
Molecular markers
NGS-general
Editorial Board Members: Josephine K. Dermawan, M.D., Ph.D., Ruta Gupta, M.D.


Last author update: 1 June 2023
Last staff update: 6 June 2023
Copyright: 2019-2024, PathologyOutlines.com, Inc.
PubMed Search: Next generation sequencing

Table of Contents
Definition / general | Essential features | Terminology | CPT codes used for NGS testing | Diagrams / tables | History | Sanger sequencing | Overview of NGS methodology | Formalin fixed paraffin embedded tissue (FFPE) | Data output and analysis | Advantages of NGS (compared with Sanger sequencing) | Disadvantages of NGS (compared with Sanger sequencing) | Platforms for NGS | Computer science | NGS applications | Clinical applications | Emerging diagnostics | Videos | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: Lindstrom M, Hartsough EM, Mroz P. NGS-general. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/molecularnextgensequencing.html. Accessed April 20th, 2024.
Definition / general
- Next generation sequencing (NGS) is a high throughput technique that allows for the sequencing of millions of nucleic acids simultaneously in parallel
Essential features
- Allows for high throughput and cost effective sequencing that facilitates clinical use
- Different methods of next generation sequencing are primarily divided into short read (pyrosequencing, sequencing by synthesis, sequencing by ligation, ion torrent) and long read (single molecule real time sequencing, nanopore sequencing)
- Clinical applications primarily include oncology panels and germline panels as well as emerging whole exome sequencing / whole genome sequencing
Terminology
- Next generation sequencing (NGS), massively parallel sequencing, whole genome sequencing (WGS), whole exome sequencing (WES), RNA sequencing (RNA seq), methylation sequencing (methyl seq) and chromatin immunoprecipitation sequencing (ChIP seq)
CPT codes used for NGS testing
Diagrams / tables
Images hosted on other servers:

Overview of major methods

Ion semiconductor sequencing
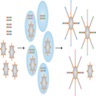
Emulsion PCR
Bridging PCR
Differences between sequencing

Sequencing pipelines and timelines

Third generation sequencing
History
- At the turn of the 20th century, many investigations strove to elucidate cell signaling pathways and cellular building blocks (Biomark Insights 2010;5:9, Int J Mol Sci 2019;20:3292, Nature 2017;550:345)
- 1921 - 1922: Frederick Banting and Charles Best discovered the peptide hormone, insulin
- 1953: Fred Sanger published the amino acid sequence of insulin, sparking efforts to determine the amino acid sequences of other proteins
- Success in determining the amino acid sequences of proteins inspired efforts to sequence genetic material (Nature 2017;550:345)
- 1965: first RNA sequences determined and published (the first sequence identified was alanine tRNA)
- DNA sequencing proved more challenging and cumbersome, until Sanger and Coulson and Maxam and Gilbert published methods of DNA sequencing with higher throughput in 1976 - 1977
- Sanger and Coulson method: chain termination using DNA primers and DNA polymerase
- Maxam and Gilbert method: restriction fragmentation and base specific chemical cleavage
- Both Sanger and Coulson's (Sanger sequencing) and Maxam and Gilbert's methods were immediately utilized and encouraged the exponential growth of new DNA sequencing projects (Nature 2017;550:345)
- Shortly after both DNA sequencing methods were published, shotgun sequencing methods (the sequencing of random clones with assembly determined by the overlap of sequences) were published in 1979 - 1980
- Growth of DNA sequencing efforts heralded advancements in bioinformatic technology and resources, including databases and search tools (such as NCBI BLAST, GenBank, etc.) (Nature 2017;550:345)
- 1980 - 1990s: massively parallel sequencing methods were described
- 1990 - 2004: The Human Genome Project, arguably the most ambitious sequencing project undertaken at the time, was performed, with a final sequence of most of the human genome published in 2004
- Ambitious nature of the project drove improvements in bioinformatic and genetic technology (it necessitated more efficient processes with higher throughput and accuracy, etc.)
- 2000s - 2010: massively parallel sequencing methods largely replaced Sanger sequencing methods and the first NGS machines were made commercially available
Sanger sequencing
- Gold standard
- Historical gold standard, given high accuracy with longer reads; however, the accuracy (99.965% validation rate), efficiency and cost effectiveness of NGS methods have largely eclipsed Sanger sequencing in clinical and research settings (Clin Chem 2016;62:647)
- Method: utilizes chain termination with modified dideoxynucleotides to determine sequence
- Advantages: can sequence any target (DNA, RNA, epigenetic changes), high quality, long read lengths
- Disadvantages: high cost (especially with longer sequences, as well as when sequencing multiple genes / loci), low throughput, time consuming and insufficient sensitivity to identify somatic variants in tumor samples and structural variants, must know sequences of interest for primer design and amplification
- Use: validation tool for NGS data; declining use as primary means of sequencing in clinical and research settings
Overview of NGS methodology
- Utilizes massively parallel sequencing methods, consisting of library preparation, amplification and sequencing
- Library (sample) preparation: random fragmentation of DNA followed by ligation of common adaptor sequences
- Sample input: DNA molecules (from blood, bone marrow, buccal swab, saliva, formalin fixed paraffin embedded tissue, etc.)
- Fragmentation: enzymatically (restriction enzyme, transposase), sonically or mechanically fragment the DNA into random sizes (typically 200 - 300 nucleotides for short read sequencing)
- Modification of target DNA
- Adaptors: unique DNA sequences are added to the ends of the fragmented DNA samples that serve multiple functions
- Provide a bar code (index) containing patient identifiers for multiplexing different samples on the same run
- Allow for hybridization of sequences to sequencing chips / beads
- Serve as universal priming sites for amplification and sequencing primers
- Adaptors: unique DNA sequences are added to the ends of the fragmented DNA samples that serve multiple functions
- Enrichment: this step is only needed if analyzing specific genomic regions (disease gene panels, exomes, etc.)
- PCR amplification (amplicon)
- Advantages: ideal for smaller genomic regions
- Disadvantages: may miss target of interest (lower sensitivity)
- Sequence capture / hybridization
- Baits (biotinylated RNA or DNA oligonucleotides) bind specific regions of DNA
- Advantages: ideal for larger genomic regions
- Disadvantages: lower enrichment for target regions due to off target capture (lower specificity)
- PCR amplification (amplicon)
- Amplification and cluster generation
- Each molecule is immobilized and amplified on a surface (chip, flow cell, beads, nanoball, etc.) utilizing multiplex PCR allowing for the PCR amplicons derived from a single template fragment to be clustered in close proximity to the original molecule (Nat Rev Genet 2016;17:333)
- Bead based systems: 1 adaptor complementary to a specific oligonucleotide fragment is fixed to a bead and amplification occurs via emulsion PCR, wherein millions of clonal DNA fragments are immobilized on a single bead
- Solid surface systems: adaptor fixes the oligonucleotide to the chip in a specific location
- Amplification occurs via bridge PCR directly on the slide, wherein forward and reverse primers are bound to the surface, which provide binding sites for complementary single stranded DNA fragments
- Flow cells: patterned solid surface system that defines precise location of primers on the slide, allowing for higher throughput
- DNA nanoballs: in solution, DNA is iteratively ligated, circularized and cleaved with 4 unique adaptor regions
- Rolling circle amplification is then used to generate DNA nanoballs, which are distributed on a patterned slide
- Each molecule is immobilized and amplified on a surface (chip, flow cell, beads, nanoball, etc.) utilizing multiplex PCR allowing for the PCR amplicons derived from a single template fragment to be clustered in close proximity to the original molecule (Nat Rev Genet 2016;17:333)
- Sequencing
- Each cluster will act as an individual sequencing reaction and libraries from multiple samples can be sequenced simultaneously in parallel
- Reads: output of sequencing; series of nucleotides which represent the sequence of the original template molecule
- Paired end reads: DNA template is sequenced from both ends, with both forward and reverse reads allowing for improved mapping, coverage and throughput (Nat Rev Genet 2016;17:333)
- Can aid in identification of structural rearrangements if there is asymmetry between forward and reverse reads
- Categorization of NGS platforms (see Platforms for NGS section for information regarding platform functions and methods)
- Generations (Nat Rev Genet 2016;17:333)
- Second generation: reliant on PCR
- Pyrosequencing (Roche 454 system)
- Sequencing by ligation (AB SOLiD system, Complete Genomics, Polonator G.007, etc.)
- Sequencing by synthesis (Illumina, Qiagen GeneReader)
- Ion semiconductor sequencing (Ion Torrent by Thermo Fisher)
- Third generation: real time sequencing without need for PCR
- Single molecule real time (SMRT) (PacBio, Roche)
- Nanopore (Oxford Nanopore Technologies)
- Second generation: reliant on PCR
- Generations (Nat Rev Genet 2016;17:333)
- Short read and long read (Int J Mol Sci 2017;18:E308)
- Short read: reads < 300 base pairs
- Examples: sequencing by synthesis, sequencing by ligation, pyrosequencing
- Advantages: low cost per Gb, high accuracy
- Disadvantages: decreased alignment, limited detection and resolution of structural rearrangements
- Long read: reads > 2.5 Kb
- Examples: singe molecule real time (SMRT), nanopore
- Advantages: improved alignment, detection of structural variations and large rearrangements, sequencing repetitive regions, discovery of novel RNA transcript isoforms, no reliance on PCR improves portability and turn around time
- Disadvantages: high cost per Gb, low accuracy
- Method and bioinformatic algorithm refinement, including increasing read depth, has increased accuracy up to 98%, although cost is still greater than short read methods (Nat Rev Genet 2020;21:597)
- Short read: reads < 300 base pairs
Formalin fixed paraffin embedded tissue (FFPE)
- One of the most common specimen types used for genetic testing
- FFPE provides a clinically convenient specimen type for NGS testing, especially as a component of the routine diagnostic workup of a new malignancy
- General workflow
- Needle core biopsies and fine needle aspirations (FNA) are often used to obtain tissue for primary tumor diagnosis
- Larger surgical specimens, such as excisional biopsies and resections, are less commonly obtained for initial diagnosis
- Tissue is fixed in formalin and embedded in paraffin wax for histopathologic evaluation
- Sectioned tissue is evaluated for morphology or immunophenotype to render a diagnosis
- Tissue remaining in the paraffin block can be utilized for ancillary testing, including molecular analysis by NGS
- Needle core biopsies and fine needle aspirations (FNA) are often used to obtain tissue for primary tumor diagnosis
- With this workflow, all essential diagnostic testing for a new cancer diagnosis can be completed on material obtained from a single, small biopsy, minimizing exposure to more invasive surgical procedures for the patient while aiding clinicians in optimizing therapeutic management
- Advantages of FFPE for NGS testing
- Archivable tissue
- DNA in FFPE tissue is relatively stable, allowing retrospective analysis of tumors
- DNA quantity and viability decrease over time at ambient conditions (in some studies, quantity has decreased to < 50% with only 11% able to be amplified after 4 - 6 years) (J Pers Med 2022;12:750)
- Storing FFPE at colder temperatures or freezing FFPE tissue has been proposed as a means to better preserve genetic material in FFPE (although more data is needed to assess this proposition, which would likely increase the cost associated with storing FFPE) (J Pers Med 2022;12:750)
- DNA in FFPE tissue is relatively stable, allowing retrospective analysis of tumors
- Histologic analysis
- In addition to diagnosis, morphologic analysis of specimens also allows for macrodissection of tumor from the remainder of nontumor specimen to maximize the final tumor percentage in the NGS specimen
- Some emerging studies also note that it may be possible to perform NGS on areas of tumor with different immunohistochemical profiles to discern genomic differences between areas of tumor with distinct immunophenotypes; however, the additional processing steps required for immunostaining have been shown in some studies to reduce DNA quantity and quality (PLoS One 2017;12:e0176280)
- Ease of transport
- FFPE tissue in blocks or sectioned on slides can be transported at ambient conditions and do not pose significant biological hazards (compared to the more stringent transportation requirements and potential infectious hazards associated with the handling of specimens such as fresh tissue, blood, bone marrow, cerebrospinal fluid, etc.)
- Archivable tissue
- Challenges associated with obtaining the ideal FFPE specimen
- Reducing pre-analytical errors
- Specimens should be removed from the patient and placed in formalin (with appropriate sectioning to allow for greater and more even penetration by formalin) as soon as possible to decrease warm and cold ischemic times
- Prolonged warm ischemia (occurs when perfusion of the specimen is reduced while being removed from the patient) and cold ischemia (occurs during the placement / storage of the specimen in formalin) are associated with a number of genetic alterations, including degradation of genetic material and potential increases and decreases in certain transcripts (RNA, given its greater instability relative to DNA, is particularly prone to potential degradation effects) (J Pers Med 2022;12:750)
- Tumor volume and tumor percentage should be optimized during sampling
- The amount of tumor available to assay in a specimen is one of the pre-analytical factors that significantly impacts the success of NGS testing (Am J Clin Pathol 2016;145:222)
- It is generally recommended that tumor represents at least 20% of the specimen submitted for testing (Am J Clin Pathol 2016;145:222)
- Many manufacturers also recommend extracting at least 10 ng of tumor DNA for greatest success with NGS testing (Am J Clin Pathol 2016;145:222)
- Platforms such as Illumina recommend at least 15,000 tumor cells per sample for hybrid capture assays (J Pers Med 2022;12:750)
- Ion Torrent platform requires 100 - 1000 tumor cells per assay (J Pers Med 2022;12:750)
- Larger specimens (excisions and resections) are more likely to yield greater tumor volume and percentage of tumor compared to core biopsies and FNA specimens
- Resection specimens and excisional biopsies are associated with greater NGS success rates (Am J Clin Pathol 2016;145:222)
- Core biopsies and FNA specimens may yield scant fragments of tumor / clusters of tumor cells or may result in specimens containing small percentages of tumor cells compared to nontumor cells (such as inflammatory cells and normal surrounding tissue)
- Low tumor cellularity may preclude meaningful genetic analyses specific to tumor cells
- When smaller or more limited specimens like core biopsies and FNA specimens are obtained, processes such as rapid onsite evaluation (ROSE) of biopsied material can aid in triaging specimen sampling to maximize tumor volume in the final specimen for both morphologic and ancillary testing (such as NGS analysis) (J Pers Med 2022;12:750)
- Specimens should be removed from the patient and placed in formalin (with appropriate sectioning to allow for greater and more even penetration by formalin) as soon as possible to decrease warm and cold ischemic times
- Adequate formalin fixation is critical to ensure optimum quality and quantity of genetic material for NGS studies
- 12 - 24 hour fixation in 10% formalin is recommended for analysis of specimens by morphology and ancillary studies such as NGS (J Pers Med 2022;12:750, Int J Mol Sci 2016;17:1579, PLoS One 2017;12:e0176280)
- Underfixation can result in enzymatic degradation of tissues and genetic material
- Overfixation can lead to the generation of more formalin fixation dependent artifacts (up to 1 modification per 500 bases), including
- Fragmentation of genetic material (i.e., generating ssDNA, short fragments (100 - 200 bp) of RNA and DNA, etc.), which can preclude useful analysis using long read sequencing methods (J Pers Med 2022;12:750, Am J Clin Pathol 2016;145:222)
- Increased volumes of ssDNA can also produce chimeric reads from noncontiguous sequences containing repetitive or similar elements, which likely form during ligation processes in the generation of NGS libraries (Nucleic Acids Res 2019;47:e12)
- High volumes of chimeric reads and ssDNA can cause false detections of copy number variations (CNVs) (Nucleic Acids Res 2019;47:e12)
- Deamination of cytosine and 5 methylcytosine bases, leading to conversion to uracil and thymine, respectively
- May be more prominent after more than 48 hours of fixation (J Pers Med 2022;12:750)
- Cross linking of bases to other molecules
- Inhibits polymerase based amplification or sequence elongation methods (J Pers Med 2022;12:750, Int J Mol Sci 2016;17:1579)
- Loss of bases
- Fragmentation of genetic material (i.e., generating ssDNA, short fragments (100 - 200 bp) of RNA and DNA, etc.), which can preclude useful analysis using long read sequencing methods (J Pers Med 2022;12:750, Am J Clin Pathol 2016;145:222)
- Additional steps during extraction of genetic material and generation of NGS libraries, such as enzymatic treatments to remove cross links and uracil bases from DNA and decrease high volumes of ssDNA fragments, can help reduce fixation artifacts and false detections of CNVs and single nucleotide variants (SNVs)
- Additional tissue processing steps (such as decalcification) should be omitted if possible
- Decalcification of bone and other significantly calcified tissues is associated with poorer NGS success (J Pers Med 2022;12:750, Am J Clin Pathol 2016;145:222)
- Decalcification requires the use of strong (hydrochloric and nitric) or weak (picric, formin, acetic) acids or chelating agents (EDTA), which degrade genetic material
- Short term exposure to weak acids and EDTA can still allow for extraction of DNA sufficient for detection of SNVs, CNVs and fusion transcripts (J Pers Med 2022;12:750)
- Generally, if a primary tumor is difficult to biopsy and has multiple metastases to bone and other tissues, obtaining a biopsy of nonbone metastases may result in procurement of tissue more likely to be successfully analyzed via NGS (Am J Clin Pathol 2016;145:222)
- Methods of genetic material extraction from FFPE can also significantly affect the quantity and quality of material used for NGS
- Studies have shown that different extraction kits (using different reagents or purification methods such as columns, magnetic beads, etc.) differ with respect to the ultimate yield, purity and quality of extracted material (Int J Mol Sci 2016;17:1579)
- Especially when working with small specimens with low tumor volume / percentage, maximizing the efficiency, quality and yield of the extraction process is essential (Int J Mol Sci 2016;17:1579)
- Reducing pre-analytical errors
- Reducing analytical errors
- Altering the base and CNV calling algorithms of the NGS pipeline to account for formalin fixation artifacts can significantly reduce false detections of genetic alterations in FFPE (BMC Med Genomics 2020;13:94, Nat Commun 2022;13:4487, Nucleic Acids Res 2019;47:e12, J Mol Pathol 2021;2:123)
- More stringent base calling algorithms can reduce false detections of SNVs (BMC Med Genomics 2020;13:94, Nat Commun 2022;13:4487, Nucleic Acids Res 2019;47:e12)
- CNV detection is particularly challenging with the use of targeted gene panels, in which exonic sequences are sequenced preferentially over intronic sequences (J Mol Pathol 2021;2:123)
- Smaller, noncontiguous sequences are more difficult for algorithms built for the analysis of whole genome sequences to analyze with respect to copy number (J Mol Pathol 2021;2:123)
- Any additional fixation artifacts, such as high volumes of chimeric reads or shortened sequences of genetic material that are difficult to map, can further complicate CNV determination, so maximizing the quality of the genetic material tested is imperative (Nucleic Acids Res 2019;47:e12, J Mol Pathol 2021;2:123)
- Altering the base and CNV calling algorithms of the NGS pipeline to account for formalin fixation artifacts can significantly reduce false detections of genetic alterations in FFPE (BMC Med Genomics 2020;13:94, Nat Commun 2022;13:4487, Nucleic Acids Res 2019;47:e12, J Mol Pathol 2021;2:123)
Data output and analysis
- Demultiplexing: pooled patient samples (sample libraries) that were sequenced simultaneously are then separated by barcodes specific to each patient sample (indices)
- Reads are physically clustered together based on sequence similarity and forward and reverse reads are aligned (paired)
- Alignment
- Resequencing: sequence reads aligned to reference sequence (the reference genome of one individual)
- De novo: sequence reads aligned to each other
- Interpretation: pathogenic, likely pathogenic, variant of uncertain significance, likely benign, benign (J Mol Diagn 2017;19:4, Genet Med 2015;17:405)
Advantages of NGS (compared with Sanger sequencing)
- High throughput / output
- Improved resolution
- Cost effective
- Algorithms are available that can detect structural variants
Disadvantages of NGS (compared with Sanger sequencing)
- Shorter read lengths
- Decreased raw accuracy in areas of homology (such as pseudogenes), repeat expansions, large indels, copy number variants (CNVs) or other structural variants
- Genetic events commonly occur in nonunique sequences (centromeric DNA, repetitive elements, etc.), which makes identification by NGS challenging
- High start up costs
Platforms for NGS
- Pyrosequencing
- First commercially successful NGS system
- Sequencing mechanism
- Beads bound with template are distributed on a plate with beads containing enzymes
- 1 of each of the 4 nucleotides is added iteratively to the sequencing reaction
- Pyrophosphate (Ppi) is released during nucleotide incorporation and the release of Ppi equals the amount of incorporated nucleotide
- Example: 454 system (Roche)
- Advantages: fast, long read length
- Disadvantages: high cost of reagents, relatively high error rate with polybase > 6, low throughput (J Biomed Biotechnol 2012;2012:251364)
- Sequencing by ligation
- Sequencing mechanism: hybridization and ligation of labeled probe to DNA strand
- Utilizes octamer oligonucleotide probes containing 2 probe specific bases and 6 degenerate bases
- Each probe will have 1 of 4 fluorescent dyes linked to the last base, a ligation site on the first base and a cleavage site on the fifth base
- Sequencing occurs by complementary binding between the probe and template and the anchor blinds complementarily to the adaptor and serves as an initiation site for ligation
- Released fluorescence is then imaged
- Fluorescent signal and last 4 nucleotides of the octamer are cleaved and the cycle continues
- After several cycles of hybridization, ligation and cleavage, the DNA strand is denatured and another sequencing primer offset by 1 base is used to repeat the reaction
- 5 total sequencing primers are used and the sequence of the fragment can be deduced after approximately 5 rounds of sequencing
- Example: AB SOLiD system (Thermo Fisher)
- Advantages: highest accuracy of second generation NGS, no reliance on a polymerase
- Disadvantages: short sequencing reads (Nat Rev Genet 2016;17:333, J Biomed Biotechnol 2012;2012:251364)
- Sequencing mechanism: hybridization and ligation of labeled probe to DNA strand
- Sequencing by synthesis (SBS)
- One of most common methods utilized in clinical applications (Arch Pathol Lab Med 2017;141:1544)
- Sequencing mechanism
- Each cycle, all 4 deoxynucleotide triphosphates (dNTPs) are added to the flow cell, which contain different cleavable fluorescent dyes and a removable blocking group
- Incorporation of the fluorescently labeled dNTP by the DNA polymerase terminates polymerization each cycle and the fluorescent signal identifies the specific nucleotide into the growing DNA strand
- Fluorescent dye and blocking group are enzymatically cleaved so a new nucleotide can be added during the next cycle
- Example: Illumina sequencing (MiSeq, HiSeq, NextSeq and NovaSeq platforms)
- Advantages: high accuracy (sequences base by base), largest output, cheapest, less susceptible to homopolymer errors than single nucleotide addition platforms (Ion Torrent, pyrosequencing)
- Disadvantages: cyclical nature leads to long sequencing times, short read assembly (Coleman: Diagnostic Molecular Pathology, 2016, Nat Rev Genet 2016;17:333, J Biomed Biotechnol 2012;2012:251364)
- Ion semiconductor sequencing
- One of most common methods utilized in clinical applications (Arch Pathol Lab Med 2017;141:1544)
- Sequencing mechanism: Ion Torrent sequencing (semiconductor sequencing)
- DNA fragments are enriched on beads, which are then arrayed onto a microwell plate so that only one bead occupies one well
- Unmodified single nucleotides are successively added to the microwell chip (sequencing by single nucleotide addition)
- Incorporation of a nucleotide by DNA polymerase causes release of a proton, which is detected by an ion sensor that is sensitive to changes in pH each time a complementary nucleotide is added
- Sequence is determined by evaluating the electrical signal intensity during each sequential nucleotide exposure
- Example: Ion Torrent (Thermo Fisher)
- Advantages: fast sequencing (no reliance on camera or fluorescence), low cost, smaller instrument size
- Useful for point of care testing and gene panels
- Disadvantages: less accurate with increased homopolymer error (Nat Rev Genet 2016;17:333)
- Single molecule real time (SMRT) sequencing
- Compared to other systems, there is no reliance on clonal amplification of target region or chemical cycling for each dNTP added
- Sequencing mechanism
- Specialized flow cell with thousands of microwells, each containing a single immobilized polymerase and a copy of the target molecule, often a circular template that can be sequenced multiple times
- Microwells are flooded with an excess of fluorescently labeled nucleotides
- Dye is cleaved during incorporation by the DNA polymerase, allowing it to diffuse away from the sensor and allowing the active site of the DNA polymerase to become available for the next dNTP
- Light detector within the microwell detects the signal in real time as the nucleotides are incorporated
- Example: Pacific Biosciences
- Advantages: fastest method, can sequence longer targets, decreases bias and error attributed to PCR, ideal for de novo genome assembly applications, real time data generation
- Disadvantages: lower accuracy and throughput (Nat Rev Genet 2016;17:333, J Biomed Biotechnol 2012;2012:251364)
- Improvements in platform performance, including increased throughput (such as up to 160 Gb per flow cell in the PacBio Sequence II platform), has allowed for increased read depth (up to 40 times genomic coverage), ultimately increasing consensus accuracy between reads up to 99.9% (Nat Rev Genet 2020;21:597)
- High fidelity reads (such as those produced by PacBio platforms) require more time and resources (including computational processing power) and result in a higher cost per genomic assembly due to the generation of multiple reads per template molecular per sequencing cell but the reads generated are both long (> 10 Kb) and accurate (> 99%) (Nat Rev Genet 2020;21:597)
- Nanopore
- Compared to other systems, this system does not monitor incorporation of nucleotides or use a secondary signal (fluorescence, pH, etc.)
- Sequencing mechanism
- Ionic current is passed through nanopore proteins
- Target DNA is then passed through a nanopore and each base alters the current
- Sequencing occurs in real time as the DNA molecule passes through the nanopore
- Hairpin library structure allows forward and reverse strands to be sequenced (e.g., Oxford Nanopore Technologies)
- Advantages: can sequence very long molecules (standard length: 10 Kb to 100 Kb; ultra long reads: > 100 Kb), lower cost, small and portable, real time data generation; de novo assembly, structural variants (Nat Rev Genet 2020;21:597)
- With increased flow cells per platform (e.g., Oxford Nanopore Technologies' PromethION, which can run up to 48 flow cells simultaneously), throughput could potentially exceed that of some short read based platforms (e.g., Illumina NovaSeq) (Nat Rev Genet 2020;21:597)
- Disadvantages: relatively high error rate (Nat Rev Genet 2016;17:333, J Biomed Biotechnol 2012;2012:251364)
- Accuracy of base calls is highly dependent on algorithms used; improvements in algorithms have increased the average accuracy of standard reads (10 - 100 Kb) to 87 - 98%, with consensus read accuracy around 97 - 98% (Nat Rev Genet 2020;21:597)
Computer science
- Overview: raw sequencing data is translated into individual sequences (reads), each read is mapped to its targeted region in the genome and this alignment then identifies differences between the sample DNA and the standardized reference
- Base calling and demultiplexing: raw format data (fluorescence intensity, electrical impulse, etc.) is converted to a nucleotide sequence assigned to each position in the target DNA sequence (e.g., binary base call (BCL) format)
- Intensity of the signal relative to the background noise will allow the base caller algorithm to generate a confidence score associated with each nucleotide
- Read data is demultiplexed by converting from BCL to FASTQ format, which contains a read identifier, nucleotide sequence and confidence scores
- FASTQ format:
- Line 1: @Sequence identifier (and optional description)
- Line 2: Raw sequence read: “ACTGACTG”
- Line 3: Spacer: ‘+’
- Line 4: Phred quality score of line 2 (probability base is incorrectly called by the sequencer): ‘!AA>**!C
- Q = -log(10)P
- Q = Phred quality score
- P = Probability of an incorrect base call (calculated by peak shape and overlap at bases)
- Example software: CASAVA (Illumina)
- Q = -log(10)P
- FASTQ format:
- Sequence alignment:
- Mapping of all individual reads to locations on the genome
- Requires read data and a reference sequence (a standard against which individual reads are compared and aligned)
- Biologic factors (benign variants and mutations) and technical factors (errors in sequencing or inaccurate base calling) contribute to imperfect alignment
- Penalties are generated for inexact alignment; at a certain penalty threshold, a read is considered unable to be aligned
- Coverage (depth): number of times a nucleotide is sequenced or the number of reads that align over a single base
- Increased coverage verifies the base call at that position and increases confidence that the base call accurately represents the original target sequence
- Twenty fold coverage (20 reads over target region, often noted as 20x) is required to be confident of the base called in a pure or germline specimen
- Five hundred to one thousand fold (500 - 1,000x) coverage is needed to identify variants in mixed samples like tumor specimens
- Alignment formats: sequence alignment / map (SAM) and binary BAM format stores alignment data indexed by reference sequence location
- Example software: BWA, Bowtie 2, MAQ, Stampy, Novoalign
- Variant calling:
- After alignment to a reference genome, single nucleotide polymorphisms (SNPs), single nucleotide variants (SNVs) and insertion / deletions (indels) can be identified
- Considers depth of coverage, variant frequency and alignment score
- File format: variant call format (VCF), mutation annotation format (MAF)
- Example software: SAM tools Mpileup, GATK
- Visualization of data:
- Example software: integrative genomics viewer (IGV), UCSC genome browser, ENSEMBL genome browser
- Reference: Arch Pathol Lab Med 2017;141:1544, Annu Rev Pathol 2020;15:97
NGS applications
- DNA sequencing
- Data analysis: identifies point mutations, insertions / deletions (indels), copy number variants (CNVs), structural variants; tumor mutation burden (TMB), mutational signature
- Whole genome sequencing
- Sequences both coding and nonncoding regions of the genome
- Advantages: library preparation does not require enrichment or amplification (high specificity)
- Disadvantages: high cost, less depth of coverage, complex data analysis and interpretation
- Whole exome sequencing
- Sequencing of only protein coding area of genome (~1 - 2%)
- Need > 20x coverage per nucleotide for sufficient specificity and sensitivity for mutation detection
- Advantages: more affordable for clinical use
- Targeted sequencing
- Utilizes enrichment with specific primers to sequence certain areas of the genome
- Advantages: allows more individual samples to be run with each sequencing reaction, improves depth of coverage of regions of interest and reduces cost
- Gene panels
- Genomic medicine
- Hereditary syndromes (e.g., Lynch syndrome, hereditary breast / ovarian cancer syndrome) or mitochondrial diseases
- Depth of 80x sufficient to detect germline variants
- Somatic mutations
- Solid tumor and hematolymphoid neoplasm panels
- > 500x coverage needed for somatic mutations
- Genomic medicine
- RNA sequencing (RNA seq)
- Utilizes reverse transcriptase PCR (RT PCR)
- Data analysis: differential expression, gene fusions, alternative splicing and RNA editing
- Other
- Methylation sequencing (methyl seq): utilizes bisulfite treated DNA to analyze DNA methylation patterns
- Useful for analyzing epigenetic changes, such as parental imprinting
- Methylation profiling is also becoming essential for CNS tumor classification (Neuro Oncol 2021;23:1231)
- Chromatin immunoprecipitation (ChIP seq): analysis of DNA protein interactions by using chromatin immunoprecipitation (Coleman: Diagnostic Molecular Pathology, 2016, Genet Med 2013;15:733)
- Assay for transposase accessible chromatin with sequencing (ATAC seq)
- Cut and run
- Methylation sequencing (methyl seq): utilizes bisulfite treated DNA to analyze DNA methylation patterns
Clinical applications
- Oncology testing
- Often utilizes specific gene panels to identify somatic variants or sequence variants specific to hematologic or solid malignancies
- Assists in diagnosis
- Offers prognostic information
- Impacts treatment options (targeted therapies)
- BRAF V600E mutation is treated with BRAF inhibitor (e.g., vemurafenib)
- Commercial testing (e.g., FoundationOne) allows for testing of hundreds of genomic regions
- Cancer screening using circulating tumor DNA (Illumina: Illumina Forms New Company to Enable Early Cancer Detection via Blood-Based Screening [Accessed 3 March 2023])
- Often utilizes specific gene panels to identify somatic variants or sequence variants specific to hematologic or solid malignancies
- Germline testing
- WES, WGS or targeted germline panels are used to identify germline DNA variants in order to
- Diagnose heritable disease
- Explain phenotypes that are likely genetic in origin
- Identify cancer predisposition (such as with BRCA1 / BRCA2 or TP53 mutations, etc.)
- Commercial testing allows for SNP detection and analysis (e.g., 23andme, Ancestry, Gene Dx, Ambry genetics, etc.)
- WES, WGS or targeted germline panels are used to identify germline DNA variants in order to
- Microbiology
- WGS and metagenomics sequencing methods are used for
- Identification of pathogens and mechanisms of therapeutic resistance
- Tracking antimicrobial resistance within and outside of healthcare institutions
- Epidemiologic monitoring of disease outbreaks by genotyping pathogens and pathogenic strains (Clin Microbiol Infect 2018;24:335, Genes (Basel) 2022;13:1566)
- WGS and metagenomics sequencing methods are used for
- Pharmacogenomics
- Link genomic variations to phenotypic differences in drug response or drug - drug interactions (Cold Spring Harb Perspect Med 2019;9:a033027)
Emerging diagnostics
- Single cell transcriptomics, spatial transcriptomics, single cell ATAC seq
- Optical genome mapping (OGM)
- Based on genome imaging techniques first introduced in 1990
- Long, linearized DNA molecules (derived from high molecular weight DNA, with single strand fragment lengths ranging from 0.15 - 2.5 Mb) are labeled with sequence motif specific fluorescent markers (generally about 15 labels per 100 kb), run through nanopore channels and analyzed via high throughput imaging
- Detected pattern of the fluorescent labels is then used to generate a de novo genome which is then compared to a reference genomic map
- Variations in label patterns relative to reference genome maps are used to detect structural variations, including insertions, deletions, inversions, translocations and chromosomal aneuploidy
- Benefits
- While NGS and comparative genomic hybridization arrays (aCGH) can detect genetic variants at the sequence level and with respect to copy number variation, OGM can better detect balanced structural variants (such as inversions and balanced translocations) as well as complex structural rearrangements, with the potential for high resolution of variant breakpoints
- Repetitive regions of chromosomes that have been historically difficult to sequence can be more readily mapped
- As many genetic events occur in nonunique / repetitive sequences, OGM has the potential to detect structural rearrangements in these areas more effectively than can currently be done with NGS and aCGH
- Insertions and deletions as small as 500 bp can be detected
- Generation of OGM data can have a shorter turnaround time relative to conventional cell culture and karyotyping, allowing for potentially faster identification of structural variants
- Limitations
- Structural and genetic variants must be large enough to be detected or resolved
- Some regions of chromosomes do not map well (for example, some studies report poor resolution of the subtelomeric region of the X chromosome with subsequent failure to detect certain structural rearrangements)
- OGM analyses can identify multiple structural variants per specimen, sometimes resulting in the identification of tens to hundreds (or more) of potential variants
- Current data filtering settings and strategies are limited given the relative newness of the technology in research and potentially clinical settings
- Analytical algorithms for copy number neutral loss of heterozygosity are lacking
- Current reference maps are derived from a relatively small cohort
- Current scale up capabilities are limited with respect to clinical applications
- Overall, OGM promises to be a useful addition to diagnostic testing (whether for primary diagnostic testing or confirmation of results from other conventional studies), pending improvements in scale up capabilities and refinement of analytic algorithms (Am J Hum Genet 2021;108:1423, Genes (Basel) 2021;12:1958)
Videos
Introduction to different NGS platforms
Sequencing by synthesis (Illumina)
Additional references
Board review style question #1
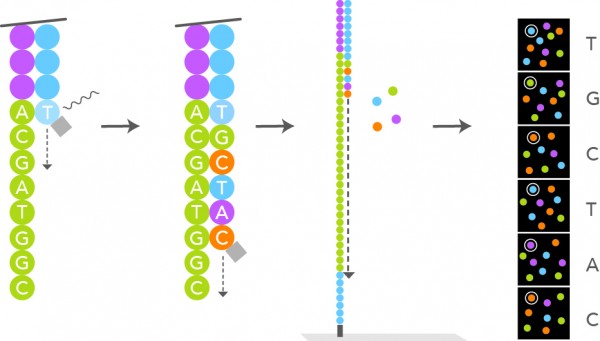
- What manner of next generation sequencing is depicted in the image above?
- Ion Torrent sequencing
- Pyrosequencing
- Sanger sequencing
- Sequencing by ligation
- Sequencing by synthesis
Board review style answer #1
E. Sequencing by synthesis. The image depicts a single, labeled base, which is incorporated into a growing sequence. The label is then removed, which allows the next labeled base to be added in a similar manner. Sanger sequencing operates via chain termination, so no additional bases could be added to the growing sequence after the incorporation of a labeled base using Sanger methods. There is no ligation of sequence fragments depicted, so sequencing by ligation is also incorrect. The image also does not depict fragments of DNA bound to beads with the release of pyrophosphate or hydrogen ions associated with base incorporation; therefore, the image depicts neither pyrosequencing nor ion semiconductor sequencing (i.e., Ion Torrent).
Comment Here
Reference: NGS-general
Comment Here
Reference: NGS-general
Board review style question #2
- Which of the following statements is true regarding long read sequencing compared with short read sequencing?
- It can more readily detect large rearrangements than short read sequencing
- It is cheaper than short read sequencing
- It is more accurate than short read sequencing
- It utilizes PCR based amplification
Board review style answer #2
A. It can more readily detect large rearrangements than short read sequencing. Depending on the methods used, long read sequencing does not depend upon PCR based amplification and it can be less accurate and (currently) more expensive than short read sequencing to perform; however, one of the benefits of long read sequencing is that it can better resolve large genetic rearrangements relative to short read sequencing. Longer read lengths (thousands to tens of thousands of base pairs in length) can cover more unique, identifying sequences than shorter read lengths and they can therefore more readily detect larger rearrangements (particularly large and complex structural variants) relative to shorter read lengths, which may fail to capture sufficient sequencing data in a few hundred base pairs to indicate and locate a large or complex structural rearrangement.
Comment Here
Reference: NGS-general
Comment Here
Reference: NGS-general

