Adrenal gland & paraganglia
Pheochromocytoma / paraganglioma
Pheochromocytoma
Editorial Board Member: Bonnie Choy, M.D.
Deputy Editors-in-Chief: Raul S. Gonzalez, M.D., Maria Tretiakova, M.D., Ph.D.


Last author update: 3 January 2023
Last staff update: 9 May 2024
Copyright: 2002-2025, PathologyOutlines.com, Inc.
PubMed Search: Pheochromocytoma adrenal
Table of Contents
Definition / general | Essential features | Terminology | ICD coding | Epidemiology | Sites | Pathophysiology | Etiology | Clinical features | Diagnosis | Laboratory | Radiology description | Radiology images | Prognostic factors | Case reports | Treatment | Clinical images | Gross description | Gross images | Frozen section description | Frozen section images | Microscopic (histologic) description | Microscopic (histologic) images | Virtual slides | Cytology images | Positive stains | Negative stains | Electron microscopy description | Electron microscopy images | Molecular / cytogenetics description | Molecular / cytogenetics images | Sample pathology report | Differential diagnosis | Practice question #1 | Practice answer #1 | Practice question #2 | Practice answer #2 | Practice question #3 | Practice answer #3Cite this page: Lehman KA, Zynger DL. Pheochromocytoma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/adrenalpheochromocytoma.html. Accessed October 2nd, 2025.
Definition / general
- Paraganglioma of the adrenal medulla composed of chromaffin cells that produce catecholamines
- Second most common entity in adrenalectomy specimens and 7% of primary adrenal tumors (J Laparoendosc Adv Surg Tech A 2012;22:22, Eur J Cancer Prev 2004;13:403)
Essential features
- Paraganglioma of the adrenal medulla composed of chromaffin cells that produce catecholamines
- Most often sporadic but associated with genetic syndromes in approximately 30 - 40% of cases and pathogenic mutations are identifiable in half of cases; therefore genetic testing in all cases may be warranted (Am J Surg Pathol 2021;45:1155)
- Malignant in approximately 10% of cases
- No single biomarker or histologic feature predicts malignancy: pheochromocytoma of the adrenal gland scaled score (PASS) provides prognosis based on histologic features (Am J Surg Pathol 2002;26:551)
- Positive for chromogranin, synaptophysin and S100
Terminology
- Pheochromocytoma may also be referred to as adrenal paraganglioma
- Distinct from paraganglioma / extra-adrenal pheochromocytoma, which arises from chromaffin cells of sympathetic ganglia
- Distinct from composite pheochromocytoma, which are tumors composed of pheochromocytoma plus ganglioneuroma, ganglioneuroblastoma, neuroblastoma or peripheral nerve sheath tumors
- Zellballen: used to describe characteristic nested architecture
- Pheochromocytoma of the adrenal gland scaled score (PASS): prognostic score based on histologic features (Am J Surg Pathol 2002;26:551)
ICD coding
Epidemiology
- Most are sporadic tumors that present in the fourth and fifth decade (J Am Coll Surg 2009;209:727)
- Approximately 33% are associated with hereditary syndromes (see Etiology, Molecular) (J Clin Oncol 2005;23:8812, Am J Surg Pathol 2021;45:1155)
- Present 1 - 2 decades earlier than sporadic
- More commonly bilateral versus sporadic (J Clin Oncol 2005;23:8812, Endocrinology 2011;152:2133)
Sites
- Adrenal gland medulla
Pathophysiology
- Normal chromaffin cell function relies on:
- 4 key enzymes: tyrosine hydroxylase (rate limiting), aromatic L amino acid decarboxylase, dopamine ß hydroxylase (makes norepinephrine) and phenylethanolamine N methyltransferase (converts norepinephrine to epinephrine)
- Storage and vesicular transport of catecholamines via vesicular monoamine transporters
- Exocytosis of storage vesicles following sympathetic neuronal stimulation and stimulatory paracrine signaling in chromaffin cells in response to stress
- Tightly regulated by neuronal stimulation (acetylcholine, pituitary adenylate cyclase activating peptide) and multiple peptides thought to participate in negative feedback and autocrine / paracrine regulation
- Alterations in components of normal catecholamine biosynthesis may drive proliferation of chromaffin cells, oversecretion of catecholamines and dysregulation of chromaffin cell division
- Chromaffin cells in pheochromocytoma release catecholamines without sympathetic innervation or neuronal stimulation
- Evidence of differences in levels of certain regulatory molecules/enzymes in normal adrenal medulla versus neoplasms (Table 1)
- 3 clusters of mutations thought to drive pathophysiology have recently been described: (Cancer Cell 2017;31:181)
- Cluster 1: induce a hypoxic response from cells in the absence of true hypoxia, resulting in excessive methylation of phenylethanolamine N methyltransferase
- Primarily germline mutations
- Phenotype: intra- or extra-adrenal; produce mainly norepinephrine, little epinephrine
- Examples: dysregulation of the TCA cycle (SDH mutations) or VHL related tumors
- Cluster 2: increased MAP kinase and P13K / AKT pathway activity, which increases cell proliferation and catecholamine synthesis
- Approximately 20% germline; primarily somatic mutations
- Phenotype: intra-adrenal, typically produce epinephrine
- Examples: RET, NF1, TMEM-127, MAX, HRAS
- Cluster 3: alter Wnt pathway signaling; little is known but attenuate phenylethanolamine N methyltransferase expression
- All known mutations are somatic
- Phenotype: intra-adrenal; produce epinephrine in quantities intermediate between cluster 1 and cluster 2
- Examples: CSDE1, MAML3
Table 1. Elements of normal catecholamine biosynthesis potentially involved in the pathophysiology of pheochromocytoma (Cancers (Basel) 2019;11:E1121)
Element Role in normal catecholamine biosynthesis Alterations in pheochromocytoma versus normal adrenal medulla Neuropeptide Y Increases tyrosine hydroxylase expression, intracellular calcium and neoangiogenesis Increased plasma levels, increased adrenal mRNA expression Adrenomedullin Increases adrenal blood flow and catecholamine release Increased plasma levels Pituitary adenylate cyclase activating peptide Increases transcription, stimulates activity of synthetic enzymes, increased expression of regulatory peptides High mRNA expression Chromogranin Protein sorting, vesicle stability and hormone storage Increased plasma levels, increased mRNA expression Tyrosine hydroxylase (rate limiting), aromatic L amino acid decarboxylase, dopamine β hydroxylase Enzymes required for catecholamine synthesis Upregulation and increased activity Carboxypeptidase E Peptide sorting, activation of prosurvival genes and increased survival of chromaffin cells in hypoxia and nutrient deprivation Increased mRNA expression, correlation with rapid growth and malignancy Galanin Stimulates catecholamine and glucocorticoid secretion Increased levels - Cluster 1: induce a hypoxic response from cells in the absence of true hypoxia, resulting in excessive methylation of phenylethanolamine N methyltransferase
Etiology
- Approximately 30% are hereditary and include the following autosomal dominant disorders:
- Von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 2 (MEN2)
- Neurofibromatosis type 1 (NF1)
- Familial paraganglioma (Cancer Genet 2012;205:1, J Clin Endocrinol Metab 2017;102:3296, Am J Surg Pathol 2021;45:1155)
- Suspect hereditary etiology when:
- Family history or symptoms of a syndrome
- Bilateral tumors
- Presentation of tumor < 45 years of age
- Paraganglioma concurrent with pheochromocytoma
Clinical features
- Classic triad of episodic headaches, sweating and tachycardia present in about 30% (Eur J Endocrinol 2004;150:681)
- Other common features: palpitations, anxiety, postural hypotension and paroxysmal hypertension (normal in 10 - 30%) (Curr Probl Cancer 2014;38:7, Exp Clin Endocrinol Diabetes 2016;124:372)
- 10 - 30% present with adrenal incidentaloma and no symptoms (subclinical pheochromocytoma) (Arch Surg 2007;142:870)
- Additional features / symptoms may be present in hereditary syndromes associated with pheochromocytoma (see Molecular / cytogenetics)
Diagnosis
- Clinical suspicion with laboratory testing and imaging for confirmation
- Rarely may be detected in a needle core needle biopsy
- Histologic appearance overlaps with normal adrenal medulla
- Diagnose or raise the consideration to prevent severe surgical complications associated with unsuspected pheochromocytoma for which patient does not receive adrenergic blockade
Laboratory
- Tumors secrete norepinephrine, epinephrine or dopamine
- Catecholamines or their metabolites detected in serum or urine via high performance liquid chromatography or tandem mass spectroscopy
- Plasma and urine free metanephrines recommended as test of choice with sensitivities of 99% and 97%, respectively (JAMA 2002;287:1427)
- If borderline elevation, follow up clonidine suppression test with measurement of plasma normetanephrine (J Clin Endocrinol Metab 2003;88:2656)
- Chromogranin A secreted by chromaffin cells detected in serum in up to 91% of cases (Clin Endocrinol (Oxf) 2006;65:287)
- Rarely, other adrenal lesions yield pheochromocytoma-like symptoms with similar borderline or elevated metanephrines (pseudopheochromocytoma)
- Reports of other catecholamine producing lesions in the adrenal gland: adenoma, hydatid cyst, oncocytoma, endothelial cyst and malignant melanoma (Ultrastruct Pathol 2012;36:287, Arch Pathol Lab Med 2002;126:1530, ScientificWorldJournal 2006;6:2420, Hormones (Athens) 2011;10:76, Pathology 2014;46:364, Hormones (Athens) 2016;15:283)
Radiology description
- CT is imaging modality of choice for visualizing lesions ≥ 0.5 cm
- MRI second line if patient has metastatic disease or contraindication to radiation (Best Pract Res Clin Endocrinol Metab 2019:101346)
- CT features that suggest malignancy versus adenoma:
- Unenhanced attenuation of ≤ 10 HU
- Spherical shape, rim enhancement and sharp necrotic borders more frequent in pheochromocytomas versus other adrenal tumors (Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2019;163:212)
- Other imaging (if negative CT with positive clinical and laboratory findings):
- 123I metaiodobenzylguanidine (MIBG) uptake scintigraphy sensitive and specific for sporadic pheochromocytoma ≥ 1 cm (Best Pract Res Clin Endocrinol Metab 2019:101346)
- PET imaging: 18F fluorodeoxyglucose (18F-FDG), 18F fluoro l dihydroxyphenylalanine (18F-DOPA), 68Ga labeled somatostatin receptor analogs (PET Clin 2014;9:43, Eur J Nucl Med Mol Imaging 2014;41:494)
Radiology images
Images hosted on other servers:

CT and scintigraphy

MRI
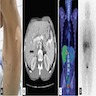
PET / CT and scintigraphy
Prognostic factors
- No specific histologic or molecular markers to differentiate benign from malignant
- See Table 2 for clinical and pathologic features associated with malignancy
- There is a TNM staging system for which pT is based on tumor size (≤ 5 cm or > 5 cm) and depth of invasion
- Worse prognosis associated with (Am J Surg Pathol 2021;45:1155)
- Hispanic ethnicity
- Larger (metastatic mean diameter ~7 - 9 cm) tumors
- Heavier (metastatic mean 280 g) tumors
- Metastasis
- Better prognosis associated with (Am J Surg Pathol 2021;45:1155)
- Family history
- Younger age
- Lack of symptoms / incidental finding
- Bilaterality
- Presence of metastases defines malignancy: most commonly to lymph nodes, bone, liver and lung (Endocr Relat Cancer 2007;14:569)
- ~10% of pheochromocytomas are metastatic (Endocr Relat Cancer 2007;14:569, J Surg Oncol 2015;112:815)
- 5 year and 10 year survival ~65% and 35%, respectively (J Surg Oncol 2015;112:815)
- 6 - 9% of excised tumors have local recurrence (Surgery 2014;156:1523)
- Median time to recurrence ~35 months (Surgery 2014;156:1523)
- 25% of recurrences do not demonstrate metanephrine elevation so surveillance should include annual CT imaging (Surgery 2014;156:1523)
Table 2. Clinical and pathologic features of pheochromocytoma associated with malignancy
| Clinical features | Younger age |
| Secretion of norepinephrine | |
| Gross features | Tumor size |
| Tumor weight | |
| Histologic features | Periadrenal adipose tissue invasion |
| Mitotic rate | |
| Atypical mitoses | |
| Necrosis | |
| Cellular spindling | |
| Marked nuclear pleomorphism | |
| Cellular monotony | |
| Large nests or diffuse growth | |
| High cellularity | |
| Capsular invasion | |
| Vascular invasion | |
| Hyperchromasia | |
| Ancillary studies | Higher Ki67 |
| SDHB, VHL, RET mutations |
Case reports
- 3 year old boy with malignant hypertension, seizures and electrolyte derangements (Case #489)
- 22 year old woman with type IIb von Hippel-Lindau (Med Princ Pract 2016;25:196)
- 30 year old man with diabetic ketoacidosis (BMJ Case Rep 2016;2016:bcr2016216961)
- 35 year old man with NF1 and asymptomatic pheochromocytoma (Korean J Intern Med 2018;33:214)
- 44 year old man with suspected myocardial infarction (BMJ Case Rep 2015;2015:bcr2015211472)
- 45 year old man with severe hyperglycemia after glucocorticoid administration (J Med Case Rep 2019;13:3)
- 64 year old man with intermittent abdominal pain (Am J Case Rep 2017;18:826)
- 69 year old man with classic symptoms (J Med Case Rep 2016;10:279)
Treatment
- Surgical resection via total adrenalectomy unless bilateral tumors
- For bilateral tumors: cortical sparing adrenalectomy to reduce complications related to lifelong glucocorticoid replacement (JAMA Netw Open 2019;2:e198898)
- Preoperative considerations: administer alpha adrenergic blockers prior to surgery to prevent intraoperative hypertensive crisis, volume expansion to counter catecholamine mediated volume contraction
- Metastatic tumor also treated with surgical resection if possible
- Though not curative, surgical excision may improve survival (J Surg Oncol 2015;112:815)
- Chemotherapy for unresectable tumor: cyclophosphamide, vincristine and dacarbazine
Clinical images
Images hosted on other servers:
Café au lait spots and axillary freckling in NF1

Intraoperative
images of giant
pheochromocytoma
Gross description
- Well circumscribed and unencapsulated
- Solid white to red-brown hemorrhagic cut surface
- Mean diameter of benign cases: 4 - 6.5 cm (Can J Surg 2008;51:276, Am J Surg Pathol 2002;26:551)
- Some tumors may be less than 1 cm (J Clin Endocrinol Metab 2017;102:3296)
- Malignant (metastatic) larger: mean diameter ~7 - 9 cm (J Urol 2011;185:1583, J Clin Endocrinol Metab 2017;102:3296, Am J Surg Pathol 2002;26:551)
- Mean weight: ~150 g for benign and 280 g for malignant (Am J Surg Pathol 2002;26:551)
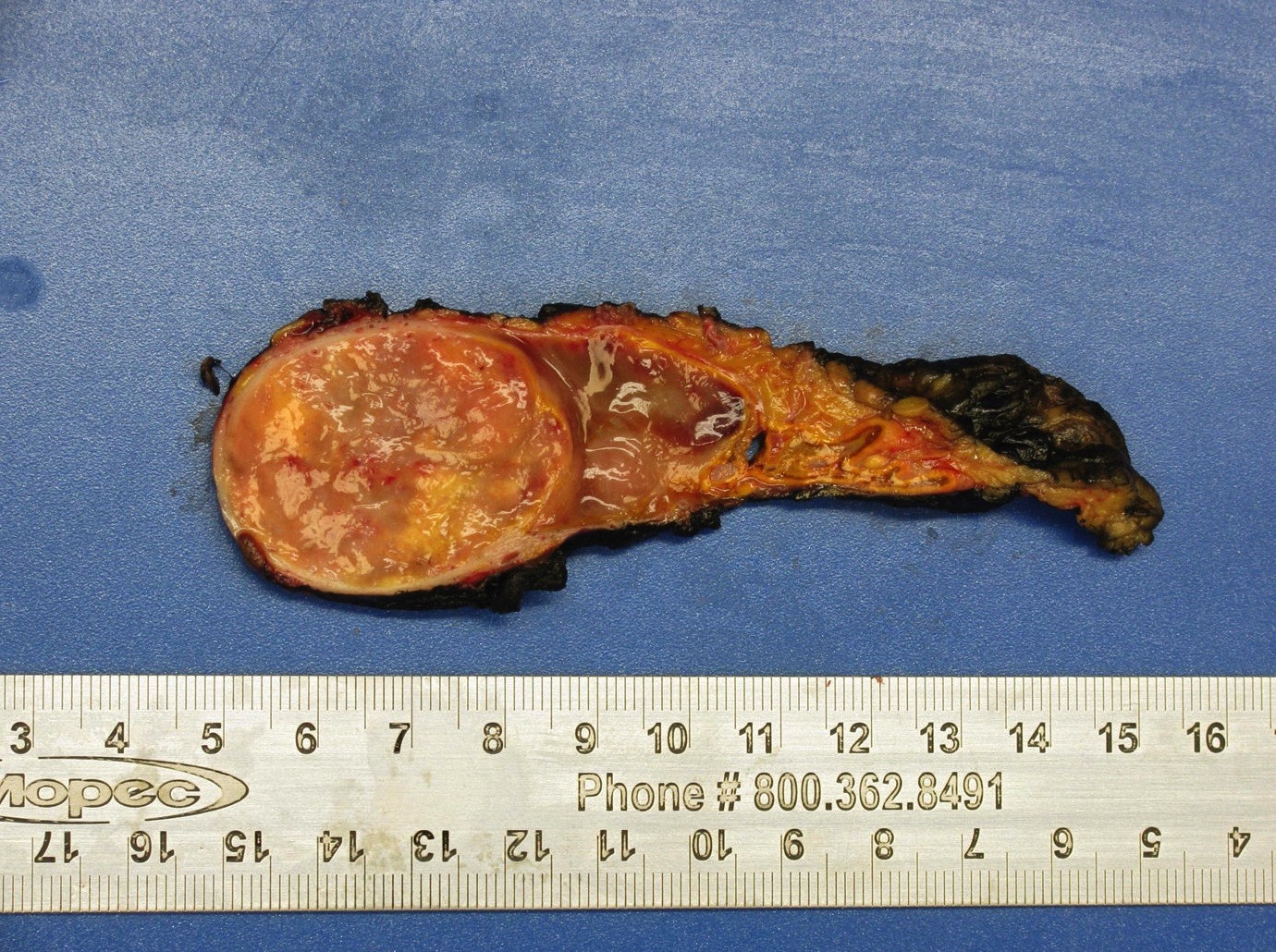
Gross images
Contributed by Debra L. Zynger, M.D. (Case #319)
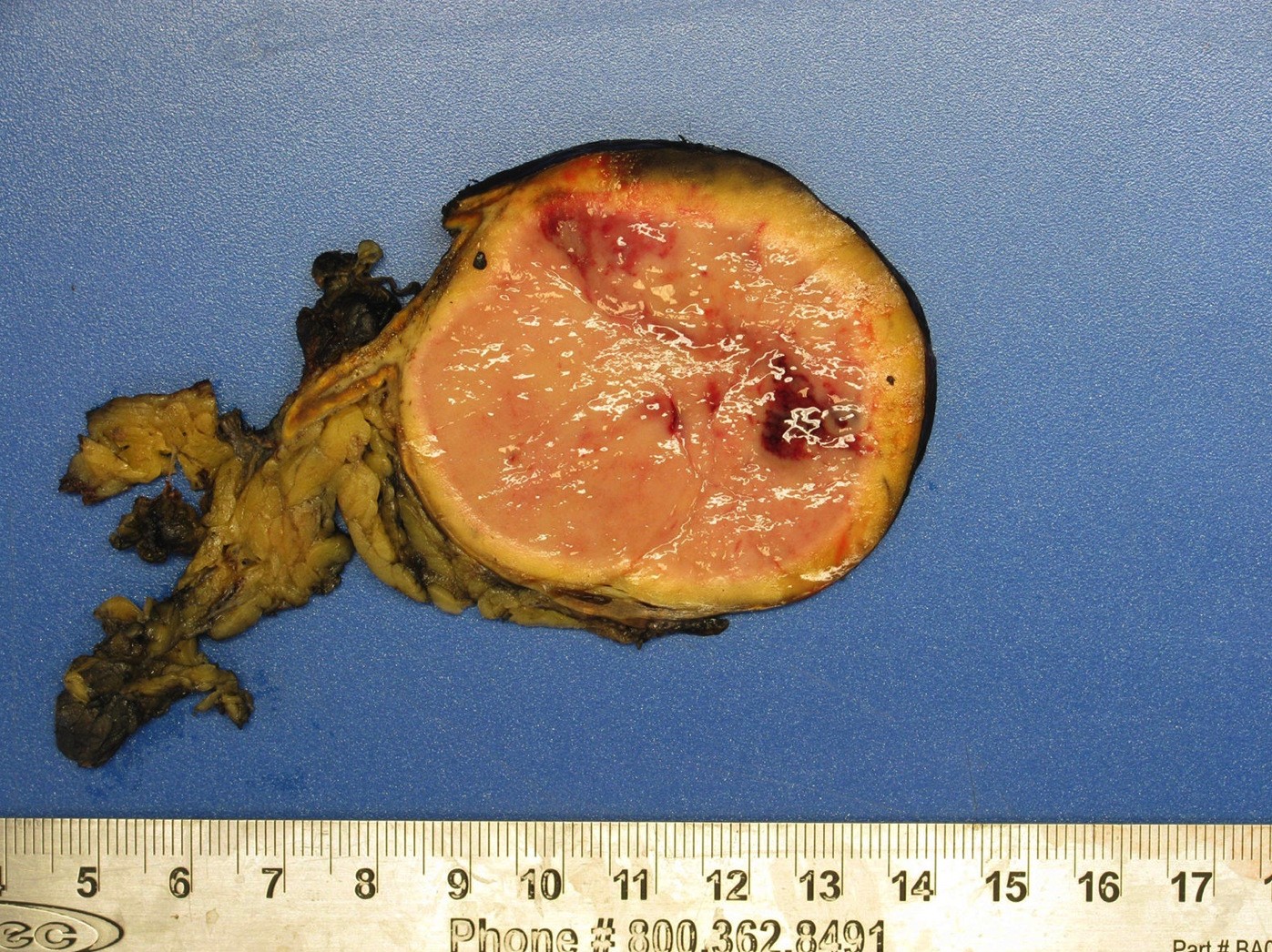
Well circumscribed tan mass
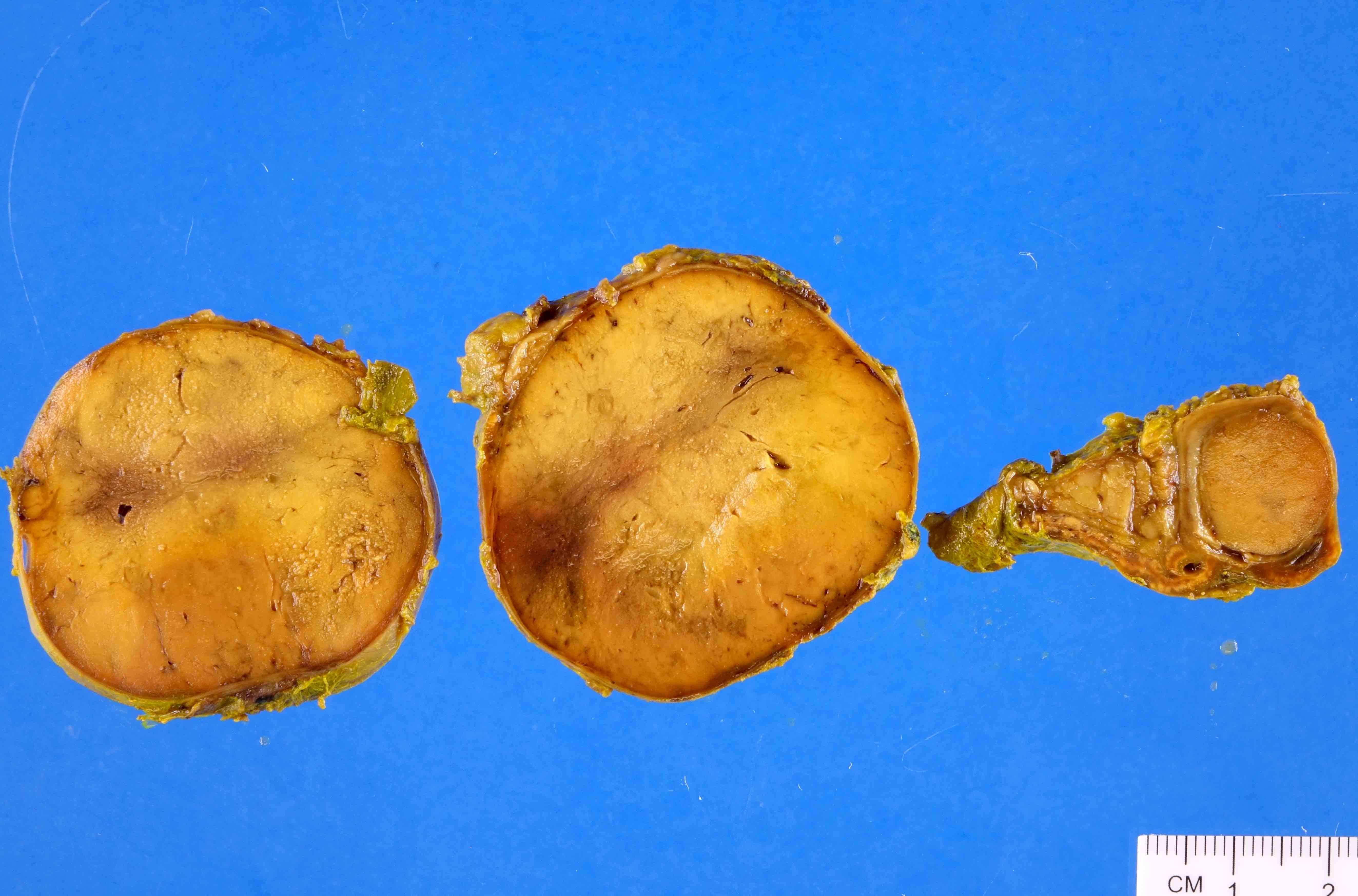
Well circumscribed tan mass
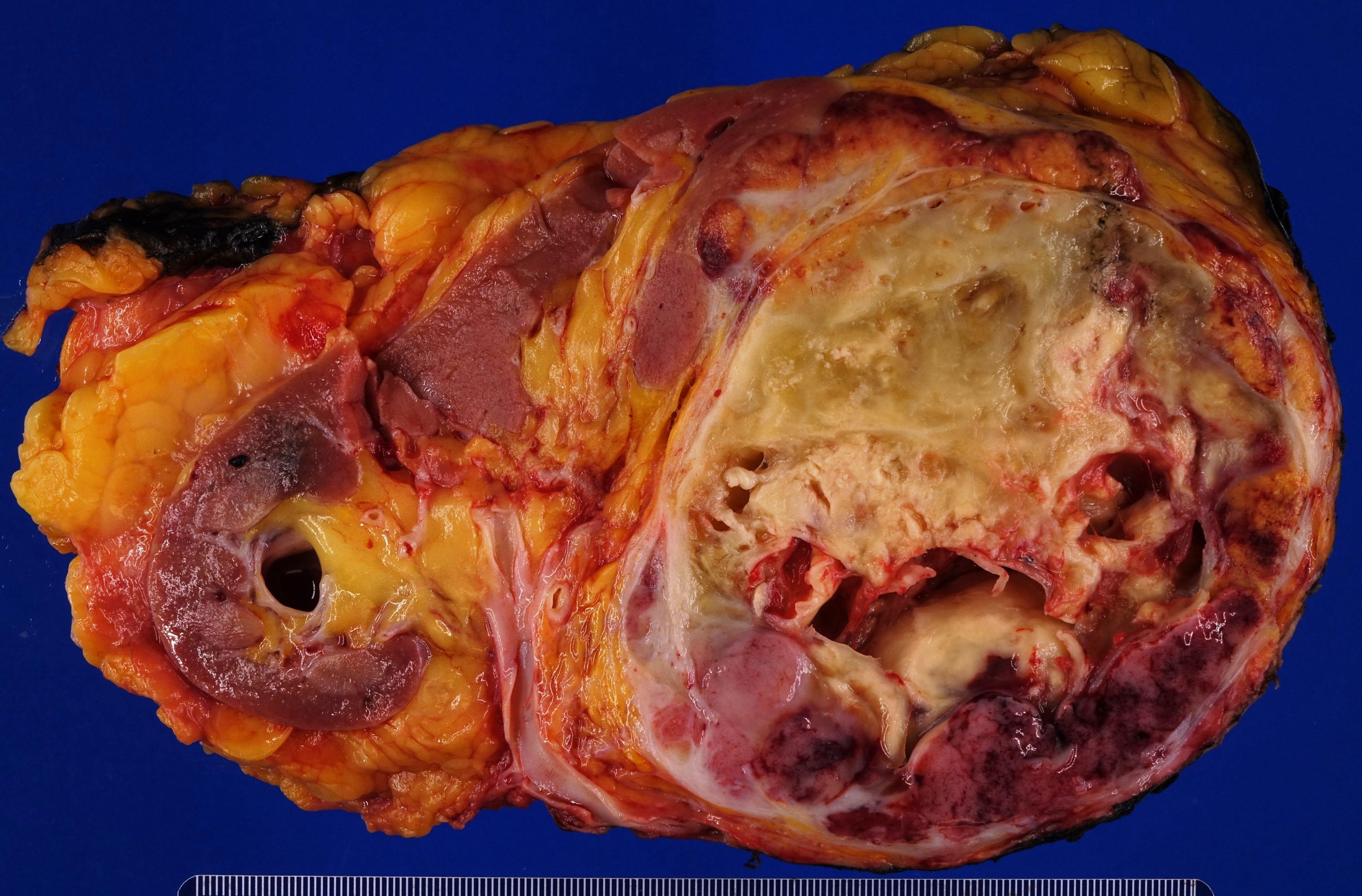
Renal invasion
Composite
pheochromocytoma
Frozen section description
- Vaguely nested architecture
- Cellular crowding
- Vesicular, overlapping nuclei
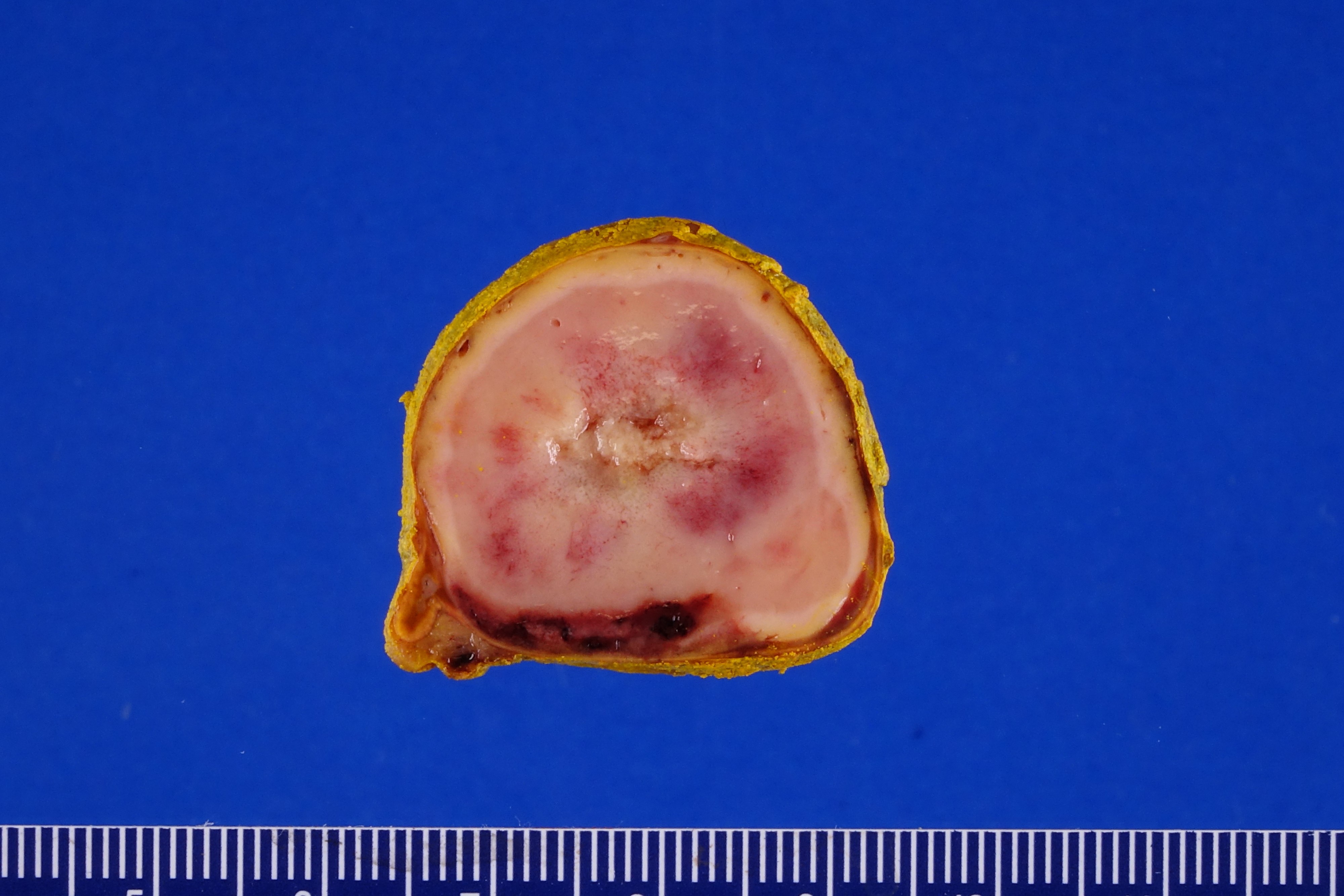
Frozen section images
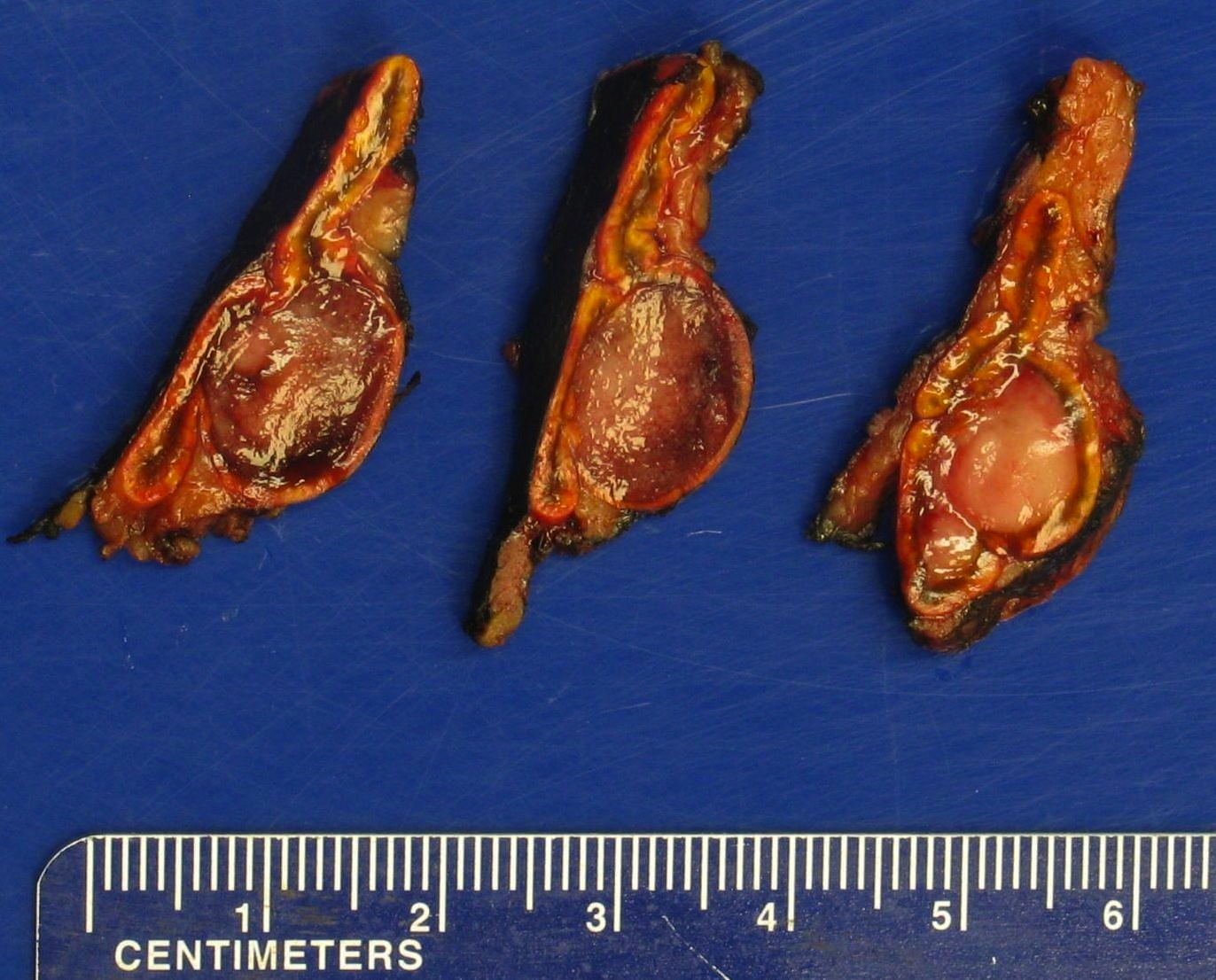
Contributed by Debra L. Zynger, M.D.

Well circumscribed tumor
Nested with cellular crowding
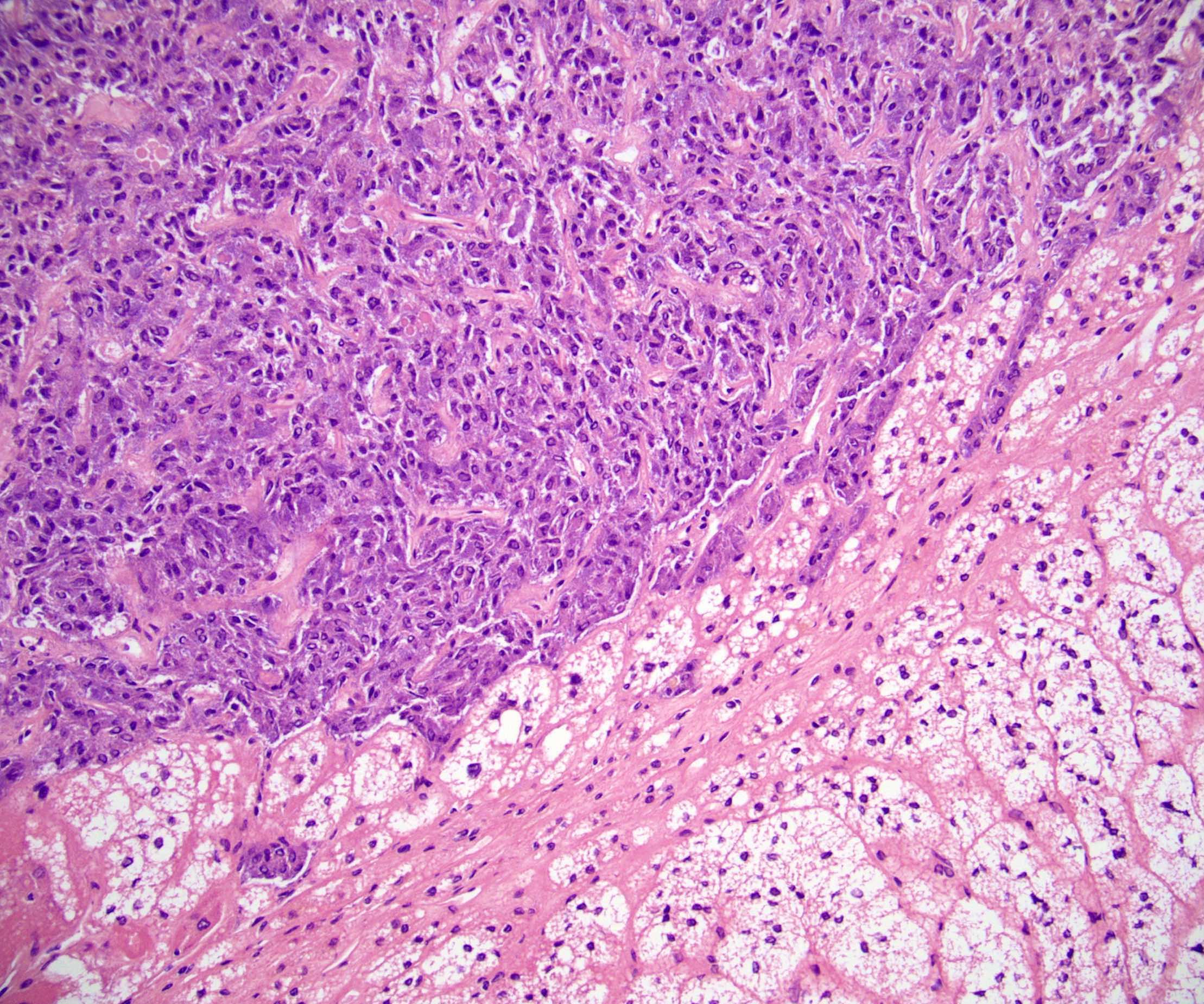
Permanent section
Microscopic (histologic) description
- Nested (zellballen), trabecular or solid arrangement
- Nests outlined with sustentacular cells best visualized with S100 immunostain
- Cells: large, polygonal, uniform or extensively vacuolated
- Cytoplasm: abundant fine, granular red-purple cytoplasm
- Pigmented granules containing hemosiderin, melanin, neuromelanin and lipofuscin may be seen
- Nuclei: may be uniform or exhibit extensive variation in size, round to oval nuclei, nucleoli prominent
- Pheochromocytoma of the adrenal gland scaled score (PASS) and grading system for adrenal pheochromocytoma and paraganglioma (GAPP) can be used to assess for malignant potential (see Table 3 and Table 4)
- Composite pheochromocytoma: pheochromocytoma with ganglioneuroma, ganglioneuroblastoma, neuroblastoma or peripheral nerve sheath tumors
Table 3. Pheochromocytoma of the adrenal gland scaled score (Am J Surg Pathol 2002;26:551)
| Histologic feature |
Score
(total ≥ 4 is concerning for malignancy) |
| Periadrenal adipose invasion | +2 |
| > 3 mitoses/10 high power fields | +2 |
| Atypical mitoses | +2 |
| Necrosis | +2 |
| Cellular spindling | +2 |
| Marked nuclear pleomorphism | +1 |
| Cellular monotony | +2 |
| Large nests or diffuse growth | +2 |
| High cellularity | +2 |
| Capsular invasion | +1 |
| Vascular invasion | +1 |
| Hyperchromasia | +1 |
Table 4. Grading system for adrenal pheochromocytoma and paraganglioma (GAPP) (Endocr Relat Cancer 2014;21:405)
| Feature |
Score (well differentiated, 0 - 2; moderately differentiated, 3 - 6; poorly differentiated, 7 - 10) |
| Histological pattern | Zellballen, 0 |
| Large and irregular cell nests, +1 | |
| Pseudorosette, +1 | |
| Cellularity (number of tumor cells in 10 mm x 10 mm square at high power magnification) | Low, < 150, 0 |
| Moderate, 150 - 250, +1 | |
| High, > 250, +2 | |
| Comedonecrosis | +2 |
| Capsular / vascular invasion | +1 |
| Ki67 % | < 1%, 0 |
| 1 - 3%, +1 | |
| > 3%, +2 | |
| Catecholamine type | Nonfunctional, 0 |
| Epinephrine or epinephrine + norepinephrine, 0 | |
| Norepinephrine or norepinephrine + dopamine , +1 |
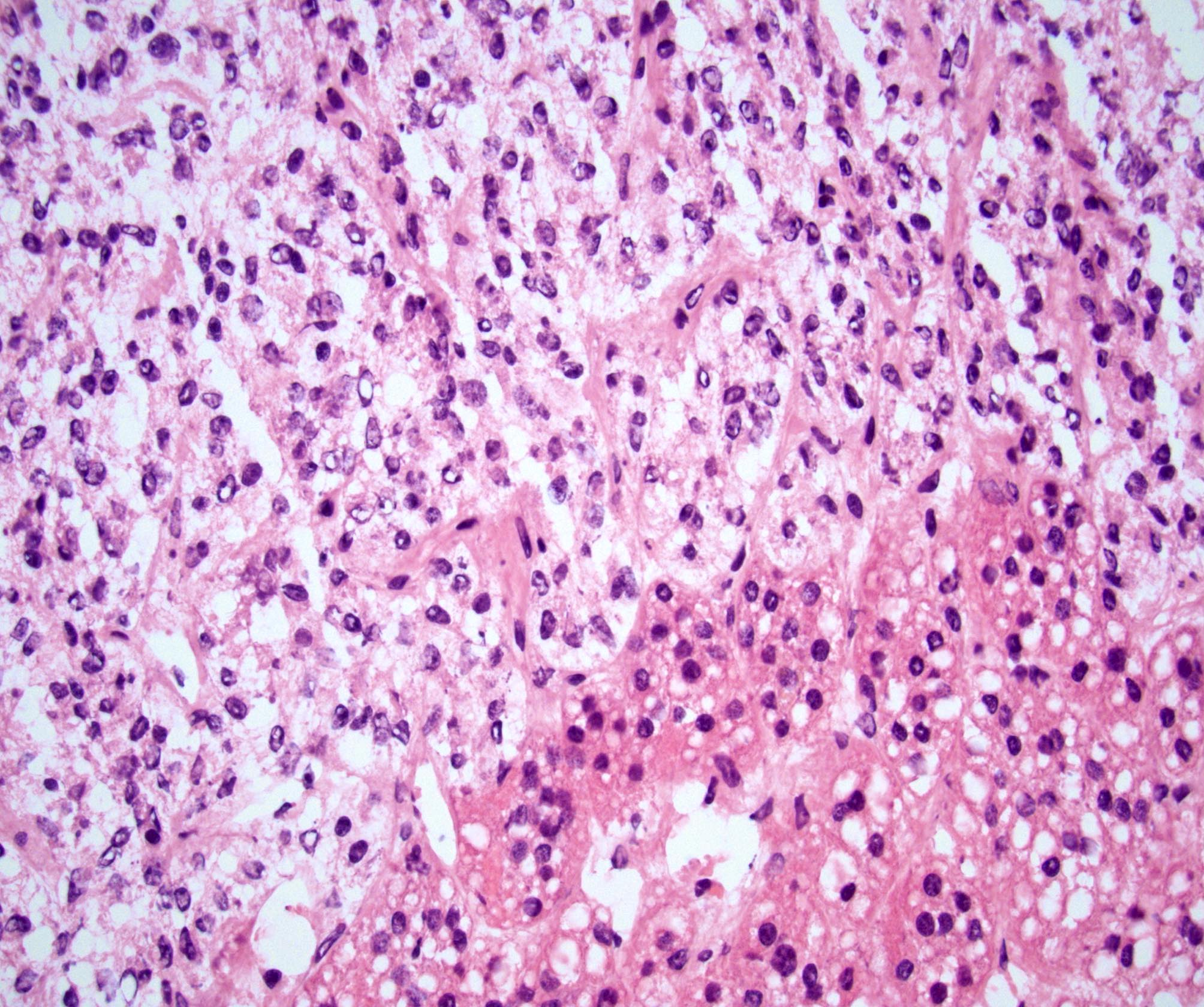
Microscopic (histologic) images
Contributed by Debra L. Zynger, M.D. (Case #319), Brian Werstein, M.D. and Nicole K. Andeen, M.D. (Case #489)
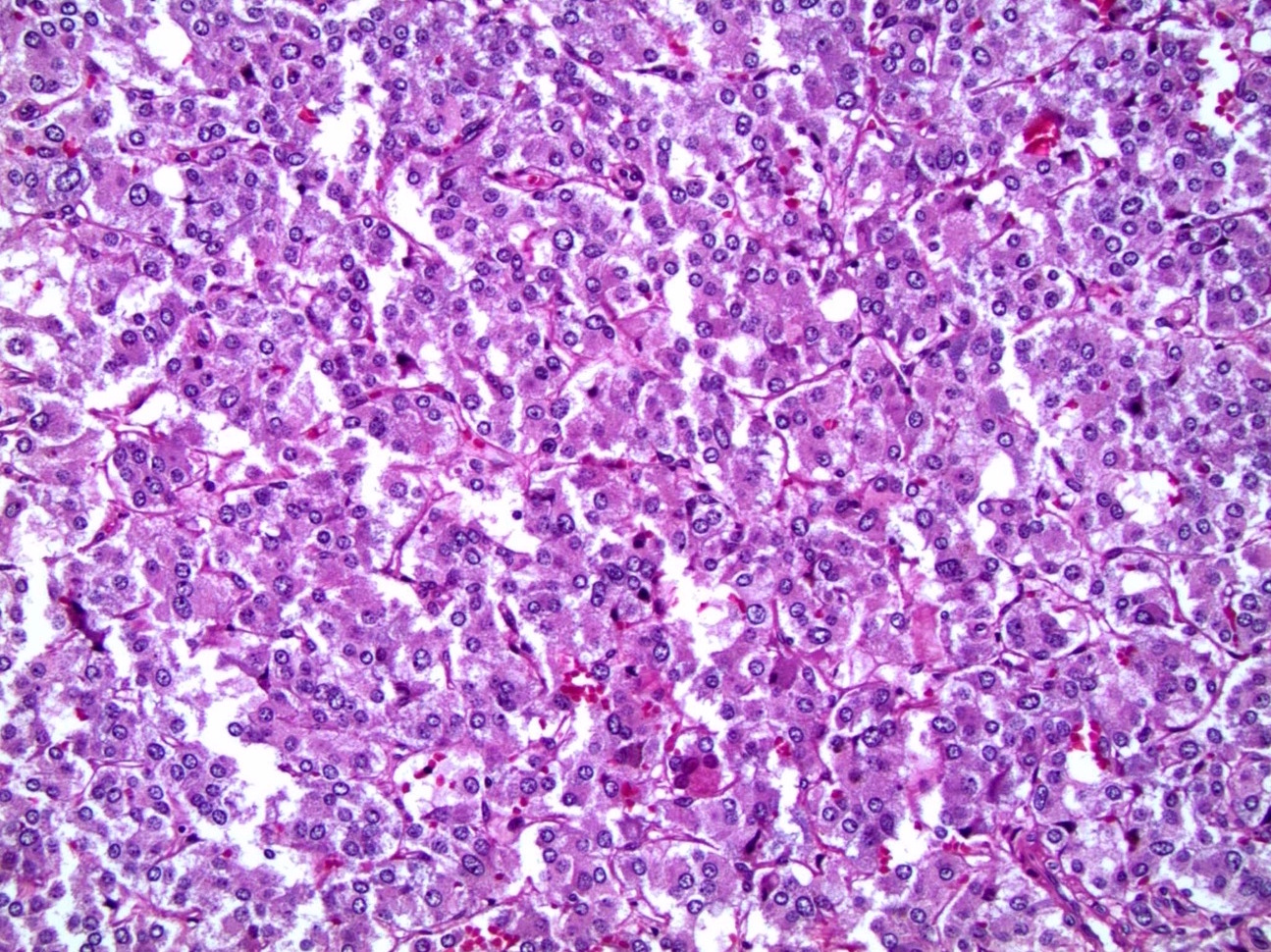
Nested architecture
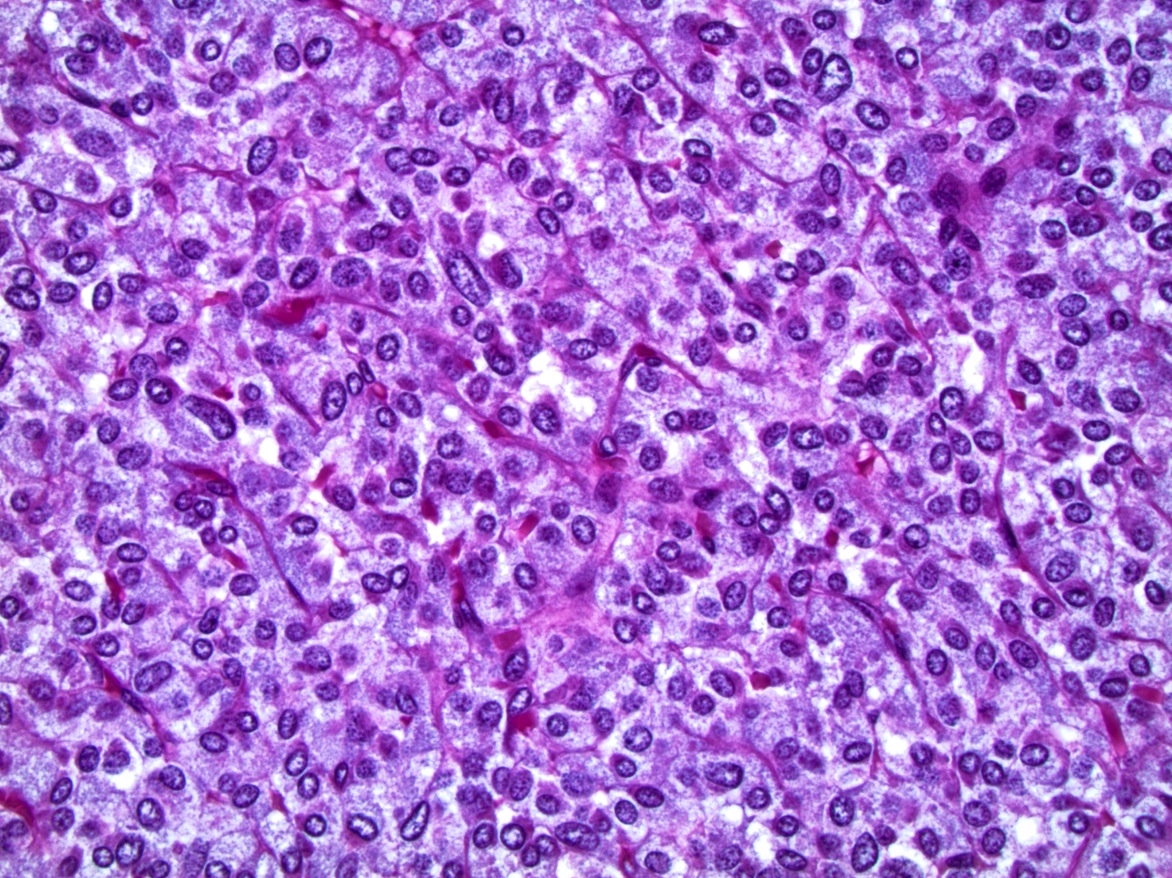
Sheets of cells
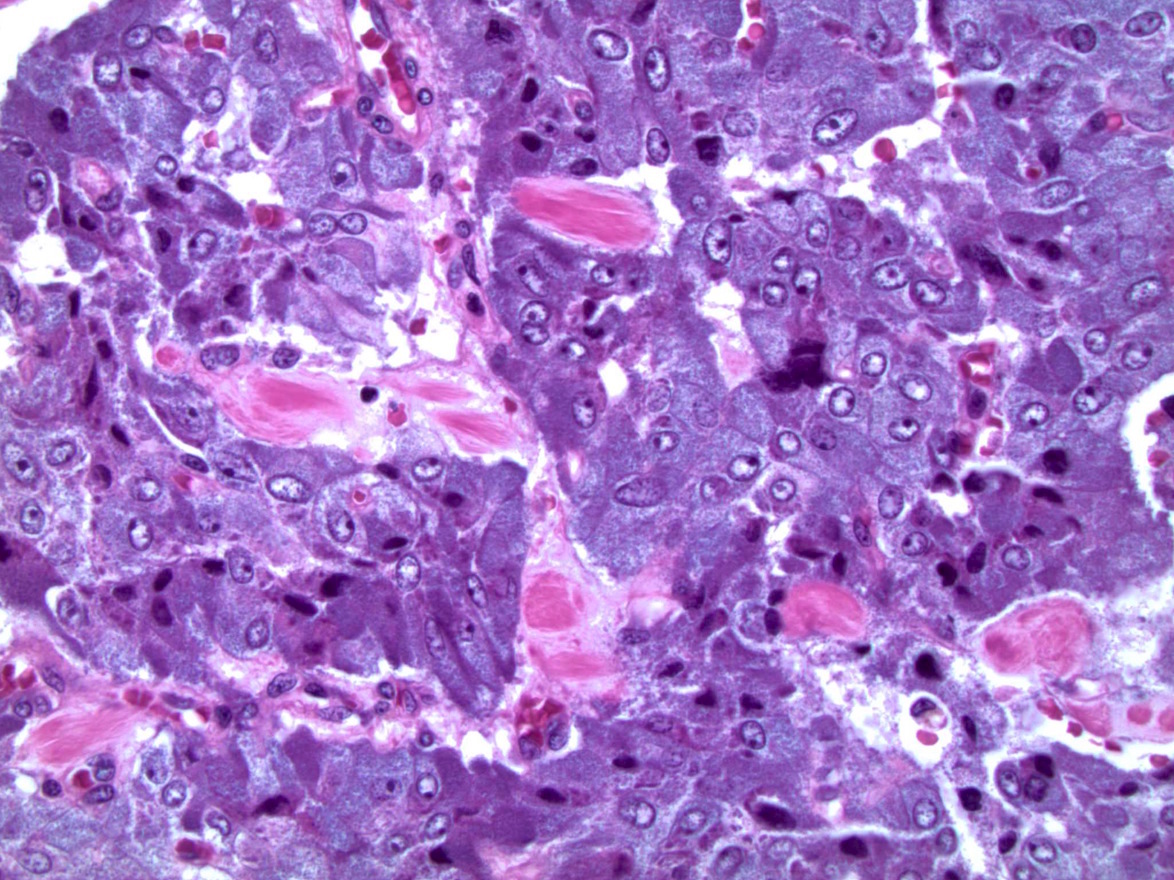
Abundant cytoplasm

Capsular invasion
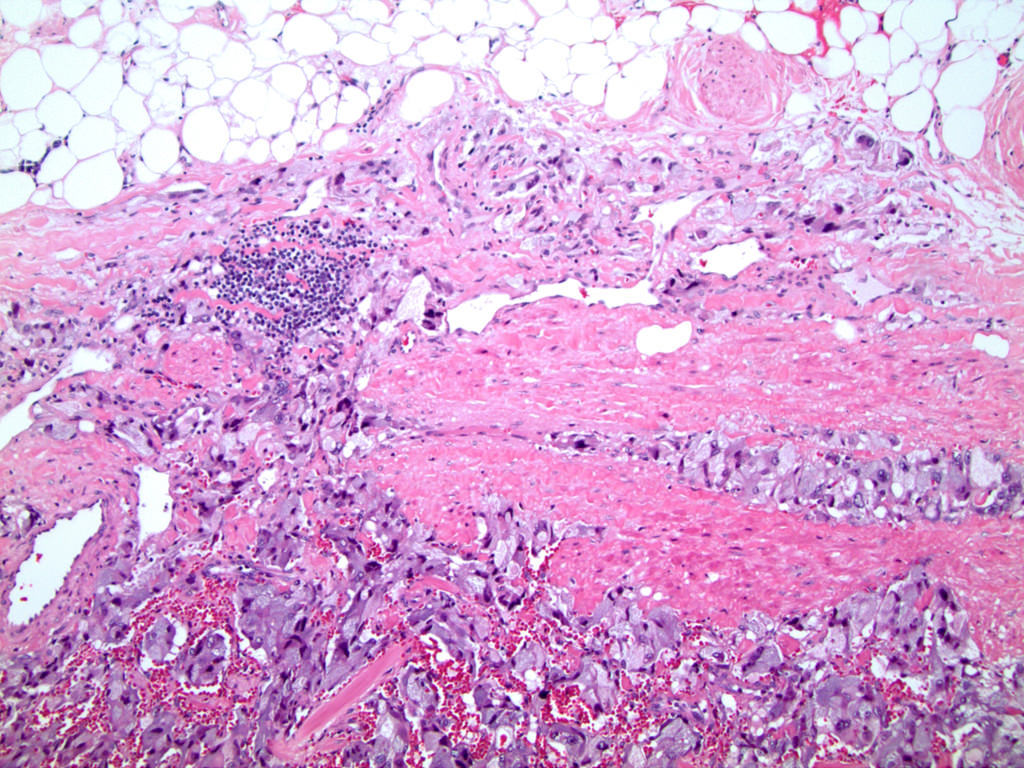
Adipose invasion

Vascular invasion
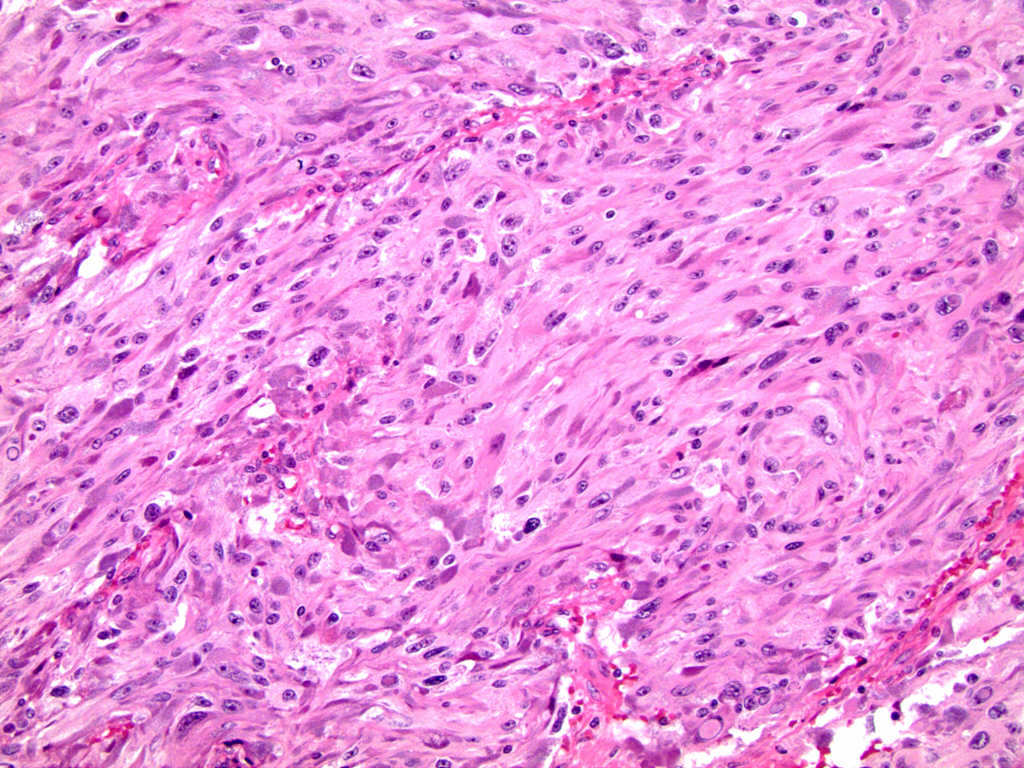
Cellular spindling
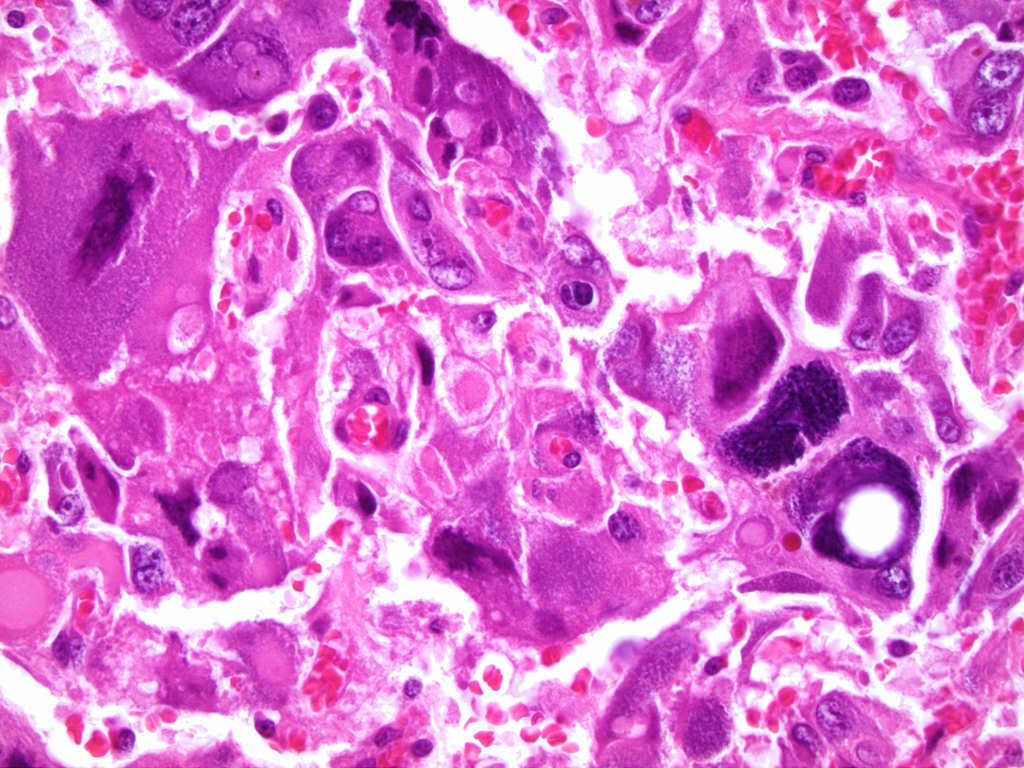
Nuclear pleomorphism
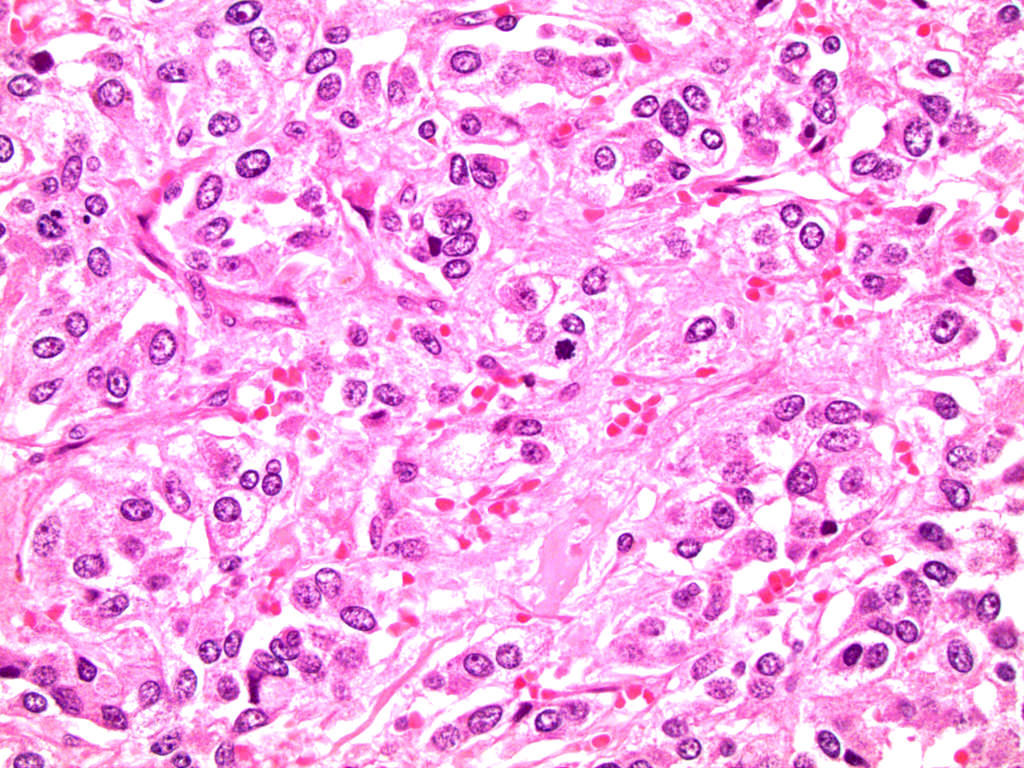
Mitotic activity
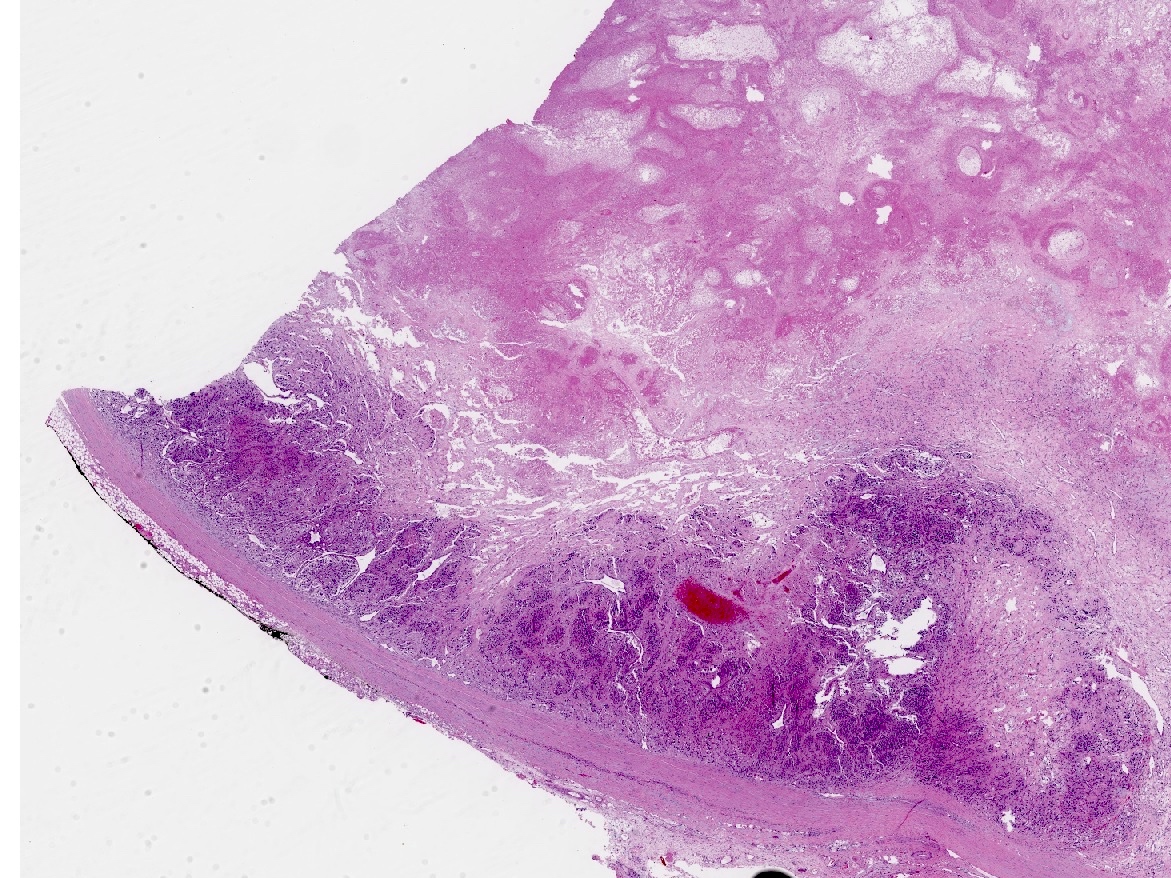
Necrosis

Large nests
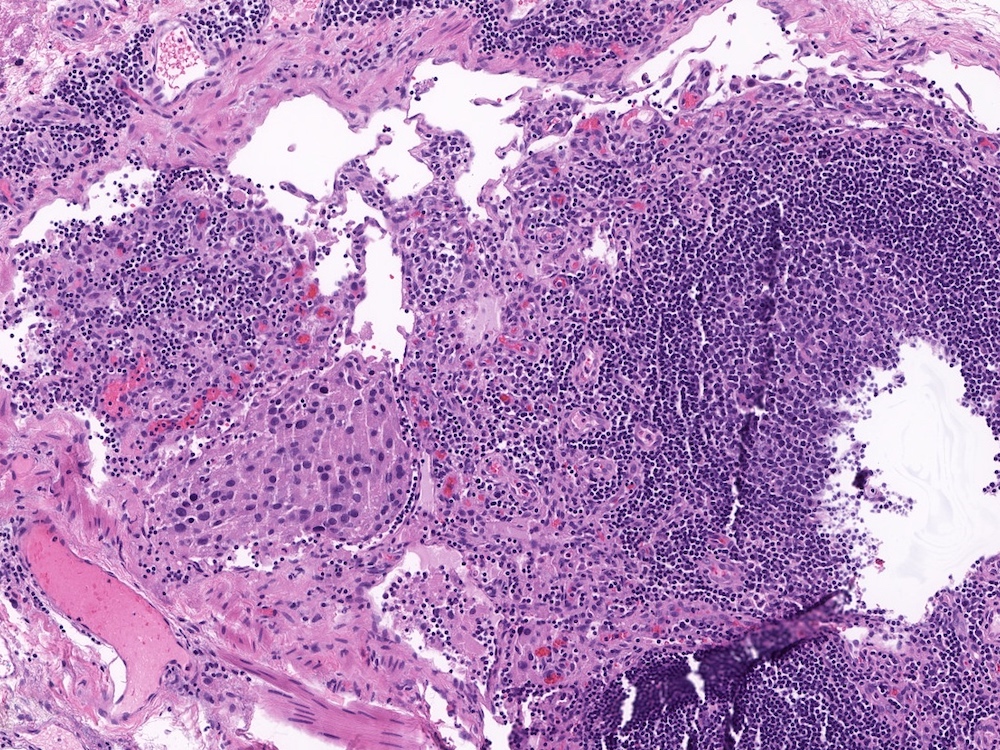
Lymph node metastasis
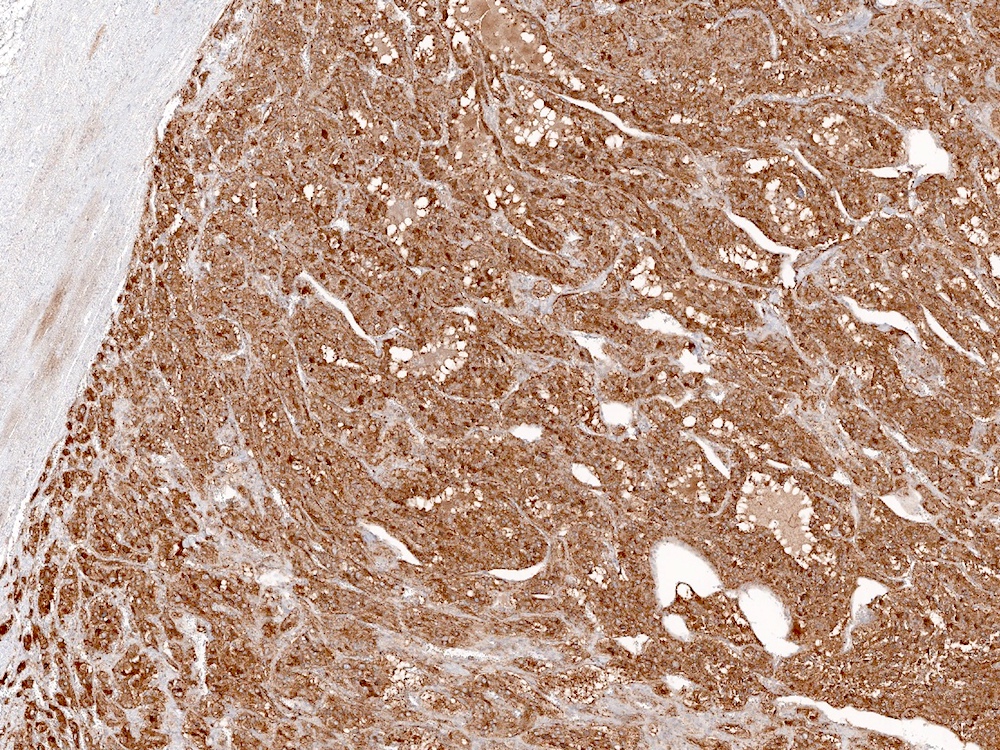
Synaptophysin
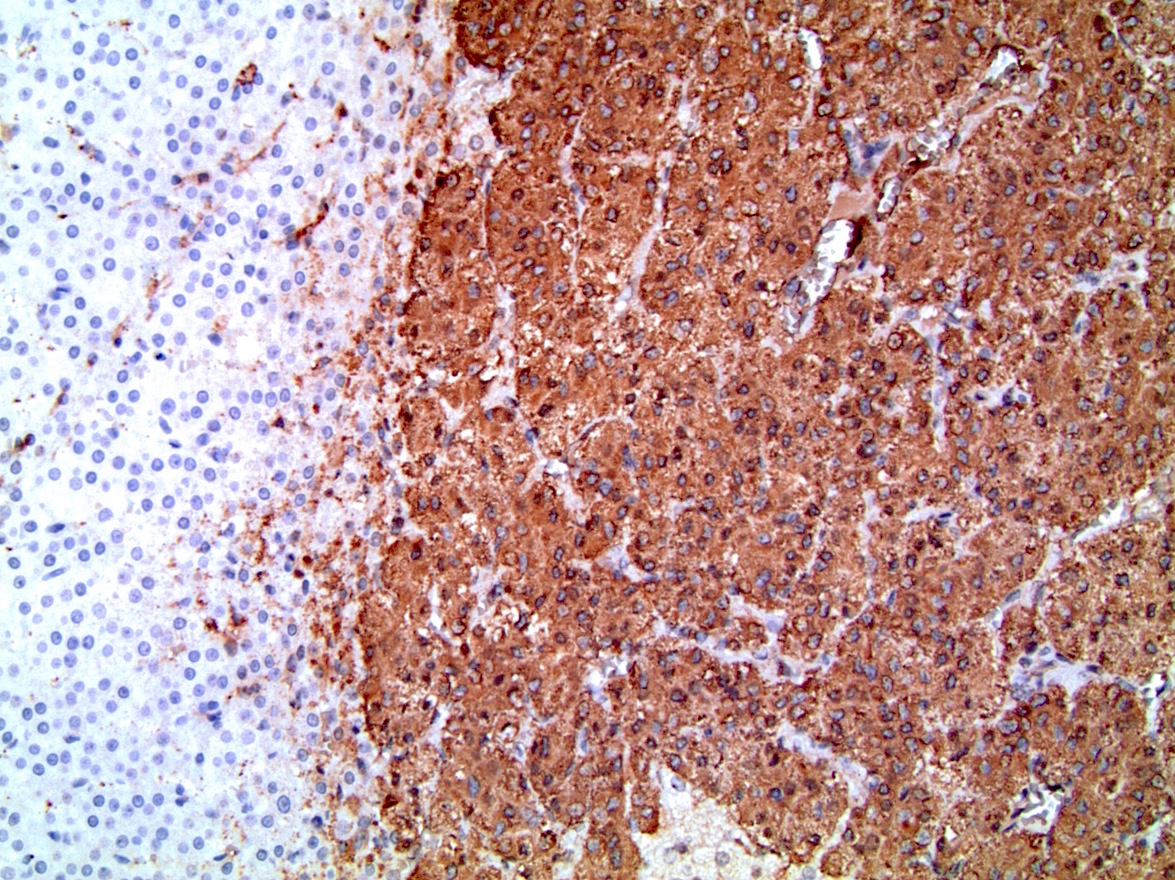
Chromogranin
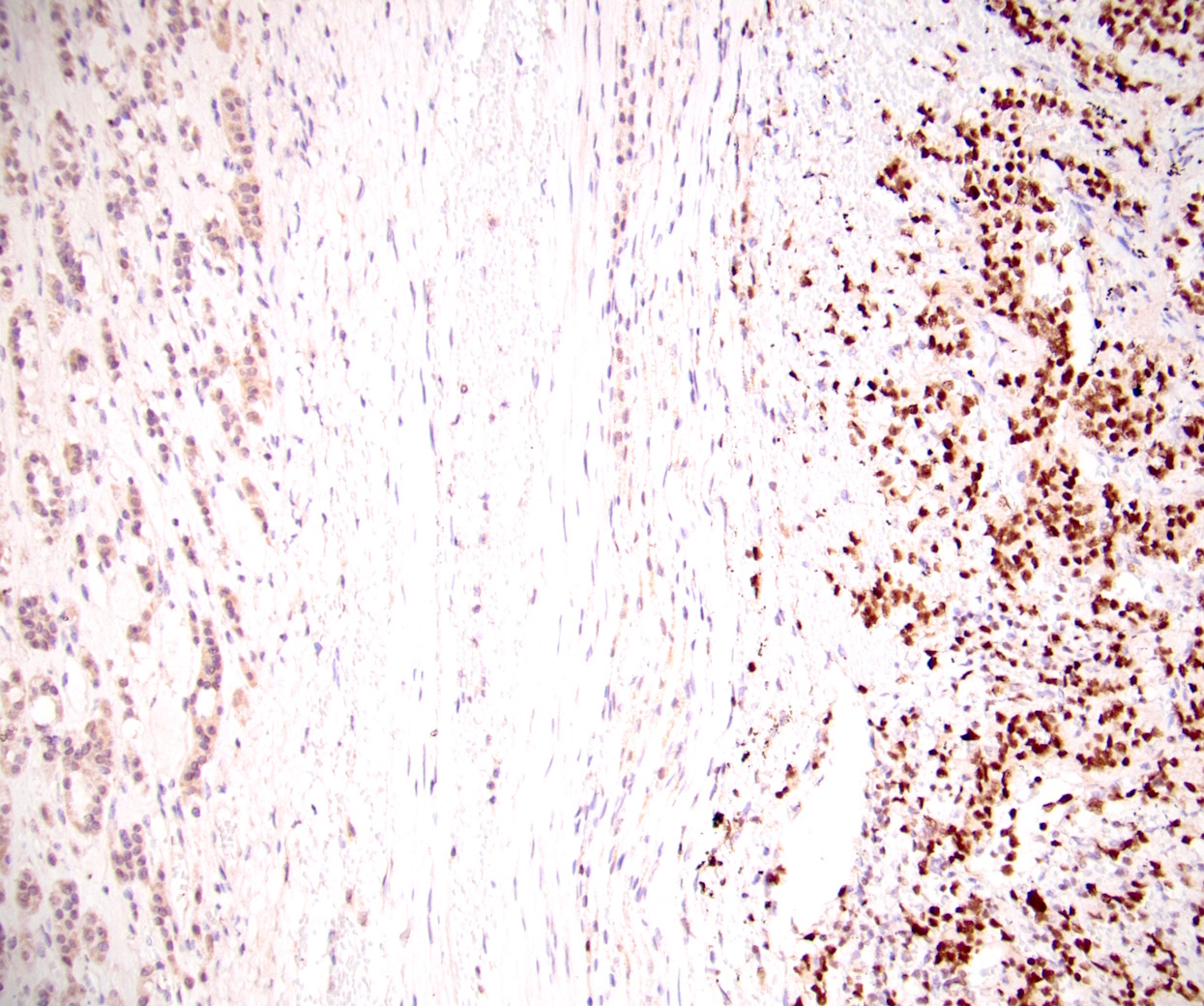
GATA3
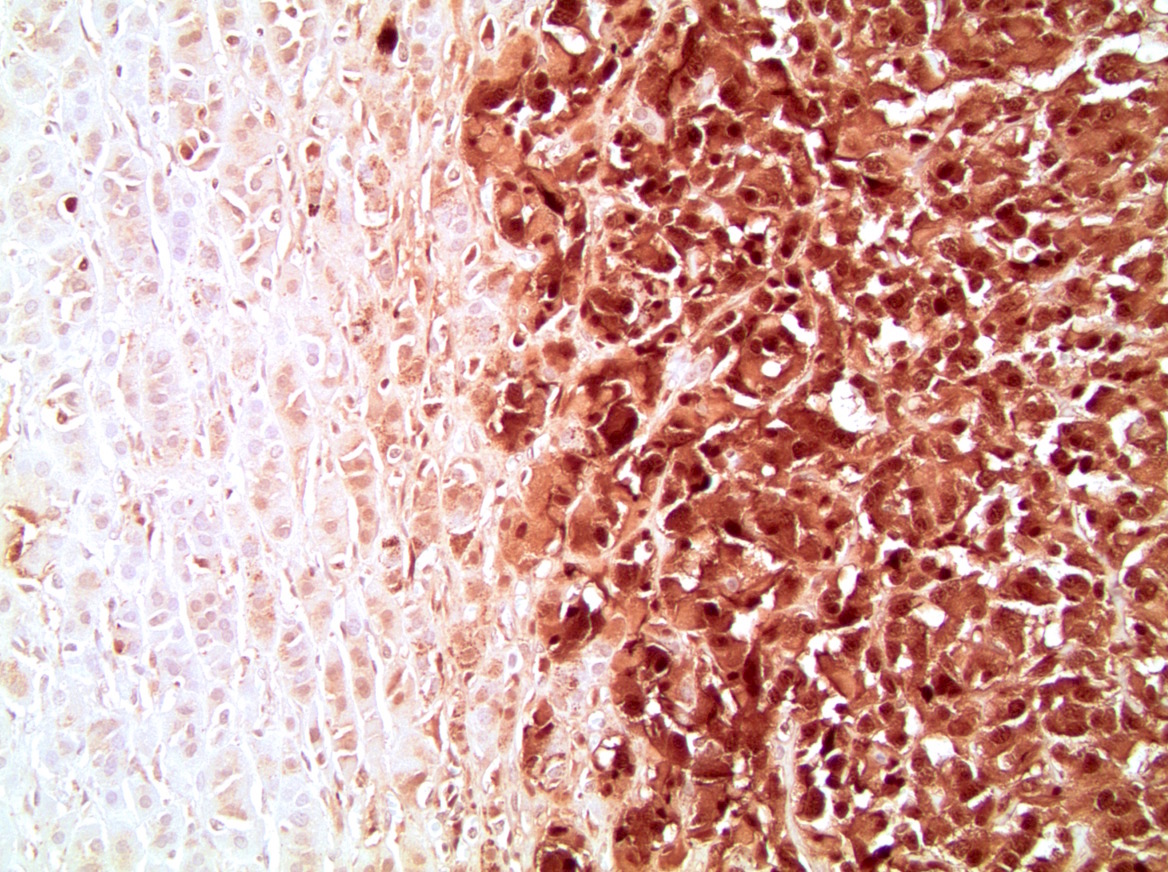
S100
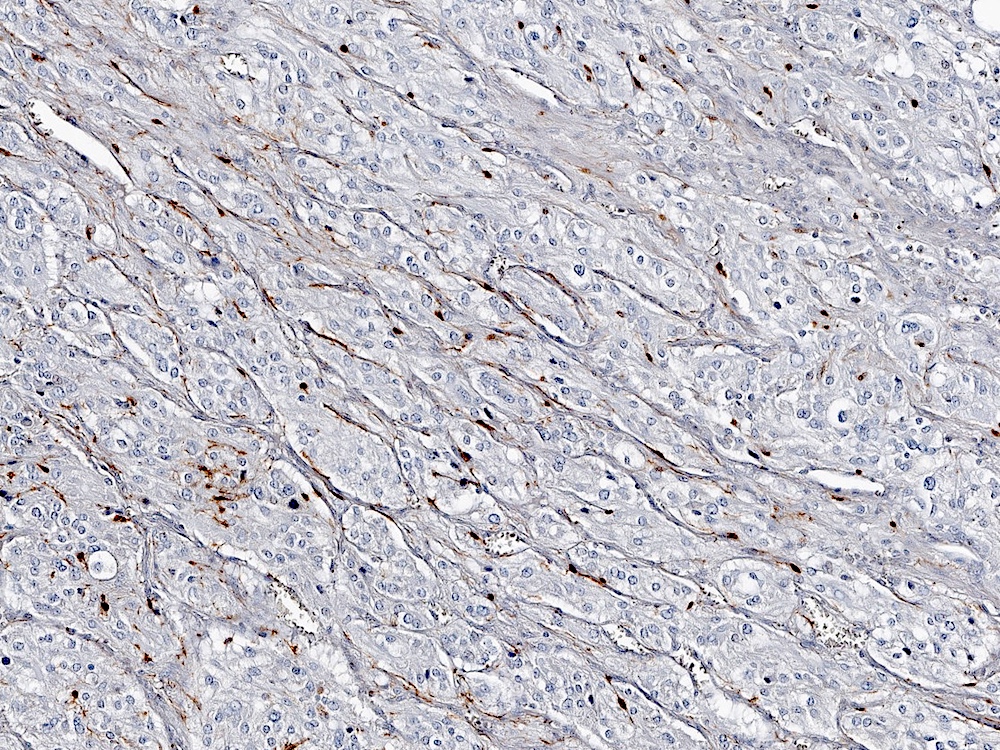
S100
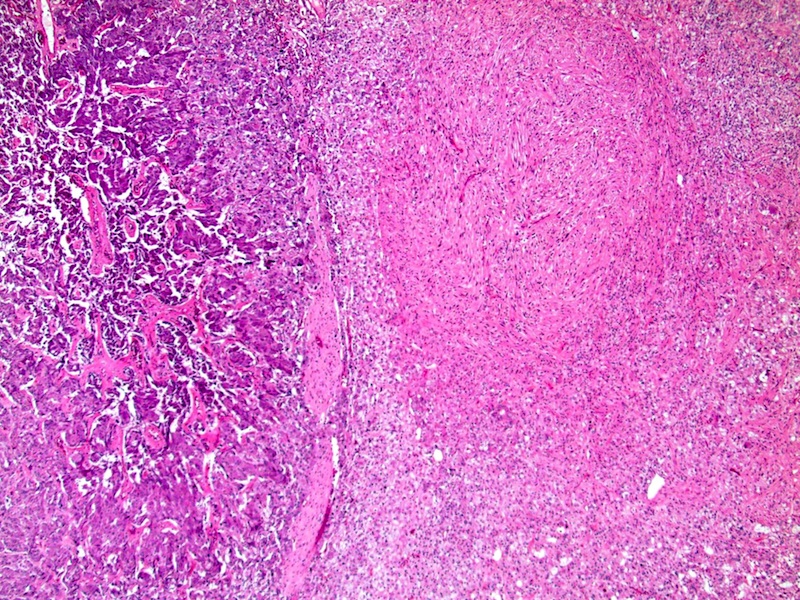
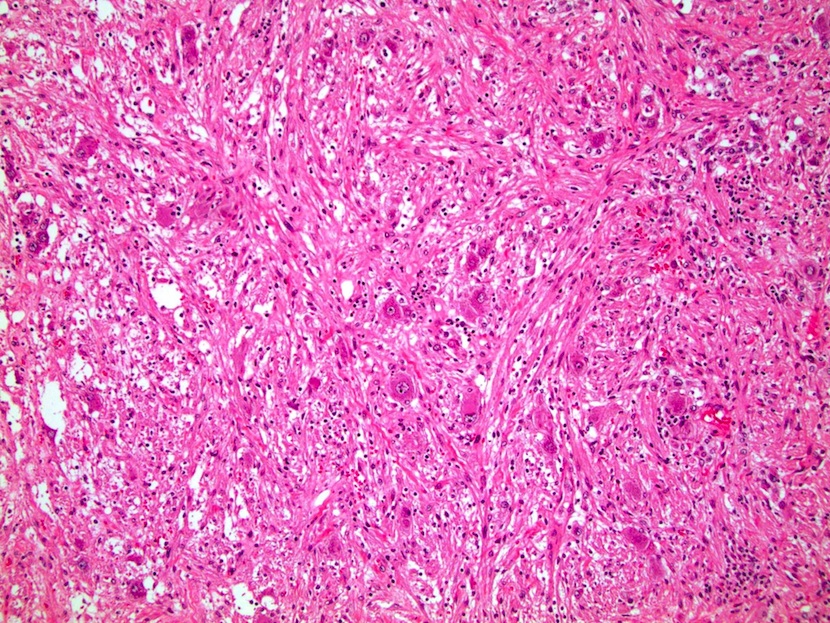
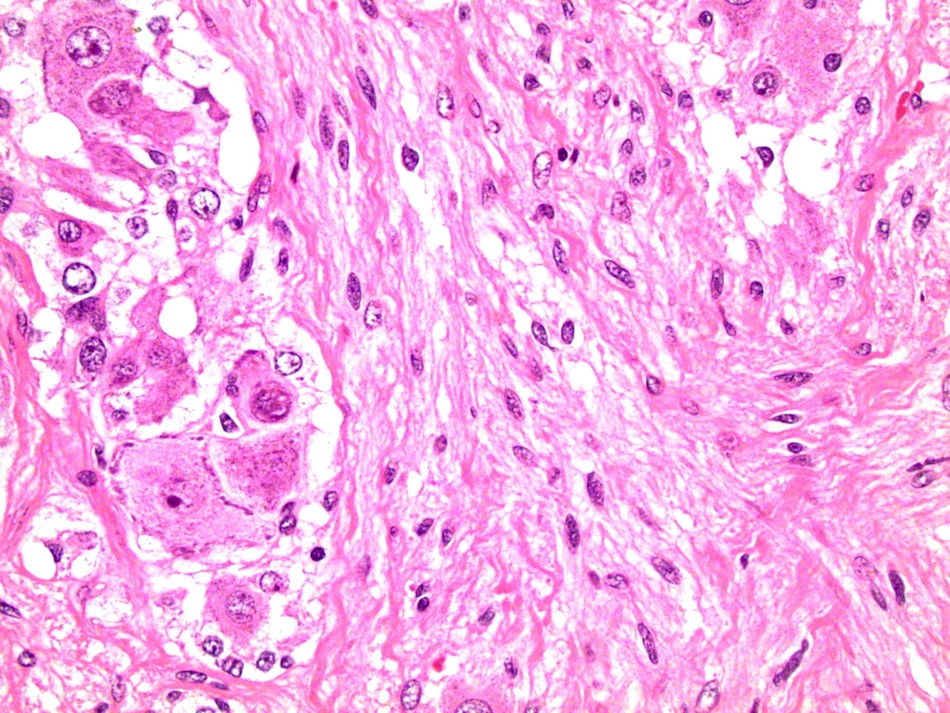

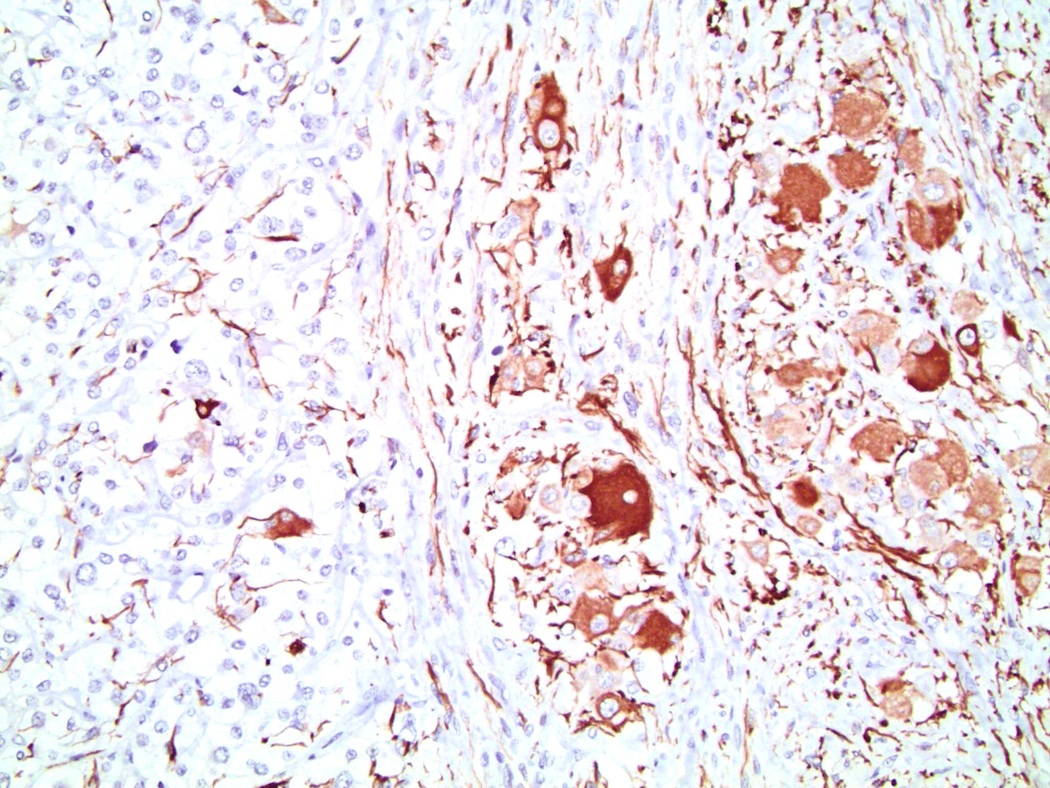
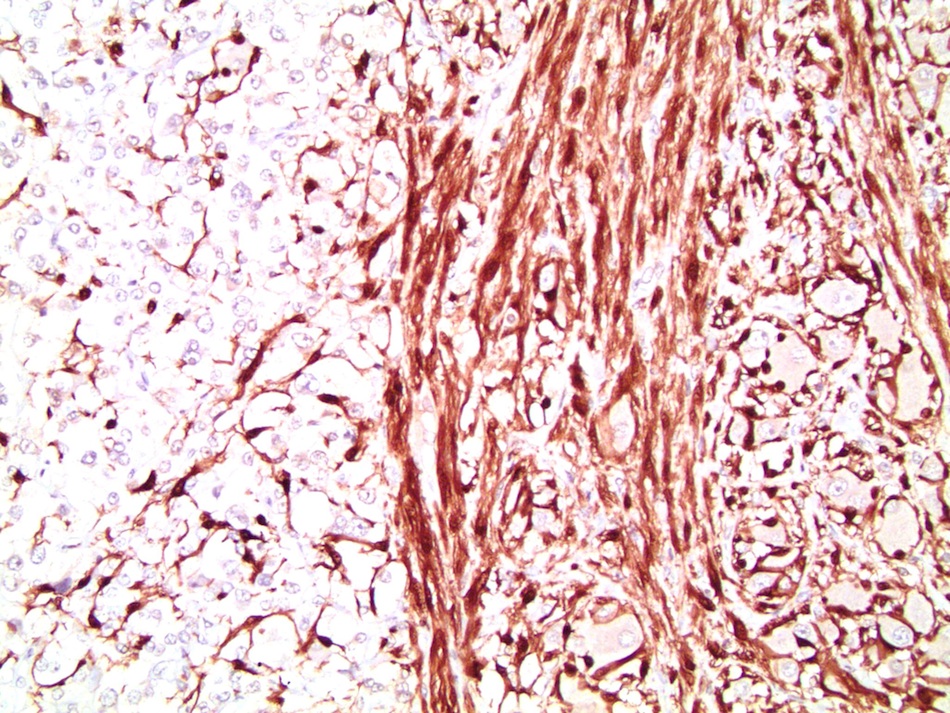
Composite pheochromocytoma with ganglioneuroma
Virtual slides
Images hosted on other servers:

Pheochromocytoma
Cytology images
Contributed by Debra L. Zynger, M.D.
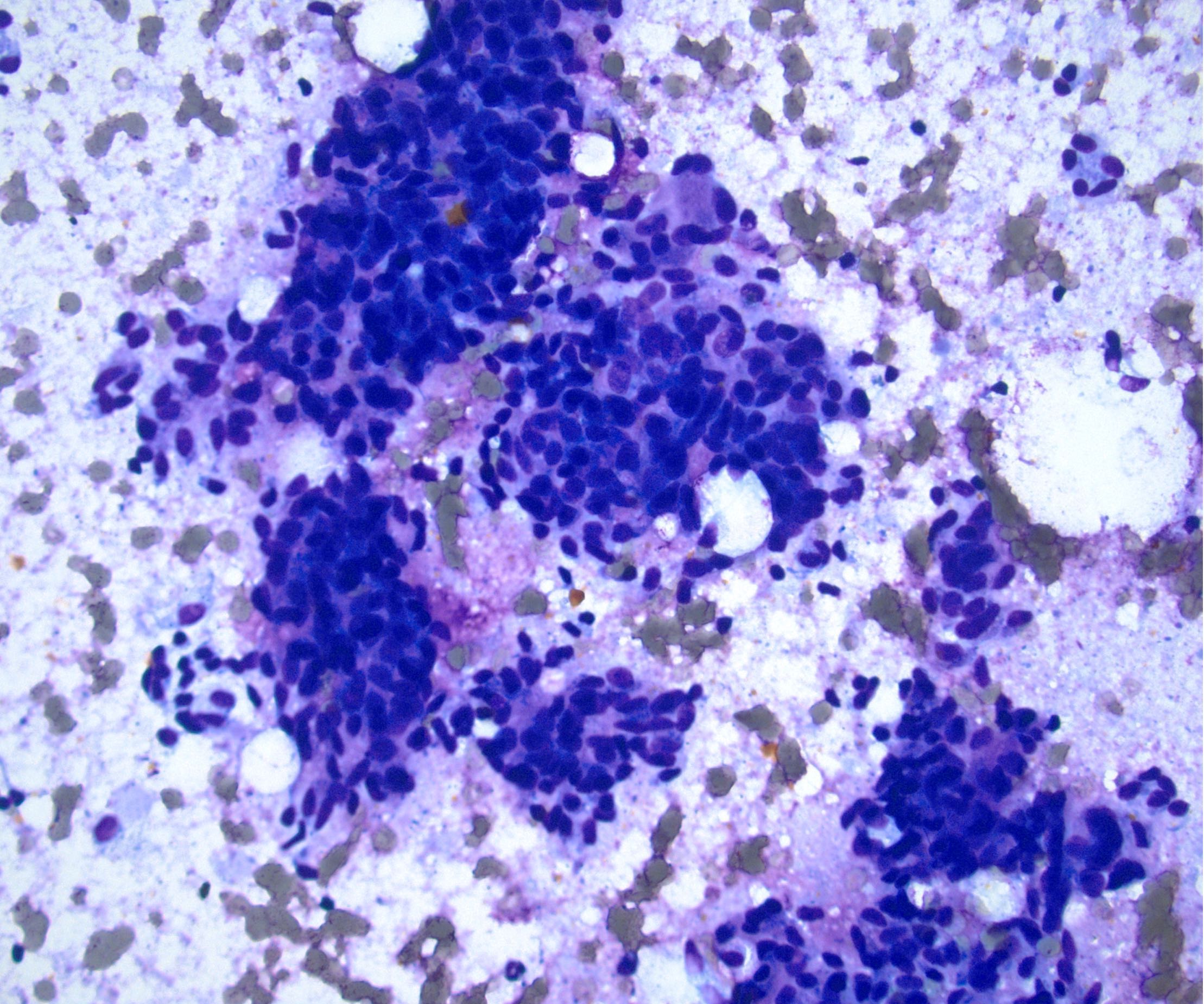
Diff-Quik
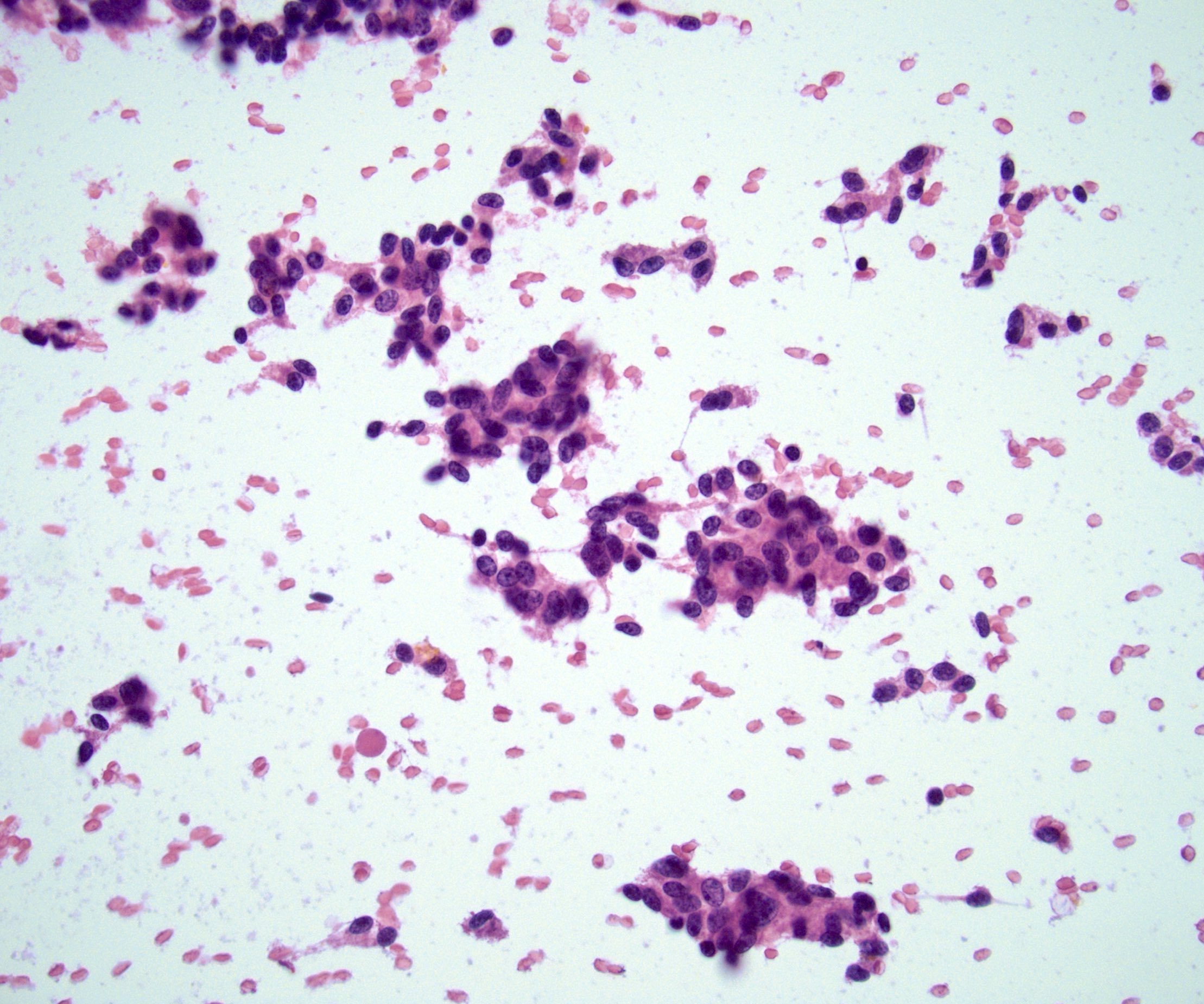
Touch preparation
Positive stains
- Chromogranin A (Am J Surg Pathol 2010;34:423)
- Synaptophysin (Am J Surg Pathol 2010;34:423)
- S100 expressed in nucleus and cytoplasm of sustentacular cells and some pheochromocytes throughout tumor (Arch Pathol Lab Med 1985;109:117, Am J Surg Pathol 2002;26:551)
- GATA3 diffuse nuclear expression in approximately 70% (Histopathology 2017;71:475)
- CD10, vimentin
- MAP2 (Am J Surg Pathol 2010;34:423)
- SDHB usually retained unless an SDHB deficient tumor, ~3% (Am J Surg Pathol 2021;45:1264)
Negative stains
- Calretinin 0 - 14% (Appl Immunohistochem Mol Morphol 2002;10:67, Am J Surg Pathol 2010;34:423)
- MelanA (Appl Immunohistochem Mol Morphol 2002;10:67, Am J Surg Pathol 2010;34:423)
- HMB45
- Inhibin A 0 - 38% (Am J Surg Pathol 2021;45:1264, J Clin Pathol 1998;51:114, Mod Pathol 1998;11:516, J Endocrinol 2000;165:223, Appl Immunohistochem Mol Morphol 2001;9:222, Appl Immunohistochem Mol Morphol 2002;10:67)
- Cytokeratin AE1 / AE3, CK7, CK20
- PAX8 (Appl Immunohistochem Mol Morphol 2011;19:293)
- SF1 (Am J Surg Pathol 2010;34:423)
- CAIX 17 - 32% membranous staining (Am J Surg Pathol 2021;45:1264, Histopathology 2011;59:1229)
Electron microscopy description
- Numerous dense cytoplasmic secretory granules 200 - 300 nm containing catecholamines
- Most have norepinephrine granules
- Epinephrine granules: centrally placed dense core that lacks a halo
- Norepinephrine granules: eccentrically placed dense core surrounded by an empty halo
Electron microscopy images
Contributed by Debra L. Zynger, M.D. and Edward Calomeni, B.S.
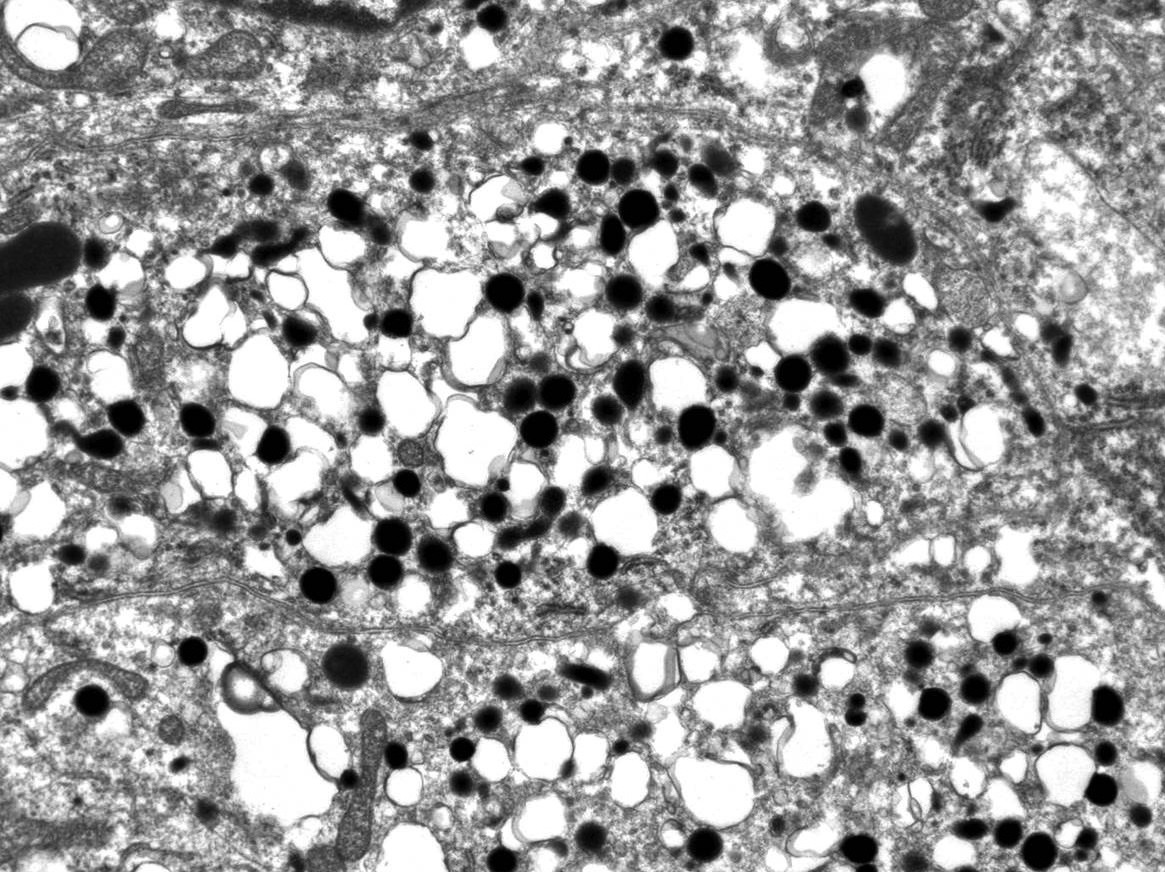
Numerous epinephrine granules
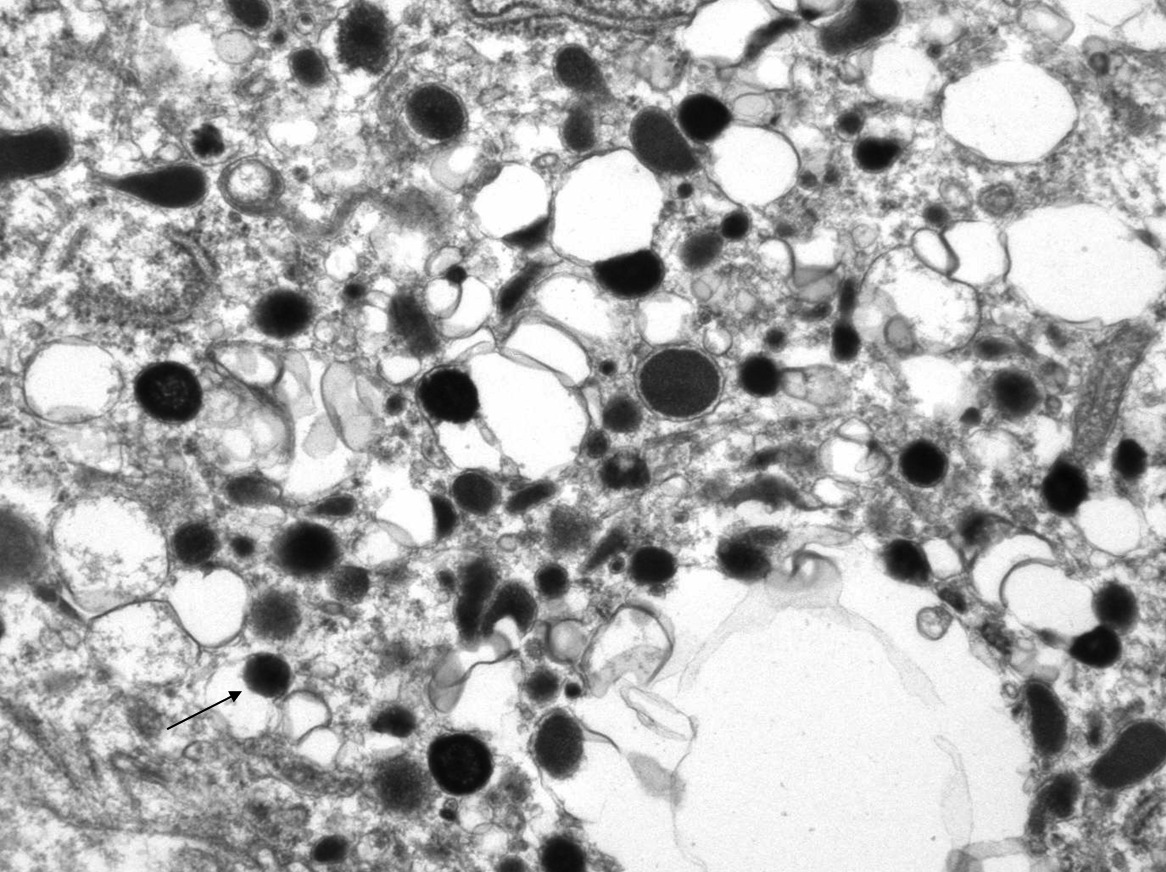
Norepinephrine granules
Images hosted on other servers:


Neurosecretory granules
Molecular / cytogenetics description
- Genetic testing is recommended for all patients with pheochromocytoma
- Germline mutations include:
- Von Hippel-Lindau syndrome: autosomal dominant truncation, deletion or missense mutation of VHL tumor suppressor gene on 3p25-26
- Pheochromocytoma in 10 - 20%
- Also hemangioblastomas of the central nervous system, renal cysts, clear cell renal cell carcinoma, pancreatic cysts and pancreatic neuroendocrine tumors, endolymphatic sac tumors and epididymal cystadenomas (Cancer Genet 2012;205:1)
- Multiple endocrine neoplasia type 2 (MEN 2): autosomal dominant activating mutations in RET proto oncogene on 10q11.2
- Pheochromocytoma in 50%
- Also medullary thyroid carcinoma (Endocr Relat Cancer 2018;25:T15)
- MEN 2a: may also present with hyperparathyroidism
- MEN 2b: may also present with Marfanoid habitus and multiple mucosal ganglioneuromas
- Neurofibromatosis type 1 (NF1): autosomal dominant inactivating mutations in NF1 tumor suppressor gene on 17q11.2 (J Clin Oncol 2005;23:8812)
- Pheochromocytoma in 5 - 7%
- Also café au lait spots, iris hamartomas, axillary or inguinal freckling, osseous dysplasia and optic pathway glioma (Cancer Genet 2012;205:1)
- Familial paraganglioma: inactivating mutations in succinate dehydrogenase tumor suppressor gene
- Associated with pheochromocytoma in 3 - 5%
- Also paraganglioma, gastrointestinal stromal tumors, breast carcinoma and papillary thyroid carcinoma (Cancer Genet 2012;205:1)
- SDHB and SDHD mutations most closely associated with pheochromocytoma (J Clin Oncol 2005;23:8812)
- SDHB mutation may increase risk of malignancy (Cancers (Basel) 2019;11. pii: E1121)
- Identifiable with immunohistochemistry staining: SDHB staining negative or weak in SDHB and SDHD mutations, respectively (Hum Pathol 2010;41:805)
- Others: mutations in TMEM127 and MYC associated factor X (MAX) (familial), HRAS (somatic) (Cancer Genet 2012;205:1, Genes Chromosomes Cancer 2016;55:452)
- Von Hippel-Lindau syndrome: autosomal dominant truncation, deletion or missense mutation of VHL tumor suppressor gene on 3p25-26
Molecular / cytogenetics images
Images hosted on other servers:

NF1 mutation

TMEM127 mutation
Sample pathology report
- Left adrenal, adrenalectomy
- Pheochromocytoma, 5.1 cm (see synoptic report)
Differential diagnosis
- Adrenal cortical carcinoma:
- Typically large and produce steroids; irregular mass with noncontrast CT attenuation ≥ 10 HU and inhomogeneous contrast enhancement with delayed washout
- Solid growth pattern, abundant eosinophilic cytoplasm with focal clear areas, variable mitoses and nuclear polymorphism depending on grade (Endocr Rev 2014;35:282)
- MelanA, inhibin A and calretinin and SF1 positive (Am J Surg Pathol 2010;34:423)
- Chromogranin and S100 negative (Am J Surg Pathol 2010;34:423)
- Ki67 usually high
- Synaptophysin variable (does not differentiate) (Am J Surg Pathol 2010;34:423)
- Adrenal medulla or adrenal medullary hyperplasia:
- Normal adrenal medulla can mimic on needle core biopsy
- Generally considered neoplastic if radiologically or grossly a discrete tumor
- IHC cannot differentiate
- Metastases:
- Metastatic renal cell carcinoma, melanoma, lung, colon, breast
- History, morphology and immunohistochemistry vary according to origin but typically lack chromogranin and S100 (unless melanoma)
- Rule out carcinoma with broad spectrum keratin such as AE1 / AE3 plus CK7 and CK20
- Adrenal cortical adenoma:
- May be nonfunctional (clinically silent) or secrete steroids; noncontrast CT shows small, round, homogeneous mass with smooth borders and attenuation typically ≤ 10 HU; contrast CT has rapid contrast washout
- Clear, compact cells without abnormal mitoses or dysplasia arranged in nests, cords or trabecular shapes within a fibrous capsule (Onco Targets Ther 2015;8:1251)
- MelanA, inhibin A and calretinin and SF1 positive (Am J Surg Pathol 2010;34:423)
- Chromogranin and S100 negative (Am J Surg Pathol 2010;34:423)
- Synaptophysin weak to negative (does not differentiate) (Am J Surg Pathol 2010;34:423)
- Composite pheochromocytoma:
- Also has a component of ganglioneuroma, ganglioneuroblastoma, neuroblastoma or peripheral nerve sheath tumors
Practice question #1
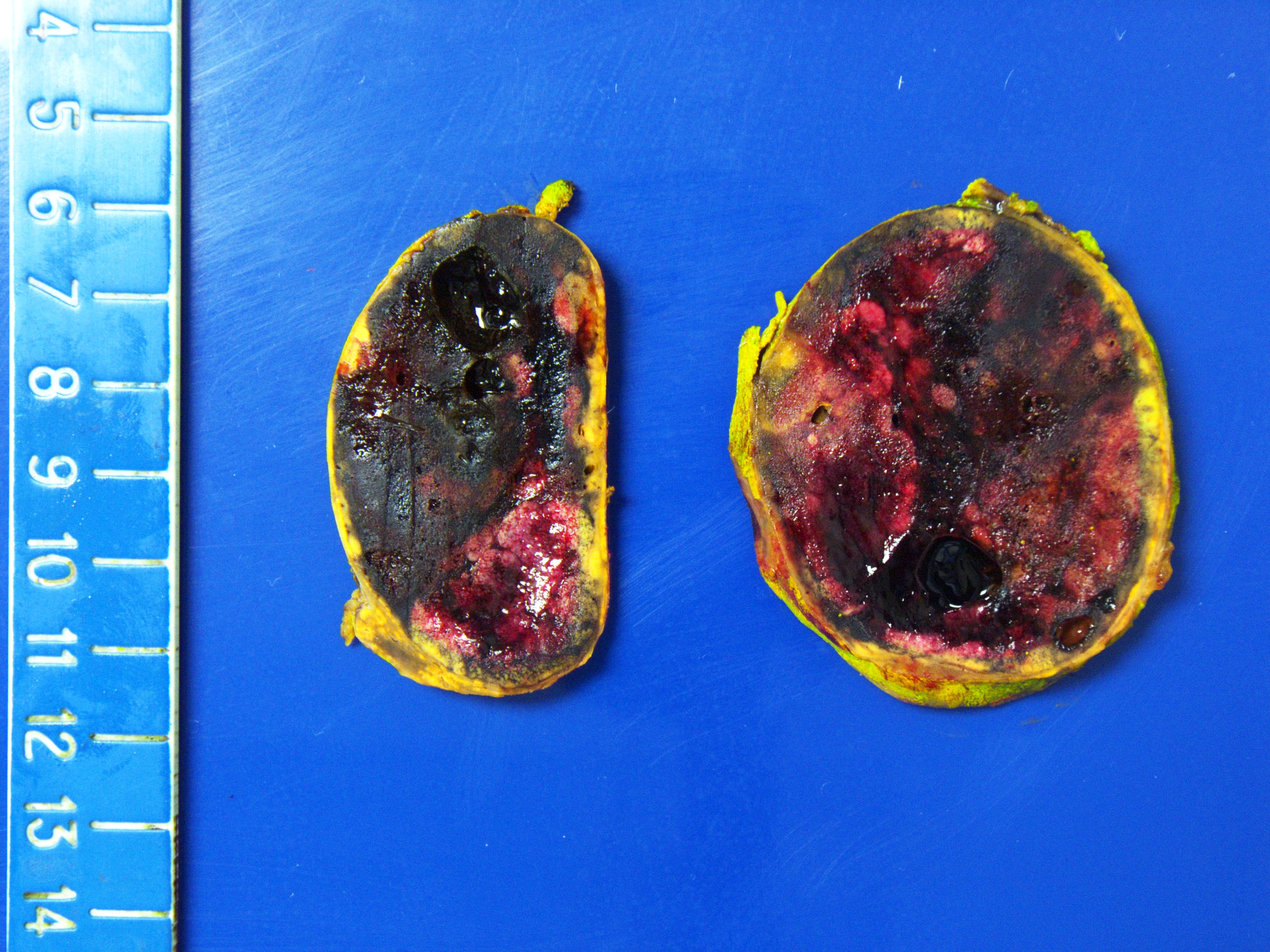
- A 38 year old woman presents to clinic complaining of intermittent episodes of headaches and sweating. Her family history is significant for VHL gene mutation in her father. On exam, she has mild hypertension and elevated plasma metanephrines. A noncontrast CT of the abdomen reveals an attenuating 8 cm tumor in the adrenal medulla. An adrenalectomy was done and is shown in the photo. Which immunostains should be expressed?
- AE1 / AE3, CK7, TTF1
- AE1 / AE3, CK7, ER
- MelanA, inhibin A, calretinin
- PAX8, PAX2, CAIX
- Synaptophysin, chromogranin, S100
Practice answer #1
E. Synaptophysin, chromogranin, S100. The clinical presentation and image are diagnostic of a pheochromocytoma. Synaptophysin, chromogranin and S100 are positive in pheochromocytoma. AE1 / AE3, CK7 and TTF1 are consistent with metastatic lung adenocarcinoma. AE1 / AE3, CK7 and ER are consistent with metastatic breast carcinoma. MelanA, inhibin A are calretinin are seen in adrenal cortical adenoma / carcinoma. PAX8, PAX2 and CAIX are associated with renal cell carcinoma, clear cell type.
Comment Here
Reference: Pheochromocytoma
Comment Here
Reference: Pheochromocytoma
Practice question #2
A 65 year old man presents with acute worsening of previously well controlled hypertension. Further investigation reveals elevated plasma metanephrines and a right sided adrenal mass. Adrenalectomy reveals a 3 cm, 120 g pheochromocytoma with large, uniform, polygonal cells and 4 mitotic figures per 10 high power fields (HPF). It is positive for chromogranin and synaptophysin. Which of the following features is associated with a more aggressive course?
- > 3 mitoses per HPF
- Age of diagnosis ≥ 45 years
- Positive for synaptophysin
- Tumor size < 5 cm
- Tumor weight < 130 g
Practice answer #2
A. > 3 mitoses per HPF. > 3 mitoses per HPF increases risk for malignant pheochromocytoma. It is part of the pheochromocytoma of the adrenal gland scaled score (PASS). A score of ≥ 4 is concerning for malignancy, and scores are based on the presence of the following features: periadrenal adipose invasion (+2), > 3 mitosis per 10 high powered fields (+2), atypical mitoses (+2), necrosis (+2), cellular spindling (+2), marked nuclear pleomorphism (+1), cellular monotony (+2), large nests or diffuse growth (+2), high cellularity (+2), capsular invasion (+1), vascular invasion (+1), hyperchromasia (+1). Age of diagnosis < 45 years is associated with more aggressive disease, rather than older age. Staining with synaptophysin is a characteristic feature of pheochromocytoma and does not predict a more aggressive disease course. Larger and heavier tumors are associated with more aggressive disease, rather than smaller and lighter tumors.
Comment Here
Reference: Pheochromocytoma
Comment Here
Reference: Pheochromocytoma
Practice question #3
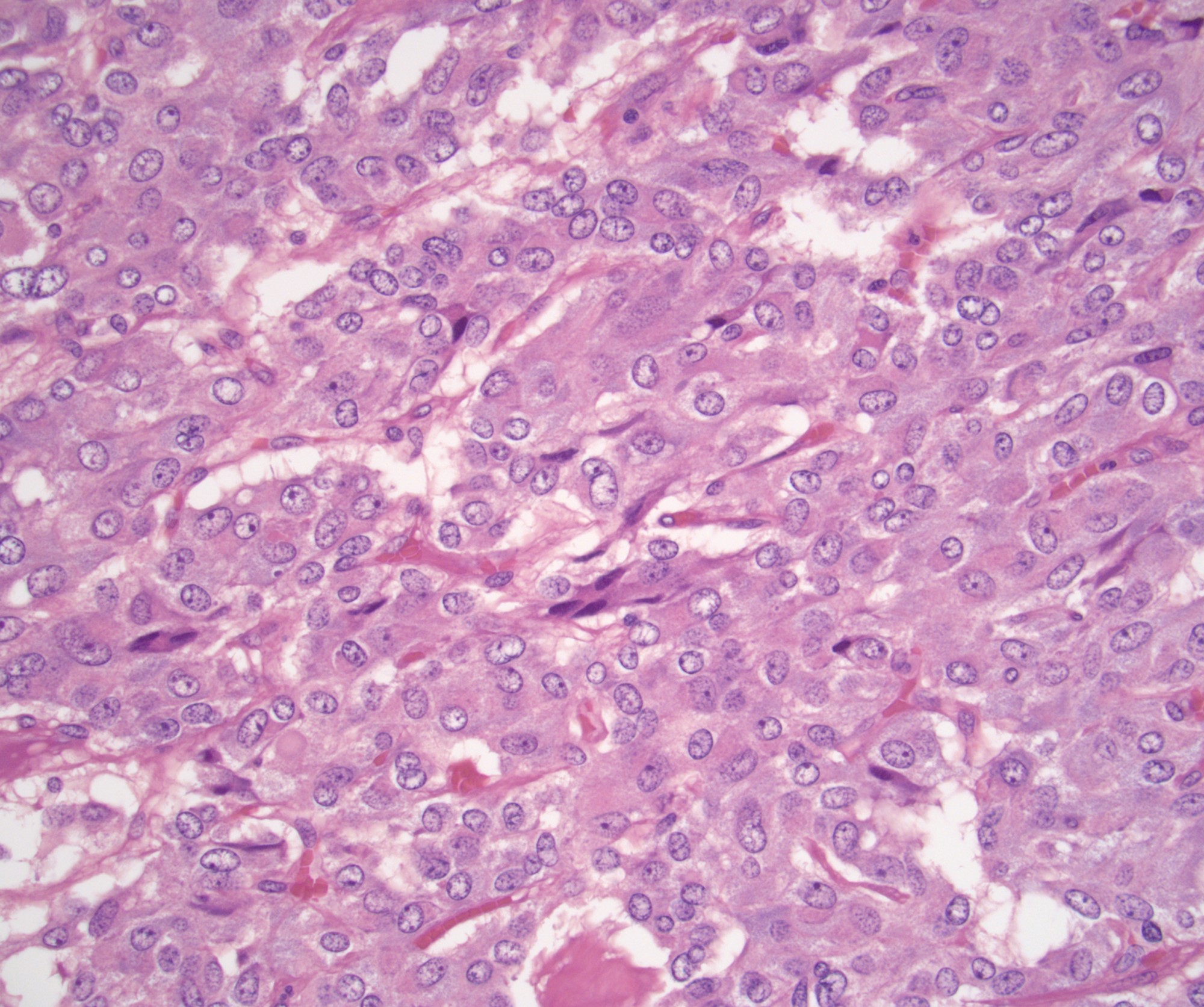
The adrenal tumor shown above was found to express chromogranin and GATA3 and was negative for AE1 / AE3. Which is the correct diagnosis?
- Adrenal cortical adenoma
- Adrenal cortical carcinoma
- Lung adenocarcinoma
- Pheochromocytoma
- Urothelial carcinoma
Practice answer #3
