
Lymphoma & related disorders
Mature B cell neoplasms
Small B cell lymphomas with lymphoplasmacytic differentiation / marginal zone lymphomas
Lymphoplasmacytic lymphoma
Authors: Sanjay Bridgelall, M.D., Ling Zhang, M.D.
Editorial Board Member: Julie Feldstein, M.D.
Deputy Editor-in-Chief: Genevieve M. Crane, M.D., Ph.D.


Last author update: 27 September 2023
Last staff update: 27 August 2024
Copyright: 2001-2025, PathologyOutlines.com, Inc.
PubMed Search: Lymphoplasmacytic lymphoma

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Flow cytometry description | Flow cytometry images | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Bridgelall S, Zhang L. Lymphoplasmacytic lymphoma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomalpl.html. Accessed September 14th, 2025.
Definition / general
- Lymphoplasmacytic lymphoma (LPL) is a B cell neoplasm of small lymphocytes, plasmacytoid lymphocytes and plasma cells
- Usually involves bone marrow, sometimes lymph nodes and spleen
- Does not fulfill the criteria for any other small B cell lymphoid neoplasm
- Waldenström macroglobulinemia (WM): lymphoplasmacytic lymphoma involving the bone marrow associated with IgM monoclonal paraprotein of any concentration in the blood
Essential features
- Lymphoplasmacytic lymphoma
- Spectrum of lymphocytes, plasmacytoid lymphocytes and plasma cells > 10% bone marrow infiltration
- Flow cytometry: monoclonal B cells with typical immunophenotype and monoclonal plasma cells
- Monoclonal IgM paraprotein by serum and urine protein electrophoresis (SPEP / UPEP), minority with IgA or IgG paraprotein
- MYD88 L265P mutation present or detection of CXCR4 somatic mutation (Virchows Arch 2016;468:259)
- Waldenström macroglobulinemia
- Serum IgM of any level
- Bone marrow involvement by small B cell lymphoma with plasmacytic differentiation
- Exclusion of other types of small B cell lymphoma
Terminology
- Lymphoplasmacytic lymphoma
- Waldenström macroglobulinemia
ICD coding
Epidemiology
- Incidence: 3 - 7 per 1,000,000 per year (U.S. and Europe) (Hemasphere 2021;5:e634)
- Median age: seventh decade
- Slight male predominance (Leuk Lymphoma 2012;53:1625)
- Higher in incidence in White people compared to other races
Sites
- Bone marrow
- Some cases involving blood, lymph node and extranodal sites manifest with splenomegaly, hepatomegaly or adenopathy, or have cutaneous, gastrointestinal, lung and central nervous system (Bing-Neel syndrome) involvement
Pathophysiology
- Genetic / molecular factors may contribute to pathogenesis of lymphoplasmacytic lymphoma
- Evidence of familial clustering in ~20% of patients
- MYD88 mutation in > 90% of cases (Blood 2014;123:1637)
- Gene at 3p22.2
- Most common mutation c794T>C results in switching of leucine to proline at codon 265 (pL265P)
- Protein MYD88 plays significant role in Toll-like receptor and interleukin 1 (IL1) receptor signaling
- Gain of function mutation product activates downstream transcription protein complex nuclear factor κ light chain enhancer of activated B cells (NFkB)
- NFkB promotes cell proliferation and survival
- MYD88 mutation is also present in significant proportion (50%) of IgM monoclonal gammopathy of unknown significance (IgM MGUS)
- Present in ~30% of nongerminal center type diffuse large B cell lymphoma
- Present in > 50% of primary cutaneous diffuse large B cell lymphoma, leg type
- In many diffuse large B cell lymphoma at immune privileged sites (e.g., central nervous system, testicle)
- 6q deletion in 40 - 60% of lymphoplasmacytic lymphoma cases
- Most common recurrent cytogenetic abnormality
- Tumor suppressor genes B lymphocyte induced maturation protein 1 (PRDM1) and TNFα induced protein 3 (TNFAIP3) are deleted
- Gene at 3p22.2
- Other cytogenetic abnormalities identified (Blood 2014;123:1637)
- Trisomy 18, 13q deletion, 17p (TP53) deletion, trisomy 4, trisomy 12 and 11q (ATM) deletion are seen in various proportions
- CXCR4 mutation in ~30% of lymphoplasmacytic lymphoma cases and 20% of IgM MGUS (Blood 2016;128:827)
- CXC chemokine receptor type 4 (CXCR4) plays critical role in lymphoplasmacytic lymphoma cells homing to bone marrow
- Almost always coexists with MYD88 mutation
- Mutation in CXCR4 gene is linked to disease progression and drug resistance
- ARID1A mutations in 17% of cases
- JAK-STAT signaling pathway may be involved
- MicroRNA-9, miRNA-155 and miRNA-206 abnormalities may contribute to pathogenesis of lymphoplasmacytic lymphoma
- CXC chemokine receptor type 4 (CXCR4) plays critical role in lymphoplasmacytic lymphoma cells homing to bone marrow
- Chronic immune stimulation
- Lymphoplasmacytic lymphoma / Waldenström macroglobulinemia risk is elevated among individuals with autoimmune disorders, particularly Sjögren syndrome and autoimmune hemolytic anemia
- Lymphoplasmacytic lymphoma / Waldenström macroglobulinemia risk is elevated in persons with hepatitis, HIV infection and rickettsiosis
- Predisposition
- MGUS of IgM type is associated with estimated annual progression rate of 1.5% to lymphoplasmacytic lymphoma
- IgM MGUS is precursor lesion to lymphoplasmacytic lymphoma
- MYD88 mutation is identified in 50% of IgM MGUS cases
- MGUS of IgM type is associated with estimated annual progression rate of 1.5% to lymphoplasmacytic lymphoma
Etiology
- Postgerminal center memory B cell is likely cell of origin
- Hepatitis C is associated in some series (Clin Lymphoma Myeloma Leuk 2020;20:e195)
- Hepatitis C associated lymphoplasmacytic proliferations are nonprogressive and may be similar to monoclonal B cell lymphocytosis
- Treating hepatitis C with antivirals may lead to regression to the lymphoplasmacytic proliferation
- Autoimmune disorders: associated with an increased risk of occurrence
Clinical features
- 20 - 30% of patients are asymptomatic at diagnosis
- Anemia related symptoms: fatigue, shortness of breath and chest pain
- Thrombocytopenia or acquired von Willebrand disease related tendency
- Constitutional symptoms, including weight loss and night sweats
- Splenomegaly and adenopathy
- High concentration of IgM may form aggregates and bind water
- IgM may cause red blood cell aggregation (rouleaux formation) and increase red cell internal viscosity
- Results in circulatory disturbances
- Most common symptoms include oronasal bleeding, headache, visual disturbances due to retinal bleeding and dizziness
- Hyperviscosity
- 15 - 30% of patients with Waldenström macroglobulinemia have hyperviscosity (in particular when IgM level reaches ≥ 6 g/dL) (Br J Haematol 2017;177:717)
- Worse clinical scenarios could occur when IgM ≥ 5 g/dL
- Hyperviscosity syndrome
- Due to IgM's large size and quantity
- Symptoms
- Visual impairment (distended and tortuous retinal veins with hemorrhage and exudates)
- Neurologic: slowing blood flow
- Bleeding: IgM binding of clotting factors and cryoglobulinemia causing Raynaud phenomena and cold urticaria
- The following findings can be associated with bone marrow infiltration, cryoprecipitation, autoimmune or nonautoimmune antibody activity
- Cytopenia
- Cold agglutinin hemolytic anemia (10% of cases) (IgM antibodies that bind at < 37 °C)
- Peripheral neuropathy
- Primary amyloidosis
- No lytic bone lesion distinguishes it from plasma cell myeloma
- May transform to diffuse large B cell lymphoma with Reed-Sternberg-like cells or immunoblasts
- Reference: Blood Rev 2015;29:301
Diagnosis
- Diagnostic criteria for Waldenström recommended by Second International Workshop (Semin Oncol 2003;30:110)
- IgM monoclonal gammopathy of any concentration
- Bone marrow infiltration by small lymphocytes, plasmacytoid lymphocytes or plasma cells
- Combined paratrabecular and interstitial pattern of bone marrow infiltration
- Frequently associated with reactive mast cells and hemosiderin laden macrophages
- Lymph node involvement by LPL usually in a diffuse pattern, loaded with monotonous small lymphoid, plasmacytoid lymphocytes and plasma cells
- IgM monoclonal gammopathy of any concentration
- Essential and desirable diagnostic criteria
- Essential
- Bone marrow infiltration by > 10% small lymphocytes with plasmacytoid or plasma cell differentiation
- Immunophenotype of LPL cells: IgM+, CD19+, CD20+, CD22+, CD25+, CD10-, CD23-, CD103-, variable CD138
- Desirable
- Detection of MYD88 (NP_002459.2:p.L265P) (93 - 97% of LPL / WM)
- Detection of CXCR4 somatic mutation
- Serum electrophoresis and immunofixation showing presence of monoclonal Ig
- Essential
- International Consensus Committee 2022 (Blood 2023;141:437)
- Diagnosis of lymphoplasmacytic lymphoma can be rendered in cases of abnormal lymphoplasmacytic aggregates in the bone marrow and evidence of clonal plasma cells and clonal B cells, even when the aggregates are < 10% of the trephine biopsy (Blood 2023;141:437)
- Immunophenotype
- Positive: IgM, CD19, CD20, CD22, CD79a, CD25 and CD38 frequent expression
- Negative: CD5, CD10, CD103, CD23; however, CD23 expression is not uncommon in some cases
- CD5 and CD10 positive but BCL6 negative in a minority of cases
- Plasma cells in lymphoplasmacytic lymphoma
- Positive: CD138, CD19, CD45 and sometimes MUM1 but can be negative
- Can be PAX5 positive
Laboratory
- IgM monoclonal paraprotein is commonly present in serum, rarely IgG or IgA
- Lymphoplasmacytic lymphoma
- IgM paraprotein not required for diagnosis
- IgA or IgG paraprotein alone or coexists with IgM in serum (rare)
- Waldenström macroglobulinemia
- IgM paraprotein required for diagnosis
- No cutoff level for IgM in serum
- Lymphoplasmacytic lymphoma
- Other abnormal tests with Waldenström macroglobulinemia
- Elevated erythrocyte sedimentation rate (ESR), cytopenia / anemia
- Elevated serum levels of lactate dehydrogenase (LDH) and beta 2 microglobulin
- Reference: Blood 2015;126:721
Prognostic factors
- Indolent clinical course but incurable
- Adverse prognostic factors (International Prognostic Scoring System) (Blood 2009;113:4163)
- > 65 years old
- Hemoglobin < 11.5 g/dL
- Platelet count < 100 x 109/L
- Monoclonal IgM > 7 g/dL
- Increased beta 2 microglobulin > 3 mg/L
- Overall survival varies from 5 - 11 years depending on the study; median survival is 5 years, 40% survive 10 years or more or die of unrelated causes (Blood 2009;113:4163, Am J Hematol 2013;88:60)
- Low risk (≤ 65 years, 0 - 1 risk factor) 5 year survival rate of 87%
- High risk (> 65 years, > 2 risk factors) 5 year survival rate of 36%
- Transformation to large cell lymphoma can occur (13%) (Blood Cancer J 2015;5:e394, Am J Clin Pathol 2003;120:246)
- Relapse: inevitable, can occur years after initial treatment
- Main disease related causes of death
- Disease progression
- Transformation to high grade lymphoma
- Complications of therapy
Case reports
- 43 year old woman with headaches; MRI revealed an abnormal homogeneously enhancing mass in the cerebellopontine angle (Medicine (Baltimore) 2016;95:e4627)
- 53 year old woman diagnosed with relapsed lymphoplasmacytic lymphoma complicated by Bing-Neel syndrome who was successfully managed with tirabrutinib (Int J Hematol 2022;115:585)
- 60 year old man with fatigue, sensory neuropathy and lab findings of anemia and elevated albumin globulin gap (Case Rep Hematol 2019;2019:4075960)
- 65 year old man who presented with fatigue and decreased renal function was diagnosed with acute kidney injury due to direct infiltration by lymphoplasmacytic lymphoma secreting IgG paraproteins (Medicine (Baltimore) 2022;101:e29449)
- 69 year old man presented with altered mental status and was diagnosed with intravascular large B cell lymphoma secondary to lymphoplasmacytic lymphoma (Int J Clin Exp Pathol 2015;8:3339)
Treatment
- No standardized treatment
- Asymptomatic patients: watch and wait
- Therapy initiated in patients with
- Constitutional symptoms: fever, weight loss, night sweats, fatigue
- Hyperviscosity
- Severe neuropathy
- Amyloidosis
- Symptomatic cryoglobinemia
- Cold agglutinin disease
- Evidence of transformation
- Treatment regimen / options (Leukemia 2014;28:1799, J Clin Oncol 2009;27:120)
- Single agent
- Combination therapy
- Monoclonal antibodies
- Proteasome inhibitors
- Immunomodulatory agents
- Signaling pathway inhibitors
- Histone deacetylase
- First line treatment
- Anti-CD20 antibody in combination with a purine analogs or alkylating agent
- Rituximab in combination with thalidomide or bortezomib and dexamethasone
- Rituximab with bendamustine
- Plasmapheresis: reduce circulating IgM in hyperviscosity patients
- Splenectomy: chemoresistant patient
- Stem cell transplant (Blood 2015;126:721)
- Autologous stem cell transplantation is feasible, safe and associated with significant cytoreduction in relapsed or refractory patient
- Allogeneic stem cell transplantation is used only in patients with advanced and refractory disease for whom no other options are available
- Investigational therapies (Blood 2015;126:721)
- Agents target MYD88, CXCR4, BCL2, CD27 / CD70 signaling
- Novel proteasome inhibitors
- Chimeric antigen receptor modified T cell therapy
- Please follow the consensus treatment recommendation from the tenth International Workshop for Waldenström macroglobulinemia (Lancet Haematol 2020;7:e827)
- Preferred treatment options
- Bendamustine plus rituximab
- Bortezomib, dexamethasone and rituximab
- Cyclophosphamide, dexamethasone and rituximab
- Ibrutinib (with or without rituximab)
- Treatment recommendations
- Avoid bortezomib and vincristine in patients with neuropathy
- Avoid carfilzomib in patients with cardiac disease or patients who are older than 65 years
- Avoid nucleoside analogues in patients who are candidates for stem cell transplantation
- Consider delaying rituximab if serum IgM concentrations are > 4000 mg/dL
- Consider ofatumumab in patients who are intolerant to rituximab
- Other treatment options
- Acalabrutinib
- Carfilzomib, dexamethasone and rituximab
- Fludarabine and rituximab
- Ixazomib, dexamethasone and rituximab
- R-CHOP
- R-CVP
- Rituximab
- Ofatumumab
Microscopic (histologic) description
- Blood
- Often shows lymphocytosis with a spectrum of lymphocytes, plasmacytoid lymphocytes and plasma cells
- Rouleaux formation of red blood cells is routinely seen
- Cold agglutinin or cryoglobulin may be present
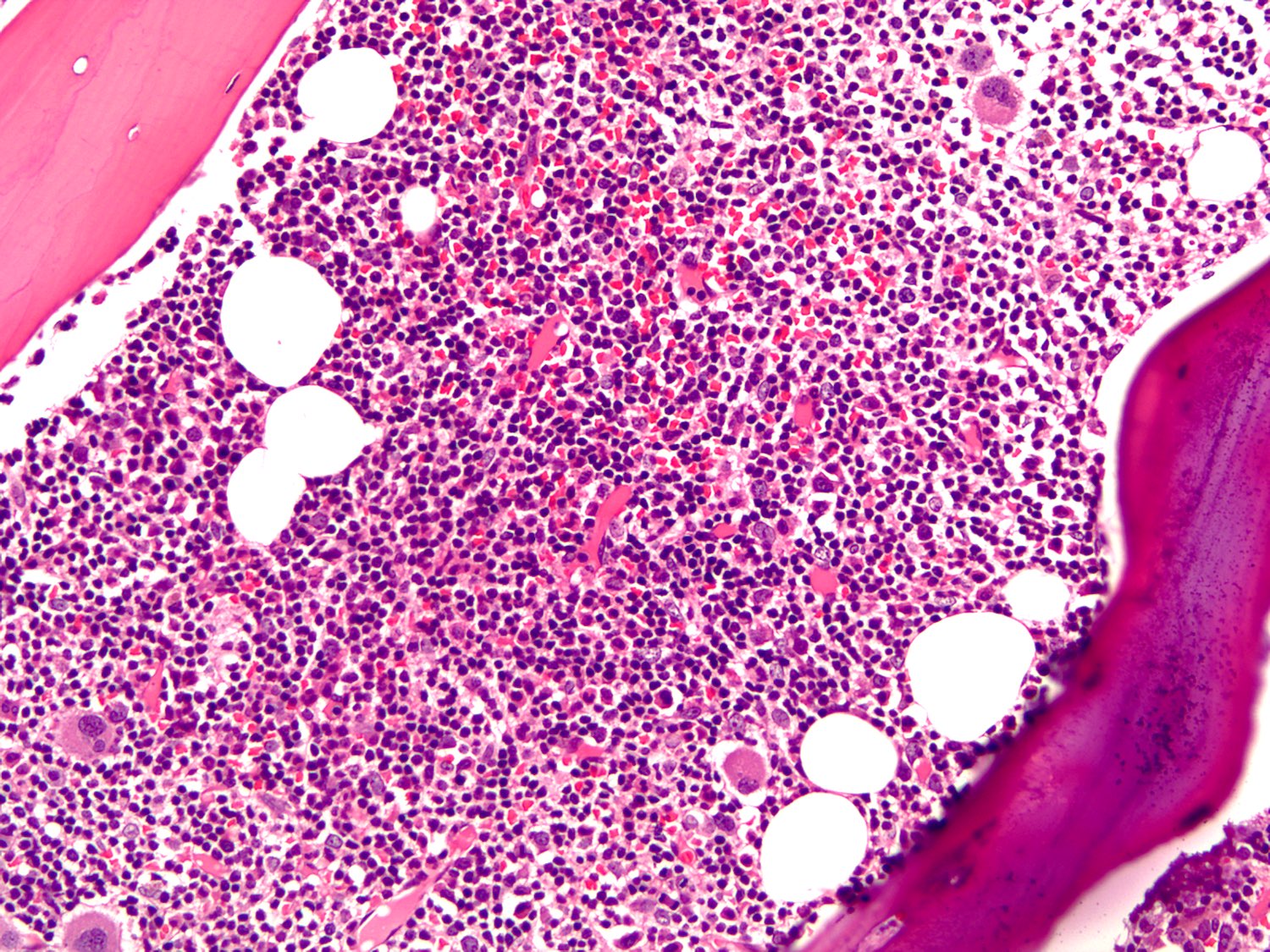
- Bone marrow (Arch Pathol Lab Med 2013;137:580)
- Involvement in almost all cases
- Aspirate is the most helpful sample in morphologic diagnosis
- Predominantly small lymphocytes with a variable number of plasmacytoid lymphocytes and plasma cells
- Mast cells are typically increased; most prominent within particles on aspirate smears
- Core biopsy (Am J Surg Pathol 2005;29:1549, Am J Clin Pathol 2015;143:797)
- Most common pattern is the combination of paratrabecular and nonparatrabecular lymphoid nodules
- Less common patterns include paratrabecular or intrasinusoidal with or without interstitial infiltrate
- Pseudointranuclear (Dutcher bodies) and intracytoplasmic inclusions (Russell bodies) are most prominent on core biopsy section
- Amyloid deposition may be present within vessel walls or interstitial deposits
- Lymph node (Arch Pathol Lab Med 2013;137:580)
- 2 main patterns of nodal involvement
- Classic pattern
- Subtotal architecture effacement
- Retention of small primary or enlarged reactive follicles
- Patent dilated sinuses
- Interfollicular areas contain monomorphous infiltrate of small lymphocytes, plasmacytoid lymphocytes and plasma cells
- Dutcher bodies, increased number of mast cells and hemosiderin deposition may be present
- Polymorphous pattern
- Complete architectural effacement
- Nodular to diffuse polymorphous infiltrate
- Small lymphocytes
- Plasmacytoid lymphocytes
- Plasma cells
- Greater numbers of large transformed cells
- Clusters of epithelioid histiocytes may be conspicuous
- Termed polymorphous immunocytoma
- Extracellular immunoglobulin in the form of amyloid may be present
- Classic pattern
- 2 main patterns of nodal involvement
- Spleen (Am J Surg Pathol 2003;27:1104)
- Not well described
- Older published series and review
- Presence of nodular and diffuse infiltrates of lymphoplasmacytic cells involving red pulp
- Spectrum of lymphoma cells analogous to bone marrow
- Sparing of white pulp with absence of marginal zone growth pattern
Microscopic (histologic) images
Contributed by Ling Zhang, M.D.

Increased lymphoid cells
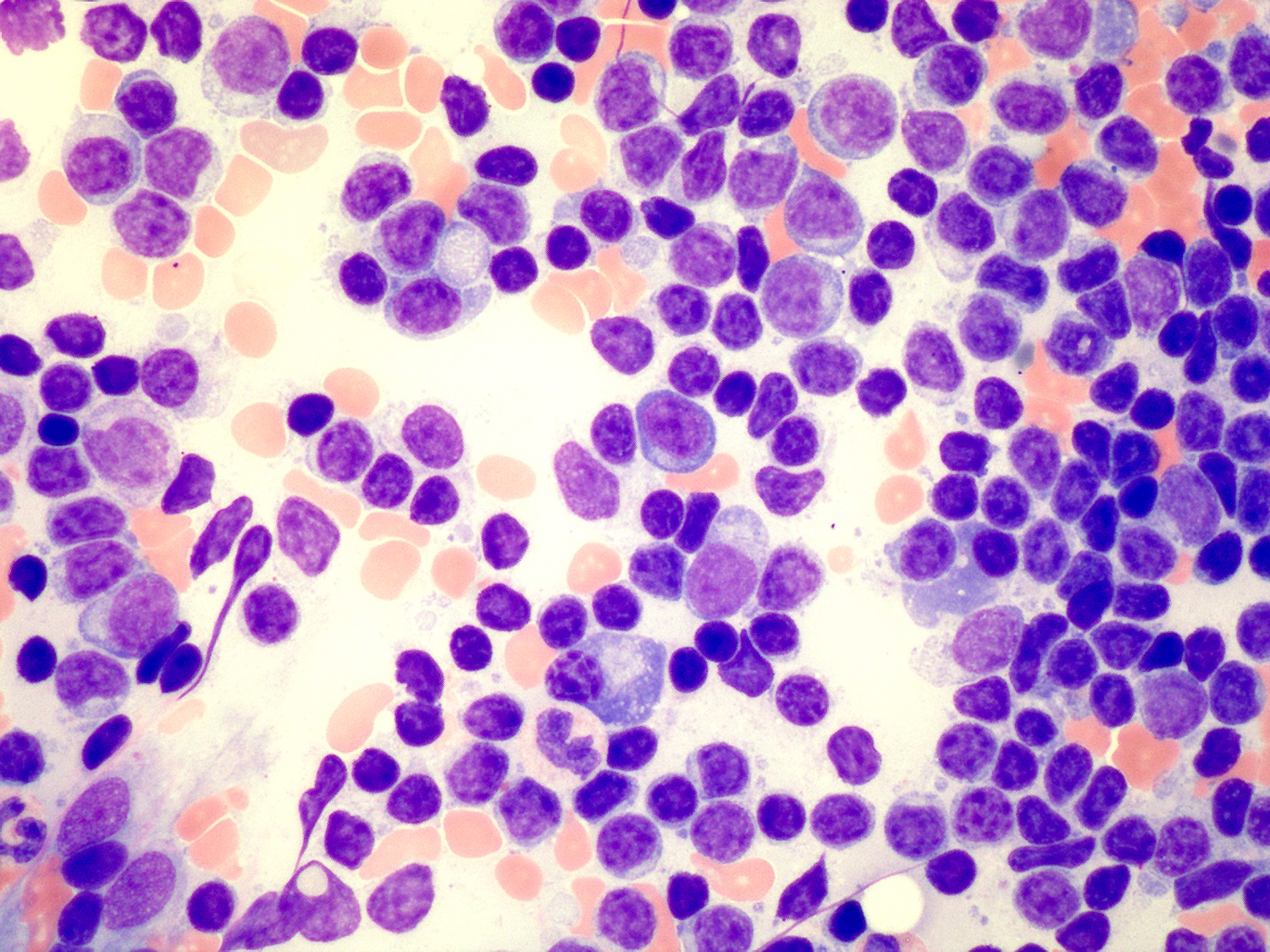
Round eccentrically located nuclei
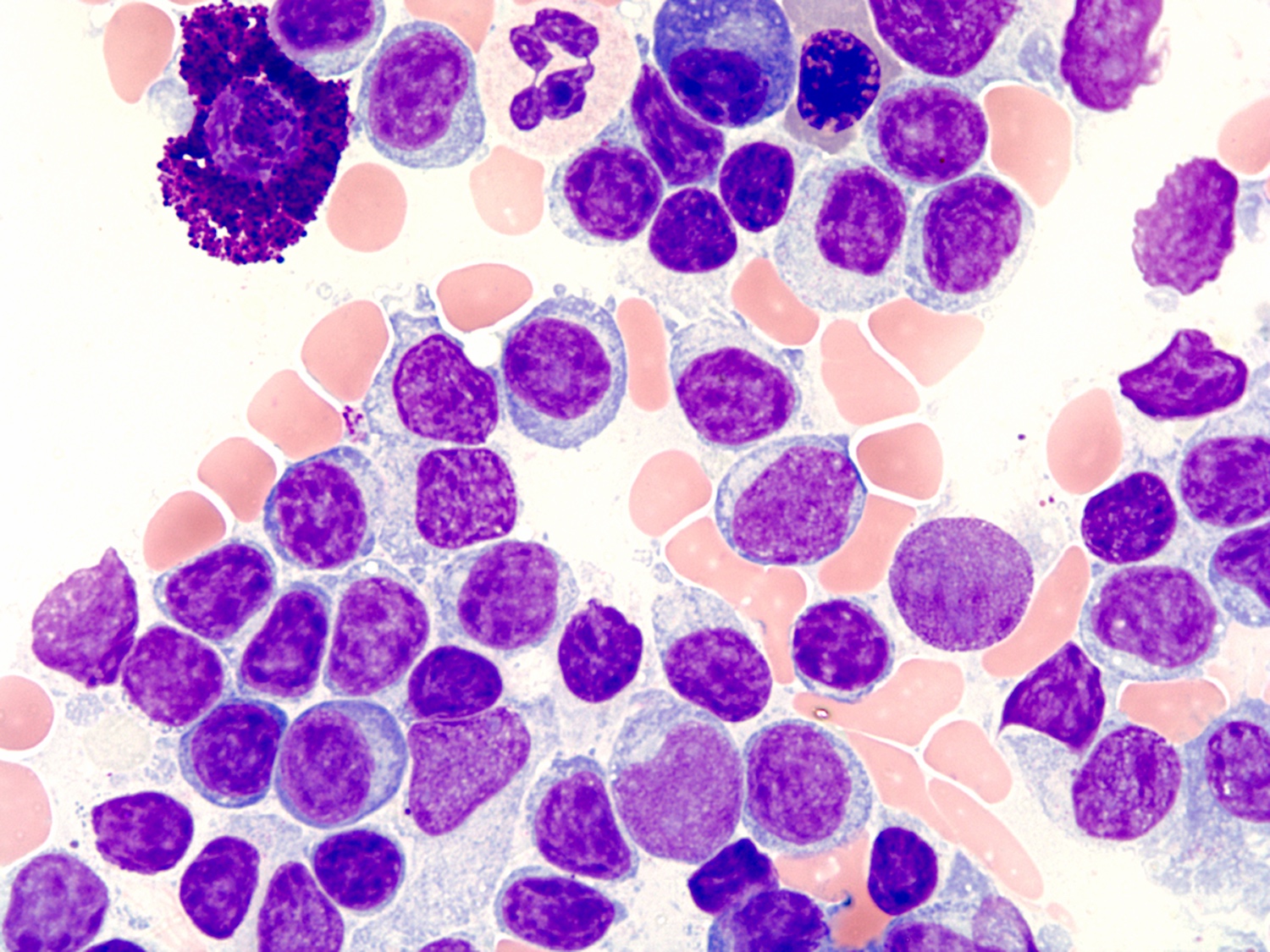
Condensed chromatin, inconspicuous nucleoli
Small mature lymphoid cells

Intermingled plasma cells
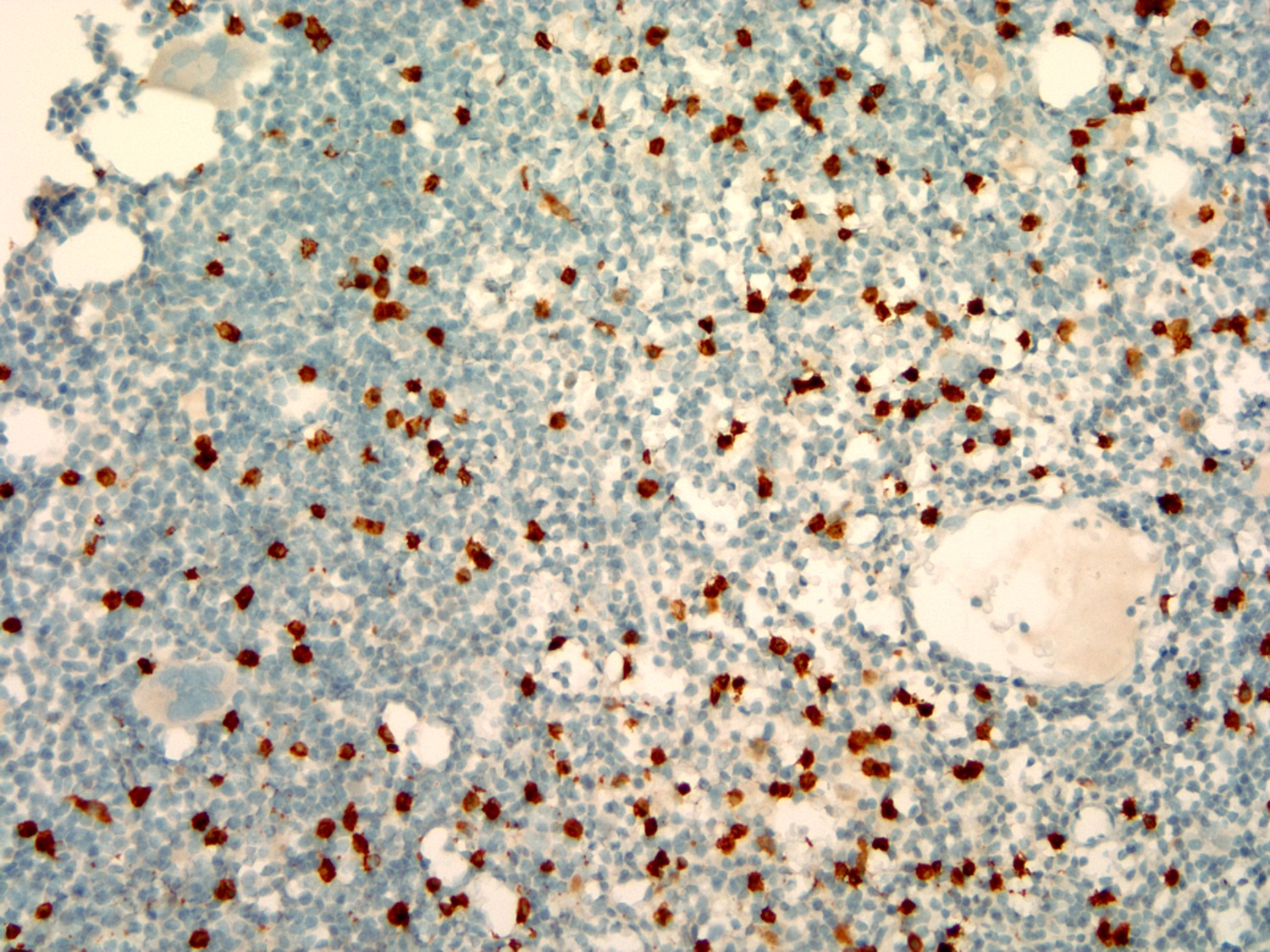
Lymphoid cells negative for CD3
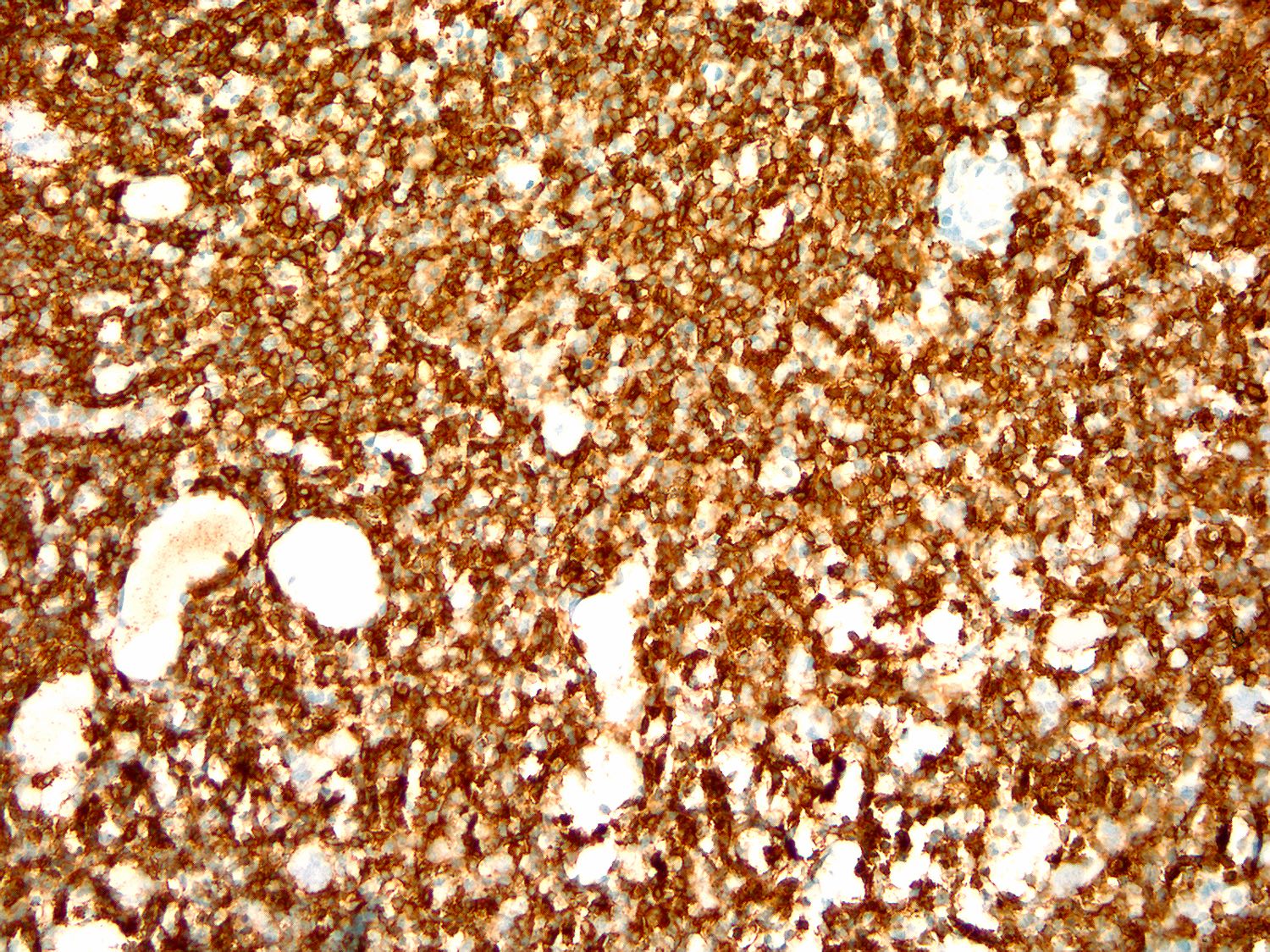
Diffuse CD20 positivity
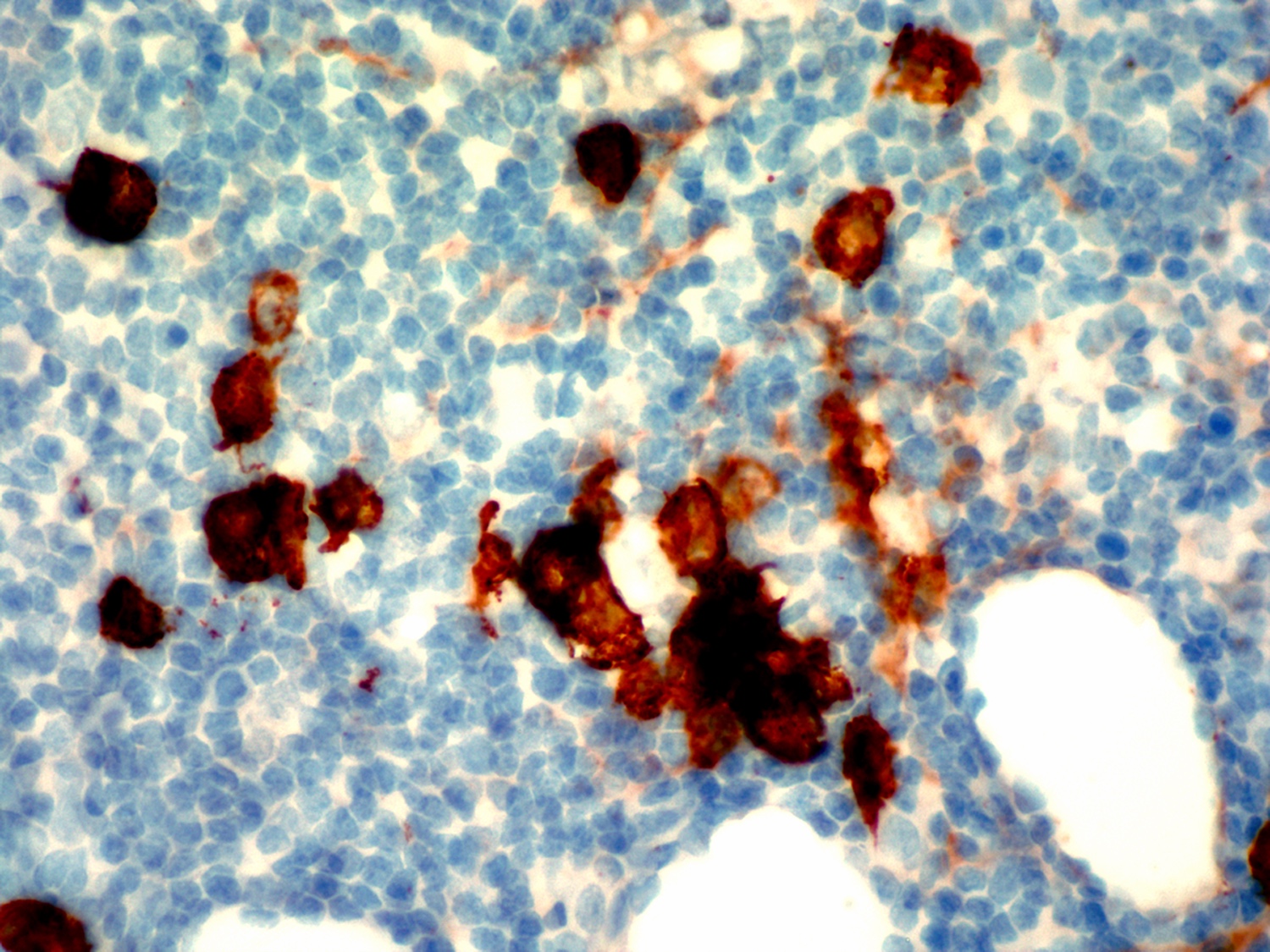
Focal plasma cell positivity (CD138)
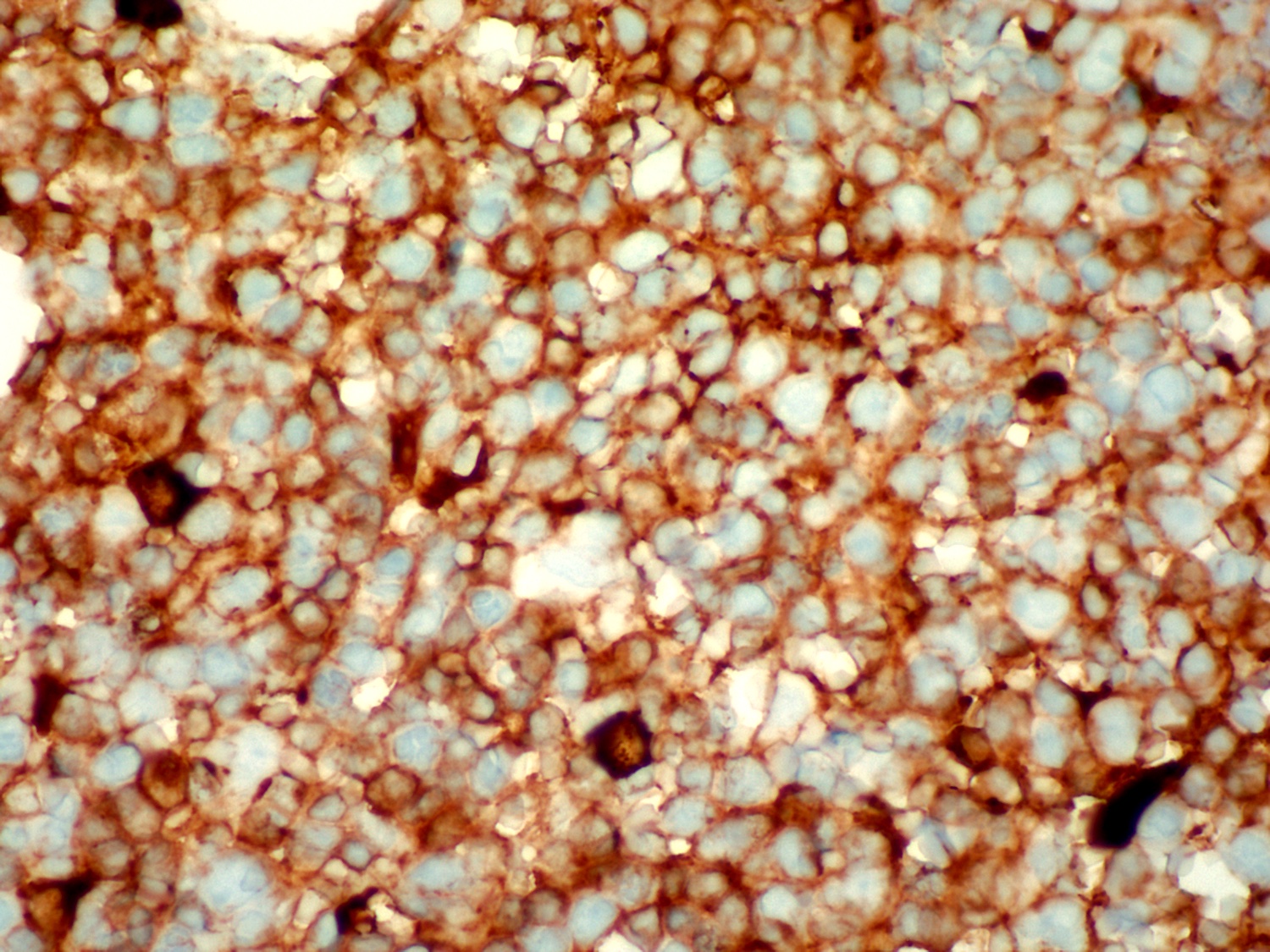
Immunoglobulin M positivity
Positive stains
Flow cytometry description
Flow cytometry images
Contributed by Ling Zhang, M.D. and Caroline An, M.D.
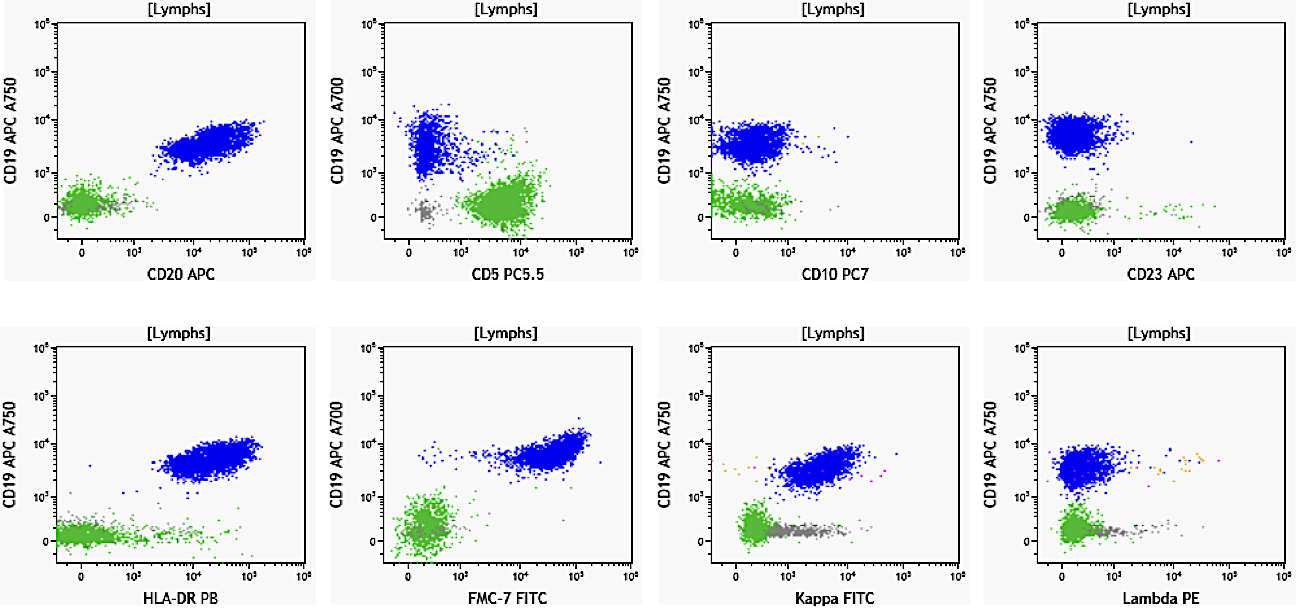
Kappa clonal population of B cells
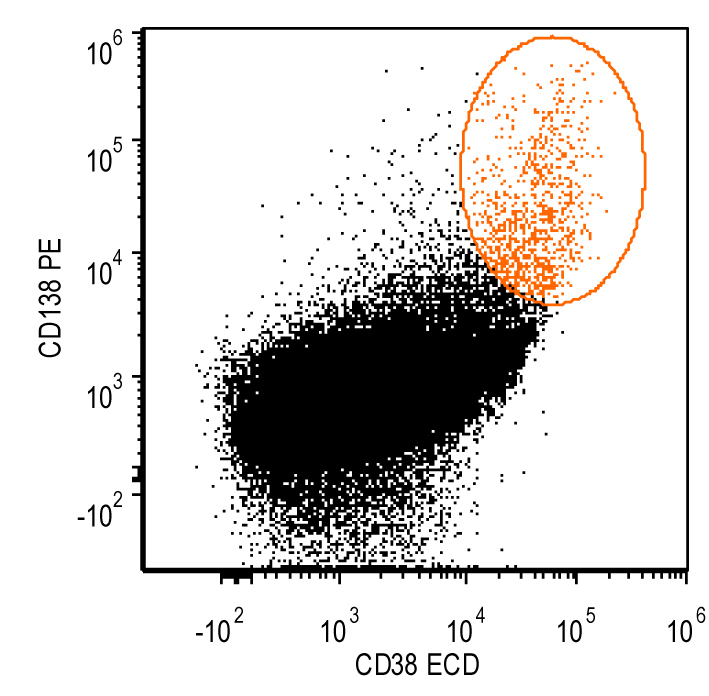
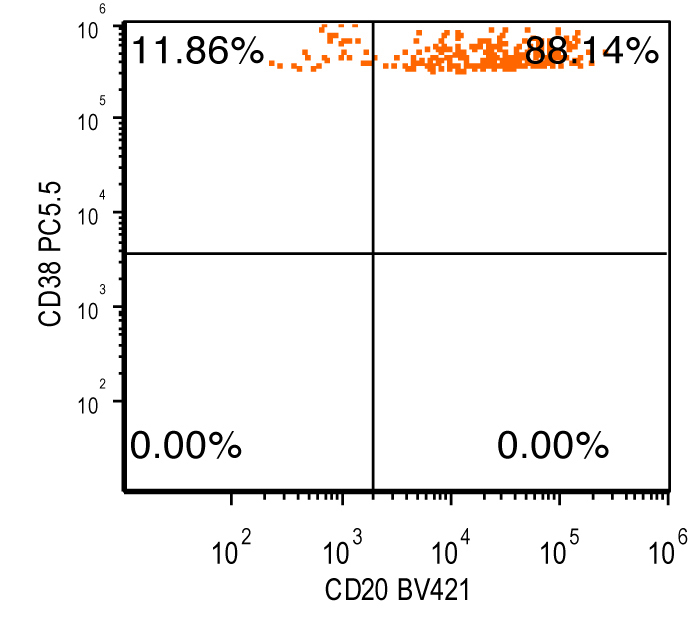
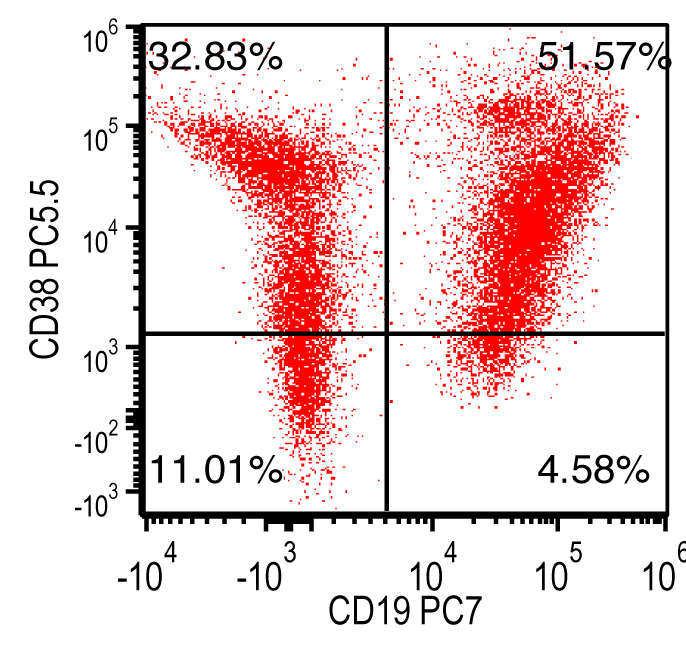
CD38+ / CD138+ plasma cells
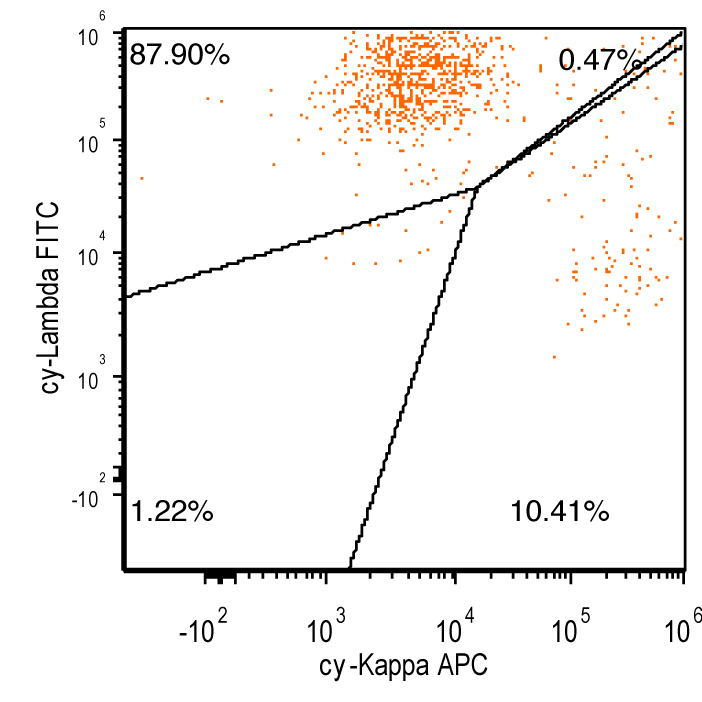
Lambda restriction
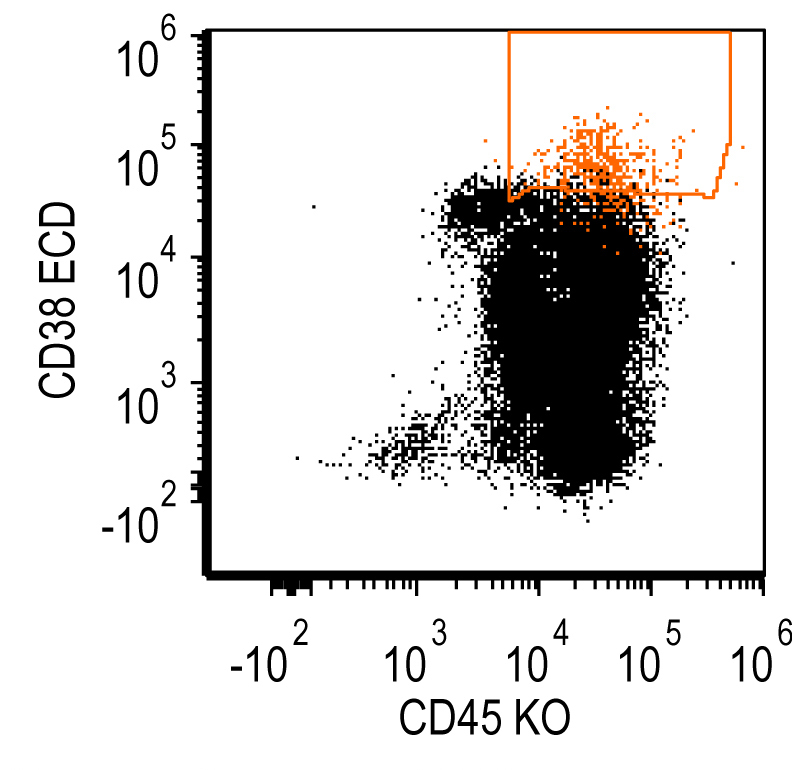
Plasma cells express CD45
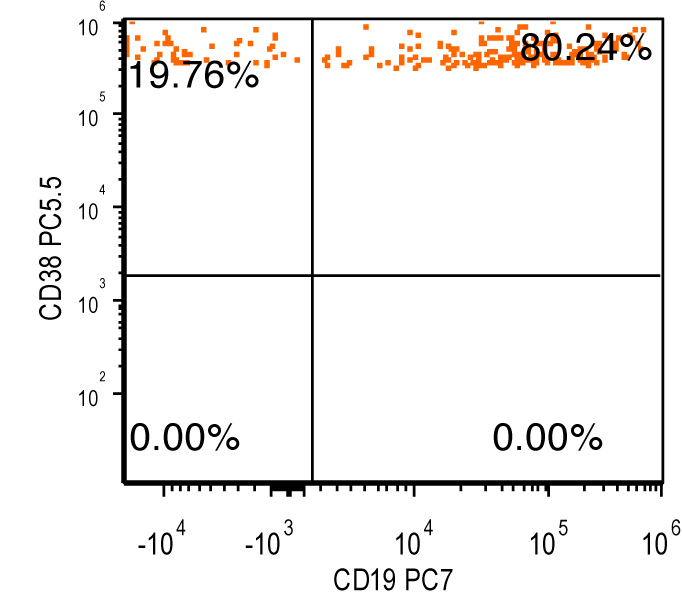
Presence of CD19
Presence of CD20 expression
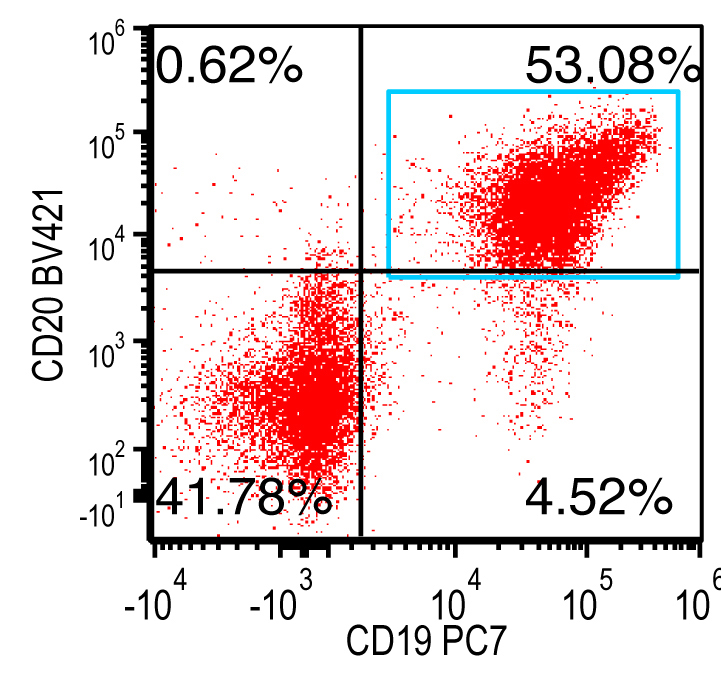
B cells expressing both CD19 and CD20
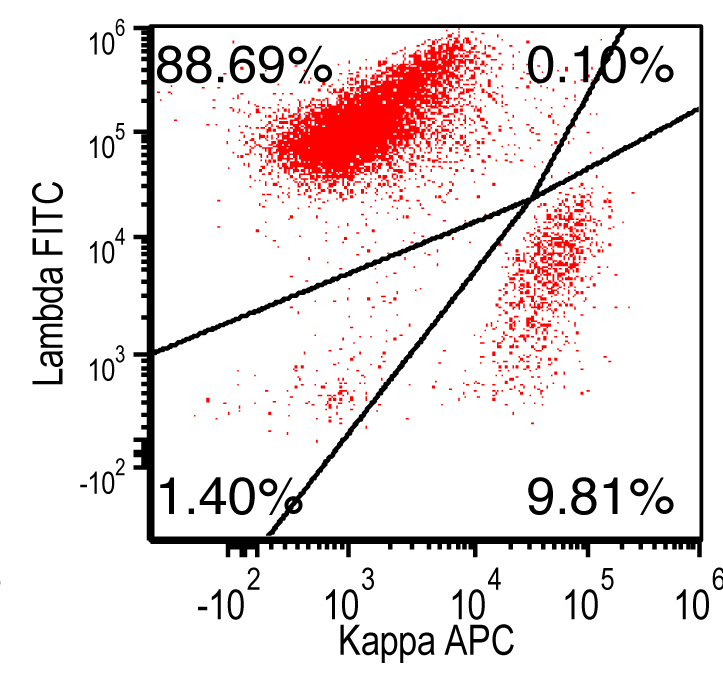
B cells are lambda monoclonal
B cells show variable expression for CD38
Molecular / cytogenetics description
- MYD88 mutation in > 90% of cases; increasingly used in diagnostic workup
- CXCR4 mutation; not routinely tested
- Cytogenetic study is not required for diagnosis; when done, 6q deletion is reported in 40 - 50% of patients
- IGH gene rearranged
- Biased VH3 and VH3-23 usage
- Reference: Blood 2014;123:1637
Molecular / cytogenetics images
Contributed by Ling Zhang, M.D.

ISH clonal kappa light chain expression
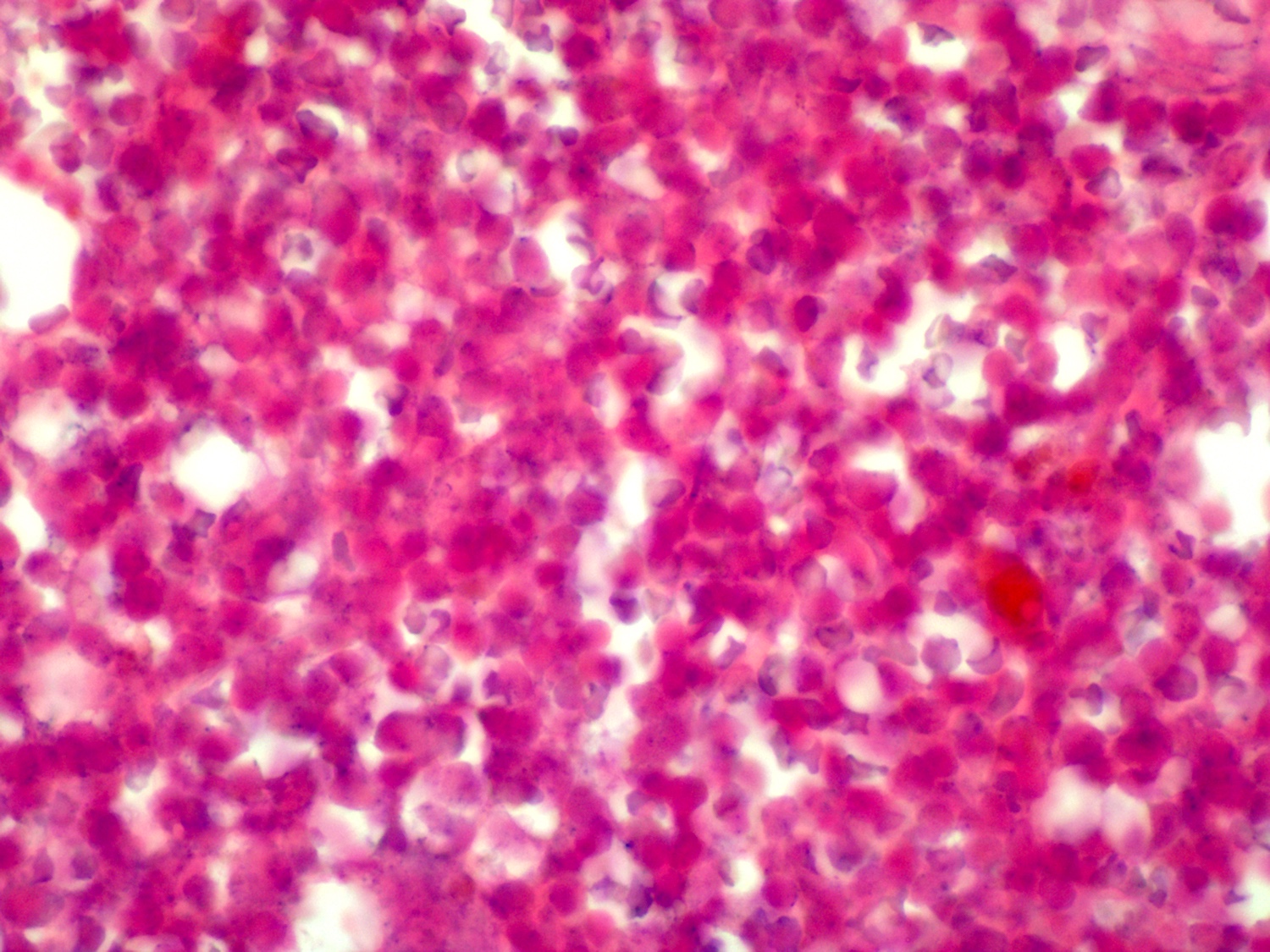
Negative lambda light chain expression
Sample pathology report
- Bone marrow, right posterior iliac crest, aspirate and biopsy:
- Peripheral blood: The red blood cells are normocytic and normochromic with mildly decreased hemoglobin level (11.8 g/dL). The reticulocyte count is adequate. There is no overt rouleaux formation. The white blood cell count is within normal range. Lymphocytes are not increased; however, few atypical lymphocytes are seen, which are mostly small in size, with slightly irregular nuclear contour and mature chromatin or plasmacytoid morphology. Platelets are mildly decreased in number with normal morphology.
- Hypercellular marrow with extensive involvement by lymphoplasmacytic lymphoma (see comment)
- Aspirate smear: The Wright stained bone marrow aspirate smears are cellular, with adequate spicules included for evaluation. Megakaryocytes are decreased in number, when identified, showing normal morphology. Myeloid and erythroid precursors show progressive maturation but are relatively decreased in number. The estimated M:E ratio is 4:1. The cellularity is composed of mostly atypical lymphocytes (84% of differential count). These atypical lymphocytes are predominantly small in size, with oval or slightly irregular nuclear contours, mature chromatin and small amount of basophilic cytoplasm. Rare atypical lymphocytes are medium in size, with plasmacytoid differentiation. Plasma cells account for 4.8% of the differential count. Scattered mature mast cells are present in the background.
- Core biopsy and cell clot: The bone marrow core biopsy includes normal appearing trabecular bone and marrow, adequate for morphologic evaluation. The marrow cellularity is very high, up to 95%. Megakaryocytes are essentially normal in number with normal morphology. Myeloid and erythroid precursors are markedly decreased in number. The marrow is diffusely infiltrated by atypical lymphocytes, accounting for approximately 80% of total cellularity, a small subset of which shows plasmacytoid differentiation. Mature plasma cells are identified. Iron stain shows adequate stainable storage iron. Reticulin stain highlights mild reticulin fibrosis. The clot section contains a few small cellular particles including predominantly atypical lymphoid cells, with findings similar to the core biopsy.
- Immunohistochemical study: Immunohistochemical stains and in situ hybridization stains are performed on the core biopsy with adequate controls. CD20 and PAX5 highlight a diffuse infiltration of neoplastic B cells, occupying 80% of total cellularity. CD3 highlights scattered interstitial T lymphocytes, ranging from 2% to 5%. CD138 highlights scattered plasma cells, focally forming small clusters, accounting for approximately 5% of the total cells. By in situ hybridization (ISH), the plasma cells are positive for kappa light chain and negative for lambda light chain.
Differential diagnosis
- IgM monoclonal gammopathy of undetermined significance (IgM MGUS):
- All 3 criteria must be met
- Serum IgM monoclonal protein < 3 gm/dL
- Bone marrow lymphoplasmacytic infiltration < 10%
- No evidence of anaemia, constitutional symptoms, hyperviscosity, lymphadenopathy or hepatosplenomegaly that can be attributed to the underlying lymphoproliferative disorder
- All 3 criteria must be met
- IgG and IgA MGUS:
- More closely related to plasma cell myeloma
- Negative for MYD88
- Chronic lymphocytic leukemia (CLL) / small lymphocytic lymphoma:
- B cell lymphoma composed of small mature forms
- Often coexpresses dim CD20, CD5 and CD23 by flow cytometry
- Frequent nodal presentation with generalized lymphadenopathy, vaguely nodular proliferation with identifiable pseudoproliferation centers; the latter is lacked in LPL / WM
- Frequent peripheral involvement, mature lymphocytosis with clumped chromatin (soccer ball chromatin pattern), > 5,000/uL neoplastic B cells, CLL phenotype is required to diagnose CLL
- Usually negative for MYD88 mutation
- Marginal zone lymphoma (MZL):
- B cell lymphoma with monocytoid or plasmacytoid differentiation
- Immunophenotypically similar to LPL / WM, triple negative for CD5, CD10 and CD23
- Bone marrow involvement: mostly interstitial and intrasinusoidal infiltration pattern
- Lymph node, spleen and extranodal involvement might not be easy to distinguish it from LPL / WM
- Presence of MYD88 L265P mutation favors LPL / WM over MZL
- Plasmacytoid differentiation with Dutcher bodies is less frequently in MZL than in LPL
Additional references
Practice question #1
Which of the following immunoglobulins is most commonly seen in lymphoplasmacytic lymphoma?
- IgA
- IgD
- IgE
- IgG
- IgM
Practice answer #1
E. IgM. More than 90% of patients with LPL / WM show an overt elevated IgM level. Answer A is incorrect because non-IgM LPL / WM, mainly IgM or IgA, is reported in ~5% of cases. Answer D is incorrect because concurrent IgM and IgG LPL is rarely reported. Answers B and C are incorrect because IgD and IgE LPL / WM might occasionally exist but have not been well reported.
Comment Here
Reference: Lymphoplasmacytic lymphoma
Comment Here
Reference: Lymphoplasmacytic lymphoma
Practice question #2
What somatic mutation is associated with lymphoplasmacytic lymphoma and can aid in diagnosis?
- BCL2
- BRAF
- KRAS
- MYD88 L265P
Practice answer #2
D. MYD88 L265P. MYD88 is considered the driver mutation and detected in 93 - 97% of patients with LPL / WM. The particular mutation leads to constitutive activation of NFkB pathway. While answer D is correct, be cautious, as the presence of MYD88 is not 100% specific for LPL / WM. The mutation has also been found in monoclonal gammopathy of unknown significance (MGUS), diffuse large B cell lymphoma, activated B cell (DLBCL ABC) type, etc. Integrating with other clinical and pathologic results is necessary for an accurate diagnosis. Answers A - C are incorrect because other mutations are infrequently reported in LPL / WM and are not helpful for diagnosis.
Comment Here
Reference: Lymphoplasmacytic lymphoma
Comment Here
Reference: Lymphoplasmacytic lymphoma
