

Kidney nontumor / medical renal
Glomerular disease
Inherited glomerular disease
Fabry disease
Author: Nikhil Sangle, M.D.

Last author update: 6 November 2017
Last staff update: 13 September 2021
Copyright: 2002-2024, PathologyOutlines.com, Inc.
PubMed Search: Fabry’s disease [title] "loattrfree+full+text"[sb]
Table of Contents
Definition / general | Clinical features | Diagnosis | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Immunofluorescence description | Positive stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Differential diagnosisCite this page: Sangle N. Fabry disease. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/kidneyfabry.html. Accessed April 26th, 2024.
Definition / general
- Also called alpha-galactosidase A deficiency, angiokeratoma corporis diffusum universale
- X linked (Xq22.1) recessive lysosomal storage disease which causes deficiency in lysosomal alpha-galactosidase A, which catabolizes neutral glycosphingolipids
- Deficiency causes intracellular accumulation of galabiosylceramide (ceramide trihexoside) and digalactosyl ceramide within skin, renal glomeruli, renal tubular epithelium, blood vessels, corneal epithelium, myocardium and ganglion cells
Clinical features
- Affects 1 per 40,000
- Highly penetrant in hemizygous males with symptoms at infancy or childhood
- Later presentation in heterozygous females, who have more variable severity due to variable lyonization of X chromosome and may have normal leukocyte alpha-galactosidase A activity
- Clinical symptoms include angiokeratomas on skin of abdomen, buttocks, lips, genitalia and upper thighs
- Also hematuria and proteinuria progressing to renal failure, corneal dystrophy and recurrent shooting pains in legs
- Death due to renal, cardiac or cerebrovascular disease at age 40+ years
Diagnosis
- Low blood or urine levels of alpha-galactosidase by enzymatic assay (may be normal in female heterozygotes)
- Elevated ceramide trihexoside in urine by thin layer chromatography
- Immunostains for ceramide trihexoside
- In women, must perform DNA mutation analysis of alpha-galactosidase A gene to exclude carrier state
- Patients may present with advanced disease identifiable only by ultrastructural studies (Ultrastruct Pathol 2010;34:307)
Case reports
- 23 year old man with congenital agammaglobulinemia (J Korean Med Sci 2011;26:966)
- 34 year old man with atypical variant (Arch Pathol Lab Med 1996;120:86)
- 42 year old woman with persistent proteinuria (Arch Pathol Lab Med 1985;109:89)
- Cases with accumulation in heart, not kidney or liver (Hum Pathol 1990;21:1067)
Treatment
- Recombinant human alpha-galactosidase A replacement therapy
Microscopic (histologic) description
-
Kidney:
- Enlarged and bubbly, clear vacuoles in visceral epithelium (demonstrated by trichrome stain), parietal epithelium, mesangial cells, endothelial cells, vascular smooth muscle and distal tubular cells
- Narrowing and thrombosis of arteries and arterioles
- Patchy tubular atrophy and interstitial fibrosis
- Progression to focal segmental and global glomerulosclerosis
Microscopic (histologic) images
Images hosted on other servers:
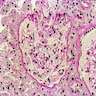
Various images including EM
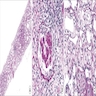
Global sclerosis,
segmental sclerosis
with moderate
interstitial fibrosis
Immunofluorescence description
- Negative
Positive stains
- PAS, Oil red O, Sudan black and Luxol fast blue (stain glycolipid and phospholipid-like material)
Electron microscopy description
- Characteristic single membrane bound intracellular inclusions (myelin-like figures, zebra bodies), that are 0.1 to 10 microns in diameter, round and lamellated with concentric electron dense layers, found in endothelial and smooth muscle cells, myocardium, fibroblasts and glomerular epithelium; deposits reduced after enzyme therapy (Clin Nephrol 2009;71:550)
- Changes also present in urine sediment (Arch Pathol Lab Med 1981;105:361)
Electron microscopy images
Images hosted on other servers:
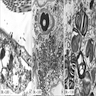
inclusion bodies in
cytoplasm of skin
fibroblasts
Molecular / cytogenetics description
- Wide molecular heterogeneity (Rev Med Interne 2010;31 Suppl 2:S275)
Differential diagnosis
- Foam cell change of Gaucher’s disease, gangliosidoses, fucosidosis, mucopolysaccharidoses (all have different intracellular distribution and ultrastructural features of inclusions, lack electron dense myeloid bodies and can detect by laboratory assays)
- Treatment with chloroquine, amiodarone or aminoglycosides (have similar myelin-like figures, Hum Pathol 2003;34:285)
