Soft tissue
Skeletal muscle
Rhabdomyosarcoma
Embryonal rhabdomyosarcoma
Author: Michael R. Clay, M.D.
Editorial Board Member: Debra L. Zynger, M.D.
Deputy Editor-in-Chief: Borislav A. Alexiev, M.D.

Last author update: 17 August 2022
Last staff update: 2 June 2025
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Embryonal rhabdomyosarcoma
See Also: Spindle cell / sclerosing rhabdomyosarcoma
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagnosis | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Pitfalls and tips | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Cytology images | Positive stains | Electron microscopy description | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Clay MR. Embryonal rhabdomyosarcoma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/softtissueembryonalrhabdo.html. Accessed September 14th, 2025.
Definition / general
- Subtype of the rhabdomyosarcoma (RMS) soft tissue cancer family whose lineage derives from the undifferentiated mesoderm
- Embryonal rhabdomyosarcoma (ERMS) is the most common RMS subtype
- Several distinct and prognostic patterns of growth have been noted, including the botryoid and anaplastic variants
Essential features
- Most common subtype of rhabdomyosarcoma in the pediatric and adolescent setting
- Displays a wide spectrum of morphologic features, including cases with spindled morphology
- Displays some degree of myogenic differentiation, including either MyoD1 or myogenin positivity
- Not associated with recurrent gene fusions but displays some recurrent copy number changes
- Diagnostic criteria are: (a) round and spindle cell morphology with scattered differentiated rhabdomyoblasts; (b) desmin positivity and heterogenous nuclear staining for myogenin or MYOD1; (c) lack of FOXO1 gene rearrangements distinguishes poorly differentiated ERMS from solid alveolar RMS
Terminology
- Botryoid embryonal rhabdomyosarcoma (sarcoma botryoides) only occurs in certain locations, specifically beneath mucosal epithelial lined viscera, such as the bladder, biliary tract, vagina or upper respiratory tract, extrahepatic bile ducts or near a space; rarely in eyelid or anal region
- Often has a grape-like (botryoid) growth pattern
- 25% of rhabdomyosarcomas, 10% of embryonal subtype
- Considered a subtype of embryonal rhabdomyosarcoma (ERMS) (Am J Surg Pathol 2001;25:856)
- Not recommended to use: sarcoma botryoides; mixed embryonal and alveolar rhabdomyosarcoma
ICD coding
- ICD-O: 8910/3 - embryonal rhabdomyosarcoma, NOS
- ICD-11: 2B55.Z & XH83G1 - rhabdomyosarcoma, unspecified primary site & embryonal rhabdomyosarcoma, NOS
Epidemiology
- Most common in children 0 - 4 years old (with a maximum reported incidence of 4 cases per 1 million children) but can be identified at any age (Cancer 2009;115:4218)
- Represents approximately 60 - 70% of childhood cases and 20% of adult cases
- Rare congenital cases have also been reported (Int J Surg Case Rep 2017;31:47)
Sites
- Most common in the head and neck region, including the nasal and oral cavities, as well as the orbit and middle ear (J Clin Oncol 1995;13:610)
- Also common in the paratesticular soft tissues and the genitourinary tract
- Extremity involvement of the somatic soft tissues is less common (< 9%)
- Metastasizes to soft tissue, serosa, lung, lymph nodes and bone marrow
Pathophysiology
- Derived from the undifferentiated mesoderm and shows phenotypic and biologic features of primitive skeletal muscle
Etiology
- Most cases occur sporadically with no genetic predisposition
- Children with predisposing familial syndromes, in which there are frequently defects in the RAS or Hedgehog pathways, are at higher risk for developing embryonal rhabdomyosarcoma (Genes Chromosomes Cancer 2009;48:975)
- Seen secondarily in syndromes including Costello syndrome (HRAS), neurofibromatosis type 1 (NF1), Noonan syndrome (PTPN11 and others), Beckwith-Wiedemann syndrome (changes to 11p15), Dicer syndrome (DICER1), Li-Fraumeni syndrome (TP53) and Gorlin syndrome (PTCH1) (Cancer Med 2015;4:781, J Biomol Tech 2013;24:S47, Mod Pathol 2012;25:602)
Diagnosis
- Clinical and morphologic features: primitive hypercellular tumor with spindled to round shaped cells; mitotic activity and necrosis are common
- Positivity for desmin, myogenin and MyoD1
- Confirmation of no evidence of FOXO1 gene fusion in challenging cases (to exclude alveolar rhabdomyosarcoma)
Prognostic factors
- Intergroup Rhabdomyosarcoma Study Group (IRSG) has subdivided rhabdomyosarcoma into low, intermediate and high risk groups for the purpose of protocol placement in pediatric patients (Pediatr Blood Cancer 2012;59:5)
- Based on clinicopathologic variables including histologic type, extent of surgical control of local disease and primary tumor site
- Botryoid variant has a particularly good prognosis, although may have late relapse (Zhonghua Bing Li Xue Za Zhi 2004;33:225, Pediatr Blood Cancer 2008;51:140)
- Other favorable prognostic factors are age (between 1 and 9 years), orbital and paratesticular location and absence of metastatic disease at the time of resection
- Anaplasia is defined similar to anaplastic Wilms tumor as a significant nuclear variation (cells that are 3x larger than background tumor cells) and the presence of atypical multipolar mitotic figures
- It was historically a negative prognostic factor but this may no longer be true (Cancer 2008;113:3242, Eur J Cancer 2021;143:127)
- Extremity involvement is associated with more relapses and lower survival
Case reports
- Newborn boy with embryonal rhabdomyosarcoma (ERMS) presenting as a mass of the urethra at birth (Urology 2015;86:805)
- Newborn girl with anaplastic rhabdomyosarcoma (Iran J Pathol 2016;11:80)
- 7 year old boy with primary ERMS of the anterior neck and thyroid (Laryngoscope 2013;123:2072)
- 11 year old boy with tonsillar mass (Case #276)
- 16 year old girl with ERMS of the cervix (J Med Case Rep 2014;8:241)
- 46 year old man with ERMS arising from a mediastinal teratoma (J Korean Med Sci 2013;28:476)
- 55 year old postmenopausal woman with ERMS arising from the uterine corpus (Diagn Pathol 2016;11:3)
- 58 year old man with cardiac ERMS (Eur J Cardiothorac Surg 2014;45:e233)
Treatment
- Surgery is the preferred local therapy, with radiotherapy typically used only in the event of residual disease, nodal disease or poor response to combined chemotherapy
- Intergroup Rhabdomyosarcoma Study Postsurgical Clinical Grouping System is recommended to plan treatment in pediatric cases (Pediatr Blood Cancer 2012;59:5)
- American Joint Committee on Cancer (AJCC) TNM staging system remains appropriate for planning treatment in adult patients (Amin: AJCC Cancer Staging Manual, 8th Edition, 2018)
- Anaplastic tumors often require more intensive treatment (Cancer 2008;113:3242)
- Botryoid variant: conservative surgery plus radiation and chemotherapy (Int J Gynecol Cancer 2008;18:190)
Gross description
- Poorly circumscribed mass, white, soft or firm, infiltrative
- Botryoid variant: resembles cluster of grapes or allergic nasal polyp, fleshy nodular polypoid projections of variable size into lumen
Gross images
Contributed by Mark R. Wick, M.D. and AFIP
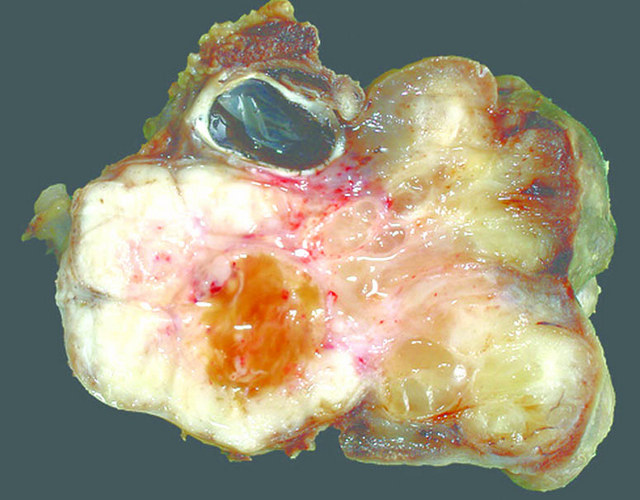
Orbital

Botryoid
Images hosted on other servers:
White tumor mass with focal necroses
Incision of the oral mucosa
Pitfalls and tips
- Anaplasia specifically requires multipolar mitotic figures
- Make sure you are not overinterpreting degenerating or apoptotic cells
- Look for organization of the division and exclude cells that appear to be exploding
- Also try to avoid interpreting overlapping cells as 1 larger cell
- Absence of a translocation does not result in the default diagnosis of embryonal rhabdomyosarcoma; tumor must adhere to the described histologic features
- Some rhabdomyosarcomas defy classification (rhabdomyosarcoma, not otherwise specified); this designation is preferable to misclassification, especially in small or otherwise limited samples
- Beware nonspecific staining (aberrant positivity for cytokeratin or S100 protein) versus true staining (S100 protein as a component of malignant peripheral nerve sheath tumor)
- Damaged skeletal muscle can express MyoD1 and myogenin; a different sarcoma that otherwise infiltrates skeletal muscle will very much mimic a rhabdomyosarcoma on immunohistochemistry staining
- Look for the MyoD1 / myogenin to be positive in tumor cells in a linear pattern, which suggests the staining is of background muscle and not the tumor
Microscopic (histologic) description
- Composed of primitive mesenchymal cells that show variable degrees of skeletal muscle differentiation
- They are moderately cellular but in the typical pattern often contain both hypocellularity and hypercellular areas with a loose, myxoid stroma
- Perivascular condensations of tumor cells in the less cellular regions are common
- Sheets of small, stellate, spindled or round cells with scant or deeply eosinophilic cytoplasm and eccentric, small oval nuclei with a light chromatin pattern and inconspicuous nucleoli
- Can occasionally identify tumor cells that contain generous amounts of eosinophilic cytoplasm, a feature of rhabdomyoblastic differentiation (so called strap cells)
- These may become more prominent with chemotherapy (chemotherapeutic induced cytodifferentiation)
- May have cells with elongated tails of cytoplasm (tadpole cells)
- If densely cellular, may resemble solid alveolar rhabdomyosarcoma (Am J Clin Pathol 2013;140:82)
- Botryoid variant frequently shows a cambium layer: a hypercellular zone immediately beneath the epithelial surface
- Cells are undifferentiated, round or spindled with minimal cytoplasm, frequent mitotic figures
- Deeper layers of the tumor are typically less cellular but overall conform to the histology of embryonal rhabdomyosarcoma (ERMS) with variation by region
- Rare morphological patterns:
- Rhabdomyosarcoma with rhabdoid features (epithelioid or rhabdoid-like RMS) (Am J Surg Pathol 2011;35:1523)
- Clear cell change (Pediatr Pathol Lab Med 1996;16:951)
- Cartilaginous metaplasia (Oral Surg Oral Med Oral Pathol Oral Radiol 2017;124:e288)
- Do not confuse with the following different entities:
- Ectomesenchymoma: melanocytic, neuroblastic / ganglionic or Schwannian differentiation in the setting of background ERMS
- Malignant triton tumor: rhabdomyosarcomatous differentiation in a malignant peripheral nerve sheath tumor; very bad prognosis
Microscopic (histologic) images
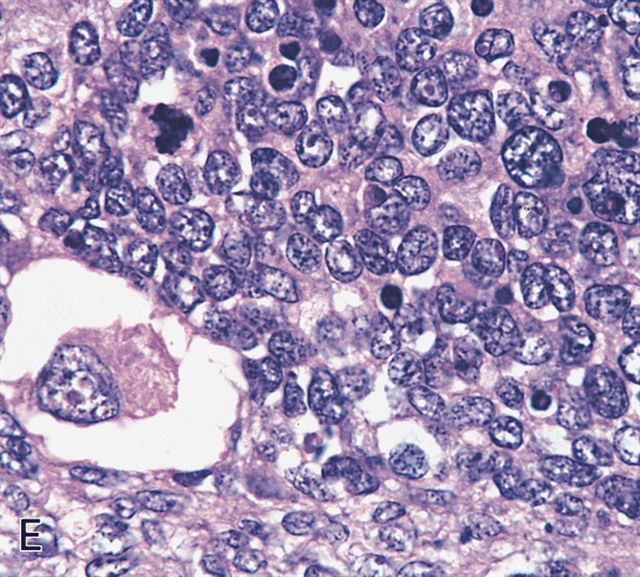
Contributed by Erdener Özer, M.D., Ph.D. and Mark R. Wick, M.D.
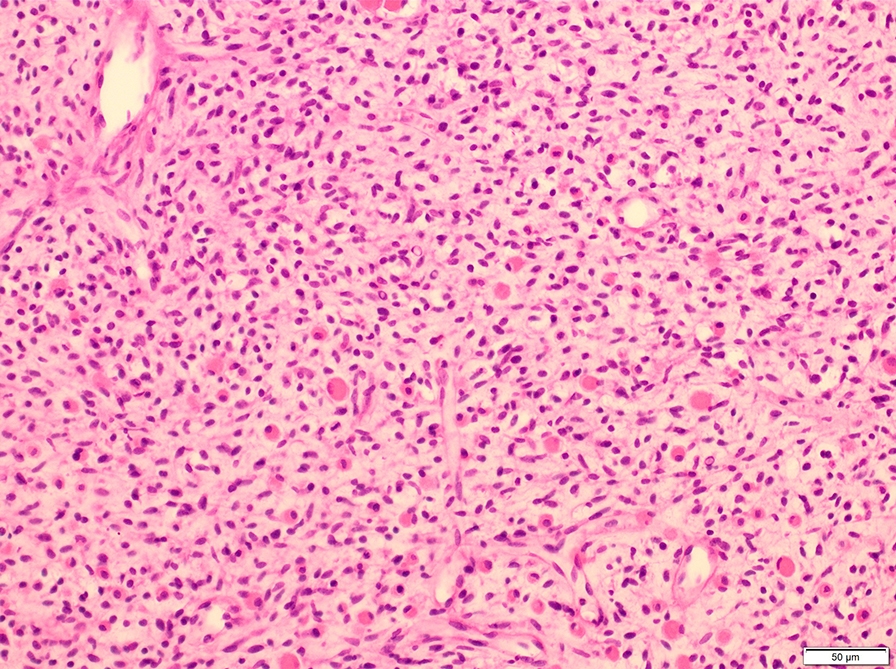

Spindled and round cells
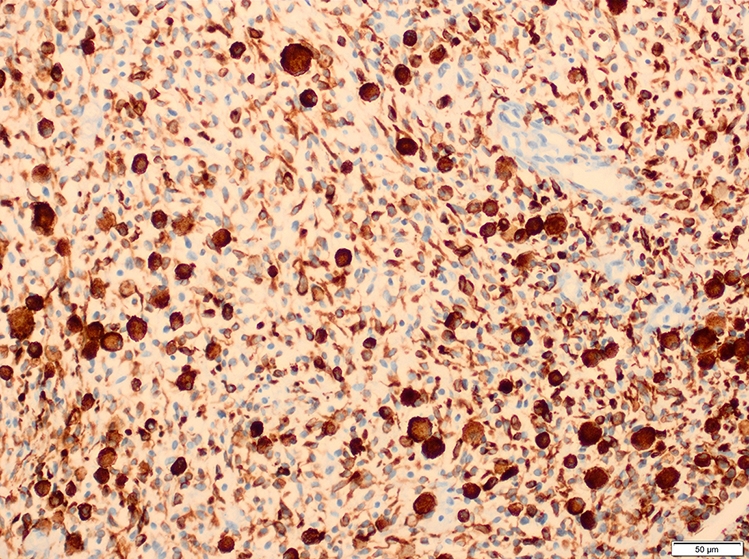
Desmin
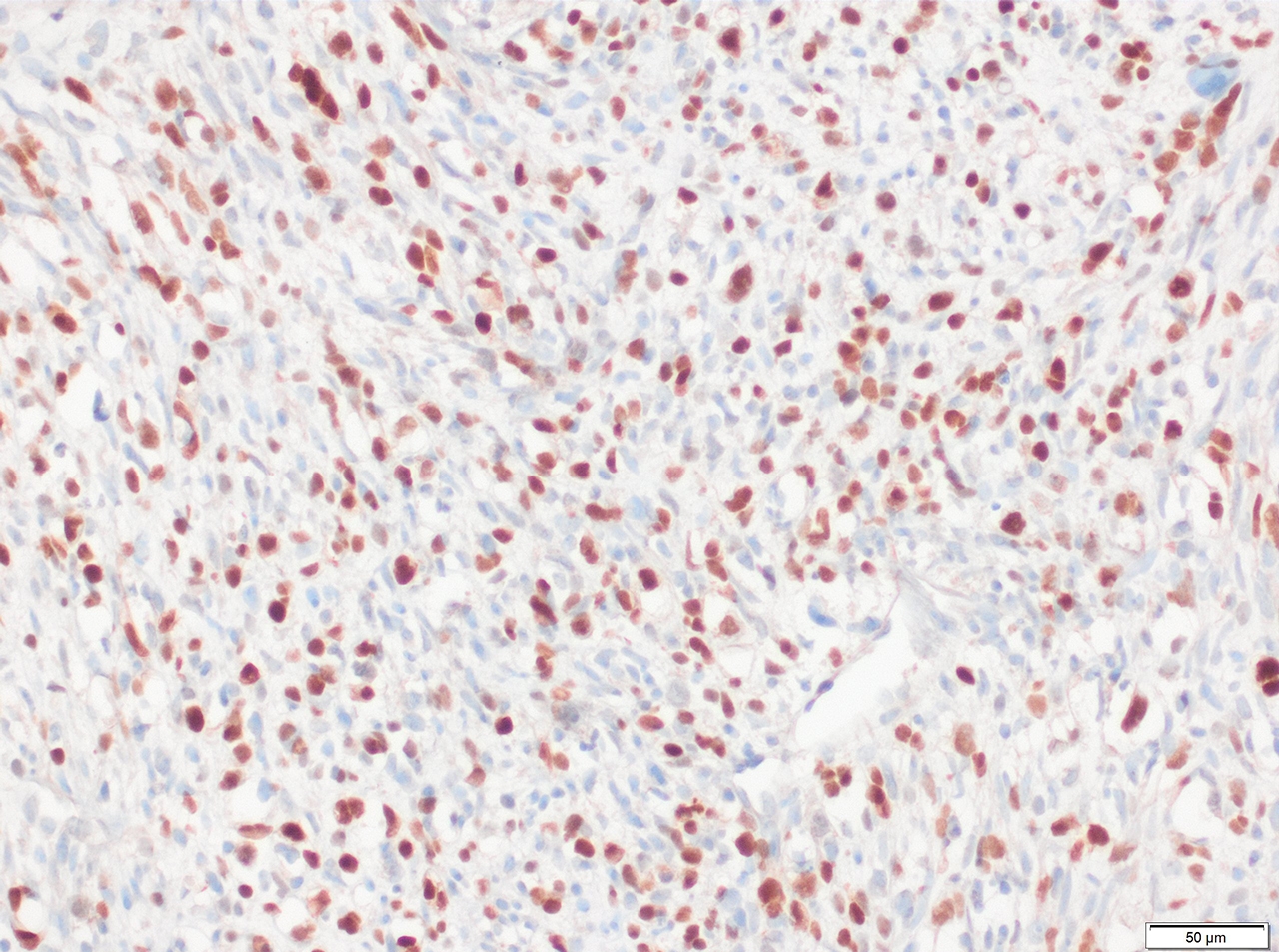
Myogenin
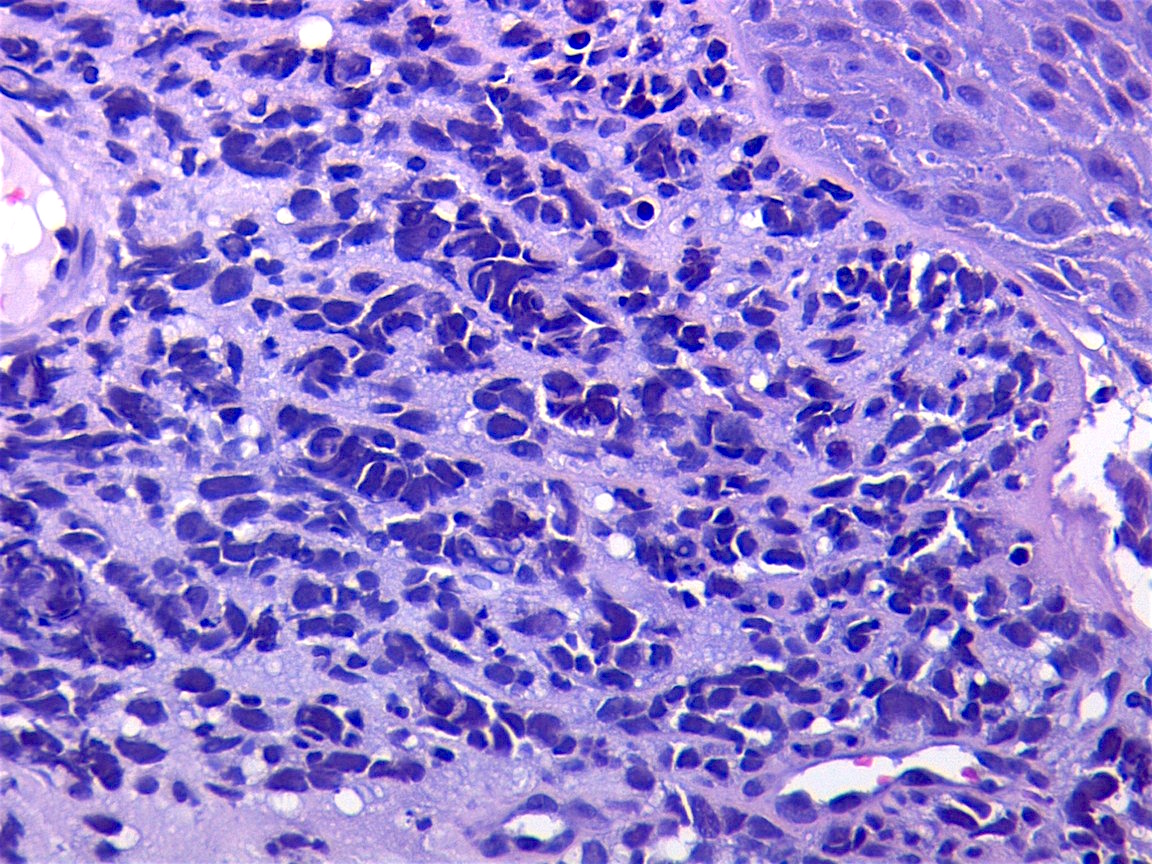
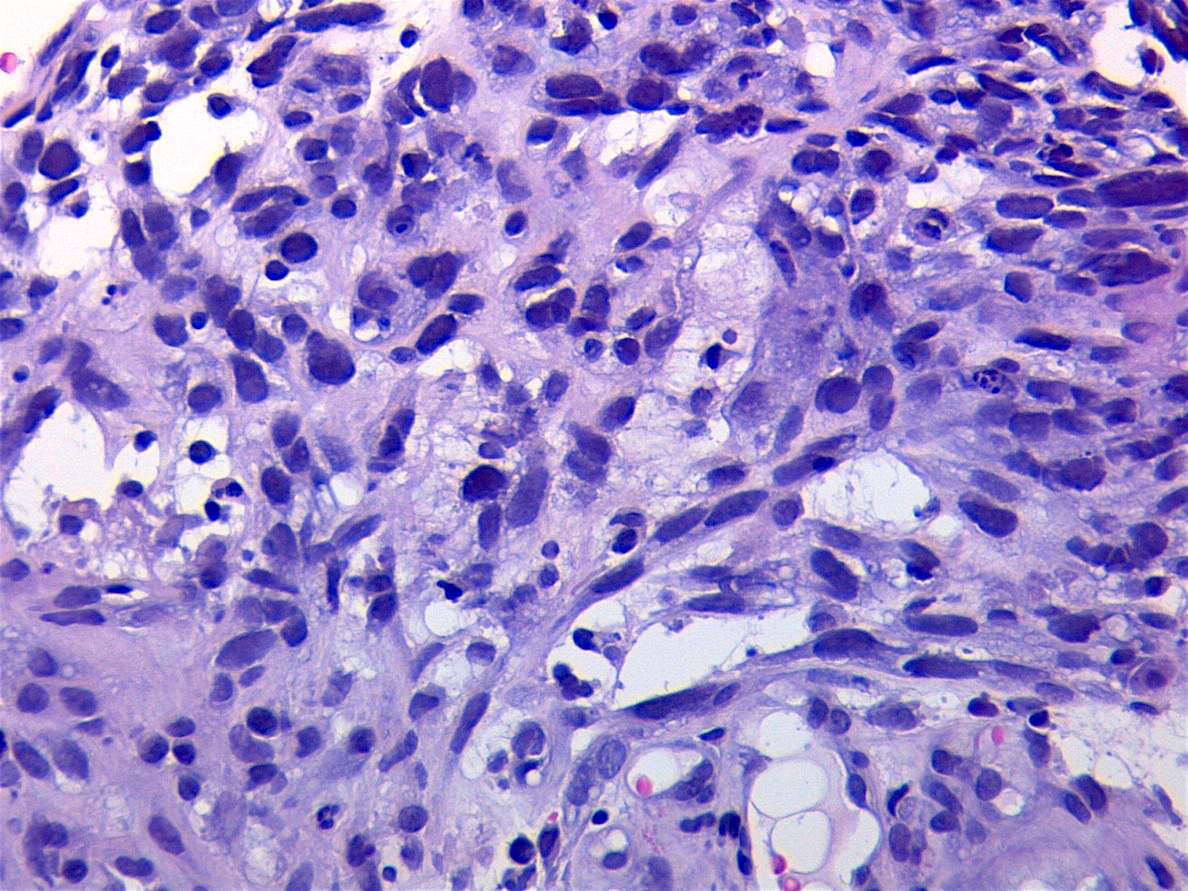
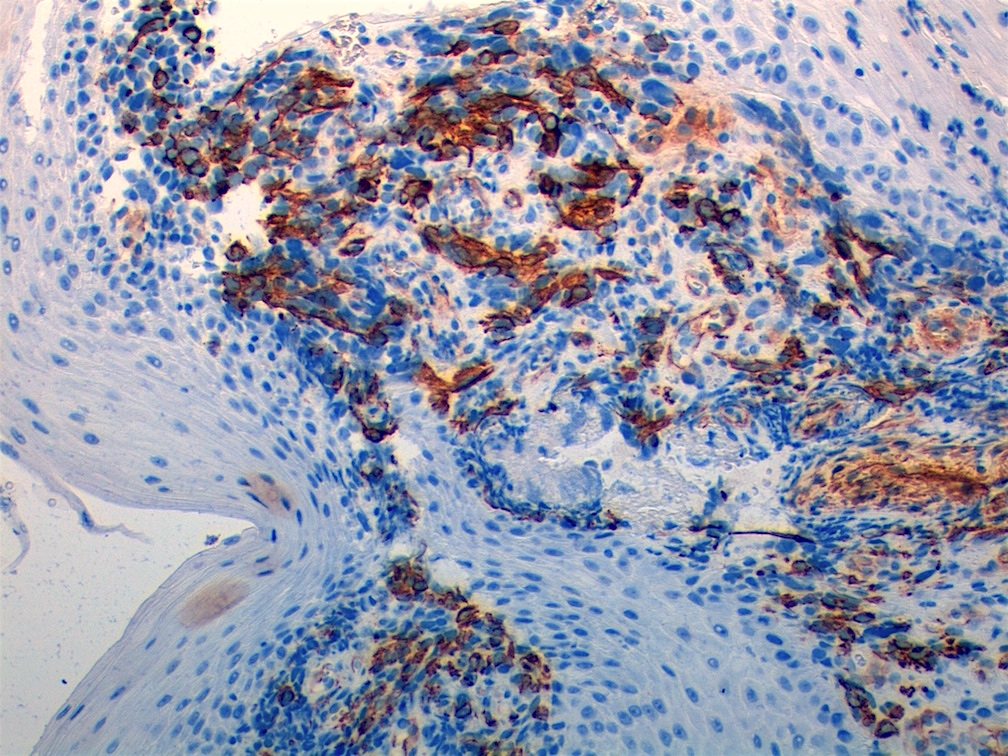
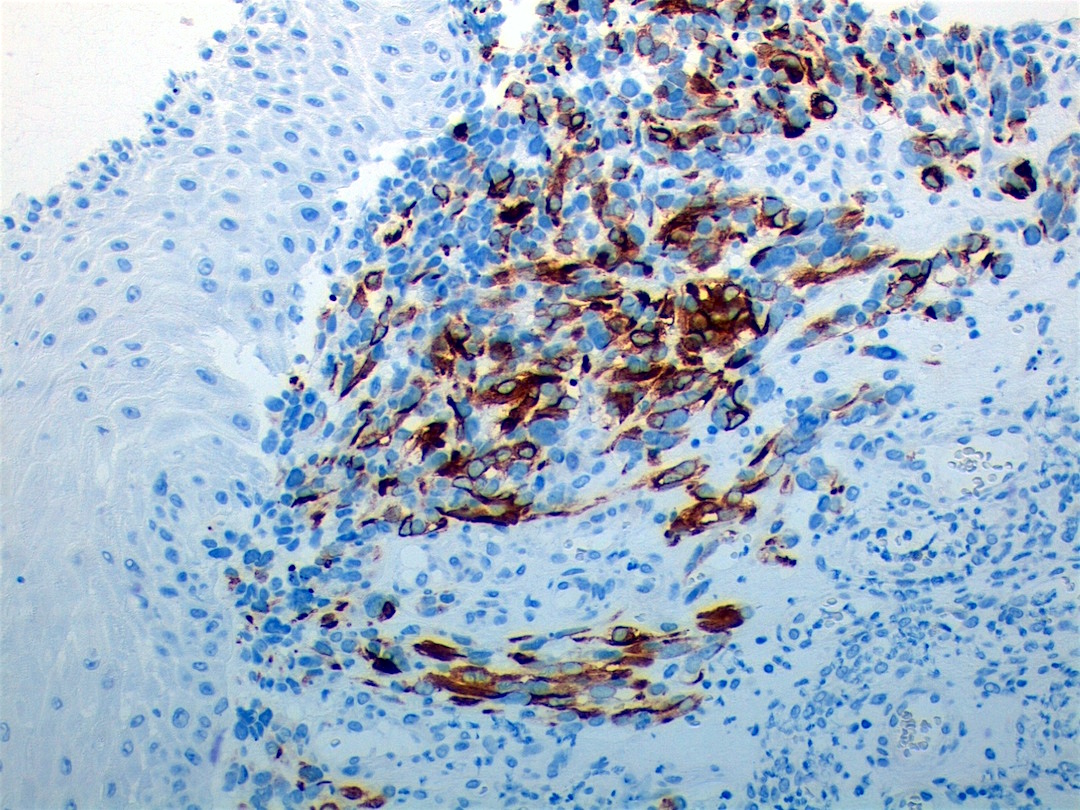
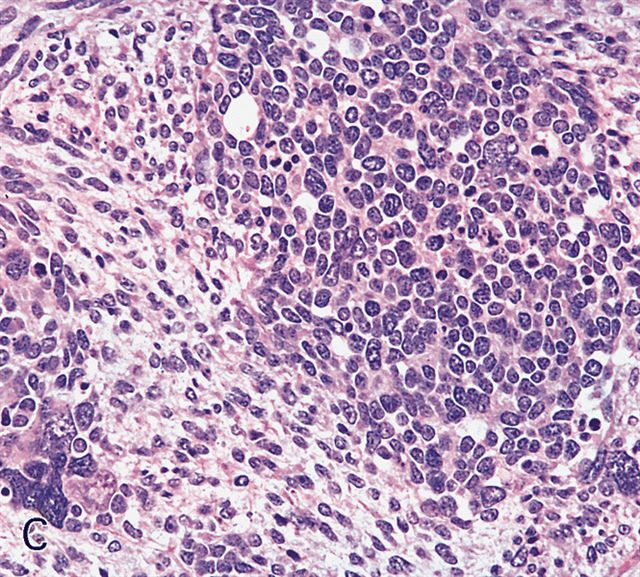
Contributed by Carolina Martinez Ciarpaglini, M.D., Ph.D. (Case #276) - tonsillar mass
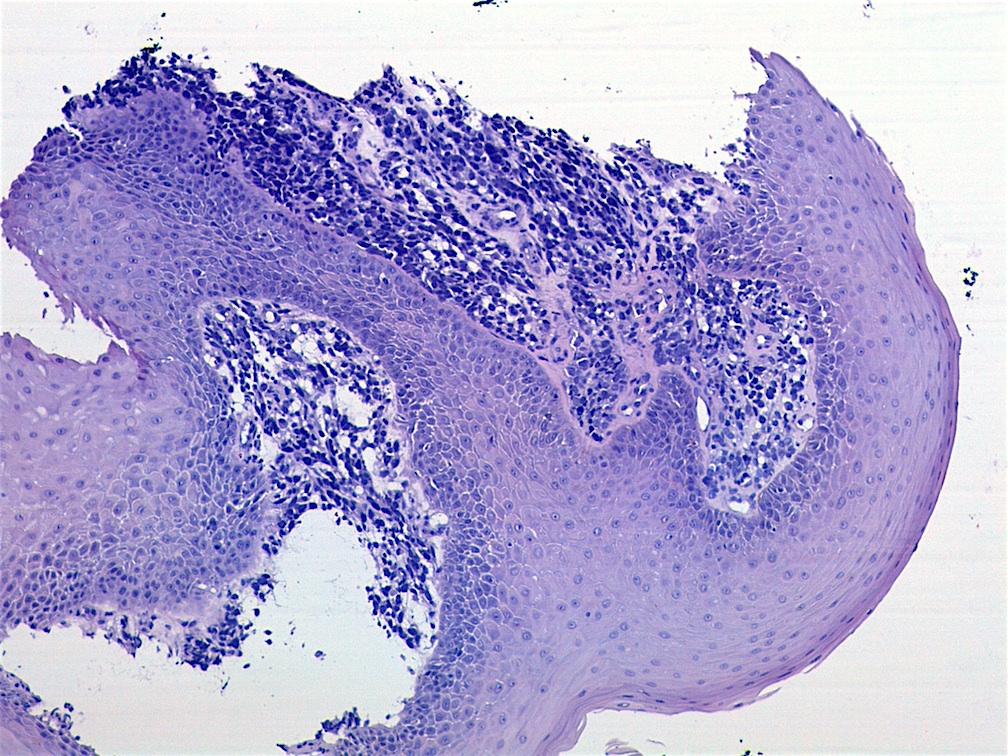
Nests and sheets
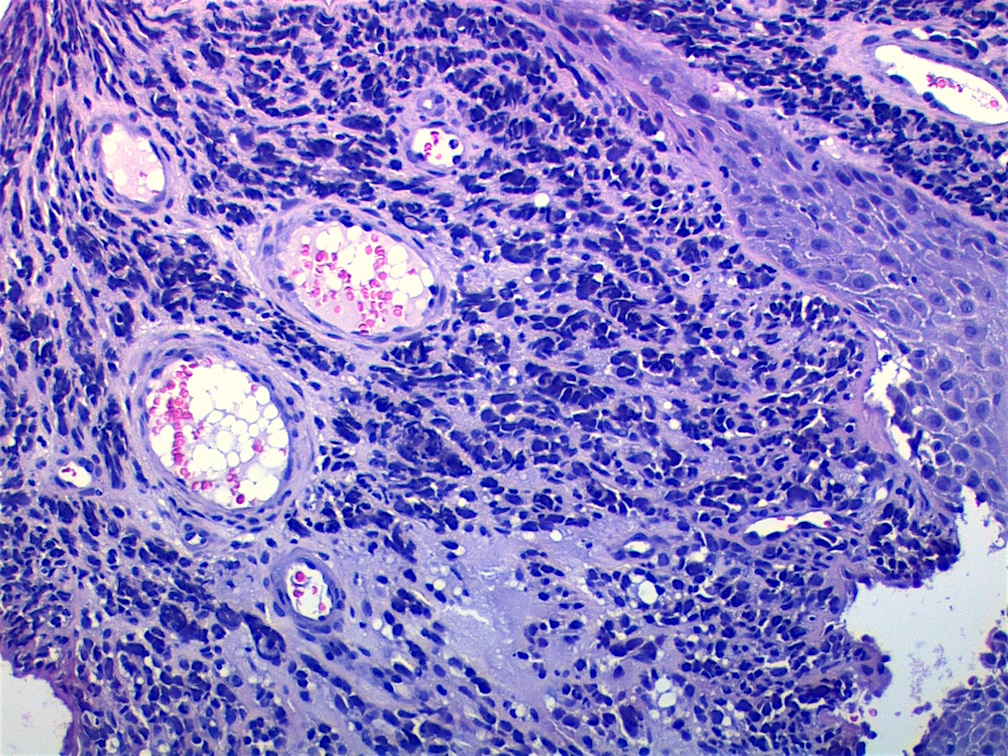
Crush artifact
Nuclear molding
Focally epithelioid
Actin
Desmin
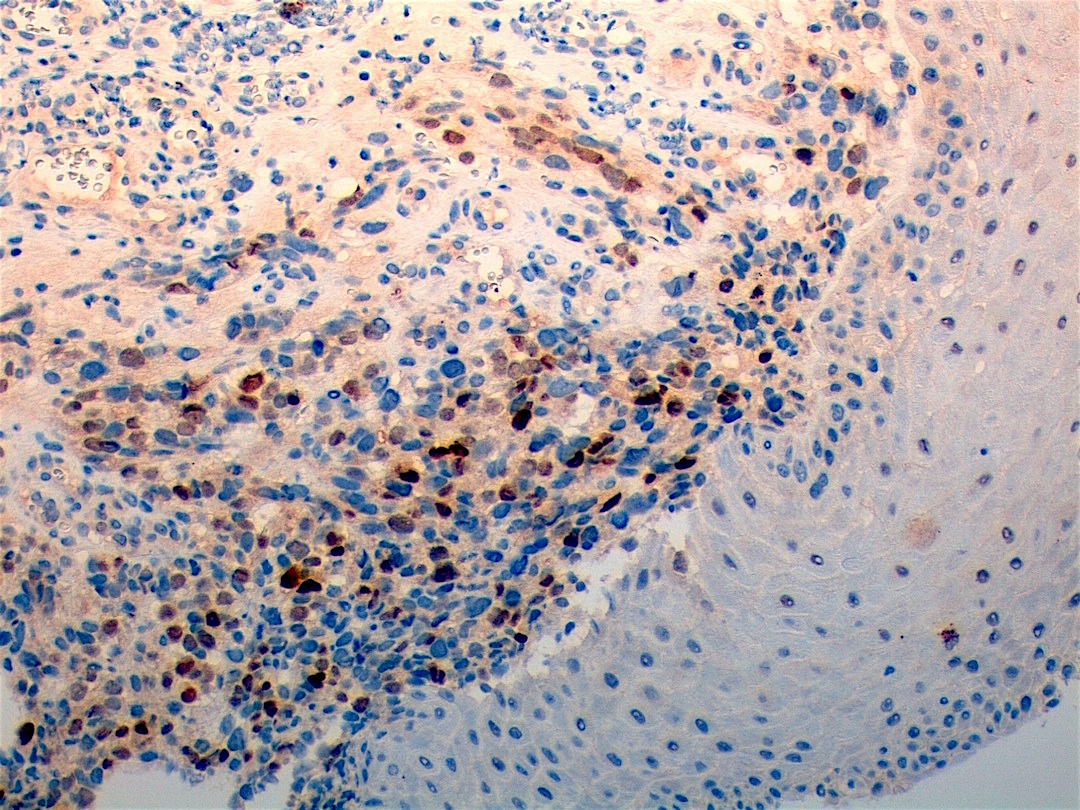
Myogenin
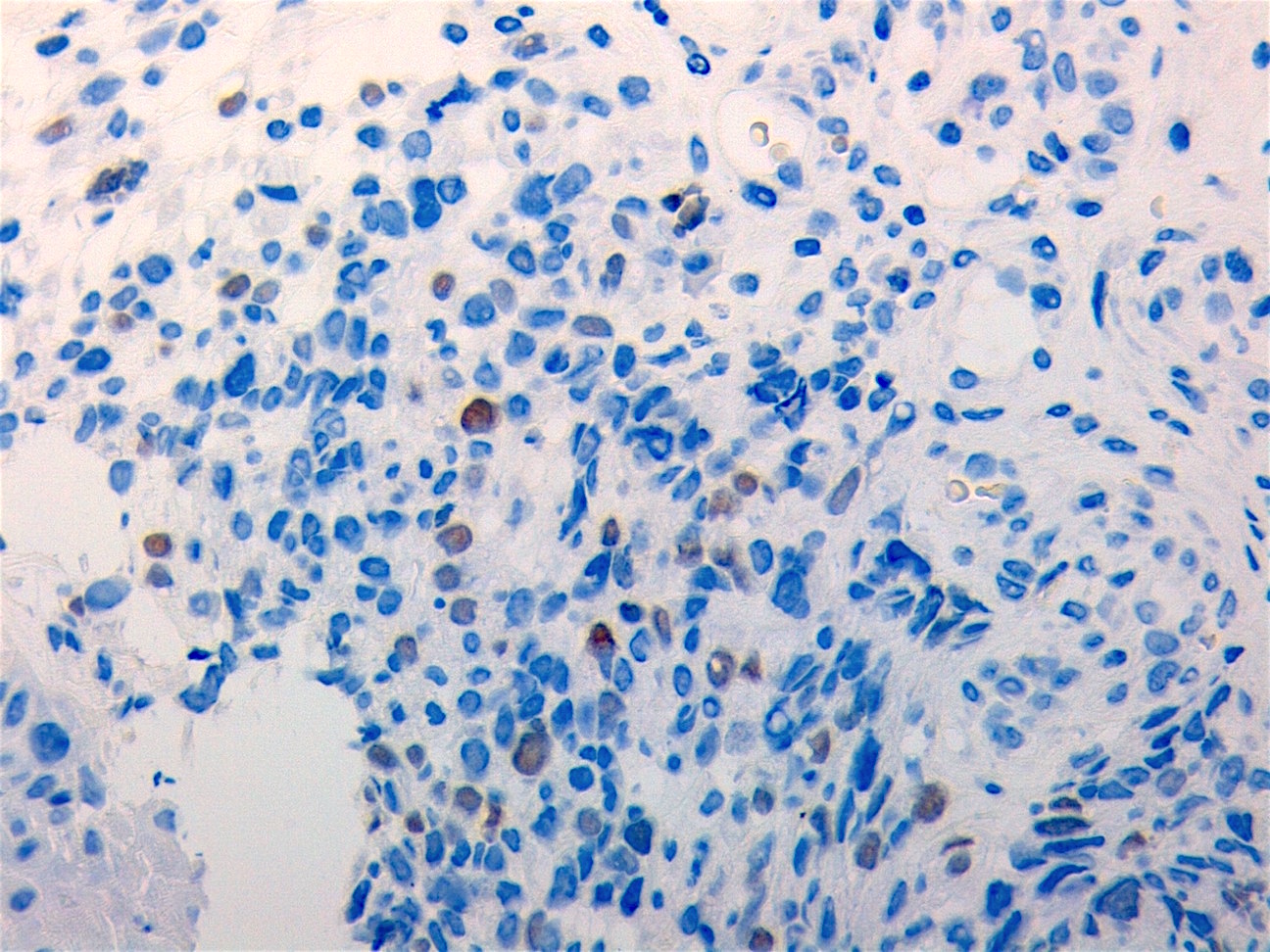
MyoD1
AFIP images - anaplastic rhabdomyosarcoma
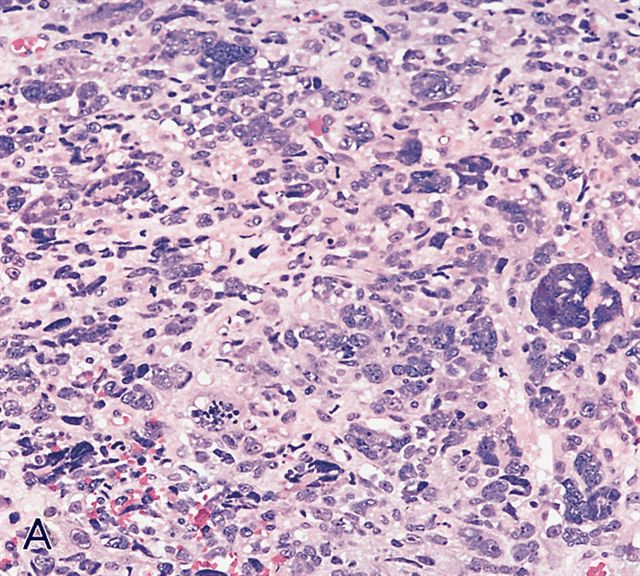
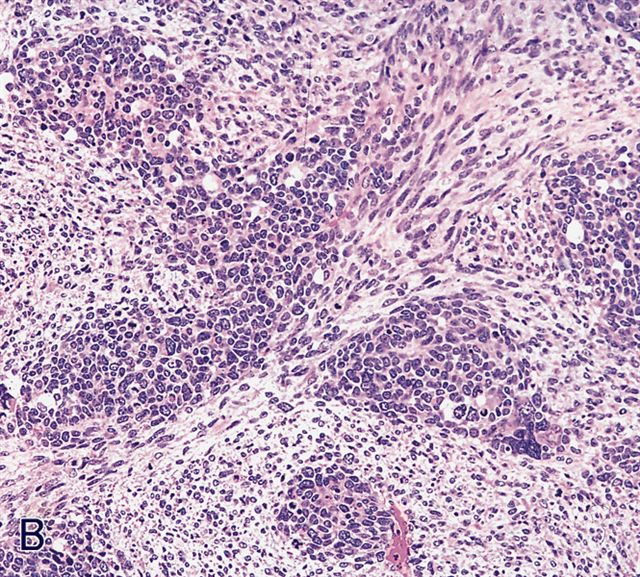
Hyperchromatic nuclei 3x larger than adjacent tumor cells
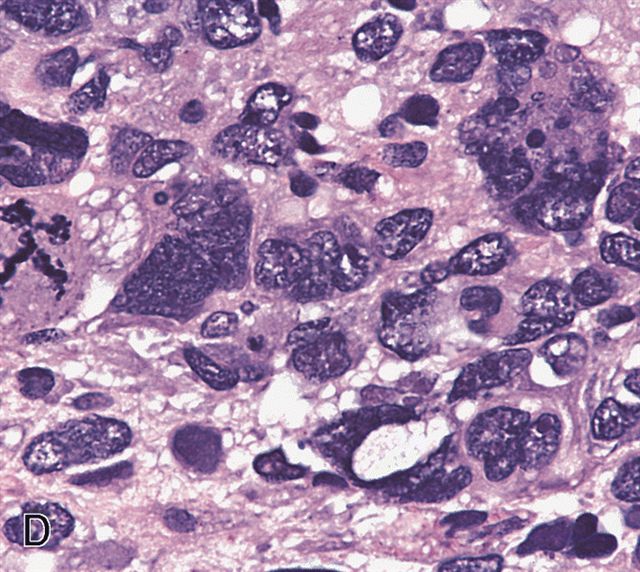
Bizarre mitotic figure
Rare muscle differentiation
AFIP images - botryoid variant
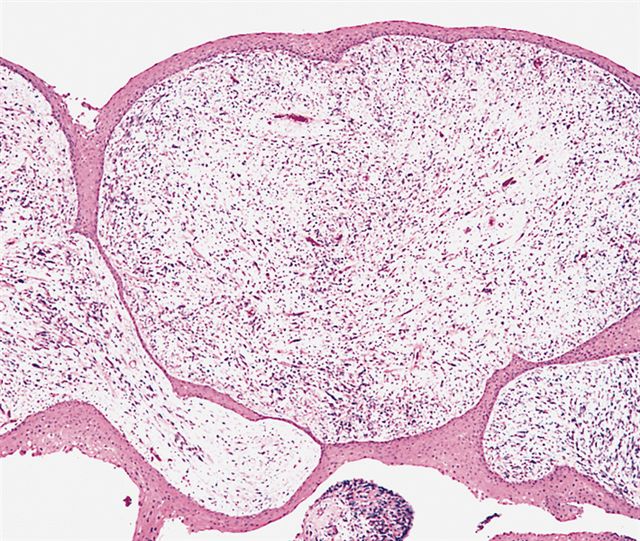
Polypoid growth
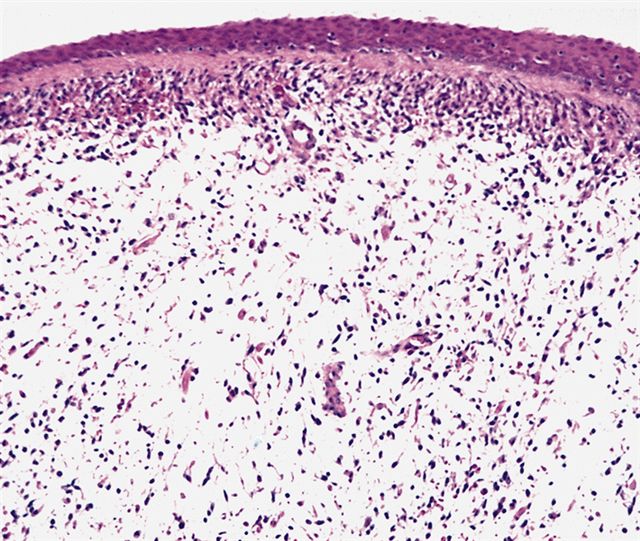
Overlying mucosa

Spindle cells
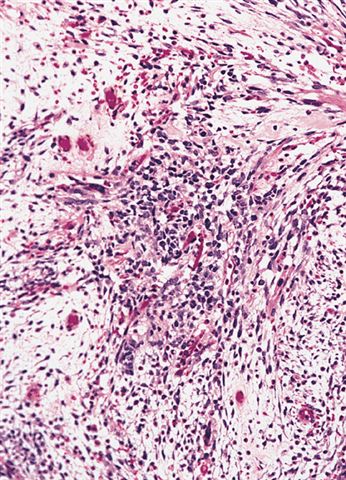
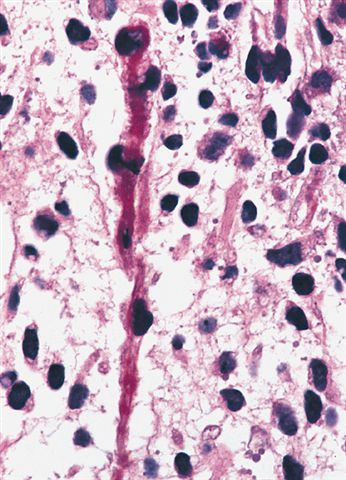
Deep foci of hypercellularity
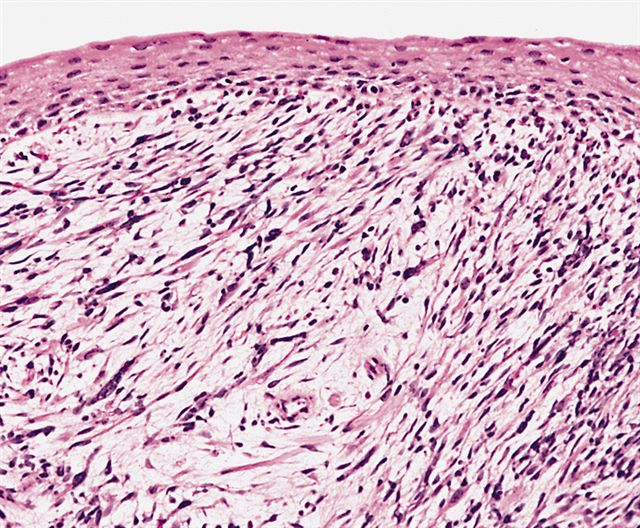
Focal cambium layer
Cytology description
- Cellular with features of a small round cell tumor
- Rarely intranuclear cytoplasmic inclusions (Diagn Cytopathol 2009;37:740)
Cytology images
Contributed by Erdener Özer, M.D., Ph.D.

Small round cell tumor
Positive stains
- Desmin, MyoD1 or myogenin are critical to document (Pathol Oncol Res 2008;14:233)
- Without myogenic differentiation (MyoD1 or myogenin), it is very difficult to diagnose embryonal rhabdomyosarcoma
- These stains will be less diffuse than in alveolar rhabdomyosarcoma and can be extremely focal
- Vimentin in all cells (even most primitive)
- Actin, myoglobin, myosin and creatine kinase M staining in more differentiated cells
- There may be aberrant staining for several antigens: occasional immunoreactivity to cytokeratin, S100 protein, neurofilaments and B cell markers such as CD20
- PAS highlights glycogen in most tumors
- Botryoid variant: Desmin, MyoD1, smooth muscle actin, muscle specific actin (Pediatr Dev Pathol 2005;8:427)
Electron microscopy description
- Developing striated muscle, thick and thin filaments
Molecular / cytogenetics description
- 11p15.5 loss of heterozygosity or loss of imprinting is a frequent finding
- Chromosomal aneuploidies are very common, especially with gains of chromosome 8 (seen in 90% of patients)
- Additional gains frequently seen include chromosomes 2, 11, 12, 13 and 20
- No diagnostic translocation found to date (see Terminology regarding spindled rhabdomyosarcoma)
- Inactivating mutations of TP53 and CDKN2A and activating mutations of RAS family genes (Genes Chromosomes Cancer 2011;50:397)
- Frequency of ALK dysregulation is debatable, as are any significant associated clinical features (Mod Pathol 2013;26:772)
Sample pathology report
- Soft tissue mass, left chest wall, excision:
- Embryonal rhabdomyosarcoma, negative for evidence of anaplasia (see comment)
- Comment: Histologic sections of this left chest wall excision demonstrate a spindle cell sarcoma comprised of hyperchromatic cells arranged into disorganized intersecting fascicles. No evidence of anaplastic features is identified. Immunohistochemical staining shows diffuse positivity for desmin, with scattered cells positive for myogenin and MyoD1. These findings are compatible with embryonal rhabdomyosarcoma.
Differential diagnosis
- Alveolar rhabdomyosarcoma (ARMS):
- Diffuse and strong nuclear staining for myogenin and MyoD1; molecular studies (including FISH) show PAX-FOXO1 fusion gene product in approximately 85% of cases (Am J Clin Pathol 2013;140:82)
- Histologically, ARMS is more uniform and shows more predictable patterns of growth (a finding which correlates with its consistent driver fusion event; it looks like a translocation sarcoma)
- Desmoplastic small round cell tumor:
- Tumor nodules on serosal surfaces, strongly keratin+ and EMA+, may be desmin+ but muscle specific actin-
- Associated with desmoplasia and histologically more closely mimic alveolar rather than embryonal rhabdomyosarcoma (ERMS)
- Will not display myogenin or MyoD1 positivity; beware false positivity if they infiltrate background skeletal muscle
- See Pitfalls and tips above
- Ewing / PNET:
- Another round cell tumor, often displays Homer Wright rosettes
- Nuclei are far more uniform and pale, not dense and hyperchromatic; CD99+, desmin-, MyoD1-, myogenin-
- Look for characteristic rearrangements including t(11;22) or t(12;22)
- Although there can be histologic overlap, if you sample the tumor enough you will find more atypia and cell to cell variability in rhabdomyosarcoma
- Large cell lymphoma:
- CD45+, B / T cell markers present, desmin-, myogenin-, MyoD1-, muscle specific actin-
- Histologically, can be very difficult to distinguish but lack of myogenic antibodies can be very helpful
- Monophasic synovial sarcoma:
- Negative for muscle markers, usually far more spindled and organized (long fascicles) when compared to ERMS
- Cytokeratin and EMA expression will show scattered and focal positivity in the spindled cells, the characteristic pattern seen in nearly all of these lesions
- Molecular or FISH testing for the t(X;18) fusion, if needed
- Myxoid liposarcoma (MLS):
- Although MLS typically is very easy to distinguish from ERMS, it can have a very similar background matrix that is loose and lightly basophilic
- Cells can focally resemble one another as well
- Look for signet ring lipoblasts in MLS, more bland histology and more uniformity when compared to ERMS
- Immunostaining and molecular features are also easily utilized to differentiate the 2 entities
- Neuroblastoma:
- Undifferentiated neuroblastoma can be very difficult to distinguish histologically; you may need to rely on the clinical presentation and the immunoprofile
- Elevated urinary catecholamines, rosettes, granular chromatin, neuropil
- S100+ (often), chromogranin+, synaptophysin+, GFAP+
- Absence of myogenic markers
- Pleomorphic rhabdomyosarcoma:
- Exclusively adults, usually in their 60s - 70s
- Usually deep soft tissue of the extremity and remarkable for its universal diffuse cytologic atypia
- Uniformly pleomorphic and does not contain elements of embryonal rhabdomyosarcoma
- Anaplastic, as opposed to ERMS, which occasionally displays anaplasia in a background of classic EMRS
- Spindle cell rhabdomyosarcoma:
- Historically, tumors with spindled morphology were regarded as a variant of ERMS but the 2020 WHO now has a separate spindle cell / sclerosing subtype category (i.e. this is not a variant of ERMS)
- ERMS can display spindled growth but there are several molecularly defined subtypes of spindle cell / sclerosing RMS, which are considered separate and will likely be better defined in the future by WHO (Am J Surg Pathol 2016;40:224)
- Wilms tumor:
- Can have rhabdomyoblastic differentiation, especially in the setting of chemotherapy
- Beware any tumor in the kidney or any metastatic site in a patient with a previous Wilms tumor
- Epithelial and blastemal components can be very focal following chemotherapy
- If a Wilms tumor displays this pattern, it usually shows abundant cytodifferentiation, as opposed to classic rhabdomyosarcoma, which is usually focal
Additional references
Practice question #1
Which FISH testing may assist in making distinction between the dense pattern of embryonal rhabdomyosarcoma and the alveolar subtype of rhabdomyosarcoma?
- ETV6::NTRK3
- EWS::FLI1
- PAX3::FOXO1
- SYT::SSX1
- TPM3::ALK
Practice answer #1
C. PAX3::FOXO1. This fusion gene product occurs in approximately 80% of alveolar rhabdomyosarcoma cases. Additionally, PAX7::FOXO1 rearrangements are present in a minority of cases.
Comment Here
Reference: Embryonal rhabdomyosarcoma
Comment Here
Reference: Embryonal rhabdomyosarcoma
Practice question #2
Which histologic features in embryonal rhabdomyosarcoma are needed to diagnose anaplasia?
- 3x nuclear size variability and tripolar mitotic figures
- Alternating hypercellular and hypocellular areas
- Coagulative necrosis
- More than 20 mitoses per 10 high power fields
- Solid growth pattern
Practice answer #2
A. 3x nuclear size variability and tripolar mitotic figures. Multipolar mitotic figures (including tripolar mitoses) and nuclear anaplasia (3x variation in nuclear size) are required to diagnose anaplastic embryonal rhabdomyosarcoma (ERMS). Solid growth pattern is irrelevant and necrosis can be seen in any subtype of rhabdomyosarcoma. ERMS typically displays the alternating pattern of hypocellularity and hypercellularity (also a buzzword for high grade malignant peripheral nerve sheath tumors). There is no requirement for a specific mitotic rate.
Comment Here
Reference: Embryonal rhabdomyosarcoma
Comment Here
Reference: Embryonal rhabdomyosarcoma
