
6 January 2010 - Case #164
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
This case was contributed by Dr. Mowafak Hamodat, Eastern Health of Newfoundland and Labrador, St. Johns, Canada.
Case #164
Clinical history:
A 51 year old man presented with chronic liver disease and feeling unwell. He had splenomegaly with thrombocytopenia and elevated liver enzymes. A liver biopsy was obtained.
Microscopic images:
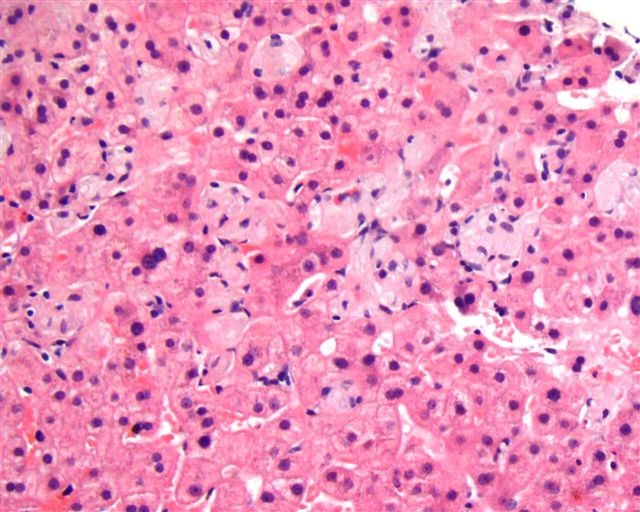
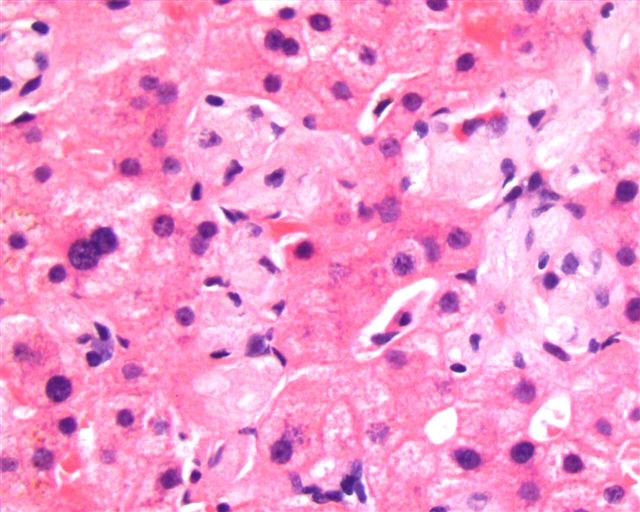
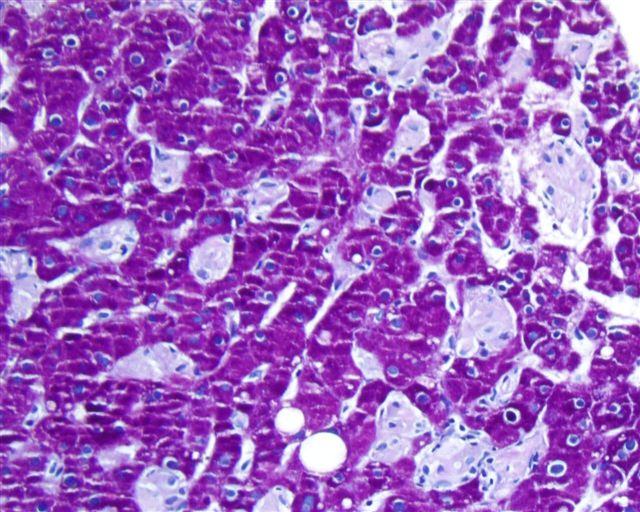
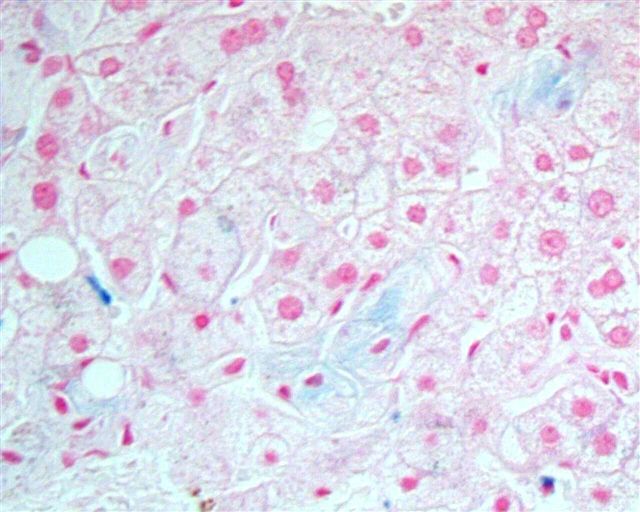
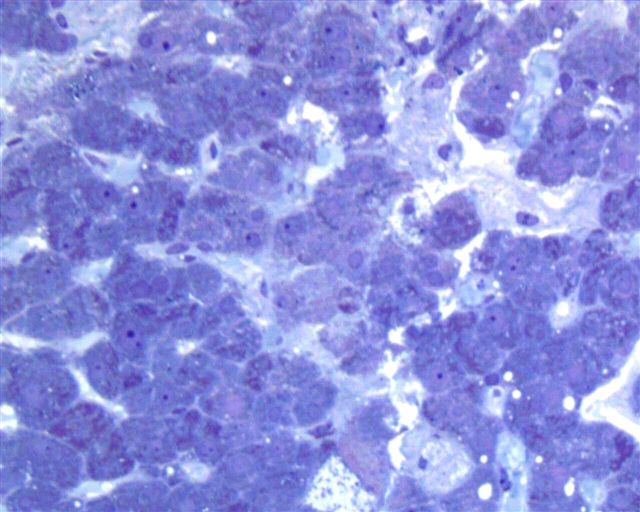

What is your diagnosis?
Diagnosis: Gaucher disease
Discussion:
The liver biopsy showed enlarged Kupffer cells and diffuse infiltration of liver parenchyma by histiocytes, which showed crinkle paper type cytoplasm. Electron microscopy showed angulated lysosomes.
Gaucher disease (preferable to Gauchers disease) is a lysosomal storage disease caused by an autosomal recessive mutation in the β glucocerebrosidase gene (also called glucosylceramidase and β glucosidase). The highest risk group are Ashkenazi Jews, in who 1 in 15 are carriers. The defective enzyme leads to an accumulation of glucocerebroside substrate in cells of the mononuclear phagocyte system, including histiocytes in the spleen, lymph nodes, bone marrow, GI and GU tracts, as well as Kupffer cells in the liver, osteoclasts in bone, microglia in the CNS and alveolar macrophages in the lungs (Arch Pathol Lab Med 2008;132:851).
The differential diagnosis includes other lipid storage disorders, such as Niemann-Pick disease (foamy and vacuolated cytoplasm, due to sphinomyelin accumulation) and Pompe disease (primarily affects skeletal and cardiac muscle). Hematologic disorders such as CML, AML and CLL may also have Gaucher-like cells. Sea blue histiocytes containing ceroid are found in CML.
The diagnosis can be confirmed by measuring glucocerebrosidase activity in peripheral blood leukocytes. A finding of < 15% of mean normal activity is diagnostic. The enzyme activity in carriers (heterozygotes) is generally half normal but may overlap with healthy controls. Molecular analysis, particular in Ashkenazi patients, may also be helpful.
Gaucher disease is divided into 3 clinical subtypes. Type I is nonneuropathic and may be mild in severity. Type II, or acute infantile neuropathic Gaucher disease, affects infants within a few months of birth and is usually fatal within 2 years. Type III is a chronic neurological variant, with onset anywhere between birth and adulthood and presents with a slowly progressing neurological decline (eMedicine: Gaucher Disease [Accessed 25 April 2024], Wikipedia: Gaucher's disease [Accessed 25 April 2024]).
Symptoms of all 3 types may include splenomegaly, hepatomegaly, hypersplenism, osteoporosis and a yellow-brown tint to the skin. Anemia is also common. Patients with types II or III may have seizures and dementia.
Enzyme replacement therapy with imiglucerase (Cerezyme), a recombinant version of β glucocerebrosidase, can help manage the disease for patients with types I or III, although the drug is very expensive (Curr Opin Pediatr 2007;19:628).
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
This case was contributed by Dr. Mowafak Hamodat, Eastern Health of Newfoundland and Labrador, St. Johns, Canada.
Case #164
Clinical history:
A 51 year old man presented with chronic liver disease and feeling unwell. He had splenomegaly with thrombocytopenia and elevated liver enzymes. A liver biopsy was obtained.
Microscopic images:
PAS
Iron stain
Thick section
Electron microscopy
What is your diagnosis?
Click here for diagnosis and discussion:
Diagnosis: Gaucher disease
Discussion:
The liver biopsy showed enlarged Kupffer cells and diffuse infiltration of liver parenchyma by histiocytes, which showed crinkle paper type cytoplasm. Electron microscopy showed angulated lysosomes.
Gaucher disease (preferable to Gauchers disease) is a lysosomal storage disease caused by an autosomal recessive mutation in the β glucocerebrosidase gene (also called glucosylceramidase and β glucosidase). The highest risk group are Ashkenazi Jews, in who 1 in 15 are carriers. The defective enzyme leads to an accumulation of glucocerebroside substrate in cells of the mononuclear phagocyte system, including histiocytes in the spleen, lymph nodes, bone marrow, GI and GU tracts, as well as Kupffer cells in the liver, osteoclasts in bone, microglia in the CNS and alveolar macrophages in the lungs (Arch Pathol Lab Med 2008;132:851).
The differential diagnosis includes other lipid storage disorders, such as Niemann-Pick disease (foamy and vacuolated cytoplasm, due to sphinomyelin accumulation) and Pompe disease (primarily affects skeletal and cardiac muscle). Hematologic disorders such as CML, AML and CLL may also have Gaucher-like cells. Sea blue histiocytes containing ceroid are found in CML.
The diagnosis can be confirmed by measuring glucocerebrosidase activity in peripheral blood leukocytes. A finding of < 15% of mean normal activity is diagnostic. The enzyme activity in carriers (heterozygotes) is generally half normal but may overlap with healthy controls. Molecular analysis, particular in Ashkenazi patients, may also be helpful.
Gaucher disease is divided into 3 clinical subtypes. Type I is nonneuropathic and may be mild in severity. Type II, or acute infantile neuropathic Gaucher disease, affects infants within a few months of birth and is usually fatal within 2 years. Type III is a chronic neurological variant, with onset anywhere between birth and adulthood and presents with a slowly progressing neurological decline (eMedicine: Gaucher Disease [Accessed 25 April 2024], Wikipedia: Gaucher's disease [Accessed 25 April 2024]).
Symptoms of all 3 types may include splenomegaly, hepatomegaly, hypersplenism, osteoporosis and a yellow-brown tint to the skin. Anemia is also common. Patients with types II or III may have seizures and dementia.
Enzyme replacement therapy with imiglucerase (Cerezyme), a recombinant version of β glucocerebrosidase, can help manage the disease for patients with types I or III, although the drug is very expensive (Curr Opin Pediatr 2007;19:628).
