Heart & vascular pathology
Systemic conditions
Amyloidosis
Authors: Victoria Walker, M.D., Jaishree Jagirdar, M.D.
Editorial Board Member: Matthew J. Cecchini, M.D., Ph.D.


Last author update: 9 January 2024
Last staff update: 12 November 2024
Copyright: 2020-2025, PathologyOutlines.com, Inc.
PubMed Search: Cardiac amyloidosis
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Gross description | Microscopic (histologic) description | Microscopic (histologic) images | Cytology description | Immunofluorescence description | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Walker V, Jagirdar J. Amyloidosis. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/heartamyloidosis.html. Accessed September 2nd, 2025.
Definition / general
- Amyloid infiltration of the myocardium
- Multiple types are named after the precursor protein of amyloid deposit; however, they are all characterized by the deposition of an eosinophilic amorphous material that stains bright red with Congo red and shows an apple green birefringence upon polarization
Essential features
- Heterogenous group of diseases caused by deposition of amyloid, an abnormal protein with a beta pleated structure composed of haphazardly arranged nonbranching fibrils measuring 8 - 10 nm in diameter
- Light microscopy shows amorphous eosinophilic deposits that show apple green birefringence under polarized light
- Main cardiac forms are transthyretin amyloidosis (including wild type and hereditary subtypes) and light chain amyloidosis
- Endomyocardial biopsy and histological analysis is the gold standard for diagnosis but less invasive techniques such as echocardiography, cardiac magnetic resonance imaging (CMR) and cardiac nuclear imaging can raise index of suspicion for this diagnosis
Terminology
- 2 main types (95% of cardiac amyloidosis cases)
- Transthyretin amyloidosis (ATTR amyloidosis)
- Wild type (wtATTR) subtype (previously senile systemic amyloidosis)
- Hereditary (hATTR) subtype
- Light chain amyloidosis (AL amyloidosis), also known as primary systemic amyloidosis (Clin Cardiol 2021;44:322)
- Transthyretin amyloidosis (ATTR amyloidosis)
- Rare types of cardiac amyloidosis include
- Serum amyloid A amyloidosis (AA)
- Hereditary apoprotein A1 (AApoA1)
- Apoprotein A4 (AApoA4) amyloidosis
ICD coding
- ICD-10
- ICD-11
- 5D00.0 - AL amyloidosis
- 5D00.1 - AA amyloidosis
- 5D00.2 - hereditary amyloidosis
- 5D00.20 - hereditary ATTR amyloidosis
- 5D00.21 - nonneuropathic heredofamilial amyloidosis
- 5D00.2Y - other specified hereditary amyloidosis
- 5D00.2Z - hereditary amyloidosis, unspecified
- 5D00.3 - dialysis associated amyloidosis
- 2A83.5 - monoclonal immunoglobulin deposition disease
- 2A83.50 - heavy chain deposition disease
- 2A83.51 - light and heavy chain deposition disease
- 2A83.52 - light chain deposition disease
- 5D00.Y - other specified amyloidosis
- 5D00.Z - amyloidosis, unspecified
Epidemiology
- Wild type transthyretin amyloidosis (wtATTR amyloidosis)
- Prevalence unknown
- Age: > 60 years
- Males: 90%
- Hereditary transthyretin amyloidosis (hATTR amyloidosis)
- Autosomal dominant, variable penetrance
- Prevalence unknown (> 120 gene variants associated with hATTR; some more prevalent in specific regions or ethnic groups and others more widely distributed) (Curr Med Res Opin 2013;29:63)
- Age: > 50 years but can be seen ranging from 30 years
- Males: 76 - 86%
- Light chain amyloidosis (AL amyloidosis)
- Rare, annual incidence 1/100,000 people in the U.S.
- Age: > 40 years
- Males: 60% (Clin Cardiol 2021;44:322)
Sites
- Amyloid can deposit in any cardiac structures: endocardium, valves, myocardium, epicardium, parietal pericardium (Clin Cardiol 2021;44:322)
- Commonly causes concentric biventricular hypertrophy, biatrial enlargement and atrial septal thickening, valvular thickening
Pathophysiology
- Extracellular (predominantly) tissue deposition of amyloid, composed of both fibril and nonfibril components
- Fibrils make up ~95% of deposits and are soluble polymers that undergo conformational change into an antiparallel beta pleated sheet configuration; this renders them insoluble and causes autoaggregation into highly ordered fibrils
- Nonfibril components make up ~5% of deposits and include serum amyloid P component (SAP), apolipoprotein E (ApoE) and glycosaminoglycans
- Infiltration of cardiac structures causes changes in calcium transport, receptor modulation, cellular metabolism and cardiomyocyte edema
- Damage via cardiomyocyte necrosis and interstitial fibrosis
- Unique to AL amyloidosis, circulating light chain toxicity is directly myotoxic (Clin Cardiol 2021;44:322)
Etiology
- Acquired or hereditary
- Wild type ATTR - precursor protein is structurally normal transthyretin (TTR)
- Mechanism of pathogenic deposition of this normally structured protein is unclear
- Hereditary ATTR - precursor protein is mutated TTR (point mutation)
- Mutated transthyretin tetrads are unstable, misfolded and prone to deposit
- Most common variants associated with cardiac involvement are Val122Ile (most common in the U.S.), Val130Met (most common globally) and Thr60A1a
- Light chain amyloidosis - precursor protein is misfolded immunoglobulin light chains
- Associated with plasma cell dyscrasias (monoclonal gammopathy of uncertain significance [MGUS], multiple myeloma, B cell lymphoma, others)
- Wild type ATTR - precursor protein is structurally normal transthyretin (TTR)
- References: Clin Cardiol 2021;44:322, UpToDate: Cardiac Amyloidosis [Accessed 16 November 2023]
Clinical features
- Presents as restrictive cardiomyopathy / diastolic dysfunction (HFpEF) with right heart failure
- Patients with symptoms of dyspnea, lower extremity edema, hepatic congestion, ascites, presyncope or syncope
- ATTRwt
- More conduction issues than hATTR, such as atrial fibrillation, first and second degree heart blocks
- Typically an isolated cardiomyopathy (J Am Coll Cardiol 2016;68:1323)
- Can be associated with carpal tunnel syndrome or lumbar spinal stenosis (ESC Heart Fail 2019;6:1128)
- hATTR
- Atrial and ventricular arrhythmias, heart blocks
- Val30Met variant often requires pacemaker (Circulation 2020;142:e7)
- Cardiac predominant, neuropathy predominant or mixed
- Can be associated with neuropathic, renal, ophthalmologic or musculoskeletal (MSK) involvement (Clin Cardiol 2021;44:322)
- AL amyloidosis
- 50 - 70% of systemic AL amyloidosis cases have cardiac infiltration and this is the main prognostic determinant
- Right sided HFpEF more severe than ATTR types (Curr Oncol Rep 2017;19:46)
- Usually sinus rhythm but can also have arrhythmias and heart blocks
- Vascular involvement is not uncommon
- Multiorgan complex: nephrotic syndrome, hepatic, neuropathic, MSK, macroglossia (Clin Cardiol 2021;44:322)
Diagnosis
- General approach: EKG, echocardiogram, cardiac MRI (CMR), serum studies (see Laboratory)
- If monoclonal protein is identified, then tissue biopsy is pursued
- AL amyloidosis
- Noncardiac biopsy with amyloid of AL type and CMR consistent with cardiac amyloidosis is sufficient to diagnose the majority of cardiac AL amyloidosis cases
- Negative findings on tissue biopsy of noncardiac organs do not rule out cardiac amyloidosis
- ATTR amyloidosis
- May or may not have monoclonal protein identified (can be present in up to 40% of ATTR cases) (Clin Cardiol 2021;44:322)
- Technetium pyrophosphate scintigraphy (PYP scan): cardiac nuclear imaging to detect cardiac transthyretin
- Endomyocardial biopsy, followed by immunohistochemistry or mass spectrometry to type the specific amyloid protein
- Genetic testing after ATTR is proven via positive scintigraphy or cardiac biopsy to differentiate between ATTRwt and hATTR
- In cases of hATTR, it may be recommended for other family members to consider genetic testing
Laboratory
- AL specific biomarkers
- Identification of a monoclonal protein via serum and urinary free light chain (FLC) measurements and immunofixation electrophoresis (IFE)
- Serum kappa / lambda FLC ratio analysis
- 90% of untreated AL cases have abnormal ratio (< 0.26 or > 1.65) (J Am Coll Cardiol 2016;68:1323)
- Note: FLC and IFE are sometimes raised in ATTRwt and in cases of MGUS, hence not specific markers of AL
- Nonspecific serum biomarkers
- B type natriuretic peptide (BNP) and N terminal proBNP (NT-proBNP) disproportionately high in cardiac amyloidosis due to direct compression of cardiomyocytes and stress caused by raised filling pressures (J Am Coll Cardiol 2016;68:1323)
- Cardiac troponin T (cTnT) reflects cardiomyocyte death and is useful as a negative prognostic indicator (BMC Cardiovasc Disord 2018;18:221)
Radiology description
- Echocardiogram: relative apical sparing of longitudinal strain, ventricular hypertrophy, biatrial dilation, thickened cardiac valves, diastolic > systolic dysfunction
- CMR: diffuse subendocardial pattern of late gadolinium enhancement (LGE) is pathognomonic (95% specificity), although transmural LGE is most common (Clin Cardiol 2021;44:322)
- Diffuse transmural: ATTR > AL
- Subendocardial: AL > ATTR
- Technetium pyrophosphate scintigraphy (PYP scan): cardiac nuclear imaging study that detects cardiac transthyretin can identify early deposition prior to onset of clinical disease and can prognosticate in some cases
Radiology images
Images hosted on other servers:
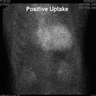
Biopsy proved ATTR cardiac amyloidosis with positive uptake
Prognostic factors
- Chronic and progressive condition
- AL amyloidosis: extent of cardiac involvement is most important predictor of survival
- ATTR amyloidosis: heart to contralateral (H/CL) ratio ≥ 1.6, calculated on PYP scintigraphy, is associated with significantly worse outcome over 5 years (Clin Cardiol 2021;44:322)
- Staging system incorporating NT-proBNP, cTnT and serum FLC (J Clin Oncol 2012;30:989)
- Stages I through IV, with higher stage conferring increased mortality
Case reports
- 50 year old woman diagnosed with AL amyloidosis demonstrated long term pathological response to chemotherapy and was able to withdraw from the heart transplant list (Eur Heart J Case Rep 2022;6:ytac130)
- 73 year old woman presented with cardiogenic shock and was subsequently diagnosed with cardiac amyloidosis with scintigraphy and endomyocardial biopsy (World J Clin Cases 2019;7:742)
- 84 year old man with new onset signs and symptoms of heart failure, diagnosed with wild type transthyretin amyloidosis (Cureus 2023;15:e33364)
Treatment
- AL amyloidosis: stem cell transplants, chemotherapy, proteasome inhibitors
- Chemotherapy care standard: cyclophosphamide, bortezomib and dexamethasone (CyBorD) and addition of daratumumab based on recent phase 3 study ANDROMEDA (Blood 2020;136:71)
- ATTR amyloidosis: goal is transthyretin stabilization and deposit removal
- Stabilizing: tafamidis and diflusinal
- Removal: doxycycline and tauroursodeoxycholic acid (Rev Esp Cardiol (Engl Ed) 2017;70:991)
- hATTR: gene silencer patisiran targets TTR mRNA to decrease liver production (Neurodegener Dis Manag 2019;9:5)
- Caution with traditional heart failure therapies
- Typical heart failure drugs such as ACEinh, ARBs, beta blockers, calcium channel blockers are detrimental in cardiac amyloidosis
- Low sodium diet, fluid restriction, loop diuretics and aldosterone inhibitors are recommended for volume management
- Transplant is the definitive treatment option
- AL amyloid: cardiac transplant
- ATTR amyloidosis: both cardiac and liver transplants needed
Gross description
- Amyloid deposits are most commonly seen in myocardium and also in atria, pericardium, endocardium and microvasculature, giving the myocardium a waxy pale appearance
- Myocardium is thickened, firm and rubbery in consistency
- Endocardial deposits may cause lining of heart to look gritty or sandpaper-like
- More than half of myocardium is involved in AL type
- Intracardiac thrombus formation is frequently seen
- Reference: J Clin Pathol 2005;58:125
Microscopic (histologic) description
- Amorphous and homogenous, with pale eosinophilic areas / hyaline deposits seen predominantly in the extracellular space
- Cardiac amyloidosis is a myocardial and a vascular / microvascular disease (Front Cardiovasc Med 2022;9:1081098)
- Myocardial interstitial patterns: 2 main patterns that can also be mixed
- Pericellular: amyloid distributed around individual cardiomyocytes, varied thickness and extent of involvement, produces lace-like aspect
- Nodular / replacement: nodular or micronodular aggregates distort architecture and replace myocardium
- Vascular / microvascular patterns
- Arteries and veins, epicardial and intramyocardial sites, more extensive involvement in mural vessels
- Deposits can partially or fully involve vessel circumference, can be localized to wall layers (intima, media) or involve full wall and can cause varying degrees of stenosis to the point of obstruction
- Capillary networks may show reduced density
- Deposits are also seen in subendocardium as nodular aggregates with or without fibrosis, as well as in the epicardial tissue (Front Cardiovasc Med 2022;9:1081098)
- Myocardial interstitial patterns: 2 main patterns that can also be mixed
- Disease specific patterns of deposition
- AL amyloidosis: usually diffuse pericellular infiltration with deposition in small blood vessels
- ATTR amyloidosis: usually nodular
- Atrophy of surrounding myocytes and fibrosis of the conduction system have been noted in relation to amyloid deposition
Microscopic (histologic) images
Contributed by Jaishree Jagirdar, M.D.
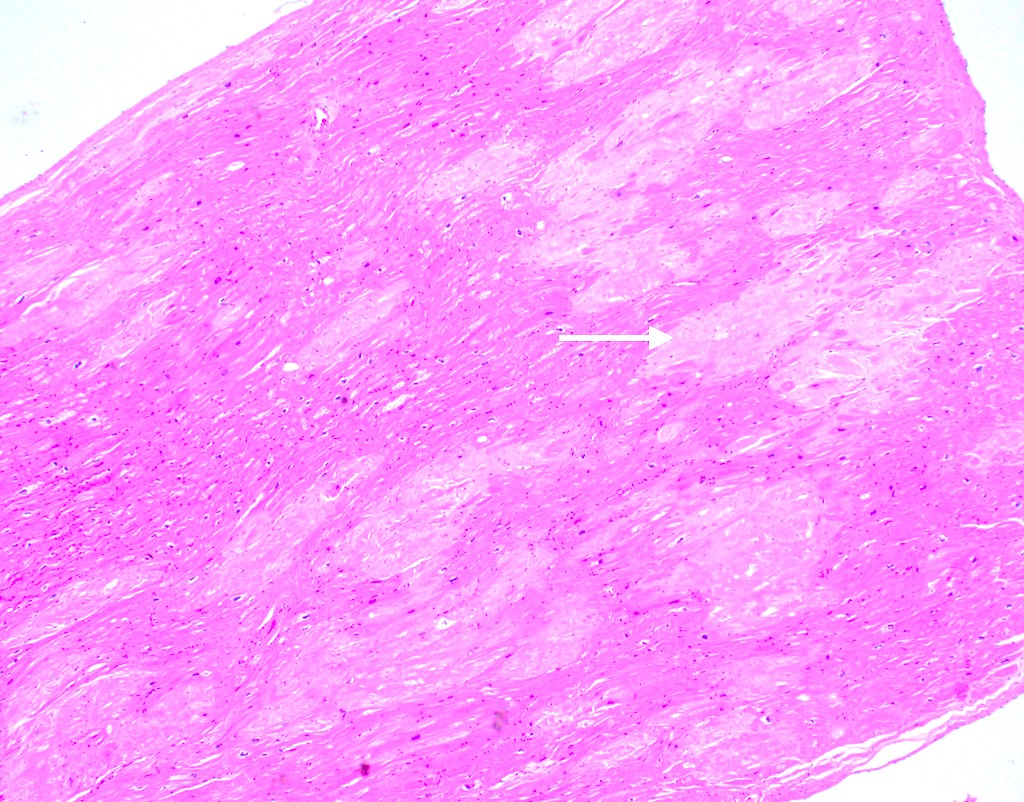
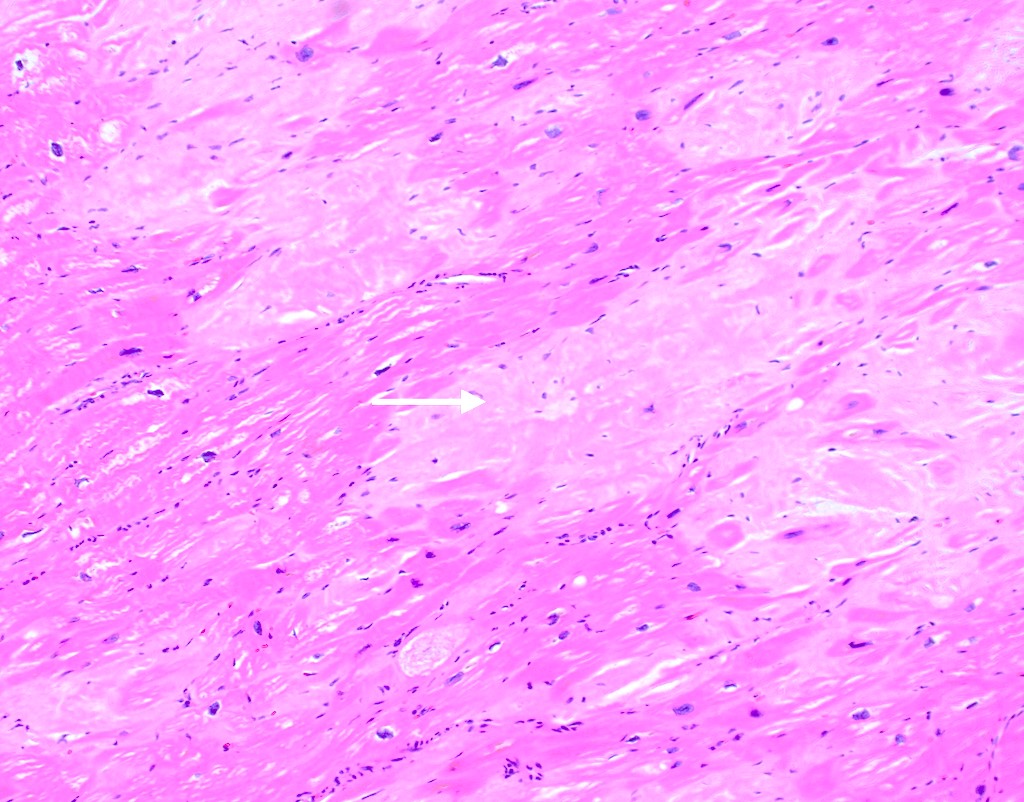
Amyloid deposition in myocardium
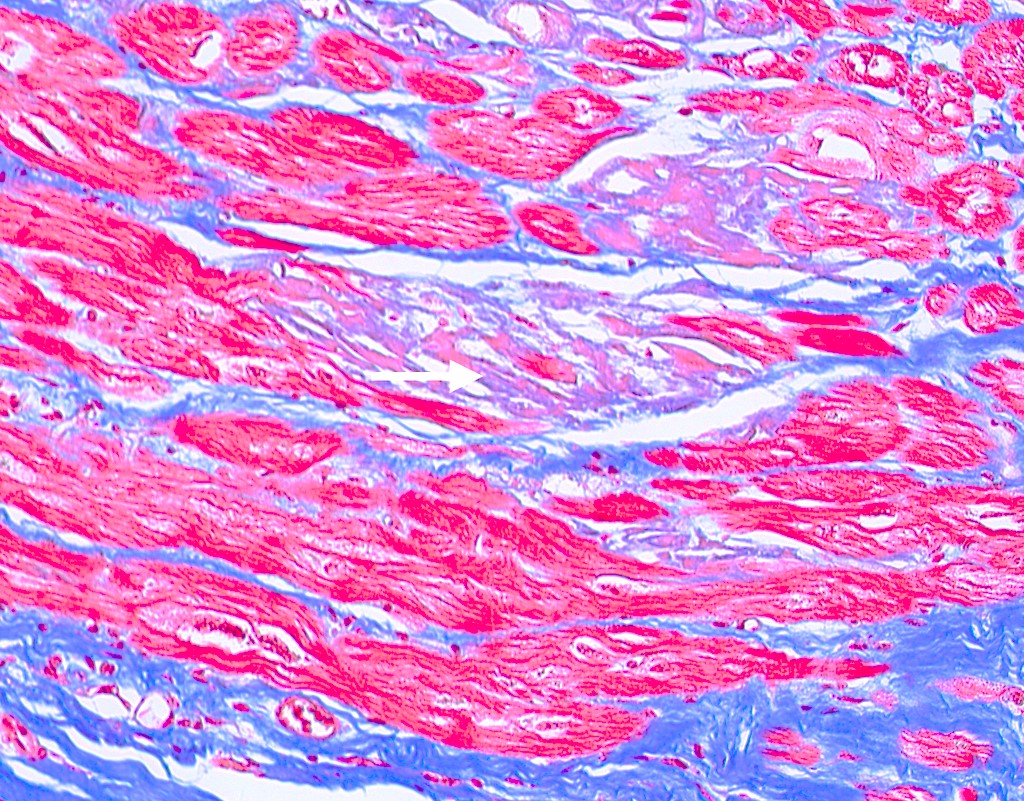
Trichrome stain
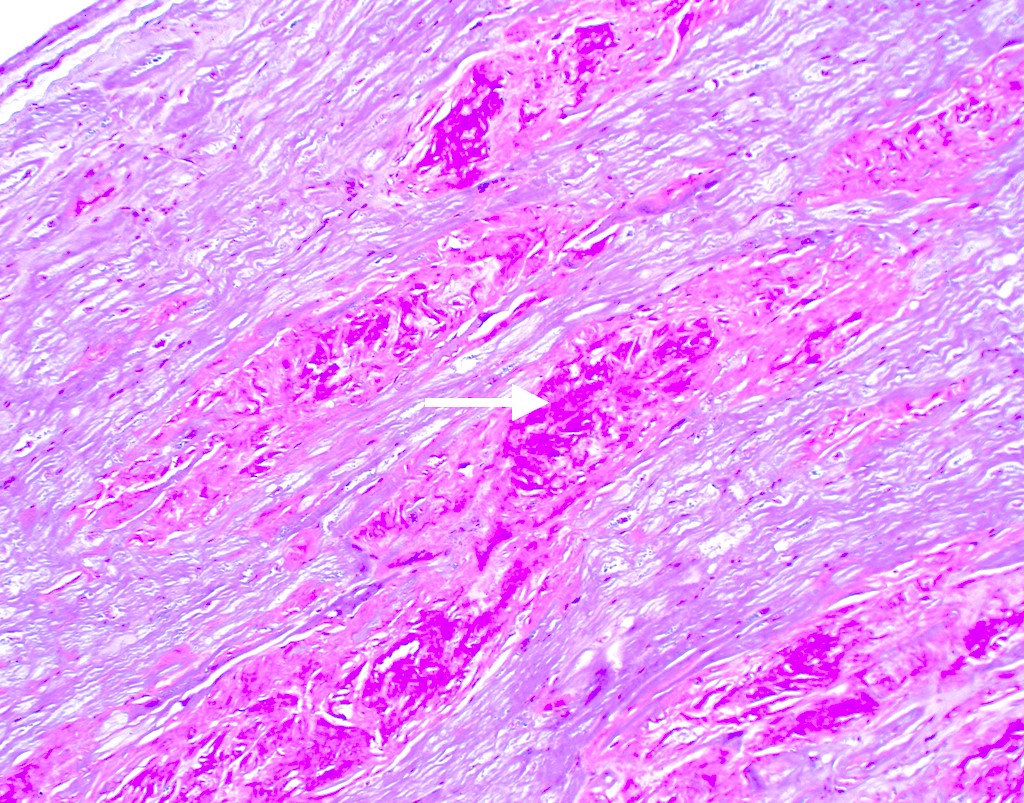
Crystal violet stain
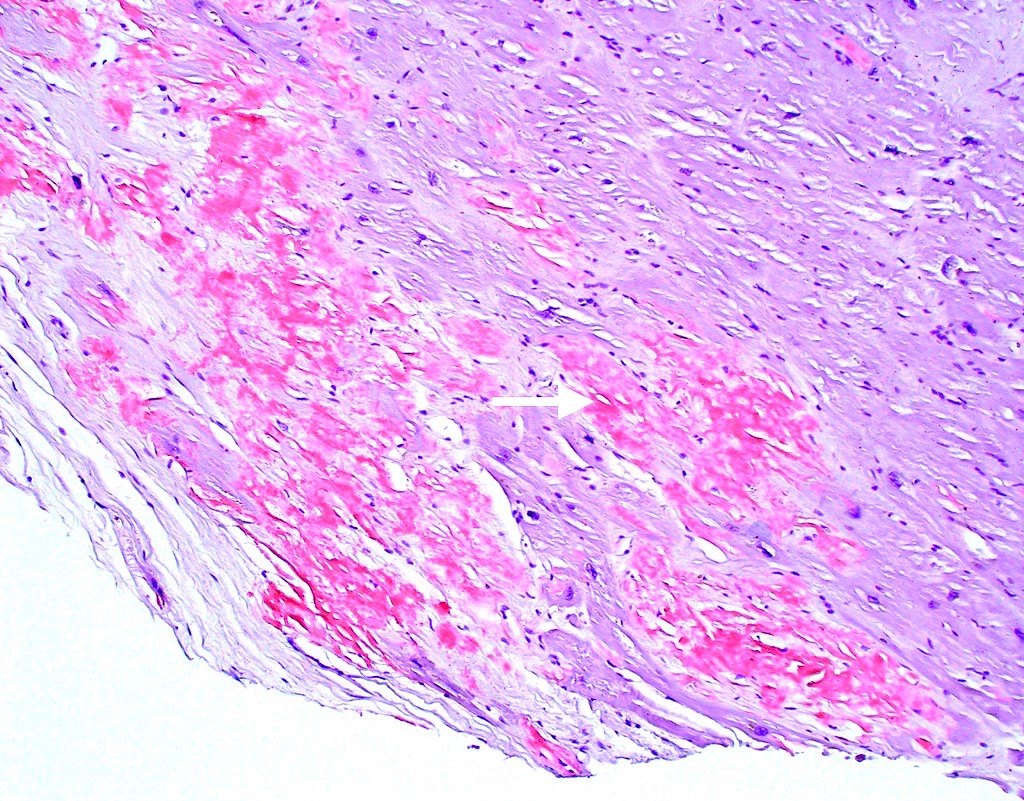
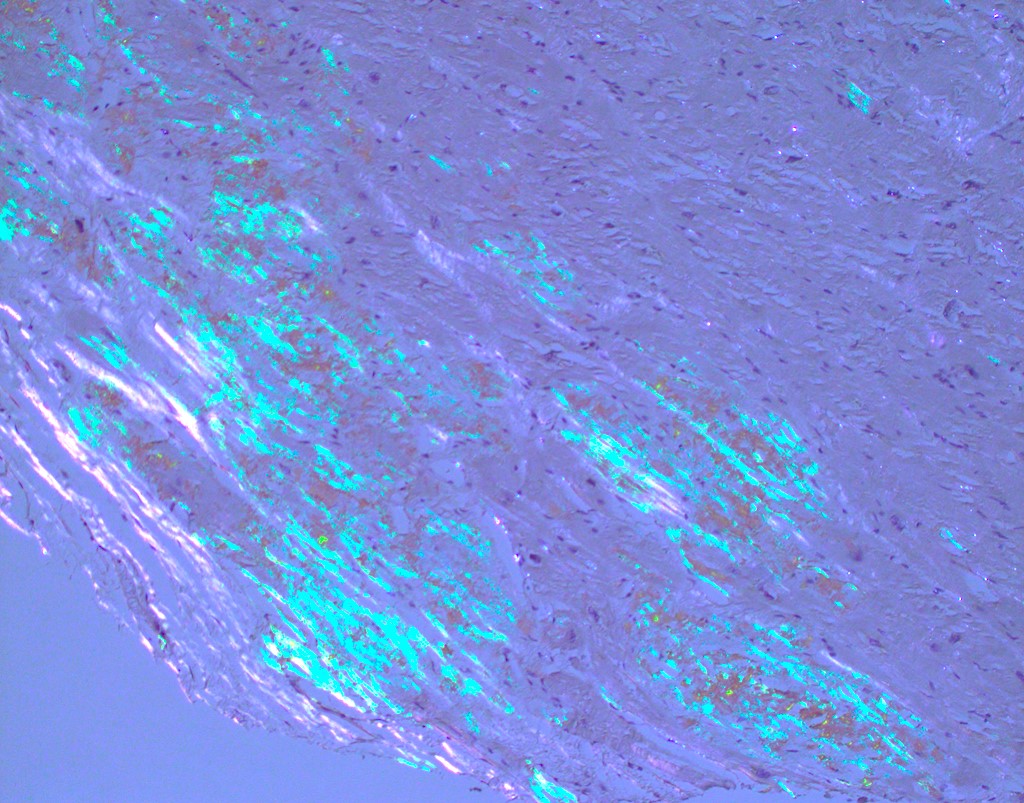
Congo red stain
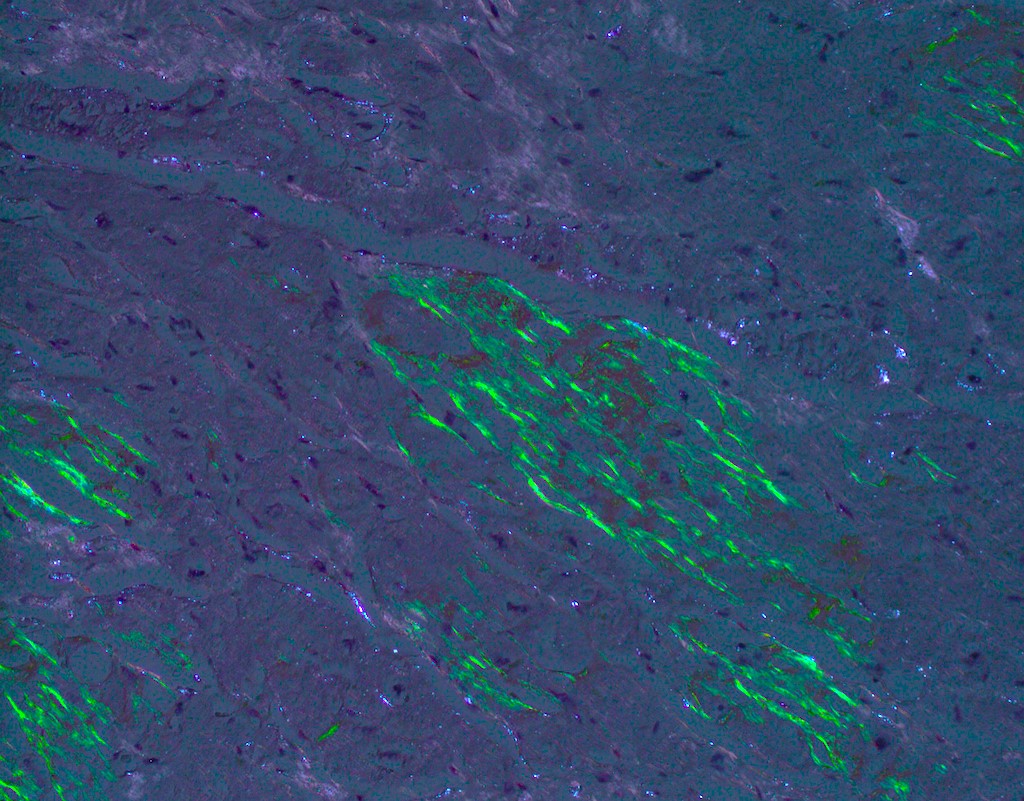
Congo red stain
Cytology description
- Limited value in routine smears
Immunofluorescence description
Positive stains
- Congo red on 8 - 10 micron sections: apple green birefringence under polarized light
- High sensitivity and specificity rate on endomyocardial biopsy
- Caveat: systemic AL amyloidosis may stain weakly or even negative
- Crystal violet: purple metachromatic staining
- Infrequently used - thioflavin T: yellow-green fluorescence
- Infrequently used - sulfated Alcian blue: amyloid stains a turquoise / green color
- Special note: pitfalls of Congo red staining
- Proper equipment is necessary to improve sensitivity (Diagn Pathol 2019;14:57)
- Use a mechanical rotating stage to view slides at variable angles
- Avoid plastic coverslips as they interfere with ability to perform crossed polarized light examination
- When a sample is deemed negative or equivocal, follow previously published modifications such as using polar mounting media or omitting the alcohol differentiation step
- Use proper stain free optics (such as a metallurgical microscope) and avoid use of polarizer with built in compensator
- Proper equipment is necessary to improve sensitivity (Diagn Pathol 2019;14:57)
Negative stains
- Trichrome: pale blue staining but no deep blue staining (differentiates from collagen deposition, which stains brilliant blue)
Electron microscopy description
- Electron microscopy is highly specific but has poor sensitivity due to the patchy nature of disease and magnification levels of electron microscopy
- Straight, unbranching fibrils 8 - 10 nm in width
- Composed of protofilaments at higher resolution
- In the myocardium, pericellular encasement nonbranching fibrils are found adjacent to the basement membrane; associated focal increase in mitochondria
- Can detect amyloid even when histochemical stains (Congo red) are negative (J Clin Pathol 2005;58:125)
Electron microscopy images
Contributed by Bradley Farris, M.D.
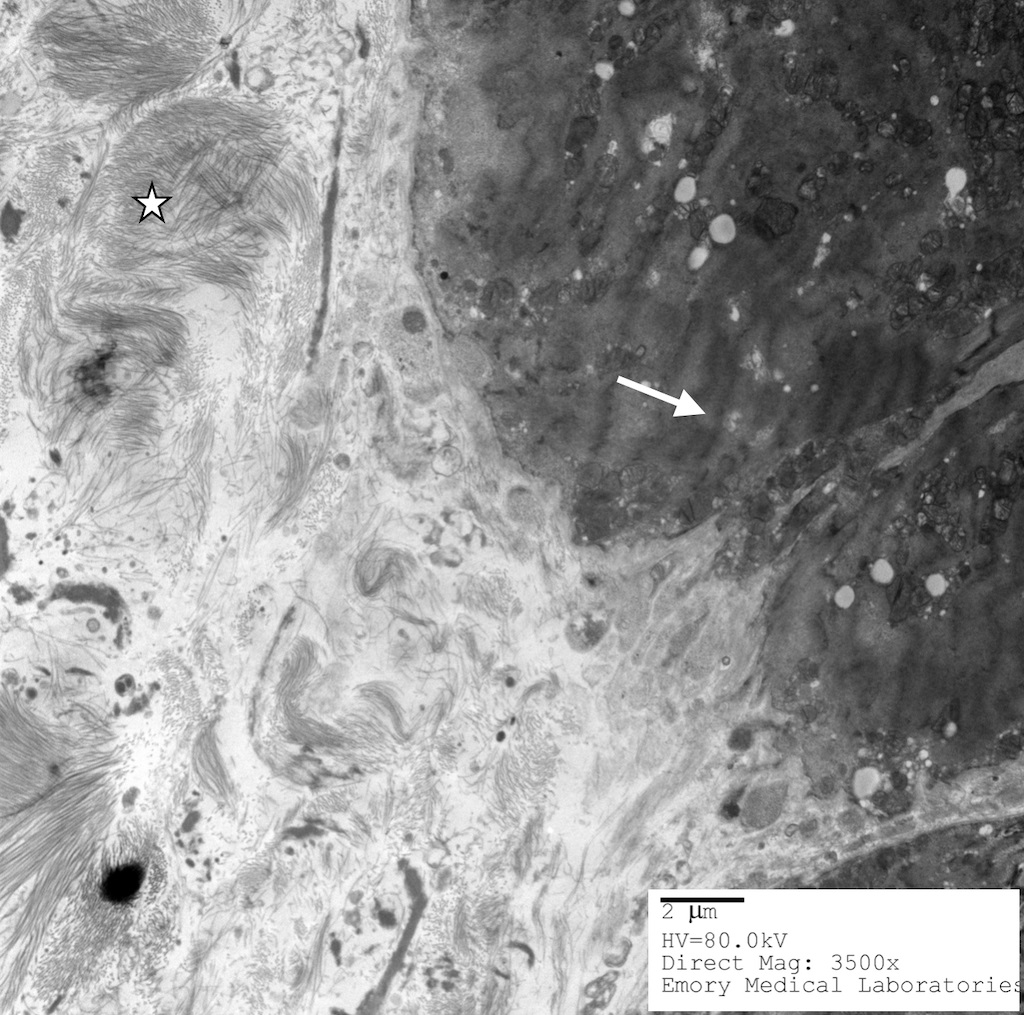

Amyloid deposition with beta pleated sheets
Molecular / cytogenetics description
- Mass spectrometry with laser microdissection - gold standard to identify precursor protein and amyloidosis type (even over immunohistochemistry, immunofluorescence and immunoelectron microscopy discussed above)
Sample pathology report
- Cardiac, core biopsy of left ventricular apex:
- Myocardium with extensive amyloid deposition in the interstitium with mild interstitial fibrosis
- Special studies: strongly positive on Congo red stain with apple green birefringence under polarized light; strongly positive crystal violet stain with purple metachromatic staining
Differential diagnosis
- Other disease processes that present with LV hypertrophy, such as
- Ischemic cardiomyopathy with fibrosis:
- Hypertrophic cardiomyopathy:
- Myocardium has a glistening leiomyoma-like appearance
- Myocytes display bizarre Y shaped forms into characteristic whorled appearance
- Often associated with family history of sudden cardiac death
- LVH secondary to hypertension:
- Most commonly concentric (less commonly eccentric) thickening of the left ventricular wall without outflow tract obstruction
- Boxcar nuclei on microscopy: enlarged nuclei with squaring of the nuclear edges
- Anderson-Fabry disease (Fabry disease):
- Rare
- Reduced concentration of serum alpha galactosidase A level or its activity is diagnostic
- Deficiency causes intracellular accumulation of galabiosylceramide and digalactosyl ceramide in myocardium (as well as in skin, vessels, kidney, eyes, ganglion cells)
- In prehypertrophy stage, the myocytes have perinuclear glycosphingolipid engorged vacuoles, which increase with extent of hypertrophy; this finally ends in necrosis and moderate fibrosis, intraluminal vessels are narrowed and thickened
Additional references
Practice question #1
A cardiac biopsy demonstrates negative Congo red staining and there is a well founded morphologic suspicion for cardiac amyloidosis. What is the next best step?
- Abdominal fat pad biopsy
- Crystal violet stain
- Provide the negative diagnosis and suggest alternative diagnoses
- Repeat Congo red stain on 2 more sections
Practice answer #1
D. Repeat Congo red (CR) stain on 2 more sections. Repeat the CR stain on 2 or more 8 - 10 micron sections in order to exclude technical problems. Use a rotating stage to evaluate various angles, avoid plastic coverslips and use proper optics (avoid polarizer with built in compensator). If CR stain is still negative, then proceed to crystal violet stain (answer B) (Diagn Pathol 2019;14:57). In practice, a repeat CR stain and crystal violet stain may be done simultaneously. Answer A is incorrect, as the patient has undergone a cardiac biopsy. An abdominal fat pad biopsy, even if positive for amyloidosis, is not diagnostic of cardiac amyloidosis. Answer C is incorrect because in a patient with a high index of suspicion and a negative CR stain, the stain should be double checked or repeated to control for possible technical errors.
Comment Here
Reference: Cardiac amyloidosis
Comment Here
Reference: Cardiac amyloidosis
Practice question #2
What is the gold standard to identify amyloid subtype and precursor protein?
- Electron microscopy
- Immunofluorescence studies
- Immunohistochemistry
- Mass spectrometry
Practice answer #2
D. Mass spectrometry. Mass spectrometry is the gold standard for identifying amyloid subtypes and precursor proteins. Answers A - C are incorrect as these methods are not the gold standard. Note that formalin fixed and paraffin embedded (FFPE) specimens can be used for a complete examination and characterizations (suitable for histologic, histochemical, immunohistochemical investigations, as well as molecular analysis). Frozen sections are only needed at centers that traditionally use immunofluorescence for amyloid typing (Front Cardiovasc Med 2022;9:1081098, Biomedicines 2022;10:3054).
Comment Here
Reference: Cardiac amyloidosis
Comment Here
Reference: Cardiac amyloidosis
