

Muscle & peripheral nerve nontumor
Muscular dystrophies
Myotonic dystrophy
Authors: Wesley Hiser, M.D., Jesse L. Kresak, M.D.


Last author update: 1 March 2018
Last staff update: 12 August 2022
Copyright: 2018-2024 PathologyOutlines.com, Inc.
PubMed Search: Myotonic dystrophy [title] muscle Review[ptyp]

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Electron microscopy description | Molecular / cytogenetics images | Differential diagnosis | Additional references | Board review style question #1 | Board review style answer #1Cite this page: Hiser W. Myotonic dystrophy. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/musclemyotonicdystrophy.html. Accessed April 17th, 2024.
Definition / general
- Inherited muscular dystrophy characterized by muscle weakness, myotonia and additional systemic manifestations including cardiac and neurologic
- Myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2) are caused by differing nucleotide repeat expansions but have similar pathophysiologic mechanisms
- DM1 is the most common type of adult onset muscular dystrophy
Essential features
- Autosomal dominant (AD) muscular dystrophy caused by expansions of different nucleotide repeats which affect RNA splicing and processing, leading to muscle weakness, myotonia and systemic effects
- Variable clinical course, from late onset of mild symptoms to death in infancy
- Frequently involves cardiac conduction system and CNS
- Significantly increased internal nuclei on histologic examination
Terminology
- Dystrophia myotonica (DM)
- Classic type DM1 was first clinically described in 1909 by German physician Hans Steinert and termed, "Steinert's disease" (Biochim Biophys Acta 2015;1852:594)
ICD coding
- ICD-10: G71.11 - myotonic muscular dystrophy
Epidemiology
- Prevalence varies by region and is most common in individuals of European descent
- DM1 is more common than DM2 in the U.S. but some studies suggest similar prevalence of DM1 and DM2 in Europe (Neurol Clin 2014;32:705)
- Combined prevalence reported as 1 in 8,000 (12.5 per 100,000) but this is likely an underestimate due to clinical heterogeneity (Curr Opin Genet Dev 2017;44:30)
Sites
- DM1:
- Involvement of distal limb muscles with preferential involvement of finger, wrist and ankle flexors
- Diaphragm involvement can occur early in disease course
- Neck flexors involved
- Facial muscles involved with wasting of temporalis muscles (hatchet appearance)
- DM2:
- Proximal musculature affected including limb girdle, neck flexors and elbow extensor muscles (Neurol Clin 2014;32:705)
- Less involvement of facial or respiratory musculature
Pathophysiology
- Both forms are a result of toxicity from abnormal mRNA caused by expanded repeats
- DM1 has been more extensively studied
- Mutant RNA with expanded repeats is not exported to cytoplasm but is retained in the nucleus where it forms multiple clumps or foci
- RNA binding proteins such as MBNL1 and DDX6 exhibit high affinity for the mutant RNA and become sequestered in the nucleus
- These proteins are normally involved in splicing, as well as mRNA transport, stability and decay
- Protein function is lost once sequestered, leading to incorrect splicing and defects of proteins including insulin receptor, dystrophin, BIN1 and ClC-1 and L type calcium channels
- Mutated RNA may also produce peptides which are directly cytotoxic (Neurol Clin 2014;32:705)
Etiology
- Autosomal dominant (AD) inheritance in both DM1 and DM2
- DM1 is caused by expansion of a CTG repeat in the 3' noncoding region of the DMPK gene on chromosome 19q13.3, which codes for myotonic dystrophy protein kinase
- Normal individuals have between 5 and 37 repeats but symptomatic patients typically have > 50 repeats
- Anticipation is frequently seen
- Symptoms appear earlier and with greater severity in successive generations
- Individuals with borderline elevated CTG repeats (> 50) may be asymptomatic but offspring are at risk
- Clinical presentation correlates with CTG repeat size (Neurol Clin 2014;32:705)
- DM2 is caused by expansion of a CCTG repeat in the first intron of the CNTB gene (previously ZNF9) on chromosome 3q21, which codes for CCHC type zinc finger nucleic acid binding protein
- Normal individuals typically have between 10 and 33 repeats but symptomatic patients usually have greater than 1,000 repeats (range, 75 to greater than 11,000)
- Anticipation is less prominent
- No correlation between repeat size and clinical presentation (Neurol Clin 2014;32:705)
Clinical features
- DM1 is typically broken down into four subtypes: congenital, childhood, classic and minimal / late onset
- Spectrum of clinical severity: from death in infancy to onset in late adulthood with extremely mild symptoms
- Congenital DM1: fetal onset, involving musculature and CNS; severe
- Prenatal features: decreased fetal movement, polyhydramnios
- Neonatal features: hypotonia with feeding or respiratory distress
- 79% require nasogastric feeding, 53% require ventilator support (Neurol Clin 2014;32:705)
- Childhood: delayed motor milestones, intellectual impairment, prominent oropharyngeal weakness with tenting of upper lip
- Degenerative features develop by second or third decade, resembling classic DM1
- More than half of mothers do not carry DM1 diagnosis so diagnosis can be delayed (Neurol Clin 2014;32:705)
- Childhood DM1: between 1 and 10 years of age
- Predominantly cognitive and behavioral issues
- Facial weakness and conduction abnormalities
- Approximately half with intellectual impairment
- Range of psychiatric disorders
- Classic DM1: onset usually between second and fourth decades
- Myotonia is the most common presenting symptom
- More pronounced after rest, improves with activity
- Involves forearms and hands (grip), tongue and jaw
- Muscle weakness of distal limbs and craniofacial muscles
- Wasting of fascial muscles with characteristic ptosis and hatchet appearance
- Respiratory distress secondary to diaphragmatic involvement
- Cardiac conduction abnormalities common
- Risk of sudden cardiac death as high as 1.1% per year
- Cataracts located on the posterior lens capsule with a multicolored, iridescent appearance on slit lamp examination (Neurol Clin 2014;32:705)
- Sleep disturbances (80% with daytime hypersomnolence)
- Gastrointestinal involvement: cholelithiasis, intestinal dysmotility
- Insulin resistance, metabolic syndrome, frontal balding and hypogonadism in men
- Evidence for increased risk of malignancy (thyroid, ovarian, colorectal, endometrial, Mayo Clin Proc 2012;87:130, JAMA 2011;306:2480)
- Myotonia is the most common presenting symptom
- Minimal / late onset DM1: small expansions (70 - 100 repeat) with mild weakness, myotonia and development of cataracts, usually after age 40 (range 20 to 70 years, Neurol Clin 2014;32:705)
- DM2: overall milder disease; most often presents in the third decade of life (range second to sixth decades)
- Presents with proximal muscle weakness as well as myotonia
- May resemble limb girdle dystrophy
- Less muscle wasting and respiratory involvement than DM1
- Frontal balding, hypogonadism, cataracts and insulin resistance
- Less cardiac and CNS involvement than DM1 (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Some patients exhibit calf and thigh hypertrophy (true hypertrophy)
- Patients often have a history of unexplained pain and may have a diagnosis of fibromyalgia (Neurol Clin 2014;32:705)
- Presents with proximal muscle weakness as well as myotonia
Diagnosis
- Molecular testing is definitive and may be the only test performed in the appropriate clinical setting
- PCR is most commonly used for detection of repeat expansion
- Southern blot is sometimes utilized in addition to PCR testing (Eur J Hum Genet 2012;20:1203)
- Muscle biopsy is infrequently performed if clinical suspicion is high
Laboratory
- Not specific
- Liver function tests (LFTs) commonly abnormal
- Creatine kinase (CK) can be normal to slightly elevated
- Electromyography (EMG) shows combination of myotonic features and myopathic changes (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
Radiology description
- No specific imaging features of the involved musculature have been identified
- Imaging of CNS may show alterations in the white matter signal intensity, most notable in the frontotemporal region
- Prenatal ultrasound in congenital DM1 patients may show borderline ventriculomegaly or talipes equinovarus (club foot) (Neurol Clin 2014;32:705)
Prognostic factors
- Classic DM1 has a slowly progressive course
- Respiratory failure is the leading cause of death in DM1, followed by sudden cardiac death (Neurol Clin 2014;32:705)
- Congenital DM1 patients may live to adulthood and typically die of cardiorespiratory complications (similar to classic DM1)
- DM2 patients typically have a milder clinical course
Case reports
- 38 year old man with myotonic dystrophy mimicking postpolio syndrome in a polio survivor (Am J Phys Med Rehabil 2009;88:161)
- 61 year old man with normal pressure hydrocephalus (Neurosurgery 2006;58:E796)
- Woman with gastric bypass surgery for obesity in DM1 (Neuromuscul Disord 2015;25:414)
- Patient with hydrocephalus and cognitive decline in myotonic dystrophy (Arch Phys Med Rehabil 1998;79:1022)
Treatment
- No curative treatment - supportive therapy only
- Many patients require nighttime respiratory support
- May require pacemaker or defibrillator placement
- Ongoing research into curative genetic therapies
Microscopic (histologic) description
- Variation in myofiber size, ranging from 10 um to 100 um
- Ring fibers and sarcoplasmic masses (dark staining regions) are frequently seen in DM1
- DM1: type 1 myofiber atrophy with type 2 hypertrophy
- DM2: greater variation in both type 1 and 2 fibers with predominantly type 2 myofiber atrophy (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
- Pyknotic nuclear clumps in atrophic fibers
- May see moth eaten or whorled fibers
Microscopic (histologic) images
Contributed by Jesse L. Kresak, M.D
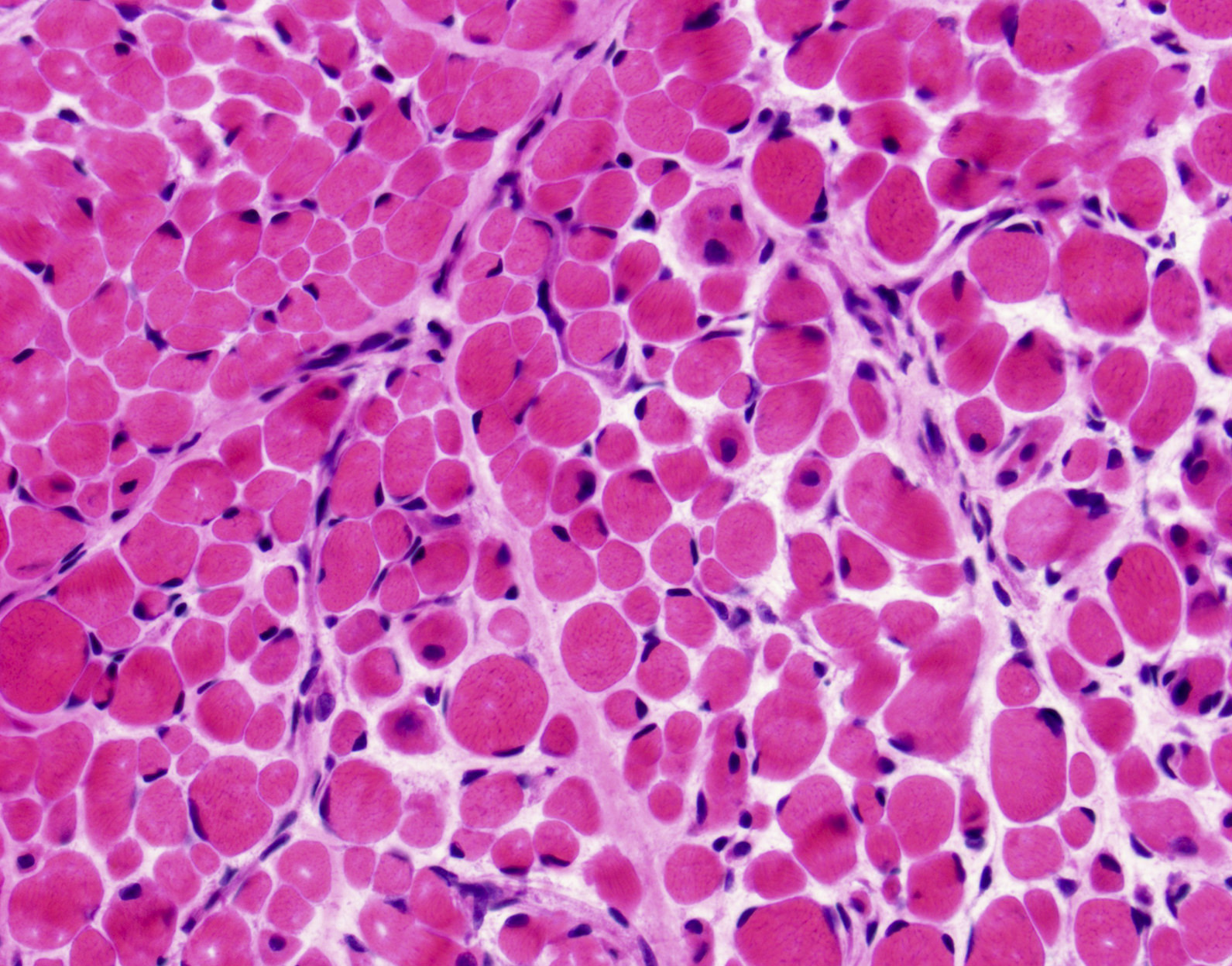
Scattered internal nuclei
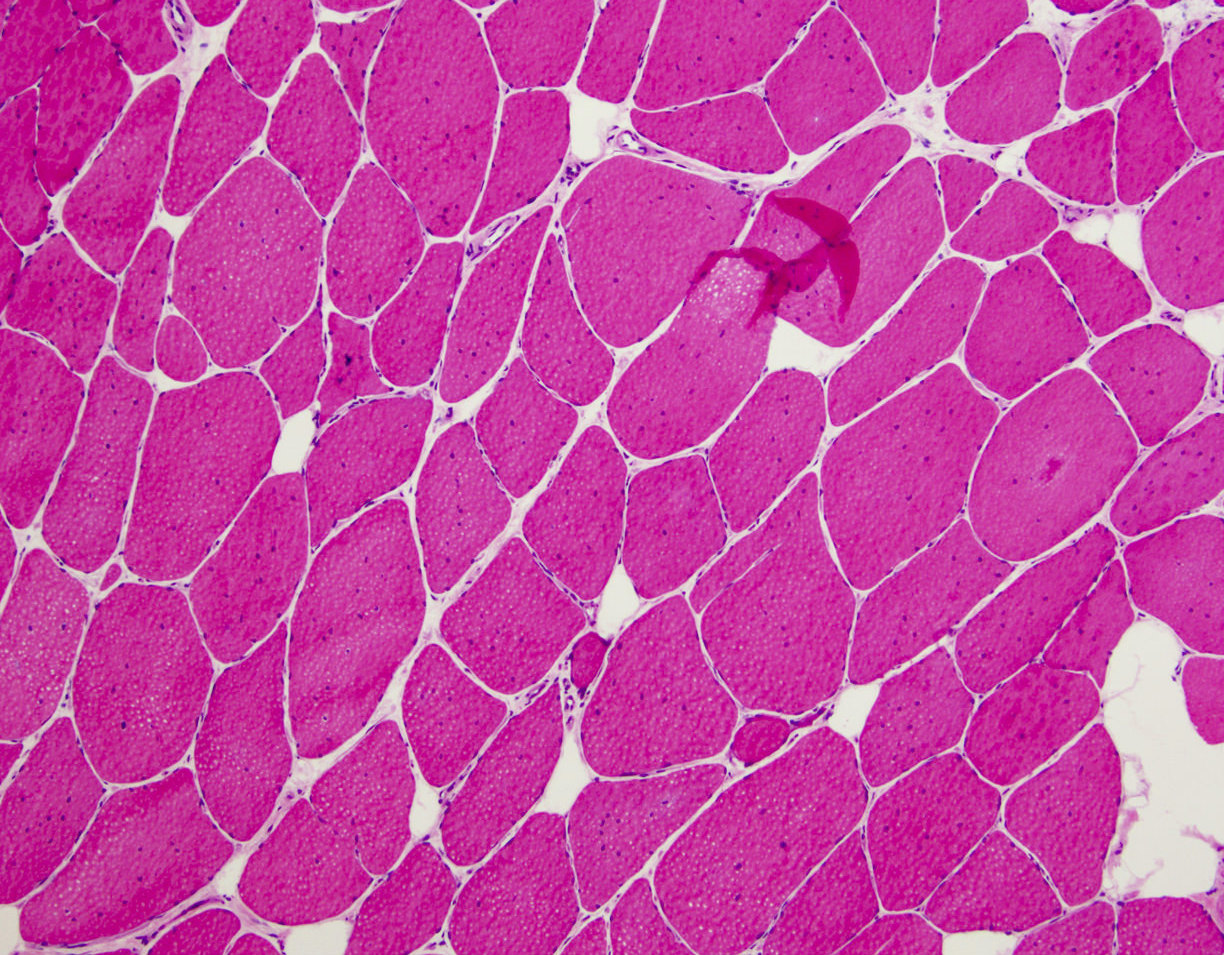
Increased internal nuclei
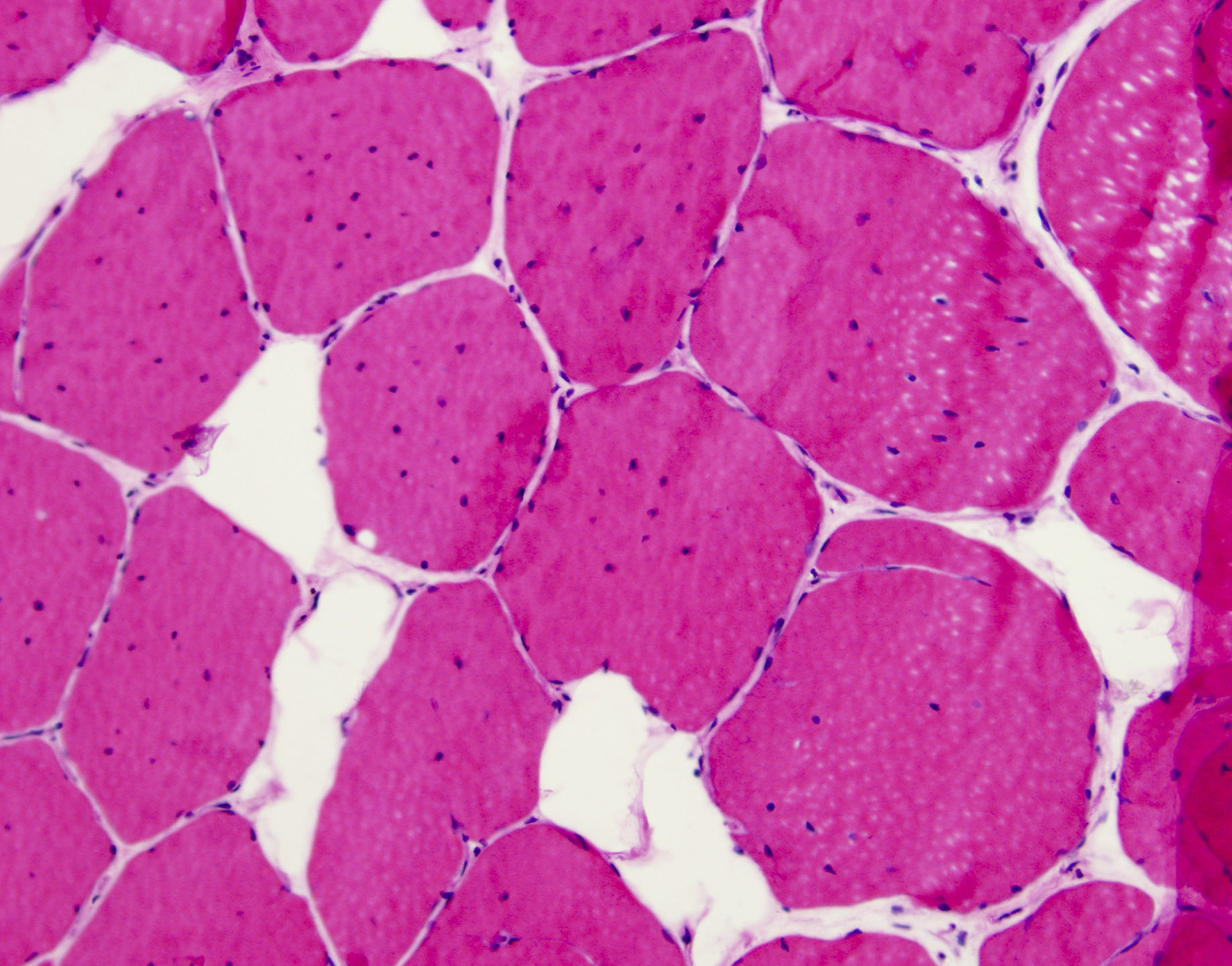
Markedly increased internal nuclei
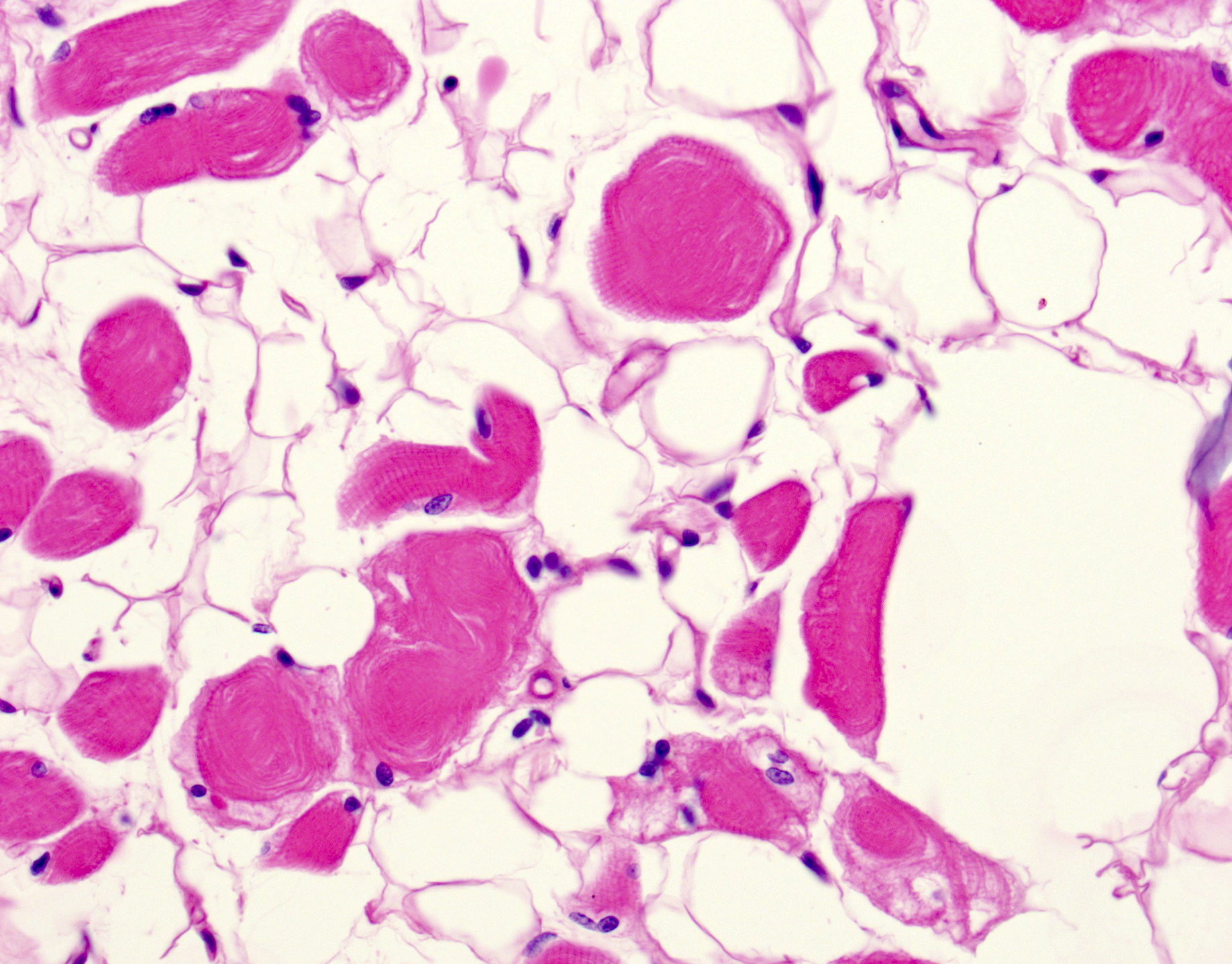
Fatty replacement and ring fibers
Positive stains
- No specific special or immunohistochemical stains are typically utilized for the diagnosis
- In situ hybridization (ISH) specific for CTG or CCTG repeats may be utilized in some laboratories to allow for direct visualization of aberrant mRNA in muscle biopsies (Acta Myol 2013;32:154)
- Staining for MBNL1 is also available in some laboratories (Neuromuscul Disord 2012;22:225)
- Involved neurons of the limbic system and brain stem contain tau protein positive neurofibrillary tangles (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
Electron microscopy description
- Sarcoplasmic masses composed of disorganized myofibrils, dilated sarcoplasmic reticulum and free ribosomes (Yachnis: Neuropathology - A Volume in the High Yield Pathology, 1st Edition, 2014)
Molecular / cytogenetics images
Images hosted on other servers:
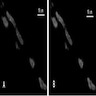
DM2 muscle biopsy: FISH and MBNL1 immunofluorescence
Differential diagnosis
- Limb girdle dystrophy (DM2): often has significantly increased CK; molecular testing and immunohistochemistry on muscle biopsy can also be utilized to make the diagnosis
- Other muscular dystrophies: molecular studies will aid in differentiation; myotonic dystrophy shows a greater number of and more consistent internal nuclei
- Myotubular myopathy (in congenital DM1): greater number of internal nuclei in DM1 without peripheral halos seen in myotubular myopathy; can also look for MTM1 gene mutation
- Myopathic conditions (i.e. inflammatory myopathies): degenerating and regenerating fibers, as well as inflammatory cell infiltrates, are not commonly seen in myotonic dystrophy
Additional references
Board review style question #1
Myotonic dystrophy type 2 (DM2) is inherited in what pattern?
- Autosomal dominant
- Autosomal recessive
- Mitochondrial
- X linked
Board review style answer #1
A. Both DM1 and DM2 are inherited in an autosomal dominant pattern.
Comment Here
Reference: Myotonic dystrophy
Comment Here
Reference: Myotonic dystrophy
