

CNS nontumor
Malformations
Meckel-Gruber syndrome
Author: Erdener Özer, M.D., Ph.D.

Last author update: 1 July 2016
Last staff update: 18 May 2022
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Meckel-Gruber syndrome

Table of Contents
Definition / general | Essential features | Terminology | Epidemiology | Pathophysiology | Etiology | Clinical features | Diagnosis | Radiology description | Prognostic factors | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Differential diagnosis | Additional referencesCite this page: Özer E. Meckel-Gruber syndrome. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/syndromesmeckelgruber.html. Accessed April 25th, 2024.
Definition / general
- Meckel-Gruber syndrome (Meckel syndrome or MKS) is a rare, lethal ciliopathic genetic disorder characterized by abnormalities affecting several organ systems
- Three classic symptoms are normally associated with MKS: occipital encephalocele, polycystic kidneys and polydactyly
- Affected children or fetuses may also have abnormalities affecting the head and face, liver, lungs, heart and genitourinary tract
- Meckel-Gruber syndrome is named for Johann F. Meckel and George B. Gruber; the first reports of the syndrome were published in 1822 by J. F. Meckel; G. B. Gruber also published reports in 1934 and gave it the name dysencephalia splanchnocystica
Essential features
-
The three phenotypic features of the classical triad are:
- Protrusion of a portion of the brain and its surrounding meninges through a defect in the back of the skull (occipital encephalocele)
- Multiple cysts on the kidneys (cystic kidneys)
- Extra fingers or toes (polydactyly)
Terminology
- Meckel-Gruber syndrome, Meckel syndrome, Gruber syndrome, dysencephalia splanchnocystica, MES, MKS
Epidemiology
- Prevalence is estimated at 1/50,000 births in Europe; worldwide prevalence is reported to be 1/13,250 to 1/140,000 live births; the live birth prevalence is significantly higher in the Finnish population (1/9,000); therefore this syndrome is suggested to be a Finnish heritage disease (J Pediatr Neurosci 2013;8:154)
- No gender or ethnic predilection is reported
Pathophysiology
- Failure of mesodermal induction has been suggested to cause Meckel-Gruber syndrome; the induction cascades of early morphogenesis involve numerous growth factors, homeobox genes and paired domain genes
- It belongs to the ciliopathies, a category of diseases thought to be caused by dysfunction of cilia and flagella; polycystic liver and kidney disease, Bardet-Biedl syndrome, Alstrom syndrome and Joubert syndrome also belong to the same group (Annu Rev Genomics Hum Genet 2006;7:125)
Etiology
- MKS is inherited as an autosomal recessive condition through thirteen genes: B9D1, B9D2, CC2D2A, CEP290, MKS1, RPGRIP1L, TCTN2, TCTN3, TMEM67, TMEM107, TMEM216, TMEM231 and TMEM237 (Eur J Hum Genet 2016;24)
- It has a recurrence risk of 25%; genetic counseling should be provided to affected families
Clinical features
- A study by Barisic et al, using data from 191 cases of MKS, as accessed through the European Surveillance of Congenital Anomalies (EUROCAT) network, found the prevalence of various characteristics of the disorder to be as follows (Eur J Hum Genet 2015;23:746):
- Polycystic dysplastic kidneys (97.7%)
- Polydactyly (87.3%)
- Occipital encephalocele (83.8%)
- Fibrotic / cystic changes of the liver (65.5%)
- Other central nervous system (CNS) anomalies (51.4%): the features include hydrocephalus, anencephaly, holoprosencephaly, the absence of olfactory lobes as well as Dandy-Walker and Arnold-Chiari malformation
- Orofacial clefts (31.8%): cleft lip and palate, microphthalmia and micrognathia may be observed
- In addition cardiac malformations including atrial septal defect, aorta coarctation, patent arterial duct and valvular pulmonary stenosis may be present; genital ambiguity secondary to incomplete development of internal and external genitalia, and cryptorchidism in males are common; urothelial atresia and bone dysplasia are rare features; bowing or shortening of the limbs are also present
Diagnosis
- Finding at least two of the three phenotypic features of the classical triad, in the presence of normal karyotype, makes the diagnosis solid; improvements in ultrasonography have enabled prenatal diagnosis as early as 10 weeks' gestation
- Alpha-fetoprotein (AFP) level from either maternal blood or amniotic fluid may help detect encephalocele; AFP can be measured in amniotic fluid after 12 weeks' gestation and in maternal blood after 15 weeks' gestation
- If anomalies are detected early in the first trimester, chorionic villus sampling (CVS) can be performed at 10-12 weeks' gestation or later in pregnancy if oligohydramnios does not permit amniocentesis; molecular genetic testing can be used to to assess prenatal diagnosis, linkage as well as carrier testing
- The level of amniotic fluid may be significantly altered or remain normal, but a normal level of fluid should not be criteria for exclusion of diagnosis
Radiology description
- Fetal ultrasonography can detect an occipital encephalocele, polydactyly and dysplastic kidneys in fetuses with Meckel-Gruber syndrome if oligohydramnios is not present
- MRI is a valuable complement to ultrasonography in assessing fetal anomalies in the presence of severe oligohydramnios; the level of amniotic fluid may be significantly altered or remain normal
- Color Doppler can assess lung perfusion in the last trimester, detect the presence of renal arteries in cases of oligohydramnios (suspected renal agenesis or hypoplasia), and assess flow in the umbilical arteries
Prognostic factors
- Fetuses affected by MKS survive only a few days to a few weeks or die in utero; pulmonary hypoplasia secondary to oligohydramnios is the leading cause of death
- Other complications are renal and liver failure
Treatment
- No treatment is currently available; the outcome is always fatal
Gross description
- Occipital encephalocele is characterized by extrusion of rhombic roof elements, cerebellar vermis, caudal third ventricle and distended fourth ventricle through a widened posterior fontanelle
- Microscopic cysts cause the kidney to enlarge 10 to 20 times
- Polydactyly may affect all four extremities and is typically postaxial or very rarely preaxial
Gross images
Images hosted on other servers:
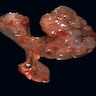
Cystic renal dysplasia
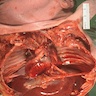
Pulmonary hypoplasia
Microscopic (histologic) description
- The primary renal abnormality appears to be failed interaction of the metanephric duct and renal blastema; the kidneys, therefore, show little corticomedullary differentiation, and the nephrons are severely deficient, causing enlargement of the kidneys; thin walled cysts appear throughout the parenchyma
- Hepatic dysgenesis and liver fibrosis are identified only at postmortem examination; in Meckel-Gruber syndrome, the plates do not atrophy and prevent reorganization by the remaining biliary cells to form tubular ducts; the resultant fibrosis can be severe enough to occlude portal veins; bile canaliculi are smaller and less well developed and have inspissated bile within the abnormal ductules
Microscopic (histologic) images
Images hosted on other servers:
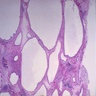
Cystic renal dysplasia
Differential diagnosis
- Chromosome analysis is essential to exclude trisomy 13, which Meckel-Gruber syndrome mimics; trisomy 13 carries a 1% recurrence risk, as opposed to the 25% recurrence rate for Meckel-Gruber syndrome
- Must also exclude Bardet-Biedl syndrome, Hydrolethalus syndrome and Smith-Lemli-Opitz syndrome
Additional references
