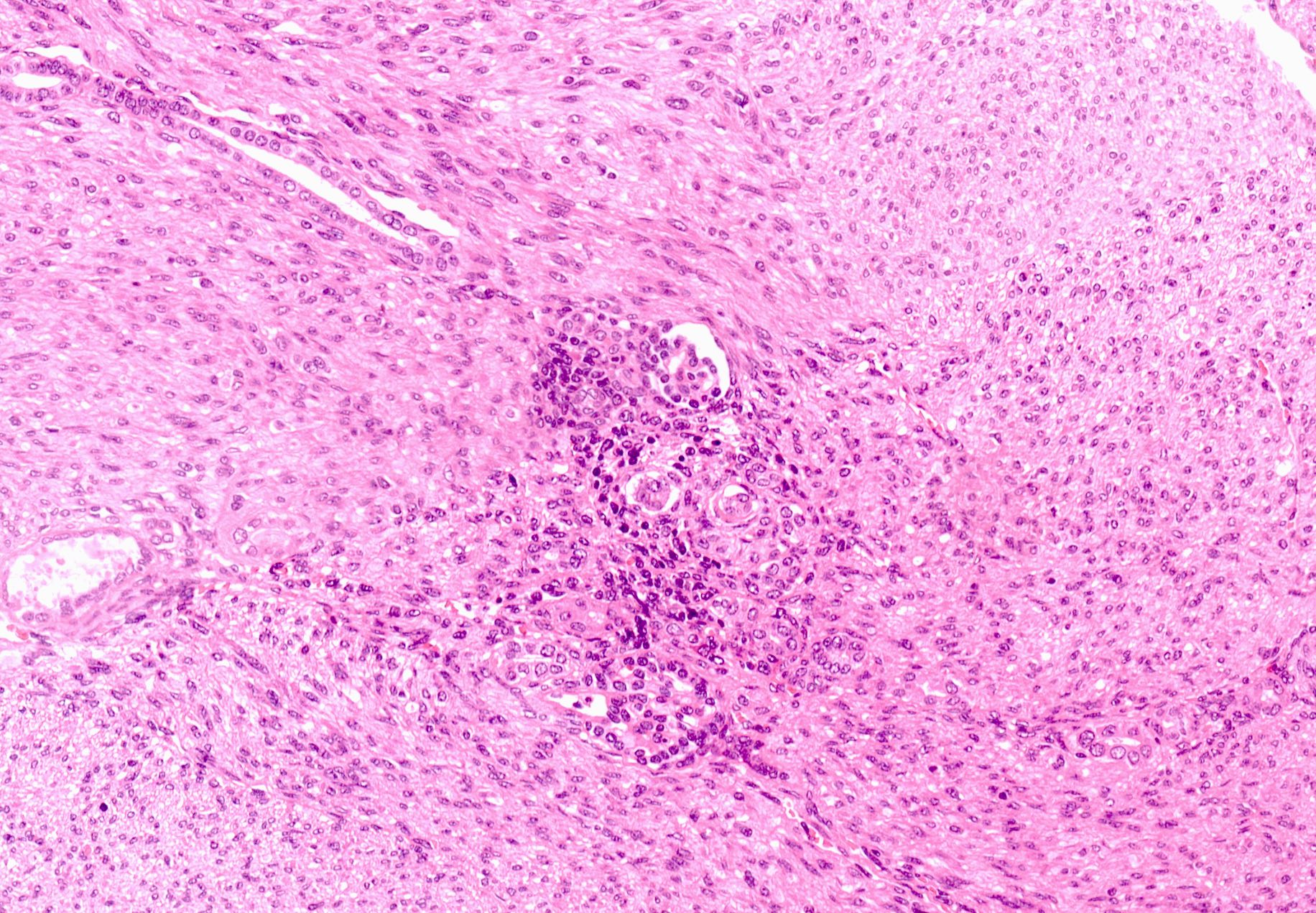
Extramedullary hematopoiesis
Islands of cartilage
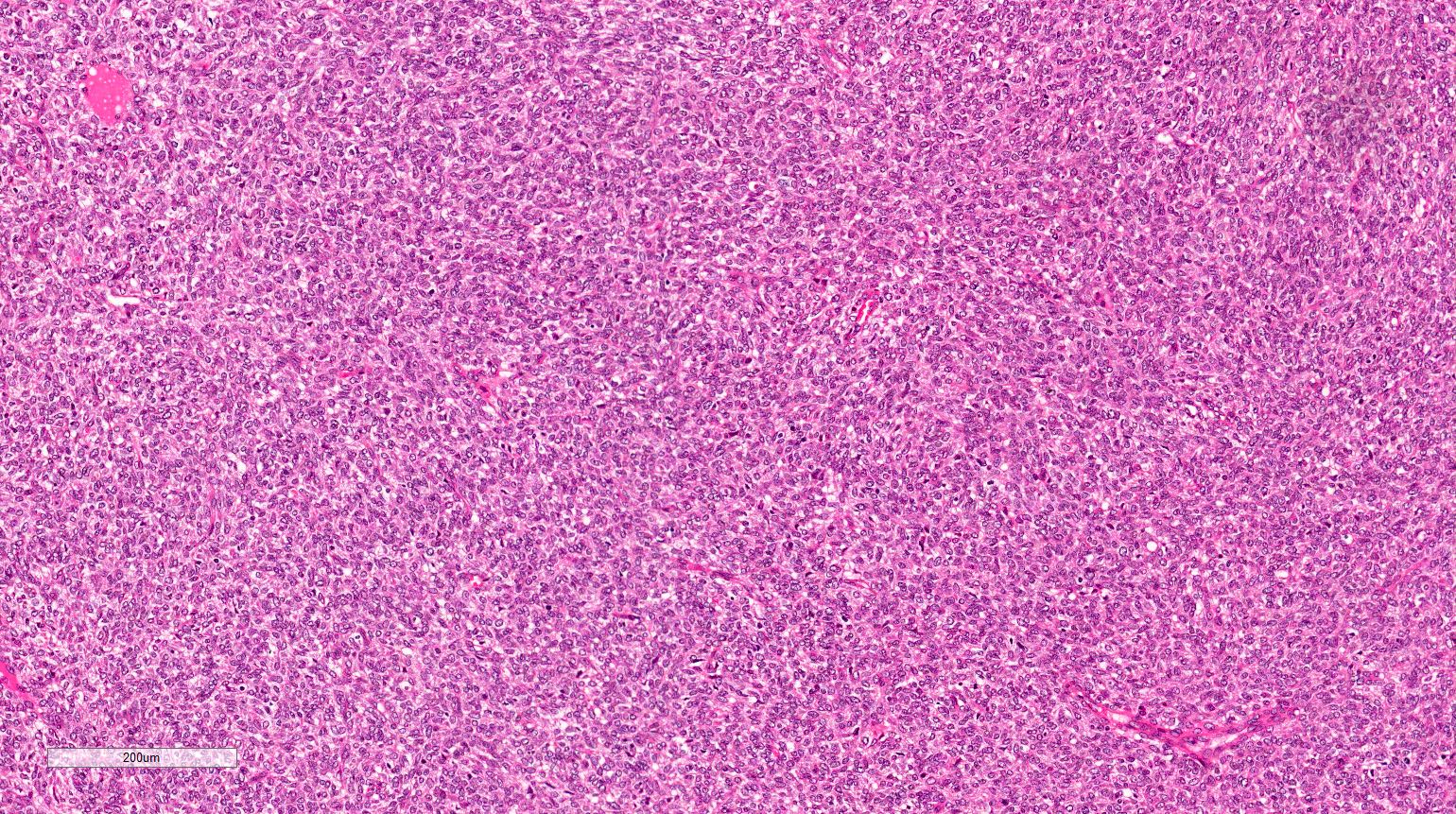
Cellular type

Cellular type
Single entrapped tubule
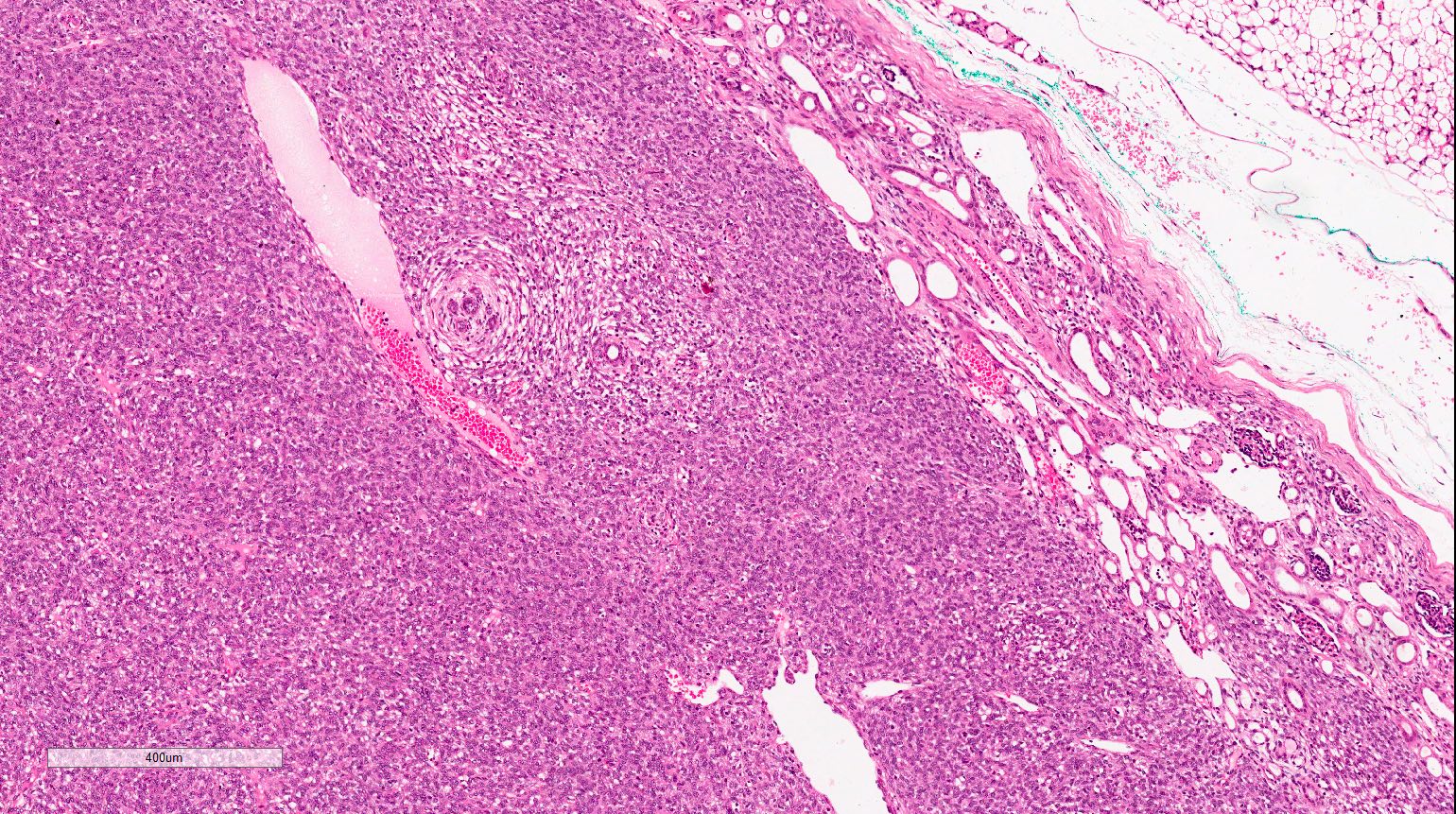
Tumor-kidney interface
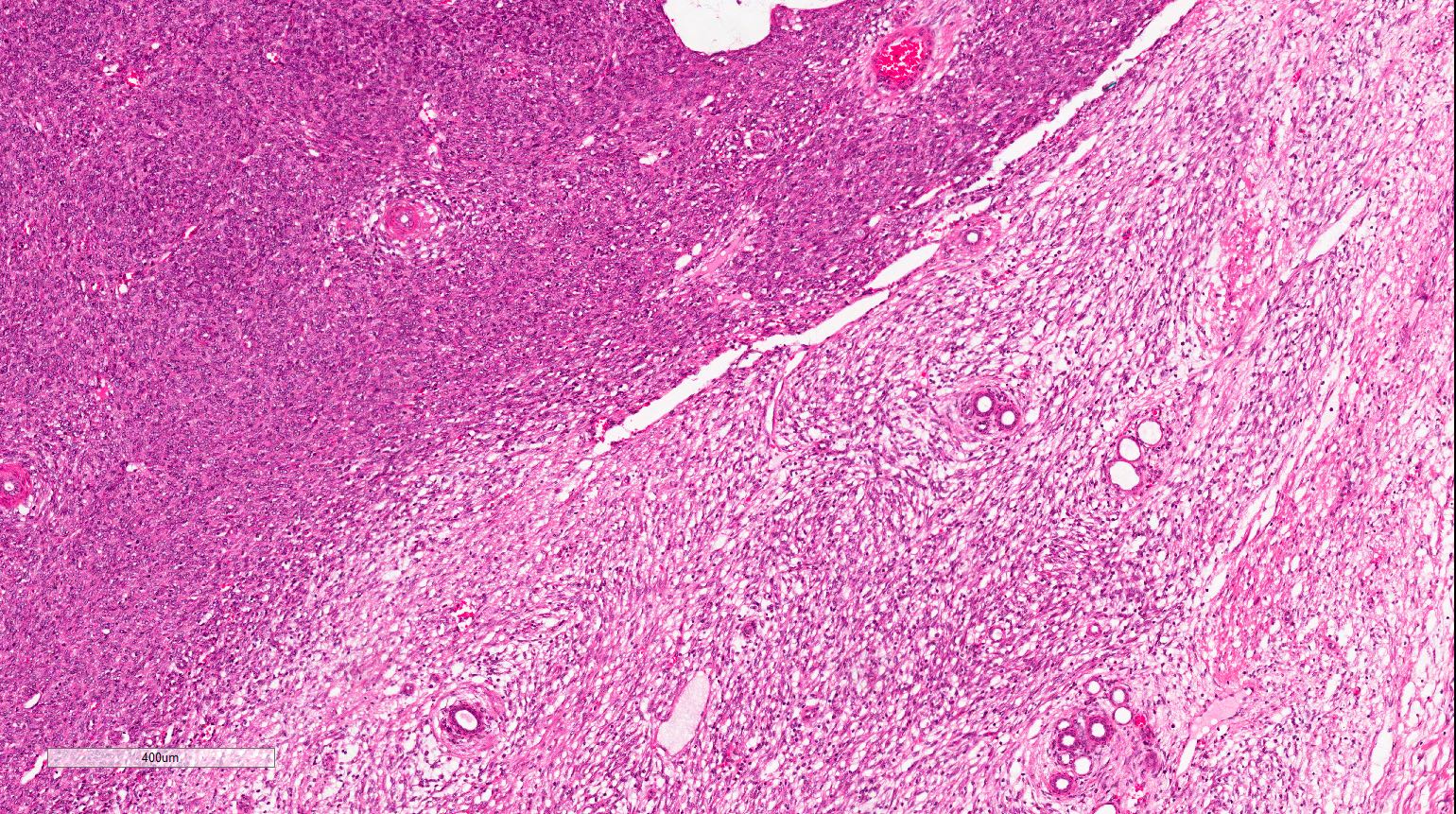
Mixed type
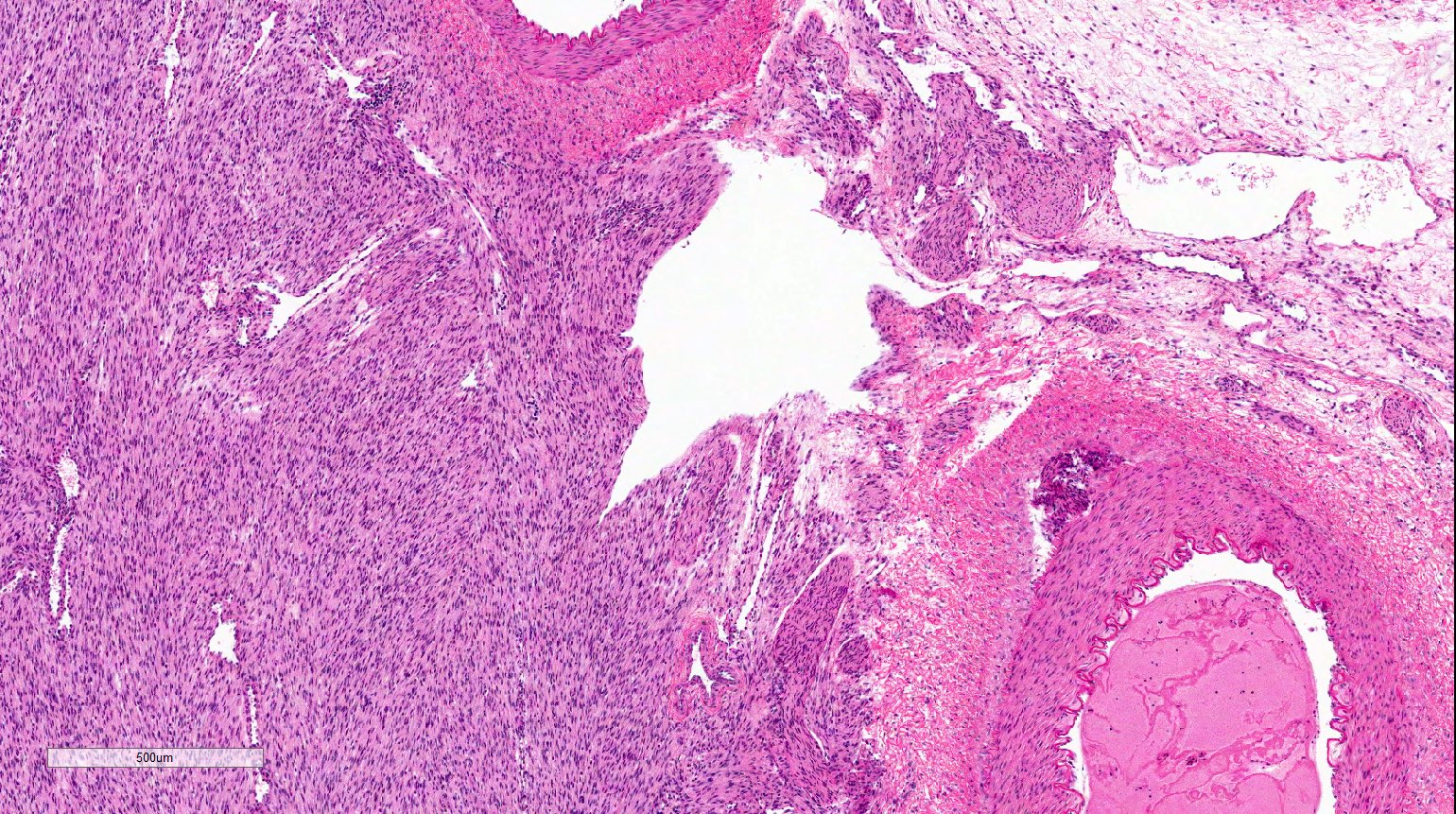
Sinus invasion
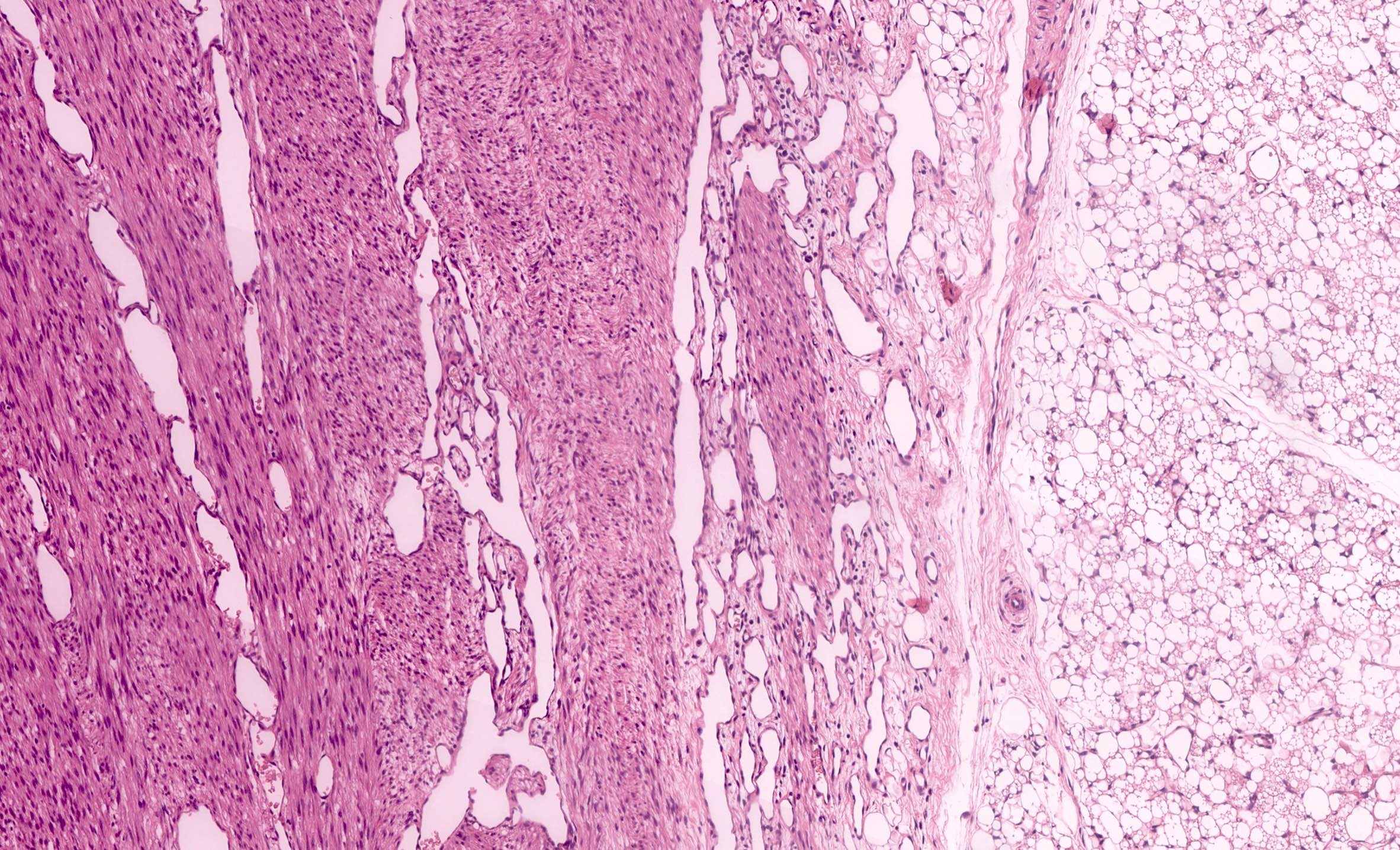
Perirenal fat invasion
Cytology description
- Fine needle aspiration is not indicated
Positive stains
- Cyclin D1: diffuse in ~70% of cases (Histopathology 2015;67:306)
- Pan-TRK: cytoplasmic or nuclear positivity in cellular type with ETV6-NTRK3 gene fusion (Am J Surg Pathol 2018;42:927)
- Alpha smooth muscle actin
- Vimentin
Negative stains
Molecular / cytogenetics description
- Cellular type:
- Specific chromosomal translocation t(12;15)(p13;q25), which results in a fusion of ETV6 and NTRK3 genes in ~70% of cases (Pediatr Blood Cancer 2017;64:e26925)
- The same translocation is found in infantile fibrosarcoma (Mod Pathol 2000;13:29, Mod Pathol 2001;14:1246)
- Few cases reported with EGFR internal tandem duplication (ITD) mutation (Nat Commun 2018;9:2378)
- Rare cases with BRAF rearrangements but not BRAF V600E mutation (Histopathology 2020;77:611, Nat Commun 2018;9:2378)
- Classic type: EGFR ITD mutation is a consistent and recurrent genetic event (Histopathology 2020;77:611)
- Mixed type: may have either EGFR ITD or ETV6-NTRK3 gene fusion
Sample pathology report
- Right kidney, total nephrectomy:
- Mesoblastic nephroma, cellular type, stage II (due to renal sinus and perirenal fat invasion) (see comment)
- Comment: Tumor shows dense cellularity with plump cells with vesicular nuclei and with a high mitotic activity. Tumor is infiltrating the renal sinus and perirenal fat but is not reaching the resection margins. Lymph nodes are free of tumor.
Differential diagnosis
- Metanephric stromal tumor:
- Alternating, nodular cellularity, angiodysplasia, onion skinning
- CD34+
- BRAF V600E mutation
- Stromal type Wilms tumor:
- Often rhabdomyoblasts and adipose tissue
- Often associated with nephrogenic rests
- More sampling might reveal epithelial and or blastemal elements
- BCL2+
- Clear cell sarcoma of the kidney:
- Characteristic arborising vascular pattern
- BCOR+
- Rhabdoid tumor of the kidney:
- INI1 loss
- Inflammatory myofibroblastic tumor:
- ALK+
Additional references
Practice question #1

An 11 month old boy presents with an abdominal mass noticed by parents. Imaging studies show a right sided renal mass. Total right nephrectomy is performed. The tumor is composed of undifferentiated cells with no particular pattern, shown above. Which finding is most characteristic for this entity?
- BCOR+
- BRAF V600E mutation
- ETV6-NTRK3 gene fusion
- INI1 loss
Practice answer #1
C. ETV6-NTRK3 gene fusion. This is a mesoblastic nephroma, cellular type.
Comment Here
Reference: Congenital mesoblastic nephroma
Comment Here
Reference: Congenital mesoblastic nephroma
Practice question #2
What is the most important prognostic factor for mesoblastic nephroma?
- Age at presentation
- Completeness of resection
- Genetic abnormalities
- Histologic type
Practice answer #2
