
11 March 2020 - Case of the Month #489
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. Brian Werstein and Dr. Nicole K. Andeen, Oregon Health and Science University, Portland, Oregon (USA), for contributing this case and to Dr. Brian Werstein for writing the discussion.
Case of the Month #489
Clinical history:
A 3 year old previously healthy boy presented with malignant hypertension, seizures and electrolyte derangements. Imaging demonstrated a left sided abdominal mass which appeared suprarenal on cross sectional imaging.
Histopathology images:
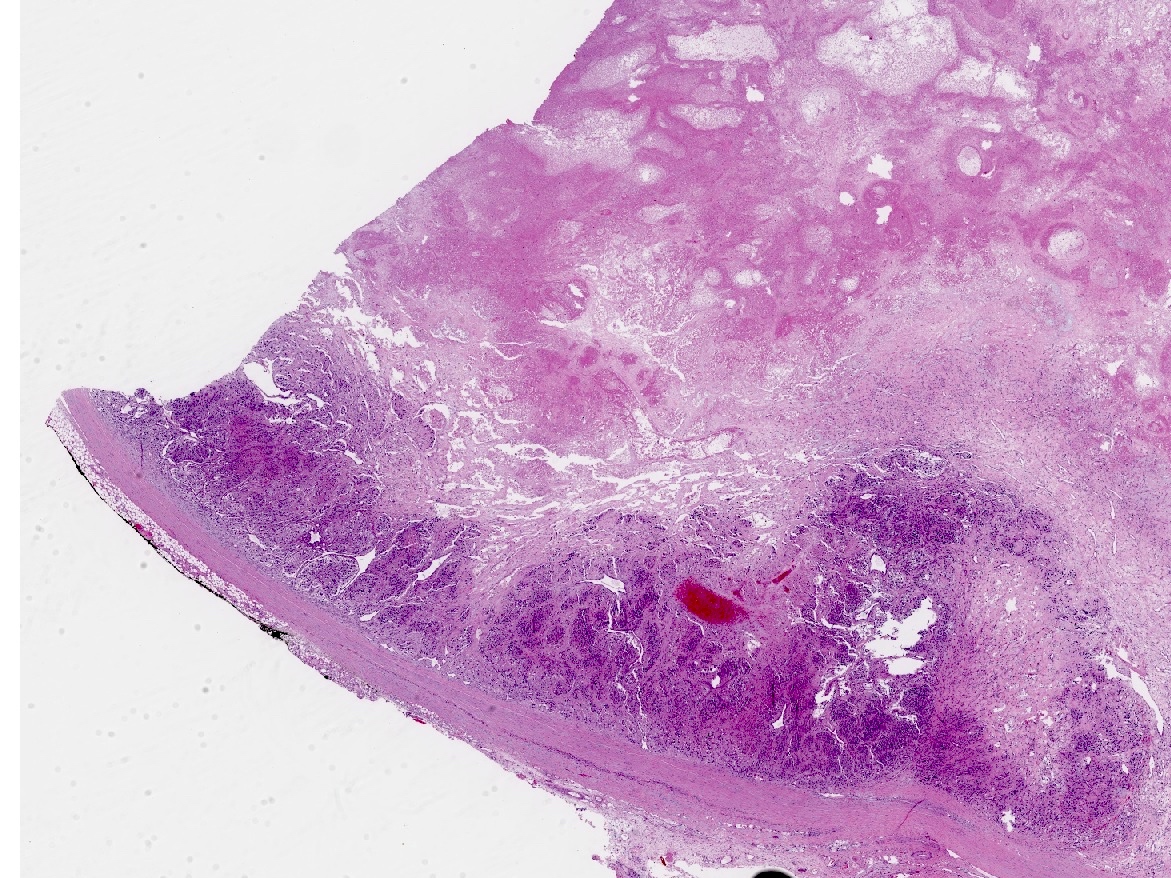
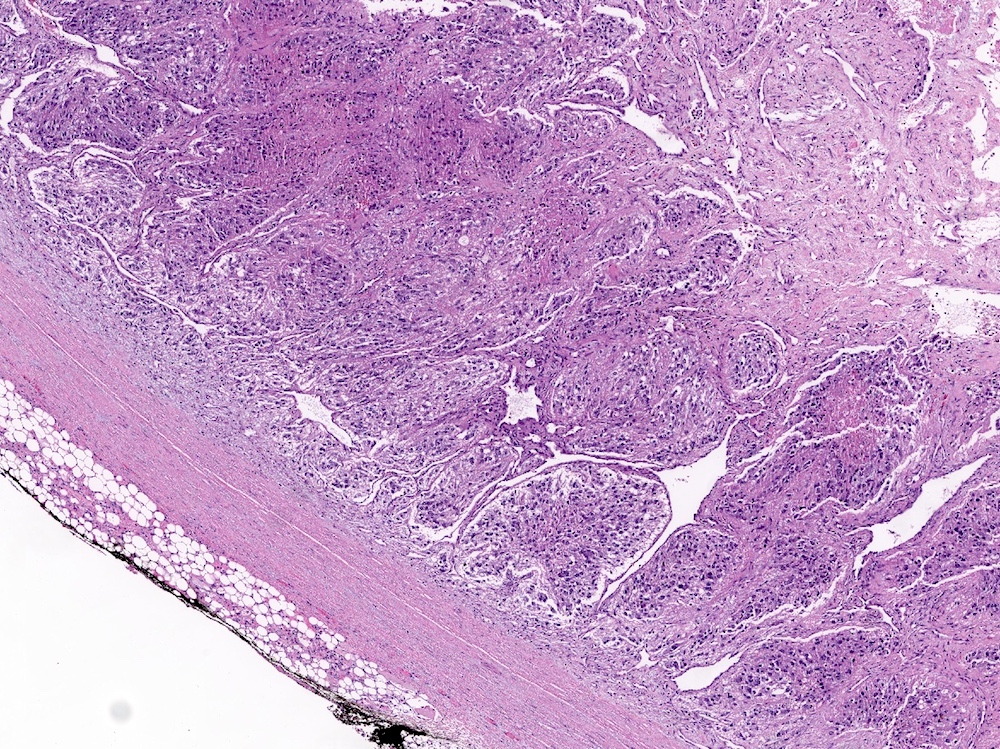

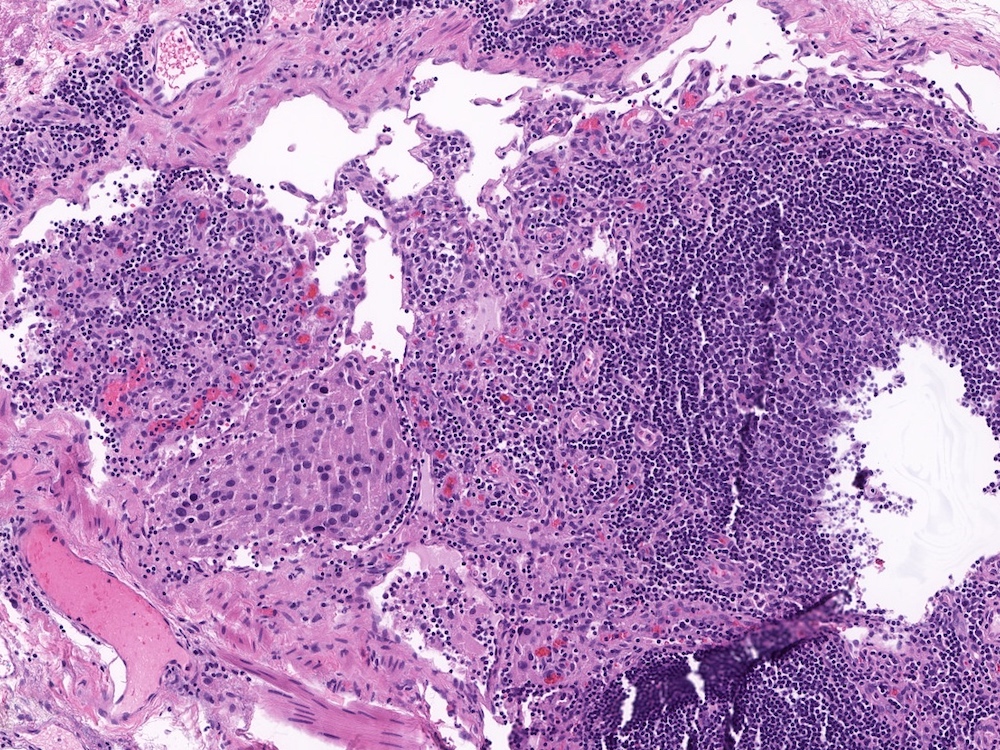
What is your diagnosis?
Diagnosis: Pheochromocytoma with aggressive histologic features; metastatic pheochromocytoma to one lymph node
Test question (answer at the end):
Which of the following genes are NOT associated with hereditary pheochromocytoma?
A. NF1
B. VHL
C. RET
D. MEN1
E. MAX
Stains:
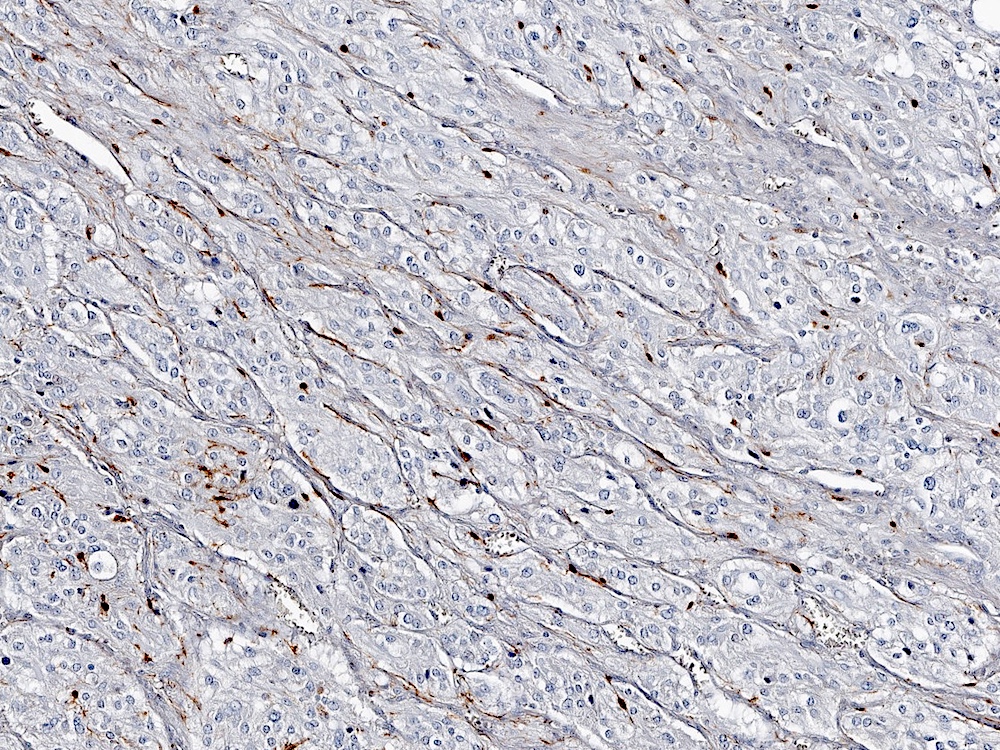
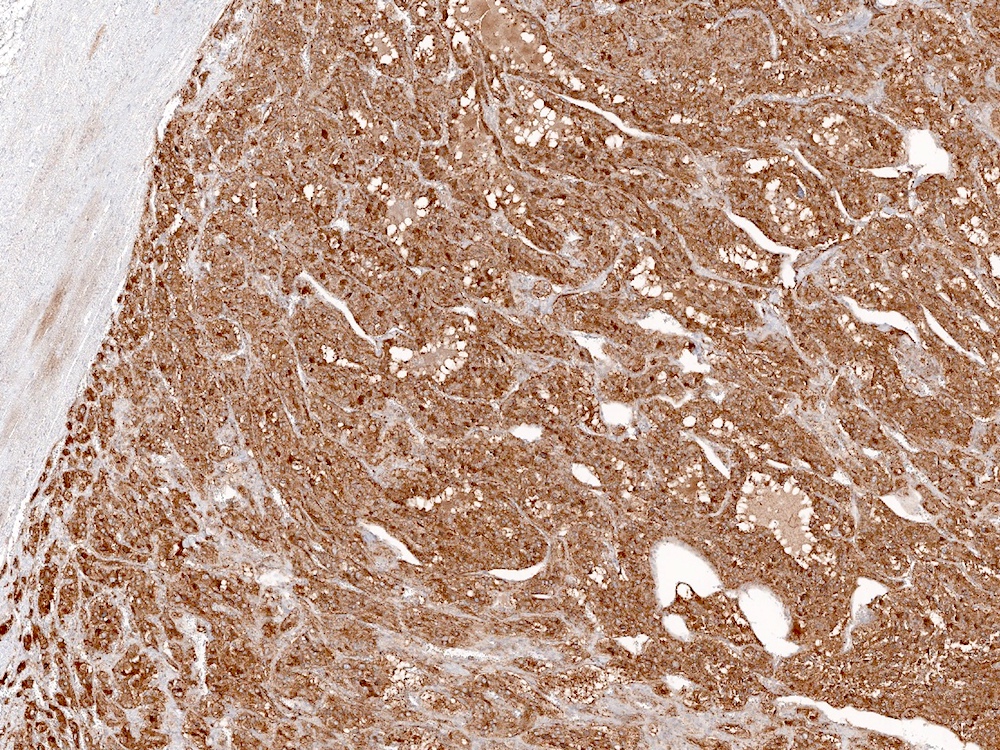
Discussion:
Pheochromocytoma is a catecholamine secreting tumor arising from the chromaffin cells of the adrenal medulla. The tumor typically presents with symptoms related to the catecholamine secretion: sweating, tachycardia, headaches and refractory hypertension. Histologically, these tumors typically have a zellballen (small nested) architecture composed of polygonal cells with basophilic to amphophilic granular cytoplasm and round to oval nuclei with prominent nucleoli and variably vesicular chromatin. Pleomorphism may be striking. The tumor cells stain positive for chromogranin and synaptophysin, while sustentacular cells surrounding the tumor nests can be highlighted with an S100 stain.
The accepted indication of malignancy is metastatic disease (the WHO no longer recommends a diagnosis of malignant or benign pheochromocytoma, but prefers metastatic pheochromocytoma). However, scoring systems have been devised to help predict the malignant potential of these tumors. One of these systems is the Pheochromocytoma of the Adrenal gland Scaled Score (PASS). The PASS score can be used to separate tumors with a potential for biologically aggressive behavior (PASS ≥ 4) from tumors that behave in a benign fashion (PASS < 4), as outlined below (Am J Surg Pathol 2002;26:551).
PASS score
Two points each for:
One point each for:
Interpretation:
In this case, for example, large nested growth, spindling, central and confluent necrosis, high mitotic rate (5/10 HPF) and vascular invasion were identified for a total PASS score of 9, indicating a tumor with aggressive behavior. This was manifested by the identification of a lymph node metastasis.
Pheochromocytomas and malignant pheochromocytomas in particular are rare in children. They have been associated with a variety of genetic conditions, including multiple endocrine neoplasia (MEN), particularly type II, von Hippel-Lindau (VHL), neurofibromatosis type I (NF1), Sturge-Weber and succinate dehydrogenase (SDH) gene mutations. Multiple other genes have also been implicated in hereditary pediatric pheochromocytoma (Front Pediatr 2017;5:155). In cases such as these, it is prudent to recommend genetic testing / counselling and, if available, perform immunohistochemistry to identify SDH deficient tumors.
Treatment for metastatic pheochromocytoma is complete surgical resection. Radioactive iodine I 131-metaiodobenzylguanidine (131I-MIBG) or other radioactive isotopes may be used, as well as chemotherapy with cyclophosphamide, vincristine and dacarbazine or radiation. In a small study in children, survival rates were 78%, 62% and 31%, at 5, 10 and 15 years, respectively (Front Pediatr 2017;5:155).
The differential diagnosis includes an adrenal cortical neoplasm and composite pheochromocytoma. Adrenal cortical neoplasms were excluded based on morphology and negative SF1, MelanA and inhibin immunostains. Composite pheochromocytoma is a tumor containing pheochromocytoma in combination with another neurogenic tumor, such as neuroblastoma, ganglioneuroma, ganglioneuroblastoma or peripheral nerve sheath tumor. In this case it was particularly important, given the patient’s age, to thoroughly sample the tumor and rule out a neuroblastoma component. Identification of neuroblastoma can also be aided with use of PHOX2B immunohistochemical stain, which was negative in this case.
Test question answer:
D. MEN1
Inherited mutations in the MEN1 gene are associated with multiple endocrine neoplasia (MEN) type 1. MEN type 1 is associated with tumors of the parathyroid glands, the pituitary gland and the pancreas, but there is no significant association with pheochromocytoma. Mutations in the rest of the listed genes are associated with hereditary pheochromocytoma (Endocr Pract 2017;23:690).
All cases are archived on our website. To view them sorted by case number, diagnosis or category, visit our main Case of the Month page. To subscribe or unsubscribe to Case of the Month or our other email lists, click here.
Thanks to Dr. Brian Werstein and Dr. Nicole K. Andeen, Oregon Health and Science University, Portland, Oregon (USA), for contributing this case and to Dr. Brian Werstein for writing the discussion.

Case of the Month #489
Clinical history:
A 3 year old previously healthy boy presented with malignant hypertension, seizures and electrolyte derangements. Imaging demonstrated a left sided abdominal mass which appeared suprarenal on cross sectional imaging.
Histopathology images:
What is your diagnosis?
Click here for diagnosis, test question and discussion:
Diagnosis: Pheochromocytoma with aggressive histologic features; metastatic pheochromocytoma to one lymph node
Test question (answer at the end):
Which of the following genes are NOT associated with hereditary pheochromocytoma?
A. NF1
B. VHL
C. RET
D. MEN1
E. MAX
Stains:
S100
Synaptophysin
Discussion:
Pheochromocytoma is a catecholamine secreting tumor arising from the chromaffin cells of the adrenal medulla. The tumor typically presents with symptoms related to the catecholamine secretion: sweating, tachycardia, headaches and refractory hypertension. Histologically, these tumors typically have a zellballen (small nested) architecture composed of polygonal cells with basophilic to amphophilic granular cytoplasm and round to oval nuclei with prominent nucleoli and variably vesicular chromatin. Pleomorphism may be striking. The tumor cells stain positive for chromogranin and synaptophysin, while sustentacular cells surrounding the tumor nests can be highlighted with an S100 stain.
The accepted indication of malignancy is metastatic disease (the WHO no longer recommends a diagnosis of malignant or benign pheochromocytoma, but prefers metastatic pheochromocytoma). However, scoring systems have been devised to help predict the malignant potential of these tumors. One of these systems is the Pheochromocytoma of the Adrenal gland Scaled Score (PASS). The PASS score can be used to separate tumors with a potential for biologically aggressive behavior (PASS ≥ 4) from tumors that behave in a benign fashion (PASS < 4), as outlined below (Am J Surg Pathol 2002;26:551).
PASS score
Two points each for:
- Large nests or diffuse growth (> 10% of tumor volume)
- Central (middle of large nests) or confluent tumor necrosis (not degenerative change)
- High cellularity
- Cellular monotony
- Tumor cell spindling (even if focal)
- Mitotic figures > 3/10 HPF
- Atypical mitotic figure(s)
- Extension into adipose tissue
One point each for:
- Vascular invasion
- Capsular invasion
- Profound nuclear pleomorphism
- Nuclear hyperchromasia
Interpretation:
- PASS score >= 4: potentially aggressive behavior
- PASS score < 4: benign behavior
In this case, for example, large nested growth, spindling, central and confluent necrosis, high mitotic rate (5/10 HPF) and vascular invasion were identified for a total PASS score of 9, indicating a tumor with aggressive behavior. This was manifested by the identification of a lymph node metastasis.
Pheochromocytomas and malignant pheochromocytomas in particular are rare in children. They have been associated with a variety of genetic conditions, including multiple endocrine neoplasia (MEN), particularly type II, von Hippel-Lindau (VHL), neurofibromatosis type I (NF1), Sturge-Weber and succinate dehydrogenase (SDH) gene mutations. Multiple other genes have also been implicated in hereditary pediatric pheochromocytoma (Front Pediatr 2017;5:155). In cases such as these, it is prudent to recommend genetic testing / counselling and, if available, perform immunohistochemistry to identify SDH deficient tumors.
Treatment for metastatic pheochromocytoma is complete surgical resection. Radioactive iodine I 131-metaiodobenzylguanidine (131I-MIBG) or other radioactive isotopes may be used, as well as chemotherapy with cyclophosphamide, vincristine and dacarbazine or radiation. In a small study in children, survival rates were 78%, 62% and 31%, at 5, 10 and 15 years, respectively (Front Pediatr 2017;5:155).
The differential diagnosis includes an adrenal cortical neoplasm and composite pheochromocytoma. Adrenal cortical neoplasms were excluded based on morphology and negative SF1, MelanA and inhibin immunostains. Composite pheochromocytoma is a tumor containing pheochromocytoma in combination with another neurogenic tumor, such as neuroblastoma, ganglioneuroma, ganglioneuroblastoma or peripheral nerve sheath tumor. In this case it was particularly important, given the patient’s age, to thoroughly sample the tumor and rule out a neuroblastoma component. Identification of neuroblastoma can also be aided with use of PHOX2B immunohistochemical stain, which was negative in this case.
Test question answer:
D. MEN1
Inherited mutations in the MEN1 gene are associated with multiple endocrine neoplasia (MEN) type 1. MEN type 1 is associated with tumors of the parathyroid glands, the pituitary gland and the pancreas, but there is no significant association with pheochromocytoma. Mutations in the rest of the listed genes are associated with hereditary pheochromocytoma (Endocr Pract 2017;23:690).
