
Muscle & peripheral nerve nontumor
Muscle atrophy
Spinal muscular atrophy (SMA)
Editorial Board Members: Meaghan Morris, M.D., Ph.D., Jared T. Ahrendsen, M.D., Ph.D.


Last author update: 26 August 2025
Last staff update: 26 August 2025
Copyright: 2020-2025, PathologyOutlines.com, Inc.
PubMed Search: Spinal muscular atrophy

Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Diagrams / tables | Clinical features | Diagnosis | Laboratory | Radiology description | Prognostic factors | Case reports | Treatment | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Negative stains | Electron microscopy description | Sample pathology report | Differential diagnosis | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Elnagdy M, Cai C. Spinal muscular atrophy (SMA). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/musclespinal.html. Accessed August 26th, 2025.
Definition / general
- Spinal muscular atrophy (SMA) is a genetic neuromuscular disorder characterized by degeneration of alpha motor neurons in the anterior horn of the spinal cord, leading to progressive muscle weakness and atrophy
- It is caused primarily by homozygous deletions or mutations in the SMN1 gene, which encodes the survival motor neuron (SMN) protein, essential for motor neuron maintenance
Essential features
- Both the SMN1 and SMN2 genes are located on chromosome 5q13 and encode the survival motor neuron (SMN) protein
- SMN1 gene produces full length, functional SMN protein (FL-SMN)
- SMN2 gene differs by a single nucleotide (C to T) in exon 7, leading to the production of a less stable, truncated SMN protein (SMNΔ7) that lacks exon 7
- SMA results from biallelic loss of the SMN1 gene, preventing sufficient production of functional FL-SMN protein; the severity of SMA is modulated by the number of SMN2 gene copies a patient has; more copies of SMN2 generally correlate with a milder disease phenotype due to increased production of SMNΔ7 protein
- SMA is clinically classified into subtypes (0 - 4) based on the age of onset and the severity of symptoms
Terminology
- SMA type 0: prenatal onset, neonatal death
- SMA type 1 (Werdnig-Hoffmann syndrome): onset < 6 months, most severe
- SMA type 2: onset 6 - 18 months, can sit but not walk
- SMA type 3 (Kugelberg-Welander syndrome): onset > 18 months, can walk initially
- SMA type 4: adult onset, milder phenotype
ICD coding
Epidemiology
- Incidence: ~1 in 6,000 - 10,000 live births (Orphanet J Rare Dis 2017;12:124)
- Carrier frequency: ~1 in 40 - 60 in most populations (Eur J Hum Genet 2012;20:27)
- SMA type 1 accounts for ~60% of all SMA cases
- Equal distribution between males and females
Sites
- Primarily affects anterior horn motor neurons in the spinal cord and motor cranial nerve nuclei in the brainstem, leading to denervation and atrophy of skeletal muscles (Cell 1995;80:155)
Pathophysiology
- SMA is caused by deficiency of the SMN protein, which is crucial for the assembly of small nuclear ribonucleoproteins (snRNPs) (essential components of the spliceosomal machinery)
- SMN protein is widely expressed but is especially critical for the health and survival of lower motor neurons in the anterior horn of the spinal cord
- Humans have 2 nearly identical SMN genes on chromosome 5q13
- SMN1: telomeric primary gene encoding full length, functional SMN protein (FL-SMN)
- SMN2: centromeric paralog with a C>T transition in exon 7 that leads to exon 7 skipping in ~90% of transcripts, producing a truncated, unstable protein (SMNΔ7)
- Disease results from biallelic mutations or deletions in SMN1, leading to reduced FL-SMN protein levels
- Gene conversion from SMN1 to SMN2 accounts for an increased copy number of SMN2 in some SMA cases
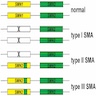
- SMN2 is the most important disease modifying gene; disease severity inversely correlated with SMN2 copy numbers (see diagram below)
- 1 - 2 copies → SMA type 1 (severe)
- 3 copies → SMA type 2
- 4 or more copies → SMA type 3 or 4 (milder phenotypes)
- Certain SMN2 mutations (e.g., c.859G>C and c.835-44A>G) produce more stable SMN transcripts that resulted in better than expected SMA subtypes. (e.g., 2 copies of SMN2 with mild SMA3 / 4 phenotypes) (Neurol Genet 2020;6:e530)
- Reference: Continuum (Minneap Minn) 2020;26:1348, Int J Mol Sci 2021;22:7896
Etiology
- Autosomal recessive genetic disorder due to SMN1 deletion or mutation
Clinical features
- Symmetric proximal muscle weakness (legs > arms)
- Hypotonia and areflexia
- It is classified based on age of onset and highest motor milestone achieved
- Type 0
- Onset at birth
- Severe hypotonia and respiratory failure
- Type 1
- Onset at < 6 months
- Never sits independently
- Respiratory weaknesses
- Type 2
- Onset at 6 - 18 months
- Sits but never walks
- Scoliosis
- Tongue fasciculations and joint contractures may also be observed
- Type 3
- Onset after 18 months to adolescence
- Patients are ambulatory initially but may lose ambulation
- Tremor, especially in the hands, is a distinguishing clinical feature in many patients
- Type 4
- Mild adult onset proximal weakness
- Type 0
- Cognition and sensation are typically normal
- Reference: J Child Neurol 2007;22:946
Diagnosis
- Genetic testing for biallelic SMN1 deletion or mutation is required for diagnosis
- Additional SMN2 copy number and mutation testing guides prognosis and treatment (Neuromuscul Disord 2018;28:208, Neurol Genet 2020;6:e530)
- SMA testing was added to the U.S. Federal recommended newborn screening panel in 2018
- Muscle biopsy is not required for diagnosis and characterized by infantile pattern denervation atrophy (Brain 1970;93:15)
Laboratory
- Creatine kinase (CK) is usually normal or mildly elevated
Radiology description
- Quantitative magnetic resonance imaging (MRI) can aid in selecting target muscles for biopsy, monitoring disease progression and treatment response (NMR Biomed 2020;33:e4357)
- Generalized atrophy and fatty infiltration of muscle
- More severe in proximal than distal muscles
- Fatty infiltration is more prominent in the anterior compartments, especially in the vastus muscles
Prognostic factors
- Prognosis is closely related to age of onset and SMN2 copy number
- Type 1: worst prognosis if untreated (death in < 2 years)
- Type 2 - 4: better outcomes with supportive care and disease modifying therapies
- U.S. Food and Drug Administration (FDA) approved therapies (e.g., nusinersen, onasemnogene abeparvovec, risdiplam) improve survival and function, especially when started early
- References: Neurol Genet 2020;6:e530, Int J Mol Sci 2023;24:11939
Case reports
- 1 year old girl with delayed diagnosis of spinal muscular atrophy type 1 (Clin Case Rep 2024;12:e8513)
- 6 and 7 year old boys with spinal muscular atrophy with progressive myoclonic epilepsy (SMA PME) (Ital J Pediatr 2023;49:64)
- 47 year old woman with late diagnosed SMA presenting with progressive muscle weakness and anemia (Cureus 2024;16:e59786)
Treatment
- Early treatment is critical; FDA approved therapies include
- Nusinersen (Spinraza): antisense oligonucleotide that increases SMN2 exon 7 inclusion (N Engl J Med 2017;377:1723)
- Onasemnogene abeparvovec (Zolgensma): AAV9 vector gene therapy delivering SMN1 (Med Lett Drugs Ther 2019;61:113)
- Risdiplam (Evrysdi): oral SMN2 pre-mRNA splicing modifier (J Neurol 2023;270:2531)
- Supportive care includes
- Respiratory support (BiPAP, cough assist)
- Nutritional support (feeding tubes if needed)
- Physical therapy and orthopedic management
- Reference: Lancet Neurol 2024;23:205
Microscopic (histologic) description
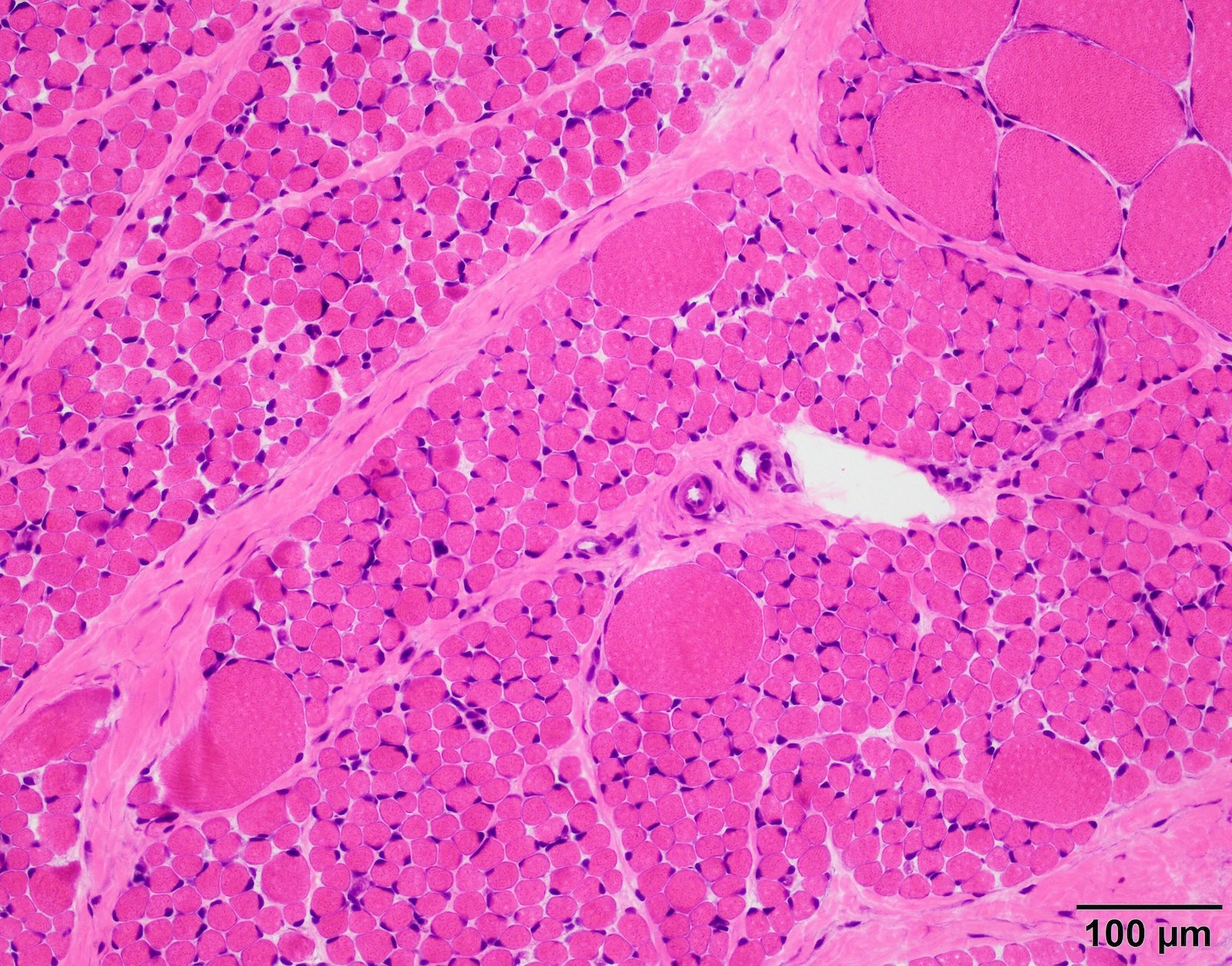
- SMA type 1 and 2: infantile pattern denervation atrophy
- Prominently hypertrophied fibers are exclusively type I fibers
- Small atrophic fibers are mixed type I and type II, mostly round rather than angulated
- Fiber type grouping
- Muscle spindles may be numerous
- Pyknotic nuclear clumps are rare
- SMA type 1 and 2
- May show typical neurogenic changes
- Fiber type grouping
- Group atrophy
- May show chronic changes difficult to differentiate from muscular dystrophy
- May show typical neurogenic changes
- References: Dubowitz: Muscle Biopsy - A Practical Approach, 5th Edition, 2020, WU Neuromuscular: Spinal Muscular Atrophy (5q) [Accessed 6 August 2025], Brain 1970;93:15
Microscopic (histologic) images
Contributed by Chunyu Cai, M.D., Ph.D.
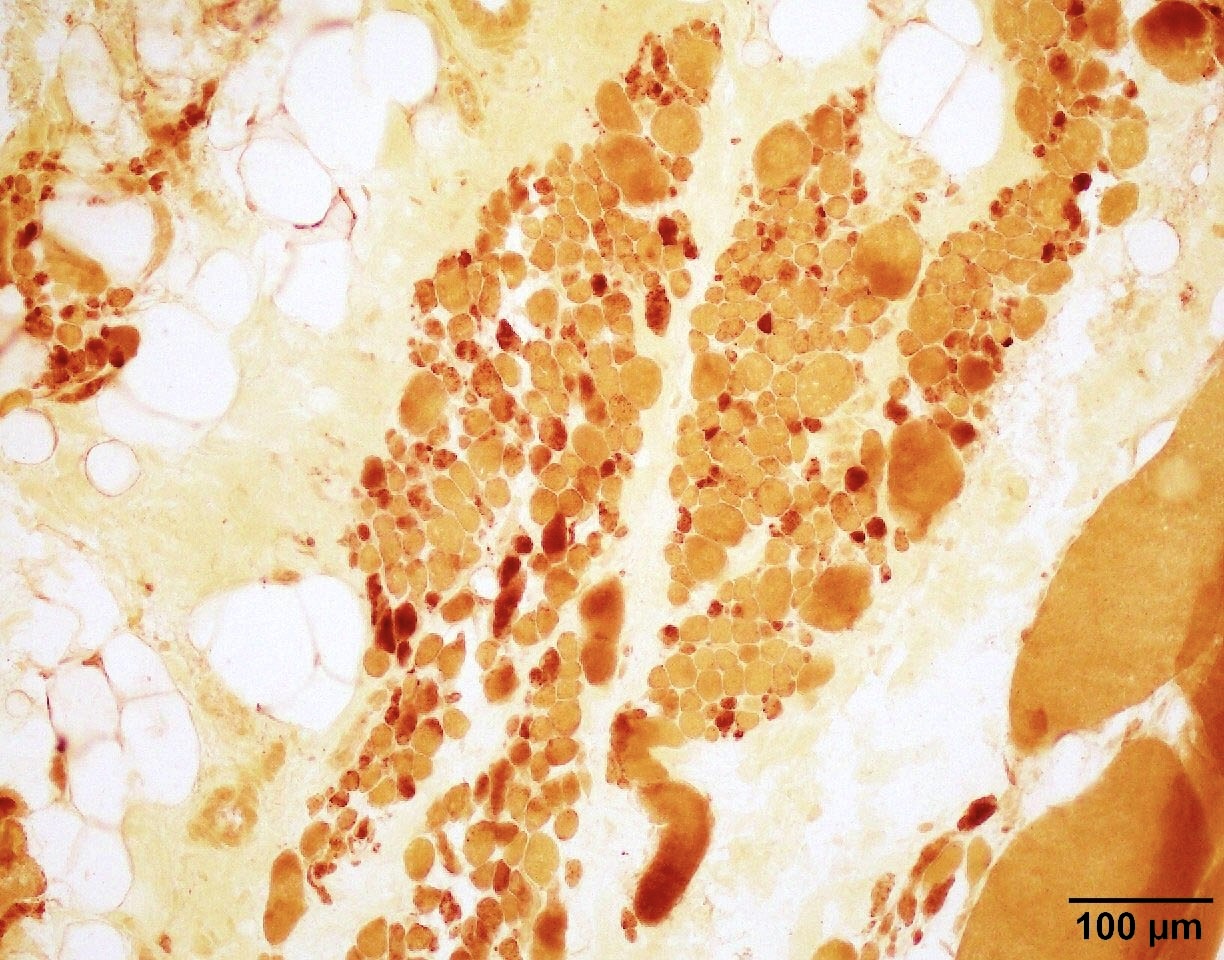
SMA type 1: 2 month old boy with failure to thrive. Genetic analysis showed homozygous deletions of SMN1 exons 7 and 8. Muscle biopsy was performed at 2 months of age. Patient died from respiratory failure at 4 months of age.
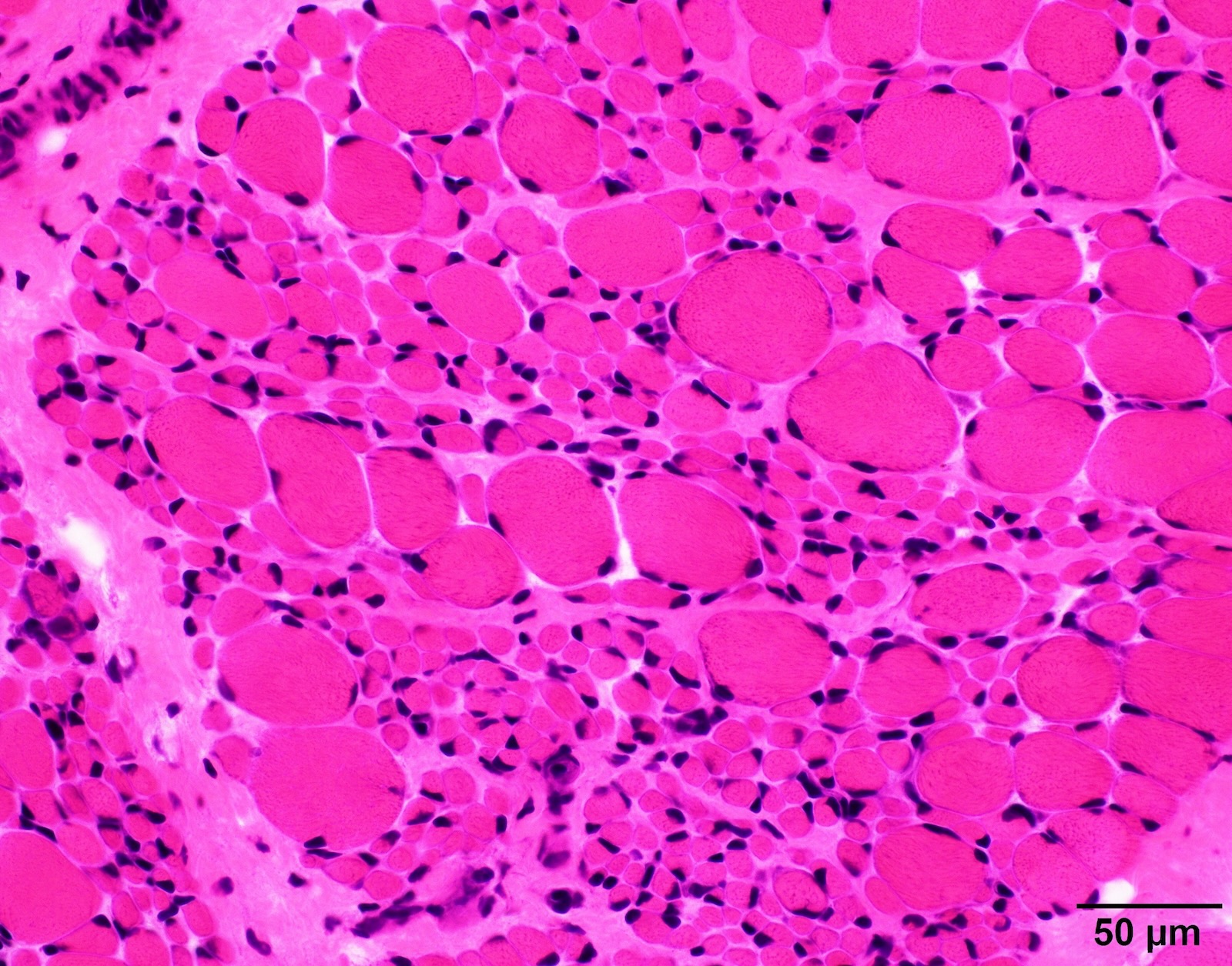
Infantile denervation atrophy
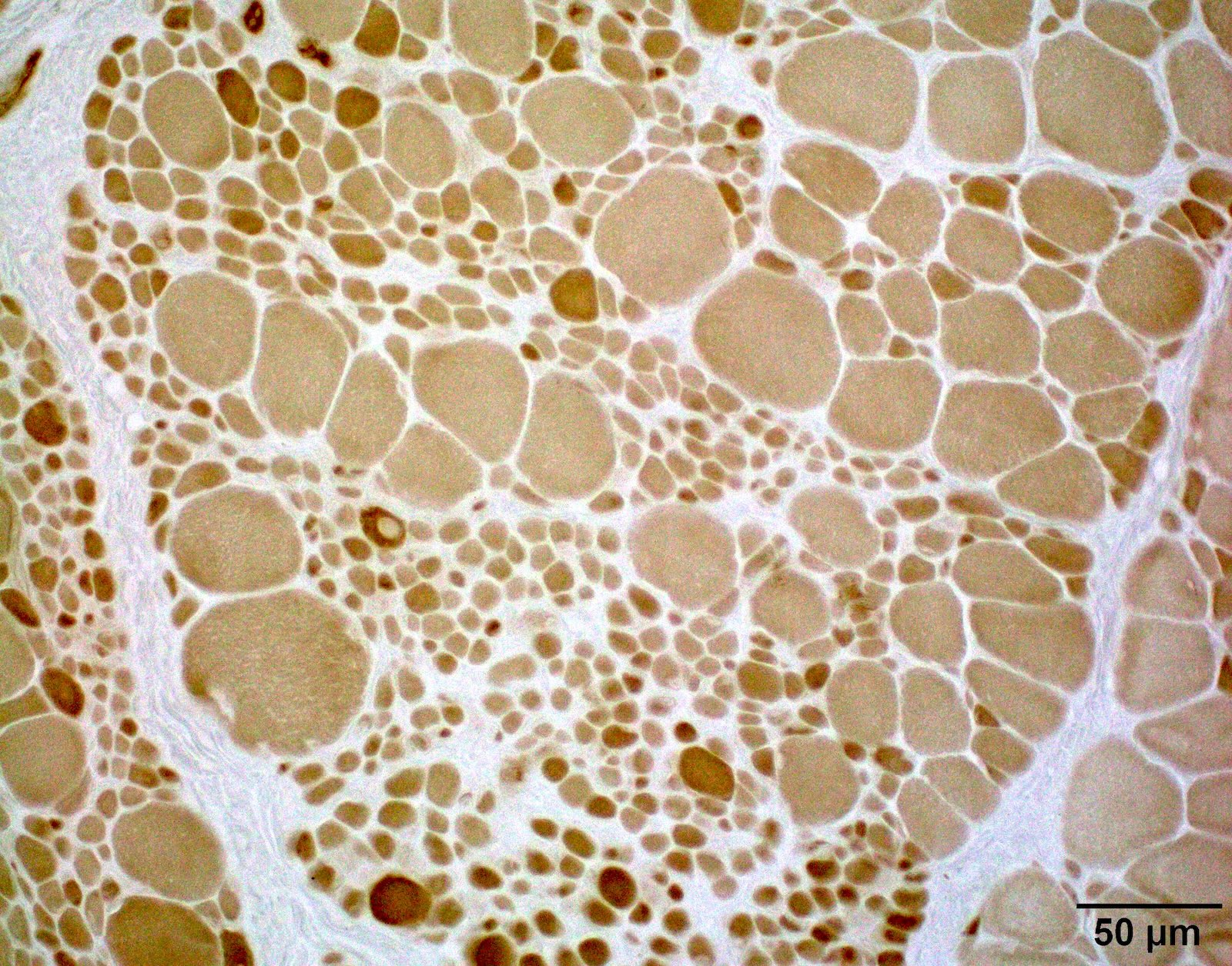
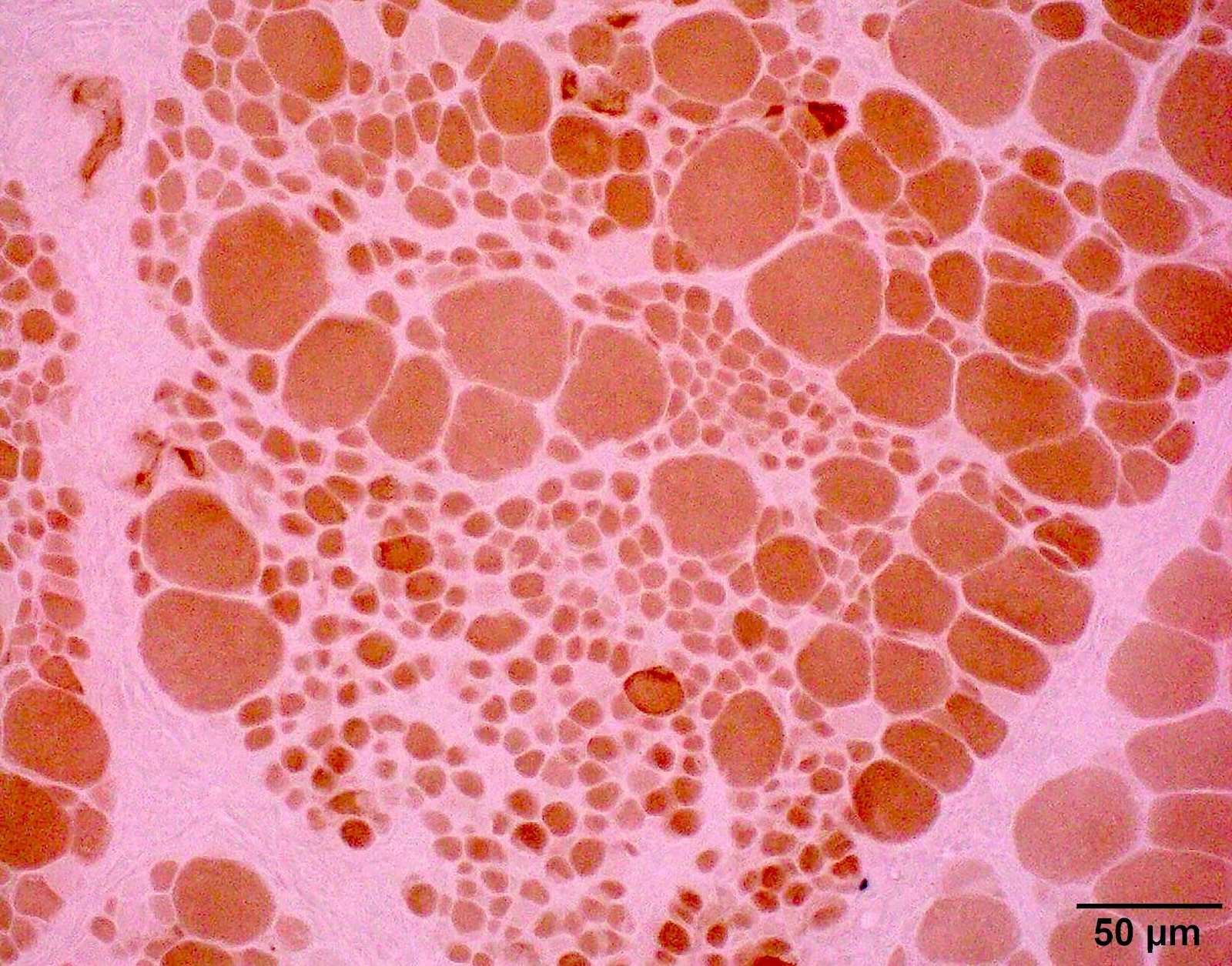
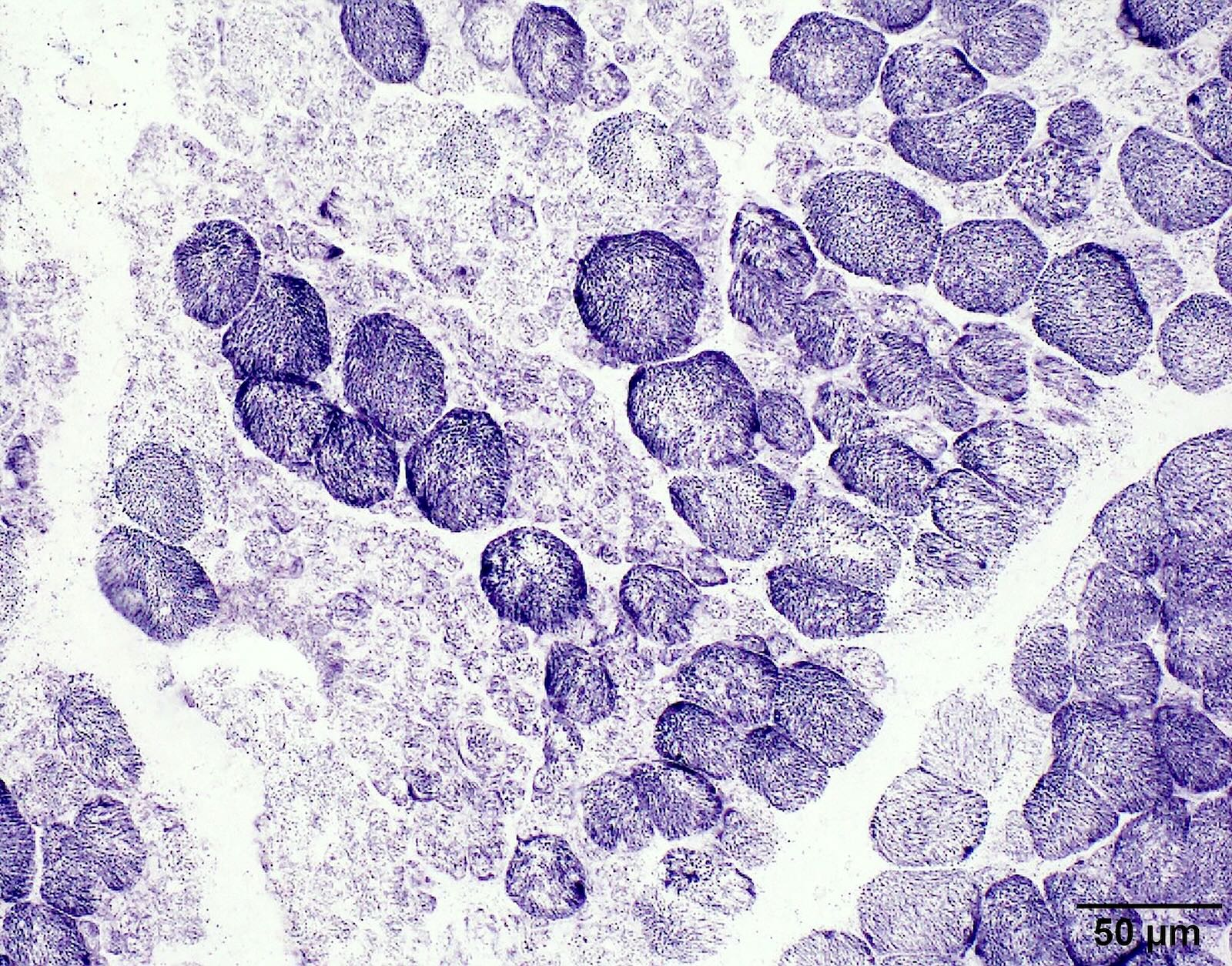
Hypertrophic fibers type I
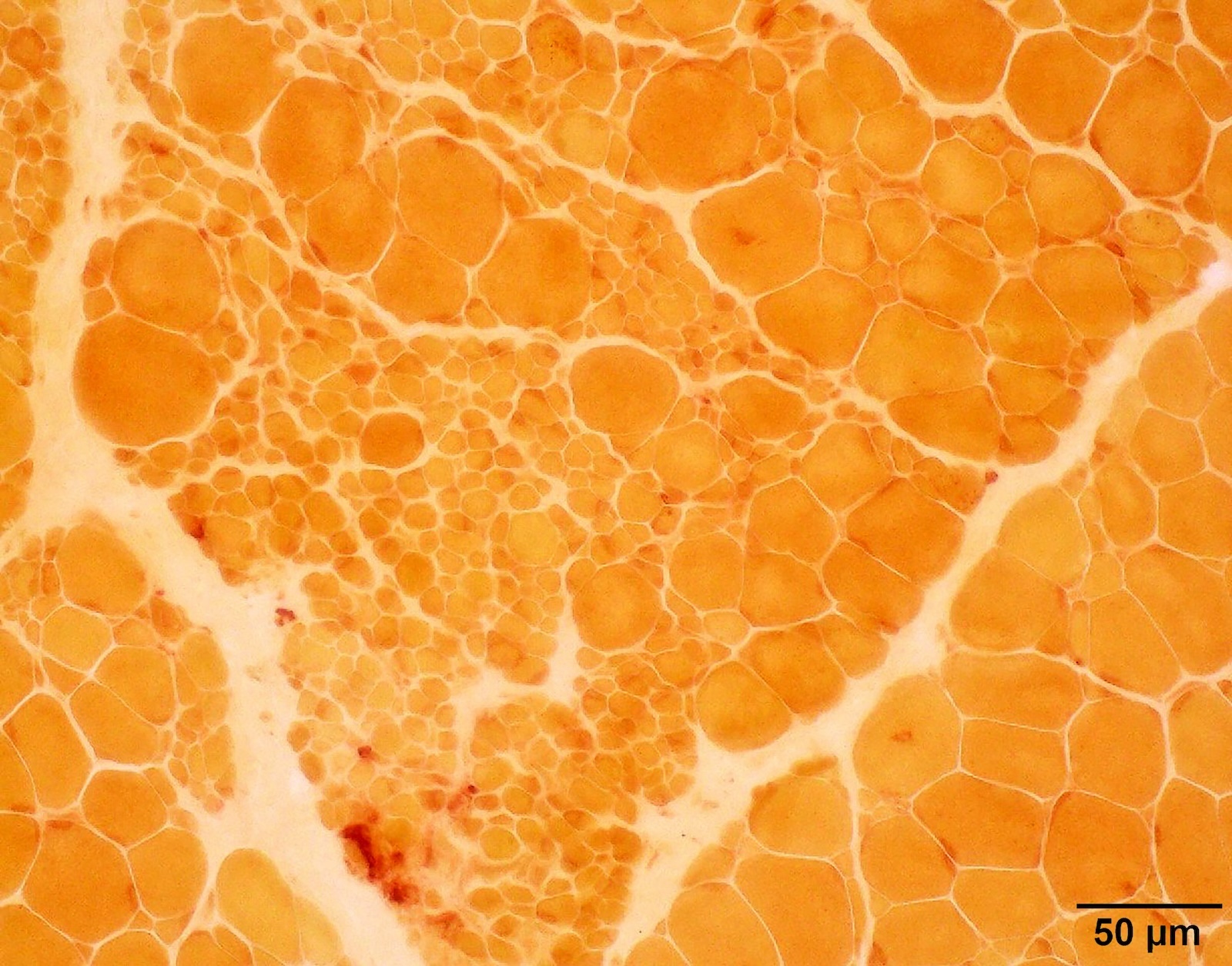
Esterase not increased
Intramuscular nerve
SDH
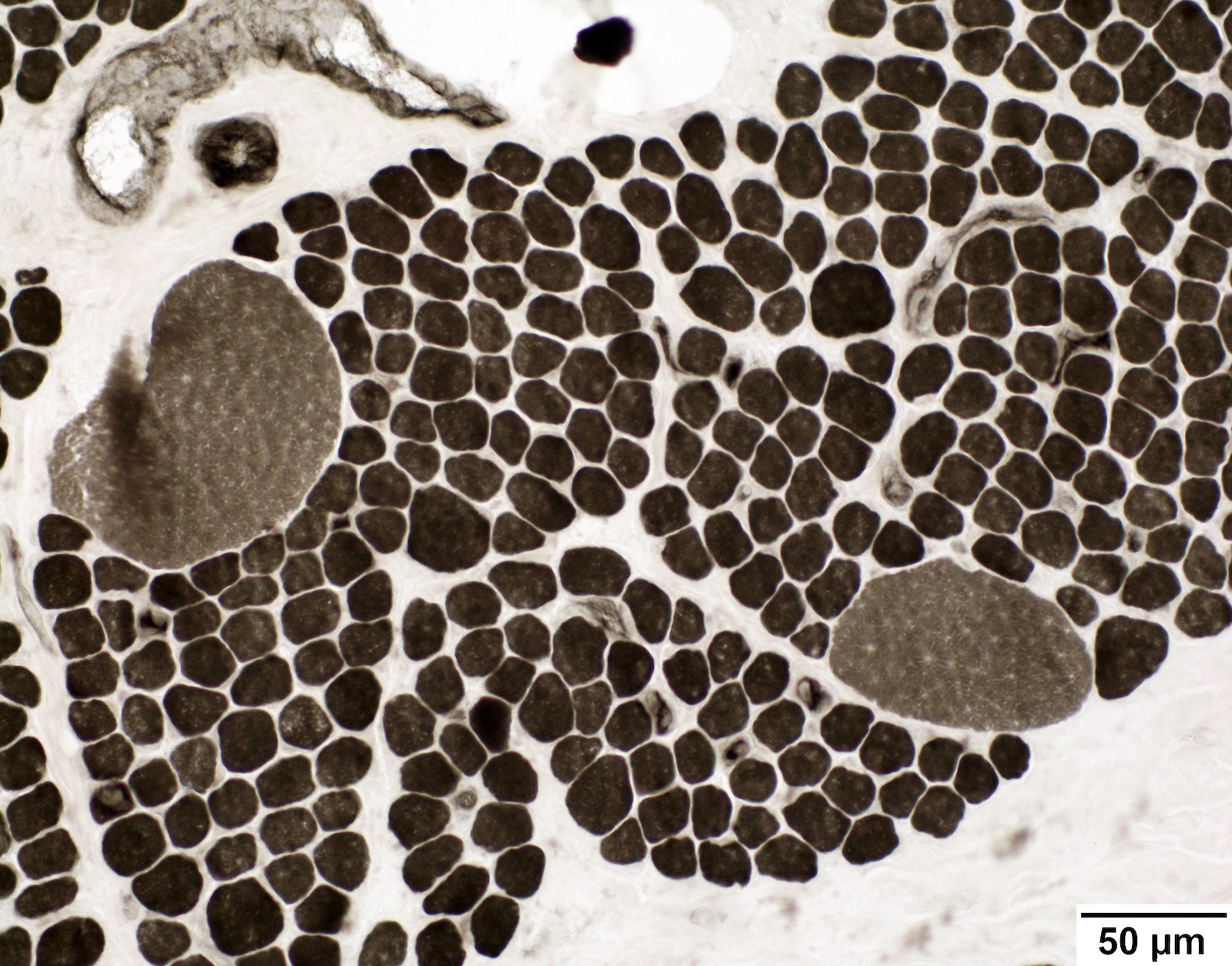
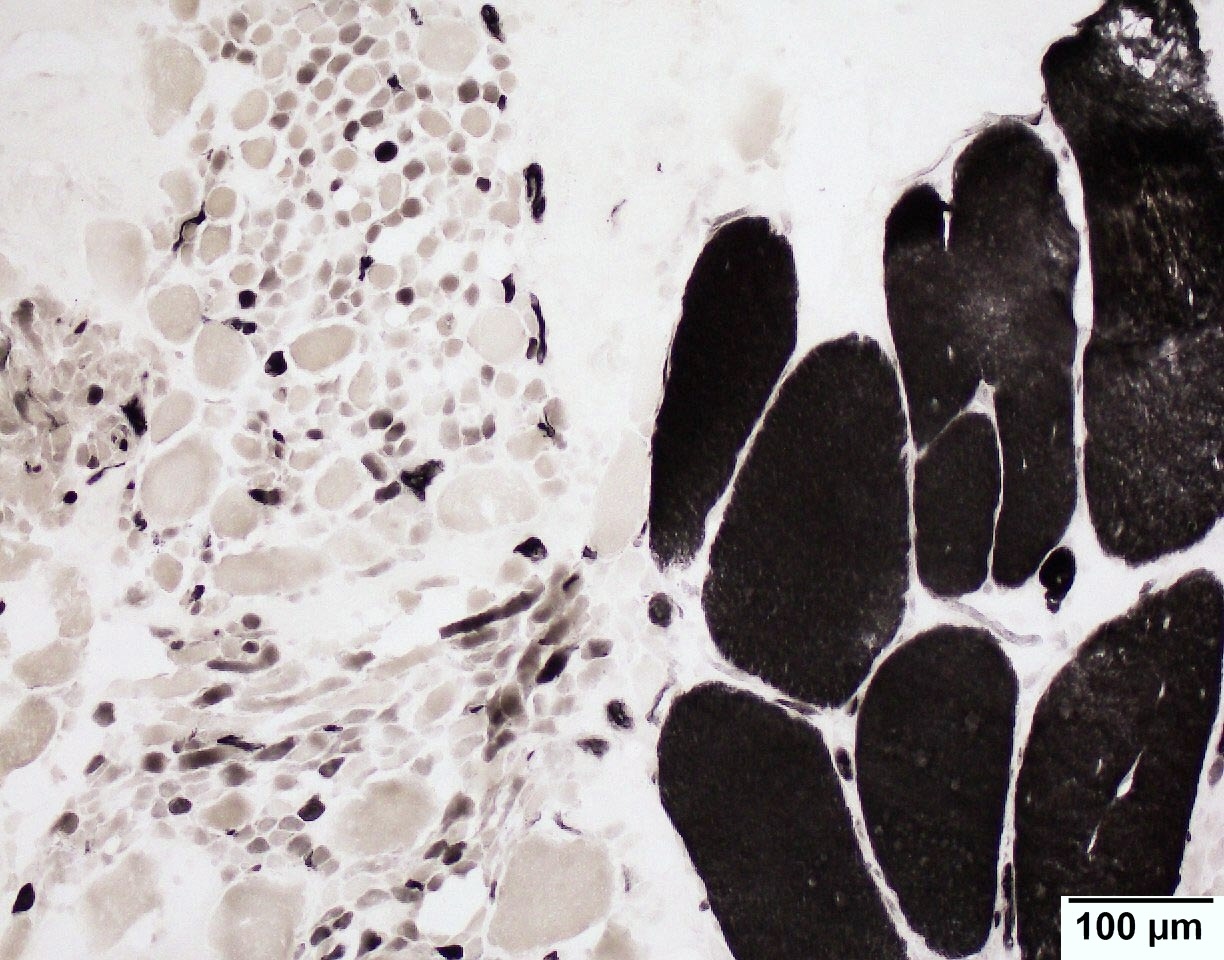
SMA type 2: 2 year old girl with congenital hypotonia and developmental delay. Genetic analysis showed homozygous deletions of SMN1 and 3 copies of SMN2.
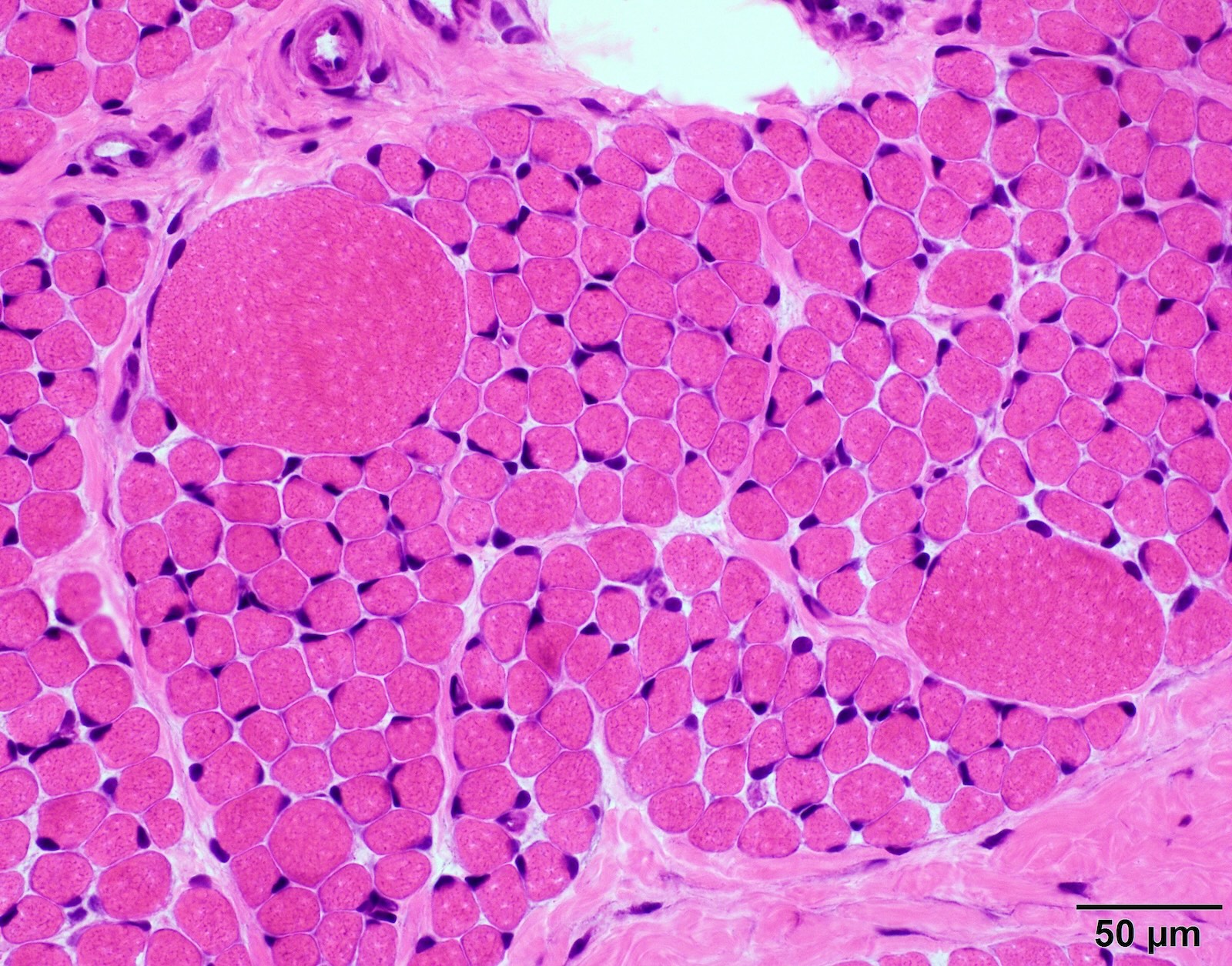
Infantile denervation atrophy
Fiber type grouping
Mixed small atrophic fibers
Hypertrophic fibers are type I
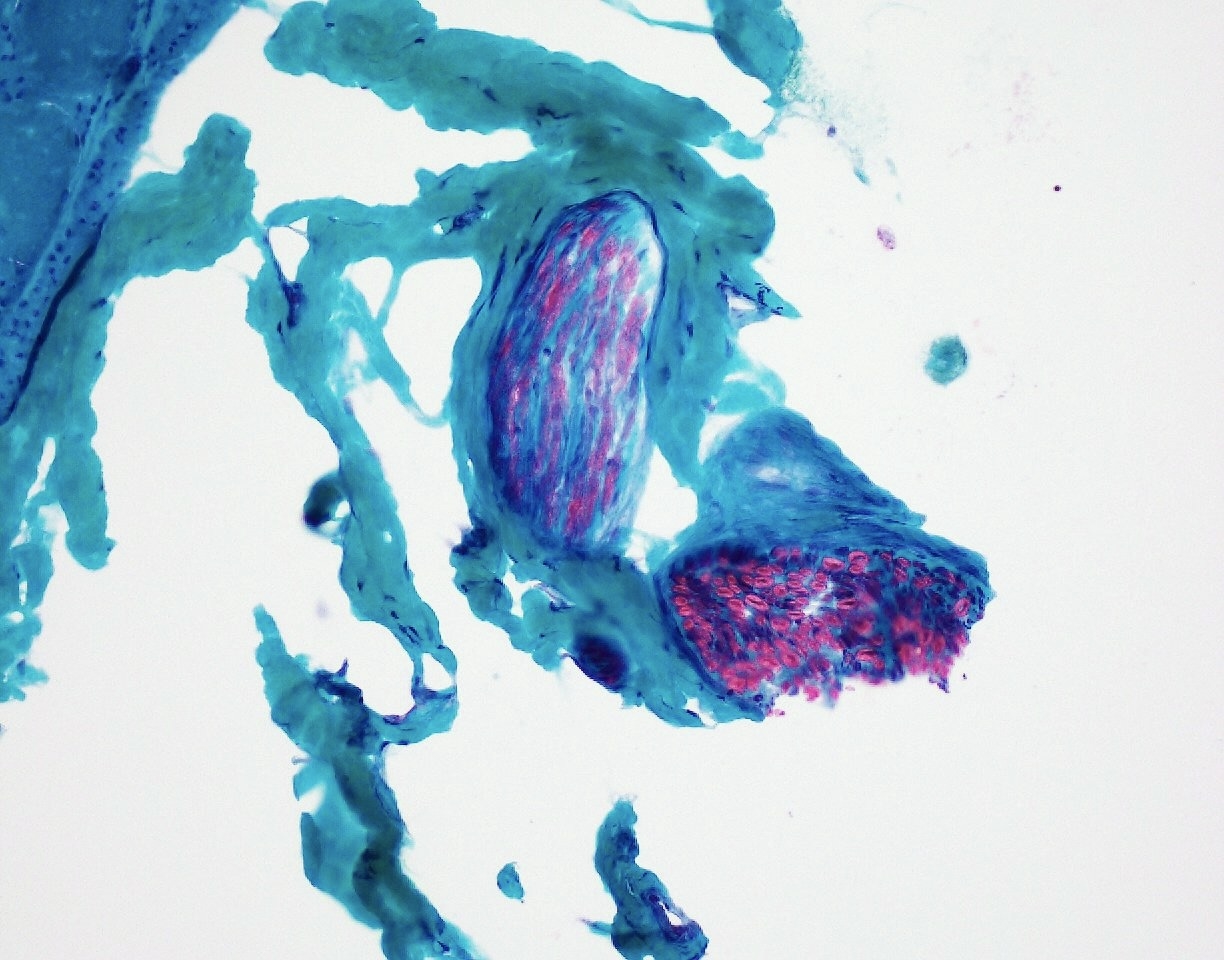
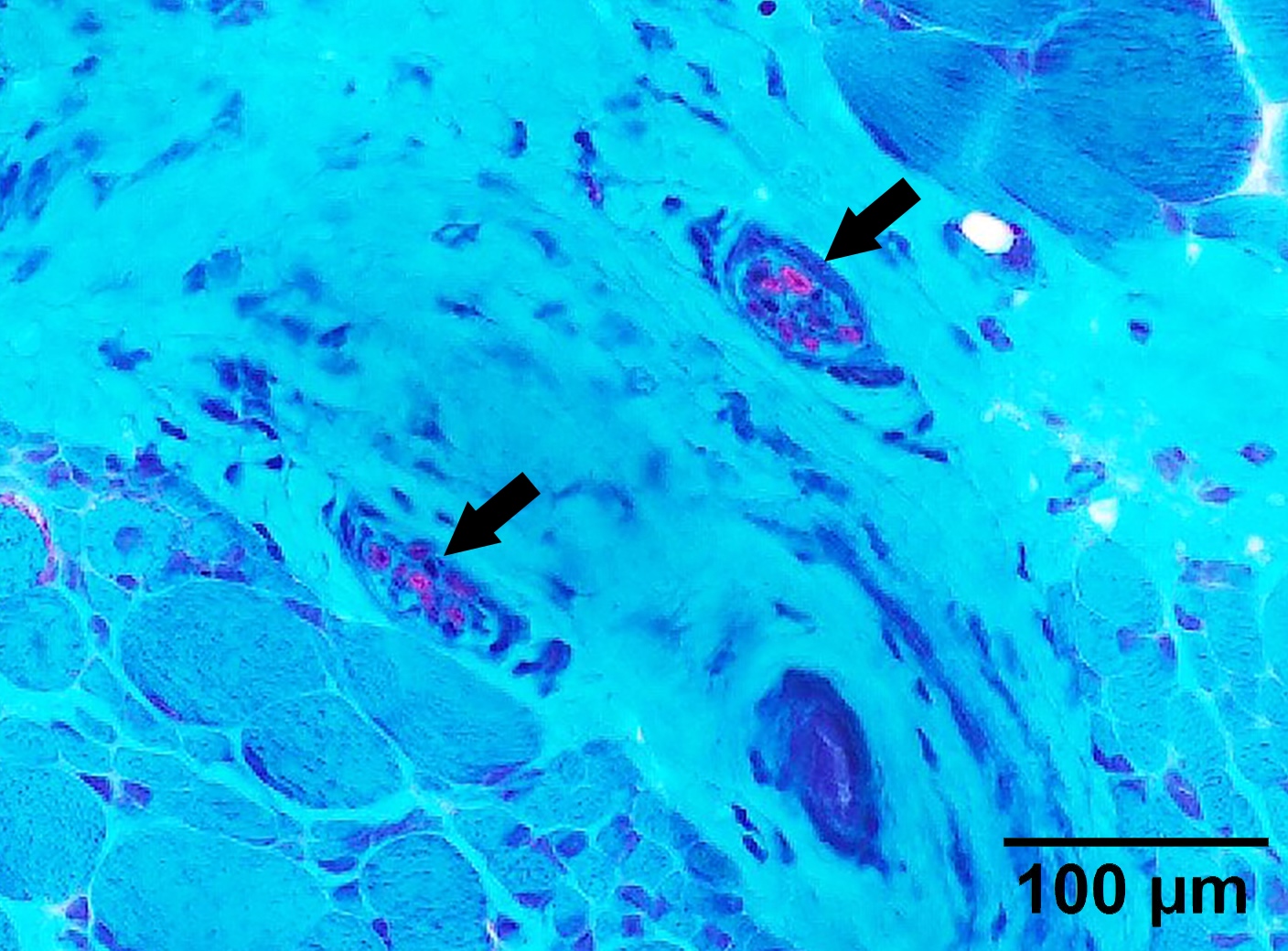
Intramuscular nerve
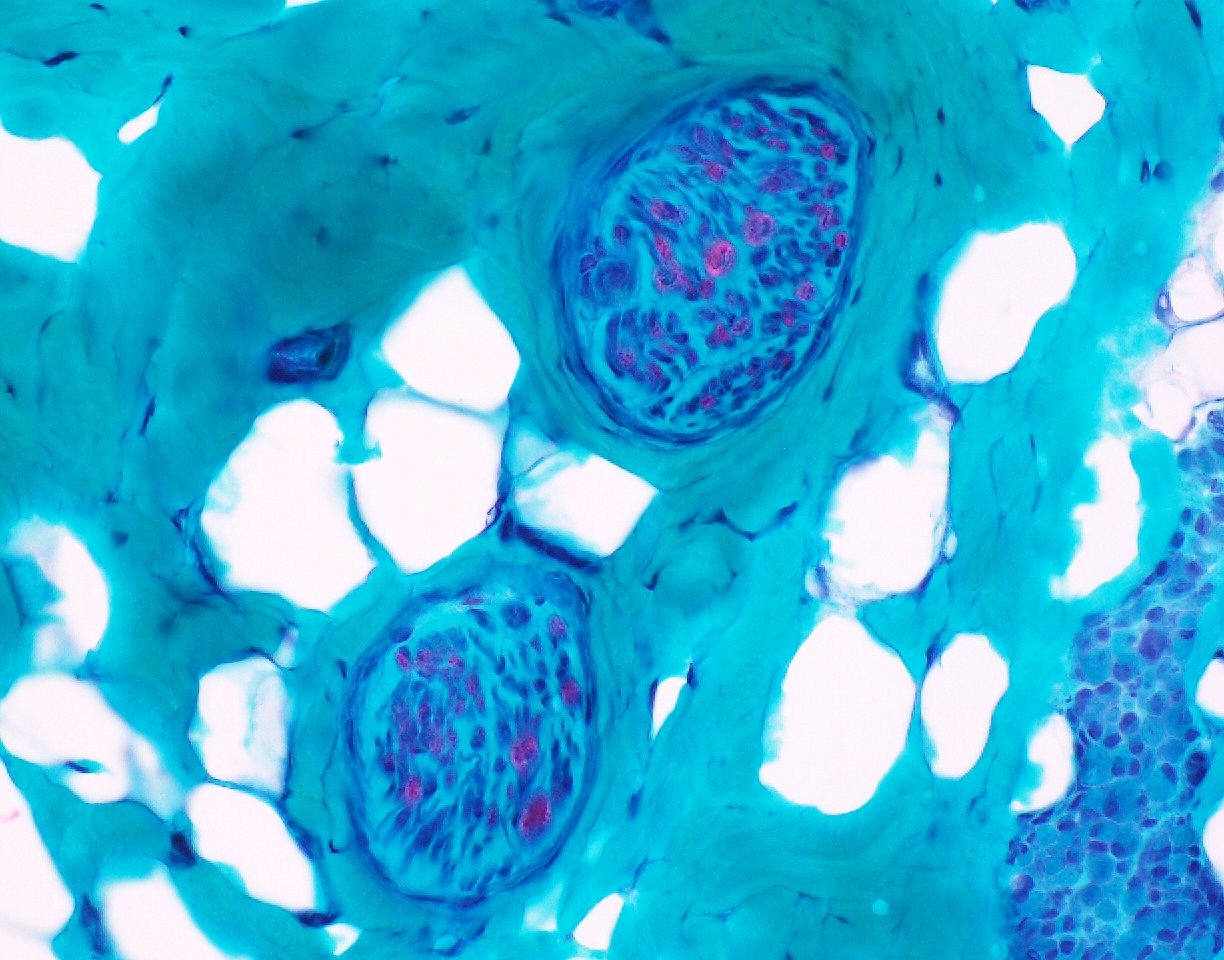
Intramuscular nerve with axon loss
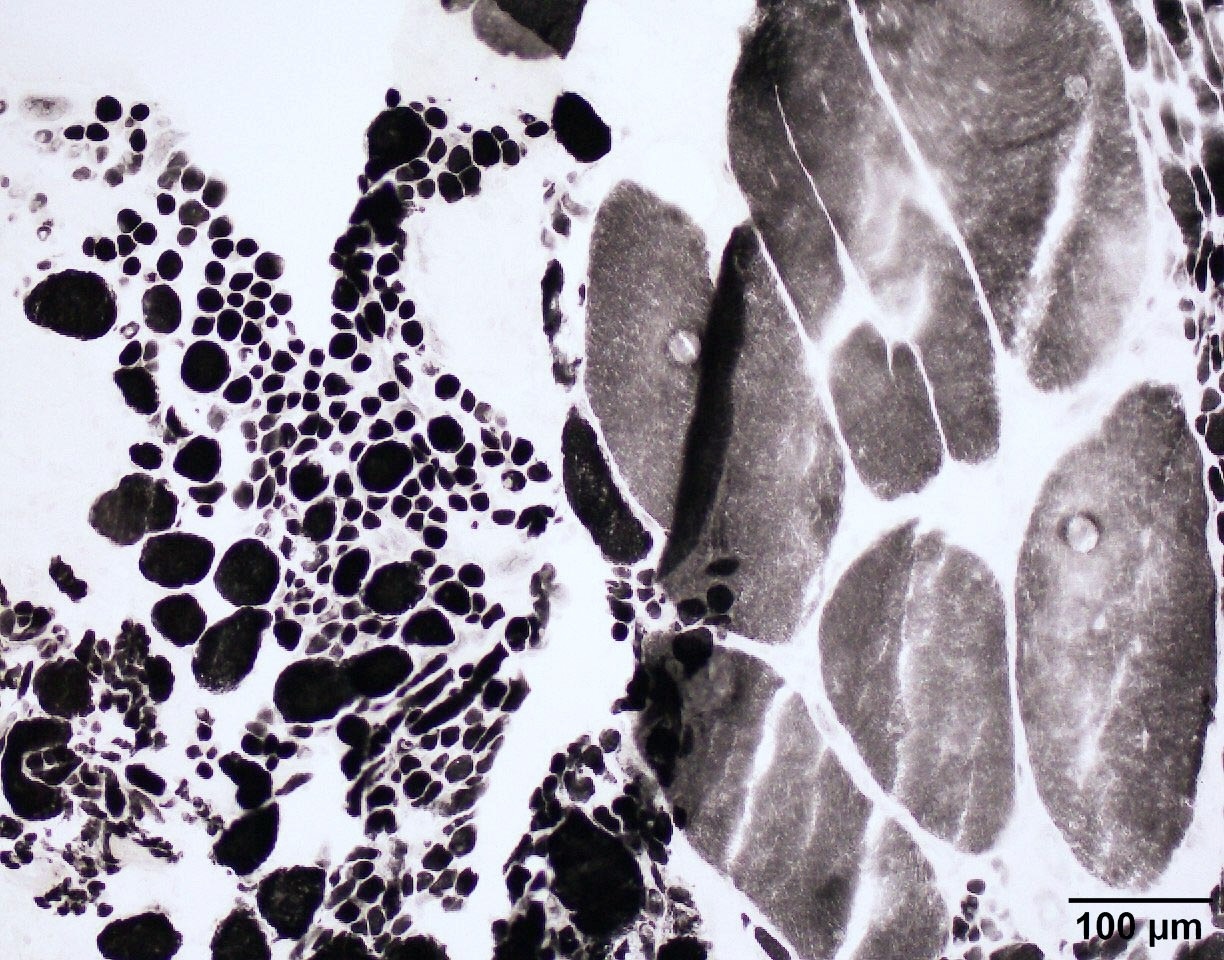
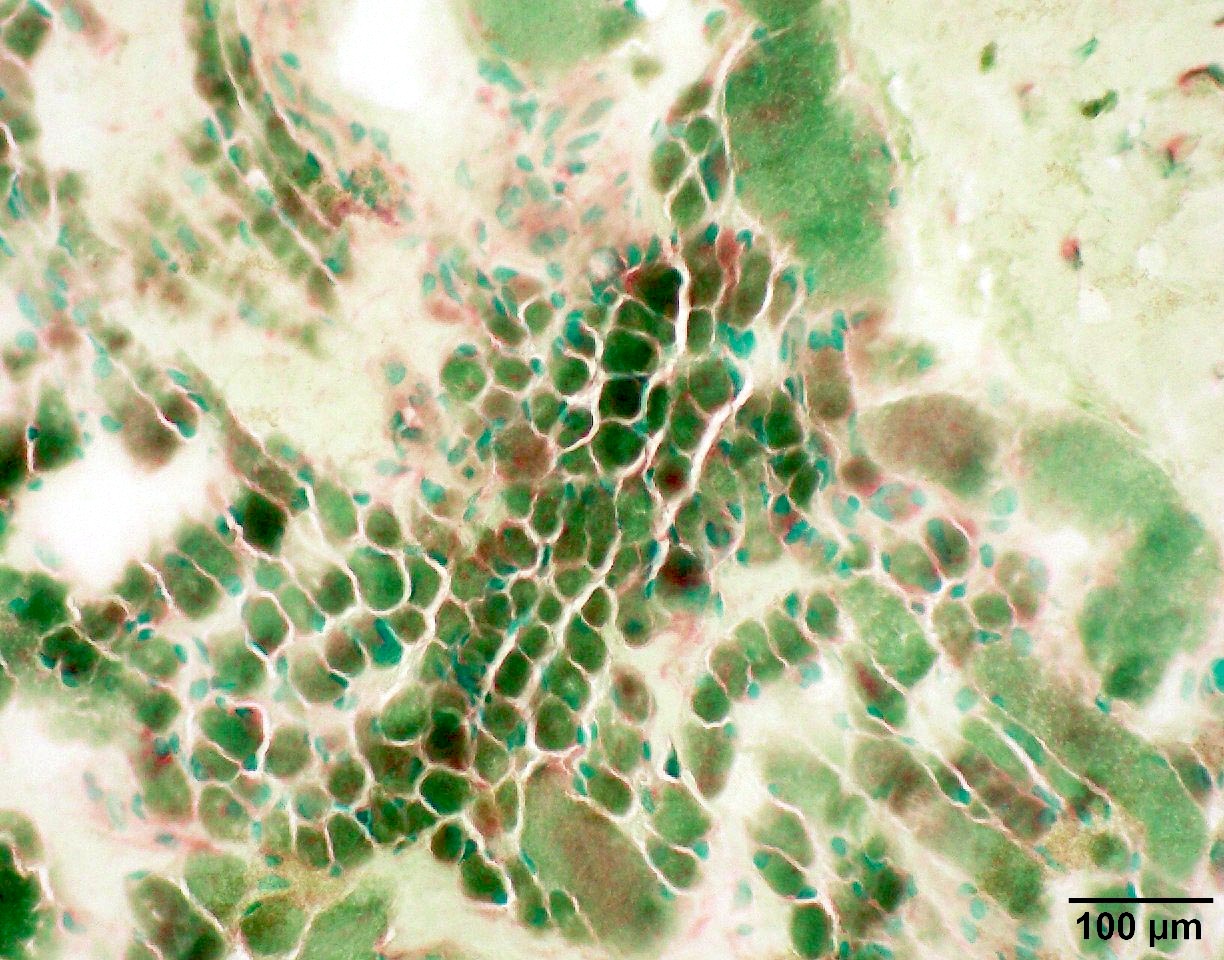
SMA type 3: 20 year old man presented with bilateral lower extremity weakness. Genetic analysis showed homozygous deletions of SMN1 and 4 copies of SMN2.
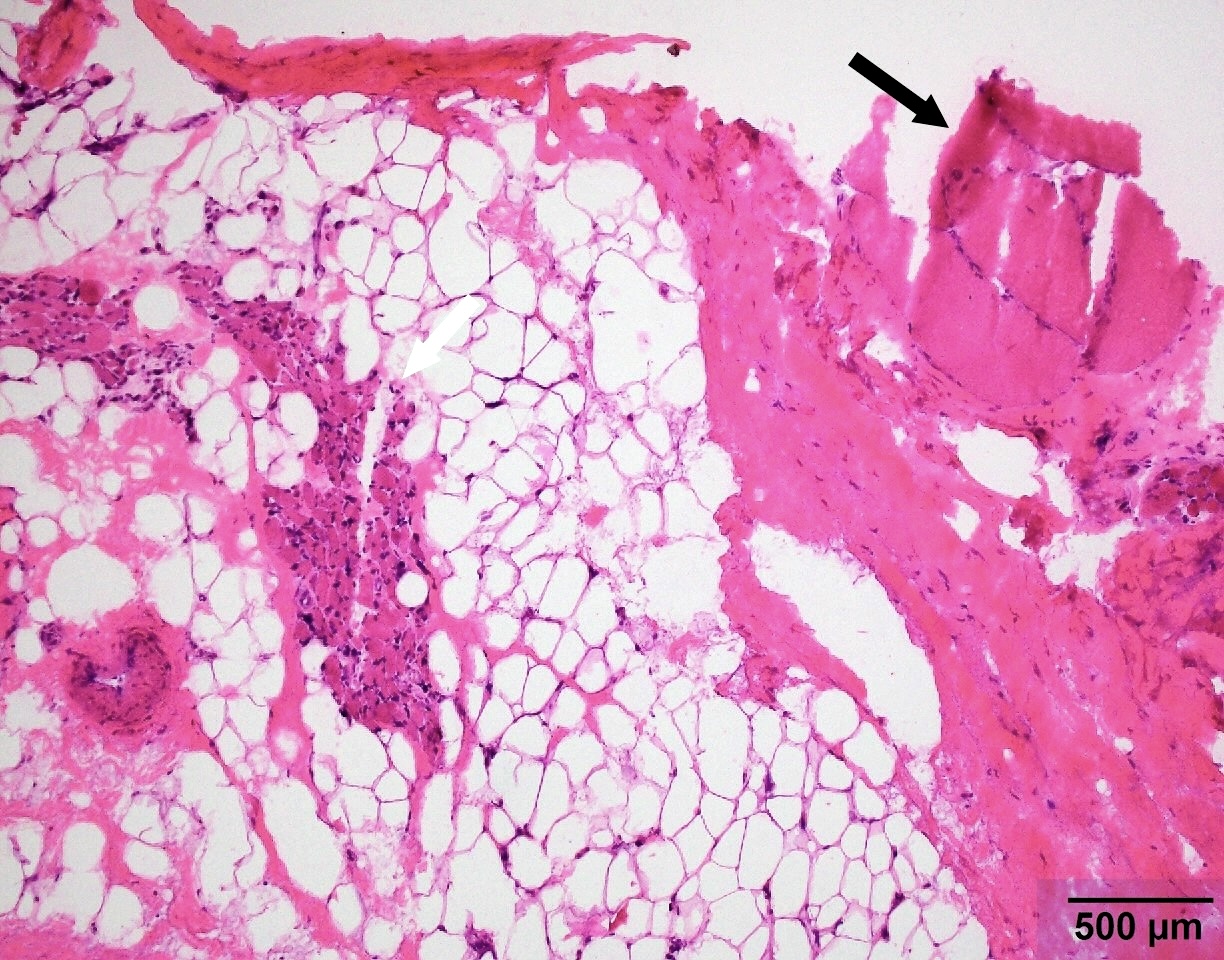
Group atrophy and fatty infiltration
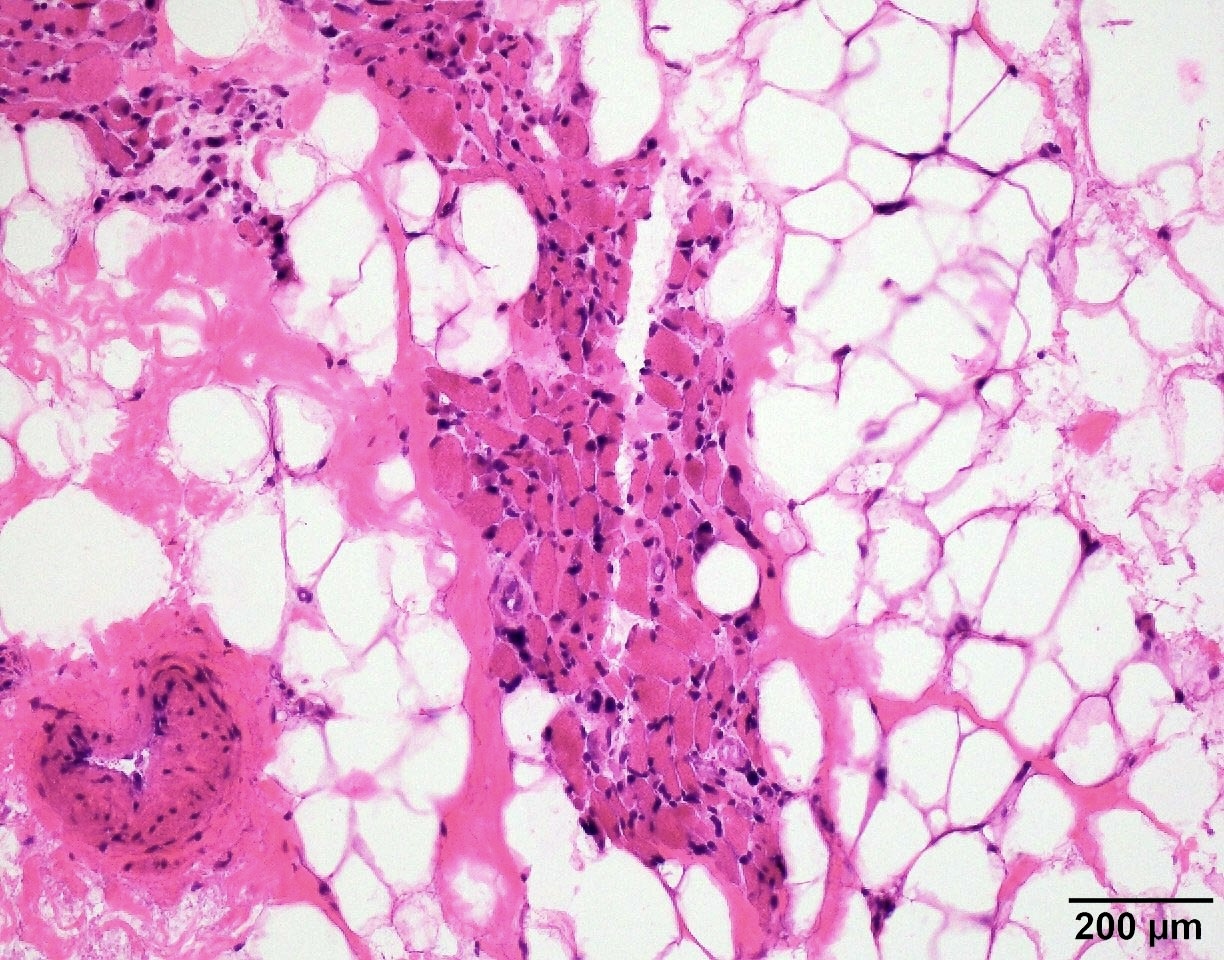
Group atrophy
Fiber type grouping
Fiber type grouping
Esterase dark fibers
Degenerating fibers
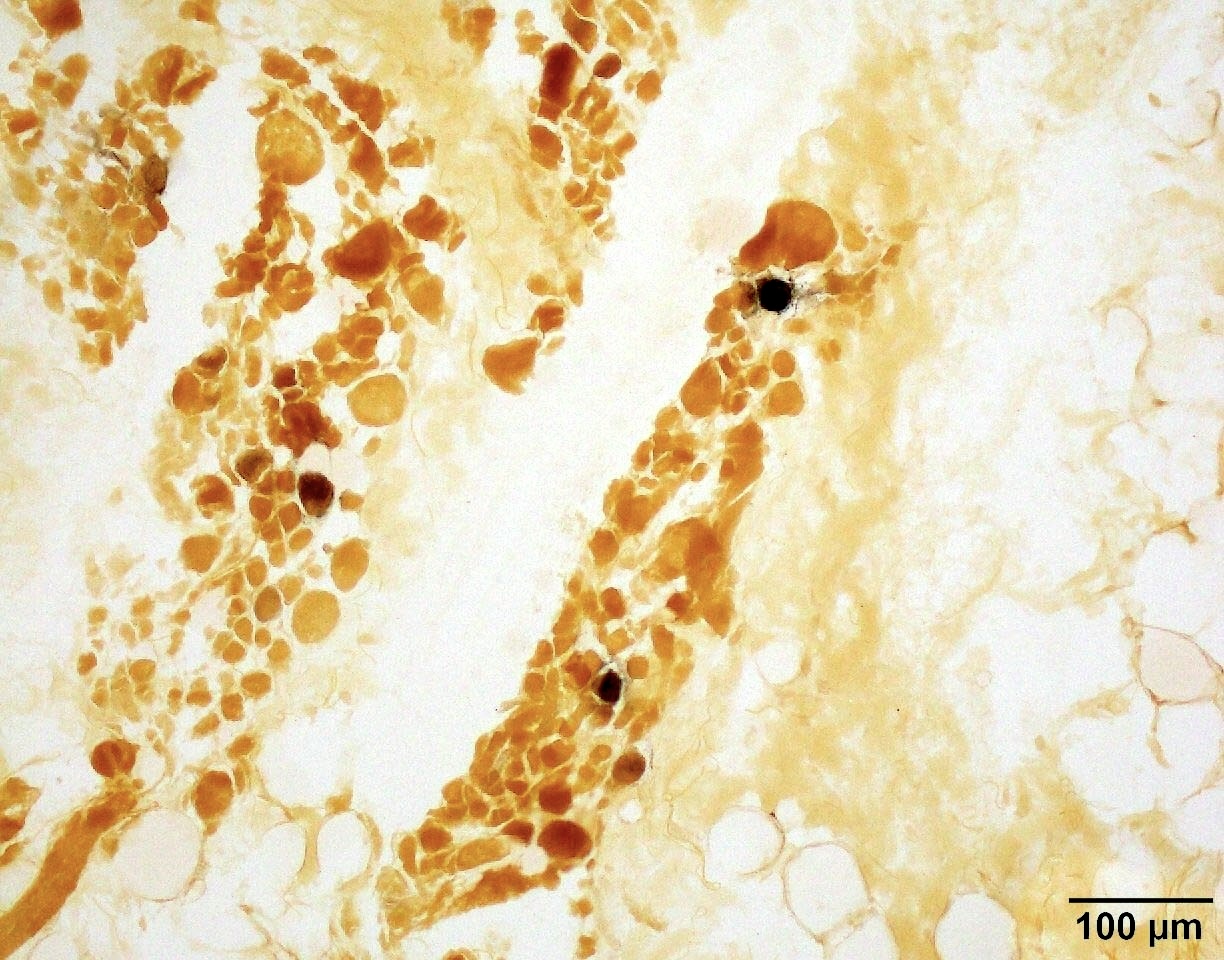
Regenerating fibers
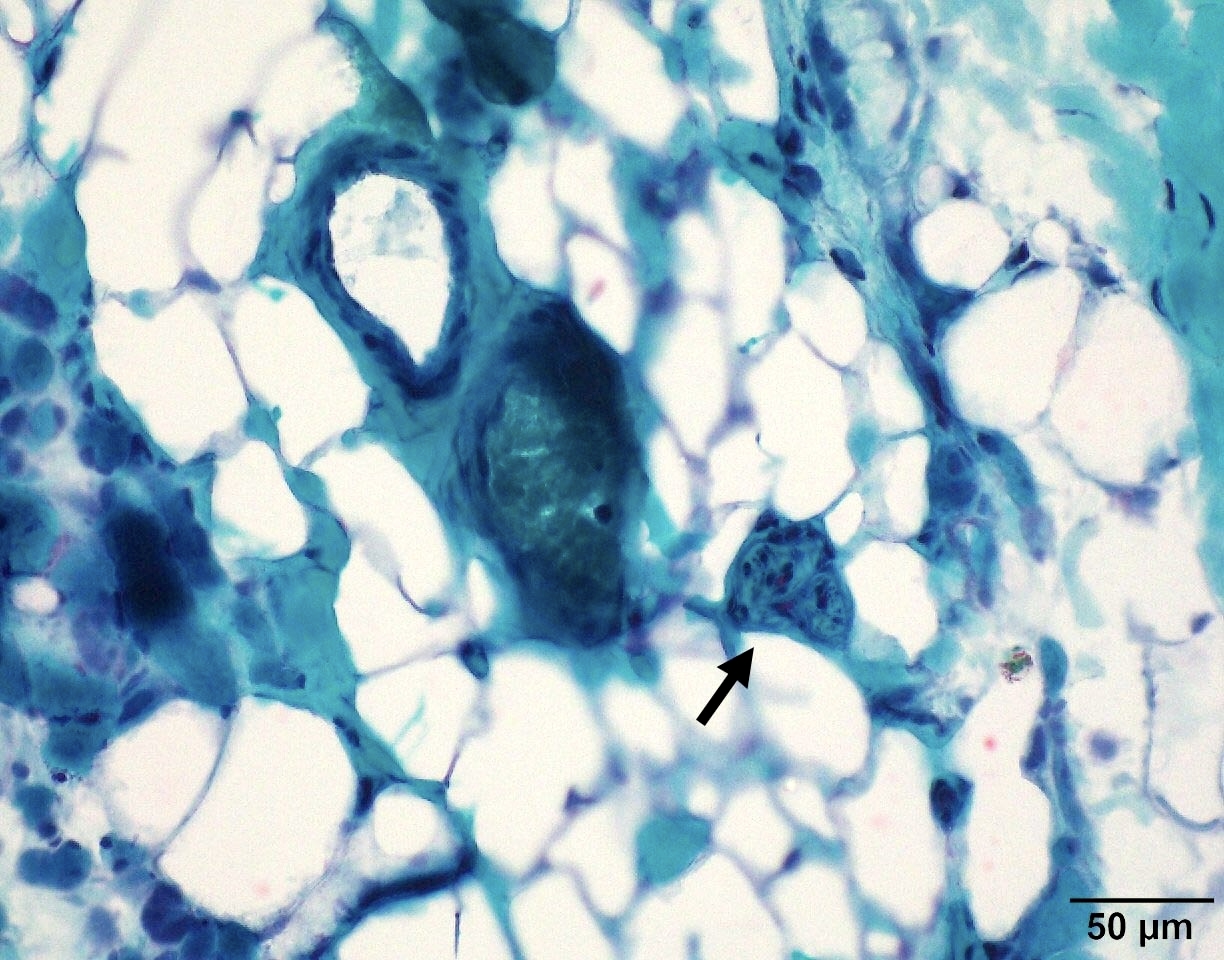
Intramuscular nerve depleted axon
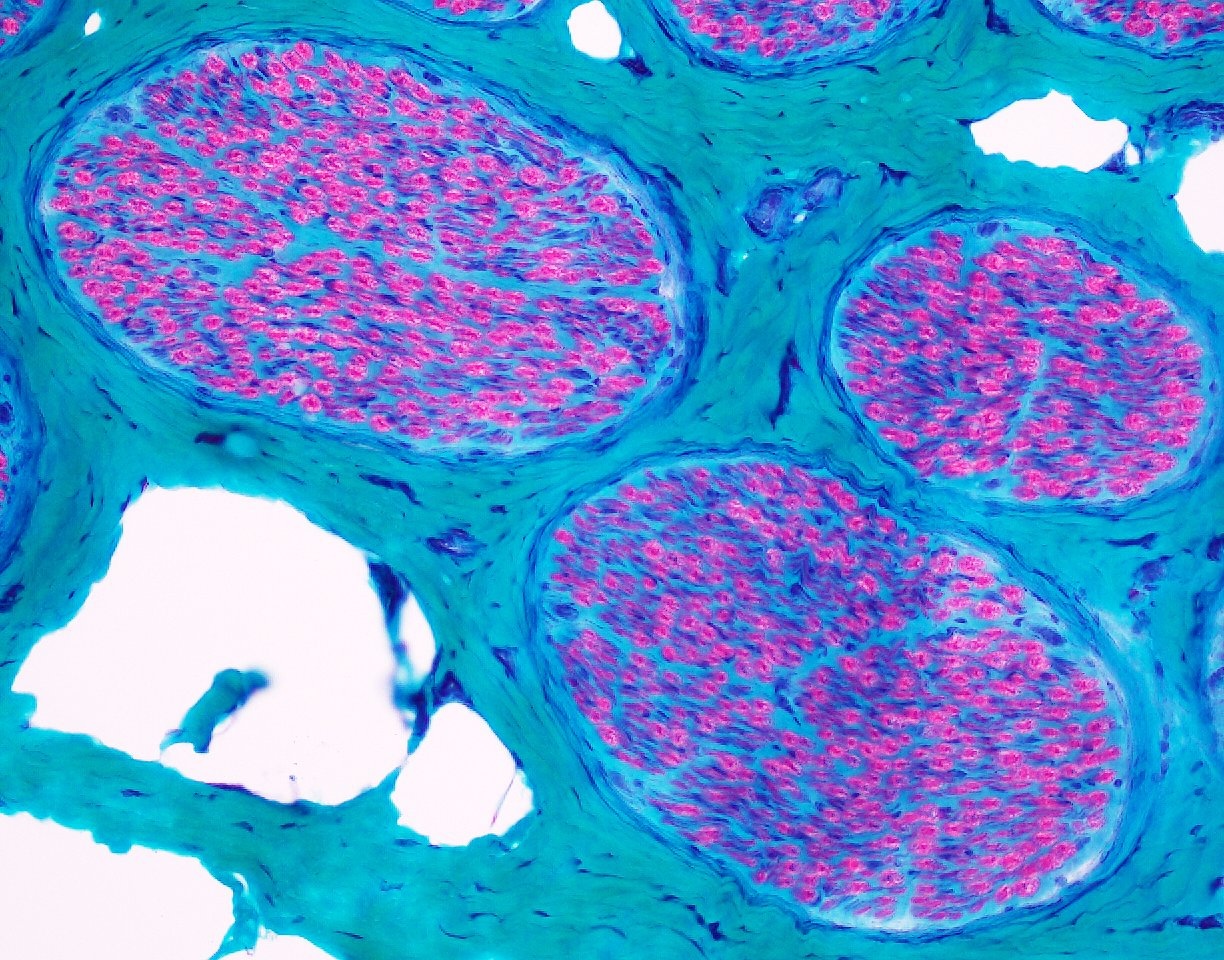
Normal sural nerve
Positive stains
- No disease specific immunostains
- ATPase or NADH histochemistry may show fiber type grouping consistent with chronic denervation
- Dystrophin immunostaining panel intact
- Reference: Dubowitz: Muscle Biopsy - A Practical Approach, 5th Edition, 2020
Negative stains
- No evidence of MHC class I upregulation (which is commonly seen in inflammatory myopathies)
Electron microscopy description
- No specific diagnostic features on electron microscopy
- No endothelial tubuloreticular inclusions or other features suggestive of inflammatory / autoimmune myopathies
Sample pathology report
- Skeletal muscle, left quadriceps, biopsy:
- Denervation atrophy, infantile pattern (see comment)
- Comment: The morphological changes of this biopsy are those of infantile pattern denervation atrophy. Although occasionally seen in patients with a disease process involving the peripheral nerves, this morphologic pattern is most suggestive of infantile spinal muscular atrophy. SMN gene analysis is recommended to confirm the diagnosis.
Differential diagnosis
- SMA type 1 and 2: needs to differentiate from other causes of congenital onset weakness (i.e., floppy baby syndrome) (Clin Neuropathol 2013;32:471)
- Congenital myopathies:
- Usually shows diffuse type I atrophy / hypotrophy, type II fibers are normal sized
- Presence of nemaline rods, cores or central nuclei
- Usually no fiber type grouping
- Congenital muscular dystrophies:
- Usually shows dystrophic changes and markedly elevated CK
- Usually no neurogenic features, such as group atrophy and fiber type grouping
- Peripheral neuropathies:
- Usually do not show prominent hypertrophy of exclusively type I fibers
- Often have sensory involvement in addition to motor involvement
- Small fibers are usually angulated and show increased esterase reactivity
- Muscular dystrophies:
- SMA type 3 or 4 may show prominent fatty replacement and fiber size variation that resemble muscular dystrophy
- SMA type 3 and 4 usually has some neurogenic changes, such as fiber type grouping and group atrophy
- Dystrophy immunostaining panel can rule in or out some cases
- Definitive diagnosis relies on genetic analysis
Practice question #1
A muscle biopsy from a 2 month old boy with a history of hypotonia and failure to thrive reveals the above findings (H&E and ATPase at pH 9.4, respectively). Genetic analysis is most likely to reveal a homozygous deletion of which of the following genes?
- DMD
- FMR1
- GAA
- MTM1
- SMN1
Practice answer #1
E. SMN1. The constellation of findings (infantile pattern denervation atrophy on muscle biopsy, with type I fiber hypertrophy and the clinical history of hypotonia and failure to thrive in a young infant) is highly suggestive of spinal muscular atrophy (SMA) type 1. SMA is caused by homozygous deletions or mutations in the SMN1 gene. Answer A is incorrect because DMD is associated with Duchenne / Becker muscular dystrophy. Answer B is incorrect because FMR1 is associated with fragile X syndrome. Answer D is incorrect because MTM1 is associated with myotubular myopathy. Answer C is incorrect because GAA is associated with Pompe disease.
Comment Here
Reference: Spinal muscular atrophy (SMA)
Comment Here
Reference: Spinal muscular atrophy (SMA)
Practice question #2
A 1 year old child presents with progressive muscle weakness. Electromyography (EMG) demonstrates chronic denervation. Muscle biopsy reveals grouped atrophy, fiber type grouping and occasional hypertrophic fibers. Genetic testing reveals homozygous deletion of the SMN1 gene and 3 copies of SMN2. Based on these findings, which of the following is the most likely classification of this patient's spinal muscular atrophy (SMA)?
- SMA type 0
- SMA type 1
- SMA type 2
- SMA type 3
- SMA type 4
Practice answer #2
C. SMA type 2. The child's presentation of progressive weakness with 3 copies of SMN2 is most consistent with SMA type 2 (onset of 6 - 18 months, can sit but not walk). Answer A is incorrect because while not explicitly stated, the scenario suggests the weakness was not present at birth (ruling out type 0). Answer B is incorrect because SMA1 usually contains 1 - 2 copies of SMN2. Answers D and E are incorrect because types 3 and 4 have 4 or more copies of SMN2 and are juvenile or adult onset, respectively.
Comment Here
Reference: Spinal muscular atrophy (SMA)
Comment Here
Reference: Spinal muscular atrophy (SMA)
