Colon
Syndromes
Familial adenomatous polyposis, classic
Last author update: 19 February 2021
Last staff update: 2 August 2023
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Familial adenomatous polyposis classic colon



Table of Contents
Definition / general | Essential features | Terminology | Epidemiology | Sites | Pathophysiology | Diagrams / tables | Diagnosis | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) images | Molecular / cytogenetics description | Videos | Sample pathology report | Differential diagnosis | Additional references | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Findeis-Hosey J, Gonzalez RS. Familial adenomatous polyposis, classic. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/colontumorfap.html. Accessed September 15th, 2025.
Definition / general
- Autosomal dominant familial polyposis syndrome due to a defect in the APC gene (5q21) which prototypically results in numerous (> 100) colonic adenomatous polyps
Essential features
- Most frequent genetic polyposis syndrome, which is due to a germline mutation in the APC gene (5q21)
- Greater than 100 colorectal adenomatous polyps
- Essentially 100% lifetime risk of colorectal adenocarcinoma
- Variants include Gardner syndrome, Turcot syndrome and attenuated familial adenomatous polyposis (AFAP)
Terminology
- Also known as familial polyposis coli or adenomatous polyposis coli
Epidemiology
- Incidence of 1 per 7 - 30,000 individuals
- Males and females are equally affected
- Colon polyps typically are present by the end of the second decade (mean age: 15.9 years)
- Essentially 100% of patients develop colonic adenocarcinoma without surgical intervention (average age: 39 years)
- 25% of patients will be diagnosed with colorectal carcinoma at time of presentation
Sites
- In addition to colonic adenomatous polyps, patients may develop polyps in the stomach (fundic gland polyps) and small intestine
- May also develop carcinoma of thyroid gland, gallbladder and adrenal gland
- Desmoid tumors are a common extraintestinal manifestation (10 - 25%) - they frequently develop in the abdominal wall following local surgery / trauma
- May develop bone lesions (i.e. osteomas), skin lesions (i.e. epidermal inclusion cysts), dental abnormalities (i.e. supranummary or absent teeth), ocular problems (i.e. congenital hypertrophy of retinal pigment epithelium [CHRPE]) or juvenile nasopharyngeal angiofibromas
- Gardner syndrome encompasses the subset of FAP patients with extraintestinal tumors
- Turcot syndrome encompasses the subset of FAP patients with brain tumors, which are typically medulloblastomas
Pathophysiology
- APC (adenomatous polyposis coli) is a tumor suppressor gene involved in cell cycle control and downregulation of beta catenin through the Wnt signaling pathway
- APC protein is normally involved in apoptosis of colonic epithelial cells
- APC mutations may cause expansion of the crypt base cell population, including crypt stem cells
- Mutant crypt stem cells may clonally expand to form adenomas and carcinomas (Am J Pathol 2004;165:1489)
- When APC is mutated, beta catenin is no longer downregulated and can function to stimulate cell growth
- As explained by the 2 hit hypothesis, patients with a germline APC mutation develop adenomatous polyps when there is inactivation of the remaining normal APC allele
- Colorectal adenocarcinomas in FAP develop in a similar fashion to sporadic type colorectal adenocarcinomas (i.e. mutations in KRAS, TP53)
- Abundance of adenomatous polyps results in the near certain development of colorectal adenocarcinoma
Diagrams / tables

Diagnosis
- Diagnostic criteria:
- 100 or more colorectal adenomatous polyps or
- Germline mutation in APC or
- Family history of FAP with colorectal adenomas (age < 30) or
- Family history of FAP and presence of at least one epidermoid cyst, osteoma or desmoid tumor
- Patients with a history of > 10 colorectal adenomas, a family history of adenomatous polyposis syndromes or a history of adenomas with FAP type extracolonic lesions should undergo assessment for adenomatous polyposis syndrome (Am J Gastroenterol 2015;110:223)
Prognostic factors
- Location of the mutation in APC has an impact on the phenotypic presentation, including number of polyps and presence or absence of desmoid tumors and congenital hypertrophy of retinal pigment epithelium
- Most common causes of noncolorectal carcinoma deaths in FAP patients are duodenal / ampullary carcinoma and complications of desmoid tumor
Case reports
- 11 and 12 year old boys with cancer (Eur J Pediatr 2005;164:306)
- 24 year old woman with FAP and small bowel GIST (Exp Oncol 2015;37:227)
- FAP in 3 generations of a single family (Case Rep Oncol 2014;7:349)
Treatment
- Prophylactic colectomy in late second to early third decade
- Must monitor rectal stump if preserved
- FAP patients who undergo consistent colorectal screening with subsequent surgery have a significantly decreased rate of mortality from colorectal malignancies
- Children at risk for FAP should undergo sigmoidoscopy every 1 - 2 years, beginning at age 10 - 12 years
- Relatives should also be screened
- NSAIDs and aspirin use have been studied to decrease polyp regrowth following subtotal colectomy
Gross description
- Most adenomatous polyps are small (< 0.5 cm) and sessile
- Polyps are more common in the left sided colon, although the entire length of the colon is typically involved
- Polyps may be flat or depressed (Int J SurgPathol 2006;14:133)
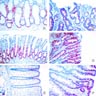
Gross images
Contributed by @Andrew_Fltv on Twitter
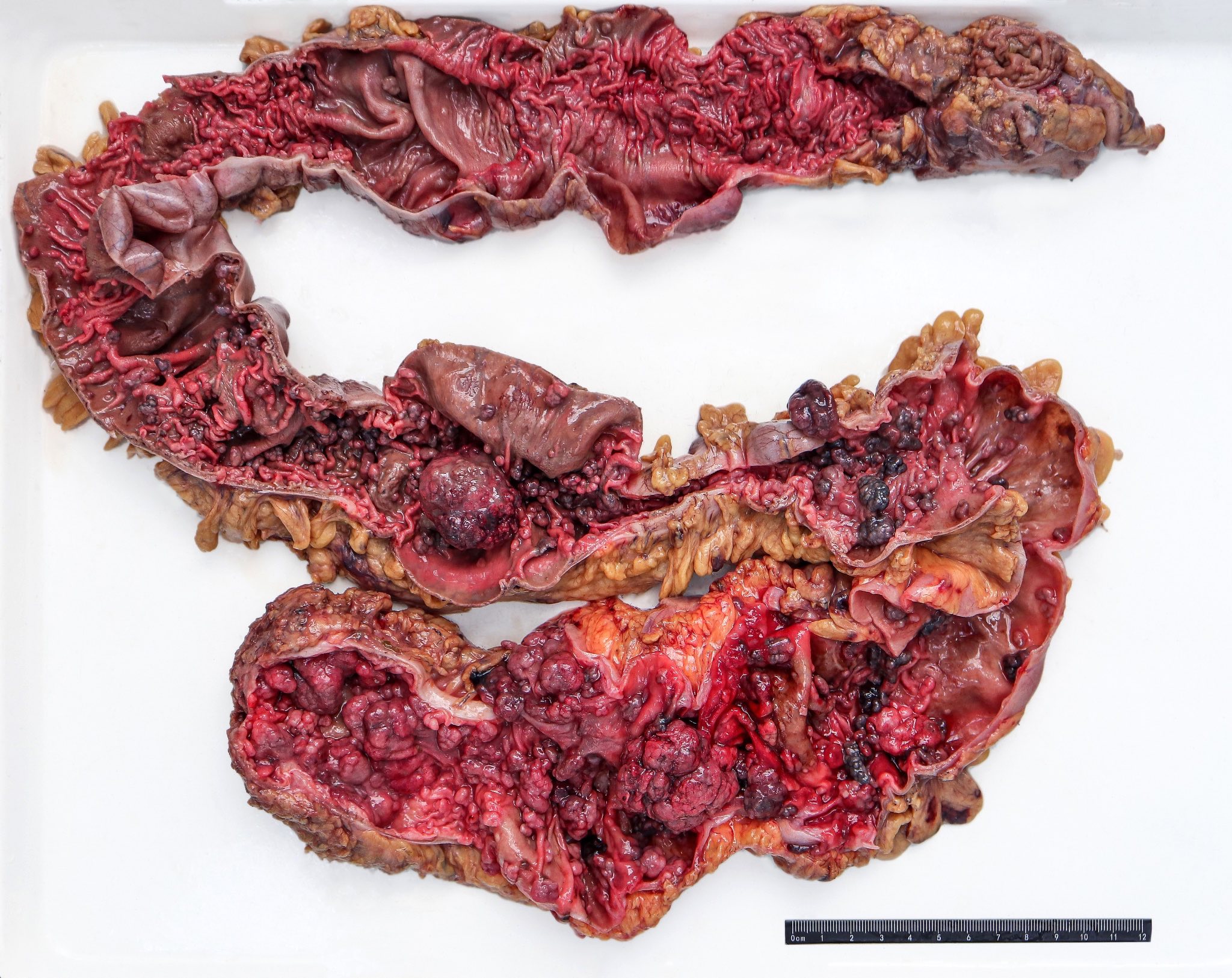
Familial adenomatous polyposis, classic
Images hosted on other servers:

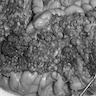
Carpet of adenomatous polyps
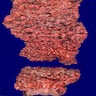

Numerous polyps
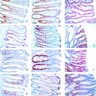
Microscopic (histologic) images
Contributed by Jennifer Findeis-Hosey, M.D. and @Andrew_Fltv on Twitter
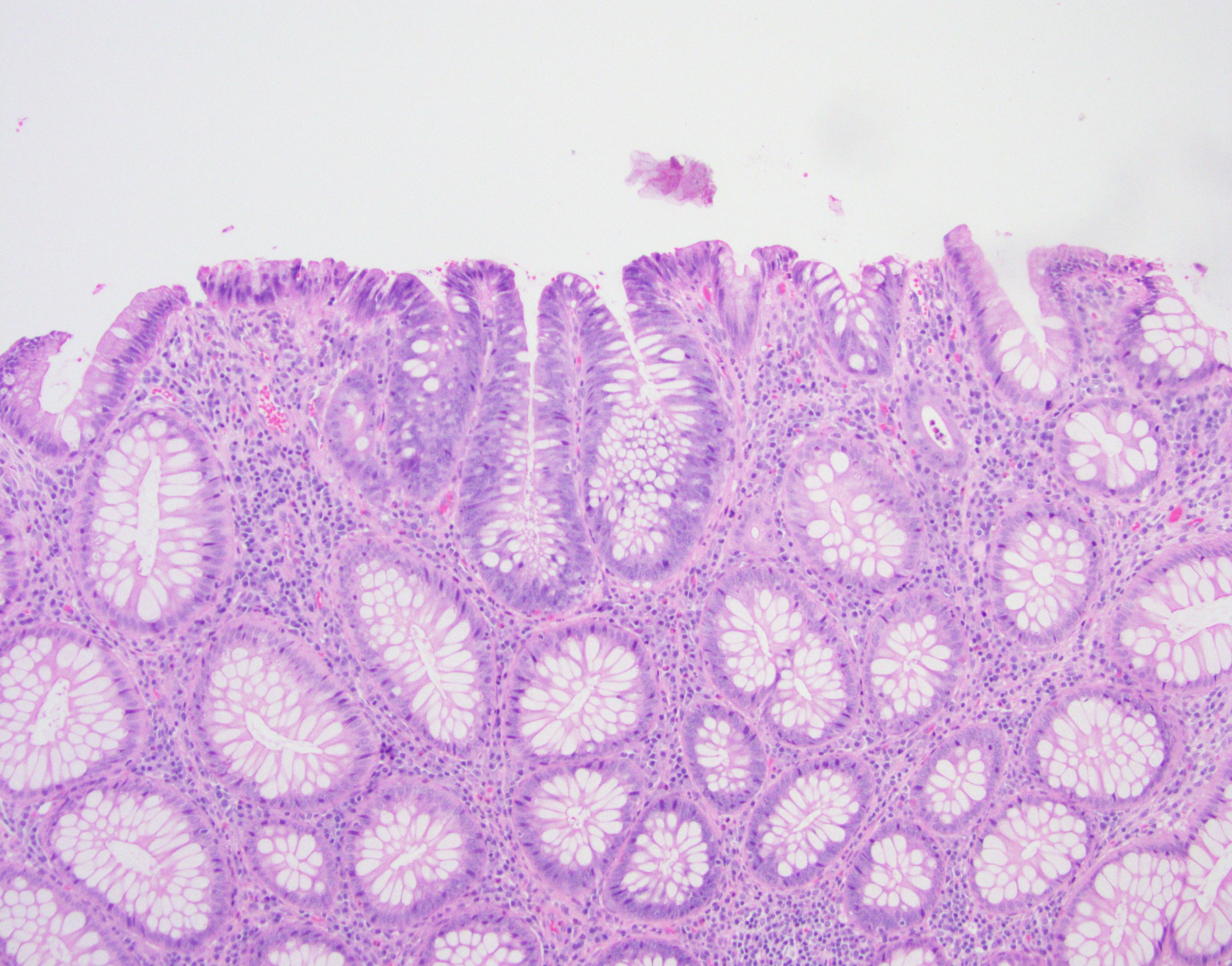
Adenoma involving few crypts
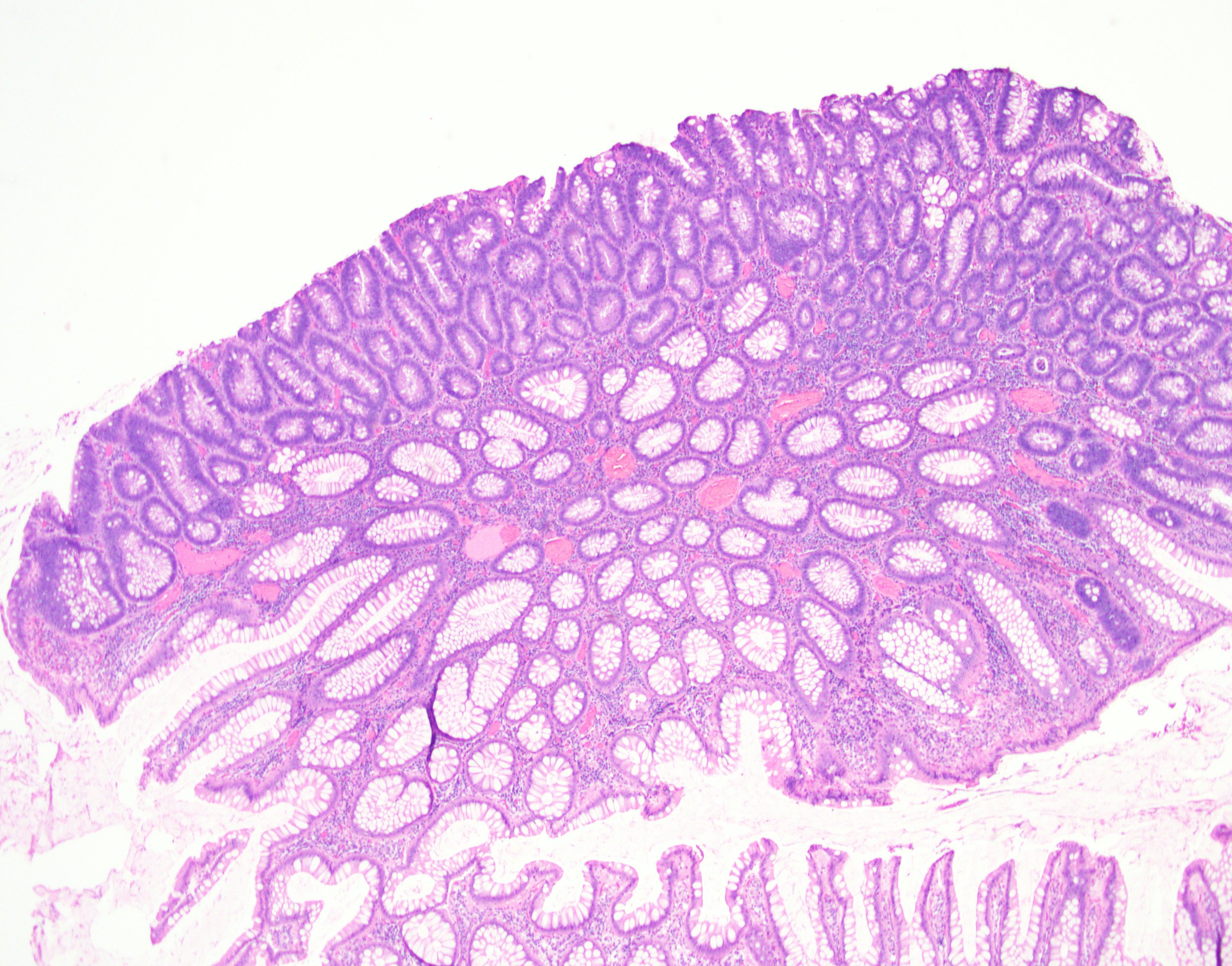
Tubular adenoma polyp, FAP patient
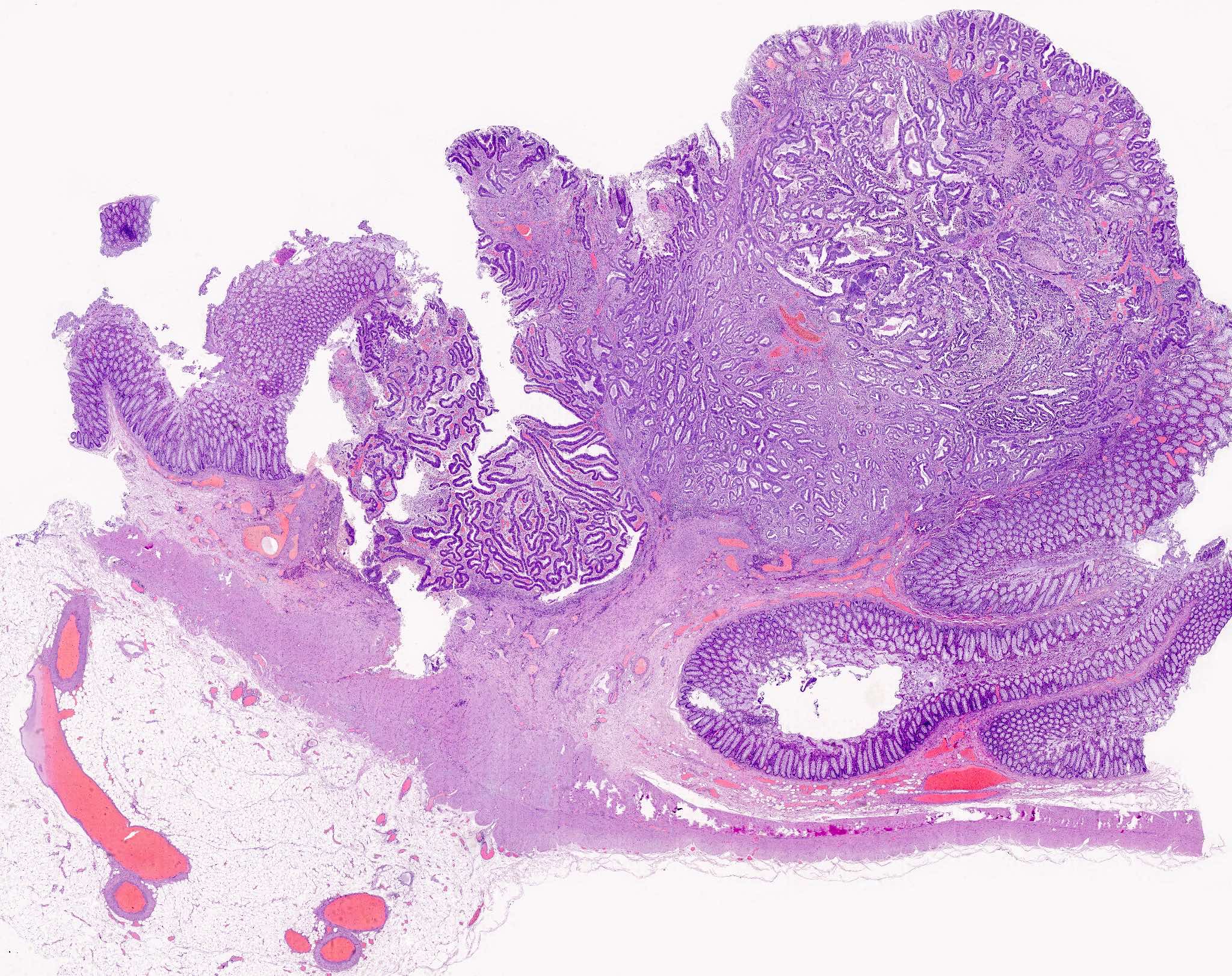
Familial adenomatous polyposis, classic
Images hosted on other servers:
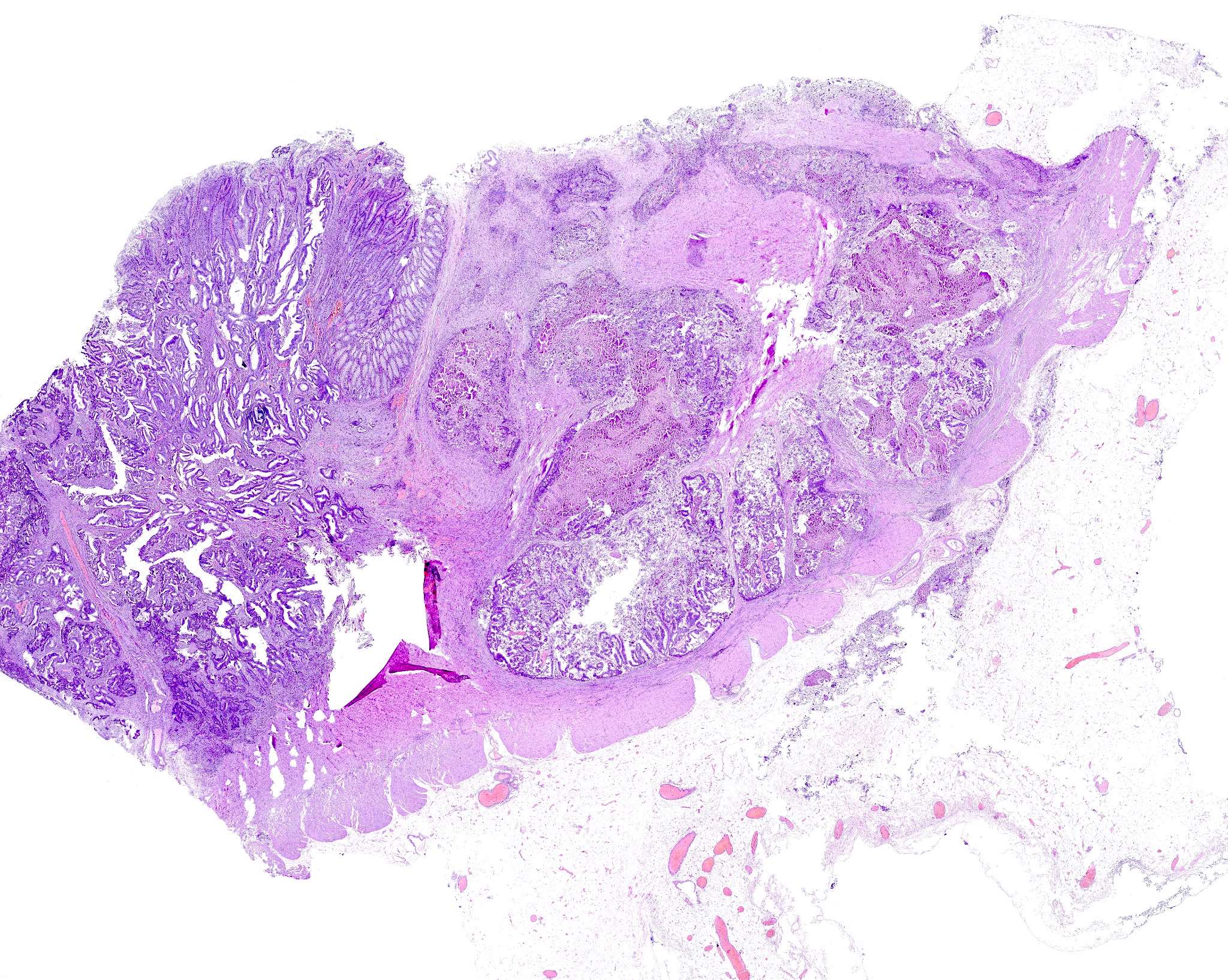
Adenoma and adenocarcinoma
FAP versus non-FAP
BCL2
Molecular / cytogenetics description
- Hereditary colorectal cancer syndromes are usually due to defect in APC gene at 5q21
- Polyposis patients without APC mutations often have mutations in MYH gene
- Autosomal dominant trait with high degree of penetrance (> 90%)
- 20% represent a new de novo APC mutation without family history
Videos
Full colonoscopy in retroflexed maneuver in a familial adenomatous polyposis coli
Sample pathology report
- Colon, total colectomy:
- Colon with innumerable tubular adenomas, some with focal high grade dysplasia (see comment)
- Negative for malignancy.
- Adenomas focally present at both proximal and distal mucosal resection margins.
- Twelve benign lymph nodes.
- Comment: The findings are consistent with the patient's reported history of familial adenomatous polyposis. Numerous grossly identifiable polyps were submitted for microscopy, including all polyps larger than 1 cm.
Differential diagnosis
- Attenuated FAP:
- Polyposis syndrome also due to an APC mutation but patients have fewer than 100 colorectal polyps
- MYH associated polyposis:
- Polyposis syndrome caused by a mutation in the MYH gene
Additional references
Practice question #1
Without surgical intervention, patient with familial adenomatous polyposis have lifetime risk of developing colorectal carcinoma of approximately
- 25%
- 50%
- 75%
- 100%
Practice answer #1
Practice question #2
Patients with Turcot syndrome develop what tumors in addition to the typical manifestations of familial adenomatous polyposis?
- Desmoid tumors (fibromatosis)
- Gastrointestinal stromal tumors
- Keratoacanthomas
- Medulloblastomas
Practice answer #2
