
Liver & intrahepatic bile ducts
Metabolic diseases
Primary hyperoxaluria
Author: Belinda Lategan, M.D.
Editorial Board Member: Raul S. Gonzalez, M.D.
Editor-in-Chief: Debra L. Zynger, M.D.

Last author update: 1 May 2018
Last staff update: 7 May 2021
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed Search: Primary hyperoxaluria[title] AND Liver AND (free full text[sb] AND Humans[Mesh])
Table of Contents
Definition / general | Essential features | Epidemiology | Pathophysiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology images | Prognostic factors | Case reports | Treatment | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Molecular / cytogenetics description | Molecular / cytogenetics images | Differential diagnosis | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Lategan B. Primary hyperoxaluria. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverprimaryhyperox.html. Accessed April 24th, 2024.
Definition / general
- Primary hyperoxaluria is a rare genetic disorder in which defective glyoxylate metabolism results in excessive oxalate production
- Excess oxalate is deposited as insoluble calcium oxalate salts in the kidneys and systemically (systemic oxalosis), including in the retina (diminished visual acuity), myocardium (conduction defects), blood vessel walls (vascular occlusion and gangrene), skin (livedo reticularis, calcinosis cutis metastatica, gangrene), bone (pain, joint immobility, anemia, fractures) and central nervous system (Curr Rheumatol Rep 2013;15:340, N Eng J Med 2013;369)
Essential features
- Birefringent oxalate crystals are noted in vessel walls and connective tissues of the portal areas of explanted livers, the kidneys (Arch Pathol Lab Med 2002;126:1250) and other affected organs
- Three most common subtypes are caused by mutations in the AGXT (type 1), GRHPR (type 2) and HOGA1 (type 3) genes
Epidemiology
- Primary hyperoxaluria is estimated to affect 1 in 58,000 individuals worldwide and accounts for 1 - 2% of children with end stage renal disease (Clin J Am Soc Nephrol 2012;7:458)
- Type 1 is the most common form, accounting for approximately 80% of cases
- Types 2 and 3 each account for about 10% of cases
- Type 1 likely remains underdiagnosed because of the wide variability in its clinical presentation and age of onset (GeneReviews: Primary Hyperoxaluria Type 1 [Accessed 8 November 2018])
- Prevalence of type 1 in Europe ranges from 1/333,000 - 1/1,000,000 (Nephrol Dial Transplant 2003;18:273, N Engl J Med 2013;369:649)
- Prevalence is higher in certain populations in Tunisia and Kuwait, in Arabs and Druze families of Israel and in Iran due to consanguinity but no common founder variant identified (Pediatr Nephrol 1996;10:479, Transplant Proc 2004;36:1788, Pediatr Nephrol 2006;21:1075, J Am Soc Nephrol 1999;10:2352, Pediatr Nephrol 2001;16:140, Am J Nephrol 2005;25:264, Am J Nephrol 2005;25:269)
Pathophysiology
- Glyoxylate metabolism, which almost exclusively occurs in hepatocytes, is defective and results in excessive oxalate production
- Oxalate excretion is almost entirely via the kidneys, predominantly as highly insoluble calcium salts
- Oversaturation of the renal tubular filtrate leads to crystallization in the renal tubules, nephrocalcinosis and urolithiasis, often with superimposed infection
- Combination of direct renal tubular toxicity and obstruction results in renal injury with subsequent end stage renal failure in the more severe subtypes
- Type 1 is typically the most severely affected of the 3 types (Kidney Int 2014;86:1197, Ann Clin Lab Sci 2013;43:328)
Diagrams / tables
Images hosted on other servers:

Diagnosis and treatment of primary hyperoxalurias
Clinical features
- Typically manifests in infancy or childhood with recurring kidney and bladder stones and other symptoms of systemic oxalosis but some patients are diagnosed in adulthood (Ann Clin Lab Sci 2013;43:328, Pediatr Nephrol 2015;30:1781)
Diagnosis
- Diagnosis is suspected on clinical (recurrent urolithiasis or nephrocalcinosis) and biochemical grounds and can be confirmed with genetic testing for the most common gene mutations (AGXT, GRHPR and HOGA1)
- Antenatal and preimplantation diagnosis is possible in affected families
- Prior to the availability of genetic testing, liver biopsy was required to demonstrate AGT deficiency in type 1
- Immunoblot assays allow analysis of the protein and immunoelectron examination illustrates the near absence of AGT in peroxisomes
Laboratory
- Markedly increased urinary oxalate excretion (> 1 mmol/1.73 m² per day [90 mg/1.73 m² per day])
- As long as glomerular filtration rates are within normal limits, plasma oxalate concentration remains normal
- Calcium oxalate renal and bladder stones
Radiology images
Images hosted on other servers:

Oxalate osteopathy

Abdomen Xray and renal ultrasonography

Systemic involvement ("oxalosis")
Prognostic factors
- Most type 1 and 2 patients require ongoing medical treatment
- Type 3 becomes clinically silent by age 6 years and does not typically progress beyond mild renal impairment
Case reports
- 10 month old girl with primary hyperoxaluria type 2 and urolithiasis (BMC Med Gene 2017;18:59)
- 21 year old man with primary hyperoxaluria type 1 and partial orthotopic liver transplant (Medicine (Baltimore) 2015;94:e1267)
- 33 year old woman with primary hyperoxaluria type 1 and systemic calcium oxalate deposition (Ann Clin Lab Sci 2013;43:328)
- 54 year old woman with end stage renal disease and primary hyperoxaluria diagnosed based on bone marrow biopsy (Case Rep Hematol 2015;2015:402947)
- 78 year old man with late diagnosis of hyperoxaluria type 3 (Ann Clin Biochem 2017;54:406)
Treatment
- In the most severely affected patients, liver transplantation is currently the only cure as this addresses the underlying causative enzyme deficiency, although the organ is otherwise functionally normal
- Combined liver and renal transplantation is often necessary due to end stage renal disease in types 1 and 2
- Renal transplantation without concomitant liver transplantation is associated with a higher rate of transplant failure due to recurrent oxalate induced renal injury (N Engl J Med 2013;369, Int J Nephrol 2011;2011:864580, Curr Rheumatol Rep 2013;15:340)
Gross images
Images hosted on other servers:

Calcium oxalate stones
Microscopic (histologic) description
- Birefringent oxalate crystals in vessel walls and connective tissues of the portal areas of explanted livers and other affected organs (Arch Pathol Lab Med 2002;126:1250)
Microscopic (histologic) images
Case #436

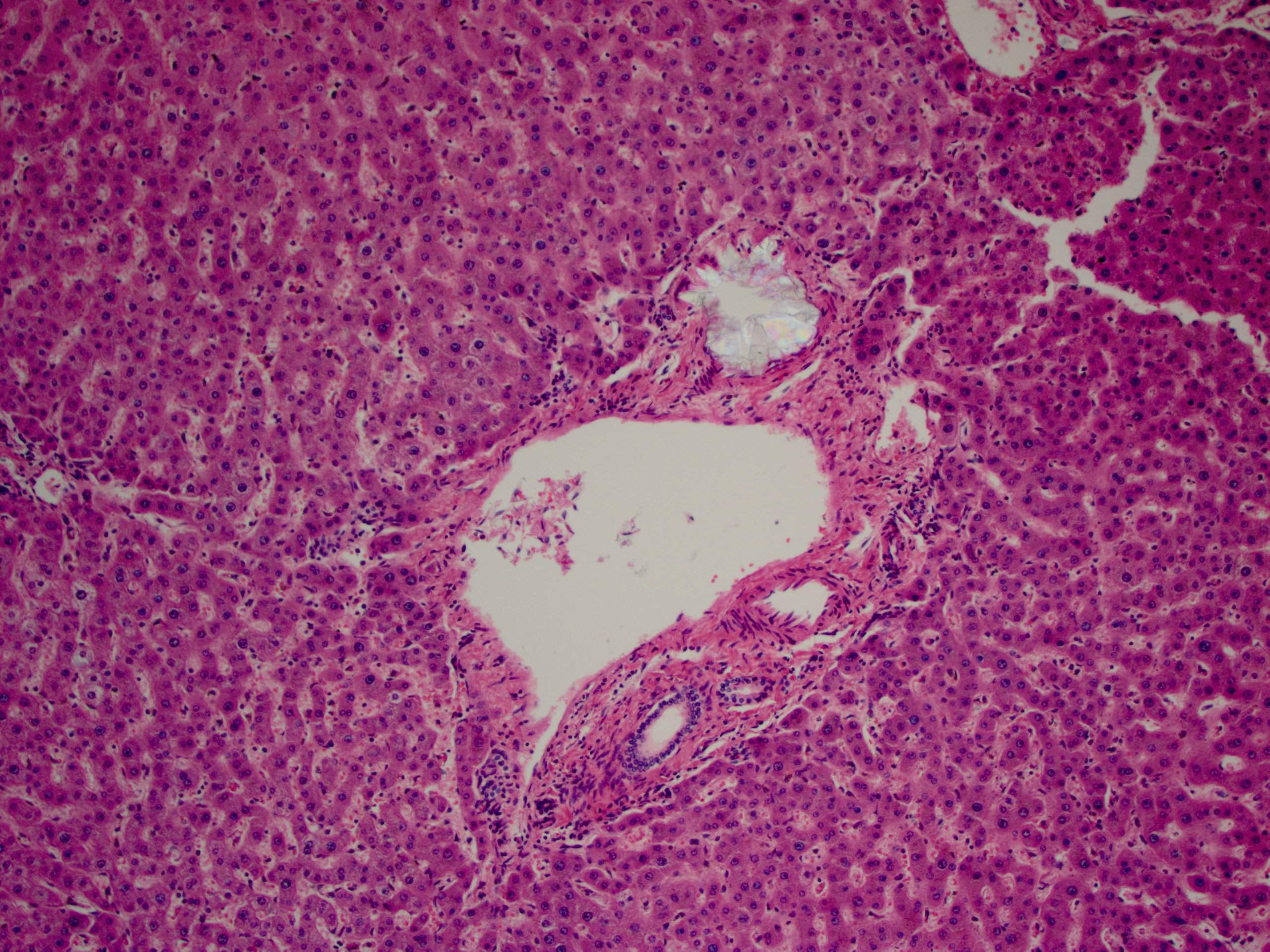

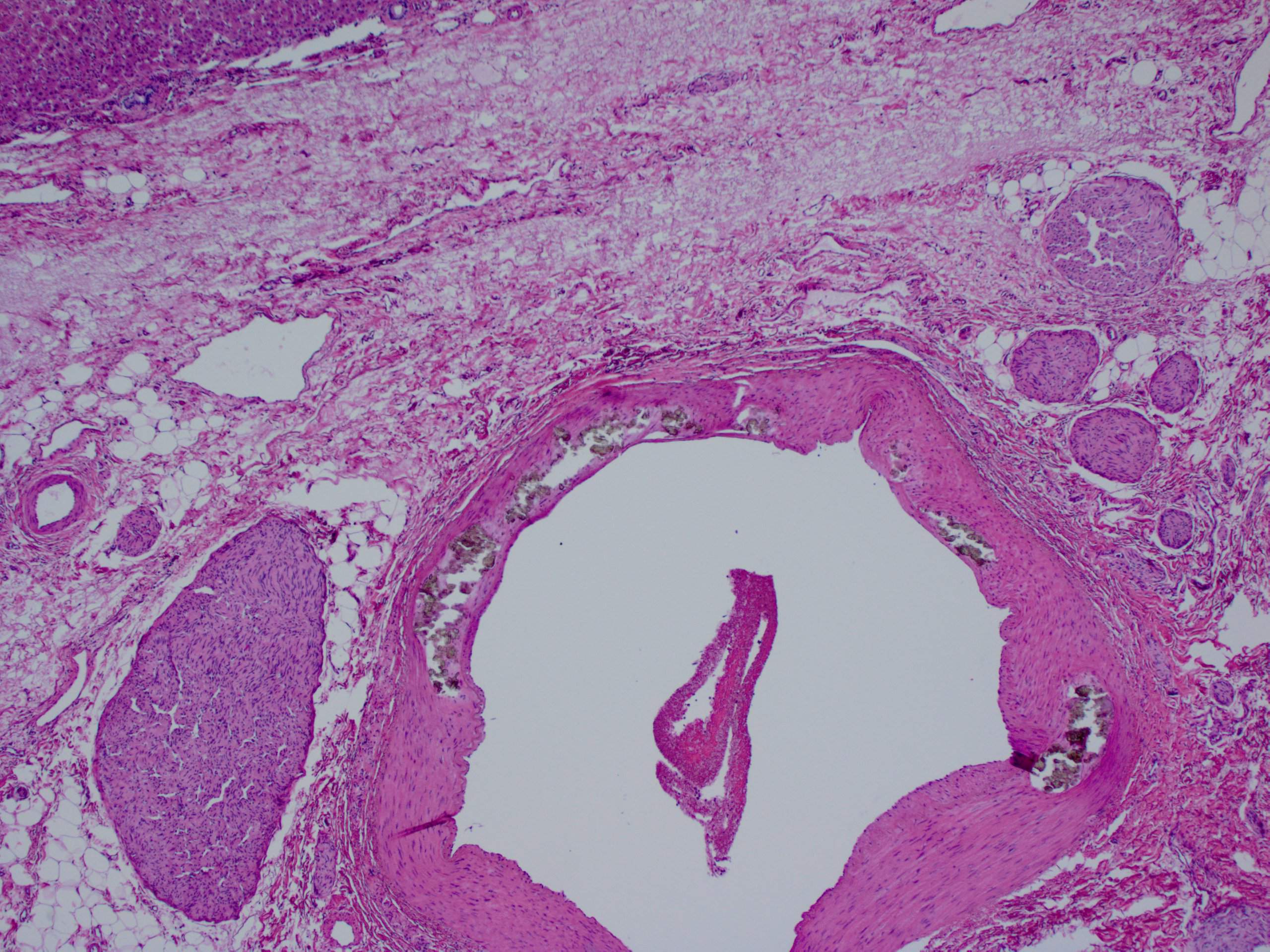
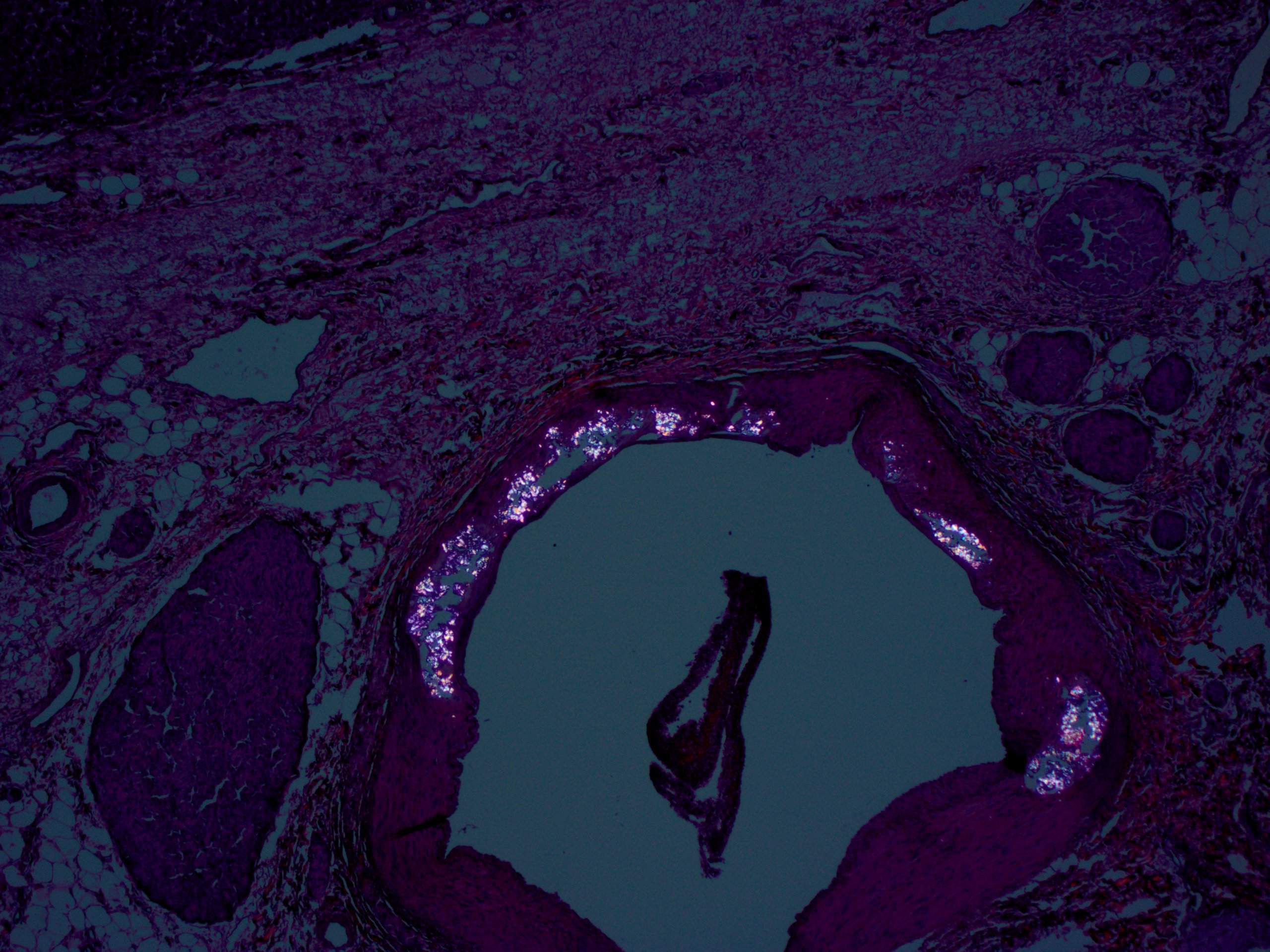
Liver with portal birefringent oxalate crystals
Molecular / cytogenetics description
- Type 1 demonstrates mutations in the AGXT gene, which encodes for the hepatic peroxismal enzyme alanine: glyoxylate aminotransferase (AGT)
- Type 2 has defects in the GRHPR gene, which encodes for GRHPR enzyme
- Type 3 demonstrates defects in the HOGA1 gene which encodes for the mitochondrial 4-hydroxy-2-oxoglutarate (HOG) aldolase enzyme
- A small subset of patients (~5%) do not have demonstrable mutations in any of these three genes (N Engl J Med 2013;369, Int J Nephrol 2011;2011:864580)
Molecular / cytogenetics images
Images hosted on other servers:

GRHPR mutation
Differential diagnosis
- Causes of secondary hyperoxaluria (Kidney Int 2009;75:1264, World J Nephrol 2015;4:235)
- Chronic inflammatory bowel disease: short bowel syndrome, cystic fibrosis, post bariatric surgery; enhanced oxalate absorption from the GI tract
- Dietary hyperoxaluria: excessive ingestion of foods high in oxalate, megadose vitamin C ingestion (World J Nephrol 2015;4:235)
- Ethylene glycol poisoning (World J Nephrol 2015;4:235)
- Methoxyflurane anesthesia: discontinued from use in North America in late 1970s, still in use in pediatric and trauma settings in Australia (Emerg Med Australas 2009;21:4)
Board review style question #1
- The type of kidney and bladder stones most often encountered in primary hyperoxaluria are
- Calcium stones
- Mixed stones
- Pyruvate stones
- Struvite stones
Board review style answer #1
A. Calcium stones. Oxalate excretion is almost entirely via the kidneys, predominantly as highly insoluble calcium salts.
Comment Here
Reference: Primary hyperoxaluria
Comment Here
Reference: Primary hyperoxaluria
Board review style question #2
- What is the most common genetic mutation and associated abnormal enzyme affecting 80% of patients with primary hyperoxaluria?
- AGXT gene and AGT enzyme
- GRHPR gene and GRHPR enzyme
- HOGA1 gene and HOG aldolase enzyme
Board review style answer #2
A. AGXT gene and AGT (hepatic peroxismal enzyme alanine: glyoxylate aminotransferase)
Comment Here
Reference: Primary hyperoxaluria
Comment Here
Reference: Primary hyperoxaluria
