

Muscle & peripheral nerve nontumor
Metabolic myopathies
Glycogen storage diseases


Copyright: 2020-2024, PathologyOutlines.com, Inc.
PubMed Search: Glycogen storage diseases

- Glycogen storage diseases (GSD) are inherited metabolic disorders of glycogen metabolism
- Occur due to a lack of specific enzymes that control the synthesis, regulation and degradation of glycogen
- After the discovery of deficient glucose 6 phosphatase in von Gierke disease (GSD I) by Carl F. Cori and Gerty T. Cori, a number of inborn errors of glycogen metabolism have been recognized (J Biol Chem 1929;81:389, J Med Biogr 2021;29:143)
- Classified numerically in the order of recognition and identification of the enzyme defect causing the disorder
- There are 15 types, most of them are autosomal recessive, except for X linked GSD IXd, phosphoglycerate kinase deficiency (J Endocrinol 2018;238:R131)
- The most common presenting symptoms are hypoglycemia and exercise intolerance
- Danon disease (lysosomal glycogen storage disease without acid maltase deficiency, pseudoglycogenosis II) caused by deficiency of lysosome associated membrane protein 2 (LAMP2), led to failure of cellular autophagy with accumulation of glycogen granules and intracytoplasmic vacuoles containing autophagic material; recently not recognized as GSD (Cell Death Dis 2017;8:e2565)
- GSD affecting the skeletal muscles are mainly involved
- GSD 0 (glycogen synthase 1 deficiency)
- GSD II (acid maltase deficiency, Pompe disease)
- GSD III (debrancher enzyme deficiency, Cori-Forbes disease)
- GSD IV (branching enzyme deficiency, Andersen disease)
- GSD V (glycogen phosphorylase deficiency, McArdle disease)
- GSD VII (phosphofructokinase deficiency, Tarui disease)
- GSD IXd (phosphorylase kinase deficiency)
- Phosphoglycerate kinase deficiency
- GSD X (phosphoglycerate mutase deficiency)
- GSD XI (lactate dehydrogenase deficiency)
- GSD XII (aldolase A deficiency)
- GSD XIII (β enolase deficiency)
- GSD XIV (phosphoglucomutase deficiency)
- GSD XV (glygogenin 1 deficiency)
- Rare
- 1 per 20,000 - 43,000 live births (Pediatrics 2000;105:e10)
- GSD I / GSD VI only cause liver disease, while GSD II / GSD V / GSD VII / GSD X / GSD XI cause predominantly muscle disease; the rest cause mixed muscle / systemic disease (Amato: Neuromuscular Disorders, 2nd Edition, 2015)
- GSD 0 / GSD II / GSD III / GSD IV / GSD XIV / GSD XV can cause cardiomyopathy; GSD IV can involve the nervous system (adult polyglucosan body disease) (Dubowitz: Muscle Biopsy - A Practical Approach, 5th Edition, 2020)
- Primary physiologic function of glycogen is to store and provide glucose
- Liver glycogen stores are utilized to maintain glucose homeostasis in the serum; the muscle glycogen stores are utilized as a source of energy for muscular activity (Semin Hematol 2002;39:103)
- Aerobically, glucose is metabolized into pyruvate and acetyl CoA, which enters the citric acid cycle to produce water, carbon dioxide and adenosine triphosphate (ATP) or is used for the synthesis of fatty acids
- Under anaerobic conditions, glucose is metabolized to lactate (Curr Mol Med 2002;2:101)
- Failure to maintain glycolytic pathway (glycogenesis, glycogenolysis or glycolysis) leads to glycogen storage diseases
- Defects in the enzymes caused by inherited mutations that are required for glycolytic pathway, such as glycogen synthase (encoded by the GYS1 and GYS2 genes), acid maltase (GAA gene), branching enzyme (GBE1 gene), debrancher enzyme (AGL gene), myophosphorylase (PYGM gene), phosphorylase (PYGM), phosphofructokinase (PFKM), phosphorylase kinase (PHKA1), phosphoglycerate kinase (PGK1), phosphoglycerate mutase (PGAM2), lactate dehydrogenase (LDHA), aldolase A (ALDOA), β enolase (ENO3), phosphoglucomutase (PGM1), glygogenin 1 (GYG1) (World J Gastroenterol 2007;13:2541)
Contributed by Truong Phan Xuan Nguyen, M.D.

Metabolic glycolytic pathway
| GSD 0 | GYS1 (skeletal muscle) or GYS2 (liver) | Glycogen synthase 1 | Autosomal recessive | Glycogen synthase 1 deficiency |
| GSD II | GAA | Acid maltase | Autosomal recessive | Acid maltase deficiency; Pompe disease |
| GSD III | AGL | Debrancher enzyme | Autosomal recessive | Debrancher enzyme deficiency; Cori-Forbes disease |
| GSD IV | GBE1 | Branching enzyme | Autosomal recessive | Branching enzyme deficiency; Andersen disease |
| GSD V | PYGM | Glycogen phosphorylase | Autosomal recessive | Glycogen phosphorylase deficiency; McArdle disease |
| GSD VII | PFKM | Phosphofructokinase | Autosomal recessive | Phosphofructokinase deficiency; Tarui disease |
| GSD IXd | PHKA1 | Phosphorylase kinase | X linked inheritance | Phosphorylase kinase deficiency |
| Phosphoglycerate kinase deficiency | PGK1 | Phosphoglycerate kinase | X linked inheritance | N/A |
| GSD X | PGAM2 | Phosphoglycerate mutase | Autosomal recessive | Phosphoglycerate mutase deficiency |
| GSD XI | LDHA | Lactate dehydrogenase | Autosomal recessive | Lactate dehydrogenase deficiency |
| GSD XII | ALDOA | Aldolase A | Autosomal recessive | Aldolase A deficiency |
| GSD XIII | ENO3 | β enolase | Autosomal recessive | β enolase deficiency |
| GSD XIV | PGM1 | Phosphoglucomutase | Autosomal recessive | Phosphoglucomutase deficiency |
| GSD XV | GYG1 | Glygogenin 1 | Autosomal recessive | Glygogenin 1 deficiency |
- Muscle cramps, exercise intolerance, muscle weakness (hypotonia), hypoglycemia and fatigue are typical symptoms of GSDs that relate to the energy deficiency in skeletal muscle
- Rhabdomyolysis and myoglobinuria may lead to renal failure in severe cases (Goebel: Muscle Disease, 2nd Edition, 2013)
- Clinical spectrum is highly variable, ranging from severe exercise intolerance or multisystem involvement in infancy to isolated progressive muscle weakness in adulthood (Gaspar: Myopathology A Practical Clinico-pathological Approach to Skeletal Muscle Biopsies, 1st Edition, 2019)
- Late onset GSD II is characterized by proximal muscle weakness and respiratory insufficiency (Aging 2020;12:15856)
- In GSD V, the second wind phenomenon is the hallmark, which is the alleviation of myalgia and exhaustion after a swift increase in physical activity (Ann Neurol 2003;54:539)
- GSD II is part of the newborn screening panel by measurement of acid α glucosidase (GAA) enzyme activity in dried blood spots for early diagnosis (J Neurol Neurosurg Psychiatry 2022 Apr 25 [Epub ahead of print], Int J Neonatal Screen 2020;6:31)
- Genetics and muscle pathology can confirm diagnosis
- Nonischemic forearm exercise shows a flat lactate response and marked hyperammonemia in GSD V, GSD VII (Ann Neurol 2002;52:153)
- Exercise to exhaustion on a cycle ergometer shows severely impaired maximal oxidative capacity in GSD V, GSD VII (Acta Neurol Scand 2018;138:301)
- Serum creatine kinase (CK) levels are high in GSDs but may be normal in late onset cases of GSD II (Gaspar: Myopathology A Practical Clinico-pathological Approach to Skeletal Muscle Biopsies, 1st Edition, 2019, Genet Med 2006;8:267)
- Cutoff in the GAA activity assay using dried blood spots is set between 0.1 and 0.5 percentile values for the population or 20 - 30% of the mean value of the population (6.5 pmol/h/disk)
- A second GAA activity assay and GAA gene analysis are performed if newborns have values that are less than the cutoff (Int J Neonatal Screen 2020;6:31)
- Chest Xray: massive cardiomegaly in GSD II infant onset (Genet Med 2006;8:267)
- MRI in GSD II:
- In late onset, T1 weighted shows fatty replacement commonly in the tongue, subscapularis, latissimus dorsi, paraspinal and abdominal muscles, psoas, glutei, adductor magnus, posterior muscles of the thigh (especially the semimembranosus) and vastus intermedius
- Vastus lateralis and medialis can be affected in the progression of the disease
- Noticeable loss of muscle volume in the psoas and adductor magnus muscles
- In infant onset, T1 weighted shows normal or identifies only mild fatty replacement, although patients may have significant muscle weakness; muscles commonly affected include the tongue, glutei and adductor magnus muscles (Muscle Nerve 2021;63:640)
- In late onset, T1 weighted shows fatty replacement commonly in the tongue, subscapularis, latissimus dorsi, paraspinal and abdominal muscles, psoas, glutei, adductor magnus, posterior muscles of the thigh (especially the semimembranosus) and vastus intermedius
- Infant onset is aggressive and can be fatal in some GSDs (Ultrastruct Pathol 2011;35:183)
- Early diagnosis and proper treatment are important for good prognosis (Ann Transl Med 2018;6:474)
- Infant boy with hypertrophic cardiomyopathy in infant onset Pompe disease (GSD II) (Medicine (Baltimore) 2017;96:e9186)
- 14 year old boy with seizures and rhabdomyolysis in GSD XII (Ital J Pediatr 2022;48:39)
- 23 year old man with several episodes of rhabdomyolysis in phosphoglycerate kinase deficiency (Intern Med 2022 May 7 [Epub ahead of print])
- 35 year old woman with cardiomyopathy after a pregnancy in late onset Pompe disease (GSD II) (JIMD Rep 2017;31:79)
- 40 year old woman with severe left forearm pain after moving furniture was diagnosed with McArdle disease (GSD V) (J Hand Surg Am 2015;40:2377)
- Most treatments involve symptomatic management and surveillance (Hum Mol Genet 2019;28:R31)
- GSD II can be treated with enzyme replacement therapy (ERT) and gene therapy (Neurotherapeutics 2018;15:928)
- GSD 0, muscle:
- Glycogen depletion in all fibers
- Mitochondrial proliferation
- Type I fiber predominance
- Phosphorylase is deficient in all muscle fibers
- GSD II:
- Histopathological features vary with the phenotypic forms: infant form, childhood form and adult form
- Marked sarcoplasmic membrane bound vacuoles containing basophilic amorphous material (glycogen) in most fibers
- Type 1 muscle fibers are more involved than type 2
- Little muscle fiber degeneration or increased connective tissue
- Abundant acid phosphatase activity (increased lysosomes)
- GSD III:
- Subsarcolemmal nonmembrane bound vacuoles with glycogen accumulation
- At a later stage, the muscle may appear dystrophic with atrophy and connective tissue infiltration and no apparent glycogen storage
- GSD IV:
- Adult form shows PAS positivity and sarcoplasmic rounded opalescent inclusions
- Perinatal forms show polyglucosan bodies which have variable size, shape and reaction to PAS
- GSD V, VII, IXd:
- Muscle pathology is variable from minor nonspecific changes to subsarcolemmal nonrimmed vacuoles and occasional fibers with accumulation
- Excess glycogen on PAS staining may be visible but may only be apparent at the periphery of the fiber
- GSD X, XIV, XV, phosphoglycerate kinase deficiency:
- May reveal variation in myofiber size with normal to mild glycogen distribution
- Reference: Goebel: Muscle Disease, 2nd Edition, 2013
Contributed by Ichizo Nishino, M.D., Ph.D.
GSD 0: no specific changes
GSD 0: glycogen depletion
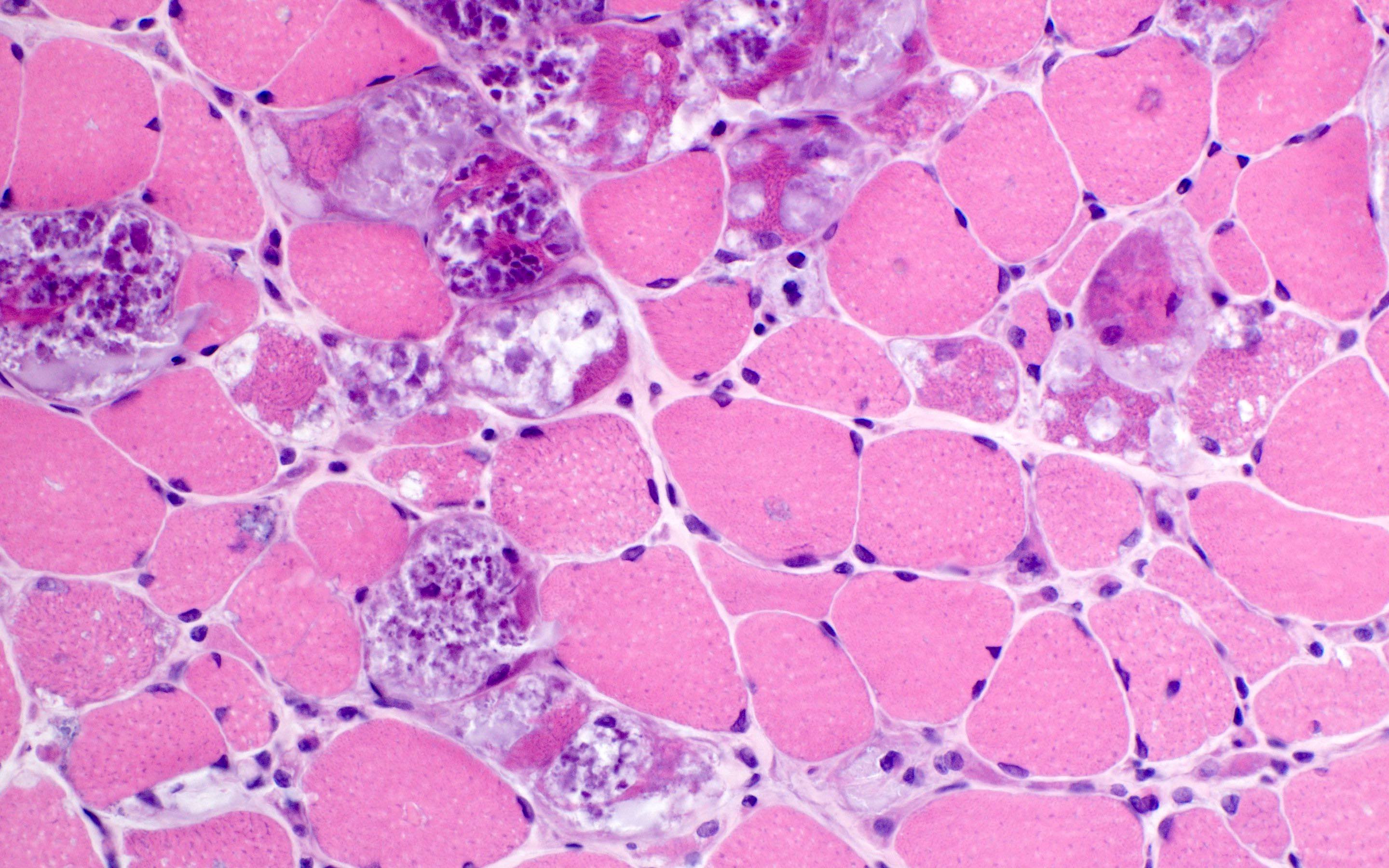
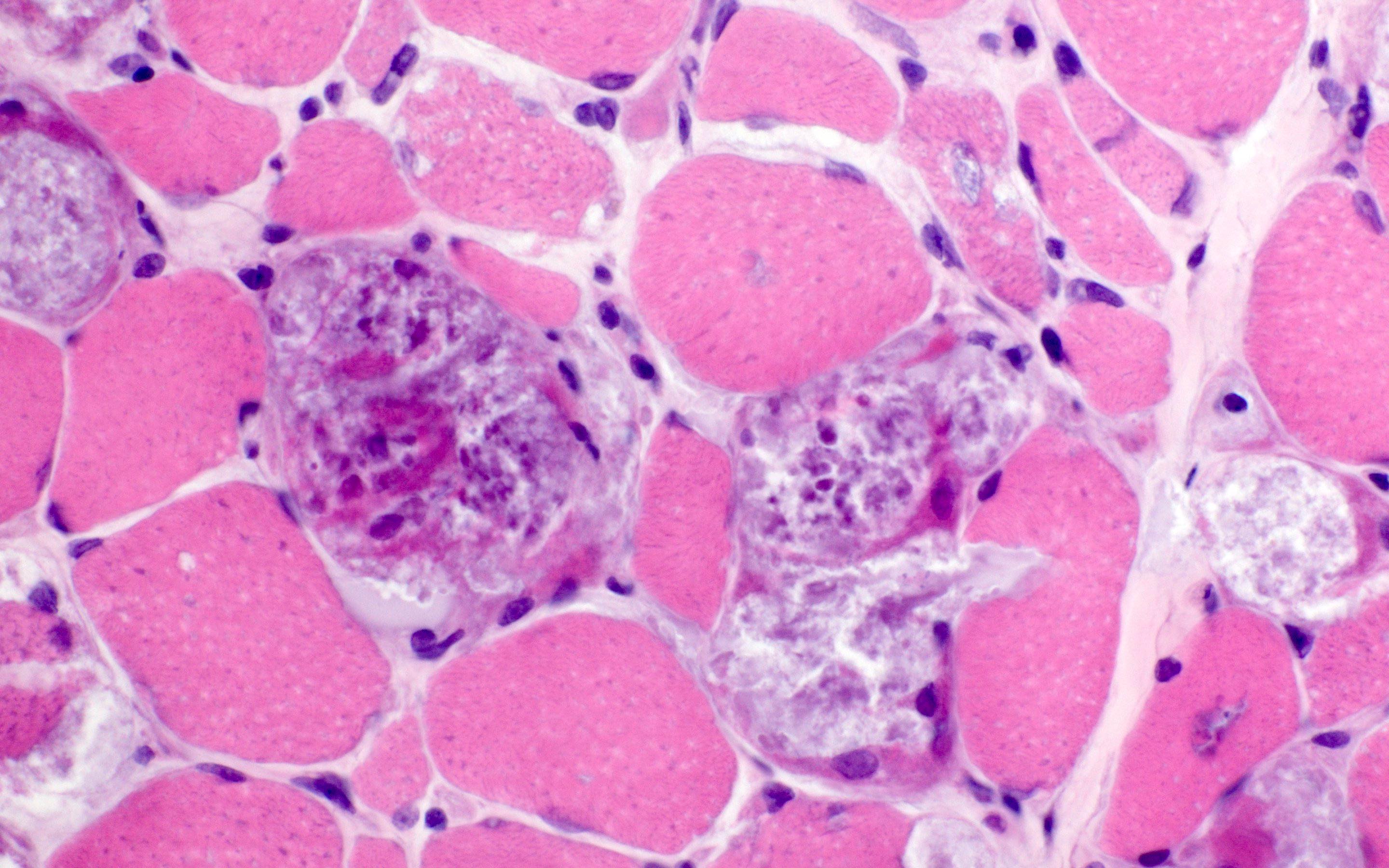
GSD II childhood: marked sarcoplasmic vacuoles
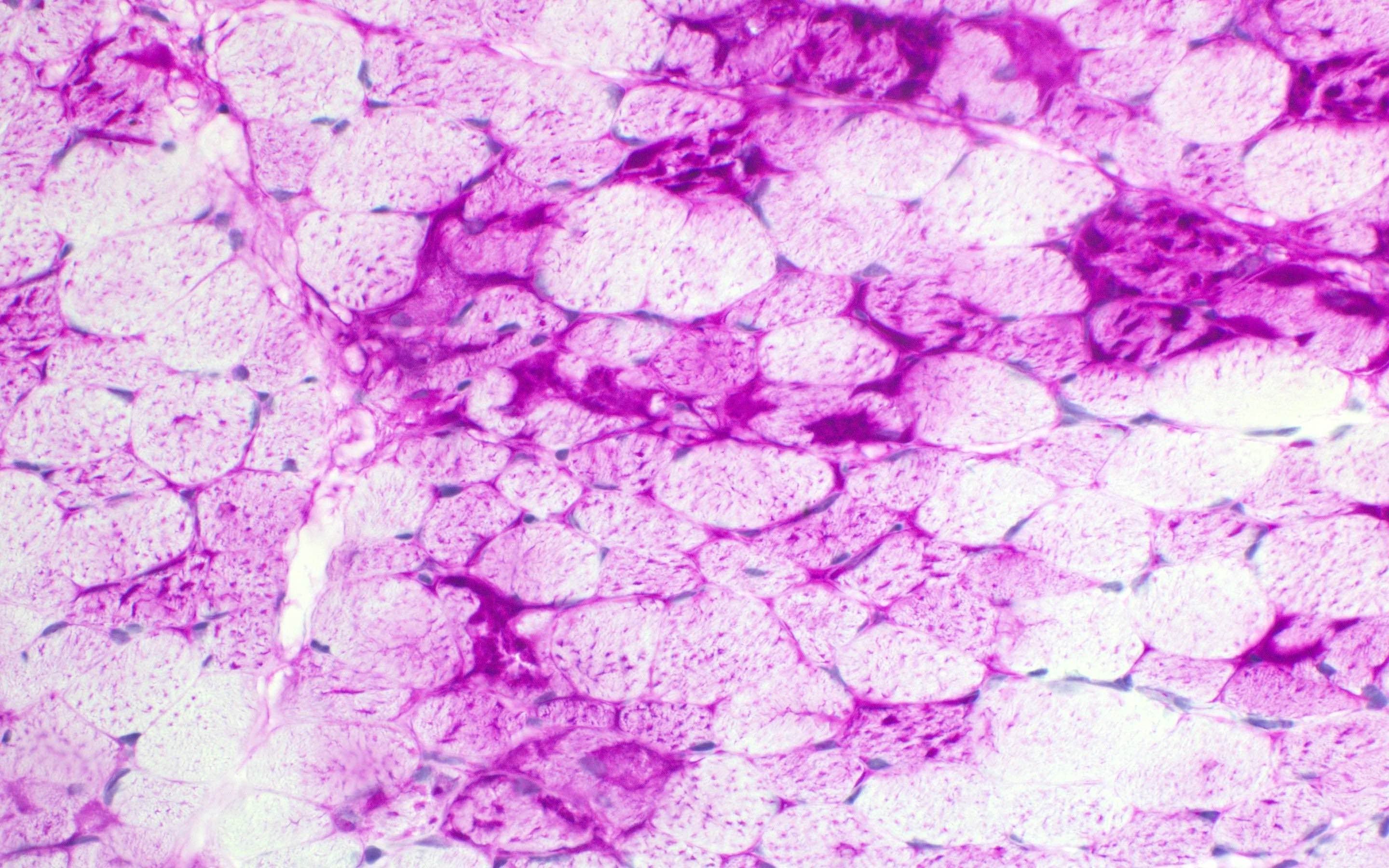
GSD II childhood: PAS

GSD II childhood: epon PAS

GSD II childhood: NADH TR

GSD II childhood:
predominant
involved
type 1 fibers
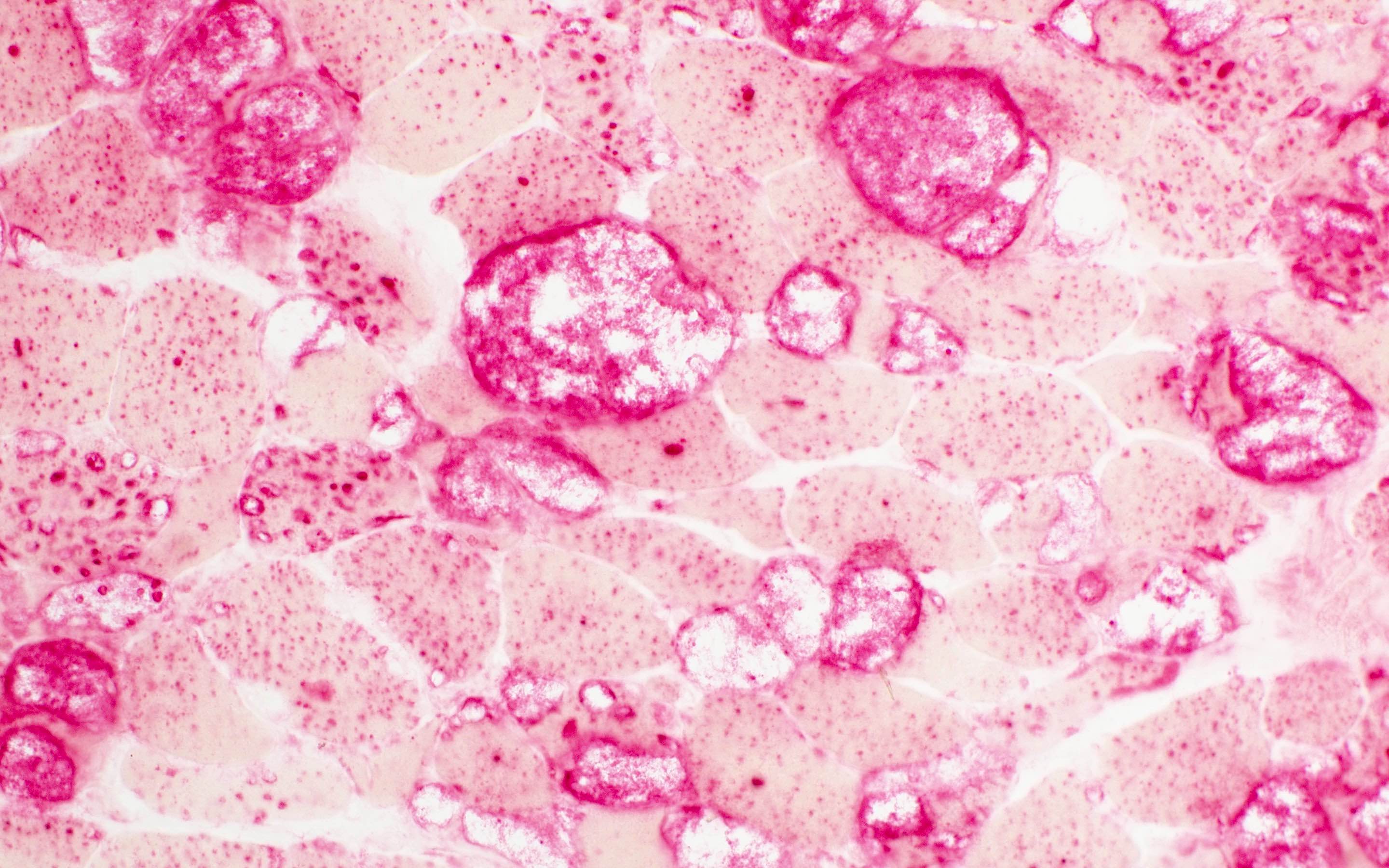
GSD II childhood:
high acid
phosphatase
activity
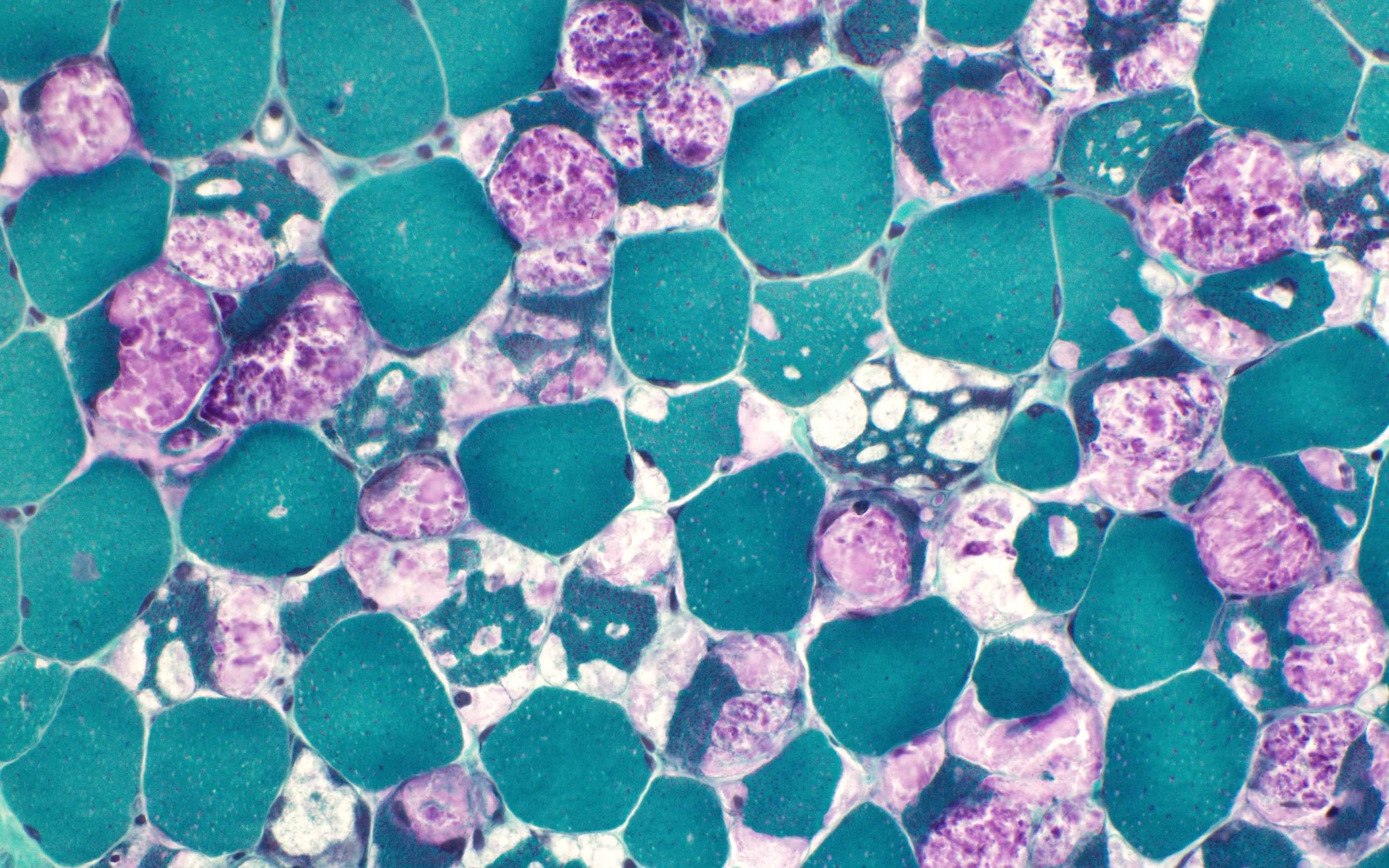
GSD II childhood: modified Gomori trichrome
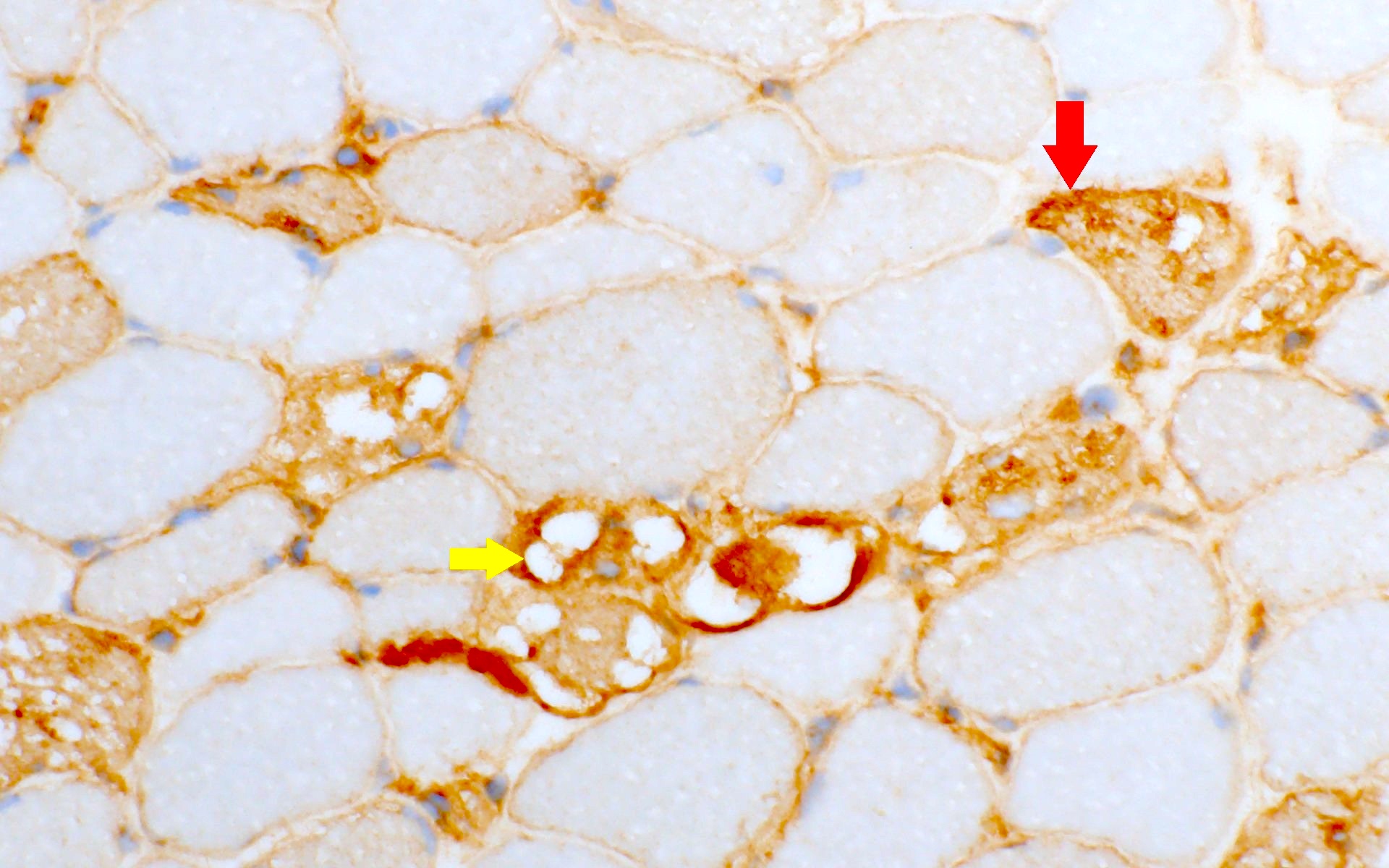
GSD II childhood:
MHC I

GSD II adult: small vacuoles
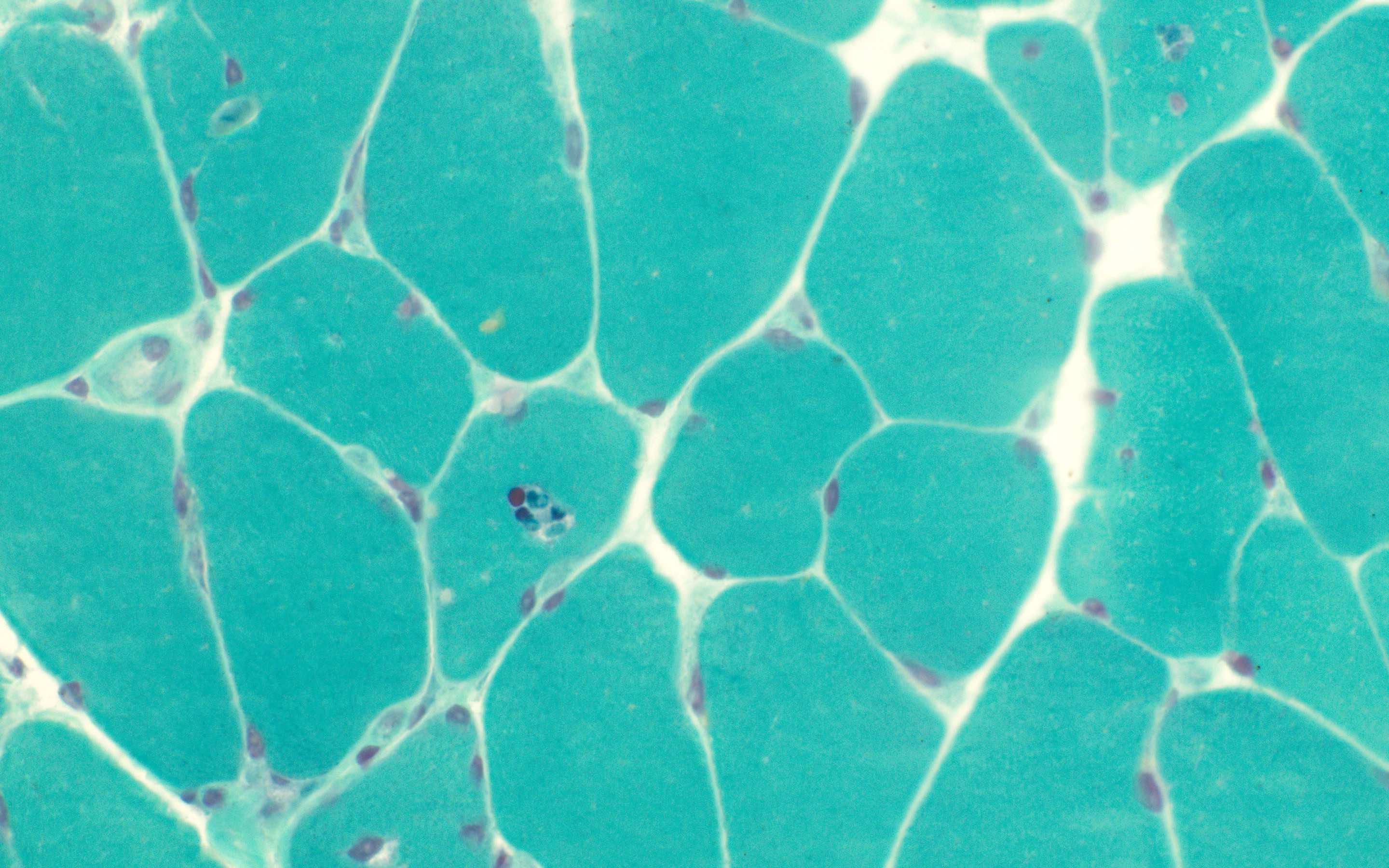
GSD II adult: modified Gomori trichrome
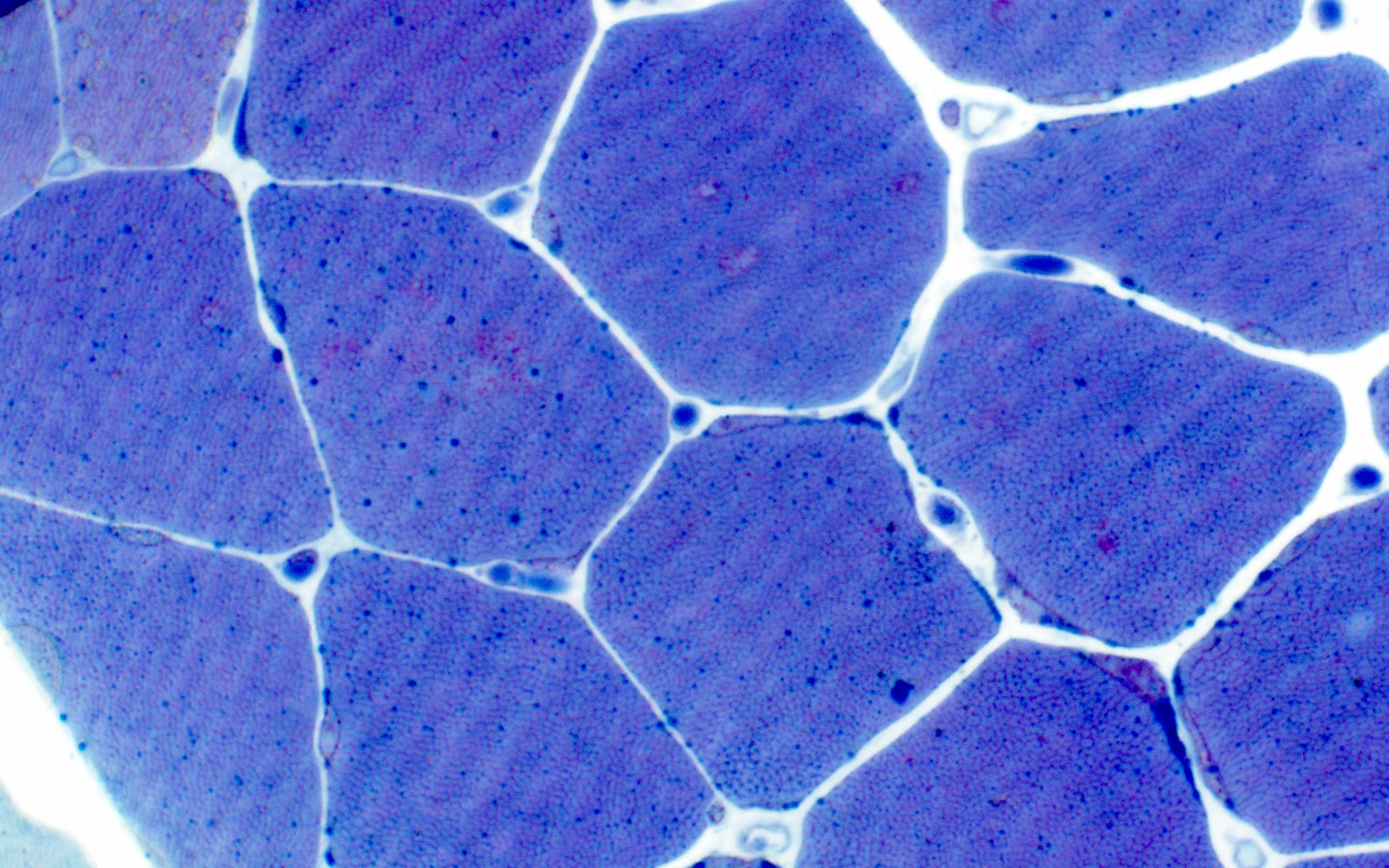
GSD II adult: epon PAS
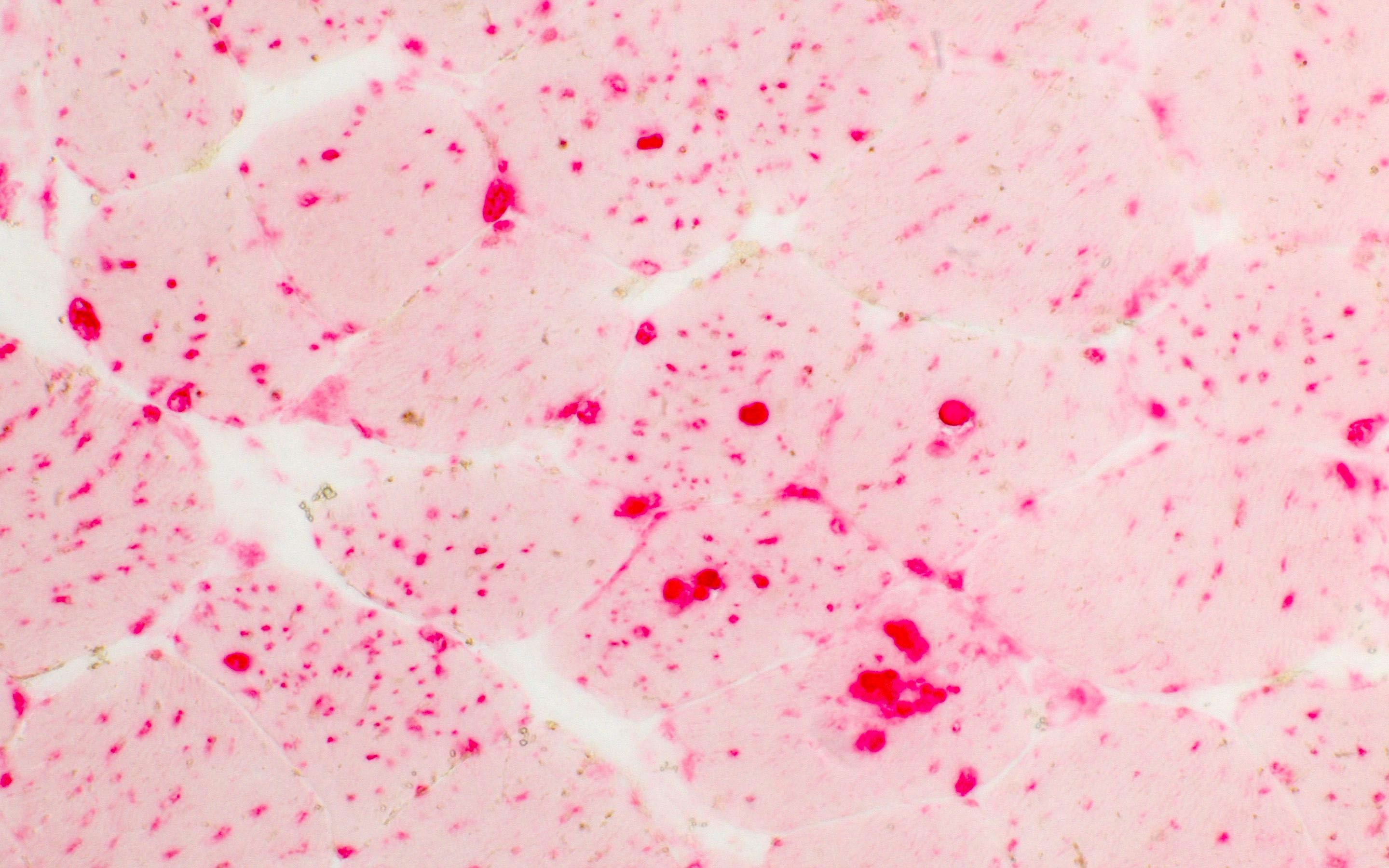
GSD II adult: increased acid phosphatase activity
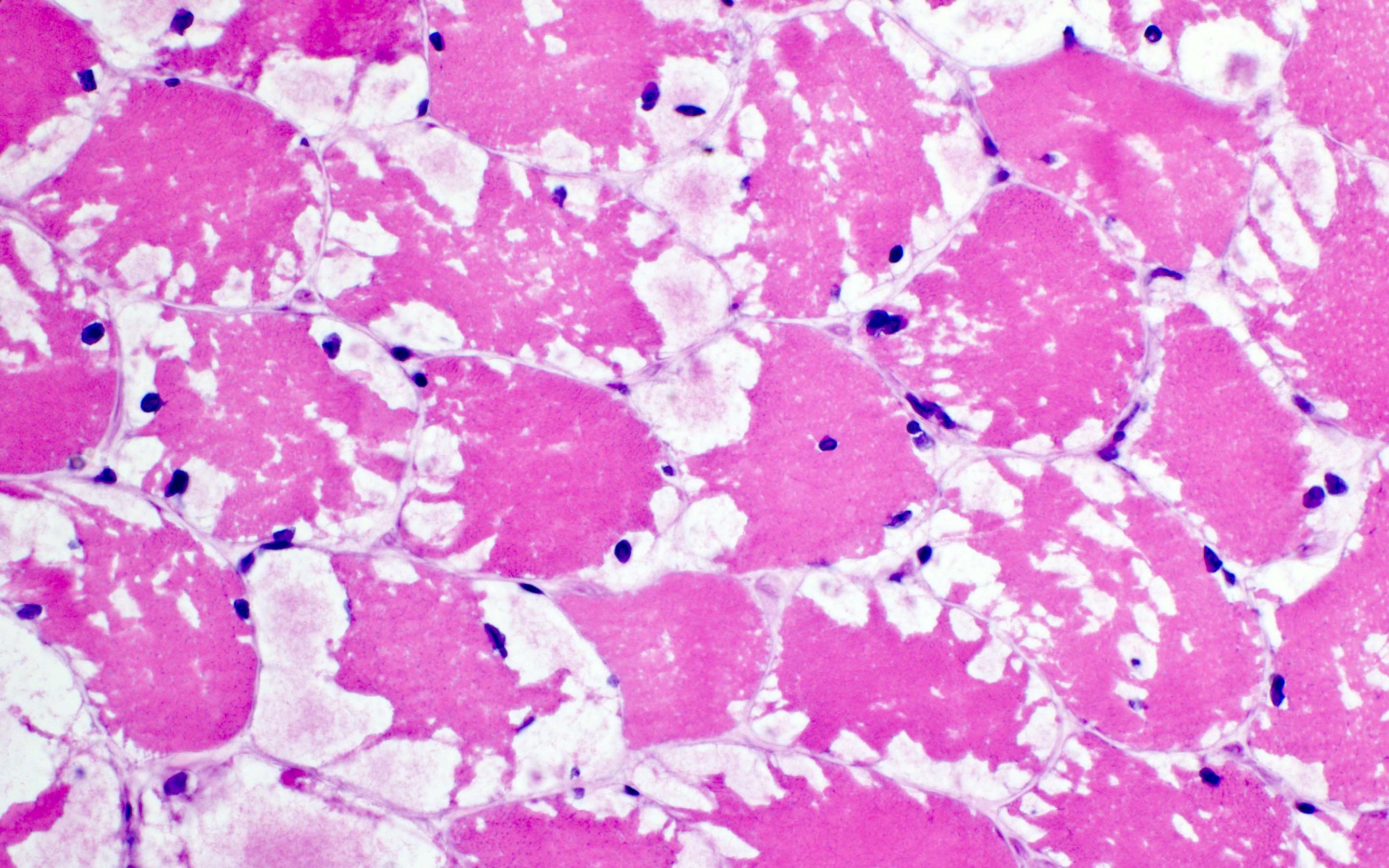
GSD III: nonmembrane bound vacuoles
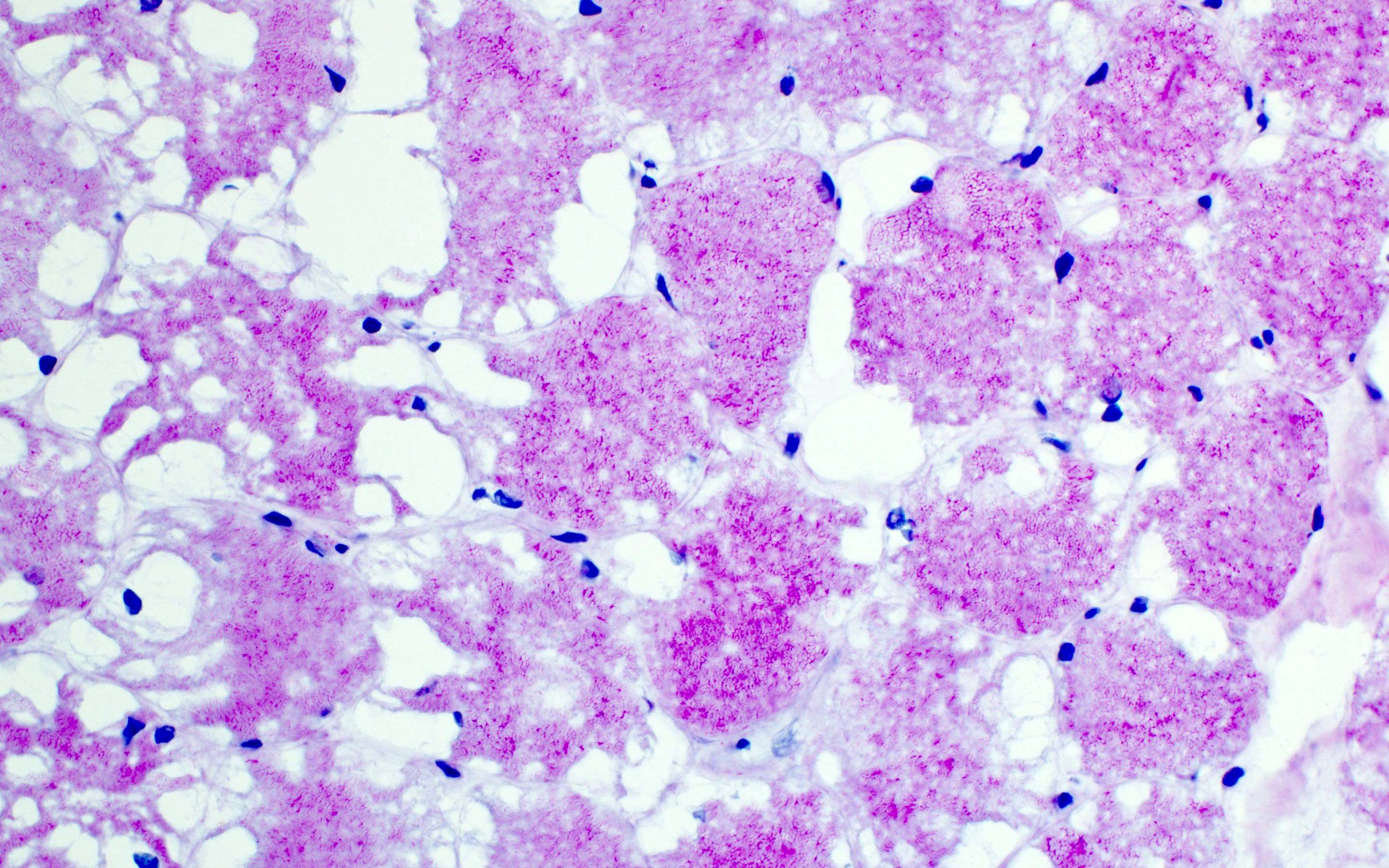
GSD III: PAS positive
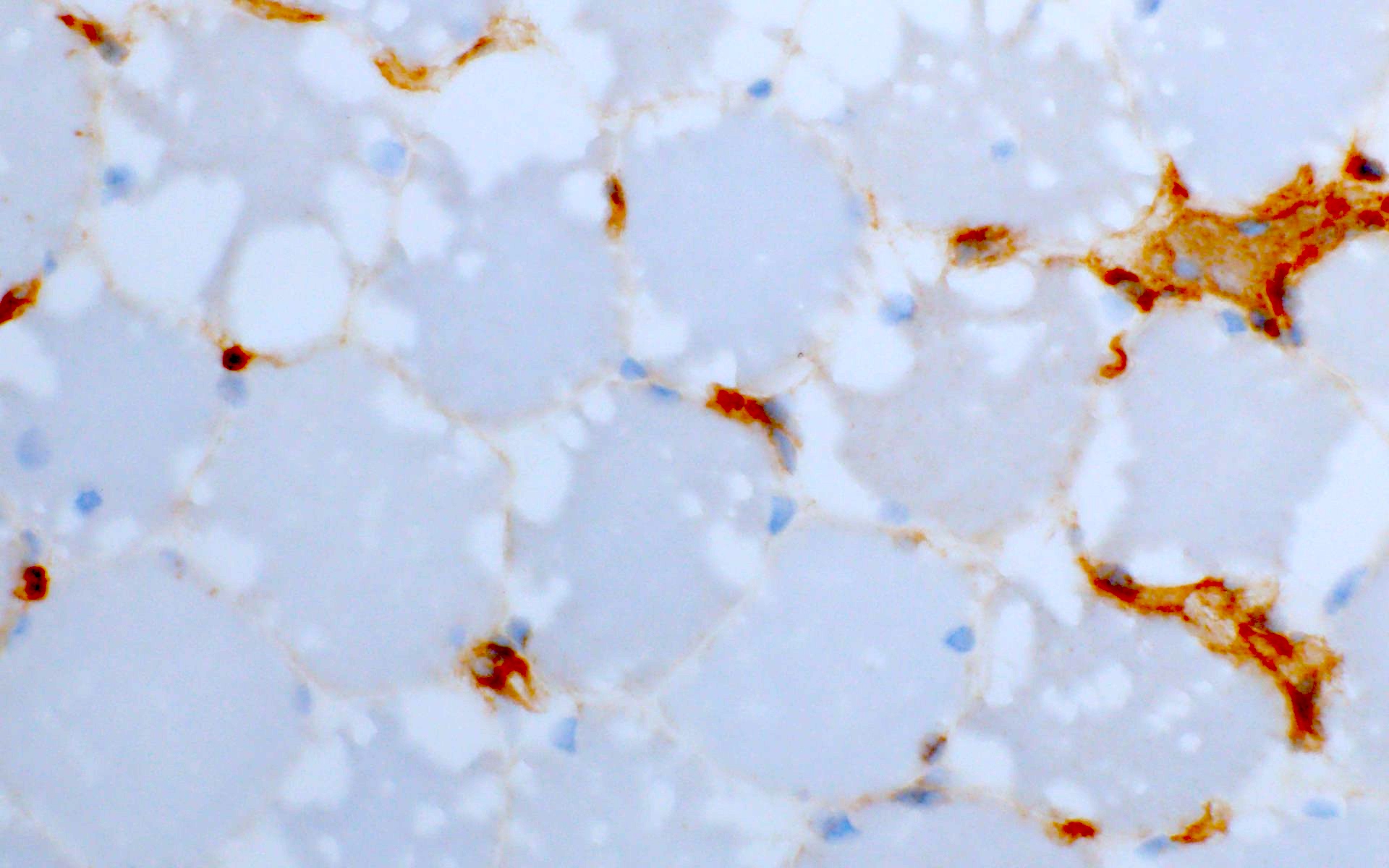
GSD III: MHC I

GSD IV: fibrosis
fibers and rounded
opalescent
inclusions

GSD IV: PAS
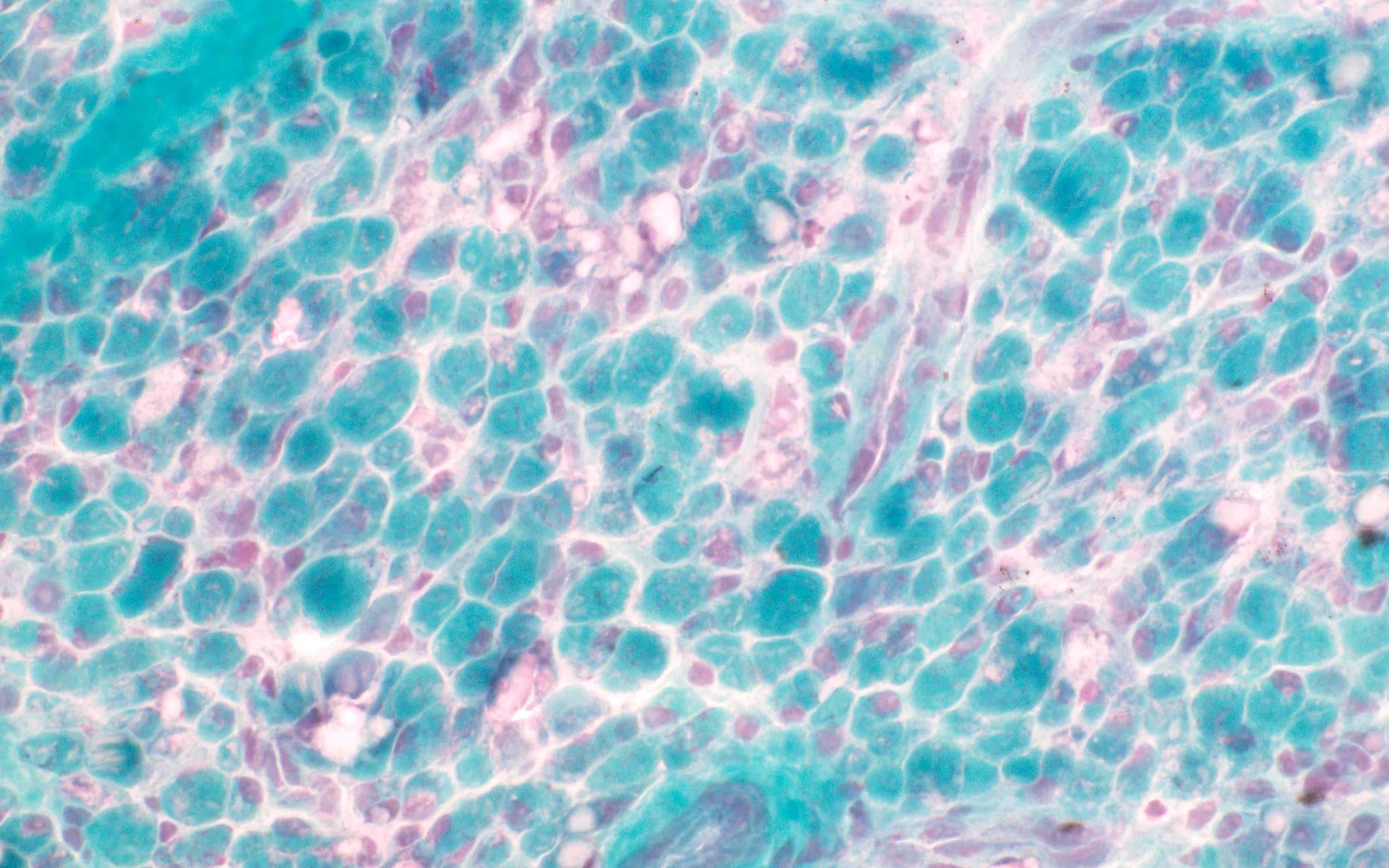
GSD IV: modified Gomori trichrome
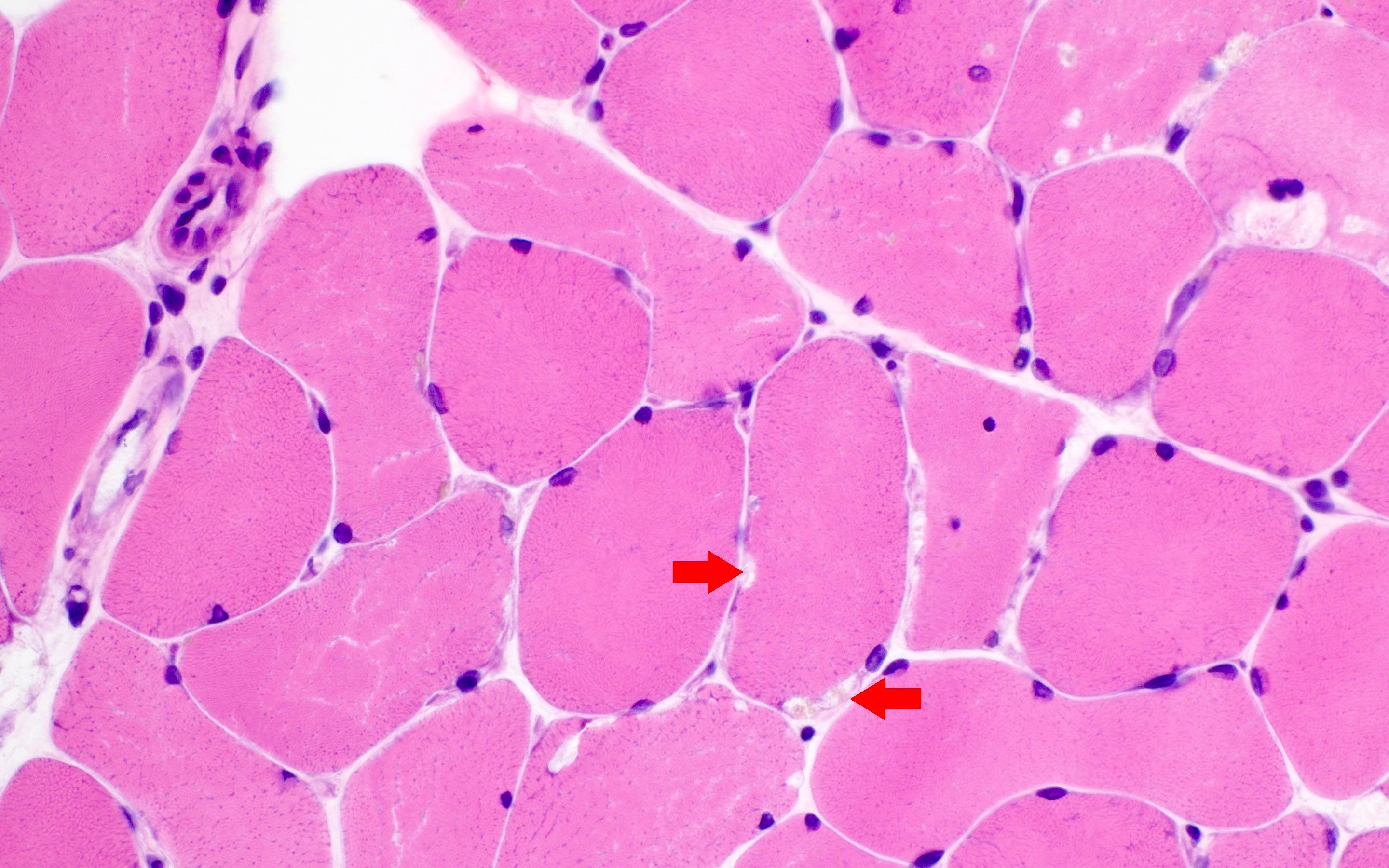
GSD V: small sarcolemmal vacuoles
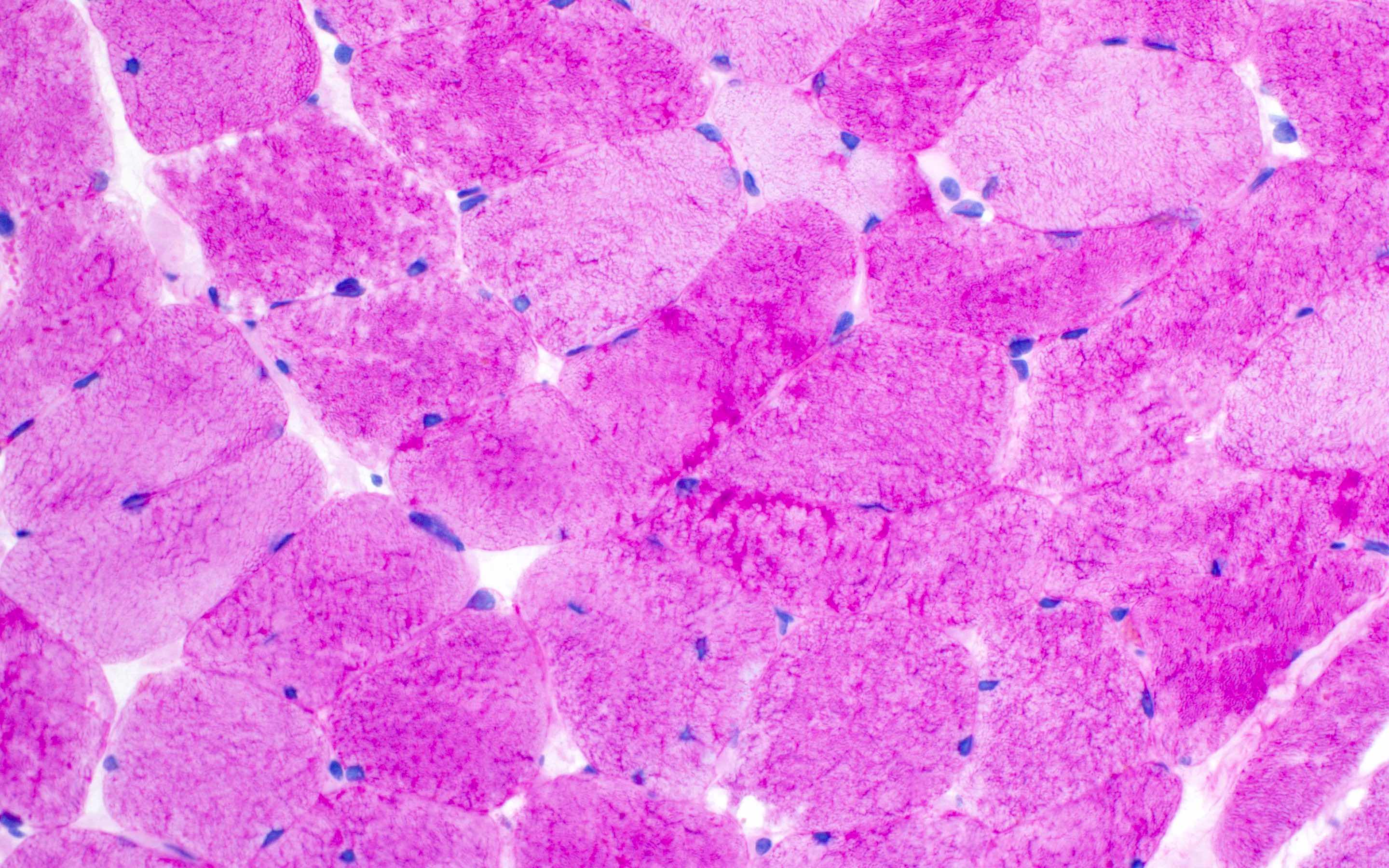
GSD V: PAS
GSD V: negative PHS
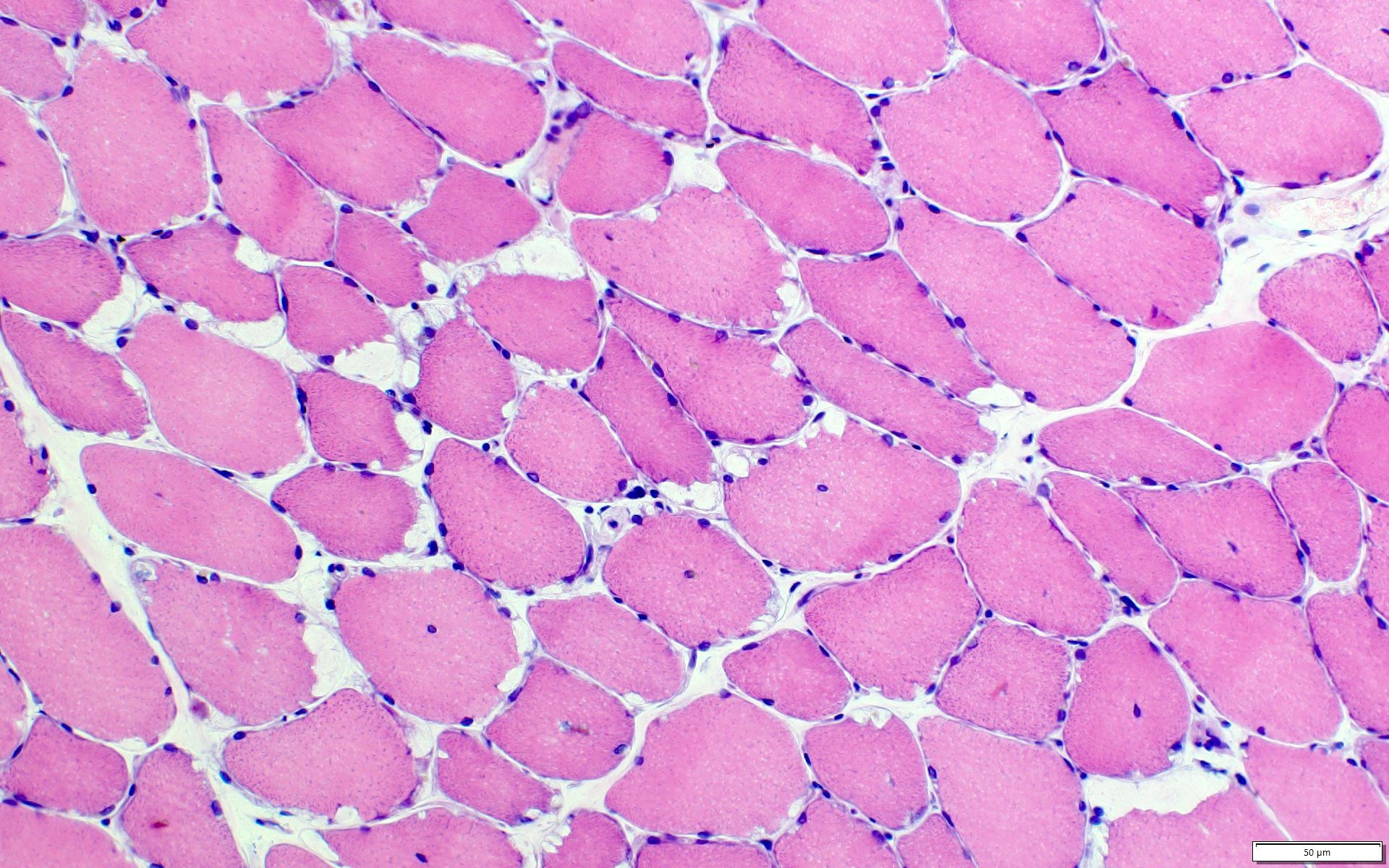
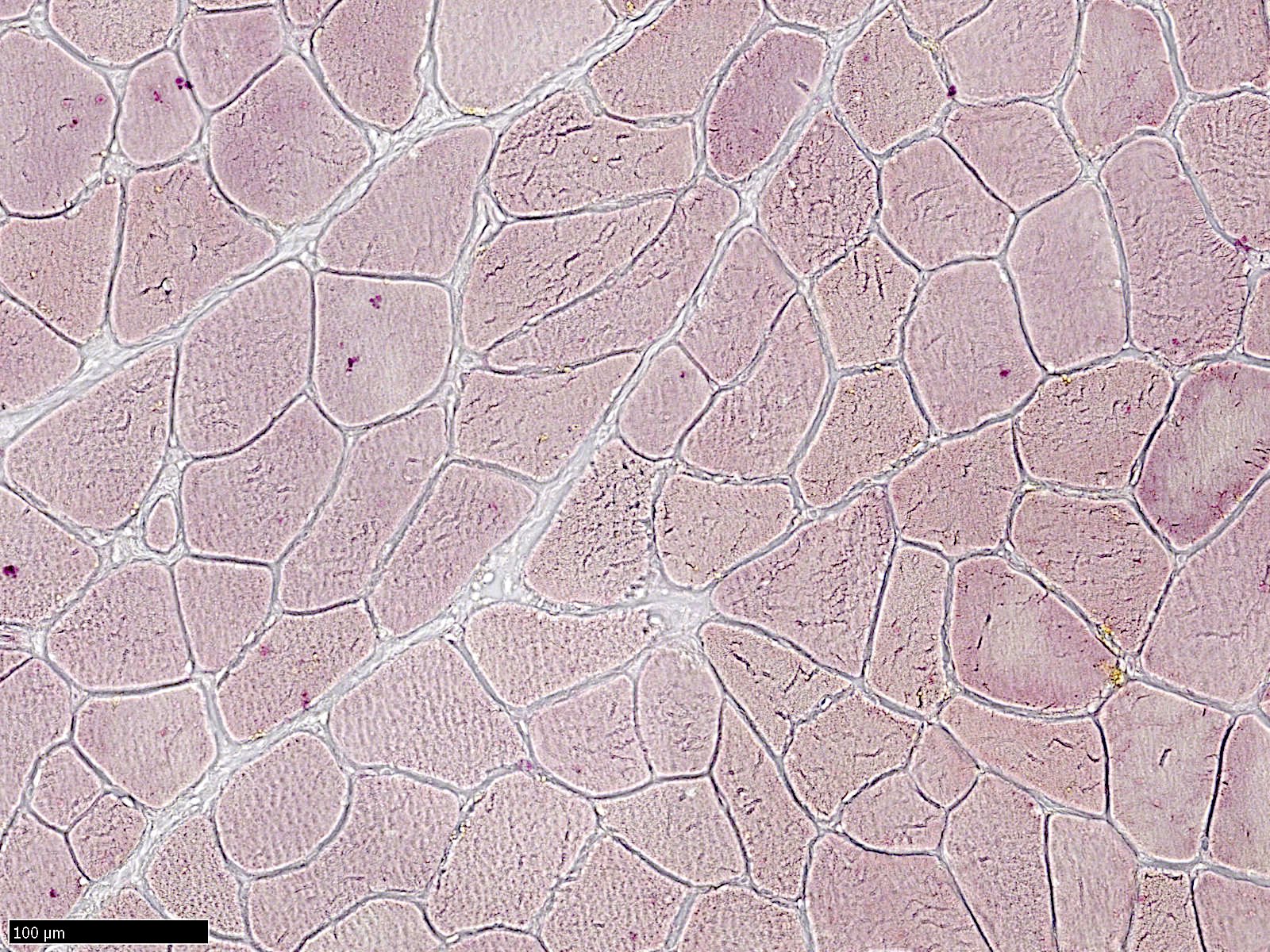
GSD VII: small sarcolemmal vacuoles

GSD VII: PAS
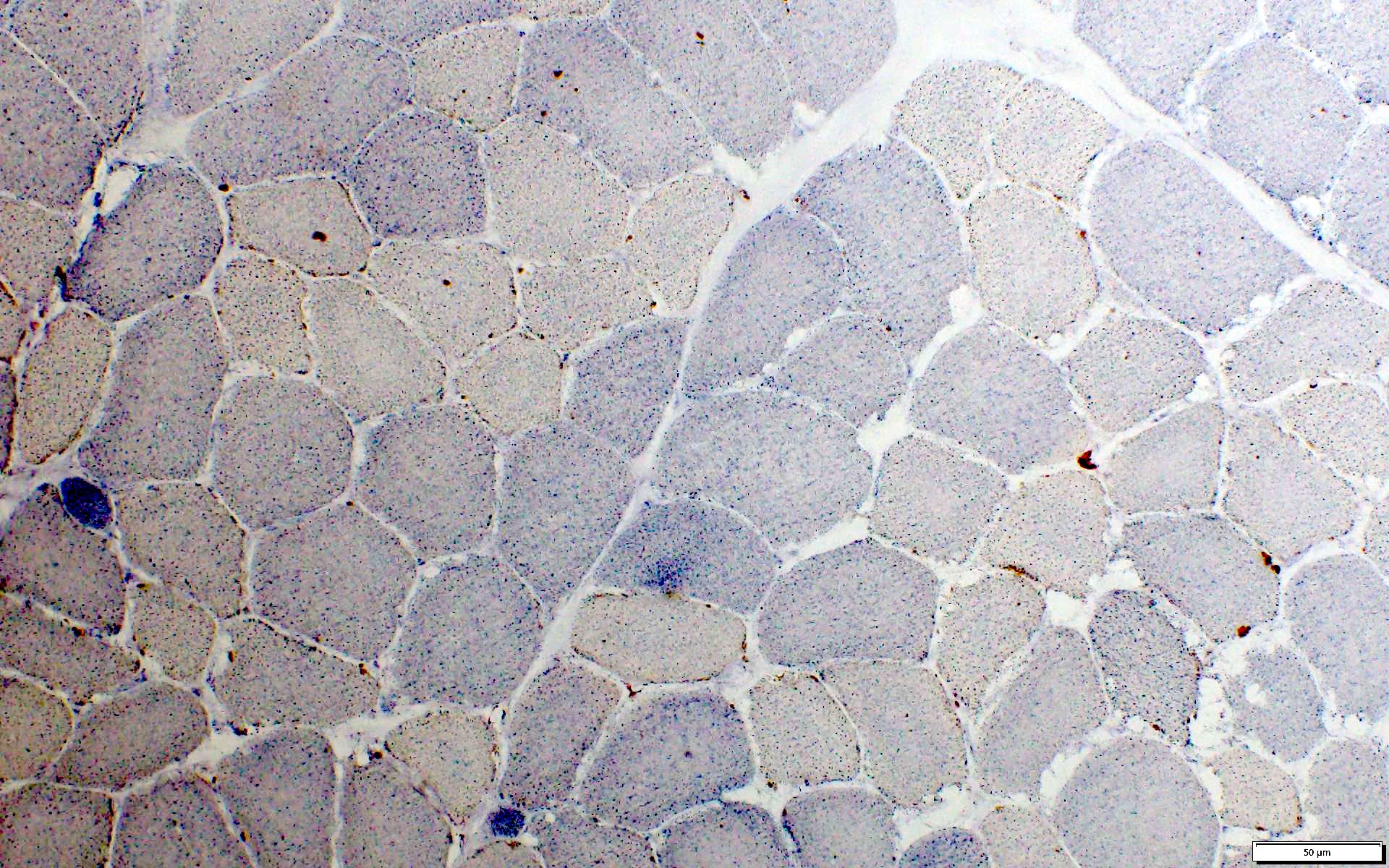
GSD VII:
phosphofructokinase
is negative
Images hosted on other servers:

McArdle disease
- PAS stain is intense positive in cytoplasm and vacuoles (except GSD 0)
- PAS stain is normal or mild positive in GSD X, XIV, XV, phosphoglycerate kinase deficiency
- ACP stain: acid phosphatase activity increases (increased lysosomes in GSD II)
- In GSD II, vacuoles are surrounded by MHC I (Goebel: Muscle Disease, 2nd Edition, 2013)
- PAS stain is reduced (in GSD 0)
- PHS stain: phosphorylase activity is deficient (in GSD V and when the GYS1 gene is mutated) (Dubowitz: Muscle Biopsy: A Practical Approach, 5th Edition, 2020, Biomedicines 2020;8:33)
- PFK stain: phosphofructokinase activity is reduced (< 10% of normal) or mildly reduced (in GSD VII) (Neuromuscul Disord 2021;31:1296)
- Inflammatory myopathy panel is negative
- GSD II: glycogen is seen within membrane bound vacuoles in the sarcoplasm of muscle fibers and disrupted myofibrils in the fiber
- Other GSDs: extensive areas of myofibrillar loss, subsarcolemmal and intermyofibrillar accumulation of accumulated nonmembrane bound glycogen
- Occasional fibers with accumulation of polyglucosan that look filamentous or amorphous (GSD IV, GSD VII) (Gaspar: Myopathology A Practical Clinico-pathological Approach to Skeletal Muscle Biopsies, 1st Edition, 2019)
Contributed by Ichizo Nishino, M.D., Ph.D.

GSD II childhood: membrane bound vacuoles

GSD II adult: membrane bound vacuoles

GSD III: nonmembrane bound glycogen
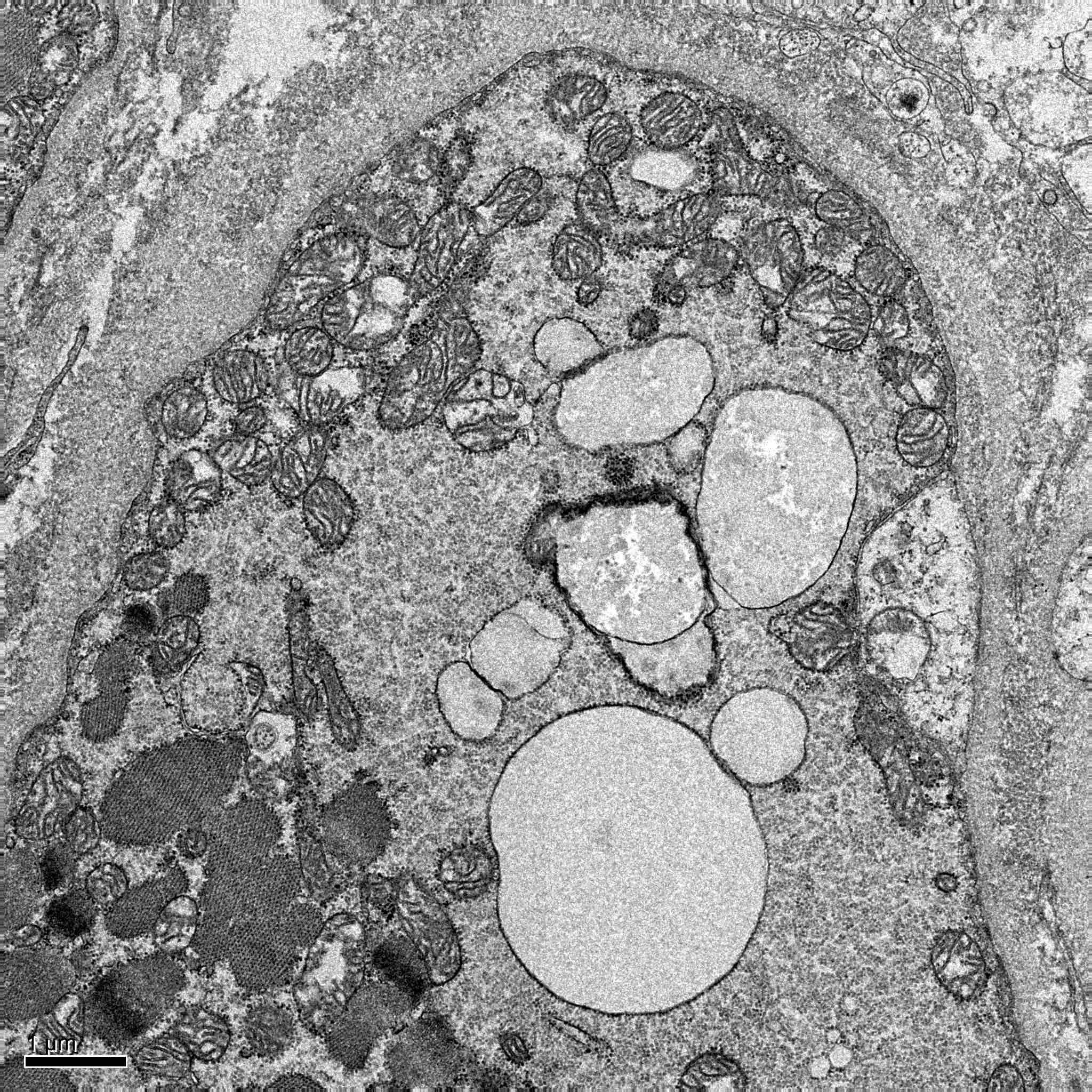
GSD IXd: sarcoplasmic glycogen deposits
- GSDs are inherited by an autosomal recessive trait, except for X linked GSD IXd and phosphoglycerate kinase deficiency (Ann Transl Med 2018;6:474)
- GSD 0: gene GYS1 (skeletal muscle) or GYS2 (liver) mutation
- GSD II: gene GAA mutation
- GSD III: gene AGL mutation
- GSD IV: gene GBE1 mutation
- GSD V: gene PYGM mutation
- GSD VII: gene PFKM mutation
- GSD IXd: gene PHKA1 (X linked) mutation
- Phosphoglycerate kinase deficiency, gene PGK1 (X linked) mutation
- GSD X: gene PGAM2 mutation
- GSD XI: gene LDHA mutation
- GSD XII: gene ALDOA mutation
- GSD XIII: gene ENO3 mutation
- GSD XIV: gene PGM1 mutation
- GSD XV: gene GYG1 mutation
- Left vastus lateralis, muscle biopsy, serial frozen sections were stained with H&E, modified Gomori trichrome (mGT) and a battery of histochemical methods:
- Compatible with glycogen storage disease (see comment)
- Comment:
- On H&E, there is mild to moderate variation in fiber size ranging from 20 to 105 microns in diameter. There are some fibers with multiple cytoplasmic vacuoles mainly in the subsarcolemmal region. No necrotic and regenerating fibers are seen. Scattered fibers with internal nuclei are seen. Mononuclear cell infiltration is not seen in endomysium. Perifascicular atrophy is not seen. Endomysial fibrosis is not seen.
- On mGT, no ragged red fibers, fibers with rimmed vacuoles or nemaline rods are seen.
- On NADH TR, intermyofibrillar networks are mildly disorganized in many fibers.
- On SDH, strongly SDH reactive blood vessels (SSVs) are not highlighted.
- On ALP, enzymatic activity is not seen in perimysium.
- On acid phosphatase, enzymatic activity is increased.
- On ATPase, moderate type 2 fiber atrophy is seen. Some type 2C fibers are seen.
- On PAS, strongly positive in vacuoles.
- On PHS, enzymatic activity is decreased.
- Other stains, including COX, AChE, NSE, AMP and MAG, show no additional abnormalities.
- The above findings are compatible with glycogen storage disease. Genetic analysis should be tested.
- Limb girdle muscular dystrophy:
- Can resemble GDS II in late onset
- Has a similar pattern of muscle involvement and disease course
- Muscle MRI: usually shows (in contrast to GSD II) fatty replacement of leg muscles
- Immunohistochemistry and genetic analysis help to differentiate
- Duchenne-Becker muscular dystrophy:
- Can resemble GDS II in late onset
- Progressive proximal muscle weakness, respiratory insufficiency and difficulty ambulating are seen
- Primarily affects males; inheritance is X linked
- Immunohistochemistry and genetic analysis help to differentiate
- Acid maltase
- Debrancher enzyme
- Glucose 6 phosphatase
- Phosphoglucomutase
- Phosphorylase
Comment Here
Reference: Glycogen storage diseases

Comment Here
Reference: Glycogen storage diseases
