Lung
Cystic disease / congenital anomalies
Congenital pulmonary airway malformation (CPAM)
Editorial Board Member: Jefree J. Schulte, M.D.


Last author update: 1 November 2022
Last staff update: 6 August 2025
Copyright: 2003-2025, PathologyOutlines.com, Inc.
PubMed search: Congenital pulmonary airway malformation
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Etiology | Clinical features | Diagnosis | Radiology description | Prognostic factors | Case reports | Treatment | Gross description | Gross images | Microscopic (histologic) description | Microscopic (histologic) images | Positive stains | Molecular / cytogenetics description | Sample pathology report | Differential diagnosis | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2Cite this page: Nelson ND, Pogoriler J. Congenital pulmonary airway malformation (CPAM). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lungnontumorcysticadenomatoid.html. Accessed September 18th, 2025.
Definition / general
- Congenital pulmonary airway malformations encompass a spectrum of cystic and noncystic lung malformations that develop in utero
Essential features
- Cysts are interconnected with each other and with alveolar spaces and typically lined by ciliated columnar epithelium; there is substantial overlap in cyst size between type 1 and 2 CPAMs
- Type 1 and 3 CPAMs are associated with mosaic activating KRAS mutations and an increased risk of metastatic mucinous adenocarcinoma if incompletely resected (Mod Pathol 2022 Jul 6 [Epub ahead of print])
- Type 2 CPAMs are associated with bronchial atresia and do not have KRAS mutations (Semin Pediatr Surg 2003;12:17, Mod Pathol 2022;35:1404)
- Terms type 0 and type 4 CPAM are now obsolete (see Terminology below)
Terminology
- Previously called congenital cystic adenomatoid malformation (CCAM) (Hum Pathol 1977;8:155)
- Most commonly used classification systems are the Stocker and Langston classification systems (Hum Pathol 1977;8:155, Semin Pediatr Surg 2003;12:17)
- Stocker type 0 and type 4 CPAMs are now recognized to represent other lung abnormalities; these terms are no longer in use
- Acinar dysplasia is the preferred term for the diffuse malformation previously described as type 0 CPAM (Am J Med Genet A 2016;170:2440, Am J Respir Crit Care Med 2019;200:1093)
- Type 4 CPAMs are now known to represent cystic pleuropulmonary blastomas and should be diagnosed as such (Pediatr Dev Pathol 2015;18:504)
- Congenital lobar emphysema due to bronchial atresia is thought to be on a spectrum with type 2 CPAMs; these findings often co-occur
ICD coding
- ICD-10: Q33.9 - congenital malformation of lung, unspecified
Epidemiology
- Congenital lung lesion
- Type 2 CPAMs are the most common but are usually asymptomatic (Am J Surg Pathol 2019;43:47)
- Clinical course is dependent upon the size of the lesion and timing of resection
- Nearly all type 1 CPAMs and some type 3 CPAMs are caused by mosaic activating KRAS mutations; these are usually symptomatic (Mod Pathol 2022 Jul 6 [Epub ahead of print])
Sites
- Most commonly involve a single lobe of the lung
- Rarely, 1 lesion may involve multiple lobes or a patient may have 2 distinct lesions
Etiology
- Type 1 CPAMs and many type 3 CPAMs result from mosaic activating KRAS mutations (Mod Pathol 2022 Jul 6 [Epub ahead of print])
- Mosaic mutations in other genes have been rarely reported in type 3 CPAMs (Mod Pathol 2022 Jul 6 [Epub ahead of print], Am J Med Genet A 2020;182:746)
- Type 2 CPAMs are believed to arise secondary to bronchial atresia and do not have KRAS mutations (Semin Pediatr Surg 2003;12:17, Arch Pathol Lab Med 2002;126:934, J Pediatr Surg 2006;41:61, Pediatr Dev Pathol 2006;9:361, Mod Pathol 2022;35:1404)
Clinical features
- Type 2 CPAMS are rarely associated with other congenital anomalies, including congenital diaphragmatic hernias (Front Pediatr 2020;8:446)
- Type 1 and 3 CPAMs have been described in patients with nevus comedonicus syndrome and Schimmelpenning syndrome (Mod Pathol 2022 Jul 6 [Epub ahead of print], Am J Med Genet A 2020;182:746)
Diagnosis
- Lesions are typically detected on prenatal ultrasound
- Definitive diagnosis requires pathologic examination following resection
Radiology description
- Ultrasound: multiple hypoechoic lung cysts (Neonatology 2016;110:101)
- Fetal MRI: T2 hyperintense lesion with cystic spaces
Prognostic factors
- Type 1 and type 3 CPAMs:
- Incomplete resection in infancy or resection later in childhood is associated with an increased risk of developing metastatic mucinous adenocarcinoma (Am J Surg Pathol 2020;44:1118)
- There are no reports of metastatic mucinous adenocarcinoma arising in CPAMs that are completely resected in infancy or early childhood
Case reports
- Neonate born at 32 weeks gestational age who developed respiratory failure and died shortly after birth (Autops Case Rep 2018;8:e2018022)
- 14 year old boy with shortness of breath and exercise intolerance (Am J Clin Pathol 2021;156:313)
- 14 year old boy with spontaneous pneumothorax (Ann Med Surg (Lond) 2021;68:102692)
Treatment
- Complete surgical resection
- Thoracoamniotic shunts may be placed to decompress type 1 CPAMs in utero until resection can be performed after birth
- Maternal steroids may be given for symptomatic patients whose cysts are too small for shunt
- Reference: Eur J Pediatr 2017;176:1559
Gross description
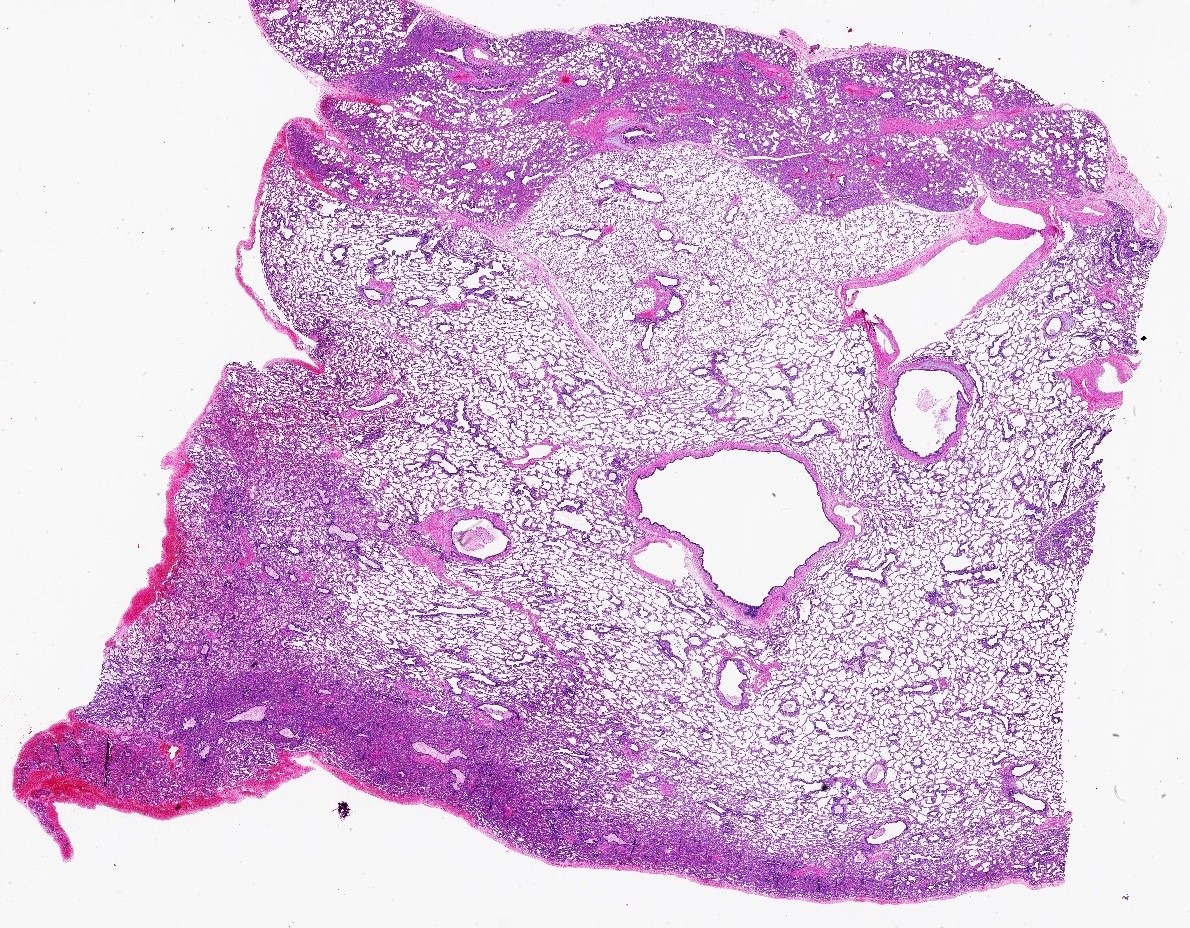
- Type 1 (large cyst) CPAM
- Grossly cystic lung lesion
- Largest cyst size is highly variable and ranges from < 0.5 cm to > 7 cm in greatest dimension (Mod Pathol 2022 Jul 6 [Epub ahead of print])
- Cysts are interspersed with relatively normal appearing lung parenchyma (alveoli are enlarged and simplified)
- No mucocele
- Type 2 (small cyst) CPAM
- Variable gross appearance
- In cystic specimens, cysts can measure up to 2.5 cm in greatest dimension and cysts are interspersed with normal appearing lung parenchyma
- Some specimens may have no grossly identifiable lesion
- May have a mucocele or grossly evident bronchial atresia
- Type 3 CPAM
- Solid / dense appearing lesion, often with a lobulated appearance
- Lesion is distinct from the uninvolved lung parenchyma
- May have very small interspersed cysts (Mod Pathol 2022 Jul 6 [Epub ahead of print])
Gross images
Contributed by Nya D. Nelson, M.D., Ph.D. and Jennifer Pogoriler, M.D., Ph.D.
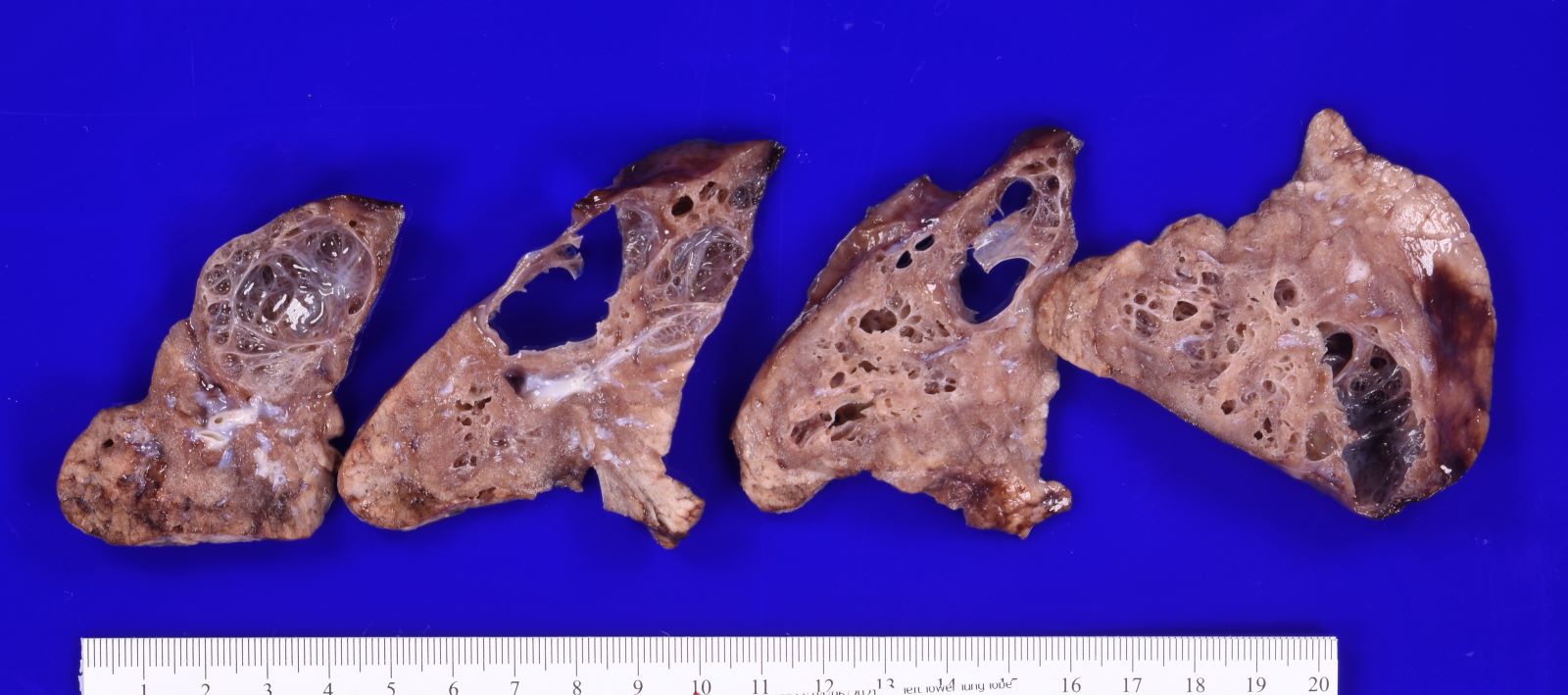
Type 1 CPAM
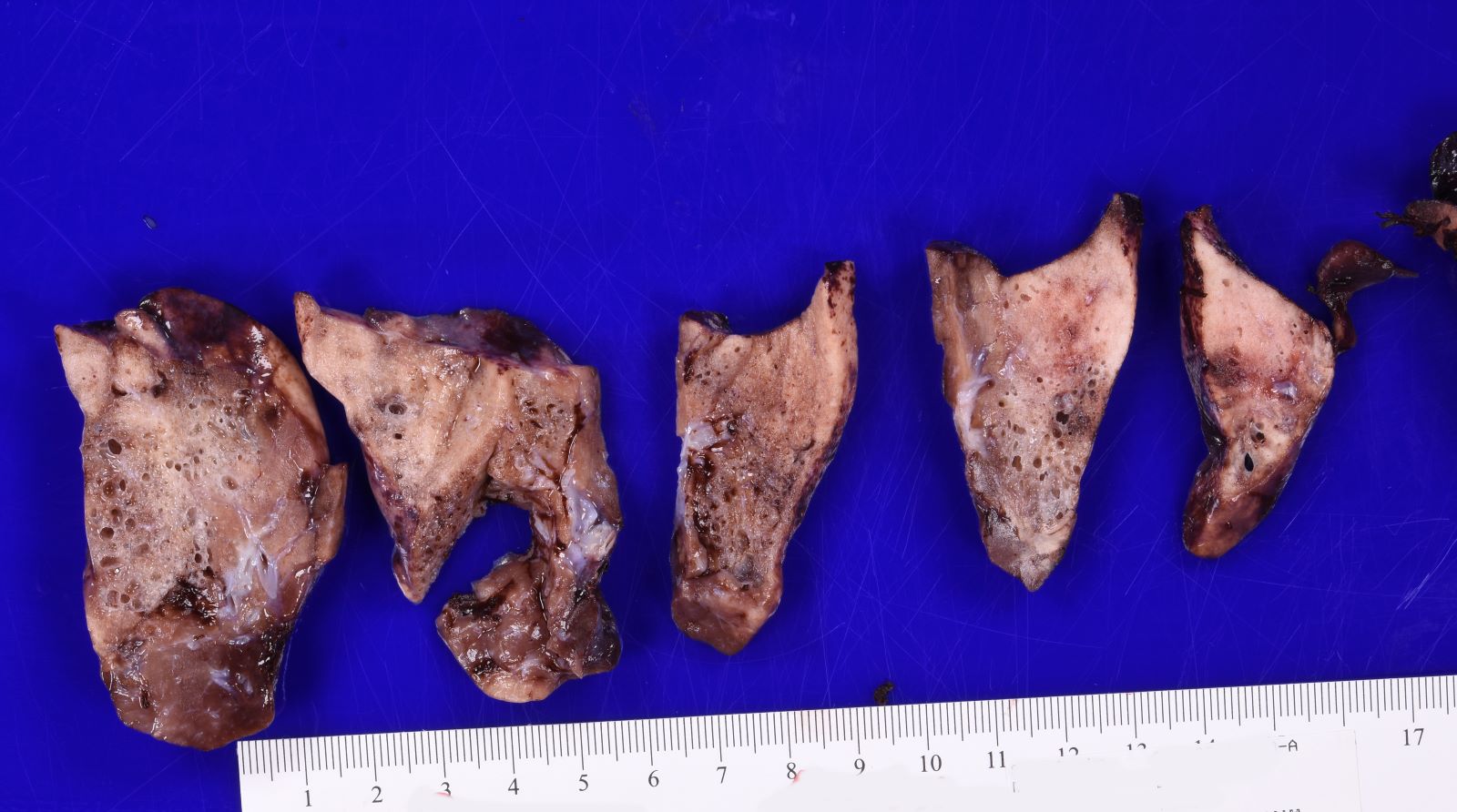
Type 2 CPAM
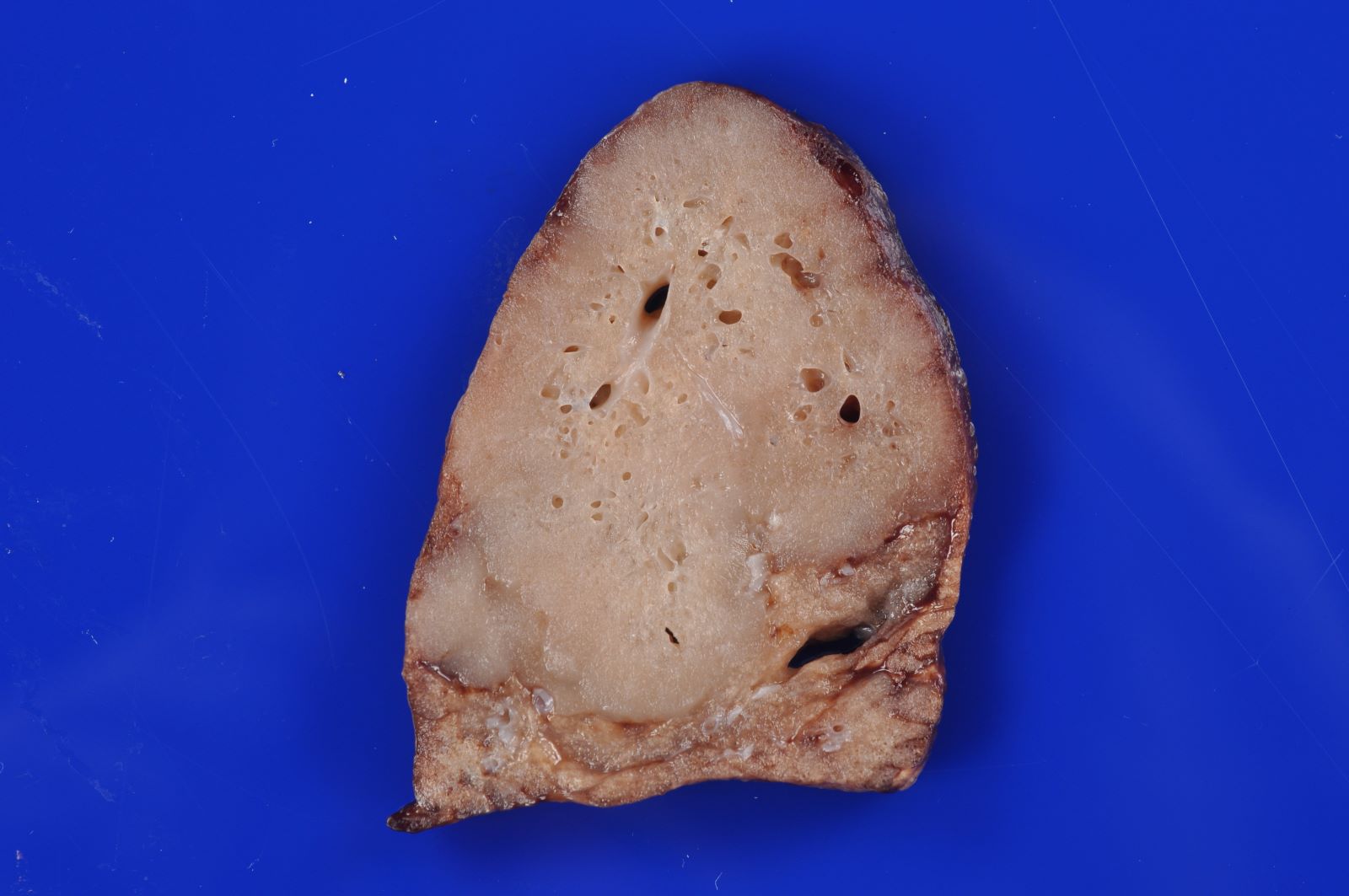
Type 3 CPAM
Microscopic (histologic) description
- Type 1 (large cyst) CPAM
- Readily identifiable cysts lined by ciliated cuboidal to stratified columnar epithelium with interspersed alveolar type spaces (Mod Pathol 2022 Jul 6 [Epub ahead of print])
- Cysts connect with adjacent alveoli with frequent transitions from thick cyst wall to adjacent alveolar wall
- Often has epithelial complexity - papillary projections, irregularly shaped small cystic spaces
- May have solid appearing areas with features of both type 1 and type 3 CPAM
- Mucinous cell clusters are seen in ~75% (when carefully searched for)
- Presence of mucinous cell clusters is specific but not entirely sensitive for a KRAS mutation
- Mucinous cell clusters in infants are often multifocal and have papillary or acinar architecture when they lepidically cover a small alveolus (Am J Surg Pathol 2020;44:1118)
- Most pediatric pathologists do not diagnose mucinous adenocarcinoma in specimens from infants when only these small clusters are present
- May have foci of cartilage within the large cyst walls or foamy appearing pneumocytes with vacuolated cytoplasm (Mod Pathol 2022 Jul 6 [Epub ahead of print])
- Thoracoamniotic shunt placement may result in reactive changes, such as squamous metaplasia
- Type 2 (small cyst) CPAM
- Spectrum of histologic changes ranging from readily identifiable cysts to mildly malformed alveolar type spaces
- Cysts may be focal with the majority of the malformation having only mild changes
- Cysts are typically round and lined by ciliated columnar epithelium
- Epithelial complexity is rare
- Often with prominent evidence of mucostasis, including pools of mucin and foamy intra-alveolar macrophages
- Transitions from cystic spaces into adjacent alveolar type spaces are rare
- Cases that are clinically considered to be CPAMs but do not have cysts show milder parenchymal maldevelopment with enlarged and simplified alveoli (formerly congenital lobar emphysema)
- Striated skeletal muscle may occasionally be seen in the septa between cysts
- Type 3 CPAM
- Consist predominantly of small irregularly shaped airway spaces lined by ciliated cuboidal to columnar epithelium (Mod Pathol 2022 Jul 6 [Epub ahead of print])
- Surrounding septa often appear thickened, with prominent mesenchyme and cuboidal epithelium
- Interspersed alveolar type spaces (with thin walls and flat pneumocytes) are rare / absent in most type 3 CPAMs
- Mucinous cell clusters are seen in ~45%
- Mucinous cell clusters are often multifocal and have papillary or acinar architecture (Am J Surg Pathol 2020;44:1118)
- Most pediatric pathologists do not diagnose mucinous adenocarcinoma in specimens from infants when only these small clusters are present
Microscopic (histologic) images
Contributed by Nya D. Nelson, M.D., Ph.D. and Jennifer Pogoriler, M.D., Ph.D.
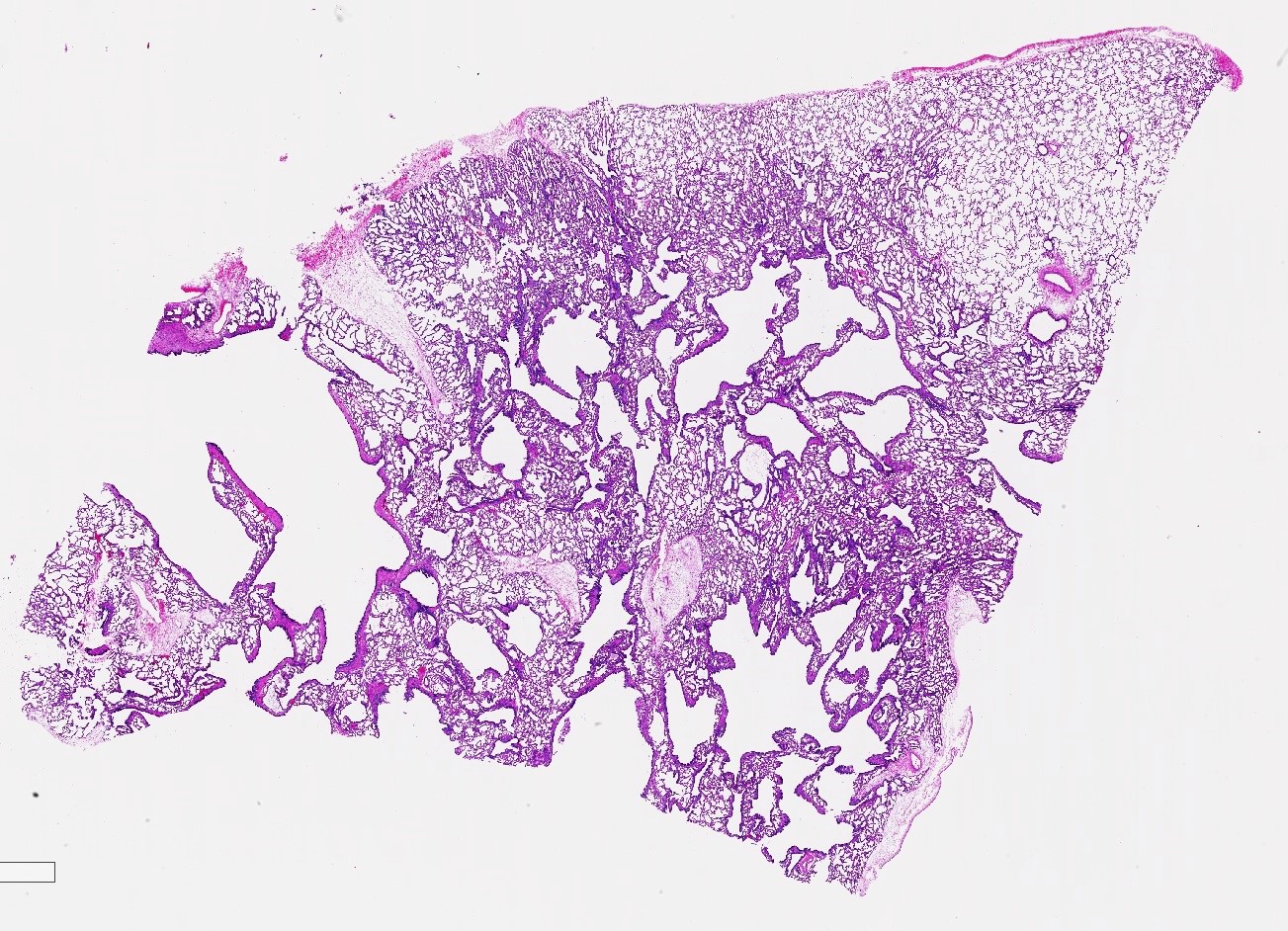
Type 1 CPAM
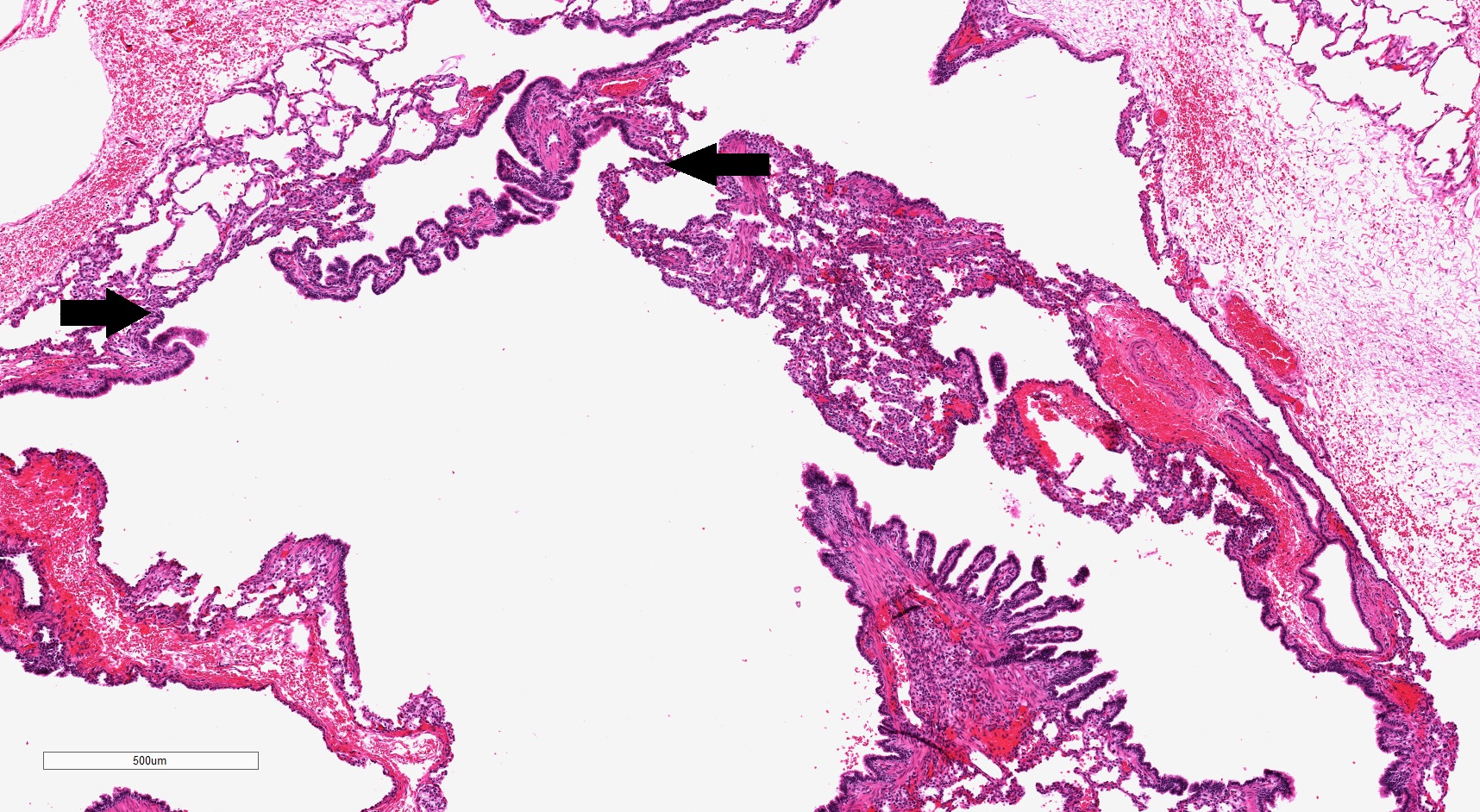
Type 1 CPAM complexity
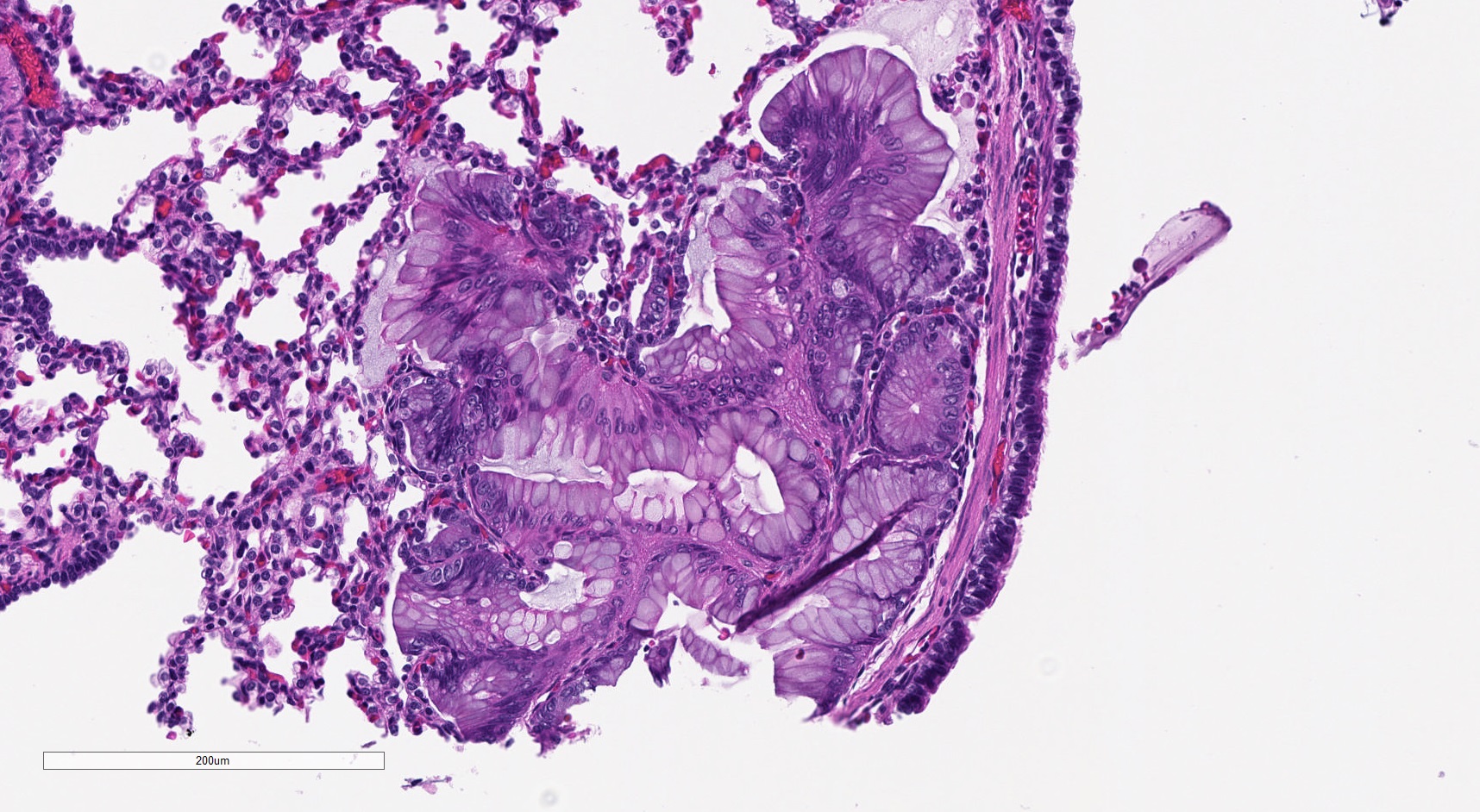
Mucinous cell clusters
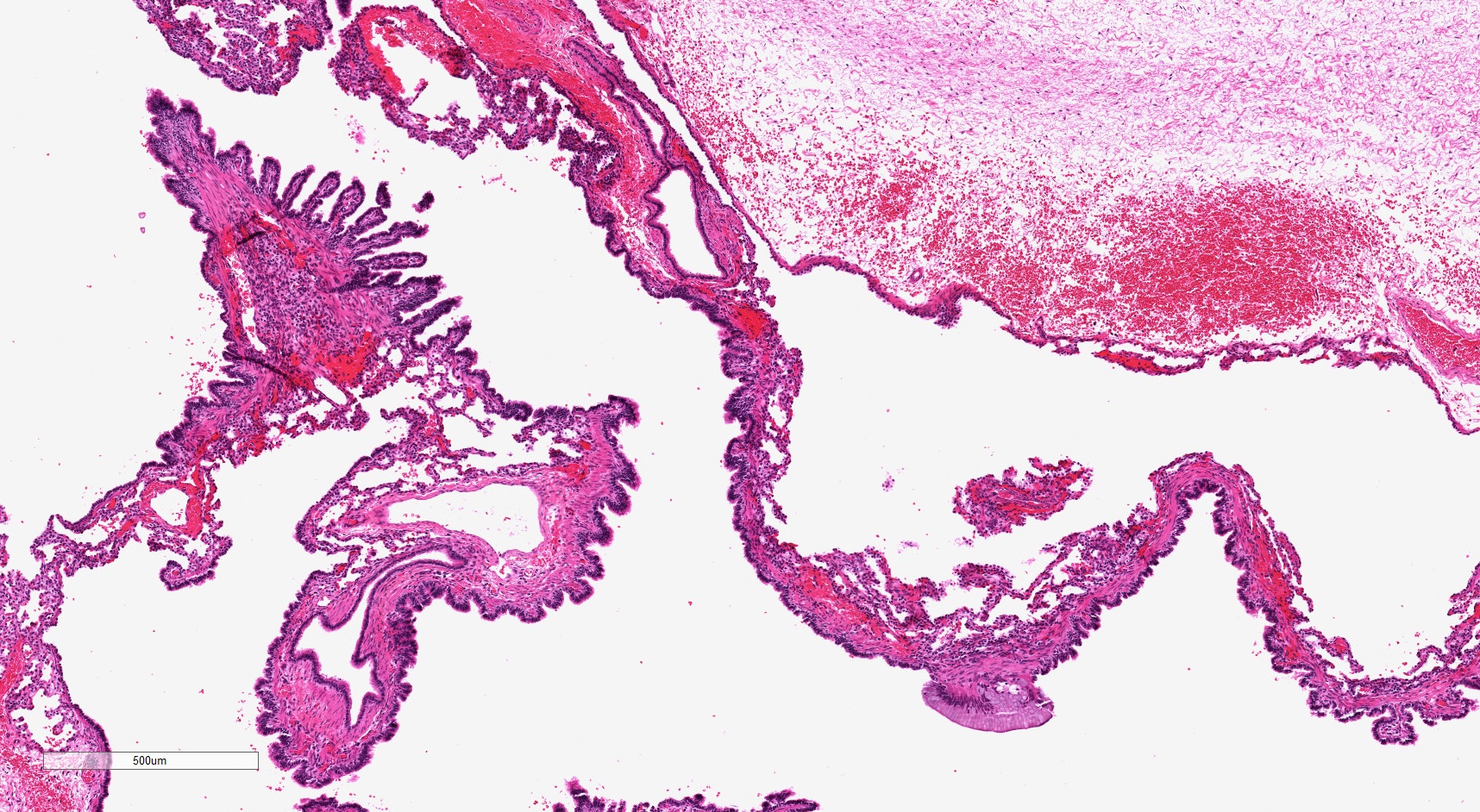
Small mucinous cell cluster
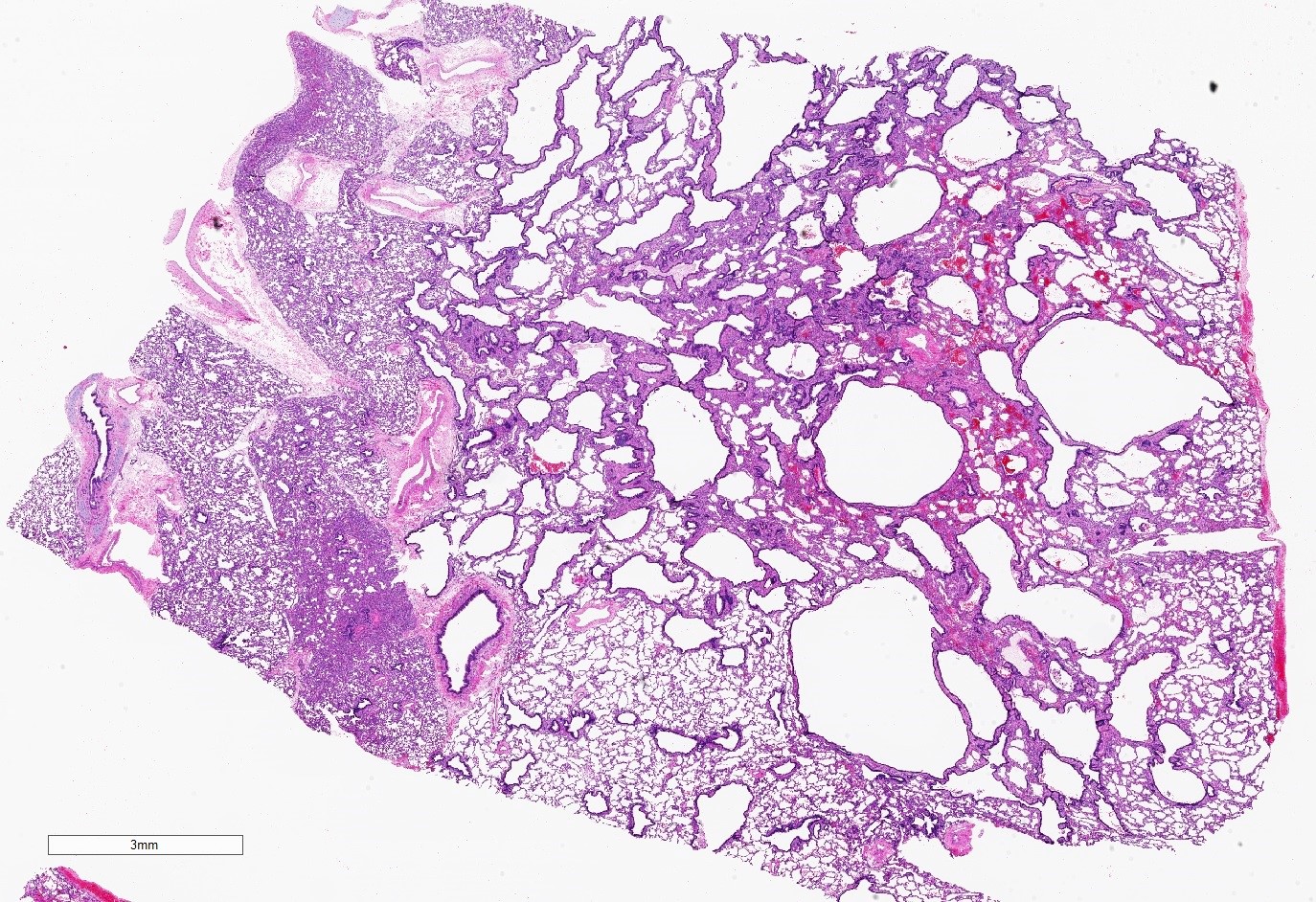
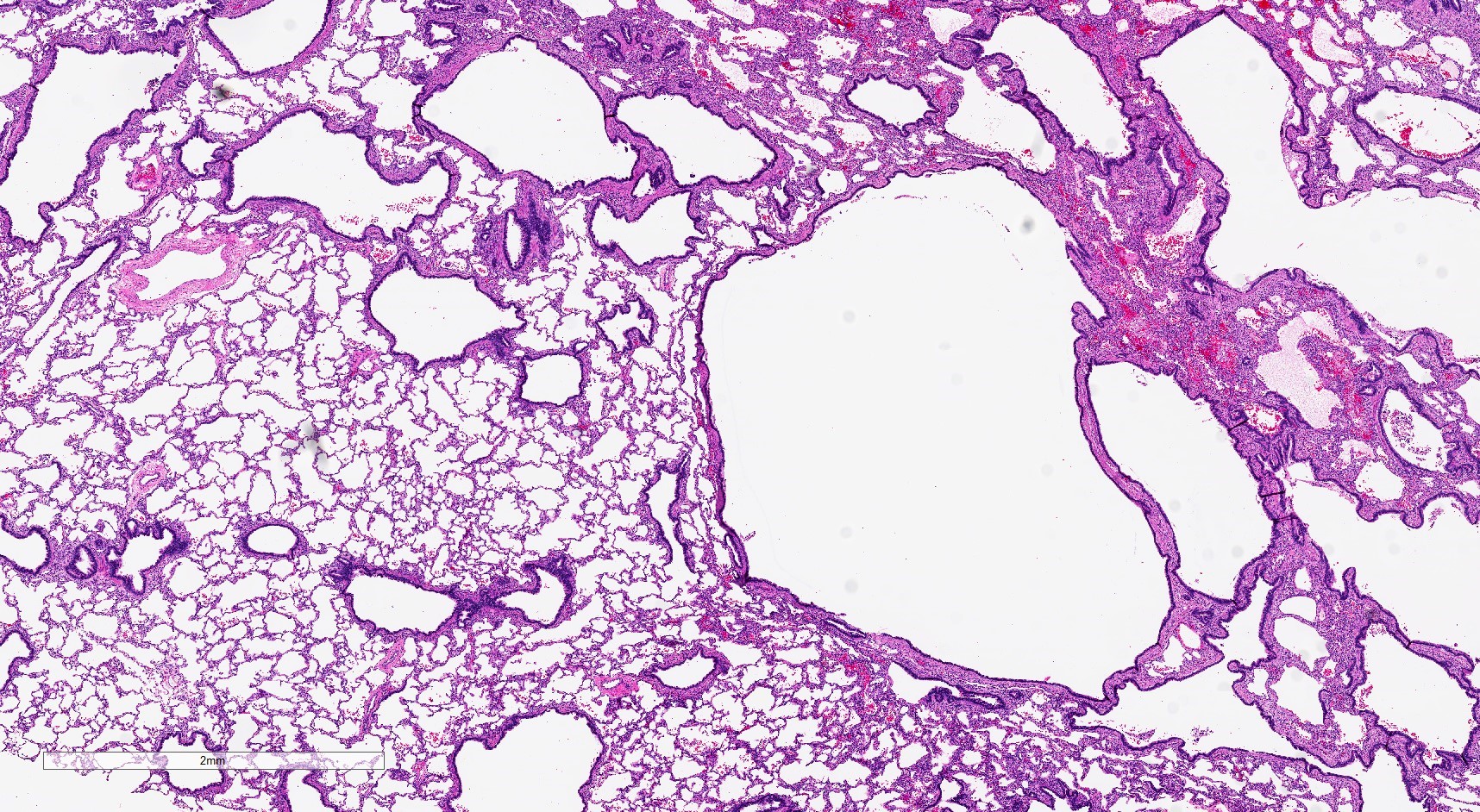
Type 2 CPAM
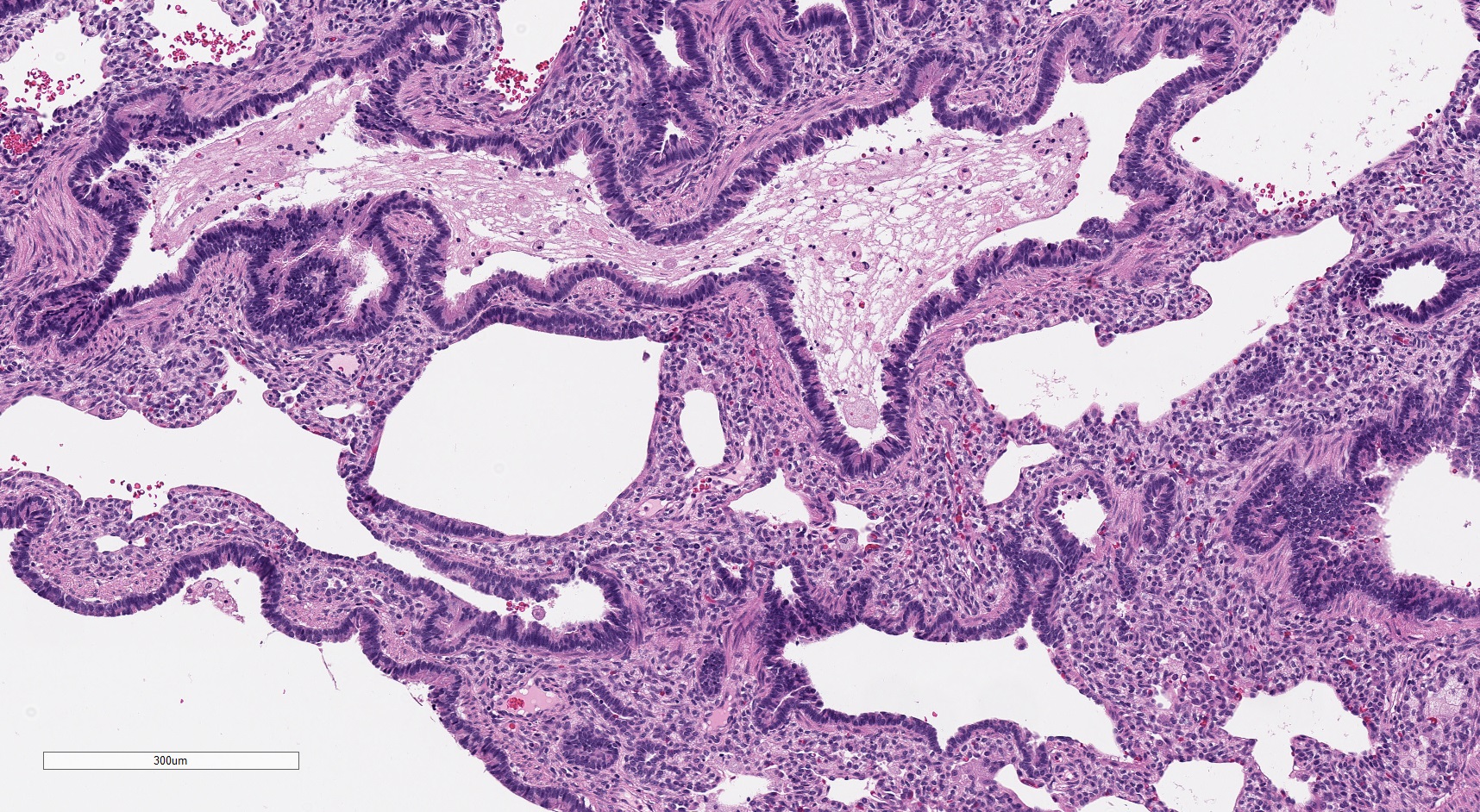
Type 2 CPAM - mucostasis
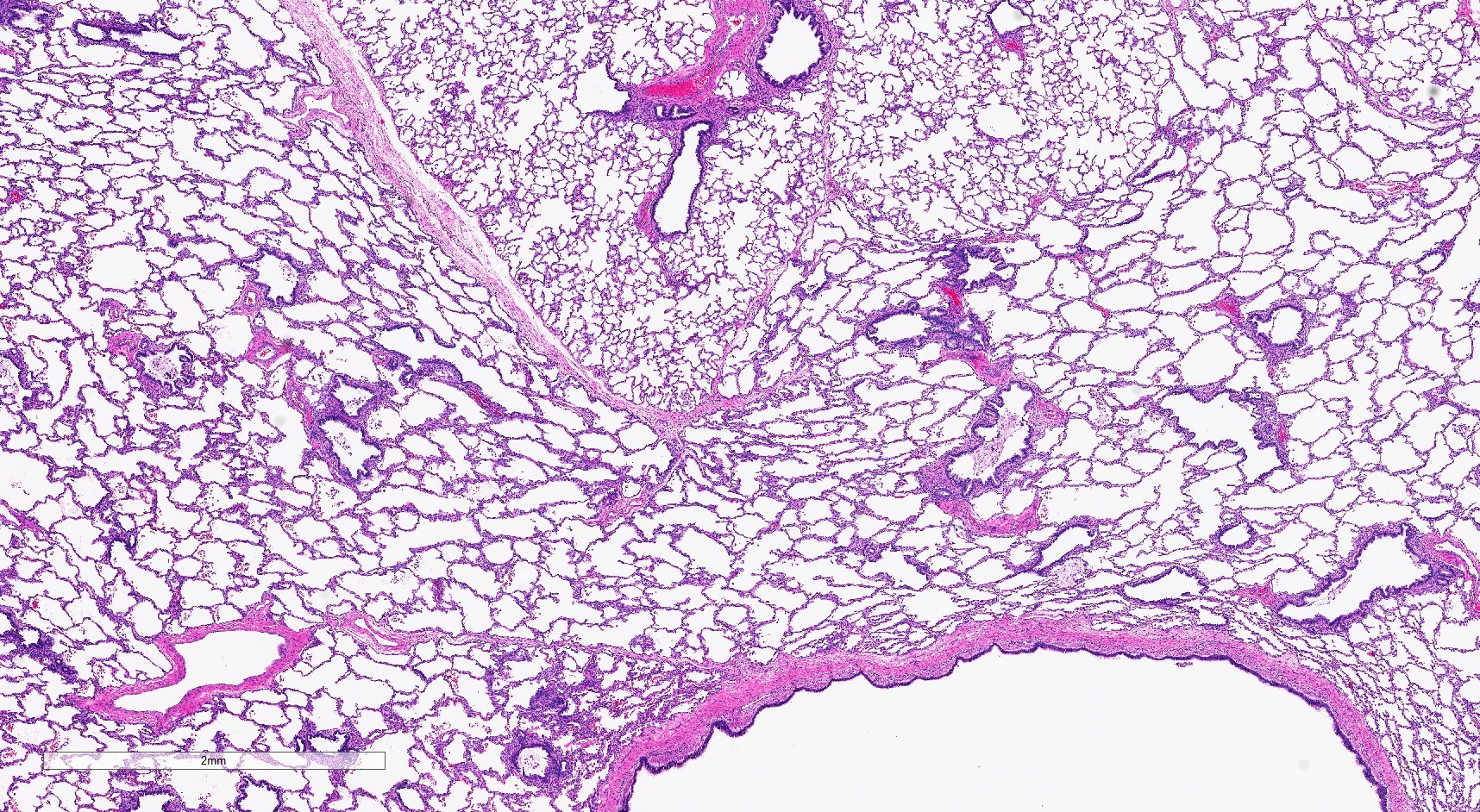
Congenital lobar emphysema

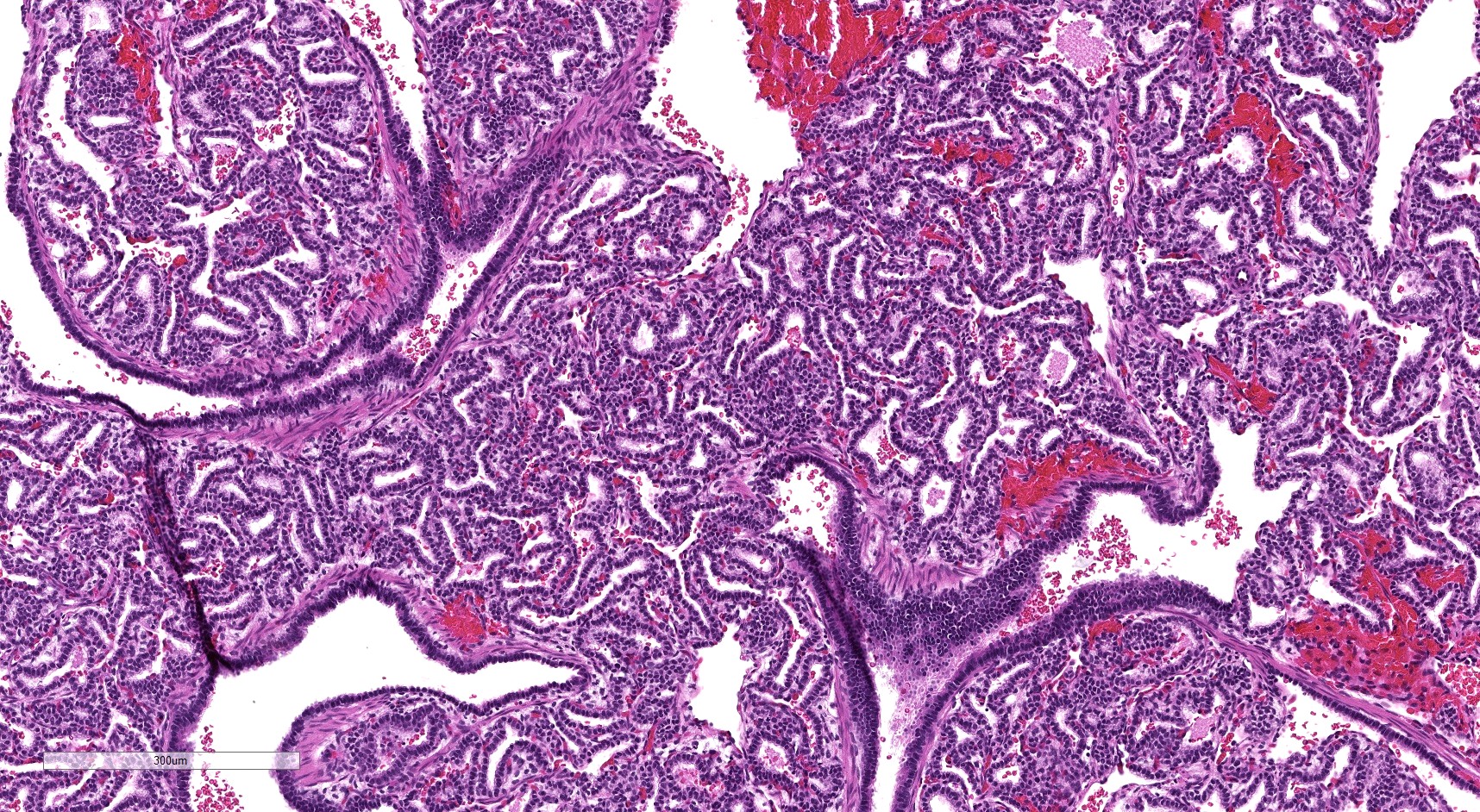
Type 3 CPAM
Positive stains
Molecular / cytogenetics description
- Nearly all classic type 1 CPAMs (> 98%) have an activating KRAS mutation in the mucinous and nonmucinous epithelium
- KRAS p.G12D is the most common mutation reported in type 1 CPAMs
- Type 3 CPAMs are associated with activating KRAS mutations or mutations in other genes involved in cell cycle regulation and growth
- 50% have a KRAS mutation and p.G12V is the most common variant
- 2 reported cases with mutations in other genes (FGFR2 and NEK9)
- Presence of mucinous cell clusters is highly specific but not entirely sensitive for a KRAS mutation
- Reference: Mod Pathol 2022 Jul 6 [Epub ahead of print]
Sample pathology report
- Lung, right upper lobe, lobectomy:
- Type 1 (large cyst) congenital pulmonary airway malformation, with atypical mucinous cells (see comment)
- Comment: Sections throughout the lobe demonstrate numerous, variably enlarged and cystically dilated airspaces lined by ciliated respiratory epithelium. Foci of epithelial complexity, including scattered foci of mucinous cells (1 - 2 mm each) and branching papillae are seen. The mucinous cells are seen in association with the epithelium of the cysts and in the adjacent alveoli. Children and young adults with unresected or incompletely resected type 1 CPAMs have rarely developed metastatic mucinous adenocarcinoma. While the mucinous cells in this lesion are histologically indistinguishable from mucinous adenocarcinoma in situ, recurrence / metastasis has not been reported following complete resection by lobectomy in infancy. Type 1 CPAMs are associated with mosaic activating KRAS mutations throughout the epithelium and should be entirely resected (Mod Pathol 2022 Jul 6 [Epub ahead of print]).
- Type 2 (small cyst) congenital pulmonary airway malformation (see comment)
- Comment: The epithelium is flat and simple with prominent mucostasis, consistent with bronchial atresia sequence.
Differential diagnosis
- Type I pleuropulmonary blastoma:
- Gross: peripherally located multiseptated cyst
- Back to back cysts without intervening alveoli
- Septa may have primitive CD56+ round cells but may be regressed
- Associated with DICER1 mutations
- Intrapulmonary bronchogenic cyst:
- Gross: single thick walled cyst
- Single cyst wall does not connect to adjacent alveolar spaces
- Usually cartilage or submucosal glands in wall
- May have type 2 CPAM changes adjacent (due to obstruction during development)
- Intrapulmonary sequestration:
- Gross: systemic feeding vessel entering away from the hilum
- Large cartilage containing airway paired with the feeding vessel (aberrant hilum)
- Parenchymal maldevelopment is similar to type 2 CPAMs ranging from mild alveolar enlargement to type 2 CPAM-like change
- Pulmonary hypertensive changes may be present in older patients
Practice question #1

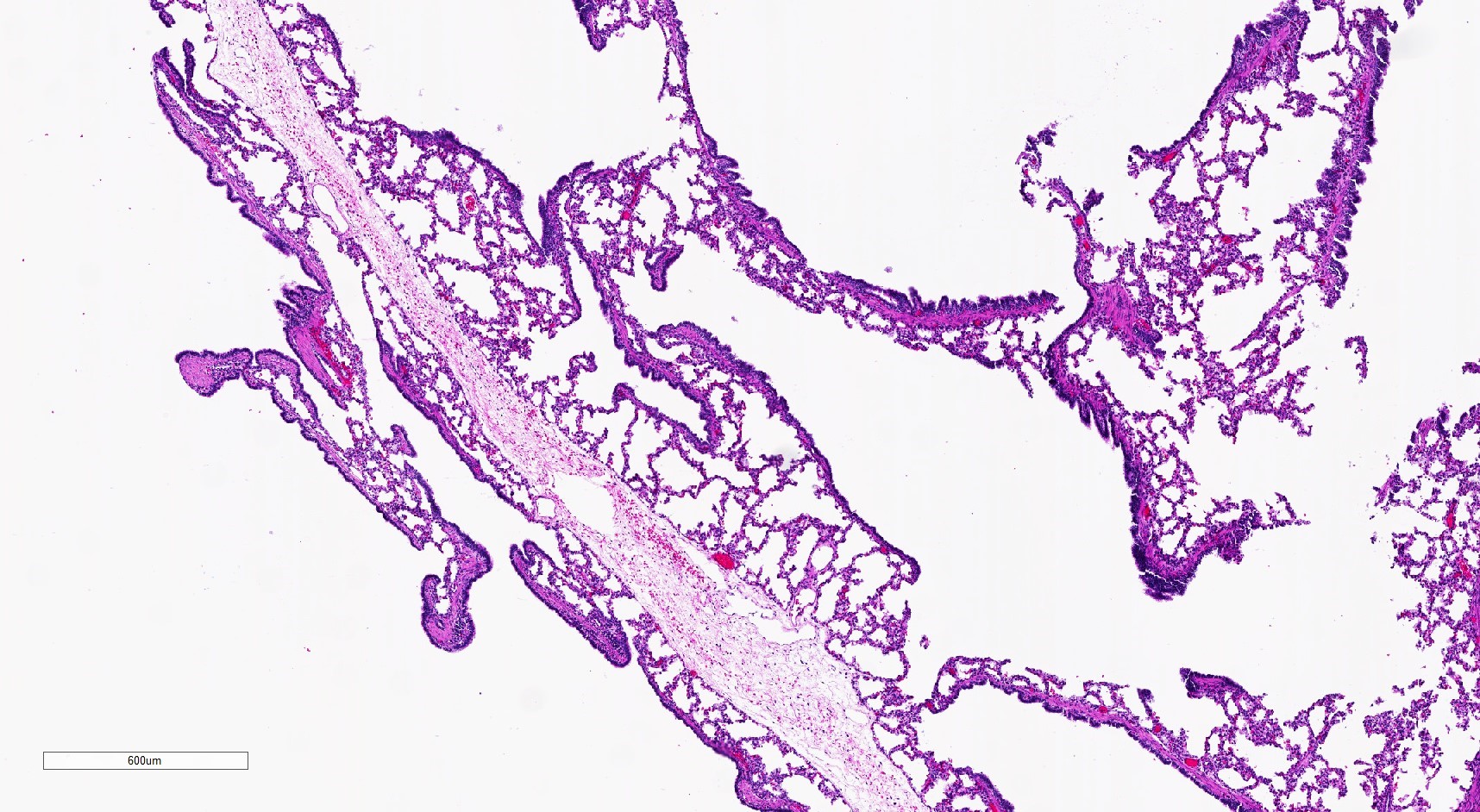
Routine prenatal imaging revealed a cystic lung lesion. The pregnancy was otherwise uncomplicated and the lesion was resected at 3 weeks of age following a term delivery. What is the most appropriate diagnosis?
- Type 1 (large cyst) CPAM
- Type 2 (small cyst) CPAM
- Type 3 CPAM
- Type I pleuropulmonary blastoma
Practice answer #1
A. Type 1 (large cyst) CPAM. At least 1 cyst is relatively large. The thicker cyst walls frequently directly transition abruptly into alveolar type spaces. Some of the cyst epithelium has prominent papillary infoldings.
Comment Here
Reference: Congenital pulmonary airway malformation (CPAM)
Comment Here
Reference: Congenital pulmonary airway malformation (CPAM)
Practice question #2
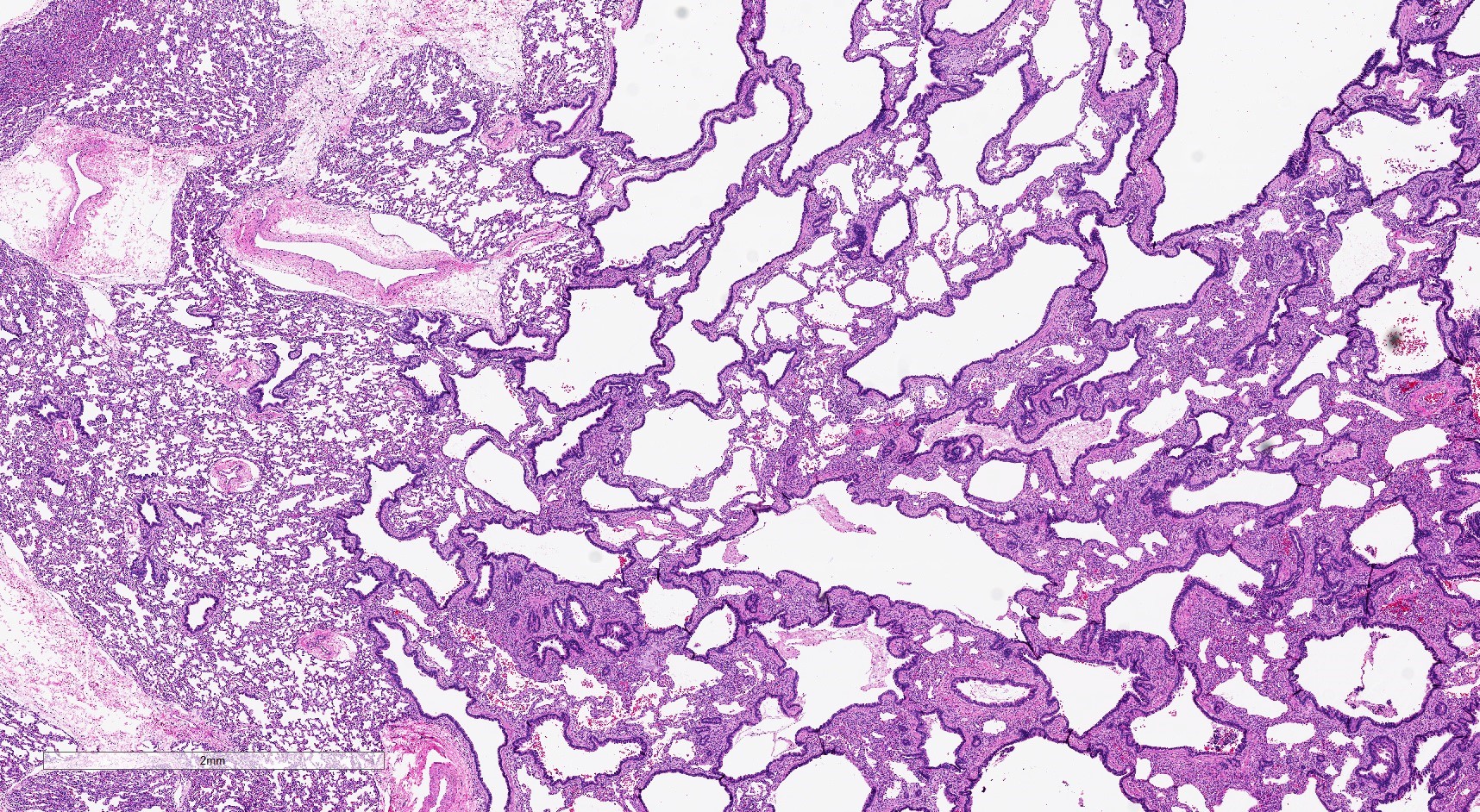
A right upper lobe resection is performed in a 2 month old baby for an asymptomatic cystic lesion identified on routine prenatal ultrasound. It is most likely associated with which of the following?
- An aberrant systemic feeding vessel
- Bronchial atresia
- DICER1 syndrome
- Mosaic activating KRAS mutations
Practice answer #2
B. Bronchial atresia. The cysts here are relatively small with a continuous wall (few transitions to alveolar type walls) and a flat epithelium without complexity. This is a type 2 CPAM, which is associated with bronchial atresia.
Comment Here
Reference: Congenital pulmonary airway malformation (CPAM)
Comment Here
Reference: Congenital pulmonary airway malformation (CPAM)
