Pancreas
Neuroendocrine neoplasms
MEN1 syndrome
Authors: R. Peter Powell, M.D., Jing He, M.D.
Editorial Board Member: Wei Chen, M.D., Ph.D.
Deputy Editor-in-Chief: Aaron R. Huber, D.O.


Last author update: 1 July 2025
Last staff update: 1 July 2025
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed Search: MEN1 syndrome

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Negative stains | Molecular / cytogenetics description | Videos | Sample pathology report | Differential diagnosis | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Powell RP, He J. MEN1 syndrome. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/pancreasmen1.html. Accessed October 2nd, 2025.
Definition / general
- Autosomal dominant syndrome characterized by inactivating MEN1 gene mutations, leading to multiple endocrine tumors (NETs) and other tumor types
- Classic triad of pituitary, parathyroid and pancreatic tumors (the 3 Ps)
Essential features
- Pathogenic MEN1 variant (chromosome 11q13), encoding the tumor suppressor protein menin, is inherited in 90% of cases or arises de novo in 10%, with 100% penetrance
- Classically associated with duodenopancreatic, parathyroid and pituitary tumors; currently, the phenotypic penetrance is appreciated in > 13 organs
- Variety of nonendocrine tumors are also part of MEN1 (meningioma, ependymoma, angiofibroma)
- Mortality is primarily due to duodenopancreatic neuroendocrine tumors (DP NETs)
Terminology
- Not recommended: Wermer syndrome, multiple endocrine adenomatosis type 1, familial Zollinger-Ellison syndrome
ICD coding
Epidemiology
- Prevalence is ~3 - 20:100,000 (Endocr Rev 2021;42:133)
- 90% familial and 10% sporadic (F1000Res 2017;6:73)
- Sex differences (Eur J Endocrinol 2011;165:97)
- Primary hyperparathyroidism: M = F
- Pancreatic NETs (PanNETs): M > F = 1.5:1
- Pituitary NET: M < F
- Thymic NET: M >>> F (Western population), M ≈ F (Eastern population)
- Geographical differences: thymic NET almost exclusively in males in Western countries
- 100% penetrance (100% with parathyroid adenomas by age 50) (F1000Res 2017;6:73)
- Penetrance depends on age, type of tumor and phenotypic expression
- Primary hyperparathyroidism is the earliest manifestation of MEN1 (58 - 75% prevalence in pediatric patients) and average age at diagnosis is ~40 years
- Most frequent cause of MEN1 related death is complications secondary to pancreatic / gastrointestinal neuroendocrine tumors (J Pediatr Genet 2016;5:89)
Sites
- Variety of neuroendocrine and nonneuroendocrine tumors that can develop in different organ sites (J Pediatr Genet 2016;5:89, Cancer Genet 2016;209:36, Endocr Relat Cancer 2017;24:T119)
- Endocrine tumors
- Parathyroid (~95% of patients)
- Pancreas / gastrointestinal tract (30 - 70% of patients)
- Anterior pituitary (30 - 40% of patients)
- Adrenal cortex
- Thymus (~3%) (J Clin Endocrinol Metab 2014;99:3325, Horm Cancer 2016;7:279)
- Nonendocrine tumors
- Facial skin (angiofibromas, collagenomas)
- Testis (epididymomas)
- Central nervous system (meningioma, ependymoma) (Clin Cancer Res 2024;30:2835)
- Nonsyndromic tumors have somatic MEN1 mutations at the following frequencies
- Glucagonoma: 60%
- VIPoma: 57%
- Nonfunctioning PanNETs: 44%
- Gastrinoma: 38%
- Bronchial carcinoid: 35%
- Parathyroid adenoma: 35%
- Lipoma: 28%
- Insulinoma: 2 - 19%
- Angiofibroma: 10%
- Anterior pituitary tumor: 3.5%
- Adrenocortical tumor: 2%
- Endocrine tumors
Pathophysiology
- MEN1 gene was first identified in 1997 and is located on chromosome 11q13 (Science 1997;276:404)
- Menin, the protein product of MEN1, is a tumor suppressor gene involved in regulating cell proliferation, apoptosis and maintaining genome integrity (F1000Res 2017;6:73)
- Menin is conserved across species, from Drosophila to humans; however, it does not show homology with any other known protein (Cancer Genet 2016;209:36)
- Truncation, frameshift, nonsense mutations or large deletions (including those in intronic areas leading to splice site defects) cause MEN1 gene loss and a lack of menin, resulting in uncontrolled endocrine cell proliferation
- 12 MEN1 gene mutations were initially identified and > 1,800 mutations have been identified to date (F1000Res 2017;6:73)
- 75% of MEN1 mutations are inactivating (F1000Res 2017;6:73)
- 5 - 20% of individuals may not harbor mutations in the MEN1 gene coding region, suggesting alternative genetic mechanisms (Cancer Genet 2016;209:36)
- 2 hit hypothesis suggests that germline MEN1 mutation combined with a somatic mutation leads to tumorigenesis
- There is no direct genotype phenotype correlation between the type of MEN1 mutation and the specific types of tumors that develop (F1000Res 2017;6:73)
Etiology
- MEN1 is inherited in an autosomal dominant manner
- ~10% of cases result from a de novo mutation
Diagrams / tables

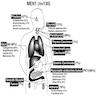

Clinical features
- Endocrine tumors are found in parathyroid, anterior pituitary, pancreas / gastrointestinal tract, adrenal cortex and thymus
- Parathyroid adenomas cause hypersecretion of parathyroid hormone (PTH), leading to hypercalcemia with symptoms such as gastrointestinal pain, cognitive dysfunction and renal stones
- Pituitary adenomas may cause visual field defects due to optic chiasm compression or extraocular muscle involvement
- Functional pituitary adenomas can cause Cushing disease (adrenocorticotropic hormone [ACTH] secreting tumors), Nelson syndrome (postadrenalectomy ACTH overproduction), acromegaly (excess growth hormone), hyperprolactinemia (prolactin secreting adenomas)
- Patients with gastrinomas, typically found in the duodenum, may have Zollinger-Ellison syndrome (excess stomach acid, severe peptic ulcers)
- Primary gastrinomas may be microadenomas but lymph node metastases can be much larger (J Pediatr Genet 2016;5:89)
- Tumors in the adrenal cortex and thymus can occur, though they are less common
- Nonendocrine tumors: skin lesions (angiofibromas, collagenomas), epididymomas, meningiomas
Diagnosis
- MEN1 may be diagnosed by clinical, familial or genetic characteristics
- Clinical: at least 2 MEN1 syndrome associated tumors
- Familial: at least 1 MEN1 syndrome associated tumor and a first degree relative with a confirmed diagnosis of MEN1 syndrome
- Genetic: 1 of the various MEN1 mutations (Neuroendocrinology 2021;111:609)
- MEN1 mutations are identified through PCR amplification of exons and splicing sites
- PCR method fails to detect MEN1 mutations in patients whose mutations lie outside the coding region (World J Exp Med 2015;5:124)
- Next generation sequencing (NGS) overcomes these limitations by enabling the detection of noncoding region mutations
- Mutational analysis should be offered to
- Asymptomatic first degree relatives of a known MEN1 patient
- Patients with at least 2 typical MEN1 tumors or 1 MEN1 tumor and a positive family history
- Patients with early onset (< 30 years) of a typical MEN1 manifestation (hyperparathyroidism, Zollinger-Ellison syndrome) (Endocr Relat Cancer 2017;24:T209)
Laboratory
- Intact parathyroid hormone measured after diagnosis of hypercalcemia (J Clin Endocrinol Metab 2014;99:3561)
- ~90% of MEN associated pituitary NETs are nonfunctional (Clin Endosc 2017;50:537)
- See Diagrams / tables regarding laboratory tests for pancreatic NETs (Ther Adv Chronic Dis 2021;12:20406223211035288)
Radiology description
- Osteoporosis, terminal tuft erosion (distal phalanges) and nephrolithiasis in patients with parathyroid adenomas and hypercalcemia
- Calvarial thickening, frontal bossing, enlarged sinuses (especially frontal sinuses), enlarged sella turcica, prognathic mandible and increased vertebral fractures in patients with acromegaly due to pituitary adenoma
- PanNETs are usually well circumscribed, solid and avidly enhancing lesions (Radiol Clin North Am 2016;54:441)
- Sensitivity of imaging technique for PanNETs: endoscopic ultrasound (EUS) ~90%, computed tomography (CT) 63 - 82%, magnetic resonance imaging (MRI) 85 - 100% (AJR Am J Roentgenol 2021;217:245, Ann Gastroenterol 2015;28:193)
Radiology images
Contributed by R. Peter Powell, M.D.
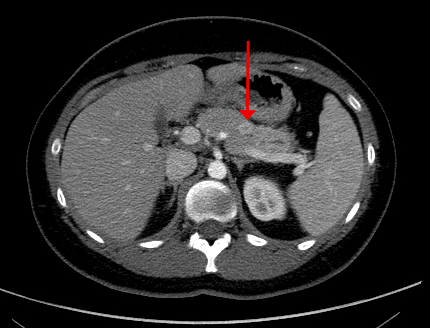
Pancreatic NET in MEN1
Prognostic factors
- Pancreatic neuroendocrine tumors (pNETs) are the most critical determinant of survival in MEN1
- Duodenopancreatic NETs and thymic NETs represent ~70% of the mortality rate (Medicine (Baltimore) 2013;92:135)
- Insulinomas: usually benign, good prognosis
- Gastrinomas: high malignant potential, risk of metastatic spread (liver, lymph nodes)
- Nonfunctional pNETs: often aggressive, found late and more likely to metastasize
- Glucagonomas / VIPomas / somatostatinomas: rare but highly malignant
- Patients with a strong family history of aggressive pNETs may have worse outcomes
- Patients with metastatic pNETs have the worst prognosis, especially if liver involvement occurs
- Parathyroid tumors
- Usually benign but can recur after surgery
- Parathyroid carcinomas are rare but have a high mortality rate
- Pituitary tumors
- Prolactinomas: often responsive to medical therapy
- Pituitary tumors rarely metastasize but can cause significant morbidity if invasive
- ACTH secreting tumors (Cushing disease): rare but associated with increased mortality
Case reports
- 16 year old girl with primary hyperparathyroidism and pituitary adenoma (Genes (Basel) 2020;11:986)
- 25 year old woman with nonfunctioning microprolactinoma and nonfunctioning pancreatic neuroendocrine tumors presented with uveal melanoma (Can J Ophthalmol 2025;60:e173)
- 40 year old man with prototypic MEN presentation with new MEN mutation (Cardiorenal Med 2016;6:129)
- 43 year old woman with insulinoma (Genes (Basel) 2021;12:512)
- 49 year old woman with pancreatic insulinoma, adrenal gland tumor and primary hyperparathyroidism (Hered Cancer Clin Pract 2022;20:10)
- 54 and 55 year old men with parathyroid carcinoma (Int J Surg 2016;31:10)
- 60 year old woman with Zollinger-Ellison syndrome and hyperparathyroidism (Case Rep Gastrointest Med 2019;2019:9605769)
Treatment
- Parathyroid tumors
- Surgical resection of the affected parathyroid glands is the main treatment
- Calcimimetics (e.g., cinacalcet) may be used to control calcium levels if surgery is not possible or preferred
- Pancreatic neuroendocrine tumors
- Surgical resection if pancreatic tumors are symptomatic or large
- Proton pump inhibitors (for gastrinomas causing Zollinger-Ellison syndrome)
- Somatostatin analogs (e.g., octreotide, lanreotide) to reduce hormone secretion (Pharmaceuticals (Basel) 2021;14:1039)
- Targeted therapies like everolimus or sunitinib for advanced disease (N Engl J Med 2011;364:501)
- Peptide receptor radionuclide therapy (PRRT) in metastatic cases
- Pituitary tumors
- Prolactinomas: treated with dopamine agonists (e.g., cabergoline, bromocriptine)
- Other pituitary tumors: may require surgery, radiation therapy or medical therapy with somatostatin analogs or hormone replacement
- Other tumors (adrenal, thymic, lung NETs, etc.): surgical removal if tumors are functional or growing; monitoring for nonfunctional tumors that do not require immediate intervention
Gross description
- Parathyroid adenomas / hyperplasia
- Enlarged parathyroid glands, typically soft and reddish brown to tan in color, measuring ~0.5 - 1.5 cm but can be larger
- Involvement may be limited to a single gland or affect all 4 parathyroid glands
- Pancreatic / duodenal neuroendocrine tumors
- Often present as multiple, well demarcated, tan to yellowish, soft or firm nodules scattered in the pancreas and duodenum (Int J Mol Sci 2021;22:4041)
- Size can vary widely, ranging from microscopic lesions to tumors several centimeters in diameter
- Pituitary adenomas
- Usually solitary, can present as a macroadenoma (> 1 cm) (J Neurosurg 2020;134:1132)
- Well circumscribed, soft and typically appear as a pale tan to gray mass
Gross images
AFIP images
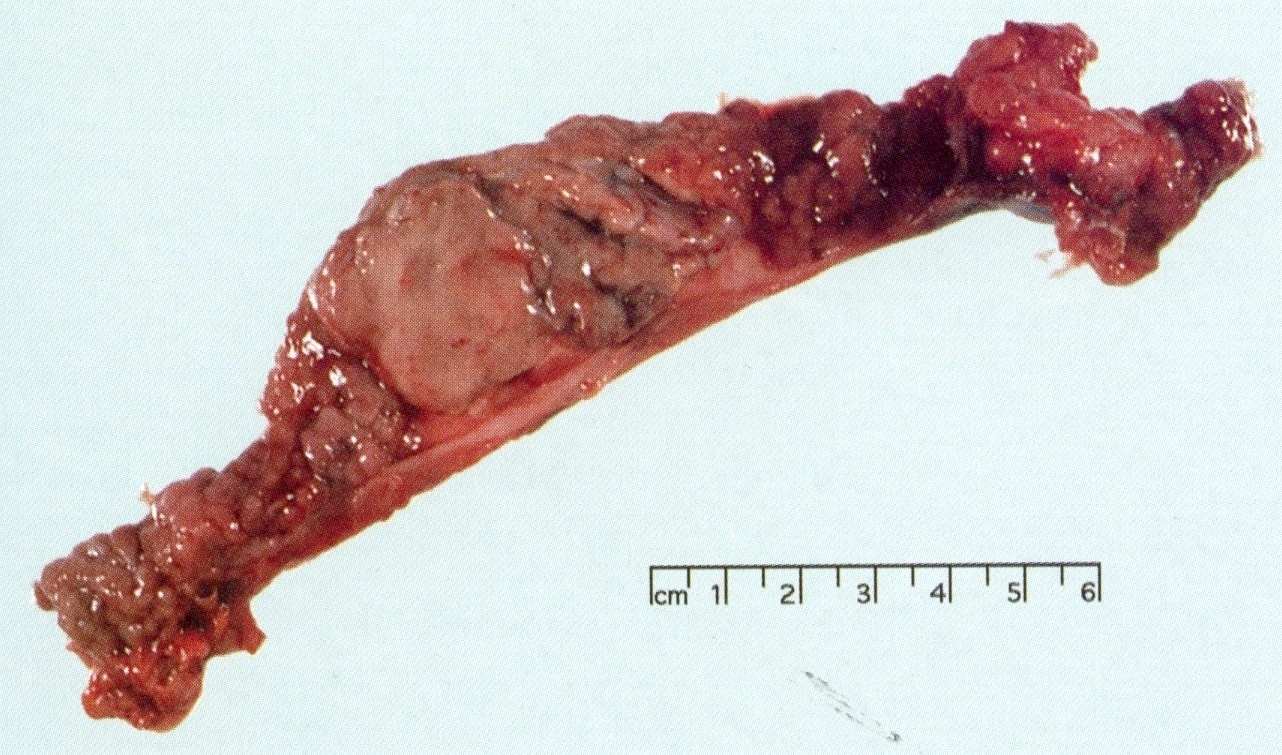
Intramucosal tumor nodules
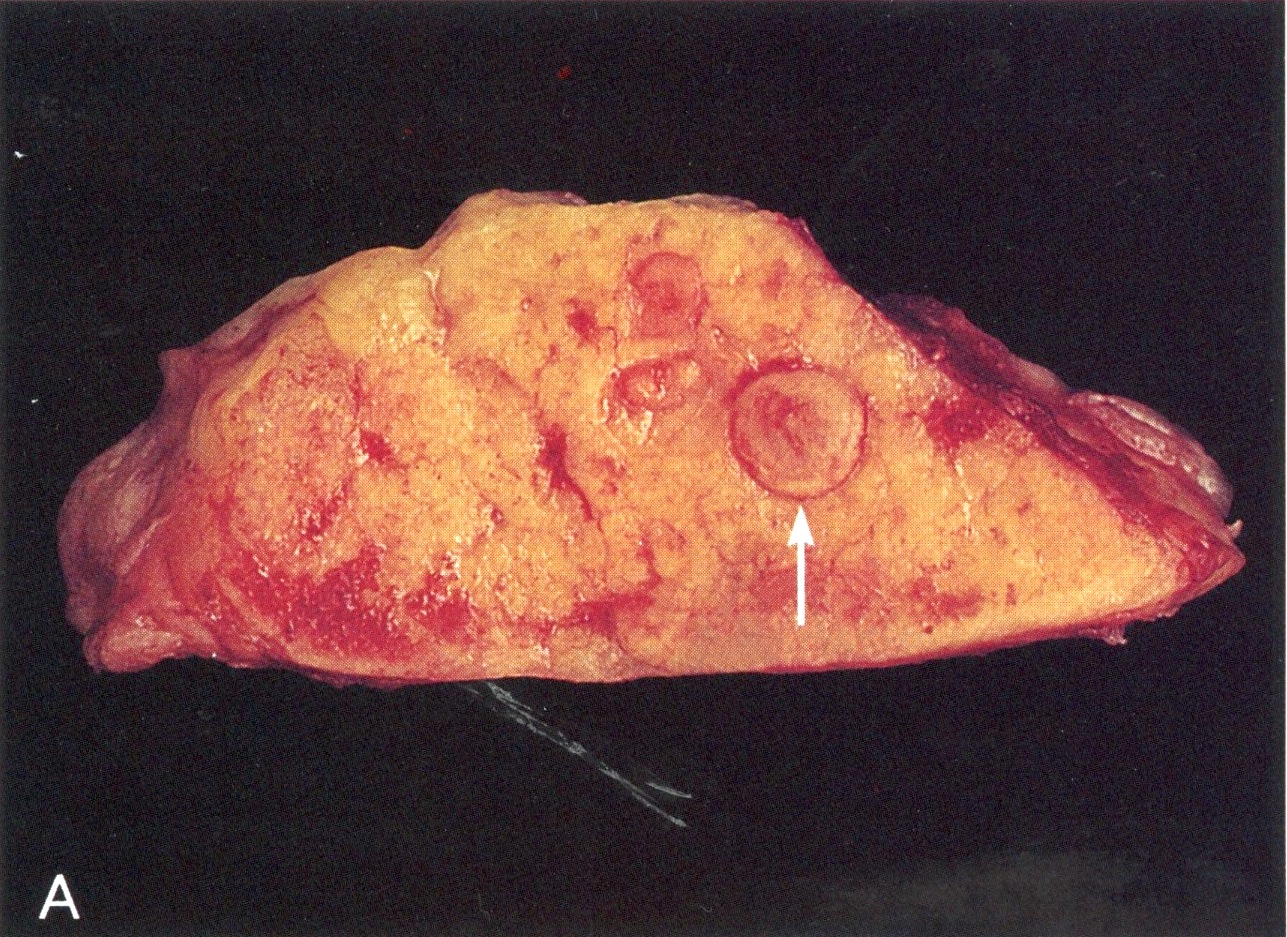
Zollinger-Ellison syndrome
Microscopic (histologic) description
- MEN1 is characterized by the development of multiple endocrine tumors, primarily affecting the parathyroid glands, pancreas and pituitary gland
- Each tumor type has distinct microscopic features
- Parathyroid adenomas / hyperplasia
- Chief cell hyperplasia with a diffuse or nodular growth pattern
- Increased oxyphil cells in some cases (Histopathology 1992;20:305, Surg Pathol Clin 2019;12:1007)
- Loss of fat content in the gland (Histopathology 1992;20:305)
- No significant nuclear atypia but may show monotonous cells with round nuclei and mild anisocytosis (Histopathology 1992;20:305)
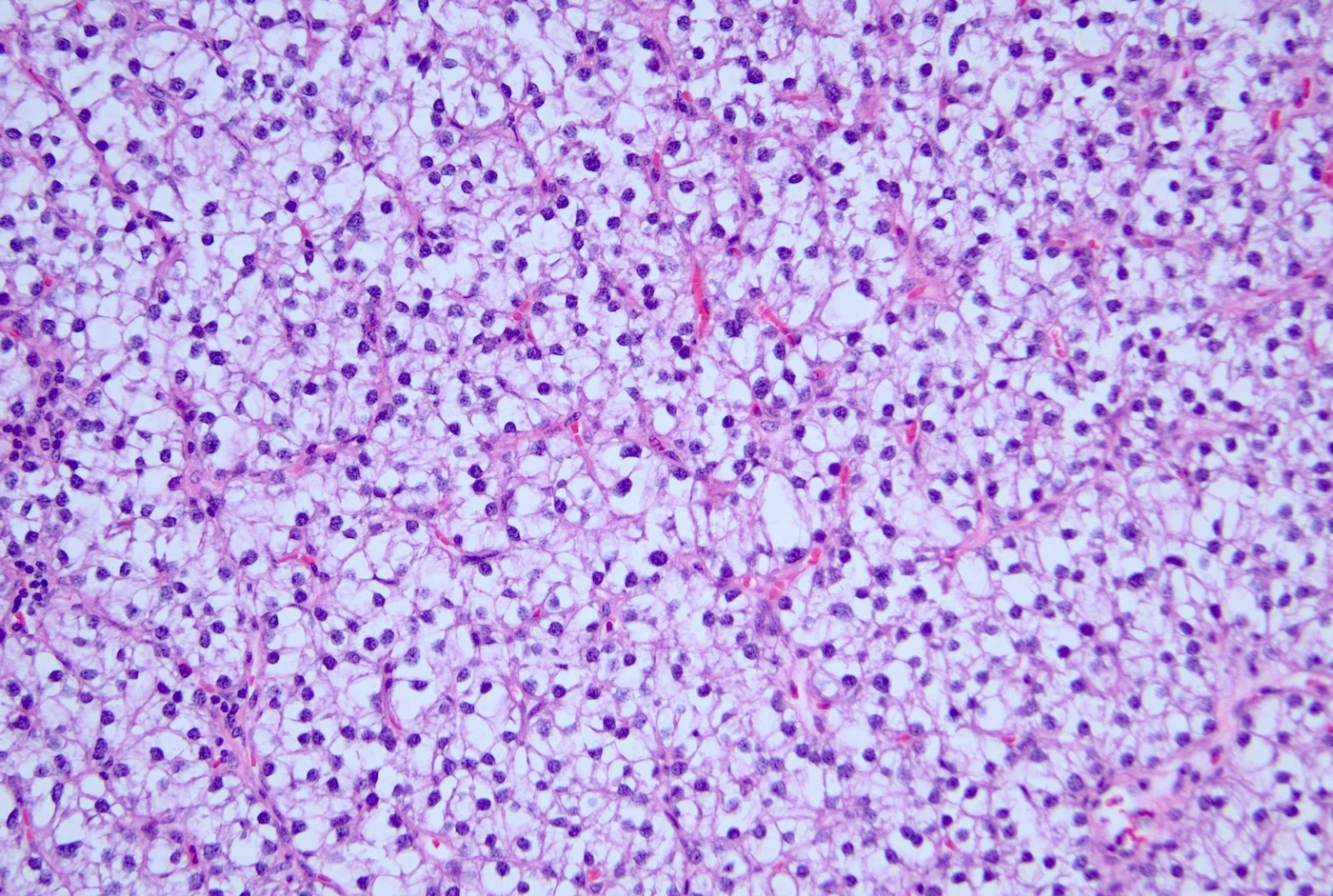
- Pancreatic neuroendocrine tumors
- Well differentiated NETs composed of uniform polygonal cells with eosinophilic or clear cytoplasm, salt and pepper chromatin (finely granular nuclear chromatin) (Arch Pathol Lab Med 2025 Mar 4 [Epub ahead of print])
- Immunohistochemical staining positive for chromogranin A, synaptophysin and neuroendocrine markers (Arch Pathol Lab Med 2025 Mar 4 [Epub ahead of print])
- Some tumors may show vascular invasion or mitotic activity, indicating more aggressive potential
- Diffuse islet cell hyperplasia occurs frequently (World J Gastroenterol 2011;17:137)
- Multiple pancreatic microadenomas or microtumors are a common feature of MEN1 associated PanNETs and serve as a useful clue to the diagnosis of MEN1
- Pituitary adenomas
- Composed of monomorphic polygonal cells arranged in nests, trabeculae or diffuse sheets (Am J Surg Pathol 2008;32:534)
- Lack of significant pleomorphism
- Immunohistochemical staining positive for hormones such as prolactin, growth hormone or ACTH depending on the functional status of the tumor (Am J Surg Pathol 2008;32:534)
- Other possible findings
- Thymic and bronchial carcinoids: small nests of neuroendocrine cells with a trabecular or organoid growth pattern (J Intern Med 1998;243:501)
- Adrenal involvement: cortical hyperplasia or adenomas with clear cell changes
- Duodenal gastrinomas: small nests of well differentiated neuroendocrine cells producing gastrin, seen in Zollinger-Ellison syndrome
- Parathyroid adenomas / hyperplasia
Microscopic (histologic) images
Contributed by R. Peter Powell, M.D. and Wei Chen, M.D., Ph.D.
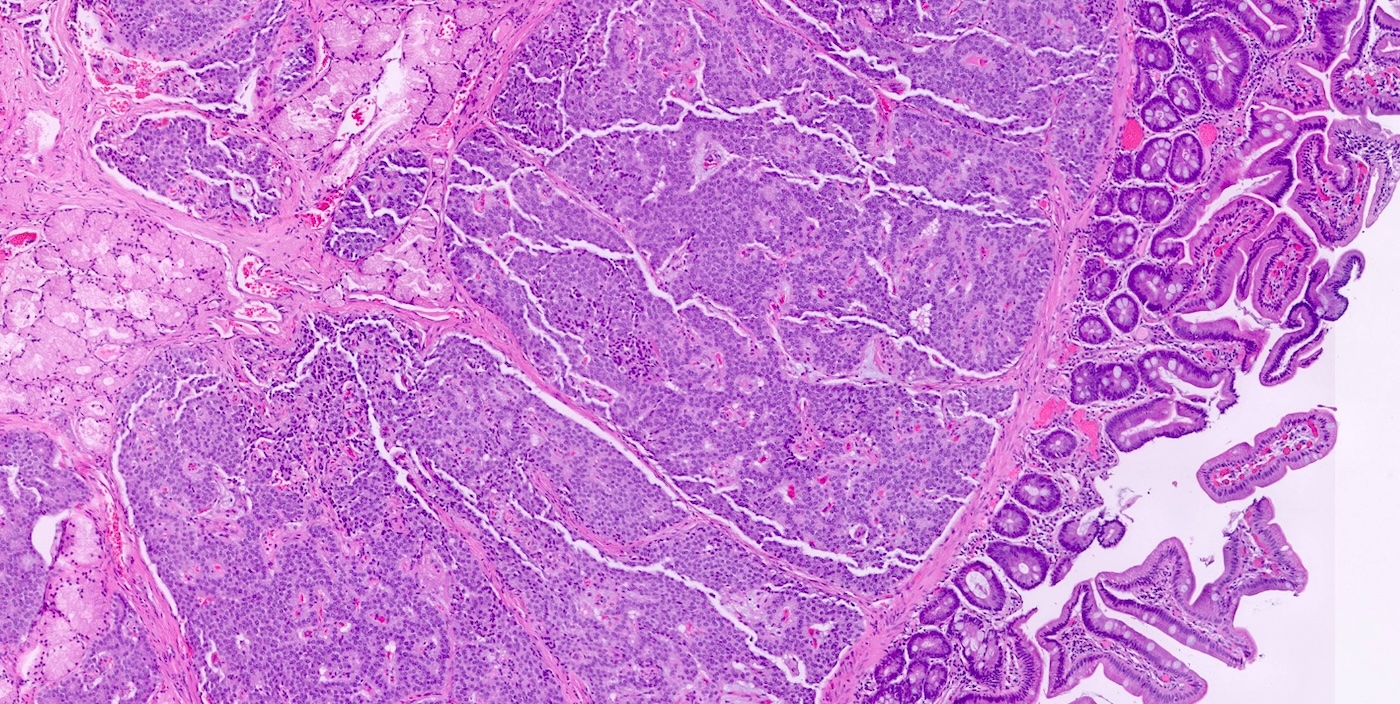
Duodenal neuroendocrine tumor
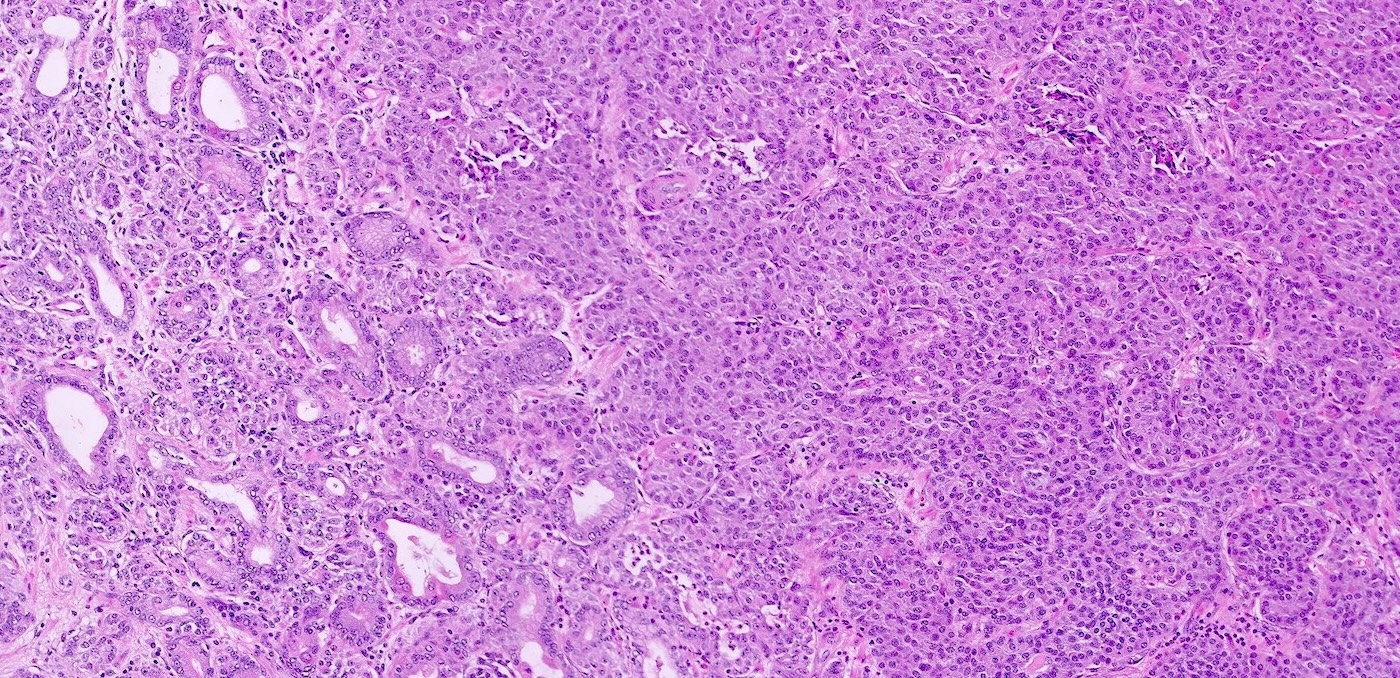
Gastric neuroendocrine tumor
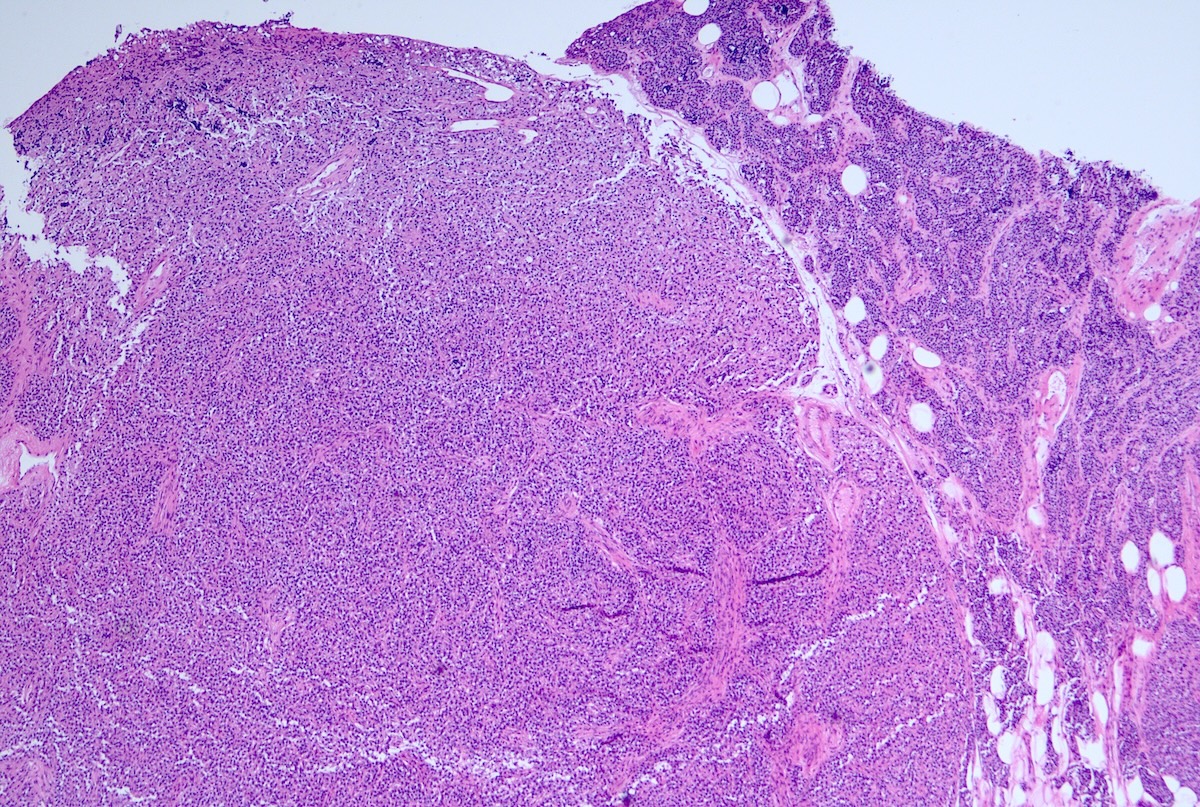
Parathyroid adenoma
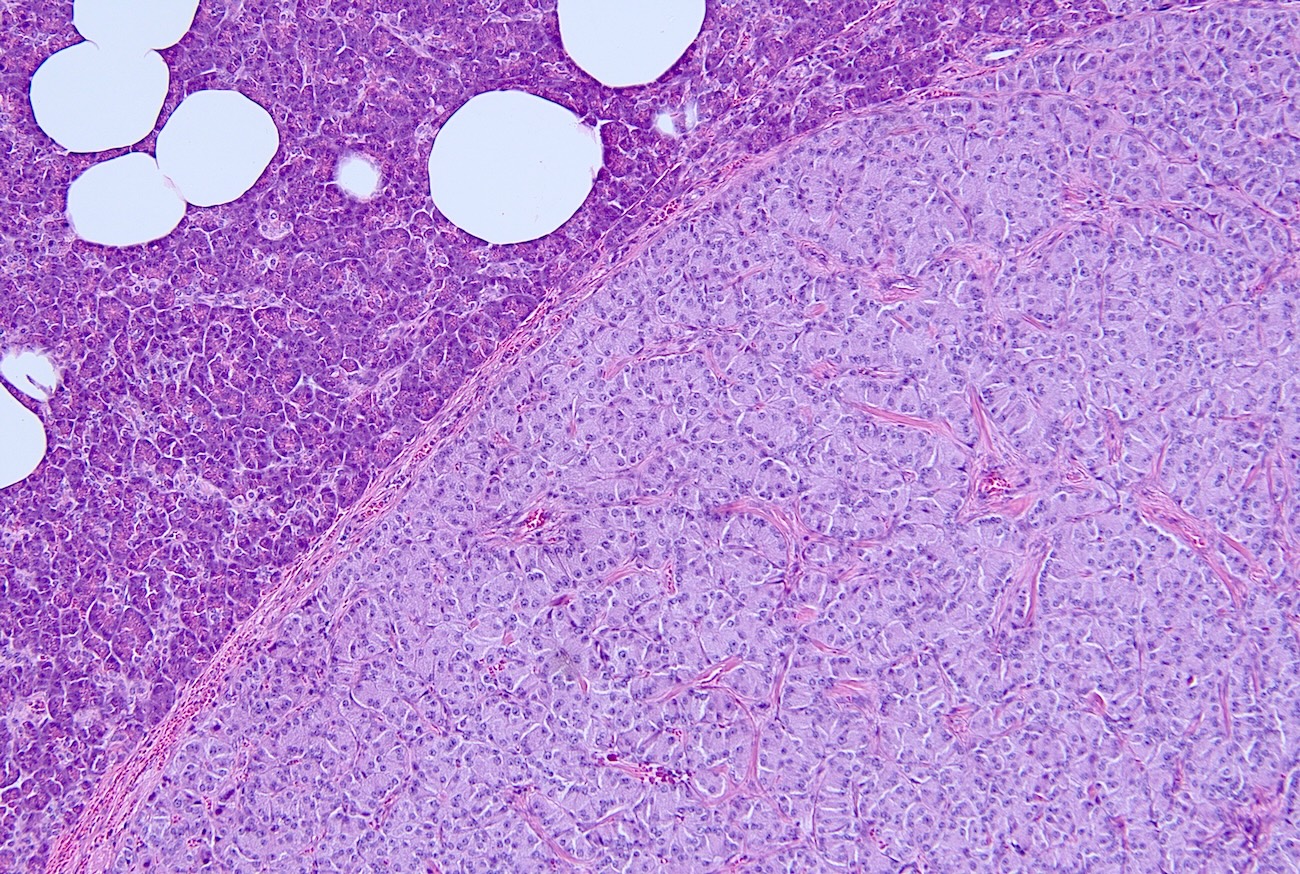
Pancreatic neuroendocrine tumor
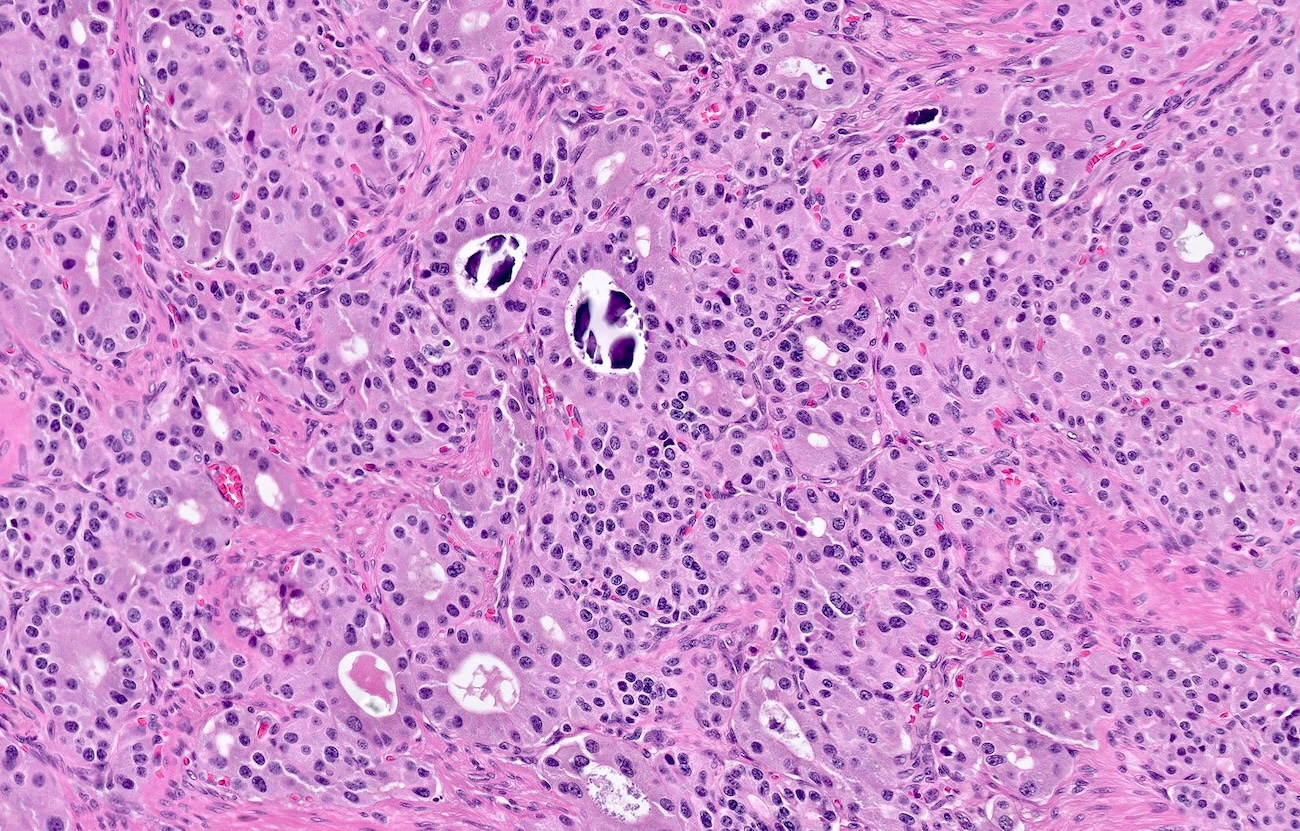
Somatostatinoma
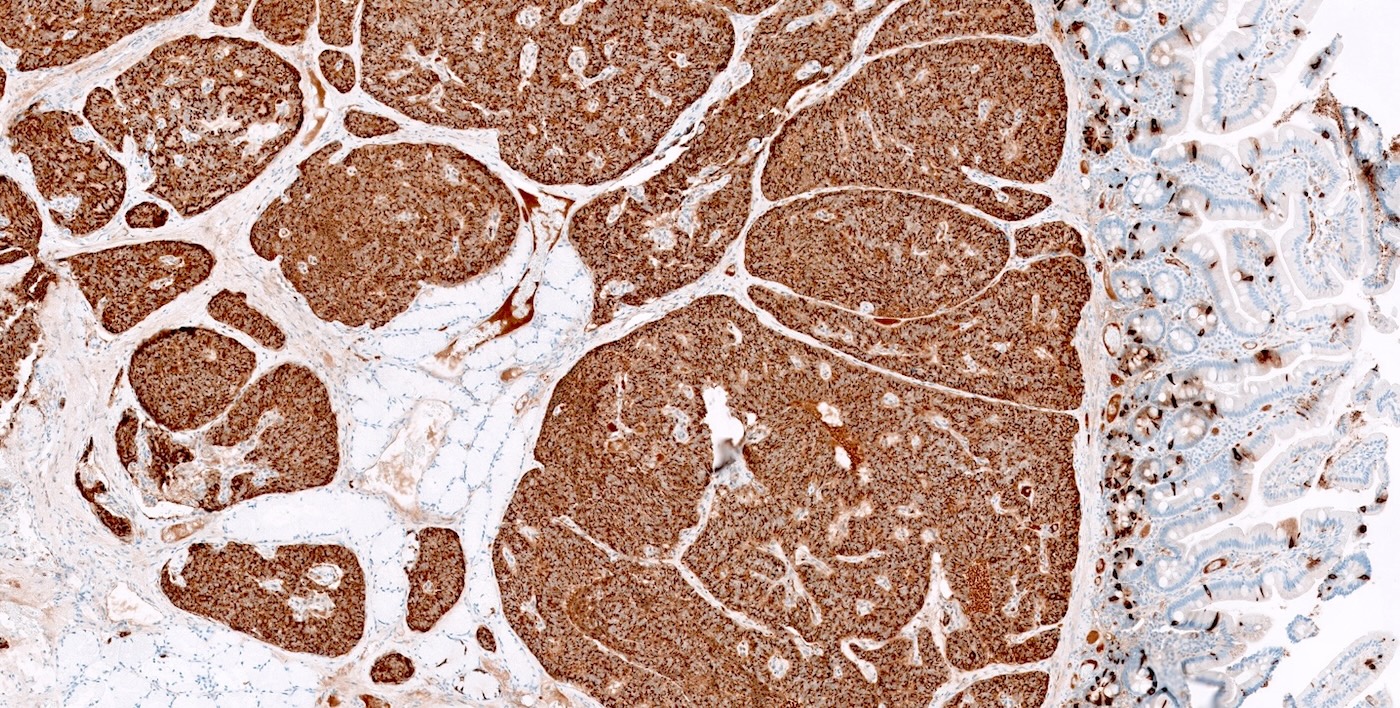
Chromogranin immunostaining in duodenal NET
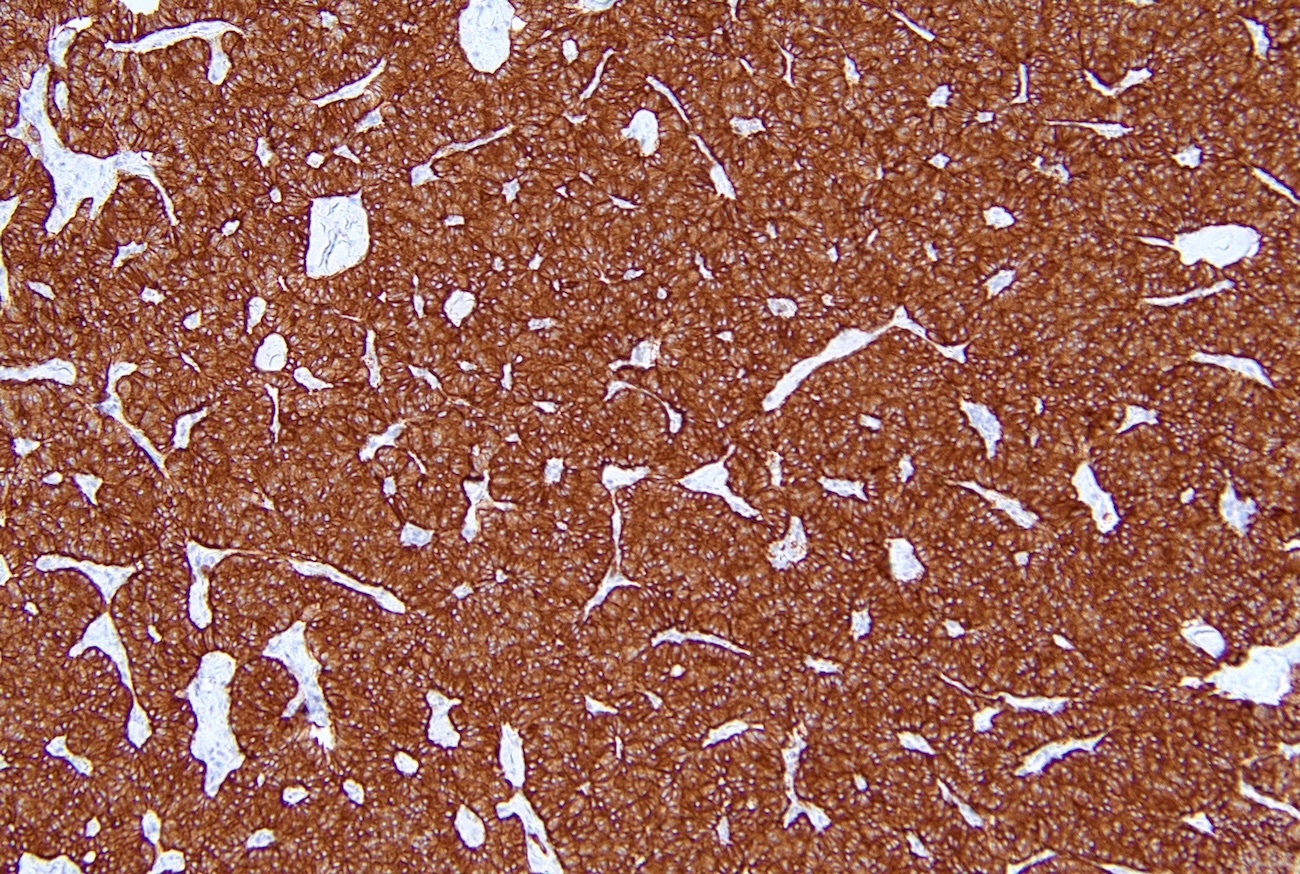
Synaptophysin immunostaining in pancreatic NET
Negative stains
- Immunohistochemical analysis may reveal nuclear menin loss in MEN1 associated tumors (Am J Surg Pathol 2023;47:785, Front Endocrinol (Lausanne) 2023;14:1221514)
Molecular / cytogenetics description
- MEN1 is an autosomal dominant disorder that is caused by mutations in the MEN1 gene, a tumor suppressor gene located on chromosome 11q13, which encodes the protein menin
- Over 1,500 germline and somatic MEN1 mutations (Neuroendocrinology 2021;111:609)
- Germline mutations (inherited): heterozygous mutations are present in all cells of affected individuals
- Somatic mutations (acquired): found in sporadic cases of parathyroid, pancreatic and pituitary tumors
- Mutation types
- Nonsense (30%) mutation leads to truncated, nonfunctional menin protein
- Frameshift (40%) mutation disrupts protein reading frame, leading to loss of function
- Missense (20%) mutation alters protein function without truncation
- Large deletions / insertions (10%) result in complete gene loss
- Other chromosome changes (e.g., translocations or amplifications in aggressive pancreatic neuroendocrine tumors)
- 2 hit hypothesis: first hit is germline MEN1 mutation; second hit is somatic deletion / mutation of the remaining normal MEN1 allele
- Fluorescence in situ hybridization (FISH), multiplex ligation dependent probe amplification (MLPA) and PCR sequencing can detect MEN1 deletions and mutations
- Diagnostic genetic testing for MEN1 mutations is recommended for family members of affected individuals or individuals with multiple MEN1 related tumors
Videos
Multiple endocrine neoplasia - Osmosis from Elsevier
Basics of multiple endocrine neoplasia -
Mayo Clinic - Dr. Mark Lewis
Multiple endocrine neoplasia - Canadian Neuroendocrine Tumour Society
Sample pathology report
- Pancreas, distal pancreatectomy:
- Neuroendocrine tumor, well differentiated, WHO grade 1 (see comment)
- Comment: The histologic findings, along with immunohistochemical studies (positive synaptophysin and chromogranin staining and a Ki67 proliferative rate of 1% in tumor cells), confirm the diagnosis of a well differentiated pancreatic neuroendocrine tumor (grade 1). The patient has a known history of parathyroid hyperplasia and a pituitary adenoma. The presence of primary hyperparathyroidism, pancreatic neuroendocrine tumors and pituitary adenoma is consistent with the classic triad of multiple endocrine neoplasia type 1 (MEN1).
Differential diagnosis
- Sporadic PanNETs:
- Usually single tumors, no other endocrine neoplasms (Mod Pathol 2023;36:100065)
- Von Hippel-Lindau (VHL) syndrome:
- Caused by mutations in the VHL gene, leading to increased risk of developing tumors and cysts in multiple organs, e.g., PanNETs, renal cell carcinoma, hemangioblastomas, pheochromocytomas, endolymphatic sac tumors of inner ear, cysts in kidneys, pancreas and reproductive organs
- Neurofibromatosis type 1 (NF1):
- Caused by mutations in the NF1 gene, symptoms include PanNETs (often somatostatinomas), café au lait spots, neurofibromas, Lisch nodules, optic gliomas, freckling in the groins or armpit, skeletal abnormalities and learning disabilities
- MEN4 (CDKN1B mutation):
- Similar to MEN1 but much rarer (Endocr Connect 2023;12:e220411, J Clin Endocrinol Metab 2022;107:2339)
Practice question #1
A 38 year old man with a history of recurrent duodenal ulcers and nephrolithiasis presents for evaluation of new onset headaches and visual disturbances. His lab results reveal elevated parathyroid hormone (PTH), hypercalcemia, increased gastrin and prolactin levels. Which of the following is the most likely explanation for his symptoms?
- Medullary thyroid carcinoma associated with RET mutation
- Patient with MEN1 mutation and no tumor development at the time of evaluation
- Patient with parathyroid adenoma, meningioma and epididymoma
- Patient with parathyroid adenoma, pituitary adenoma and duodenopancreatic neuroendocrine tumor
- Pituitary adenoma secreting adrenocorticotropic hormone (ACTH)
Practice answer #1
D. Patient with parathyroid adenoma, pituitary adenoma and duodenopancreatic neuroendocrine tumor. This patient's symptoms are most consistent with multiple endocrine neoplasia type 1 (MEN1), which is characterized by tumors of the parathyroid glands, pituitary gland and pancreatic / duodenal neuroendocrine tumors. The elevated PTH and hypercalcemia point toward a parathyroid tumor. The gastrin elevation and duodenal ulcer suggest a gastrinoma (duodenopancreatic neuroendocrine tumor). The elevated prolactin level suggests a pituitary adenoma. Answer E is incorrect because ACTH secreting pituitary adenomas lead to Cushing syndrome, not elevated prolactin level. Answer A is incorrect because medullary thyroid carcinoma (MTC) can be seen in MEN2, not MEN1. Answer B is incorrect because the patient's clinical signs and elevated gastrin, prolactin and PTH indicate the presence of tumors. Answer C is incorrect because meningiomas and epididymomas are not characteristic features of MEN1 meningiomas.
Comment Here
Reference: MEN1 syndrome
Comment Here
Reference: MEN1 syndrome
Practice question #2
A 45 year old woman presents with a history of recurrent kidney stones, fatigue and constipation. Laboratory studies show hypercalcemia, low phosphate levels and an elevated parathyroid hormone (PTH). Imaging evaluation shows an enlargement of the parathyroid gland and a well circumscribed pancreatic mass. Genetic testing confirms a mutation in the MEN1 gene. Which of the following best describes the function of the gene mutated in this patient?
- DNA repair gene
- Gene encoding a protein involved in calcium sensing
- Oncogene encoding a receptor tyrosine kinase
- Proto-oncogene involved in cell cycle progression
- Tumor suppressor gene involved in transcription regulation
Practice answer #2
E. Tumor suppressor gene involved in transcription regulation. The MEN1 gene encodes menin, a tumor suppressor protein that regulates transcription and is involved in cellular proliferation and survival. Mutations in MEN1 lead to the development of endocrine tumors, commonly in the parathyroid glands, pancreas and pituitary glands. The patient's symptoms, including hypercalcemia, elevated PTH, enlargement of the parathyroid gland and a pancreatic mass, are consistent with MEN1 and its associated tumors, such as parathyroid adenomas and pancreatic neuroendocrine tumors. Answer D is incorrect because the MEN1 gene is a tumor suppressor gene. MEN1 mutations result in the loss of tumor suppression. Answer C is incorrect because oncogenes typically encode proteins that promote cell division or survival via receptor tyrosine kinases or other growth factors. However, MEN1 is a tumor suppressor gene. Answer B is incorrect because the MEN1 gene does not directly encode a protein involved in calcium sensing. Answer A is incorrect because the MEN1 gene is not directly involved in DNA repair.
Comment Here
Reference: MEN1 syndrome
Comment Here
Reference: MEN1 syndrome
