

Lung
Cystic disease / congenital anomalies
Cystic fibrosis
Author: Elliot Weisenberg, M.D.

Last author update: 1 August 2011
Last staff update: 17 July 2020
Copyright: 2003-2024, PathologyOutlines.com, Inc.
PubMed search: cystic fibrosis [title] pulmonary lung

Table of Contents
Definition / general | Etiology | Associated Infections | Diagrams / tables | Clinical features | Gross description | Differential diagnosisCite this page: Weisenberg E. Cystic fibrosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lungnontumorcysticfibrosis.html. Accessed April 26th, 2024.
Definition / general
- Autosomal recessive disorder
- 1 in 20 Caucasians in US are carriers; much lower incidence in African Americans, Asians, Hispanics
- In US, approximately 1 in 2500 live births have disease
- Over 1300 known mutations of cystic fibrosis transmembrane conductance regulator / CFTR, most common mutation (seen in 70% in UK) is #508 (Eur J Hum Genet 2002;10:583)
Etiology
- Mutations cause reduced chloride ion in secretions and thicker respiratory secretions
- Primary defect is abnormal function, deficiency or absence of cystic fibrosis transmembrane conductance regulator / CFTR that regulates chloride channel in epithelial cells
- Allelic variation correlates with some aspects of disease, but lung function, neonatal intestinal obstruction, diabetes and anthropometry display strong genetic control independent of CFTR, and candidate gene studies have revealed genetic modifiers underlying these traits (Ann NY Acad Sci 2010;1214:57)
Associated Infections
- Allergic bronchopulmonary aspergillosis: increased incidence
- Burkholderia cepacia: unique to cystic fibrosis, seen in 20% of patients; causes rapid deterioration of pulmonary status and death; transmitted person to person, has marked social impact as those infected are excluded from social functions (camps) and ineligible for transplant; treat with chloramphenicol and trimethoprim-sulfamethoxazole
- Hemophilus influenza: common
- Mycobacteria (atypical): common
- Pseudomonas aeruginosa: bacteria produces alginate, a capsular protein that mediates adherence; mucoid phenotype is unique to cystic fibrosis; bacteria is never eradicated from lung; treat with ceftazidime (BMC Med 2011 Apr 4;9:32)
- Staphylococcus aureus: infection persists despite treatment
- Stenotrophomonus maltophilia: aerobic Gram-negative rod, multidrug resistant, smells like onions; treat with trimethoprim-sulfamethoxazole, resistant to imipenim
- Also anaerobes, although difficult to detect (New Microbiol 2010;33:185)
- Note: many taxonomic changes have occured (Clin Microbiol Rev 2010;23:299)
Diagrams / tables
Images hosted on other servers:
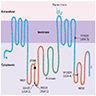

CFTR
Clinical features
- Disease usually manifests in young children, but also prenatally or in adolescents
- Recurrent infections cause chronic lung disease, the most severe manifestation of the disease; recurrent upper respiratory infections occur, late sequela are pancreatic insufficiency, steatorrhea, malnutrition, cirrhosis
- Mutations also cause defective cilia and infertility
- Meconium ileus seen in 5% - 10% of patients; also intussusception
- Heterozygotes (carriers) also have higher incidence of respiratory and pancreatic disease than general population
Gross description
- Emphysema, bronchiectasis, abscess, fibrosis
Differential diagnosis
- Kartegeners (defective cilia syndrome)
