Lung
Interstitial lung diseases
UIP / IPF
Editorial Board Member: Jefree J. Schulte, M.D.


Last author update: 21 March 2023
Last staff update: 19 February 2025
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed Search: Usual interstitial pneumonia / idiopathic pulmonary fibrosis
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy description | Videos | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Sathirareuangchai S, Yoshikawa A, Bychkov A. UIP / IPF. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lungnontumoruip.html. Accessed October 4th, 2025.
Definition / general
- Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, fibrosing interstitial lung disease (ILD) of unknown cause, associated with histologic and radiological features of usual interstitial pneumonia (UIP) (Am J Respir Crit Care Med 2022;205:e18)
- Progressive pulmonary fibrosis (PPF) is a relatively new clinical term introduced in 2022 and used to describe a patient with an ILD other than IPF with worsening clinical course (at least 2 of 3: worsening symptoms, radiological progression and physiological progression)
Essential features
- Chronic and progressive respiratory failure due to lung fibrosis
- One of the most common interstitial lung diseases and the most lethal entity
- Characterized by bibasilar patchy fibrosis and honeycomb change
Terminology
- Usual interstitial pneumonia (UIP) is the name of morphological pattern in histology and radiology and is also seen in other etiologies, such as connective tissue diseases and hypersensitivity pneumonitis
- IPF was previously called chronic interstitial pneumonitis or cryptogenic fibrosing alveolitis
- PPF is not a pathologic diagnosis and its definition is agnostic to the underlying condition
ICD coding
Epidemiology
- First or second most common (17% - 86%) interstitial lung disease (Clin Epidemiol 2013;5:483)
- Incidence increases with age, common in the sixth to seventh decades (Am J Respir Crit Care Med 2018;198:e44)
- In patients younger than 50 years old, consider familial IPF and connective tissue disease
- Male predominance
- U.S. incidence 93.7 per 100,000, prevalence 494.5 per 100,000 in population ≥ 65 years (Lancet Respir Med 2014;2:566)
- Lower incidence and prevalence in Asia (Am J Respir Crit Care Med 2014;190:773, Respirology 2008;13:926, Respir Med 2012;106:1566)
- Currently increasing globally (Respirology 2016;21:427, Clin Epidemiol 2013;5:483)
Sites
- May diffusely involve bilateral lungs but typically shows lower lobe, subpleural and basilar predominance
Pathophysiology
- Damage of alveolar epithelium (or pneumocytes) is a key event
- Aberrant Wnt / β catenin signaling pathway may be related to abnormal remodeling (Am J Pathol 2003;162:1495)
- Increased TGFβ1 activity likely has role in abnormal fibrosis (Annu Rev Pathol 2014;9:157)
Etiology
- Unknown etiology in IPF
- Several potential risk factors:
- Genetic factors, including several mutations (Proc Am Thorac Soc 2011;8:158, Respirology 2016;21:712):
- SFTPC (surfactant protein C)
- SFTPA2 (surfactant protein A2)
- TERT, encoding protein component of telomerase
- TERC, encoding RNA component of telomerase
- MUC5B (mucin 5B)
- Cigarette smoking (Eur Respir Rev 2015;24:428)
- Gastroesophageal reflux (Am J Med 2010;123:304)
- Genetic factors, including several mutations (Proc Am Thorac Soc 2011;8:158, Respirology 2016;21:712):
Diagrams / tables
Images hosted on other servers:
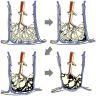
Schema of UIP pattern

IPF diagnosis by lung biopsy and HRCT
Clinical features
- Manifestations of chronic respiratory syndrome:
- Chronic dyspnea
- Dry cough
- Fatigue
- Digital clubbing
- Abnormal chest auscultation
- End inspiratory fine crackles in bibasilar lung
- Restrictive pattern in pulmonary function tests
- Decreased total lung capacity (TLC)
- Decreased forced vital capacity (FVC)
- Decreased diffusing capacity of the lung for carbon monoxide (DLCO)
- Reduced diffusing capacity is mainly due to ventilation - perfusion mismatch from ventilation of lung tissue with capillary destruction and perfusion of under ventilated alveoli; smaller component due to reduced diffusion across fibrotic alveolar septa
Diagnosis
- Diagnosis of UIP / IPF is often challenging and strict criteria are still being discussed
- Based on clinical features (including laboratory tests), high resolution computed tomography (HRCT) and surgical lung biopsy (SLB)
- It is necessary to rule out other etiologies such as particle exposure, connective tissue diseases and hypersensitivity pneumonia
- Transbronchial lung biopsy has limited role in establishing diagnosis of UIP
- Cryobiopsy has been shown to produce high diagnostic accuracy and good interobserver agreement for UIP pattern (Respiration 2019;98:421)
- Guidelines suggest multidisciplinary decision by physicians, radiologists and pathologists, especially when there is a discrepancy between them (Am J Respir Crit Care Med 2022;205:e18)
- Current guidelines do not recommend lung biopsy for cases with typical UIP pattern on HRCT; nevertheless, histopathology remains an important diagnostic modality (Eur J Radiol 2014;83:20, Am J Respir Crit Care Med 2004;170:904, Eur Respir J 2010;35:1322, Thorax 2003;58:143)
Laboratory
- Negative serum antibodies of connective tissue diseases
- Negative serum antibodies of hypersensitivity pneumonitis
Radiology description
- Chest Xray can detect lesions; however, it is usually not sensitive enough to render the diagnosis
- Honeycomb change: clustered cystic airspaces, 3 to 10 mm in diameter (Eur J Radiol 2014;83:27)
- UIP pattern on HRCT (Am J Respir Crit Care Med 2022;205:e18)
- Distribution
- Subpleural and basal predominant
- Often heterogeneous (areas of normal lung interspersed with fibrosis)
- Occasionally diffuse
- May be asymmetric
- Features
- Honeycombing with or without traction bronchiectasis / bronchiolectasis
- Presence of irregular thickening of interlobular septa
- Usually superimposed with a reticular pattern, mild ground glass opacity (GGO)
- May have pulmonary ossification
- Distribution
- Probable UIP pattern on HRCT
- Distribution
- Subpleural and basal predominant
- Often heterogeneous (areas of normal lung interspersed with reticulation and traction bronchiectasis / bronchiolectasis)
- Features
- Reticular pattern with traction bronchiectasis / bronchiolectasis
- May have mild GGO
- Absence of subpleural sparing
- Distribution
- Indeterminate for UIP
- Diffuse distribution without subpleural predominance
- CT features of lung fibrosis that do not suggest any specific etiology
Radiology images
Images hosted on other servers:
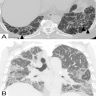
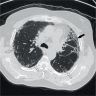
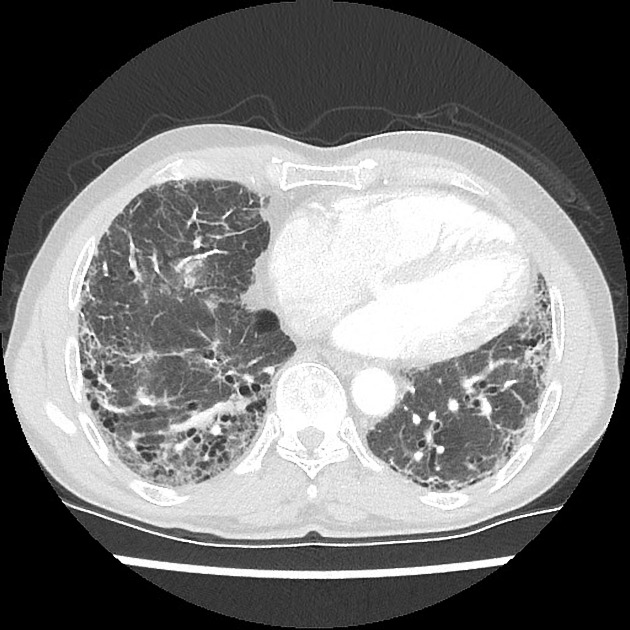
Usual interstitial pneumonia (UIP) pattern
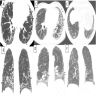
UIP and possible UIP pattern
Prognostic factors
- 5 year survival rate is 20% to 30% (Am J Respir Crit Care Med 2011;183:431)
- Median survival is 2 to 3 years
- Acute exacerbation (Am J Respir Crit Care Med 2016;194:265)
- Incidence of 4.1 per 100 patient years
- Precedes up to 46% of the deaths
- Median survival of patients with IPF who experience an acute exacerbation is 3 to 4 months
- Low forced vital capacity is the most consistent risk factor
- Severity of elastofibrosis on histology (Respir Med 2013;107:1608)
- Comorbidity:
- Pulmonary hypertension (Chest 2006;129:746)
- Lung cancer (Chest 2015;147:157)
- Emphysema: in this case, patient may be diagnosed as combined pulmonary fibrosis and emphysema (Chest 2012;141:222, Eur Respir J 2005;26:586)
- Prognosis of PPF is comparable to IPF (Eur Respir J 2020;55:2000085)
Case reports
- 45 year old man with IPF and adenocarcinoma (Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2011;155:403)
- 58 year old woman with acute exacerbation of IPF associated with interferon α therapy for hepatitis C (Springerplus 2013;2:101)
- 58 year old man with IPF and polyarteritis nodosa (Fukushima J Med Sci 2003;49:141)
- 66 year old woman with diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) presenting with UIP pattern on HRCT (Pneumonol Alergol Pol 2016;84:174)
- 69 year old man with IgG4 related disease and lung fibrosis showing UIP pattern (Intern Med 2022;61:2637)
- 79 year old male man died of tracheobronchomegaly (Mounier-Kuhn syndrome) caused by IPF (Eur J Intern Med 2017;42:e7)
Treatment
- No treatment has been found to substantially improve its prognosis
- Recommended by guidelines (Am J Respir Crit Care Med 2015;192:e3):
- Pirfenidone, an antifibrotic drug reducing TGFβ induced myofibroblast differentiation (N Engl J Med 2014;370:2083)
- Nintedanib, an inhibitor targeting multiple tyrosine kinases (N Engl J Med 2014;370:2071)
- Edematous changes in the interlobular septum could be associated with poor response to Nintedanib (PLoS One 2021;16:e0245147)
- Lung transplantation
Gross description
- Fibrotic changes in lower lobes
- Shrunken lung with hobnailed pleura due to scarring
- Elastic hard consistency
- Diffuse (relatively subpleural dominant) destruction of lung mesenchyme
- Multiple air cysts due to honeycomb change
- Traction bronchiectasis can be seen
Gross images
Images hosted on other servers:
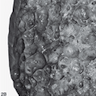
Honeycomb lung

Cobblestone appearance of the pleura
Microscopic (histologic) description
- Major findings are consistent with UIP pattern (Am J Respir Crit Care Med 2022;205:e18)
- Patchy dense fibrosis with architectural distortion
- Lesion representing UIP pattern is mainly composed of hyalinized collagen
- Often destructs alveolar architecture
- Smooth muscle and elastofibrosis are often present
- Only mild infiltration by inflammatory cells should be seen (usually near areas of honeycomb change)
- Honeycombing: cystic spaces lined by bronchiolar epithelium and fibrotic wall
- Predilection for subpleural and paraseptal lung parenchyma
- Spatial heterogeneity (dense fibrosis alternating with areas of the uninvolved lung) is evident on low power
- Normal lung parenchyma can remain in the center of the lobule
- Fibroblastic foci
- Active fibrotic lesions composed of myofibroblasts
- Aggregation of spindle cells with gray to pale purple matrix adjacent to dense fibrosis
- Absence of features that suggest an alternate diagnosis
- Patchy dense fibrosis with architectural distortion
- Clinical guidelines suggest 4 levels of certainty for pathologic diagnosis (Am J Respir Crit Care Med 2018;198:e44)
- Definite UIP pattern
- Dense fibrosis with architectural distortion (i.e., destructive scarring or honeycombing)
- Predominant subpleural or paraseptal distribution of fibrosis
- Patchy involvement of lung parenchyma by fibrosis
- Fibroblast foci
- Absence of features to suggest an alternate diagnosis
- Probable UIP pattern
- Some histologic features of UIP are present but to an extent that precludes a definite diagnosis of UIP / IPF
- Absence of features to suggest an alternate diagnosis
- Or honeycombing only
- Indeterminate for UIP
- Fibrosis with or without architectural distortion, with features favoring either a pattern other than UIP or features favoring UIP secondary to another cause
- Some histologic features of UIP but with other features suggesting an alternative diagnosis
- Features of an alternative diagnosis: a cellular inflammatory infiltrate away from areas of honeycombing; prominent lymphoid hyperplasia, including secondary germinal centers; and a distinctly bronchiolocentric distribution that could include extensive peribronchiolar metaplasia
- Alternative diagnosis
- Features of other histologic patterns of idiopathic interstitial pneumonia (e.g., absence of fibroblast foci or loose fibrosis) in all biopsies
- Histologic findings indicative of other diseases; hypersensitivity pneumonitis, Langerhans cell histiocytosis, sarcoidosis, lymphangioleiomyomatosis (LAM)
- Some criteria of these findings (e.g., how many granulomas to be considered significant) are still not clear
- Definite UIP pattern
- Additional findings
- Squamous metaplasia: squamoid epithelium without cilia, indicator of acute exacerbation (Eur Respir J 2003;22:821)
- Interstitial emphysema: a cystic space without epithelial lining, predictor of pneumothorax (Am J Surg Pathol 2014;38:339)
- Smoking related changes including respiratory bronchiolitis, accumulation of smoker’s macrophages and desquamative interstitial pneumonia-like reaction
- Pulmonary alveolar proteinosis (PAP)-like change: eosinophilic and fine granular proteinaceous material in airspace (Respir Investig 2016;54:272)
- Centriacinar emphysema, especially in smokers
- Peribronchiolar metaplasia, replacement of bronchiolar epithelium in alveolar walls and terminal bronchioles (Am J Surg Pathol 2005;29:948)
- Fat metaplasia
- Bone metaplasia
- Kuhn hyaline (Mallory hyaline): dense, waxy, eosinophilic clumps within cytoplasm of reactive type 2 pneumocytes (Hum Pathol 1980;11:59)
- Fibrin deposition
Microscopic (histologic) images
Contributed by Akira Yoshikawa, M.D. and Yale Rosen, M.D.
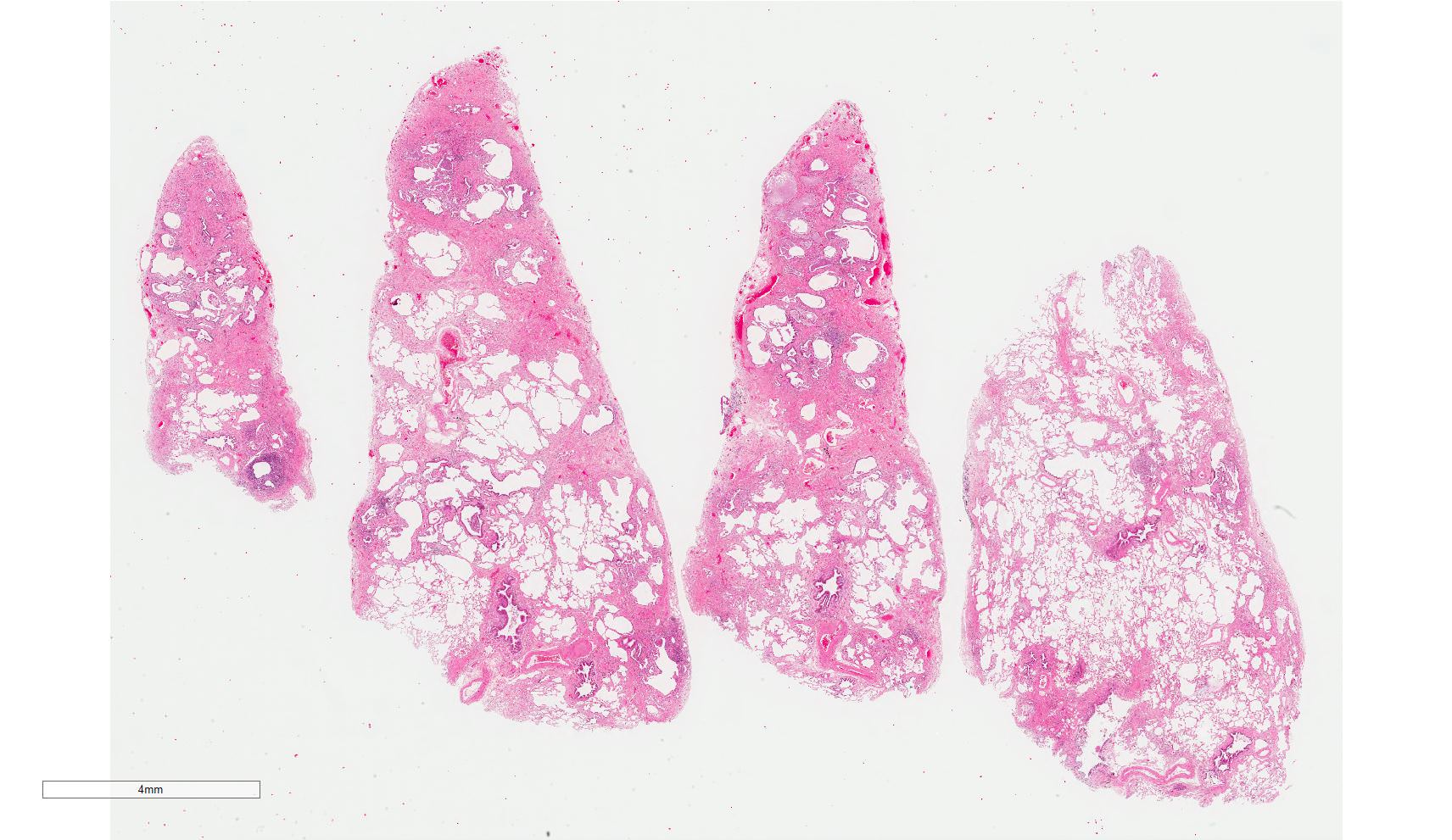
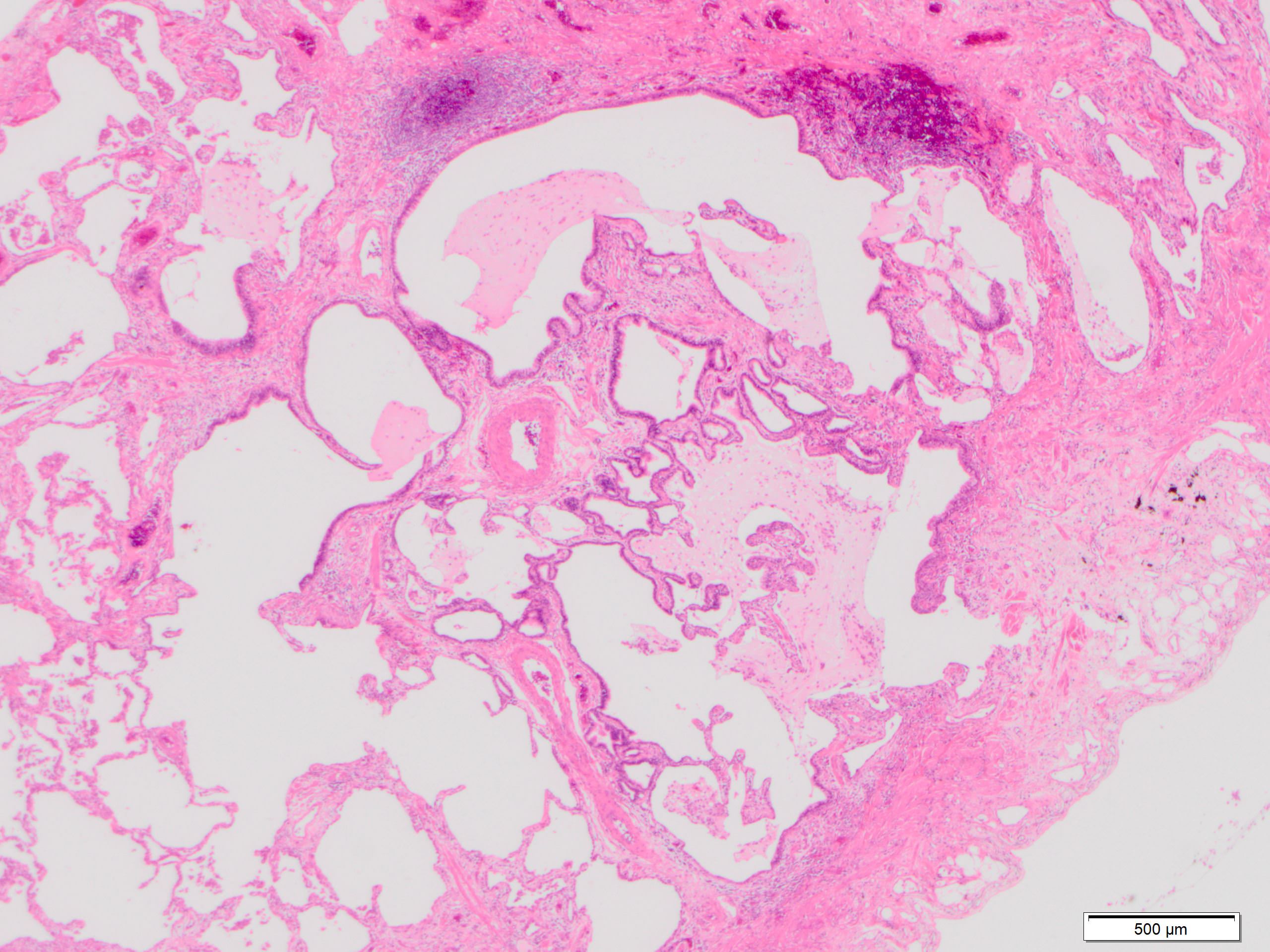
Spatial heterogeneity
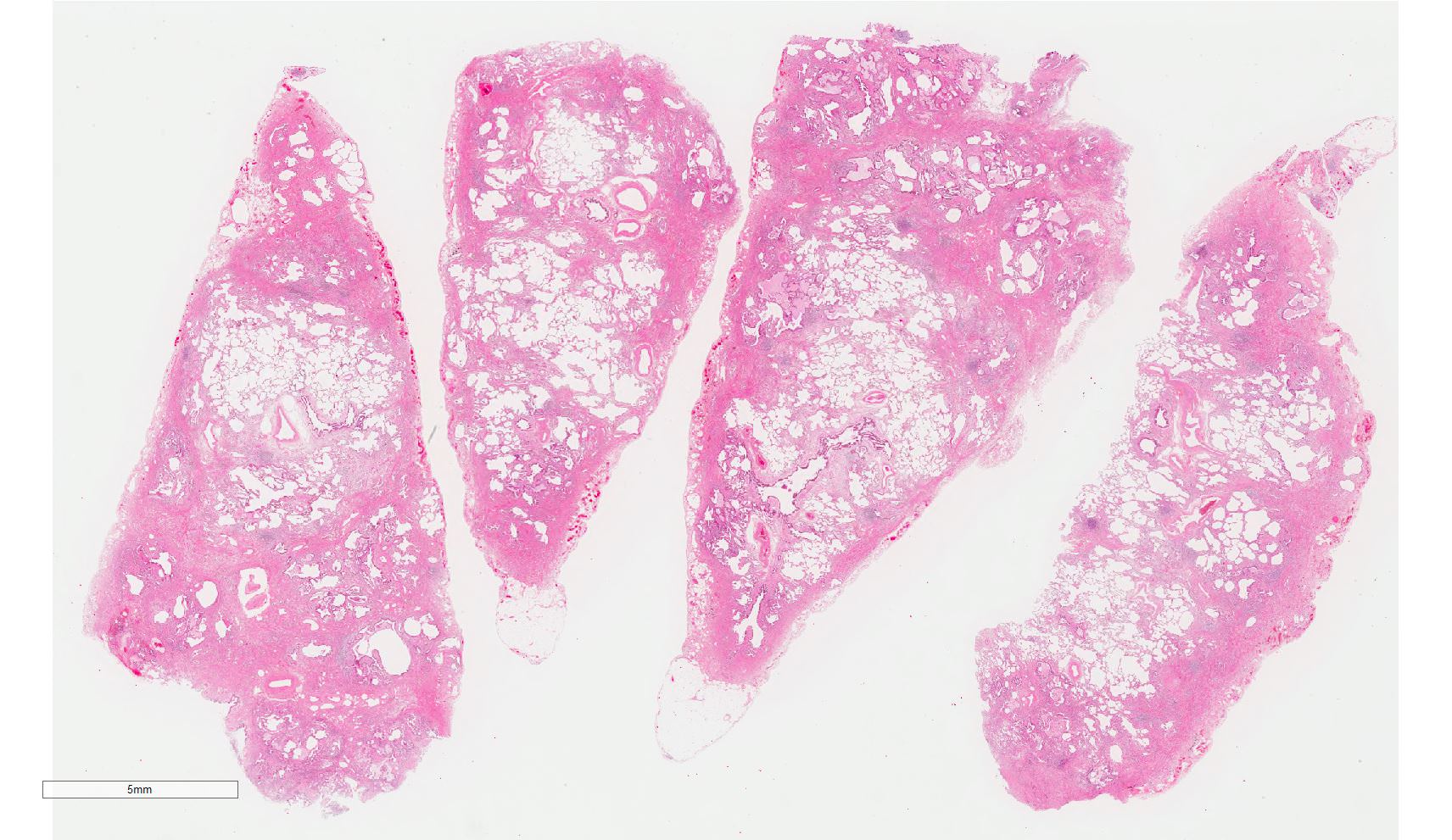
Spatial heterogeneity
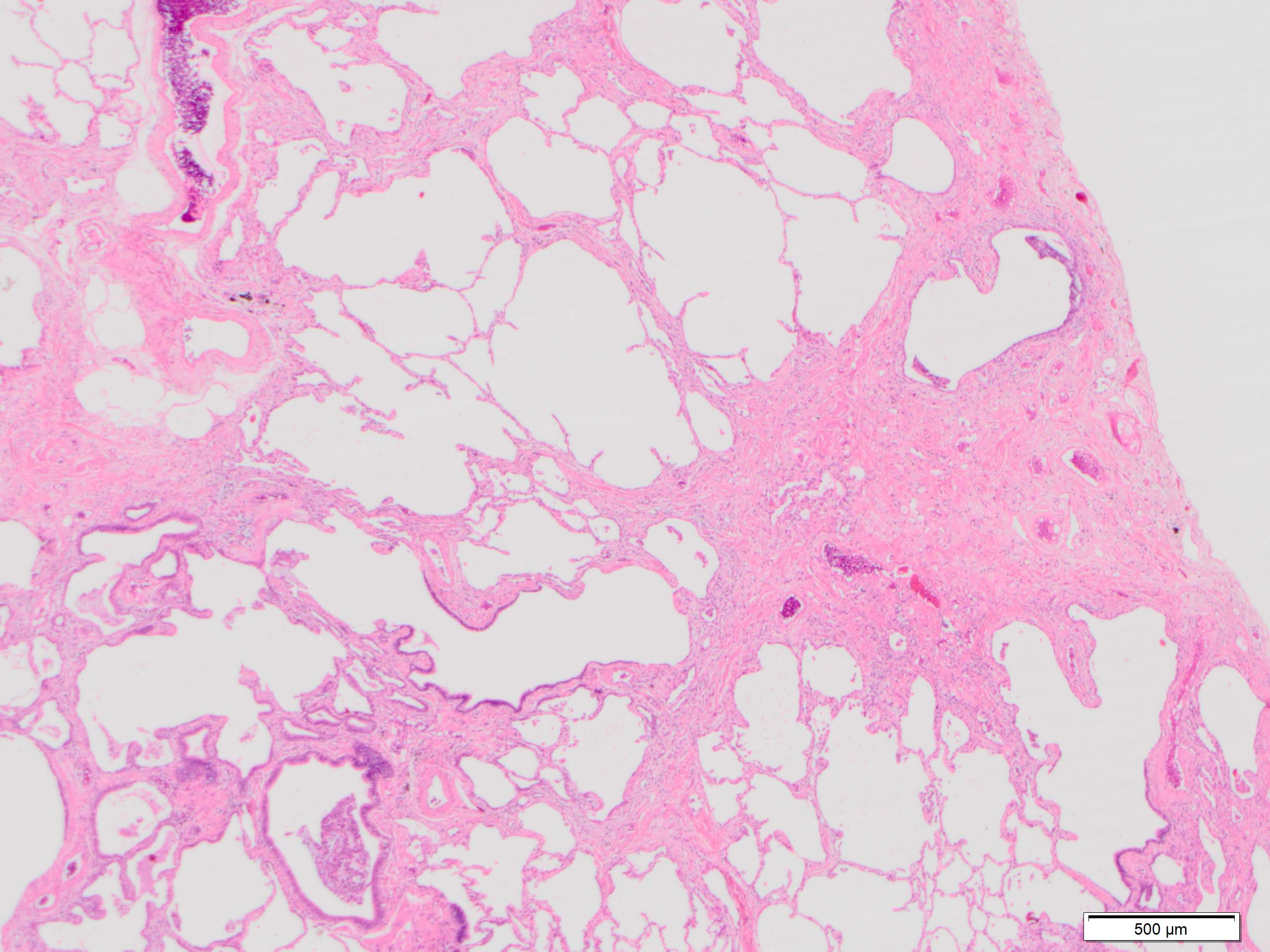
Subpleural fibrosis
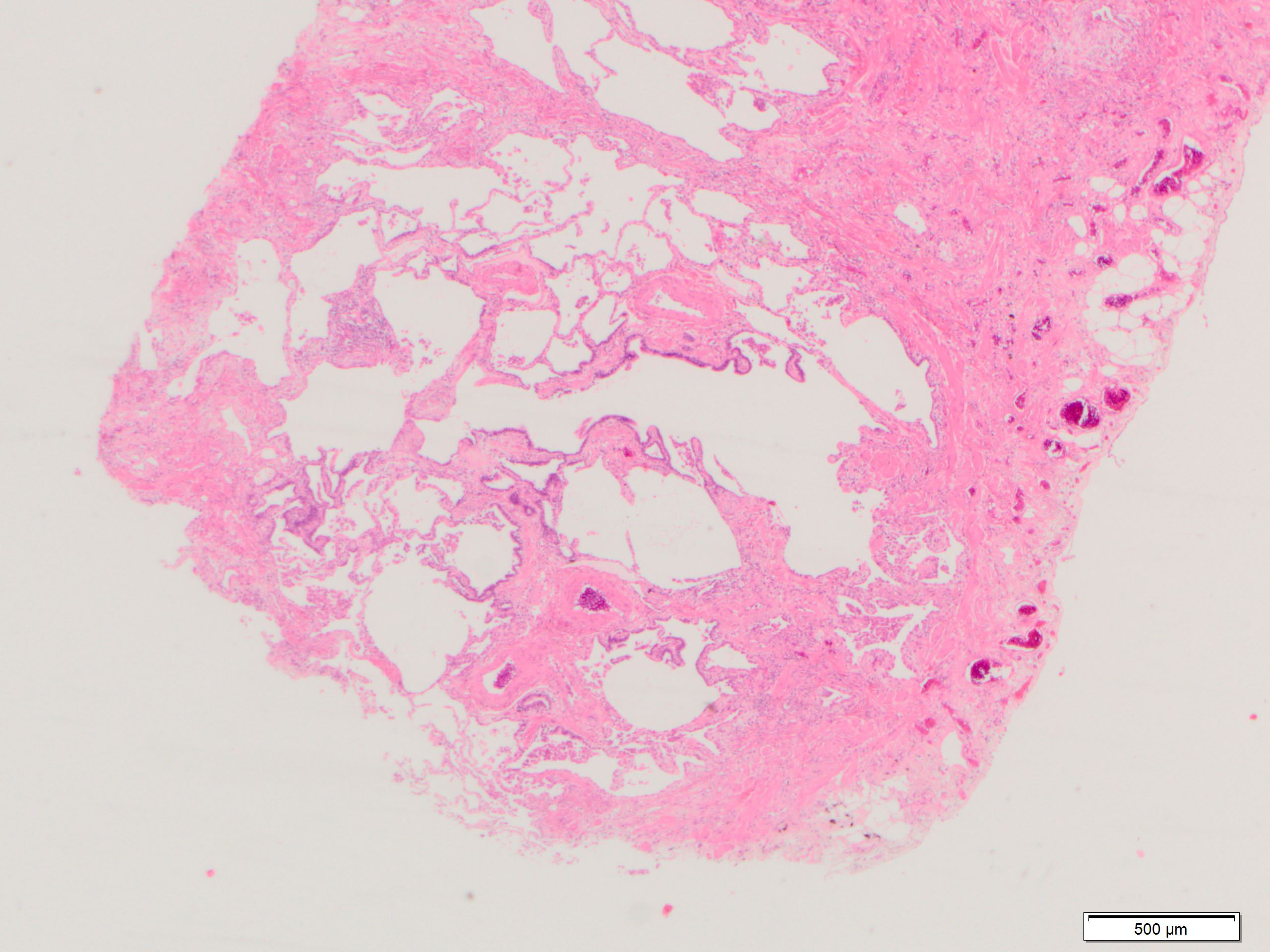
Subpleural and paraseptal fibrosis
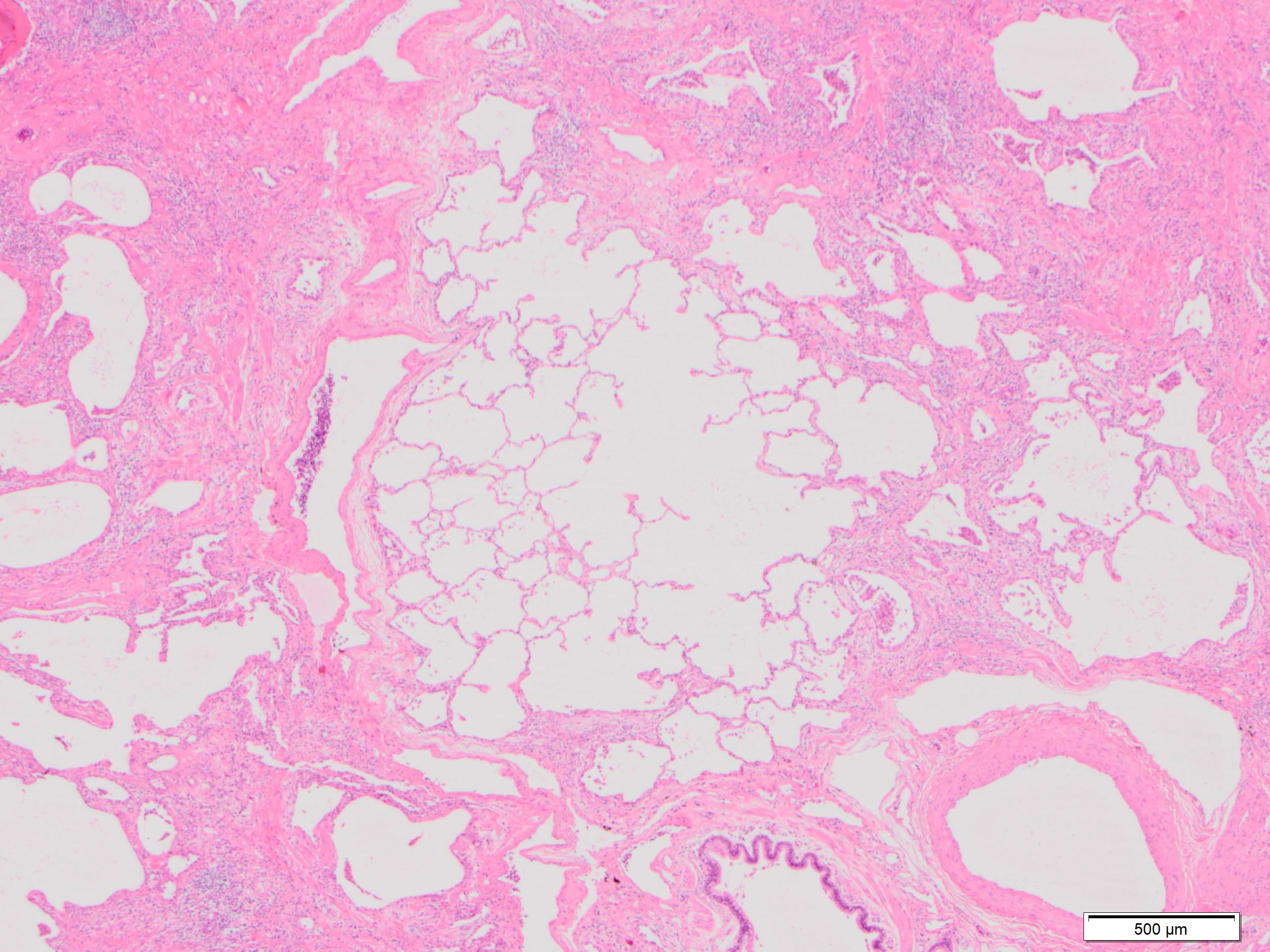
Paraseptal fibrosis
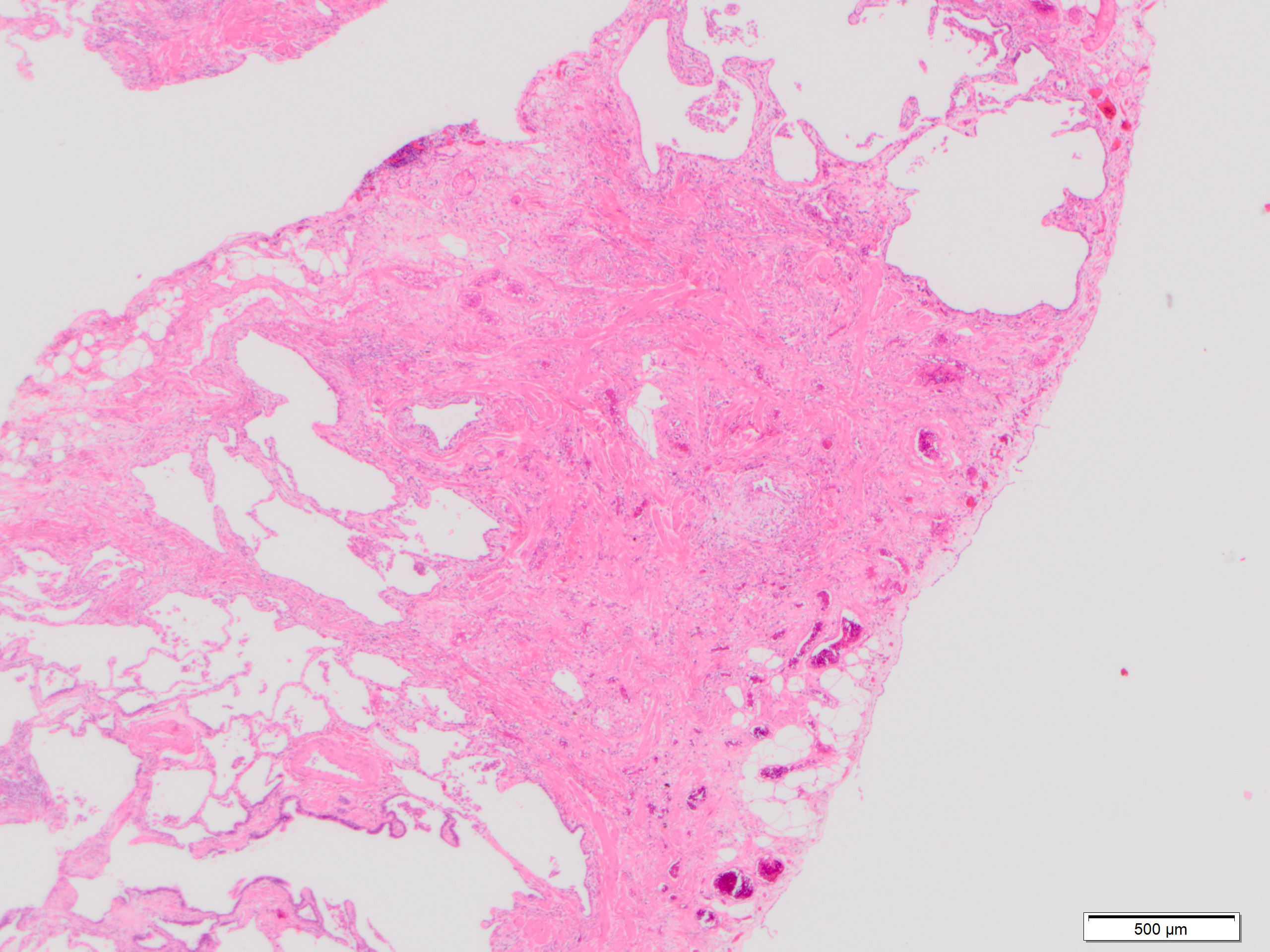
Dense fibrosis
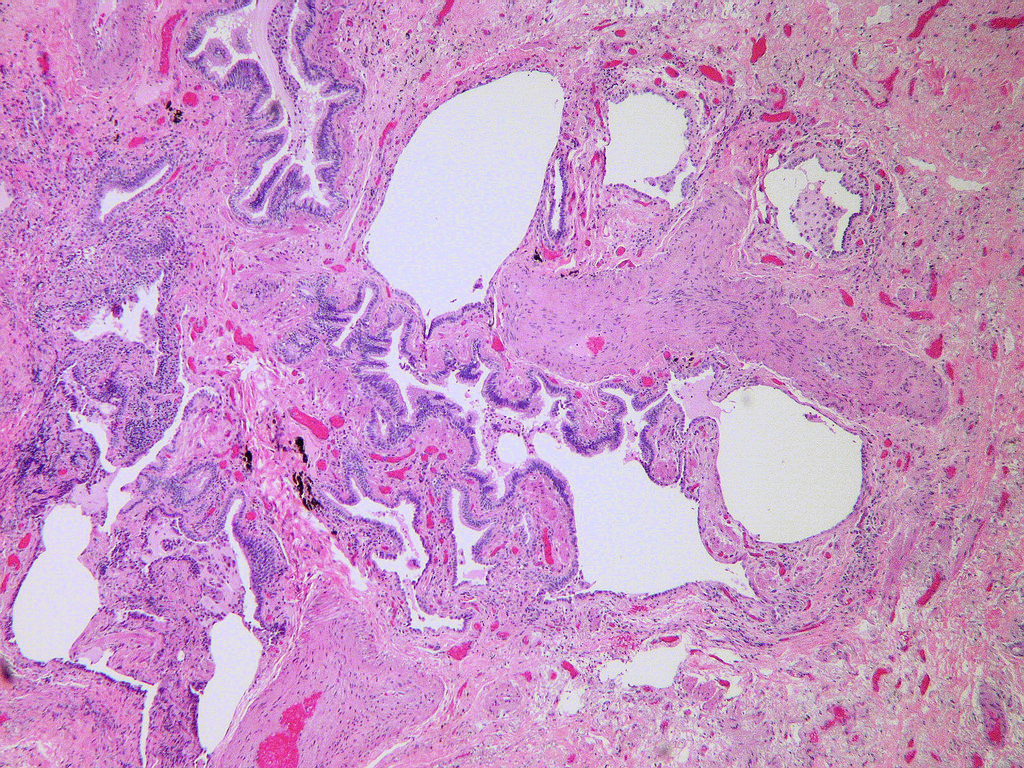
Dense fibrosis with architectural destruction
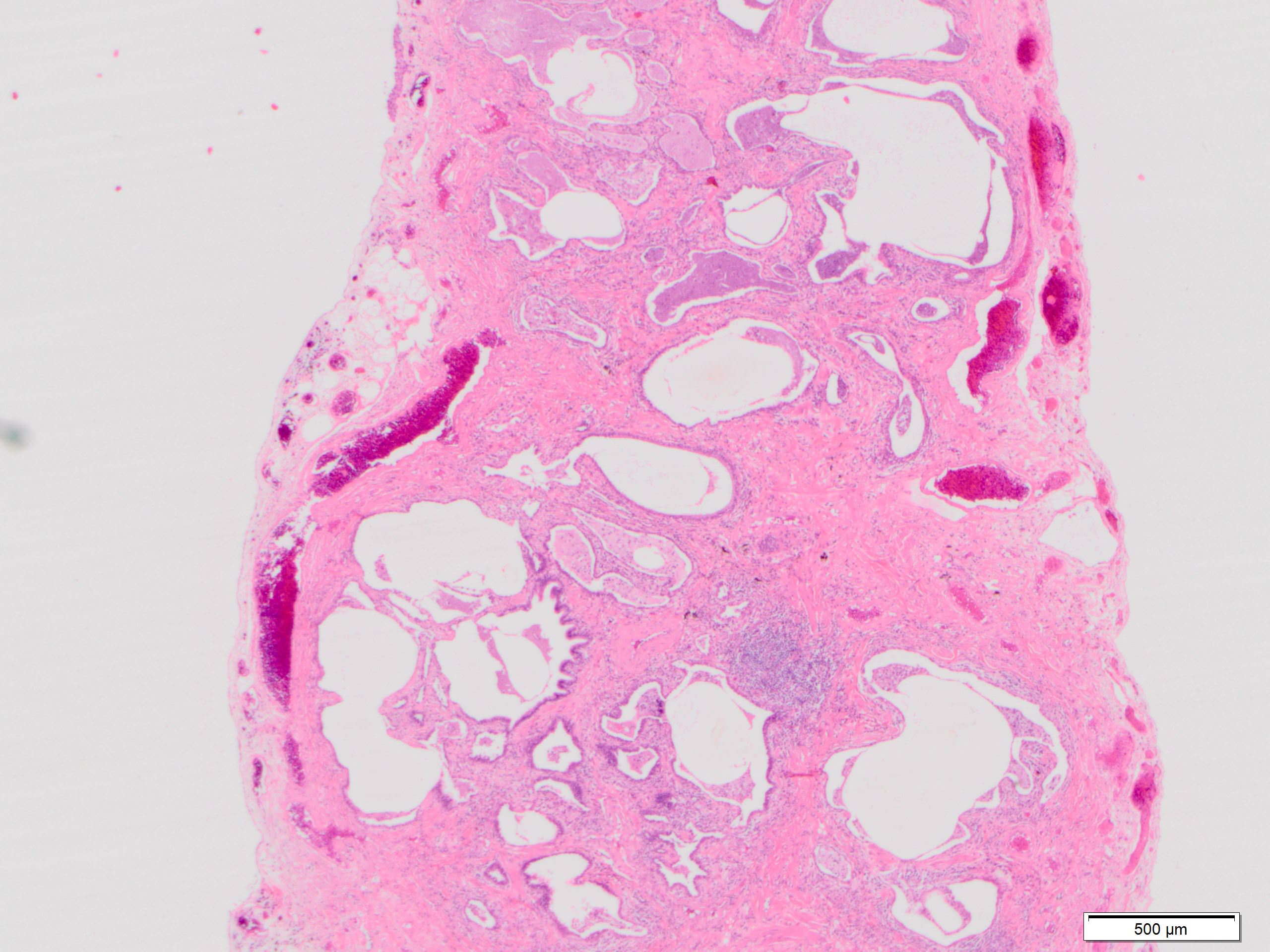
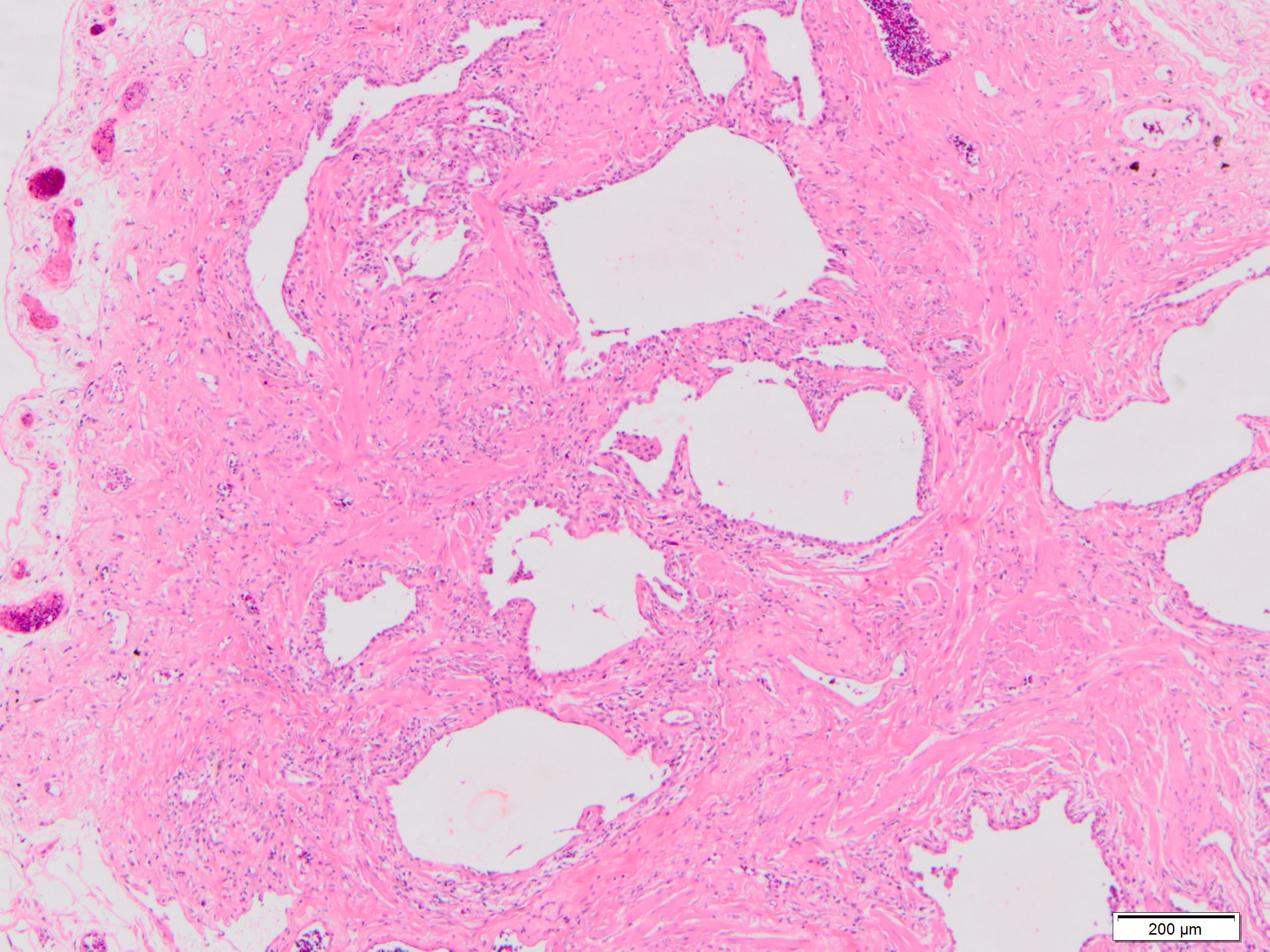
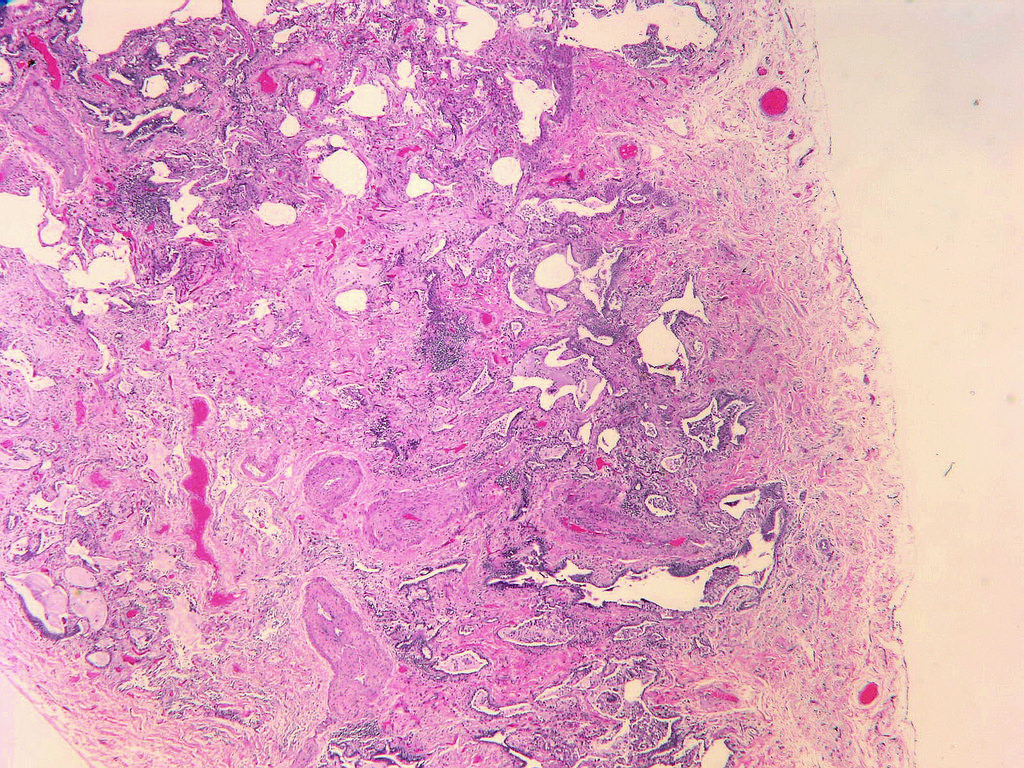
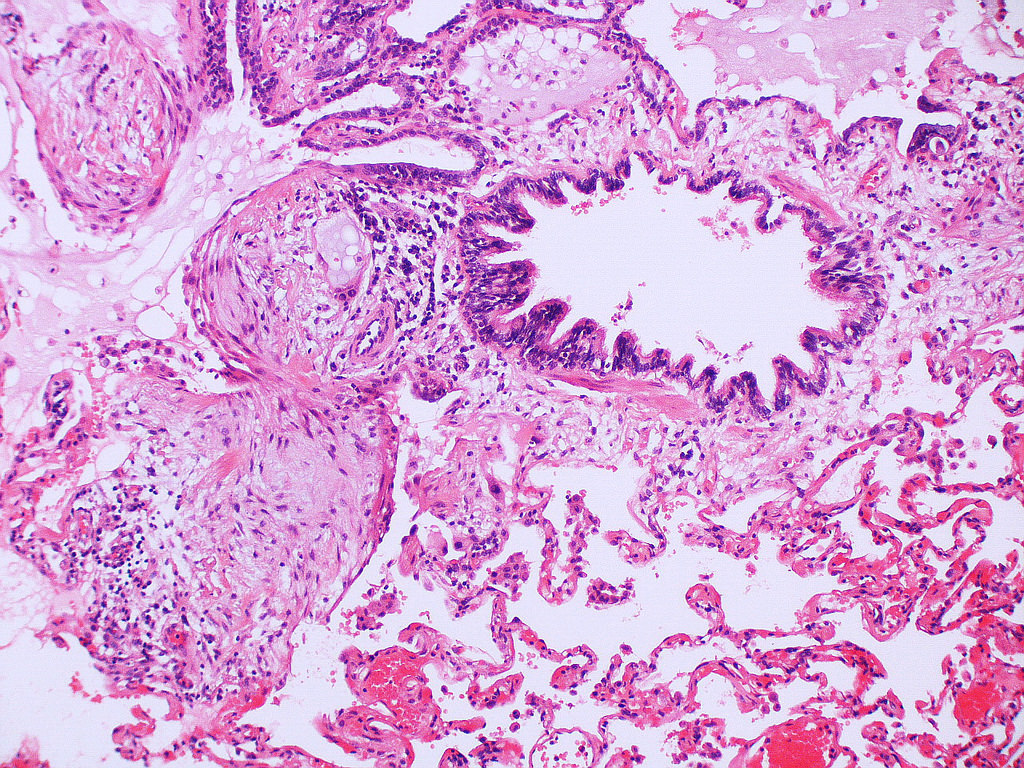
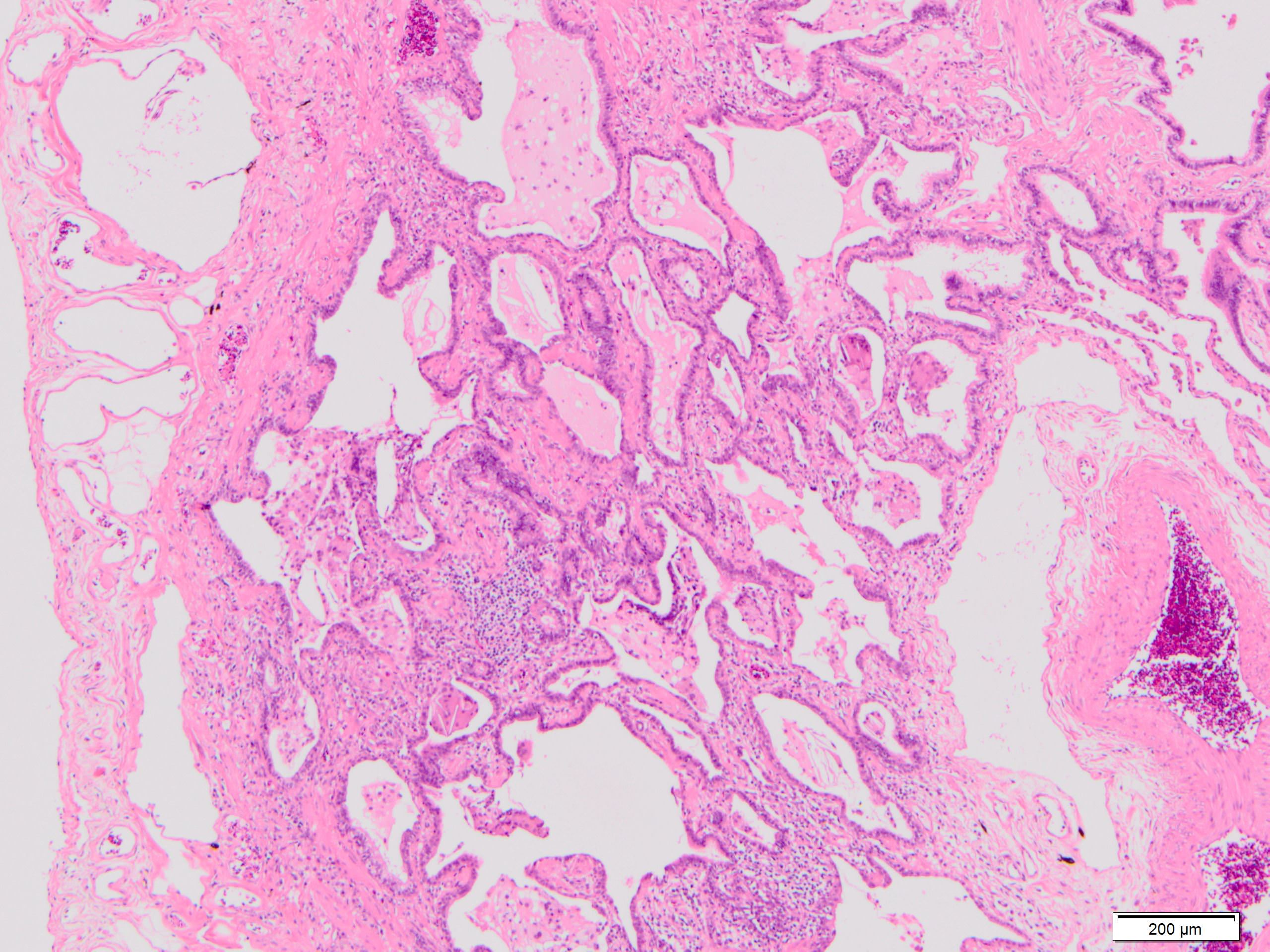
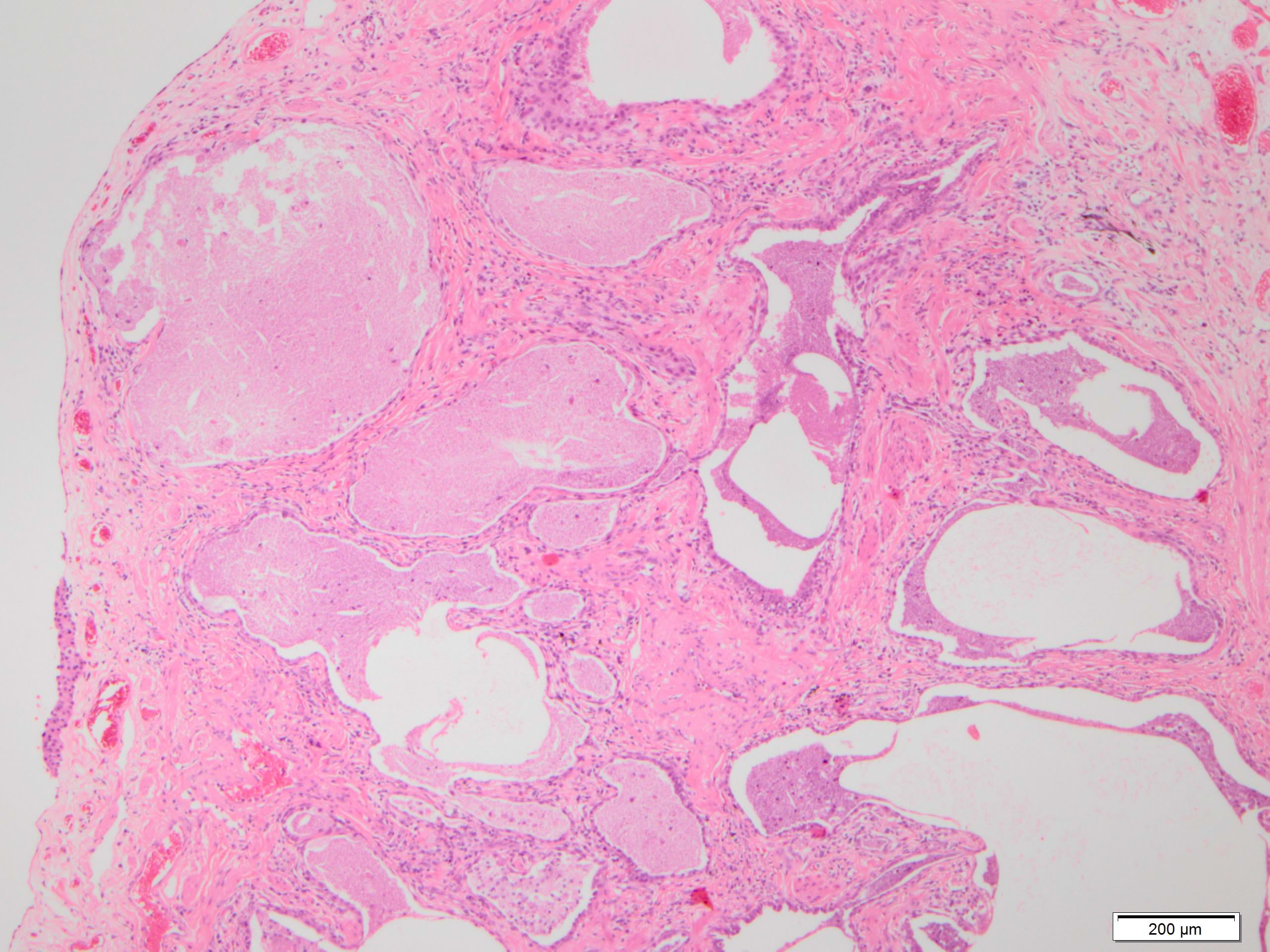
Honeycomb change
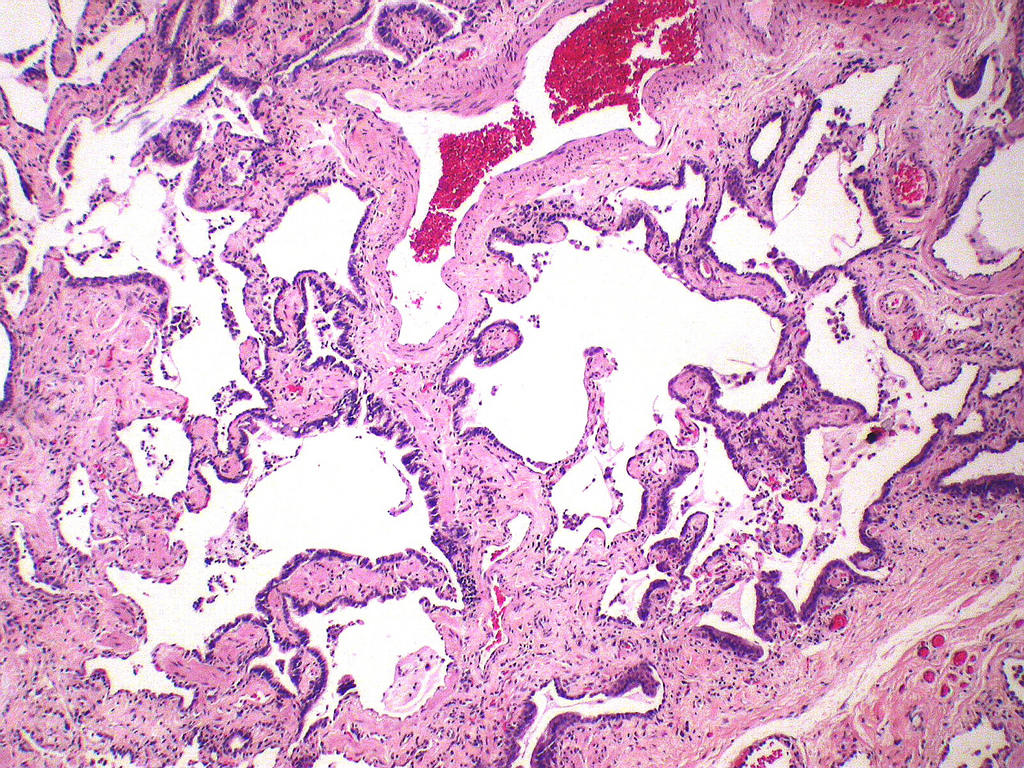
Honeycomb change
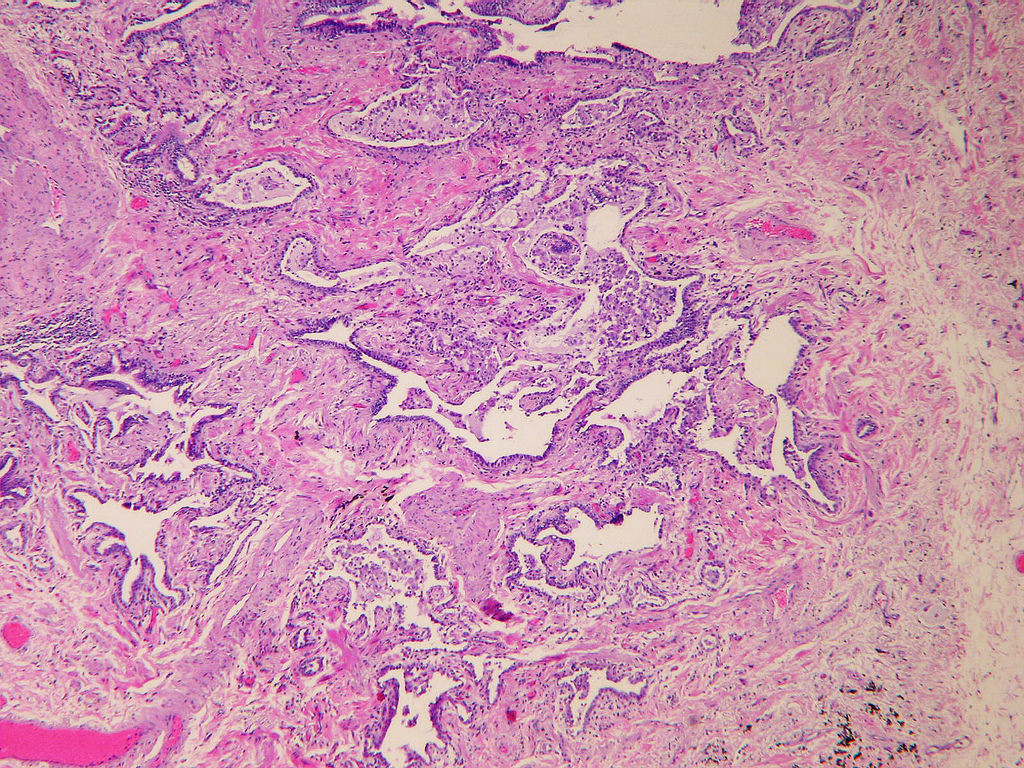
Honeycomb change
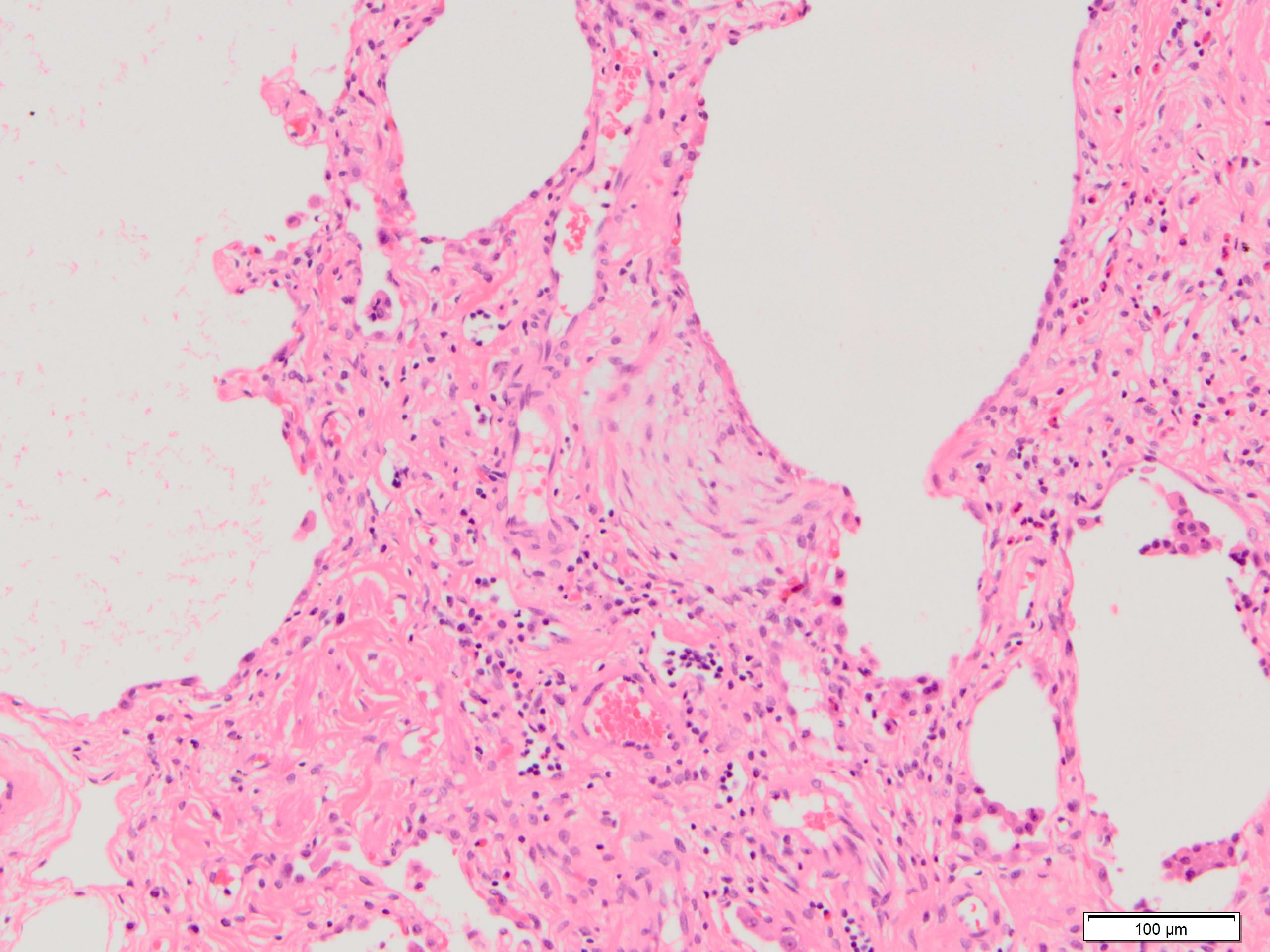
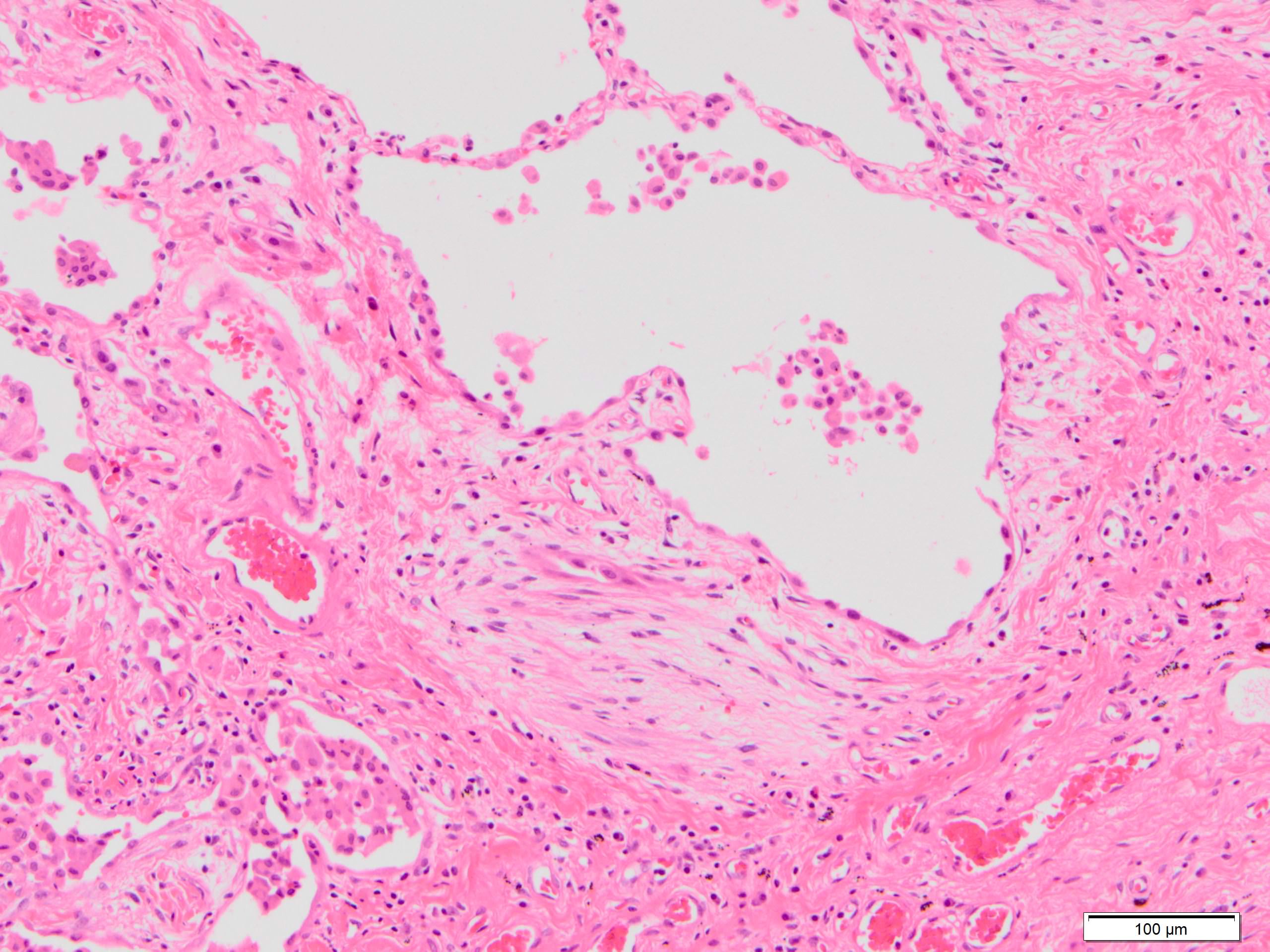
Fibroblast focus
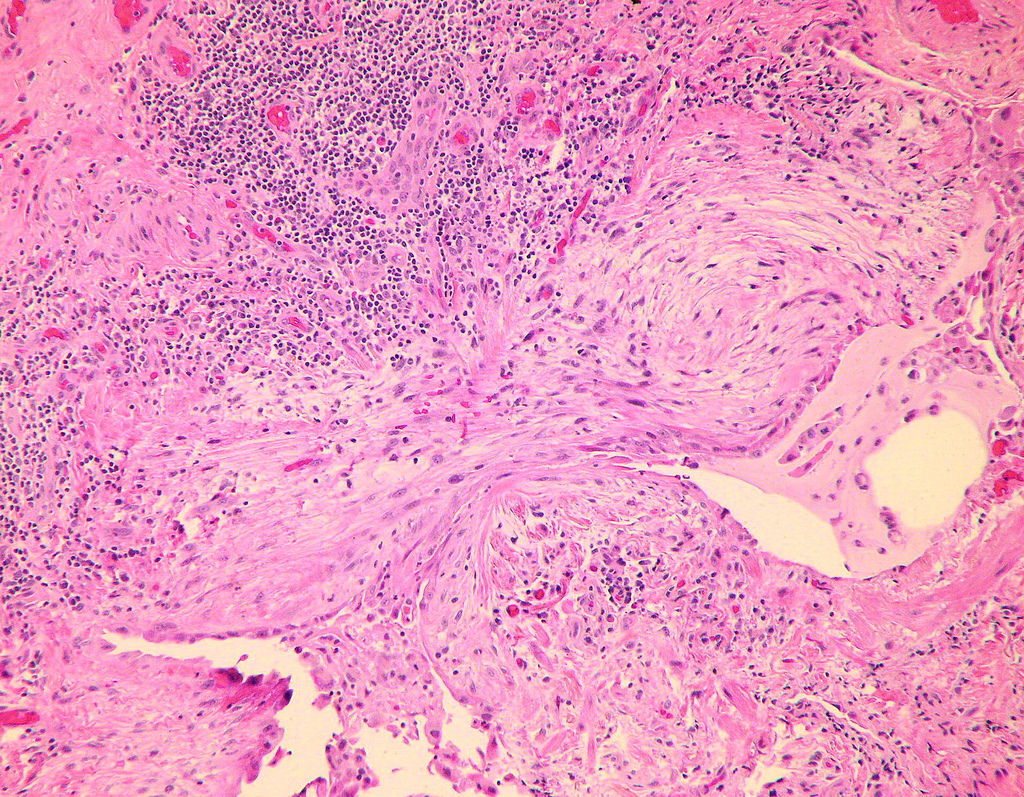
Fibroblast focus
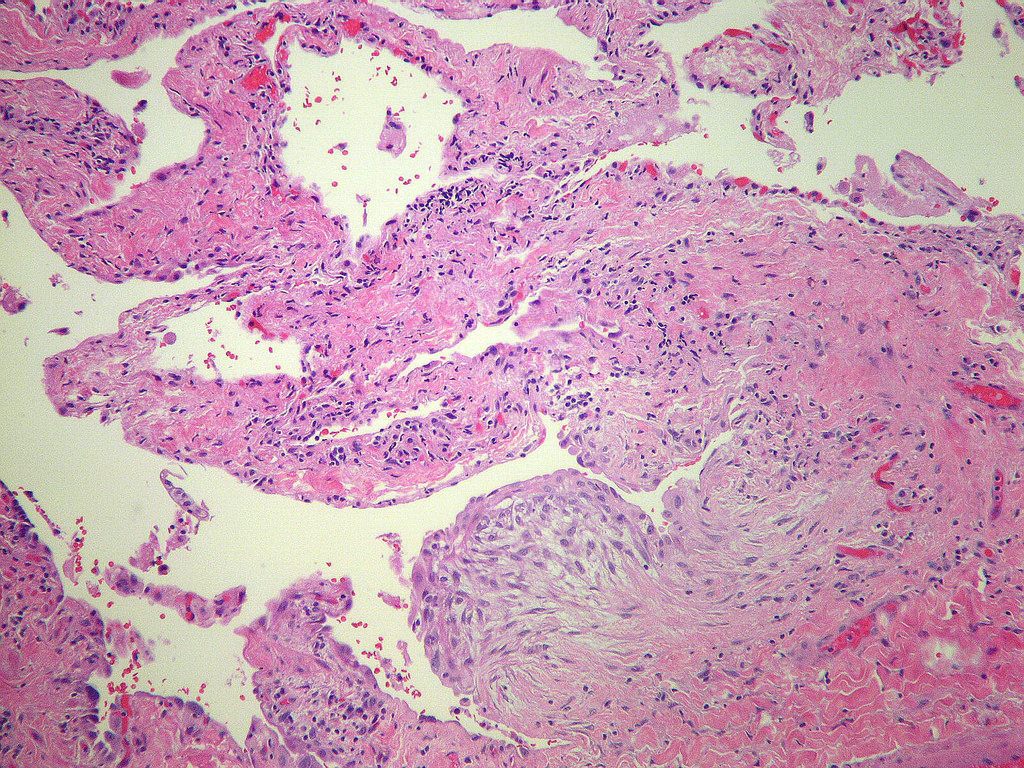
Fibroblast focus
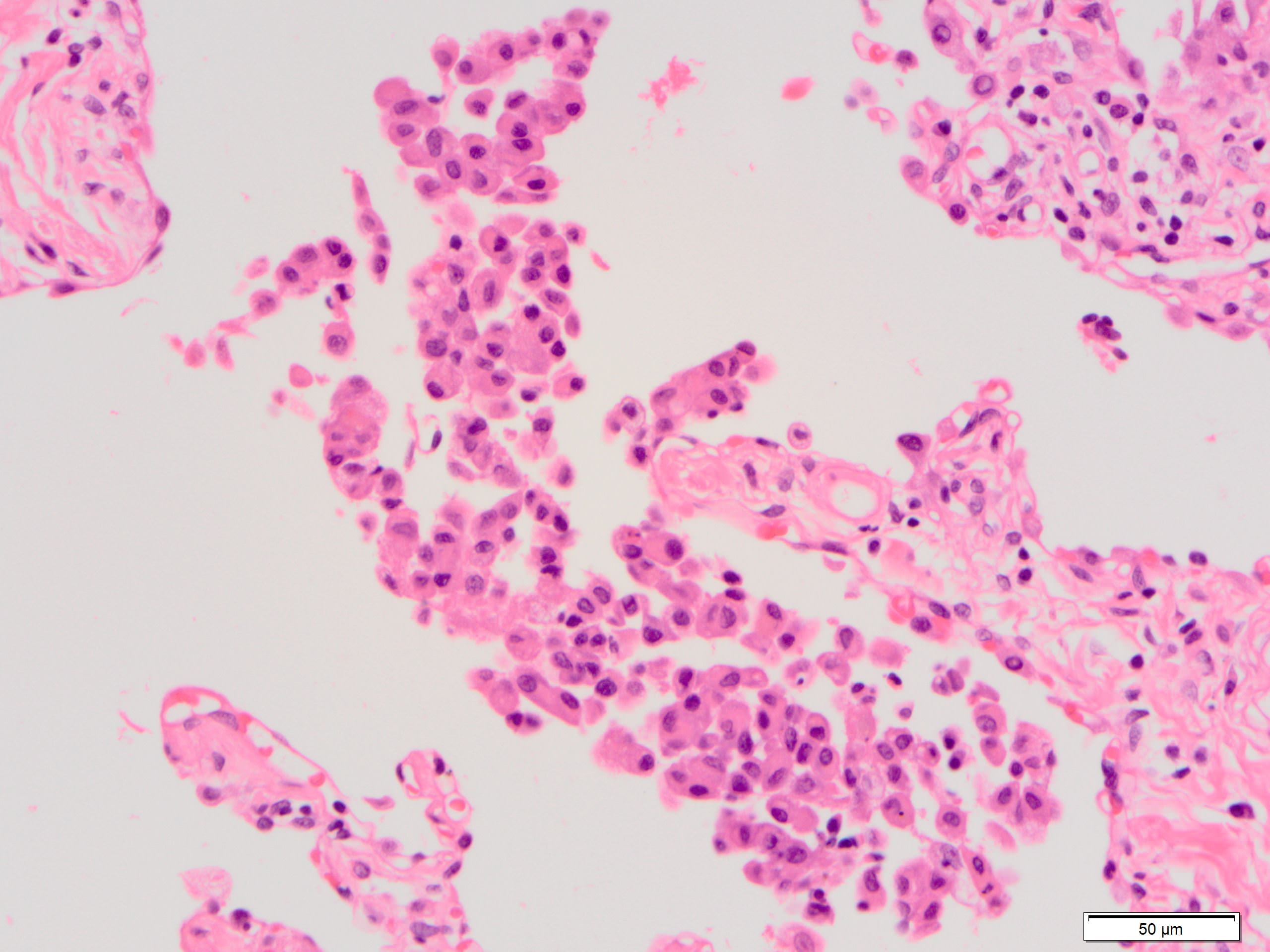
Peribronchiolar metaplasia
PAP-like change
Respiratory bronchiolitis
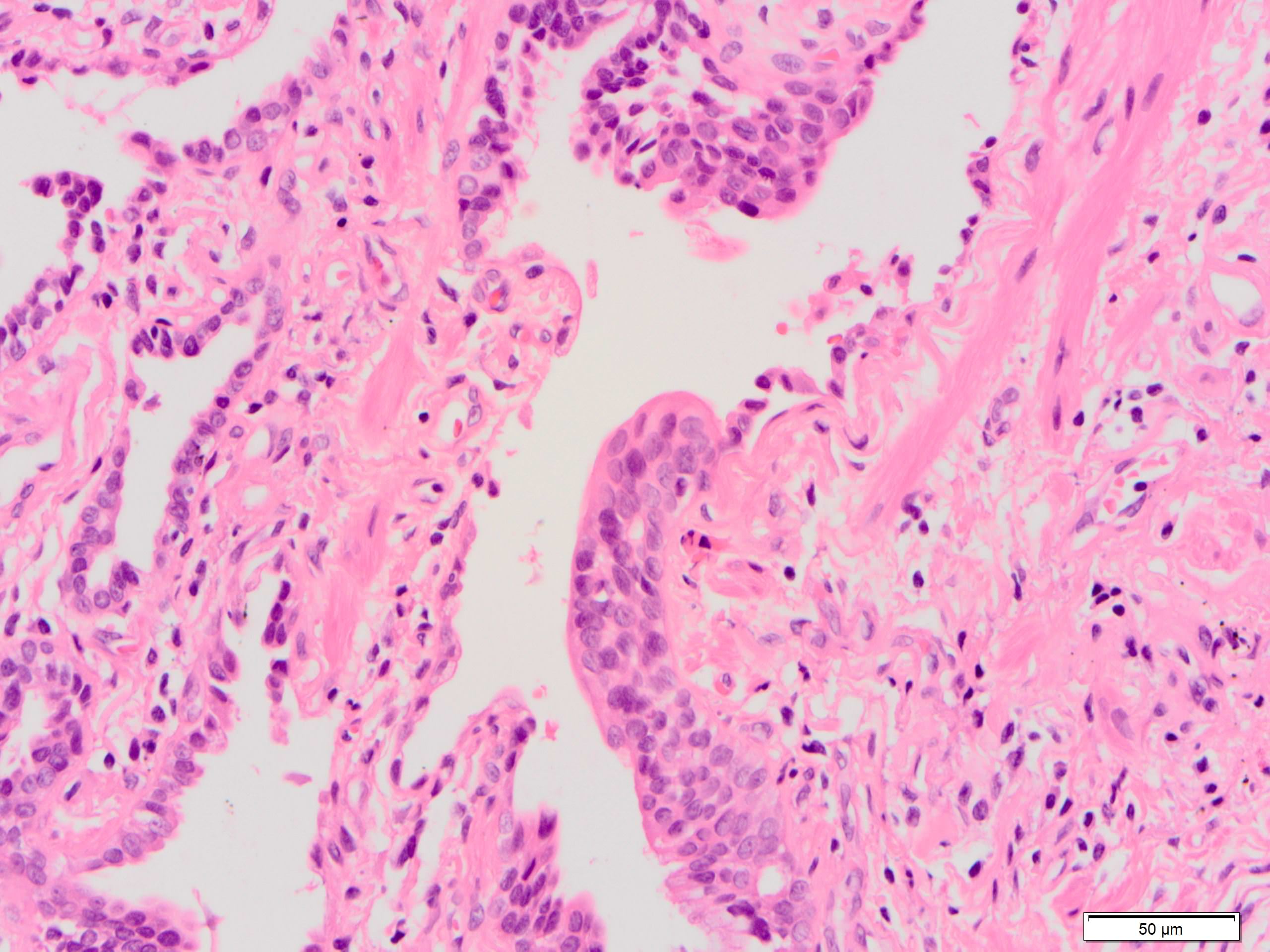
Squamous metaplasia
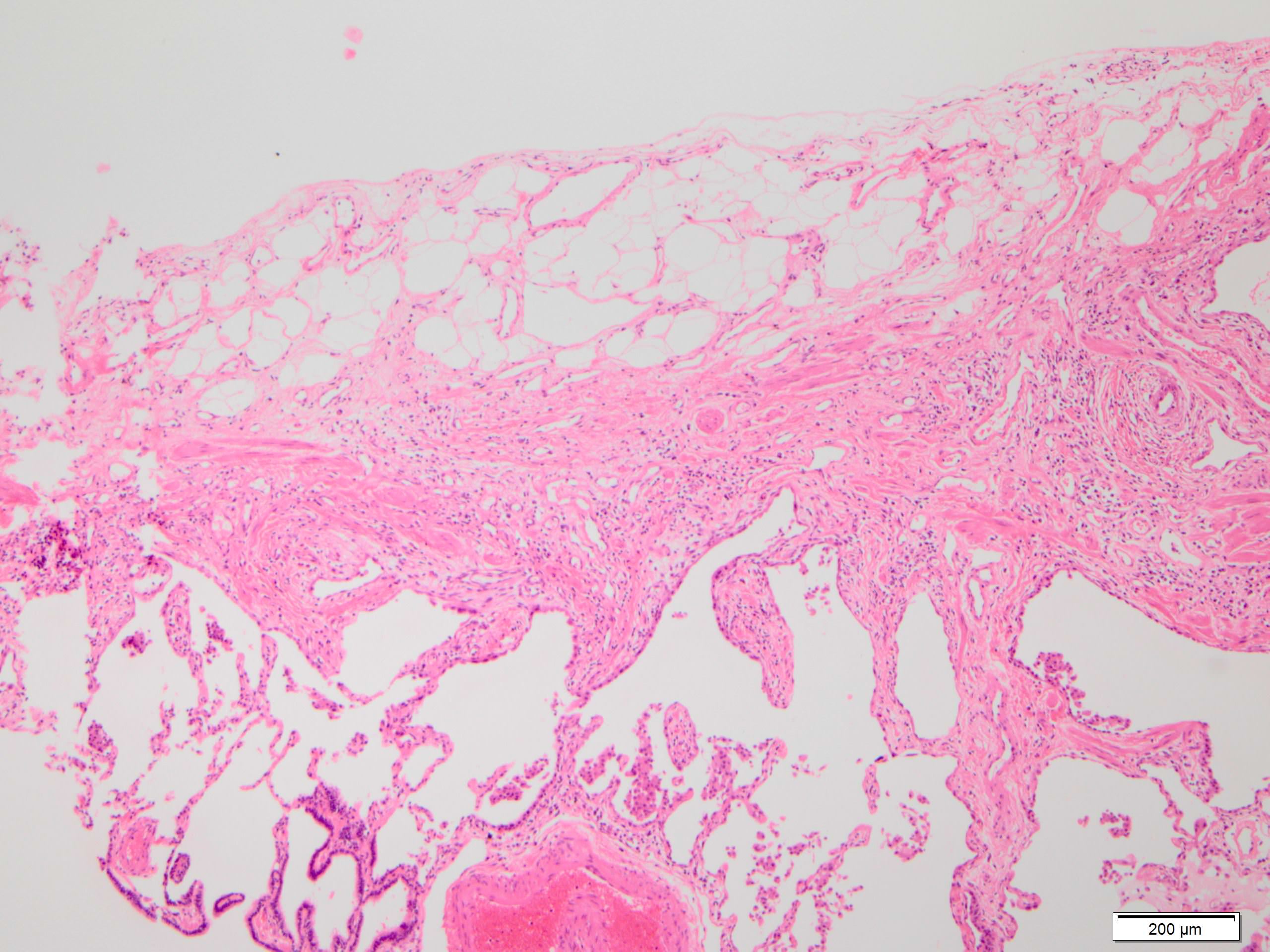
Fat metaplasia

Bone metaplasia
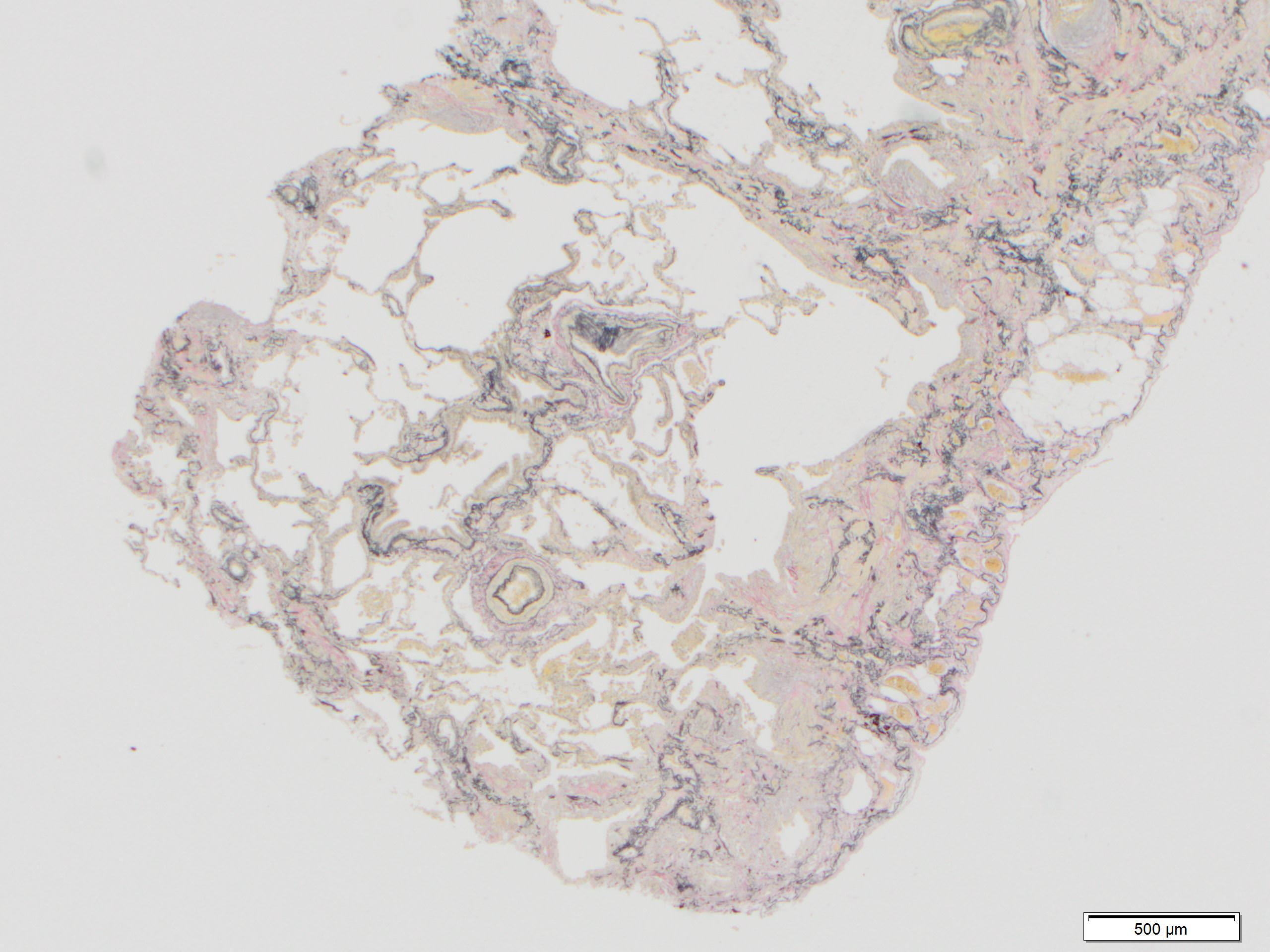
Elastosis
Positive stains
- Elastica van Gieson - highlight elastic fibers
Electron microscopy description
- Fibrosis due to migration of activated mesenchymal cells through defects in epithelial lining and its basement membrane from interstitial to intraluminal compartments
- Replacement of alveolar type I cells by hyperplastic alveolar type II cells
Videos
Usual interstitial pneumonia
Usual interstitial pneumonia
Usual interstitial pneumonia
Sample pathology report
- Lung, right upper, middle and lower lobes, wedge biopsy:
- Usual interstitial pneumonia (UIP) (see comment)
- Comment: Sections from the lung wedge biopsies show a fibrosing interstitial pneumonia, characterized by predominantly honeycombing and dense fibrosis. The areas of fibrosis are primarily located in the peripheral portions of the specimen. There appears to be a temporally variegated appearance, with areas of relatively uninvolved lung, along with areas showing patchy interstitial fibrosis. Fibroblast foci are frequently identified. Mild chronic inflammatory infiltrate is noted. There is no evidence of opportunistic microorganisms, granulomatous inflammation and vasculitis.
Differential diagnosis
- Nonspecific interstitial pneumonia:
- Diffuse thickening of the alveolar septa, with or without a cellular infiltrate and preserved alveolar architecture
- Connective tissue disease associated interstitial lung disease (CTD ILD):
- Lymphoid follicles with germinal centers, lymphoplasmacytic infiltration, pleuritis (Eur Respir J 2015;46:976)
- Hypersensitivity pneumonitis:
- Airway centered changes, poorly formed granuloma, giant cells, peribronchiolar metaplasia (Am J Respir Crit Care Med 2012;186:314, Histopathology 2012;61:1026)
- Diffuse alveolar damage:
- Hyaline membranes, intra-alveolar hemorrhage
- Cryptogenic organizing pneumonia:
- Organizing pneumonia
- Pleuroparenchymal fibroelastosis (PPFE):
- Diffuse elastofibrosis in upper lung
- Pneumoconiosis:
- Foreign particles such as carbon, silica and asbestos
- Sarcoidosis:
- Well formed, nonnecrotizing granuloma, consists of epithelioid histiocytes and multinucleated giant cells
- Combined pulmonary fibrosis and emphysema (CPFE):
- Centriacinar emphysema in upper lobes, UIP pattern in lower lobes
Additional references
Practice question #1
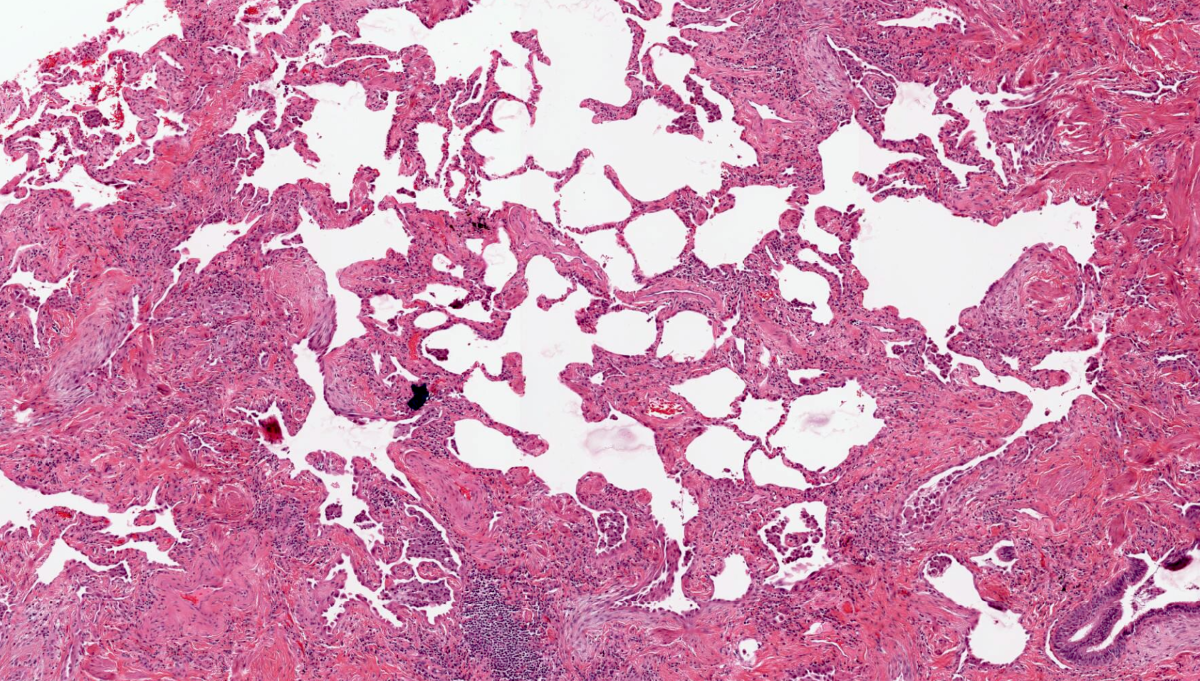
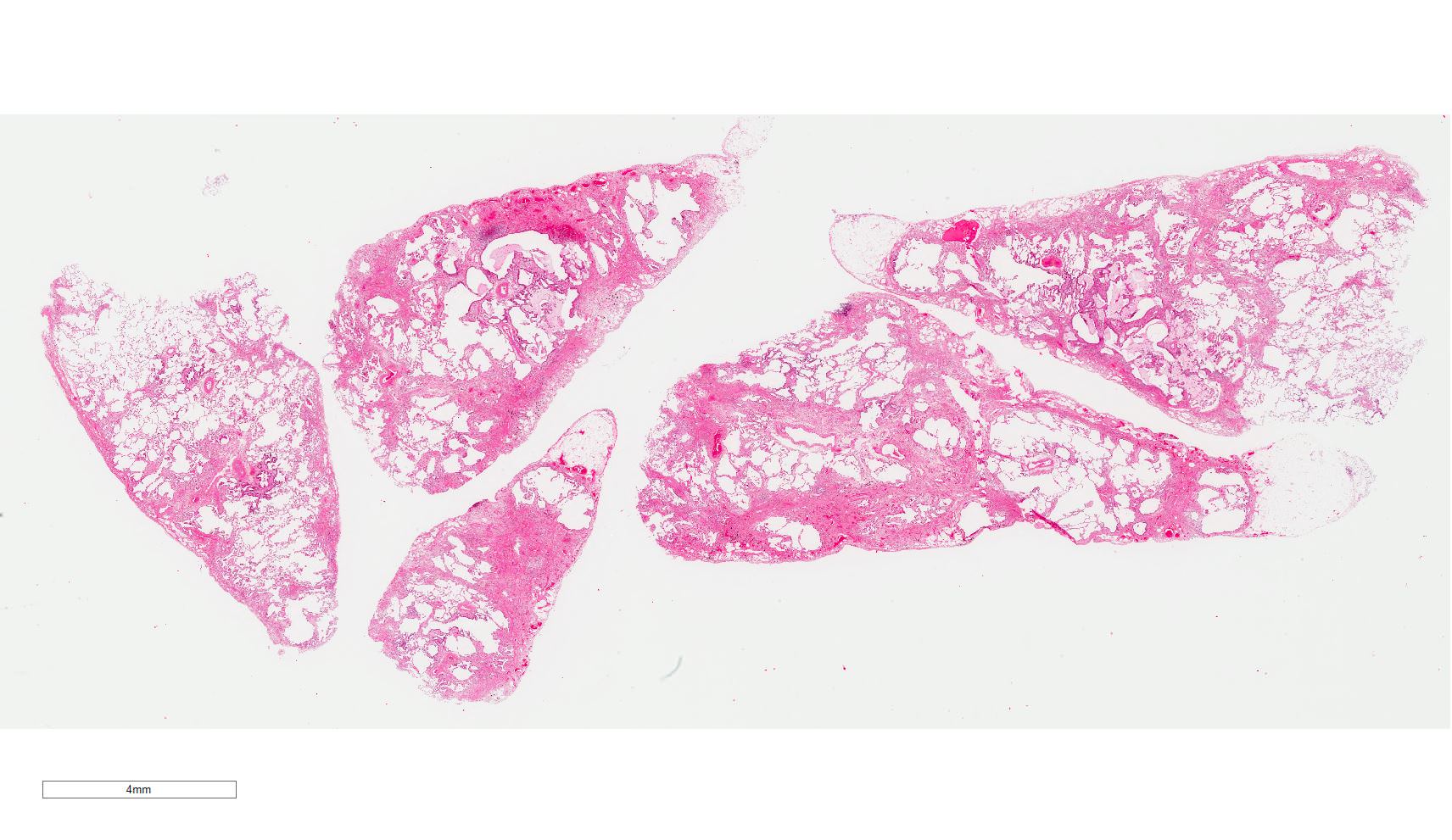
A lung wedge biopsy of a 55 year old man is shown. What is the most likely diagnosis?
- Diffuse alveolar damage
- Emphysema
- Nonspecific interstitial pneumonia
- Organizing pneumonia
- Usual interstitial pneumonia
Practice answer #1
E. Usual interstitial pneumonia. Sections show patchy fibrosis adjacent to the preserved alveoli. Fibroblast foci are also present.
Comment Here
Reference: UIP / IPF
Comment Here
Reference: UIP / IPF
Practice question #2
A 70 year old man presented with progressive dyspnea over the past few years. Which of the following findings, if present, would suggest an alternative diagnosis other than idiopathic pulmonary fibrosis (IPF)?
- Fibroblastic foci
- Granuloma
- Honeycomb change
- Subpleural and paraseptal fibrosis
- Temporal heterogeneity
Practice answer #2
B. Granuloma. The presence of granuloma is not a feature of UIP / IPF and would be suggestive of an alternative diagnosis.
Comment Here
Reference: UIP / IPF
Comment Here
Reference: UIP / IPF
