
Coagulation
Hereditary bleeding disorders
Factor VIII deficiency (hemophilia A)


Last author update: 1 October 2010
Last staff update: 28 February 2023
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Factor VIII deficiency[title] AND Hemophilia A [title]
Table of Contents
Definition / general | Terminology | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Laboratory | Prognostic factors | Case reports | Treatment | Differential diagnosis | Additional referencesCite this page: Crookston K, Rosenbaum LS, Gober-Wilcox J. Factor VIII deficiency (hemophilia A). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/coagulationfactorVIIIdef.html. Accessed May 14th, 2024.
Definition / general
- Factor VIII deficiency (hemophilia A) is the most common congenital bleeding disorder that is inherited as an X-linked recessive trait
- It is characterized by mild, moderate or severe bleeding episodes
Terminology
- Factor VIII is also known as the anti-hemophilic factor
Epidemiology
- Almost exclusively affects males
- 1 in every 10,000 births or 1 in 5000 male births
- Rarely affects females (see etiology)
- Female carriers are unaffected
Sites
- Bleeding into muscle, soft tissue or joints (hemarthrosis), GI / GU tract bleeding, easy bruising, excessive bleeding after surgery, trauma, dental procedures or circumcision; epistaxis, poor wound healing, intracranial hemorrhage, scalp hematoma, development of pseudotumors with repetitive hematoma formation, menorrhagia
- Incidence of sites of bleeding:
- Hemarthrosis: 70 - 80%
- Muscle/soft tissue: 10 - 20%
- Other major bleeds: 5 - 10%
- Central nervous system: < 5%
- Incidence of bleeding into joints:
- Knee: 45%
- Elbow: 30%
- Ankle: 15%
- Shoulder: 3%
- Wrist: 3%
- Hip: 2%
- Other: 2%
Pathophysiology
- Factor VIII is a plasma protein produced in the liver and by the reticuloendothelial system (Wikipedia)
- It circulates mainly bound to von Willebrand factor protein
- It functions as a cofactor along with activated factor IX to activate factor X, which in turn with its cofactor, factor Va, activates thrombin
- Normal plasma activity is 50 - 150% (0.5-1.5 IU/mL)
- Biologic half life is 8 - 12 hours
Etiology
- Inherited as an X-linked recessive trait, however 30% are due to spontaneous mutations
- The gene for factor VIII is a large gene located on a fragile region of the X chromosome
- The most common mutation involves inversion of intron 22; less common mutations include missense, large deletions, small point mutations and insertions/deletions
- Hemophilic females develop disease due to:
- High degree of X-inactivation in carriers
- Hemizygosity of the X chromosome in females with Turner syndrome (XO karyotype)
- Homozygosity in female progeny of a hemophilia A carrier and an affected hemophilic male
Diagrams / tables
Images hosted on other servers:
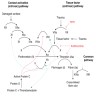
Coagulation cascade
Clinical features
- Clinical severity is dependent on factor levels:
- Mild (> 5% activity; > 0.05 IU/mL); occurs in 30 - 40% and typically presents with bleeding after surgery or trauma
- Moderate (1 - 5% activity; 0.01 - 0.05 IU/mL); occurs in 10% and presents with bleeding after surgery or trauma, less commonly with spontaneous bleeding
- Severe (< 1% activity; < 0.01 IU/mL); occurs in 50% and presents with spontaneous bleeding into joints, muscles and with life-threatening hemorrhage
- 30% of cases are due to spontaneous mutations, and have no family history of bleeding
- 35% of patients with severe hemophilia A will develop alloantibody inhibitors to factor VIII after replacement therapy (Factor VIII inhibitor)
- Formation of factor VIII alloantibodies is highest in individuals with intron 22 inversions, large deletions and nonsense mutations (Blood Coagul Fibrinolysis 2003;14:S17)
Laboratory
- Prolonged PTT with correction after mixing study (at 0 and 2 hr)
- Normal PT and bleeding time
- Measure both factor VIII and IX activity by functional plasma clot-based assay or chromogenic substrate-based assay
- Rule out vWD by vWF antigen and ristocetin cofactor activity
- Bethesda assay for quantitation of inhibitor
- Candidates for genetic testing include patients who have a diagnosis of hemophilia A or B, at-risk women who are related to an affected man (proband) who has a known mutation, and female carriers of hemophilia A or B seeking prenatal diagnosis
- First-line testing involves identification of the inversion of intron 22
- Obtain linkage analysis using restriction fragment length polymorphism (RFLP) if no inversion is detected or family members are available for testing
- Cord blood sampling for measurement of factor VIII activity in male fetus of known carrier
Prognostic factors
- Chronic complications of hemophilia include musculoskeletal problems (e.g.chronic synovitis, arthropathy, fractures, contractures), inhibitor formation (which complicates treatment), and transfusion-related infections (e.g. HIV, HBV, HCV, etc.)
Case reports
- Intraosseous hematoma in a newborn (AJNR Am J Neuroradiol 2000;21:308)
- Intracranial pseudotumor (Br J Neurosurg 2009;23:455))
Treatment
- Need 80 - 100% of normal factor VIII levels for surgical hemostasis with major surgery or major bleeding, 30 - 50% postoperatively or to prevent minor bleeding
- Use plasma-derived or recombinant factor VIII concentrates (1 unit/kg raises levels in vivo by 2%):
- Major surgery/bleeding - 40 - 50 units factor VIII concentrate/kg every 12 hours as necessary, usually for 7 - 10 days
- Postoperatively - 15 - 25 units/kg every 12 hours as necessary, usually for 7 - 10 days
- Minor bleeding - 15 - 20 units/kg every 12 - 24 hours as necessary
- Mild / moderate bleeding - DDAVP (if patients respond to DAVP)
- Prophylaxis in severe hemophilia A 25 - 40 units/kg three times weekly
- Antifibrinolytic and topical agents (e.g. epsilon-aminocaproic acid, tranexamic acid, fibrin sealants) as adjuvant therapy
- Treatment of acute bleeding episodes in patients with inhibitors:
- For low-titer inhibitors: high dose factor VIII (to overwhelm inhibitor), porcine factor VIII (if no cross reactivity with inhibitor)
- For high-titer inhibitors: factor VIII bypassing agents (prothrombin complex concentrates, FEIBA, recombinant factor VIIa)
Differential diagnosis
- Acquired hemophilia A: autoantibody against factor VIII
- Factor IX deficiency (hemophilia B)
- Familial combined factor deficiencies: typically have an autosomal recessive pattern
- Other factor deficiencies: XI, XII
- von Willebrand Disease: particularly type 2N or type 3
