
Bone marrow neoplastic
Bone marrow - plasma cell and lymphoid neoplasms
Other
Primary amyloidosis
Author: Meenakshi Garg Bansal, M.D.

Last author update: 1 April 2018
Last staff update: 22 November 2023
Copyright: 2001-2024, PathologyOutlines.com, Inc.
PubMed Search: Primary amyloidosis lymphoma
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Bansal M. Primary amyloidosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomaprimaryamyloid.html. Accessed April 24th, 2024.
Definition / general
- Light chain (AL) amyloidosis is the most common form of systemic amyloidosis (Clin J Am Soc Nephrol 2006;1:1331)
- Monoclonal immunoglobulin deposition disease with visceral and soft tissue Ig deposition resulting in organ dysfunction
- Underlying plasma cell dyscrasia or (less commonly, 5 - 7%) lymphoplasmacytic neoplasm (Expert Rev Hematol 2018;11:117)
- Fibrils consist of whole or fragments of immunoglobulin light chains
- Form beta pleated sheets (AL amyloid) in tissue that bind Congo red dye and show characteristic apple green birefringence in polarized light (Clin J Am Soc Nephrol 2006;1:1331)
Essential features
- M protein is found in the serum or urine in > 80% of patients
- Diagnostic criteria for myeloma met in a subset of cases (12 - 15%), typically Ig molecule accumulates before there is a large tumor burden (N Engl J Med 2003;349:583)
- Primary amyloidosis and light chain and heavy chain deposition disease (nonamyloid immunoglobulin deposition in tissues) appear to be chemically different manifestations of similar pathologic processes
- Rare and may present with a broad range of symptoms, some vague, making it challenging to recognize (Clin J Am Soc Nephrol 2006;1:1331)
Terminology
- Accepted nomenclature for Amyloid is AX, where A stands for Amyloid and X represents the protein in the fibril
- Historically light chain amyloidosis (AL) referred to as primary amyloidosis
- Overall, > 30 different proteins can cause amyloidosis (Blood 2009;114:4957, Clin J Am Soc Nephrol 2010;5:2180)
- Those involving kidney recently grouped into the category of monoclonal gammopathies of renal significance (MGUS) to separate them from MGUS based on need for treatment (Blood 2012;120:4292)
ICD coding
Epidemiology
- Incidence: approximately 1 case per 100,000; approximately 3,000 cases in the U.S. per year
- Most common type of systemic amyloidosis in developed countries (75%) (Blood 2016;128:159)
- Median age 64, > 95% are older than 40
- Predominant in men (65 - 70%)
Sites
- Protean clinical presentation due to widespread organ involvement, involving all except possibly the brain (Expert Rev Hematol 2018;11:117)
- Survival mostly determined by extent of cardiac involvement (Blood 2016;128:159)
Pathophysiology
- Abnormal folding of a protein that is normally soluble as a result of a proteolytic event or mutation that renders it unstable / prone to self aggregation (Clin J Am Soc Nephrol 2006;1:1331)
- Aggregates form protofilaments that associate into amyloid fibrils
- Interactions with the extracellular environment may result in proteolytic cleavage and matrix components such as glycosaminoglycans and collagen may serve as scaffolds to facilitate aggregation and fibril buildup (Sci Rep 2016;6:29096)
- Only a subset of Ig light chains are amyloidogenic (12 - 15% of myeloma) (N Engl J Med 2003;349:583)
- Repertoire of variable region germline genes involved in systemic amyloidosis is restricted and certain gene usage may affect organ targeting; e.g. use of IGVL1-44 had 5 fold odds of dominant cardiac involvement (Blood 2012;119:144)
- Amyloid precursor proteins directly impair cardiac function independent of fibril formation, invoking oxidative stress, cellular dysfunction, apoptosis (Proc Natl Acad Sci U S A 2010;107:4188)
Etiology
- Clonal expansion of plasma cells or low grade lymphoproliferative disorder secreting monoclonal immunoglobulin light chains which misfold, aggregate and deposit as interstitial fibrils of amyloid
Clinical features
- ~30% present with advanced, irreversible organ damage (Blood 2016;128:159)
- General: dyspnea, fatigue, weight loss
- GI: macroglossia (highly suggestive of AL but only present in 10 - 15%), hepatomegaly (25 - 30%), malabsorption (5%)
- Renal: nephrotic syndrome (28%)
- Heart: restrictive cardiomyopathy, congestive heart failure (17%), amyloid precursor proteins with direct cardiotoxic effect (Proc Natl Acad Sci U S A 2010;107:4188)
- Neural: peripheral sensory neuropathy (17%), carpal tunnel syndrome (21%), autonomic dysfunction
- Bone pain (5%), periorbital or facial purpura
- Hemorrhage: can result from factor X binding to amyloid (Blood 2001;97:1885)
- Patients with underlying IgM-secreting lymphoplasmacytic lymphoma are typically older, higher prevalence of neuropathy and lymph node involvement and lower level of cardiac involvement than non IgM AL amyloidosis (Expert Rev Hematol 2018;11:117)
Diagnosis
- Tissue diagnosis is the gold standard
- Laser microdissection with analysis by mass spectrometry is the most reliable method of characterizing amyloid type (Blood 2009;114:4957)
- Abdominal fat pad aspirate: most common and easily accessible tissue (positive in more than 80% of patients with primary amyloidosis)
- Rectal biopsies diagnostic in 80% of cases
- Biopsy of kidney, heart, liver
- Bone marrow: 60% of cases show amyloid; detection improved if adequate sized vessels present
Laboratory
- Serum and urine protein electrophoresis and immunoelectrophoresis identifies M spike (99% of cases); IgG most frequent (followed by light chain only, IgA, IgM and IgD); 80% lambda (Expert Rev Hematol 2018;11:117)
- Critical to test for free light chain as some patients lack circulating intact immunoglobulin
- Nephrotic range proteinuria and hypoalbuminemia with renal involvement
- Elevated erythrocyte sedimentation rate (ESR)
- Elevated brain natriuretic peptide (BNP), proBNP and troponin in cardiac involvement
- Cholestasis with elevated alkaline phosphatase with liver involvement
- AL / primary amyloidosis may also present with hypothyroidism / hypoadrenalism or hypopituitarism
- Scintigraphy with radiolabeled serum amyloid P(SAP) can be used to test for amyloid where SAP binds to the amyloid and reveals its presence (Annu Rev Med 2006;57:223)
Radiology description
- Bone lesions restricted to patients with myeloma
- Uncommon to see splenomegaly or lymphadenopathy
Prognostic factors
- Extent of cardiac involvement a major determinant of outcome and most frequent cause of death (40% of cases)
- Higher risk: monoclonal plasma cells > 10% bone marrow involvement, high serum free light chain, elevated beta-2-microglobulin, multiple organ involvement, elevated uric acid level
- Median survival time of 12 months if untreated
- Multiple staging systems have been proposed based on cardiac markers or renal function (Blood 2016;128:159)
Case reports
- 18 year old man with primary AL (kappa light chain) amyloidosis manifesting as peripheral neuropathy (Clin Neuropathol 2005;24:118)
- 27 year old woman with primary systemic amyloidosis (Cent Eur J Immunol 2014;39:61)
- 70 year old woman with undiagnosed primary amyloidosis unmasked during surgical treatment (J Endocr Soc 2018;2:112)
Treatment
- Antiplasma cell chemotherapy to reduce concentration of toxic light chains based on myeloma regimens
- Myeloma treatment regimens are discussed in the NCCN guidelines (J Natl Compr Canc Netw 2016;14:389)
- Not solely a hematologic malignancy with amyloid related organ dysfunction limiting access to aggressive treatment; clinical trials are recommended with close monitoring of response to treatment (Blood 2016;128:159)
- Common regimens include melphalan or dexamethasone; thalidomide and its analogues may also be used
- May have increased sensitivity to proteasome inhibitors such as bortezomib due to presence of misfolded light chain (Blood 2017;129:2132)
- Autologous stem cell transplant may also be considered with caution for transplant related mortality; not solely a hematologic malignancy with amyloid related organ dysfunction limiting access to aggressive treatment (Blood 2016;128:159)
- Targeting amyloid deposits to interfere with organ damage is a newer strategy: the small molecule anthracycline 49-iodo-49-deoxy-doxorubicin and related molecule doxycycline (antibiotic) may disrupt amyloid fibrils and improve clinical status (Proc Natl Acad Sci U S A 1995;92:2959, Blood Cancer J 2017;7:e546) and other approaches as (Blood 2016;128:159)
Clinical images
Images hosted on other servers:

Systemic AL amyloidosis
Gross description
- Amyloidosis may or may not be apparent on macroscopic examination
- Classic gross appearance porcelain-like or waxy
- Involved organ may appear enlarged, gray, firm and waxy when amyloid accumulates in larger amounts
Microscopic (histologic) description
- Pale pink, extracellular, glassy, hyaline material on H&E staining, mostly deposited in vascular or perivascular location
- Characteristic cracking artifact
- Macrophages and foreign body giant cells may be found around deposits
Microscopic (histologic) images
Contributed by Meenakshi Bansal, M.D.
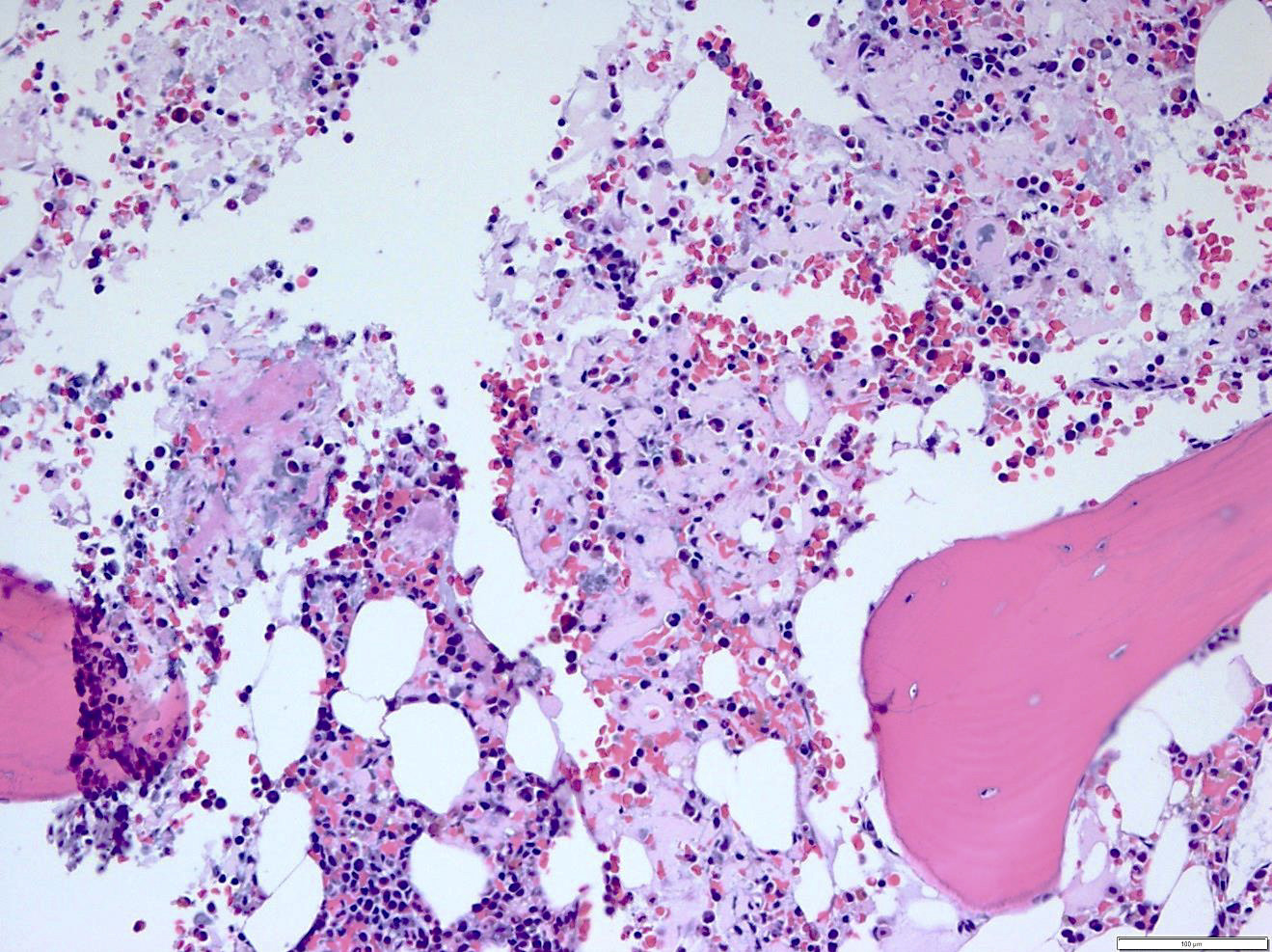
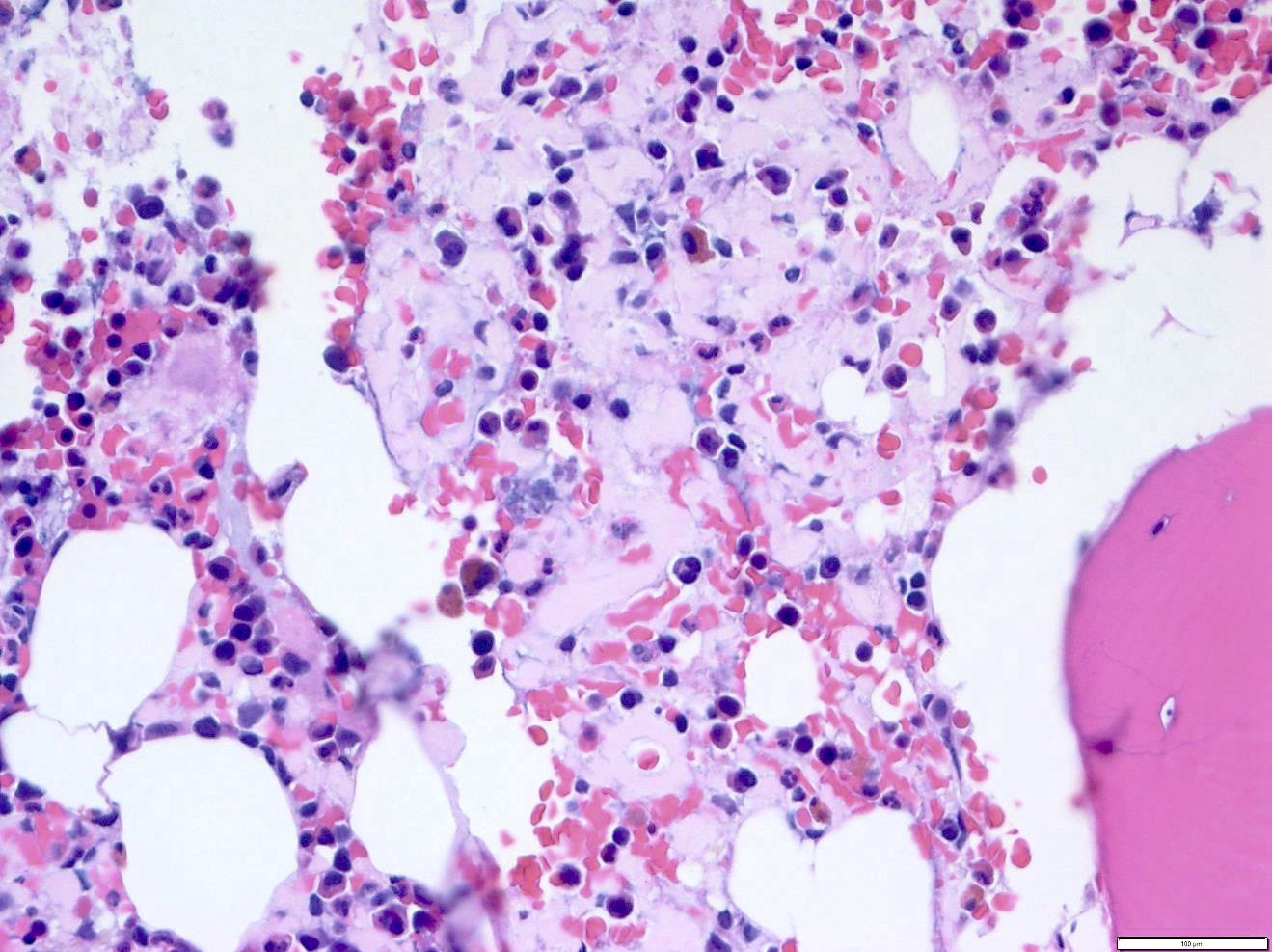
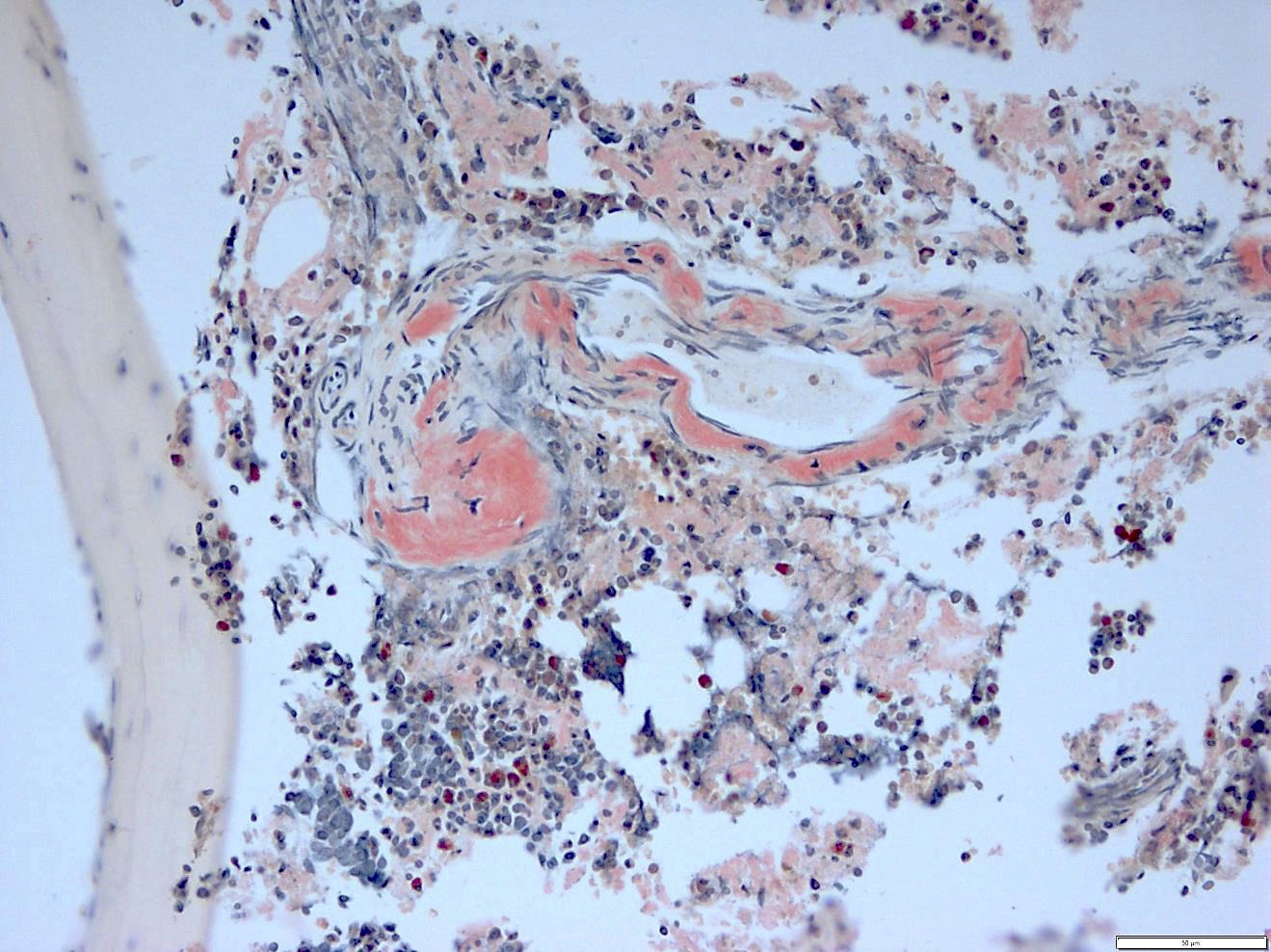
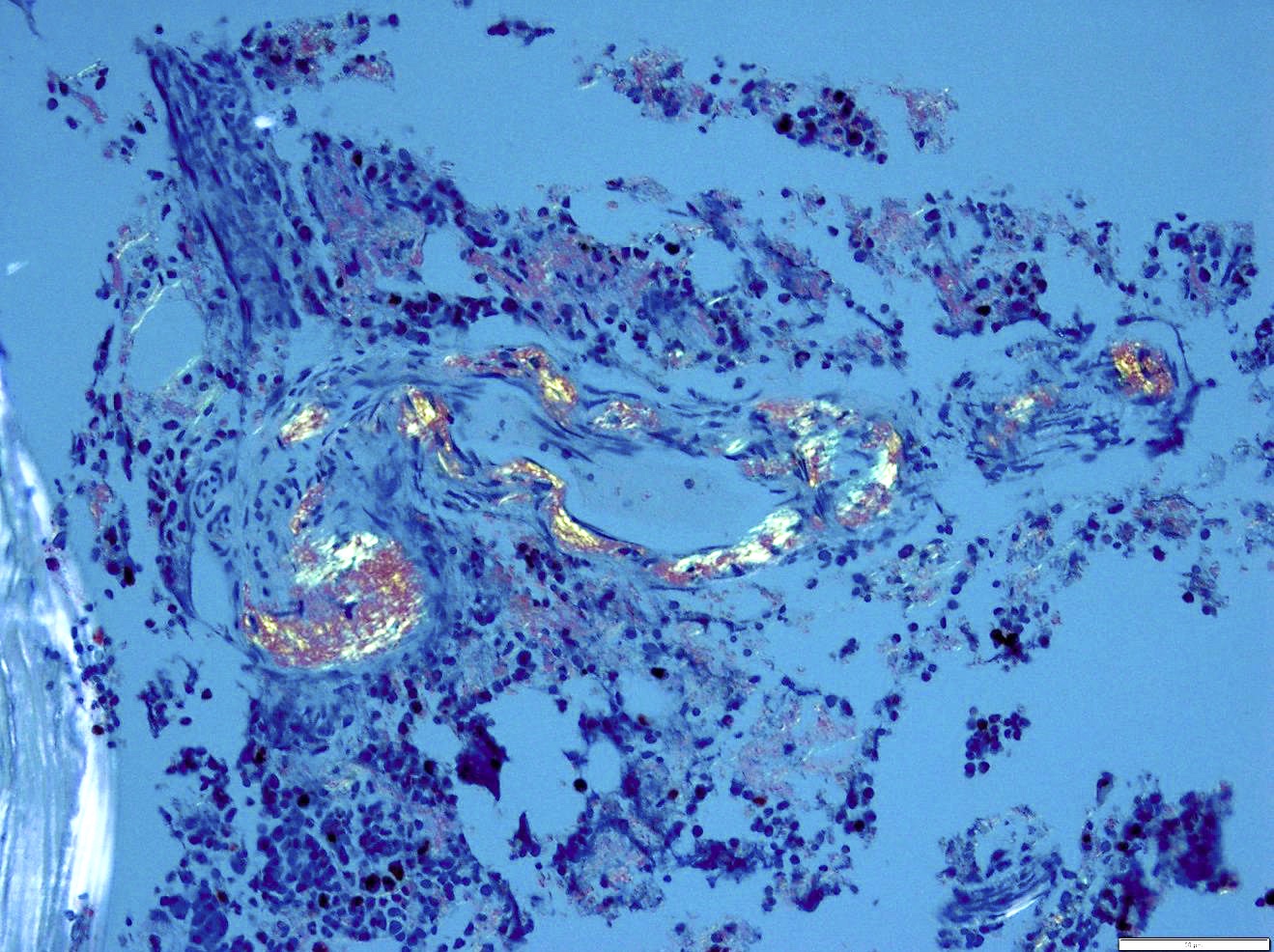
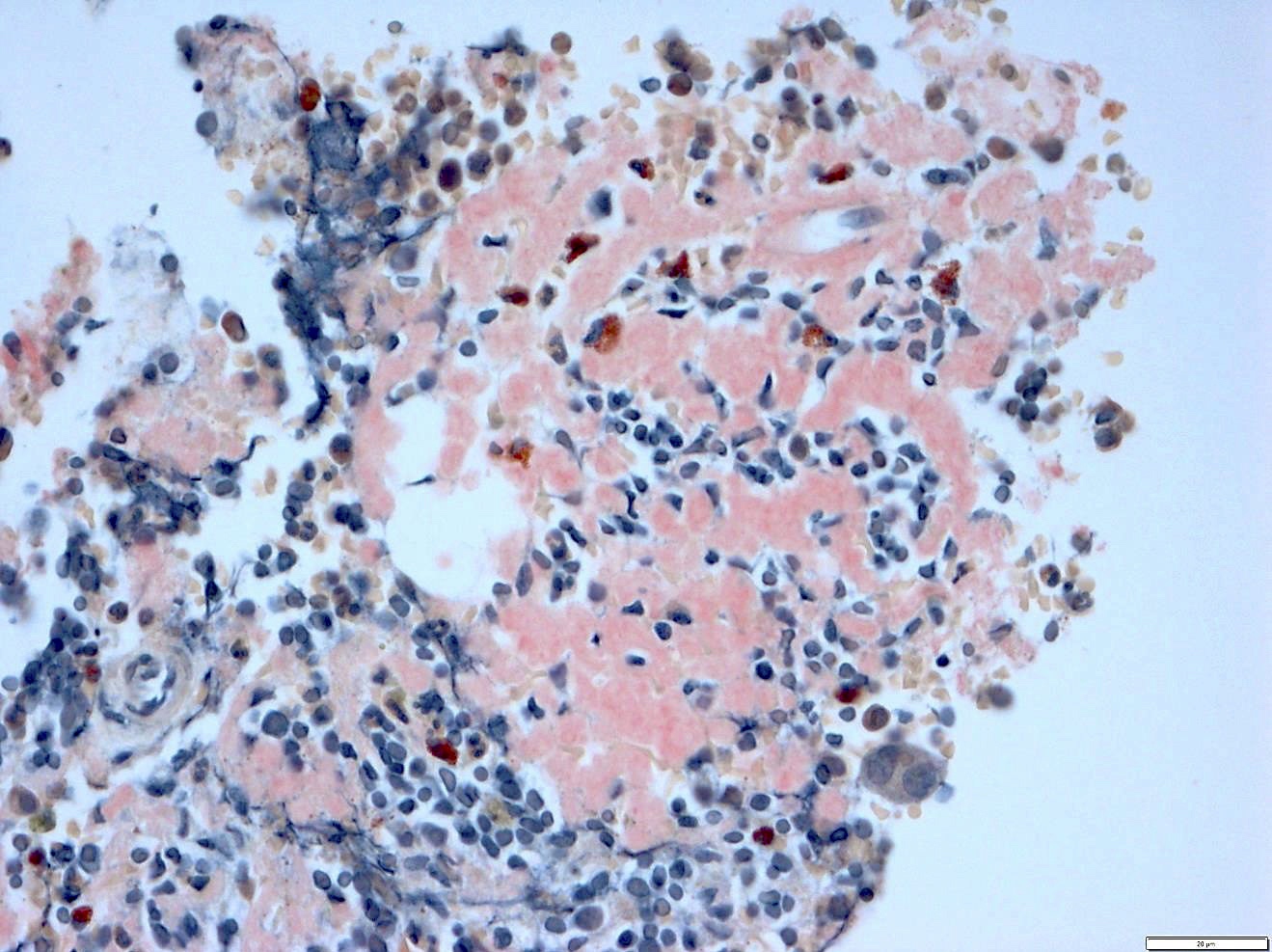
Bone marrow with extensive involvement by amyloid
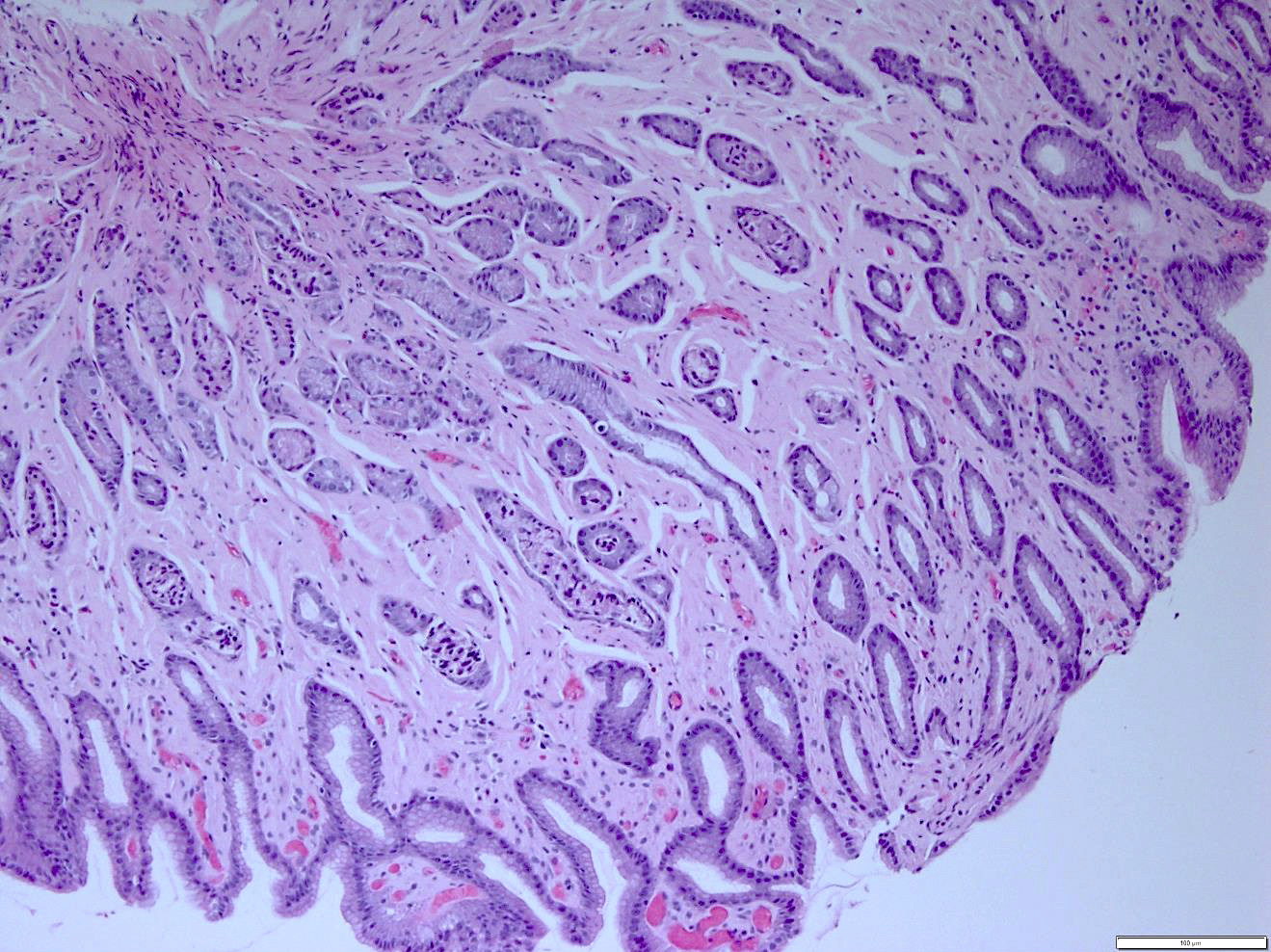

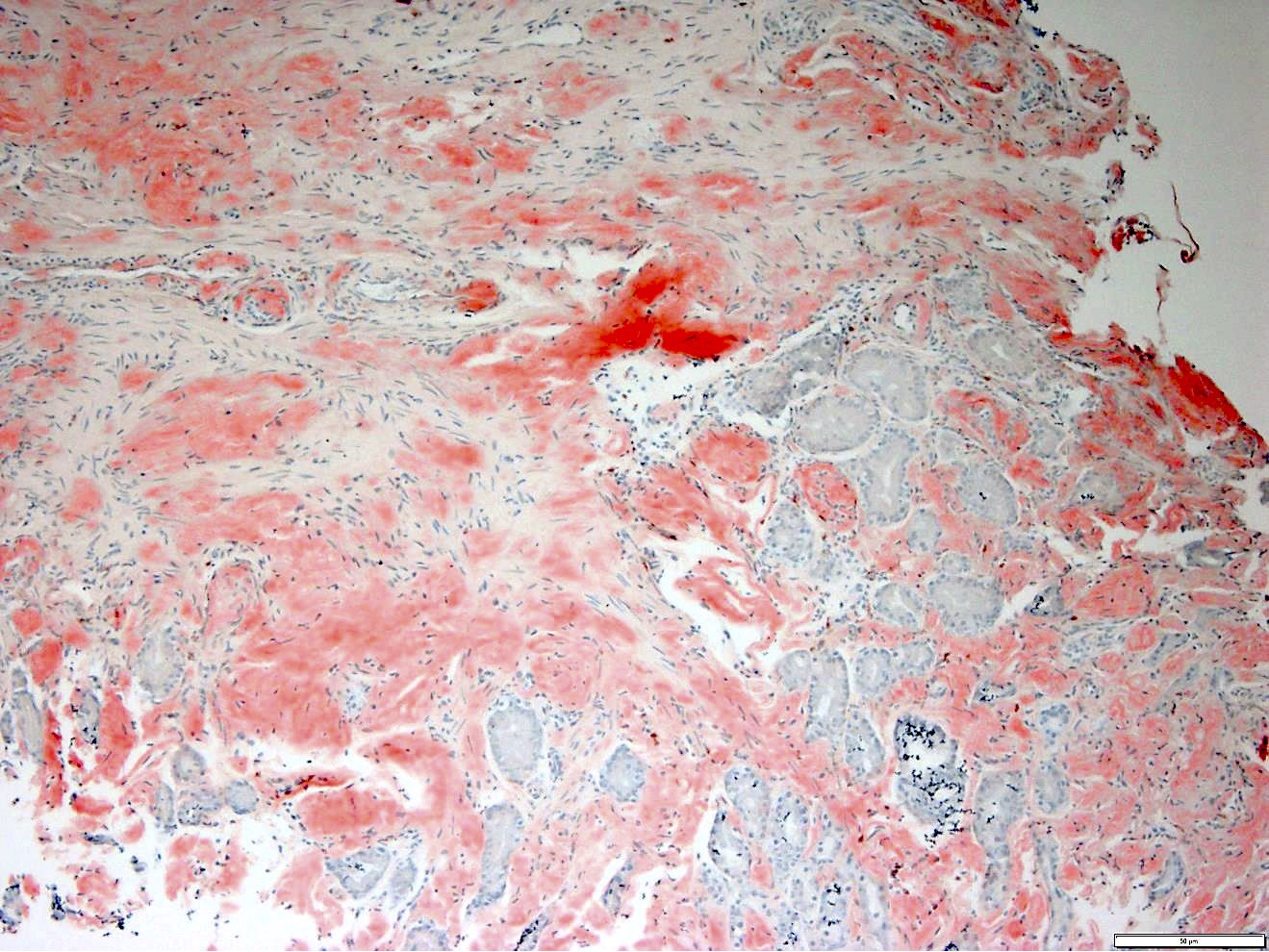

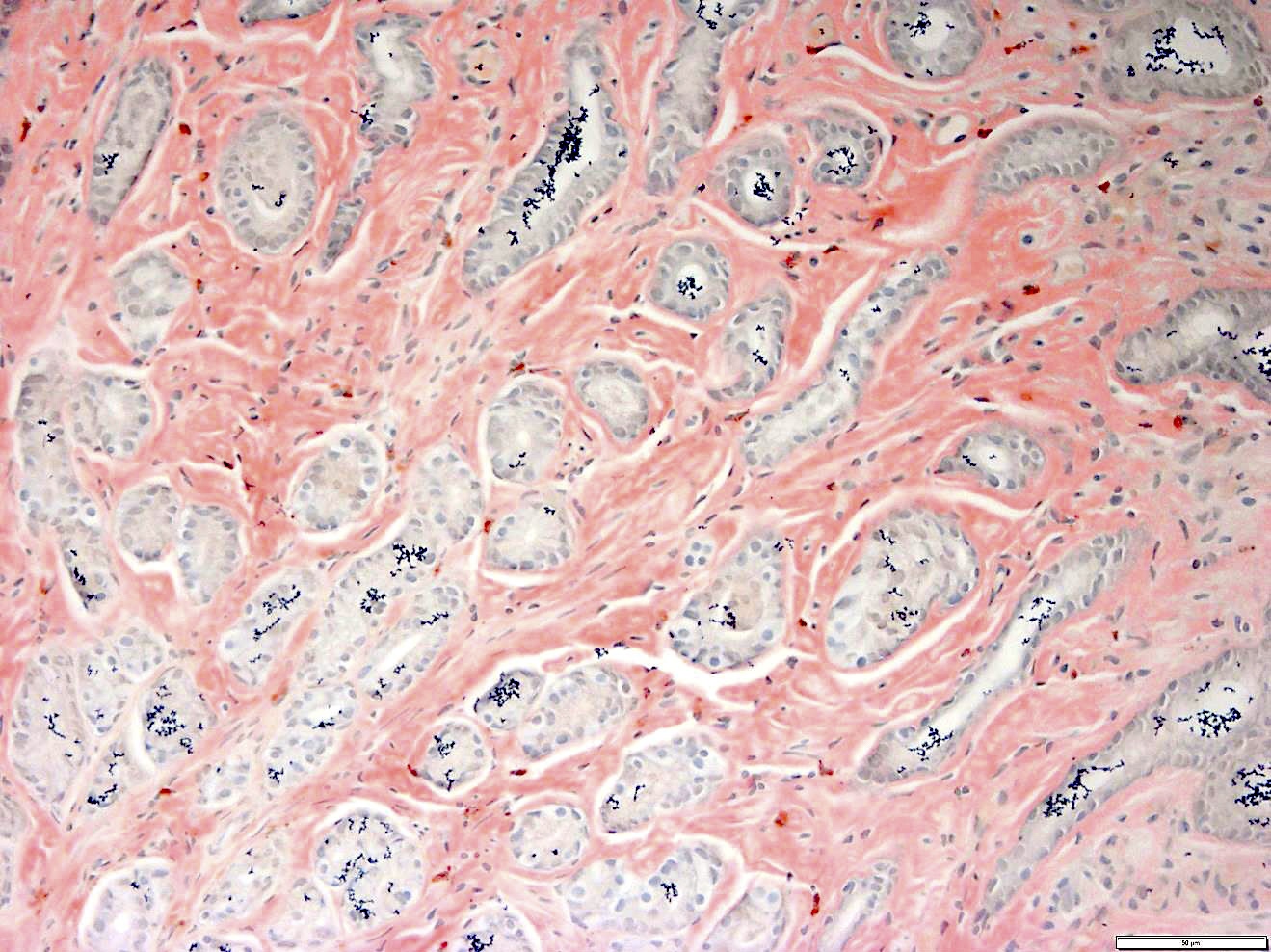
Gastric biopsies from the same patient showing extensive involvement by amyloid
Cytology description
- Marrow aspirate smears typically contain > 10% plasma cells
- Plasma cells may show a range of morphologies including those seen in myeloma, vacuolated as seen in mu heavy chain disease may be seen
- If extensive amyloid deposition, proteinaceous clumps (lightly eosinophilic to basophilic) may be present
Positive stains
- Congo red: ordinary light imparts a pink to red color to amyloid deposits; the beta pleated sheet structure of amyloid produces an apple green birefringence under polarized microscopy when stained by Congo red or sirius red stain
- PAS: moderately positive
- Iodine: stains deep brown but turns blue after treatment with concentrated sulfuric acid ("iodine sulphuric reaction" historical review Histochemistry 1976;49:131)
- Crystal violet and toluidine blue: metachromatic staining
- Thioflavin T and thioflavin S: exhibits fluorescence
- Clonal plasma cells can also be identified
Negative stains
- Trichrome for collagen
Electron microscopy description
- Extracellular, amorphous, straight nonbranching thin fibrils of 4 - 9 nm
Electron microscopy images
Contributed by Karen Vanderbilt and Bruce Goldman, M.D.
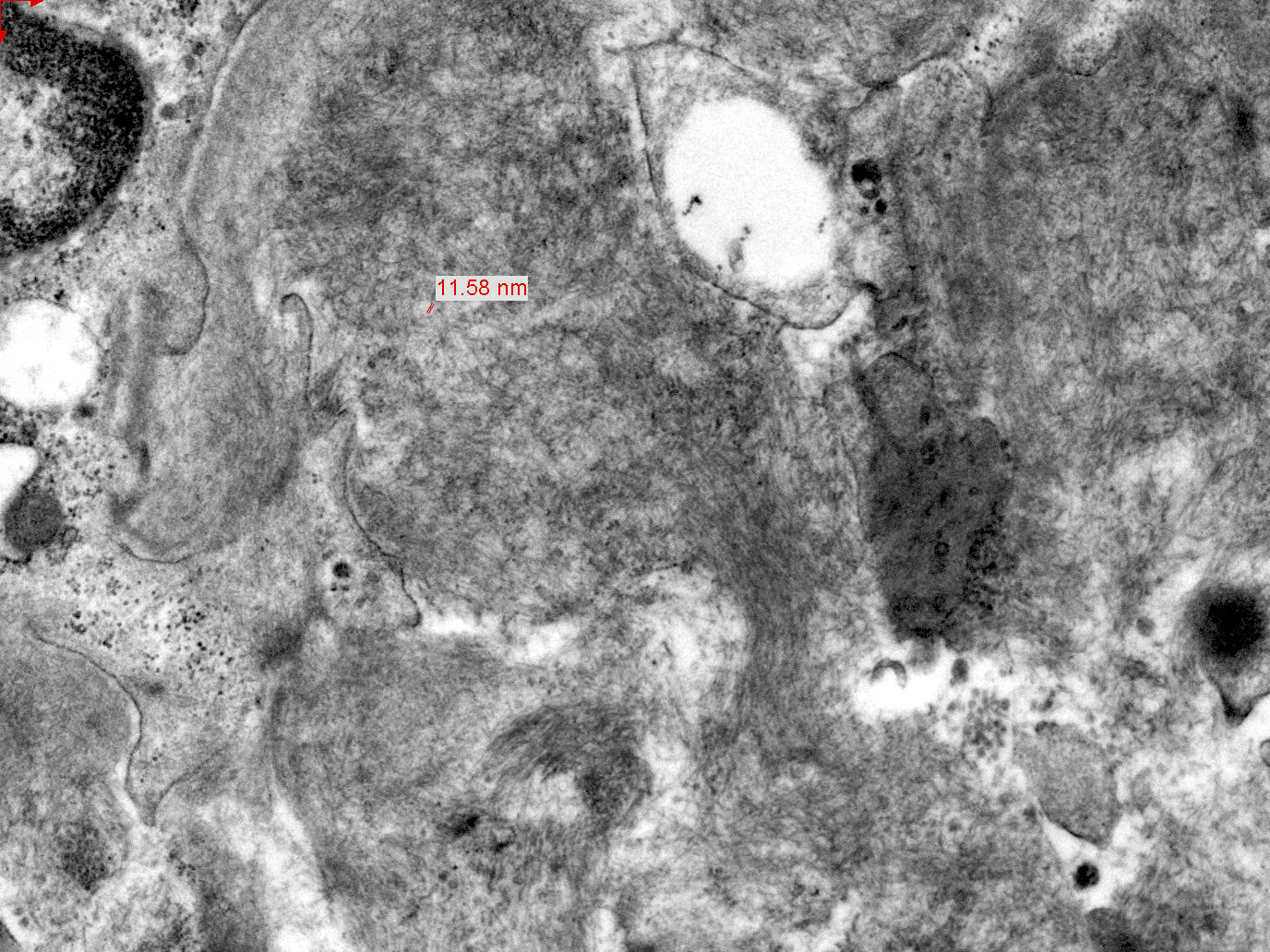
Amyloid by electron micrograph
Molecular / cytogenetics description
- Genetic abnormalities similar to non-IgM MGUS; e.g. 13q14 deletion, 1q21 gain
- t(11;14) overrepresented (present > 40% of cases) and may be associated with worse survival in contrast to the favorable prognosis in myeloma (Hematology Am Soc Hematol Educ Program 2010;2010:287)
- Not typically seen are t(4;14) and del 17p
Differential diagnosis
- Light chain or heavy chain deposition disease: plasma cell tumors that secrete abnormal light or heavy chain which deposit in tissues and cause organ dysfunction
- Do NOT have amyloid beta pleated sheets or bind Congo red
- Often do have monoclonal gammopathy (75%), most often kappa (80%), variable myeloma
- Secondary or hereditary amyloidosis: laser microdissection and analysis by mass spectrometry most reliable to distinguish; IHC with anti-amyloid fibril antibodies can also be used; presence of MGUS does not exclude concurrent secondary or familial amyloidosis (Engl J Med 2002;346:1786)
- Serous fat atrophy: extensive extravascular deposits may resemble serous fat atrophy (clinical history, Congo red staining will help distinguish)
Additional references
Board review style question #1
Which parameter is most closely linked to a poorer prognosis for primary amyloidosis?
- Extent of cardiac involvement
- Liver involvement
- Macroglossia
- Nephrotic range proteinuria
- Presence of myeloma
Board review style answer #1
A. The best answer is extent of cardiac involvement, which also results in 40% of deaths due to primary amyloidosis. Increased bone marrow plasma cells (> 10%) and involvement of multiple organs are also associated with a worse prognosis.
Comment Here
Reference: Primary amyloidosis
Comment Here
Reference: Primary amyloidosis
Board review style question #2
Which genetic alteration is most overrepresented in primary amyloidosis compared to plasma cell myeloma?
- del 17p
- Hyperdiploidy
- Monosomy 13q14
- t(11;14)
- TP53 mutation
Board review style answer #2
D. t(11;14)
While all of these alterations have been described in plasma cell myeloma and may be seen in primary amyloidosis, the t(11;14) translocation is particularly overrepresented in primary amyloidosis (> 40% of cases).13q14 deletion is also common but del 17p is rarely seen.
Comment Here
Reference: Primary amyloidosis
While all of these alterations have been described in plasma cell myeloma and may be seen in primary amyloidosis, the t(11;14) translocation is particularly overrepresented in primary amyloidosis (> 40% of cases).13q14 deletion is also common but del 17p is rarely seen.
Comment Here
Reference: Primary amyloidosis
