

Muscle & peripheral nerve nontumor
Muscular dystrophies
Becker and Duchenne muscular dystrophy
Resident / Fellow Advisory Board: Meaghan Morris, M.D., Ph.D.
Editorial Board Member: Chunyu Cai, M.D., Ph.D.


Last author update: 10 June 2021
Last staff update: 6 July 2021
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Becker[title] and Duchenne[title] muscular dystrophy pathology

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1 | Board review style question #2 | Board review style answer #2Cite this page: De Los Santos Y, Kresak JL. Becker and Duchenne muscular dystrophy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/musclebeckerduchennemusculardystrophy.html. Accessed April 20th, 2024.
Definition / general
- Becker muscular dystrophy (BMD) is caused by dystrophin (DMD) gene mutations on chromosome Xp21, which decreases / alters dystrophin production and causes variable progressive proximal weakness in childhood, progressing to paralysis by adulthood
- Duchenne muscular dystrophy (DMD) is also caused by DMD gene mutations, which causes severe progressive muscle weakness, progressive cardiorespiratory compromise in adulthood and death
Essential features
- X linked muscular dystrophies include DMD and BMD
- Both are caused by mutations in the dystrophin gene
- DMD is caused by frameshift mutations which disrupt the normal reading frame, therefore little to no dystrophin protein is produced
- BMD is caused by mutations which do not alter the reading frame and some protein is produced
- Clinically, DMD is a more severe, lethal disorder with earlier onset and more rapid deterioration
- Histologically, both show myopathic changes and decreased staining with dystrophin
- DMD shows more severe changes
Terminology
- X linked dystrophinopathy includes both BMD and DMD
ICD coding
- ICD-10: G71.0: muscular dystrophy
Epidemiology
- Both BMD and DMD are X linked and therefore are seen almost exclusively in males
- DMD is the most common muscular dystrophy, with an incidence of 1:5,000 live male births (Curr Opin Neurol 2019;32:722)
Sites
- X linked typically affects proximal muscle groups, especially the lower extremities, with variable cardiomyopathy
Pathophysiology
- Mutations in the DMD gene lead to reduced production of a truncated dystrophin protein, which normally functions to stabilize the sarcolemmal membrane (Continuum (Minneap Minn) 2019;25:1619)
Etiology
- Because dystrophin is located on the X chromosome, dystrophinopathies are X linked
- Men are affected; women can be carriers and exhibit cardiomyopathy (Pediatr Clin North Am 2015;62:723)
Clinical features
- Heterogenous presentation, typically proximal muscle weakness affecting lower extremities more than upper extremities
- Course is more severe for DMD
- Symptoms begin at earlier age (mean age of diagnosis is 4 years)
- Delayed motor milestones
- Waddling gait
- Difficulty rising from floor (Gower maneuver)
- Difficulty climbing stairs, running and jumping
- Hypertrophy of calf muscles
- Wheelchair needed by age 10
- Contractures
- Most patients expire by third decade of life (Semin Neurol 2015;35:369)
- BMD is less severe
- Symptoms begin between 5 - 15 years of age
- Wheelchair needed after age 16
- More severe cardiomyopathy (Continuum (Minneap Minn) 2019;25:1619)
Diagnosis
- Relies on clinical features in combination with genetic testing and creatinine kinase levels
- Timed function tests (patient asked to walk for 6 minutes)
- Muscle biopsy is less frequently performed but is useful for assessment of dystrophin expression (Pediatr Clin North Am 2015;62:723)
Laboratory
- DMD has extremely elevated creatinine kinase levels, usually 50 - 100 times normal
- Elevation may decrease over course of disease
- Up to 70% of carriers may have elevated creatinine kinase levels
- BMD has elevated creatinine kinase levels
- Highest levels seen in 10 - 15 years of age
- Alanine transaminase (ALT) and aspartate transaminase (AST) may be elevated
- Can see occasional myoglobinuria following strenuous activity (Pediatr Clin North Am 2015;62:723)
Radiology description
- Skeletal muscle ultrasound and magnetic resonance imaging can show amount of fibrosis and atrophy in muscle groups (Continuum (Minneap Minn) 2019;25:1619)
Prognostic factors
- DMD is progressive and invariably fatal, with death typically occurring in the third or fourth decade of life
- BMD has an extremely variable prognosis with some patients following a course like DMD and some having only mild muscle weakness (Semin Neurol 2015;35:369)
Case reports
- Symptomatic Duchenne muscular dystrophy carrier (J Neurol Sci 2014;336:36)
- 13 year old boy with a mutation in DMD causing Becker muscular dystrophy associated with intellectual disability (J Dev Behav Pediatr 2016;37:239)
- 15 year old boy with Duchenne muscular dystrophy (BMJ Case Rep 2014;2014:bcr2014205296)
- 18 year old man with a case of Becker muscular dystrophy with early manifestation of cardiomyopathy (Korean J Pediatr 2012;55:350)
Treatment
- No curative treatment for the X linked dystrophinopathies
- Corticosteroids are the primary therapy in DMD (N Engl J Med 1989;320:1592)
- Intensive physical therapy is the primary treatment in BMD
- Corticosteroids are used in severe cases
- Numerous clinical trials for gene therapy are currently in progress (Pediatr Clin North Am 2015;62:723)
- One of the most promising is adeno-associated virus (AAV) based gene replacement therapy (Curr Opin Neurol 2019;32:722)
Microscopic (histologic) description
- X linked dystrophinopathies include similar histologic changes with difference in severity
- DMD has more pronounced changes than BMD
- Variation in myofiber size with small atrophic fibers admixed with large, rounded, hypertrophic fibers
- Increased internal nuclei
- Myofiber splitting, necrosis, phagocytosis and regeneration
- Increased endomysial fibrosis and fatty replacement of muscle (more prominent later in disease course)
- May have inflammation (macrophages, T cells) in association with necrosis
- Architectural changes (whorled fibers, moth eaten fibers) may be seen (Neurology 2011;76:346)
- Carriers of DMD may demonstrate histologic abnormalities as well as a mosaic pattern of dystrophin expression
Microscopic (histologic) images
Contributed by Jesse L. Kresak, M.D.
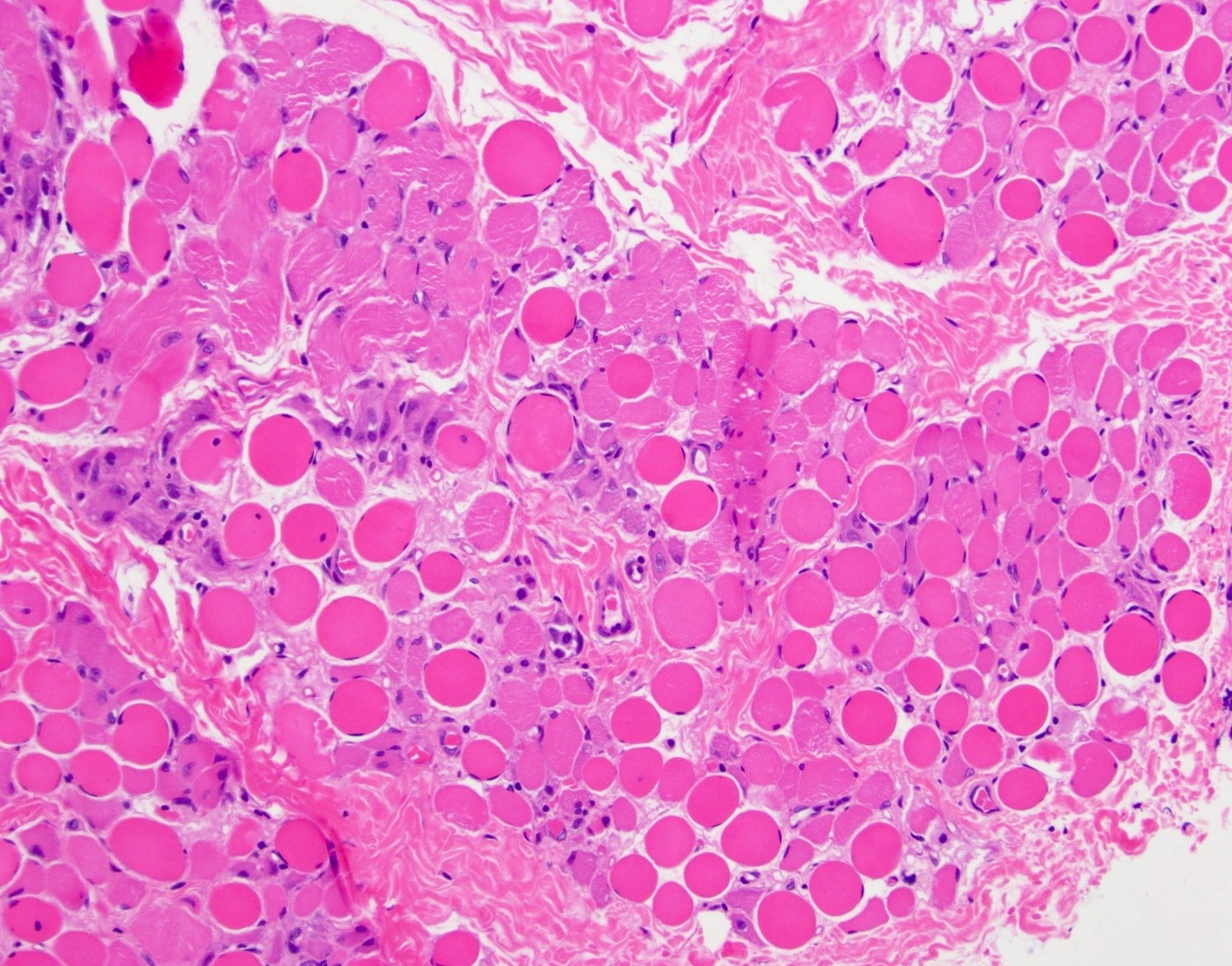
Myopathic changes
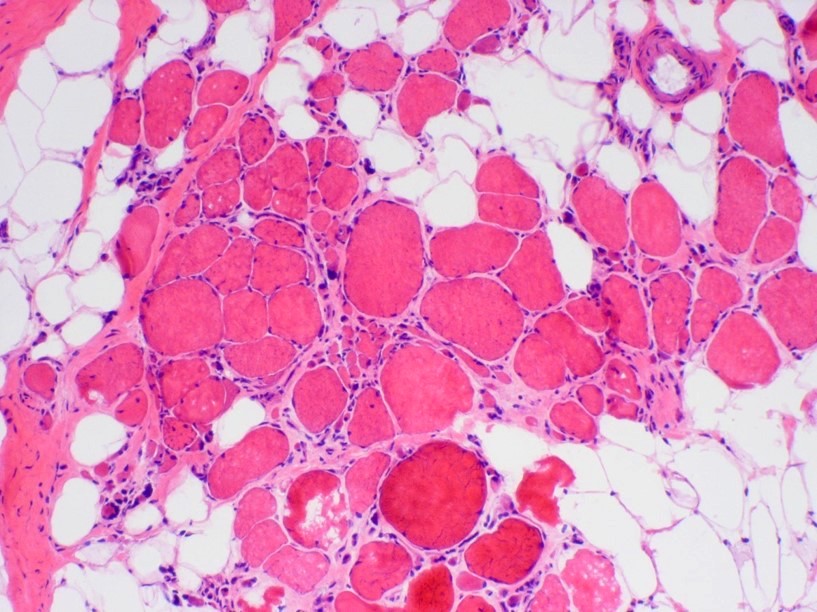
Fatty replacement

End stage muscle
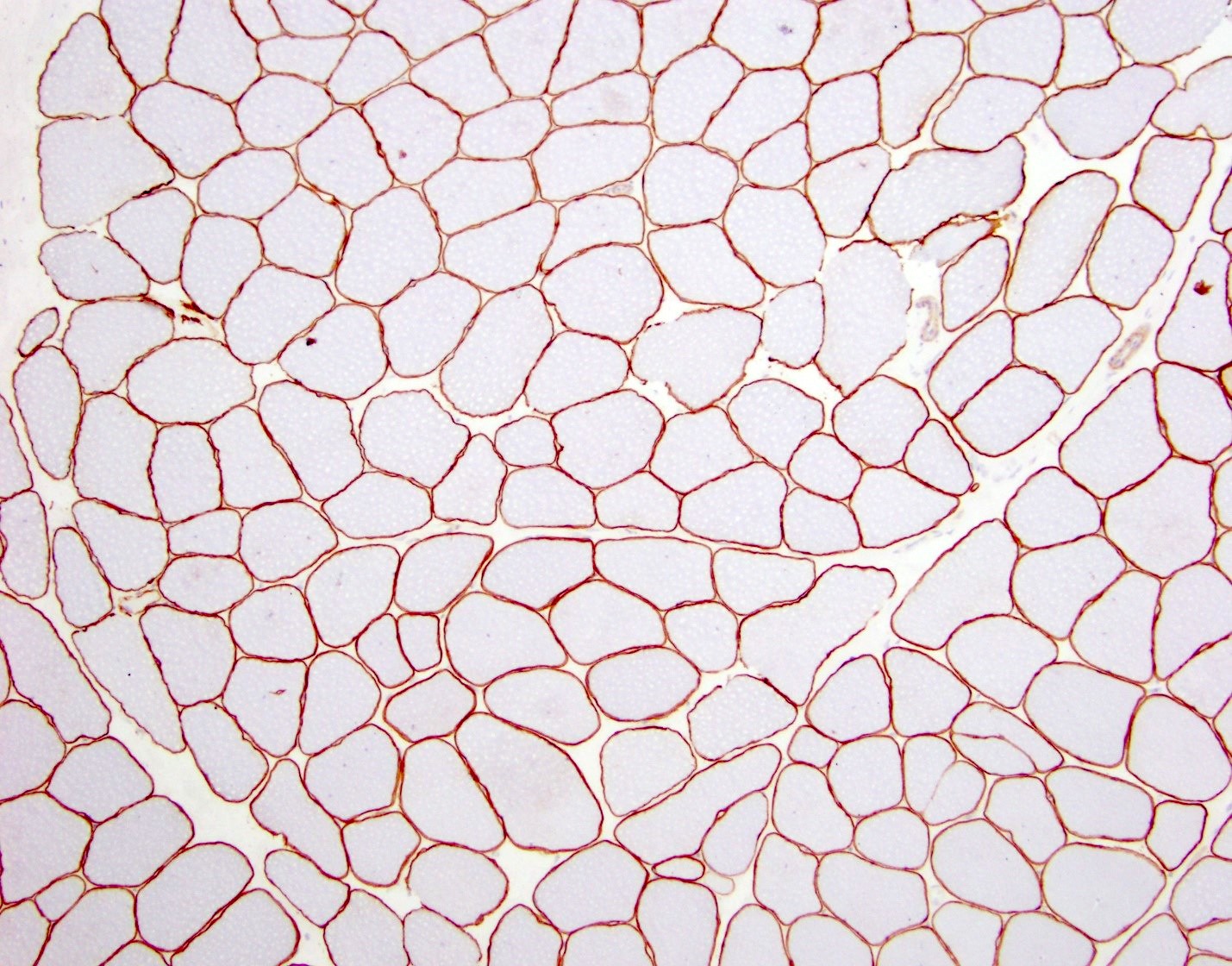
Dystrophin IHC control
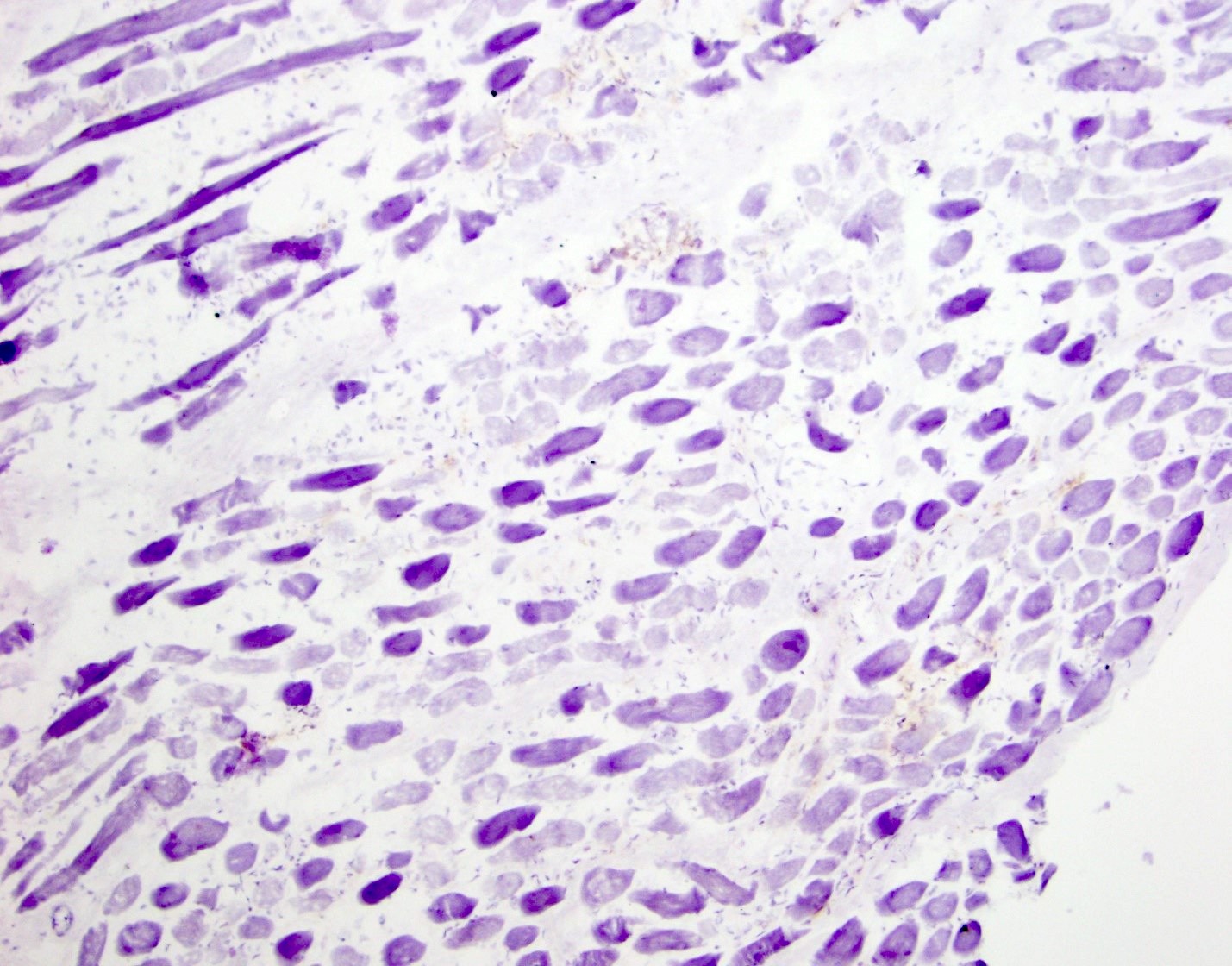
Dystrophin IHC in Duchenne muscular dystrophy
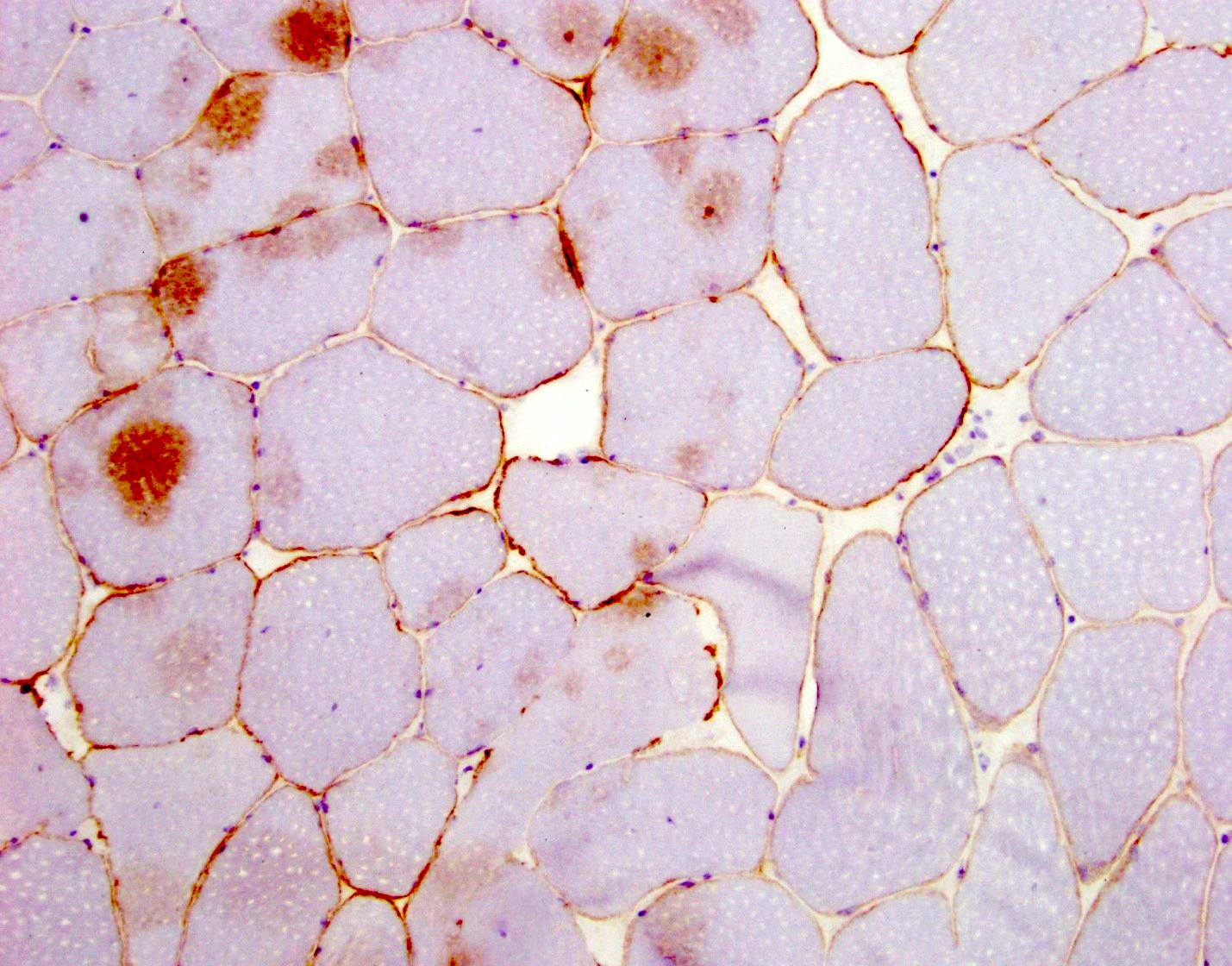
Dystrophin IHC in Becker muscular dystrophy
Positive stains
- NADH and SDH oxidative stains may show nonspecific myofibrillar changes, such as moth eaten, lobulated or whorled fibers
- Expression of other proteins is sometimes increased including utrophin and fetal myosins (Neuromuscul Disord 1992;2:177)
Negative stains
- 3 antibodies which recognize epitopes in different domains are used to assess dystrophin expression (DYS1 - rod domain, DYS2 - C terminal, DYS3 - N terminal)
- In general, most cases of DMD show an absence of the C terminus, while in most BMD cases it is preserved (Dubowitz: Muscle Biopsy - A Practical Approach, 5th Edition, 2020)
- In DMD, staining for dystrophin will show absent to markedly reduced expression in sarcolemma of myofibers
- 95 - 100% of fibers will be negative
- May have secondary reduced expression of proteins in the dystrophin associated complex, such as dystroglycan and sarcoglycans to variable degrees
- BMD will show reduced dystrophin expression
- 50 - 80% of fibers will be negative
- Female carriers can have greater than 60% of fibers that stain for dystrophin (Semin Neurol 2015;35:369)
Molecular / cytogenetics description
- Both X linked dystrophinopathies are caused by mutations in the dystrophin gene on Xp21
- In DMD, mutations commonly disrupt the reading frame, which severely reduces or eliminates normal protein production
- In BMD, mutations commonly do not alter the reading frame and some protein is still produced (Pediatr Clin North Am 2015;62:723)
Sample pathology report
- Quadriceps, muscle biopsy:
- Skeletal muscle with late stage myopathy, consistent with dystrophinopathy (see comment)
- Comment: H&E stained sections demonstrate skeletal muscle with features of myopathy, including marked myofiber size variation ranging from minute, atrophic myofibers to hypertrophic forms exhibiting splitting, increased myofibers with internal nuclei and scattered degenerating / regenerating myofibers. The endomysium is expanded by collagen deposition and is replaced by adipose tissue in areas, indicative of a chronic process evolving into end stage muscle changes. A targeted immunohistochemical panel for muscular dystrophy reveals loss of expression of dystrophin 1, dystrophin 2 and dystrophin 3 proteins, with reduced / attenuated expression of sarcoglycan B and sarcoglycan D proteins. Considering the results of this panel together with the advanced stage changes within this patient's skeletal muscle, these findings are consistent with muscular dystrophy, specifically a dystrophinopathy. The differential diagnosis includes Duchenne and Becker muscular dystrophies. Definitive determination of muscular dystrophy type is based on genetic study results. Clinicopathologic correlation is highly suggested.
Differential diagnosis
- Congenital muscular dystrophies:
- Heterogenous group of disorders that present in neonates and infants with hypotonia, muscle weakness, atrophy and joint deformities
- Limb girdle muscular dystrophy:
- Heterogeneous group of inherited muscular dystrophies, which can clinically present like BMD / DMD
- May require molecular testing to differentiate
- Spinal muscular atrophy
- Group of autosomal recessive disorders due to degeneration of spinal anterior horn motor neurons
- Clinically will have tongue fasciculations
- Congenital myopathies such as central core disease, centronuclear myopathy, nemaline myopathy:
- Frequently have earlier onset
- Characteristic histologic findings and staining pattern based on disease type
- Metabolic myopathy:
- Heterogenous group of disorders in cellular energy metabolism that range for severe infantile disease to adult onset mild disease
Additional references
Board review style question #1
Mutations in what gene, whose product is shown in this immunohistochemical stain, are responsible for the Duchenne and Becker muscular dystrophies?
- BKR
- DMD
- MTM1
- NEB
- RYR1
Board review style answer #1
Board review style question #2
What inheritance pattern do Duchenne and Becker muscular dystrophies have?
- Autosomal dominant
- Autosomal recessive
- De novo
- Mitochondrial
- X linked recessive
Board review style answer #2
