Liver & intrahepatic bile ducts
Developmental anomalies / cysts
Alagille syndrome
Author: Kalyani R. Patel, M.D.

Last author update: 1 November 2016
Last staff update: 16 May 2025 (update in progress)
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Alagille syndrome liver
Table of Contents
Definition / general | Essential features | Pathophysiology | Clinical features | Microscopic (histologic) description | Microscopic (histologic) imagesCite this page: Patel K. Alagille syndrome. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liveralagillessyndrome.html. Accessed August 22nd, 2025.
Definition / general
- Also called arteriohepatic dysplasia
- Represents the etiology behind syndromic paucity of bile ducts
Essential features
- Genetic disorder with vascular, biliary and other anomalies
- Absence of intrahepatic bile ducts with clinical severity ranging from severe neonatal cholestasis mimicking biliary atresia to childhood intermittent jaundice
- Progression to cirrhosis is rare
Pathophysiology
- 2 distinct genetic mechanisms:
- Vast majority (ALGS1) are autosomal dominant, due to mutations in Jagged1 gene on chromosome 20p12, which encodes a ligand for NOTCH1 and plays a role in epithelial mesenchymal interactions (Nat Genet 1997;16:235)
- Gene penetrance is high but expression is variable; 50 - 70% patients have new mutations
- Second genetic abnormality (ALGS2) accounting for small proportion of cases, is related to the mutations in the gene encoding NOTCH2 on chromosome 1p13 and has more severe renal disease (Am J Hum Genet 2006;79:169)
- Vast majority (ALGS1) are autosomal dominant, due to mutations in Jagged1 gene on chromosome 20p12, which encodes a ligand for NOTCH1 and plays a role in epithelial mesenchymal interactions (Nat Genet 1997;16:235)
Clinical features
- Reported incidence is 1:30,000 of live births
- Common clinical features: abnormal inverted triangular facies, posterior embryotoxon in the eye (Digital Reference of Ophthalmology: Cornea & External Diseases [Accessed 25 October 2017]), pulmonary stenosis or more severe congenital heart disease, butterfly vertebrae or other vertebral arch anomalies, other skeletal anomalies such as short distal phalanges or clinodactyly
- Several renal abnormalities such as tubulointerstitial nephropathy, membranous nephropathy, mesangiolipidosis, renovascular hypertension, etc. have been reported
- Liver findings are characterized by progressive loss of bile ducts with or without hypoplasia of extrahepatic bile ducts, hypoplasia of gallbladder and cholelithiasis, leading to jaundice and pruritus (Nat Genet 1997;16:235, Nat Genet 1997;16:235)
- It can mimic other causes of high GGT cholestasis, particularly biliary atresia
Microscopic (histologic) description
- Portal tracts are devoid of bile ducts; the ratio of bile ducts to portal tracts is 0 to 0.4 (normal 0.9 to 1.8) (Pediatr Pathol 1988;8:1)
- Ductular reaction is typically absent but rare examples present with ductular reaction in early infancy (Nat Genet 1997;16:235)
- Giant cell transformation and periportal copper deposits can be seen
- Progression to cirrhosis is rare
- Liver transplantation is reserved for patients with progressive liver disease or intractable pruritus
- Mortality is related to liver disease, cardiac abnormalities and intracranial bleeding
- Patients may survive into adulthood but with increased risk of hepatic failure and hepatocellular carcinoma
Microscopic (histologic) images
Contributed by Kalyani R. Patel, M.D.
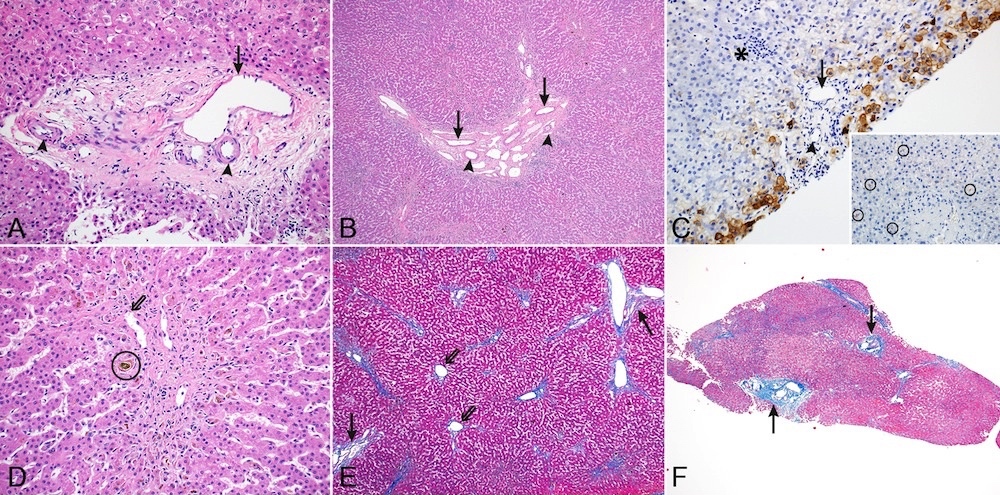
Liver biopsies
