Liver & intrahepatic bile ducts
Metabolic diseases
Glycogen storage diseases
Authors: Nigar Anjuman Khurram, M.D., Monika Vyas, M.D.
Editorial Board Member: Kimberley J. Evason, M.D., Ph.D.
Deputy Editor-in-Chief: Aaron R. Huber, D.O.


Last author update: 24 August 2023
Last staff update: 18 July 2024
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Glycogen storage diseases
Table of Contents
Definition / general | Essential features | ICD coding | Type 0 | Type I | Type Ia | Type Ib | Type Ic and Id | Type III | Type IV | Type VI | Type IX | Type XI | Sample pathology report | Practice question #1 | Practice answer #1Cite this page: Khurram NA, Vyas M. Glycogen storage diseases. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/liverglycogenstoragedisease.html. Accessed August 23rd, 2025.
Definition / general
- Called GSD, also glycogenosis and dextrinosis
- Groups of diseases caused by various enzyme deficiencies that result in abnormal glycogen synthesis or glycolysis, typically within the muscles or liver cells (EXCLI J 2019;18:30, Genet Med 2013;15:106)
- Overall GSD incidence is estimated to be 1 case per 20,000 - 43,000 live births (World J Gastroenterol 2007;13:2541)
- 23 types of GSD have been recognized, involving different organs (Genet Med 2016;18:1037)
- Liver, skeletal muscle, heart and the central nervous system are the most common organs involved (Genet Med 2016;18:1037)
- Classified by the organ affected and the enzyme deficiency involved
- GSD types I, III, 0, XI, IX, VI and IV affect the liver (with 80% of hepatic GSD being type I, III or IX) (Ultrastruct Pathol 2011;35:183)
- Some GSD types can affect both the liver and muscles (III and IXb) (Ultrastruct Pathol 2011;35:183)
- Associated with hypoglycemia and hepatomegaly (Genet Med 2013;15:106)
Essential features
- Rare disorder
- Group of rare inborn disorders of glycogen metabolism
- Variable expressivity and most patients present with hepatomegaly and hypoglycemia
- Increased incidence of hepatic adenoma and hepatocellular carcinoma
ICD coding
Type 0
Definition / general
Diagnosis
Treatment
Microscopic (histologic) description
- Also called aglycogenosis
- Rare, autosomal recessive
- Deficiency in glycogen synthase (chromosome 12p12.2)
- Clinically, hyperglycemia, glycosuria, hyperlactic acidemia, which alternate with hypoglycemia and hyperketonemia during fasting
- Due to the hyperglycemia and hyperuricemia, the child may be thought to have an early stage of diabetes, especially considering that the liver is not enlarged
- Symptoms of GSD type 0 are those associated with hypoglycemia
- Developmental delay may be seen in some children
Diagnosis
- Liver biopsy
- Enzymatic evaluation of the liver tissue reveals low to absent glycogen synthase activity
- Genetic / molecular testing can be performed with either liver or muscle tissue to further confirm that there is mutation of the glycogen synthase gene (chromosome 12p12.2)
- Currently, gene mutational analysis is performed to provide a definitive diagnosis (World J Gastroenterol 2007;13:2541)
Treatment
- Symptoms ameliorate with protein rich meals and bedtime feeding of uncooked cornstarch in low fat or skim milk
Microscopic (histologic) description
- Small amounts of glycogen and moderate steatosis
Type I
Definition / general
Microscopic (histologic) description
Microscopic (histologic) images
Contributed by Nigar Anjuman Khurram, M.D.
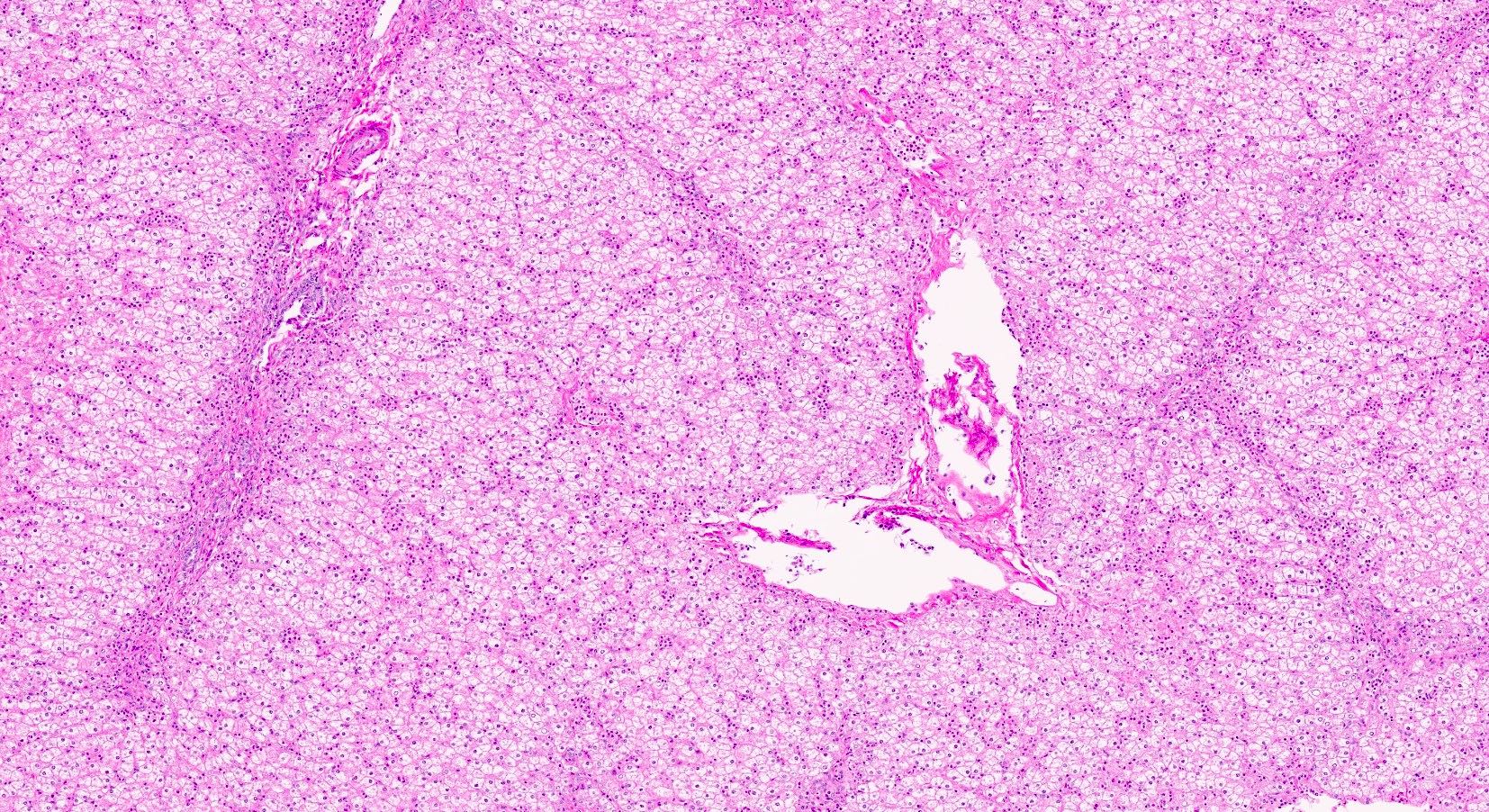
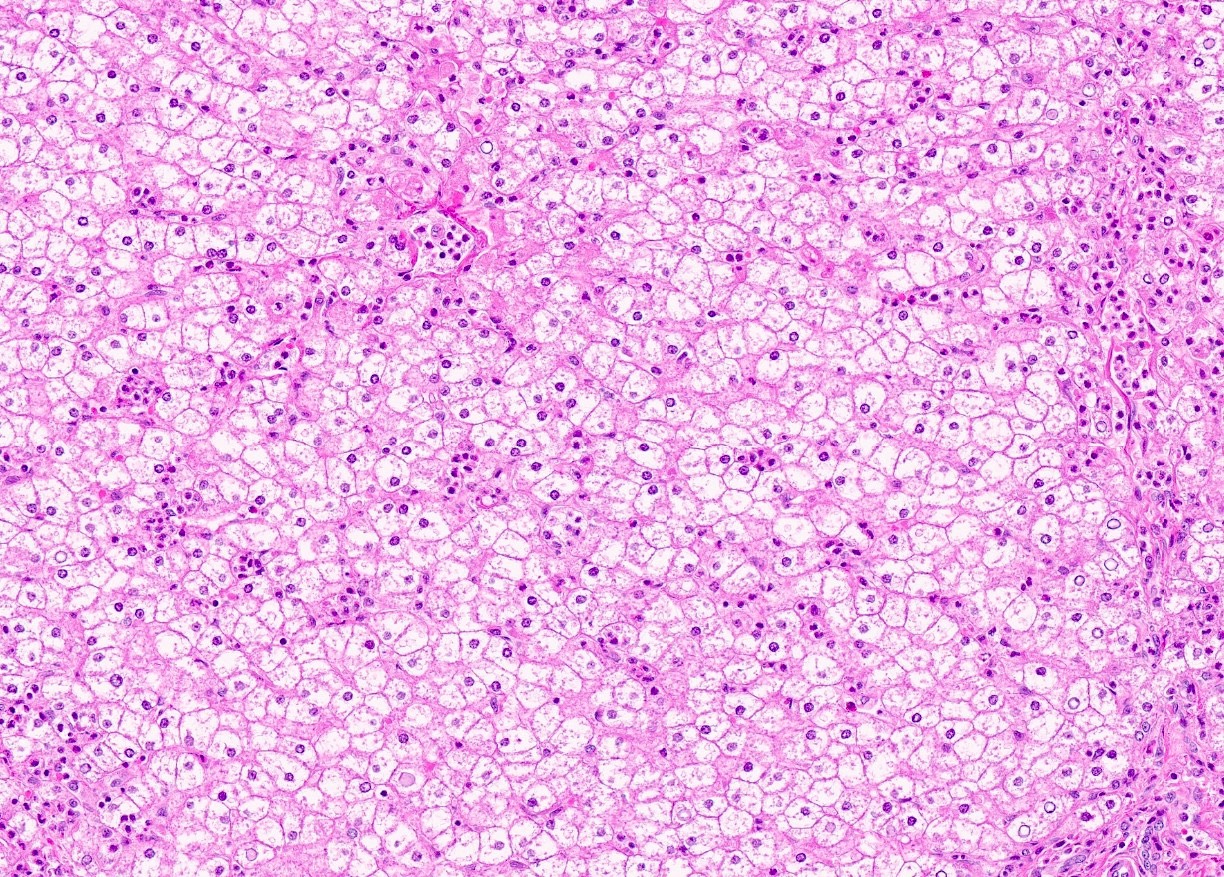
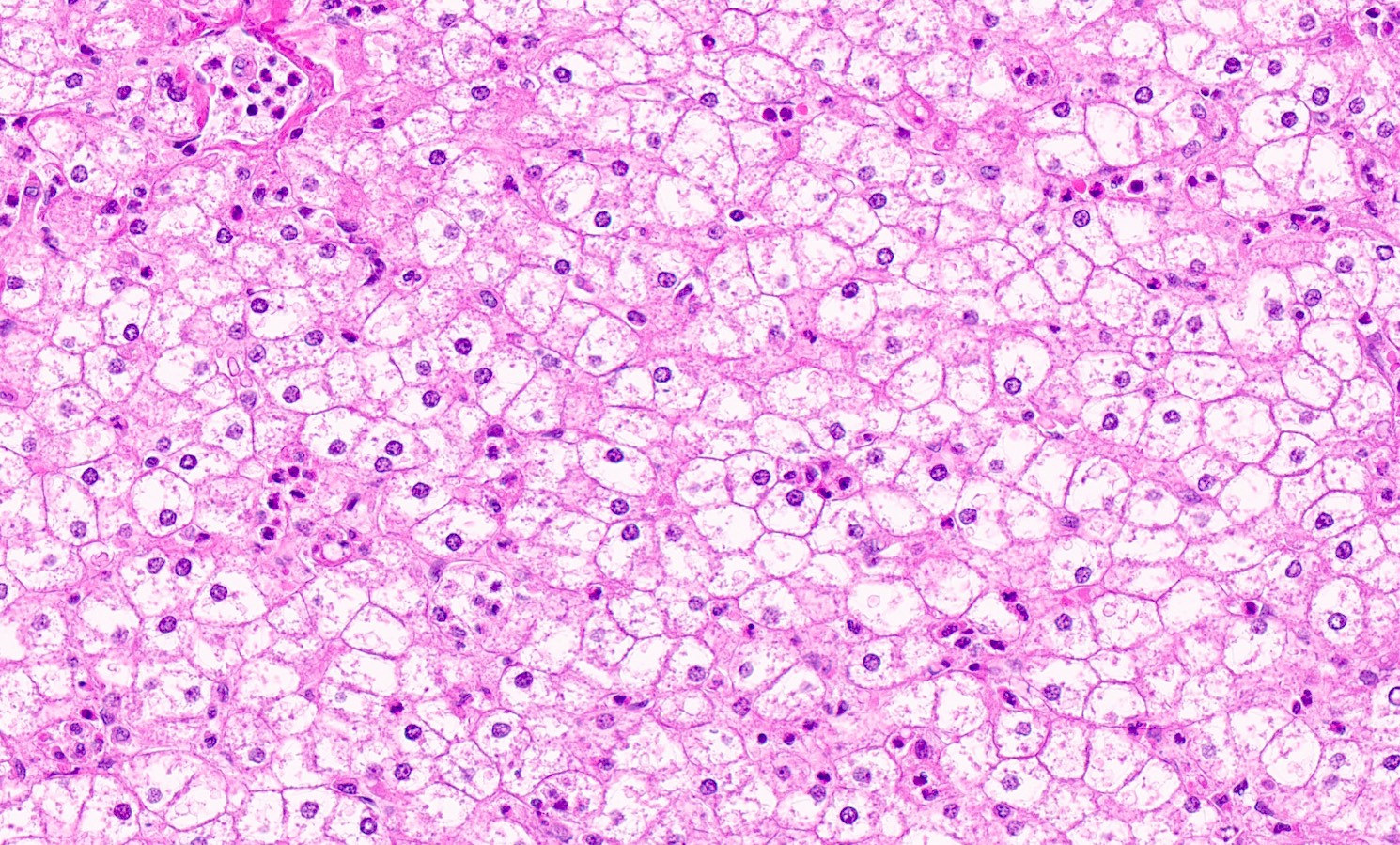
Electron microscopy description
- Also known as von Gierke disease or hepatorenal glycogenosis
- Occurs in an autosomal recessive pattern
- Type Ia: glucose-6-phosphatase (G6Pase) deficiency
- Type Ib: translocase T1 deficiency
- Type Ic: translocase T2 deficiency (carries inorganic phosphates from microsomes into cytosol and pyrophosphates from cytosol into microsomes)
- Type Id: deficiency in transporter that translocates free glucose molecules from microsomes
- Incidence of GSD type Ia is 1 per 100,000 - 400,000 births per year in the White population
- GSD types Ib and Ic are even less frequent
- Incidence of 1 in 20,000 in the Ashkenazi Jewish population (Ultrastruct Pathol 2011;35:183)
- May present as severe hypoglycemia and increased lactic acid, triglycerides and uric acid shortly after birth
- Older children have short stature and may develop eruptive xanthomas, rickets, anemia, chronic renal disease due to hyperuricemia and renal stones
- Tissue type affected is different with the GSD type I subtypes: type Ia - liver, kidney, intestine; type Ib - liver; and type Ic - liver
- Diagnosis of GSDI established in a proband by identification of biallelic pathogenic variants in either G6PC1 (GSDIa) or SLC37A4 (GSDIb)
- If molecular genetic testing is inconclusive, hepatic enzyme activity analysis is only available for glucose-6-phosphatase catalytic activity (GSDIa) (GeneReviews: Glycogen Storage Disease Type I [Accessed 14 July 2023])
Microscopic (histologic) description
- Distended liver cells with a uniform distribution of glycogen
- Liver cells arranged in mosaic pattern with plant like appearance
- Steatosis with hyper glycogenated nuclei
- Fibrosis may also be present
Microscopic (histologic) images
Contributed by Nigar Anjuman Khurram, M.D.
Uniform hepatic parenchyma
Hepatocyte mosaics with cytoplasmic clearing
Enlarged hepatocytes with pale cytoplasm
Electron microscopy description
- Abundant and uniform distribution of glycogen within enlarged hepatocytes
- Increased cytoplasmic glycogen, enlarged organelles, lipid droplets, glycogenated nuclei
Type Ia
Definition / general
Diagnosis
Case reports
Treatment
Gross description
Microscopic (histologic) description
- Rare
- Caused by the deficiency of glucose-6-phosphatase (G6Pase) catalytic activity
- Laboratory findings include hypoglycemia, deranged liver function tests (LFTs), lactic acidosis, hyperuricemia and hyperlipidemia
- Clinical presentation: hypoglycemia and marked hepatomegaly in first year of life; later in life, short stature, chronic lactic acidosis, focal segmental glomerulosclerosis, hepatic adenomas, iron deficiency, enterocolitis, gout, osteoporosis
Diagnosis
- Biallelic pathogenic variants in G6PC1 (GSDIa) or SLC37A4 (GSDIb) on molecular genetic testing
- Deficient hepatic enzyme activity (glucose-6-phosphatase catalytic activity [GSDIa]) from a liver biopsy specimen
Case reports
- 17 year old woman with hepatocellular carcinoma with glycogen storage disease type 1a (Pediatr Int 2020;62:744)
- 28 year old woman with multiple hepatic adenomas (Arch Pathol Lab Med 2003;127:e402)
Treatment
- Continuous dietary nasogastric infusion of glucose or frequent oral uncooked cornstarch at 3 - 6 hour intervals during the day and at night
- Liver transplantation may be necessary if not compliant with dietary restrictions or with refractory hyperglycemia or hepatocellular carcinoma (World J Gastroenterol 2007;13:2541)
- Gene therapy using adenovirus vectors has been evaluated in murine models of GSD type I (World J Gastroenterol 2007;13:2541)
Gross description
- Massive hepatomegaly
- May have variable sized tumor nodules
Microscopic (histologic) description
- Large hepatocytes with prominent cell membrane and glycogenated nuclei
- PAS+ accumulated glycogen
- Rarely contains Mallory hyaline and steatosis in hepatic adenomas
Type Ib
Definition / general
- Caused by a defect in glucose-6-phosphate exchanger SLC37A4 (transporter)
- Normal G6Pase activity in frozen tissue and lowered activity in fresh specimens
- Recurrent infections, neutropenia and neutrophil dysfunction are recognized as distinctive features (J Pediatr 2000;137:187)
- Another distinctive feature of GSD-Ib is the observation of inflammatory bowel disease (Crohn's)-like colitis (J Pediatr 1986;109:55, Turk J Pediatr 2005;47:180)
- Clinical presentation: young children may have frequent otitis, gingivitis, boils, intermittent diarrhea; although it is rare, terminal kidney disease may develop and kidney transplantation may be necessary
- Treatment includes dietary therapy, granulocyte colony stimulating factor (G-CSF) therapy may restore myeloid functions and liver transplantation to prevent malignant transformation of hepatic adenomas and for refractory hypoglycemia
Type Ic and Id
Definition / general
- Liver microsomal transport of phosphate and glucose is deficient in GSD-Ic and GSD-Id
Type III
Definition / general
Diagnosis
Case reports
Treatment
Gross description
Microscopic (histologic) description
Electron microscopy description
- Also called Cori disease
- Autosomal recessive
- Rare disease of variable clinical severity affecting primarily the liver, heart and skeletal muscle
- Deficiency of debrancher enzyme amylo-1,6-glucosidase due to various mutations causes hypoglycemia (Hum Mol Genet 2009;18:2045)
- Most individuals with GSD III survive into adulthood
- GSD IIIa: most common subtype (85%), liver and muscle involvement
- GSD IIIb: 15%; liver involvement only
Diagnosis
- Demonstration of excessive and structurally abnormal glycogen accumulation with shorter outer branches and deficient debranching enzyme activity in frozen liver or muscle biopsy samples
- Deficient identification of pathogenic mutations in the AGL gene on both alleles
Case reports
- 56 year old woman known to be affected by GSD III and liver cirrhosis complicated by hepatocellular carcinoma (J Clin Gastroenterol 2000;31:80)
Treatment
- Individuals with GSD III and advanced liver disease should be considered for liver transplant
Gross description
- Hepatomegaly
Microscopic (histologic) description
- Liver or muscle biopsy demonstrate a vacuolar accumulation of nonmembrane bound glycogen primarily in the cytoplasm
- Lipid vacuoles are less frequent in GSD III (compared to GSD I)
- Steatosis, hepatocyte ballooning
- Presence of fibrosis, ranging from minimal periportal fibrosis to micronodular cirrhosis, is noted in GSD III (not in GSD I)
- Stored material in GSD III is periodic acid-Schiff positive and diastase sensitive within the cytoplasm
Electron microscopy description
- Increased cytoplasmic glycogen, lipid droplets, glycogenated nuclei
Type IV
Definition / general
Diagnosis
Case reports
Treatment
Microscopic (histologic) description
Electron microscopy description
Differential diagnosis
- Also called Andersen disease, Brancher deficiency, amylopectinosis, glycogen branching enzyme deficiency
- Rare, autosomal recessive, caused by deficiency of glycogen branching enzyme on 3p14, leading to excessive deposition of structurally abnormal, amylopectin-like glycogen in affected tissues, causing irreversible tissue and organ damage
- Clinically, extremely heterogeneous, owing in part to variation in tissue involvement
- Fatal perinatal neuromuscular subtype: in utero with fetal akinesia, polyhydramnios and fetal hydrops; death usually in the neonatal period
- Congenital neuromuscular subtype: newborn period with profound hypotonia, respiratory distress and dilated cardiomyopathy; death usually in early infancy
- Classic (progressive) hepatic subtype: may appear normal at birth but rapidly develop failure to thrive
- Hepatomegaly, liver dysfunction and progressive liver cirrhosis, hypotonia and cardiomyopathy
- Without liver transplantation, death from liver failure usually occurs by age 5
- Nonprogressive hepatic subtype: hepatomegaly, liver dysfunction, myopathy and hypotonia; likely to survive without progression of the liver disease and may not show cardiac, skeletal muscle or neurologic involvement
- Childhood neuromuscular subtype: rare, variable course, ranging from onset in the second decade with a mild disease course to a more severe, progressive course resulting in death in the third decade
Diagnosis
- Demonstration of glycogen branching enzyme (GBE) deficiency in liver, muscle or skin fibroblasts
- Identification of biallelic pathogenic variants in GBE1 on molecular genetic testing (GeneReviews: Glycogen Storage Disease Type IV [Accessed 19 July 2023])
Case reports
- 10 month old boy with massive hepatomegaly (Arch Pathol Lab Med 2002;126:630)
Treatment
- Liver transplantation
- Liver transplantation is helpful also for muscular involvement due to systemic microchimerism after liver allotransplantation and amelioration of pancellular enzyme deficiencies (Eur J Pediatr 1999;158:S43, N Engl J Med 1991;324:39)
Microscopic (histologic) description
- Hepatocellular periodic-acid Schiff positive, diastase resistant inclusions of the abnormal glycogen deposits
Electron microscopy description
- Filamentous nonbranching cytoplasmic aggregates
Differential diagnosis
- Lafora disease:
- Autosomal recessive progressive myoclonus epilepsy due to mutations in the EPM2A (laforin) and EPM2B (malin) genes, with no substantial genotype / phenotype differences between the two (Epileptic Disord 2016;18:38)
- Patients are typically older than patients with glycogen storage diseases and may have epilepsy, myoclonus or dementia (see case report of 17 year old girl with generalized myoclonus epilepsy following a seizure, Case #361)
- Inclusions are distinctive polyglucosans (abnormal glycogen), also called Lafora bodies, typically found in brain, periportal hepatocytes, skeletal and cardiac myocytes, eccrine duct and apocrine myoepithelial cells of sweat glands (Epileptic Disord 2016;18:38)
Type VI
Definition / general
Diagnosis
Treatment
Microscopic (histologic) description
Microscopic (histologic) images
Contributed by Miguel Reyes-Mugica, M.D. and Nigar Anjuman Khurram, M.D.
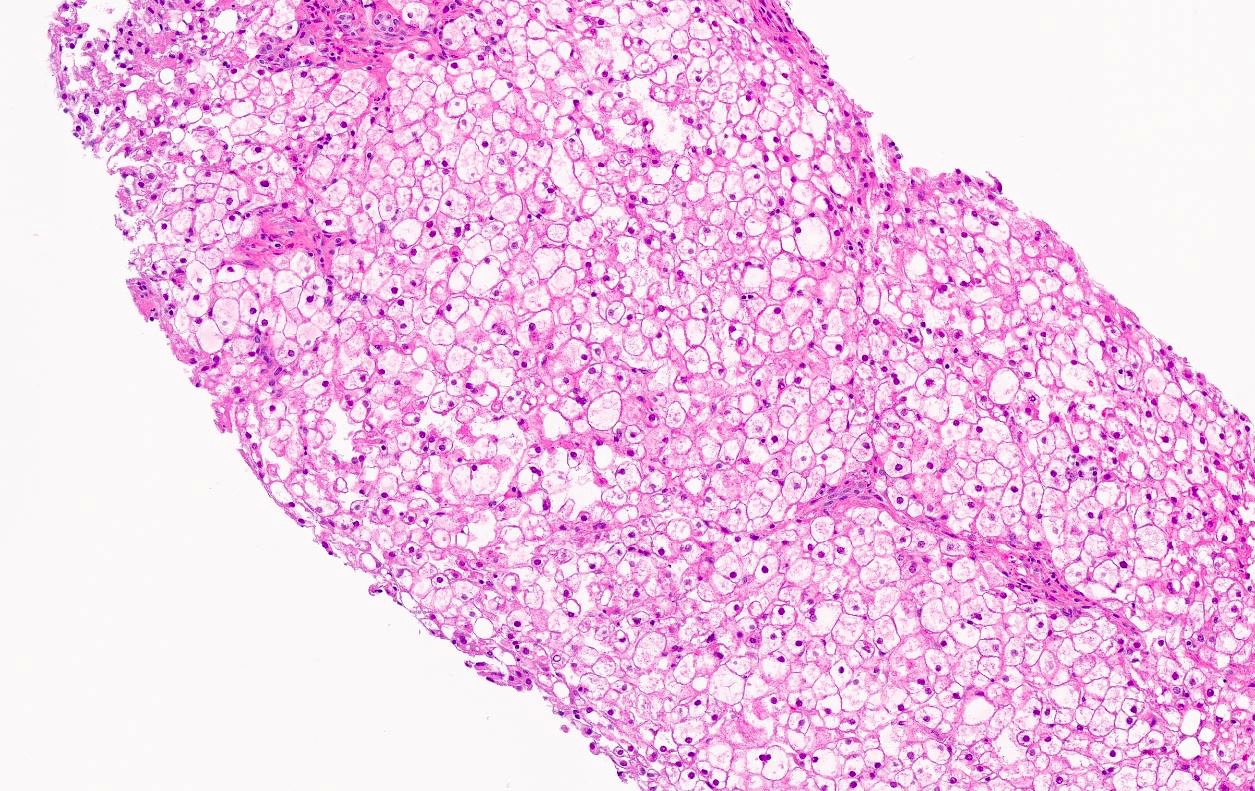
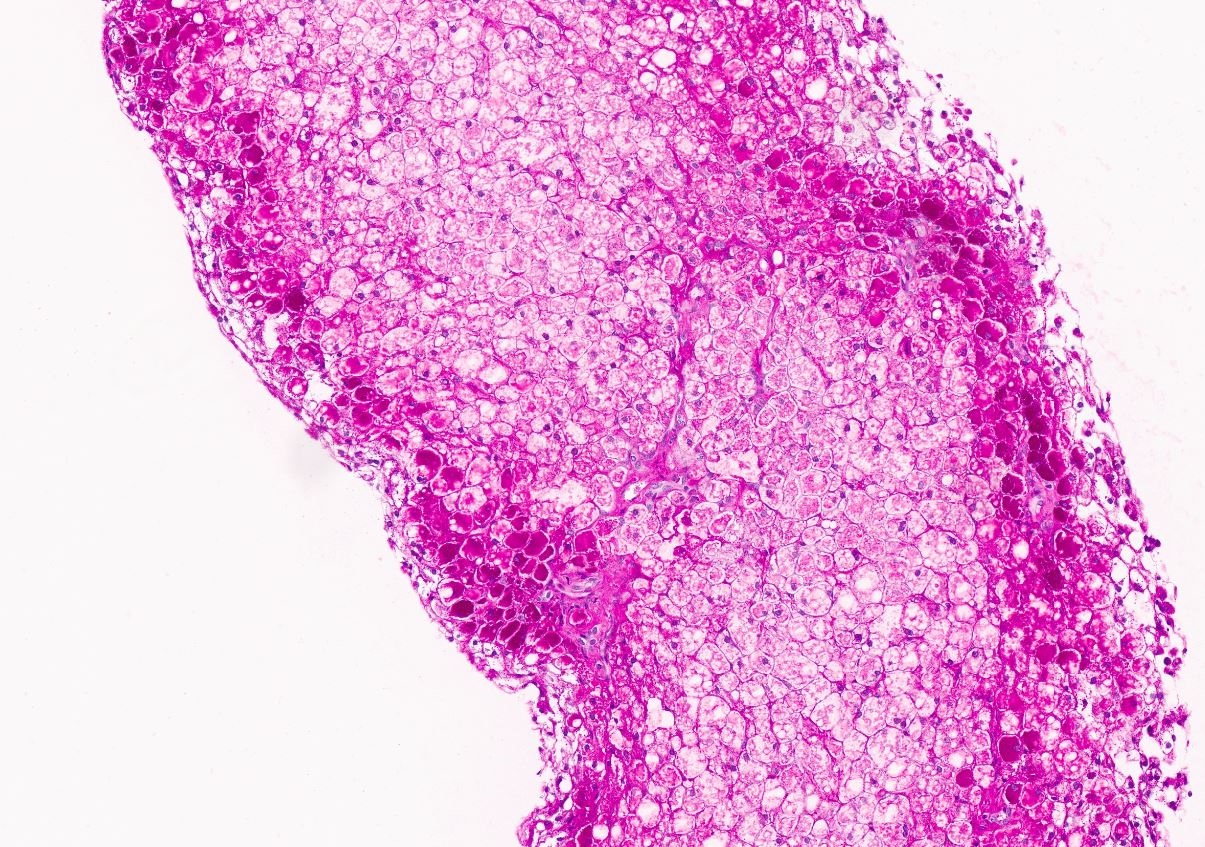
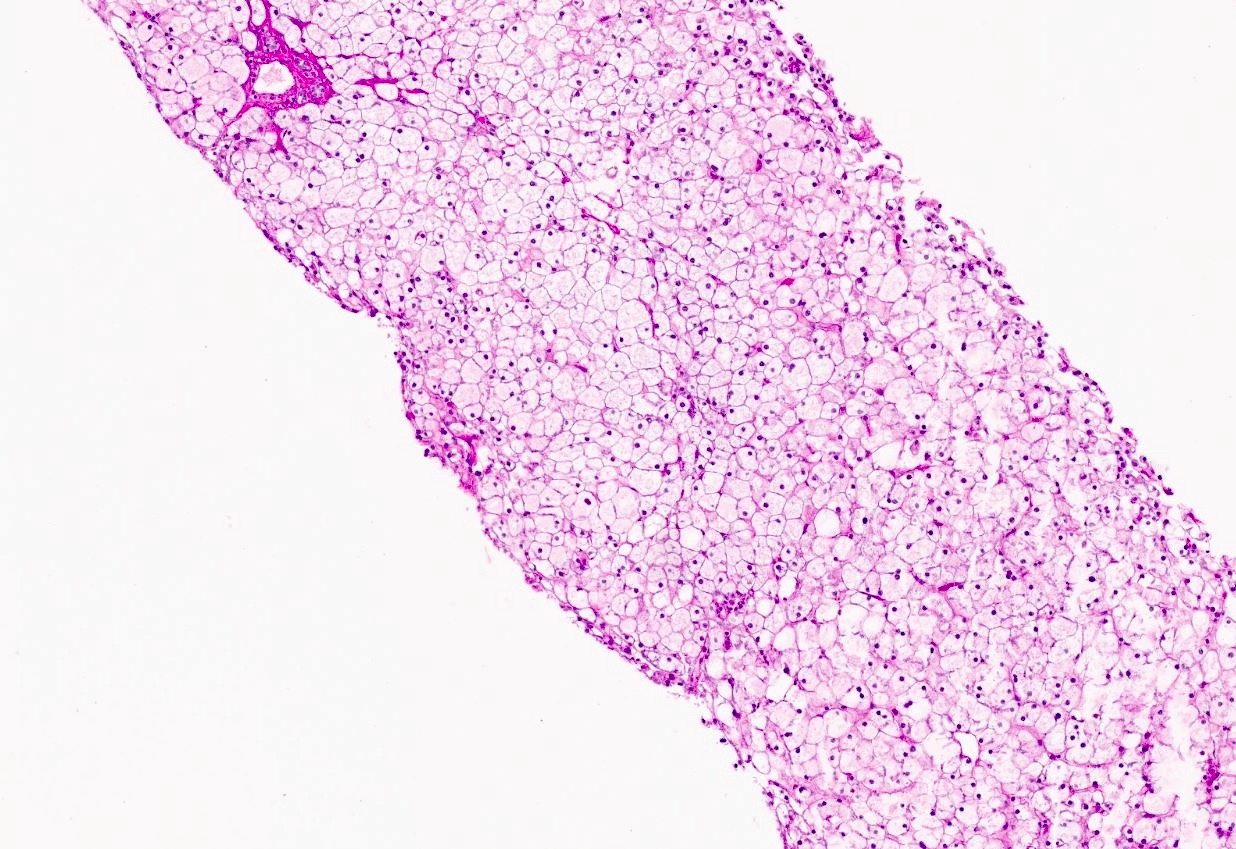
Electron microscopy description
Electron microscopy images
Contributed by Miguel Reyes-Mugica, M.D. and Nigar Anjuman Khurram, M.D.
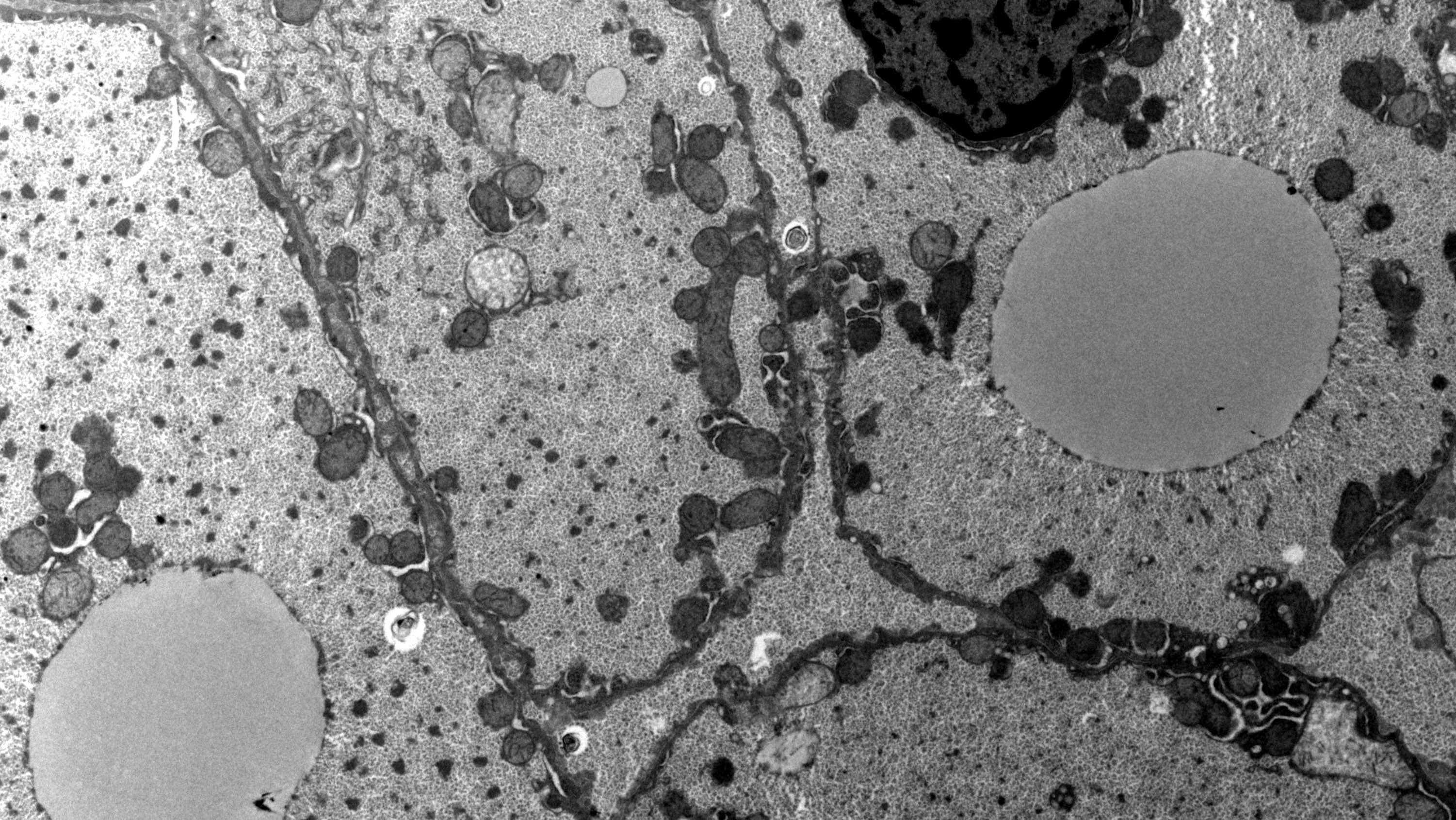
Additional references
- Also called Hers disease; liver glycogen phosphorylase deficiency
- Rare, autosomal recessive
- Most commonly affected group is the Mennonite community
- Clinically, asymptomatic hepatomegaly and growth retardation
- Usually has a benign course with mild to moderate hypoglycemia
Diagnosis
- Enzyme assay using liver, leukocytes and erythrocytes
- Gene mutation analysis can also be performed
Treatment
- Avoiding prolonged fasting and ingestion of a bedtime snack to avoid early morning hypoglycemia
Microscopic (histologic) description
- Small mosaic pattern and irregular distension of the hepatocytes due to glycogen deposition
- Microsteatosis, mild periportal fibrosis and septal formation may be present
Microscopic (histologic) images
Contributed by Miguel Reyes-Mugica, M.D. and Nigar Anjuman Khurram, M.D.
Plant cell-like morphology
Intracytoplasmic glycogen
Digestion of glycogen stores
Electron microscopy description
- Large pools of monoparticulate glycogen interspersed with glycogen rosettes and lipid vacuoles with glycogen embedded
- Hepatocytes have irregularly shaped aggregates of low density granular material scattered within the glycogen, giving a starry sky appearance
Electron microscopy images
Contributed by Miguel Reyes-Mugica, M.D. and Nigar Anjuman Khurram, M.D.
Intracytoplasmic free glycogen
Additional references
Type IX
Definition / general
Diagnosis
Treatment
Microscopic (histologic) description
Microscopic (histologic) images
Contributed by @RaulSGonzalezMD on Twitter
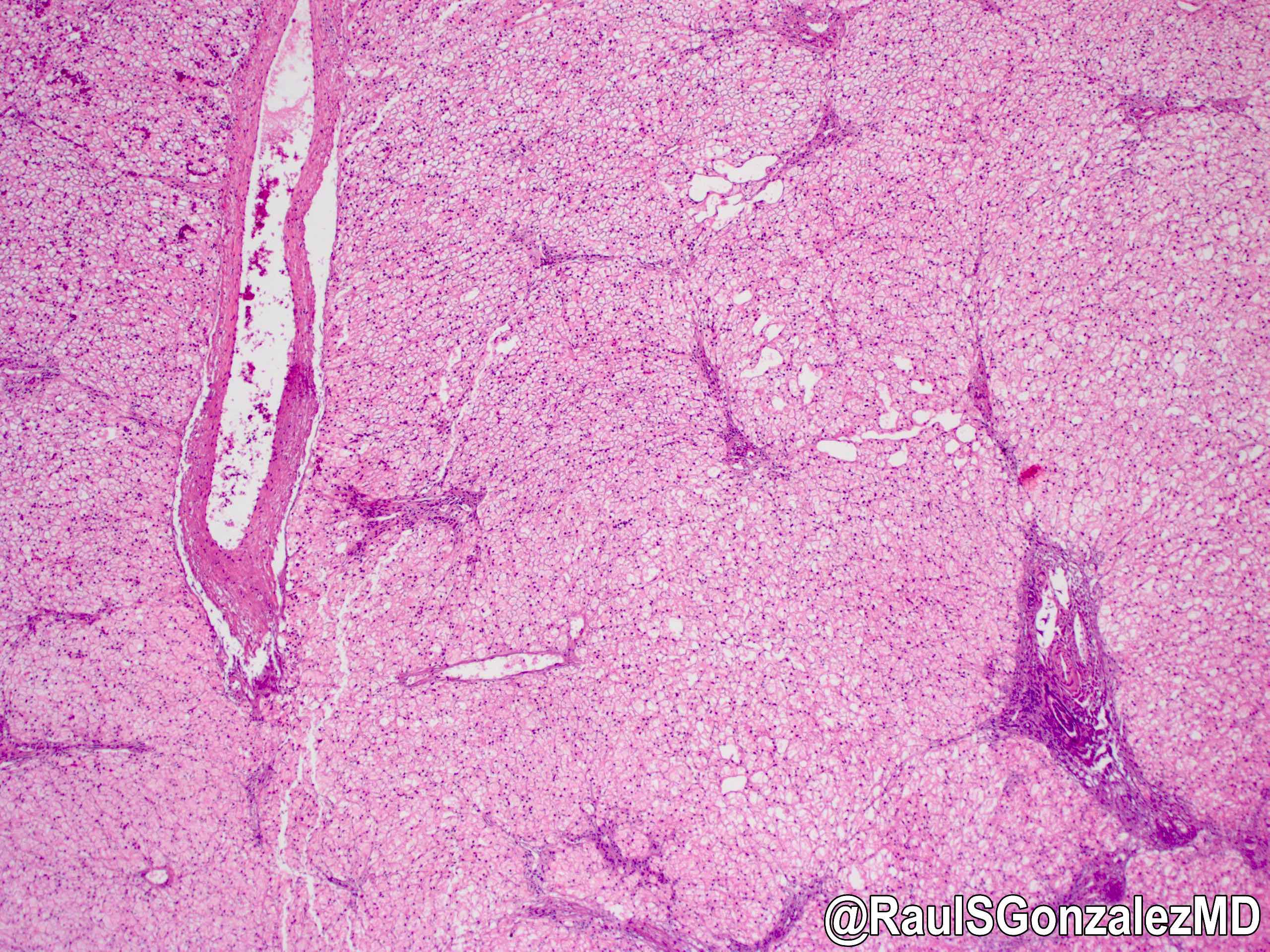
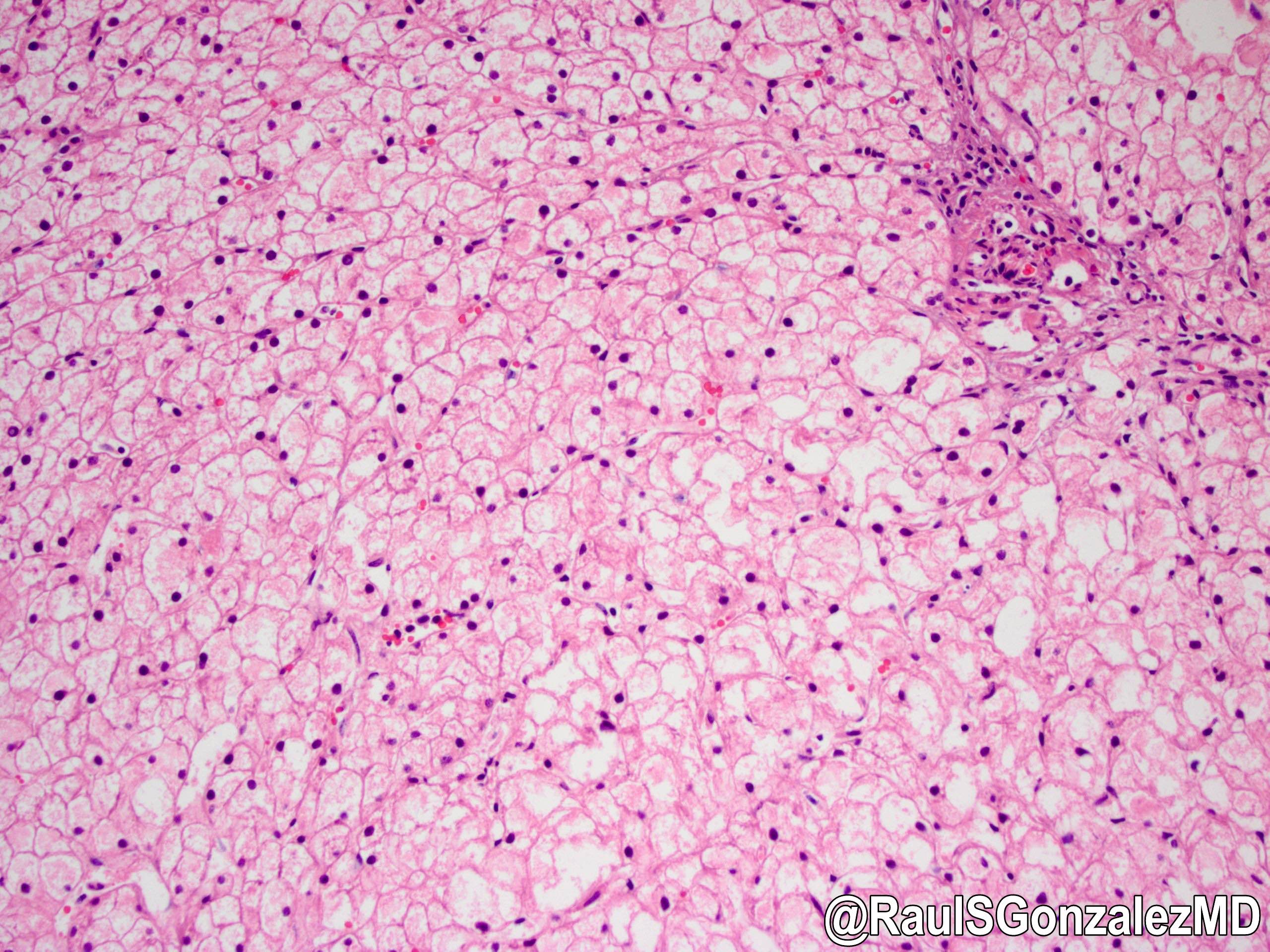
Electron microscopy description
Additional references
- Deficiency in phosphorylase kinase (PhK)
- Types involving the liver are mainly classified into 2 forms: the X linked liver form (there are 2 subtypes; XLG-I and XLG-II) and autosomal recessive form
- X linked liver phosphorylase kinase (alpha subunit) deficiency (PHKA2 related GSD type IX)
- Mildest GSD with low phosphorylase activity in the absence of adenosine monophosphate
- Most common signs and symptoms present between 1 and 5 years and include hepatomegaly, growth retardation (68%), hypotonia, deranged liver function tests, fasting hyperketosis, hypoglycemia
- Hepatomegaly and growth retardation usually resolve during puberty
- Splenomegaly and cirrhosis are very rare
- Autosomal liver and muscle phosphorylase kinase (beta subunit) deficiency (PHKB related GSD type IX)
- Autosomal recessive
- Deficiency in the beta subunit of phosphorylase kinase
- Hepatomegaly with abdominal distension, mild growth retardation and lipidemia
- When symptoms are present, the liver is primarily affected with the potential for cirrhosis
- Autosomal liver phosphorylase kinase (gamma subunit) deficiency (PHKG2 related GSD type IX)
- Autosomal recessive
- Deficiency in the gamma subunit of liver phosphorylase
- Associated with liver cirrhosis, renal tubular acidosis or neurologic disorders
Diagnosis
- Diagnosis can be made with enzyme analysis of liver tissue and by gene mutation analysis
Treatment
- Symptoms ameliorate with protein rich meals and bedtime feeding of uncooked cornstarch in low fat or skim milk
Microscopic (histologic) description
- X linked liver phosphorylase kinase (alpha subunit) deficiency (PHKA2 related GSD type IX)
- Biopsy shows irregular distension of hepatocytes with glycogen
- Hepatocytes are organized into a mosaic pattern
- Microsteatosis may be seen
- Autosomal liver and muscle phosphorylase kinase (beta subunit) deficiency (PHKB related GSD type IX): both liver and muscle biopsies show glycogen accumulation by light and electron microscopy
Microscopic (histologic) images
Contributed by @RaulSGonzalezMD on Twitter
Glycogen storage disease
Electron microscopy description
- X linked liver phosphorylase kinase (alpha subunit) deficiency (PHKA2 related GSD type)
- Extensive monoparticulate glycogen, glycogen rosettes and frequent lipid vacuoles containing glycogen particles
- Starry sky pattern attributed to scattered areas with finely granular organelle free clear zones alternating with densely packed glycogen particles
- Mitochondria tend to be decreased in size and numbers; skeletal muscle is normal
Additional references
Type XI
Definition / general
Diagnosis
Case reports
Treatment
Microscopic (histologic) description
- Also called Fanconi-Bickel syndrome
- Rare, autosomal recessive
- Responsible gene is glucose transport 2 gene (GLUT2), localized to 3q26.1-q26
- Defective transport of monosaccharide across the membranes
- Clinically, hepatorenal glycogen accumulation, fasting hypoglycemia, postprandial hyperglycemia and hypergalactosemia, proximal renal tubular dysfunction, marked stunted growth, dwarfism in older patients, hypercholesterolemia, hyperlipidemia, pancreatitis, generalized osteopenia
Diagnosis
- Detected by neonatal screening for galactose
Case reports
- 2 siblings with familial Fanconi syndrome with malabsorption and galactose intolerance, normal kinase and transferase activity (Acta Paediatr Scand 1981;70:527)
Treatment
- Liver transplantation
- Liver transplantation helpful also for muscular involvement due to systemic microchimerism after liver allotransplantation and amelioration of pancellular enzyme deficiencies (Eur J Pediatr 1999;158:S43, N Engl J Med 1991;324:39)
Microscopic (histologic) description
- Excessive glycogen accumulation with steatosis
Sample pathology report
- Liver, needle biopsy:
- Hepatic parenchyma with features suggestive of glycogen storage disease (see comment)
- Comment: The morphologic features are classic for glycogen storage disease (GSD), with a plant cell-like morphology, intracytoplasmic glycogen on PAS staining, which disappears after digestion with diastase. There is also mild steatosis and no significant nuclear glycosylation. The electron microscopic analysis also shows massive amounts of intracytoplasmic free glycogen. The main variants of GSD with features of this appearance are I, III, VI and IX. However, these diseases have overlapping histopathologic features and therefore are best distinguished enzymatically and genetically. Given the lack of significant hypoglycemia, types VI and IX are favored. Further genetic / enzymatic analysis is required to definitively establish the specific subtype of GSD. Glycogen storage disease enzyme assay is pending and will be reported in an addendum.
Practice question #1
Deficiency of amylo-1,6-glucosidase debranching enzyme results in which of the following glycogen storage disease (GSD) types?
- GSD type 0, aglycogenosis
- GSD type I, hepatorenal glycogenosis / von Gierke disease
- GSD type III, Cori disease
- GSD type IV, Andersen disease
- GSD type XI, Fanconi-Bickel syndrome
Practice answer #1
C. GSD type III, Cori disease.
In GSD type III / Cori disease, deficiency of debrancher enzyme amylo-1,6-glucosidase due to various mutations causes hypoglycemia. Answer A is incorrect because GSD type 0 / aglycogenosis is caused by deficiency of glycogen synthase. Answer B is incorrect because GSD type 1, hepatorenal glycogenosis / von Gierke disease is caused by glucose-6 phosphatase deficiency. Answer D is incorrect because GSD type IV / Andersen disease is caused by a deficiency of glycogen branching enzyme on 3p14. Answer E is incorrect because GSD type XI / Fanconi-Bickel syndrome is caused by glucose transport 2 gene.
Comment Here
Reference: Glycogen storage diseases
Comment Here
Reference: Glycogen storage diseases
