Part 1 of this series discussed the basics of metastatic disease.
Part 2 discussed features of human biology that are important in understanding cancer and metastases.
Part 3 discussed how these features are altered during the development of cancer and metastases.
In Part 3a, we discuss how the malignant properties of normal cells are switched on, beginning with rapid or otherwise inappropriate cell division. This essay discusses 4 mechanisms: KRAS mutation, p53 mutation, Helicobacter pylori infection and Epstein-Barr virus infection. Click here for the summary for nonscientists.
Rapid cell division occurs normally during embryogenesis and fetal and childhood growth, as well as in adults to repair damaged tissue and to replace cells in organs with rapid cell turnover. Unlike the operation of a machine, this process involves a complex, nonlinear series of events that cannot be easily understood with a 2 dimensional diagram.
Rapid cell division during the onset of embryogenesis
At fertilization:
- The egg and sperm combine and their DNA is fused.
- The fertilized egg and its daughter cells (constituting the embryo) use only the mRNA and proteins present in the egg before it was fertilized; the embryo is unable to use its own DNA because this process is repressed.
- The fertilized egg and early embryonic cells have a small nuclear to cytoplasmic ratio because they have abundant cytoplasm.
After fertilization, the cleavage stage begins:
- The cleavage stage is characterized by extremely rapid cell division that oscillates between DNA synthesis (DNA is created but not used by the embryo at this time) and mitosis (Siefert 2015, Brantley 2021). Each cell divides in half, splitting its cytoplasm between the daughter cells.
- There is no time to produce more cytoplasm between the cell divisions.
- There is no time for the checkpoints that are usually part of the cell cycle to function.
- Note: The malignant process may mimic certain aspects of this process, namely rapid cell division (although malignant cells use their own DNA to control the process) and the inability of the DNA damage checkpoint to operate correctly in this limited time, which allows DNA errors to accumulate.
- The next phase is the midblastula transition, which causes the embryo to start using its own DNA to produce mRNA and proteins.
- The cause of the midblastula transition is controversial (Strong 2020). It may be due to (A) an increase in the nuclear to cytoplasmic ratio of the embryonic cells that occurs as each cell divides without producing more cytoplasm or (B) increases in the concentration of cell division limiting factors (Collart 2013).
- During the midblastula transition, the cell cycle slows down, which allows the DNA damage checkpoint to have time to verify that DNA is copied correctly and that there are no mutations.
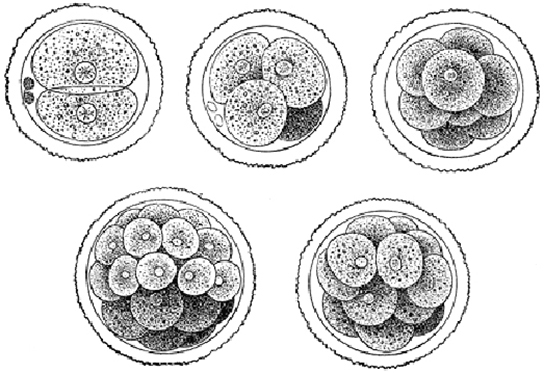
The newly fertilized egg rapidly divides into multiple cells that have smaller sizes and a higher nuclear to cytoplasmic ratio (University of Minnesota).

Movie of cell division in Xenopus (frog); a similar process occurs in humans (Collart 2013)
Cell division at the midblastula stage now requires activation of a trigger and progression through checkpoints
After the midblastula transition, cell division requires both the activation of a trigger and progression through cellular checkpoints:
- Activation of a trigger typically consists of both turning on the enzyme cyclin dependent kinase 1 (Cdk1) and turning off opposing phosphatase enzymes (Crncec 2019).
- Cells must also progress through the G2 cellular checkpoint (DNA damage checkpoint) that tests whether irreparable DNA damage has occurred, which could ultimately lead to malignancy. If not present, the checkpoint allows cell division to progress. If this damage is present, this checkpoint directs cells to exit from the cell cycle or promotes apoptosis (programmed cell death, Matthews 2021).
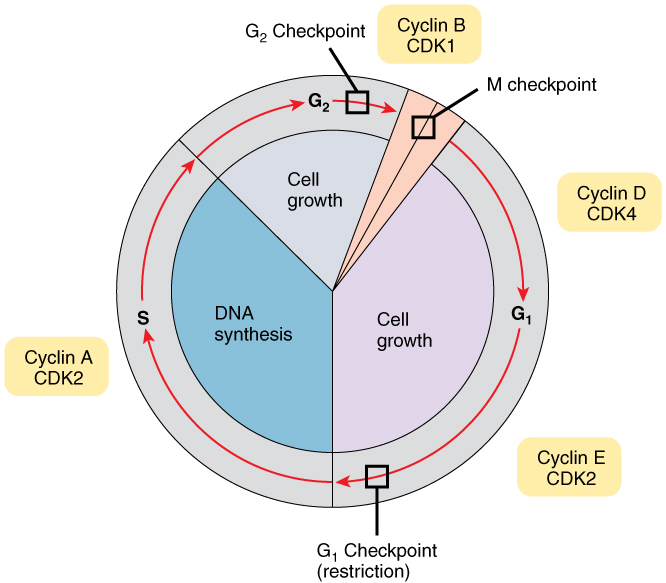
Cellular checkpoints: The G2 checkpoint (DNA damage checkpoint) determines whether cells can divide (mitosis) or not, based on the evaluation of errors in the DNA synthesis step (Wikipedia).
- The activation and cellular checkpoint steps are controlled by many networks with specific functions related to allowing or halting cell division. Since the networks' genes and proteins often also regulate other functions, it is often difficult to understand precisely how they work together in cell division.

A simplified version of the cell cycle (click here for larger image) shows numerous levels of network involvement. Each of these genes and proteins is also controlled by a web of other genes and proteins, which makes the processes very difficult to understand (Suski 2021).
- The activation and cellular checkpoint pathways are robust and can withstand simple mutations; however, the accumulation of mutations can disrupt their usual function and cause cell division to occur continuously instead of only when needed.
- Driver mutations are mutations capable of causing cancer. Scientists have identified 568 to 619 driver mutations that often affect cell division. This includes KRAS and p53, described below. Note that genes are italicized in the medical literature, but the proteins which they produce are not.
- Nonmalignant cells with faster cell cycling times, or their precursors, may be more prone to malignancy in the presence of carcinogens (Chen 2019).
KRAS
Cancer risk factors may create driver mutations in KRAS and other genes that ultimately lead to cancer. For example, tobacco smoke contains over 70 carcinogens, including some that interfere with DNA replication and produce KRAS mutations (Veluswamy 2021). Normally, the KRAS protein functions as an on-off switch for cell division; however, the mutant KRAS protein may be stuck in the "on" position, causing uncontrolled cell division. KRAS mutations are considered driver mutations in lung, pancreas and colorectal cancer. Smoking may cause KRAS mutations and cancer through this process:
- Tobacco leaf, when burned during cigarette smoking, produces benzo[a]pyrene, nitrosamines and other carcinogens.
- Inhaling tobacco smoke causes these carcinogens to enter the lungs and bloodstream.
- Benzo[a]pyrene is metabolized by the liver to form water soluble BPDE, which promotes its excretion into the urine, but this is not instantaneous.
- Before it is excreted, water soluble forms of BPDE enter cells and their nuclei, bind to DNA, and form bulky attachments called DNA adducts that change the DNA structure, affecting KRAS and other genes when DNA is duplicated.
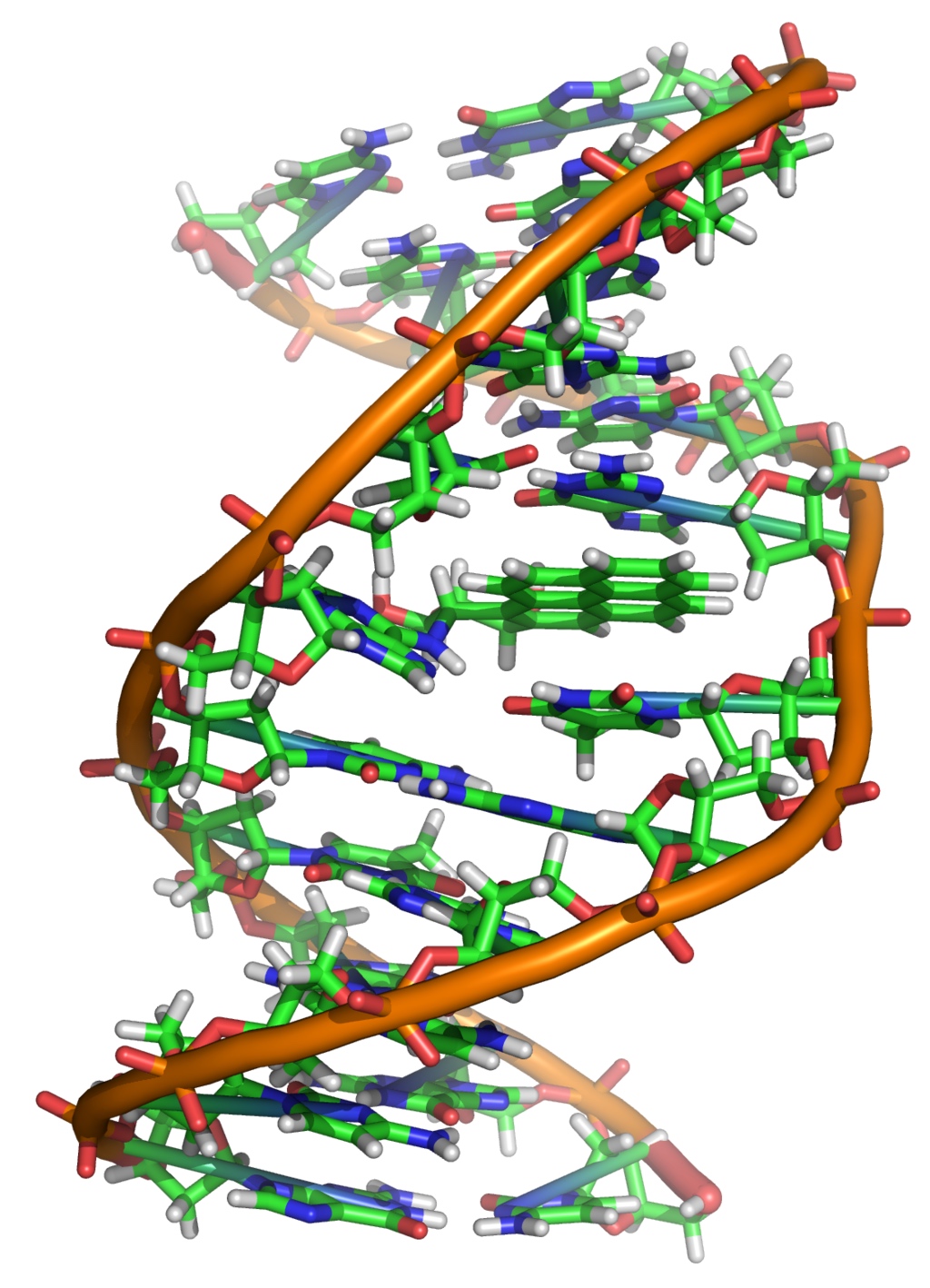
A BPDE metabolite forms a DNA adduct, shown by the green linear structures in the center of the image (Wikipedia).
The DNA repair process typically corrects errors before DNA is duplicated:
- Replication of DNA's 3 billion base pairs is a central part of cell division, but accuracy is crucial.
- Our cells have robust processes to reduce the error rate of DNA replication, including over 200 genes that participate in or influence DNA repair (Wood 2020).
- These genes reduce the error rate from 1 per 100,000 to an estimated 1 per 100 million base pairs (Pray 2008); however, this still allows an average of 30 errors during 1 round of DNA replication.
- DNA repair enzymes remove most but not all of the DNA adducts.
- When DNA adducts evade the repair process, the DNA may replicate with a higher error rate.
- A hallmark of smoking-induced damage is the replacement of a cytosine nucleotide base with an adenine base opposite a guanine base. This is called a G to T transversion and commonly affects the KRAS and p53 genes.
- This mutated KRAS gene may become a permanent part of the cell's DNA and direct the cell to divide continuously, particularly when associated with inflammation (Katajima 2016); however, this may take years to occur.
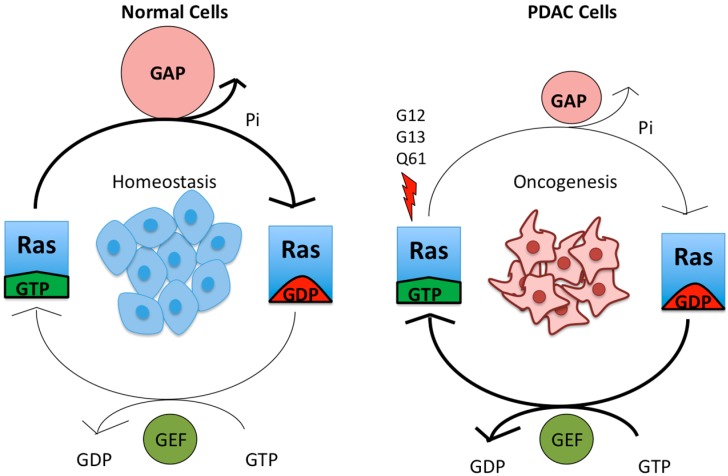
KRAS mutations. In normal cells, the KRAS (Ras) gene is active when combined with GTP (left side of left drawing) and inactive when combined with GDP (right side). The active gene quickly becomes inactive (thick arrow at top of drawing). In the pancreas, lung and colon (right drawing), G12, G13 and Q61 mutations (left side of right drawing with the lightning bolt) prevent the Ras gene from becoming inactive (thin arrow), so the gene is always active, leading to continuous cell division (Zeitouni 2016).
p53 (TP53)
p53 is a tumor suppressor gene and the most commonly mutated gene in human cancer. About half of all cancers have p53 mutations or deletions and the rest may inactivate p53 through other mechanisms (Engeland 2022). The p53 protein ensures that cells repair any damaged DNA before cell division by inducing cell cycle arrest (stoppage) to allow time for DNA repair to occur. If the DNA damage is not fixed, the p53 protein forces the cell to undergo apoptosis or to leave the cell cycle.
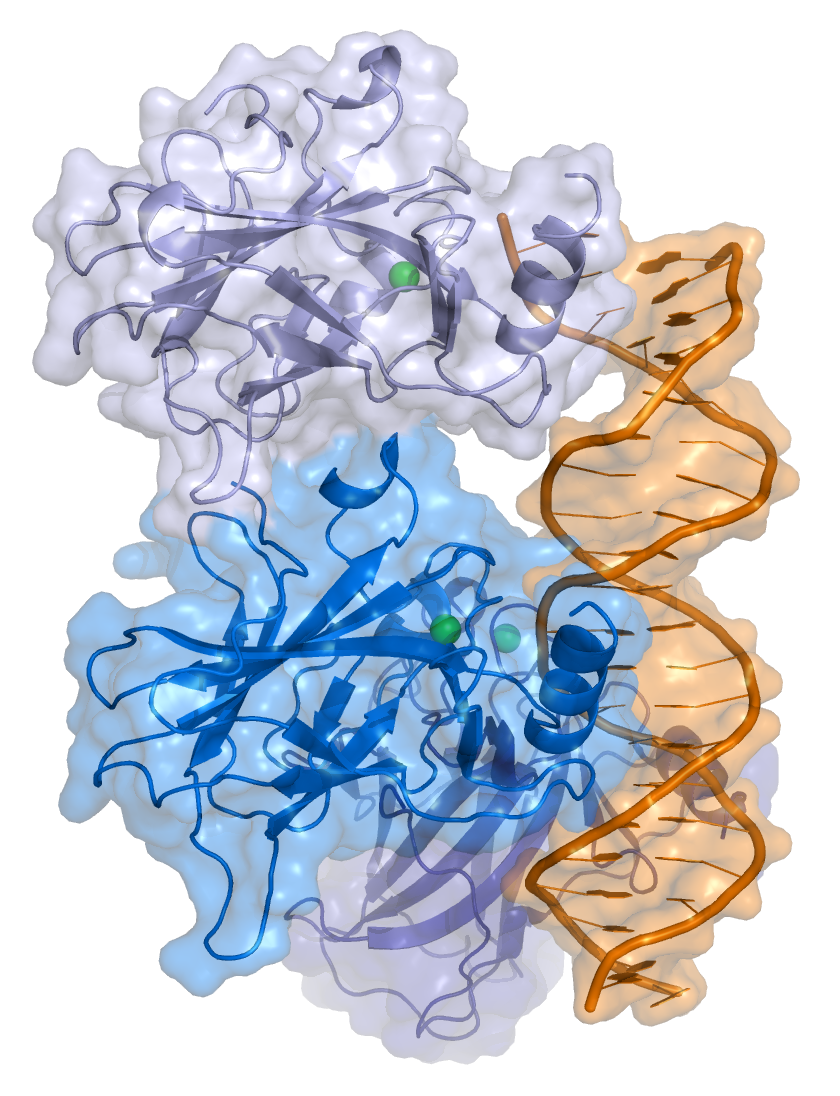
p53 mutations promote cell division in 2 ways (Marei 2021). First, cells attempting to divide but with damaged DNA typically will die via apoptosis or exit the cell cycle. But cells with p53 mutations survive, so there is effectively more cell division. Second, cells with p53 mutations accumulate additional mutations, including those that affect the cell cycle, ultimately leading to an increased rate of cell division. Recent studies suggest that preventing apoptosis and exit from the cell cycle is an important cause of continuous cell division, not just mutations that drive uncontrolled cell division, such as KRAS (Matthews 2022).
Bacteria and viruses can also directly cause cancer, although this is less common in Western countries.
Helicobacter pylori stimulates lymphocytes to become malignant

Stomach: An upper GI (gastrointestinal) tract endoscopy shows Helicobacter pylori gastritis; the organisms appear dark brown based on the Warthin-Starry stain (PathologyOutlines.com).
The bacteria Helicobacter pylori (H. pylori) directly stimulates lymphocytes that attempt to kill the bacteria, which may ultimately cause gastric MALT lymphoma, a low grade non-Hodgkin lymphoma with malignant B lymphocytes (B cells). It typically occurs in the stomach through a process called antigen driven lymphoproliferation:
- H. pylori infection of the stomach is common and affects half the world's population.
- The infection attracts B cells, T cells and neutrophils to the stomach, but these immune system cells usually cannot kill this bacteria.
- The persistence of the bacteria causes chronic immune system stimulation and proliferation of the B cells (lymphoproliferation), which are inherently unstable and occasionally transform into malignant B cells.
- This process is typically reversible; antibiotics can kill the bacteria, end the chronic immune system stimulation and usually cause regression of the cancer, although it may take up to 2 years.
- Untreated MALT lymphoma may transform into diffuse large B cell lymphoma, a more aggressive lymphoma, which may also respond to antibiotic therapy.
- H. pylori also causes gastric (stomach) cancer but through a different mechanism.
Epstein-Barr virus can stimulate lymphocytes to become malignant
Epstein-Barr virus (EBV) infection directly promotes the proliferation of B cells but through a different process than H. pylori. An EBV protein mimics a protein that provides a growth signal to B cells. Although 95% of the world's population is infected by EBV, EBV related lymphomas are rare because an intact immune system keeps this infection in check and prevents these B cells from developing malignant properties.
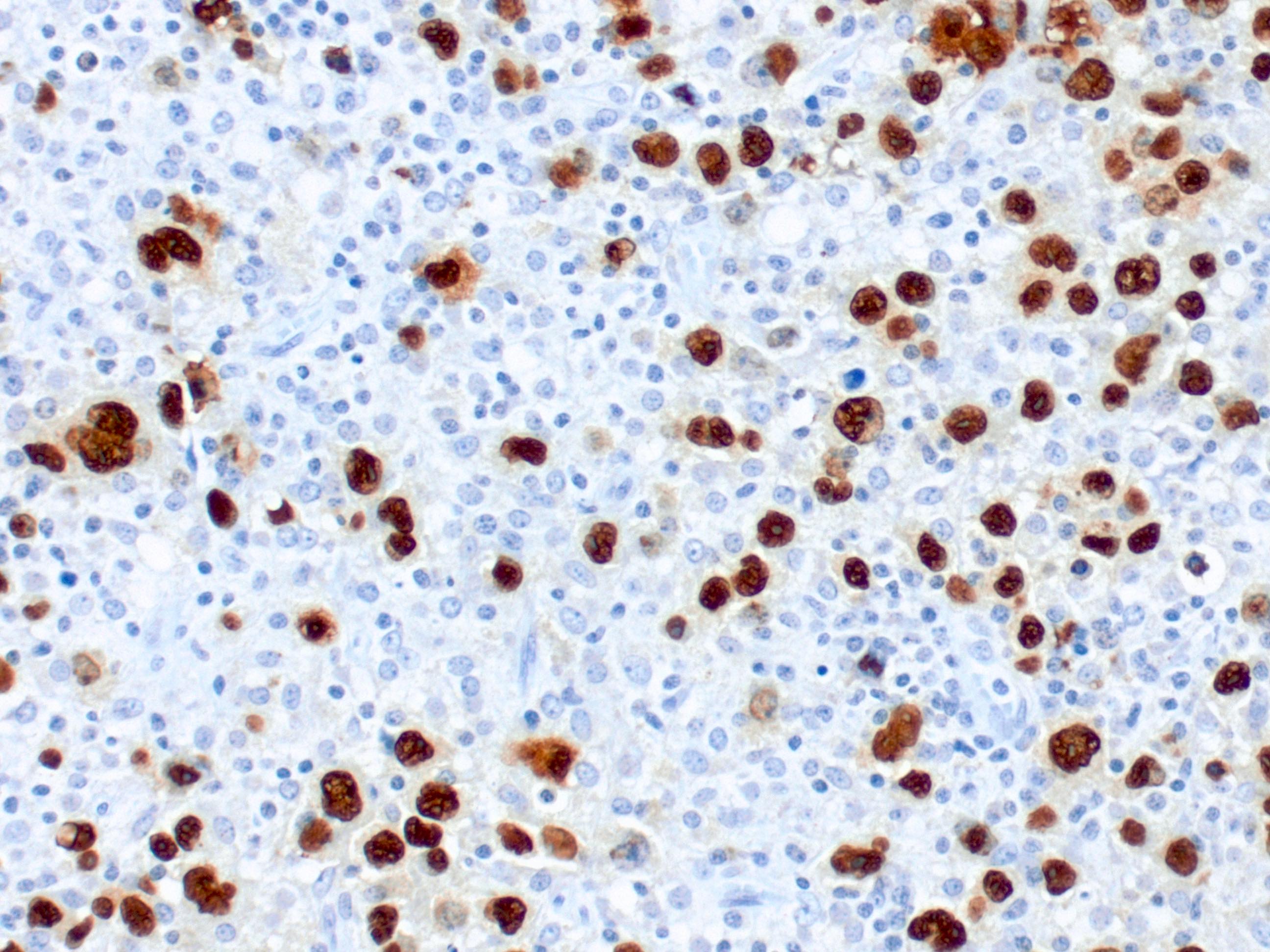
Diffuse large B cell lymphoma due to EBV infection. The EBER protein produced by the virus stains dark brown in the tumor cells (PathologyOutlines.com).
Part 3b will discuss how the migration property of normal cells is switched on during malignancy.
