
Liver & intrahepatic bile ducts
Developmental anomalies / cysts
Benign recurrent intrahepatic cholestasis
Author: Zaid Mahdi, M.D., Ph.D.
Editorial Board Member: Naziheh Assarzadegan, M.D.
Deputy Editor-in-Chief: Aaron R. Huber, D.O.

Last author update: 27 July 2023
Last staff update: 27 July 2023
Copyright: 2019-2024, PathologyOutlines.com, Inc.
PubMed Search: Benign recurrent intrahepatic cholestasis liver
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Mahdi Z. Benign recurrent intrahepatic cholestasis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverbric.html. Accessed April 18th, 2024.
Definition / general
- Benign recurrent intrahepatic cholestasis (BRIC) is a rare autosomal recessive disease of hepatocanalicular protein transporters characterized by recurrent episodes of jaundice and pruritis that resolve spontaneously without subsequent liver damage
- Considered to be a known clinical entity in the spectrum of primary familial intrahepatic cholestasis (PFIC)
- First described by Summerskill and Walshe in 1959 (Z Gastroenterol 1998;36:379)
Essential features
- Cholestasis during attacks but no histologic progression to fibrosis
- Autosomal recessive disease
- Subclassified into 2 types (BRIC1 and BRIC2), which are caused by mutations of ATP8B1 and ABCB11 genes, respectively (the same genes that are mutated in progressive familial intrahepatic cholestasis [PFIC]) (Case Rep Gastroenterol 2022;16:110)
- BRIC and its subtypes can be considered as a mild form of the more severe progressive familial intrahepatic cholestasis (PFIC)
- Mainly affects patients in early life or before their twenties (World J Hepatol 2020;12:64)
- Diagnosis is based on clinical suspicion after excluding other causes of cholestasis and can be confirmed by genetic testing
Terminology
- Formerly known as Summerskill-Walshe-Tygstrup syndrome
- Benign recurrent intrahepatic cholestasis (BRIC)
ICD coding
Epidemiology
- Incidence is ~1 in 50,000 - 100,000 people worldwide (J Clin Exp Hepatol 2014;4:25)
- Age at diagnosis is typically ≤ 20 years of age
- M = F
- Genetic mutation: autosomal recessive inheritance (see Etiology)
Sites
- Liver
Pathophysiology
- Genetic mutations that lead to impaired bile salt excretion, which causes impaired functioning of the canalicular pump, leading to cholestasis (see Etiology and Molecular / cytogenetics description)
Etiology
- Autosomal recessive disorder is associated with mutation of ATP8B1 and ABCB11 genes
- Nonsense and missense mutations in those genes will lead to partial protein expression for bile canalicular protein transport (complete deletion of protein expression will show a severe form of cholestasis, which is known as primary familial intrahepatic cholestasis [types 1 and 2]) (Hepatology 2009;50:1597)
- Defective ATP8B1 gene is located on chromosome 18 and encodes F1C1, which causes impaired bile salt excretion, while defective ABCB11 gene is located on chromosome 2, which causes impaired functioning of the canalicular pump, leading to cholestasis
Clinical features
- Patients will experience spontaneous resolution within a few weeks to some months
- Presents without liver parenchymal involvement toward chronic liver disease
- Episodes are precipitated by stress, infections, medications or oral contraceptives and pregnancy (Front Med (Lausanne) 2022;9:897108)
Diagnosis
- Clinical picture and history after excluding other forms of cholestasis
- Genetic testing can be confirmatory
- Luketic and Shiffman diagnostic criteria includes (J Clin Diagn Res 2016;10:OD01)
- At least 2 episodes of jaundice with free interval of months to years
- Laboratory investigations suggestive of intrahepatic cholestasis
- Cholestasis induced severe pruritus
- Cholangiography showing normal intra and extrahepatic bile ducts
- Liver histology suggests cholestasis with no advanced fibrosis
- Absence of other causes of cholestasis
Laboratory
- Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) are usually normal
- Mildly elevated bilirubin concentrations
- Mildly elevated gamma glutamyl transferase (GGT) values
Radiology description
- Unremarkable biliary tree by cholangiography
Prognostic factors
- Favorable; prognosis is good and does not usually lead to fibrosis
Case reports
- 21 year old man presented with pruritis and jaundice (Case Rep Gastrointest Med 2020;2020:2894293)
- 27 year old man presented with complaints of jaundice for 2 weeks (J Clin Diagn Res 2016;10:OD01)
- 27 year old Hispanic woman presented with jaundice, pruritus and weakness for 4 weeks (ACG Case Rep J 2020;7:e00412)
- 29 year old man presented with severe jaundice (World J Hepatol 2020;12:64)
Treatment
- Treatment is symptomatic with antipruritics, ursodiol (ursodeoxycholic acid) and rifampicin (Scand J Gastroenterol 2023;58:512)
Microscopic (histologic) description
- Canalicular and intracellular cholestasis with associated hepatocyte rosettes
- No associated inflammation, bile duct loss or marked ductular reaction
- No advanced fibrosis
- Reference: Case Rep Gastrointest Med 2020;2020:2894293
Microscopic (histologic) images
Contributed by Pierre Russo, M.D.
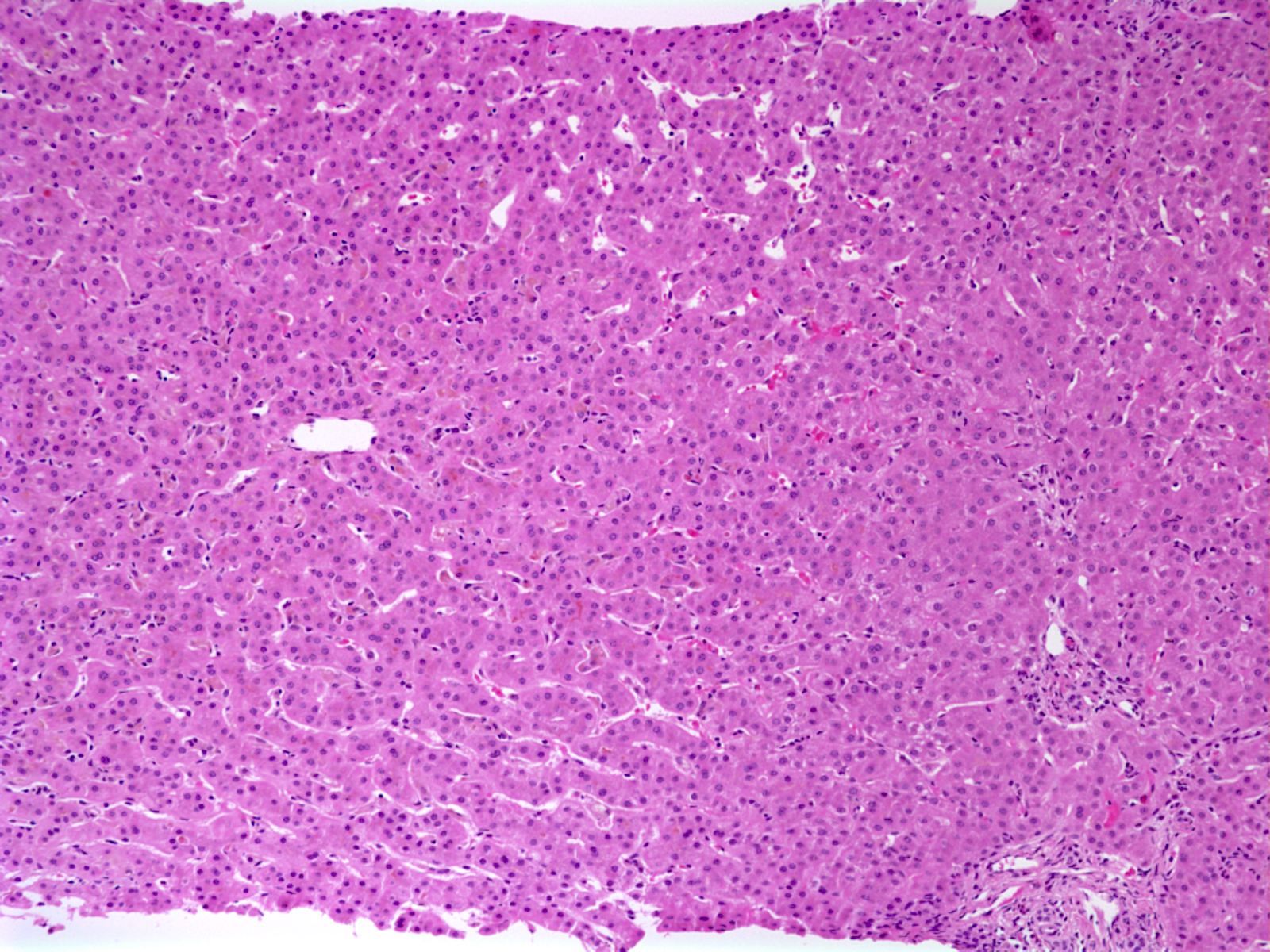
Mild cholestasis
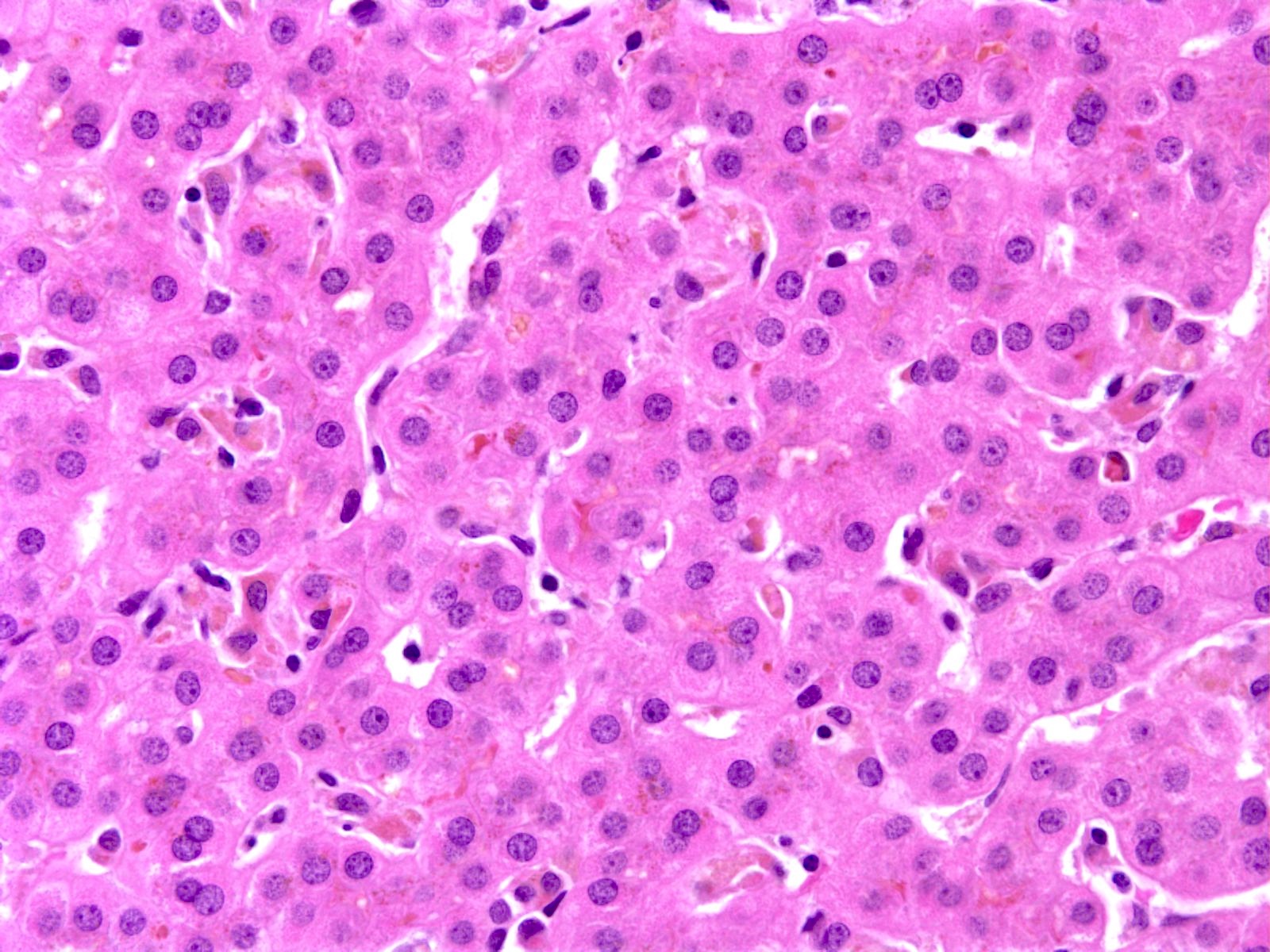
Mild cholestasis (canalicular and intracytoplasmic)
Molecular / cytogenetics description
- Mutation in ATP8B1 gene that is located on chromosome 18 and encodes F1C1, which causes impaired bile salt excretion due to partial protein expression
- Mutation in ABCB11 gene is located on chromosome 2, which causes impaired functioning of the canalicular pump, leading to cholestasis
- Rare cases have been associated with TJP2 gene mutation (JPGN Rep 2021;2:e087)
Sample pathology report
- Liver, biopsy:
- Liver parenchyma with mild cholestasis (canalicular and intracellular); no fibrosis (see comment)
- Comment: The patient's history of recurrent attacks of jaundice and pruritis are noted. Sections show liver core biopsies with bland cholestasis (both canalicular and intracytoplasmic). No associated inflammation, bile ductular reaction or bile duct loss is seen. No fibrosis is seen.
Differential diagnosis
- Diagnosis is usually by excluding other causes of cholestasis in young patients with disease recurrence
- Progressive familial intrahepatic cholestasis (Clin Res Hepatol Gastroenterol 2019;43:20):
- Severe form of cholestasis that leads to chronic liver disease
- Autosomal recessive disease
- Mutations in the same genes involved in BRIC (genes are either deleted or have frameshift mutations rather than missense mutations leading to complete loss of canalicular protein pump synthesis and manifested in a severe clinical presentation)
- Intrahepatic cholestasis of pregnancy (StatPearls: Pregnancy Intrahepatic Cholestasis [Accessed 26 June 2023]):
- Associated with pregnancy
- Symptoms and enzymes return to normal following delivery and recur with subsequent pregnancies
- Extrahepatic biliary atresia (J Pediatr Surg 1999;34:1706):
- Presents as neonatal cholestasis
- Ultrasound: absence of the gallbladder and the triangular cord sign
- Cholangiography is confirmatory
- Late infancy with pruritus:
- Bile acid synthetic defects:
- Present with cholestasis and normal GGT
- Serum bile acid concentration is low or absent
- Pruritus is mild or absent
- Response to bile acid therapy is good
- Drug induced cholestasis:
- Antimicrobials or acetaminophen may impair bile flow through hepatocellular damage, resulting in episodic cholestasis
Additional references
Board review style question #1
What statement is true regarding benign recurrent intrahepatic cholestasis (BRIC)?
- Biliary tree usually shows severe strictures by radiology
- Frameshift mutation or deletion in ATP8B1 gene or ABCB11 genes
- Liver parenchyma with cirrhosis
- Normal serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST)
- Usually associated with cystic fibrosis
Board review style answer #1
D. Normal serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Serum ALT and AST are usually normal in cases of BRIC. Answers A and C are incorrect because BRIC is not usually associated with advanced liver disease / cirrhosis or abnormal biliary tree by imaging. Answer B is incorrect because the mutated genes show missense mutations and not frameshift mutations. Answer E is incorrect because BRIC is not usually associated with cystic fibrosis.
Comment Here
Reference: Benign recurrent intrahepatic cholestasis
Comment Here
Reference: Benign recurrent intrahepatic cholestasis
