Liver & intrahepatic bile ducts
Metabolic diseases
Wilson disease
Editorial Board Member: Monika Vyas, M.D.
Deputy Editor-in-Chief: Catherine E. Hagen, M.D.

Last author update: 15 March 2022
Last staff update: 23 April 2024
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Wilson disease liver pathology[TIAB]

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Nalbantoglu I, DiDomenico P. Wilson disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverwilsonsdisease.html. Accessed September 12th, 2025.
Definition / general
- Increased / toxic copper deposition within liver, cornea, kidney and central nervous system
- Due to ATP7B mutation (absence or dysfunction of copper transporting ATPase)
- Autosomal recessive
Essential features
- Also known as hepatolenticular degeneration
- Autosomal recessive inheritance (ATP7B mutation)
- Toxic accumulation of copper in liver, kidneys, cornea (Kayser-Fleischer ring) and central nervous system
- No pathognomonic histologic features; can present with a variety of histologic patterns
- Psychiatric and neurological symptoms
- References: Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011, Am J Clin Pathol 1980;73:12, Am J Pathol 1968;53:883, Semin Liver Dis 2011;31:239
Terminology
- Hepatolenticular degeneration (Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011)
Epidemiology
- Autosomal recessive inheritance
- Incidence of 1:30,000 individuals
- Geographic variations exist
- References: Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011, Hepatology 2008;47:2089
Sites
- Liver, cornea, kidneys and brain
Pathophysiology
- Copper is absorbed by intestinal cells
- Plasma copper gets transported to hepatocytes
- Copper is trafficked within hepatocyte and transported to canaliculi for biliary excretion by ATP7B
- Liver secretes ceruloplasmin, which is important in copper transportation, as well as iron hemostasis (Clin Liver Dis (Hoboken) 2021;17:61)
- Mutations in ATP7B gene (more than 300 mutations) causes accumulation of copper in hepatocytes (basal ganglia, cornea and kidneys), resulting in cell damage
- Altered copper metabolism (low ceruloplasmin) eventually causes increased iron stores and iron accumulation in liver (Clin Liver Dis (Hoboken) 2021;17:61)
- No reliable phenotype genotype correlation, since the majority of patients are compound heterozygotes
- References: Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011
Etiology
- Oxidative damage to hepatocytes due to toxic accumulation of copper
Diagrams / tables
Images hosted on other servers:
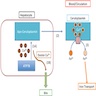
Normal copper and iron metabolism
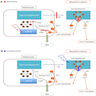
Impaired copper and iron metabolism
Clinical features
- Varying clinical presentations
- Acute fulminant hepatitis with liver failure with or without hemolytic anemia (Coombs negative)
- Unexplained chronic hepatitis / elevations in liver enzymes
- Cirrhosis
- Liver symptoms usually precede the neurologic findings
- References: Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011, Hepatology 2008;47:2089
Diagnosis
- Suspect Wilson disease in children and adults with unexplained liver enzyme elevations
- Diagnosis may be challenging (Hepatology 2008;47:2089)
- Low serum ceruloplasmin levels (< 50 mg/L)
- Increased 24 urine copper (> 100 µg in symptomatic, > 40 µg in others)
- Liver copper concentration (dry weight) > 250 µg/g
- Kayser-Fleischer (KF) rings in cornea (slit lamp examination); absence of Kayser-Fleischer ring does not exclude Wilson
- Mutational analysis for ATP7B (difficult since most patients are compound heterozygotes): gene alterations can be found in Wilson disease
Laboratory
- Elevations of alanine transaminase (ALT) and aspartate transaminase (AST)
- Low serum alkaline phosphatase (ALP)
- Serum ceruloplasmin levels < 50 mg/L
- Increased 24 urine copper > 100 µg in symptomatic, > 40 µg in others
- Liver copper concentration (dry weight) > 250 µg/g
- Positive antismooth muscle or antinuclear antibodies with elevated gamma globulin levels in some patients (may mimic autoimmune hepatitis)
- Reference: Hepatology 2008;47:2089
Radiology description
- No pathognomonic findings
- Attenuation abnormalities may be seen diffusely in the liver on CT (hyperdense liver can be seen); however, this is finding is variable and can be due to other causes of metal deposition in the liver, such as iron overload due to hemochromatosis (Eur J Radiol 2007;61:25)
- Parenchymal heterogeneity on ultrasound has also been reported though nonspecific (Eur J Radiol 2007;61:25)
- Morphologic changes of cirrhosis including regenerative nodules and surface contour nodularity may be seen and are typically nonspecific as to underlying cause; however, MRI findings of hyperintense nodules on T1 weighted imaging and hypointense nodules on T2 weighted imaging in the setting of Wilson disease have been reported (Radiographics 2010;30:e38)
Case reports
- 32 and 37 year old sisters with Wilson disease (BMC Med Genet 2020;21:128)
- 37 year old man with Wilson disease and the value of liver biopsy (Intern Med 2020;59:77)
- 37 year old woman with drug induced hepatitis mimicking Wilson disease (BMC Gastroenterol 2019;19199)
- 64 year old woman with clinically diagnosed late onset fulminant Wilson disease without cirrhosis (World J Gastroenterol 2018;24:290)
Treatment
- D penicillamine and trientine: oral chelating agents
- Zinc salts: inhibit absorption of copper
- Liver transplantation: phenotype correction reserved for patients with cirrhosis or fulminant liver failure
- Other experimental treatments (Transl Gastroenterol Hepatol 2021;6:21)
Gross description
- No pathognomonic gross characteristics of Wilson disease
- Gross findings vary based on the disease histology and phase
- Cases with fulminant hepatitis with parenchymal necrosis will have a small shrunken liver
- Cases with chronic long term disease who developed cirrhosis will exhibit features of cirrhosis on gross examination
Gross images
Images hosted on other servers:
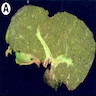
Liver with cirrhosis
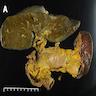
Liver with necrosis and parenchymal collapse
Microscopic (histologic) description
- Histologic features vary based on clinical presentation and stage (Am J Clin Pathol 1980;73:12, Am J Pathol 1968;53:883, Semin Liver Dis 2011;31:239, Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011)
- Acute hepatitis:
- Lobular and portal inflammation with lymphocytes and plasma cells
- Can mimic autoimmune hepatitis
- Hepatocyte ballooning mostly in periportal hepatocytes
- Acidophil bodies
- Acute cholestasis
- Fulminant hepatitis:
- Massive hepatocyte necrosis with parenchymal collapse
- Hard to distinguish from other causes of fulminant hepatitis
- Copper can be present in Kupffer cells or portal macrophages
- Chronic hepatitis:
- Nonzonal, patchy steatosis
- Lipofuscin pigment deposition
- Mild chronic portal inflammation (predominantly lymphocytic with some plasma cells) with interface activity
- Acidophil bodies
- Periportal glycogenated nuclei
- Periportal Mallory-Denk bodies (MDB)
- Copper deposition within periportal hepatocytes
- Hemosiderin pigment deposition, including Kupffer cells in a subset
- Cirrhosis:
- Nodular liver (micro or macronodular)
- Steatosis
- Hepatocyte anisonucleosis
- Acidophil bodies
- Hepatocyte ballooning and Mallory-Denk bodies
- Satellitosis in some cases
- Central vein fibrosis
- Copper deposition is variable and patchy (Am J Pathol 1968;53:883)
Microscopic (histologic) images
Contributed by ILKe Nalbantoglu, M.D. and Zafar Ali, M.D. (Case #388)
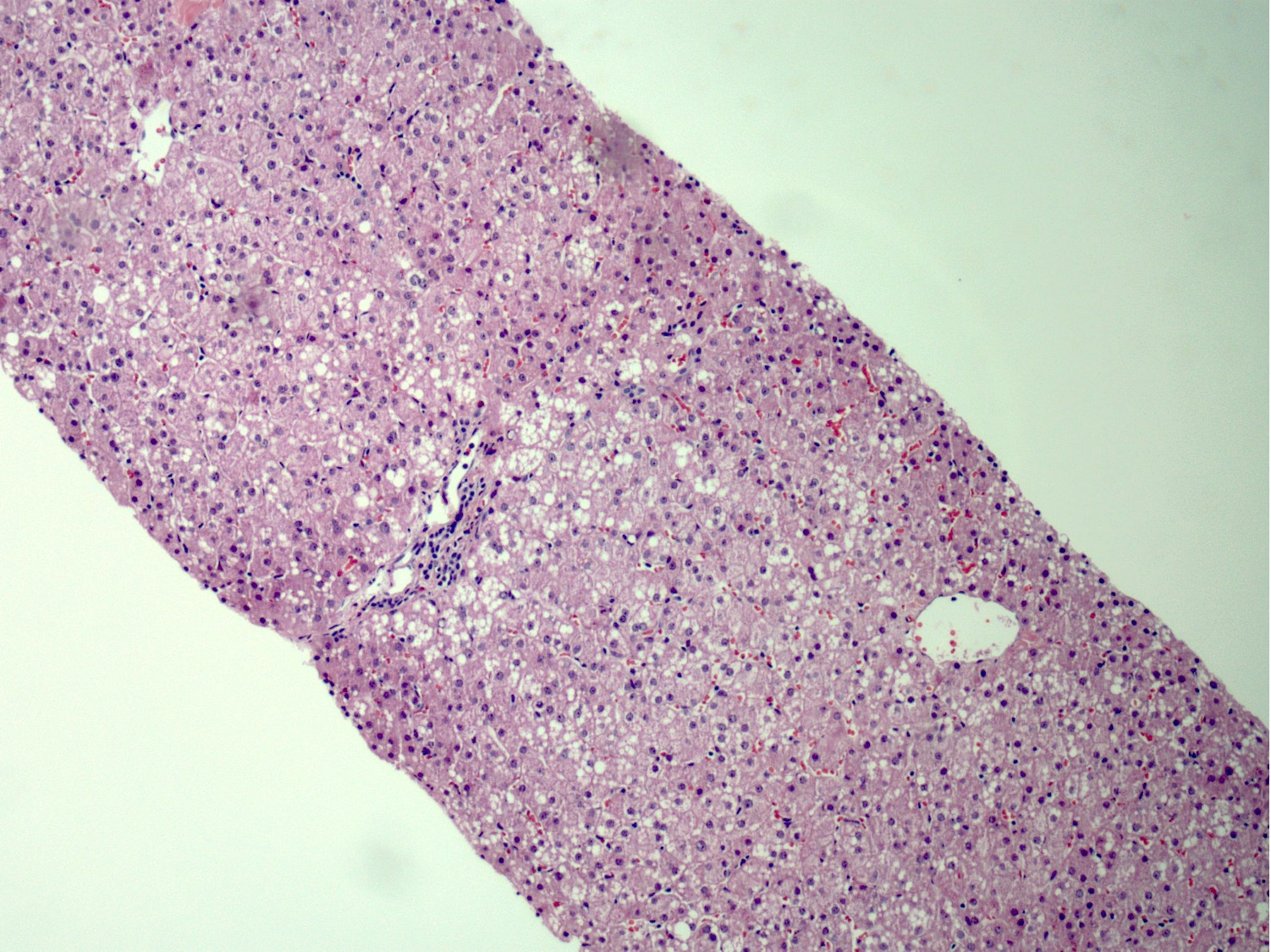
Normal architecture, patchy steatosis
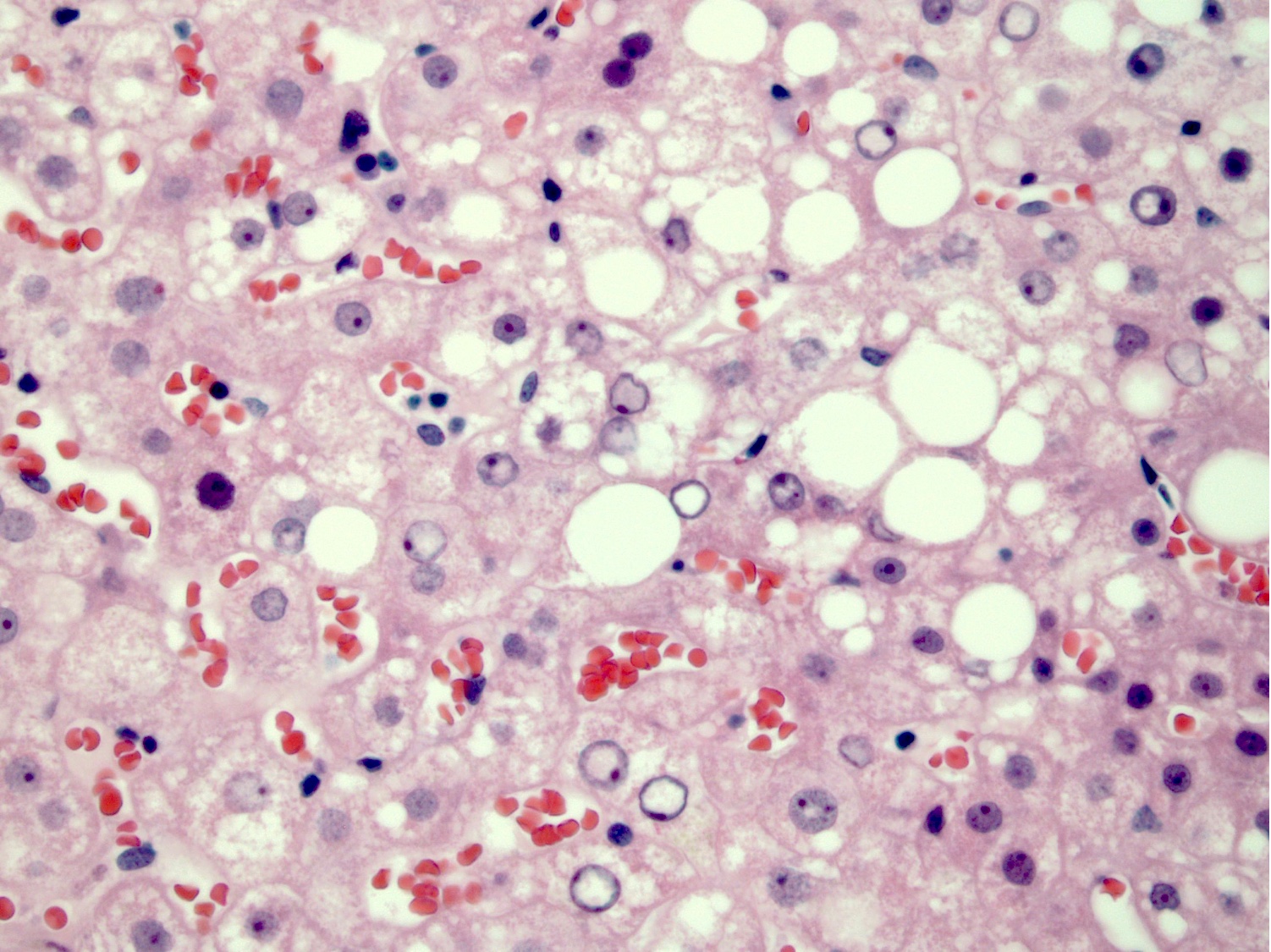
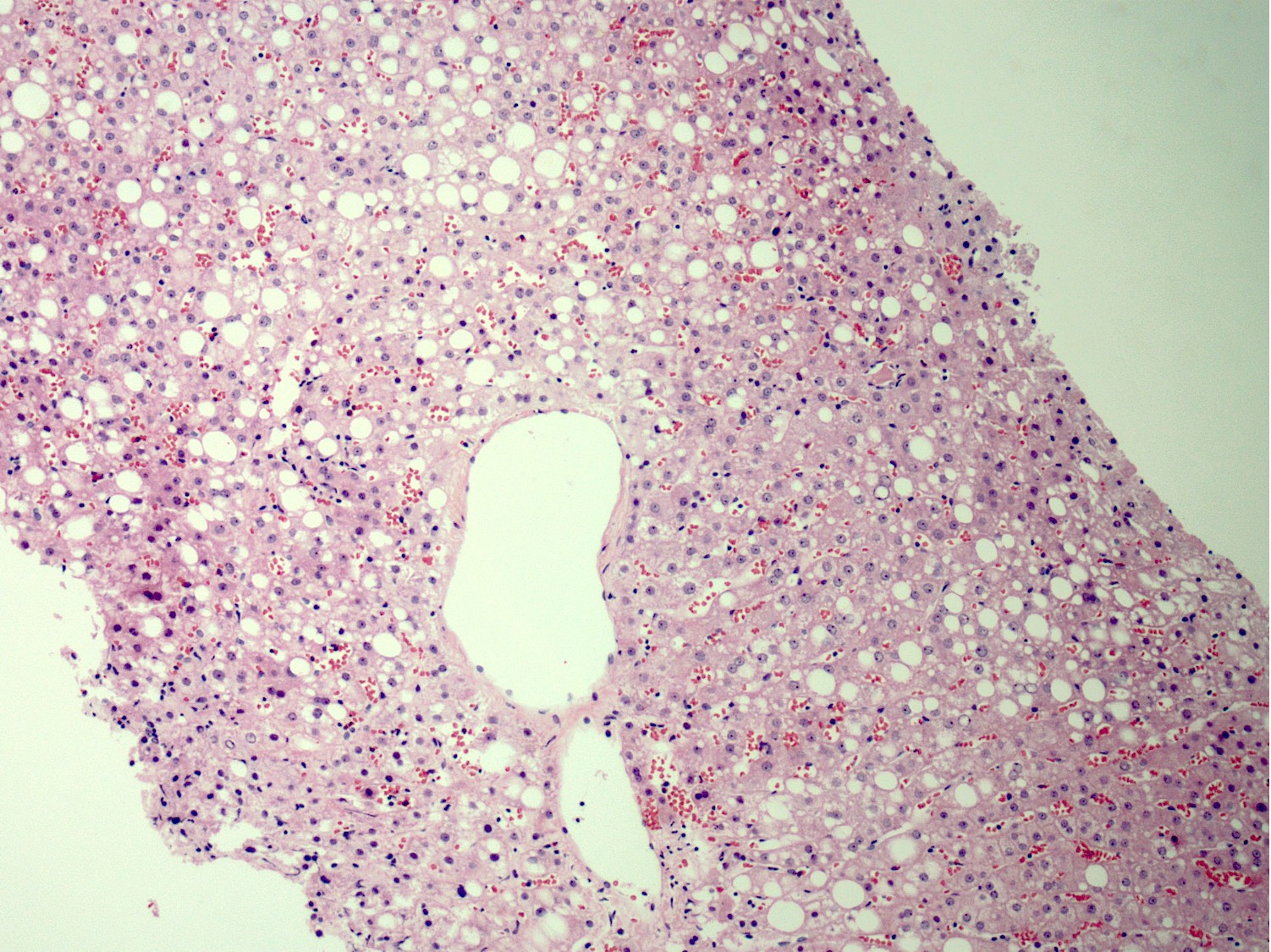
Steatosis and glycogenated nuclei
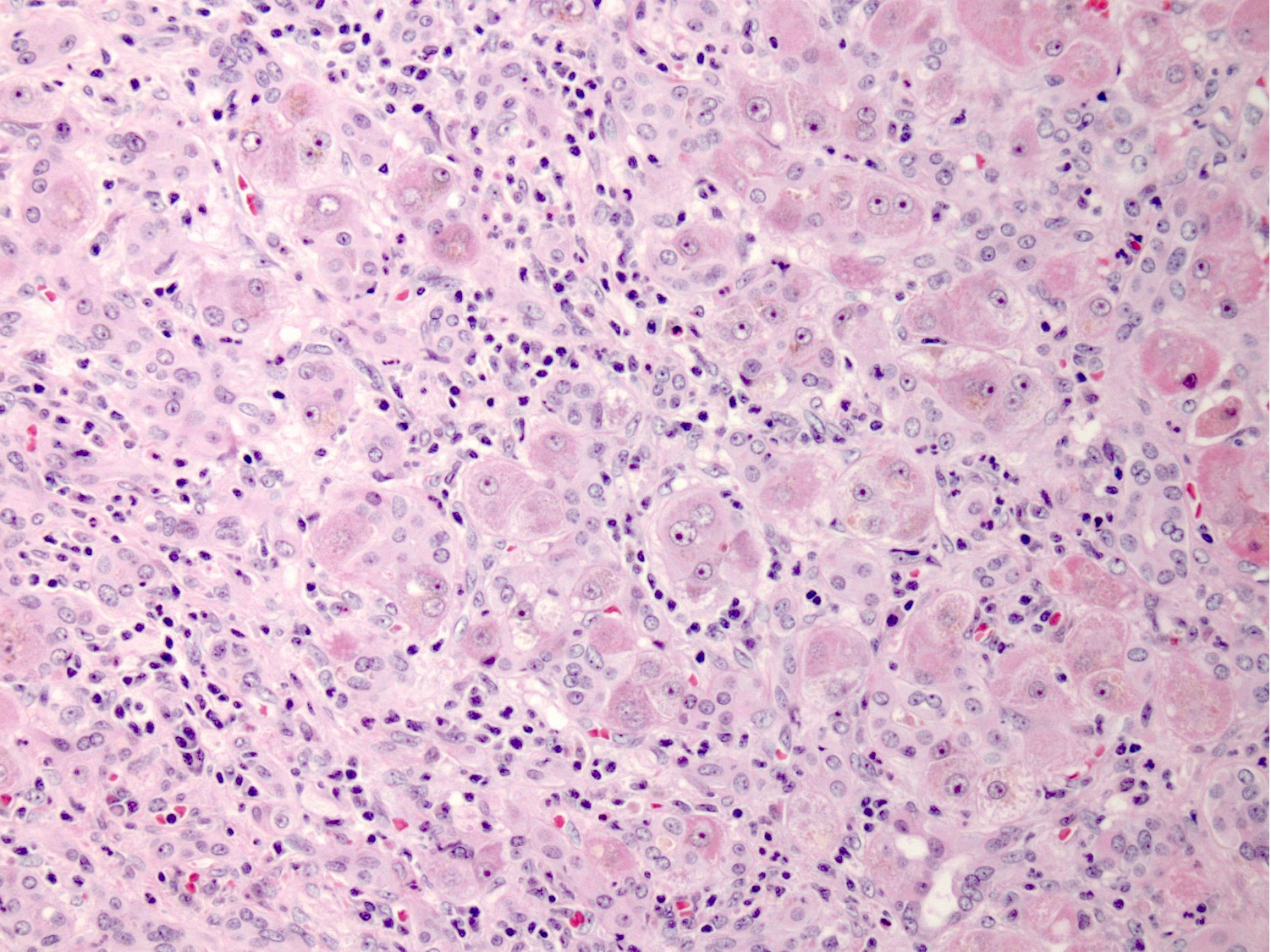
Acute hepatitis
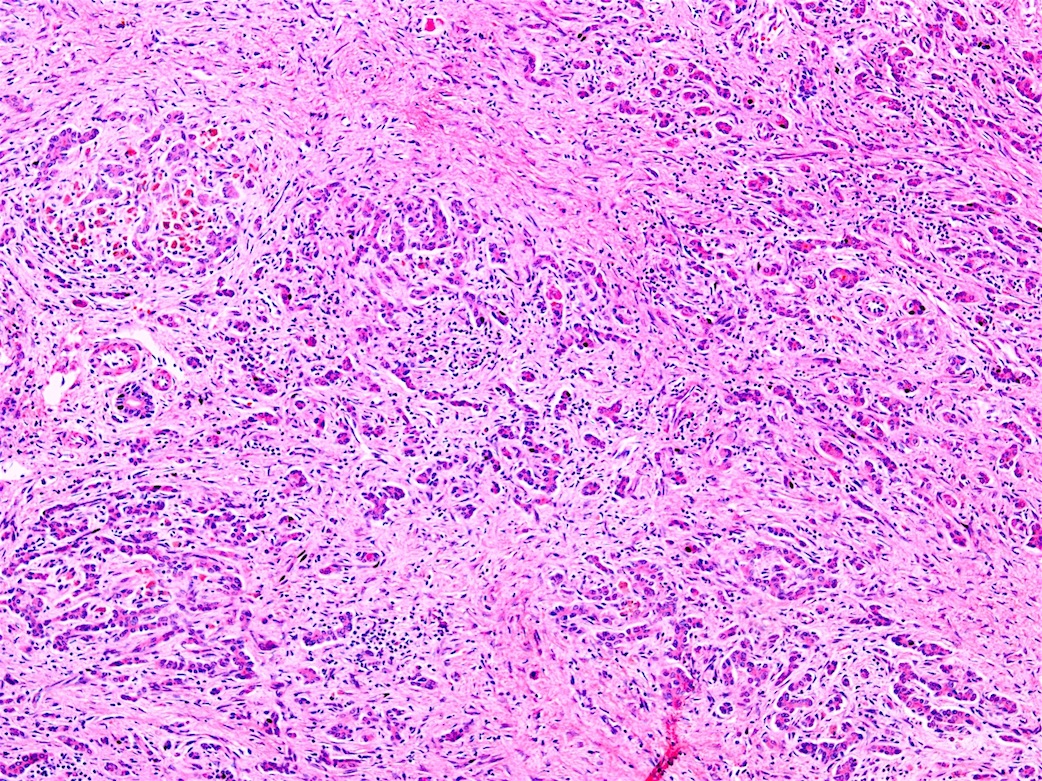
Fulminant hepatitis

Hepatitis
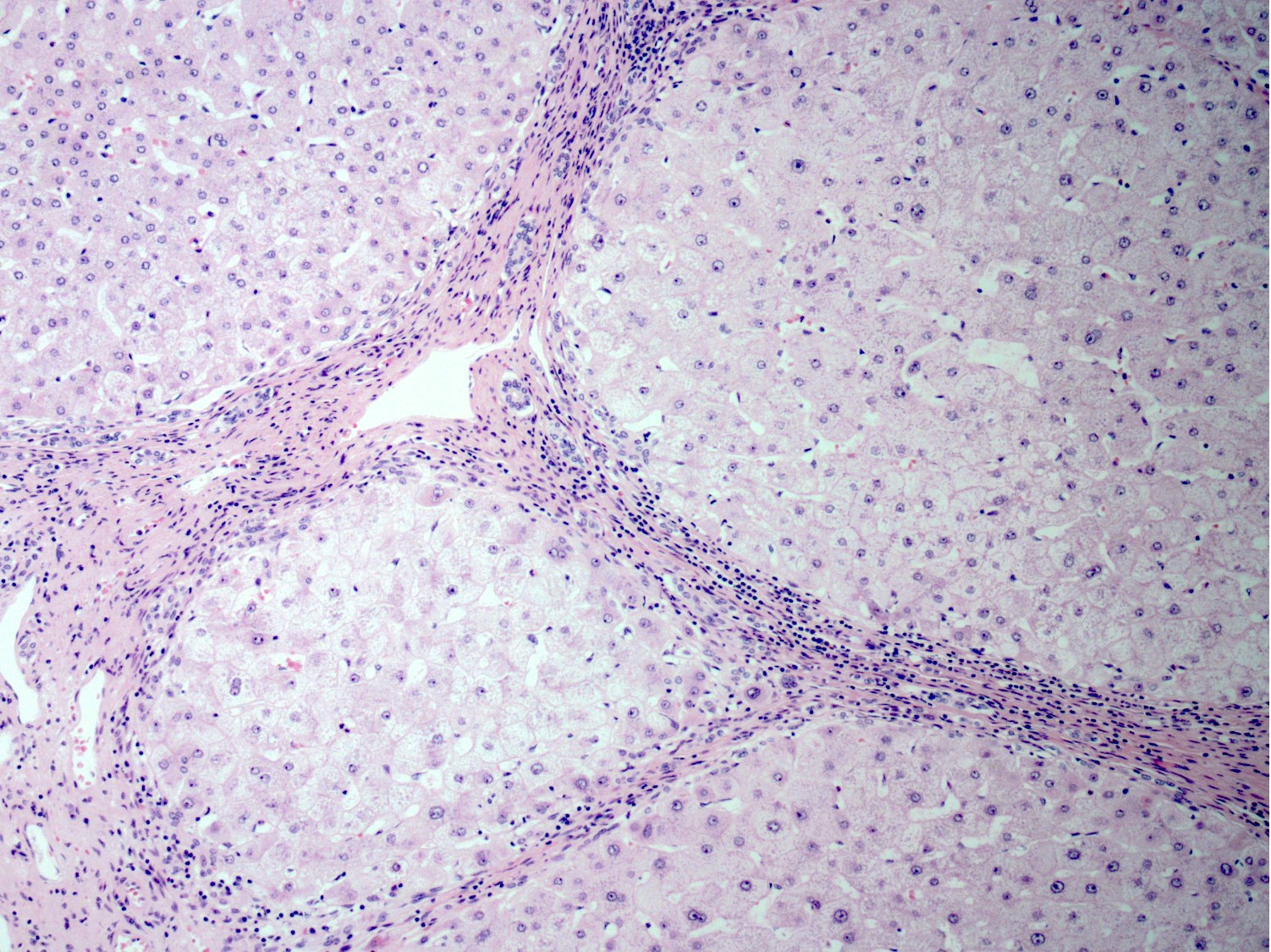
Cirrhosis
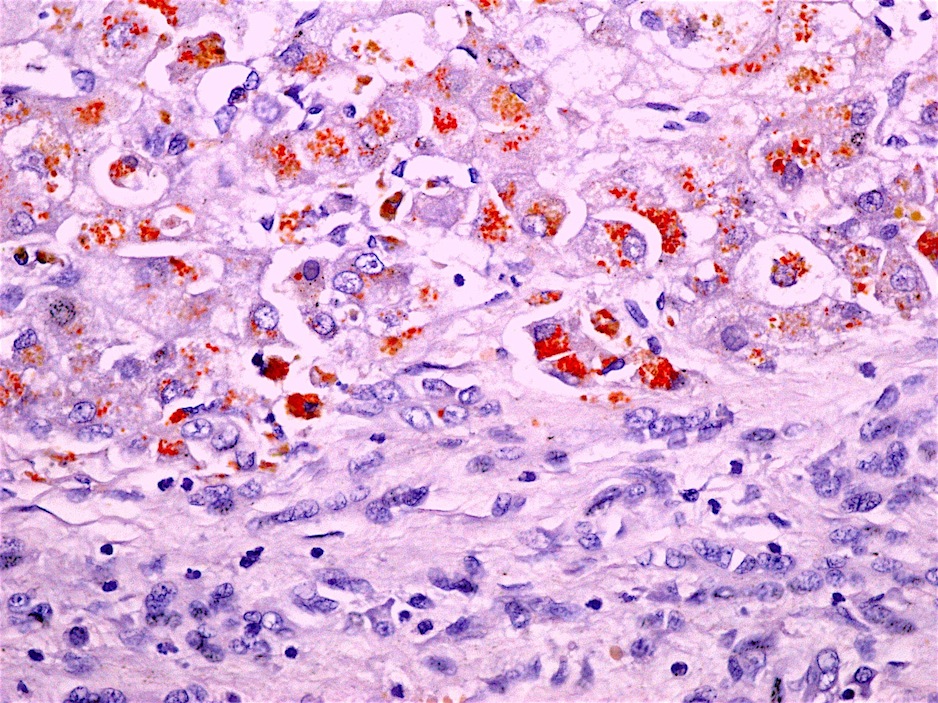
Copper stain (rhodamine)
Positive stains
- No diagnostic stains
- Copper stain is positive in hepatocytes and in Kupffer cells and histiocytes (latter is seen in acute / fulminant hepatitis)
- Positive copper stain can be seen in other cholestatic diseases (primary biliary cholangitis, MDR3 deficiency, cirrhosis, primary sclerosing cholangitis, etc.)
- CK8 / 18, p62 and ubiquitin are positive in Mallory-Denk bodies
- Other copper stains include orcein, Timm’s silver and rubeanic acid (Histopathology 1998;33:28)
- References: Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Saxena: Practical Hepatic Pathology - A Diagnostic Approach, 1st Edition, 2011, Hepatology 2008;47:2089
Negative stains
- Negative copper stain does not exclude Wilson disease
Electron microscopy description
- Not pathognomonic for Wilson disease (Burt: MacSween's Pathology of the Liver, 7th Edition, 2017, Am J Pathol 1968;53:883)
- Abnormal mitochondria with heterogeneity in shape and size, inclusions and enlarged intercristal spaces (Am J Pathol 1968;53:883)
- Increase in size of peroxisomes
- Lipofuscin
Molecular / cytogenetics description
- ATP7B mutation (more than 300 mutations)
- Mutational analysis can be difficult since most patients are compound heterozygotes
Sample pathology report
- Liver, needle core biopsy:
- Mild steatosis with inflammation
- Focal portal fibrosis (see comment)
- Comment: Sections of the liver show a preserved architecture. Steatosis comprising approximately 10% of the liver parenchyma is seen in a nonzonal distribution. Hepatocyte ballooning and Mallory-Denk bodies are not appreciated. Glycogenated nuclei are seen. Lipofuscin pigment deposition in zone 3 hepatocytes is identified. Lobules show mild lobular inflammation with rare acidophil bodies. Portal tracts have minimal inflammation and preserved bile ducts. Reticulin stain confirms preserved architecture. Trichrome stain shows focal portal fibrosis. PASD is negative for diastase resistant globules within hepatocytes. Iron stain is negative.
- The histologic findings are not diagnostic of a specific entity. The findings of steatosis and inflammation may be in keeping with fatty liver disease in the proper clinical setting. However, other etiologies, including drugs and metabolic diseases (such as Wilson disease), are in the differential diagnosis. Clinical correlation is recommended.
Differential diagnosis
- Since the histologic features can mimic any liver injury, clinical suspicion is the key for diagnosis of Wilson disease
- Some of the differential diagnoses include but are not limited to:
- Fatty liver disease:
- No definitive distinguishing features
- Hepatocyte ballooning, Mallory-Denk bodies in pericentral hepatocytes and zone 3 pericellular fibrosis are features of fatty liver disease
- Autoimmune hepatitis:
- No definitive distinguishing features
- Chronic hepatitis:
- No definitive distinguishing features
- Wilson disease should be suspected in any patient with unexplained liver enzyme elevations
- Drug induced hepatitis:
- No definitive distinguishing features
- Clinical history of medication exposure can help
- Fatty liver disease:
Additional references
Practice question #1
Which of the following is a feature of Wilson disease?
- ATP7B mutations are found in these patients
- Autosomal dominant
- Disease affects liver only
- Patients will have high serum ceruloplasmin
- Patients will have low hepatic copper
Practice answer #1
Practice question #2
Which of the following is true regarding the morphologic findings in Wilson disease?
- Chronic hepatitis is almost never seen in these patients
- Electron microscopy findings are pathognomonic for Wilson disease
- Histologic and serologic findings can mimic autoimmune hepatitis
- Positive copper stain confirms the diagnosis of Wilson disease
- Steatosis is rarely seen in these patients
Practice answer #2
C. Histologic and serologic findings can mimic autoimmune hepatitis
Comment Here
Reference: Wilson disease
Comment Here
Reference: Wilson disease
